

Abstract
A catalytic system for intramolecular C(sp2)–H and C(sp3)–H amination of substituted tetrazolopyridines has been successfully developed. The amination reactions are developed using an iron-porphyrin based catalytic system. It has been demonstrated that the same iron-porphyrin based catalytic system efficiently activates both the C(sp2)–H and C(sp3)–H bonds of the tetrazole as well as azide-featuring substrates with a high level of regioselectivity. The method exhibited an excellent functional group tolerance. The method affords three different classes of high-value N-heterocyclic scaffolds. A number of important late-stage C–H aminations have been performed to access important classes of molecules. Detailed studies (experimental and computational) showed that both the C(sp2)–H and C(sp3)–H amination reactions involve a metalloradical activation mechanism, which is different from the previously reported electro-cyclization mechanism. Collectively, this study reports the discovery of a new class of metalloradical activation modes using a base metal catalyst that should find wide application in the context of medicinal chemistry, drug discovery and industrial applications.
A catalytic system for intramolecular C(sp2)–H and C(sp3)–H amination of substituted tetrazolopyridines has been successfully developed.
Introduction
Over the past few decades, first-row transition metal complexes of porphyrins, especially cobalt and iron, have been utilized to catalyze a wide range of fundamentally and practically important chemical transformations. In particular, iron porphyrin complexes have recently received extensive attention from the scientific community. This is because (i) they mimic the catalytic activity of enzymes containing an iron-heme unit,1–3 (ii) iron is a cheap, eco-friendly and very active metal4 and (iii) the low-toxicity porphyrin ligand can be structurally modified to fine tune the catalytic performance. Among several metalloporphyrin catalyzed C–H functionalization reactions, C–H bond amination of saturated C–H bonds both intramolecularly and intermolecularly with porphyrin complexes has witnessed substantial advancement toward the construction of high-value N-heterocyclic scaffolds. For example, the groups of Zhang, de Bruin and many others have published landmark reports on Co(ii) and Fe(ii)-based C(sp3)–H amination of unactivated C–H bonds by the generation of an α-metalloaminyl radical derived from different organic azides for intramolecular radical amination of different C–H bonds, leading to the formation of N-heterocycles of variable ring size with a high control of reactivity and selectivity (Chart 1A).5–16
Chart 1. Previous work and new challenge.

Although significant efforts have been made in C(sp3)–H amination via metalloporphyrin catalysis, in comparison, C(sp2)–H bond amination using a porphyrin-based catalyst is less explored.
In this context, Che (with FeIII porphyrin using aryl azide) and Singh et al. (with FeIII porphyrin using hydroxyl amine) have reported intramolecular aryl C(sp2)–H amination for the synthesis of nitrogen containing molecules.17,18 But using 1,2,3,4-tetrazoles (as azide surrogates)19 as a metal nitrene in the presence of a metalloporphyrin to activate the C(sp2)–H bond is underdeveloped. Therefore, the development of reaction conditions using earth abundant first row transition metals especially iron would be advantageous for the cost effective and efficient synthesis of N-heterocycles.
In the last couple of years, we have been exploring the unique metal carbene and nitrene reactivity of 1,2,3-triazole and 1,2,3,4-tetrazole using a transition metal catalyst towards the synthesis of various N-heterocyclic scaffolds.20–25 Inspired by the seminal work of Wentrup26–29 in nitrene–nitrene rearrangement of 1,2,3,4-tetrazole under flash vacuum pyrolysis, we were able to show a new method to access N-pyridyl metal nitrenes using an Cp*Ir(iii) cationic complex through an electrocyclization pathway (Chart 1B).20 Subsequently, we reported a method to activate the strong C(sp3)–H bond intramolecularly using the metalloradical concept to construct important azaindoline derivatives (Chart 1B).22 Motivated by this Fe-porphyrin based C(sp3)–H bond amination report, we became curious to see whether or not this catalytic system could activate the C(sp2)–H bonds as well. If successful, then it would be the first report of metalloporphyrin catalyzed intramolecular C(sp2)–H amination of 1,2,3,4-tetrazole. Moreover, this method will be superior to the previously developed Ir-catalyzed reaction, as it is a less expensive, non-toxic and biocompatible catalyst. Furthermore, it would be mechanistically very different from the Ir-catalyzed electrocyclization mechanism. Thus, intrigued by this anticipation, herein, we present the concept of an Fe-catalyzed radical activation of 1,2,3,4-tetrazole and its application in a catalytic intramolecular denitrogenative annulation reaction.30 We have demonstrated that the annulation underwent via the C(sp2)–H amination while the aryl group attached with the tetrazole having no functionalizable C(sp3)–H bonds, however, the same annulation reaction proceeded via the C(sp3)–H amination while the tetrazole bearing an aryl group having a functionalizable C(sp2)–H bond (Chart 1C). Collectively, a single catalytic system has exhibited its dual role for the C(sp2)–H and C(sp3)–H amination reactions with high selectivity leading to the access of various high-value N-heterocyclic scaffolds in high yields. The developed catalytic system has also been employed to access various biomedically important N-heterocycles from the corresponding tetrazole-featured starting materials. Detailed mechanistic investigations as well as DFT calculations have revealed that the mechanism follows a radical activation mechanism that is completely different from our previously developed mechanism.
Results and discussion
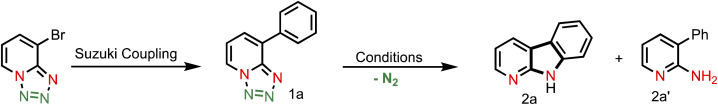
In order to develop the reaction conditions for intramolecular C(sp2)–H amination, we choose substrate (1a) (synthesized by Suzuki coupling) as a model substrate.
As our goal was to develop a metalloporphyrin based catalytic system, at first, we examined several synthesized metalloporphyrin based catalysts like Mn(TPP)Cl, Co(TPP), and Co(F20TPP), but it resulted in no reaction (Table 1, entries 1–2). The employment of other additives (e.g., AgSbF6 and AgBF4) and reductants (e.g., Zn) also failed to activate the C(sp2)–H bond (entries 3 and 4). At an elevated temperature (entry 5), 10% free amine was observed. Next, we turned our attention towards the Fe-porphyrin based catalyst. First we performed a reaction using Fe(TPP)Cl without any additives, which resulted in no reaction (entry 6). Subsequently, the same reaction was conducted in the presence of various additives (entry 7 and others, see the ESI† for details), but it showed no reactivity, and only the starting material remained unreacted. After that, we moved to our previously developed Fe(ii)-based metalloradical activation strategy for C(sp3sp3)–H bond activation. Accordingly, when the reaction was performed using 5.0 mol% Fe(TPP)Cl in conjugation with Zn dust as the reductant in PhH at 120 °C (entry 8), an aminated product (2a, 10% NMR conversion) along with a free amine (2a′, 5% NMR conversion) was observed. Increasing the reaction temperature and changing the solvent resulted in moderate product conversion along with the byproduct amine (entries 9 and 10). We reasoned that the low product conversion could be due to the incomplete formation of the Fe(ii) active catalyst. Thus, we performed an amination reaction with an increased Zn-dust loading (entries 11–12, see the ESI† for details) and we observed more product conversion. Finally, using 60 mol% Zn-dust and 5.0 mol% Fe(TPP)Cl in PhH solvent at 120 °C, we achieved 95% product conversion (2a) along with 5% free amine (2a′). The employment of other types of reductants (e.g., NaBH4, Mn, and LiAlH4) was found to be ineffective for amination (entry 14). As expected without Fe(TPP)Cl no reaction occurred (entries 15 and 16).
Reaction optimization for the intramolecular C(sp2)–H bond aminationa.
| |||||||
|---|---|---|---|---|---|---|---|
| # | Catalyst (mol%) | Reductant (mol%) | Additive (mol%) | Solvent | T (°C) | Time (h) | 2a/2a′ |
| 1 | Mn(TPP)CI (5) | — | — | PhH/PhMe | 120 | 24 | 0/0 |
| 2 | Co(TPP)/Co(F20TPP) (5) | — | — | PhH/PhMe | 120 | 24 | 0/0 |
| 3 | Mn(TPP)CI (5) | — | AgSbF6/AgBF4(20) | PhH/PhMe | 120 | 24 | 0/0 |
| 4 | Mn(TPP)CI (5) | Zn (10) | — | PhH/PhMe | 120 | 24 | 0/0 |
| 5 | Mn(TPP)CI (5) | Zn (10) | — | PhH/PhMe | 150 | 24 | 0/10 |
| 6 | Fe(TPP)CI (5) | — | — | PhH/PhMe | 120 | 24 | 0/0 |
| 7 | Fe(TPP)CI (5) | — | AgSbF6/AgBF4(20) | PhH/PhMe | 120 | 24 | 0/0 |
| 8 | Fe(TPP)CI (5) | Zn (10) | — | PhH | 120 | 24 | 10/5 |
| 9 | Fe(TPP)CI (5) | Zn (10) | — | PhH | 150 | 24 | 15/10 (3%)b |
| 10 | Fe(TPP)CI (5) | Zn (10) | — | PhMe | 120 | 24 | 20/50 (11%)b |
| 11 | Fe(TPP)CI (5) | Zn (30) | — | PhH | 120 | 24 | 20/5 (12%)b |
| 12 | Fe(TPP)CI (5) | Zn (40) | — | PhH | 120 | 24 | 40/5 (33%)b |
| 13 | Fe(TPP)CI (5) | Zn (60) | — | PhH | 120 | 24 | 95/5 (92%)b |
| 14 | Fe(TPP)CI (5) | NaBH4/Mn/LiAIH4 | — | PhH | 120 | 24 | 0/0 |
| 15 | — | Zn (10) | — | PhH | 120 | 24 | 0/0 |
| 16 | — | Zn (60) | — | PhH | 120 | 24 | 0/7 |
Reactions were conducted on a 0.25 mmol scale and NMR conversions were determined using 1,3,5-trimethoxybenzene as the internal standard.
In the parentheses, the isolated yield is given. For details see the ESI.
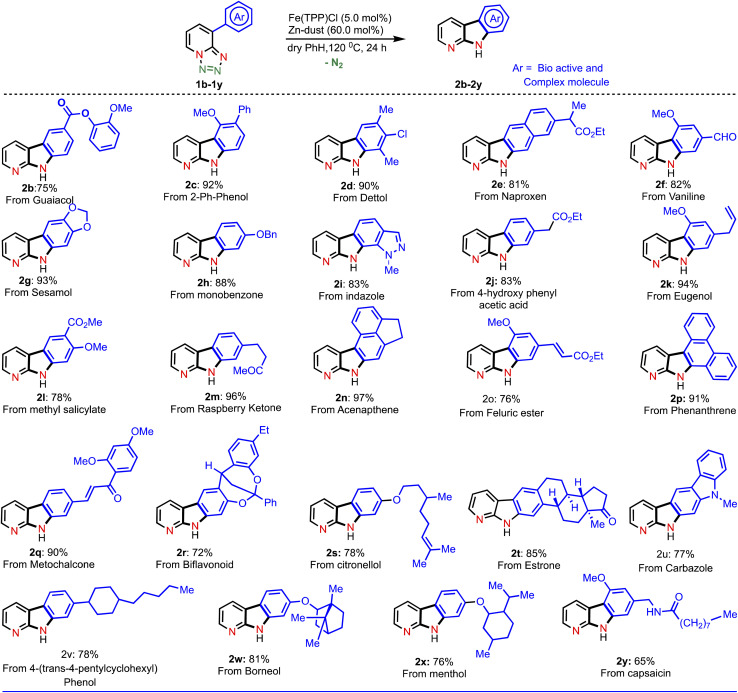
With this established reaction conditions in hand, we decided to showcase the synthetic utility of this amination reaction. Accordingly, we connected many pharmaceutically and medicinally important molecules that possess aromatic bromide or a phenol moiety, which were first transformed into the corresponding triflate derivatives to couple with tetrazoles and then performed a denitrogenative amination reaction (Table 2). Various medicinally important and complex molecules such as guaiacol (2b, phenolic natural product), 2-phenyl phenol (2c, agricultural fungicide), dettol (2d, antiseptic agent), naproxen (2e, NSAID, nonsteroidal anti-inflammatory drug), vanillic ester (2f, used as a flavoring agent), sesamol (2g, antioxidant), monobenzone (2h, used for skin disease), indazole (2i, an important alkaloid), 4-hydroxy phenyl acetic ester (2j, used for the treatment of chest pain), eugenol (2k, as a flavoring agent), methyl salicylate (2l, wintergreen oil), raspberry ketone (2m, found in raspberries), acenaphthene (2n, used as a precursor of dye and optical brighteners), ferulic ester (2o, used as an antioxidant), phenanthrene (2p, used to make dyes), metochalcone (2q, use as a choleretic and diuretic agent), bioflavonoid analogue (2r, used as an anti-inflammatory agent), citronellol (2s, a flavoring agent), estrone (2t, important metabolite), carbazole (2u, for the treatment of human psoriasis), 4-(trans-4-pentylcyclohexyl) phenol (2v, intermediate of liquid crystals), borneol (2w, insect repellent) and menthol (2x, used as a pain reliever), and capsaicin (2y, used for postherpetic neuralgia) were shown to be highly compatible with our developed reaction conditions. Notably, tetrazole (1i) reacted differently from other tetrazoles, where we observed nitrene radical attack on the more hindered site (for better understanding, see ESI†). We believe that these late-stage aminated products could be useful as a potential building block unit for further structural diversity of drug molecules in the pharmaceutical industry as it resulted in bioactive α-carboline derivatives.
Substrate scopea.
|
Reactions were conducted on a 0.5 mmol scale; isolated yields were reported after purification.
Next, we explored the substrate scope with respect to various substituted tetrazoles (Table 3A).
Substrate scopea.
|
Reactions were conducted on a 0.25 mmol scale; isolated yields were reported after purification.
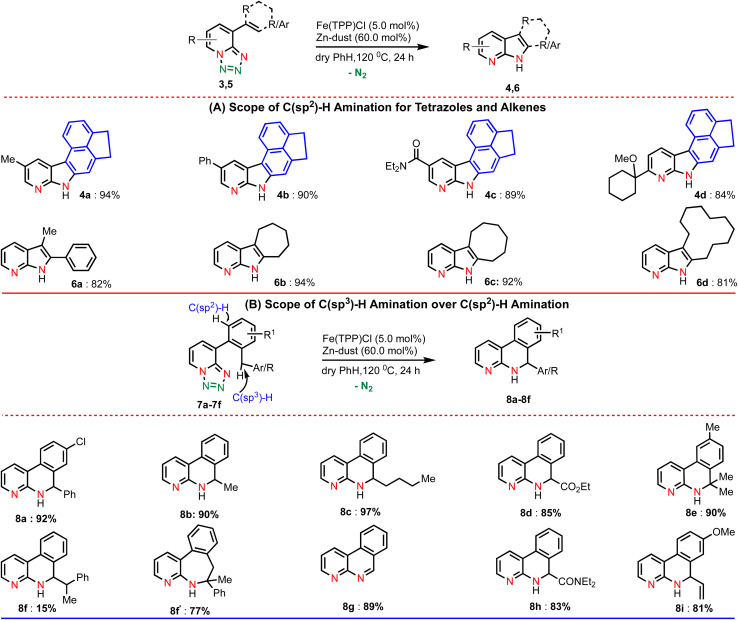
For example, different substituents at the pyridine ring (such as methyl, phenyl, amide and cyclohexyl) underwent the amination reaction smoothly and produced the corresponding aminated products in excellent yields (4a–4d). We were also interested to extend this amination method towards C8-substituted alkene derivatives (Table 3A). Gratifyingly, our developed amination method was proved to be equally efficient with numerous alkenes and the results are illustrated in Table 3A. Performing the reaction with the di-substituted alkene resulted in the desired di-substituted azaindole (6a) in good yield.
Pleasingly, the amination of cyclic alkenes (ring size 7–12) bearing 1,2,3,4-tetrazoles was also found to be compatible delivering the corresponding azaindoles in excellent yields (6b–d). To demonstrate further the utility of this amination method, we designed a substrate in such a way that a competitive C(sp3)–H bond is present in the ortho position of the arene ring along with a C(sp2)–H bond (Table 3B). We anticipated that if C(sp3)–H amination is favoured over the C(sp2)–H amination, then it would lead to important N-heterocycle dihydropyridine. Accordingly, we tested a reaction using the same reaction conditions. To our delight, we obtained only the C(sp3)–H aminated product (8a) in high yield instead of the competitive C(sp2)–H aminated product. Substrates featuring aliphatic secondary and tertiary C–H bonds afforded only C(sp3)–H aminated products (8b–8e) in excellent yields. Interestingly, on substrates with two distinct reactive C–H bonds (secondary and tertiary), the amination reaction preferred the tertiary C–H bond and resulted in a seven-membered heterocycle (8f′) as the major product along with a six-membered product (8f) as the minor one. The substrate (7g) with a coordinating methoxy group smoothly underwent the amination reaction affording the aminated product (8g) in good yield. Notably, the methoxy group was eliminated after annulation. Moreover, the C–H bond close to the amide group (7h) and allylic C–H bond (7i) smoothly participated in the reaction.
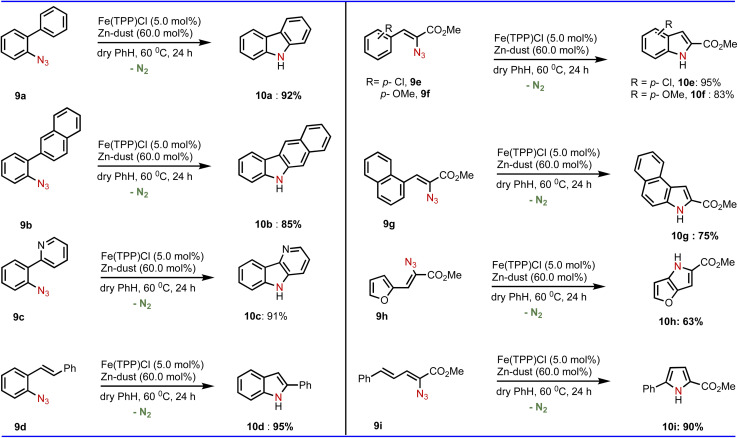
To this end, we investigated the application of this Fe-catalyzed C(sp2)–H amination reaction towards various ortho-substituted phenyl azide systems (Table 4). To our delight, our developed method was observed to be equally applicable that produced the corresponding aminated products such as carbazole (10a and 10b), δ-carboline (10c) and indole (10d). In addition to phenyl azide, various vinyl azides (Table 4) can be aminated smoothly employing our developed conditions of the amination reaction.
Scope of organic azidesa.
|
Reactions were conducted on a 0.25 mmol scale; isolated yields were reported after purification. Active iron-catalyst preparation was done at 120 °C and the amination was performed at 60 °C. For details, see the ESI.
Next, we studied the reaction mechanism of this amination reaction. The product formation might take place through any of the five mechanisms as depicted in Chart 2(A), such as (i) electrocyclization viaTS-I, (ii) C–H activation viaTS-II, (iii) C–H insertion viaTS-III, (iv) electrophilic aromatic substitution (EAS) viaTS-IV and (v) radical activation pathway viaTS-V. To gain insights into the mechanism of this Fe(ii)-catalyzed C–H amination, a set of mechanistic experiments were conducted (Chart 2(B)). At first, we decided to analyze potential kinetic isotope effects by using di-deuterated (intermolecular) and mono-deuterated (intramolecular) tetrazoles (Chart 2(B), eqs. 1 & 2). We observed a secondary kinetic isotope effect (KIE = 1.0) intermolecularly and intramolecularly. These KIE values suggested that the electrocyclization pathway (generally KIE = 1) might be involved in our amination reaction.
Chart 2. Mechanistic investigations: control experiments.

Importantly, since for the electrocyclization process, conjugation is necessary, we designed a substrate (15), where conjugation is disturbed by one N–H group. We hypothesized that if our amination reaction follows an electrocyclization pathway, then the substrate (15) should not produce an aminated product as continuous conjugation is essential for the electrocyclization process. Accordingly, we performed a reaction using this substrate (15) under our developed conditions. Surprisingly, we observed a dehydrogenative aromatized aminated product (16, eq. 3) in 50% isolated yield, which clearly discarded the possibility of the electrocyclization pathway. The driving force behind dehydrogenation is the gaining of aromaticity (see the ESI† for the formation of product 16). To verify the involvement of the C–H activation and C–H insertion pathway, we performed another experiment with the substrate (17) (Z-isomer), where the C–H bond is away from the reaction center. We anticipated that due to the geometric constrain, annulation should not occur with the Z-isomer (17). But, to our surprise, the substrate (17) smoothly underwent the amination reaction affording quantitative product conversion (eq. 4), which ruled out the possibility of the C–H activation and C–H insertion mechanism (TS-II & TS-III). Next, we conducted a competitive experiment under standard reaction conditions. The substrate with an electron donating group (19b) and electron withdrawing group (19a) produced the same product conversion under our reaction conditions (Chart 2(B), eq. 5). This indicated that there is no beneficial electronic effect, further suggesting that the EAS mechanism is unlikely. Finally, performing the reaction in the presence of a radical scavenger (TEMPO), we observed no product conversion (Chart 2(B), eq. 6), which strongly points to the involvement of a radical pathway in our amination reaction.
In light of the observations made in the above-mentioned set of experiments, we have examined two of the most likely mechanisms in greater detail, namely, the electrophilic aromatic substitution (EAS) and metalloradical activation (MRA) to identify the most preferred catalytic pathway leading to the desired product.
Toward this key objective, density functional theory computations at the SMD(benzene)/B3LYP-D3/6-31G**, SDD(Fe) level of theory were undertaken.31 First, computations on the EAS mechanism were carried out in the closed-shell singlet (CSS) spin state. In the case of the metalloradical activation (MRA) mechanism via the intermediacy of a nitrene radical, the likely involvement of different spin states at the SMD(benzene)/UB3LYP-D3/6-31G**, SDD(Fe) level of theory was examined.32 Depending on the arrangement of the unpaired electrons between the Fe and the nitrene radical, three different scenarios such as an open-shell singlet (OSS), triplet, and quintet spin states were considered.33–36 Although, all these possibilities are examined in detail, for the sake of better clarity, we present only the most favorable mechanistic pathway in the following sections while the remaining details can be readily found in the ESI.†37 In Fe-porphyrin catalyzed reactions, the participation of species with different spin states is known, which typically helps in avoiding higher activation barriers.38–48
In the present example, the triplet state was found to be the ground state of the active catalyst 21 (FeII(TPP)). Hence, the catalytic cycle proceeding through the MRA pathway in the triplet state is presented in Scheme 1, whereas the details of the CSS and OSS are placed in the ESI.†49 In line with the cue collected from our experiments that the participation of radicals was highly likely in this reaction, our computations revealed that the triplet pathway was energetically lower than the open-shell singlet or the quintet alternatives (vide infra). The reaction can be considered as starting with the reduction of the pre-catalyst 21A [FeIII(TPP)Cl] by the action of Zn dust to form the active catalyst 21 [FeII(TPP)]. The ground state spin multiplicity of 21A (FeIII) and 21 (FeII) was respectively sextet and triplet. The Gibbs free energies of alternative spin states for the active catalyst, i.e., 521 and OSS21, were respectively found to be higher by 7.2 and 11.9 kcal mol−1 as compared to 321.50
Scheme 1. The key mechanistic steps in the Fe-porphyrin catalyzed intramolecular C(sp2)–H amination. The spin multiplicities are shown in italic superscript.

Given that the key substrate 1a, a fused 1,2,3,4-tetrazole, can exist in equilibrium with its open form 1a′,51 it can develop a weak coordination with the active catalyst 21 to form a catalyst–substrate complex denoted as (22).52 The elimination of N2 from intermediate 22 through the transition state (22-23)‡ can then generate the FeIII-nitrene intermediate (23). In the ensuing step, a radical addition to the adjacent aryl C C bond forms the desired C–N bond via the transition state (23-24)‡, with a concomitant reduction of FeIII to FeII to generate another intermediate 24. A weak coordination of the hydropyridoindole to the Fe center of the catalyst is likely in intermediate 24. In the next step, the product precursor 25 will depart from the active catalyst prior to a 1,5-H shift via the transition state (25-2a)‡ to form the final product 2a.
The catalytic steps were examined in more detail to identify the energetically most preferred pathway. Key insights into various intermediates and transition states are as follows. In the formation of the catalyst–substrate complex 322, we have considered the open form of the fused 1,2,3,4-tetrazole substrate denoted as 1a′, although the Gibbs free energy of 1a is 4.5 kcal mol−1 lower than that of 1a′. A weak coordination of 1a′ to the active catalyst 321 was evident from the Fe–N distance of 2.53 Å noted in 322, which is longer than a standard Fe–N single bond distance (∼1.86 Å). The spin density of 2.1 on Fe in the catalyst–substrate complex 322 is commensurate with a triplet spin state (Fig. 1). In the next step of N2 elimination, the spin densities in 322 depleted from 2.1 to 1.2 on Fe and increased from 0.0 to 0.8 on N, as the formation of FeIII-nitrene radical intermediate 323 is complete. This suggested that one unpaired electron from the Fe got coupled ferromagnetically with the unpaired electron on the nitrene moiety in the transition state 3(22-23)‡, leading to 323. The optimized geometries of the important transition states are shown in Fig. 2.53 As we proceed from intermediate 323, where the spin densities were localized on Fe and N, toward the C–N bond formation transition state 3(23-24)‡, the spin density got more delocalized over the adjacent conjugated aryl framework (Fig. 1). These changes of the spin density distribution became more complete in intermediate 324, wherein the Fe center was reduced to FeII. The spin density of 2.1 on Fe is more characteristic of 324. On the other hand, the spin density on the ring N dwindled to 0.0 in intermediate 324.
Fig. 1. Spin density iso-surface plots for important intermediates and transition states (the iso-surface value is 0.05). Mulliken spin density distribution of Fe and N is shown (the carbon atom involved in the new C–N bond formation in 324 with a value of 0.21 is not shown for improved clarity).

Fig. 2. The optimized geometries of important transition states in the triplet spin state and the corresponding Gibbs free energies (in kcal mol−1) with respect to the separated reactants. Distances are shown in Å.

The combined Gibbs free energy profile for the MRA mechanism involving different spin states is provided in Fig. 3. The relative Gibbs free energies were calculated with respect to the ground state of the active catalyst 321 [FeII(TPP)] and fused 1,2,3,4-tetrazole 1a. The profile can broadly be viewed as consisting of N2 elimination, C–N bond formation, and 1,5-H shift. In the N2 elimination step leading to the generation of the nitrene radical, the triplet 3(22-23)‡ was found to be of the lowest energy. The Gibbs free energy of this TS was 5.0 kcal mol−1 lower than that of OSS(22-23)‡ and 8.3 kcal mol−1 lower than that of 5(22-23)‡. Since the quintet pathway was of much higher energy in the very first step of N2 elimination, this high spin state was not considered for the ensuing steps such as the C–N bond formation. The C–N bond formation transition state 3(23-24)‡ was 2.0 kcal mol−1 lower in energy than OSS(23-24)‡, indicating that the triplet pathway was more favored over the alternative OSS pathway. While the mechanistic steps are similar up to the generation of intermediate 24, the triplet and OSS cases follow different routes for the 1,5-H shift that followed. In the triplet pathway, the active catalyst was regenerated as we proceed from intermediate 24 to 25, whereas in the OSS pathway, the active catalyst regeneration took place after the 1,5-H shift. In the triplet pathway, the formation the final product through a 1,5-H shift via the transition state (25-2a)‡ occurred outside the catalytic cycle after the release of the active catalyst 321. The elementary step barrier for (25-2a)‡ leading to the final product 2a was 7.5 kcal mol−1. In the OSS pathway, the 1,5-H shift took place in the presence of the Fe(ii)-porphyrin system via the transition state OSS(25-21)‡ with an elementary triplet spin state of lower energy among all the other pathways. The application of the energetic span model54 in the lower energy route conveyed that active catalyst 321 was the turn-over determining intermediate (TDI) and the N2 elimination transition state 3(22-23)‡ was the turn-over determining transition state (TDTS) in the MRA pathway. The computed energetic span (δE) was 28.5 kcal mol−1 and the turnover frequency (TOF) of the catalytic cycle at 393 K was found to be 1.16 × 10−3 s−1.
Fig. 3. Gibbs free energy (kcal mol−1) profile obtained at the SMD(Benzene)/UB3LYP-D3/6-31G**, SDD(Fe) level of theory for the Fe-porphyrin catalyzed intramolecular C(sp2)–H amination of fused 1,2,3,4-tetrazole. The color codes used for distinguishing various spins are brown (close-shell singlet), red (open-shell singlet), dark blue (triplet), and black (quintet).

The alternative EAS pathway was found to proceed through a higher energy route with a δE as high as 43.9 kcal mol−1.55 Thus, on the basis of the δEs of the MRA and EAS pathways, it is evident that the reaction follows the MRA pathway for the C(sp2)–H amination of fused 1,2,3,4-tetrazole, in agreement with our experimental observations.
C(sp2)–H vs. C(sp3)–H amination
An interesting substrate 7a (Scheme 2), as borne out through an important set of experimental observations as presented in Table 3B, was examined next. Here, the ortho C(sp3)–H bond of the aryl ring or the benzylic C(sp2)–H bond could undergo a competitive C–H functionalization. The C(sp3)–H amination involves three important steps: N2 elimination, hydrogen atom abstraction, and C–N bond formation (Scheme 2).6,56,57
Scheme 2. The key mechanistic steps involved in the Fe-catalyzed intramolecular C(sp3)–H amination. 524a and 525a are conformers that differ in the orientation of the benzyl group formed as a rotation around the benzylic C–C bond.

The Gibbs free energy profiles involving different spin states for the competitive C(sp2)–H/C(sp3)–H functionalizations are provided in Fig. 6.58 On the basis of the overall energetic advantage that would become clear in the following paragraphs, here we focus on the quintet active catalyst and the quintet pathway. Examining the ortho C(sp3)–H bond activation (shown toward the right side of the profile) reveals that the N2 elimination transition state 5(22a-23a)‡ was of the lowest energy, with an activation barrier of 21.5 kcal mol−1 with respect to 521. The Gibbs free energy for OSS(22a-23a)‡ and 3(22a-23a)‡ is 33.7 and 29.3 kcal mol−1 respectively with an activation barrier of 21.8 and 29.3 kcal mol−1 calculated respectively with respect to OSS21 and 321. Since the triplet pathway requires a higher activation barrier for N2 elimination as compared to the quintet pathway, an intersystem crossing (ISC) from 321 to 521 is quite likely. The spin crossover to a high spin Fe(ii)-porphyrin complex (521) should occur before the N2 elimination. The alternative singlet pathway requires an ISC from the ground state triplet to access a 11.9 kcal mol−1 higher state. In the next step, the nitrene radical abstracts the hydrogen atom from the adjacent C(sp3)–H bond through the transition state (23a-24a)‡ and then generates another intermediate 24a. The H-atom abstraction transition state 5(23a-24a)‡ was 6.3 and 11.3 kcal mol−1 lower than 3(23a-24a)‡ and OSS(23a-24a)‡ respectively. In the next step, an intramolecular addition of the benzylic radical via the C–N bond formation transition state (25a-8a)‡ results in product 8a. The spin density on Fe in 5(25a-8a)‡ was 3.6, indicating a high spin quintet state (Fig. 4). Importantly, the Gibbs free energy of the quintet spin state was lower compared to the open-shell singlet and triplet states for the N2 elimination as well as the H-atom abstraction steps. In the C–N bond formation step, the triplet spin state was of comparable energy with the quintet state. Thus, the overall features of the Gibbs energy profile suggest that the reaction is likely to follow the quintet pathway for the benzylic C(sp3)–H activation.
Fig. 6. Gibbs free energy (kcal mol−1) profile obtained at the SMD(Benzene)/UB3LYP-D3/6-31G**, SDD(Fe) level of theory for Fe-catalyzed intramolecular C–H amination of 1,2,3,4-tetrazole. The color codes used for distinguishing various spins are brown (closed-shell singlet), red (open-shell singlet), dark blue (triplet), and black (quintet).

Fig. 4. Spin density iso-surface plots generated with an iso-surface value of 0.05 for important intermediates and transition states. Mulliken spin density distribution only on Fe, N and benzylic carbon is given for improved clarity (wherever applicable).

The triplet and quintet pathways were considered for the C(sp2)–H and C(sp3)–H activations in our energetic span calculation. The application of the energetic span model conveyed that the active catalyst 21 is the turn-over determining intermediate (TDI) and the transition state (22a-23a)‡ or (22b-23b)‡ for the N2 elimination step, leading to the formation of the nitrene radical (23a or 23b), is the turn-over determining transition state (TDTS). The higher energy triplet and singlet pathways for the C(sp3)–H amination exhibited a higher δE of 29.3 and 26.4 kcal mol−1 respectively as compared to the δE of 21.5 kcal mol−1 for the quintet pathway, indicating that the quintet pathway was more favored over the triplet and singlet pathways. The δE was 25.6 and 21.5 kcal mol−1 respectively for the C(sp2)–H and C(sp3)–H activations of substrate 7a and the turn over frequency (TOF) of the catalytic cycle at 393 K was 4.8 × 10−2 s−1 and 9.06 s−1 respectively.59 The computed energetic span therefore indicated that the C(sp3)–H activation was more likely over the C(sp2)–H activation and effective catalytic transformation under our experimental conditions as employed. The optimized geometries of important transition states are shown in Fig. 5.
Fig. 5. The optimized geometries of important transition states in the quintet spin state and the corresponding Gibbs free energies (in kcal mol−1) with respect to the separated reactants obtained at the SMD(Benzene)/UB3LYP-D3/6-31G**, SDD(Fe) level of theory. Distances are shown in Å.

Conclusion
In conclusion, we have developed an operationally simple base metal-catalyzed method for intramolecular C(sp2)–H and C(sp3)–H amination reactions that can be employed to access various high-value N-heterocyclic molecules. The developed catalytic method exhibited high reactivity and regioselectivity for a wide range of complex substrates. The method has also been applied for the late-stage C–H amination of numerous biomedically significant molecules. Importantly, depending on the nature of the substrates featuring either C(sp2)–H or C(sp3)–H bonds, the amination underwent with a high level of selectivity with excellent yields of various product classes. Apart from tetrazole-bearing substrates, the reaction also took place smoothly for those substrates bearing simple organic azides.
Detailed mechanistic investigations using a combination of experimental and computational tools suggested that both the C(sp2)–H and C(sp3)–H amination reactions followed a metalloradical activation mechanism. The most favorable pathway for the C(sp2)–H amination was found to proceed through a triplet spin state, whereas the quintet spin state was preferred for the competitive C(sp3)–H amination. The triplet to quintet intersystem crossing (ISC) was identified as more likely before the formation of the nitrene radical intermediate in the case of benzylic C(sp3)–H amination. The application of the energetic span model on the Gibbs free energy profile indicated that the formation of the nitrene radical was the turn over determining step of the catalytic cycle and that the C(sp3)–H amination was more favored by 4.1 kcal mol−1 than the C(sp2)–H amination. Collectively, this study highlights the discovery of a new class of metalloradical activation methods using base metal catalysis that should find broad application in the context of medicinal chemistry, drug discovery and industrial deployment.
Data availability
Experimental data including experimental procedures, characterization data, and NMR spectra for new compounds; mechanistic experiments; detailed computational studies, calculated structures, and energies are accessible in the ESI.†
Author contributions
S.K.D. and S.D. discovered and developed the reactions. S.K.D., S.D. and S.R. performed the experiments. S.G. and M.P. performed the DFT calculation. B.C. and R.B.S. supervised the research. All authors revised and approved the manuscript and ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This work was supported by a CSIR-EMR grant (02/0396/21/EMR-II). SKD, SD, SG and SR thank CSIR for their SRF. We also thank CBMR for research facilities and SpaceTime super-computing at IIT Bombay.
Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03505g
References
- Khade R. L. Zhang Y. J. Am. Chem. Soc. 2015;137:7560–7563. doi: 10.1021/jacs.5b03437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessjohann L. A. Brandt W. Thiemann T. Chem. Rev. 2003;103:1625–1648. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- Chakraborty J. Nath I. Verpoort F. Chem. Rev. 2016;326:135–163. [Google Scholar]
- Enthaler S. Junge K. Beller M. Angew. Chem., Int. Ed. 2008;47:3317–3321. doi: 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]
- Lang K. Torker S. Wojtas L. Zhang X. P. J. Am. Chem. Soc. 2019;141:12388–12396. doi: 10.1021/jacs.9b05850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M. Lankelma M. van der Vlugt J. I. Bruin B. de. Angew. Chem., Int. Ed. 2020;59:11073–11079. doi: 10.1002/anie.202002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagh B. Broere D. L. J. Sinha V. Kuijpers P. F. van Leest N. P. de Bruin B. Demeshko S. Siegler M. A. van der Vlugt J. I. J. Am. Chem. Soc. 2017;139:5117–5124. doi: 10.1021/jacs.7b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lang K. Li C. Kim I. P Zhang X. J. Am. Chem. Soc. 2020;142:20902–20911. doi: 10.1021/jacs.0c11103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu Y. Lang K. Li C. Gill J. B. Kim I. Lu H. Fields K. B. Marshall M. Cheng Q. Cui X. Wojtas L. Zhang X. P. J. Am. Chem. Soc. 2019;141(45):18160–18169. doi: 10.1021/jacs.9b08894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li C. Lang K. Lu H. Hu Y. Cui X. Wojtas L. P Zhang X. Angew. Chem., Int. Ed. 2018;57:16837–16841. doi: 10.1002/anie.201808923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Lang K. Jiang H. Wojtas L. Zhang X. P. Chem. Sci. 2016;7:6934–6939. doi: 10.1039/C6SC02231F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Li C. Jiang H. Lizardi C. L. Zhang X. P. Angew. Chem., Int. Ed. 2014;53:7028–7032. doi: 10.1002/anie.201400557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Jiang H. Wojtas L. Zhang X. P. Angew. Chem., Int. Ed. 2010;49:10192–10196. doi: 10.1002/anie.201005552. [DOI] [PubMed] [Google Scholar]
- Shing K.-P. Liu Y. Cao B. Chang X.-Y. You T. Che C.-M. Angew. Chem., Int. Ed. 2018;57:11947–11951. doi: 10.1002/anie.201806059. [DOI] [PubMed] [Google Scholar]
- Hennessy E. T. Betley T. A. Science. 2013;340:591–595. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]
- Paradine S. M. White M. C. J. Am. Chem. Soc. 2012;134:2036–2039. doi: 10.1021/ja211600g. [DOI] [PubMed] [Google Scholar]
- Alt I. T. Guttroff C. Plietker B. Angew. Chem., Int. Ed. 2017;56:10582–10586. doi: 10.1002/anie.201704260. [DOI] [PubMed] [Google Scholar]
- Kweon J. Chang S. Angew. Chem., Int. Ed. 2021;60:2909–2914. doi: 10.1002/anie.202013499. [DOI] [PubMed] [Google Scholar]
- Liu Y. Wei J. Che C.-M. Chem. Commun. 2010;46:6926–6928. doi: 10.1039/C0CC01825B. [DOI] [PubMed] [Google Scholar]
- Prasanthi A. V. G. Begum S. Srivastava H. K. Tiwari S. K. Singh R. ACS Catal. 2018;8:8369–8375. doi: 10.1021/acscatal.8b02939. [DOI] [Google Scholar]
- Chattopadhyay B. Rivera Vera C. I. Chuprakov S. Gevorgyan V. Org. Lett. 2010;12:2166–2169. doi: 10.1021/ol100745d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S. K. Roy S. Khatua H. Chattopadhyay B. J. Am. Chem. Soc. 2018;140:8429–8433. doi: 10.1021/jacs.8b05343. [DOI] [PubMed] [Google Scholar]
- Roy S. Khatua H. Das S. K. Chattopadhyay B. Angew. Chem., Int. Ed. 2019;58:11439–11443. doi: 10.1002/anie.201904702. [DOI] [PubMed] [Google Scholar]
- Das S. K. Roy S. Khatua H. Chattopadhyay B. J. Am. Chem. Soc. 2020;142:16211–16217. doi: 10.1021/jacs.0c07810. [DOI] [PubMed] [Google Scholar]
- Khatua H. Das S. K. Roy S. Chattopadhyay B. Angew. Chem., Int. Ed. 2021;60:304–312. doi: 10.1002/anie.202009078. [DOI] [PubMed] [Google Scholar]
- Roy S. Das S. K. Khatua H. Das S. Singh K. N. Chattopadhyay B. Angew. Chem., Int. Ed. 2021;60:8772–8780. doi: 10.1002/anie.202014950. [DOI] [PubMed] [Google Scholar]
- (a) Roy S. Das S. K. Khatua H. Das S. Chattopadhyay B. Acc. Chem. Res. 2021;54:4395–4409. doi: 10.1021/acs.accounts.1c00563. [DOI] [PubMed] [Google Scholar]; (b) Roy S. Das S. K. Chattopadhyay B. Angew. Chem., Int. Ed. 2018;57:2238–2243. doi: 10.1002/anie.201711209. [DOI] [PubMed] [Google Scholar]
- Harder R. Wentrup C. J. Am. Chem. Soc. 1976;98:1259–1260. doi: 10.1021/ja00421a035. [DOI] [Google Scholar]
- Wentrup C. Winter H. W. J. Am. Chem. Soc. 1980;102:6159–6161. doi: 10.1021/ja00539a039. [DOI] [Google Scholar]
- Evans R. A. Wong M. W. Wentrup C. J. Am. Chem. Soc. 1996;118:4009–4017. doi: 10.1021/ja9541645. [DOI] [Google Scholar]
- Kvaskoff D. Vosswinkel M. Wentrup C. J. Am. Chem. Soc. 2011;133:5413–5424. doi: 10.1021/ja111155r. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay B. Gevorgyan V. Angew. Chem., Int. Ed. 2012;51:862–872. doi: 10.1002/anie.201104807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Computational details are provided in the Computational methods section in the ESI.†; The Gibbs free energies of important stationary points are provided in Table S6 in the ESI.†
- (a) Gérard E. F. Yadav V. Goldberg D. P. de Visser S. P. J. Am. Chem. Soc. 2022;144:10752–10767. doi: 10.1021/jacs.2c01375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yeh C.-C. G. Ghafoor S. Satpathy J. K. Mokkawes T. Sastri C. V. de Visser S. P. ACS Catal. 2022;12:3923–3937. doi: 10.1021/acscatal.1c04029. [DOI] [Google Scholar]; (c) Ali H. Z. de Visser S. P. Chem.–Eur. J. 2022;28:e2021041. doi: 10.1002/chem.202104167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Daru A. Hu X. Harvey J. N. ACS Omega. 2020;5:1586–1594. doi: 10.1021/acsomega.9b03578. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Louka S. Barry S. M. Heyes D. J. Mubarak M. Q. E. Ali H. S. Alkhalaf L. M. Munro A. W. Scrutton N. S. Challis G. L. de Visser S. P. J. Am. Chem. Soc. 2020;142:15764–15779. doi: 10.1021/jacs.0c05070. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Coin G. Patra R. Rana S. Biswas J. P. Dubourdeaux P. Clémancey M. de Visser S. P. Maiti D. Maldivi P. Latour J.-M. ACS Catal. 2020;10:10010–10020. doi: 10.1021/acscatal.0c01427. [DOI] [Google Scholar]
- Li X. Dong L. Liu Y. Inorg. Chem. 2020;59:1622–1632. doi: 10.1021/acs.inorgchem.9b02216. [DOI] [PubMed] [Google Scholar]
- Sharon D. A. Mallick D. Wang B. Shaik S. J. Am. Chem. Soc. 2016;138:9597–9610. doi: 10.1021/jacs.6b04636. [DOI] [PubMed] [Google Scholar]
- Conradie J. Ghosh A. Inorg. Chem. 2010;49:243–248. doi: 10.1021/ic901897w. [DOI] [PubMed] [Google Scholar]
- Rovira C. Ballone P. Parrinello M. Chem. Phys. Lett. 1997;271:247–250. doi: 10.1016/S0009-2614(97)00492-2. [DOI] [Google Scholar]
- The mechanistic details of the higher energy EAS pathway (Schemes S2 and Fig. S1†) and the corresponding optimized geometries of all TSs and intermediates (Fig. S3) are provided in the ESI.†
- Holland P. L. Acc. Chem. Res. 2015;48:1696–1702. doi: 10.1021/acs.accounts.5b00036. [DOI] [PubMed] [Google Scholar]
- Sun Y. Tang H. Chen K. Hu L. Yao J. Shaik S. Chen H. J. Am. Chem. Soc. 2016;138:3715–3730. doi: 10.1021/jacs.5b12150. [DOI] [PubMed] [Google Scholar]
- Shaik S. Hirao H. Kumar D. Acc. Chem. Res. 2007;40:532–542. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]
- Poli R. Harvey J. N. Chem. Soc. Rev. 2003;32:1–8. doi: 10.1039/B200675H. [DOI] [PubMed] [Google Scholar]
- Sharma A. K. Sameera W. M. C. Jin M. Adak L. Okuzono C. Iwamoto T. Kato M. Nakamura M. Morokuma K. J. Am. Chem. Soc. 2017;139:16117–16125. doi: 10.1021/jacs.7b05917. [DOI] [PubMed] [Google Scholar]
- Fledt M. Martin-Fernandez C. Harvey J. N. Phys. Chem. Chem. Phys. 2020;22:23908–23919. doi: 10.1039/D0CP04401F. [DOI] [PubMed] [Google Scholar]
- Ricciarelli D. Belpassi L. Harvey J. N. Belanzoni P. Chem.–Eur. J. 2020;26:3080–3089. doi: 10.1002/chem.201904314. [DOI] [PubMed] [Google Scholar]
- Lin Y.-T. Ali H. S. de Visser S. P. Chem.–Eur. J. 2022:e202103982. doi: 10.1002/chem.202103982. [DOI] [PubMed] [Google Scholar]
- Wolny J. A. Paulsen H. McGarvey J. J. Diller R. Schunemann V. Toftlund H. Phys. Chem. Chem. Phys. 2009;11:7562–7575. doi: 10.1039/B905328J. [DOI] [PubMed] [Google Scholar]
- Gutlich P. Garica Y. Goodwin H. A. Chem. Soc. Rev. 2000;29:419–427. doi: 10.1039/B003504L. [DOI] [Google Scholar]
- Mondal P. Ishigami I. Gerard E. F. Lim C. Yeh S.-R. de Visser S. P. Wijeratne G. B. Chem. Sci. 2021;12:8872–8883. doi: 10.1039/D1SC01952J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Scheme S2 and S3 in the ESI† for mechanistic details of CSS and OSS pathways respectively
- The relative Gibbs free energies of Fe(II)-TPP and Fe(III)-TPP complexes involving different spin states of Fe are shown in Table S1 in the ESI.†
- The barrier for 1a to 1a′ interconversion leading to tetrazole ring opening is 21.7 kcal mol−1. The corresponding barrier is 22.5 kcal mol−1, when the Fe of the catalyst in 321 is coordinated with the azide-N atom of 1a.
- The coordination of Fe in 321 through a pyridine N-atom was less favored by about 2 kcal mol−1 than that through the azide-N atom.
- The optimized geometries of TSs and intermediates for the MRA pathway are provided in Fig. S2 in the ESI.†
- Kozuch S. Shaik S. Acc. Chem. Res. 2011;44:101–110. doi: 10.1021/ar1000956. [DOI] [PubMed] [Google Scholar]
- To obtain maximum δE, different likely combinations of the stationary points involved in the catalytic cycle are considered. The energetic span calculations are shown in Tables S2 and S5 in the ESI† for MRA and EAS pathways respectively.
- Kuijpers P. F. Tiekink M. J. Breukelaar W. B. Broere D. L. J. van Leest N. P. van der Vlugt J. I. Reek J. N. H. de Bruin B. Chem. – Eur. J. 2017;23:7945–7952. doi: 10.1002/chem.201700358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Grotenhuis C. Das B. G. Kuijpers P. F. Hageman W. Trouwborst M. de Bruin B. Chem. Sci. 2017;8:8221–8230. doi: 10.1039/C7SC03909C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The optimized geometries of TSs and intermediates for C(sp3)–H amination involving the MRA pathway are provided in Fig. S4 in the ESI.†
- See Tables S3 and S4 in the ESI† for the comparison of energetic span between competitive C(sp2)–H and C(sp3)–H amination pathways respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental data including experimental procedures, characterization data, and NMR spectra for new compounds; mechanistic experiments; detailed computational studies, calculated structures, and energies are accessible in the ESI.†