

Abstract
Mutations in PTEN-induced kinase 1 (PINK1) contribute to autosomal recessive Parkinson's disease with cognitive and neuropsychiatric comorbidities. Disturbances in dendritic and spine architecture are hallmarks of neurodegenerative and neuropsychiatric conditions, but little is known of the impact of PINK1 on these structures. We used Pink1−/− mice to study the role of endogenous PINK1 in regulating dendritic architecture, spine density, and spine maturation. Pink1−/− cortical neurons of unknown sex showed decreased dendritic arborization, affecting both apical and basal arbors. Dendritic simplification in Pink1−/− neurons was primarily driven by diminished branching with smaller effects on branch lengths. Pink1−/− neurons showed reduced spine density with a shift in morphology to favor filopodia at the expense of mushroom spines. Electrophysiology revealed significant reductions in miniature EPSC (mEPSC) frequency in Pink1−/− neurons, consistent with the observation of decreased spine numbers. Transfecting with human PINK1 rescued changes in dendritic architecture, in thin, stubby, and mushroom spine densities, and in mEPSC frequency. Diminished spine density was also observed in Golgi–Cox stained adult male Pink1−/− brains. Western blot study of Pink1−/− brains of either sex revealed reduced phosphorylation of NSFL1 cofactor p47, an indirect target of PINK1. Transfection of Pink1−/− neurons with a phosphomimetic p47 plasmid rescued dendritic branching and thin/stubby spine density with a partial rescue of mushroom spines, implicating a role for PINK1-regulated p47 phosphorylation in dendrite and spine development. These findings suggest that PINK1-dependent synaptodendritic alterations may contribute to the risk of cognitive and/or neuropsychiatric pathologies observed in PINK1-mutated families.
SIGNIFICANCE STATEMENT Loss of PINK1 function has been implicated in both familial and sporadic neurodegenerative diseases. Yet surprisingly little is known of the impact of PINK1 loss on the fine structure of neurons. Neurons receive excitatory synaptic signals along a complex network of projections that form the dendritic tree, largely at tiny protrusions called dendritic spines. We studied cortical neurons and brain tissues from mice lacking PINK1. We discovered that PINK1 deficiency causes striking simplification of dendritic architecture associated with reduced synaptic input and decreased spine density and maturation. These changes are reversed by reintroducing human PINK1 or one of its downstream mediators into PINK1-deficient mouse neurons, indicating a conserved function, whose loss may contribute to neurodegenerative processes.
Keywords: cortical neuron, dementia, dendritic architecture, dendritic spines, NSFL1C, PTEN-induced kinase 1
Introduction
PTEN-induced kinase 1 (PINK1) has been implicated in the pathogenesis of familial and sporadic neurodegeneration. Loss-of-function mutations in the PINK1 gene represent the second most common cause of recessive Parkinson's disease (PD; Kumazawa et al., 2008). Interestingly, PINK1-linked PD patients often present with cognitive and affective symptoms in addition to motor symptoms (Li et al., 2005; Ephraty et al., 2007); PINK1 mutation shows the highest rates of cognitive impairment among the monogenic forms of PD (Piredda et al., 2020). Significant deficits in cognitive function are observed not only in homozygote patients but also in 62% of heterozygote carriers (Ricciardi et al., 2014). Radiologically, limbic and frontal cortical degeneration is present in 70% of heterozygote PINK1 mutation carriers (Reetz et al., 2008). Moreover, expression of wild-type (WT) PINK1 is reduced by chronic complex I inhibition in a model of environmental PD (Verma et al., 2020). Reduced PINK1 expression is also observed in the cortex of sporadic Alzheimer's disease (AD) patients (George et al., 2010; Du et al., 2017) and in mouse models of AD (George et al., 2010; Du et al., 2017; Manczak et al., 2018), whereas restoring PINK1 expression ameliorates AD pathology and cognitive dysfunction in vivo (Du et al., 2017; Jiang et al., 2021). Together, these studies suggest that reduced PINK1 function contributes to cognitive impairment, yet the underlying substrate for this deficit is unclear.
Loss of PINK1 function has been associated with deficits in long-term potentiation and long-term depression in striatal slice cultures (Kitada et al., 2007), although there is little to no overt neurodegeneration in Pink1 knock-out (KO) mice (Kitada et al., 2009; Zhi et al., 2019). Striatal slice cultures from young Pink1 KO rats show enhanced presynaptic glutamate release that disappears with aging (Creed et al., 2021). In Drosophila, loss of PINK1 function leads to reduced ability to mobilize the reserve pool of synaptic vesicles (Morais et al., 2009). PINK1 has been heavily studied for its central role in one of several pathways that leads to damage-induced mitophagy (Chu, 2018; Killackey et al., 2020), although its role in neuronal mitophagy remains controversial (Cummins and Gotz, 2017). PINK1 also acts to regulate mitochondrial transport (Liu et al., 2012; Das Banerjee et al., 2017), phosphorylation of mitochondrial complex I (Morais et al., 2014), and signaling through the protein kinase B (Akt) and protein kinase A (PKA) pathways (Murata et al., 2011; Wang et al., 2018). We have recently shown that increased expression of PINK1 promotes dendritic complexity, with striking protection against dendritic retraction elicited by the Parkinsonian toxins MPP+ and 6-hydroxydopamine (Wang et al., 2018; Liu et al., 2020).
The goal of the present study is to test the hypothesis that loss of endogenous PINK1 disrupts cortical dendritic architecture, spine density, and morphology. Using primary neurons from Pink1 KO mice, we observed that PINK1 is necessary for full dendritic arborization. Moreover, loss of mouse PINK1 reduces spine density in vitro and in vivo, shifting the dendritic spine population to a more immature state. These changes can be reversed by reintroduction of human PINK1 indicating a conserved function. The p47 protein, also known as NSFL1C, is a mediator of spinogenesis that is phosphorylated by PKA in a PINK1-dependent manner (Shih and Hsueh, 2016; Wang et al., 2018). We found that Pink1 KO cortices exhibit reduced p47 phosphorylation and that transfection of Pink1 KO neurons with a p47 phosphomimetic restored dendritic arborization and significantly ameliorated changes in spine density. Given that synaptic loss is a major structural correlate of cognitive impairment in PD, AD, and related disorders, understanding how the loss of PINK1 dysregulates neuronal architecture may lead to new insights into early stages of disease pathogenesis.
Materials and Methods
PINK1 knock-out mice
Animal support facilities are provided by the University of Pittsburgh Laboratory Animal Resources, which is fully accredited by the American Association for the Accreditation of Laboratory Animal Care. The Pink1−/− (KO) mouse line (C57BL/6 × 129/SvEv) has been previously characterized (Dagda et al., 2011; Zhi et al., 2019). KNS-25 (5′-ATG AGA TGG AGG GGA GTC-3′), KNS-27 (5′-GGA AGG AGG CCA TGG AAA TTG-3′), and Neo3a (5′-GCA GCG CAT CGC CTT CTA TC-3′) primers are used to distinguish wild-type (WT) and KO littermates. To facilitate the generation of primary neuron cultures, the Pink1 KO line was backcrossed into a C57BL/6 background and maintained as a homozygous line. WT and KO mice were housed under standard controlled temperature, humidity, and light/dark cycles.
All procedures for tissue/cell harvest were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. In brief, primary E14–16 cortical neurons were isolated from cerebral cortices as described previously (Cherra et al., 2013; Dagda et al., 2014) and plated at 150,000 cells/cm2 in 12-mm-diameter coverslips (Ted Pella) coated with poly-L-ornithine (0.1 mg/ml). They were maintained in antibiotic-free Neurobasal medium supplemented with 2% B27, 1% N2, and 1 mm Glutamax (Invitrogen). The media was refreshed every third day. Neurons from embryos of unknown sex were pooled before plating for primary neuron cultures. Because of known effects of estrogen cycle and parity history on spine morphology (Luine and Frankfurt, 2013), only male mice were used for Golgi-Cox studies. For 2D-gel studies, no trends to suggest sex-related differences were observed, and data from both sexes were combined for analysis.
Plasmid transfections
Mouse primary cortical neurons were transfected at 7 d in vitro (DIV7) with GFP (eGFPc-1; Clontech) or cotransfected with a half-plasmid dose of GFP plus the equivalent amount of PINK1-GFP (Genecopoeia) using Lipofectamine 2000 as described previously. For some studies, primary neurons were transfected with GFP and pGW1-2b-myc-rp47(S176D; p47D) or pGW1-2b-myc-rp47(S176A; p47A), which were previously described (Wang et al., 2018). For dendritic length and spine morphometry, neurons were fixed and stained 7 d after transfection (DIV14).
Indirect immunofluorescence and neuronal morphometry
Mouse primary cortical neurons were fixed in 4% paraformaldehyde (2% final; Sigma-Aldrich) at room temperature for 15 min, and permeabilized with 0.1% Trition X-100 in PBS. After incubation in Superblock Buffer (Thermo Scientific), cultures were stained with chicken anti-GFP (Table 1) and anti-chicken Alexa 488 secondary antibodies (diluted 1:1000; Invitrogen). Neurons were imaged on a Nikon A1R confocal microscope at 20× using a 1.4 NA objective at a step size of 0.75 µm, with an 8 s dwell time per plane, and a total of 5–7 planes. Dendrite analysis of pyramidal neurons, defined by triangular shape and presence of apical dendrite, was performed in National Institutes of Health ImageJ software, supplemented with the SNT plug-in. Analysis parameters include a continuous radius of 1 μm step size. The apical dendrite was identified by a thick trunk generally emanating from the apex of the soma and distinguished from the basal arbor. Equivalent bifurcating branches are defined as sister dendrites if the summated diameter of each individual branch is approximately equal to the parent dendrite (Rall, 1962). Individual, higher-order branches emanating off a parent branch were identified if the branch diameter is less than half the diameter of the parent. The area under the (Sholl) curve (AUC) was calculated as |a∫b f(x)dx|, where f(x) equals the number of intersections at a specific radius using the whole neuron from a = 0 to b = 400 μm for each Sholl profile (Rall, 1962). Branching index (Garcia-Segura and Perez-Marquez, 2014) was adapted from (Bird and Cuntz, 2019) where only positive values are summed, and calculated as with Intr defined as the number of intersections at the indicated radius n, with a radial count of 400.
Table 1.
Primary antibodies used
| Antigen | Species | Company | Catalog/lot no. | Dilution (use) | RRID no. |
|---|---|---|---|---|---|
| GFP | Chicken | Abcam | ab13970 | 1:1000 | AB_300798 |
| NSFL1C | Rabbit | Novus Biologicals | NBP2-13677 | 1:500 | NA |
| Alpha tubulin | Rabbit | Abcam | ab15246 | 1:2000 | AB_301787 |
| GAPDH | Rabbit | Abcam | ab37168 | 1:5000 | AB_732652 |
Details of primary antibodies, dilutions, and sources are shown.
For dendritic spine analysis, dendrites were imaged using a 60 × 1.4 NA oil immersion objective with a 2× digital zoom and saved as 32-bit multidimensional ND2 files. The entire depth of the dendrite was acquired at a step size of 0.25 μm. File names were coded to conceal group identity and the number and morphology of dendritic spines in clearly visualizable segments (interquartile range, 125–300 μm) of primary or secondary dendrite were manually analyzed and quantified in a blinded fashion. Mushroom spines were defined as showing a head-to-neck width ratio >2 and stubby spines with a length-to-width ratio ≤ 1, as previously described (Qiao et al., 2016). Thin spines showed a high length-to-width ratio with no more than a slight expansion at the tip. Filopodia were identified as extremely thin, generally 3- to 10-µm-long structures that frequently contained bends (Qiao et al., 2016).
Electrophysiology
Whole-cell recordings were performed in primary cortical neuronal cultures either derived from Pink1 WT or Pink1 KO mice at DIV13–DIV14. Pyramidal neurons were identified by their apical dendrites and triangular somata. Patch electrodes (2–10 MΩ open-tip resistance) were filled with a solution containing the following (in mm): 120 Cs gluconate, 10 HEPES, 10 BAPTA, 10 NaCl, 4 Mg-ATP, 0.3 GTP balanced to pH 7.2 ± 0.05 with CsOH; solution osmolality was 280 ± 10 mOsm. mEPSCs were recorded at −70 mV in an external solution containing the following (in mm): 140 NaCl, 2.8 KCl, 1 CaCl2, 10 HEPES, 0.01 EDTA, 1 MgSO4, 0.001 tetrodotoxin (to block action potentials), and 0.01 glycine, balanced to pH 7.2 ± 0.05 with NaOH, and osmolality raised to 290 ± 10 mOsm with sucrose. Voltage-clamp recordings were performed using an Axon Axopatch 200B amplifier (Molecular Devices). Signals were filtered at 2 kHz and acquired at a sampling rate of 10 kHz using Clampex 10.2 software (Molecular Devices). Access resistance typically was 10–20 MΩ and remained relatively stable during experiments (≤30% increase). Corrections were made for liquid junction potential (−11 mV). Miniature events were analyzed and averaged using the MiniAnalysis Program (Synaptosoft).
Golgi–Cox Staining
Brains were harvested from male 6-month-old Pink1 KO and Pink1 WT littermates and stained using the FD Rapid GolgiStain Kit (FD Neurotechnologies). Brains were rinsed with double distilled water and then immersed in a 1 : 1 mixture of FD Solution A/B for 2 weeks at room temperature in the dark. Brains were transferred to FD Solution C for 72 h and kept in the dark. After 24 h the whole brain was mounted onto a cryostat chuck in optimal cutting temperature compound and stored at −80°C until sectioning. Coronal sections of 50 µm thickness were cut and transferred onto drops of FD Solution C on gelatin-coated slides labeled with a code to conceal the genotype of the mouse. After allowing sections to dry at room temperature in the dark, slides were stained using the directions of the manufacturer and coverslipped in Permount (Fisher). Neurons from the somatosensory cortex were selected for imaging from five to eight sections of similar location along the rostral-caudal axis based on clear pyramidal morphology, solid staining, and relative lack of crossing neurites or debris. Z-stack images with a step size of 0.31 µm were collected using a 60× UPlanSApo 1.35 NA oil immersion objective on an Olympus IX83 microscope fitted with a DP80 camera. Only secondary apical dendrites that were >15 µm away from the soma and >70 µm in length were selected for manual counting of spine projections along the entire focusable length of the dendrite. Spine counts were divided by the length of the segment quantified to yield spine density.
Immunoblotting and 2D gel analysis
Cortical tissue was harvested from male and female mice from Pink1 KO and WT littermates. For isoelectric focusing, equivalent amounts of lysed protein were precipitated and resuspended in a rehydration buffer containing 7 m urea, 10 ml water, 2 m Thiourea, 1 g CHAPS, and 25 mm DTT. Using the PROTEAN IEF cell from Bio-Rad, the protein was actively rehydrated onto 7 cm, pH3–pH6, IPG ReadyStrips (Bio-Rad), and isoelectric focusing was performed according to the preset protocol. The gel strip was then washed for 10 min using Equilibration Buffer I (catalog #1632107, Bio-Rad), followed by Equilibration Buffer II (catalog #1632108, Bio-Rad), and subjected to SDS-PAGE for the second dimension.
Experimental design and statistical analysis
Images were coded before analysis by individuals with no knowledge of the treatment conditions. Sholl curves were generated and data analyzed by repeated-measures ANOVA using GraphPad Prism 9 software. Sholl curves show mean ± SEM. Violin plots show median values and the interquartile range. Outliers were identified using ROUT with Q set at 0.1%. Parametric data were analyzed by Welch's t test for two-group comparisons. For multiple groups, data were analyzed using Brown–Forsythe ANOVA with Dunnett's T3 multiple comparisons test or one-way ANOVA with Tukey post hoc test for homoscedastic data, using GraphPad Prism 9 (version 9.0.0).
Results
PINK1 is necessary for full dendritic arborization
Primary cortical neurons from Pink1 WT and Pink1 KO mice were transfected with GFP to visualize neuronal processes and imaged at DIV14 (Fig. 1A). Pink1 KO neurons exhibited decreased dendritic complexity, as quantified by reduced intersections with circles of increasing radii in Sholl analysis, particularly in the 30–270 µm range (Fig. 1B). However, no changes in the maximum extension radius were observed. AUC analysis revealed a significant decrease in the KO neurons (Fig. 1C). There were no differences in somal size (data not shown). Calculations of branching index (see above, Materials and Methods) revealed a striking reduction in KO neurons relative to WT controls (Fig. 1D), with more modest reduction in the average branch length/neuron (Fig. 1E). These data suggest that endogenous PINK1 may play a greater role in controlling dendritic branching relative to extension.
Figure 1.
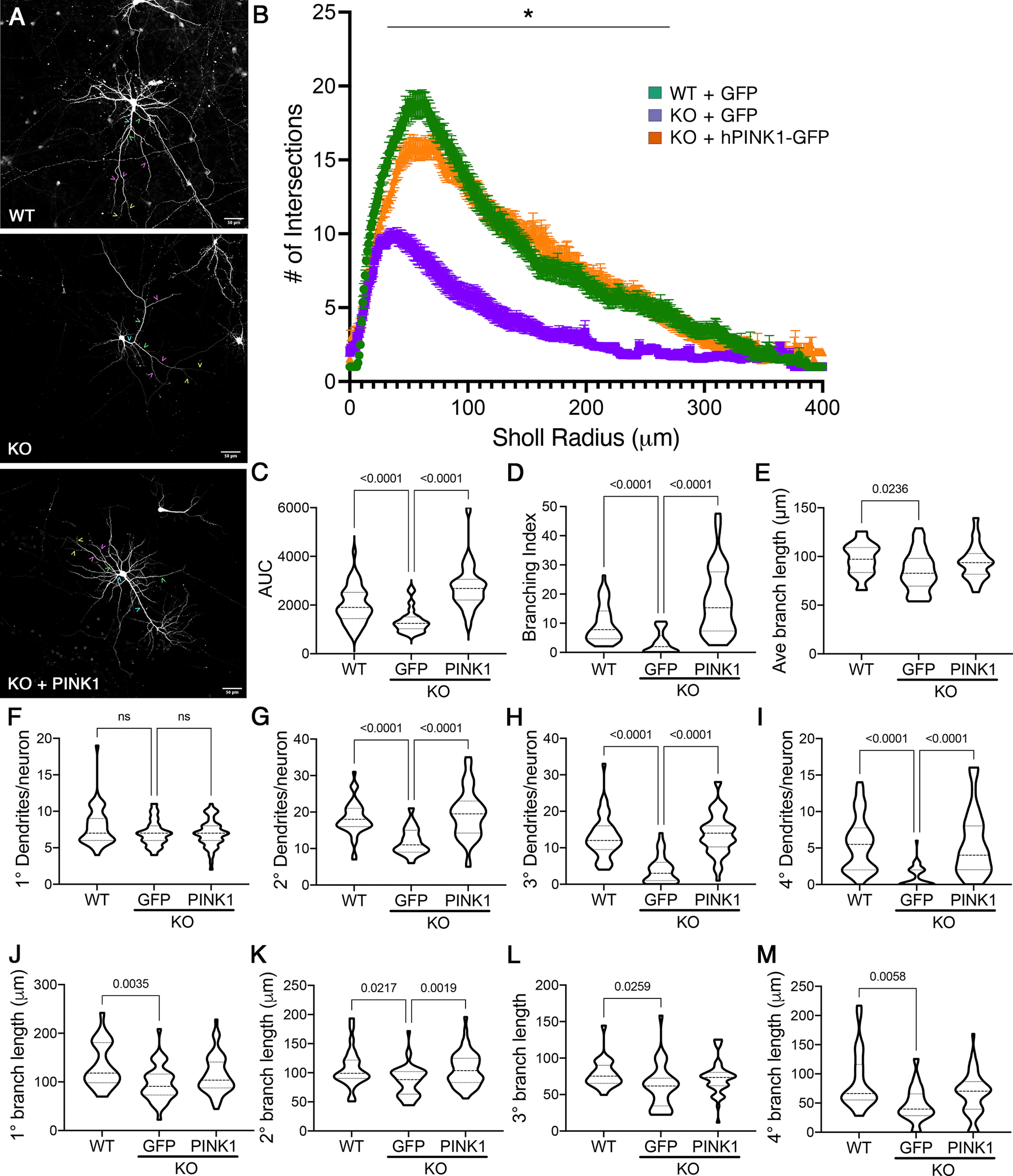
Loss of endogenous PINK1 elicits dendritic simplification in primary cortical neurons, which is reversed by introduction of human PINK1. A, Primary cortical neurons from Pink1−/− (KO) mice were transfected with GFP or GFP+PINK1-GFP at low efficiency to enable visualization of their arbors and compared with WT controls transfected with GFP. Scale bar, 50 µm. Representative examples of 1° dendrites are marked with teal carets, 2° with green carets, 3° with pink carets, and 4° with yellow carets. B, Sholl analysis of dendritic arbors expressed as mean ± SEM; *radii with significant differences for KO versus WT and KO versus KO plus hPINK1 (multiple-comparison testing following two-way repeated-measures ANOVA; Table 2). C–E, AUC, branching index, and average branch lengths, respectively. F–I, The number of primary, secondary, tertiary, and quaternary branches per neuron. J–M, The average branch lengths of primary, secondary, tertiary, and quaternary dendrites per neuron. Data are expressed as violin plot probability densities with the median and interquartile range indicated. Adjusted p values following post hoc Dunnett's T3 multiple comparisons test are shown (n = 25–52 neurons per condition compiled from 3–4 independent experiments; Table 2). ns - not significant.
Table 2.
ANOVA F and p values
| Figure | Brief description | F (DFn, DFd) | p value | Test |
|---|---|---|---|---|
| 1 B | Sholl interaction | 13.78 (800, 47600) | <0.0001 | Repeated-measures ANOVA |
| Effect of genotype | 35.95 (2, 119) | <0.0001 | ||
| Effect of radius | 279 (3.887, 462.5) | <0.0001 | ||
| 1 C | AUC | 33.47 (2.000, 79.20) | <0.0001 | Brown–Forsythe ANOVA |
| 1 D | BI | 27.42 (2.000, 51.29) | <0.0001 | Brown–Forsythe ANOVA |
| 1 E | Average branch length | 4.818 (2.000, 99.16) | 0.0101 | Brown–Forsythe ANOVA |
| 1 F | No. primary | 2.271 (2.000, 130.4) | 0.1073 | Brown–Forsythe ANOVA |
| 1 G | No. secondary | 29.66 (2.000, 121.5) | <0.0001 | Brown–Forsythe ANOVA |
| 1 H | No. tertiary | 50.37 (2.000, 125.9) | <0.0001 | Brown–Forsythe ANOVA |
| 1 I | No. quaternary | 24.60 (2.000, 102.9) | <0.0001 | Brown–Forsythe ANOVA |
| 1 J | Primary length | 6.186 (2.000, 89.19) | 0.003 | Brown–Forsythe ANOVA |
| 1 K | Secondary length | 6.585 (2.000, 78.70) | 0.0023 | Brown–Forsythe ANOVA |
| 1 L | Tertiary length | 4.348 (2.000, 86.41) | 0.0159 | Brown–Forsythe ANOVA |
| 1 M | Quaternary length | 5.719 (2.000, 57.26) | 0.0054 | Brown–Forsythe ANOVA |
| 2 A | Sholl interaction | 4.745 (800, 39600) | <0.0001 | Repeated-measures ANOVA |
| Effect of genotype | 17.22 (2, 99) | <0.0001 | ||
| Effect of radius | 79.46 (5.756, 569.9) | <0.0001 | ||
| 2 B | Sholl interaction | 8.047 (800, 39200) | <0.0001 | Repeated-measures ANOVA |
| Effect of genotype | 27.96 (2, 98) | <0.0001 | ||
| Effect of radius | 244.0 (3.891, 381.3) | <0.0001 | ||
| 2 C | Apical AUC | 16.17 (2.000, 80.88) | <0.0001 | Brown–Forsythe ANOVA |
| 2 D | No. 2° apical | 3.564 (2.000, 118.6) | 0.0314 | Brown–Forsythe ANOVA |
| 2 E | No. 3° apical | 19.58 (2.000, 125.5) | <0.001 | Brown–Forsythe ANOVA |
| 2 F | No. 4° apical | 9.391 (2.000, 101.6) | 0.0002 | Brown–Forsythe ANOVA |
| 2 G | Apical 1° length | 2.245 (2.000, 89.78) | 0.1118 | Brown–Forsythe ANOVA |
| 2 H | Apical 2° length | 1.696 (2.000, 97.36) | 0.1888 | Brown–Forsythe ANOVA |
| 2I | Apical 3° length | 11.27 (2.000, 80.40) | <0.0001 | Brown–Forsythe ANOVA |
| 2 J | Apical 4° length | 6.161 (2.000, 30.34) | 0.0057 | Brown–Forsythe ANOVA |
| 2 K | Basal AUC | 21.59 (2.000, 75.32) | <0.0001 | Brown–Forsythe ANOVA |
| 2 L | No. 2° basal | 38.39 (2.000, 132.9) | <0.0001 | Brown–Forsythe ANOVA |
| 2 M | No. 3° basal | 29.53 (2.000, 124.5) | <0.0001 | Brown–Forsythe ANOVA |
| 2 N | No. 4° basal | 43.84 (2.000, 127.6) | 0.0712 | Brown–Forsythe ANOVA |
| 2 O | Basal 1° length | 15.54 (2.000, 84.77) | <0.0001 | Brown–Forsythe ANOVA |
| 2 P | Basal 2° length | 8.867 (2.000, 97.49) | 0.0003 | Brown–Forsythe ANOVA |
| 2 Q | Basal 3° length | 3.199 (2.000, 78.06) | 0.0462 | Brown–Forsythe ANOVA |
| 2 R | Basal 4° length | 2.259 (2.000, 47.83) | 0.1155 | Brown–Forsythe ANOVA |
| 3 B | Spine density | 69.04 (2.000, 33.19) | <0.0001 | Brown–Forsythe ANOVA |
| 3 C | Filopodia density | 3.065 (2.000, 60.97) | 0.0539 | Brown–Forsythe ANOVA |
| 3 D | Stubby density | 44.74 (2.000, 44.09) | <0.0001 | Brown–Forsythe ANOVA |
| 3 E | Thin density | 25.11 (2.000, 44.88) | <0.0001 | Brown–Forsythe ANOVA |
| 3 F | Mushroom density | 57.25 (2.000, 38.94) | <0.0001 | Brown–Forsythe ANOVA |
| 5 B | Amplitude | 8.218 (2.000, 14.00) | 0.0044 | One-way ANOVA |
| 5 C | Frequency | 5.626 (2.000, 14.00) | 0.0161 | One-way ANOVA |
| 5 D | Decay constant | 1.410 (2.000, 14.00) | 0.2767 | One-way ANOVA |
| 5 E | Interevent interval | 12.93 (2.000, 5.809) | 0.0073 | Brown–Forsythe ANOVA |
| 7 B | Sholl interaction | 7.367 (1200, 38800) | <0.0001 | Repeated-measures ANOVA |
| Effect of genotype | 23.51 (3, 97) | <0.0001 | ||
| Effect of radius | 214.9 (3.206, 311.0) | <0.0001 | ||
| 7 C | p47 AUC | 16.81 (3.000, 45.00) | <0.001 | Brown–Forsythe ANOVA |
| 7 D | p47 BI | 12.25 (3.000, 33.94) | <0.0001 | Brown–Forsythe ANOVA |
| 7 E | p47 total spine | 30.18 (3.000, 35.15) | <0.0001 | Brown–Forsythe ANOVA |
| 7 F | p47 mushroom | 73.22 (3.000, 22.37) | <0.0001 | Brown–Forsythe ANOVA |
| 7 G | p47 thin/stubby | 13.03 (3.000, 30.74) | <0.0001 | Brown–Forsythe ANOVA |
| 7 H | p47 filopodia | 12.84 (3.000, 41.89) | <0.0001 | Brown–Forsythe ANOVA |
ANOVA test statistics for individual figures. Figure legends provide two-group test statistics. Dfn, degree of freedom for the numerator of the F ratio; DFd, degree of freedom for the denominator.
As increased branching increases the potential synaptic density around the neuron, we further examined how the loss of endogenous PINK1 influences higher-order dendrites of neurons (Fig. 1F–M). Loss of PINK1 had no impact on the number of primary dendrites (Fig. 1F); however, significant reductions were observed in the number of secondary, tertiary, and quaternary structures (Fig. 1G–I). Pink1 KO exhibited slightly shorter overall average segment lengths/neuron than WT controls (Fig. 1E). There were small, but statistically significant, decreases when primary and higher-order dendrites were analyzed separately (Fig. 1J–M).
To establish whether these changes can be reversed by reintroduction of PINK1, a subset of Pink1 KO neuron culture wells were cotransfected with human PINK1-GFP. The deficits in AUC, branching index, and the number of higher-order dendrites were all completely rescued following the restoration of PINK1 expression (Fig. 1C–D, G–I), suggesting that the effects of PINK1 on these structures are conserved between mice and humans. However, with the exception of secondary branch length, transfection with human PINK1 elicited only a modest upward trend for average branch lengths (Fig. 1J–M). Together, these data indicate that endogenous PINK1 plays a major role in regulating dendritic branching with a lesser effect on dendritic extension.
PINK1 regulates morphology of both apical and basal dendrites
Dendrites on neocortical pyramidal neurons can be segregated into apical, basal, and proximal integration zones, which show different properties in creating sequence memory (Hawkins and Ahmad, 2016). To determine whether PINK1 expression differentially regulates apical and basal domains, we analyzed apical and basal arbors separately by Sholl analysis. We found that PINK1 loss decreased Sholl AUC of both regions, and these deficits were reversed by reintroduction of PINK1 (Fig. 2A–C,K). In the apical tree, deficits induced by loss of PINK1 were observed in tertiary and higher-order branches of the apical tree (Fig. 2E–F, I–J). In the basal arbor, Pink1 KO showed the greatest effects on secondary and tertiary branch numbers (Fig. 2L,M) and primary and secondary branch lengths (Fig. 2O,P). Each of these deficits was significantly reversed by transfection with human PINK1.
Figure 2.
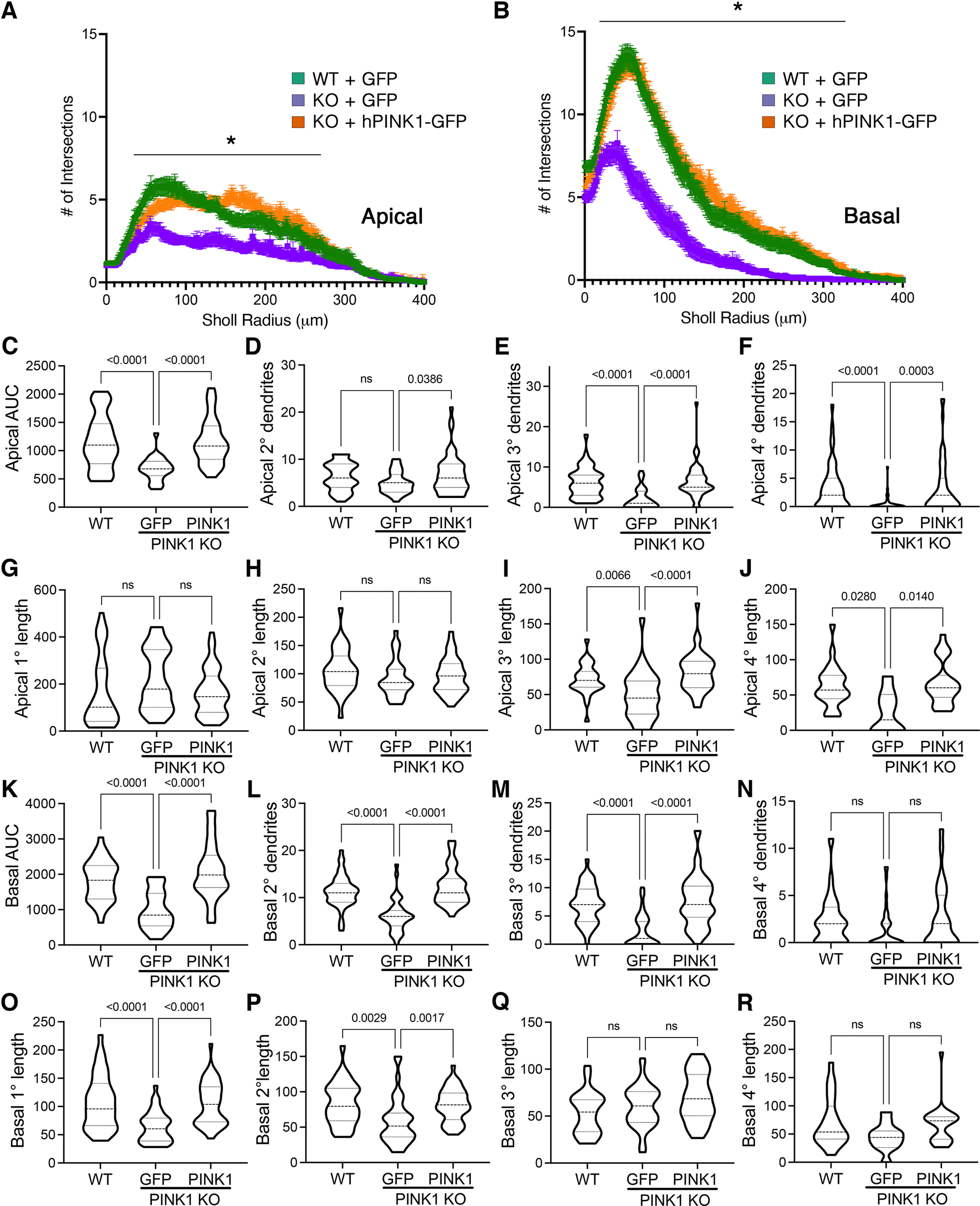
Loss of PINK1 shows greater effects on higher-order apical dendrites and 2°/3° basal dendrites. A, B, Sholl curves for apical (A) and basal (B) dendritic arbors of KO mouse cortical neurons compared with WT neurons and following rescue with human PINK1. Mean ± SEM; *radii with significant differences for KO versus WT, KO versus KO + hPINK1 (multiple-comparison testing following two-way repeated-measures ANOVA; Table 2). C, AUC analysis for apical dendrites. D–F, The number of secondary, tertiary, and quaternary branches off the apical dendrite per neuron. G–J, The average branch lengths of primary, secondary, tertiary, and quaternary apical dendrites per neuron. K, AUC analysis for basal dendrites. L–N, The number of secondary, tertiary, and quaternary basilar dendrites per neuron. O–R, The average basilar branch lengths for primary, secondary, tertiary, and quaternary dendrites. Data are expressed as violin plot probability densities with the median and interquartile range indicated. Adjusted p values following post hoc Dunnett's T3 multiple comparisons test are shown (n = 25–52 neurons per condition compiled from three to four independent experiments; Table 2). ns - not significant.
Loss of PINK1 elicits reduced synaptic density and altered spine structure
Excitatory synapses are largely localized to dendritic spines, structures altered in numerous neuropsychiatric and neurodegenerative diseases. To determine the effect of PINK1 loss on dendritic spine density, we analyzed at DIV14 primary cortical neurons that had been transfected with GFP to highlight their morphology using confocal microscopy. We observed significant decreases in spine density of Pink1 KO neurons compared with WT (Fig. 3A,B).
Figure 3.

Pink1 KO cortical neurons exhibit decreased spine density with relative increases in filopodia at the expense of mushroom spines, reversed by introduction of human PINK1. A, Representative images of primary and secondary dendrites in GFP-transfected neurons. Scale, 5 µm. B, Overall spine density. C–F, Density of filipodia, stubby spines, thin spines, and mushroom spines, respectively. G, Pie charts showing relative distribution of spine morphologies in the primary and secondary dendrites of WT versus KO cortical neurons with rescue in KO neurons transfected with hPINK1. Data in B–F are expressed as violin plots with the median and interquartile range indicated. Adjusted p values following post hoc Dunnett's T3 multiple comparisons test are shown (n = 26–39 neurons per condition compiled from three to four independent experiments; Table 2). ns - not significant.
To study the effects of Pink1 KO on spine morphology, we classified dendritic protrusions into the following morphologic classes: filopodia, stubby, thin, or mushroom (Peters and Kaiserman-Abramof, 1970). Filopodia are immature projections, whereas stubby, thin, and mushroom spines are functional structures thought to be associated with varying degrees of maturity or plasticity (Harris et al., 1992; Bourne and Harris, 2007). This examination revealed a reduction in the three functional spine types (Fig. 3D–F), whereas the decrease in filopodia density was not statistically significant (Fig. 3C). Transfection of KO neurons with human PINK1 not only reversed the deficits in stubby, thin, and mushroom spine numbers (Fig. 3D–F) but also increased the number of stubby and mushroom spines relative to WT controls (adjusted p values, stubby p < 0.0001; mushroom p = 0.0004).
During neuronal development, dendritic spines shift from immature filopodia that lack postsynaptic densities to stubby morphologies (Harris et al., 1992). Thin and mushroom spines increase in prevalence in mature animals, with mushroom spines showing the greatest stability consistent with a role in memory (Bourne and Harris, 2007). To investigate the potential impact of PINK1 on spine maturation, we analyzed the relative distribution of different spine morphologies in primary and secondary dendrites of WT and KO neurons (Fig. 3G). We found that among the remaining spines observed on Pink1 KO neurons, the proportion of filopodia increased from 26 to 49% on primary dendrites and from 22 to 34% on secondary dendrites. In contrast, mushroom spines decreased from 34 to 12% on primary dendrites and from 30 to 23% on secondary dendrites. The distribution of thin spines was decreased only on secondary dendrites, from 36 to 29%. Restoration of PINK1 expression rescued these changes in spine maturation (Fig. 3G).
PINK1 loss of function reduces synaptic function
To determine whether there was a functional impact from the structural changes observed in Pink1 KO dendritic spines, we studied the effects of PINK1 loss on endogenous glutamatergic synaptic activity. Whole-cell voltage-clamp recordings of dissociated neurons from untransfected primary cortical neurons from Pink1 KO and WT mice (Fig. 4A) revealed a reduced mEPSC frequency in the KO neurons (Fig. 4C) without significantly affecting mEPSC amplitude or decay kinetics (Fig. 4B,D). Cumulative probability plots reveal significantly increased interevent intervals in the KO neurons (Fig. 4E,F). These electrophysiological changes are consistent with the reduction in mature dendritic spines observed in the Pink1 KO neurons and suggest that this spine loss functionally affects excitatory synaptic transmission.
Figure 4.

Pink1 KO neurons exhibit significantly reduced frequency of mEPSCs compared with WT neurons. A, Representative traces illustrating mEPSCs recorded at −70 mV in WT or Pink1 KO primary neuronal cultures (left) and averaged mEPSCs (right). B–D, Amplitude (B), frequency (C), and decay time constant (D; τ) of the mEPSCs recorded from WT and Pink1 KO neuronal cultures (n = 5 neurons per genotype; ***p = 0.0001; t = 6.739 using Student's unpaired t test). E, Representative interevent intervals cumulative probability curve. F, Interevent intervals for mEPSCs compared between WT and Pink1 KO neuronal cultures (n = 5 neurons per genotype; ****p < 0.000001; F = 2638 using F test).
To determine whether introduction of human PINK1 into Pink1 KO neurons would rescue the electrophysiological changes, we compared WT neurons transfected with GFP, KO neurons transfected with GFP, and KO neurons transfected with hPINK1-GFP. Similar to experiments using untransfected neurons (Fig. 4), GFP-transfected Pink1 KO neurons showed reduced mEPSC frequency and increased mEPSC interevent intervals relative to WT controls (Fig. 5C,E). Introduction of human PINK1 increased mEPSC amplitude in KO neurons and rescued mEPSC frequency and interevent interval (Fig. 5B,C,E).
Figure 5.
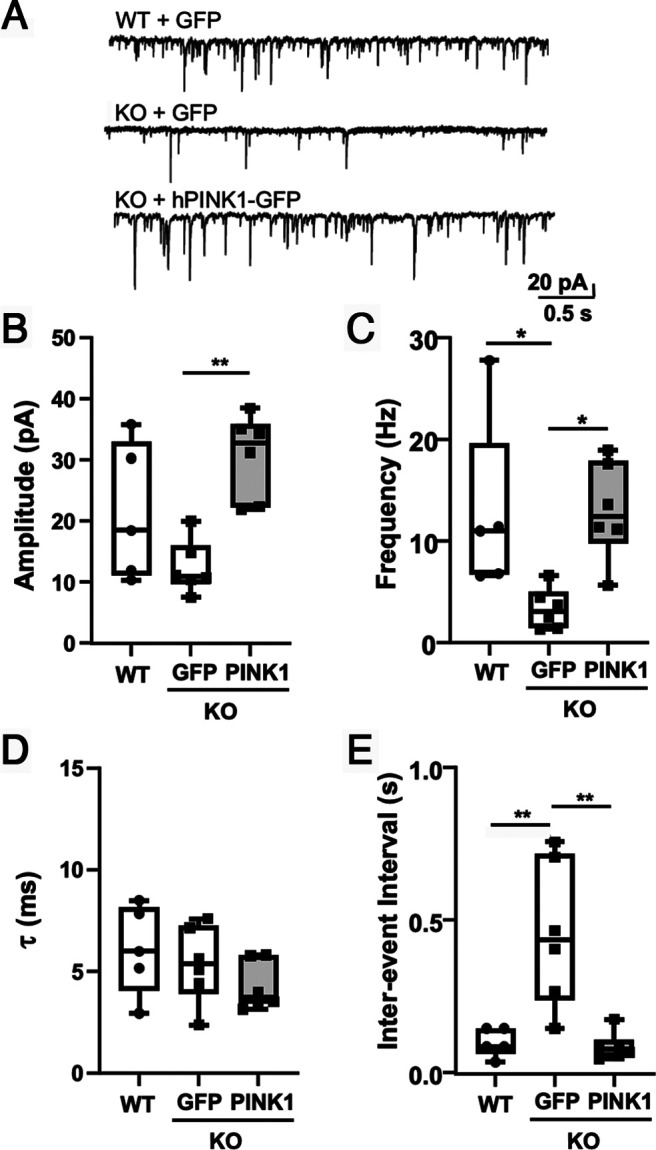
Introduction of human PINK1 significantly increases amplitude and frequency of mEPSCs in Pink1 KO neurons. A, Representative traces illustrating mEPSCs recorded at −70 mV in primary cortical neurons. B, mEPSC amplitude shows significant difference between KO+GFP and KO+hPINK1 (p = 0.0032). C, mEPSCs frequency shows significant difference between WT+GFP and KO+GFP (p = 0.0385) and between KO+GFP and KO+hPINK1 (p = 0.0246). D, mEPSC decay time constant (τ) shows no difference among groups. E, mEPSC interevent interval shows significant difference between WT+GFP and KO+GFP (p = 0.0036) and between KO+GFP and KO+hPINK1 (p = 0.0018). Values compared with ANOVA and Tukey post hoc test (n = 5–6 neurons per condition; Table 2).
Loss of PINK1 reduces spine density in vivo
To examine the importance of PINK1 in regulating spine architecture in vivo, we used the Golgi–Cox impregnation stain to highlight the architecture of cortical neurons (Fig. 6). We quantified spine density in secondary apical dendrites of pyramidal neurons in the somatosensory cortex of 6-month-old Pink1 KO and WT mice. Pink1 KO mice (Fig. 6B,D,F) exhibited significantly decreased spine densities compared with WT littermates (Fig. 6G).
Figure 6.

Pink1 KO mice show reduced spine densities in vivo. A, B, Representative coronal images of the somatosensory cortices of 6-month-old WT (A) and KO (B) littermates stained using the Golgi–Cox method. Scale bars: 200 μm. C, D, Neurons in the boxed areas of A and B are shown at higher magnification for (C) WT and (D) KO mice, respectively. Secondary apical dendrites are indicated with arrowheads. Scale bars: 50 μm. E, F, Representative images of secondary apical dendrites from a different pair of (E) WT and (F) KO mice. Scale bars: 10 μm. G, Quantification of spine density (n = 28–34 neurons compiled from two WT and three KO mice; p < 0.0001, t = 4.918 using Student's unpaired t test with Welch's correction).
Administration of the p47 phosphorylation mimic significantly restores dendritic complexity and spine densities in Pink1 KO neurons
We have previously found that PINK1 promotes the PKA-mediated phosphorylation of p47 (Wang et al., 2018), a protein implicated in regulating spinogenesis (Shih and Hsueh, 2016). To determine whether changes in p47 may contribute to the dendritic architectural changes observed in PINK1-deficient neurons, we studied p47 expression level and phosphorylation state in brains from Pink1 KO mice versus WT controls. There were no significant differences in p47 expression (data not shown). However, we did find a significant decrease in the more acidic p47 species by isoelectric focusing in the KO mouse cortex compared with WT (Fig. 7A), consistent with decreased p47 phosphorylation.
Figure 7.

The p47 phosphomimetic p47D reverses dendritic branching deficits with partial restoration of spines in Pink1 KO neurons. A, 2D isoelectric focusing/gel electrophoresis analysis reveals a significant reduction in the ratio of more acidic (Spot 1) and less acidic (Spot 2) forms of p47 consistent with reduced phosphorylation in the cortex of KO mice (n = 3 mice/group). Data are expressed as violin plots with the median and interquartile range indicated; p value from Welch's t test is shown. B–I, Primary cortical neurons from Pink1 KO mice were transfected with GFP and the phosphomimetic p47D or nonphosphorylatable p47A, and compared with neurons from WT controls transfected with GFP. Sholl curves show greater effects of p47D compared with p47A (B). Mean ± SEM; *radii with significant differences for KO versus KO+p47A; **radii with significant differences for KO versus WT, KO versus KO+p47D (multiple-comparison testing following two-way repeated-measures ANOVA; Table 2). AUC (C) and branching index analysis of Sholl data (D). Total spine density (E), mushroom spine density (F), and thin/stubby spine density (G) were significantly elevated by p47D in KO neurons but not the density of filopodia (H). Pie charts (I) showing the effects of p47D and p47A on the relative distribution of spine morphologies. Data in A and C–H are expressed as violin plots with the median and interquartile range indicated. Adjusted p values following post hoc Dunnett's T3 multiple comparisons test are shown (n = 12–43 neurons per condition compiled from three independent experiments; Table 2). ns - not significant.
Next, we transfected KO neurons with two different forms of p47 to determine whether either could rescue the effects of PINK1 deficiency on dendritic structures. Although the nonphosphorylatable p47A showed a partial rescue of intersections in the 20–50 μm region (Fig. 7B), this did not result in restoration of AUC or branching index (Fig. 7C,D). In contrast, transfection with the phosphomimetic p47D restored dendritic complexity accompanied by rescue of AUC and branching (Fig. 7B–D), similar to the effects of PINK1 (Fig. 1B–D). Like PINK1 itself, p47D did not significantly affect filopodia density (Fig. 7H) but was able to significantly ameliorate the effects of PINK1 loss on total, mushroom, and thin/stubby spine densities (Fig. 7E–G).
In contrast to human PINK1, p47D showed only partial rescue of mushroom spine densities (Fig. 7F), consistent with only a partial restoration of the mushroom to filipodia ratio (Fig. 7I). Although the effects on spine density appeared to be dependent on p47 phosphorylation (Fig. 7E–G), p47A was as effective as p47D in the partial effects on spine maturation (Fig. 7I). Together, these data suggest that endogenous PINK1 regulates dendritic branching and spine density in part through p47 phosphorylation but that additional mechanisms may also contribute to its effects on spine density and maturation.
Discussion
Loss of PINK1 function is associated with cognitive/executive dysfunction in recessive familial PD (Li et al., 2005; Ricciardi et al., 2014; Piredda et al., 2020) and in sporadic AD and its mouse models (George et al., 2010; Du et al., 2017; Manczak et al., 2018; Jiang et al., 2021). Here, we demonstrate that loss of endogenous mouse PINK1 elicits dendritic simplification of cortical pyramidal neurons, accompanied by reductions in dendritic spine density in vitro and in vivo. These changes are accompanied by decreased mEPSC frequency. Introducing human PINK1 or a p47 phosphomimetic significantly rescued these dendritic and spine alterations in KO neurons.
Prior work showing that Pink1 mutation impairs neurite outgrowth in Caenorhabditis elegans (Samann et al., 2009) and Pink1 KO neurons have shorter overall dendrite lengths (Dagda et al., 2014) support the generality of our findings, although dendritic spines, branch order, and effects on subdivisions of the dendritic tree have not been previously studied. Apical and basal dendrites mediate distinct feedback and feedforward inputs, with basal dendrites showing a relative dominance in determining orientation selectivity in the visual cortex (Park et al., 2019). We found that loss of PINK1 affects higher-order branches of both basal and apical arbors. Neurons with shorter and more simplified dendritic arbors tend to exhibit more irregular firing patterns in vivo (Montero et al., 2021). Whereas stimulation of proximal dendrites is more likely to trigger action potentials, computational studies predict that higher-order dendrites are necessary for time-based sequence memory (Hawkins and Ahmad, 2016). Interestingly, sequential working memory is impaired in nondemented patients with mild PD (Zhang et al., 2021).
We previously found that PINK1 interacts with valosin-containing protein (VCP), and knock down of either VCP or p47 blocks the ability of overexpressed PINK1 to promote overall dendritic complexity (Wang et al., 2018). The current data implicate p47 downstream of endogenous PINK1 in regulating spine density/maturation. Phosphorylation of p47 at S176, which is mediated by PKA (Wang et al., 2018), is important as the nonphosphorylatable p47A mutant showed reduced ability to complement the loss of PINK1.
VCP is a multifunctional ATPase linked to frontotemporal dementia and other diseases of protein homeostasis (Nalbandian et al., 2011; Darwich et al., 2020). The VCP-cofactor p47 regulates endoplasmic reticulum (ER) and Golgi remodeling, membrane tethering, and fusion (Buchberger et al., 2015). VCP interacts with neurofibromin to regulate spine density, and p47 is present in the complex (Wang et al., 2011). Neurofibromin is the major Ras regulator in dendritic spines, and its loss causes an activity-dependent spine loss (Oliveira and Yasuda, 2014). One of the functions of p47 during spine formation is to promote localized protein synthesis (Shih and Hsueh, 2016). The potential role of p47 phosphorylation in regulating localized protein synthesis or ER-Golgi biology remains unknown.
Introduction of human PINK1 to mouse KO neurons rescued each of the dendritic and spine parameters studied, indicating conserved PINK1 function in mouse and human. Given the low transfection efficiency, which aids in visualizing individual neuronal arbors, rescue by either PINK1 or p47D likely occurs through postsynaptic mechanisms. Although there are pharmacological agents to increase endogenous PINK1 expression or activity (Osgerby et al., 2017; Liu et al., 2020), these agents would not be effective in the KO setting. It would be interesting to assess the potential impact of these agents in induced pluripotent stem cell (iPSC) and mouse models of AD and PD that preserve at least some level of wild-type or mutant PINK1 expression.
Our current analysis of spine distribution, although statistically robust, does not differentiate decreased spine maturation from developmental delay or either activity-dependent or degenerative changes in mushroom spines. Loss of PINK1 has been linked to neural stem cell deficits in the hippocampus (Agnihotri et al., 2017), suggesting a possible role in neurodevelopment. Future studies to probe issues of development and spine dynamics would be highly interesting, given that Pink1 KO mice show abnormal developmental patterns of spontaneous excitatory postsynaptic currents (Pearlstein et al., 2016).
Interestingly, expression of p47D only partially rescued mushroom spine density and did not fully restore the relative distribution of mature and immature spine morphologies (Fig. 7F,I). These findings indicate additional pathways downstream of PINK1 that contribute to its ability to increase mushroom spines. Given greater shifts in the percentage of spines with thin and mushroom morphology compared with stubby morphology and filopodia that are observed in KO neurons (Fig. 3G), PINK1 may promote spine maturation through distinct mechanisms from p47 phosphorylation, as p47 has currently been implicated mainly in spine formation (Shih and Hsueh, 2016). It is possible that both mitochondrial and cytosolic functions of PINK1 are involved, given that the dendritic distribution of mitochondria is important for spine plasticity (Li et al., 2004).
There are multiple mitochondrial mechanisms by which PINK1 could regulate dendritogenesis, spinogenesis, spine maturation, or maintenance. Pink1 KO neurons exhibit increased mitochondrial fragmentation, reduced mitochondrial membrane potential, and reduced mitochondrial density within dendrites (Dagda et al., 2014; Das Banerjee et al., 2017). Localized energy production by nearby mitochondria is critically important for stabilizing emerging dendritic structures and supporting synaptogenesis (Chang et al., 2006; Fukumitsu et al., 2016). PINK1 is known to regulate mitochondrial dynamics and transport (Deng et al., 2008; Dagda et al., 2009; Weihofen et al., 2009; Shlevkov et al., 2016). Full-length PINK1 is implicated in retrograde axonal transport with cleaved, cytosolic PINK1 enhancing anterograde transport and the percentage of stationary mitochondria (Matenia et al., 2012). PINK1 also enhances complex I function (Liu et al., 2011; Morais et al., 2014) and mitochondrial calcium homeostasis (Marongiu et al., 2009; Kostic et al., 2015; Huang et al., 2017). Given the heavy demand for appropriate postsynaptic calcium handling, it is possible that these other roles of PINK1 contribute to regulating spine morphology and function.
Another mechanism by which PINK1 could regulate synaptic biology is through PKA, a critical regulator of neuronal development, synaptogenesis, and plasticity (Kandel, 2012). PINK1 is capable of phosphorylating the PKA catalytic subunit in vitro (Wang et al., 2018), and numerous studies have found that PINK1 promotes PKA-regulated processes such as mitochondrial fission, mitochondrial calcium efflux, mitochondrial transport, and BDNF secretion (Sandebring et al., 2009; Kostic et al., 2015; Das Banerjee et al., 2017; Soman et al., 2021).
Studies of other brain regions in Pink1 KO mice add further support to the importance of PINK1 for maintaining cognitive function. Pink1 KO mice show deficits in learning and memory and contextual discrimination, associated with deficits in dopamine receptor signaling (Maynard et al., 2020). KO mice also show compromised maturation of hippocampal neurons positive for doublecortin, a marker of adult neurogenesis, in the dentate gyrus (Agnihotri et al., 2017). Acute silencing of PINK1 in hippocampal cultures decreases PSD95, Shank, glutamate receptors, and actin regulatory protein expression (Hernández et al., 2019). Amyloid pathology is associated with reduced hippocampal expression of PINK1 (Du et al., 2017; Manczak et al., 2018), and overexpression of PINK1 rescues pathology and behavioral deficits in both amyloid and tau-based AD models (Manczak et al., 2018; Jiang et al., 2021). These observations raise the possibility that some of the synaptic deficits observed in AD models may be related to the impact of PINK1 on spine density and maturation. It would thus be interesting to study the role of endogenous PINK1 on hippocampal neuron structure and function.
The cognitive deficits observed in patients with familial PINK1 mutations involve frontal-executive dysfunction, working memory, and attention, with psychiatric changes of anxiety and depression (Ephraty et al., 2007; Ricciardi et al., 2014; Piredda et al., 2020). In PD patients, deficits in sequential working memory are observed early in the disease process (Kawashima et al., 2021; Zhang et al., 2021). Both prefrontal and hippocampal atrophy can be detected at early stages before development of frank dementia (Brück et al., 2004). Cortical and limbic atrophy and abnormal executive function are also reported in heterozygote PINK1 mutation carriers with low risk of developing PD (Reetz et al., 2008; Ricciardi et al., 2014). Discovering how deficits in PINK1 expression and function affect the dendritic architecture of cortical neurons, including spine density and morphology, represents an important advance in understanding pathways by which PINK1 normally functions to promote neuron health and function.
The data in the current study demonstrate the effects of PINK1 deficiency on dendritic branching and elongation in cortical neurons, as well as spine density and subtypes. Branching and segment length deficits predominantly affect higher-order dendrites. Synaptic activity on more distal branches is believed to be important for sequential working memory (Hawkins and Ahmad, 2016), a deficit observed early in PD patients (Zhang et al., 2021). Moreover, our data implicate endogenous PINK1 in regulating spine density and the distribution of mature and immature spines. The effects on dendritic morphology and spine density may be mediated in part through p47 phosphorylation, although other mechanisms undoubtedly contribute to the ability of PINK1 to regulate spine morphology. These findings add to the increasing number of reports that PINK1 functions outside of mitophagy to regulate multiple processes important to neurons, including the dendritic and synaptic architecture of cortical neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS101628, AG026389, and AG065594, and a gift from the Helen Mendel Fund. We thank Jason Callio for technical assistance, Dr. Robert Sweet and Susan Erickson for providing training for the Golgi–Cox silver stain, Dr. Chenjian Li (current affiliation, Peking University, China) for the Pink1 knock-out mice, originally on a C57/129 background.
References
- Agnihotri SK, Shen R, Li J, Gao X, Büeler H (2017) Loss of PINK1 leads to metabolic deficits in adult neural stem cells and impedes differentiation of newborn neurons in the mouse hippocampus. FASEB J 31:2839–2853. 10.1096/fj.201600960RR [DOI] [PubMed] [Google Scholar]
- Bird AD, Cuntz H (2019) Dissecting Sholl analysis into its functional components. Cell Rep 27:3081–3096.e5. 10.1016/j.celrep.2019.04.097 [DOI] [PubMed] [Google Scholar]
- Bourne J, Harris KM (2007) Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol 17:381–386. 10.1016/j.conb.2007.04.009 [DOI] [PubMed] [Google Scholar]
- Brück A, Kurki T, Kaasinen V, Vahlberg T, Rinne JO (2004) Hippocampal and prefrontal atrophy in patients with early non-demented Parkinson's disease is related to cognitive impairment. J Neurol Neurosurg Psychiatry 75:1467–1469. 10.1136/jnnp.2003.031237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Schindelin H, Hänzelmann P (2015) Control of p97 function by cofactor binding. FEBS Lett 589:2578–2589. 10.1016/j.febslet.2015.08.028 [DOI] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ (2006) Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26:7035–7045. 10.1523/JNEUROSCI.1012-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra IIS, Steer E, Gusdon AM, Kiselyov K, Chu CT (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182:474–484. 10.1016/j.ajpath.2012.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT (2018) Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis 122:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed RB, Roberts RC, Farmer CB, McMahon LL, Goldberg MS (2021) Increased glutamate transmission onto dorsal striatum spiny projection neurons in Pink1 knockout rats. Neurobiol Dis 150:105246. 10.1016/j.nbd.2020.105246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins N, Gotz J (2017) Shedding light on mitophagy in neurons: what is the evidence for PINK1/Parkin mitophagy in vivo? Cell Mol Life Sci 75:1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855. 10.1074/jbc.M808515200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ 3rd, Chu CT (2011) Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson's disease. Cell Death Differ 18:1914–1923. 10.1038/cdd.2011.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem 128:864–877. 10.1111/jnc.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwich NF, et al. (2020) Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science 370:eaay8826. 10.1126/science.aay8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Banerjee T, Dagda RY, Dagda M, Chu CT, Rice M, Vazquez-Mayorga E, Dagda RK (2017) PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J Neurochem 142:545–559. 10.1111/jnc.14083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A 105:14503–14508. 10.1073/pnas.0803998105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF, Tieu K, Yan SS (2017) PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer's disease. Brain 140:3233–3251. 10.1093/brain/awx258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephraty L, Porat O, Israeli D, Cohen OS, Tunkel O, Yael S, Hatano Y, Hattori N, Hassin-Baer S (2007) Neuropsychiatric and cognitive features in autosomal-recessive early Parkinsonism due to PINK1 mutations. Mov Disord 22:566–569. 10.1002/mds.21319 [DOI] [PubMed] [Google Scholar]
- Fukumitsu K, Hatsukano T, Yoshimura A, Heuser J, Fujishima K, Kengaku M (2016) Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol Cell Neurosci 71:56–65. 10.1016/j.mcn.2015.12.006 [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Perez-Marquez J (2014) A new mathematical function to evaluate neuronal morphology using the Sholl analysis. J Neurosci Methods 226:103–109. 10.1016/j.jneumeth.2014.01.016 [DOI] [PubMed] [Google Scholar]
- George AJ, Gordon L, Beissbarth T, Koukoulas I, Holsinger RM, Perreau V, Cappai R, Tan SS, Masters CL, Scott HS, Li QX (2010) A serial analysis of gene expression profile of the Alzheimer's disease Tg2576 mouse model. Neurotox Res 17:360–379. 10.1007/s12640-009-9112-3 [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B (1992) Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci 12:2685–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins J, Ahmad S (2016) Why neurons have thousands of synapses, a theory of sequence memory in neocortex. Front Neural Circuits 10:23. 10.3389/fncir.2016.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández CJ, Báez-Becerra C, Contreras-Zárate MJ, Arboleda H, Arboleda G (2019) PINK1 silencing modifies dendritic spine dynamics of mouse hippocampal neurons. J Mol Neurosci 69:570–579. 10.1007/s12031-019-01385-x [DOI] [PubMed] [Google Scholar]
- Huang E, Qu D, Huang T, Rizzi N, Boonying W, Krolak D, Ciana P, Woulfe J, Klein C, Slack RS, Figeys D, Park DS (2017) PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun 8:1399. 10.1038/s41467-017-01435-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XJ, Wu YQ, Ma R, Chang YM, Li LL, Zhu JH, Liu GP, Li G (2021) PINK1 alleviates cognitive impairments via attenuating pathological tau aggregation in a mouse model of tauopathy. Front Cell Dev Biol 9:736267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2012) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5:14. 10.1186/1756-6606-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima S, Shimizu Y, Ueki Y, Matsukawa N (2021) Impairment of the visuospatial working memory in the patients with Parkinson's disease: an fMRI study. BMC Neurol 21:335. 10.1186/s12883-021-02366-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killackey SA, Philpott DJ, Girardin SE (2020) Mitophagy pathways in health and disease. J Cell Biol 219:e202004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A 104:11441–11446. 10.1073/pnas.0702717104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Karouani M, Haburcak M, Martella G, Tscherter A, Platania P, Wu B, Pothos EN, Shen J (2009) Impaired dopamine release and synaptic plasticity in the striatum of parkin-/- mice. J Neurochem 110:613–621. 10.1111/j.1471-4159.2009.06152.x [DOI] [PubMed] [Google Scholar]
- Kostic M, Ludtmann MH, Bading H, Hershfinkel M, Steer E, Chu CT, Abramov AY, Sekler I (2015) PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep 13:376–386. 10.1016/j.celrep.2015.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumazawa R, Tomiyama H, Li Y, Imamichi Y, Funayama M, Yoshino H, Yokochi F, Fukusako T, Takehisa Y, Kashihara K, Kondo T, Elibol B, Bostantjopoulou S, Toda T, Takahashi H, Yoshii F, Mizuno Y, Hattori N (2008) Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch Neurol 65:802–808. 10.1001/archneur.65.6.802 [DOI] [PubMed] [Google Scholar]
- Li Y, Tomiyama H, Sato K, Hatano Y, Yoshino H, Atsumi M, Kitaguchi M, Sasaki S, Kawaguchi S, Miyajima H, Toda T, Mizuno Y, Hattori N (2005) Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset Parkinsonism. Neurology 64:1955–1957. 10.1212/01.WNL.0000164009.36740.4E [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. 10.1016/j.cell.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, Takahashi R, Hattori N, Imai Y, Lu B (2012) Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet 8:e1002537. 10.1371/journal.pgen.1002537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Acín-Peréz R, Geghman KD, Manfredi G, Lu B, Li C (2011) Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A 108:12920–12924. 10.1073/pnas.1107332108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lear TB, Verma M, Wang KZ, Otero PA, McKelvey AC, Dunn SR, Steer E, Bateman NW, Wu C, Jiang Y, Weathington NM, Rojas M, Chu CT, Chen BB, Mallampalli RK (2020) Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight 5:e131834. 10.1172/jci.insight.131834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luine V, Frankfurt M (2013) Interactions between estradiol, BDNF and dendritic spines in promoting memory. Neuroscience 239:34–45. 10.1016/j.neuroscience.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet 27:1332–1342. 10.1093/hmg/ddy042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E (2009) Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson's disease by disturbing calcium flux. J Neurochem 108:1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matenia D, Hempp C, Timm T, Eikhof A, Mandelkow EM (2012) Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at Thr-313, a mutation site in Parkinson disease: effects on mitochondrial transport. J Biol Chem 287:8174–8186. 10.1074/jbc.M111.262287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard ME, Redell JB, Kobori N, Underwood EL, Fischer TD, Hood KN, LaRoche V, Waxham MN, Moore AN, Dash PK (2020) Loss of PTEN-induced kinase 1 (Pink1) reduces hippocampal tyrosine hydroxylase and impairs learning and memory. Exp Neurol 323:113081. 10.1016/j.expneurol.2019.113081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero T, Gatica RI, Farassat N, Meza R, González-Cabrera C, Roeper J, Henny P (2021) Dendritic architecture predicts in vivo firing pattern in mouse ventral tegmental area and substantia nigra dopaminergic neurons. Front Neural Circuits 15:769342. 10.3389/fncir.2021.769342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais V, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D, Van Coster R, Wurst W, Scorrano L, De Strooper B (2009) Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med 1:99–111. 10.1002/emmm.200900006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, Aerts L, Overbergh L, Grünewald A, Seibler P, Klein C, Gevaert K, Verstreken P, De Strooper B (2014) PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344:203–207. 10.1126/science.1249161 [DOI] [PubMed] [Google Scholar]
- Murata H, Sakaguchi M, Jin Y, Sakaguchi Y, Futami J, Yamada H, Kataoka K, Huh NH (2011) A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem 286:7182–7189. 10.1074/jbc.M110.179390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Donkervoort S, Dec E, Badadani M, Katheria V, Rana P, Nguyen C, Mukherjee J, Caiozzo V, Martin B, Watts GD, Vesa J, Smith C, Kimonis VE (2011) The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget's disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci 45:522–531. 10.1007/s12031-011-9627-y [DOI] [PubMed] [Google Scholar]
- Oliveira AF, Yasuda R (2014) Neurofibromin is the major ras inactivator in dendritic spines. J Neurosci 34:776–783. 10.1523/JNEUROSCI.3096-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osgerby L, Lai YC, Thornton PJ, Amalfitano J, Le Duff CS, Jabeen I, Kadri H, Miccoli A, Tucker JHR, Muqit MMK, Mehellou Y (2017) Kinetin riboside and its ProTides activate the Parkinson's disease associated PTEN-induced putative kinase 1 (PINK1) independent of mitochondrial depolarization. J Med Chem 60:3518–3524. 10.1021/acs.jmedchem.6b01897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Papoutsi A, Ash RT, Marin MA, Poirazi P, Smirnakis SM (2019) Contribution of apical and basal dendrites to orientation encoding in mouse V1 L2/3 pyramidal neurons. Nat Commun 10:5372. 10.1038/s41467-019-13029-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlstein E, Michel FJ, Save L, Ferrari DC, Hammond C (2016) Abnormal development of glutamatergic synapses afferent to dopaminergic neurons of the pink1(-/-) mouse model of Parkinson's disease. Front Cell Neurosci 10:168. 10.3389/fncel.2016.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR (1970) The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat 127:321–355. 10.1002/aja.1001270402 [DOI] [PubMed] [Google Scholar]
- Piredda R, Desmarais P, Masellis M, Gasca-Salas C (2020) Cognitive and psychiatric symptoms in genetically determined Parkinson's disease: a systematic review. Eur J Neurol 27:229–234. 10.1111/ene.14115 [DOI] [PubMed] [Google Scholar]
- Qiao H, Li MX, Xu C, Chen HB, An SC, Ma XM (2016) Dendritic spines in depression: what we learned from animal models. Neural Plast 2016:8056370. 10.1155/2016/8056370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W (1962) Electrophysiology of a dendritic neuron model. Biophys J 2:145–167. 10.1016/s0006-3495(62)86953-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reetz K, Lencer R, Steinlechner S, Gaser C, Hagenah J, Büchel C, Petersen D, Kock N, Djarmati A, Siebner HR, Klein C, Binkofski F (2008) Limbic and frontal cortical degeneration is associated with psychiatric symptoms in PINK1 mutation carriers. Biol Psychiatry 64:241–247. 10.1016/j.biopsych.2007.12.010 [DOI] [PubMed] [Google Scholar]
- Ricciardi L, Petrucci S, Guidubaldi A, Ialongo T, Serra L, Ferraris A, Spanò B, Bozzali M, Valente EM, Bentivoglio AR (2014) Phenotypic variability of PINK1 expression: 12 years' clinical follow-up of two Italian families. Mov Disord 29:1561–1566. 10.1002/mds.25994 [DOI] [PubMed] [Google Scholar]
- Samann J, Hegermann J, Gromoff EV, Eimer S, Baumeister R, Schmidt E (2009) Caenorhabditis elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J Biol Chem 284:16482–16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, Miller DW, Zambrano I, Cowburn RF, Behbahani H, Cedazo-Mínguez A, Cookson MR (2009) Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One 4:e5701. 10.1371/journal.pone.0005701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih YT, Hsueh YP (2016) VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat Commun 7:11020. 10.1038/ncomms11020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL (2016) Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci U S A 113:E6097–E6106. 10.1073/pnas.1612283113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soman SK, Tingle D, Dagda RY, Torres M, Dagda M, Dagda RK (2021) Cleaved PINK1 induces neuronal plasticity through PKA-mediated BDNF functional regulation. J Neurosci Res 99:2134–2155. 10.1002/jnr.24854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Zhu J, Wang KZQ, Chu CT (2020) Chronic treatment with the complex I inhibitor MPP(+) depletes endogenous PTEN-induced kinase 1 (PINK1) via up-regulation of Bcl-2-associated athanogene 6 (BAG6). J Biol Chem 295:7865–7876. 10.1074/jbc.RA119.010474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HF, Shih YT, Chen CY, Chao HW, Lee MJ, Hsueh YP (2011) Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J Clin Invest 121:4820–4837. 10.1172/JCI45677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KZQ, Steer E, Otero PA, Bateman NW, Cheng MH, Scott AL, Wu C, Bahar I, Shih Y-T, Hsueh Y-P, Chu CT (2018) PINK1 Interacts with VCP/p97 and activates PKA to promote NSFL1C/p47 phosphorylation and dendritic arborization in neurons. eNeuro 5:ENEURO.0466-18.2018. 10.1523/ENEURO.0466-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ (2009) Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48:2045–2052. 10.1021/bi8019178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Ma J, Chan P, Ye Z (2021) Tracking response dynamics of sequential working memory in patients with mild Parkinson's disease. Front Psychol 12:631672. 10.3389/fpsyg.2021.631672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi L, Qin Q, Muqeem T, Seifert EL, Liu W, Zheng S, Li C, Zhang H (2019) Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol Aging 75:1–10. 10.1016/j.neurobiolaging.2018.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]