

Abstract
Persistent firing is commonly reported in both cortical and subcortical neurons under a variety of behavioral conditions. Yet the mechanisms responsible for persistent activity are only partially resolved with support for both intrinsic and synaptic circuit-based mechanisms. Little also is known about physiological factors that enable epochs of persistent firing to continue beyond brief pauses and then spontaneously terminate. In the present study, we used intracellular recordings in rat (both sexes) neocortical and hippocampal brain slices to assess the ionic mechanisms underlying persistent firing dynamics. Previously, we showed that blockade of ether-á-go-go-related gene (ERG) potassium channels abolished intrinsic persistent firing in the presence of low concentrations of muscarinic receptor agonists and following optogenetic activation of cholinergic axons. Here we show the slow dynamics of ERG conductance changes allows persistent firing to outlast the triggering stimulus and even to initiate discharges following ∼7 s poststimulus firing pauses. We find that persistent firing dynamics is regulated by the interaction between ERG conductance and spike afterhyperpolarizations (AHPs). Increasing the amplitude of spike AHPs using either SK channel activators or a closed-loop reactive feedback system allows persistent discharges to spontaneously terminate in both neocortical neurons and hippocampal CA1 pyramidal cells. The interplay between ERG and the potassium channels that mediate spike AHPs grades the duration of persistent firing, providing a novel, generalizable mechanism to explain self-terminating persistent firing modes observed behaving animals.
SIGNIFICANCE STATEMENT Many classes of neurons generate prolonged spiking responses to transient stimuli. These discharges often outlast the stimulus by seconds to minutes in some in vitro models of persistent firing. While recent work has identified key synaptic and intrinsic components that enable persistent spiking responses, less is known about mechanisms that can terminate and regulate the dynamics of these responses. The present study identified the spike afterhyperpolarizations as a potent mechanism that regulates the duration of persistent firing. We found that amplifying spike afterpotentials converted bistable persistent firing into self-terminating discharges. Varying the spike AHP amplitude grades the duration of persistent discharges, generating in vitro responses that mimic firing modes associated with neurons associated with short-term memory function.
Keywords: afterhyperpolarization, cholinergic modulation, ether-á-go-go-related K+ current, intrinsic physiology, persistent activity
Introduction
Most neurons respond only transiently to brief excitatory stimuli, generating spike discharges that subside at, or before, the end of the stimulus. However, in a minority of neurons, brief stimuli trigger prolonged epochs of spiking that outlast the stimulus. Neurons capable of persistent firing in neocortex in vitro are found in the deep layers of prefrontal cortex (Haj-Dahmane and Andrade, 1998), sensorimotor cortex (Rahman and Berger, 2011), parietal cortex (Gnadt and Andersen, 1988; Pesaran et al., 2002), and temporal association (TeA) cortex (Cui and Strowbridge, 2018, 2019; present study). Intrinsic persistent firing modes in cortical neurons are typically revealed following treatment with a modulator that enhances excitability and do not require fast synaptic circuit activity. Although a variety of neuromodulators can reveal persistent firing in cortical neurons, cholinergic modulation of intrinsic persistent firing has been studied most extensively. Neurons capable of intrinsic persistent firing modes are often densely innervated by cholinergic projections from the basal forebrain (Rye et al., 1984) and express muscarinic acetylcholine receptors (mAChRs; Mash et al., 1988; Volpicelli and Levey, 2004). Muscarinic receptor-initiated signaling reduces a variety of intrinsic conductances expressed in cortical pyramidal cells (PCs) including ether á-go-go-related gene (ERG) channels (Saganich et al., 2001; Papa et al., 2003; Cockerill et al., 2007). Our group recently reported that several highly specific ERG channel antagonists abolished persistent firing in layer 5 (L5) neocortical pyramidal cells (Cui and Strowbridge, 2018, 2019), suggesting that intrinsic persistent activity may reflect an increase in neuronal excitability associated with a reduction in ERG leak K+ current.
Neuronal discharges that continue past a triggering stimulus also occur in vivo and have been extensively characterized in the context of working memory (WM) tasks. Classic studies identified a subclass of prefrontal cortical neurons that discharged selectively during the delay period in delay–response WM tasks (Fuster and Alexander, 1971; Funahashi et al., 1989; Takeda and Funahashi, 2002). Activity in “delay” neurons was selective for a particular visual field area and predicted WM performance (Funahashi et al., 1989). Despite intensive experimental and theoretical investigations, the mechanistic basis of persistent firing during WM tasks remains unclear. Deep-layer neocortical pyramidal cells can be interconnected (Douglas et al., 1995), potentially providing a substrate for recurrent circuit activity that could sustain stimulus-triggered persistent spiking. While computational studies suggest persistence via recurrent networks (Seung, 1996; Wang, 1999; Fellous and Sejnowski, 2003) or feedforward networks (Goldman, 2009) is possible, whether synaptic interconnections are frequent and strong enough to create persistently active networks is unclear (for review, see Zylberberg and Strowbridge, 2017).
Behavioral studies also provided indirect evidence that biophysical mechanisms may contribute to delay cell activity. Working memory performance depends on subjects maintaining attention during the task and periods of heightened attention often are correlated with increased firing in cortically projecting basal forebrain cholinergic neurons (Gill et al., 2000; Broussard et al., 2009). Pharmacological blockade of muscarinic receptors required for cholinergic-stimulated persistent firing in vitro (Cui and Strowbridge, 2018) also impairs WM performance (Zhou et al., 2011; Major et al., 2015; but see Vijayraghavan et al., 2018), suggesting that many of the intrinsic physiological changes studied in vitro following muscarinic stimulation may also contribute to persistent firing modes associated with behavioral tasks such as WM.
Despite potentially similar biophysical modulation mechanisms, persistent activity recorded in vivo and in vitro have dramatically different kinetics. Delay cell activity during WM tasks is irregular and can diminish during long delay periods (Fuster and Alexander, 1971; Funahashi et al., 1989; Takeda and Funahashi, 2002), while most intrinsic persistent firing modes recorded using standard patch-clamp methods are bistable and long lasting. Unlike delay cell firing, which is typically terminated when the subject makes an eye saccade to terminate the task, cholinergic modulation of neocortical neurons promotes tonic persistent firing that continues 10s of seconds, than behaviorally relevant WM delay intervals. Whether the termination of persistent firing in vivo requires synaptic input (e.g., inhibition coinciding with the eye saccade response) that is absent in brain slice recordings remains an open question. Intriguingly, early investigators into this question had success in capturing examples of persistent firing that spontaneously terminated using sharp electrode recordings in brain slices (Schwindt et al., 1988; Egorov et al., 2002, 2006; Reboreda et al., 2007)—a method that disrupts intracellular signaling less dramatically than whole-cell recordings. Those early studies raise the possibility that physiologically relevant forms of persistent firing could be generated intrinsically in cortical neurons if normal intracellular signaling mechanisms could be maintained in vitro. However, little is known about which particular signaling pathways, and their channel targets, are required to generate naturalistic persistent firing modes.
The present study addressed this question and identified SK K+ channels as a key regulator of persistent firing dynamics in cortical neurons. Spontaneously terminating persistent firing reliably occurred in L5 neocortical pyramidal cells if spike afterhyperpolarizations (AHPs) were amplified using a SK positive modulator or a reactive feedback controller. Persistent firing dynamics were regulated by negative feedback only when the exogenous hyperpolarizing current injections were timed to overlap with the endogenous spike AHP. Finally, we demonstrate that both SK channel attenuation and reactive feedback amplification of spike AHPs also regulated persistent firing dynamics in hippocampal CA1 pyramidal cells, suggesting that in vivo-like graded and self-terminating persistent firing responses can be generated through biophysical mechanisms in a variety of cortical neurons.
Materials and Methods
Slice preparation.
Neocortical brain slices were prepared from postnatal day 14 (P14) to P25 Sprague Dawley rats of either sex using a vibratome (model 1200, Leica) as described previously (Cui and Strowbridge, 2018, 2019). In brief, rats were anesthetized with ketamine and decapitated, and then the brain was dissected and transferred into ice-cold artificial CSF (ACSF) composed of the following (in mm): 124 NaCl, 2.5 KCl, 1.23 NaH2PO4, 3 MgSO4, 26 NaHCO3, 10 dextrose, and 1 CaCl2, equilibrated with 95% O2/5% CO2. Slices were incubated at 30°C for ∼30 min and then maintained at room temperature (∼25°C) until use. All experiments were conducted under guidelines approved by the Case Western Reserve University Animal Care and Use Committee.
Electrophysiology.
Intracellular recordings were performed in a submerged chamber maintained at 30°C and perfused continuously (∼2 ml/min) with ACSF containing the following (in mm): 124 NaCl, 3 KCl, 1.23 NaH2PO4, 1.2 MgSO4, 26 NaHCO3, 10 dextrose, and 2.5 CaCl2, equilibrated with 95% O2/5% CO2. Whole-cell recordings were made using a patch clamp amplifier (model Axopatch 1D, Molecular Devices/Molecular Dynamics). Current-clamp intracellular recordings were implemented using the same methods described in our previous publications (Cui and Strowbridge, 2018, 2019). Membrane potentials presented in the text and figures were not corrected for the liquid junction potential (estimated to be +10 mV).
We recorded from L5 neocortical pyramidal cells from TeA region that generated “regular spiking” responses to 2 s duration depolarizing currents, as defined by previous neocortical studies (Connors and Gutnick, 1990; Schubert et al., 2001; Dégenètais et al., 2002). Recordings with resting membrane potential more depolarized than −60 mV or input resistance (Rin) <40 MΩ were excluded.
Standard salts and ACSF chemicals were obtained from Sigma-Aldrich. Unless noted, all receptor and channel blockers/modulators were obtained from Tocris Bioscience. All drugs were applied by switching the bath perfusion reservoir. Carbamoylcholine chloride (CCh; or carbachol; used at 2 µm except where noted) was purchased from Abcam and Tocris Bioscience; 10 mm stock solutions of CCh in water were prepared each day. N-(3,4-difluorophenyl)-N′-(3-methyl-1-phenyl-1H-pyrazol-5-yl)urea (ML297; GIRK activator, used at 0.67 µm) was prepared in ethanol at 10 mm. Terfenadine (used at 10 µm) was prepared in DMSO at 10 mm. 6,7-Dichloro-1H-indole-2,3-dione 3-oxime (NS309; SK channel positive modulator used between 0.5 and 2 µm; Pedarzani et al., 2005) was prepared in DMSO at 10 mm. N-[(1R)−1,2,3,4-tetrahydro-1-naphthalenyl]-1H-benzimidazol-2-amine hydrochloride (NS8593; SK channel negative modulator, used at 10 µm) was prepared in ethanol at 10 mm. 6,12,19,20,25,26-Hexahydro-5,27:13,18:21,24-trietheno-11,7-metheno-7H-dibenzo [b,n] [1,5,12,16]tetraazacyclotricosine-5,13-diium dibromide (UCL 1684; SK channel blocker, used at 1 µm) prepared in DMSO at 5 mm. 4-chloro-N-(6-chloropyridin-3-yl)benzamide (ICA-110381; KCNQ channel opener, used at 10 µm; Alomone Labs) was prepared in DMSO at 10 mm.
Photometric measurements of intracellular epifluorescent Ca2+ transients were acquired using a silicon photodiode and a 30-mm-diameter condenser lens attached to the wide-field video port of an upright microscope (model Axioskop FS1, Zeiss). We restricted the area illuminated by the LED light source (470 nm) to include the cell body region using the microscope epi-illumination field stop and a 63× water-immersion objective (numerical aperture, 0.9; Zeiss). Excitation light was blocked from the photodiode detection system using a 505 nm long-pass dichroic mirror (Omega Optical) and a BG39 emission filter. Photocurrents were converted into voltage using a custom transimpedance amplifier based on an op amp (model OPA111, Burr Brown/Texas Instruments) and a 1 GΩ feedback resister. The output of this initial amplifier stage was low-pass filtered (500 Hz cutoff; model FLA-01 Bessel filter, Cygnus Technology) and further amplified by 1–5× before being digitized on the ITC-18 data acquisition interface simulatenously with the electrophysiological signals. Single neurons were loaded with a Ca2+-sensitive indicator (Cal520 K+ salt, 150 µm; AAT Bioquest) through the patch-clamp recording electrode. Exogenous Ca2+ buffers (normally, 0.2 mm EGTA) were omitted from the internal solution in these experiments.
Closed-loop system for augmenting spike AHPs.
Spike-triggered hyperpolarizing current injections (see Figs. 6, 7, 8H) were generated using a microcontroller-based feedback system that interfaced with the patch-clamp amplifier (model AxoPatch 1D). An Arduino-compatible high-speed microcontroller (model ChipKit uC32, Digilent) operated a 16 bit analog-to-digital converter (ADC)/digital-to-analog converter (DAC) subsystem (Analog Shield, Digilent) that monitored the intracellular voltage output of the AxoPatch amplifier using one ADC channel. The microcontroller system looked for threshold crossings that signaled the rising phase of an action potential (AP; threshold set at −30 mV) only when an external digital gating signal was enabled (set by a transistor–transistor logic output on the ITC-18 interface). This digital gating signal from the ITC-18 was used to block spike-triggered hyperpolarizations during the step response. Once triggered, the uC32 system waited 2 ms to match the approximate AP duration and then generated a steady negative voltage corresponding to 50–150 pA injected current for 2 ms before decaying with a predetermined exponential time constant (1 s in most experiments). The delay before initiating the feedback AHP was increased to 100 ms in one set of experiments described in the text. Subsequent APs reset the microcontroller system DAC output to the same maximal level for 2 ms before decaying again (i.e., AP-triggered current injections did not summate beyond the maximal feedback AHP amplitude preset for that experiment).
Figure 6.
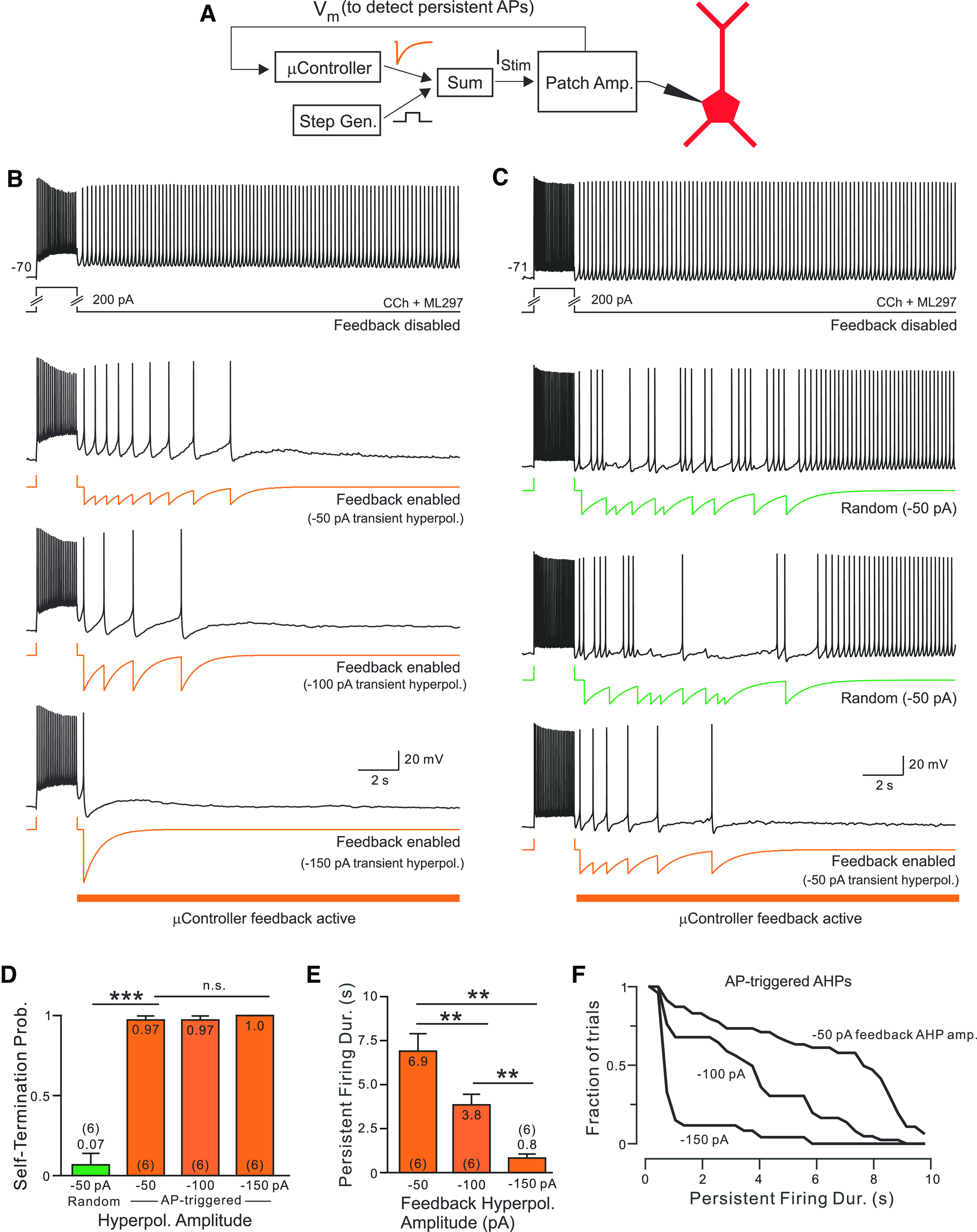
Spike-triggered hyperpolarizations regulate the duration of persistent discharges. A, Diagram of microcontroller-based responsive feedback system used to augment endogenous spike AHPs. The analog output of the microcontroller produces exponentially decaying hyperpolarizing transients that are added to the step generator to generate the patch-clamp amplifier current input. Analog signal summation (“Sum”) was mediated by a two-channel unity gain buffer amplifier. A digital control signal (data not shown) disabled the responsive feedback system during the step stimulus. B, Persistent discharges triggered by depolarizing current steps in an L5 pyramidal cell alone (Control) and spike AHPs augmented via responsive feedback (bottom three panels with orange current traces). Grading the amplitude AP-linked hyperpolarizations modulated the duration of self-terminating discharges (maximal injected current varied between −50 and −150 pA). C, Two different randomly timed trains of similar hyperpolarizing transients failed to terminate persistent firing (middle panels with green current traces). Control bistable response (top panel) and self-terminating response with AP-linked feedback hyperpolarizations (same −50 pA peak current amplitude; bottom panel) recorded from the same neuron. D, Summary of probability of self-terminating activity with AP-linked (orange) and randomly timed hyperpolarizations (green). All decay τ, 1 s: ***p = 2e-7; T = 14.10, df = 5; paired t test. E, Summary of reduction in the duration of persistent activity by different amplitude AP-linked hyperpolarizations (all decay τ, 1 s). All comparisons: **p < 0.002, T > 4.587, df = 5; paired t tests. Analysis restricted to self-terminating trials. F, Survival plots of duration of self-terminating discharges with different responsive feedback current injection maximal amplitudes (−50 vs −100 pA: p < 0.0005, T = 0.498; −50 vs −150 pA: p < 3.33e-09, T = 0.755; −100 vs −150 pA: p < 3e-05, T = 0.562; K-S tests). All recordings with CCh and ML297 are shown. Horizontal orange bars indicate times when the responsive feedback system was operational.
Figure 7.
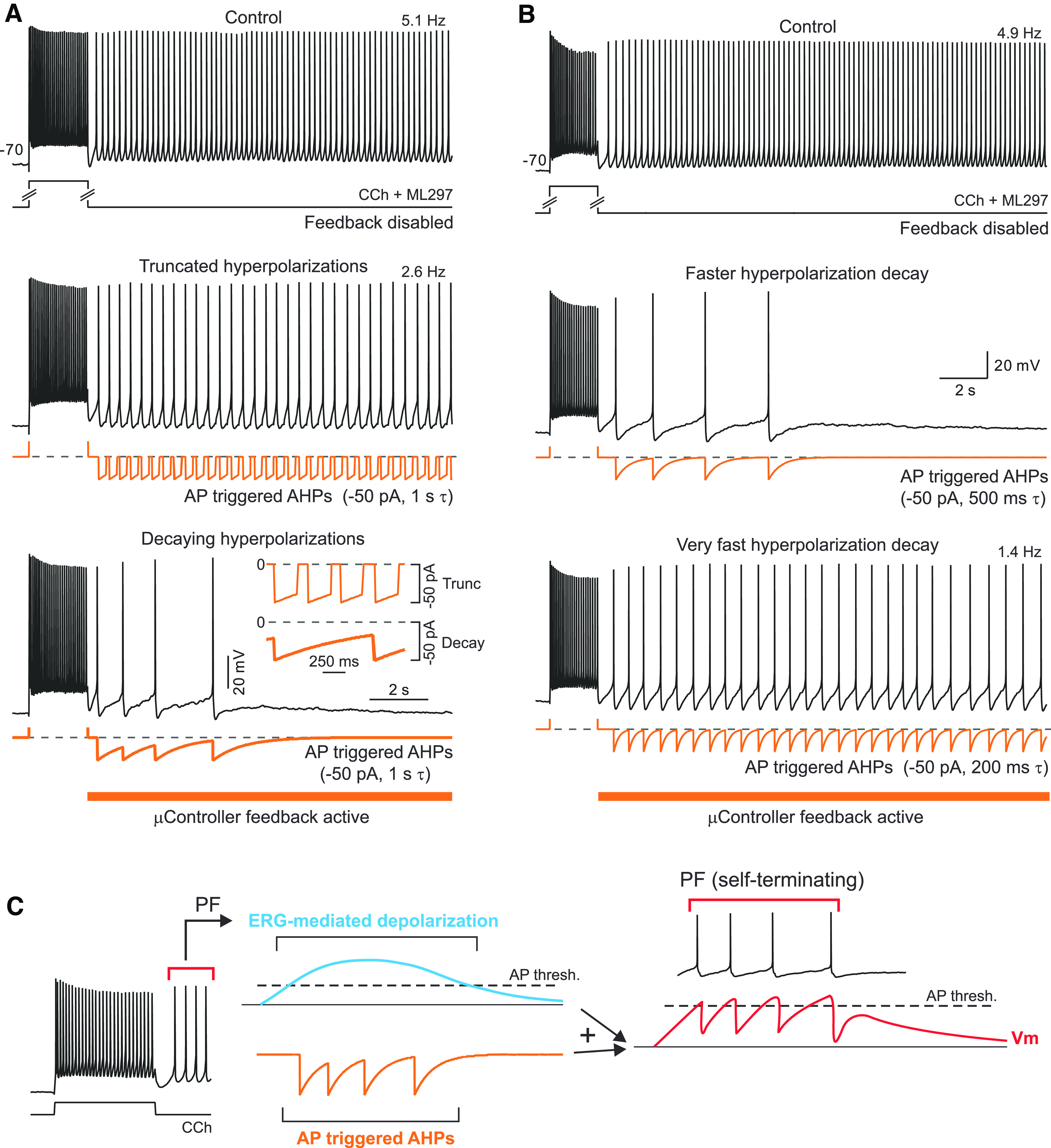
Bistable persistent activity prevented when spike afterhyperpolarizations slow initial discharge frequency. A, Truncating slow-feedback AHP decays (middle panel, τ = 1 s truncated to 0 pA after 250 ms) fails to convert bistable persistent firing into self-terminating discharges. Allowing the same feedback AHP to continue to decay exponentially generated self-terminating responses in the same neuron (bottom panel). Inset, Expansion of microcontroller-generated current waveforms when feedback AHPs were truncated at 250 ms (thin trace) and when feedback AHPs were allowed to exponentially decay until the next AP (thick trace) in the same neuron. B, Persistent discharges spontaneously terminate even when feedback AHP decay kinetics is accelerated to prevent steady-state hyperpolarizing current injection (τ = 500 ms; middle panel, vs 1 s τ used elsewhere in study). Self-termination does require temporal summation of feedback AHPs since injected current decayed to <5% of peak during interspike intervals (all with >3 decay time constants). Accelerating feedback AHP decay kinetics further (to 200 ms τ; bottom panel) generated long-lasting bistable persistent firing. Dashed lines indicate zero microcontroller-injected current level. C, Diagram illustrating hypothesized mechanism of self-terminating persistent discharges. See text for details.
Figure 8.

Spike afterhyperpolarizations regulate persistent firing modes in hippocampal CA1 pyramidal cells. A, Activation of muscarinic receptors with CCh (2 μm) enables a persistent discharge following a step stimulus in a CA1 pyramidal cell (blue trace). B, The same CCh treatment increase failed to trigger persistent firing in a CA3 pyramidal cell. C, Summary plot of probabilities of triggering persistent firing in CCh in CA1 and CA3 hippocampal pyramidal cells and TeA L5 neocortical pyramidal cells. Probabilities of triggering persistence estimated from two different holding potentials (−65 and −70 mV) in each cell type. D, Persistent firing in CA1 pyramidal neurons exposed to CCh is blocked by terfenadine (10 μm; orange trace). E, Attenuating ERG conductance with terfenadine had little effect on the step response in a CA3 pyramidal cell. Inset, Enlargement of the initial phase of the afterhyperpolarization before and after terfenadine treatment. F, Summary plot of change in apparent input resistance of CA1 pyramidal cells during persistent activity using the same methodology as the experiment shown in Figure 2A. Input resistance was estimated in CCh (blue plot) and in response to similar depolarizing step stimuli in control conditions (black plot) and after persistent firing was abolished by terfenadine (orange plot). Magnitude of maximal change in input resistance plotted in inset: *p = 0.013, T = 4.227, df = 4; paired t test. G, Attenuating SK conductances with NS309 (2 μm) converted persistent firing into self-terminating discharges in CA1 pyramidal cells (blue traces). H, Applying microcontroller-based closed-loop feedback to selectively enhance spike afterhyperpolarizations terminated persistent firing in a CA1 pyramidal cell. Hyperpolarizing current transients commanded by the microcontroller shown in orange trace. As with the neocortical neuron experiment in Figure 6B, the closed-loop system only modulated the magnitude of the spike afterhyperpolarization during the persistent firing phase, not during the depolarizing step stimulus.
The DAC output of the microcontroller system (generated by the Analog Shield add-on board) was combined with the DAC output of the ITC18 data acquisition system used to generate the depolarizing current step using a 1× summing voltage amplifier (model FLA-01, Cygnus Technologies). The microcontroller system DAC output scaling was adjusted to generate 50–150 pA current injections applied to the neuron.
The microcontroller system was programmed in C using the standard Ardunio IDE (version 1.8.9) and libraries supporting the uC32 controller and analog shield interface supplied by the manufacturers. Current stimulus waveforms containing randomly timed transient hyperpolarizations (containing the same 2 ms constant period followed by exponential decays) were applied to neurons through the ITC-18 data acquisition interface.
Experimental design and statistical analysis.
Throughout the study, we defined persistent activity as continuous firing beyond the offset of triggering depolarizing stimulus. We refer to persistent firing responses that lasted at least 10 s as “long lasting,” in contrast to persistent discharges that ended after <10 s of firing (termed “self-terminating”). Following the acquisition of episodes containing long-lasting persistent firing, we manually terminated persistent discharges by applying a negative holding current sufficient to suppress firing for at least 60 s. We then slowly removed the added hyperpolarizing current until the initial holding potential was re-established. In most experiments, we coapplied a low concentration of a GIRK channel activator (ML297; 0.67 µm) with CCh. We previously demonstrated (Cui and Strowbridge, 2018) that using ML297 to slightly enhance tonic GIRK currents reduces the propensity of pyramidal cells to spike spontaneously when mAChRs are activated by CCh. Several experiments were conducted without GIRK activators (indicated in figure legends). We also indicate the drug conditions under each example recording trace in the illustrations.
Dynamic estimates of input resistance changes were computed using the same protocol used in our previous studies (Cui and Strowbridge, 2018, 2019). The values reported reflect the difference from the input resistance expected at that particular membrane potential. Calibration experiments were conducted in the same pharmacological conditions (e.g., CCh and ML297), immediately before recording responses to depolarizing stimuli to determine the expected input resistance at different membrane potentials (0.25 mV increments). We applied a DC hyperpolarization to suppress persistent firing in experiments to measure input resistance changes. Trials to assay input resistance were interleaved with trials without added DC hyperpolarization to ensure that the depolarizing stimulus reliably triggered persistent firing.
Unless noted, data were expressed as the mean ± SEM. Significance level with p < 0.05 was used. Multiple comparisons were Bonferroni corrected. Statistical tests were performed in Python and R. We used survival curves (1 – cumulative distributions) to compare the distributions of self-terminating persistent discharges and used the Kolmogorov–Smirnov (K-S) test to determine whether two cumulative probability curves reflected the same distribution. We fit a Boltzmann function () to the plot of probability of persistent firing versus hyperpolarization duration (see Fig. 2B2), where k is the slope of the sigmoid and t1/2 is the corresponding duration that resulted in persistent firing in 50% of trials at that particular pause duration.
Figure 2.
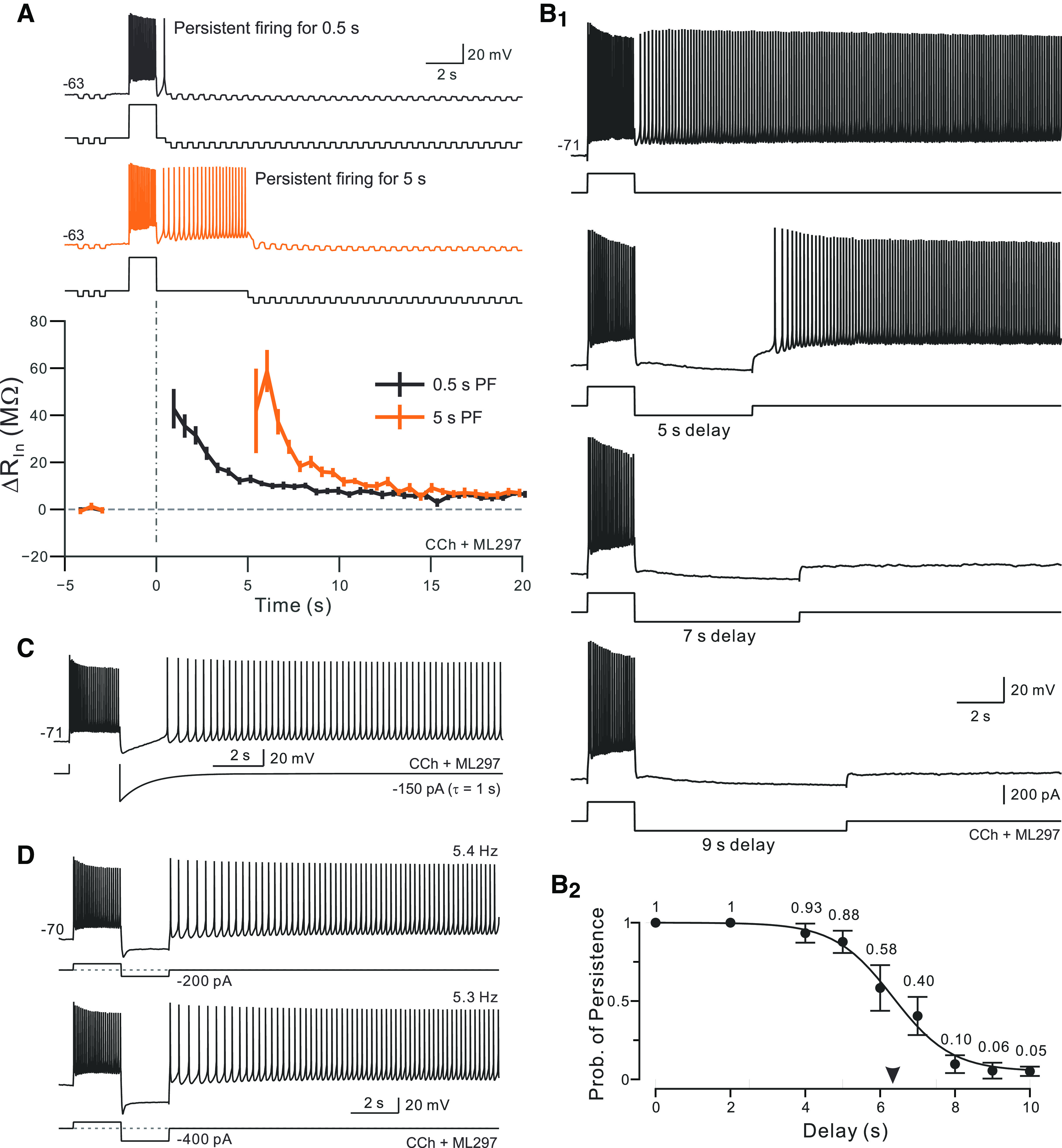
Bistable persistent activity is abolished by long-duration firing pauses. A, Example episode used to estimate change in input resistance associated with the current underlying persistent firing. Continuous hyperpolarization applied 0.5 s following step stimulus offset to prevent persistent firing along with brief hyperpolarizing test pulses were used to create continuous Rin estimates. Without DC hyperpolarization, the same depolarizing stimulus reliably triggered bistable persistent firing (data not shown). Orange trace from same neuron but with a longer initial period of persistent firing before the DC hyperpolarizing current was applied (5 s). Bottom plots mean ± SEM of change in input resistance from expected Rin at that membrane potential from five experiments with alternating trials in which persistent firing was allowed to continue for 0.5 s (black plot) or 5 s (orange plot). Dashed horizontal line indicates no change from expected Rin. B1, Example trials with different duration hyperpolarizing pulses applied immediately following the depolarizing step stimulus. Bistable persistent firing was abolished in trials with 7 and 9 s hyperpolarizing pulses. B2, Summary plot of the probability of bistable persistent firing versus the duration of the hyperpolarizing pulse applied immediately following the step stimulus computed from six experiments. Arrowhead on y-axis indicates a delay required to produce bistable firing on half the trials (6.34 s; slope = −0.78) calculated from Boltzmann function fit (continuous curve). C, Injecting an exponentially decaying hyperpolarizing transient (amplitude, −150 pA; 1 s τ) failed to abolish bistable persistent firing. D, Injecting large-amplitude hyperpolarizing steps (top trace, −200; bottom trace, −400 pA; duration, 2 s for both) also failed to abolished bistable persistent firing. Experiments were conducted using 2 μm CCh without ML297.
Results
The present study builds on results presented in two recent publications from our group (Cui and Strowbridge, 2018, 2019) that support the hypothesis that slow changes in the K+ leak conductance mediated by ERG are responsible for much of the intrinsic hyperexcitability underlying cholinergic persistent firing. The present study extends this work by first asking whether persistent firing is influenced by brief firing interruptions. This experiment is a key test to determine whether persistent firing is maintained by elevated residual Ca2+ from recent preceding spikes or, instead, is maintained by slow changes in ERG conductance triggered by the brief initial intracellular Ca2+ transient. The second part of the present study uses three independent approaches to ask which conductances regulate persistent firing dynamics.
Mechanism for maintenance of persistent firing in neocortical pyramidal cells.
We first tested whether persistent firing (Fig. 1A, left) reflected continuous modulation of an intrinsic current or a state change by applying a weak hyperpolarizing step immediately following the stimulus. If persistent firing is maintained by a current that is continuously sensing intracellular Ca2+, then delaying persistent firing should allow the initial Ca2+ influx to subside, weakening the modulatory change underlying persistent firing (Fig. 1B, top). By contrast, if a depolarizing stimulus triggered a long-lasting state change that modulated intrinsic currents, then interrupting the intracellular Ca2+ stimulus for 4 s should have little effect because the channel modulation already started during the stimulus (Fig. 1B, bottom).
Figure 1.

Bistable persistent firing reflects a neuronal state change. A, Long-lasting persistent firing triggered by a depolarizing step is abolished by the ERG antagonist ErgTx1 (50 nm; red trace). Recording from a L5 TeA neocortical PC in a slice bathed in 2 μm CCh with 0.67 μm GIRK activated ML297 added to help stabilize the membrane potential, as in the study by Cui and Strowbridge (2018). B, Diagrams of two proposed models of underlying cellular mechanism (right top and bottom). See text for details. C, Hyperpolarizing current injection for 4 s delays but does not abolish persistent activity (orange trace at top). Instantaneous firing rates from the same two responses shown below traces. D, Plot of rate-persistent firing frequency increased following stimulus offset in the same trials: p > 0.05, T = 0.045; paired t test. Mean values are indicated at the top, and N values are indicated at the bottom of each bar. E, Increasing the duration of the depolarizing step stimulus does not affect persistent firing frequency. Example responses evoked by 500 and 2 s depolarizing steps recorded from the same neuron and at the same membrane potential. F, Summary plot of average persistent firing frequency evoked by 500 ms and 2 s depolarizing steps in 6 PCs: p > 0.05; paired t test using same cell comparisons. Experiments illustrated in C–F were conducted using 2 μm CCh without ML297.
We adjusted the amplitude of a 4 s hyperpolarizing step in each neuron to be just large enough to transiently suppress persistent firing (typically, −50 pA). As shown in the example experiment illustrated in Figure 1C, 4-s-long firing pauses generated by these hyperpolarizing steps had very little effect on the persistent firing response other than introducing a delay. Pausing firing for 4 s failed to abolish persistent activity in eight of eight experiments. Both the steady-state persistent firing rate and the rate that the instantaneous firing frequency increased as persistent firing began were similar whether or not firing was delayed following the depolarizing stimulus (Fig. 1C, bottom, black and orange scatter plots). Over eight experiments with interleaved control and 4 s hyperpolarizing steps, there were no statistically significant differences in either the steady-state persistent firing rate (Fig. 1D) or firing acceleration time constant (p > 0.05).
These results suggest that the depolarizing stimulus evoked a long-lasting modulation of intrinsic currents (Fig. 1B, bottom, state-change model) rather than a continuous modulation that is prolonged by Ca2+ influx from each spike. We also found further support for the state-change model when we assayed the effects of changing the depolarizing stimulus. Varying the duration of the depolarizing stimulus between 500 ms and 2 s had no effect on the probability of triggering persistent firing (both near 1 in all six experiments) and did not alter the frequency of persistent firing (Fig. 1E,F). Similarly, doubling the amplitude of the depolarizing step or including a second depolarizing step had no effect on the frequency of persistent firing (both p > 0.05). These results suggest that each pyramidal cell generated persistent activity at a relatively constant frequency despite changes in the stimulus. We found more variation in the frequency of persistent firing between different cells than in repeated trials in any one cell. Overall, the average persistent firing frequency spanned between 2 and 12 Hz across 25 L5 pyramidal cells (mean, 4.8 Hz). The coefficient of variation of persistent firing frequency was four times higher when comparing different cells than when comparing repeated trials recorded in the same neuron (p = 0.0001, T = 23.8, df = 24, paired t test). These results are consistent with the hypothesis that the steady-state firing rates achieved are stereotyped on repeated trials in each neuron despite the large variation in persistent firing dynamics across cells.
We next asked whether pausing firing long enough for the transient reduction in ERG K+ current to recover would abolish persistent firing. We first measured the time course of the input resistance increase to estimate the time course of the transient ERG attenuation. As shown in Figure 2A, the increase in input resistance following the stimulus was similar in control trials (in 2 µm CCh and with persistent firing suppressed 500 ms following the stimulus by DC hyperpolarization; black trace) and in interleaved trials where persistent firing was allowed to continue for 5 s (orange trace; maximal increase in input resistance 43.9 MΩ with 0.5 s delay vs 42.5 MΩ with 5 s persistent firing; p > 0.05; T = 0.367, paired t test). We have previously reported that the increase in input resistance following depolarizing step stimuli in CCh reflected a reduction in ERG conductance since it was blocked by ERG antagonists (Cui and Strowbridge, 2018). The rate of input resistance relaxation following the peak elevation also was similar whether or not persistent firing was allowed to continue for 5 s following the stimulus (Fig. 2A, bottom, black and orange plots). The similar transient increases in input resistance in these two conditions suggests that the time course of ERG-mediated hyperexcitability is relatively stereotyped and lasts for ∼10 s.
Over six experiment, we applied a graded series of hyperpolarizing pulses from 2 to 10 s in duration (Fig. 2B1, examples). Long-duration hyperpolarizing pulses reliably blocked persistent firing (in >90% of trials with 8 or 10 s pauses) while hyperpolarizations lasting <6 s rarely blocked persistent firing. Figure 2B2 summarizes these results and predicts that 6.4-s-long hyperpolarizations would abolish persistent firing in 50% of trials (Fig. 2B2, midpoint of the Boltzmann curve fit). The similarity in the time courses of ERG-mediated increases in input resistance (Fig. 2A, bottom) and the sensitivity of persistent activity to long-duration firing pauses (Fig. 2B2) suggests that the short-term mechanisms underlying persistent activity can reverse within 10 s.
The inability of short-duration hyperpolarizing pulses to abolish persistent firing was not related to recruitment of additional IH current. Hyperpolarizing steps were unable to abolish persistent firing even after the IH current was attenuated using ZD7288 (10 µm; tested with 4 s steps). Similarly, blocking firing with an exponentially relaxing current injection immediately following the step offset (τ = 1 s; Fig. 2C) also failed to abolish persistent firing.
In principle, long-duration hyperpolarizing pulses could abolish persistent firing by either accelerating the recovery of inactivated K+ currents or by simply delaying spiking long enough for the modulation of ERG currents to reverse. We tested whether hyperpolarizing steps functioned to deinactivate K+ currents by applying a graded amplitude series of 2-s-duration hyperpolarizing steps immediately after the depolarizing stimulus (Fig. 2D). If long-duration hyperpolarizing pulses were effective because they recovered inactivated K+ currents, we expected to find that large-amplitude 2 s pulses would abolish persistent firing. Instead, 2-s-duration hyperpolarizing pulses never abolished persistent firing even at very large amplitudes (−400 pA; also tested but not shown: −100, −200, and −300 pA). These results suggest that long-duration hyperpolarizing pulses were effective in abolishing persistent firing primarily because they paused firing long enough for the modulation of ERG current to reverse.
The results thus far established that the critical pause duration necessary to disrupt persistent firing (delays of ∼6 s abolish persistent firing in 50% of trials) is similar to the time required for the ERG-mediated increase in input resistance to relax back to near baseline conditions (input resistance reduced to ∼25% of peak elevation in 6 s; Fig. 2A, bottom). We next asked how these relatively slow dynamics related to the time course of intracellular Ca2+ changes in response to similar stimuli. The continuous modulation model (Fig. 1B, top) predicts that some residual intracellular Ca2+ elevation should remain following brief (duration, ∼4 s) firing pauses to allow Ca2+-dependent inward currents to trigger persistent firing. By contrast, if persistent firing is governed by a state change triggered by the depolarizing stimulus (e.g., a slowly relaxing modulation in ERG conductance; Fig. 1B, bottom), intracellular Ca2+ dynamics need not be coupled to changes in intrinsic physiology.
We used Ca2+ photometry to measure somatic Ca2+ transients following depolarizing stimuli and to determine whether 4 s firing pauses were long enough to dissipate the initial Ca2+ transient evoked by the stimulus. Wide-field photometric measurements from a single Cal520-filled neuron enabled high resolution measurements of both spike-evoked Ca2+ transients (Fig. 3A) and the kinetics of intracellular Ca2+ decay following depolarizing stimuli. In both control conditions (without CCh) and in 2 µm CCh, the Ca2+ transient evoked by the depolarizing stimulus decayed monotonically, reaching basal levels within 3 s (Fig. 3B; persistent firing prevented by setting the initial membrane potential to −75 mV). The Ca2+ indicator decay time constant following the depolarizing step response was 740 84.8 ms (N = 4), approximately four times slower than the decay time constant following single isolated spikes (207 ± 24 ms; N = 4). Figure 3C summarizes the Ca2+ dynamics following standard step stimuli in control conditions and in CCh. These results demonstrated that the 4 s firing pauses used in Figure 1, C and D, were long enough to dissipate the somatic Ca2+ accumulation triggered by the depolarizing stimulus. The resumption of persistent firing following the pause was, therefore, unlikely to be triggered by elevated intracellular Ca2+ as postulated in the continuous modulation model shown in Figure 1B (top).
Figure 3.

Dynamics of intracellular calcium transients evoked by depolarizing steps. A, Example photometric recording of Ca-related epifluorescence in a TeA L5 PC filled with the calcium indicator dye Cal520. Top trace shows fluorescent transients associated with each AP evoked by the depolarizing step. B, Example photometric recording in a different PC over a longer-sweep window reveals rapid decay kinetics of the Cal520 dye signal in the cell body region. Intracellular Cal520 transients following depolarizing step stimuli are eliminated after 3 s. Decay responses were similar in ACSF and CCh conditions (example response recorded in CCh). C, Summary plot of Cal520 fluorescence assayed photometrically from the soma at different time points following depolarizing step offset. Fluorescence levels normalized to peak response immediately following step stimulus in each experiment. Separate experiments assayed the rate of Ca2+ transient decay in control conditions (gray bars) and in 2 μm CCh (black bars). Initial membrane potential set to −75 mV to prevent persistent firing.
Spike afterhyperpolarizations regulate persistent firing duration
We identified three experimental manipulations that revealed a latent capacity for persistent firing to terminate after <10 s in neocortical pyramidal cells. All three mechanisms we identified appeared to function by amplifying the endogenous hyperpolarizations that followed each AP within the persistent discharge. This similarity suggests that the modulation of spike afterhyperpolarizations may be a general mechanism for regulating the duration of persistent discharges.
The first manipulation we found that enabled self-termination was treatment with the positive SK channel modulator NS309. Applying NS309 in control conditions decreased the number of APs evoked by the depolarizing current steps in neurons held at a constant membrane potential (Fig. 4A), suggesting that L5 pyramidal cells express SK currents that are recruited by spiking discharges. Over seven experiments, 5 µm NS309 reduced the average firing rate during the depolarizing stimulus from 16.0 to 4.8 Hz (p = 0.0001; T = 15.6, df = 6; N = 5, paired t test). The effects of NS309 appeared to be specific to SK channels as we found the opposite change with NS8593, a negative SK channel modulator (NS8593 increased firing frequency during depolarizing steps in seven of seven cells tested). Enhancing SK-mediated K+ currents with NS309 increased spike AHP amplitude (Fig. 4B,C) and slowed the AHP decay kinetics from 162 ± 63 to 276 ± 67 ms (p = 0.005, T = 4.93, df = 5; N = 6; paired t test), suggesting that firing slowing occurred because of larger and slower spike AHPs. We found a similar increase in spike AHP amplitude when NS309 was tested in slices exposed to 2 µm CCh (from −0.91 0.77 to −4.65 0.97 mV; p = 0.02, T = 3.355, df = 6; N = 7; paired t test), demonstrating the activation of muscarinic receptors with CCh did not occlude SK modulation by NS309. The SK negative modulator NS8593 had the opposite behavior and reduced, rather than increased, spike AHP amplitude (data not shown; N = 6).
Figure 4.

Pharmacologically enhancing SK currents converts bistable persistent firing into self-terminating discharges. A, Enhancing SK currents with NS309 (blue trace) decreased the step-evoked firing rate from control conditions (black). B, Enhancing SK currents with NS309 also increased spike AHP amplitude and slowed AHP kinetics. Horizontal dashed line at membrane potential before depolarizing pulse stimulus that triggered the AP. Action potentials were truncated. C, Summary of spike AHP amplitude before (Ctrl) and after NS309. **p = 0.016, T = 3.58, df = 5; paired t test. D, Low concentrations of NS309 (0.5–1 μm) convert bistable persistent firing responses into self-terminating discharges. Example self-terminating discharge responses recorded with 0.5 μm (middle panel, same cell as control trace) and 1 μm NS309 (different cell). Stimulus amplitude increased in NS309 traces to compensate for reduction in step-evoked firing frequency. E, Summary of persistent firing response dynamics in control experiments without NS309 and following bath application of 0.5 or 1 μm NS309 (middle and right bars, respectively). Response types categorized as either bistable (persistent firing for >10 s following stimulus offset; black), discharges that spontaneously terminated within 10 s (blue), or trials with no spiking following stimulus (teal). F, Summary of the duration of self-terminating discharges triggered in either 0.5 μm (thin line; N = 29 trials) or 1 μm NS309 (thick line; N = 22 trials). The two distributions are statistically different (p < 0.015, d = 0.357; K-S test). G, Ability of NS309 to promote self-terminating discharges is reversed by the SK channel blocker UCL1684 (1 μm; bottom trace). Blue traces indicate responses recorded with 0.5 μm NS309 present. H, The SK modulator NS309 promotes self-terminating firing in ACSF containing 1.3 mm Ca2+. Control and NS309 conditions include 4 μm CCh.
Enhancing SK currents using NS309 converted bistable persistent firing into self-terminating discharges (Fig. 4D). The average duration of self-terminating trials varied with the concentration of NS309 applied (5.25 ± 1.1 s in 0.5 µm vs 3.16 ± 0.33 s in 1 µm NS309; N = 7). As shown in Figure 4E, the proportion of self-terminating discharges also increased with higher concentrations of NS309 as did the fraction of trials with no persistent firing (all cells showed reliable bistable persistent firing before testing NS309). When self-terminating discharges occurred in NS309, they always lasted <10 s (Fig. 4F; survival curves significantly different in 0.5 and 1 µm NS309). The absence of very long-duration self-terminating discharges (>10 s) suggests that a similar ERG-related mechanism governed persistent firing dynamics in these experiments since ERG modulation appears to reverse within 10 s in the absence of sustained persistent firing. Presumably, the relatively fast kinetics of m1 receptor-mediated ERG modulation sets an upper limit on the duration of self-terminating persistent discharges.
While NS309 is commonly used to activate SK channels (Pedarzani et al., 2005; Nam et al., 2017; Sun et al., 2020), this compound also modulates intermediate conductance Ca2+-activated K+ channels (Hougaard et al., 2007). We used the SK channel-selective blocker UCL1684 (Hilgers and Webb, 2007) to test whether the ability of NS309 to promote self-terminating firing depended on modulating SK channels. In three of three experiments in which NS309 application converted long-lasting persistent firing into self-terminating discharges, the subsequent addition of 1 µm UCL1684 reverted the step responses back to bistable firing. An example of the effect of UCL1684 reversing self-terminating firing is shown in Figure 4G. These results suggest that the modulation of SK channels regulates persistent firing dynamics.
We also verified that NS309 promoted self-terminating firing in ACSF containing a lower Ca2+ concentration (1.3 mm), similar to the Ca2+ concentration found in CSF. We tested 0.5 µm NS309 in 5 L5 pyramidal cells that showed long-lasting persistent firing in 4 µm CCh. Activating SK channels with NS309 converted bistable responses into self-terminating discharges in three of five experiments (mean discharge duration, 3.8 1.9 s; N = 3; Fig. 4H). In one experiment, NS309 completely abolished persistent firing and in another experiment it failed to abolish bistable firing.
By enhancing spike AHPs, the SK positive modulator NS309 likely slowed persistent firing sufficiently to prevent triggering long-term mechanisms that reinforce persistent activity, leaving transient ERG attenuation as the primary persistence-generating mechanism. The ability of SK modulators to slow and, in most experiments, stop persistent firing was limited to NS309. Over seven experiments, testing the negative SK modulator NS8593 (10 µm) under the same conditions, persistent firing frequency increased and persistent discharges never spontaneously terminated in <10 s (zero of seven cells tested). We also tested the M current activator ICA110381 (Boehlen et al., (2013), used at 10 µm) in four experiments. In four of four recordings, activating M current with ICA110381 abolished persistent firing without revealing self-terminating discharges.
Complementary experiments also suggest that NS309 converted bistable persistence into self-terminating discharges by enhancing spike AHPs during the poststimulus period. Increasing extracellular Ca2+ from 2.5 to 4 mm is another treatment that would be expected to enhance SK currents, albeit indirectly by increasing transmembrane Ca2+ currents. While this experimental manipulation did not lead to self-terminating discharges similar to responses that were similar to what we observed in NS309, persistent firing frequency slowed in all five cells tested with 4 mm Ca2+ to 76% of the control rate (p = 0.037; T = 3.087, df = 4; paired t test; Fig. 5A). The same manipulation reduced the number of APs triggered during the depolarizing step (p = 0.034, T = 3.160, df = 4; paired t test) and increased the amplitude of the spike AHP (Fig. 5B). These results suggest that the net effect of Ca2+ currents may be to reduce excitability via activation of Ca2+-activated K+ currents like SK, as shown previously in Purkinje neurons (Swensen and Bean, 2003).
Figure 5.

Reduction in afterhyperpolarization amplitude with elevated extracellular calcium. A, Increasing extracellular Ca concentration from 2.5 to 4 mm (orange trace) slows persistent firing frequency. B, Plot of step AHP amplitude in standard (2.5 mm) and high (4 mm) iCa2+ concentration conditions. **p = 0.006, T = 7.358, df = 4; paired t test.
A central problem in testing the hypothesis that spike AHPs regulate persistent firing dynamics is that the manipulations tested thus far affected afterpotentials triggered by all APs—those evoked during the step stimulus and during the persistent discharge. While we tried to disambiguate the effects on the stimulus and the persistent firing response (e.g., by adjusting the step amplitude to normalize the number of APs evoked), it is difficult with global manipulations to isolate the key functional effect to one component of the response. To circumvent this problem, we created a microcontroller/neuron interface system that enabled a small computer (an Ardunio microcontroller) to detect each spike during the persistent discharge and then inject an exponentially decaying hyperpolarizing current transient into the neuron (Fig. 6A; for details, see Materials and Methods).
Our approach was inspired by related work using closed-loop feedback systems (Sharp et al., 1993; Bettencourt et al., 2008), including a recent report that used a similar microcontroller to create a dynamic clamp system (Desai et al., 2017). We could not use traditional dynamic clamp system in this experiment since the goal was to mimic the functional effect of Ca2+-gated (not voltage-gated) K+ currents and because we wanted to limit the feedback-generated spike AHPs to the persistent discharge, leaving the spikes directly evoked by step stimulus unaffected. In our experiments, the reactive feedback system generated predefined hyperpolarizing current transients that were delayed by 2 ms from the triggering event on the AP rising phase. This timing enabled the outward current waveform generated by the microcontroller to temporally overlap with (and augment) the endogenous spike AHP.
Enhancing the spike AHP during the persistent firing phase with AP-triggered current injections converted bistable persistent activity into self-terminating persistent firing. In 10 of 10 experiments using the microcontroller-based feedback system, we could reliably trigger self-terminating persistent discharges in the same neuron when the feedback spike AHP system was enabled (50–150 pA hyperpolarizing transients injected that decayed with 1 s time constants). Moreover, the duration of self-terminating persistent firing was graded with the amplitude of the feedback spike AHP current (Fig. 6B). In the example recording shown in Figure 6B, a single precisely timed 150 pA hyperpolarizing current injection abolished persistent firing following the initial poststep spike. As shown above (Fig. 2C), the same hyperpolarizing current waveform injected immediately following the step stimulus (instead of following the first poststimulus spike) failed to block persistent firing. In all experiments with self-terminating discharges, disabling the microcontroller feedback reverted the response to long-lasting bistable firing (Fig. 6B,C, top traces).
These initial results suggest that the timing of the externally generated hyperpolarizations was critical for controlling bistable persistent firing. We tested this hypothesis by constructing three different “frozen” current waveforms with a series of randomly timed hyperpolarizations. Each hyperpolarizing transient included in the random stimulus pattern matched the amplitude and kinetics of the feedback current waveforms that effectively terminated bistable persistent firing when applied immediately following each persistent spike (50 pA, 1 s decay time constants). In six experiments, all three random stimulus patterns failed to abolish bistable persistent firing, although they created long (>1 s) pauses in the poststep discharge. In six of six experiments, we could alternate between all three experimental conditions (no hyperpolarizations, AP-linked hyperpolarizations, or randomly timed hyperpolarizations) in the same neuron and demonstrate that only spike-triggered hyperpolarizations terminated persistent firing (Fig. 6C). The average probability of terminating bistable persistent firing using random and spike-linked external AHPs in six experiments is summarized in Figure 6D. The ineffectiveness of randomly timed hyperpolarizations was not because of presenting weaker inputs: the number of hyperpolarizations presented in all random waveform trials was greater than in precisely timed trials, and the overall hyperpolarizing charge injected in random trials also was greater than in AP-linked trials. Also supporting the pivotal role of spike-timed AHPs, we found that injecting one precisely timed large-amplitude hyperpolarizing transient could stop persistent discharges after the first spike (Fig. 6B, bottom trace) while injecting the same inhibitory transient immediately following the step stimulus did not abolish long-lasting firing (Fig. 2C).
On average, the duration of persistent firing varied from ∼1–7 s as the amplitude of spike-linked feedback AHPs was increased from 50–150 pA (Fig. 6E; N = 6 experiments). As shown in the cumulative plots in Figure 6F, grading the amplitude of feedback AHPs resulted in different dynamics with persistent firing up to ∼7.5 s in most trials with low-amplitude (−50 pA) external spike AHPs while moderately large spike AHPs (−100 pA) evoked a near linear decrease in firing probability throughout the initial 8 s poststep period.
Unlike highly precise spike-timing-dependent mechanisms of long-term plasticity (Markram et al., 1997), the timing requirement for feedback AHPs was looser as both 2 and 100 ms AP-to-AHP latencies were effective in converting long-lasting persistent firing into self-terminating discharges (N = 4 in each condition). We could not test negative AP-to-AHP latencies since our responsive feedback system was triggered by spontaneous APs. Self-terminating persistent discharges recorded using the external feedback system appeared to rely on the same ERG modulation mechanism previously demonstrated for long-lasting bistable persistent firing (Cui and Strowbridge, 2019). Bath application of ERG blocker terfenadine (10 µm) blocked self-terminating discharges acquired when the feedback system was operational (data not shown). By contrast, attenuating SK currents with NS8593 increased the firing rate during the depolarizing step response but failed to block persistent discharges whether or not the feedback AHP system was operational (N = 3). The ability of the responsive feedback system to terminate persistent firing even in NS8593 provides evidence suggesting that AP-linked hyperpolarizing current transients were not functioning to enhance SK current activation via increasing Ca2+ tail currents through voltage-dependent Ca2+ channels (VDCCs).
Together, these results suggest that the feedback AHPs enable self-terminating discharges by generating slow hyperpolarizations that counteract the ERG-mediated afterdepolarization. The combination of the slow depolarization and enhanced spike-triggered AHPs divides the time when the net depolarization is above firing threshold into discrete temporal windows. We first tested this model by modifying the feedback microcontroller to truncate each AP-linked AHP current injection after it had decayed for 250 ms. This manipulation tests whether the most important component of the feedback AHP is the initial fast transient or the slow decay phase. Aborting the prolonged exponential AHP decay after 250 ms prevented feedback AHPs from terminating persistent discharges (Fig. 7A; five of five experiments). In the same recording, interleaved trials in which feedback AHP that decayed continuously until the next spontaneous AP occurred generated self-terminating discharges (Fig. 7A, bottom trace). These results demonstrate that the critical phase of the AHP required to terminate persistent discharges was not the initial hyperpolarization but, instead, the slow, prolonged decay of the AHP response. This result provides additional evidence that feedback AHPs did not function to enhance VDCC tail currents (and thereby recruit SK-mediated endogenous AHPs). Rather, feedback AHPs provided a prolonged source of hyperpolarization that helped dampen excitability after each AP.
Feedback AHPs triggered during self-terminating discharges typically overlapped (Fig. 7A, inset), leading to a sustained DC hyperpolarization. However, continuous hyperpolarization was not required for persistent discharges to terminate since accelerating AHP decay kinetics (by reducing the exponential decay time constant from 1 s to 500 ms) still produced self-terminating discharges without generating AHP summation (Fig. 7B). In the example shown in Figure 7B, feedback AHPs evoked by the second and subsequent spikes decayed almost completely (to <5% of the initial current injected) during the interspike intervals. Over five experiments, we found no difference in the mean duration of self-terminating discharges (all between 4.0 and 4.3 s; p > 0.05) or the distribution of persistent firing durations (all p > 0.05, d = 0.29–0.313 for the three comparisons; K-S tests) when feedback AHP decay kinetics were varied among 1 s, 750 ms, and 500 ms. All three decay kinetics were effective in converting bistable firing into self-terminating discharges (in five of five cells tested) with the fastest decays (500 ms τ) generating almost no temporal summation of injected hyperpolarizing current transients. Further accelerating feedback AHPs to 200 ms decay τ resulted in bistable firing (Fig. 7B, bottom trace), though at a slower frequency than in control conditions without the feedback-augmented AHPs.
These results suggest that the magnitude of spike AHPs regulates persistent firing dynamics. The primary stimulus for persistent firing during the initial 10 s period appears to be a temporary negative modulation of the leak K+ current mediated by ERG (Cui and Strowbridge, 2018, 2019; Fig. 7C). This effective inward current (a reduced steady-state outward current) is partially offset by brief hyperpolarizations generated by spike AHPs (Fig. 7C, orange trace), slowing the persistent firing rate and likely preventing the triggering of long-term mechanisms supporting bistable firing modes. Persistent firing frequency can be reduced further when spike AHPs are enhanced either pharmacologically by applying a positive SK modulator or externally using the microcontroller feedback system. One consequence of this “AHP sculpting” mechanism is that the timing of individual APs within the persistent discharge can be constrained within relatively narrow windows determined by when the AHP generated by preceding spike has subsided sufficiently to allow the net current (ERG-mediated depolarization plus spike AHP) to become inward.
Persistent firing dynamics in hippocampal pyramidal neurons
Finally, we asked whether ERG also regulates intrinsic persistent firing in neurons in other brain areas. We focused on CA1 and CA3 pyramidal cells, two well studied excitatory cell types in the hippocampus. Bath application of CCh increases neuronal excitability in both cell types. The number of spikes evoked by our standard depolarizing step stimulus increased 1.8-fold in CCh in CA1 PCs and 2.1-fold in CA3 PCs (p < 0.005, T > 4.46 in both comparisons; t test). However, in the same 2 µm concentration of CCh, depolarizing steps only triggered persistent firing in CA1 PCs (in 17 of 18 experiments; Fig. 8A) and never in CA3 PC recordings at either membrane potential (0 of 7 experiments; Fig. 8B). The ability of depolarizing step stimuli to trigger persistent activity in three different pyramidal cell types is summarized in Figure 8C. Once triggered, the persistent firing frequency was higher in CA1 neurons than in neocortical neurons by 1.4-fold (p = 0.015, T = 2.71, df = 35, N = 25 TeA neocortical cells and 12 CA1 pyramidal cells; t test), suggesting that persistent firing is robust in CA1 neurons even at the low CCh concentration used in this study.
We next asked whether ERG modulation underlies persistent firing in CA1 pyramidal cells as it does in L5 neocortical pyramidal cells (Cui and Strowbridge, 2018, 2019). In the hippocampus, ERG1 expression varies sharply in different subregions. Pyramidal cells in CA1 strongly express ERG1, while ERG markers are low in neighboring CA3 pyramidal cells (Saganich et al., 2001; Papa et al., 2003). The ERG1 expression difference paralleled the sharp differences we observed in the ability of depolarizing step stimuli to trigger persistent firing in CCh, consistent with the hypothesis that ERG modulation underlies intrinsic persistent firing in CA1 neurons but not in CA3 pyramidal cells.
We tested whether terfenadine affected three different intrinsic physiological properties of CA1 and CA3 neurons to determine whether the differential ERG expression result reported in previous publications affects neuronal function. Attenuating ERG conductance with terfenadine increased the number of spikes triggered by our standard depolarizing step stimulus by 1.7-fold in CA1 pyramidal cells but had no effect in CA3 neurons (<3% change, p > 0.05, paired t test). Similarly, terfenadine increased input resistance only in CA1 neurons (83.4–107 MΩ; p = 0.0081, T = 4.90, df = 4, N = 5) and not in CA3 pyramidal cells (<2 MΩ change, p = 0.91, T = 0.125, N = 5; paired t tests in both). And finally, terfenadine abolished the afterhyperpolarization that normally followed the step response in CA1 neurons, revealing a small afterdepolarization (poststep response amplitude shifted from −2.4 to 0.5 mV, p = 0.040, T = 2.99, df = 4, N = 5; paired t test). The same treatment had no effect on the poststep response in CA3 pyramidal cells (response amplitude, −1.7 vs −1.6 mV; p = 0.94, T = 0.084, N = 5; paired t test).
The differential responsiveness to terfenadine in CA1 and CA3 we find in control conditions (without using CCh to activate muscarinic receptors) suggests that ERG contributes to the resting conductance in CA1 but not in CA3 neurons, consistent with differences in published ERG1 expression markers across hippocampal subfields (Saganich et al., 2001). Regional differences in ERG expression also likely underlie the differential ability of low concentrations of CCh to increase neuronal excitability in CA1 pyramidal cells but not in CA3 neurons. [Persistent firing in CA3 pyramidal cells can occur with higher concentrations of CCh; 10 µm CCh was used in studies by Jochems and Yoshida (2013, 2015).]
If the primary target of muscarinic receptor modulation in our hippocampal experiments is ERG, then the absence of persistent firing in CA3 neurons could be explained by the relatively low levels of ERG1 expression in these neurons. Consistent with the hypothesis that ERG modulation underlies intrinsic persistence in the hippocampus, we find that persistent firing was abolished by terfenadine in four of four CA1 pyramidal cells tested (Fig. 8D), paralleling our results in L5 neocortical pyramidal cells that express ERG (Cui and Strowbridge, 2018, 2019). Terfenadine has little effect on responses to depolarizing steps in CA3 neurons treated with CCh (Fig. 8E), including the poststep response (Fig. 8E, inset). In CA1 neurons, input resistance increased immediately following depolarizing stimuli presented in CCh and then recovered with a slow time constant (3.2 0.16 s, N = 5; Fig. 8F). Attenuating ERG conductance with terfenadine abolished the transient increase in input resistance following depolarizing stimuli in CA1 neurons (Fig. 8F, inset), consistent with the hypothesis that ERG modulation underlies the persistent firing mode in our CA1 experiments.
In neocortical neurons, we find that the duration of poststimulus firing is regulated by the amplitude of spike afterpolarizations with large-amplitude AHPs converting long-lasting persisting firing into self-terminating discharges (Figs. 4–7; see above). Our results suggest that a similar mechanism operates in CA1 pyramidal cells. Spike AHPs evoked by the depolarizing step were increased by the positive SK channel modulator NS309 (from 4.8 1.4 to −2.2 1.3 mV; p = 0.007, T = 5.204, df = 4, N = 5 CA1 neurons; paired t test. Enhancing spike AHPs by applying NS309 significantly reduced the initial persistent firing frequency from 5.4 1.1 to 2.4 0.7 Hz; p = 0.005, T = 6.02, df = 4, N = 5; paired t test). In five of five CA1 cells tested, 2 µm NS309 converted long-lasting persisting firing into self-terminating discharges such as the example episodes shown in Figure 8G. All self-terminating discharges terminated in <10 s following the offset of the depolarizing stimulus, suggesting that ERG-mediated hyperexcitability subsides within that ∼10 s time frame in both neocortical and hippocampal CA1 neurons (Fig. 8F). Finally, we tested whether enhancing only the poststimulus spike AHPs using closed-loop feedback also revealed self-terminating persistent firing modes in CA1 pyramidal cells. This approach was successful in five of five experiments including the example shown in Figure 8H and demonstrate that persistent firing duration can be regulated by spike AHP amplitude in both neocortical and hippocampal CA1 neurons.
Discussion
We make three principal conclusions in this study. First, we extend previous work on ERG-mediated mechanisms of persistent firing (Cui and Strowbridge, 2018, 2019) by demonstrating that the slow kinetics of ERG modulation in neocortical pyramidal cells enables persistent firing to continue after pauses of up to ∼8 s. ERG-mediated hyperexcitability outlasts transient increases in intracellular Ca2+ concentration, making this mechanism of intrinsic persistent activity more robust to perturbations than most previously studied persistence mechanisms, including bistability associated with high expression of low-threshold voltage-gated Ca2+ channels (Williams et al., 1997) and classical synfire chain networks.
The slow kinetics of ERG-mediated hyperexcitability that follows depolarizing stimuli likely functions to reinforce and stabilize behaviorally relevant network states defined by patterns of synaptic connections (Koulakov et al., 2002; Goldman et al., 2003). The combination of slow intrinsic plasticity mediated by ERG channels and recurrent synaptic connections may be especially effective in creating self-sustaining network states because cholinergic modulation of ERG selectively amplifies the late phase of neuronal discharges without altering the initial response (Cui and Strowbridge, 2019). The ability of ERG plasticity to selectively affect only a small subset of neurons—those with the largest and longest responses to a stimulus—likely enables intrinsic plasticity to coexist within recurrent networks without disrupting the precise collection of synaptic connection strengths necessary for generating persistently active network states (Seung, 1996).
The present study also defines a novel biophysical mechanism that regulates the duration of persistent discharges: modulating the amplitude of spike afterhyperpolarizations. A central question regarding most in vitro approaches to define persistent activity mechanisms has been the behavioral relevance of the extremely long duration of tonic discharges often reported, typically lasting >20 s. Our results provide one of the first demonstrations of self-terminating discharges triggered by transient stimuli in acute brain slices using standard whole-cell patch-clamp recordings. We found variable duration epochs of persistent spiking using two independent experimental approaches: using a SK channel positive modulator to augment all spike AHPs; and using a closed-loop negative feedback system to selectively amplify only AHPs associated with persistent spikes. This mechanism for regulating persistence also explains our counterintuitive finding that increasing extracellular calcium concentration slows, rather than increases, the frequency of persistent firing. Presumably, in the neocortical neurons we studied, changes in intracellular Ca2+ concentration enhance currents mediated by SK channels, increasing the magnitude of spike AHPs.
And finally, we demonstrate that ERG-based intrinsic plasticity is not restricted to neocortical neurons since the blockade of ERG channels with terfenadine abolishes persistent firing in hippocampal CA1 pyramidal cells. In the hippocampus, we find the ability of cholinergic stimulation to promote intrinsic persistent firing modes follows previously established patterns of ERG1 expression (relatively high in CA1 pyramidal cells and low in CA3 neurons; Saganich et al., 2001; Papa et al., 2003). Our work is also consistent with previous studies demonstrating that ERG contributes to regulating CA1 neuron excitability (Fano et al., 2012; Xiao et al., 2018; Carver and Shapiro, 2019). Persistent firing in CA1 neurons is mediated by conductance changes with slow kinetics, similar to the transient modulation of ERG current following depolarizing stimuli we demonstrated previously in neocortical neurons (Cui and Strowbridge, 2018, 2019) and is regulated by the same spike AHP mechanism.
Mechanism and phases of ERG-mediated persistent firing
Defining the principal mechanisms that mediate intrinsic persistent firing in cortical neurons has been a challenging problem, in part, because of uncertainty about the role of intracellular Ca2+ transients. Most investigators who approached this problem found filling pyramidal cells with Ca2+ chelators, such as EGTA or BAPTA, abolished persistent firing. But that result did not discriminate between a requirement for an initial Ca2+ transient to initiate a modulatory cascade and the subsequent Ca2+ transients driven by APs during the persistent discharge. Our results suggest that only the initial Ca2+ transient is required since the stimulus-triggered intracellular Ca2+ transient dissipated back to basal levels during 4 s firing pauses. Since persistent firing typically resumes following 4 s pauses, a latent increase in excitability likely remains to restart persistent firing even after intracellular Ca2+ levels have returned to basal levels. While the wide-field photometric measurements we used are dominated by somatic fluorescent changes, this measurement should reveal the slowest Ca2+ indicator kinetics since our work using more spatially localized two-photon photometry indicated that Cal520 transients decayed faster in dendritic compartments than in the cell body region in a variety of neuron types (R. Pressler and B. Strowbridge, unpublished observations).
Our results are consistent with prior work that demonstrated past intrinsic persistent firing in entorhinal neurons could continue past ∼4 s interruptions (Fransén et al., 2006). These authors proposed that channels mediating calcium-activated nonselective cation (Ican) currents could be modulated into a persistently active form, leading to a long-lasting source of depoarization. While the molecular identity, and modulatory mechanisms, of channels that mediate Ican remain uncertain, likely candidates are transient receptor potential cation (TRPC) channels (Yan et al., 2009; Zhang et al., 2011). However, the association between TRPC channels and intrinsic persistent activity in cortical neurons was challenged by recent molecular studies that demonstrated persistent firing even after genetic deletion of different combinations of TRPC channels (Dasari et al., 2013; Egorov et al., 2019). In addition, our findings that ∼4 s pauses are long enough to allow the stimulus-evoked intracellular Ca2+ transient to subside does not support the simpler Ican model in which firing is maintained by periodic influxes of Ca2+ associated with persistent spiking.
Our results following the transient changes in input resistance and testing of ERG antagonists both suggest that a transient attenuation of ERG conductance contributes to this latent hyperexcitability. However, pyramidal cells do not immediately begin to fire at their fastest tonic rate. Instead, the persistent firing rate typically ramps up over the initial 10 s. A similar ramp up in firing rates is often observed in vivo where neocortical neurons active during the delay period of WM tasks often do not reach steady-state firing rates until several seconds following the stimulus (>5 s: Fuster and Alexander, 1971; Fuster and Jervey, 1981; >1–3 s: Funahashi et al., 1989; Takeda and Funahashi, 2002). In our experiments, this gradual increase in firing frequency appears to reflect the interplay between the slow development of ERG-mediated hyperexcitability and the poststimulus AHP. The depolarizing stimuli likely triggers a variety of K+ currents that contribute to the initial AHP, including SK channels, as indicated by the reduced latency to persistent firing following attenuation of SK channels using NS8593. Presumably, these K+ currents are recruited maximally immediately following the stimulus, contributing to the initial slow rate of persistent firing observed in both in vitro studies (Yoshida and Hasselmo, 2009; Rahman and Berger, 2011; Zhang et al., 2011; Jochems and Yoshida, 2013) and in the in vivo studies cited above.
In vivo, persistent firing frequency is often graded with stimulus type or amplitude. Integrator neurons within the vestibulo-ocular reflex circuit that regulate eye position, for example, are capable of firing persistently over a wide frequency span (Dalezios et al., 1998; Major et al., 2004). By contrast, most previous in vitro studies of intrinsic persistent firing that used whole-cell recording found a bistable, all-or-none firing mode that was unaffected by changes in stimulus amplitude or duration (Yoshida and Hasselmo, 2009; Rahman and Berger, 2011; Zhang et al., 2011; Knauer et al., 2013). Our results are consistent with these prior reports and found no significant changes in persistent firing rates when we varied the triggering stimulus under control conditions. Persistent firing frequency, however, was altered by relatively small additional hyperpolarizations timed to coincide with endogenous spike AHPs (Fig. 6B). A similar conversion from bistable to graded persistent firing occurred when we enhanced SK currents using NS309 (Fig. 4), suggesting that intracellular dialysis associated with whole-cell recordings likely attenuated endogenous spike AHPs and thereby promoted bistable firing. These results may also help explain why the relatively few studies that found graded persistent firing used sharp electrode recordings (Schwindt et al., 1988; Egorov et al., 2002, 2006; Reboreda et al., 2007) which generate less intracellular dialysis than the whole-cell recording configuration. The greater variety of persistent firing modes found using sharp electrodes could reflect larger amplitude spike AHP responses as well as potentially different intracellular Ca2+ transients during the triggering stimulus.
During WM tasks, delay period activity was typically terminated when the subject made an eye saccade response (Funahashi et al., 1989; Takeda and Funahashi, 2002). Before the response movement, persistent firing recorded in delay neurons was rarely tonic, including during the ramp-up phase. Although not often highlighted, multiple studies found delay cell firing frequency decreased gradually from the peak rate until the response movement (Fuster and Alexander (1971; Funahashi et al., 1989; Young et al., 1997; Zhou et al., 2011) or even terminated before the response (Oshio et al., 2006; Watanabe and Funahashi, 2007, 2014; Liu et al., 2014).
Replicating these self-terminating forms of persistent activity under in vitro conditions has proved challenging, especially in studies using whole-cell recordings. In vivo, termination may occur because of strong inhibitory input or depression of recurrent excitatory synaptic responses. However, the present study demonstrates that spontaneous termination of intrinsic persistent firing does not require GABAergic or glutamatergic input. Instead, our results suggest that both graded-frequency persistent firing and self-termination can reflect the interplay between ERG and SK conductances. Both of these common features of delay cell responses were revealed in our brain slice recordings when we enhanced spike AHPs using a pharmacological modulator or a reactive feedback system. Our experiments also revealed a timing requirement for effective feedback AHPs that is reminiscent of spike timing-dependent plasticity (Markram et al., 1997), though the coincidence requirement between the APs and feedback AHPs extended to at least 100 ms.
Together, these findings raise the possibility that artificially attenuated SK currents under whole-cell recording conditions promoted long-lasting bistable persistent firing modes at the expense of graded and self-terminating responses. Intriguingly, several groups reported that attenuating SK currents pharmacologically can improve WM performance in rodents (Stackman et al., 2002; Brennan et al., 2008), perhaps by prolonging the duration of persistent firing. Dynamic changes in cholinergic input also can contribute to self-terminating responses, as demonstrated using optogenetic modulation of activity in cholinergic fibers (Joshi et al., 2016; Baker et al., 2018; Cui and Strowbridge, 2019). These studies indicate that neuronal hyperexcitability reverses gradually when ACh release is terminated, perhaps mimicking the reduction in cortical modulator release when attention lapses during WM tasks. Future studies will be required to assess the degree to which these biophysical mechanisms (recovery of ERG leak conductance, enhanced SK conductance, reduced cholinergic activity) influence the duration of persistent firing and WM performance in vivo. Defining the consequences of modulating ERG and SK channel activity during WM tasks is likely to be especially informative since highly specific pharmaceutical agents are available for these channels. Our work predicts that the blockade of ERG channels (e.g., using ErgToxin1) should reduce the prevalence of persistent firing in cortical neurons in vivo and impair WM performance, while enhancing SK conductances using NS309 should prematurely terminate epochs of persistent firing, reducing the duration of delay cell activity. Our results also raise the possibility that other mechanisms that enhance postspike hyperpolarization, such as feedback GABAergic inhibition, may regulate persistent firing dynamics through the same mechanism we demonstrate for SK channel modulators.
Footnotes
This work was supported by National Institutes of Health Grant R01-DC-04285 to B.W.S. We thank Drs. Dominique Durand and Chris Ford for helpful discussions related to this project.
The authors declare no competing financial interests.
References
- Baker AL, O'Toole RJ, Gulledge AT (2018) Preferential cholinergic excitation of corticopontine neurons. J Physiol 596:1659–1679. 10.1113/JP275194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt JC, Lillis KP, Stupin LR, White JA (2008) Effects of imperfect dynamic clamp: computational and experimental results. J Neurosci Methods 169:282–289. 10.1016/j.jneumeth.2007.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlen A, Schwake M, Dost R, Kunert A, Fidzinski P, Heinemann U, Gebhardt C (2013) The new KCNQ2 activator 4-chlor-N-(6-chlor-pyridin-3-yl)-benzamid displays anticonvulsant potential. Br J Pharmacol 168:1182–1200. 10.1111/bph.12065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AR, Dolinsky B, Vu M-AT, Stanley M, Yeckel MF, Arnsten AF (2008) Blockade of IP3-mediated SK channel signaling in the rat mediated medial prefrontal cortex improves spatial working memory. Learn Mem 15:93–96. 10.1101/lm.767408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard JI, Karelina K, Sarter M, Givens B (2009) Cholinergic optimization of cue-evoked parietal activity during challenged attentional performance. Eur J Neurosci 29:1711–1722. 10.1111/j.1460-9568.2009.06713.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, Shapiro MS (2019) Gq-coupled muscarinic receptor enhancement of KCNQ2/3 channels and activation of TRPC channels in multimodal control of excitability in dentate gyrus granule cells. J Neurosci 39:1566–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill SL, Tobin a B, Torrecilla I, Willars GB, Standen NB, Mitcheson JS (2007) Modulation of hERG potassium currents in HEK-293 cells by protein kinase C. Evidence for direct phosphorylation of pore forming subunits. J Physiol 581:479–493. 10.1113/jphysiol.2006.123414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ (1990) Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci 13:99–104. 10.1016/0166-2236(90)90185-d [DOI] [PubMed] [Google Scholar]
- Cui ED, Strowbridge BW (2018) Modulation of ether-à-go-go related gene (ERG) current governs intrinsic persistent activity in rodent neocortical pyramidal cells. J Neurosci 38:423–440. 10.1523/JNEUROSCI.1774-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui ED, Strowbridge BW (2019) Selective attenuation of Ether-à-go-go related K currents by endogenous acetylcholine reduces spike-frequency adaptation and network correlation. Elife 8:e44954. 10.7554/eLife.44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalezios Y, Scudder CA, Highstein SM, Moschovakis AK (1998) Anatomy and physiology of the primate interstitial nucleus of Cajal. II. Discharge pattern of single efferent fibers. J Neurophysiol 80:3100–3111. 10.1152/jn.1998.80.6.3100 [DOI] [PubMed] [Google Scholar]
- Dasari S, Abramowitz J, Birnbaumer L, Gulledge AT (2013) Do canonical transient receptor potential channels mediate cholinergic excitation of cortical pyramidal neurons? Neuroreport 24:550–554. 10.1097/WNR.0b013e3283621344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Gray R, Johnston D (2017) A dynamic clamp on every rig. eNeuro 4:ENEURO.0250-17.2017. 10.1523/ENEURO.0250-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dégenètais E, Thierry A-M, Glowinski J, Gioanni Y (2002) Electrophysiological properties of pyramidal neurons in the rat prefrontal cortex: an in vivo intracellular recording study. Cereb Cortex 12:1–16. [DOI] [PubMed] [Google Scholar]
- Douglas RJ, Koch C, Mahowald M, Martin KA, Suarez HH (1995) Recurrent excitation in neocortical circuits. Science 269:981–985. 10.1126/science.7638624 [DOI] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA (2002) Graded persistent activity in entorhinal cortex neurons. Nature 420:173–178. 10.1038/nature01171 [DOI] [PubMed] [Google Scholar]
- Egorov AV, Unsicker K, von Bohlen und Halbach O (2006) Muscarinic control of graded persistent activity in lateral amygdala neurons. Eur J Neurosci 24:3183–3194. 10.1111/j.1460-9568.2006.05200.x [DOI] [PubMed] [Google Scholar]
- Egorov AV, Schumacher D, Medert R, Birnbaumer L, Freichel M, Draguhn A (2019) TRPC channels are not required for graded persistent activity in entorhinal cortex neurons. Hippocampus 29:1038–1048. 10.1002/hipo.23094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fano S, Çalişkan G, Heinemann U (2012) Differential effects of blockade of ERG channels on gamma oscillations and excitability in rat hippocampal slices. Eur J Neurosci 36:3628–3635. 10.1111/ejn.12015 [DOI] [PubMed] [Google Scholar]
- Fellous JM, Sejnowski TJ (2003) Regulation of persistent activity by background inhibition in an in vitro model of a cortical microcircuit. Cereb Cortex 13:1232–1241. 10.1093/cercor/bhg098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransén E, Tahvildari B, Egorov AV, Hasselmo ME, Alonso AA (2006) Mechanism of graded persistent cellular activity of entorhinal cortex layer V neurons. Neuron 49:735–746. 10.1016/j.neuron.2006.01.036 [DOI] [PubMed] [Google Scholar]
- Funahashi S, Bruce CJ, Goldman-Rakic PS (1989) Mnemonic coding of visual space in the monkey's dorsolateral prefrontal cortex. J Neurophysiol 61:331–349. 10.1152/jn.1989.61.2.331 [DOI] [PubMed] [Google Scholar]
- Fuster JM, Alexander GE (1971) Neuron activity related to short-term memory. Science 173:652–654. 10.1126/science.173.3997.652 [DOI] [PubMed] [Google Scholar]
- Fuster JM, Jervey JP (1981) Inferotemporal neurons distinguish and retain behaviorally relevant features of visual stimuli. Science 212:952–955. 10.1126/science.7233192 [DOI] [PubMed] [Google Scholar]
- Gill TM, Sarter M, Givens B (2000) Sustained visual attention performance-associated prefrontal neuronal activity: evidence for cholinergic modulation. J Neurosci 20:4745–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnadt JW, Andersen RA (1988) Memory related motor planning activity in posterior parietal cortex of macaque. Exp Brain Res 70:216–220. 10.1007/BF00271862 [DOI] [PubMed] [Google Scholar]
- Goldman MS (2009) Memory without feedback in a neural network. Neuron 61:621–634. 10.1016/j.neuron.2008.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MS, Levine JH, Major G, Tank DW, Seung HS (2003) Robust persistent neural activity in a model integrator with multiple hysteretic dendrites per neuron. Cereb Cortex 13:1185–1195. 10.1093/cercor/bhg095 [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R (1998) Ionic mechanism of the slow afterdepolarization induced by muscarinic receptor activation in rat prefrontal cortex. J Neurophysiol 80:1197–1210. 10.1152/jn.1998.80.3.1197 [DOI] [PubMed] [Google Scholar]
- Hilgers RH, Webb RC (2007) Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 292:H2275–H2284. 10.1152/ajpheart.00949.2006 [DOI] [PubMed] [Google Scholar]
- Hougaard C, Eriksen BL, Jørgensen S, Johansen TH, Dyhring T, Madsen LS, Strøbaek D, Christophersen P (2007) Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br J Pharmacol 151:655–665. 10.1038/sj.bjp.0707281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochems A, Yoshida M (2013) Persistent firing supported by an intrinsic cellular mechanism in hippocampal CA3 pyramidal cells. Eur J Neurosci 38:2250–2259. 10.1111/ejn.12236 [DOI] [PubMed] [Google Scholar]
- Jochems A, Yoshida M (2015) A robust in vivo-like persistent firing supported by a hybrid of intracellular and synaptic mechanisms. PLoS One 10:e0123799. 10.1371/journal.pone.0123799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Kalappa BI, Anderson CT, Tzounopoulos T (2016) Cell-specific cholinergic modulation of excitability of layer 5B principal neurons in mouse auditory cortex. J Neurosci 36:8487–8499. 10.1523/JNEUROSCI.0780-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer B, Jochems A, Valero-Aracama MJ, Yoshida M (2013) Long-lasting intrinsic persistent firing in rat CA1 pyramidal cells: a possible mechanism for active maintenance of memory. Hippocampus 23:820–831. 10.1002/hipo.22136 [DOI] [PubMed] [Google Scholar]
- Koulakov AA, Raghavachari S, Kepecs A, Lisman JE (2002) Model for a robust neural integrator. Nat Neurosci 5:775–782. 10.1038/nn893 [DOI] [PubMed] [Google Scholar]
- Liu D, Gu X, Zhu J, Zhang X, Han Z, Yan W, Cheng Q, Hao J, Fan H, Hou R, Chen Z, Chen Y, Li CT (2014) Medial prefrontal activity during delay period contributes to learning of a working memory task. Science 346:458–463. 10.1126/science.1256573 [DOI] [PubMed] [Google Scholar]
- Major AJ, Vijayraghavan S, Everling S (2015) Muscarinic attenuation of mnemonic rule representation in macaque dorsolateral prefrontal cortex during a pro- and anti-saccade task. J Neurosci 35:16064–16076. 10.1523/JNEUROSCI.2454-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major G, Baker R, Aksay E, Seung HS, Tank DW (2004) Plasticity and tuning of the time course of analog persistent firing in a neural integrator. Proc Natl Acad Sci U|S|A 101:7745–7750. 10.1073/pnas.0401992101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B (1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275:213–215. 10.1126/science.275.5297.213 [DOI] [PubMed] [Google Scholar]
- Mash DC, White WF, Mesulam MM (1988) Distribution of muscarinic receptor subtypes within architectonic subregions of the primate cerebral cortex. J Comp Neurol 278:265–274. 10.1002/cne.902780209 [DOI] [PubMed] [Google Scholar]
- Nam YW, Orfali R, Liu T, Yu K, Cui M, Wulff H, Zhang M (2017) Structural insights into the potency of SK channel positive modulators. Sci Rep 7:17178. 10.1038/s41598-017-16607-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshio KI, Chiba A, Inase M (2006) Delay period activity of monkey prefrontal neurones during duration-discrimination task. Eur J Neurosci 23:2779–2790. 10.1111/j.1460-9568.2006.04781.x [DOI] [PubMed] [Google Scholar]
- Papa M, Boscia F, Canitano A, Castaldo P, Sellitti S, Annunziato L, Taglialatela M (2003) Expression pattern of the ether-a-gogo-related (ERG) K+ channel-encoding genes ERG1, ERG2, and ERG3 in the adult rat central nervous system. J Comp Neurol 466:119–135. 10.1002/cne.10886 [DOI] [PubMed] [Google Scholar]
- Pedarzani P, McCutcheon JE, Rogge G, Jensen BS, Christophersen P, Hougaard C, Strøbaek D, Stocker M (2005) Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current I(AHP) and modulates the firing properties of hippocampal pyramidal neurons. J Biol Chem 280:41404–41411. 10.1074/jbc.M509610200 [DOI] [PubMed] [Google Scholar]
- Pesaran B, Pezaris JS, Sahani M, Mitra PP, Andersen RA (2002) Temporal structure in neuronal activity during working memory in macaque parietal cortex. Nat Neurosci 5:805–811. 10.1038/nn890 [DOI] [PubMed] [Google Scholar]
- Rahman J, Berger T (2011) Persistent activity in layer 5 pyramidal neurons following cholinergic activation of mouse primary cortices. Eur J Neurosci 34:22–30. 10.1111/j.1460-9568.2011.07736.x [DOI] [PubMed] [Google Scholar]
- Reboreda A, Raouf R, Alonso A, Séguéla P (2007) Development of cholinergic modulation and graded persistent activity in layer v of medial entorhinal cortex. J Neurophysiol 97:3937–3947. 10.1152/jn.01233.2006 [DOI] [PubMed] [Google Scholar]
- Rye DB, Wainer BH, Mesulam MM, Mufson EJ, Saper CB (1984) Cortical projections arising from the basal forebrain: a study of cholinergic and noncholinergic components employing combined retrograde tracing and immunohistochemical localization of choline acetyltransferase. Neuroscience 13:627–643. 10.1016/0306-4522(84)90083-6 [DOI] [PubMed] [Google Scholar]
- Saganich MJ, Machado E, Rudy B (2001) Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J Neurosci 21:4609–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Staiger JF, Cho N, Kötter R, Zilles K, Luhmann HJ (2001) Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J Neurosci 21:3580–3592. 10.1523/JNEUROSCI.21-10-03580.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Chubb MC, Crill WE (1988) Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol 59:450–467. 10.1152/jn.1988.59.2.450 [DOI] [PubMed] [Google Scholar]
- Seung HS (1996) How the brain keeps the eyes still. Proc Natl Acad Sci U|S|A 93:13339–13344. 10.1073/pnas.93.23.13339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AA, O'Neil MB, Abbott LF, Marder E (1993) Dynamic clamp: computer-generated conductances in real neurons. J Neurophysiol 69:992–995. 10.1152/jn.1993.69.3.992 [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T (2002) Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci 22:10163–10171. 10.1523/JNEUROSCI.22-23-10163.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Liu Y, Baudry M, Bi X (2020) SK2 channel regulation of neuronal excitability, synaptic transmission, and brain rhythmic activity in health and diseases. Biochim Biophys Acta Mol Cell Res 1867:118834. 10.1016/j.bbamcr.2020.118834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP (2003) Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci 23:9650–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Funahashi S (2002) Prefrontal task-related activity representing visual cue location or saccade direction in spatial working memory tasks. J Neurophysiol 87:567–588. 10.1152/jn.00249.2001 [DOI] [PubMed] [Google Scholar]
- Vijayraghavan S, Major AJ, Everling S (2018) Muscarinic M1 receptor overstimulation disrupts working memory activity for rules in primate prefrontal cortex. Neuron 98:1256–1268.e4. 10.1016/j.neuron.2018.05.027 [DOI] [PubMed] [Google Scholar]
- Volpicelli LA, Levey AI (2004) Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog Brain Res 145:59–66. 10.1016/S0079-6123(03)45003-6 [DOI] [PubMed] [Google Scholar]
- Wang XJ (1999) Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci 19:9587–9603. 10.1523/JNEUROSCI.19-21-09587.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Funahashi S (2007) Prefrontal delay-period activity reflects the decision process of a saccade direction during a free-choice ODR task. Cereb Cortex 17 [Suppl 1]:88–100. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Funahashi S (2014) Neural mechanisms of dual-task interference and cognitive capacity limitation in the prefrontal cortex. Nat Neurosci 17:601–611. 10.1038/nn.3667 [DOI] [PubMed] [Google Scholar]
- Williams SR, Tóth TI, Turner JP, Hughes SW, Crunelli V (1997) The “window” component of the low threshold Ca2+ current produces input signal amplification and bistability in cat and rat thalamocortical neurones. J Physiol 505:689–705. 10.1111/j.1469-7793.1997.689ba.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Sun Z, Jin X, Ma W, Song Y, Lai S, Chen Q, Fan M, Zhang J, Yue W, Huang Z (2018) ERG3 potassium channel-mediated suppression of neuronal intrinsic excitability and prevention of seizure generation in mice. J Physiol 596:4729–4752. 10.1113/JP275970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H-D, Villalobos C, Andrade R (2009) TRPC channels mediate a muscarinic receptor-induced afterdepolarization in cerebral cortex. J Neurosci 29:10038–10046. 10.1523/JNEUROSCI.1042-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Hasselmo ME (2009) Persistent firing supported by an intrinsic cellular mechanism in a component of the head direction system. J Neurosci 29:4945–4952. 10.1523/JNEUROSCI.5154-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BJ, Otto T, Fox GD, Eichenbaum H (1997) Memory representation within the parahippocampal region. J Neurosci 17:5183–5195. 10.1523/JNEUROSCI.17-13-05183.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Reboreda A, Alonso A, Barker PA, Séguéla P (2011) TRPC channels underlie cholinergic plateau potentials and persistent activity in entorhinal cortex. Hippocampus 21:386–397. 10.1002/hipo.20755 [DOI] [PubMed] [Google Scholar]
- Zhou X, Qi X-L, Douglas K, Palaninathan K, Kang HS, Buccafusco JJ, Blake DT, Constantinidis C (2011) Cholinergic modulation of working memory activity in primate prefrontal cortex. J Neurophysiol 106:2180–2188. 10.1152/jn.00148.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylberberg J, Strowbridge BW (2017) Mechanisms of persistent activity in cortical circuits: possible neural substrates for working memory. Annu Rev Neurosci 40:603–627. 10.1146/annurev-neuro-070815-014006 [DOI] [PMC free article] [PubMed] [Google Scholar]