

Abstract
The midbrain periaqueductal gray (PAG) plays a central role in pain modulation via descending pathways. Opioids and cannabinoids are thought to activate these descending pathways by relieving intrinsic GABAergic inhibition of PAG neurons which project to the rostroventromedial medulla (RVM), a process known as disinhibition. However, the PAG also receives descending extrinsic GABAergic inputs from the central nucleus of the amygdala (CeA) which are thought to inhibit PAG GABAergic interneurons. It remains unclear how opioids and cannabinoids act at these different synapses to control descending analgesic pathways. We used optogenetics, tract tracing and electrophysiology to identify the circuitry underlying opioid and cannabinoid actions within the PAG of male and female rats. It was observed that both RVM-projection and nonprojection PAG neurons received intrinsic-PAG and extrinsic-CeA synaptic inputs, which were predominantly GABAergic. Opioids acted via presynaptic µ-receptors to suppress both intrinsic and extrinsic GABAergic inputs onto all PAG neurons, although this inhibition was greater in RVM-projection neurons. By contrast, cannabinoids acted via presynaptic CB1 receptors to exclusively suppress the direct descending GABAergic input from the CeA onto RVM-projection PAG neurons. These findings indicate the CeA controls PAG output neurons which project to the RVM via parallel direct and indirect GABAergic pathways. While µ-opioids indiscriminately inhibit GABAergic inputs onto all PAG neurons, cannabinoids selectively inhibit a direct extrinsic GABAergic input from the amygdala onto PAG projection neurons. These differential actions of opioids and cannabinoids provide a flexible system to gate the descending control of analgesia from the PAG.
SIGNIFICANCE STATEMENT The disinhibition hypothesis of analgesia states that opioids activate the midbrain periaqueductal gray (PAG) descending pathway by relieving the tonic inhibition of projection neurons from GABAergic interneurons. However, the PAG also receives extrinsic GABAergic inputs and is the locus of action of cannabinoid analgesics. Here, we show the relative sensitivity of GABAergic synapses to opioids and cannabinoids within the PAG depends on both the origin of presynaptic inputs and their postsynaptic targets. While opioids indiscriminately inhibit all GABAergic inputs onto all PAG neurons, cannabinoids selectively inhibit a direct extrinsic GABAergic input from the amygdala onto PAG descending projection neurons. These differential actions of opioids and cannabinoids provide a flexible system to gate PAG descending outputs.
Keywords: cannabinoid, electrophysiology, opioid, optogenetic, pain, synaptic
Introduction
The midbrain periaqueductal gray (PAG) plays a pivotal role in integrating a range of analgesic, behavioral and autonomic responses to threat, stress, and pain (Keay and Bandler, 2001). Of particular interest is the ventrolateral column of the PAG which forms part of an endogenous analgesic system that projects via the rostroventromedial medulla (RVM) to the spinal dorsal horn where it modulates ascending nociceptive transmission (Fields and Basbaum, 1978; Heinricher and Fields, 2013). In conjunction with this physiological role, the PAG is a major site of the analgesic actions of endogenously released and exogenously applied opioids and cannabinoids (Yeung et al., 1977; Martin et al., 1999; Hohmann et al., 2005; Lane et al., 2005).
Opioids and cannabinoids are thought to activate descending analgesic pathways by relieving GABAergic inhibition of PAG projection neurons, a process known as disinhibition (Moreau and Fields, 1986; Depaulis et al., 1987). Cellular studies have shown that opioids act via presynaptic µ-opioid receptors to inhibit GABAergic synaptic transmission within the ventrolateral PAG, particularly onto RVM-projecting neurons (Vaughan and Christie, 1997; Chiou and Huang, 1999; Vaughan et al., 2003; Park et al., 2010; Lau et al., 2020). Likewise, cannabinoids are known to act via presynaptic CB1 receptors to suppress GABAergic synaptic transmission within the ventrolateral PAG, however their relative actions on RVM-projection versus nonprojection PAG neurons are unclear (Vaughan et al., 2000; Drew et al., 2008, 2009).
It has long been thought that the inhibitory control of PAG descending outputs is derived from GABAergic interneurons within this brain structure (Barbaresi and Manfrini, 1988; Reichling and Basbaum, 1990a, b; Park et al., 2010; Samineni et al., 2017). However, the PAG also receives extrinsic GABAergic inputs from higher centers such as the central nucleus of the amygdala (CeA; Rizvi et al., 1991; Oka et al., 2008). Recent optogenetic studies have shown that GABAergic CeA neurons specifically synapse onto GABAergic neurons within the PAG, and in turn, these intrinsic GABAergic neurons synapse onto glutamatergic PAG neurons which are presumably projection neurons (Reichling and Basbaum, 1990b; Tovote et al., 2016; Avegno et al., 2018; Yin et al., 2020). These observations indicate that the CeA activates PAG descending projection neurons via disynaptic GABAergic disinhibition. However, it is unknown how this wide range of inhibitory synapses is targeted by opioids and cannabinoids. This information is essential as it defines how opioid and cannabinoid disinhibition regulates descending analgesic outputs from the PAG. We therefore used a combination of electrophysiology, optogenetics and tract tracing to identify the circuitry underlying opioid and cannabinoid modulation of the descending analgesic output from the PAG.
Materials and Methods
Experiments were conducted on male and female Sprague Dawley rats in accordance with guidelines set by the National Health and Medical Research Council Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. All experiments were approved by the Royal North Shore Hospital Animal Ethics Committee (protocols 1311-013A, RESP-17-94, RESP-18-208). Pregnant female rats were obtained from the Animal Resources Center (Canning Vale) and were housed in the Kolling Institute Facility. After weaning [postnatal day (P)21], animals of the same gender were housed in groups of two to four in individually ventilated cages under controlled light (12/12 h light/dark cycles) and temperature (23 ± 1°C, 70% humidity) with ad libitum access to water and food pellets. Cages were enriched with a house igloo, tissues for nesting, and straws or paddle pop sticks on alternate weeks.
Retrograde tracer and channelrhodopsin-2 (ChR2) microinjections
For brain microinjections, three- to five-week-old rats were anaesthetized (1–2% isoflurane in O2), placed in a stereotaxic frame and the dura exposed by trephination over the RVM, PAG, and CeA. Microinjections were made via a glass micropipette (tip diameter 30 µm, Drummond Nanoject). For retrograde tracing, red-orange fluorescent microspheres (Invitrogen) were microinjected into the RVM (adult coordinates: −1.2 mm posterior and −10.8 mm ventral from λ; total volume: 80–180 nl). For optogenetics, AAV8-hSyn-ChR2(H134R)-GFP (1.9 × 1013 vg/ml, Addgene, RRID: Addgene_58880) was unilaterally microinjected into the ventrolateral/lateral PAG (adult coordinates: +1.0 mm anterior, ±0.62 mm medial/lateral, −6.25 mm ventral to λ; total volume: 70–150 nl; injection site randomized equally between left and right hemispheres), or bilaterally into the CeA (adult coordinates: −2.0 mm posterior, ±4.15 mm medial/lateral, −8.05 ventral to bregma; total volume: 50–130 nl). The coordinates for all injections were adjusted using the juvenile: adult ratio of the bregma-λ distance, regardless of gender. The burr hole was then filled with bone wax and the incision was irrigated with saline and closed with box stitches using 4.0 or 6.0 silk sutures. The wound was treated with antiseptic (1% w/v iodine solution) and animals received an analgesic (buprenorphine 0.05 mg kg−1, s.c.) immediately after surgery. Animals were then recovered from anesthesia in a clean cage warmed by a heating pad before being returned to their holding room. If required, animals received an antibiotic (Benacillin 64 mg kg−1, i.p.) 4–7 d after surgery. Animals were used for ex vivo slice experiments at 8–12 weeks postsurgery and tracer/ChR2 injections were examined post hoc to verify correct placement within the RVM, ventrolateral PAG, and CeA.
Electrophysiology
For ex vivo slice experiments, animals were deeply anaesthetized with isoflurane, decapitated and coronal slices (300 μm) containing the PAG, amygdala (CeA and BLA), and RVM were cut using a vibratome (VT1200S, Leica Microsystems) in ice-cold artificial CSF (ACSF) of composition (in mm): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3; equilibrated with 95% O2 and 5% CO2. Slices were then incubated for 10 min at 34°C in a N-Methyl-D-Glucamine (NMDG) recovery solution (in mm): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 3 sodium pyruvate, 2 thiourea, 10 MgCl2, and 0.5 CaCl2 (pH 7.4). The slices were then maintained in a submerged chamber containing ACSF for at least 1 h at room temperature (RT) before recording or fixation. For recording, PAG or CeA containing slices were individually transferred to a chamber on an upright fluorescence microscope (Olympus BX51) and superfused continuously with ACSF (34°C, flow rate 2.5 ml min−1). Neurons were visualized with a 40× water-immersion objective using Dodt gradient contrast optics. In animals that received retrograde tracer or ChR2 injections, the presence/absence of red-orange microspheres and/or ChR2-GFP in recorded neurons was detected under brief fluorescent illumination (460- to 495- or 530- to 550-nm wavelengths) before recording.
Whole-cell voltage-clamp recordings (Axopatch 200B, or 700B, Molecular Devices) of synaptic currents (holding potential, –65 mV) were made using a CsCl-based internal solution containing (in mm): 140 CsCl, 0.2 EGTA, 10 HEPES, 1 MgCl2, 2 MgATP, 0.3 NaGTP, and 0.1% biocytin (pH 7.3; osmolarity, 280–285 mOsm l−1). Whole-cell current-clamp recordings of synaptic potentials were made using a K-gluconate-based internal solution containing (in mm): 135 K-gluconate, 4 NaCl, 0.5 EGTA, 10 HEPES, 1 MgCl2, 2 MgATP, 0.3 NaGTP, and 0.1% biocytin (pH 7.3; osmolarity, 280–285 mOsm l−1). Series resistance (<25 MΩ) was continuously monitored during experiments.
Electrically evoked currents were elicited using nickel-chromium bipolar electrodes placed 200–500 µm away from the recording electrode. Optically evoked currents were elicited using a 473-nm fiber-coupled laser (100 mW, Ikecool), or an LED (17 mW, Thorlabs) via the microscope objective. Electrical and optically evoked current recordings were conducted at twice the intensity required to elicit a stable response above noise threshold. IPSCs were pharmacologically isolated in the presence of the AMPA/kainate receptor antagonist, 2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX; 5 μm), the glycine receptor antagonist, strychnine (3 μm) and the NMDA receptor antagonist, dl-2-amino-5-phosphonopentanoic acid (AP5; 50 μm); the role of GABAA receptors was confirmed with the GABAA receptor antagonist gabazine (10 μm). The actions of opioids were examined with maximal concentrations of the µ-opioid agonist DAMGO (3 μm) and the µ-opioid antagonist CTAP (1 μm), and cannabinoids with maximal concentrations of the pan-cannabinoid agonist WIN55212-2 (1 μm) and the CB1 selective antagonists AM251 and AM281 (1 μm, results pooled).
In recordings, drug testing was conducted after obtaining a stable baseline recording for at least 5 min. To obtain stable, steady-state actions, synaptic transmission agents and opioids were applied for periods of 5–6 min, and cannabinoids for 8–12 min. All recordings were filtered (3- to 5-kHz low-pass filter) and sampled (20 kHz) for on-line and later off-line analysis using AxographX (Axograph Scientific Software, RRID: SCR_014284). The peak amplitude of electrically and optically evoked PSCs were measured relative to a 2-ms baseline preceding the stimulus. Drug effects on electrophysiological parameters were measured at fixed time points: over the last 2 min before, and during drug application. Drug effects on PSC parameters in each recording were calculated as a percentage of the predrug value. Experimenters were not blinded to the drug, or neuron type being tested because (1) opioids and cannabinoids had to be applied for different durations because of their differing lipophilicity (see above), (2) identification of neurons as RVM-retrolabeled or unlabeled was required for each recording, and (3) intra-CeA versus intra-PAG AAV-ChR2-injected animals could easily be distinguished by the intensity and distribution of GFP in each slice before recording. Neuron recordings were only excluded from analysis if (1) the RVM or CeA injection site was incorrect, (2) series resistance varied by >25% during a recording, or (3) evoked synaptic currents/potentials during the preagonist, baseline period changed by >20%. Data outliers were not excluded.
Immunohistochemistry and confocal imaging
For post hoc staining following electrophysiology (PAG and amygdala), slices (300 µm) containing neurons filled with 0.1% biocytin during whole-cell recordings were fixed overnight at 4°C in 4% PFA in 0.1 m phosphate buffer (PB). Similarly, to check and image injection locations, after recovery (10 min in NMDG at 34°C, then >1 h in ACSF at RT), all slices containing the amygdala and RVM were fixed overnight in 4% PFA at 4°C. In all cases, slices were then washed three times for 10 min with 0.1 m PB and stored either in PB (less than two weeks) or 0.2% sodium azide in PB (less than three months) at 4°C before staining. For staining, slices were washed three times (5 min) to remove azide and then incubated for 1 h at RT in 5% horse serum, 1% BSA and 0.3% Triton X-100 in PB. To amplify GFP signals in slices isolated from animals that received intra-PAG or intra-CeA injections of AAV-ChR2, the primary antibody anti-GFP (1:500) was diluted in 1% BSA/0.1% Triton X-100 in PB and incubated overnight at 4°C. Slices were then washed 4 times (10 min) at RT with PB before secondary antibody incubation. The secondary antibody donkey anti-chicken-488 (1:500) and Steptavidin Avidin-647 (1:1000, ThermoFisher) were co-diluted in 1% BSA/0.1% Triton X-100 in PB and slices were incubated for 2 h at RT (light protected). In all cases, the nuclear stain, DAPI (1:2000, Sigma), was added for the last 30 min of the final incubation period. Slices were then washed four times (10 min) with PB and mounted onto slides using Fluoromount-G (SouthernBiotech).
All sections were visualized using a Leica TCS SP5 confocal microscope (lasers: 405, 488, 561, and 633 nm) and images were captured with LAS AF (Leica) software. Images were taken sequentially with different lasers using either 10× (NA 0.4) or 20× (NA 0.7) dry objectives or a 63× (NA 1.4) oil immersion objective. To image larger fields of view, tile-scanned images (10× or 20× objectives) were taken and stitched (10% overlap) using LAS AF software. Z-stacks and tiled images were collected where indicated and images were processed using ImageJ software (NIH).
Experimental design and statistical analysis
Based on our prior electrophysiology studies in PAG projection neurons (effect size of 1.5 with α/β errors of 0.05), it was estimated that 8 neuron recordings were required per group for the opioid and cannabinoid experiments (Drew et al., 2009; Lau et al., 2020). To ensure this minimum sample size, we prepared 10 animals per experimental group as (1) there was initially an 80% success rate with the combined tracer/AAV injections, and (2) one neuron recording was obtained per slice and between two and four slices obtained per animal. While only one neuron recording was usually obtained per animal for each experiment group, in some cases two neuron recordings were obtained from different slices in one animal. This, plus a higher tracer/AAV success rate led to the variable number of neuron recordings per opioid/cannabinoid experimental group (n = 8–11 neurons from 8 animals per group). Higher numbers of neurons were obtained in the initial experiments characterizing optically evoked currents in Figures 1–3 as the frequency of the connections was unknown; these experiments were first conducted as individual experimental groups, and then in the same neurons used for multiple opioid/cannabinoid experiment groups.
Figure 1.

The CeA projects to the midbrain PAG. A, Schematic of experimental configuration; optically evoked currents were recorded in CeA and BLA neurons from animals which received intra-CeA AAV-ChR2 injection, and in some cases an intra-RVM retrograde tracer injection. Bi, Low-power (10×), stacked image (z = 52 µm) displaying AAV-ChR2 injection site; 488 and BF channels overlaid to illustrate different amygdala regions. Bii, High-power (40×), stacked image (z = 4 µm) of ROI illustrated in Bi, arrows indicate cell surface labeling of AAV-ChR2-GFP. C, Single traces of (i) currents in CeA neurons evoked by focal optical stimulation of increasing duration (0.2, 0.5, 1, 5, and 20 ms) in voltage-clamp mode, and (ii) optically evoked action potential in current-clamp mode. D, Single traces of (i) currents in BLA neurons evoked by focal optical stimulation of increasing duration (0.2, 0.5, 1, 5, and 20 ms) in voltage-clamp mode, and (ii, iii) in current-clamp mode showing action potential generation during a current step, but not during optical stimulation. Ei, Eii, Low-power (20×), tiled (4 × 4) stacked image (z = 38 µm), showing distribution of (i) AAV-ChR2-GFP terminal labeling and (ii) retrolabeling, inset displays injection location in RVM. Eiii, High-power (63×), stacked image (z = 26 µm) of ROI illustrated in Eii, arrows indicate retrograde beads surrounded by terminal AAV-ChR2-GFP labeling. Scale bars: 200 µm (Bi, Ei, Eii) and 50 µm (Bii, Eiii).
Figure 3.
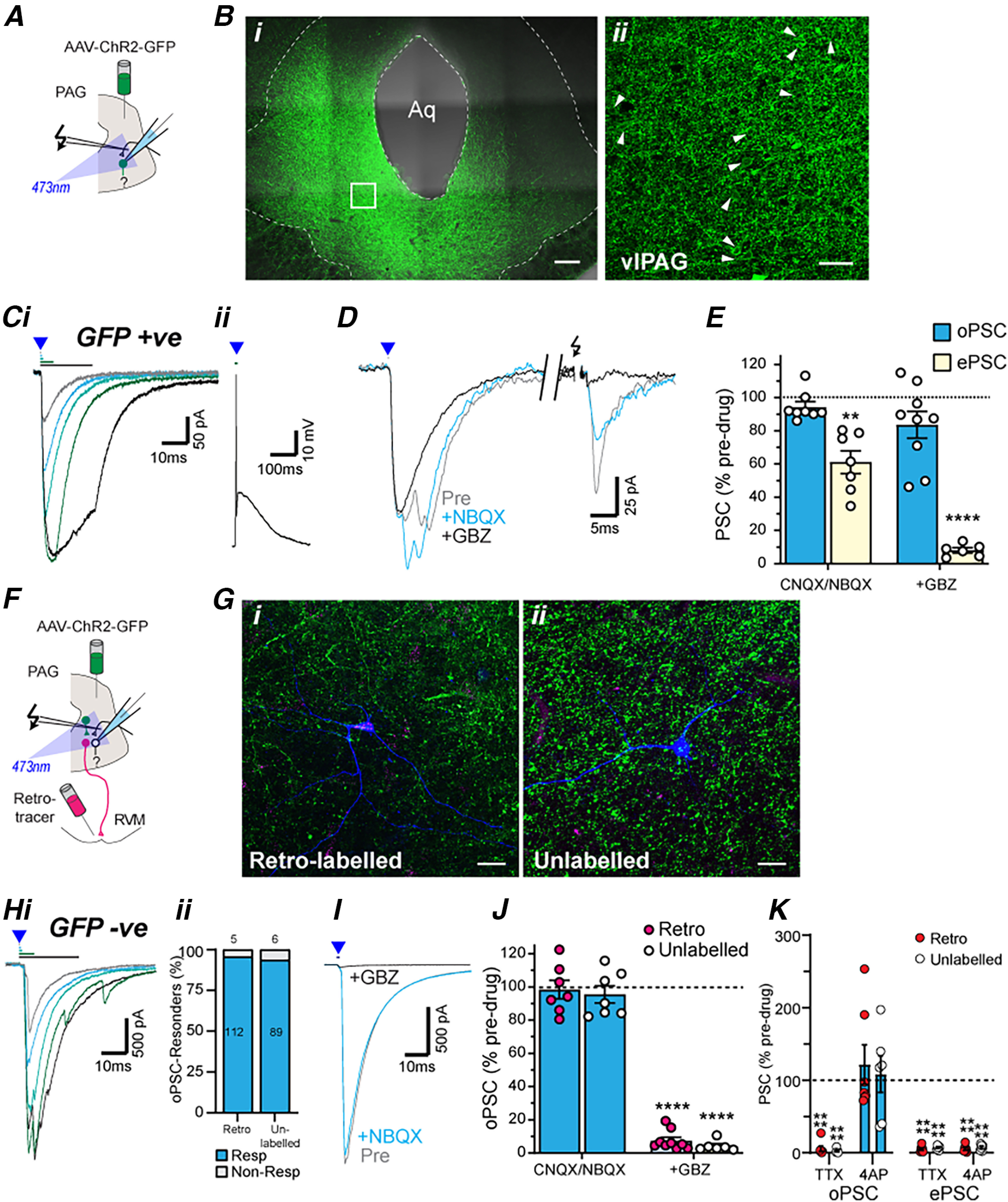
PAG projection and nonprojection neurons receive intrinsic monosynaptic GABAergic inputs from within PAG. A, Schematic of experimental configuration for panels B–E; optically evoked currents were recorded in GFP positive PAG neurons from animals which received an intra-PAG AAV-ChR2-GFP injection. Bi, Low-power (20×) tiled (4 × 3), stacked (z = 40 µm) image of AAV-ChR2 injection site within the PAG. Bii, High-power (40×), stacked (z = 5 µm) image of the dotted region of interest. Arrows depict examples of neurons encased by ChR2-GFP (green), indicating cell surface expression of ChR2-GFP. C, Single traces in GFP-positive PAG neurons of (i) optically evoked PSCs with increasing stimulation durations (0.2, 0.5, 1, 5, and 20 ms) in voltage-clamp mode and (ii) a current-clamp trace of an optically evoked action potential. D, Single traces of optically (blue triangle) and electrically (black arrows) evoked currents in the neuron from (C) before and during application of NBQX and then gabazine (GBZ). E, Scatter plots of the effect of NBQX (or CNQX) and GBZ on optically and electrically evoked currents in GFP-positive PAG neurons. F, Schematic of experimental configuration for panels G–K; optically evoked currents were recorded in GFP-negative PAG neurons from animals which received an intra-PAG AAV-ChR2-GFP injection and an intra-RVM tracer injection. G, High-power (63×) stacked images of biocytin filled (i) RVM-retrolabeled (magenta; z = 50 µm) or (ii) unlabeled (z = 77 µm) PAG neurons surrounded by ChR2-GFP positive terminals. Hi, Single traces of optically evoked currents in a GFP-negative RVM-retrolabeled PAG neuron with increasing stimulation durations (0.2, 0.5, 1, 5, and 20 ms). Hii, The frequency of occurrence of these optically evoked currents. I, Single traces of optically evoked currents in the neuron from (C) before and during application of NBQX and then gabazine (GBZ). J, Scatter plots of the effect of NBQX (or CNQX) and GBZ on optically evoked currents in RVM-retrolabeled and unlabeled GFP-negative PAG neurons. K, Scatter plots of the effect of TTX and 4AP on normalized optically and electrically evoked PSCs (oPSC, ePSC) in RVM-retrolabeled and unlabeled PAG neurons. In E, J, K, **p < 0.01, ****p < 0.0001 for pre versus drug. Scale bars: 200 µm (Bi), 50 µm (Bii), and 25 µm (Gi, Gii).
Individual drug effects on optically and electrically evoked PSCs were first made using Student's paired t test (pre vs drug). These drug effects were then compared between retrograde tracer neuronal types (RVM-labeled, unlabeled) and AAV-injection site (intra-CeA, -PAG AAV-ChR2) using two-way independent ANOVA, with post hoc comparisons made using Sidak's adjustment for multiple comparisons (data satisfied Levene's test for homogeneity of variance). Comparisons of proportions of retrolabeled versus unlabeled neuron responders to optical stimulation were made using Fisher's exact test. Statistical analysis was performed using Prism (RRID:SCR_002798) and SPSS (RRID:SCR_002865). All numerical data are expressed as mean ± SEM, and differences were considered significant if p < 0.05. The number of male and female animals was balanced across experimental groups, but gender was not considered as a factor for statistical analysis.
Drugs and reagents
AAV-ChR2s were obtained from the University of North Carolina Vector Core or Addgene (RRID: SCR_002448; RRID: Addgene_58880, respectively); all other reagents were obtained from Sigma-Aldrich, Abcam, and Tocris Bioscience. Stock solutions of neurochemicals were made in distilled water, or dimethyl sulfoxide, then diluted to working concentrations in ACSF (≤1:3000 solvent) immediately before use and applied by bath superfusion. The slice perfusion apparatus was washed in ethanol (70–80%) following the completion of cannabinoid recordings to remove any potential drug residue that might affect subsequent recordings.
Results
Extrinsic GABAergic inputs from the CeA target both RVM-projection and nonprojection neurons within PAG
The descending CeA-PAG-RVM pathway forms part of an endogenous analgesic system that modulates nociception within the spinal cord. To examine the amygdala-PAG circuitry involved in this analgesic system, ChR2 containing viral constructs (AAV-hSyn-ChR2(H134A)-GFP) were stereotaxically injected into the CeA (Fig. 1A). At 8–12 weeks following intra-CeA injection of AAV-ChR2-GFP, GFP-positive cell body and punctate staining was observed throughout the CeA, but not within the adjacent basolateral amygdala (BLA; Fig. 1B).
To examine whether this intra-CeA injection of AAV-ChR2-GFP led to functional expression of ChR2 in CeA neurons, we conducted electrophysiological recordings from amygdala slices. In identified GFP positive CeA neurons, focal optical stimulation (473 nm, 0.1–20 ms in duration) evoked currents which had an immediate onset, increased in amplitude with stimulus duration, and displayed only partial desensitization during long duration stimuli (Fig. 1Ci, n = 16). In current-clamp mode, optical stimulation led to depolarization and the generation of action potentials in these neurons (Fig. 1Cii). Within the adjacent BLA, focal optical stimulation did not evoke any currents, or depolarization in voltage-clamp and current-clamp mode, but the neurons readily fired action potentials when injected with current via the patch pipette (Fig. 1Di–Diii, n = 6). Thus, intra-CeA injection of AAV-ChR2 led to functional ChR2 expression within CeA neurons.
In these intra-CeA AAV-ChR2-injected animals, punctate GFP staining was observed throughout the lateral and ventrolateral columns of PAG (Fig. 1Ei), consistent with prior studies (Rizvi et al., 1991; Oka et al., 2008; Tovote et al., 2016; Yin et al., 2020). Previous studies using cre-dependent genetic strategies indicate GABAergic CeA neurons selectively project to GABAergic neurons within the PAG, which are presumed interneurons (Tovote et al., 2016; Yin et al., 2020), but it is unclear whether RVM-projection neurons also receive direct CeA inputs. We therefore examined whether CeA afferents target PAG neurons which project to the RVM by injecting retrograde tracer (red-orange fluorescent microspheres) into the RVM. RVM tracer injection led to retrograde cell body labeling in subpopulations of neurons throughout the lateral and ventrolateral PAG columns (Fig. 1Eii). In PAG slices, punctate GFP staining was detected around, but not in the cell bodies and dendrites of both RVM-retrolabeled and unlabeled neurons within the ventrolateral PAG (Figs. 1Eiii, 2B).
Figure 2.
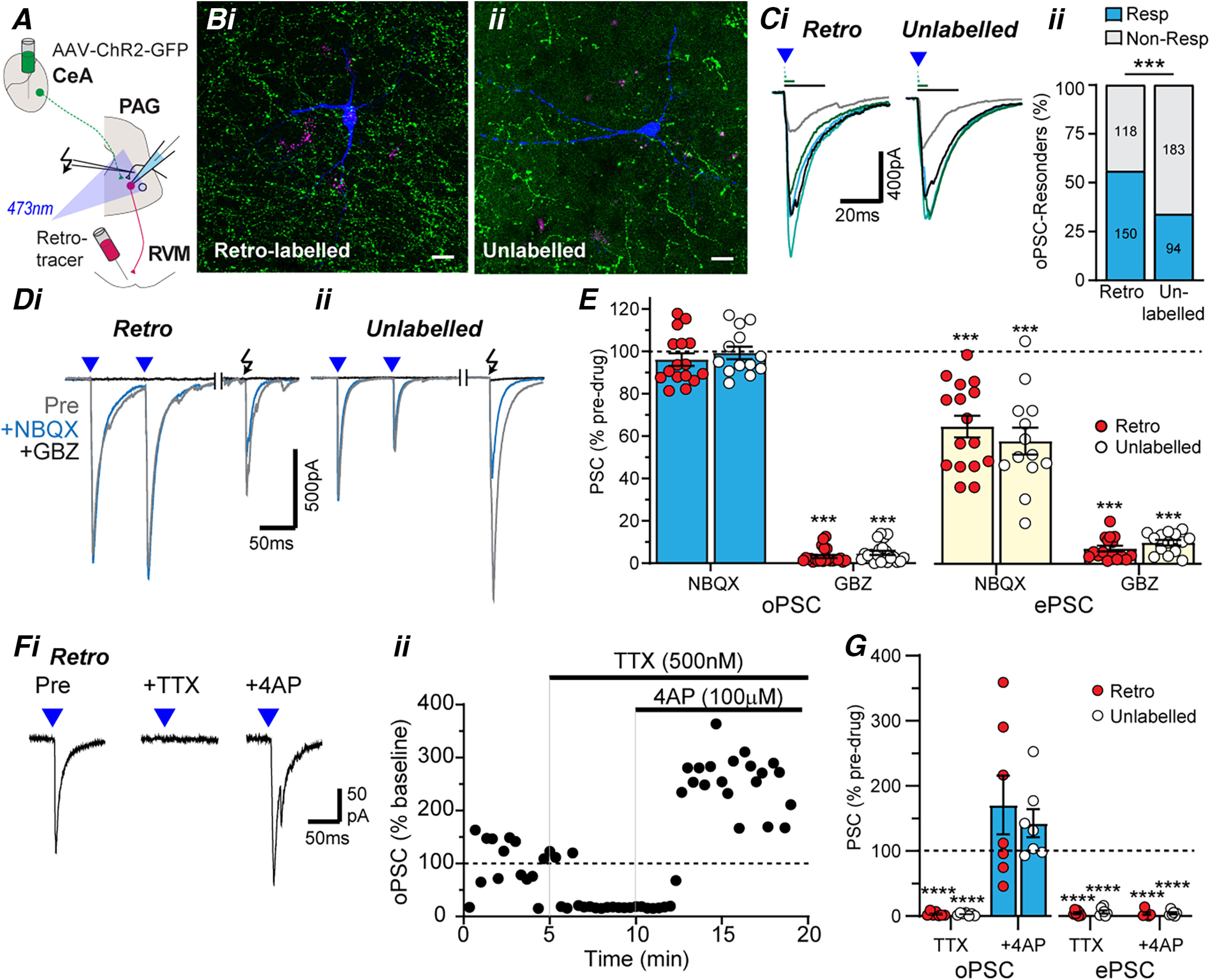
PAG projection and nonprojection neurons receive monosynaptic GABAergic inputs from CeA. A, Schematic of experimental configuration; optically evoked currents were recorded in retrolabeled and unlabeled PAG neurons from animals which received intra-RVM tracer injection and intra-CeA AAV-ChR2 injection. B, High-power (63×) stacked confocal images of biocytin- filled PAG neurons (blue) that were (i) retrogradely labeled (magenta; z = 61 µm) or (ii) unlabeled (z = 37.5 µm) from the RVM, surrounded by ChR2/GFP-positive fibers (green). Scale bars: 25 µm. Ci, Single traces of PSCs (oPSCs) in RVM-retrolabeled and unlabeled PAG neurons evoked by focal optical stimulation of increasing duration (0.2, 0.5, 1, 5, and 20 ms), and (Cii) the frequency of occurrence of these optically evoked PSCs. D, Averaged traces optically (blue triangles) and electrically (black arrow) evoked currents in (i) RVM-retrolabeled and (ii) unlabeled PAG neurons, before (Pre) and during application of NBQX, then gabazine (GBZ). E, Scatter plots of the effect of NBQX and GBZ on optically and electrically evoked currents in RVM-retrolabeled and unlabeled PAG neurons. F, Averaged traces (i) and normalized time plot (ii) of the amplitude of optically evoked currents in an RVM retrolabeled PAG neuron, before and during application of TTX and 4AP. G, Scatter plots of the effect of TTX and 4AP on normalized optically and electrically evoked PSCs (oPSC, ePSC) in RVM-retrolabeled and unlabeled PAG neurons. In C, ***p < 0.001 retrolabeled versus unlabeled; E, G, ***p < 0.001, ****p < 0.0001 for pre versus drug.
We next examined the specific ventrolateral PAG neurons targeted by the CeA using electrophysiology in PAG slices from animals that received both an intra-CeA AAV-ChR2 injection and an intra-RVM retrograde tracer injection (Fig. 2A). Focal optical stimulation evoked currents in subpopulations of RVM-retrolabeled and unlabeled neurons which had a delayed onset, increased in amplitude with stimulus duration, but rapidly desensitized regardless of the stimulus duration (Fig. 2Ci). These optically evoked currents were observed in a greater proportion of RVM-retrolabeled compared with unlabeled PAG neurons (Fig. 2Cii; χ(1)2 = 26.8, p = 0.0000002, 56% vs 34%, n = 150/268 and 94/277 in retrolabeled and unlabeled, Fisher's test).
The optically evoked PSCs in both RVM-retrolabeled and unlabeled PAG neurons were unaffected by NBQX (Fig. 2D,E, pre vs NBQX: t(15) = 1.3, p = 0.21 and t(12) = 0.24, p = 0.81; paired t tests for retrolabeled and unlabeled, n = 16, 13), but were abolished by gabazine (Fig. 2D,E, pre vs gabazine: t(22) = 133, p < 1 × 10−9, t(20) = 96, p < 1 × 10−9; paired t tests for retrolabeled and unlabeled neurons, n = 23, 21). Unlike optically evoked PSCs, electrically evoked PSCs in these neurons were reduced by NBQX and subsequently abolished by co-application of gabazine (Fig. 2D,E, pre vs NBQX: t(15) = 6.9, p = 5 × 10−6 and t(12) = 6.6, p = 3 × 10−5, n = 16, 13; pre vs gabazine: t(15) = 69, p < 1 × 10−9, t(13) = 69, p < 1 × 10−9, n = 16, 14, paired t tests for retrolabeled and unlabeled neurons). The effect of the blockers, NBQX and gabazine, did not differ between RVM-retrolabeled and unlabeled PAG neurons for both optically and electrically evoked PSCs (opto: F(1,69) = 1.9, p = 0.2; electrical: F(1,55) = 0.2, p = 0.62, ANOVA).
Finally, these optically evoked currents were abolished by tetrodotoxin (TTX), and this inhibition was recovered by the K+-channel blocker 4-aminopyridine (4AP; 100 μm; Fig. 2F,G, pre vs TTX: t(6) = 74, p = 4 × 10−9 and t(6) = 124, p = 2 × 10−11; pre vs TTX + 4AP: t(6) = 1.1, p = 0.32, t(6) = 2.0, p = 0.094, paired t tests for retrolabeled and unlabeled neurons, n = 7, 7), indicating the response was monosynaptic (Cho et al., 2013). By contrast, electrically evoked currents were abolished by TTX, and this inhibition was not recovered by 4AP (Fig. 2G, pre vs TTX: t(5) = 65, p = 2 × 10−8 and t(5) = 35, p = 3 × 10−7; pre vs TTX + 4AP: t(5) = 46, p = 1 × 10−7, t(5) = 58, p = 3 × 10−8, paired t tests; for retrolabeled and unlabeled neurons, n = 6, 6). The effect of the blockers, TTX and 4AP, did not differ between RVM-retrolabeled and unlabeled PAG neurons for both optically and electrically evoked PSCs (opto: F(1,24) = 0.3, p = 0.58; electrical: F(1,22) = 0.4, p = 0.56, ANOVA). Together with the NBQX insensitivity, these observations indicate the CeA sends monosynaptic GABAergic synaptic inputs onto both RVM-projection and unlabeled neurons within PAG, with a higher proportion of these extrinsic inputs targeting RVM-projection neurons.
Intrinsic GABAergic inputs target both RVM-projection and nonprojection neurons within PAG
According to the disinhibition hypothesis, PAG output neurons are thought to be directly regulated by local GABAergic interneurons. To address this, AAV-ChR2-GFP was unilaterally injected into the PAG (Fig. 3A). Bilateral GFP positive staining was observed throughout the PAG, although at higher levels on the side of the injection (Fig. 3B). Within the PAG, both cell body and punctate staining was observed, with staining in cell bodies localised around their surface rather than throughout the cytoplasm, indicating preferential cell surface expression of ChR2-GFP (Fig. 3B).
To examine whether intra-PAG injection of AAV-ChR2-GFP led to functional expression of ChR2 in PAG neurons, we conducted electrophysiological recordings from PAG slices (Fig. 3A). In PAG neurons identified as having GFP in their cell bodies, focal optical stimulation evoked currents which had an immediate onset and displayed partial desensitization during long duration stimuli (Fig. 3Ci, n = 12). In current-clamp mode, optical stimulation led to depolarization and the generation of action potentials (Fig. 3Cii). In these neurons, application of NBQX and subsequent addition of gabazine did not have a significant effect on optically evoked currents (Fig. 3D,E, pre vs NBQX: t(7) = 1.8, p = 0.11, n = 8; pre vs gabazine: t(8) = 2.0, p = 0.076, n = 9, paired t tests). By contrast, NBQX reduced and gabazine abolished electrically evoked currents in these neurons (Fig. 3D,E; pre vs NBQX: t(6) = 5.7, p = 0.001, n = 7; pre vs gabazine: t(5) = 60, p = 2 × 10−8, n = 6, paired t tests). This indicates that intra-PAG injection of AAV-ChR2 led to functional postsynaptic ChR2 expression within the PAG.
To identify the intrinsic synaptic inputs onto RVM-projection and unlabeled PAG neurons, we performed electrophysiological recordings in GFP-negative neurons from animals that received intra-PAG AAV-ChR2 and intra-RVM retrograde tracer injections (Fig. 3F). In these PAG slices, GFP-labeled axons and terminals were identified that were apposed to the cell bodies and dendrites of both RVM-retrolabeled and unlabeled GFP-negative neurons (Fig. 3G). Focal optical stimulation evoked currents in RVM-retrolabeled and unlabeled GFP-negative PAG neurons which had a delayed onset, increased in amplitude with stimulus duration, and rapidly desensitized regardless of the stimulus duration (Fig. 3Hi). The success rate for obtaining optically evoked PSCs was similarly high in RVM-retrolabeled and unlabeled in GFP-negative PAG neurons (Fig. 3Hii; χ(1)2 = 0.44, p = 0.51, 96% vs 94%, n = 112/117 and 89/95, Fisher's test in retrolabeled and unlabeled). These optically evoked currents were unaffected by NBQX but were abolished by gabazine in both RVM-retrolabeled and unlabeled neurons (Fig. 3I,J; pre vs NBQX: t(6) = 0.3, p = 0.8 and t(6) = 0.9, p = 0.42, n = 7, 7; pre vs gabazine: t(8) = 48, p < 1 × 10−9, t(5) = 73, p = 1 × 10−8, n = 9, 6 paired t tests for retrolabeled and unlabeled neurons, respectively). In these neurons, electrically evoked PSCs were reduced by NBQX and subsequently abolished by co-application of gabazine (Fig. 3I,J, pre vs NBQX: t(5) = 2.8, p = 0.038 and t(7) = 3.8, p = 0.0067, n = 6, 8; pre vs gabazine: t(5) = 107, p = 1 × 10−9, t(5) = 71, p = 1 × 10−8, n = 6, 6, paired t tests for retrolabeled and unlabeled neurons). The effect of the blockers, NBQX and gabazine, did not differ between RVM-retrolabeled and unlabeled PAG neurons for both optically and electrically evoked PSCs (opto: F(1,25) = 0.6, p = 0.44; electrical: F(1,2) = 0.1, p = 0.78, ANOVA).
Finally, these gabazine-sensitive currents were abolished by TTX and this inhibition was recovered by addition of 4AP (Fig. 3K, pre vs TTX: t(6) = 26, p = 2 × 10−7 and t(5) = 85, p = 4 × 10−9; pre vs TTX + 4AP: t(6) = 0.5, p = 0.45, t(5) = 0.8, p = 0.75, n = 7, 6, paired t tests for retrolabeled and unlabeled neurons). By contrast, electrically evoked currents were abolished by TTX, and this inhibition was not recovered by 4AP (Fig. 3K, pre vs TTX: t(5) = 110, p = 1 × 10−9 and t(5) = 44, p = 1 × 10−7; pre vs TTX + 4AP: t(5) = 107, p = 1 × 10−7, t(5) = 40, p = 2 × 10−7, n = 6, 6, paired t tests; for retrolabeled and unlabeled neurons). The effect of the blockers, TTX and 4AP, did not differ between RVM-retrolabeled and unlabeled PAG neurons for both optically and electrically evoked PSCs (opto: F(1,22) = 0.2, p = 0.67; electrical: F(1,20) = 1.4, p = 0.26, ANOVA). Together with the NBQX insensitivity, these observations indicate both RVM-retrolabeled and unlabeled PAG neurons receive intrinsic monosynaptic inputs from GABAergic interneurons.
Opioids presynaptically inhibit intrinsic and extrinsic GABAergic inputs onto all PAG neurons
Opioids are thought to activate the descending PAG-RVM analgesic pathway by relieving intrinsic GABAergic inhibition of PAG neurons which project to the RVM (Lau et al., 2020). However, the above experiments indicate that in addition to intrinsic inputs, PAG neurons receive extrinsic GABAergic inputs from the CeA, which appear to preferentially target RVM-projecting neurons. The µ-opioid sensitivity of these amygdala inputs to the PAG are unknown. We therefore investigated the effect of the µ-opioid agonist DAMGO on optically evoked IPSCs in PAG neurons from animals that received an intra-RVM retrograde tracer together with either an intra-PAG or an intra-CeA injection of AAV-ChR2 (Fig. 4A–D). The level of DAMGO-sensitivity was compared between the two distinct GABAergic inputs and between RVM-retrolabeled and unlabeled neurons.
Figure 4.
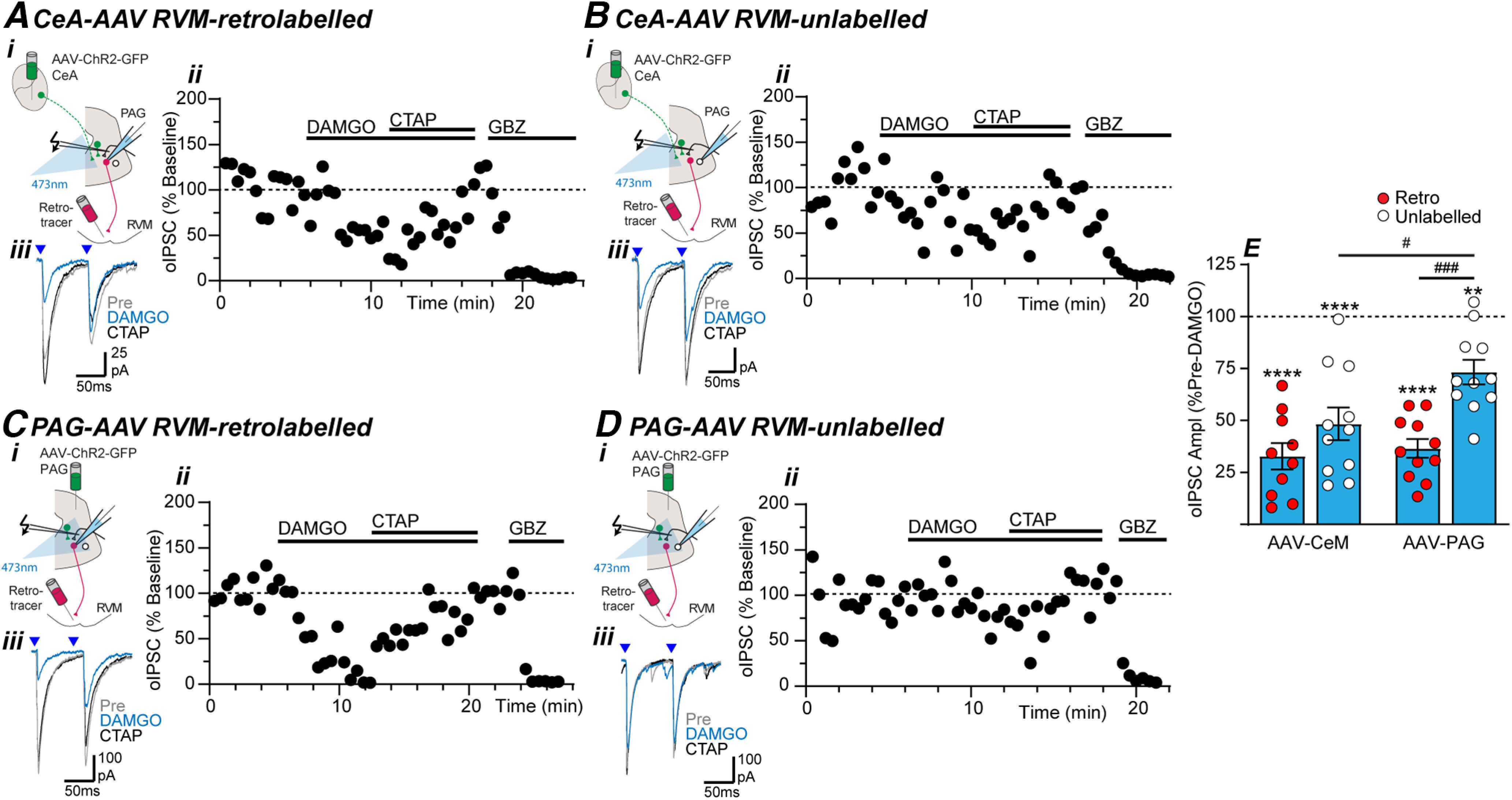
µ-Opioids suppress intrinsic and extrinsic-CeA GABAergic inputs onto RVM-projection and nonprojection PAG neurons. Effect of DAMGO, CTAP, and gabazine (GBZ) on optically evoked IPSCs (oIPSCs) in (A, C) RVM-retrolabeled and (B, D) unlabeled PAG neurons from (A, B) intra-CeA and (C, D) intra-PAG AAV-ChR2-injected animals. In panels A–D, (i) the experiment configuration in animals with an intra-RVM retrograde tracer injection plus an intra-CeA, or intra-PAG AAV-ChR2 injection; (ii) time plots of the normalized amplitude of evoked IPSCs (oIPSCs) from individual neurons, and (iii) averaged traces of paired optically evoked PSCs (blue arrows) from the same neuron. E, Scatter plots of the effect of DAMGO on the amplitude of optically evoked IPSCs (oIPSCs) in RVM-retrolabeled and unlabeled PAG neurons from intra-CeA and intra-PAG AAV-ChR2-injected animals. In E, **p < 0.01, ****p < 0.0001 (pre vs DAMGO paired t tests); #p < 0.05, ###p < 0.001 (post hoc Sidak comparisons).
In PAG slices from intra-CeA AAV-ChR2-injected animals, DAMGO produced a reduction in the amplitude of optically evoked IPSCs in both RVM-retrolabeled and unlabeled neurons which was reversed by the µ-opioid antagonist CTAP (Fig. 4A,B,E, pre vs DAMGO: t(9) = 11, p = 2 × 10−6 and t(10) = 6.5, p = 7 × 10−5, n = 10, 11, paired t tests in retrolabeled and unlabeled). Similarly, DAMGO produced a CTAP-sensitive inhibition of optically evoked IPSCs in both RVM-retrolabeled and unlabeled PAG neurons from intra-PAG AAV-ChR2-injected animals (Fig. 4C–E, pre vs DAMGO: t(10) = 14, p = 6 × 10−8 and t(10) = 4.6, p = 0.001, n = 11, 11, paired t tests in retrolabeled and unlabeled). However, the effect of DAMGO on optically evoked IPSCs differed between RVM-retrolabeled versus unlabeled PAG neurons (F(1,39) = 17.4, p = 0.0002, ANOVA), and between intra-PAG versus intra-CeA AAV-ChR2-injected animals (F(1,39) = 5.2, p = 0.029, ANOVA). Post hoc analysis revealed that the DAMGO-induced inhibition of optically evoked IPSCs was greater in RVM-retrolabeled PAG neurons compared with unlabeled neurons in slices from intra-PAG AAV-ChR2-injected animals (Fig. 4E; p = 0.0003 Sidak's post hoc comparison). By contrast, the DAMGO-induced inhibition of optically evoked IPSCs was not significantly different between RVM-retrolabeled and unlabeled neurons in slices from intra-CeA AAV-ChR2-injected animals (Fig. 4E; p = 0.17 Sidak's post hoc comparison). Further while the level of DAMGO-inhibition in unlabeled neurons differed between intra-PAG and intra-CeA-injected animals (Fig. 4E; p = 0.03, Sidak's post hoc comparison), there was no difference in the level of DAMGO-inhibition between the two inputs in RVM-retrolabeled neurons (Fig. 4E; p = 0.9, Sidak's post hoc comparison).
We next identified the locus of action of DAMGO by examining its effect on the paired-pulse ratio of evoked IPSCs, a well characterised form of presynaptic short-term plasticity (Zucker and Regehr, 2002). In these recordings, paired optical stimulation at a short interstimulus interval (70 ms) led to both paired pulse inhibition and facilitation for both extrinsic and intrinsic GABAergic synaptic inputs (Fig. 5A–E). There was no significant difference in the baseline paired pulse ratio of optically evoked IPSCs between RVM-retrolabeled and unlabeled neurons from intra-PAG and intra-CeA AAV-ChR2-injected animals (Fig. 5E, F(3,39) = 0.16, p = 0.92, ANOVA). DAMGO increased the paired-pulse ratio of optically evoked IPSCs in RVM-retrolabeled neurons in slices from both intra-CeA and intra-PAG AAV-ChR2-injected animals (Fig. 5A,C,E, pre vs DAMGO: t(9) = 2.4, 4.8, p = 0.041, 0.0007, n = 10, 10 for intra-CeA and intra-PAG; paired t tests). By contrast, DAMGO did not have a significant effect on the paired-pulse ratio of optically evoked IPSCs in unlabeled neurons in slices from intra-CeA and intra-PAG AAV-ChR2-injected animals (Fig. 5B,D,E, pre vs DAMGO: t(10) = 0.7, 0.9, p = 0.52, 0.4, n = 11, 11; paired t tests).
Figure 5.
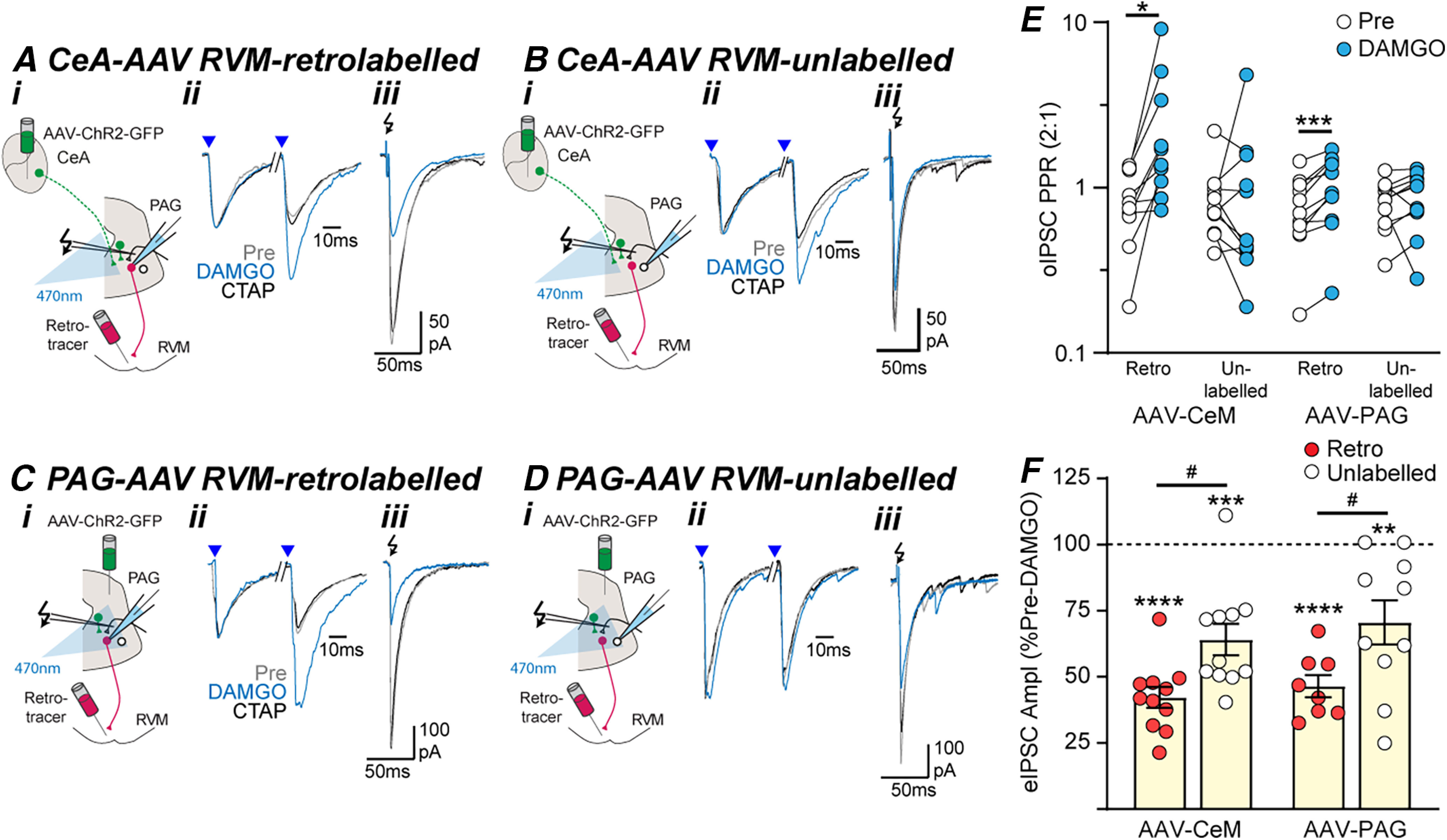
Opioid inhibition of GABAergic inputs onto PAG output neurons is presynaptic. Effect of DAMGO and CTAP on optically and electrically evoked IPSCs (oIPSCs, eIPSCs) in (A, C) RVM-retrolabeled and (B, D) unlabeled PAG neurons from (A, B) intra-CeA and (C, D) intra-PAG AAV-ChR2-injected animals. In panels A–D, (i) the experiment configuration in animals with an intra-RVM retrograde tracer injection plus and intra-CeA, or PAG AAV-ChR2 injection; (ii) averaged traces of paired optically evoked IPSCs (blue arrows) normalized to the first IPSC; (iii) and electrically (black arrows) evoked IPSCs (from the corresponding neurons in Fig. 4A–D). Scatter plots of the effect of DAMGO on (E) the paired-pulse ratio of oIPSCs and (F) the amplitude of eIPSCs in RVM-retrolabeled and unlabeled PAG neurons from intra-CeA and intra-PAG AAV-ChR2-injected animals. In E, F, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (pre vs DAMGO, paired t tests). In F, #p < 0.05 (post hoc Sidak comparisons).
Like optically evoked IPSCs, DAMGO produced a CTAP-sensitive reduction in the amplitude of electrically evoked IPSCs in RVM-retrolabeled and unlabeled PAG neurons (Fig. 5A–D,F, pre vs DAMGO: t(10) = 15, p = 5 × 10−8 and t(10) = 6.1, p = 1 × 10−4, n = 11, 11, paired t tests in intra-CeA AAV-ChR2 animals; t(7) = 13, p = 4 × 10−6 and t(9) = 3.5, p = 0.006, n = 8, 10, paired t tests in intra-PAG AAV-ChR2 animals). Unlike optically evoked IPSCs, while the DAMGO-induced inhibition of electrically evoked IPSCs differed between RVM-retrolabeled and unlabeled PAG neurons (F(1,36) = 15, p = 0.0005, two-way ANOVA, main effect), but there was no difference between intra-CeA and intra-PAG AAV2-injected animals (F(1,36) = 0.8, p = 0.38, two-way ANOVA, main effect) and no interaction between input and retrograde labeling (F(1,36) = 0.03, p = 0.9. two-way ANOVA, interaction; Fig. 5F). Thus, the DAMGO-induced inhibition of electrically evoked IPSCs was greater in RVM-retrolabeled PAG neurons compared with unlabeled neurons in slices from both intra-PAG and intra-CeA AAV-ChR2-injected animals (Fig. 5F, p = 0.02, 0.022 Sidak's post hoc comparisons).
Together, these data indicate intrinsic GABAergic inputs from putative PAG interneurons, are more sensitive to DAMGO presynaptic inhibition if they postsynaptically target RVM-projection neurons. In contrast, extrinsic GABAergic inputs from the CeA are equally sensitive to DAMGO-inhibition regardless of their target. This difference in DAMGO sensitivity was not identified in the electrical stimulation experiments, presumably because of the nonselective recruitment of extrinsic and intrinsic inputs.
Cannabinoids selectively inhibit GABAergic inputs from extrinsic CeA neurons onto RVM-projection neurons in PAG
We used a similar approach to determine the synapses targeted by cannabinoids within the descending PAG pathway. Intriguingly, the pan-cannabinoid receptor agonist WIN55212-2 only inhibited oIPSCs in RVM-projecting neurons from animals that received intra-CeA AAV-ChR2 injections (Fig. 6A–D). Thus, when the level of WIN55212-2 inhibition at all these synapses was compared, there were main effects of input (intra-CeA vs intra-PAG: F(1,35) = 13.0, p = 0.001, two-way ANOVA) and retrolabeling (retrolabeled vs unlabeled: F(1,35) = 7.7, p = 0.009, two-way ANOVA), plus an interaction between input and retrolabeling (F(1,35) = 12.29, p = 0.001, two-way ANOVA). Post hoc analysis revealed that WIN55212-2 produced a reduction in the amplitude of optically evoked IPSCs in RVM-retrolabeled but not unlabeled neurons in PAG slices from intra-CeA AAV-ChR2-injected animals (Fig. 6A–B,E, retro vs unlabeled: p = 0.0002 Sidak's post hoc comparisons; pre vs WIN55212-2 paired t tests: t(9) = 14, p = 2 × 10−7, n = 10 for retrolabeled and t(8) = 2.0 p = 0.081, n = 9 for unlabeled). By contrast, WIN55212-2 did not affect optically evoked IPSCs in either RVM-retrolabeled, or unlabeled PAG neurons in slices from intra-PAG AAV-ChR2-injected animals (Fig. 6C–E; retro vs unlabeled: p = 0.84 Sidak's post hoc comparison; pre vs WIN55212-2 paired t tests: t(9) = 2.0, p = 0.075, n = 10 for retrolabeled and t(9) = 2.0, p = 0.072, n = 10 for unlabeled). Addition of the CB1-cannabinoid receptor antagonist AM251 reversed the WIN55212-2 induced inhibition of optically evoked IPSCs in all RVM-retrolabeled neurons from intra-CeA AAV-ChR2-injected animals (Fig. 6A). Addition of AM251 had no effect on optically evoked IPSCs at the other synapses where WIN55212-2 was without effect (Fig. 6B–D).
Figure 6.
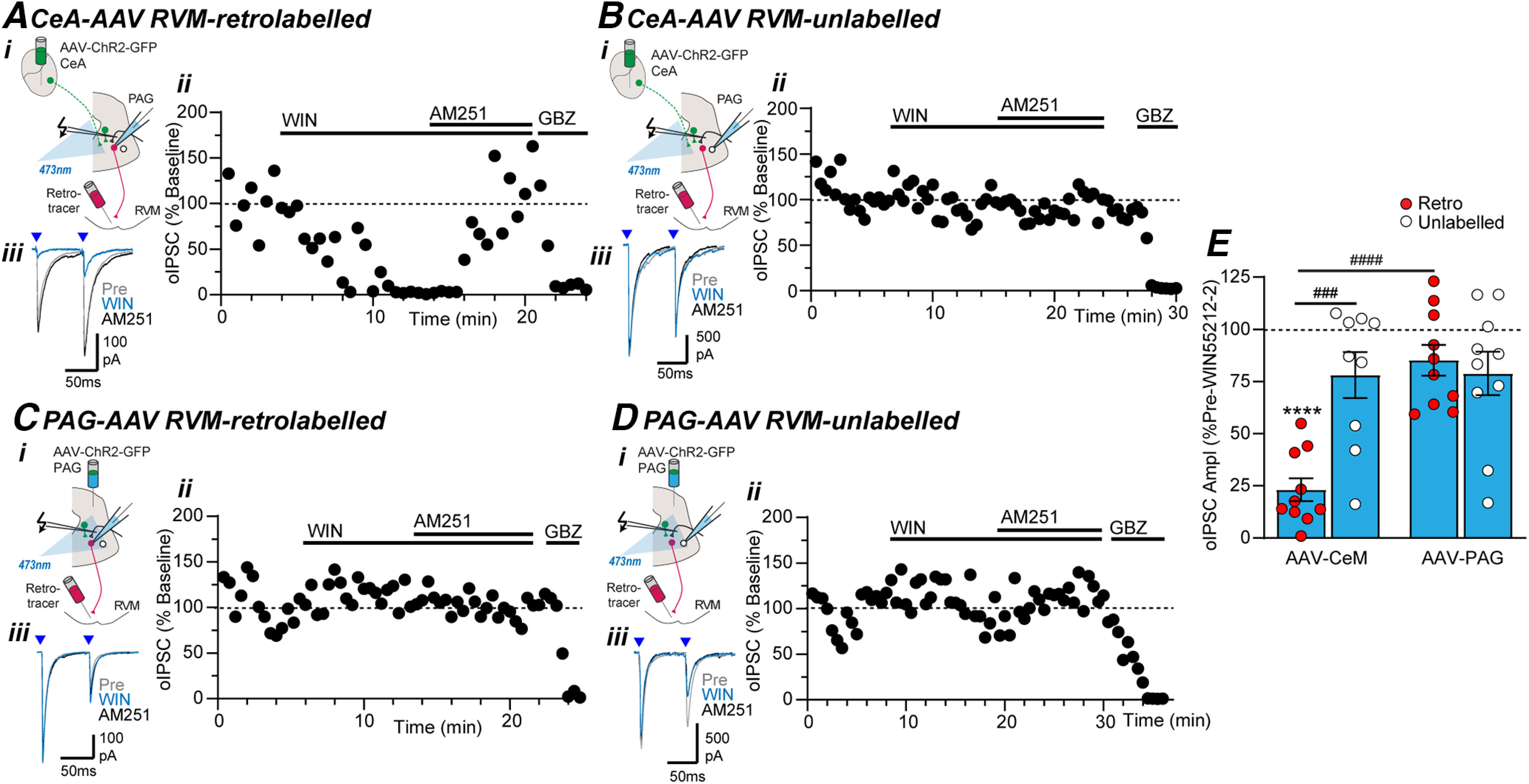
Cannabinoids selectively suppress extrinsic GABAergic inputs from CeA onto RVM-projection neurons in PAG. Effect of WIN55212-2 (WIN), AM251, and gabazine (GBZ) on optically evoked IPSCs (oIPSCs) in (A, C) RVM-retrolabeled and (B, D) unlabeled PAG neurons from (A, B) intra-CeA and (C, D) intra-PAG AAV-ChR2-injected animals. In panels A–D, (i) the experiment configuration in animals with an intra-RVM retrograde tracer injection plus an intra-CeA, or intra-PAG AAV-ChR2 injection, (ii) time plots of the normalized amplitude of evoked IPSCs (oIPSCs) from individual neurons, and (iii) averaged traces of paired optically evoked PSCs (blue arrows) from the same neuron. E, Scatter plots of the effect of WIN55212-2 on the amplitude of optically evoked IPSCs (oIPSCs) in RVM-retrolabeled and unlabeled PAG neurons from intra-CeA and intra-PAG AAV-ChR2-injected animals. In E, ****p < 0.0001 (pre vs WIN55212-2 paired t tests); ###p < 0.001, ####p < 0.0001 (post hoc Sidak comparisons).
As observed in the DAMGO experiments, there was no significant difference in the baseline paired pulse ratio of optically evoked IPSCs between RVM-retrolabeled and unlabeled neurons from intra-PAG and intra-CeA AAV-ChR2-injected animals (Fig. 7E, F(3,33) = 1.1, p = 0.37, ANOVA). The WIN55212-2 induced inhibition of optically evoked IPSCs in RVM-retrolabeled neurons from intra-CeA AAV-ChR2-injected animals was associated with an increase in paired-pulse ratio (Fig. 7A,E, pre vs WIN55212-2: t(9) = 4.9, p = 0.0008, n = 8, paired t test). WIN55212-2 did not have a significant effect on the paired-pulse ratio of optically evoked IPSCs in unlabeled neurons from intra-CeA AAV-ChR2-injected animals (Fig. 7B,E, pre vs WIN55212-2: t(8) = 0.4, p = 0.96, n = 9, paired t test), or in RVM-retrolabeled and unlabeled neurons from intra-PAG AAV-ChR2-injected animals (Fig. 7C–E, pre vs WIN55212-2: t(9) = 0.1, 0.7, p = 0.93, 0.48, n = 10, 10; paired t tests). Thus, WIN55212-2 acts via presynaptic CB1 receptors to selectively target extrinsic GABAergic inputs from the CeA onto RVM-projection PAG neurons.
Figure 7.
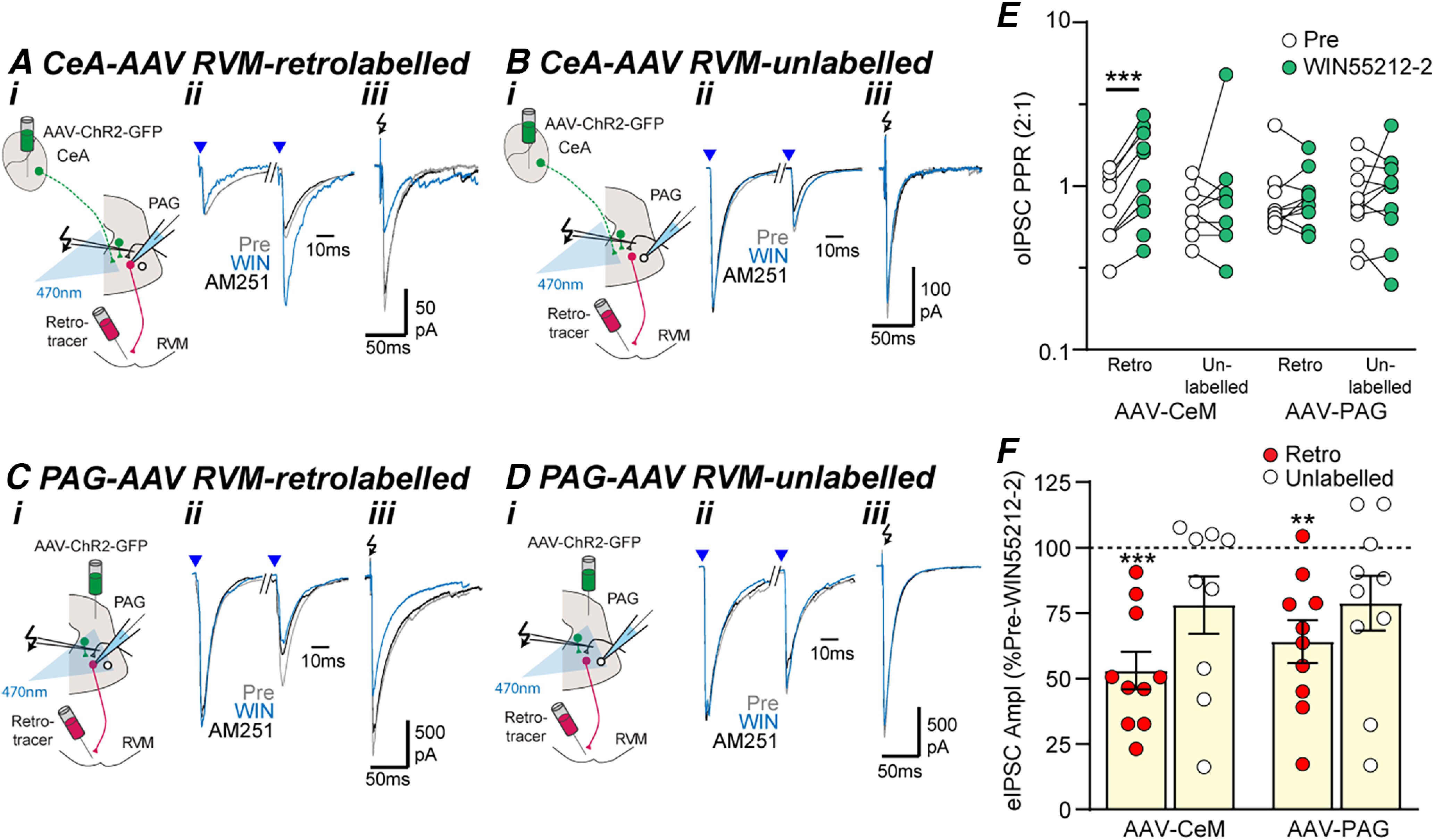
Cannabinoid inhibition of extrinsic GABAergic inputs onto PAG output neurons is presynaptic. Effect of WIN55212-2 (WIN) and AM251 on optically and electrically evoked IPSCs (oIPSCs, eIPSCs) in (A, C) RVM-retrolabeled and (B, D) unlabeled PAG neurons from (A, B) intra-CeA and (C, D) intra-PAG AAV-ChR2-injected animals. In panels A–D, (i) experiment configuration in animals with an intra-RVM retrograde tracer injection plus and intra-CeA, or PAG AAV-ChR2 injection, (ii) averaged traces of paired optically evoked IPSCs (blue arrows) normalized to the first IPSC, (iii) and electrically (black arrows) evoked IPSCs (from the corresponding neurons in Fig. 6A–D). Scatter plots of the effect of WIN55212-2 on (E) the paired-pulse ratio of oIPSCs and (F) the amplitude of eIPSCs in RVM-retrolabeled and unlabeled PAG neurons from intra-CeA and intra-PAG AAV-ChR2-injected animals. In E, F, **p < 0.01, ****p < 0.001 (pre vs WIN55212-2, paired t tests).
The effect of WIN55212-2 on electrically evoked IPSCs differed between RVM-retrolabeled and unlabeled PAG neurons, but not between animals which received an intra-PAG versus an intra-CeA injection of AAV-ChR2 (Fig. 7F, F(1,35) = 4.7, 0.41, p = 0.038, 0.52 two-way ANOVA main effects). WIN55212-2 inhibited electrically evoked IPSCs in RVM-retrolabeled, but not unlabeled PAG neurons from intra-CeA AAV-ChR2-injected animals (Fig. 7A–B,F, pre vs WIN55212-2: t(9) = 6.6, p = 0.0001, t(8) = 2.0, p = 0.081, n = 10, 9, paired t tests for retrolabeled and unlabeled) and from intra-PAG AAV-ChR2-injected animals (Fig. 7C,D,F, pre vs WIN55212-2: t(9) = 4.4, p = 0.002, t(9) = 2.0, p = 0.072, n = 10, 10, paired t tests for retrolabeled and unlabeled). Addition of the cannabinoid CB1 receptor antagonist AM251 reversed the WIN55212-2 induced inhibition of electrically evoked IPSCs in RVM-retrolabeled neurons, but had no effect in the WIN55212-2 insensitive unlabeled PAG neurons (Fig. 7A–Diii).
Discussion
The present study has demonstrated that within the midbrain PAG there is a complex integration of extrinsic and local GABAergic inputs onto output neurons which project along the descending analgesic pathway. It was observed that extrinsic CeA and local GABAergic inputs directly target both RVM-projection neurons and nonprojection neurons within the PAG. While local intrinsic GABAergic inputs targeted all PAG neurons indiscriminately, GABAergic inputs originating from the CeA preferentially innervated RVM-projection neurons. Further, these GABAergic synapses displayed differential sensitivity to opioids and cannabinoids. Opioids globally inhibited GABAergic synapses within the PAG, regardless of their source, although they had a greater impact on neurons projecting to the RVM. By contrast, cannabinoids exclusively controlled descending extrinsic GABAergic inputs from the CeA onto RVM-projection neurons. These findings suggest that the CeA controls RVM-projecting PAG neurons via a direct inhibitory pathway and a parallel indirect disinhibitory interneuron pathway. Unlike opioids, cannabinoids selectively target the direct inhibitory pathway, thereby acting as a disinhibitory gate for PAG descending outputs.
Intrinsic and extrinsic GABAergic inputs provide monosynaptic and disynaptic control of PAG output neurons
In the present study, optically evoked synaptic currents were observed in PAG neurons from animals which received an injection of AAV-ChR2 into the CeA, or the PAG under neuronal promoters. These optically evoked synaptic currents were abolished by GABAA, but not non-NMDA receptor antagonists, and were abolished and reinstated by TTX and 4AP, as observed previously (Michael et al., 2020). These observations indicate that PAG neurons receive monosynaptic intrinsic and extrinsic inputs from the CeA which are both primarily GABAergic, as observed previously (Michael et al., 2020). This is also consistent with recent optogenetic studies which have described extrinsic and intrinsic GABAgeric synaptic inputs using virally targeted delivery of ChR2 to GABAergic neurons (Tovote et al., 2016; Avegno et al., 2018; Yin et al., 2020). Unlike optically evoked synaptic currents, those induced by electrical stimulation contained both GABAA and non-NMDA receptor-mediated components, as observed previously (Vaughan and Christie, 1997). It is likely that the glutamatergic synaptic inputs onto PAG neurons are derived from extrinsic sources such as the prefrontal cortex, parabrachial nucleus, and spinal cord (Chen et al., 2017; Huang et al., 2019; Phelps et al., 2021).
The nature of CeA-PAG GABAergic circuitry detected in the present study was more complex than that described in prior optogenetic studies (Fig. 8A,B). The observation that PAG neurons that project to the RVM received a direct intrinsic GABAergic input is consistent with local inhibition of descending analgesic pathways, as proposed by Fields and Basbaum (1978). The observation that intrinsic PAG nonprojection neurons, which were likely to be GABAergic interneurons, received extrinsic GABAergic inputs from the CeA is similar to that reported in recent optogenetic studies (Tovote et al., 2016; Yin et al., 2020). Together, these findings are consistent with the idea that GABAergic CeA output neurons indirectly activate PAG output neurons via intrinsic GABAergic interneurons (Fig. 8A). Unlike recent optogenetic studies, a direct monosynaptic GABAergic input from CeA neurons onto RVM-projecting PAG output neurons was also detected in the present study (Fig. 8B; Tovote et al., 2016; Michael et al., 2020; Yin et al., 2020). Interestingly, we found that RVM-projecting PAG neurons received a higher proportion of GABAergic CeA inputs than unlabeled nonprojection PAG neurons. The lack of identification of a direct monosynaptic pathway in prior optogenetic studies may have been because of methodological and/or species differences (Tovote et al., 2016; Michael et al., 2020; Yin et al., 2020). Thus, while prior cre-lox mouse studies identified PAG neurons by their GABAergic versus glutamatergic phenotype, the present rat study identified PAG neurons by projection target. Alternatively, it is possible that tonic endocannabinoid silencing may have limited detection of the direct pathway in prior studies (see below), as observed at some central synapses (Winters and Vaughan, 2021). Overall, the present findings suggest that the CeA has the potential to control RVM-projection PAG neurons via parallel monosynaptic inhibitory and disynaptic disinhibitory pathways (Fig. 8B). While the phenotype of PAG output neurons was not examined in the present study, it might be noted that the RVM receives both glutamatergic and GABAergic inputs from the PAG, which is suggestive of even complexity in the downstream components of this pathway (Reichling and Basbaum, 1990b; Morgan et al., 2008).
Figure 8.

Models of GABAergic disinhibitory analgesic circuitry within the midbrain PAG. A, Prior optogenetic studies have identified an indirect disynaptic GABAergic pathway from the CeA to PAG output neurons. In this model, activation of CeA GABAergic output neurons inhibits tonically active PAG GABAergic interneurons, thereby disinhibiting, or activating PAG output neurons (CeA Stim in A). B, The current study has identified an additional parallel direct monosynaptic GABAergic pathway from the CeA onto PAG output neurons. In this model, activation of CeA GABAergic output neurons would inhibit PAG output neurons via the direct monosynaptic pathway. Thus, activation of CeA GABAergic output neurons would produce lesser activation of PAG output neurons because this direct inhibition would oppose the indirect disinhibition (CeA Stim in B vs A). The current study indicates that opioids disinhibit, and thereby activate PAG output neurons by inhibiting both their intrinsic and extrinsic-CeA GABAergic inputs; cannabinoids would produce less activation of PAG output neurons because they only act on extrinsic-CeA GABAergic inputs (OP, CB via µ and CB1 receptors in B). Furthermore, cannabinoids would facilitate CeA-induced activation of PAG output neurons via the indirect disynaptic disinhibitory pathway because they selectively silence the direct inhibitory pathway (CB + CeA Stim in B).
Differing global opioid and selective cannabinoid control of RVM-projection PAG output neurons in terms of their inputs and outputs
In the present study, the opioid and cannabinoid agonists, DAMGO and WIN55212-2, produced a suppression of optically evoked IPSCs in subpopulations of PAG neurons which was associated with an increase in their paired pulse ratio. The actions of DAMGO and WIN55212-2 were reversed by addition of CTAP and AM251, respectively. These observations are consistent with presynaptic µ-opioid and cannabinoid CB1 receptor-mediated inhibition of GABA release, as demonstrated in prior PAG electrical stimulation studies (Chieng and Christie, 1994; Vaughan and Christie, 1997; Vaughan et al., 1997, 2000, 2003; Chiou and Huang, 1999; Drew et al., 2008, 2009; Liao et al., 2011).
Opioids and cannabinoids, however, acted on distinct synapses within the PAG descending pathway. While opioids inhibited GABAergic synaptic transmission at all synapses, this inhibition was greater in RVM-projection neurons than nonprojection neurons, as demonstrated in a recent electrical stimulation study (Lau et al., 2020). However, this opioid presynaptic inhibition did not differ between intrinsic and extrinsic CeA inputs, in RVM-projection and nonprojection neurons. These observations are consistent with the intrinsic presynaptic disinhibition hypothesis of opioid induced analgesia and extends this to include disinhibition of extrinsic amygdala GABAergic inputs to the PAG (Fields and Basbaum, 1978; Lau and Vaughan, 2014). By contrast, cannabinoid presynaptic inhibition varied not only between RVM-projection and nonprojection PAG neurons, but also between intrinsic and extrinsic GABAergic inputs. Thus, cannabinoids inhibited extrinsic CeA GABAergic inputs onto RVM-projection neurons but not nonprojection neurons. Furthermore, cannabinoids had no effect on intrinsic GABAergic inputs onto RVM-projection and nonprojection neurons. These findings indicate that, unlike opioids, cannabinoids produce disinhibition within the CeA-PAG-RVM pathway by acting exclusively on extrinsic descending inputs onto RVM-projection neurons.
Implications for the analgesic properties of CeA-PAG descending inputs
A key question arising from these findings is “what is the primary function of CeA inputs to RVM-projecting PAG neurons and how do opioids and cannabinoids modulate this pathway?” As mentioned above, recent optogenetic and chemogenetic circuit mapping strategies have identified an indirect disynaptic GABAergic pathway from the CeA to PAG glutamatergic/RVM-projection neurons (Fig. 8A; Tovote et al., 2016; Yin et al., 2020). Activation of this indirect CeA descending GABAergic pathway would inhibit GABAergic interneurons, and thereby disinhibit or activate PAG output projection neurons (Fig. 8A, CeA Stim). This is consistent with both early studies and more recent findings suggesting activation of the CeA is involved in various functions such as analgesia, freezing and vocalisation, which require activation of PAG output neurons (Manning and Mayer, 1995; Oliveira and Prado, 2001; Tovote et al., 2016; Yin et al., 2020). Intriguingly, however, we also identified a prominent direct monosynaptic pathway from the CeA to PAG RVM-projection neurons (Fig. 8B). Activation of this direct CeA descending pathway would inhibit PAG RVM-projection neurons and consequently reduce the excitatory disynaptic disinhibition of RVM-projection neurons via the indirect pathway (Fig. 8B, CeA Stim). We propose that selective cannabinoid silencing of the direct GABAergic pathway would facilitate CeA-induced activation of RVM-projecting PAG neurons via the indirect disynaptic pathway (Fig. 8B, CB+CeA Stim). Therefore, these parallel monosynaptic and disynaptic GABAergic pathways within the PAG provide a flexible system which can potentially activate or inhibit the descending PAG-RVM analgesic pathway, depending on the prevailing state of these GABAergic synapses. This complex modulatory system provides a basis for the distinct roles of cannabinoids and opioids in stress-induced analgesia within the CeA-PAG-RVM descending pathway (Akil et al., 1976; Lewis et al., 1980; Valverde et al., 2000; Hohmann et al., 2005; Atwal et al., 2020; Yin et al., 2020).
In conclusion, it has long been thought that opioids activate descending analgesic systems from the midbrain PAG by relieving intrinsic GABAergic inhibition of PAG output neurons. The present study has expanded this by demonstrating that opioids nonselectively disinhibit PAG output neurons and interneurons by acting on both intrinsic and extrinsic GABAergic inputs. Unlike opioids, cannabinoids selectively disinhibited PAG output neurons by targeting extrinsic GABAergic inputs from the amygdala. Thus, while opioids have a widespread role in controlling GABAergic activity within the amygdala-PAG-RVM descending pathway, cannabinoids exclusively target direct GABAergic amygdala inputs onto RVM-projecting PAG neurons. This suggests that cannabinoid and opioid systems will have distinct functional actions, as has been observed for stress-induced analgesia from within this brain region.
Footnotes
This work was supported by the Ernst Heine Family Foundation and the Pain Foundation.
The authors declare no competing financial interests.
References
- Akil H, Mayer DJ, Liebeskind JC (1976) Antagonism of stimulation-produced analgesia by naloxone, a narcotic antagonist. Science 191:961–962. 10.1126/science.1251210 [DOI] [PubMed] [Google Scholar]
- Atwal N, Winters BL, Vaughan CW (2020) Endogenous cannabinoid modulation of restraint stress-induced analgesia in thermal nociception. J Neurochem 152:92–102. 10.1111/jnc.14884 [DOI] [PubMed] [Google Scholar]
- Avegno EM, Lobell TD, Itoga CA, Baynes BB, Whitaker AM, Weera MM, Edwards S, Middleton JW, Gilpin NW (2018) Central amygdala circuits mediate hyperalgesia in alcohol-dependent rats. J Neurosci 38:7761–7773. 10.1523/JNEUROSCI.0483-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaresi P, Manfrini E (1988) Glutamate decarboxylase-immunoreactive neurons and terminals in the periaqueductal gray of the rat. Neuroscience 27:183–191. 10.1016/0306-4522(88)90229-1 [DOI] [PubMed] [Google Scholar]
- Chen Q, Roeder Z, Li MH, Zhang Y, Ingram SL, Heinricher MM (2017) Optogenetic evidence for a direct circuit linking nociceptive transmission through the parabrachial complex with pain-modulating neurons of the rostral ventromedial medulla (RVM). eNeuro 4:ENEURO.0202-17.2017. 10.1523/ENEURO.0202-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ (1994) Inhibition by opioids acting on mu-receptors of GABAergic and glutamatergic postsynaptic potentials in single rat periaqueductal gray neurones in vitro. Br J Pharmacol 113:303–309. 10.1111/j.1476-5381.1994.tb16209.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou LC, Huang LY (1999) Mechanism underlying increased neuronal activity in the rat ventrolateral periaqueductal grey by a mu-opioid. J Physiol 518:551–559. 10.1111/j.1469-7793.1999.0551p.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Deisseroth K, Bolshakov VY (2013) Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron 80:1491–1507. 10.1016/j.neuron.2013.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis A, Morgan MM, Liebeskind JC (1987) GABAergic modulation of the analgesic effects of morphine microinjected in the ventral periaqueductal gray matter of the rat. Brain Res 436:223–228. 10.1016/0006-8993(87)91665-9 [DOI] [PubMed] [Google Scholar]
- Drew GM, Mitchell VA, Vaughan CW (2008) Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signaling. J Neurosci 28:808–815. 10.1523/JNEUROSCI.4876-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew GM, Lau BK, Vaughan CW (2009) Substance P drives endocannabinoid-mediated disinhibition in a midbrain descending analgesic pathway. J Neurosci 29:7220–7229. 10.1523/JNEUROSCI.4362-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI (1978) Brainstem control of spinal pain-transmission neurons. Annu Rev Physiol 40:217–248. 10.1146/annurev.ph.40.030178.001245 [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Fields HL (2013) Central nervous system mechanisms of pain modulation. In: Wall and Melzack's textbook of pain, Ed 6 (McMahon SB, Koltzenburg M, Tracey I, Turk D, eds), pp 129–142. Philadelphia: Elsevier. [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112. 10.1038/nature03658 [DOI] [PubMed] [Google Scholar]
- Huang J, Gadotti VM, Chen L, Souza IA, Huang S, Wang D, Ramakrishnan C, Deisseroth K, Zhang Z, Zamponi GW (2019) A neuronal circuit for activating descending modulation of neuropathic pain. Nat Neurosci 22:1659–1668. 10.1038/s41593-019-0481-5 [DOI] [PubMed] [Google Scholar]
- Keay KA, Bandler R (2001) Parallel circuits mediating distinct emotional coping reactions to different types of stress. Neurosci Biobehav Rev 25:669–678. 10.1016/s0149-7634(01)00049-5 [DOI] [PubMed] [Google Scholar]
- Lane DA, Patel PA, Morgan MM (2005) Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neuroscience 135:227–234. 10.1016/j.neuroscience.2005.06.014 [DOI] [PubMed] [Google Scholar]
- Lau BK, Vaughan CW (2014) Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr Opin Neurobiol 29:159–164. 10.1016/j.conb.2014.07.010 [DOI] [PubMed] [Google Scholar]
- Lau BK, Winters BL, Vaughan CW (2020) Opioid presynaptic disinhibition of the midbrain periaqueductal grey descending analgesic pathway. Br J Pharmacol 177:2320–2332. 10.1111/bph.14982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Cannon J, Liebeskind J (1980) Opioid and nonopioid mechanisms of stress analgesia. Science 208:623–625. 10.1126/science.7367889 [DOI] [PubMed] [Google Scholar]
- Liao HT, Lee HJ, Ho YC, Chiou LC (2011) Capsaicin in the periaqueductal gray induces analgesia via metabotropic glutamate receptor-mediated endocannabinoid retrograde disinhibition. Br J Pharmacol 163:330–345. 10.1111/j.1476-5381.2011.01214.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BH, Mayer DJ (1995) The central nucleus of the amygdala contributes to the production of morphine antinociception in the rat tail-flick test. J Neurosci 15:8199–8213. 10.1523/JNEUROSCI.15-12-08199.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM (1999) Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Res 822:237–242. 10.1016/s0006-8993(98)01368-7 [DOI] [PubMed] [Google Scholar]
- Michael V, Goffinet J, Pearson J, Wang F, Tschida K, Mooney R (2020) Circuit and synaptic organization of forebrain-to-midbrain pathways that promote and suppress vocalization. Elife 9:e63493. 10.7554/eLife.63493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau JL, Fields HL (1986) Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission. Brain Res 397:37–46. 10.1016/0006-8993(86)91367-3 [DOI] [PubMed] [Google Scholar]
- Morgan MM, Whittier KL, Hegarty DM, Aicher SA (2008) Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain 140:376–386. 10.1016/j.pain.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Tsumori T, Yokota S, Yasui Y (2008) Neuroanatomical and neurochemical organization of projections from the central amygdaloid nucleus to the nucleus retroambiguus via the periaqueductal gray in the rat. Neurosci Res 62:286–298. 10.1016/j.neures.2008.10.004 [DOI] [PubMed] [Google Scholar]
- Oliveira MA, Prado WA (2001) Role of PAG in the antinociception evoked from the medial or central amygdala in rats. Brain Res Bull 54:55–63. 10.1016/s0361-9230(00)00420-2 [DOI] [PubMed] [Google Scholar]
- Park C, Kim JH, Yoon BE, Choi EJ, Lee CJ, Shin HS (2010) T-type channels control the opioidergic descending analgesia at the low threshold-spiking GABAergic neurons in the periaqueductal gray. Proc Natl Acad Sci U S A 107:14857–14862. 10.1073/pnas.1009532107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps CE, Lumb BM, Donaldson LF, Robinson ES (2021) The partial saphenous nerve injury model of pain impairs reward-related learning but not reward sensitivity or motivation. Pain 162:956–966. 10.1097/j.pain.0000000000002177 [DOI] [PubMed] [Google Scholar]
- Reichling DB, Basbaum AI (1990a) Contribution of brainstem GABAergic circuitry to descending antinociceptive controls: II. Electron microscopic immunocytochemical evidence of GABAergic control over the projection from the periaqueductal gray to the nucleus raphe magnus in the rat. J Comp Neurol 302:378–393. 10.1002/cne.903020214 [DOI] [PubMed] [Google Scholar]
- Reichling DB, Basbaum AI (1990b) Contribution of brainstem GABAergic circuitry to descending antinociceptive controls: I. GABA-immunoreactive projection neurons in the periaqueductal gray and nucleus raphe magnus. J Comp Neurol 302:370–377. 10.1002/cne.903020213 [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM, Shipley MT (1991) Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. J Comp Neurol 303:121–131. 10.1002/cne.903030111 [DOI] [PubMed] [Google Scholar]
- Samineni VK, Grajales-Reyes JG, Copits BA, O'Brien DE, Trigg SL, Gomez AM, Bruchas MR, Gereau RW 4th (2017) Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. eNeuro 4:ENEURO.0129-16.2017. 10.1523/ENEURO.0129-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Esposito MS, Botta P, Chaudun F, Fadok JP, Markovic M, Wolff SB, Ramakrishnan C, Fenno L, Deisseroth K, Herry C, Arber S, Lüthi A (2016) Midbrain circuits for defensive behaviour. Nature 534:206–212. 10.1038/nature17996 [DOI] [PubMed] [Google Scholar]
- Valverde O, Ledent C, Beslot F, Parmentier M, Roques BP (2000) Reduction of stress-induced analgesia but not of exogenous opioid effects in mice lacking CB1 receptors. Eur J Neurosci 12:533–539. 10.1046/j.1460-9568.2000.00929.x [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ (1997) Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J Physiol 498:463–472. 10.1113/jphysiol.1997.sp021872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor M, Christie MJ (1997) How opioids inhibit GABA mediated neurotransmission. Nature 390:611–4614. 10.1038/37610 [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Connor M, Bagley EE, Christie MJ (2000) Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol Pharmacol 57:288–295. [PubMed] [Google Scholar]
- Vaughan CW, Bagley EE, Drew GM, Schuller A, Pintar JE, Hack SP, Christie MJ (2003) Cellular actions of opioids on periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR-1. Br J Pharmacol 139:362–367. 10.1038/sj.bjp.0705261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BL, Vaughan CW (2021) Mechanisms of endocannabinoid control of synaptic plasticity. Neuropharmacology 197:108736. 10.1016/j.neuropharm.2021.108736 [DOI] [PubMed] [Google Scholar]
- Yeung JC, Yaksh TL, Rudy TA (1977) Concurrent mapping of brain sites for sensitivity to the direct application of morphine and focal electrical stimulation in the production of antinociception in the rat. Pain 4:23–40. [DOI] [PubMed] [Google Scholar]
- Yin W, Mei L, Sun T, Wang Y, Li J, Chen C, Farzinpour Z, Mao Y, Tao W, Li J, Xie W, Zhang Z (2020) A central amygdala-ventrolateral periaqueductal gray matter pathway for pain in a mouse model of depression-like behavior. Anesthesiology 132:1175–1196. 10.1097/ALN.0000000000003133 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]