

Abstract
Despite the increasing knowledge about factors shaping the human microbiome, the host genetic factors that modulate the skin-microbiome interactions are still largely understudied. This contrasts with recent efforts to characterize host genes that influence the gut microbiota. Here, we investigated the effect of genetics on skin microbiota across three different skin microenvironments through meta-analyses of genome-wide association studies (GWAS) of two population-based German cohorts. We identified 23 genome-wide significant loci harboring 30 candidate genes involved in innate immune signaling, environmental sensing, cell differentiation, proliferation and fibroblast activity. However, no locus passed the strict threshold for study-wide significance (P < 6.3 × 10−10 for 80 features included in the analysis). Mendelian randomization (MR) analysis indicated the influence of staphylococci on eczema/dermatitis and suggested modulating effects of the microbiota on other skin diseases. Finally, transcriptional profiles of keratinocytes significantly changed after in vitro co-culturing with Staphylococcus epidermidis, chosen as a representative of skin commensals. Seven candidate genes from the GWAS were found overlapping with differential expression in the co-culturing experiments, warranting further research of the skin commensal and host genetic makeup interaction.
Subject terms: Microbiome, Genetics research
Microbiome composition is influenced by genetics, although the specific host genetic factors responsible are not well known. Here, the authors performed a genome-wide meta-analysis to discover host genetic effects on skin microbiota and finding potential causal effects of microbiota composition on skin diseases.
Introduction
Human-associated microbial communities show individual-specific variation shaped by a multitude of factors1,2. For skin in particular, the bacterial community composition is strongly influenced by host characteristics, such as skin microenvironment, sex, age and body mass index (BMI), and to a lesser extent by lifestyle and environmental expositions3. The genetic influence of the host on skin microbiome composition and diversity was suggested by findings indicating heritability of up to 56.4% for single taxonomic branches of skin commensals in twins4. Furthermore, host genetics and skin microbiota interactions haven been suggested by studies including targeted genes4 and in the context of inflammatory diseases, such as atopic dermatitis5. Nevertheless, the influence of host genetics on the skin microbiome is largely understudied and no dedicated genome-wide association study (GWAS) of host genetics and the bacterial community inhabiting the skin has been performed so far. This strongly contrasts with what is known about the human gut microbiota, where a variety of associated genomic loci and pathways has been identified by large GWAS6,7. Together, these gut microbiome-based GWAS have not only suggested how human molecular mechanisms modulate the microbiome but also indicated the consequences of such modulation to the host health and disease.
Therefore, we aimed to study the effects of genetics on skin microbiota across skin microenvironments through meta-analyses of GWAS of two German cohorts. To investigate the putative influence of the skin microbiota in skin diseases we applied Mendelian randomization (MR) analysis. Finally, putative effects of the skin microbiome members on the expression of candidate genes identified by GWAS were tested using normal human epidermal keratinocytes cultured with the common skin bacterium Staphylococcus epidermidis.
Results and discussion
A total of 1656 skin samples from participants of two cross-sectional, population-based German cohorts, KORA FF4 (nIndividuals = 324) and PopGen (nIndividuals = 273)8,9 were analyzed. Skin samples were taken from dry [dorsal and volar forearm (PopGen)], moist [antecubital fossa (KORA FF4 and PopGen)] and sebaceous [retroauricular fold (KORA FF4) and forehead (PopGen)] skin microenvironments (Fig. 1a–c, Supplementary Table 1). Microbial community profiles were obtained from sequencing of the V1-V2 regions from the 16 S ribosomal RNA (rRNA) gene (see Methods). Genome-wide association analyses were conducted on univariate relative abundances of individual bacteria (amplicon sequence variants; ASVs) and non-redundant taxonomic groups ranging from genus to phylum levels (79 in total; see Methods). Additionally, multivariate community composition (i.e., beta diversity as captured by Bray-Curtis dissimilarity) was analyzed for association with host genetic variation. The umbrella term “microbial feature” will henceforth be used in this article for all 80 analyzed input data.
Fig. 1. Characteristics of KORA FF4 and PopGen cohorts.

a Female (orange) and male (blue) composition of cohorts. b age and, c body mass index (BMI) distribution in cohorts. d ordination of skin microbiome profiles based on Bray-Curtis dissimilarity and principal coordinates analysis. Samples (n = 1,656) were colored by the skin site and represent dry [dorsal (D.) forearm (n = 260) and volar (V.) forearm (n = 251)], moist [antecubital (A.) fossa (n = 318 in KORA FF4, n = 258 in PopGen)] and sebaceous [seb.; forehead (n = 252) and retroauricular (R.) fold (n = 317)] microenvironments. Cohort names were abbreviated, PopGen (P) and KORA FF4 (K). Marginal boxplots are shown to visualize sample distributions along axes. The boxplot area represents the interquartile range (IQR) divided by the median. Lines extend to a maximum of 1.5 × IQR beyond the area. Points are outliers. Percentage of variation explained by each axis is shown in parentheses.
We tested the association of microbial features with variation in 4,685,714 human autosomal single nucleotide polymorphisms (SNP), accounting for main confounders of the skin microbiota (age, sex and BMI) and genetic background of study participants (see Methods)3,7. Cohort-wise association results were combined in a meta-analysis framework according to skin microenvironment, justified by the observed similarity of the microbiota profiles of samples from the same microenvironment (Fig. 1d). To assure robustness of association results, only loci with genome-wide significance (PMeta < 5 × 10−8) and with nominal significance in both cohorts (P < 0.05) were further considered (see Methods for details).
A total of 23 loci showed a genome-wide significant association with skin microbial features, of which 22 were linked to univariate features (Table 1 and Fig. 2a). However, none of these passed the strict threshold for study-wide significance (P < 6.3 × 10−10 for 80 features included in the analysis, see Methods). Most of the associations were found in moist skin microenvironment (n = 11), followed by dry (n = 7) and sebaceous (n = 5) (Fig. 2b). There was a tendency for a higher number of associations found in deeper taxonomic levels: the highest number of significant associations were found at the ASV level (n = 8), followed by genus level (n = 6; Fig. 2c). Of all microbial features deeper than family level, features within the genus Staphylococcus were associated with most loci (n = 5; Fig. 2d). Bayesian fine-mapping or linkage disequilibrium (LD) structure prioritized 462 genetic variants as potentially causal (Supplementary Data 1). A total of 30 genes were found of interest for containing potentially causal variants and/or because these variants were significantly associated with the gene expression in skin tissue from the GTEx portal10 (Table 1). Of these, 27 were protein coding genes, one an rRNA pseudogene and two long-non coding RNA (lncRNA) genes (Supplementary Data 2). Most of the protein coding genes were expressed in skin tissue and found expressed in different cell types in skin in datasets from previous studies (see Methods for details11,12) (Fig. 3). In the next section, we will explore the genes of interest with functional roles related to the host-microbiome interface.
Table 1.
Results summary of GWAS for microbial features
| ID | Chr | Position | rsID | Effect allele | EAF | Other allele | Microenv. | Feature | N (total) | Beta ± s.e. | P value | Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 17323400 | rs1396075 | T | 0.76 | A | Moist | c.Gammaproteobacteria | 563 | 0.37 ± 0.067 | 3.4×10−08 | ■ |
| 2 | 2 | 43812831 | rs12466030 | A | 0.66 | G | Moist | a.ASV070 [Veillonella (unc.)] | 226 | 0.583 ± 0.103 | 1.7×10-08 | THADA |
| 3 | 3 | 9164097 | rs2664121 | T | 0.73 | G | Dry | g.Micrococcus | 402 | −0.33 ± 0.112 | 4.3×10-09 | ENSG00000269886,SRGAP3 |
| 4 | 3 | 12514124 | rs709165 | G | 0.54 | A | Moist | a.ASV006 [S. hominis] | 398 | 0.379 ± 0.069 | 4.0×10-08 | MKRN2,MKRN2OS,RAF1,RNA5SP123,TSEN2 |
| 5 | 4 | 3266916 | rs2159173 | A | 0.93 | T | Sebaceous | a.ASV093 [Staphylococcus (unc.)] | 276 | −0.957 ± 0.159 | 1.8×10-09 | HTT,MSANTD1,RGS12 |
| 6 | 4 | 55057749 | rs55702239 | G | 0.77 | A | Dry | o.Bacteroidales,g.Bacteroides | 349 | −0.43 ± 0.103 | 3.5×10-08 | FIP1L1,PDGFRA |
| 7 | 5 | 14584609 | rs152620 | A | 0.79 | T | Moist | g.Acinetobacter | 454 | −0.444 ± 0.081 | 3.7×10-08 | OTULINL |
| 8 | 6 | 69060156 | rs9445997 | T | 0.62 | C | Sebaceous | g.Staphylococcus | 569 | −0.328 ± 0.06 | 4.0×10-08 | ■ |
| 9 | 6 | 93109029 | rs2757026 | C | 0.51 | T | Moist | f.Clostridiales_Incertae_Sedis_XI | 363 | −0.404 ± 0.072 | 2.2×10-08 | ■ |
| 10 | 6 | 144022040 | rs9484795 | T | 0.77 | C | Dry | g.Anaerococcus | 352 | −0.41 ± 0.132 | 3.4×10-08 | PHACTR2 |
| 11 | 7 | 57369680 | rs11762959 | A | 0.55 | G | Moist | o.Lactobacillales | 489 | 0.348 ± 0.064 | 4.5×10-08 | ■ |
| 12 | 7 | 57369974 | rs7791487 | A | 0.56 | T | Moist | f.Streptococcaceae | 458 | 0.378 ± 0.064 | 3.4×10-09 | ■ |
| 13 | 8 | 125763112 | rs59379063 | T | 0.92 | A | Sebaceous | a.ASV093 [Staphylococcus (unc.)] | 279 | −0.733 ± 0.133 | 3.2×10-08 | ■ |
| 14 | 9 | 126919126 | rs10121400 | T | 0.64 | A | Moist | o.Burkholderiales | 509 | −0.354 ± 0.063 | 2.4×10-08 | ■ |
| 15 | 11 | 106233170 | rs17105612 | G | 0.94 | A | Moist | a.ASV013 [S. epidermidis] | 361 | −0.928 ± 0.165 | 1.8×10-08 | ■ |
| 16 | 12 | 96938142 | rs12423627 | T | 0.93 | C | Sebaceous | a.ASV002 [Staphylococcus (unc.)] | 568 | 0.653 ± 0.113 | 6.9×10-09 | CFAP54 |
| 17 | 12 | 101194600 | rs4764996 | G | 0.93 | A | Dry | a.ASV013 [S. epidermidis] | 348 | −1.2 ± 0.204 | 2.1×10-08 | ANO4 |
| 18 | 13 | 33581388 | rs1543797 | T | 0.75 | C | Dry | Beta-diversity | 511 | ■ | 3.2×10-08 | ■ |
| 19 | 13 | 38067575 | rs12583353 | A | 0.89 | G | Dry | g.Paracoccus | 372 | −0.94 ± 0.144 | 5.6×10-10 | ■ |
| 20 | 14 | 33629140 | rs17100281 | G | 0.94 | A | Moist | a.ASV021 [Micrococcus (unc.)] | 331 | −0.969 ± 0.176 | 3.7×10-08 | NPAS3 |
| 21 | 16 | 28056516 | rs8049083 | C | 0.72 | A | Sebaceous | a.ASV004 [Corynebacterium (unc.)] | 505 | −0.378 ± 0.068 | 2.8×10-08 | GSG1L |
| 22 | 17 | 5341050 | rs2472614 | G | 0.89 | C | Dry | a.ASV086 [A. johnsonii] | 255 | −1.08 ± 0.186 | 4.7×10-08 | C1QBP,DERL2,DHX33,ENSG00000263272,MIS12,NUP88,RABEP1,RPAIN |
| 23 | 19 | 48742067 | rs6509364 | C | 0.65 | T | Moist | f.Rhodobacteraceae | 377 | −0.415 ± 0.072 | 8.5×10-09 | CARD8,TMEM143,ZNF114 |
Single variant association tests were performed for each sample type and each microbial feature. Tests were adjusted for age, sex, body mass index (BMI) and genetic background (first ten genetic principal components). Positions are given as in genome assembly hg19 (GRCh37). Effect allele frequency (EAF) and total sample number (N) for the meta-analysis (sample pairs for dry) are shown. Results from moist and sebaceous skin sites were combined by meta-analysis (using METAL software for beta diversity and METASOFT software for univariate microbial features) and considered significant when PMeta value were genome-wide significant (PMeta < 5 × 10−8) and data sets were nominal significant (P < 0.05). Loci from dry data sets were considered significant when at least one data set resulted in genome-wide significance (lowest P value shown as PMeta) and the other in nominal significance. Meta-analyses were weighted by sample size for multivariate microbial feature and by inverse variance for univariate features. Effect sizes (β) and its standard error (s.e.) from meta-analyses are shown for moist and sebaceous. Effect size and standard error from tests with volar forearm are shown for dry skin. Tests were two-sided. Candidate causal variants were identified by fine-mapping or based on LD > 0.6 to the lead genetic variant. Genes with variants within their region (no formatted font) or with variants associated with their expression (italic font) are shown. Genes are shown in bold font when both conditions are met. Microbial features are prefixed with their level, amplicon variant sequence (a.), genus (g.), family (f.), order (o.), class (c.) or phylum (p.). Association with rs55702239 in dry sites have been identified with the non-redundant features o.Bacteroidales and g.Bacteroides. For simplicity, only statistics related to the genus level is shown in this case. ENSG00000263272 is a novel transcript, antisense to RPAIN and ENSG00000269886 is a novel transcript, antisense to TTLL3.
Fig. 2. Results from the GWAS.

a Manhattan plot of per skin microenvironment meta-analysis. Lowest P value of each position is shown and identified by locus ID and rsID. Meta-analysis P values were obtained using the software METAL and METASOFT or by combining P values from data sets that originated from dry skin sites, see Methods. Significant positions are colored according to skin microenvironment and listed, where leading genetic variant, protein coding genes selected by fine-mapping as containing possible causal variants and microbial features are reported. Table 1 contains the list of loci characteristics and genes. b Count of significantly associated loci per microenvironment. c Level of microbial features with highest number of significant associations. d Sub-family features with the highest number of significant associations.
Fig. 3. Expression of human genes associated with the skin microbiome in public databases.

Candidate protein coding genes were selected by GWAS in skin. Upper panel shows the normalized transcriptional expression of genes in skin tissue. Data are from Human Protein Atlas version 20.111, which additionally includes data sets from the GTEx10 and the Functional annotation of the mammalian genome (FANTOM5)68 projects. Bottom panel shows candidate gene expression in different skin cell types. Single-cell expression was normalized by cell type. Genes differently expressed in each cell type in comparison with the others are highlighted. Displayed log-normalized gene expression data and differential expression analyses are retrieved from Solé-Boldo et al.12. Candidate genes were mapped by gene symbol.
Host functions associated with the human skin microbiota
Genetic variants associated with the skin bacteria were localized in genes related to pathogen sensing and regulation of response to pathogens. C1QBP (locus id: 22, lead variant rs2472614, PMeta = 4.7 × 10−8, associated with ASV086 [Acinetobacter johnsonii]), for instance, encodes the complement component 1, q subcomponent binding protein (C1qBP, a.k.a. gC1q-R/p33) and is abundantly expressed in keratinocytes (Fig. 312). C1qBP is an ubiquitous, multi-ligand, multifunctional and multicompartmental protein, which also acts as endothelial receptor to plasma proteins from the complement and kinin/kallikrein systems and is a marker for epithelial cell proliferation13,14. C1qBP binds to microbial proteins15, including Staphylococcus aureus protein A16, and therefore, is suggested to play a role in both the response to and pathogenesis of microbes17. Additionally, DHX33, (locus id: 22, same locus containing gene C1QBP), and CARD8 (locus id: 23, rs6509364, PMeta = 8.5 × 10−09, associated with the Rhodobacteraceae family) encode proteins which regulate inflammasome activity, which in turn regulate innate immunity caspase 1 activation18. DHX33 activates the NLRP3 inflammasome after sensing cytosolic RNA derived from viruses, bacteria or achaea19,20. CARD8 is structurally related to NLRP1, a sensor component of the NLRP1 inflammasome, and has been shown to activate caspase 1 activity in resting T cells and is a negative regulator of NLRP3 inflamasome21,22. Together, these results suggest that innate immune components carrying out sensing and regulatory activities may be involved in shaping the human skin microbiota.
Associated genetic variants were also localized at genes HTT (locus id: 5, rs2159173, PMeta = 1.8 × 10−09, associated with ASV093 [Staphylococcus (uncl.)]) and CFAP54 (locus id: 16, rs12423627, PMeta = 6.9 × 10−09, associated with ASV002 [Staphylococcus (uncl.)]), which encode proteins required for cilia formation in mammalian cells23,24 and expressed in different cell types in skin (Fig. 3). Further, we found SNPs that were associated with the expression of the transcript ENSG00000269886 (locus id: 3, rs2664121, PMeta = 4.3 × 10−09, associated with the genus Micrococcus). Interestingly, the effector alleles of all of these (rs2664121, rs2075337, rs2543492, rs1300250) were associated with the decrease in both tissue expression of ENSG00000269886 and relative abundance of the genus Micrococcus (the GTEx portal10 and Supplementary Data 1). ENSG00000269886 is an lncRNA antisense to the gene TTLL3, which regulates cilia assembly across eukaryotes25,26. Skin cells do not have motile cilia. Thus, it is likely that these genes are related to primary cillium, an organelle at the cell surface that senses extracellular signals, such as chemo-mechanical signals, osmolarity, pH, oxygen and light27. Primary cillium is found in various skin cells such as keratinocytes, fibroblasts, melanocytes and Langerhans cells28. Its formation is influenced by the dynamics of the actin cytoskeleton29, which is regulated by SRGAP3 encoded protein (locus id: 3)30. Together, these results suggest that extracellular sensing through primary cilium may be involved in the regulation of the skin microbiota.
Additional associations were observed with SNPs located in genes involved in cellular differentiation and proliferation. These were RAF1 (locus id: 4, rs709165, PMeta = 4.0 × 10−08, associated with ASV006 [Staphylococcus hominis])31–33 and RGS12 (locus id: 5, rs2159173)34–36, the latter found abundantly expressed in keratinocytes12 (Fig. 3). Furthermore, SNPS in PDGFRA (locus id: 6, rs55702239, PMeta = 3.5 × 10−08) were associated with order Bacteroidales and genus Bacteroides. PDGFRA is abundantly expressed in fibroblasts12 (Fig. 3) and participates in cellular maintenance37 and extracellular matrix production38. Keratinocyte proliferation, differentiation and function as well as innate immune signaling are major forces contributing to the complex function of the skin barrier. Therefore, it is conceivable that the discovered GWAS associations may represent links between the skin barrier and members of the skin microbiota.
Expression of candidate genes by keratinocytes co-cultured with Staphylococcus epidermidis
To gain insights in the putative participation of the identified candidate genes in the molecular interaction with the skin bacteria, we analyzed the in vitro transcriptional profile of normal human epidermal keratinocytes co-cultured with S. epidermidis, an abundant commensal in human skin39. Transcriptional profiles (six replicates) of keratinocytes from the foreskin of a 0-year-old male donor co-cultured with the S. epidermidis ATCC 14990 strain clearly differed from the profiles of controls, keratinocytes that were not co-cultured with bacteria (Fig. 4a). The S. epidermidis ATCC 14990 strain is a well characterized laboratory strain which is close to the strains found in the skin of the participants of the two cohorts studied. This proximity is suggested by the observation of 100% overlap and identity with the full length of ASV002 [Staphylococcus (uncl.)] amplicon sequence (307 base pairs), the second most abundant ASV in the whole database (~10% of rarefied sequences) and the most abundant ASV assigned to Staphylococcus genus.
Fig. 4. GWAS genes expressed by keratinocytes co-cultured with Staphylococcus epidermidis.

a Bacteria were added to two-dimension keratinocyte cultures, which were cultivated in six replicates, two per weekly batch. First and second dimensions of principal component analysis of gene expression after variance stabilizing transformation (vst) are shown. Differential expression analysis was performed to compare the expression of keratinocyte genes in culture with and without S. epidermidis. b Enrichment of biological processes mapped to Gene Ontology (GO) was performed on differentially expressed genes [q < 0.05 (derived from Wald test on negative binomial generalized linear models) and absolute logarithmic (log2) fold change >1]. Top ten lowest adjusted P values (Fisher exact test) of each up and down regulated processes are shown, ordered by number of detected genes. Large subunit ribosomal ribonucleic acid (LSU-rRNA), transfer RNA (tRNA) and noncoding RNA (ncRNA) are abbreviated. c Change in the transcription of GWAS selected genes which were differentially expressed are shown. Approximate posterior estimation for generalized linear model (apeglm78) shrinkage was applied to effect size (log2 fold change). Error bars represent posterior standard deviation.
A total of 4134 genes were differentially regulated (Supplementary Data 3), suggesting a strong transcriptional response of human keratinocytes to S. epidermidis ATCC 14990 strain in vitro. According to pathway enrichment analysis (Supplementary Data 4), the most significant biological processes upregulated were related to immune response, including cytokine-mediated and innate immune responses, as well as response to virus and symbionts (Fig. 4b). On the other hand, ribosomal biogenesis, processing of ribosomal RNA and non-coding RNA were among the most significantly down regulated biological processes. In this scenario, a quarter of the candidate genes (n = 7) were differentially expressed (q < 0.05 and absolute log2 fold change >1) when comparing cultures with and without S. epidermidis ATCC 14990 (Fig. 4c).
Based on knockout mouse macrophage cells, the deficiency of C1QBP protein increases the DNA sensor cyclic GMP-AMP (cGAMP) synthase-induced innate immune response40. Here, we observed the downregulation of C1QBP transcription associated with the upregulation of genes belonging to innate immune response (Fig. 4b, c), which sides with our GWAS suggestion that this gene may play a role in the regulation of skin bacteria via innate immunity. On the other hand, DHX33 was downregulated, contrasting to its role in innate immunity via activation of NLRP3, which transcript was upregulated (Fig. 4c and see Supplementary Data 3). It is thus likely that the reduced expression of DHX33 in our assays may be associated with the role of DHX33 in rRNA synthesis via positive regulation of transcription by RNA polymerase I41, being both pathways downregulated (Fig. 4b, c, Supplementary Data 4).
Genes coding for SRGAP3 and TTLL3, of which the lncRNA antisense gene was implied by GWAS, were upregulated (Fig. 4c and Supplementary Data 3). These observations support our discovered association of primary cilium and skin bacteria. However, it is important to bear in mind that the encoded proteins are not exclusively related to primary cilium, and their expression in our assays may also be related to other structures, e.g., cytoskeleton in the case of SRGAP330, and processes, e.g., proliferation in the case of TTLL326. Finally, the know role of PDGFRA in fibroblast activity are not directly translated to keratinocytes37,38. Therefore, the consequences of S. epidermidis-induced in vitro upregulation of PDGFRA in keratinocytes remain to be investigated.
Our in vitro experiment is explorative in nature and is limited to its reductionist approach: it consists of two-dimensional co-cultures of isolated keratinocytes and a single S. epidermidis laboratory strain. It is well known that the immunomodulatory effects of S. epidermidis depend on the specific strain, and that there is a large S. epidermidis strain level variation. Thus, it is not possible to directly extrapolate our preliminary functional results to an eventual keratinocyte response to skin commensals in vivo. A panel of commensal strains as well as in vitro models closer to the skin physiology, such as three-dimensional human skin models42, are necessary to uncover the functional dynamics of the host-commensal cellular interactions. Nevertheless, our assays allowed for the observation of the transcriptional regulation of several GWAS selected genes, being a starting point for functional investigations of the roles of these genes in the interaction with the skin microbiota.
Influence of skin microbiota on non-infectious skin diseases
Summary statistics of univariate microbial features were used as exposures in 2-sample mendelian randomization (MR; see Methods) to assess their influence on non-infectious skin diseases. A total of eight comparisons passed the per-trait suggestive threshold (q(trait) value <0.05, Fig. 5), although no comparison passed the global threshold (q(global) value <0.05; Supplementary Data 5). MR results indicated the influence of staphylococci in dermatitis/eczema (Staphylococcus genus, β = 1.5 × 10−03), and further, modulating roles of Flavobacteriaceae in two allergy-related traits with microenvironment-specific effect direction (βMoist = 8.8 × 10−04; βDry = 1.1 ×10−03). Additional results suggested involvement of Staphylococcus ASVs in psoriasis (ASV012 [Staphylococcus hominis], β = 4.0 × 10−04), seborrhoeic keratosis (ASV010 [Staphylococcus (uncl.)], β = 7.2 × 10−04) and vitiligo (ASV012 [Staphylococcus hominis], β = 1.2 × 10−04). Potential protective effects of staphylococci in allergic rhinitis were also suggested (ASV012 [Staphylococcus hominis], β = −1.1 × 10−03). It is noteworthy that these are likely coagulase-negative staphylococci, which are typical members of the skin microbiota43. However, the ASV-level signals from the MR were only weakly or inconclusively supported by the sensitivity analysis (see Methods, Supplementary Data 5). Together, our findings suggest that members of the microbiota may modulate the health-disease balance in skin.
Fig. 5. Results from Mendelian Randomization analysis.
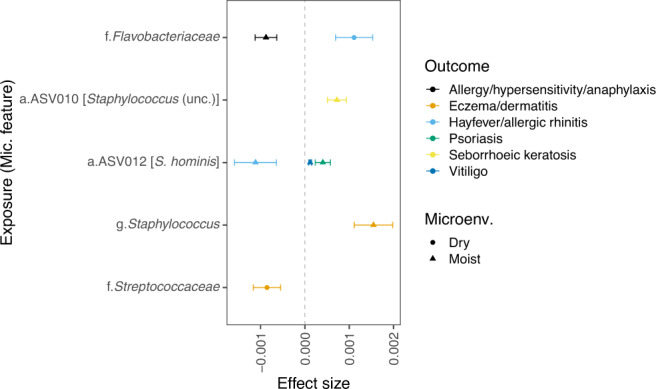
All exposure-to-outcome pairs with qtrait) <0.05 in the inverse-variance weighted 2-sample MR are shown. Error bars represent standard error. Microbial features are prefixed with their level, amplicon variant sequence (a.), genus (g.) and family (f.). Additional details and sensitive analyses can be found in Supplementary Data 5.
In summary, we conducted the first genome-wide association analysis dedicated to the human skin microbiota and identified 23 genome-wide significant loci. The combination of samples from different skin microenvironments of participants from two independent German cohorts allowed for robust results, despite the rather small number of included participants. The candidate genes have functions related to innate immune signaling, environmental sensing, cell differentiation, proliferation and fibroblast activity. Keratinocyte cultures challenged with a laboratory strain of S. epidermidis indicated regulation of seven candidate genes identified by GWAS, providing preliminary evidence that GWAS selected genes may be transcriptionally regulated by skin commensals. MR analysis further supported that specific skin microbiota features might have causal roles in the development of atopic dermatitis, but also suggested modulation of other non-infections skin diseases.
It needs to be considered that, despite our efforts to integrate information from different molecular levels and databases to understand the exact mechanisms by which the variants influence candidate gene function(s) and or expression and how this influences the skin microbiome, further and advanced experiments are needed. Likewise, it would be important to systematically establish differences in cutaneous gene expression with skin type, skin physiology and across age groups. Nevertheless, our results suggest a close interaction of the host genetic makeup and associated skin microbiomes. Furthermore, our findings point to the skin microbiota as a target for disease prevention and management, with potential for the development of personalized treatments for non-infectious, inflammatory skin conditions.
Methods
Cohorts’ description, genotyping, imputation and harmonization
PopGen cohort participants were randomly recruited via the local population registry in Kiel, Germany, and as blood donors of the University Hospital Schleswig-Holstein, Campus Kiel9. Genotypes derived from the Affymetrix Genome-Wide Human single nucleotide polymorphism (SNP) Array 6.0 were quality controlled following a previously established protocol44 and using the IKMB GWAS Quality Control Pipeline (https://github.com/ikmb/gwas-qc). Briefly, variants with excess missing data (>2%) and/or that deviated from Hardy-Weinberg Equilibrium [HWE, False Discovery Rate (FDR45) P value <10−5] were excluded. Samples with high missing data (>2%), high overall increased/decreased heterozygosity rates (i.e., ±5 standard deviation from the sample mean) and related individuals with a PLINK46 PI_HAT score >0.1875 were removed. To assess population structure, we performed a principal components analysis (PCA) including individuals of the 1000 Genomes Phase3 ref. 47 and removed outlier individuals not matching a European ancestry. Imputation was performed with the Michigan Imputation Server48 (Reference Panel: HRCr1.1 2016 (GRCh37/hg19); Array Build: GRCh37/hg19; rsq filter: off; Phasing Eagle 2.4 (phased output); Population: EUR; Mode: Quality Control & Imputation) and was followed by removal of monomorphic variants. These steps were performed following the miQTL cookbook instructions (https://github.com/alexa-kur/miQTL_cookbook#chapter-2-genotype-imputation).
KORA FF4 cohort participants from the youngest age group (39-48 years) that were previously genotyped as part of KORA S4 Survey were recruited from the southern German city of Augsburg and its two surrounding counties8. Genotyping and genotyping imputation were performed by the KORA Study Center. Briefly, genotypes were derived from the Affymetrix Genome-Wide Human SNP Array 6.0 (KORA F4). Samples with missing data (>3%), mismatch with phenotypic and genetic gender and high heterozygosity rates (i.e., ±5 standard deviation from the sample mean) were removed. Samples were also checked for European ancestry, population outliers and compared with other existing genotype data of the same individual within the KORA cohort. Variants with excess missing data (>2%), deviating from HWE (P value <5 × 10−10) and Minor allele frequency (MAF) (<2%) were removed. Prephasing was done with SHAPEIT v249 and imputation with IMPUTE v2.350 (reference panel: 1000 Genomes Phase 3 integrated variant set release in NCBI build 37).
To harmonize both genotype datasets, resulting VCF (PopGen) and IMPUTE output (KORA FF4) files were converted to PLINK format using PLINK v1.946. Participants that had their skin microbiota profiled (see section below) were selected and variants with MAF < 5% were removed. Genotype Harmonizer v 1.4.23 was used to update the KORA FF4 allele reference based on the PopGen data. SNPs with missingness >10% and non-biallelic SNPs were removed from PopGen data using PLINK v2.0-alpha-avx2-20200217. PopGen SNPs were references to set alleles in KORA F4 data, which also underwent removal of variants with missingness <10% and non-biallelic SNPs. Data sets were merged into PLINK files using PLINK v1.9 which contained SNPs available in both cohorts. Lastly, a principal component analysis (PCA) was produced with PLINK v1.9 to summarize the genetic population structure.
Written informed consent was obtained from all study participants. All protocols were approved by the ethics committees of the Medical Faculty of Kiel University (PopGen) and of the Bavarian Medical Association (KORA). We have complied with all relevant ethical regulations.
Sampling collection and microbial profiling
Skin microbiota was sampled as described previously3. Briefly, skin swabs were taken with Catch-All Sample Collection Swab (Epicentre Biotechnologies, Madison, WIS) soaked in specimen collection fluid (SCF-1) from 4 cm2 area of the skin site. Skin sites were selected to represent moist skin (antecubital fossa in both PopGen and KORA FF4), sebaceous skin (retroauricular fold in KORA FF4 and forehead in PopGen), and dry skin (volar and dorsal forearm in PopGen). Skin swabs were stored at -80 °C until DNA extraction using the QIAamp UCP Pathogen Mini Kit on an automated QIAcube system (QIAGEN GmbH, Hilden, Germany) for PopGen and the PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA) for KORA FF4.
Bacterial profiles were based on the V1 and V2 variable regions of the gene coding for 16 S ribosomal RNA (rRNA). Briefly, V1-V2 regions were amplified with PCR performed with the primer pair 27F-338R. Pooled amplicon libraries were sequenced with MiSeq Reagent Kit v3 on the Illumina MiSeq (Illumina Inc., San Diego, CA). Sequencing reads were processed with DADA2 v1.1051, resulting in an amplicon sequence variant (ASV) table, which records the number of times each exact ASV was observed in each sample52. ASV is a finer scale analogue of the operational taxonomic unit (OTU), which resolves the sequenced region variant down to a single-nucleotide difference level. ASVs were taxonomically classified down to genus level using RDP classifier algorithm based on Ribosomal Database Project (RDP) version 16 release with 50% confidence53,54. Species-level annotations were added to ASV sequences based on exact matches to the RDP database, using the function addSpecies() from DADA2 R package. Species-level abundances were not considered in the GWAS, as these are likely incomplete and possibly inaccurate55, however annotations can still serve as proxies for sub-genus level placement of ASVs. Therefore, their species-level annotations were carried as part of the ASV annotation throughout the manuscript using square brackets, i.e. ASV001 [Propionibacterium acnes] or ASV001 [P. acnes]. Finally, sequences were filtered to remove chloroplasts, mitochondria and low abundant ASVs (less than 0.1% of total sequence counts of a given skin site). Samples were removed if taken from a site with apparent skin abnormality or in which corticosteroids or antibiotics were applied in the last seven days before collection. Microbiota data was manipulated in R 3.6.2 using the Phyloseq package v1.34.056,57. Details on sequencing, read processing and ASV filtering are provided in our previous study with the same dataset3. Finally, only samples from participants with genotype data were kept for downstream analysis.
Association of microbial features with host SNPs
The association of SNPs was tested for multivariate (for inferences on the bacterial community; beta diversity), and univariate microbial features (for inferences on individual bacterial clades). Beta diversity was inferred from Bray-Curtis dissimilarities of rarefied amplicon variant (ASV) table (5,000 sequences per sample), calculated in R version 3.6.2 using the Vegan package v2.5-558. Bacterial clades included ASVs and taxonomic groups ranging from genus to phylum. Taxonomic groups were obtained by merging the ASV sequence counts that had the same taxonomy at a certain rank, using the Phyloseq function tax_glom(). For each skin site, univariate features with a median sequence count higher than 50 and that were present in more than 100 participants were kept. In addition, univariate features were kept only if present in both sites of the same skin microenvironment, i.e., moist, sebaceous or dry. This effort resulted in 103 bacterial clades. To avoid redundancy, these clades were clustered together based on a 0.985 Spearman correlation cut-off. Clustering of clades were performed in each skin microenvironment separately because skin microenvironments have distinct bacterial profiles3. This effort resulted in a total of 79 bacterial features to be tested: 3 phyla, 4 classes, 7 order, 7 families, 15 genera and 43 ASVs.
Statistical tests were conducted for each microbial feature in each skin site from a single cohort following the framework established previously7. Because this process generates subsets of the whole data, additional variant inclusion criteria were implemented when necessary prior association tests. Accordingly, genetic variants were filtered (MAF > 5%) and coded into numeric features (0 = homozygous for reference allele; 1 = heterozygous; and 2 = homozygous for alternative allele). Only non-monomorphic variants were considered for testing. All tests were performed on the alternative allele as effect allele.
For tests with multivariate microbial features, distance-based redundancy analysis was performed with the vegan function capscale() with age, sex, BMI and the first ten genetic principal components (PCs) as covariates. The variables were selected because they were found as main confounders of the skin microbiota3 and to account for the influence of the genetic background. The variance left unexplained by these covariates was extracted using the R residuals() function. The effect of genetic variants was estimated from the residual matrix with a distance-based F-test using moment matching59. For tests with univariate microbial features, zero-truncated non-rarefied count abundances were used. Outliers were filtered based on rarefied counts to account for uneven sequencing depths between samples. Samples were considered outliers when they deviated more than 5× the interquartile range (IQR) from the median abundance. Finally, count abundances (non-rarified) were fit with the Mvabund v4.1.660 function manyglm() in generalized linear models with negative binomial distributions and the covariates above mentioned as predictors. The logarithm of the total sequence counts of each sample was used as offset. Unexplained variance was extracted using residuals() function, which extracts from manyglm() models residuals that are normal61. The effect of genetic variants was estimated from the residuals using linear model. P value was calculated using the R summary function, which performs a two-sided t-test.
Microenvironment-wise meta-analysis
Genomic inflation (λGC) was calculated for all tests using the regression method as implemented in GenABEL v1.8-0 R package62. All values were below 1.02, indicating no genomic inflation. Because skin microbiota profiles are distinctive between microenvironments3, meta-analyses were performed combining data sets that originated from skin sites of the same microenvironment. Therefore, results from moist skin sites were merged into one meta-analysis and results from sebaceous skin sites into another, because skin sites from these microenvironments are from different cohorts. Because the distance-based F-test applied to multivariate features do not produce beta values, a fixed effect meta-analysis was performed with METAL release 2011-03-2563, with meta-analysis P values (PMeta) and sample size based weighting. For univariate features, an inverse-variance weighted fixed effect meta-analysis was performed with METASOFT v264 on beta values and their standard errors. Meta-analysis results were reported significant if genome-wide significance (PMeta < 5 × 10−8) was achieved and the association was found nominally significant in the two skin sites (P < 0.05). Because samples from the two dry skin sites came from a single cohort (PopGen), results were combined and considered significant if the P value of at least one skin site was genome-wide significant (P < 5 × 10−8) with at least nominal significance at the other skin site (P < 0.05). In this case, the lowest P value was reported as the PMeta value. The study-wide significance threshold was calculated considering the number of microbiota features tested (PMeta < 5 × 10−8/80 = 6.3 × 10−10).
Fine-mapping and gene prioritization
Genes were considered of interest when containing potentially causal variants and/or these variants were significantly associated with the gene expression in skin tissue. Fine-mapping was performed to explore the most likely causal set of variants using shotgun stochastic search algorithm implemented in FINEMAP v1.465. For moist and sebaceous microenvironments, fine-mapping was performed with summary statistics (beta values and their standard errors) from meta-analysis. For dry microenvironment, beta values and their standard errors from volar forearm were used for fine-mapping. Genes were reported when intersecting with the range of the 95% posterior credible SNP set assuming one causal variant as input parameter for the algorithm. If fine-mapping did not find a credible set (<50 variants), or for beta-diversity results, genes with variants with LD > 0.6 to the lead SNP were reported. SNPs and genes were annotated using the R package biomaRt v2.48.066.
To investigate whether genetic variants could affect gene expression in skin tissues, prioritized variants selected by fine-mapping or in LD > 0.6 were mapped to Genotype-Tissue Expression (GTEx) Project10 database v8 (lower leg and suprapubic skin tissues). Briefly, chromosomal positions in genome assembly hg19 (GRCh37) were converted to hg38 (GRCh38) using LiftOver from the human genome browser at the University of California Santa Cruz (UCSC)67. These positions were then mapped to single-tissue cis-quantitative trait locus (QTL) data downloaded (11/06/2021) from the GTEx portal10, specifically the file GTEx_Analysis_v8_eQTL.tar, which contains genes of which expression are significantly associated with genetic variants based on permutations. Only data from skin tissues (suprapubic non-sun-exposed and lower leg sun-exposed) were used in this analysis.
Expression of genes in skin tissues and cell types
Consensus transcriptional expression of genes in skin tissue were retrieved from the Human Protein Atlas version 20.111, which additionally includes data sets from the GTEx10 and the Functional annotation of the mammalian genome (FANTOM5)68 projects. Single-cell RNA-Seq data of skin from healthy individuals (n = 5) were retrieved from a recent study by Solé-Boldo et al.12.
Mendelian randomization
Mendelian randomization (MR) was performed using summary statistics of univariate association analyses as ‘exposures’ and six selected skin-related traits as ‘outcomes’ (allergy/hypersensitivity/anaphylaxis, seborrheic keratosis, eczema/dermatitis, hay fever/allergic rhinitis, psoriasis, vitiligo). Outcome summary statistics were retrieved from UK Biobank using the R package TwoSampleMR v0.5.569. UK Biobank originated from the IEU Open GWAS Project database70. All variants in the microbial ‘exposures’ with an association P value <10-5 were included in the analyses. After harmonization with the exposure data, only independent variants were retained using the clump_data() function with default parameters. Additionally, variants with an F statistic <10 were excluded from the analysis to avoid weak instrument bias71. In case of more than two independent retained variants, inverse variance weighted (IVW) MR analysis was performed as primary analysis, otherwise Wald-ratio was calculated. For exposures with more than two instrument variables, weighted mean, and MR Egger regression were performed for sensitivity analysis. MR Egger regression with non-significant beta values (P > 0.05) and weighted median MR results with significant (P < 0.1) and concordant effect direction to the IVW MR analysis were regarded as supporting. P values of the primary MR analysis (IVW or Wald-ratio) were corrected for multiple testing using per-trait and global FDR correction36. All MR analyses were conducted in R v3.6.1.
Keratinocytes co-culture with Staphylococcus epidermidis
Normal human epidermal keratinocytes (NHEKs) (foreskin of a 0-year-old male Caucasian donor; Promocell, Heidelberg, Germany, Lot number 407Z001) were cultured in Keratinocyte Growth Medium (KGM; Lonza Biosciences, Walkersville, USA) + supplements + CaCl2 + penicillin/streptomycin at 37 °C and 5% CO2. Cells were used at passages 4-6. Keratinocytes were seeded into 6-well plates and grown until confluency. Staphylococcus epidermidis (Winslow and Winslow) Evans (ATCC 14990) was cultured in Tryptic Soy Broth (TSB; Thermo Fischer, Waltham, USA) medium at 37 °C. For co-cultivation with keratinocytes, bacteria were centrifuged, resuspended in KGM + CaCl2, and added to the keratinocytes at an optic density (OD) of 0.1 in KGM + CaCl2. A total of six replicates of each condition, with and without the addition of S. epidermidis, were performed, two replicates per weekly batch. Plates were centrifuged at 350 x g for 5 min to allow bacteria to settle on the bottom. After 3 h incubation, plates were washed, and KGM + CaCl2 with gentamycin was added for a further incubation of 23 h at 37 °C and 5% CO2. Plates were washed twice before RNA isolation using Trizol (Thermo Fischer, Waltham, USA) as per manufacturer’s instruction.
Sequencing libraries were prepared using TrueSeq Stranded mRNA kit (Illumina Inc., San Diego, USA). Sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, USA) with 2 × 50 base pairs length. Raw sequences were processed using the nf-core/rnaseq pipeline v3.072,73, which includes adapter quality trimming with Trim Galore (https://github.com/FelixKrueger/TrimGalore), removal of ribosomal RNA with SortMeRNA74, alignment with STAR75 and transcript quantification with Salmon76. The human genome assembly hg19 was used as reference. Differentially expressed genes were detected with the R package DESeq2 v.1.30.077. Wald test was performed with negative binomial generalized linear models, which included the weekly batch and whether S. epidermidis was added to the culture or not (~batch + condition). P were corrected for multiple testing using the FDR method45. Approximate posterior estimation for generalized linear model (apeglm78) shrinkage was applied to logarithmic (log2) fold change (LFC). Results were considered significant based on q values (<0.05) and LFC (absolute LFC > 1). Enrichment of expressed genes up and down regulated were performed using the R package enrichR v3.0 and the GO_Biological_Process_2021 database79. Enriched pathways were considered significant based on q values (<0.05; Fisher exact test). To get an overview of the effect of the S. epidermidis addition to keratinocyte cultures, transcriptional profiles were visualized through principal component analysis (PCA). First, variance stabilizing transformation (VST) from the R package DESeq2 v.1.30.077 was applied to the transcriptional data. PCA was performed as implemented in the R package PCAtools v. 2.4.080.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We are grateful to all participants and study staff from the Biobank PopGen (Dr Gunar Jacobs and team) and the KORA Studienzentrum (Dr Margit Heier and team). We thank the staff from UKSH Dermatology laboratory (particularly, Anke Rose), the IKMB microbiome laboratory, the IKMB DNA laboratory and the IKMB sequencing laboratory for their excellent support. We are grateful to Dr Sören Franzenburg and Eike Matthias Wacker for assistance and troubleshooting. We thank Martin Schulzky for the design of skin site icons. The project leading to this application has received funding from the Deutsche Forschungsgemeinschaft (DFG) Grant no. WE2678/14-1 (granted to S.W.) and the Innovative Medicines Initiative 2 Joint Undertaking under Grant Agreement no. 821511 (BIOMAP, granted to S.W.). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA. This publication reflects only the author’s view and the JU is not responsible for any use that may be made of the information it contains. This work was also supported by the Deutsche Forschungsgemeinschaft (DFG) Collaborative Research Center 1182 ‘Origin and Function of Metaorganisms’ (grant no. SFB1182, Project A2 granted to A.F.). The study received infrastructure support from the DFG research unit “miTarget” (Projektnummer 426660215; EL 831/5-1 granted to A.F.). The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria.
Author contributions
Study was designed by L.M.S., F.K., E.R., H.E., F.U.W., D.E., A.F., S.W. and M.C.R. Sample, data collection and processing were done by E.R., H.E., L.T., W.L., C.G., A.P., C.B. Analysis was performed by L.M.S., F.D., H.E., S.J., L.M., F.U.W., D.E., N.S., H.B. and M.C.R. Manuscript was written by L.M.S., F.D., E.R., H.E., F.U.W., A.F., S.W. and M.C.R. All authors revised the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
Raw 16 S rRNA gene amplicon sequences of PopGen participants were deposited at the European nucleotide archive (ENA) under accession code PRJEB41215. GWAS summary statistics generated in this study are available at GWAS catalogue under accession codes GCST90133164-GCST90133313. Phenotype data from PopGen individuals can be accessed through the Material Data Access Form from the PopGen Biobank (Schleswig-Holstein, Germany). Information about the Material Data Access Form and how to apply can be found at http://www.uksh.de/p2n/Information+for+Researchers.html. KORA data are available at https://www.helmholtz-munich.de/en/kora/for-scientists/cooperation-with-kora/index.html upon request by means of a project agreement. In addition, the following public database and resources were used: 1000 Genomes Phase3 ref. 47, Ribosomal Database Project (RDP) version 1654, Genotype-Tissue Expression (GTEx) Project database v810, Functional annotation of the mammalian genome (FANTOM5)68, Skin single-cell data from by Solé-Boldo et al.12, UK Biobank and the IEU Open GWAS Project database70.
Code availability
Code used in the analysis are available at https://github.com/LucasMS/skin.mgwas.pub81.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Andre Franke, Stephan Weidinger, Malte Christoph Rühlemann.
Contributor Information
Andre Franke, Email: a.franke@mucosa.de.
Stephan Weidinger, Email: sweidinger@dermatology.uni-kiel.de.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-33906-5.
References
- 1.Zhernakova A, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–569. doi: 10.1126/science.aad3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falony G, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–564. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 3.Moitinho-Silva, L. et al. Host traits, lifestyle and environment are associated with the human skin bacteria. Br J Dermatol, 10.1111/bjd.20072 (2021). [DOI] [PubMed]
- 4.Si J, Lee S, Park JM, Sung J, Ko G. Genetic associations and shared environmental effects on the skin microbiome of Korean twins. BMC Genomics. 2015;16:992. doi: 10.1186/s12864-015-2131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baurecht H, et al. Epidermal lipid composition, barrier integrity, and eczematous inflammation are associated with skin microbiome configuration. J. Allergy Clin. Immunol. 2018;141:1668–1676.e1616. doi: 10.1016/j.jaci.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Kurilshikov A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021;53:156–165. doi: 10.1038/s41588-020-00763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rühlemann MC, et al. Genome-wide association study in 8,956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 2021;53:147–155. doi: 10.1038/s41588-020-00747-1. [DOI] [PubMed] [Google Scholar]
- 8.Holle R, Happich M, Löwel H, Wichmann H, Group MKS. KORA - a research platform for population based health research. Das. Gesundheitswesen. 2005;67:19–25. doi: 10.1055/s-2005-858235. [DOI] [PubMed] [Google Scholar]
- 9.Nöthlings U, Krawczak M. PopGen. Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz. 2012;55:831–835. doi: 10.1007/s00103-012-1487-2. [DOI] [PubMed] [Google Scholar]
- 10.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlen M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 12.Solé-Boldo L, et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020;3:188. doi: 10.1038/s42003-020-0922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dembitzer FR, et al. gC1qR expression in normal and pathologic human tissues: differential expression in tissues of epithelial and mesenchymal origin. J. Histochem Cytochem. 2012;60:467–474. doi: 10.1369/0022155412440882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghebrehiwet B, Peerschke EI. cC1q-R (calreticulin) and gC1q-R/p33: ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol. Immunol. 2004;41:173–183. doi: 10.1016/j.molimm.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Braun L, Ghebrehiwet B, Cossart P. gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. EMBO J. 2000;19:1458–1466. doi: 10.1093/emboj/19.7.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen T, Ghebrehiwet B, Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect. Immun. 2000;68:2061–2068. doi: 10.1128/IAI.68.4.2061-2068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peerschke EI, Ghebrehiwet B. The contribution of gC1qR/p33 in infection and inflammation. Immunobiology. 2007;212:333–342. doi: 10.1016/j.imbio.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitoma H, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity. 2013;39:123–135. doi: 10.1016/j.immuni.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vierbuchen T, Bang C, Rosigkeit H, Schmitz RA, Heine H. The human-associated archaeon methanosphaera stadtmanae is recognized through its RNA and Induces TLR8-dependent NLRP3 inflammasome activation. Front Immunol. 2017;8:1535. doi: 10.3389/fimmu.2017.01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linder A, et al. CARD8 inflammasome activation triggers pyroptosis in human T cells. EMBO J. 2020;39:e105071. doi: 10.15252/embj.2020105071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito S, Hara Y, Kubota T. CARD8 is a negative regulator for NLRP3 inflammasome, but mutant NLRP3 in cryopyrin-associated periodic syndromes escapes the restriction. Arthritis Res Ther. 2014;16:R52. doi: 10.1186/ar4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenzie CW, et al. CFAP54 is required for proper ciliary motility and assembly of the central pair apparatus in mice. Mol. Biol. Cell. 2015;26:3140–3149. doi: 10.1091/mbc.e15-02-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keryer G, et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Invest. 2011;121:4372–4382. doi: 10.1172/JCI57552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wloga D, et al. TTLL3 Is a tubulin glycine ligase that regulates the assembly of cilia. Dev. Cell. 2009;16:867–876. doi: 10.1016/j.devcel.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 26.Rocha C, et al. Tubulin glycylases are required for primary cilia, control of cell proliferation and tumor development in colon. EMBO J. 2014;33:2247–2260. doi: 10.15252/embj.201488466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019;15:199–219. doi: 10.1038/s41581-019-0116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toriyama, M. & Ishii, K. J. Primary cilia in the skin: functions in immunity and therapeutic potential. Front. in Cell and Developmental Biol.9, 10.3389/fcell.2021.621318 (2021). [DOI] [PMC free article] [PubMed]
- 29.Smith CEL, Lake AVR, Johnson CA. Primary cilia, ciliogenesis and the actin cytoskeleton: a little less resorption, a little more actin please. Front Cell Dev. Biol. 2020;8:622822. doi: 10.3389/fcell.2020.622822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bacon C, Endris V, Rappold GA. The cellular function of srGAP3 and its role in neuronal morphogenesis. Mech. Dev. 2013;130:391–395. doi: 10.1016/j.mod.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl Acad. Sci. USA. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samuel DS, et al. Raf-1 activation stimulates proliferation and inhibits IGF-stimulated differentiation in L6A1 myoblasts. Horm. Metab. Res. 1999;31:55–64. doi: 10.1055/s-2007-978699. [DOI] [PubMed] [Google Scholar]
- 33.Rubiolo C, et al. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood. 2006;108:152–159. doi: 10.1182/blood-2005-09-3866. [DOI] [PubMed] [Google Scholar]
- 34.Schroer AB, et al. A role for Regulator of G protein Signaling-12 (RGS12) in the balance between myoblast proliferation and differentiation. PLoS One. 2019;14:e0216167. doi: 10.1371/journal.pone.0216167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willard MD, et al. Selective role for RGS12 as a Ras/Raf/MEK scaffold in nerve growth factor-mediated differentiation. EMBO J. 2007;26:2029–2040. doi: 10.1038/sj.emboj.7601659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, et al. Regulator of G protein signaling protein 12 (Rgs12) controls mouse osteoblast differentiation via calcium channel/oscillation and galphai-ERK signaling. J. Bone Min. Res. 2019;34:752–764. doi: 10.1002/jbmr.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am. J. Physiol. Heart Circ. Physiol. 2019;317:H330–H344. doi: 10.1152/ajpheart.00054.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horikawa S, et al. PDGFRalpha plays a crucial role in connective tissue remodeling. Sci. Rep. 2015;5:17948. doi: 10.1038/srep17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh J, et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song K, et al. Leaked mitochondrial C1QBP inhibits activation of the DNA sensor cGAS. J. Immunol. 2021;207:2155–2166. doi: 10.4049/jimmunol.2100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Forys JT, Miceli AP, Gwinn AS, Weber JD. Identification of DHX33 as a mediator of rRNA synthesis and cell growth. Mol. Cell Biol. 2011;31:4676–4691. doi: 10.1128/MCB.05832-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emmert H, Rademacher F, Glaser R, Harder J. Skin microbiota analysis in human 3D skin models-“Free your mice”. Exp. Dermatol. 2020;29:1133–1139. doi: 10.1111/exd.14164. [DOI] [PubMed] [Google Scholar]
- 43.Becker K, Heilmann C, Peters G. Coagulase-negative staphylococci. Clin. Microbiol Rev. 2014;27:870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Severe Covid GG, et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 2020;383:1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995;57:289–300. [Google Scholar]
- 46.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Genomes Project C, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das S, et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2011;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 50.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Callahan BJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Callahan, B. DADA2 Pipeline Tutorial (1.16), https://benjjneb.github.io/dada2/tutorial.html (2021).
- 53.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cole JR, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson JS, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019;10:5029. doi: 10.1038/s41467-019-13036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McMurdie PJ, Holmes S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R Core Team. R: A Language and Environment for Statistical Computing. https://www.R-project.org/ (2020).
- 58.Oksanen, J. et al. vegan: Community Ecology Package. https://CRAN.R-project.org/package=vegan (2019).
- 59.Rühlemann MC, et al. Application of the distance-based F test in an mGWAS investigating β diversity of intestinal microbiota identifies variants in SLC9A8 (NHE8) and 3 other loci. Gut microbes. 2018;9:68–75. doi: 10.1080/19490976.2017.1356979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Naumann U, Wright ST, Warton DI. mvabund– an R package for model-based analysis of multivariate abundance data. Methods Ecol. Evolution. 2012;3:471–474. doi: 10.1111/j.2041-210X.2012.00190.x. [DOI] [Google Scholar]
- 61.Dunn PK, Smyth GK. Randomized Quantile Residuals. J. Computational Graph. Stat. 1996;5:236–244. [Google Scholar]
- 62.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 63.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benner C, et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Durinck S, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 67.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lizio M, et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015;16:22. doi: 10.1186/s13059-014-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife7, 10.7554/eLife.34408 (2018). [DOI] [PMC free article] [PubMed]
- 70.Elsworth, B. et al. The MRC IEU OpenGWAS data infrastructure. bioRxiv, 2020.2008.2010.244293, 10.1101/2020.08.10.244293 (2020).
- 71.Stock JH, Wright JH, Yogo M. A Survey of Weak Instruments and Weak Identification in Generalized Method of Moments. J. Bus. Economic Stat. 2002;20:518–529. doi: 10.1198/073500102288618658. [DOI] [Google Scholar]
- 72.nf-core/rnaseq: nf-core/rnaseq v3.0 - Silver Shark v. 3.0 (Zenodo, 2020).
- 73.Ewels PA, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 74.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 75.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2018;35:2084–2092. doi: 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blighe, K. & Lun A. PCAtools: Everything Principal Components Analysis. R package version 2.8.0. https://github.com/kevinblighe/PCAtools (2022).
- 81.Moitinho-Silva, L. Host genetic factors related to innate immunity, environmental sensing and cellular functions influence human skin microbiota, https://github.com/LucasMS/skin.mgwas.pub, 10.5281/zenodo.7047733. (2022). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Raw 16 S rRNA gene amplicon sequences of PopGen participants were deposited at the European nucleotide archive (ENA) under accession code PRJEB41215. GWAS summary statistics generated in this study are available at GWAS catalogue under accession codes GCST90133164-GCST90133313. Phenotype data from PopGen individuals can be accessed through the Material Data Access Form from the PopGen Biobank (Schleswig-Holstein, Germany). Information about the Material Data Access Form and how to apply can be found at http://www.uksh.de/p2n/Information+for+Researchers.html. KORA data are available at https://www.helmholtz-munich.de/en/kora/for-scientists/cooperation-with-kora/index.html upon request by means of a project agreement. In addition, the following public database and resources were used: 1000 Genomes Phase3 ref. 47, Ribosomal Database Project (RDP) version 1654, Genotype-Tissue Expression (GTEx) Project database v810, Functional annotation of the mammalian genome (FANTOM5)68, Skin single-cell data from by Solé-Boldo et al.12, UK Biobank and the IEU Open GWAS Project database70.
Code used in the analysis are available at https://github.com/LucasMS/skin.mgwas.pub81.