

Abstract
RNA contains more than 170 types of chemical modifications, and these modified nucleosides are recognized, installed and removed by their reader, writer, and eraser (RWE) proteins, respectively. Here, we employed a parallel-reaction monitoring (PRM)-based targeted proteomic method, in conjunction with stable isotope labeling by amino acids in cell culture (SILAC), to examine comprehensively the differential expression of epitranscriptomic RWE proteins in a matched pair of primary/metastatic colorectal cancer (CRC) cells, namely SW480/SW620. We were able to quantify 113 nonredundant epitranscriptomic RWE proteins; among them, 48 and 5 were up-and down-regulated by >1.5-fold in SW620 over SW480 cells, respectively. Some of those proteins with marked up-regulation in metastatic CRC cells, including NAT10, hnRNPC, and DKC1, were documented to assume important roles in the metastasis of CRC and other types of cancer. Interrogation of the Clinical Proteomic Tumor Analysis Consortium data revealed the involvement of DUS1L in the initiation and metastatic transformation of CRC. It can be envisaged that the PRM method can be utilized, in the future, to identify epitranscriptomic RWE proteins involved in the metastatic transformations of other types of cancer.
Keywords: cancer metastasis, colorectal cancer, epitranscriptomics, parallel-reaction monitoring, SILAC, targeted quantitative proteomics
1 |. INTRODUCTION
Epigenetic modifications of DNA and histones are well studied; how-ever, much less is investigated about the roles of RNA modifications in cellular processes. Over 170 types of chemical modifications exist in RNA [1]; most of these modifications are found in tRNAs, which contain an average of 13 modifications per molecule [2]. The most abundant internal modification in eukaryotic mRNA, N6-methyladenosine (m6A) [3], has drawn substantial attention after high-throughput sequencing revealed its widespread occurrence in the transcriptome [4]. Cellular proteins have been uncovered for the installation (“writers”) [5–9], recognition (“readers”) [10–12], and removal (“erasers”) [13–15] of m6A in mRNA. Aside from m6A, mRNA also contains N1-methyladenosine (m1A) [16–18], 5-methylcytidine (m5C) [19–21], N7-methylguanosine (m7G) [22], pseudouridine (ψ) [23, 24], and 2’-O-methylated nucleosides [25, 26].
Recent studies showed that genetic depletions of some of the epitranscriptomic reader, writer and eraser (RWE) proteins confer embryonic lethality and/or other developmental abnormalities, and their mutations and/or aberrant expressions result in the initiation and progression of cancer, impaired antiviral response, and defective neurogenesis [27–30]. For instance, Li et al. found that fat mass and obesity-associated protein (FTO) facilitates leukemo-genesis by modulating the expression of mRNAs of ASB2 and RARA genes, which play crucial roles in leukocyte proliferation [31]. Ma et al. observed that methyltransferase 14 (METTL14) inhibits the metastatic transformation of hepatocellular carcinoma through promoting the binding of DiGeorge syndrome critical region 8 (DGCR8) to pri-miR-126 and enhancing the maturation of miR-126 [32]. These studies provided insights into the roles of individual epitranscriptomic RWE proteins in cancer development; nonetheless, to our knowledge, there is no systematic investigation about how aberrant expression of epitranscriptomic RWE proteins modulates cancer progression.
CRC is well known for its high occurrence and mortality, with approximately 1.93 million newly diagnosed cases and 0.94 million deaths in 2021 [33]. The 5-year survival rate for CRC patients with distant metastasis is as low as 5%, with an average of 13 months of survival after diagnosis [34].
Scheduled parallel-reaction monitoring (PRM) is a targeted proteomic method where predefined m/z values of precursor ions of peptides and their retention time information are incorporated into an inclusion list for MS/MS analyses [35]. The PRM method capitalizes on the high resolution, accurate-mass-measurement abilities of an Orbitrap or time-of-flight mass analyzer, which facilitates unambiguous identification and robust quantification of peptides in complex sample matrices [36]. PRM, coupled with stable isotope labeling by amino acids in cell culture (SILAC) [37], also enables highly accurate and reproducible quantification of proteins in cultured mammalian cells [38, 39].
In this study, we applied our recently established LC-PRM method [40], together with SILAC, to assess the expression differences in epitranscriptomic RWE proteins in a matched pair of primary/metastatic colorectal cancer cell lines (SW480/SW620) derived from the same patient (Figure 1A). We uncovered a number of differentially expressed epitranscriptomic RWE proteins and explored the involvement of some of these proteins in CRC progression.
FIGURE 1.
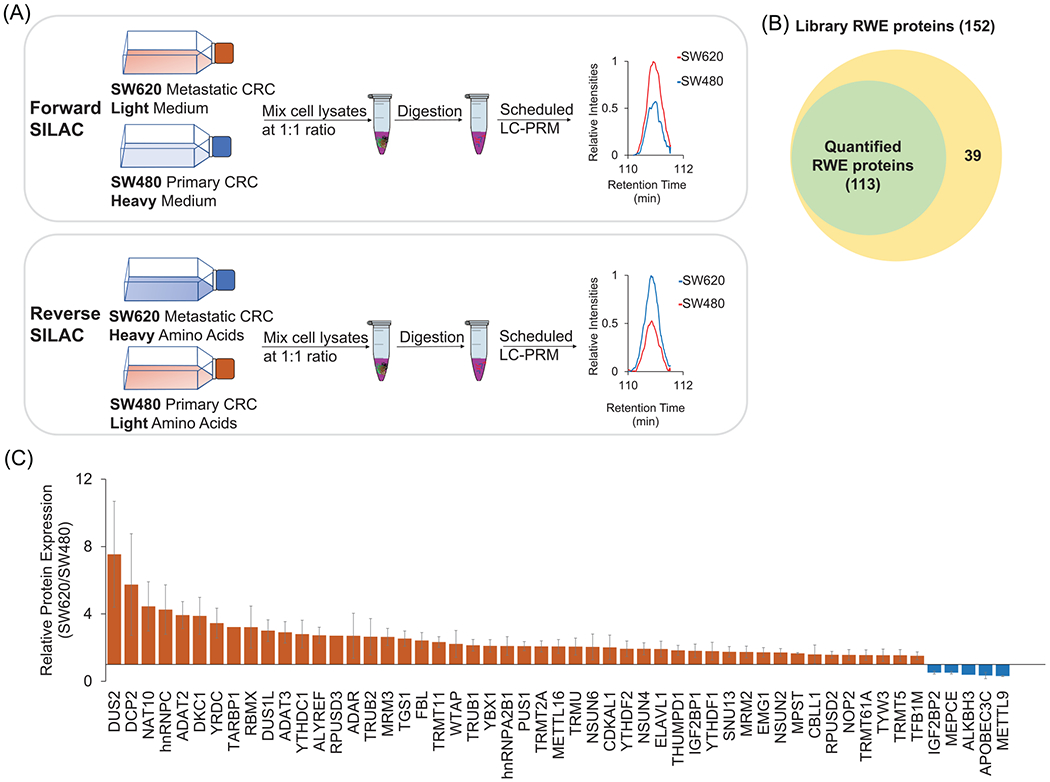
(A) A SILAC- and LC-PRM-based workflow for targeted quantifications of epitranscriptomic RWE proteins in SW620 (metastatic) and SW480 (primary) CRC cells. (B) A Venn diagram showing the number of quantified RWE proteins in SW480/SW620 cells in comparison with that of RWE proteins deposited in the PRM library. (C) A bar graph depicting the relative expression levels of differentially expressed RWE proteins in SW620 versus SW480 CRC cells. Red and blue bars designate those proteins with expression ratios in SW620/SW480 cells being >1.5 and <0.67, respectively. Error bars represent SD of results obtained from a total of four SILAC experiments after determining the protein expression ratio in each replicate based on LC-PRM results of all component peptides of the protein
2 |. MATERIALS AND METHODS
2.1 |. Tryptic digestion of whole-cell protein lysate
Details of cell culture procedures can be found in the supporting information. Light- or heavy-isotope labeled SW480 and SW620 cells were collected, and lysed on ice for 30 min with CelLytic M lysis buffer (Sigma) supplemented with 1% protease inhibitor cocktail (Sigma). After centrifugation at 16,100 × g for 30 min at 4°C, Bradford assay (Bio-Rad) was conducted to quantify total proteins in the supernatant. In forward SILAC experiments, the total protein lysate of SW480 cells cultured in the heavy medium and that of SW620 cells cultured in the light medium were combined at 1/1 ratio by mass. The reverse SILAC experiments were conducted in the opposite way. Samples from four biological replicates, with two forward and two reverse SILAC experiments, were prepared. Proteins in the cell lysates were denatured, reduced, and alkylated before digestion with MS-grade trypsin (Pierce) in 50 mM NH4HCO3, pH 8.0, at 37°C for 16 h according to the filter-aided sample preparation (FASP) procedure [41]. The tryptic peptides were collected by centrifugation and desalted using OMIX C18 pipet tips (Thermo Fisher Scientific) prior to LC-PRM analysis.
2.2 |. LC-PRM data acquisition
The LC-PRM data were acquired on a Q Exactive Plus quadrupole-Orbitrap mass spectrometer and a Dionex UltiMate 3000 RSLCnano UPLC system was employed for peptide separation. Prior to analyzing the aforementioned SILAC samples, a tryptic digestion mixture of bovine serum albumin (BSA) was analyzed under the same instrument conditions to define the linear relationship between normalized retention time (iRT) and RT, with the iRTs of BSA peptides being predefined. The RTs of tryptic peptides for RWE proteins were predicted from their iRTs in the PRM library and the above-described iRT-RT relationship for BSA peptides. By using our recently developed Skyline PRM library [40], we were able to monitor the precursor ions representing the light and heavy forms of 444 unique tryptic peptides of 152 epitran-scriptomic RWE proteins in three separate LC-MS/MS runs, where the inclusion list encompassed the m/z value and RT window (7 min in width) for each precursor ion of interest.
The analytical column was packed in-house using 3 μm Reprosil-Pur C18-AQ resin (Dr. Maisch GmbH HPLC) in a 25-cm fused silica column (75 μm i.d.). The trapping column was also prepared in-house with 5 μm Reprosil-Pur C18-AQ resin (Dr. Maisch GmbH HPLC) in a 4-cm long fused silica column (150 μm i.d.). Peptides from 500 ng of SILAC protein lysate were resolved using the analytical column with a 125-min linear gradient of 6%–43% mobile phase B (80% acetonitrile in 0.1% formic acid) in mobile phase A (0.1% formic acid in Milli-Q water), where the flow rate was 300 nl/min. The voltage for electrospray was set at 1.8 kV. The precursor ions were isolated in the quadrupole and fragmented in the HCD collision cell, where the isolation window was 1.0 m/z and the normalized collision energy (NCE) was 28. MS/MS were recorded with the resolution, automated gain control (AGC) target and maximum accumulation time being 17,500, 1 × 105 and 50 ms, respectively.
2.3 |. LC-PRM data analysis
The raw LC-MS/MS files were processed in Skyline to plot the extracted-ion chromatograms for peak integration. For a positive detection of precursor ion of interest, 4–6 most abundant y ions from the same precursor ion in light and heavy forms should share the same elution time, and the relative abundances of fragment ions from the acquired MS/MS should match those in the MS/MS in the library, which is gauged by dot product (dotp) value [42]. A dotp value of >0.7, ideally above 0.9, was considered highly similar. We manually excluded the potential interfering fragment ion if it does not overlay with other fragment ions. Quantification was based on the sum of peak areas for the 4–6 fragment ions with overlaid elution profiles. The ratio of peak areas for the light and heavy forms of the peptide, provided by Skyline, reflects the relative abundance of that peptide, and by extension, the expression ratio of the corresponding protein, in SW620 over SW480 cells.
2.4 |. Bioinformatic analyses
Gene Ontology (GO) analysis on biological process of genes encoding all up-regulated RWE proteins was carried out using Database for Annotation, Visualization, and Integrated Discovery (DAVID, version 6.8; https://david.ncifcrf.gov/). Gene set enrichment analysis (GSEA) was performed in GSEA 4.1.0 software (http://www.broad.mit.edu/gsea). The Cancer Genome Atlas–Colon Adenocarcinoma (TCGA-COAD) dataset (n = 512) was downloaded from https://gdc.xenahubs.net/download/TCGA-COAD.htseq_fpkm.tsv.gz. The mRNA expression of each gene encoding the top 10 up-regulated RWE proteins obtained from the LC-PRM analysis was first stratified using its median value. GSEA was then conducted between the stratified TCGA-COAD and the hallmark gene sets (h.all.v7.4.symbols.gmt).
The relative expression levels of RWE proteins between colon cancer tissues and normal adjacent tissues of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) samples [43] were retrieved using the UALCAN online tool (http://ualcan.path.uab.edu/) [44]. The mRNA expression levels of genes encoding RWE proteins in normal, primary tumor, and metastatic tumor tissues of liver were retrieved from GSE41258 by using GEO2R [45]. The outliers of each tissue group were identified and removed, where box-whisker plots were generated using an online tool (https://www.statskingdom.com/boxplot-maker.html) and replotted using Excel.
3 |. RESULTS
3.1 |. Scheduled LC-PRM analysis reveals differentially expressed epitranscriptomic RWE proteins in metastatic SW620 over primary SW480 CRC cells
Our goal of this study was to interrogate systematically the contributions of epitranscriptomic RWE proteins in CRC metastasis. Toward this objective, we began with assessing differential expression of epitranscriptomic RWE proteins in SW480 primary CRC cells, derived from the primary tumor site of a colon adenocarcinoma patient, and SW620 metastatic CRC cells, derived from a lymph node metastatic site of the same parent. By employing our recently developed LC-PRM method [40], in combination with SILAC, we were able to quantify 113 distinct epitranscriptomic RWE proteins in this pair of CRC cells; these proteins represent 74.3% of RWE proteins in the PRM library (Figure 1B, Table S1). Positive identification was considered achieved when 4–6 transitions (i.e., product ions) from the same precursor ion exhibit the same retention time and a dot product (dotp) value [42] of >0.7. In this regard, the dotp value gauges the similarities in relative abundances of fragment ions between the acquired MS/MS and the reference MS/MS in the PRM library that were acquired from previous shotgun proteomic analysis [46]. Of the detected epitranscriptomic RWE proteins, 48 were up-regulated, and 5 were down-regulated by >1.5-fold in the metastatic SW620 relative to the primary SW480 CRC cells. Among them, dihydrouridine synthase 2 (DUS2), decapping mRNA 2 (DCP2), N-acetyltransferase 10 (NAT10), and heterogeneous nuclear ribonucleoprotein C (hnRNPC) were markedly up-regulated by more than 4-fold in SW620 over SW480 cells (Figure 1C, and those proteins with expression ratios between 0.67 and 1.5-fold are shown in Figure S1).
3.2 |. Scheduled LC-PRM analysis affords highly reproducible and accurate quantifications of epitranscriptomic RWE proteins
We next examined the reproducibility of the PRM method by comparing the data acquired from the four SILAC replicates. In this vein, 95 RWE proteins were commonly detected from the two forward and two reverse SILAC experiments (Figure 2A). The log10-transformed protein expression ratios obtained from the averaged ratios of two forward and those of two reverse SILAC labeling experiments exhibited an excellent linear fit (Figure 2B). Moreover, we calculated the replicate ratio based on LC-PRM results of all component peptides from each replicate, and we found that the mean relative standard deviation (RSD) of ratio of the RWE proteins from the four replicates of SILAC experiments was 21.8%. These results together support the consistency of quantification results obtained from four SILAC experiments. We also determined the ratios of all detected tryptic peptides of each RWE protein from the LC-PRM data of the four SILAC replicates, and determined that the mean RSD of expression ratio for each protein from those of its component peptides was 9.1% (Table S1). This result reveals the relatively small variations among the different quantified peptides from the same protein. Together, the PRM method affords reproducible quantifications of RWE proteins.
FIGURE 2.
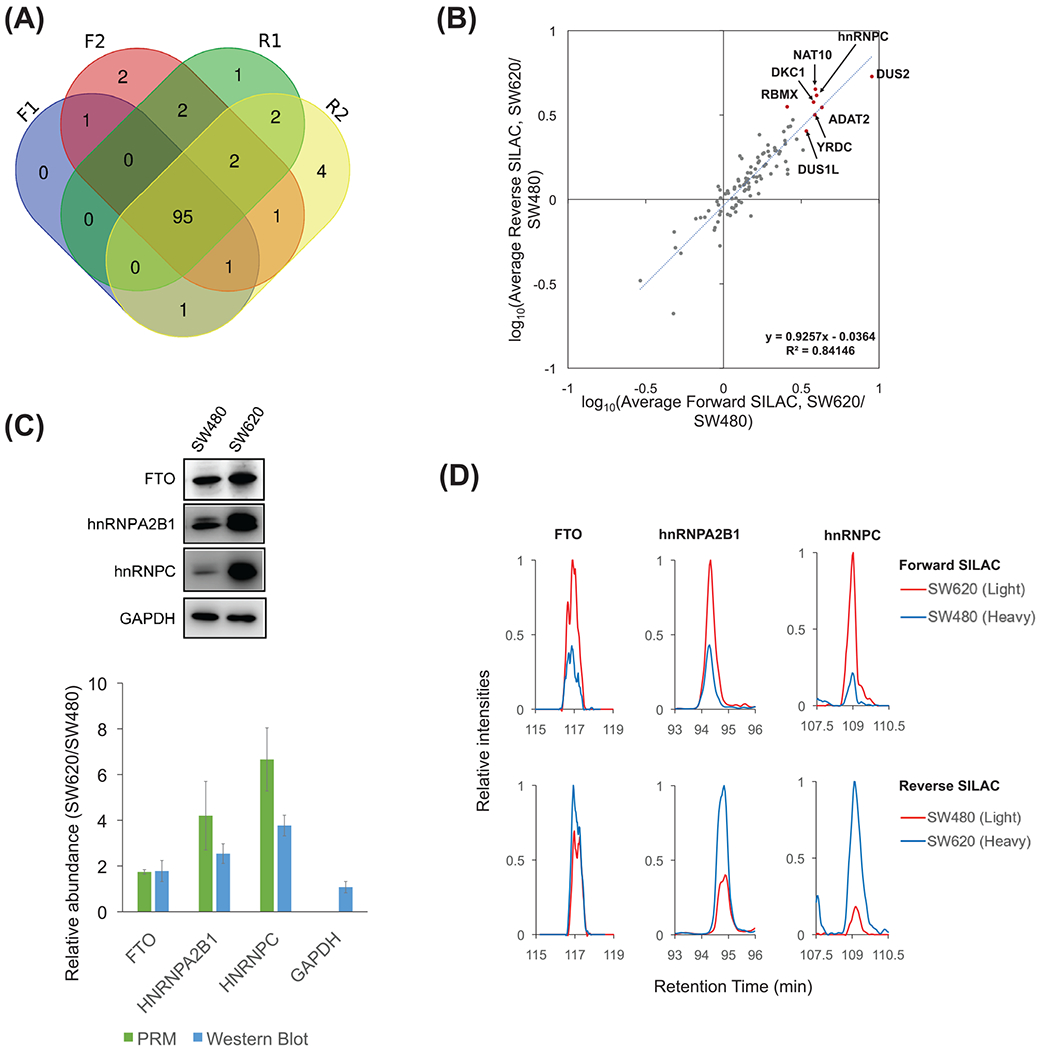
(A) A Venn diagram illustrating the numbers of RWE proteins quantified in four SILAC experiments. (B) Log10(Ratio) of the average protein expression levels in SW620 over SW480 cells obtained from two forward and two reverse SILAC experiments. One outlier (VIRMA, its quantification results from forward and reverse SILAC experiments display a large discrepancy) was excluded. The top 10 up-regulated RWE proteins were labeled in red dots, except DCP2 and TARBP1, which were quantified only in forward SILAC experiments. (C) Western blot analyses of FTO, hnRNPA2B1, and hnRNPC proteins in SW480 and SW620 cells, and the comparison of the quantification data obtained from Western blot (n = 3) and PRM analyses (n = 4). (D) Extracted-ion chromatograms of representative peptides of FTO (LFTVPWPVK), hnRNPA2B1 (IDTIEIITDR), and hnRNPC (MIAGQVLDINLAAEPK), obtained from LC-PRM analysis in one forward (SW_F1) and one reverse SILAC experiment (SW_R1)
LC-PRM also offers accurate quantification of peptides of target proteins since the quantification is based on their unique amino acid sequences derived from the proteins of interest. We further performedWestern blot analyses for three proteins, that is, FTO, heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1), and hnRNPC (Figure 2C), and the results are in agreement with what we obtained from quantitative proteomic experiments, underscoring the accuracies of the PRM method. Figures 2D and S2 illustrate the extracted-ion chromatograms for the component peptides of FTO, hnRNPA2B1 and hnRNPC, and MS/MS of the component peptide of hnRNPC, respectively.
3.3 |. Analysis of the up-regulated RWE proteins from LC-PRM analysis using the CCLE database, GO, and GSEA
We queried the RNA-Seq data in the Cancer Cell Line Encyclopedia (CCLE) database for the mRNA expression levels of the top 10 up-regulated RWE proteins in SW620 over SW480 cells, as revealed from LC-PRM analysis. We uncovered that the mRNA levels of DUS2, NAT10, ADAT2, DKC1, TARBP1, RBMX, and DUS1L genes were also up-regulated by over 1.5-fold in SW620 relative to SW480 cells, suggesting that the up-regulation of these proteins in metastatic cells arises mainly from elevated transcription of the corresponding genes. On the other hand, the mRNA levels of DCP2, hnRNPC, and YRDC genes differed slightly between SW480 and SW620 cells (Figure 3A and B). The reason of the augmented abundance of DCP2, hnRNPC, and YRDC in the metastatic CRC cells remains unclear, and it could arise from post-transcriptional regulation, for example, through increased translational efficiency and/or diminished protein turnover.
FIGURE 3.
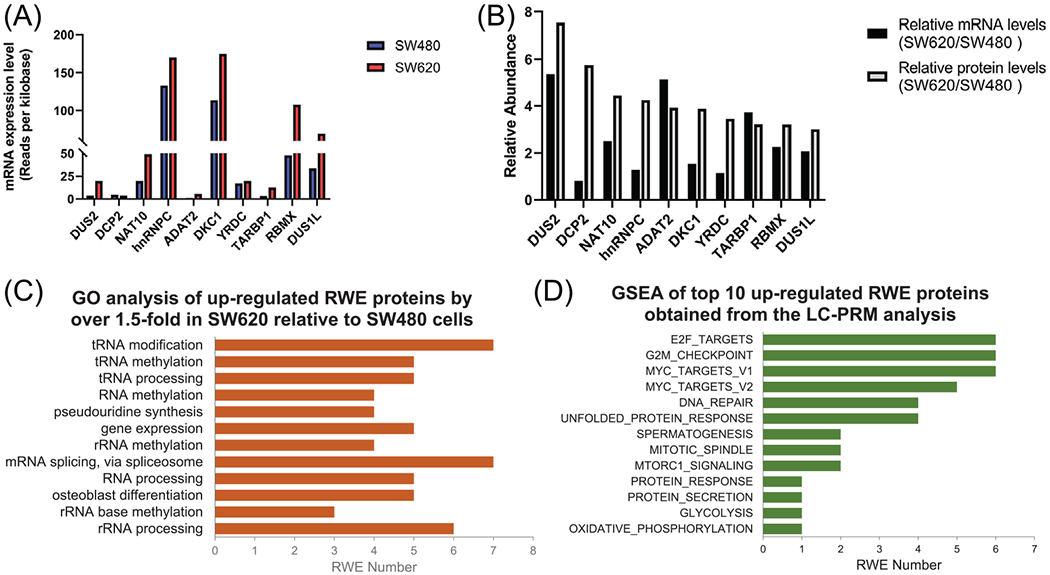
(A) mRNA expression levels of the top 10 up-regulated RWE genes identified from LC-PRM analysis. The data were retrieved from the CCLE database. (B) The comparison of relative mRNA levels obtained from the CCLE database and relative protein levels of the top 10 up-regulated RWE genes in SW620 versus SW480 cells, as obtained from LC-PRM analysis. (C) A bar graph illustrating GO analysis on biological process (BP) of all up-regulated RWE genes. The BP results were sorted by Benjamini FDR from smallest to largest using 0.01 as a cutoff. (D) A bar graph showing GSEA results of the sum of particular enriched gene sets of top 10 up-regulated RWE genes identified from LC-PRM analysis. TCGA-COAD dataset was first stratified to high- and low-expression groups using the median value of mRNA expression of a specific RWE protein among all patient tissues. The significance level in GSEA analysis was defined as nominal p-value < 0.01 and FDR q-value < 0.25. The number of permutations was set at 1000
We also conducted GO analysis on those RWE proteins that were up-regulated in metastatic over primary CRC cells by at least 1.5-fold. The result showed that these proteins are mainly associated with tRNA modification, methylation and processing, RNA methylation, and ψ synthesis (Figure 3C). GSEA identifies cumulative expression changes of multiple genes within a priori defined gene set displaying statistically significant difference in two phenotypes [47]. GSEA of the top 10 up-regulated RWE proteins from the LC-PRM analysis was performed individually for the corresponding genes after stratifying the results to high- and low-mRNA expression groups according to their median values in the TCGA-COAD dataset, against the hallmark gene sets provided by the GSEA Molecular Signatures Database [47]. Notably, we observed significantly (p < 0.01 and FDR < 0.25) enriched hallmark gene sets in the high-expression group of all the top 10 up-regulated RWE proteins except ADAT2 and TARBP1 (Table S2). Specifically, E2F targets, G2-M checkpoint, and MYC target V1 were among the most frequently enriched gene sets, where six out of eight RWE proteins display enrichment with those gene sets in the high-expression group of the protein (Figure 3D). Figure S3A depicts enrichment plots of DUS2. Those hallmark gene sets are related to cancer cell proliferation and tumor metastasis [48]. Notably, E2F transcription factors 1 and 7 (E2F1 and E2F7) were found to modulate colon cancer metastasis and development [49]. Some E2F targets, including enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) and bone morphogenetic protein 4 (BMP4), could mediate melanoma and breast cancer metastasis [50, 51]. Moreover, it was shown that ER+/HER2- breast cancer with high G2-M checkpoint pathway genes activity is more likely to metastasize [52]. Furthermore, even though the association between c-Myc and CRC metastasis remained controversial [53], c-Myc targets, such as YTHDF1 and AP4, were associated with CRC metastasis [54, 55]. It is also worth noting that the mRNA expression levels of DKC1 and NAT10 are positively associated with those of MYC in the TCGA-COAD dataset (Figure S3B).
Several differentially expressed RWE proteins identified in this study were found to be associated with cancer metastasis. For instance, DUS2, a tRNA-dihydrouridine synthase, is up-regulated in nonsmall cell lung carcinomas (NSCLC), and a higher level of DUS2 is accompanied with a poorer prognosis of lung cancer patients [56]. In addition, NAT10, an acetyltransferase, promotes gastric cancer metastasis through inducing the formation of N4-acetylcytidine in mRNA of COL5A1 gene, and glycogen synthase kinase 3β (GSK-3/β) was found to promote the invasion of CRC cells through modulating the subcellular redistribution of NAT10 [57, 58]. Moreover, hnRNPC, an m6A reader protein, is involved in CRC progression since its elevated level in SW620 cells drives alternative cleavage and polyadenylation of MTHFD1L mRNA, a potential therapeutic target for CRC [59]. Furthermore, DKC1, a ψ synthase, plays essential roles in angiogenesis and CRC metastasis by activating the transcription of HIF-1α, and may serve as a therapeutic target for CRC [60]. Additionally, YRDC, an N6-threonyl-carbamoyl-adenosine (t6A) writer, promotes hepatocellular carcinoma by activating MEK/ERK signaling pathway [61]. These studies of DUS2, NAT10, hnRNPC, DKC1, and YRDC support our proteomic results that these proteins may promote metastatic transformation of CRC.
3.4 |. DUS1L is accompanied with CRC initiation and metastasis
Vasaikar et al. analyzed the global protein expression in matched tumor and tumor-adjacent normal tissues from 110 colon cancer patients from the CPTAC [43]. For comparison, 75% of RWE proteins in the PRM library were quantified in our LC-PRM analysis, and CPTAC analysis allowed for the quantification of 66% of the RWE proteins included in the same library (Figure 4A).
FIGURE 4.
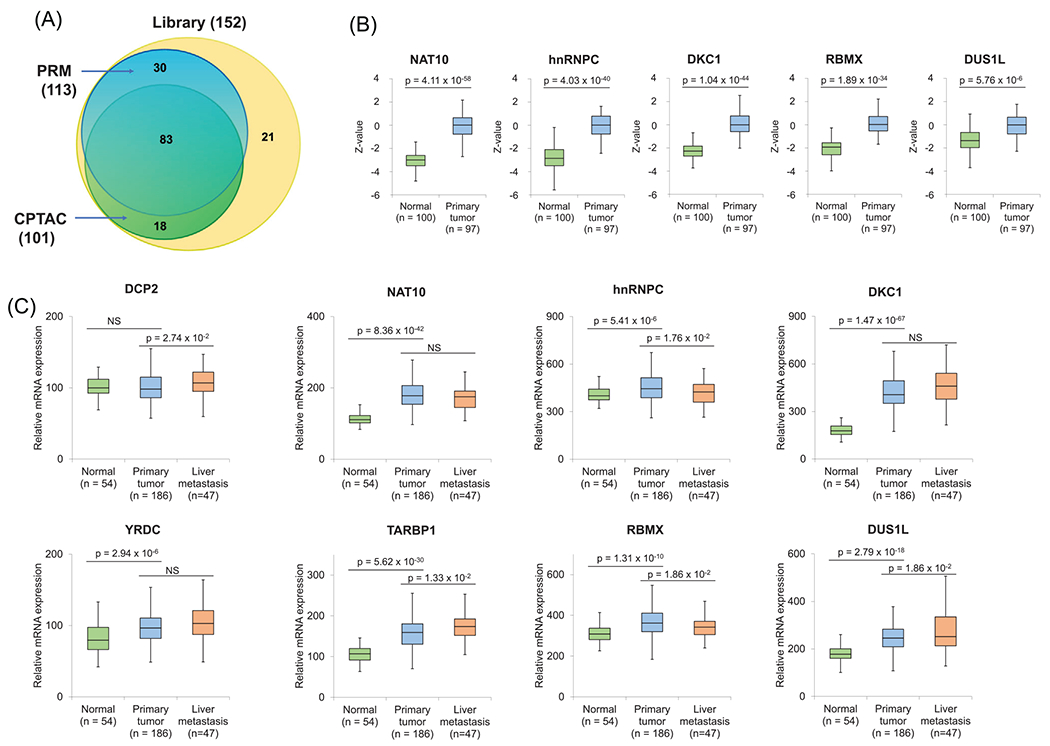
(A) A Venn diagram illustrating the number of RWE proteins quantified from the PRM analysis in this study and the previously reported CPTAC analysis, in comparison with the total number of RWE proteins deposited in the PRM library. (B) Relative protein expression levels of NAT10, hnRNPC, DKC1, RBMX, and DUS1L in primary colon tumor tissues and tumor-adjacent normal tissues in the CPTAC samples. Z-values represent SD from the median across samples. (C) Relative mRNA expression levels of DCP2, NAT10, HNRNPC, DKC1, YRDC, TARBP1, RBMX, and DUS1L genes in normal tissues, primary colon tumor tissues, and liver metastasis tissues in GSE41258. The p values were calculated using an unpaired two-tailed Student’s t test. For (B-C), the horizontal edges and inner line of the box illustrate the upper/lower quartiles and median, respectively. The top/bottom ends of the whisker denote the maximum/minimum values
We further investigated if the top 10 RWE proteins up-regulated in SW620 relative to SW480 cells also exhibit differential protein expression in colon tumor tissue compared with tumor-adjacent normal tissues. Our results showed that five differentially expressed RWE proteins, namely, NAT10, hnRNPC, DKC1, RBMX, and DUS1L, displayed pronounced differences in expression between tumor and tumor-adjacent normal tissues in the CPTAC samples (Figure 4B). This result indicates that elevated expressions of these proteins are positively associated with both the initiation and metastatic transformation of CRC. Further experiments are needed to determine if augmented expression of these proteins drives the initiation and metastatic transformation of CRC. On the other hand, DUS2, TARBP1, and YRDC were either not significantly associated or slightly down-regulated in primary tumor tissues relative to tumor-adjacent normal tissues. These differences are not surprising, considering that tumor initiation and metastasis may also involve distinct molecular pathways, and that our PRM data were acquired from cell lines derived from a single patient. In addition, DCP2 and ADAT2 were quantified in SW480/SW620 cells in our PRM analysis, but not in CPTAC samples.
Apart from examining the protein expression of the top 10 up-regulated genes in the CPTAC samples, we explored GSE41258 [45], a microarray dataset for mRNA expression in normal, primary tumor, and liver metastasis tissues collected from colon cancer patients. Such analysis revealed markedly higher mRNA expressions of NAT10, HNRNPC, DKC1, YRDC, TARBP1, RBMX, and DUS1L genes in primary tumor tissues compared with normal tissues (Figure 4C), which is in keeping with the CPTAC analysis. Moreover, DCP2, TARBP1, and DUS1L genes display significantly higher mRNA expression in liver metastasis tissues relative to primary colon tumor tissues (Figure 4C), indicating that they may also be positively associated with CRC metastasis. Hence, the CPTAC and GSE41258 data showed that DUS1L exhibits higher mRNA and protein expression in primary colon cancer tissues than normal tissues, and it also displays a higher mRNA expression in the metastatic tissues to the liver relative to primary colon tumor tissues, suggesting a dual role of DUS1L in the initiation and metastatic transformation of colorectal cancer (CRC).
4 |. DISCUSSION
We employed a scheduled LC-PRM method for highly sensitive, robust, and high-throughput profiling of epitranscriptomic RWE proteins accompanied with CRC metastasis. Our LC-PRM approach facilitated reproducible and accurate quantifications of 95 epitranscriptomic RWE proteins quantified in all four SILAC experiments, with 48 and 5 of these proteins being up- and down-regulated, respectively, by >1.5-fold in SW620 metastatic CRC cells relative to SW480 primary CRC cells. NAT10, hnRNPC, and DKC1 exhibit pronounced up-regulations in the metastatic over primary CRC cells, and the roles of these proteins in CRC metastasis are known [57, 59, 60], which is in keeping with our LC-PRM analysis. Interrogation of publicly available data of CRC patients unveiled the potential involvement of DUS1L in both the initiation and metastatic transformation of CRC. We envision that the LC-PRM method developed herein can also be harnessed for future investigations about how epitranscriptomic modulators contribute to the metastatic transformations of other types of cancer.
Supplementary Material
Significance Statement.
Several reports unveiled the roles of individual epitranscriptomic RWE proteins in cancer metastasis; however, to our knowledge, there is no systematic investigation about how aberrant expression of epitranscriptomic RWE proteins contributes to cancer progression. CRC is known for its high occurrence and mortality rates, and effective therapeutic targets for treating CRC are urgently needed. Here, we applied our recently developed LC-PRM method to explore the differential expression of epitranscriptomic RWE proteins in metastatic SW620 versus primary SW480 CRC cells. We were able to reproducibly and accurately quantify a total of 95 nonredundant epitranscriptomic RWE proteins; among them, 48 and 5 were up- and down-regulated by over 1.5-fold in SW620 relative to SW480 cells, respectively. We also discussed the potential roles of some of these differentially expressed proteins in the initiation and metastatic transformation of CRC. Thus, this represents the first application of a PRM approach for interrogating the alterations in expression levels of epitranscriptomic RWE proteins accompanied with CRC metastasis.
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (R35 ES031707).
Abbreviations:
- RWE
reader, writer, and eraser
- SILAC
stable isotope labeling by amino acids in cell culture
- CRC
colorectal cancer
- m6A
N6-methyladenosine
- m1A
N1-methyladenosine
- m5C
5-methylcytidine
- m7G
N7-methylguanosine
- ψ
pseudouridine
- PRM
parallel-reaction monitoring
- BSA
bovine serum albumin
- iRT
normalized retention time
- dotp
dot product
- GO
gene ontology
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- GSEA
gene set enrichment analysis
- TCGA-COAD
The Cancer Genome AtlasColon Adenocarcinoma
- CPTAC
the Clinical Proteomic Tumor Analysis Consortium
- RSD
relative standard deviation
- CCLE
the Cancer Cell Line Encyclopedia
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online https://doi.org/10.1002/pmic.202200059 in the Supporting Information section at the end of the article.
DATA AVAILABILITY STATEMENT
All the LC-MS/MS raw files and Skyline PRM library were deposited to the ProteomeXchange Consortium via the PRIDE [62] partner repository with the dataset identifier PXD030907.
REFERENCES
- 1.Kadumuri RV, & Janga SC (2018). Epitranscriptomic code and its alterations in human disease. Trends in Molecular Medicine, 24, 886–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan T (2018). Modifications and functional genomics of human transfer RNA. Cell Research, 28, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roundtree IA, Evans ME, Pan T, & He C (2017). Dynamic RNA modifications in gene expression regulation. Cell, 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, & Rechavi G (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485, 201–206. [DOI] [PubMed] [Google Scholar]
- 5.Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, Hess D, Andrade-Navarro MA, Biggiogera M, Helm M, Soller M, Bühler M, & Roignant J-Y (2018). Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes & Development, 32, 415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, & He C (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nature Chemical Biology, 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, & Conrad NK (2017). The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell, 169, 824–835 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ping X-L, Sun B-F, Wang L,Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, Chen Y-S, Zhao X, Li A, Yang Y, Dahal U, Lou X-M, Liu X, Huang J, Yuan W-P, Zhu X-F, … Yang Y-G (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Research, 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, Wang F, Wang X, Shen B, Wang Y, Feng X, He C, & Liu J (2018). VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discovery, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, Qi M, Lu Z, Shi H, Wang J, Cheng Y, Luo G, Dai Q, Liu M, Guo X, Sha J, Shen B, & He C (2017). Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Research, 27, 1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li A, Chen Y-S, Ping X-L, Yang X, Xiao W, Yang Y, Sun H-Y, Zhu Q, Baidya P, Wang X, Bhattarai DP,Zhao Y-L, Sun B-F, & Yang Y-G (2017). Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Research, 27, 444–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, & He C (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature, 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G, & He C (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature Chemical Biology, 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur J-J, Chen Q, Gross SS, Elemento O, Debart F, Kiledjian M, & Jaffrey SR (2017). Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature, 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H, Lu Z, Bosmans RPG, Dai Q, Hao Y-J, Yang X, Zhao W-M, Tong W-M, Wang X-J, … He C. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell, 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, Zheng G, Pan T, Solomon O, Eyal E, Hershkovitz V, Han D, Doré LC, Amariglio N, Rechavi G, & He C (2016). The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature, 530, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, & Yi C (2016). Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nature Chemical Biology, 12, 311–316. [DOI] [PubMed] [Google Scholar]
- 18.Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, Erlacher M, Rossmanith W, Stern-Ginossar N, & Schwartz S (2017). The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature, 551, 251–255. [DOI] [PubMed] [Google Scholar]
- 19.Dai X, Gonzalez G, Li L, Li J, You C, Miao W, Hu J, Fu L, Zhao Y, Li R, Li L, Chen X, Xu Y, Gu W, & Wang Y (2020). YTHDF2 binds to 5-methylcytosine in RNA and modulates the maturation of ribosomal RNA. Analytical Chemistry, 92, 1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, &Preiss T (2012). Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Research, 40, 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X, Yang Y, Sun B-F,Chen Y-S,Xu J-W, Lai W-Y, Li A, Wang X, Bhattarai DP, Xiao W, Sun H-Y, Zhu Q, Ma H-L, Adhikari S, Sun M, Hao Y-J, Zhang B, Huang C-M, Huang N, … Yang Y-G (2017). 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Research, 27, 606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, & Gregory RI (2018). Mettl1/Wdr4-mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Molecular Cell, 71, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlile TM, Rojas-Duran MF,Zinshteyn B, Shin H, Bartoli KM, & Gilbert WV (2014). Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature, 515, 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G, & Regev A (2014). Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell, 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darzacq X (2002). Cajal body-specific small nuclear RNAs: a novel class of 2 ’-O-methylation and pseudouridylation guide RNAs. EMBO Journal, 21, 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rebane A, Roomere H, & Metspalu A (2002). Locations of several novel 2 ’-O-methylated nucleotides in human 28S rRNA. BMC Molecular Biology, 3, Article 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frye M, Harada BT, Behm M, & He C (2018). RNA modifications modulate gene expression during development. Science, 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, & Liu T (2019). The critical role of RNA m6A methylation in cancer. Cancer Research, 79, 1285–1292. [DOI] [PubMed] [Google Scholar]
- 29.Tirumuru N, Zhao BS, Lu W, Lu Z, He C, & Wu L (2016). N6-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife, 5, e15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon K-J, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, Su Y, Kim N-S, Zhu Y, Zheng L, Kim S, Wang X, Doré LC, Jin P, Regot S,Zhuang X, Canzar S, He C, Ming G-L, & Song H (2017). Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell, 171, 877–889.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, Qin X, Tang L, Wang Y, Hong G-M, Huang H, Wang X, Chen P, Gurbuxani S, Arnovitz S, … Chen J. (2017). FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell, 31, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma J-Z, Yang F, Zhou C-C, Liu F, Yuan J-H, Wang F, Wang T-T, Xu Q-G, Zhou W-P, & Sun S-H (2017). METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6)-methyladenosine-dependent primary MicroRNA processing. Hepatology, 65, 529–543. [DOI] [PubMed] [Google Scholar]
- 33.Xi Y,& Xu P (2021). Global colorectal cancer burden in 2020 and projections to 2040. Translational Oncology, 14, 101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zacharakis M, Xynos ID, Lazaris A, Smaro T, Kosmas C, Dokou A, … Tsavaris N (2010). Predictors of survival in stage IV metastatic colorectal cancer. Anticancer Research, 30, 653–660. [PubMed] [Google Scholar]
- 35.Peterson AC, Russell JD, Bailey DJ, Westphall MS, & Coon JJ (2012). Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Molecular & Cellular Proteomics: MCP, 11, 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weng LW, Guo LL, Vachani A, Mesaros C, & Blai IA (2018). Quantification of serum high mobility group Box 1 by liquid chromatography/high-resolution mass spectrometry: Implications for its role in immunity, inflammation, and cancer. Analytical Chemistry, 90, 7552–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, & Mann M (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- 38.Miao W, Guo L, & Wang Y (2019). Imatinib-induced changes in protein expression and ATP-binding affinities of kinases in chronic myelocytic leukemia cells. Analytical Chemistry, 91, 3209–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao W, Li L, &Wang Y (2018). Identification of helicase proteins as clients for HSP90. Analytical Chemistry, 90, 11751–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi TF, Miao W, & Wang Y (2022). Targeted profiling of epitranscriptomic reader, writer, and eraser proteins accompanied with radioresistance in breast cancer cells. Analytical Chemistry, 94, 1525–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.WiŚniewski JR, Zougman A, Nagaraj N, & Mann M (2009). Universal sample preparation method for proteome analysis. Nature Methods, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- 42.De Graaf EL, Altelaar AFM, Van Breukelen B, Mohammed S, & Heck AJR (2011). Improving SRM assay development: A global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. Journal of Proteome Research, 10, 4334–4341. [DOI] [PubMed] [Google Scholar]
- 43.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, Gritsenko MA, Zimmerman LJ, Mcdermott JE, Clauss TR, Moore RJ, Zhao R, Monroe ME, Wang Y-T, Chambers MC, … Clinical Proteomic Tumor, A. (2019). Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell, 177, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK, & Varambally S (2017). UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia, 19, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, & Domany E (2009). Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America, 106, 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miao W, Li L, & Wang Y (2018). A targeted proteomic approach for heat shock proteins reveals DNAJB4 as a suppressor for melanoma metastasis. Analytical Chemistry, 90, 6835–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, & Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gong Y, Liu Y, Wang T, Li Z, Gao L, Chen H, Shu Y, Li Y, Xu H, Zhou Z, & Dai L (2021). Age-associated proteomic signatures and potential clinically actionable targets of colorectal cancer. Molecular & Cellular Proteomics, 20, 100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang Z, Lin M, Li C, Liu H, & Gong C (2020). A comprehensive review of the roles of E2F1 in colon cancer. American Journal of Cancer Research, 10, 757–768. [PMC free article] [PubMed] [Google Scholar]
- 50.Hollern DP, Honeysett J, Cardiff RD, & Andrechek ER (2014). The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Molecular and Cellular Biology, 34, 3229–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.White JR, Thompson DT, Koch KE, Kiriazov BS, Beck AC, Van Der Heide DM, Grimm BG, Kulak MV, & Weigel RJ (2021). AP-2α-mediated activation of E2F and EZH2 drives melanoma metastasis. Cancer Research, 81, 4455–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oshi M,Takahashi H,Tokumaru Y,Yan L, Rashid OM, Matsuyama R, Endo I, & Takabe K (2020). G2M cell cycle pathway score as a prognostic biomarker of metastasis in estrogen receptor (ER)-positive breast cancer. International Journal of Molecular Sciences, 21, 2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He W-L, Weng X-T, Wang J-L, Lin Y-K, Liu T-W, Zhou Q-Y, Hu Y, Pan Y, & Chen X-L (2018). Association between c-Myc and colorectal cancer prognosis: A meta-analysis. Frontiers in Physiology, 9, 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jackstadt R, Röh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A, & Hermeking H (2013). AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. Journal of Experimental Medicine, 210, 1331–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, Takahashi H, Nishida N, Haraguchi N, Sakai D, Kudo T, Hata T, Matsuda C, Mizushima T, Satoh T, Doki Y, Mori M, & Ishii H (2017). Oncogene c-Myc promotes epitranscriptome m(6)A reader YTHDF1 expression in colorectal cancer. Oncotarget, 9, 7476–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato T, Daigo Y, Hayama S, Ishikawa N, Yamabuki T, Ito T, Miyamoto M, Kondo S, & Nakamura Y (2005). A novel human tRNA-dihydrouridine synthase involved in pulmonary carcinogenesis. Cancer Research, 65, 5638–5646. [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Hou W, Wang H-L, Liu H-J, Jia X-Y, Zheng X-Z, Zou Y-X, Li X, Hou L, Mcnutt MA, & Zhang B (2014). GSK-3β-regulated N-acetyltransferase 10 is involved in colorectal cancer invasion. Clinical Cancer Research, 20, 4717–4729. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Jing Y, Wang Y, Tang J, Zhu X, Jin W-L, Wang Y, Yuan W, Li X, & Li X (2021). NAT10 promotes gastric cancer metastasis via N4-acetylated COL5A1. Signal Transduction and Targeted Therapy, 6, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischl H, Neve J, Wang Z, Patel R, Louey A, Tian B, & Furger A (2019). hnRNPC regulates cancer-specific alternative cleavage and polyadenylation profiles. Nucleic Acids Research, 47, 7580–7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou P, Shi P, Jiang T, Yin H, Chu S, Shi M, Bai J, & Song J (2020). DKC1 enhances angiogenesis by promoting HIF-1 alpha transcription and facilitates metastasis in colorectal cancer. British Journal of Cancer, 122, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang S, Zhu P, Sun B, Guo J, Zhou H, Shu Y, & Li Q (2019). Modulation of YrdC promotes hepatocellular carcinoma progression via MEK/ERK signaling pathway. Biomedicine & Pharmacotherapy, 114, 108859. [DOI] [PubMed] [Google Scholar]
- 62.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz Ş, Tiwary S, Cox J, Audain E, Walzer M, … Vizcaíno JA (2019). The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Research, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the LC-MS/MS raw files and Skyline PRM library were deposited to the ProteomeXchange Consortium via the PRIDE [62] partner repository with the dataset identifier PXD030907.