

Abstract
The small heat shock protein HspB1, also known as Hsp25/27, is a ubiquitously expressed molecular chaperone that responds to mechanical cues. Uniaxial cyclic stretch activates the p38 mitogen-activated protein kinase (MAPK) signaling cascade and increases the phosphorylation of HspB1. Similar to the mechanosensitive cytoskeletal regulator zyxin, phospho-HspB1 is recruited to features of the stretch-stimulated actin cytoskeleton. To evaluate the role of HspB1 and its phosphoregulation in modulating cell function, we utilized CRISPR/Cas9-edited HspB1-null cells and determined they were altered in behaviors such as actin cytoskeletal remodeling, cell spreading, and cell motility. In our model system, expression of WT HspB1, but not nonphosphorylatable HspB1, rescued certain characteristics of the HspB1-null cells including the enhanced cell motility of HspB1-null cells and the deficient actin reinforcement of stretch-stimulated HspB1-null cells. The recruitment of HspB1 to high-tension structures in geometrically constrained cells, such as actin comet tails emanating from focal adhesions, also required a phosphorylatable HspB1. We show that mechanical signals activate posttranslational regulation of the molecular chaperone, HspB1, and are required for normal cell behaviors including actin cytoskeletal remodeling, cell spreading, and cell migration.
INTRODUCTION
Mechanical forces are sensed by cells and transduced into biochemical signals that drive changes in gene expression and influence cell fate (Discher et al., 2009; Wozniak and Chen, 2009; Mammoto et al., 2012). This ability to sense and respond to mechanical cues is essential for morphogenetic movements during embryonic development and normal adult physiological functions such as required in cardiovascular and respiratory systems, while disturbances in mechanical homeostasis are associated with myriad disease phenotypes (Deanfield et al., 2007; Chen, 2008; Fournier et al., 2010; Ateshian and Humphrey, 2012). In order for a mechanical signal to influence biology, it must be converted to a chemical signal that is capable of regulating cell behavior and function; this is the process of mechanotransduction. Central elements of a mechanotransduction pathway from the cell surface to the nucleus include cell surface adhesion receptors such as cadherins and integrins (Heisenberg and Bellaiche, 2013; Low et al., 2014; Benham-Pyle et al., 2015; Shivashankar et al., 2015), mechanosensors that change conformation in response to physical cues or detect these force-induced changes (McGough et al., 1997; Sawada et al., 2006; del Rio et al., 2009; Shimozawa and Ishiwata, 2009; Ehrlicher et al., 2011; Hayakawa et al., 2011; Uyeda et al., 2011; Galkin et al., 2012), and nuclear signaling factors that regulate gene expression in response to mechanical cues (Naumanen et al., 2008; Halder et al., 2012; Smith et al., 2013; Hoffman et al., 2020). A major challenge in cell biology is to ascertain the detailed molecular mechanisms that underlie mechanotransduction.
Actin plays a central role in transcellular communication of mechanical cues
Essential for maintaining tensional homeostasis, actin stress fibers (SFs) are myosin-associated, contractile bundles of filamentous actin (F-actin) (Peterson et al., 2004, Naumanen et al., 2008, Smith et al., 2013). SFs are prominent in cultured cells, particularly fibroblasts, and also assemble in vivo when cells experience mechanical stress (Wong et al., 1983; Byers et al., 1984). Tensed actin SFs anchored at integrin-based focal adhesions (FA) provide mechanoaccumulation sites for several mechanosensitive LIM-domain proteins (Sun et al., 2020, Winkelman et al., 2020, Anderson et al., 2021, Sala and Oakes, 2021). Many of these proteins were also identified in tension-dependent focal adhesion proteomes (Kuo et al., 2011, Schiller et al., 2011) and consensus integrin-based adhesomes (Horton et al., 2015). The small heat shock protein HspB1 (Hsp25/27) was found in the tension-dependent focal adhesion proteomes and the integrin-based adhesome. Characteristics of HspB1 suggested an intriguing mechanosensitivity that we decided to explore further.
HspB1 has been implicated in multiple diseases, including cancers, neuropathologies, and even COVID-19 pathology (Kampinga and Garrido, 2012; Zoubeidi and Gleave, 2012; Calderwood and Gong, 2016; O’Brien and Sandhu, 2020; Vendredy et al., 2020). Cancer cell survival and development of drug resistance may be mediated by some heat shock proteins (Kampinga and Garrido, 2012; Calderwood and Gong, 2016). Therapeutic approaches targeting HspB1 as a cancer treatment have been developed and implemented (Zoubeidi and Gleave, 2012; Rosenberg et al., 2018; Shevtsov et al., 2019); thus a greater understanding of HspB1’s impact on cell physiology is warranted.
The small heat shock proteins play a cytoprotective role through their molecular chaperone activity and their response in cells experiencing mechanical stress (Collier and Benesch, 2020). HspB1 is phosphorylated in a p38-dependent manner in fibroblasts subjected to uniaxial cyclic stretch (Chaudhuri and Smith, 2008; Hoffman et al., 2017). Here we have developed a cell-based model system to study HspB1 function and phosphoregulation. CRISPR-Cas9 engineered HspB1-null cells are altered in several physiological responses including actin remodeling following stretch stimulation, and cell spreading and motility. Re-expression of Wild-type (WT) HspB1 and phosphomutant HspB1s revealed that only phosphorylatable HspB1, which displayed mechanoaccumulation to tensed actin SFs, restored activities lost in HspB1-null cells including actin remodeling, cell spreading and cell motility. We report here that posttranslational regulation by phosphorylation of HspB1 is critical to the mechanotransduction response in these cells.
RESULTS
p38-dependent phosphorylation of HspB1 in response to mechanical stimulation
Cells activate multiple mitogen-activated protein kinase (MAPK) signaling cascades in response to mechanical stimulation, resulting in phosphorylation of target proteins and cytoskeletal changes (Li et al., 1996; Nguyen et al., 2000; Sawada et al., 2001; Chaudhuri and Smith, 2008). Murine HspB1 (Figure 1A) contains a C-terminal alpha-crystallin domain common in heat shock proteins and two conserved serine phosphorylation sites (S15 and S86), both of which are established mitogen-activated protein kinase-activated protein kinase 2 (MK2) targets (Stokoe et al., 1992). The baseline phosphorylation of HspB1 present in unstimulated fibroblast cells is elevated by exposure of cells to uniaxial cyclic stretch stimulation (Figure 1B) a 1.9-fold increase of the phospho-HspB1 signal (Supplemental Table S1). We previously used cells null for MK2 with paired MK2-rescue cells (Ronkina et al., 2007; Sousa et al., 2007) to identify MK2 as the p38 pathway kinase responsible for the stretch-stimulated HspB1 phosphorylation (Hoffman et al., 2017). In unstimulated cells HspB1 exhibits a diffuse cytoplasmic distribution and phospho-HspB1 is barely detectable by indirect immunofluorescence microscopy (Figure 1C). On mechanical stimulation HspB1 associates with linear elements that correspond to actin SFs, with phospho-HspB1 concentrated at discrete actin SF domains (Figure 1C). Further examination of the distribution of phospho-HspB1 in stretch-stimulated cells revealed that phospho-HspB1 is concentrated at the ends of actin SFs (yellow arrowheads) where the actin cytoskeleton is anchored to integrin-based complexes known as FA and also can be found across the nucleus at structures suggestive of transmembrane actin nuclear lines (Luxton et al., 2011) (Figure 1D). The well-characterized mechanosensitive cytoskeletal adaptor zyxin (Yoshigi et al., 2005; Hoffman et al., 2012) decorates the entire actin cytoskeleton following stretch stimulation, and the molecular chaperone phospho-HspB1 overlaps with zyxin only at more discrete regions at the ends of actin SFs (Figure 1D). The stretch-stimulated phosphorylation and cytoskeletal localization of HspB1 is blocked by preincubation with the p38 inhibitor SB203580 (Figure 1E), a decreased phospho-HspB1 signal from 2.7-fold to 0.7-fold (Supplemental Table S1), indicating that p38 MAPK signaling is required for the targeting modification.
FIGURE 1:

p38-dependent HspB1 phosphorylation and cytoskeletal response. (A) Diagram of murine HspB1 depicting phosphorylation targets of Ser15 and Ser86, and the alpha-crystallin domain. (B) Western immunoblot analysis of fibroblast cells subjected to 60 min uniaxial cyclic stretch shows increased phosphorylation of Ser 86-HspB1, with Total HspB1 and vinculin loading control, quantified in Supplemental Table S1. (C) Immunolocalization of Phospho-Ser86 HspB1 and Total HspB1 in unstretched and stretch-stimulated cells shows stretch-stimulated increase in phospho-HspB1 along with cytoskeletal distribution of HspB1. Uniaxial stretch vector in the horizontal direction is indicated by double-headed arrow of 50 µm scale. (D) Fluorescence confocal microscopy (top row) of F-actin (phalloidin, magenta) and phospho-HspB1 (green) following stretch-stimulation of a WT fibroblast shows accumulation of phospho-HspB1 at the ends of actin SFs (yellow arrowhead). Immunofluorescence microscopy (bottow row) of zyxin (magenta) and phospho-HspB1 (green) in a stretch-stimulated cell shows partial overlap of zyxin and HspB1 (yellow arrowhead). (E) Western immunoblot analysis of phospho-Ser86-HspB1 stretch response (0–15 m–60 m) in control cells and in cells preincubated with 10 µM p38 inhibitor SB203580, with vinculin loading controls. Stretch-stimulated phospho-HspB1 is blocked by the p38 inhibitor. (F) WT cells transfected with zyxin-GFP (green), seeded on fibronectin-coated coverslips and incubated with SiR-actin dye (magenta) were acquired on a spinning disk confocal microscope and are shown as maximum intensity projections from deconvolved images. Inset is magnified zyxin-GFP signal with boxed region of interest and yellow arrowhead indicating start point. (G) Kymograph of the 20-micron boxed region at 2-min intervals shows zyxin-GFP flux in WT cell over 10-min time frame.
The phospho-HspB1 distribution at the FA-proximal termini of SFs is reminiscent of structures present at sites of cytoskeletal tension that are referred to as actin “comet tails” (Sechi et al., 1997; Guo and Wang, 2007; Anderson et al., 2008; Yamashiro and Watanabe, 2014). Actin comet tails have been shown to be sites of retrograde actin flow or flux driven by actin assembly, as evidenced by the centripetal movement of actin from cell edges toward the cell interior that can be observed by light microscopy. The focal adhesion protein zyxin is a useful marker to measure retrograde actin flow as it has been shown to flow along the actin emerging from FA (Guo and Wang, 2007). Using live cell microscopy of transfected zyxin-GFP and the cell-permeable dye SiR-actin in cells plated on glass coverslips, we determined that, like phospho-HspB1, zyxin is associated with actin “comet tails” (Figure 1F) and these zyxin-GFP sites represent regions of retrograde actin flux, as shown at 2-min intervals (Figure 1G). We measured a mean velocity of 17.3 nm/s, consistent with previous measurements of retrograde actin flux at SF anchorage sites (Guo and Wang, 2007; Anderson et al., 2008; Thievessen et al., 2013; Yamashiro and Watanabe, 2014). These findings raised the possibility that cytoskeletal tension might be the trigger that promotes local accumulation of phospho-HspB1.
Geometric constraints on micropattern islands induce HspB1 subcellular distribution to high-tension areas
One way to generate intracellular tension at specific subcellular areas in cells is to force cells into certain geometric shapes. Cells constrained to square or rectangular shapes develop high traction stress at corners and edges (Parker et al., 2002; Oakes et al., 2014; Chang et al., 2019). The cytoskeletal protein zyxin is a mechanosensitive protein that localizes to high-tension areas, including actin comet tails, in cells constrained in a square geometry (Guo and Wang, 2007; Hoffman et al., 2017). Using immunofluorescent microscopy on WT cells stained with a zyxin-specific antibody, we screened through WT cells adhered on multiple square and rectangular shapes of varying aspect ratio (length to width 7 to 1) and determined that a 47 µm × 47 µm square reliably elicited zyxin localization at cell corners, edges, and actin comet tails (Figure 2A). Similar to zyxin distribution in cells on micropatterned substrates, HspB1 accumulated at the high-tension areas of square cells, specifically corners, edges, and probable actin comet tails (Supplemental Figure S1). To put the HspB1 distribution into subcellular context, using confocal microscopy HspB1 was localized along with the FA protein vinculin and F-actin and nuclei. HspB1 could be found in linear elements emerging from some cell corners (Figure 2B). To better illustrate this subcellular distribution, an intensity line profile is presented for one of these structures (Figure 2C). HspB1 (green solid line), F-actin (yellow dashed line), and vinculin (magenta dashed line) signals show that the most intense HspB1 signal is along actin filaments emerging from the vinculin-rich FA at cell corners, consistent with actin comet tails (Figure 2C). This distribution suggested HspB1 has an affinity for the ends of actin filaments under tension.
FIGURE 2:

Zyxin and HspB1 response in cells on micropatterned substrates and with actomyosin contractility. (A) Immunolocalization of cytoskeletal protein Zyxin in WT cells on micropatterned fibronectin squares and rectangles shows distribution to high-tension corners and edges, especially on a 47 µm × 47 µm square (2209 µm2 island). Insets show a higher magnification view of lower left corner of cells. (B) Confocal microscopy of immunolocalized HspB1 (green) in WT cell adhered to 47 µm × 47 µm square fibronectin island and co-stained for vinculin (magenta) and F-actin (yellow), and including the Merged color image. Insets show the entire cell with magnified corner (boxed in yellow). (C) Intensity line profile (white bracket on Merge image) going from cell exterior toward the cell center in 1-µm increments is presented to illustrate the subcellular distribution. HspB1 (solid green line) is shifted away from vinculin focal adhesion (dashed magenta line) and coincident with the actin filament (dashed yellow line). (D) Confocal microscopy of Zyxin distribution (magenta, top row) and HspB1 distribution (green, middle row) in cells Untreated, Blebbistatin-treated (50 µM 30 min), and Blebbistatin wash-out 3 h Recovery conditions. Almost all Recovery cells acquired in 3 datasets had Zyxin cytoskeletal distribution (125/130 acquired cells) while evident HspB1 localization was detected in a more limited distribution and a smaller number of Recovery cells (38/114 cells). (E) Top inset shows a Zyxin distribution (magenta) to apparent comet tails with no detectable HspB1 co-localization and Bottom inset shows a Zyxin (magenta) distribution to apparent comet tails with detectable HspB1 (green) co-localization. In the merged image magenta and green overlap makes a white signal. Scale bar 20 µm.
Contractile actomyosin networks in fibroblast cells include many cytoskeletal proteins and play pivotal roles in mechanical feedback, regulation, and force transmission (Murrell et al., 2015). Intracellular actomyosin contractility can be manipulated by the myosin inhibitor Blebbistatin to disrupt the actin cytoskeleton in treated cells, and the effects can be reversed by washing out the Blebbistatin inhibitor (Aratyn-Schaus et al., 2011). We suspected that zyxin and HspB1 were responding to actomyosin contractility and the tension generated in cells. To test this possibility, we treated cells with Blebbistatin to disrupt actomyosin contractility, then removed Blebbistatin and allowed the cells to recover and re-establish their intracellular tension as described (Aratyn-Schaus et al., 2011). Typical cytoskeletal localization of zyxin at FAs and along actin SFs in untreated cells was dispersed in Blebbistatin-treated cells, which exhibit long tails and wide lamellipodia and do not have localized zyxin (Figure 2D, top row). The diffuse cytoplasmic distribution of HspB1 was seen in both untreated and blebbistatin-treated cells (Figure 2D, middle row). Cells allowed to recover their actomyosin contractility by washing out Blebbistatin had a complete restoration of zyxin localization to FA and the actin cytoskeleton, and in a smaller subset of cells, HspB1 distribution was detected along apparent actin comet tails as defined by zyxin distribution (Figure 2D). In recovered cells the zyxin localization was widespread at FAs and across the actin cytoskeleton, but the HspB1 localization appeared at actin structures adjacent to FAs. Zyxin distribution to cytoskeletal structures was evident in almost all cells while codistribution with HspB1 was detected in a subset of cytoskeletal structures (Figure 2E). This difference in distribution may reflect the difference between a cytoskeletal regulator (zyxin) maintaining cellular homeostasis and a stress-responsive molecular chaperone (HspB1) responding to acute molecular stress.
The results for HspB1 distribution in response to uniaxial cyclic stretch stimulation, geometric constraint of cell adhesion, and intracellular actomyosin contractility are consistent with the view that HspB1 responds to mechanical cues. To further explore this possibility we developed reagents to test the role of HspB1 phosphoregulation in mechanically regulated physiological processes including cell spreading and motility.
Establishment of a cell-based model system to probe the function of HspB1
To test the role of HspB1 phosphorylation in directing HspB1 to sites of cytoskeletal tension, we targeted Ser15 and Ser86 by site-directed mutagenesis. We generated a nonphosphorylatable alanine double mutant S15,86A HspB1 to block phosphorylation and a glutamate double mutant S15,86E HspB1 which could act as a possible phosphomimetic HspB1 due to the charged amino acids at these sites. Using a previously engineered CRISPR construct to disrupt the HspB1 gene and eliminate HspB1 protein expression in mouse fibroblasts (Hoffman et al., 2017), we “rescued” those HspB1-null cells with constructs to drive expression of WT HspB1, S15,86A HspB1, or S15,86E HspB1 proteins. To evaluate expression of the transgenes, immunoblots of equivalent amounts of cell lysates from WT parent cells, CRISPR-HspB1-null cells, and null cells with HspB1 constructs for WT, S15,86A, and S15,86E HspB1 were probed with an HspB1-specific antibody (Figure 3A). While the HspB1-null cell lysates lacked detectable HspB1, the engineered constructs expressed HspB1 levels comparable to the endogenous HspB1 in lysates from the parental WT cells. By indirect immunofluorescence microscopy of cells plated on glass coverslips, both WT and mutant variants of HspB1 displayed a diffuse cytoplasmic distribution (Figure 3B) similar to what has been reported previously for WT HspB1 in unstimulated cells (Lavoie et al., 1995; Clarke and Mearow, 2013; Hoffman et al., 2017). The F-actin cytoskeleton signal is shown below the HspB1 signal for these cells (Figure 3B). In cells geometrically constrained on 47 µm × 47 µm-square islands, WT HspB1 accumulated adjacent to the vinculin-containing FAs, presumably at sites of actin cytoskeletal tension (Figure 3C). Interestingly, when we probed the distribution of the mutant HspB1 variants in geometrically constrained cells, the nonphosphorylatable S15,86A HspB1 displayed a diffuse cytoplasmic distribution (although the vinculin-FAs were present), and the S15,86E HspB1 was detected at the high-tension edges and corners (Figure 3C). Intensity line profiles are presented to illustrate the subcellular distribution in the higher magnification corners and they indicate that WT and S15,86E HspB1s are in lines slightly displaced from the FA, and that S15,86A HspB1 does not accumulate there (Figure 3D). These findings suggested that phosphorylation of HspB1 is required for the protein’s tension-dependent subcellular distribution.
FIGURE 3:

Cell-based model system for evaluating HspB1 function. (A) HspB1 immunoblot of parental WT and CRISPR/Cas9 HspB1-null cells, followed by “rescue” constructs of WT HspB1, nonphosphorylatable Ser15,86A and phosphomimetic Ser15,86E HspB1s expressed in the HspB1-null cells, with vinculin loading control. (B) Widefield microscopy of immunofluorescent localization of the 3 HspB1 rescue constructs in cells on fibronectin-coated coverslips detects diffuse cytoplasmic distribution of HspB1. F-actin images (phalloidin) of the same cells are shown below. (C) Maximum intensity projections of confocal microscopy images of HspB1 immunolocalization (HspB1, green) and vinculin (magenta) in HspB1-null cells expressing the three rescue constructs of WT HspB1 and phosphomutant S15,86A and S15,86E HspB1s, on 47 µm × 47 µm micropattern islands. Insets show zoom Merge image (lower left boxed corner) cytoskeletal distribution of HspB1 detectable in WT and S15,86E HspB1 but not with S15,86A HspB1. Cytoskeletal distribution of HspB1 observed in 40% of WT HspB1 rescue cell images, 3% of S15,86A HspB1 rescue cell images, and 38% of S15,86E HspB1 rescue cell images. (D) Intensity line profiles from cell exterior toward interior (brackets) of vinculin (dashed magenta line) and HspB1 (solid green line). Scale bar 20 microns.
HspB1 phosphorylation is required for normal cell spreading
HspB1 promotes cell spreading (Hoffman et al., 2017), but the potential role of phosphorylation has not been explored. To assess the role of HspB1 in the cell spreading response we employed a classic cell biology approach (Zhang et al., 2008). Cells were seeded onto coverslips and grown overnight, then fixed and immunostained for HspB1 and the focal adhesion protein vinculin, and cell areas were measured. The HspB1-null cells spread less well than the parent WT cells expressing endogenous HspB1 (Figure 4A). Re-expression of the WT HspB1 in the null cells led to a restoration of normal cell spreading (Figure 4, A and B). To evaluate the role of HspB1 phosphorylation in cell spreading, HspB1-null cells, along with cells expressing WT HspB1, and phosphomutant S15,86A and S15,86E HspB1s were seeded onto coverslips, followed by immunostaining for HspB1 and vinculin (Figure 4C). Cell areas in HspB1-null cells and S15,86A cells were not appreciably different (Figure 4D). In contrast, cells expressing WT HspB1 or S15,86E HspB1 were significantly more spread than the null cells (Figure 4D), suggesting that the ability to phosphorylate HspB1 is critical to normal cell spreading.
FIGURE 4:

HspB1 affects cell spreading in a phosphodependent manner. (A) immunofluorescence microscopy of cells spread on glass coverslips coated with 10 µg/ml fibronectin. Subcellular distribution of HspB1 (top row, cytoplasmic) and vinculin (bottom row, FA) in WT and HspB1-null cells, and in null cells expressing the WT HspB1 rescue construct. (B) Graph of cell area measurements shows the decreased cell spread in HspB1-null cells is rescued by expressing WT HspB1 rescue construct. (C) immunofluorescence localization of HspB1 (top row) in HspB1-null cells, and in null cells expressing the rescue constructs for WT HspB1 and nonphosphorylatable S15,86A HspB1 and phosphomimetic S15,86E HspB1. Vinculin immunofluorescent localizations in same cells (bottom row). (D) Graph of cell area measurements show increased cell spreading in cells expressing WT and S15,86E HspB1, but no difference between HspB1-null cells and cells expressing S15,86A HspB1. Scale bar of 20 microns for widefield fluorescent images. Graphs are mean with standard deviations and unpaired t tests were used to determine p-values of **p < 0.01, ***p < 0.001.
HspB1 phosphorylation is required to elicit a stretch-stimulated actin cytoskeletal reinforcement response
We previously reported that actin cytoskeletal reinforcement occurs in response to mechanical stimulation (Yoshigi et al., 2005). To test the contribution of HspB1 to the mechanical stress-induced actin response, we employ a uniaxial cyclic stretch of cells adhered to a mixture of collagen I and fibronectin on a flexible silicone membrane. On exposure to stretch, cells orient perpendicular to the uniaxial stretch vector and reinforce their actin cytoskeletons (Yoshigi et al., 2005). Stretch stimulation activates the p38 MAPK signaling pathway resulting in increased phosphorylation of HspB1 and accumulation of phospho-HspB1 at actin cytoskeletal anchorage sites (see Figure 1, B and C), raising the possibility that HspB1 phosphorylation may play a critical role in the stretch response.
To rule out the possibility that HspB1 has some direct effect on p38 activation, we compared the activation of p38 signaling in stretch-stimulated WT parental cells and HspB1-null cells by monitoring the p38 phosphorylation state. While p38 levels remained stable during the stretch protocol, increased phosphorylation of p38 is observed in stretch-stimulated cells even when the downstream target, HspB1, is undetectable (Figure 5A). These findings are consistent with the view that the phenotypes we observe in HspB1-null cells are not due to changes in the upstream p38 pathway activation.
FIGURE 5:

Uniaxial cyclic stretch elicits actin remodeling in cells that express WT but not S15,86A HspB1. (A) Western immunoblot analysis of stretch-stimulated (15%, 0.5 Hz, 15 and 60 min) WT and HspB1-null cells. Phospho-p38 and PhosphoS86-HspB1 antibody signals are above the corresponding total antibody signals and a vinculin loading control. (B) Immunoblot analysis of unstretched and stretch-stimulated (15% 0.5 Hz 60 min) HspB1-null cells and cells expressing the rescue constructs for WT; S15,86A; and S15,86E HspB1s. PhosphoS86-HspB1 is elevated in stretch-stimulated WT HspB1 and is not detectable in the phosphomutant HspB1s, although they are all comparably expressed as detected by Total HspB1 antibody. Vinculin is shown as loading control. Immunoblot quantification is included in Supplemental Table S1. (C) Phalloidin-stained stretch-stimulated (15% 0.5 Hz 60 min) HspB1-null cells and cells expressing the rescue constructs for WT; S15,86A; and S15,86E HspB1s. Reorientation of actin perpendicular to the stretch vector (20 µm double-headed arrow) is maintained but actin thickening is variable. (D) Graph of SFTI measurements on the phalloidin-stained cells show the most robust actin SFs in cells expressing the WT and phosphomimetic S15,86E HspB1. Actin response between HspB1-null cells and cells expressing the nonphosphorylatable S15,86A HspB1 was similar. Graphs are mean with standard deviations and unpaired t tests were used to determine p values of **p < 0.01, ***p < 0.001. (E) Confocal microscopy of HspB1 immunolocalization in stretch-stimulated HspB1-null cells expressing WT; S15,86A; and S15,86E HspB1s. Cytoskeletal distribution of HspB1 is detectable in WT and S15,86E HspB1 (arrowheads) but not with S15,86A HspB1. In this imaging data set HspB1 cytoskeletal distribution was detected in 53% WT rescue cells (20/38), 10% S15,86A rescue cells (2 possible/20), and 47% S15,86E rescue cells (8/17). Double-headed arrow of 20 micron scale shows uniaxial stretch vector in the horizontal direction.
Next we explored whether blocking HspB1 phosphorylation was sufficient to impair the stretch response. HspB1-null cells and cells expressing the WT HspB1 and phosphomutant S15,86A and S15,86E HspB1s were unstimulated or stretch-stimulated for 60 min. The HspB1-null cells have no detectable HspB1, total or phosphorylated; the WT HspB1 rescue construct expressed in the HspB1-null cells was phosphorylated in response to mechanical stimulation (Figure 5B). The phospho-HspB1 antibody failed to detect either of the phosphomutant HspB1 constructs, although both proteins are clearly expressed and detected by the Total HspB1 antibody (Figure 5B). We examined the role of HspB1 phosphorylation in actin cytoskeletal reinforcement downstream of exposure of cells to uniaxial cyclic stretch by F-actin imaging and quantitative measurement of the SF Thickness Index (SFTI) (Yoshigi et al., 2005). In both stretch-stimulated HspB1-null cells and null cells rescued with WT HspB1, actin-rich SFs become oriented perpendicular to the stretch vector (Figure 5C, double-headed arrow); however, the actin SFs are more robust in null cells engineered to re-express WT HspB1 (Figure 5, C and D), illustrating a contribution of HspB1 to the actin reinforcement response. HspB1-null cells engineered to express the nonphosphorylatable S15,86A HspB1 fail to properly thicken their actin SFs (Figure 5, C and D), revealing a requirement for HspB1 phosphorylation in the cellular response to mechanical stress. Consistent with this view, cells expressing the phosphomimetic S15,86E HspB1 exhibit an enhanced SFTI relative to the S15,86A HspB1 variant. WT and S15,86E HspB1s are recruited to cytoskeletal elements in stretch-stimulated cells (Figure 5E, arrowheads). In contrast the nonphosphorylatable S15,86A HspB1 failed to accumulate along the actin cytoskeleton and remained diffuse in the cytoplasm, revealing that phosphorylation both directs cytoskeletal localization and promotes actin reinforcement in response to stretch stimulation.
To determine if the two serines in HspB1 influenced each other’s phosphorylation or subcellular distribution, site-directed mutagenesis was employed to make single-site mutants S15A HspB1 and S86A HspB1 and evaluate their behavior when expressed in HspB1-null cells. Immunoblot analysis showed similar expression levels for WT, S15A, and S86A HspB1s (Figure 6A). Stretch-stimulated Ser86 phosphorylation that is detectable on WT HspB1 persisted in the S15A mutant illustrating that pS86 is not dependent on Ser15 phosphorylation. Control proteins, such as p38 and zyxin, are phosphorylated in response to stretch and show the typical up-regulation of phosphorylation in each of the HspB1-expressing cells (Figure 6A). Stretch-stimulated phospho-Ser86 signal on actin comet tails was detectable for WT HspB1s and for S15A HspB1 (yellow arrowheads) but not for S86A HspB1 (Figure 6B). Using a Total HspB1 antibody to immunolocalize HspB1 in stretch-stimulated cells revealed that cytoskeletal distribution (yellow arrowheads) was detectable in WT and S15A HspB1s but not in S86A HspB1 (Figure 6C). The behavior of the single-site mutants points to Ser86 being the primary determinant of stretch-stimulated cytoskeletal distribution.
FIGURE 6:

Stretch response of single-site mutants S15A and S86A HspB1. (A) Western immunoblot analysis of HspB1-null cells expressing WT and single-site mutants S15A and S86A HspB1. The stretch-stimulated (15% 0.5 Hz 60 min) phosphorylation of Ser86 HspB1 persists in S15A mutant and is not detected in S86A mutant. Stretch-stimulated phosphorylation response is intact for other proteins (p38 and zyxin) in the single-site mutant S15A and S86A cells. Vinculin control shows equal loading. (B) Immunofluorescence localization of stretch-stimulated pS86-HspB1 (top row) to the cytoskeleton (yellow arrowhead) is detected in WT parental cells and in HspB1-null cells expressing WT and S15A HspB1 and is absent for S86A HspB1. Merged images (bottom row) of pS86-HspB1 (green), F-actin (magenta), and DAPI (blue) are included for these maximum intensity projections. In imaging data set pS86-HspB1 cytoskeletal distribution was detected in 37% of WT (57 cells), 30% of WT rescue (37 cells), 32% of S15A rescue (34 cells), and none detected in S86A rescue (22 cells). (C) Immunofluorescence localization of Total HspB1 signal in stretch-stimulated cells. Cytoskeletal distribution of WT HspB1s and S15A HspB1 (yellow arrowhead). Only diffuse cytoplasmic distribution was detected for S86A HspB1. Merged images (bottom row) of Total-HspB1 (green), F-actin (magenta), and DAPI (blue) are included for these maximum intensity projections from a stack of confocal images. In imaging data set Total HspB1 cytoskeletal distribution was detected in 36% of WT (47 cells), 29% of WT rescue (31 cells), 20% of S15A rescue (25 cells), and none detected in S86A rescue (20 cells). Uniaxial stretch vector is presented in the horizontal direction, designated with a double-headed arrow of 20-µm scale.
Phosphorylatable HspB1 is required for cell motility regulation
Cell motility is a physiological behavior regulated by and contributing to mechanical signals (Chang et al., 2019). HspB1-null cells are more motile than their WT parent cell, and this enhanced motility phenotype can be rescued by re-expression of WT HspB1 (Hoffman et al., 2017). Enhanced motility is reminiscent of our previously reported zyxin-null cells (Hoffman et al., 2006). To evaluate whether HspB1 acts in the same pathway as zyxin or acts in a parallel pathway, we examined single-null and HspB1/zyxin double-null cells in a cell motility assay (Supplemental Figure S2). We found that HspB1/zyxin double-null cells were more migratory than either of the single-null cells. The additive effects of HspB1 and zyxin deletions suggest that HspB1 and zyxin are working in parallel pathways to influence cell motility.
To evaluate the role of HspB1 phosphorylation in regulation of cell motility, a time-lapse video microscopy assay was employed to test directional cell migration with HspB1-null cells expressing either WT or phosphomutant variants of HspB1. Cells were grown to confluence in a 2-well chamber on a glass-bottom dish, then the chamber was removed, the cells were placed on the microscope, and image acquisition started. Representative images of the cells at the start of edge migration and at the end of the 18-h time course are shown for HspB1-null cells and for cells expressing the WT and phosphomutant HspB1 rescue constructs (Figure 7A). Corresponding time-lapse movies are included in the Supplemental Material. Measurements of the distance traveled at 18 h confirmed the HspB1-null cells were more migratory than the cells expressing the WT HspB1 rescue construct (Figure 7B), as previously reported (Hoffman et al., 2017). Over the 18-h period, the HspB1-null cells migrated an average of 422 μm, whereas re-expression of WT HspB1 reduced the distance traveled to 329 μm. In contrast, the nonphosphorylatable S15,86A variant did not impact the distance traveled (average distance 424 μm), consistent with the view that HspB1’s ability to influence cell motility in this assay depends on phosphorylation. The S15,86E variant of HspB1 also failed to significantly alter the distance traveled by the cells (average distance 401 μm) relative to HspB1-null cells. As the nonphosphorylatable S15,86A HspB1 and the phosphomimetic S15,86E mutants both failed to significantly affect motility, these data suggest that bona fide phosphorylation of those serine residues is critical for HspB1’s effect on cell motility. An alternative explanation for failure of both phosphomutants to rescue cell motility is that the mutant proteins were no longer expressing sufficiently. However, Western immunoblot analysis made from the cells seeded for the motility assays showed the proteins were expressing at a similar level across the cell types (Figure 7C). Thus expression of the WT HspB1 protein in the HspB1-null cells restored normal cell motility while the phosphomutant HspB1 proteins did not, illustrating the importance of phosphorylation in cell motility regulation by HspB1.
FIGURE 7:

HspB1-dependent regulation of cell motility requires phosphorylation. (A) Brightfield images of cells at the beginning and end of 18 h edge migration time course for HspB1-null cells alone or expressing WT HspB1 and phosphomutant S15,86A and S15,86E HspB1s. Border of starting edge and of migrated cells is demarcated with a white dashed line. Scale bar 100 microns. (B) Graph of migration distance at 18 h is shown for HspB1-null cells and cells expressing WT HspB1 and phosphomutant S15,86A and S15,86E HspB1s. Graph is mean with standard deviations and unpaired t tests were used to determine p values. ***p < 0.001 or n.s., not statistically significant. (C) Western immunoblot of cells seeded for migration assays shows expression of the three rescue construct HspB1s, with vinculin loading control.
Movie S1.
Cell motility HspB1-null cells. 10 min intervals for 18 hrs.
Movie S2.
Cell motility HspB1-null cells with WT HspB1 construct. 10 min intervals for 18 hrs.
Movie S3.
Cell motility HspB1-null cells with non-phosphorylatable S15,86A HspB1 mutant construct. 10 min intervals for 18 hrs.
Movie S4.
Cell motility HspB1-null cells with possible phospho-mimetic S15,86E HspB1 mutant construct. 10 min intervals for 18 hrs.
DISCUSSION
The small heat shock protein HspB1 (also known as Hsp25/27) is a ubiquitous mechanosensitive protein with actin regulatory and molecular chaperone activities (Collier and Benesch, 2020) and is phosphorylated following a variety of stimulations (Kostenko and Moens, 2009). How HspB1 responds to mechanical input and functions in the context of cell physiology and actin cytoskeletal reinforcement is not well understood. We propose that phosphorylation of HspB1 (downstream of the p38 MAPK signal transduction pathway) is key to its role in mechanotransduction. We developed a cell-based model system to evaluate HspB1 function and the requirement for phosphorylation. We determined that phospho-HspB1 is required for normal response to mechanical input used in cell spreading and motility and in geometrically constrained and stretch-stimulated cells.
For many years, HspB1 was described as having a diffuse cytoplasmic distribution in cultured cells (Collier and Schlesinger, 1986; Lavoie et al., 1995; Clarke and Mearow, 2013), a feature that provided no insight regarding possible HspB1 cellular functions. Given the reports in the literature, we were surprised to observe a striking cytoskeletal distribution of phospho-HspB1 in cells stimulated by uniaxial cyclic stretch. We provide evidence that HspB1 associates with mechanically stimulated actin comet tails that emanate from integrin-based FA. These are regions of elevated tension (Oakes et al., 2014) where the LIM domain protein, zyxin, also concentrates (Guo and Wang, 2007). Several LIM domain proteins have recently been shown to recognize sites of actin filament strain (Sun et al., 2020; Winkelman et al., 2020; Anderson et al., 2021; Sala and Oakes, 2021). We did not observe HspB1 at SF strain sites where zyxin accumulates (Smith et al., 2010, 2013) and the zyxin-HspB1 codistribution is primarily along limited actin filaments emerging from FA.
HspB1 was originally identified as an inhibitor of actin polymerization that copurified with the FA protein vinculin (Miron et al., 1988, 1991). It later became clear that phosphorylation of HspB1 shifted its activity to promote actin polymerization (Benndorf et al., 1994). It was estimated that HspB1 oligomers could sequester actin monomers at a ratio of 1 HspB1 to 1-2 globular monomeric actin (G-actin) monomers and that phosphorylation of HspB1 released aggregated G-actin for assembly into actin filaments (During et al., 2007). p38 MAPK signal transduction pathway activation results in MK2 kinase phosphorylation of HspB1, a posttranslational modification which fails in cells lacking MK2 kinase (Ronkina et al., 2007; Sousa et al., 2007; Damarla et al., 2009; Hoffman et al., 2017). In mouse HspB1 the 2 relevant MK2 sites are Ser 15 and Ser 86. Here we showed that HspB1 mutant protein that converts Ser15 and Ser86 to alanines loses its ability to accumulate at the ends of actin SFs in this tension-dependent distribution. Many stimuli have been reported to induce phosphorylation of HspB1 (Kostenko and Moens, 2009). Our data that mechanical input (stretch-stimulation, geometric constraint, actomyosin contractility) enhances HspB1 recruitment to actin filaments where they terminate at FAs, following activation of the p38 MAPK cascade and subsequent HspB1 phosphorylation and is disrupted in nonphosphorylatable HspB1, suggests that HspB1 displays highly regulated association with the actin cytoskeleton.
When we deployed CRISPR/Cas9 genome editing to disrupt HspB1 expression in cells, we noticed that the HspB1-null cells were smaller than the WT parental cells in culture. Re-expression of WT HspB1 restored the normal cell spreading but the S15,86A HspB1 did not, suggesting that phospho-HspB1 regulates cell spreading. Cell spreading reflects the mechanical work of a cell (Oakes et al., 2014). Others have reported that overexpression of HspB1 increased cell adhesion and spreading while limiting cell motility (Lee et al., 2008). At the time we reported that HspB1 acted as a negative regulator of fibroblast cell motility (Hoffman et al., 2017), it was noted that transfected and overexpressed Hsp27 reduced PDGF-BB migration in an osteoblastlike cell line (Kainuma et al., 2017). Phosphorylatable WT Hsp27 reduced PDGF-BB cell migration more than a nonphosphorylatable-mutant Hsp27 did. Consistent with these reports, we find that expression of WT HspB1, but not phosphomutant HspB1, reduces cell motility and thereby “rescues” the enhanced motility of HspB1-null cells. Although an HspB1-dependent motility phenotype suggests some possible developmental consequences, mice engineered with a deletion of HspB1 are viable, fertile, and display no obvious phenotype (Crowe et al., 2013). However in a skin biopsy punch assay, the HspB1-deficient mice displayed both impaired wound healing (decreased cell migration) and increased neutrophil infiltration (Crowe et al., 2013), highlighting the complicated role of HspB1 in cell migration in vivo.
Cellular protein homeostasis is maintained by molecular chaperones, including HspB1 (Arrigo 2017). It is possible that part of the tension-dependent characteristics of HspB1 are due to its chaperone activity. An example of a molecular chaperone protective mechanism following mechanical stress is the molecular interaction between HspB1 and the actin-binding protein filamin C (FLNC) (Collier and Benesch 2020). The interaction between FLNC and HspB1 is dependent on HspB1 phosphorylation and it is suggested that FLNC is a client protein for the molecular chaperone to alleviate adverse consequences of mechanical stress (Collier et al., 2019). A genetic model for a mechanically stressed heart with serious cardiovascular consequences is the MLP (Muscle LIM Protein) knockout (KO) mouse (Arber et al., 1997). MLP KO mouse hearts have up-regulated levels of FLNC and increased HspB1 and phospho-HspB1 levels as well (Collier et al., 2019). The inducible and phosphorylation-dependent interaction between HspB1 and the mechanosensitive paxillin family member Hic-5 (Jia et al., 2001; Srinivasan et al., 2008) may represent another client-chaperone example. The HspB1 behavior in response to mechanical inputs reported here may also be due to chaperone activity of this small heat shock protein trying to protect cell function in the midst of mechanical stress, perhaps recognizing denatured actin at sites of cytoskeletal strain. Future efforts to identify additional client proteins for this mechanosensitive molecular chaperone may illuminate its role in cytoprotection.
Retrograde flux of actin emanating from FA is promoted by mechanical signals such as those engendered by substrate stiffness or stretching force and is inversely correlated with cell migration (Guo and Wang, 2007; Chang et al., 2019). Stationary cells, such as those immobilized on square micropattern islands, have high traction stress at corners and edges, while actively migrating cells have reduced traction stress. These findings led to the proposal that migration speed is an active regulator of traction force (in a negative feedback loop) (Oakes et al., 2014; Chang et al., 2019). We show that phospho-HspB1 accumulates at sites of retrograde actin flux, and examination of the role of HspB1 in actin flux from FA will be the focus of future investigations.
We report here that the molecular chaperone HspB1 is a mechanically responsive protein that is regulated by phosphorylation and localizes to cytoskeletal elements following stretch stimulation, geometric constraint, and recovery from inhibition of actomyosin contractility. We recognize (and have previously reported) parallel characteristics in the cytoskeletal adaptor and actin regulator zyxin (Yoshigi et al., 2005, Hoffman et al., 2012). As others have reported (Sala and Oakes, 2021), zyxin is a good positive control for a mechanoresponse, and we employed it in this report of HspB1 response. On stretch stimulation, zyxin is phosphorylated and redistributes from FA to entire lengths of remodeled actin SFs (Hoffman et al., 2012). On stretch stimulation, HspB1 is phosphorylated and redistributes from diffuse cytoplasmic distribution to particular features of actin SFs, generally at the ends of SFs where they are anchored to FAs (Hoffman et al., 2017). Phosphorylation of HspB1 is key to directing this response. While the zyxin recruitment to the cytoskeleton is robust and detectable in almost all cells, the HspB1 response is limited to fewer cells and fewer regions within the same cells. These differences are not unexpected given that zyxin is part of the cytoskeletal homeostasis mechanism, and HspB1 is a stress-responsive protein. Although both proteins respond to mechanical cues, their degree of response appears tailored to their function/role inside the cell.
Incorporating published data and ideas from others, in combination with our own findings, we present a working model for the role of HspB1 in response to mechanical stress (Figure 8). Under conditions of high mechanical tension, the p38 MAPK signal transduction pathway is activated, resulting in the MK2-dependent phosphorylation of HspB1, similar to that proposed for cytokine and growth factor stimulation of cells (Gerthoffer and Gunst, 2001; Salinthone et al., 2008; Wettstein et al., 2012). It has previously been reported that phosphorylation of HspB1 leads to disruption of HspB1 oligomers and release of monomeric actin (G-actin) to promote actin filament polymerization (Lavoie et al., 1995; During et al., 2007). Based on these findings, it is plausible that the stretch-stimulated phosphorylation of HspB1 also disrupts the protein’s oligomerization. This HspB1 oligomer disruption by phosphorylation is predicted to have a twofold effect. First, the phospho-HspB1 is freed to accumulate at sites of cytoskeletal tension, such as integrin-based FAs that anchor actin filaments (F-actin), where HspB1 could serve as a tension-sensitive molecular chaperone to alleviate the molecular strain induced by mechanical stress. Such a client-chaperone interaction has been described for the actin-binding protein FLNC and HspB1 (Collier et al., 2019). Second, the release of monomeric actin from the HspB1 oligomers could serve to feed the actin polymerization that occurs at these adhesion sites (Hirata et al., 2008). Both possibilities are consistent with our results of mechanically stimulated phospho-HspB1 cytoskeletal distribution and actin remodeling.
FIGURE 8:
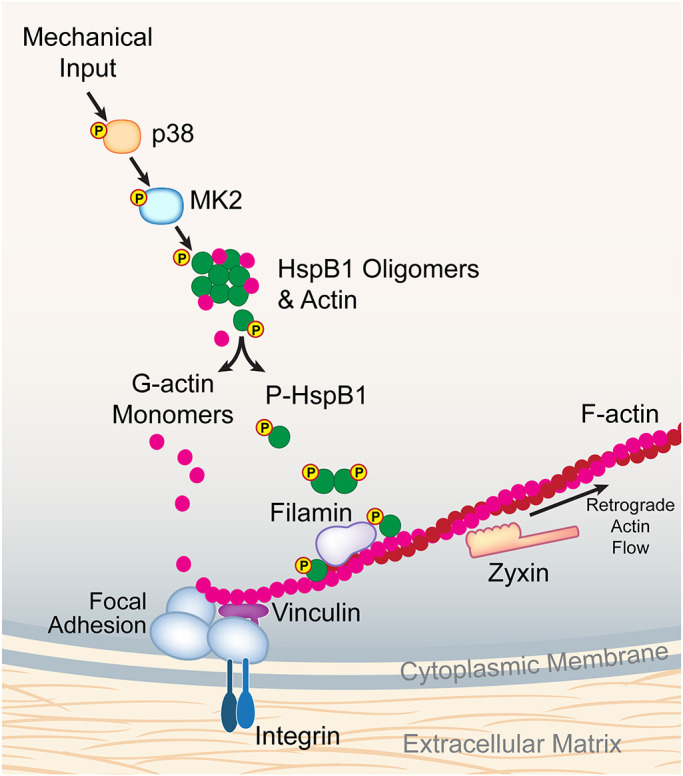
Model of mechanical stress stimulation on HspB1, FA, and F-actin filaments. Using our own data and building on published data from others (see Discussion), we propose a molecular model for HspB1 in the context of mechanical stress. Mechanical input activates the p38 MAPK pathway resulting in phosphorylation of HspB1, which disrupts oligomers of HspB1 with monomeric actin. G-actin monomers are then available for incorporation into focal adhesion-anchored F-actin filaments. Phospho-HspB1 is recruited to tension-dependent cytoskeletal structures and may interact with FilaminC. Also included are vinculin as a foundational component of integrin-based FA and zyxin as a fiducial marker moving out of FA along actin filaments in a retrograde flow.
HspB1 regulates behaviors sensitive to mechanical input like cell spreading and motility and actin reinforcement. The recruitment of HspB1 to subcellular structures like actin SFs, FA, and dynamic comet tails, in addition to high-tension cell edges, requires phosphorylation and is a key feature of HspB1 activity, reflecting both actin regulatory and molecular chaperone function. Our work identifies HspB1 as a member of a growing class of mechanosensitive proteins that associate with the actin cytoskeleton and influence actin-dependent cell behaviors.
MATERIALS AND METHODS
Antibodies and labels
Hsp B1 (also known as Hsp25/27) antibodies were from Enzo Life Sciences (ADI-SPA-801 rabbit) and Cell Signaling Technology (HspB1 #2442 rabbit, phospho-s82[human]HspB1 #9709 rabbit). Total p38 (#9212 rabbit) and phospho-p38 (#4511 rabbit) and phospho-zyxin (#4863 #8467 rabbit) antibodies were from Cell Signaling Technology. Vinculin antibodies were from Sigma (V-9131 mouse); zyxin antibody B72 (Hoffman et al., 2003) is available from Millipore (#1387 rabbit). Alexa Fluor (647, 568, 488)–conjugated secondary antibodies, Alexa Fluor Phalloidin (647, 568, 488), and nuclear stains DAPI (#D-1306) and Hoechst (#H-1399) were obtained from Molecular Probes/Invitrogen. SiR-actin was obtained from Cytoskeleton (#CY-sc001). See also Supplemental Table S2, Reagents and Resources.
Cells and expression constructs
Fibroblast cells were cultured in high-glucose DMEM supplemented with pyruvate, glutamine, penicillin, streptomycin (from Invitrogen), and 10% fetal bovine serum (FBS; Hyclone Labs, Logan UT). WT mouse fibroblasts (Hoffman et al., 2006) and CRISPR/Cas9-disrupted HspB1-null cells with WT HspB1 re-expression (Hoffman et al., 2017) were previously described. To generate HspB1 constructs to express in HspB1-null cells, a caspase 9-resistant construct was made by mutating the PAM site sequence 5′ to the start of the guide sequence (Hoffman et al., 2017) from CGG (Arg) to ACG (Arg). Site-directed mutagenesis (QuikChange II XL Site-Directed Mutagenesis Kit, Agilent Technologies #200521) was used to mutate MK2 target Serines 15 and 86 in single and double sequences. The peptide sequence LLRSPS15WEPFRD was changed to LLRSPA15WEPFRD, and the peptide sequence ALNRQLS86SGVSEI was changed to ALNRQLA86SGVSEI. After DNA sequencing to confirm the mutations, Gateway cloning into a pLenti6.3/V5 plasmid and transfection into 293FT cells for viral production, the lentiviruses were used to transduce the HspB1-null cells. The virally transduced cells were selected for resistance in 2.5 µg/ml Blasticidin (Invitrogen) for 1 wk, then protein expression was confirmed by immunoblot analysis of cell lysates. Western immunoblots used in the figures were quantified using Fiji ImageJ and protein expression levels were compared with WT parent cell levels and provided in Supplemental Table S1.
Zyxin-GFP construct was transfected into fibroblasts with Lipofectamine 2000 (#11668-019 Invitrogen) as directed by the manufacturer and live images were acquired between 48 and 96 h posttransfection.
Immunofluorescence staining
Fibroblast cells were seeded onto glass coverslips (round 18 mm #1.5) in 12-well plates and grown overnight in DMEMc+10% FBS, briefly washed in warm phosphate-buffered saline, and then fixed 15 min in 3.7% formaldehyde then 5 min permeabilization in 0.5% TritonX-100. Prior to antibody application, cells were blocked 1 h in 5% normal goat serum. Primary and secondary antibodies were incubated on cells for up to 2 h at 37°C or overnight at 4°C. Coverslips were mounted to slides in Mowiol (Sigma #81381) with DABCO anti-fade solution (Sigma #D2522). Image acquisition is described in the Microscopy section. Cells on fibronectin-coated (10 µg/ml 1 h 37°C) coverslips were treated in media containing 50 µM Blebbistatin (Millipore Sigma CAS 856925-71-8) for 30 min to 1 h, then the media were replaced with fresh DMEMc+10% FBS for cell recovery, and cells were fixed at time points (30 min–3 h) and stained as described above. Blebbistatin experiment was performed three times, with multiple staining combinations and the use of both Nikon widefield microscope and Leica SP8 confocal microscope. Image fields acquired in Blebbistatin recovery experiments were tallied and scored for zyxin cytoskeletal distribution (125/130 cells = 96%) and for HspB1 cytoskeletal distribution (38/114 cells = 33% cells).
Uniaxial cyclic stretch and immunoblots
Using a custom-designed system (Yoshigi et al., 2003, 2005), uniaxial cyclic stretch (15% 0.5 Hz 15–30–60 min as described) was applied to cells adhered to 26 mm × 33 mm flexible silicone membranes precoated with collagen I (25 µg/ml) and fibronectin (2 µg/ml) (Hoffman et al., 2017). For inhibitor experiments, cells were preincubated with 10 µM p38 inhibitor SB203580 (Cell Signaling Technology #5633) for 60 min prior to stretch stimulation. For Western immunoblot analysis, cell proteins (25 µg) were electrophoresed (denaturing 15% polyacrylamide gel) and transferred to nitrocellulose filters for detection by enhanced chemiluminescence (GE Healthcare) on film. For immunofluorescence localization, stretch-stimulated cells were fixed in 3.7% formaldehyde (15 min room temperature), permeabilized (0.5% Triton X-100 5 min room temperature), blocked (5% normal goat serum 1 h room temperature), then stained with antibodies or fluorescent probes as specified (Hoffman et al., 2017). SFTI analysis of phalloidin-stained stretch-stimulated cells (acquired with identical settings) used a custom erosion/brightness decay software written in LabView (National Instruments) (Yoshigi et al., 2003, 2005). Cells were preincubated with p38 inhibitor SB203580 (Cell Signaling Technology #5633) at 10 μm for 60 min prior to stretch stimulation in indicated experiments. Stretch stimulation experiments were performed at least three times to acquire representative images shown. All Western immunoblot film images in these figures were quantified using Fiji ImageJ (NIH) gel analysis feature and the intensity numbers and x-fold changes relative to either unstimulated cell counterpart or to WT parent cell are provided in Supplemental Table S1. In the imaging data set used in Figure 1D, stretch-stimulated cells were examined for codistribution of P-HspB1 and zyxin on cytoskeletal elements (19/175 cells = 11%), with cytoskeletal distribution of P-HspB1 at 27% and zyxin at 93% of the cells in the acquired images. In the imaging data set used in Figure 5E, HspB1 cytoskeletal distribution was detected in 53% WT rescue (20/38) cells, 10% S15,86A rescue (2 possible/20) cells, and 47% S15,86E rescue (8/17) cells.
Cell motility
For directional edge migration assays, cells (23,000) were seeded into each half of 2-well culture chambers (Ibidi #80209) on #1.5 glass bottom 12-well plates (Cellvis, Invitro Scientific). After cell growth overnight, the chambers were removed and the media were changed to CO2-independent DMEM/F-12 media (Life Technologies #11039-021, No Phenol Red) with 10% FBS. The 12-well plate was placed on the microscope stage with OkoLabs heating chamber at 37°C. A Nikon Ti Eclipse inverted widefield microscope with 10× objective (NA 0.45, PlanApo) and Perfect Focus System was used for DIC imaging (with Kohler illumination) and acquisition with Nikon Elements v4.6 software and Andor NeoZyla camera. Time-lapse acquisition at 10-min intervals for 18 h. Distance measurements were made on 10× images calibrated at 0.65 µm/px using Nikon Elements software. Three regions each in six fields/cell type were measured (n = 18) at the farthest migrating cell, checking time-lapse movies to make sure only migrating cells were included, also measured by a blinded analysis, and the migration experiment was performed four times. Western immunoblots of cells seeded for migrations were used to confirm expressions; a representative blot used in Figure 7 was quantified by Fiji Image J and is provided in Supplemental Table S1.
Micropatterns
Single cell suspensions were seeded onto fibronectin-coated micropattern islands of 12 square and rectangular shapes of increasing aspect ratios (1 to 11 and areas of 1200 to 2300 µm2 REC200 × 12FN, CYTOO, Grenoble, France) in a 6-well dish at 75,000 cells/well. Cells were incubated with the CYTOOchips for 4 h at 37°C in DMEMc with 10% FBS, then formaldehyde was gently diffused into the media to a final concentration of 3.7%. Fixed cells were then permeabilized and stained, followed by image acquisition on inverted Leica SP8 confocal microscope with 63× objective (NA 1.40 PlanApo) and HyD detectors, LASX v3.5.7 software with Lightning deconvolution. For HspB1 distribution, six data sets of WT cells seeded on Cytoo chips were imaged with both the Nikon widefield and Leica SP8 confocal microscopes for a total of 54 square cells acquired, 19 square cells with HspB1 cytoskeletal distribution is 35% of the acquired cells. For rescue constructs, four imaging data sets were scored for cytoskeletal distribution of HspB1 in 47 µm × 47 µm-square cells: WT rescue 40% (40 cells), S15,86A 3% (29 cells), and S15,86E 38% (26 cells).
Retrograde actin flow
Cells transfected with zyxin-GFP and seeded onto 25-mm #1.5 glass coverslips (precoated with 10 µg/ml fibronectin) were incubated with cell-permeable FarRed-SiR-actin (1:3000 CY-sc001, Cytoskeleton) and Hoechst stain (0.25 µg/ml Molecular Probes # H-1399) for 1 h prior to acquisition. Coverslips were positioned in a magnetic chamber (Quorom Technologies) and cells were maintained at 37°C (OkoLabs stage heater) in CO2-independent DMEM/F-12 media (Life Technologies #11039-021, No Phenol Red) with 10% FBS. Cell images (488-zyxin-GFP, 633-SiRactin) were acquired with PMT and HyD detectors and LASX software v.3.5.7 on a Leica SP8 spinning disk confocal microscope (63× oil objective NA 1.40 HC PL APO CS2), 0.25-µm z slices for minimal intervals (10–30 s) up to 30 min. Retrograde flow of zyxin was measured for distance and time using LASX software, >10 cells/data set, in a total of four experiments.
Microscopy
An inverted widefield Nikon Ti Eclipse microscope, Andor NeoZyla camera and Nikon Elements v4.60 acquisition software was used with a 60× (NA 1.40 PlanApo) oil objective for immunofluorescent cell imaging in micropattern, Blebbistatin, area, and stretch experiments and with a 10× (NA 0.45, PlanApo) objective for motility experiments. Area measurements were performed with Nikon Elements Analysis v4.6 software (binary auto-detect to ROI measurements) with 60× images calibrated at 0.11 µm/px. An upright widefield Zeiss Axioskop2 mot plus microscope (40× NeoFluor 0.75NA dry objective) was used with Zeiss AxioCamMRm camera and Zeiss AxioVision v4.8.1 software. Confocal images were acquired on a Leica SP8 DMi8 inverted microscope (Leica 63× oil objective NA 1.40 HC PL APO CS2) with PMT and hybrid detectors and LASX v3.5.7 software. Images are presented as maximum intensity projections of a confocal stack of Lightning deconvolved images. Leica acquisition was used for stretch, micropattern, Blebbistatin, and retrograde flow experiments.
Statistical analysis
Graph Pad Prism (version 9) was used for graphing and statistical analysis of quantitative measurements. P values were determined by unpaired Student’s t test and are designated ***p < 0.001, **p < 0.01.
Figure preparation
Adobe Photoshop CC software was used to process images which were assembled into figures using Adobe Illustrator CC.
Supplementary Material
Acknowledgments
We acknowledge Masaaki Yoshigi for providing expertise in mechanical stimulation and data analysis and Diana Lim for figure design and preparation. Elizabeth Blankman and Mark Smith provided constructive feedback during the course of this research. The University of Utah Cell Imaging Core Facility and Michael Bridge provided expertise and microscopes critical to this research (Nikon Ti Eclipse widefield microscope and Leica SP8 confocal microscope). This work was supported by the National Institute of Health (R01-GM50877 to M.C.B.), and the Huntsman Cancer Foundation. The Cancer Center Support Grant (2-P30-CA042014) awarded to the Huntsman Cancer Institute provided developmental funds and supported shared resources critical to this research.
Abbreviations used:
- FA
focal adhesion
- F-actin
filamentous actin
- FBS
fetal bovine serum
- FLNC
filamin C
- G-actin
globular monomeric actin
- KO
knockout
- MAPK
mitogen-activated protein kinase
- MLP
Muscle LIM Protein
- SF
stress fiber
- SFTI
stress fiber thickness index
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E22-02-0057) on June 29, 2022.
REFERENCES
- Anderson CA, Kovar DR, Gardel ML, Winkelman JD (2021). LIM domain proteins in cell mechanobiology. Cytoskeleton (Hoboken) 78, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TW, Vaughan AN, Cramer LP (2008). Retrograde flow and myosin II activity within the leading cell edge deliver F-actin to the lamella to seed the formation of graded polarity actomyosin II filament bundles in migrating fibroblasts. Mol Biol Cell 19, 5006–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aratyn-Schaus Y, Oakes PW, Gardel ML (2011). Dynamic and structural signatures of lamellar actomyosin force generation. Mol Biol Cell 22, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Hunter JJ, Ross J Jr, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P (1997). MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88, 393–403. [DOI] [PubMed] [Google Scholar]
- Arrigo AP (2017). Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones 22, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ateshian GA, Humphrey JD (2012). Continuum mixture models of biological growth and remodeling: past successes and future opportunities. Annu Rev Biomed Eng 14, 97–111. [DOI] [PubMed] [Google Scholar]
- Benham-Pyle BW, Pruitt BL, Nelson WJ (2015). Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science 348, 1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G (1994). Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J Biol Chem 269, 20780–20784. [PubMed] [Google Scholar]
- Byers HR, White GE, Fujiwara K (1984). Organization and function of stress fibers in cells in vitro and in situ. A review. Cell Muscle Motil 5, 83–137. [DOI] [PubMed] [Google Scholar]
- Calderwood SK, Gong J (2016). heat shock proteins promote cancer: It’s a protection racket. Trends Biochem Sci 41, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SS, Rape AD, Wong SA, Guo WH, Wang YL (2019). Migration regulates cellular mechanical states. Mol Biol Cell 30, 3104–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri S, Smith PG (2008). Cyclic strain-induced HSP27 phosphorylation modulates actin filaments in airway smooth muscle cells. Am J Respir Cell Mol Biol 39, 270–278. [DOI] [PubMed] [Google Scholar]
- Chen CS (2008). Mechanotransduction—a field pulling together? J Cell Sci 121(Pt 20), 3285–3292. [DOI] [PubMed] [Google Scholar]
- Clarke JP, Mearow KM (2013). Cell stress promotes the association of phosphorylated HspB1 with F-actin. PLoS One 8, e68978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier MP, Alderson TR, de Villiers CP, Nicholls D, Gastall HY, Allison TM, Degiacomi MT, Jiang H, Mlynek G, Furst DO, et al., (2019). HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci Adv 5, eaav8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier MP, Benesch JLP (2020). Small heat-shock proteins and their role in mechanical stress. Cell Stress Chaperones 25, 601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier NC, Schlesinger MJ (1986). The dynamic state of heat shock proteins in chicken embryo fibroblasts. J Cell Biol 103, 1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe J, Aubareda A, McNamee K, Przybycien PM, Lu X, Williams RO, Bou-Gharios G, Saklatvala J, Dean JL (2013). Heat shock protein B1-deficient mice display impaired wound healing. PLoS One 8, e77383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damarla M, Hasan E, Boueiz A, Le A, Pae HH, Montouchet C, Kolb T, Simms T, Myers A, Kayyali US, et al. (2009). Mitogen activated protein kinase activated protein kinase 2 regulates actin polymerization and vascular leak in ventilator associated lung injury. PLoS One 4, e4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deanfield JE, Halcox JP, Rabelink TJ (2007). Endothelial function and dysfunction: testing and clinical relevance. Circulation 115, 1285–1295. [DOI] [PubMed] [Google Scholar]
- del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP (2009). Stretching single talin rod molecules activates vinculin binding. Science 323, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Mooney DJ, Zandstra PW (2009). Growth factors, matrices, and forces combine and control stem cells. Science 324, 1673–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During RL, Gibson BG, Li W, Bishai EA, Sidhu GS, Landry J, Southwick FS (2007). Anthrax lethal toxin paralyzes actin-based motility by blocking Hsp27 phosphorylation. EMBO J 26, 2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlicher AJ, Nakamura F, Hartwig JH, Weitz DA, Stossel TP (2011). Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature 478, 260–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier MF, Sauser R, Ambrosi D, Meister JJ, Verkhovsky AB (2010). Force transmission in migrating cells. J Cell Biol 188, 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Egelman EH (2012). Actin filaments as tension sensors. Curr Biol 22, R96–R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerthoffer WT, Gunst SJ (2001). Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol (1985) 91, 963–972. [DOI] [PubMed] [Google Scholar]
- Guo WH, Wang YL (2007). Retrograde fluxes of focal adhesion proteins in response to cell migration and mechanical signals. Mol Biol Cell 18, 4519–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S (2012). Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol 13, 591–600. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Tatsumi H, Sokabe M (2011). Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J Cell Biol 195, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg CP, Bellaiche Y (2013). Forces in tissue morphogenesis and patterning. Cell 153, 948–962. [DOI] [PubMed] [Google Scholar]
- Hirata H, Tatsumi H, Sokabe M (2008). Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J Cell Sci 121(Pt 17), 2795–2804. [DOI] [PubMed] [Google Scholar]
- Hoffman L, Jensen CC, Yoshigi M, Beckerle M (2017). Mechanical signals activate p38 MAPK pathway-dependent reinforcement of actin via mechanosensitive HspB1. Mol Biol Cell 28, 2661–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, Beckerle MC (2012). Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol Biol Cell 23, 1846–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Kloeker S, Wang CL, Yoshigi M, Beckerle MC (2006). Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J Cell Biol 172, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Nix DA, Benson B, Boot-Hanford R, Gustafsson E, Jamora C, Menzies AS, Goh KL, Jensen CC, Gertler FB, et al. (2003). Targeted disruption of the murine zyxin gene. Mol Cell Biol 23, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Smith MA, Jensen CC, Yoshigi M, Blankman E, Ullman KS, Beckerle MC (2020). Mechanical stress triggers nuclear remodeling and the formation of transmembrane actin nuclear lines with associated nuclear pore complexes. Mol Biol Cell 31, 1774–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Fremillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D, et al. (2015). Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 17, 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Ransom RF, Shibanuma M, Liu C, Welsh MJ, Smoyer WE (2001). Identification and characterization of hic-5/ARA55 as an hsp27 binding protein. J Biol Chem 276, 39911–39918. [DOI] [PubMed] [Google Scholar]
- Kainuma S, Tokuda H, Yamamoto N, Kuroyanagi G, Fujita K, Kawabata T, Sakai G, Matsushima-Nishiwaki R, Kozawa O, Otsuka T (2017). Heat shock protein 27 (HSPB1) suppresses the PDGF-BB-induced migration of osteoblasts. Int J Mol Med 40, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Garrido C (2012). HSPBs: small proteins with big implications in human disease. Int J Biochem Cell Biol 44, 1706–1710. [DOI] [PubMed] [Google Scholar]
- Kostenko S, Moens U (2009). Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci 66, 3289–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JC, Han X, Hsiao CT, Yates JR 3rd, Waterman CM (2011). Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol 13, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J (1995). Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol 15, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Kwak HJ, Lee JJ, Kim YN, Lee JW, Park MJ, Jung SE, Hong SI, Lee JH, Lee JS (2008). HSP27 regulates cell adhesion and invasion via modulation of focal adhesion kinase and MMP-2 expression. Eur J Cell Biol 87, 377–387. [DOI] [PubMed] [Google Scholar]
- Li S, Piotrowicz RS, Levin EG, Shyy YJ, Chien S (1996). Fluid shear stress induces the phosphorylation of small heat shock proteins in vascular endothelial cells. Am J Physiol 271(3 Pt 1), C994–C1000. [DOI] [PubMed] [Google Scholar]
- Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M (2014). YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett 588, 2663–2670. [DOI] [PubMed] [Google Scholar]
- Luxton GW, Gomes ER, Folker ES, Worman HJ, Gundersen GG (2011). TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus 2, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A, Mammoto T, Ingber DE (2012). Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci 125(Pt 13), 3061–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGough A, Pope B, Chiu W, Weeds A (1997). Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol 138, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron T, Vancompernolle K, Vandekerckhove J, Wilchek M, Geiger B (1991). A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. J Cell Biol 114, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron T, Wilchek M, Geiger B (1988). Characterization of an inhibitor of actin polymerization in vinculin-rich fraction of turkey gizzard smooth muscle. Eur J Biochem 178, 543–553. [DOI] [PubMed] [Google Scholar]
- Murrell M, Oakes PW, Lenz M, Gardel ML (2015). Forcing cells into shape: the mechanics of actomyosin contractility. Nat Rev Mol Cell Biol 16, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumanen P, Lappalainen P, Hotulainen P (2008). Mechanisms of actin stress fibre assembly. J Microsc 231, 446–454. [DOI] [PubMed] [Google Scholar]
- Nguyen HT, Adam RM, Bride SH, Park JM, Peters CA, Freeman MR (2000). Cyclic stretch activates p38 SAPK2-, ErbB2-, and AT1-dependent signaling in bladder smooth muscle cells. Am J Physiol Cell Physiol 279, C1155–C1167. [DOI] [PubMed] [Google Scholar]
- O’Brien ER, Sandhu JK (2020). Sex differences in COVID-19 mortality: opportunity to develop HSP27 (HSPB1) immunotherapy to treat hyper-inflammation? Cell Stress Chaperones 25, 725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes PW, Banerjee S, Marchetti MC, Gardel ML (2014). Geometry regulates traction stresses in adherent cells. Biophys J 107, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KK, Brock AL, Brangwynne C, Mannix RJ, Wang N, Ostuni E, Geisse NA, Adams JC, Whitesides GM, Ingber DE (2002). Directional control of lamellipodia extension by constraining cell shape and orienting cell tractional forces. FASEB J 16, 1195–1204. [DOI] [PubMed] [Google Scholar]
- Peterson LJ, Rajfur Z, Maddox AS, Freel CD, Chen Y, Edlund M, Otey C, Burridge K (2004). Simultaneous stretching and contraction of stress fibers in vivo. Mol Biol Cell 15, 3497–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB (2007). The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol 27, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg JE, Hahn NM, Regan MM, Werner L, Alva A, George S, Picus J, Alter R, Balar A, Hoffman-Censits J, et al. (2018). Apatorsen plus docetaxel versus docetaxel alone in platinum-resistant metastatic urothelial carcinoma (Borealis-2). Br J Cancer 118, 1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala S, Oakes PW (2021). Stress fiber strain recognition by the LIM protein testin is cryptic and mediated by RhoA. Mol Biol Cell 32, 1758–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinthone S, Tyagi M, Gerthoffer WT (2008). Small heat shock proteins in smooth muscle. Pharmacol Ther 119, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Nakamura K, Doi K, Takeda K, Tobiume K, Saitoh M, Morita K, Komuro I, De Vos K, Sheetz M, Ichijo H (2001). Rap1 is involved in cell stretching modulation of p38 but not ERK or JNK MAP kinase. J Cell Sci 114(Pt 6), 1221–1227. [DOI] [PubMed] [Google Scholar]
- Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP (2006). Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 127, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller HB, Friedel CC, Boulegue C, Fassler R (2011). Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep 12, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi AS, Wehland J, Small JV (1997). The isolated comet tail pseudopodium of Listeria monocytogenes: a tail of two actin filament populations, long and axial and short and random. J Cell Biol 137, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsov M, Multhoff G, Mikhaylova E, Shibata A, Guzhova I, Margulis B (2019). Combination of Anti-Cancer Drugs with Molecular Chaperone Inhibitors. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa T, Ishiwata S (2009). Mechanical distortion of single actin filaments induced by external force: detection by fluorescence imaging. Biophys J 96, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivashankar GV, Sheetz M, Matsudaira P (2015). Mechanobiology. Integr Biol (Camb) 7, 1091–1092. [DOI] [PubMed] [Google Scholar]
- Smith MA, Blankman E, Deakin NO, Hoffman LM, Jensen CC, Turner CE, Beckerle MC (2013). LIM domains target actin regulators paxillin and zyxin to sites of stress fiber strain. PLoS One 8, e69378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Blankman E, Gardel ML, Luettjohann L, Waterman CM, Beckerle MC (2010). A zyxin-mediated mechanism for actin stress fiber maintenance and repair. Dev Cell 19, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AM, Liu T, Guevara O, Stevens J, Fanburg BL, Gaestel M, Toksoz D, Kayyali US (2007). Smooth muscle alpha-actin expression and myofibroblast differentiation by TGFbeta are dependent upon MK2. J Cell Biochem 100, 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Forman S, Quinlan RA, Ohanian J, Ohanian V (2008). Regulation of contractility by Hsp27 and Hic-5 in rat mesenteric small arteries. Am J Physiol Heart Circ Physiol 294, H961–H969. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M (1992). Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett 313, 307–313. [DOI] [PubMed] [Google Scholar]
- Sun X, Phua DYZ, Axiotakis L Jr, Smith MA, Blankman E, Gong R, Cail RC, Espinosa de Los Reyes S, Beckerle MC, Waterman CM, Alushin GM (2020). Mechanosensing through Direct Binding of Tensed F-Actin by LIM Domains. Dev Cell 55, 468–482 e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thievessen I, Thompson PM, Berlemont S, Plevock KM, Plotnikov SV, Zemljic-Harpf A, Ross RS, Davidson MW, Danuser G, Campbell SL, Waterman CM (2013). Vinculin-actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. J Cell Biol 202, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda TQ, Iwadate Y, Umeki N, Nagasaki A, Yumura S (2011). Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS One 6, e26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendredy L, Adriaenssens E, Timmerman V (2020). Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones 25, 679–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein G, Bellaye PS, Micheau O, Bonniaud P (2012). Small heat shock proteins and the cytoskeleton: an essential interplay for cell integrity? Int J Biochem Cell Biol 44, 1680–1686. [DOI] [PubMed] [Google Scholar]
- Winkelman JD, Anderson CA, Suarez C, Kovar DR, Gardel ML (2020). Evolutionarily diverse LIM domain-containing proteins bind stressed actin filaments through a conserved mechanism. Proc Natl Acad Sci USA 117, 25532–25542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AJ, Pollard TD, Herman IM (1983). Actin filament stress fibers in vascular endothelial cells in vivo. Science 219, 867–869. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Chen CS (2009). Mechanotransduction in development: a growing role for contractility. Nat Rev Mol Cell Biol 10, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, Watanabe N (2014). A new link between the retrograde actin flow and focal adhesions. J Biochem 156, 239–248. [DOI] [PubMed] [Google Scholar]
- Yoshigi M, Clark EB, Yost HJ (2003). Quantification of stretch-induced cytoskeletal remodeling in vascular endothelial cells by image processing. Cytometry A 55, 109–118. [DOI] [PubMed] [Google Scholar]
- Yoshigi M, Hoffman LM, Jensen CC, Yost HJ, Beckerle MC (2005). Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J Cell Biol 171, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP (2008). Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol 10, 1062–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoubeidi A, Gleave M (2012). Small heat shock proteins in cancer therapy and prognosis. Int J Biochem Cell Biol 44, 1646–1656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.