

SUMMARY
Serotonin (5-hydroxytryptamine; 5-HT) 5-HT2-family receptors represent essential targets for Lysergic acid diethylamide (LSD) and all other psychedelic drugs. Although the primary psychedelic drug effects are meditated by 5-HT2A serotonin receptor (HTR2A), 5-HT2B serotonin receptor (HTR2B) has been used as a model receptor to study the activation mechanisms of psychedelic drugs due to its high expression and similarity to HTR2A. In this study, we determined the cryoEM structures of LSD-bound HTR2B in the transducer-free, Gq protein-coupled and β-arrestin-1-coupled states. These structures provide distinct signaling snapshots of LSD’s action ranging from the transducer-free partially active state to the transducer-coupled fully active states. Insights from this study will both gain comprehensive molecular insights into the signaling mechanisms of the prototypical psychedelic LSD and accelerate the discovery of novel psychedelic drugs.
Keywords: Psychedelic, LSD, HTR2B, signaling transduction, Gq protein, β-arrestin-1, structural biology
eToc Blurb:
LSD is the prototypical psychedelic drug which profoundly alters human sensation, perception and mood. Can et al. determined the high-resolution cryoEM structures of LSD-bound HTR2B in transducer-free, G protein-coupled and β-arrestin-1 coupled states, which reveal the molecular bases underlying the differential engagement of downstream signaling transducers for LSD.
Graphical Abstract
INTRODUCTION
Lysergic acid diethylamide (LSD) is the prototypical psychedelic drug which profoundly alters human sensation, perception and mood (Nichols, 2016, McClure-Begley and Roth, 2022). LSD was first synthesized by Swiss chemist Albert Hoffman in 1938 (Hofmann, 1979) and subsequently found to be a potent hallucinogenic compound with an active dose as low as 25 μg in humans (Hoffer, 1965). When taken orally, the psychedelic effects of LSD can begin within 30 min and may last 12 hours or more (Passie et al., 2008).
Pharmacologically, LSD is a promiscuous drug which binds most biogenic amine G protein-coupled receptors (GPCRs) and exhibits high affinities for nearly every one of the 14 distinct serotonin receptors (Kroeze et al., 2015, Roth et al., 2002). The psychedelic effect of LSD and many other psychedelic drugs are primarily mediated by HTR2A (McClure-Begley and Roth, 2022). LSD, as well as other psychedelic drugs, may also have therapeutic potential for many neuropsychiatric diseases (McClure-Begley and Roth, 2022, Kuypers, 2020). However, chronically administrated LSD, even with microdosing, has the potential to cause drug-induced valvular heart disease in humans through the activation of HTR2B (Roth, 2007, Kuypers et al., 2019). The 5-HT2 family of serotonin receptors couples to both Gq and β-arrestins (Wacker et al., 2017). While both the G protein and β-arrestin pathways can be activated upon agonist stimulation, some agonists can preferentially stabilize a receptor conformation which favors one pathway over another (Urban et al., 2007), resulting in functional selectivity or biased signaling (Kenakin, 2011). Importantly, LSD potentially activates the β-arrestin mediated pathway for many biogenic amine GPCRs (Kroeze et al., 2015).
In our previous studies, we solved the LSD-bound HTR2B (Wacker et al., 2017) and LSD-bound HTR2A (Kim et al., 2020) structures in transducer-free states by X-ray crystallography. However, the molecular bases underlying the differential engagement of downstream signaling pathways for LSD and other ligands at most GPCRs remain largely unclear. Given the convenience of a previously described HTR2B Fab (Ishchenko et al., 2017) and the structural similarity between HTR2A-LSD complex and HTR2B-LSD complex (Wacker et al., 2017, Kim et al., 2020), we used the HTR2B as a model system to investigate the structural basis underlying the signaling mechanisms of LSD. To gain a comprehensive insight into LSD’s action at the molecular level, we determined the LSD-bound HTR2B structures in three different states—transducer-free, Gq-coupled and β-arrestin-1-coupled state, respectively, via single-particle cryo-electron microscopy (cryoEM) (Table S1).
RESULTS
Structure determination of HTR2B
The HTR2B is a class A GPCR with a molecular weight of 54 kDa, of which around 20 kDa is composed of largely unstructured extracellular and intracellular loops (Figure S1A). Thus, we used a previously reported HTR2B extracellular Fab P2C2 (Ishchenko et al., 2017) as a fiducial marker to facilitate our cryoEM work (Figure S1B). For the HTR2B-Gq complex, we assembled the LSD-bound HTR2B with miniGq (hereinafter referred to as Gq) for structural determination in a similar manner as described for the HTR2A-Gq complex (Figure S1C) (Kim et al., 2020). Purified protein complexes were applied to size-exclusion chromatography to isolate the monomers for cryoEM study (Figures S1B and S1C), leading to a 2.7 Å and 2.9 Å map for the LSD-bound transducer-free HTR2B (Figure S2) and LSD-bound HTR2B-Gq complex (Figure S3; Table S1), respectively.
As with other reported GPCR-arrestin complexes (Kang et al., 2015, Yin et al., 2019, Huang et al., 2020, Staus et al., 2020, Lee et al., 2020), the HTR2B-β-arrestin-1 complex required considerable engineering to achieve a stable complex. We noted that the HTR2B contains both a large intracellular loop 3 (ICL3) and a long C terminal tail (C-tail) (Figure S1A) (Pandy-Szekeres et al., 2018). For many GPCRs, both the ICL3 and C-tail have the potential to mediate the arrestin recruitment via phosphorylation-dependent (Yang et al., 2017, Mayer et al., 2019, Kuhn et al., 1984, Lohse et al., 1990) and independent means (Eichel et al., 2018, Krumm and Roth, 2018, Gelber et al., 1999, Gurevich and Gurevich, 2006). To determine which portions of the HTR2B are responsible for arrestin recruitment, we first performed the BRET1 β-arrestin-1 recruitment assay using different ICL3 and C-tail truncation constructs (Figures 1A and 1B). The results show that C-tail truncation construct CT464 (residues 1–464) did not impair agonist-stimulated β-arrestin-1 recruitment of HTR2B (Figure 1B; Table S2). By contrast, the maximum effect (Emax) of CT464 is approximately 50% higher than WT. The BRET assay we used here is sensitive to both the distance and orientation between the Rluc donor on receptor and the Venus acceptor on β-arrestin-1. Considering the Rluc tag is fused to the flexible C-terminus of HTR2B, the increased Emax value observed in CT464 and other C-tail truncation constructs are probably due to the closer distance between Rluc and Venus in the phosphorylated C-tail bound receptor-β-arrestin-1 complex. In contrast, the HTR2B construct CT453 (residues 1–453) and CT426 (residues 1–426), despite shorter C-tails, greatly decreased the Emax of 5-HT stimulated β-arrestin-1 recruitment (Figure 1B; Table S2) indicating the C-tail residues between I453 and L464 are critical for β-arrestin-1 recruitment. Compared with 5-HT, LSD is a partial agonist for HTR2B (McCorvy et al., 2018). To enhance the β-arrestin recruitment by LSD, we mutated the ionic lock formed by K2475.68 and E3196.30 (superscripts denote Ballesteros–Weinstein numbering (Ballesteros and Weinstein, 1995)) which stabilizes the transducer-free HTR2B structure (Figure 1C). The double mutation “K2475.68V + E3196.30L” significantly increased both the basal and maximum LSD-stimulated β-arrestin-1 recruitment to levels comparable to that of wild-type (WT) HTR2B stimulated by full agonist 5-HT (Figure 1D; Table S3).
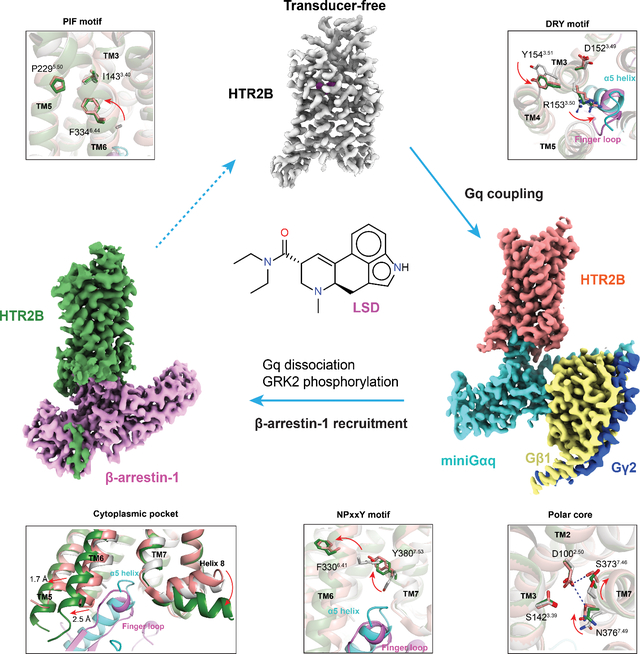
Figure 1. Construct optimization and structures of HTR2B in different states.
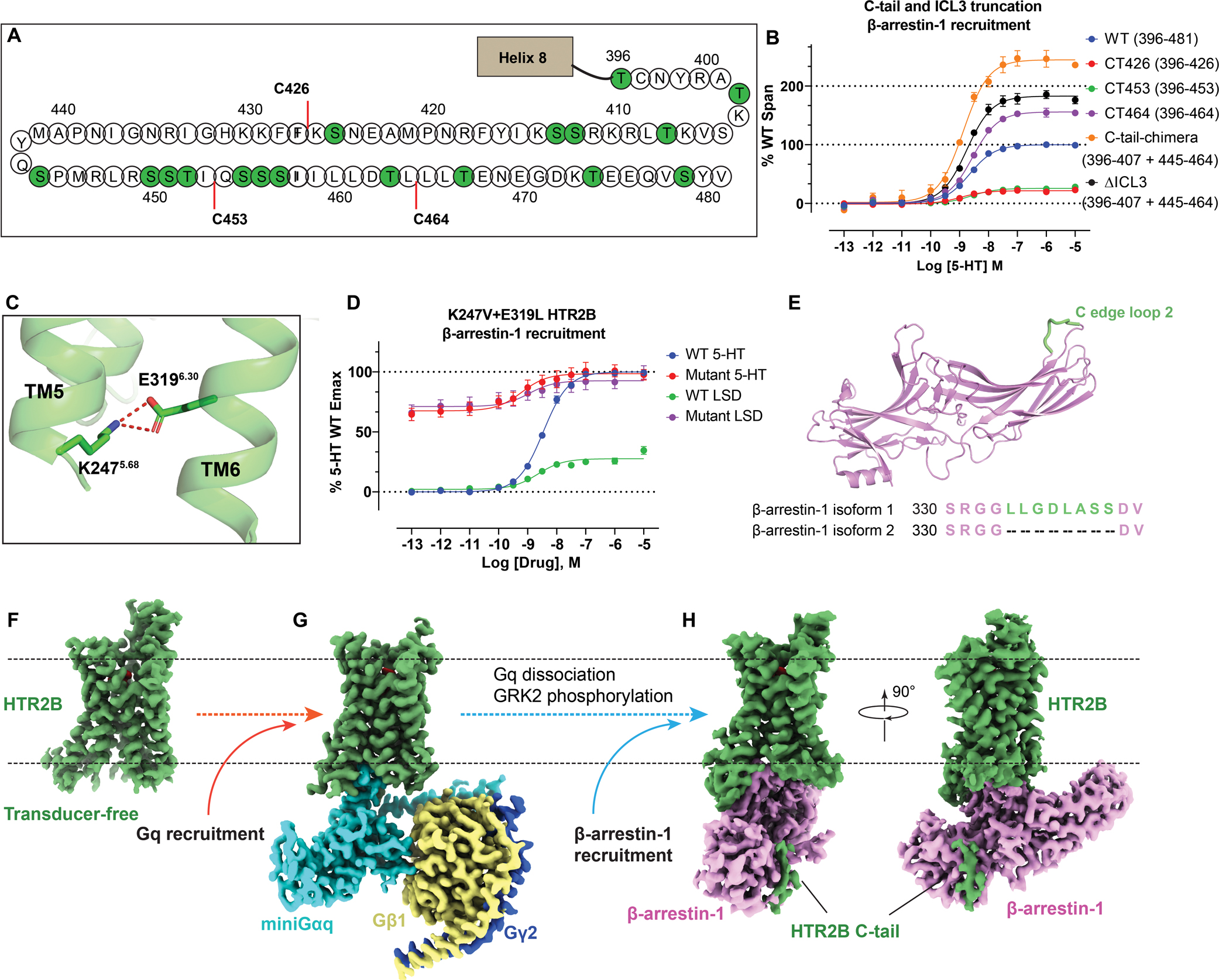
(A) The C-tail of HTR2B. Serines and threonines are highlighted by green circles. C-tail truncations C426, C453 and C464 are highlighted by red lines. See Figure S1. (B) β-arrestin-1 recruitment by different C-tail and ICL3 truncations HTR2B constructs. The remaining C-tail residues of each HTR2B truncation construct are shown in the brackets. Data represent mean ± SEM of n = 6 biological replicates. See Table S2 for fitted parameter values. (C) K2475.68 and E3196.30 form an ionic lock to restrict HTR2B (PDB 5TVN) in a partial active state. Hydrogen bonds are depicted as red dashed lines. (D) Breakage of K2475.68 and E3196.30 ionic lock by double mutations K2475.68V+E3196.30L greatly strengthened both the basal and LSD-stimulated β-arrestin-1 recruitment to a level comparable to the Emax of the full-agonist 5-HT. Data represent mean ± SEM of n = 4 biological replicates. See Table S3 for fitted parameter values. (E) Cartoon representation of β-arrestin-1 (PDB 4JQI) with the additional hydrophobic C loop 2 presented in the isoform 1 colored green. The sequence alignment of β-arrestins (residues 330–313) is also shown here to highlight the difference. (F-H) cryoEM maps of LSD-bound transducer-free (F), Gq-coupled (G) and β-arrestin-1 (H) coupled HTR2B. The maps are arranged in a sequential manner to highlight the signaling process of HTR2B. See Figures S1–S4 and Table S1 for details of protein expression and cryoEM data-processing.
We then fused the constitutively active human β-arrestin-1 with R169E mutation to the truncated C-terminus of HTR2B and the thermostabilized apocytochrome b562 RIL (BRIL) was added before residue T36 of HTR2B N-terminus to facilitate receptor protein expression. The C-tail of β-arrestin-1 was then truncated to L368 to remove self-inhibition.. Unlike all previous studies, β-arrestin-1 isoform 2, instead of the isoform 1, without the C-edge membrane anchoring loop (Staus et al., 2020) was used (Figure 1E). We used this β-arrestin-1 isoform 2 because we considered that since nanodiscs were not used in this study, the receptor-arrestin complex, without the hydrophobic C-edge loop, would be more stable in a detergent environment. To further stabilize the phosphorylated receptor-arrestin complex, an engineered single-chain Fab30 (Shukla et al., 2013), namely scFv30, was fused to the C-tail of β-arrestin-1. The engineered HTR2B-β-arrestin-1-scFv30 protein initially did not express well in Sf9 cells. Accordingly, we truncated the ICL3 (construct ΔICL3) and the middle part (L408-S444) of the C-tail (construct C-tail-chimera) (see Methods for details), both of which do not appear to be involved in arrestin recruitment (Figure 1B; Table S2). Phosphorylation of HTR2B was achieved by co-expression of HTR2B-β-arrestin-1-scFv30 chimera protein with GPCR kinase 2 (GRK2). The HTR2B-β-arrestin-1-scFv30 complex was then purified with Fab P2C2 to facilitate our cryoEM studies (Figure S1D), ultimately leading to a 3.3 Å reconstructed map with clear densities for the LSD and most side chains of receptor and β-arrestin-1, (Figure S4; Table S1). We now consider each state in turn.
HTR2B in transducer-free state
Although several transducer-free HTR2B structures have been reported previously (Wacker et al., 2013, Wacker et al., 2017, McCorvy et al., 2018, Ishchenko et al., 2017), they were all determined via X-ray crystallography. Our transducer-free HTR2B structure determined by cryoEM provided a snapshot of HTR2B close to its native state in solution as opposed to the tightly packed crystalline state. Compared to the HTR2B/LSD complex determined by crystallography (Wacker et al., 2017), some unique features were observed in the transducer-free cryoEM structure. First, the intracellular ends of transmembrane helix 5 (TM5) and TM6 are not resolved in the cryoEM structure of transducer-free HTR2B, whereas they are well-resolved in the crystal structure (Figure 2A). Second, the cryoEM HTR2B does not have the lipid molecule, which is used for crystallization in lipidic cubic phase, inserted between the TM5 and TM6. resulting in large rotations of intracellular TM6 residues (Figure 2A). Lastly, compared with our previous HTR2B crystal structure, the extracellular ends of TM1, TM2, TM4, TM5 and TM7 of the transducer-free cryoEM HTR2B display 0.9–2.5 Å outward movements (Figure 2B) and have a loose contact with LSD, which results in a slightly larger orthosteric ligand pocket for LSD binding (Figure 2C). Considering that large-scale docking is sensitive to the minor difference in the ligand binding pocket, the transducer-free cryoEM HTR2B may provide an alternative template for structure-based drug discovery.
Figure 2. Structural comparison of the transducer-free HTR2B cryoEM structure with the HTR2B crystal structure (PDB: 5VTN).
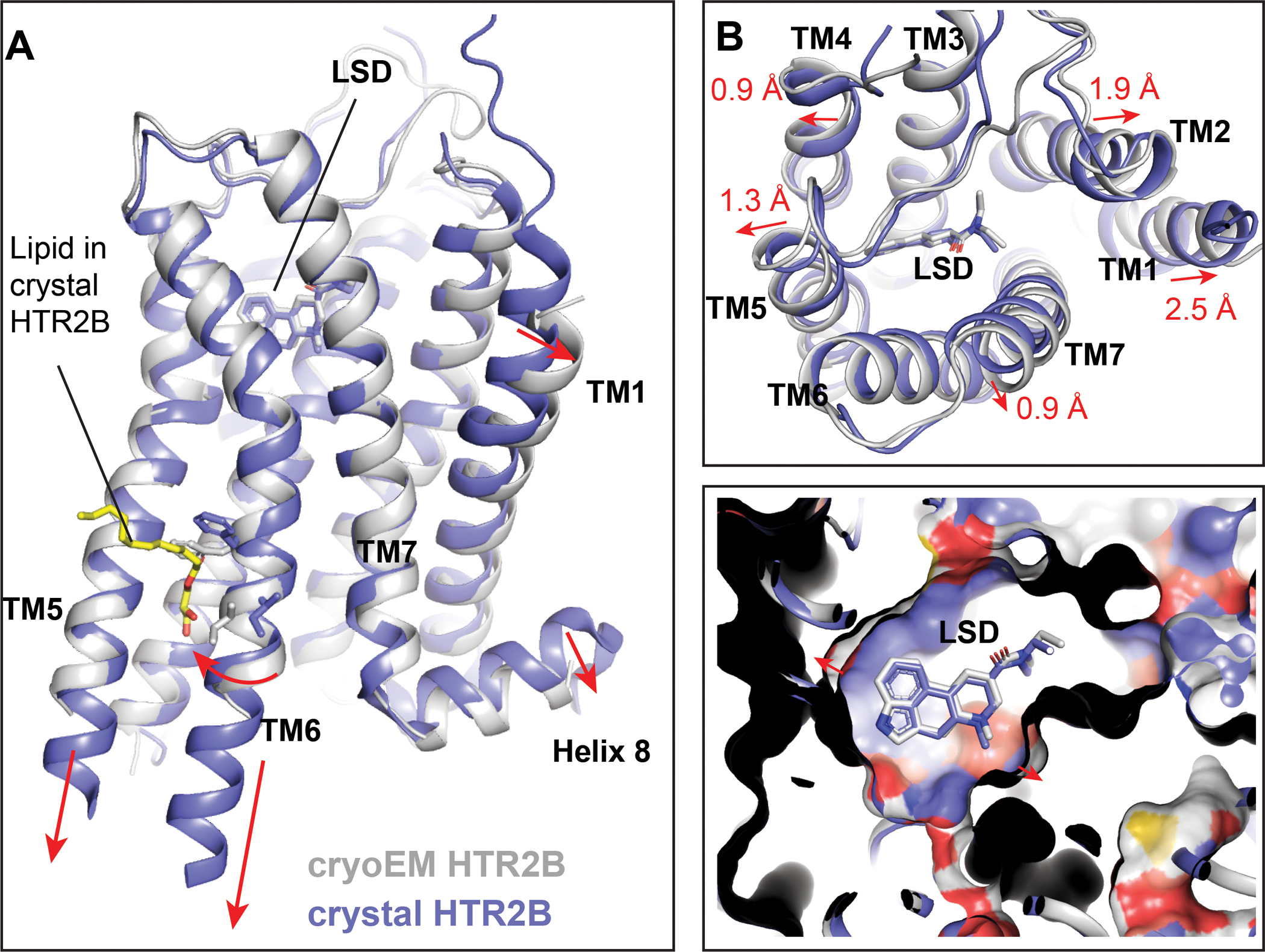
(A) Side view to show the differences in the intracellular tips of TM5 and TM6. The relatively conformational differences observed in the HTR2B cryoEM structures are indicated by red arrows. Several TM6 residues are shown as sticks to highlight the conformational changes between the cryoEM structure and the crystal structure. (B) Extracellular view of LSD binding pocket, highlighting a loose contact of LSD with surrounding helices in the transducer-free cryoEM structure of HTR2B. The relatively outward movements of helices observed in the HTR2B cryoEM structure are indicated by red arrows. (C) Side view of LSD binding pocket, showing a larger pocket in the HTR2B cryoEM structure.
Gq engagement of HTR2B
In the cryoEM structure of the LSD-bound HTR2B-Gq complex (Figure 3A), extensive interactions were observed: (1) between the cytoplasmic core of HTR2B and α5 helix of Gq (Figure 3B); (2) between the ICL2 of HTR2B and the αN helix, β2-β3 loop and α5 helix of Gq (Figure 3C). In detail, the α5 helix of Gq inserted into the cytoplasmic cavity of HTR2B primarily formed by TM2, TM3, TM5, TM6, TM7 and ICL2. Residues L236H5.16, L240H5.20 and L245H5.25 (superscription is CGN numbering system (Flock et al., 2015)) of Gq engage the interface of TM3-TM5-TM6 of HTR2B through strong hydrophobic interactions, whereas Q237H5.17, E242H5.22 and N244H5.24 of Gq form polar interactions with residues E3196.30, N16434.54 and N3848.47 of HTR2B (Figure 3B), respectively. Outside the cytoplasmic core, residue I16134.51 in the ICL2 binds into a hydrophobic grove formed by L34S1.02, V79S3.01, F228H5.08 and F235H5.15 of Gq (Figure 3C), a binding mode frequently seen in other GPCR-G protein complexes (Kim et al., 2020, Krishna Kumar et al., 2019). Additionally, ICL2 residue Q16534,55 further interacts with the αN helix of Gq through a hydrogen bond with the carbonyl group of R32 (Figure 3C). In the HTR2A-Gq structure, the equivalent interaction is mediated by R18534.55 of HTR2A with R32 of Gq (Figure 3D). Due to the variance in residue composition, the ICL2 of HTR2B has a 2 Å inward displacement compared with that of HTR2A (Figure 3D). Consequently, there is also an inward movement of Gq αN helix upon coupling to HTR2B (Figure 3D). By contrast, the α5 helix coupling modes are almost identical in both HTR2A and HTR2B (Figure 3E) as their residues in the cytoplasmic cavity are highly conserved.
Figure 3. Gq engagement of HTR2B.
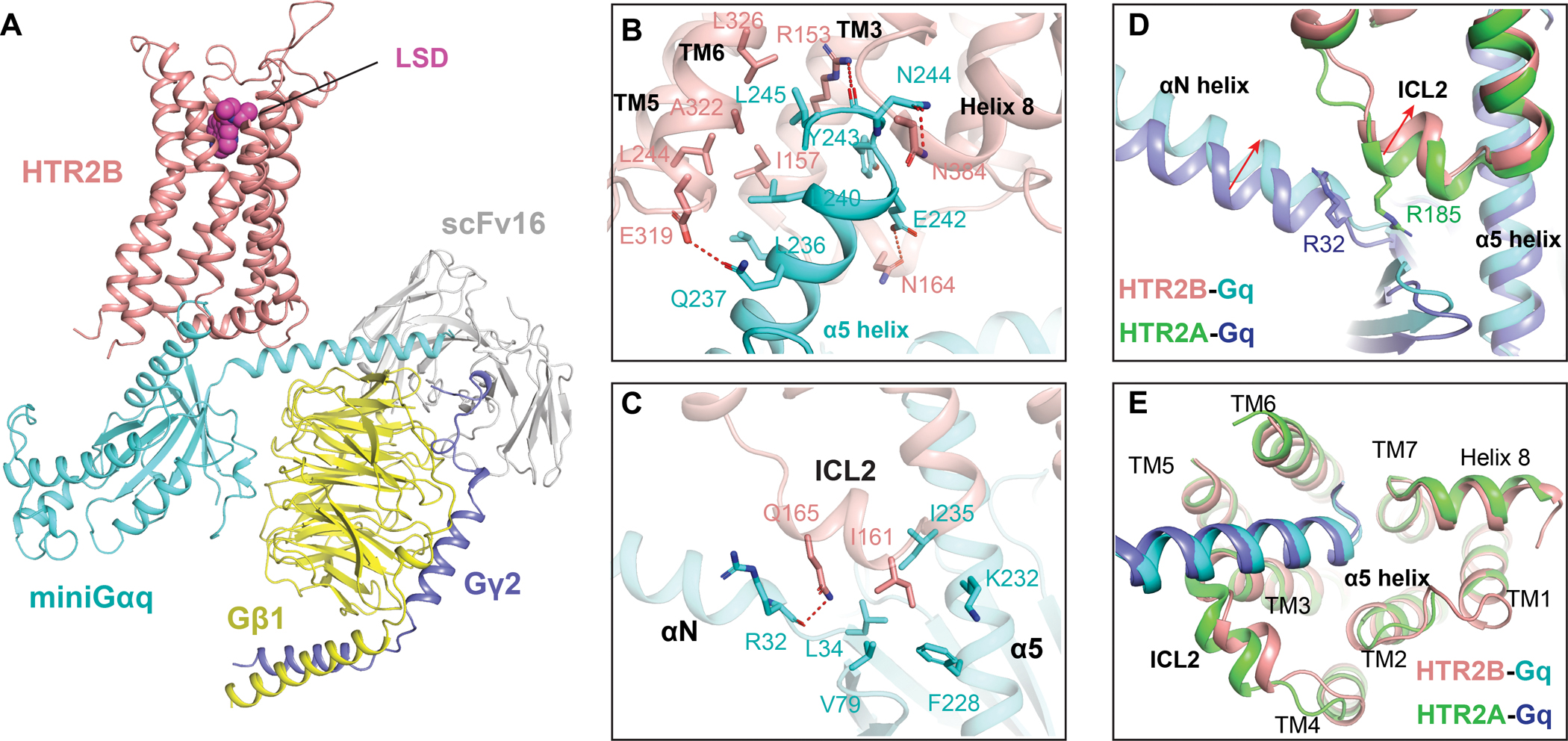
(A) The overall structure of HTR2B in complex with agonist LSD and miniGq. HTR2B and miniGq are coloured as red and cyan, respectively. (B) The interactions between the α5 helix of Gq and the cytoplasmic cavity of HTR2B. Key residues involved in interactions are shown as sticks. Polar interactions are highlighted by red dashed lines. (C) Interactions between the ICL2 of HTR2B and Gq, showing I16134.51 of HTR2B forms strong hydrophobic interactions with Gq. (D) Structural comparison of the HTR2B-Gq complex with HTR2A-Gq complex reveals inward displacements of both the ICL2 of HTR2B and the αN helix of Gq in HTR2B-Gq complex structure. (E) Structural comparison of the HTR2B-Gq complex with HTR2A-Gq complex reveals almost identical conformations of Gq α5 helix.
β-arrestin-1 Coupling of HTR2B
In the structure of HTR2B-β-arrestin-1 complex, arrestin engages the receptor core in a way more similar to the structures of rhodopsin-visual arrestin (Kang et al., 2015), β1AR-β-arrestin-1 (Lee et al., 2020) and M2R-β-arrestin-1 (Staus et al., 2020) (Figure S5A), than the NTSR1-β-arrestin-1 (Huang et al., 2020, Yin et al., 2019) structures (Figure S5B). However, β-arrestin-1 couples to HTR2B with an approximately 50° rotation to its position in NTSR1-β-arrestin-1 structure, resembling a unique intermediate conformation between M2R-β-arrestin-1 and NTSR1-β-arrestin-1 (Figure S5B). Additionally, β-arrestin-1 engages HTR2B in a way that is approximately 25° less tilted towards the receptor compared with its conformation in NTSR1-β-arrestin-1 structure (Figure S5B), resembling a conformation similar to M2R-β-arrestin-1 and β1AR-β-arrestin-1 structures solved in lipid nanodiscs (Figure S5A).
Generally, β-arrestin-1 engages HTR2B extensively at the receptor core (Figure 4A), ICL2 (Figure 4B) and C-tail (Figure 4C) regions. In the cytoplasmic receptor core, the finger loop of β-arrestin-1 adopts a helical conformation to interact with TM2, TM3, TM5, TM6 and TM7 of HTR2B through both hydrophobic and electrostatic interactions (Figure 4A). Specifically, residues L68, L71, L73 and F75 in the C-terminus of finger loop engage the hydrophobic inner face of TM3, TM5 and TM6. At the N-terminus of finger loop, R65 and D67 form electrostatic interactions with N16434.54 and N16734.57 of HTR2B, respectively (Figure 4A). An intra-helix hydrogen bond formed between R65 and D67 may reduce the net charge of the finger loop to achieve a stable engagement at the cytoplasmic core of HTR2B. Mutations of L71, L73 and R65 on β-arrestin-1 significantly affected the efficacy of LSD-stimulated β-arrestin-1 recruitment, demonstrating the importance of the finger loop for β-arrestin-1 coupling (Figure 4D; Table S4).
Figure 4. β-arrestin-1 coupling of HTR2B.
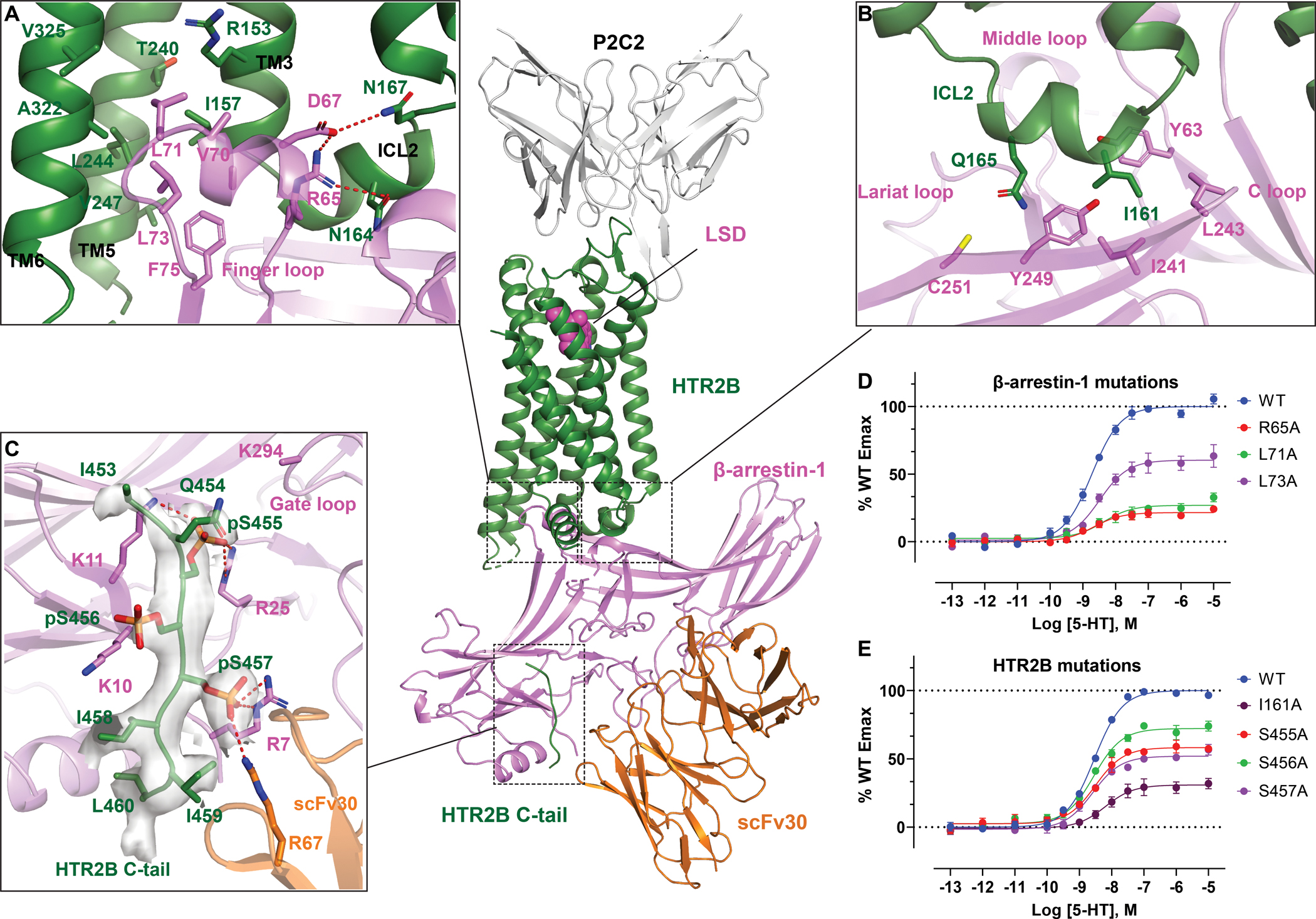
(A-C) Interactions of cytoplasmic core (A), ICL2 (B) and C-tail (C) of HTR2B with β-arrestin-1, respectively. HTR2B and β-arrestin-1 are colored as green and magenta, respectively. Key residues involved in interactions are shown as sticks. Polar interactions are depicted as red dashed lines. CryoEM Map for HTR2B C-tail is shown in panel C. See Figure S5. See also Table S5 for the phosphorylation probabilities of HTR2B C-tail residues. (D) Finger loop mutations impair of β-arrestin-1 recruitment of HTR2B. Data represent mean ± SEM of n = 3 biological replicates. See Table S4 for fitted parameter values. (E) Alanine substitutions of HTR2B ICL2 residue I16134.51 and C-tail phosphorylation residues S455, S456 and S457 impair β-arrestin-1 recruitment of HTR2B. Data represent mean ± SEM of n = 3 biological replicates. See Table S4 for fitted parameter values.
Outside the receptor cytoplasmic cavity, the ICL2 of HTR2B adopts a helical structure and sits in a hydrophobic cleft formed by the C-loop, lariat loop and middle loop of β-arrestin-1 (Figure 4B), as observed in Rhodopsin-visual arrestin (Kang et al., 2015), β1AR-β-arrestin-1 (Lee et al., 2020) and M2R-β-arrestin-1 (Staus et al., 2020) structures (Figure S5A). Notably, I16134.51 forms extensive hydrophobic interactions with Y249, I241, L243, and Y63 of β-arrestin-1 (Figure 4B). The hydrophobic interactions mediated by I16134.51 seem to be crucial for β-arrestin-1 engagement as the I16134.51A mutation reduced the Emax of agonist-stimulated β-arrestin-1 recruitment by 70% compared with the WT (Figure 4E; Table S4). In the NTSR1-β-arrestin-1 structure, ICL1, instead of ICL2, sits in the same hydrophobic cleft, resulting in a large rotation of β-arrestin-1 (Figure S5B). When the β-arrestin-1 molecules are aligned, various conformations of the finger loop can be observed upon coupling to different receptors (Figure S5D). The flexibility of the finger loop, as well as associated rotation of the arrestin core, could be essential for arrestins to adapt their conformation to engage the various cytoplasmic cavities across the GPCR superfamily. In contrast, the α5 helix of G protein apparently adopts a similar rigid conformation upon coupling to various GPCRs.
In the HTR2B-β-arrestin-1 complex structure, densities corresponding to the phosphorylated C-tail were observed (Figures 1H, 4C and S4D). As noted above, we have demonstrated through C-tail truncation that HTR2B residues between I453 and L464 are critical for β-arrestin-1 recruitment with only four residues in this range that can get phosphorylated, i.e., S455, S456, S457 and T463 (Figure 1A). Considering that arrestin recruitment usually requires a spatially arranged phosphorylation barcode with more than 1 phosphorylated residue (Zhou et al., 2017, Nobles et al., 2011, Yang et al., 2015), S455, S456 and S457 are likely the residues that were phosphorylated to recruit β-arrestin-1. Individual alanine mutations of these three residues reduced the Emax of HTR2B mediated β-arrestin-1 recruitment by 30–50% (Figure 4E; Table S4), suggesting potential phosphorylation of these residues are important for β-arrestin-1 recruitment. Mass-spectrometry successfully detected phosphate groups at S455 and S456 (Figure 5E–F; Table S5), and a slightly low phosphorylation probability is observed for S457 (Table S5). In the cryoEM map, a clear phosphate group density has been observed for the residue that interacts with R7 of β-arrestin-1 (Figure 4C). In addition, phosphorylation at the “key site” that interacts with the K294 of β-arrestin-1 gate loop is critical for the binding and activation of arrestins in both rhodopsin (Mayer et al., 2019) and V2R (He et al., 2021, Latorraca et al., 2020, Dwivedi-Agnihotri et al., 2020). Based on these structural observations, functional data and the previously studies, a possible way to place the HTR2B C-tail has been shown in Figure 4C, in which both R7 and K294 could be bound by the phosphorylated residues. Compared with the β-arrestin-1-V2Rpp-Fab30 complex (Shukla et al., 2013), the C-tail of HTR2B overlaid well with the most frequently used V2R tail (Figure S5D). However, residue R7 of β-arrestin-1 has rotated over 100° to engage pS457 in HTR2B (Figure S5D), suggesting a C-tail phosphorylation pattern different from V2R is used in HTR2B,
Figure 5. Structural comparison of the Gq- and β-arrestin-1-coupled states of HTR2B with its transducer-free state.
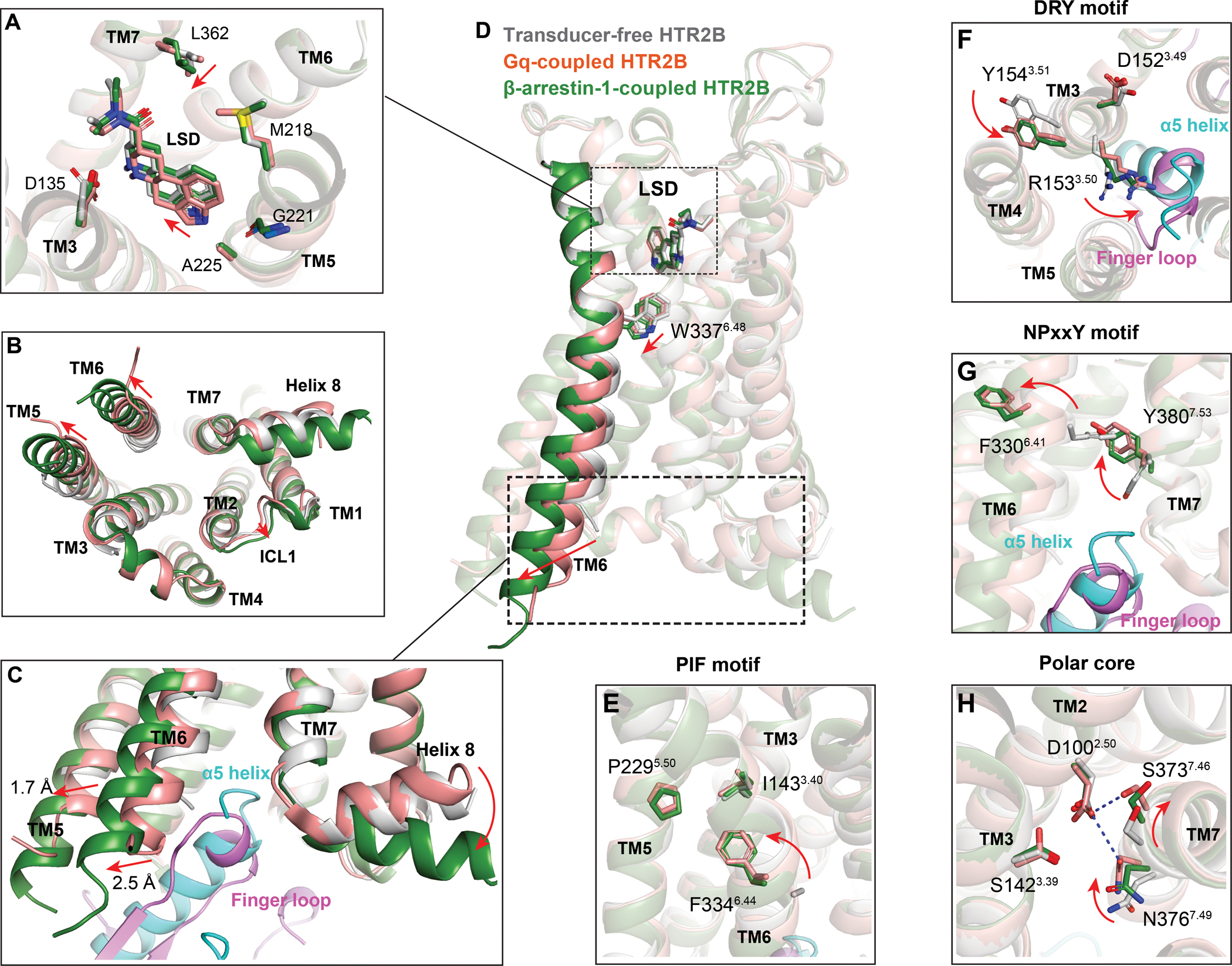
(A) Close-up view of LSD binding pocket in different states. The HTR2B in transducer-free state, Gq-coupled state and β-arrestin-1 coupled state are colored by gray, red and green, respectively. The relative movements of LSD and residues of β-arrestin-1-coupled HTR2B related to its Gq-coupled state are indicated by red arrows. (B) Intracellular view of HTR2Bs to show conformational changes from transducer-free state to transducer-coupling states. Movements of loop and helices are indicated by red arrows. (C) Structural comparison of the engagement of Gq and β-arrestin-1 to the cytoplasmic core of HTR2B. Conformational changes in TM5, TM6 and helix 8 of β-arrestin-1-coupled HTR2B related to its Gq-coupled state are depicted as red arrows. See Figure S7. (D) Side view of HTR2Bs to show the extra downward shift of the toggle switch W3376.48 along with a more prominent TM6 outward displacement in β-arrestin-1 coupled HTR2B. Movements of residues and helices are indicated by red arrows. See Figure S7. (E-H) Conformational changes of PIF motif (E), DRY motif (F), NPxxY motif (G) and polar core (H) upon transducer coupling. Movements of HTR2B residues upon transducer coupling are indicated by red arrows. See Figure S7.
Differential coupling of Gq and β-arrestin-1
Gq and β-arrestins are the two major downstream signaling transducers for the 5-HT2R subfamily. Structural comparisons of the Gq-coupled HTR2B-LSD complex with the β-arrestin-1-coupled HTR2B-LSD complex reveal structural differences that may contribute to the differential coupling of Gq and β-arrestin-1 by LSD. At the extracellular ligand binding pocket, LSD has a looser contact with TM5 of β-arrestin-1-coupled HTR2B than Gq-coupled HTR2B although their overall poses are similar (Figure 5A). In both Gq- and β-arrestin-1-coupled structures, most of the binding pocket residues adopt nearly identical side chain conformations upon binding to LSD with only subtle conformational changes observed on residues M2185.39 and L3627.35. Notably, L3627.35 adopts a different rotamer state which has a closer contact with the diethylamide group of LSD upon coupling to β-arrestin-1 (Figure 5A), demonstrating our previous finding that the strong hydrophobic contact between L3627.35 and LSD is crucial for β-arrestin recruitment (McCorvy et al., 2018)
On the intracellular side, both the Gq- and β-arrestin-1-coupled HTR2B structures display an active conformation with obvious TM6 outward displacements compared with the transducer-free state (Figures 5B and 5C). Distinct from the rhodopsin, M2R and NTSR1 arrestin states, which adopt similar TM6 outward movements upon coupling to their cognate Gi family proteins and arrestins (Figures S6A–6C), the cytoplasmic end of TM5 and TM6 in the HTR2B-β-arrestin-1 structure have an additional outward displacement of 1.7 Å and 2.5 Å, respectively, compared with their positions in the HTR2B-Gq structure (Figures 5C and S6D). These extra movements of TM5 and TM6 seem to be required to accommodate the bulkier helical finger loop of β-arrestin-1 (Figure 5C). By contrast, TM6 of β1AR displayed a significantly smaller outward movement in the β-arrestin-1-coupled state than its conformation in the Gs-coupled state (Figure S6E), suggesting the intracellular conformational changes for the selective coupling of transducers are different in Gq- and Gs-coupled receptors. In addition, TM7 of HTR2B has a less inward shift in β-arrestin-1-coupled structure. Other conformational differences, including an outward movement of ICL1 and a downward rotation of helix 8, were also observed in β-arrestin-1-coupled HTR2B (Figures 5C). Both TM7 and helix 8 have been implicated to involve in arrestin-biased signaling (Rahmeh et al., 2012, Liu et al., 2012). The structural differences observed in our structures may reflect the receptor plasticity of HTR2B that is essential for accommodating distinct transducers.
The HTR2B structures also displayed substantial conformational rearrangement in several key motifs, including CWxP motif, PIF motif, DRY motif and NPxxY motif, and the central polar core upon coupling to different transducers (Figures 5D–5H). Specifically, the downward movement of the “toggle switch” W3376.48 (Figure 5D) along with a rearrangement of the PIF motif (Figure 5E) are observed in both transducer-coupled structures. Of note, W3376.48 has an additional downward shift in β-arrestin-1-coupled HTR2B, which is in line with a larger outward displacement of TM6 upon β-arrestin-1 coupling (Figure 5D). At the DRY motif, counterclockwise rotations of the side chains of R1533.50 and Y1543.51 are observed in transducer-coupled states (Figure 5F). The rotation of R1533.50 seems to be essential for it to engage with the α5 helix of Gq or the finger loop of arrestin (Figure 5F). We also noticed that R1533.50 adopts an extended conformation to hydrogen-bond with Gq C-terminal residues N244H5.24 (Figure 3B), while in the β-arrestin-1-coupled HTR2B, R1533.50 bends downwards to the finger loop and forms non-polar interactions with L71 of β-arrestin-1 (Figures 4A and S6I). Similar conformational change of R1533.50 side chain was also observed in the β-arrestin-1-coupled structures of M2R, β1AR and NTSR1, suggesting the rotamer flexibility of R3.50 is essential to maintain a proper intracellular cavity to accommodate the variable shape of finger loop (Figures S6F–S6J). In the NPxxY motif, Y3807.53 flips its side chain upward and moves into a position previously occupied by F3306.41 when it transits to active conformations (Figure 5G). The receptor polar core near the sodium pocket has also been implicated to involve in the selective coupling of transduces (Wingler et al., 2020). In the structures of HTR2B, clockwise rotations of N3767.49 and S3737.46 upon transducer coupling rearrange the polar core (Figure 5H). Specifically, residues N3767.49 and S3737.46 rotate toward TM2 and form an electrostatic interaction with D1002.50 in Gq-coupled HTR2B. By contrast, these two residues are less rotated towards D1002.50 in β-arrestin-1-coupled HTR2B (Figures 5H and S6N), resembling an intermediate state between transducer-free and Gq-coupling. A similar side chain conformation of N7.49 was also observed in the arrestin-coupled structures of rhodopsin, M2R, β1AR, and NTSR1 (Figures S6K–S6M and S6O), suggesting that the polar interaction between D2.50 and N7.49 is crucial for G protein activation but not for the activation of arrestin. This is further supported by a previous study showing that D742.50N mutation of angiotensin II type 1 receptor (AT1R) impaired Gq activation but displayed a more potent β-arrestin recruitment (Bonde et al., 2010).
The transducer coupling modes of HTR2B further support the previously proposed desensitization mechanism that β-arrestin sterically hinders the G protein activation by overlapping with the G protein binding site (Figure 6A) (Thomsen et al., 2016, Szczepek et al., 2014). With an additional TM6 outward displacement of the receptor, β-arrestin-1 not only just overlaps with the Gq binding site but also occupies a larger intracellular cavity that could block Gq binding (Figures 6A–6C). Apart from the conformational changes in helices and motifs, structural analysis of the detailed interaction between HTR2B and transducers reveals that helix 8 residue N3848.47 may play distinct roles in Gq and β-arrestin-1 coupling. Notably, N3848.47 forms a hydrogen bond with Gq α5 helix residue N244H5.24 (Figure 6D), whereas it rotates 90° to stack on the top of the V70 of the hydrophobic finger loop of β-arrestin-1 with no strong interaction observed (Figure 6E). To investigate the potential role of N3848.47 in transducer coupling, we measured the direct recruitment of miniGq and β-arrestin-1 through BRET1 recruitment assays for both the WT and N3848.47A HTR2B (Figures 6F–6I; Table S6). Consistent with the structural observations, the N3848.47A mutation reduced the Emax of Gq recruitment by 40% (Figure 6H), whereas the Emax of LSD-stimulated β-arrestin-1 recruitment was increased at the N3848.47A mutant as favorable hydrophobic interactions were introduced (Figure 6I). Notably, the potency of agonist-stimulated β-arrestin-1 recruitment is slightly lower than that of the Gq recruitment (Figures 6F and 6G; Table S6), consistent with a more prominent TM6 outward movement in β-arrestin-1-coupled HTR2B, which might indicate a higher energy state.
Figure 6. Comparison of Gq- and β-arrestin-1-coupled HTR2B.
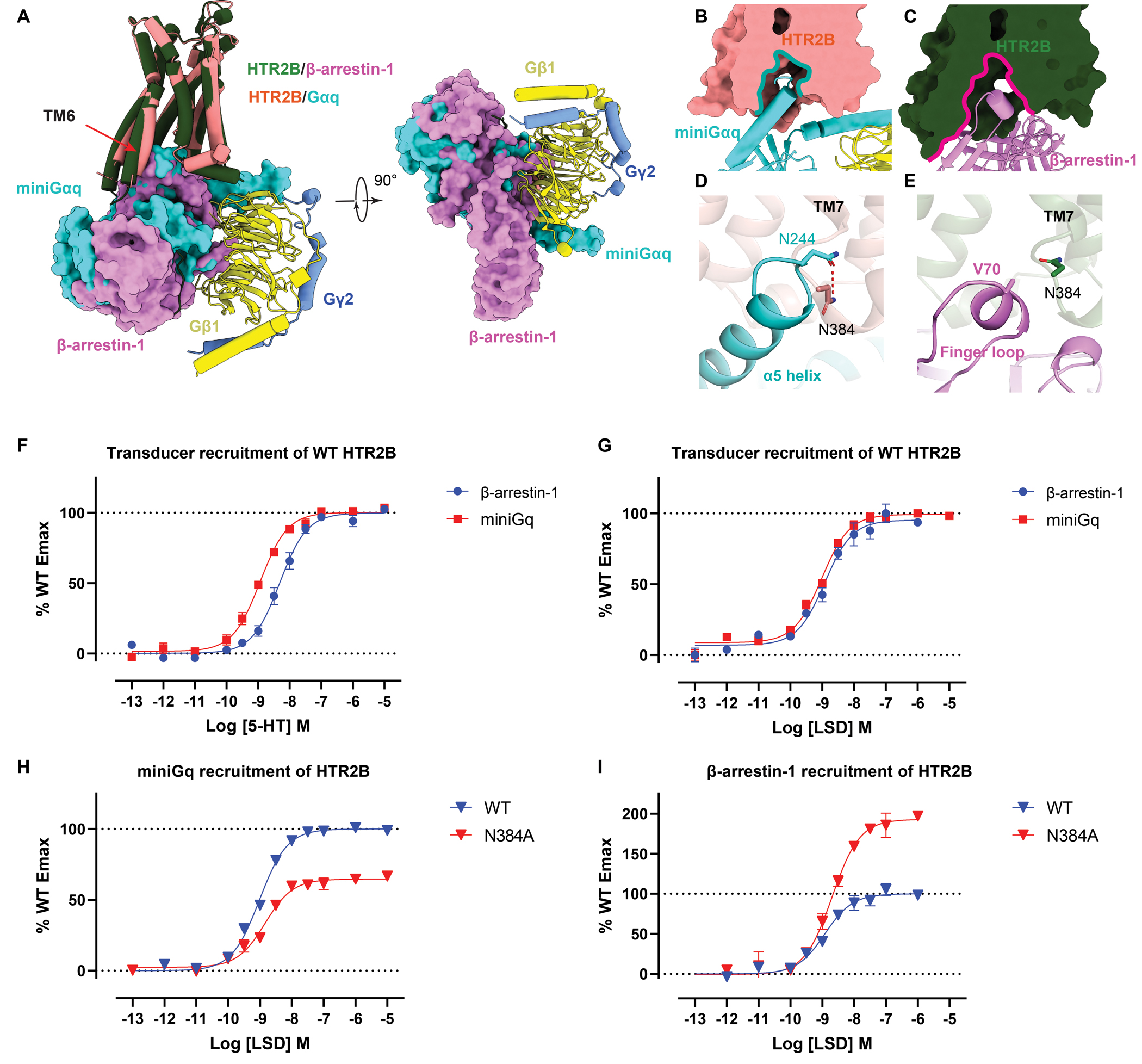
(A) Structural comparison of the overall binding mode of Gq and β-arrestin-1 to HTR2B. see Figure S7. (B-C) Intracellular cavities of HTR2B for Gq (B) and β-arrestin-1 (C) coupling. (D-E) Interactions of Gq α5 helix (D) and β-arrestin-1 finger loop (E) with the HTR2B helix 8 residue N3848.47, showing N3848.47 forms a strong hydrogen-bond with Gq but not with β-arrestin-1. (F-G) miniGq and β-arrestin-1 recruitment of WT HTR2B stimulated by 5-HT (F) and LSD (G). Data represent mean ± SEM of n = 3 biological replicates. See Table S6 for fitted parameter values. (H) miniGq recruitment of WT and N3848.47 of HTR2B stimulated by LSD. Data represent mean ± SEM of n = 3 biological replicates. See Table S6 for fitted parameter values. (I) β-arrestin-1 recruitment of WT and N3848.47 of HTR2B stimulated by LSD. Data represent mean ± SEM of n = 3 biological replicates. See Table S6 for fitted parameter values.
Mechanism for transducer coupling of HTR2B
In our transducer-free cryoEM structure of LSD-bound HTR2B, W3376.48 adopts a conformation similar to its position in Gq-coupled structure although a less obvious TM6 displacement was observed (Figure 5D), revealing an agonist-bound partially active state. Indeed, the side chain of F3336.44 in PIF motif is not resolved in the transducer-free state, suggesting multiple intermediate conformations exist. A unique feature in the transducer-free HTR2B cryoEM structure is that the intracellular tips of TM5 and TM6 outside the detergent micelle are not resolved (Figures 7A and 7B). By contrast, they are clearly resolved in the transducer-coupled HTR2B cryoEM structures (Figures 7A, 7C and 7D). A possible explanation is that the intracellular tips of TM5 and TM6 are structurally unstable with dynamic conformations presented in the solvent (Nygaard et al., 2013), which can only be stabilized through the coupling of downstream transducers (Figures 7C and 7D). As LSD alone cannot fully stabilize the active conformation of HTR2B, the dynamic intracellular tips of TM5 and TM6 may occasionally open the intracellular cavity to provide space for the initial coupling of transducers. Indeed, in molecular dynamics (MD) simulations initiated from the transducer-free structure, the transducer-binding pocket occasionally opens sufficiently to allow transducer binding (Figure 7E). Moreover, the receptor conformations sampled in the transducer-free simulations substantially overlap those observed in simulations with the G protein or arrestin bound to the receptor (Figure 7F). Taken together, these observations suggest that the LSD-bound transducer-free receptor transiently adopts conformations that could accommodate binding of either G protein or β-arrestin-1. Significantly, when removing G protein or arrestin from the receptor in simulation, the receptor adopts a conformation matching that of the transducer-free receptor (Figure 7G).
Figure 7. Activation mechanism of HTR2B.
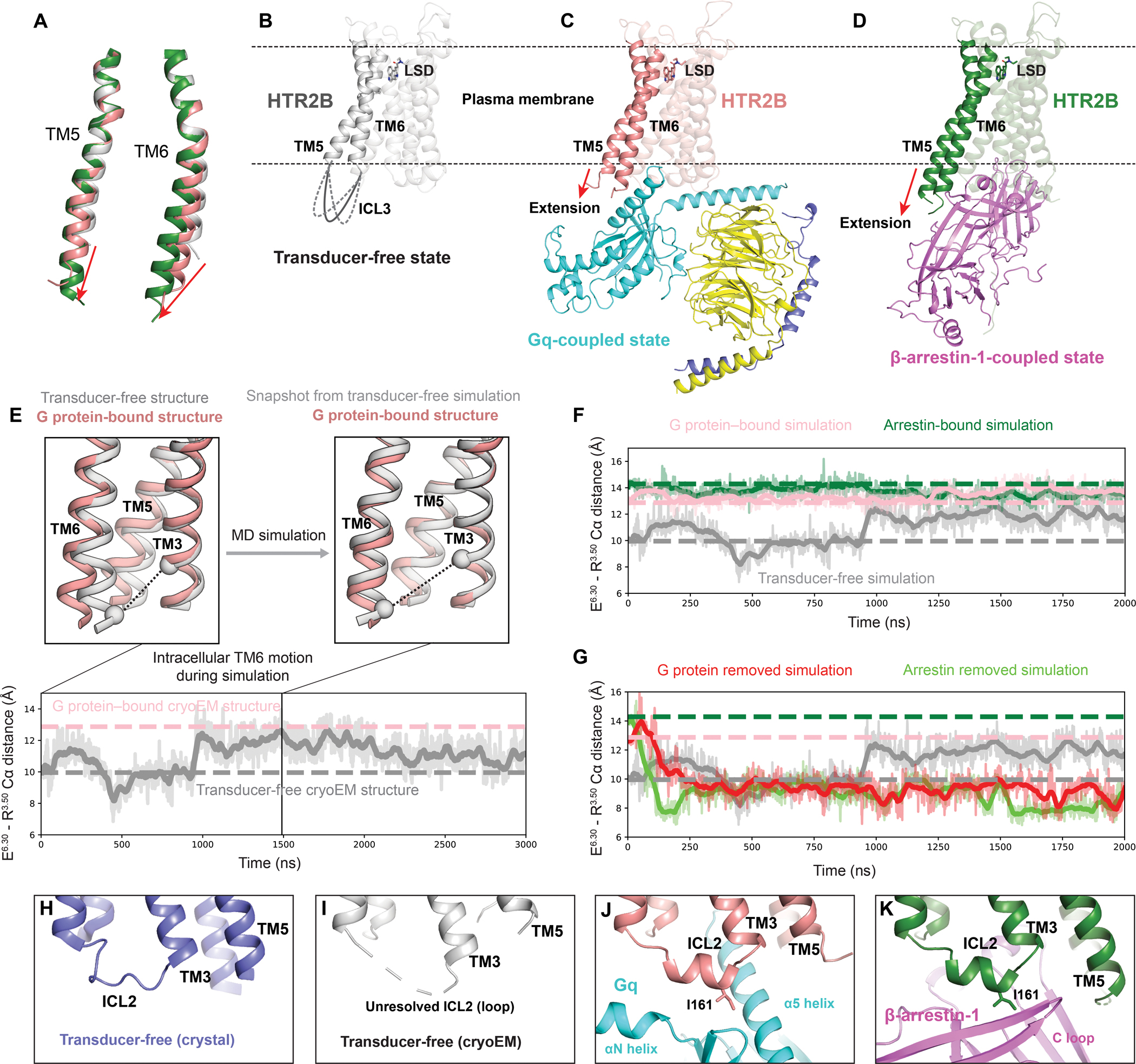
(A) Alignment of TM5 and TM6 of HTR2B in the transducer-free, Gq-coupled and β-arrestin-1-coupled states. The red arrows show the extensions and outward movements of TM5 and TM6. (B-D) Overall structures of the transducer-free state (B), Gq-coupled state (C) and β-arrestin-1-coupled state (D) of HTR2B, highlighting the further extension and outwards movement of the intracellular tips of TM5 and TM6 upon coupling to downstream transducers. (E) In simulation, TM6 of the transducer-free receptor samples intracellular conformations matching that seen in the G protein–bound structure. Traces show the distance between the Cα carbons of ionic lock residues E3196.30 and R1533.50 (black dashed lines in renderings) in a representative simulation. Thick traces represent smoothed values (i.e., moving averages); transparent traces represent original, unsmoothed values. Pink and gray dashed horizontal lines indicate values for the G protein–bound and transducer-free experimental structures, respectively. (F) Receptor conformations in transducer-free simulation overlap with those from transducer-bound simulations. Pink, green, and gray dashed horizontal lines indicate values for the G protein–bound, arrestin–bound, and transducer-free cryoEM structures, respectively. (G) In simulations initiated from transducer-bound structures but with the transducer removed, the receptor relaxes to conformations similar to the transducer-free cryo-EM structure. Pink, green, and gray dashed horizontal lines indicate values for the G protein–bound, arrestin–bound, and transducer-free cryoEM structures, respectively. (H-K) The conformation of ICL2 changes from the loop in both the crystal transducer-free state (H) and cryoEM transducer-free state (J) to α-helix in Gq-coupled state (J) and β-arrestin-1-coupled state (K), positioning HTR2B ICL2 residue I16134.51 to interact with transducers.
One possible explanation for the foregoing is that the transducers may synergistically stabilize the active state of receptor together with LSD. Consistent with this idea, F3336.44 in HTR2B has a large rotation towards TM5 to engage P2295.50 and I1433.40 upon coupling to either Gq or β-arrestin-1, forming a fully active PIF motif configuration (Figure 5E). The conformational changes of PIF motif are accompanied by the repositioned side chain of Y3807.53 in the NPxxY motif (Figure 5G), further opening the cytoplasmic cavity of HTR2B to accommodate the α5 helix of Gq or the finger loop of β-arrestin-1. Notably, the transducer-coupling process is also accompanied by a conformational change of ICL2 from loop to helical turns (Figures 7H–7K), which further stabilize the transducer-coupling states. The helical conformation of ICL2 seems to be critical for directing I16134.51 to a proper position to interact with the hydrophobic grooves of transducers for effective coupling (Figures 3C and 4B), as mutations in this position dramatically affect the coupling of both β-arrestin-1 (Figure 4E; Table S4) and G protein to receptors (Staus et al., 2020, Kim et al., 2020).
DISCUSSION
Here we provide the first structural insights into the mechanisms responsible for G protein vs arrestin signaling at a single receptor by the prototypical psychedelic LSD. To our knowledge, these represent the first structures of the same receptor with the same ligand in the transducer-free, G-protein- and arrestin-engaged states thereby providing important insights into the modes of differential transducer engagement at the identical receptor. Extensive changes in both the intracellular sides of helices and key motifs involved in HTR2B activation were observed. Of note, a larger TM6 outward movement is observed in β-arrestin-1 coupled HTR2B than G protein-coupled HTR2B, which is quite different from previously reported GPCR-transducer complex structures (Figures S6A–S6E). In addition, the molecular details provided by the high-resolution cryoEM structures clearly reveal the detailed interactions between HTR2B and transducers, enabling us to identify a residue, namely N3848.47, which plays distinct roles in Gq and β-arrestin-1 coupling. Finally, we discovered several β-arrestin-1 specific motif conformations, including the downward bending of R3.50 in the DRY motif and a weakened polar interaction between D2.50 and N7.49 in the polar core, which resembles an intermediate state between transducer-free and Gq-coupled states. Interestingly, we previously found that, at AT1R, biased ligands alone—i.e., with no G protein or arrestin present—also favor distinct R3.50 conformations, and that these conformations were allosterically coupled to the region surrounding D2.50 and N7.49 (Suomivuori et al., 2020).
G proteins and β-arrestins are the two key transducers of GPCRs. Biased agonists with functional selectivity towards a specific transducer have been proposed to be safer and more effective drugs at certain GPCRs (Smith et al., 2018, Violin et al., 2014). The cryoEM structures of HTR2B in transducer-free and transducer-coupling states reveals the signaling mechanisms of distinct transducers for the prototypical psychedelic LSD and these findings could provide insight into the structure-based biased agonist discovery at serotonin receptors. For example, considering the TM6 outward movement, which is an energy-driven process, is smaller in Gq-coupled HTR2B, one possible way to discover Gq biased agonist is to design a partial agonist with lower efficacy. In addition, the structural comparisons with other receptors solved with both G protein and arrestin reveal that the intracellular conformational changes, especially TM6, for the selective coupling of downstream transducers are quite distinct for Gi-, Gs- and Gq-coupled receptors, suggesting the mechanism of functional selectivity could be different across GPCR subfamilies.
Limitations of the study
To obtain a stable HTR2B-β-arrestin-1 complex, we have engineered the receptor with truncations and mutations, and directly fused a constitutively active β-arrestin-1 to the C-terminus of the receptor. Although the engineered construct did not change the interface residues of the receptor-β-arrestin-1 complex, we cannot rule out the possibility that the removed intracellular loop regions of HTR2B may partly affect the way β-arrestin-1 engages HTR2B. In addition, the binding of scFv30 may require phosphorylation sites at specific C-tail positions (Shukla et al., 2013). Thus, the HTR2B-β-arrestin-1 structure obtained here may represent a scFv30 preferred C-tail phosphorylation pattern and β-arrestin-1 conformation. Lastly, we co-expressed the HTR2B with the GRK2 to facilitate receptor phosphorylation. Other C-tail phosphorylation patterns, especially these with few phosphorylation sites or the sites could only be phosphorylated by other GRKs, may not be purified. The cryoEM data-processing may also class out some minor populations of the HTR2B-β-arrestin-1 complexes with different conformations. These technical limitations, although important, could not be addressed in this study. Better cryoEM technology development in the future combined with the in vivo functional assays may be helpful to obtain GPCR-arrestin complexes with more cellular and physiological relevance.
STAR METHOD
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bryan L.Roth (bryan_roth@med.unc.edu)
Materials Availability
All the plasmids and cells generated from this study could be obtained directly from the Lead Contact with a completed Materials Transfer Agreement if there is potential for commercial application. All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
All data generated or analyzed in this study are included in this article and the Supplementary Information. The cryo-EM density maps and corresponding coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB), respectively, under the following accession codes: EMD-25403 and 7SRS (HTR2B/β-arrestin-1), EMD-25402 and 7SRR (HTR2B-LSD/miniGq) and EMD-25401 and 7SRQ (HTR2B-LSD). The mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD030752.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Two eukaryotic cell lines, Spodoptera frugiperda (Sf9, Expression systems) cells and HEK293T cells (ATCC), were used in this study. Sf9 cells suspension in ESF-921 medium were purchased from Expression systems and used for protein expression of HTR2B for structural study. Sf9 cells were grown in ESF-921 medium at 27°C, 120 rpm without further validation. HEK293T cells were purchased from the American Type Culture Collection (ATCC, CRL-11268) and used for Gq and β-arrestin-1 recruitment assay in this study. HEK293T cells were grown in a humidified 37°C incubator with 5% CO2 using DMEM medium (VWR, #45000) supplemented with 10% (v/v) fetal bovine serum (FBS, VWR, #89510–186) and 100 I.U./mL penicillin and 100 mg/mL streptomycin. Before cell plating, the DMEM medium were changed from 10% FBS to 1% (v/v) dFBS to remove serotonin. HEK293T cells were authenticated by the supplier (ATCC) using morphology, growth characteristics and STR profiling.
METHODS DETAILS
Constructs for structural studies
To get the structure of HTR2B in complex with β-arrestin-1, the ICL3 and C-terminus of HTR2B were truncated based on BRET1 recruitment assay as noted in the main text. In detail, residues A248-V313 in the ICL3 of HTR2B were removed and replaced with a 7 residues linker “RLLSGSR”. For the C-terminus, the middle part (L408-S444) and the last 17 residues (L465-V481) were removed to make a C-tail chimera. Mutations K2475.68V and E3196.30L were added to the construct to increase the β-arrestin-1 recruitment by LSD. A modified thermostabilized apocytochrome b562RIL (BRIL) as a fusion partner was fused before T36 of HTR2B with M1443.41W mutation adapted from the previous construct (Wacker et al., 2013). β-arrestin-1 isoform 2 with the R169E constitutive mutation was then fused to the C-terminus of HTR2B with a 4 × GSA linker. At last, an engineered scFv30 was directly fused after L368 of β-arrestin-1 to achieve a tandem expression. The HTR2B-β-arrestin-1-scFv30 chimera was cloned into a pFastBac1 vector containing a haemagglutinin (HA) signal sequence followed by FLAG-tag, His10-tag and TEV protease site at the N-terminus.
The HTR2B construct used for transducer-free HTR2B has the same sequence as the receptor portion used for HTR2B-β-arrestin-1-scFv30 chimera, except that it does not have the E3196.30L mutation and the C-terminus is further truncated to C405 according to a previous crystallography construct (Wacker et al., 2013). Fab P2C2 was cloned into a pFastBac-Dual vector with a honeybee signal peptide at the N-terminus of the light chain and a GP67 signal peptide at the N-terminus of the heavy chain (Ishchenko et al., 2017). A 6 × His-tag was added to the C-terminus of heavy chain to facilitate protein purification.
The HTR2B construct used for the HTR2B-Gq complex study contains residues 36–405 and a M1443.41W mutation. A N-terminal BRIL as a fusion partner was added before T36 of HTR2B. The N-bril fused receptor portion was then subcloned into a same vector used for HTR2B-β-arrestin-1-scFv30 complex. Constructs of scFv16 and the heterotrimeric mini-Gαq protein complex were the same as we used in our previous HTR2A-Gq structure (Kim et al., 2020).
Expression and purification of Fab P2C2 and scFv16
Bac-to-Bac expression system was used to generate the baculovirus for all the protein expressions in this study. Before infection, virus titer was determined by flow-cytometric analysis using gp64-PE antibody (Expression systems) stained cells. For Fab P2C2, Sf9 cells at a density of 2 million cells per ml were infected with P1 virus at a multiplicity of infection (MOI) of 3. Supernatant containing the secreted P2C2 was collected at 96 h post-infection. Tris powder was then added to adjust the medium to pH 7.8. Chelating agents were quenched by addition of 1 mM nickel chloride and 5 mM calcium chloride and stirring in cold room for 1 hour. After another centrifugation, 1 ml His60 Ni Superflow Resin (Takara) was added to the supernatant for overnight binding at 4°C. The resin was collected next day and washed with 20 column volumes buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 10 mM imidazole. The protein was eluted with a same buffer containing 250 mM imidazole and further purified by size exclusion chromatography using a Superdex 200 16/60 column (GE healthcare). The peak fractions were collected and concentrated to 1 mg ml-1 for future use. scFv16 was expressed and purified using the same protocol.
Protein expression and purification of HTR2B-β-arrestin-1-scFv30 complex
For HTR2B-β-arrestin-1-scFv30 chimera, Sf9 cells at a density of 2 million cells per were infected with P1 virus of HTR2B-β-arrestin-1-scFv30 and GRK2 at a multiplicity of infection (MOI) ratio of 3:1.5. Cells were harvested by centrifugation at 48 h post-infection. The cell pellet was then washed with a low-salt buffer containing 10 mM HEPES pH 7.5, 10 mM MgCl2 and 20 mM KCl and proteinase inhibitor containing 500 mM AEBSF, 1 mM E-64, 1 mM Leupeptin and 0.15 mM Aprotinin. Subsequently, two rounds of high-salt wash were performed using a buffer containing 10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl and 500 mM NaCl to remove membrane associated proteins. Purified cell membranes were resuspended in low-salt buffer and incubated with 10 μM LSD and 200 μM TCEP for 30 min at room temperature. After additional 1 hour incubation in cold room, the cell membranes were solubilized using solubilization buffer at a final concentration of 30 mM HEPES, 100 mM NaCl, 10 % glycerol, 5 μM LSD, 100 μM TCEP and 0.6% (w/v) n-dodecyl-beta-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma) at 4°C for 2.5 h. The solubilized proteins in the supernatants were isolated by ultra-centrifugation at 40,000 rpm for 30 min, and then incubated at 4°C overnight with 1 ml TALON IMAC resin (Clontech) and 20 mM imidazole. The resin was collected next day and washed with 10 column volumes buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 30 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% glycerol, 1 μM LSD and 100 μM TCEP. The resin was then incubated with 20 mM HEPES pH 7.5, 100 mM NaCl, 0.8% Lauryl Maltose Neopentyl Glycol (LMNG, Anatrace), 0.27% GDN (Anatrace), 0.08% CHS, 5% glycerol, 1 μM LSD and 100 μM TCEP to exchange the detergent from DDM/CHS to LMNG/GDN/CHS. The resin was washed with additional 15 column volumes buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 0.01% LMNG, 0.033% GDN, 0.001% CHS, 5% glycerol, 1 μM LSD, 100 μM TCEP and 30 mM imidazole. The protein was then eluted with 4.5 column volumes buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 0.01% LMNG, 0.033% GDN, 0.001% CHS, 5% glycerol, 5 μM LSD, 100 μM TCEP and 250 mM imidazole. Eluted protein was concentrated to 400 μl and incubated with 100 μl P2C2 Fab (1 mg/ml) at 4°C for 2 h before subjected to size-exclusion chromatography on a Superdex 200 Increase 10/300 column (GE Healthcare) that was pre-equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, 1 μM LSD, 100 μM TCEP, 0.001% (w/v) MNG, 0.00033 (w/v) GDN and 0.0001% (w/v) CHS. Peak fractions were then collected and concentrated to 3.5 mg ml-1 to make the cryoEM grids.
Protein expression and purification of HTR2B-P2C2 complex
For the transducer-free HTR2B-P2C2 complex, Sf9 cells at a density of 2 million cells per were infected with receptor P1 virus at a multiplicity of infection (MOI) of 3. Cells were harvested by centrifugation at 48 h post-infection. Followed by a low-salt wash, the cell membranes were washed twice by high-salt buffer containing 10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl and 1 M NaCl. The membrane was solubilized and binding overnight using a same protocol as HTR2B-β-arrestin-1-scFv30, except TCEP was not added. The resin was collected next day and washed with 25 column volumes buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 30 mM imidazole, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% glycerol and 1 μM LSD. The protein was then eluted using a same buffer containing 250 mM imidazole. Eluted protein was concentrated to 350 μl and incubated with 150 μl P2C2 Fab (1 mg/ml) at 4°C for 2 h before subjected to size-exclusion chromatography on a Superdex 200 Increase 10/300 column (GE Healthcare) that was pre-equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, 1 μM LSD, 0.001% (w/v) MNG, 0.00033 (w/v) GDN and 0.0001% (w/v) CHS. Peak fractions were then collected and concentrated to 6.5 mg ml−1 to make the cryoEM grids.
Protein expression and purification of HTR2B-Gq complex
The cell pellet of HTR2B and Gq complex from 2 L culture was thawed at room temperature and resuspended in law salt buffer containing 10 mM HEPES pH 7.5, 50 mM NaCl, 20 mM KCl, and protease inhibitor 500 μM AEBSF, 1 μM E-64, 1 μM Leupeptin and 0.15 μM Aprotinin. The HTR2B and Gq complexes were formed on the membrane in the presence of 5 μM LSD, followed by incubation for 1.5 h at room temperature. Cell membranes were collected by ultra-centrifugation at 40,000 rpm for 25 min. The membranes were then resuspended and solubilized in solubilization buffer containing 20 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 20 mM imidazole, 0.1 mM TCEP, 10%(v/v) glycerol, 0.5% DDM, 0.1%(w/v) CHS, 10 μM LSD, and protease inhibitor cocktail at 4°C. After 3 h incubation, the supernatant was isolated by centrifugation at 70,000 rpm for 50 min and then incubated overnight with pre-equilibrated TALON resin (CLONTECH) at 4°C. The resin was collected and washed with 30 column volumes with washing buffer 20 mM HEPES, pH 7.5, 100 mM NaCl, 30 mM imidazole, 0.1 %(w/v) LMNG, 0.01 % (w/v) CHS and 10 μM LSD. The protein was then eluted using the same buffer supplemented with 300 mM imidazole. 500 μl of protein sample was applied to PD MiniTrap G-25 columns (GE Healthcare) to remove imidazole and change the detergent to LMNG with buffer containing 20 mM HEPES, pH 7.5, 100 mM NaCl, 0.5% (w/v) LMNG, 0.05% (w/v) CHS, 0.00025% (w/v) GDN, 100 μM TCEP, and 20 μM LSD. The N-terminal BRIL protein was removed by the addition of His-tagged PreScission protease (Genescript) and incubation overnight at 4 °C. Unnecessary his-tagged proteins such as protease, BRIL, and free his tag were removed by equilibrated TALON resin, and the flow-through was collected. The eluate protein was concentrated and subjected to size-exclusion chromatography on a Superdex 200 Increase 10/300 column (GE Healthcare) that was pre-equilibrated with 20 mM HEPES, pH 7.5, 100 mM NaCl, 10 μM LSD (agonist), 0.00075% (w/v) LMNG, 0.00025% (w/v) GDN, and 0.000075% (w/v) CHS. The peak fractions of complex were pooled and concentrated to 8.5 mg/ml for electron microscopy experiments.
Cryo-EM data acquisition and processing for HTR2B-LSD complexes
Particles from two cryo-EM data collections of the same sample vitrified on UltrAuFoil holey gold (Quantifoil, Au300-R1.2/1.3) or Quantifoil holey carbon (R1.2/1.3) grids contributed to the HTR2B/β-arrestin-1 complex reconstruction presented here. For this, 3 μl of purified HTR2B/β-arrestin-1 complex at a concentration of 3.5 mg/mL were applied to the glow-discharged (45–50 seconds at 10–15 mA) grids in 100% humidity at 18°C. Samples were blotted for 1 second and plunged-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Cryo-EM imaging was performed on a Titan Krios (ThermoFisher) electron microscope operated at 300 kV with a K3 Summit direct electron detector (Gatan) at a magnification of 57,050 × (0.8521 Å/pixel) in counting mode using SerialEM (Mastronarde, 2005). The collection on holey gold or carbon grids generated 6,217 movies and 1,533 movies, respectively, dose fractioned over 50 frames, recorded for 0.05 sec/frame for a total dose of 68 electrons/Å2 in super-resolution mode with a defocus range of 0.8–1.8 μm, for a total of 7,750 movies. Cryo-EM data processing was performed with cryoSPARC (Punjani et al., 2017). 3 μl of purified HTR2B-LSD/miniGq or HTR2B-LSD/Fab complexes at concentrations of 8.5 mg/mL or 6.5 mg/mL, were applied to glow-discharged (45–50 seconds at 10–15 mA) UltrAuFoil holey gold grids (Quantifoil, Au300-R1.2/1.3) in 100% humidity at 18°C or 4°C, respectively. Samples were blotted for 1 second and plunged-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). Cryo-EM imaging was performed on a Titan Krios (ThermoFisher) electron microscope operated at 300 kV with a K3 Summit direct electron detector (Gatan) at a magnification of 55,000 × (0.8677 Å/pixel) in counting mode using SerialEM (Mastronarde, 2005). For the HTR2B-LSD/miniGq/i complex, 3,493 movies, dose fractioned over 57 frames, were recorded for 0.044 sec/frame for a total dose of 61 electrons/Å2 in super-resolution mode with a defocus range of 0.8–1.8 μm. For HTR2B-LSD/Fab, 12,303 movies dose fractioned over 50 frames, were recorded for 0.05 sec/frame for a total dose of 61.5 electrons/Å2 in super-resolution mode with a defocus range of 0.7–1.8 μm.
Initial sets of 8,491,352 and 3,214,021 particles were selected and subjected to multiple 2D and 3D classification rounds for HTR2B-LSD/Fab or HTR2B-LSD/miniGq complexes, respectively, using cryoSPARC (Punjani et al., 2017). Subsets of 665,475 and 772,614 particles contributing to the HTR2B-LSD/Fab and HTR2B-LSD/miniGq reconstructions, went through global CTF refinement, non-uniform and homogeneous refinement. The HTR2B-LSD/Fab map was further refined locally obtaining a 2.7 Å resolution map for the LSD bound receptor. The HTR2B-LSD/miniGq global map was refined locally obtaining separate 2.9 Å resolution maps for the LSD bound receptor and the G protein. Maps resulting from the local refinements for the HTR2B-LSD or HTR2B-LSD/miniGq complexes were sharpened using DeepEMhancer (Sanchez-Garcia et al., 2021). A total of 4,415,425 particles, 3,544,745 from holey gold and 870,680 from holey carbon grids, were extracted from the corrected 7,750 micrographs for the HTR2B/β-arrestin-1 complex. After 2D and 3D classification, a subset of 126,485 particles were subjected to homogeneous refinement followed by local refinements of the LSD bound receptor (with an scFv fragment of the HTR2B Fab P2C2 used for particle alignment) and β-arrestin-1 at a resolution of 3.3 Å. Maps resulting from local refinements for HTR2B-LSD/miniGq or HTR2B/β-arrestin-1 were combined in Chimera (Pettersen et al., 2004). Flowcharts describing data processing steps are presented in the Figures S2–S4.
Model building and refinement
Coordinates for HTR2B-LSD (PDB ID: 5TVN) (Wacker et al., 2017), miniGq (PDB: 6WHA) (Kim et al., 2020) and β-arrestin-1 (PDB: 6UP7) (Huang et al., 2020) were used as an initial models for docking into the EM density maps using Chimera (Pettersen et al., 2004). Models were subjected to iterative rounds of manual refinement in Coot (Emsley et al., 2010) and real-space refinement in Phenix. Validation of cryo-EM maps and models was performed with Phenix (Liebschner et al., 2019) comprehensive cryo-EM validation. Model statistics were validated with Molprobity (Chen et al., 2010) and final refinement statistics are provided in Supplemental Table 1. Map/model visualizations and figure preparation were done in UCSF Chimera (Pettersen et al., 2004) and ChimeraX (Pettersen et al., 2021).
Bioluminescence resonance energy transfer assays (BRET1)
For the HTR2B-mediated b-arrestin1 and miniGq recruitment assays, HEK293T cells were co-transfected with C-terminal RLuc8-tagged human HTR2B, and either N-terminal Venus-tagged β-arrestin-1 in a 1:8 ratio or N-terminal Venus-tagged miniGq in a 1:4 ratio. After 8 hours, transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in plating media (DMEM + 1% (v/v) dialyzed FBS) at a density of 25–50,000 cells in 200 μl per well and incubated overnight. The next day, media was decanted and cells were washed with 60 μl of drug buffer (1xHBSS, 20 mM HEPES, 0.1% (w/v) BSA, 0.01% (w/v) ascorbic acid, pH 7.4). Afterward, 60 μl of drug buffer and 30 μl of the drug (3X) were added per well, and then the plates were transferred to the 37 °C incubator and kept for 20 minutes. Before reading, 10 μl of the coelenterazine h (Promega, final concentration is 5 mM) was added per well and the plate was incubated for an additional 10 minutes to allow for the substrate diffusion. Plates were read for both luminescence at 475 nm and fluorescent eYFP emission at 535 nm for 1 s per well using a PHERAstar FSX multimode microplate reader. The ratio of eYFP/RLuc was calculated per well and analyzed in Graphpad Prism 9 (Graphpad Software Inc., San Diego, CA). Mutagenesis data were normalized to WT stimulation and reanalyzed using nonlinear regression ‘log(agonist) vs. response’ in GraphPad Prism 9. The protein expression of WT and mutant HTR2B measured by the Rluc8 counts of each construct are shown in Figure S7.
Mass spectrometry analysis
For the protein digestion, 100 μg HTR2B-β-arrestin-1-scFv30 protein was denatured in 2 M urea, 100 mM Tris-HCl (pH 8.0), followed by reduction in 5 mM dithiothreitol (DTT) at 37°C for 30min and alkylation in 10 mM iodoacetamide in the dark for 30 min. The protein mixture was digested with 2 μg Trypsin at 37°C for 6 h and additional 1 μg Asp-N for overnight digestion at 37°C. The digestion reaction was quenched by a final 0.5% trifluoroacetic acid (TFA) and desalted using SepPak C18 cartridges (Waters, Milford, MA). To achieve Phosphopeptide enrichment, 100 μl (20 μL per sample) of Ni-NTA magnetics beads (QIAGEN) was washed with 3 × 100 μl H2O, incubated with 100 μl of 50 mM EDTA (pH 8.0) for 30 min, washed with 3 × 100 μl H2O, incubated with 100 μl of 50 mM FeCl3 for 30 min, and washed with 3 × 100 μl 0.1% TFA in 80% acetonitrile (ACN). Beads were resuspended in 150 μl of 80% ACN/0.1% TFA. 100 μg protein digests were resuspended in 150 μl 80% ACN/0.1% TFA and incubated with equilibrated beads for 30 min. Beads were washed with 3 × 150 μl 80% ACN/0.1% TFA. Phosphopeptides were eluted with 50 μl of 50% ACN/0.75% ammonium hydroxide and acidified with 30 μl of 75% ACN/10% formic acid. The samples were then analyzed on an Orbitrap Exploris 480 mass spectrometry system (Thermo Fisher Scientific) equipped with an Easy nLC 1200 ultrahigh pressure liquid chromatography system (Thermo Fisher Scientific). Digested peptide samples were loaded onto a C18 reverse phase column (15 cm × 75 μm packed with BEH 1.7 μm particles). Mobile phase A consisted of 0.1% formic acid (FA) and mobile phase B consisted of 0.1% FA/80% ACN. Peptides were separated by an organic gradient from 4% to 16% mobile phase B over 30 minutes, followed by an increase to 28% B over 20 minutes and 44% B over 10 min, then held at 90% B for 8 minutes at a flow rate of 300 nl/min. FTMS survey scans of peptide precursors from 350 to 1250 m/z were performed in the Orbitrap at 120K resolving power with a normalized AGC target of 300%, an RF lens setting of 40%, and auto maximum injection time. Using a data-dependent acquisition mode, the 20 most abundant ions at charge states 2–6 were fragmented by higher energy collisional dissociation (HCD) with an isolation width of 1.6 Da, a normalized collision energy (NCE) of 30%, a normalized AGC target of 250% at a resolving power of 15,000 with a maximum injection time of 40 ms. The proteomic data was searched against the human UniProt database augmented with the sequence of beta-arrestin 1 fused to HTR2B. Peptide and protein identification were searched using the default setting in MaxQuant (version 1.6.12.0) with variable modification of methionine oxidation and phosphorylation of serine, threonine, and tyrosine, and static modification of cysteine carbamidomethylation. All peptide and protein identification were filtered to a 1% false discovery rate. The phosphorylation probabilities of HTR2B C-tail residues are shown in Table S5. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD030752.
Molecular Dynamics (MD) Simulations
Simulation setup:
We performed simulations in 5 distinct conditions: (A) the HTR2B/β-arrestin-1/LSD complex (12 independent simulations, roughly 2 μs each), (B) the HTR2B/LSD transducer-free receptor (12 independent simulations, roughly 2 μs each), (C) the HTR2B/Gq/LSD complex (12 independent simulations, roughly 3 μs each), (D) the HTR2B/LSD receptor from the HTR2B/β-arrestin-1/LSD complex, but with β-arrestin-1 removed (12 independent simulations, roughly 2 μs each) and (E) the HTR2B/LSD receptor from the HTR2B/Gq /LSD complex, but with Gq removed (12 independent simulations, roughly 2 μs each). For all conditions, the initial structures were based on the three cryoEM structures reported in this paper and were prepared using Maestro (Schrödinger, LLC).
Condition A was prepared as follows. The scFv30 and Fab nanobodies were removed from the initial structure. The HTR2B construct mutations M1443.41W, K2475.68V, and E3196.30L as well as the β-arrestin-1 construct mutation R169E were reverted to WT. The unresolved residues in the ICL3 replacement (RLLSGSR) of HTR2B construct G252 and S253, the unresolved residues that connect helix 8 with the resolved part of the C-tail in the HTR2B construct (NYRATKSVKTPMRLRSST) as well as the unresolved residue G137 in β-arrestin-1 were modeled using Maestro’s ‘crosslink’ tool.
Condition B was prepared as follows. The Fab P2C2 nanobody used for structure determination was not included in the initial structure. The HTR2B construct mutations M1443.41W and K2475.68V were reverted to WT. The unresolved residues of ICL2 (I16134.51-Q16234.52-A16334.53-N16434.54-Q16534.55-Y16634.56-N16734.57) were modeled based on the HTR2B/LSD crystal structure (PDB ID: 5TVN) (Wacker et al., 2017). To achieve a smooth transition between the intracellular tips of TM3 and TM4 with ICL2, K1593.56-P16034.50 of TM3 and S1684.38-R1694.39 of TM4 in the transducer-free cryoEM structure were replaced by the corresponding residues in the HTR2B/LSD crystal structure (PDB ID: 5TVN). The unresolved residues of the intracellular tips of TM5 and TM6 as well as the connecting ICL3 replacement (RLLSGSR) were modeled as follows: L2445.65-Q2455.66-K2465.67 and N3186.29-E3196.30-Q3206.31-R3216.32 were modeled based on the HTR2B/LSD crystal structure (PDB ID: 5TVN). To achieve a smooth transition, A2435.64 and A3226.33-S3236.34 in the transducer-free cryoEM structure were replaced by the corresponding residues in the HTR2B/LSD crystal structure (PDB ID: 5TVN). The residues K2475.68-R248-L249-L250-S251-G252-S253-R254-Q3146.25-T3156.26-I3166.27-S3176.28, which include the ICL3 replacement (RLLSGSR) were modeled based on the HTR2B/LSD/β-arrestin-1 cryoEM structure (including modeling G252 and S253 with Maestro’s ‘crosslink’ tool and reverting the construct mutation K2475.68V to WT). The unresolved C-terminal residues I3958.58-T396-C397 of helix8 were modeled based on the HTR2B/LSD crystal structure (PDB ID: 5TVN).
Condition C was prepared as follows. The scFv16 nanobody was removed from the initial structure. The missing parts of the N-terminus of the Gα subunit and the C-terminus of the Gγ subunit were modeled in and lipidated as follows: G2 of the Gα subunit was myristoylated, C3 of the Gα subunit was palmitoylated, and C68 of the Gγ subunit was prenylated (Wedegaertner et al., 1995). The unresolved residues G88-Q89 in the Gα subunit were modeled with Maestro’s ‘crosslink’ tool. The HTR2B construct mutation M1443.41W was reverted to WT. The HTR2B/LSD-Gq construct contains the WT ICL3, which is not resolved in the cryoEM map. For consistency, we included the ICL3 replacement also in the HTR2B/LSD-Gq complex for simulations and modeled it based on the HTR2B/LSD-β-arrestin-1 structure. More specifically, Q2455.66-K2465.67-K2475.68-R248-L249-L250-S251-G252-S253-R254-Q3146.25-T3156.26-I3166.27-S3176.28-N3186.29, were modeled based on the HTR2B/LSD/β-arrestin-1 cryoEM structure (including modeling G252 and S253 with Maestro’s ‘crosslink’ tool and reverting the construct mutation K2475.68V to WT). Moreover, the C-terminal residues of helix 8 (G3928.55-R3938.56-Y3948.57-I3958.58-T396-C397) were also modeled based on the HTR2B/LSD/β-arrestin-1 cryoEM structure to include the palmitoylated membrane anchor residue C397.
Condition D was obtained from condition A by removing β-arrestin-1.
Condition E was obtained from condition C by removing Gq.
For all simulation conditions, a palmitoyl group was added to residue C397 of the receptor using Maestro (Schrödinger, LLC). Missing amino acid side chains were modeled using Prime (Schrödinger, LLC). Neutral acetyl and methylamide groups were added to cap the N- and C-termini, respectively, of the protein chains, except for the N-myristoylated N-terminus of the Gα subunit and the natural C-termini of the Gα and Gβ subunit, which were carboxylated. Titratable residues were kept in their dominant protonation state at pH 7, except for D1002.50 and D1523.49, which were protonated to their neutral form, as studies indicate that these conserved residues are protonated in active class-A GPCRs (Ghanouni et al., 2000, Ranganathan et al., 2014). Histidine residues were modeled as neutral, with a hydrogen atom bound to either the delta or epsilon nitrogen depending on which tautomeric state optimized the local hydrogen-bonding network. Dowser (Zhang and Hermans, 1996) was used to add water molecules to protein cavities. The LSD tertiary amine nitrogen was protonated, corresponding to the dominant protonation state at pH 7.0 and enabling formation of the conserved salt bridge with neighboring D1353.32. The receptor in the HTR2B/LSD transducer-free system and the HTR2B/LSD-β-arrestin-1 system was aligned on the receptor in the crystal structure of HTR2B/LSD (PDB ID: 5TVN) in the Orientation of Proteins in Membranes (OPM) database (Lomize et al., 2006), the receptor of the HTR2B/LSD-Gq system was aligned on the receptor in the HTR2A/NBOH-Gq cryoEM structure (PDB ID: 6WHA) (Kim et al., 2020) in the OPM database. The aligned structures were inserted into a pre-equilibrated palmitoyloleoyl-phosphatidylcholine (POPC) membrane bilayer using Dabble (RM, 2017). Sodium and chloride ions were added to neutralize each system at a concentration of 150 mM.
The final system of condition A comprised 218,598 atoms, including 365 lipid molecules and 52,858 water molecules (initial system dimensions: 120 Å × 120 Å × 150 Å). The final system of condition B comprised 62,370 atoms, including 150 lipid molecules and 12,505 water molecules (initial system dimensions: 99 Å × 69 Å × 93 Å). The final system of condition C comprised 290,075 atoms, including 464 lipid molecules and 70,953 water molecules (initial system dimensions: 139 Å × 123 Å × 166 Å). The final system of condition D comprised 59,036 atoms, including 141 lipid molecules and 11,797 water molecules (initial system dimensions: 79 Å × 84 Å × 92 Å). The final system of condition E comprised 54,146 atoms, including 126 lipid molecules and 10,839 water molecules (initial system dimensions: 74 Å × 82 Å × 92 Å).
Simulation protocols:
For each simulation, initial atom velocities were assigned randomly and independently. We employed the CHARMM36m force field for protein molecules, the CHARMM36 parameter set for lipid molecules and salt ions, and the associated CHARMM TIP3P model for water (Huang et al., 2017, Klauda et al., 2010). Parameters for LSD were taken from (Wacker et al., 2017). Simulations were run using the AMBER20 software (D.A. Case, 2021) under periodic boundary conditions with the Compute Unified Device Architecture (CUDA) version of Particle-Mesh Ewald Molecular Dynamics (PMEMD) on one GPU (Salomon-Ferrer et al., 2013).
After energy minimization, the systems were first heated over 12.5 ps from 0 K to 100 K in the NVT ensemble using a Langevin thermostat with harmonic restraints of 10.0 kcal∙mol−1∙Å−2 on the non-hydrogen atoms of the lipids, protein, and ligand. Initial velocities were sampled from a Boltzmann distribution. The systems were then heated to 310 K over 125 ps in the NPT ensemble. Equilibration was performed at 310 K and 1 bar in the NPT ensemble, with harmonic restraints on the protein and ligand non-hydrogen atoms tapered off by 1.0 kcal∙mol−1∙Å−2 starting at 5.0 kcal∙mol−1∙Å−2 in a stepwise manner every 2 ns for 10 ns, and finally by 0.1 kcal∙mol−1∙Å−2 every 2 ns for an additional 18 ns. All restraints were completely removed during production simulation. Production simulations were performed at 310 K and 1 bar in the NPT ensemble using the Langevin thermostat and Monte Carlo barostat. Lengths of bonds to hydrogen atoms were constrained using SHAKE, and the simulations were performed using a timestep of 4.0 fs while using hydrogen mass repartitioning (Hopkins et al., 2015). Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method with an Ewald coefficient (β) of approximately 0.31 Å and B-spline interpolation of order 4. The PME grid size was chosen such that the width of a grid cell was approximately 1 Å. Snapshots of the trajectory were saved every 200 ps.
Simulation analysis protocols
The AmberTools17 CPPTRAJ package (Roe and Cheatham, 2013) was used to reimage trajectories at 1 ns per frame, Visual Molecular Dynamics (VMD) (Humphrey et al., 1996) was used for visualization and analysis, and PyMOL (The PyMOL Molecular Graphics System, Schrödinger, LLC) was used for renderings.
Plots of time traces from representative simulations were generated with Matplotlib (Hunter, 2007) and show both original, unsmoothed traces (transparent lines) and traces smoothed with a moving average (thick lines), using an averaging window of 20 ns. All traces include the initial equilibration with harmonic restraints on the protein and ligand non-hydrogen atoms.
QUANTIFICATION AND STATISTICAL ANALYSIS
For the β-arrestin-1 and miniGq recruitment assay, the data was analyzed in Graphpad Prism 9.0 using nonlinear regression “log(agonist) vs. response”. Data in figures and tables are reported as mean ± standard error of the mean (SEM) with the number of biological and technical replicates indicated in the figure and table legends where “n” represents the number of biological replicates performed.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| gp64-PE antibody | Expression Systems | Cat#97-201, RRID:AB_2922960 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AEBSF | GoldBio | Cat#A-540-5 |
| Leupeptin | Sigma | Cat#L2884 |
| E-64 | AG Scientific | Cat# E-2030-25MG |
| Aprotinin | GoldBio | Cat#A-655-100 |
| n-dodecyl-beta-D-Maltopyranoside (DDM) | Anatrace | Cat#D310 |
| Lauryl maltose neopentyl glycol (LMNG) | Anatrace | Cat#NG310 |
| Glyco-Diosgenin (GDN) | Anatrace | Cat#GDN101 |
| Cholesteryl Hemisuccinate Tris Salt | Anatrace | Cat#CH210 25GM |
| His-tagged PreScission protease | GenScript | Cat#Z0392-500 |
| Tris HCl | VWR | Cat#0497 |
| HEPES | Fisher Scientific | Cat#BP310 |
| Glycerol | Fisher Scientific | Cat#BP229 |
| Imidazole | Sigma | Cat#I0250 |
| Sodium Chloride | Fisher | Cat#BP358 |
| Potassium Chloride | Sigma | Cat#793590 |
| Magnesium Chloride | Sigma | Cat#M0250 |
| TALON IMAC resin | Clontech | Cat#635507 |
| Coelenterazine-h | Promega | Cat#S2011 |
| Poly-L-lysine | Sigma | Cat#P2636 |
| Penicillin/Streptomycin | Invitrogen | Cat#15140-122 |
| Sf-900 II SFM | Invitrogen | Cat#10902096 |
| ESF921 | Expression Systems | Cat#96-001-01 |
| DMEM | Sigma | Cat# D6429-6X50 |
| USDA FBS | Omega Scientific | Cat#2463 FB-01 |
| Dialyzed FBS | Omega Scientific | Cat#2459 FB-03 |
| 10xHBSS | Invitrogen | Cat#14065-056 |
| BSA, Free Fatty Acid | Akron | Cat#AK8909 |
| Ascorbic Acid | Sigma | Cat#A5960 |
| Cellfectin II Reagent | Invitrogen | Cat#10362-100 |
| TransIT-2020 | Mirus | Cat#MIR5400 |
| Critical Commercial Assays | ||
| Bac-to-Bac Baculovirus Expression system | Invitrogen | Cat#A11100 |
| pcDNA 3.1 | Thermo Fisher | Cat#V79020 |
| Deposited Data | ||
| HTR2B/β-arrestin-1 | This paper | PDB: 7SRS |
| HTR2B-LSD/miniGq | This paper | PDB: 7SRR |
| HTR2B-LSD | This paper | PDB: 7SRQ |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda Sf9 cells | Expression Systems | Cat#94-001S |
| HEK293T | ATCC | Cat#CRL-11268 |
| Recombinant DNA | ||
| Human HTR2B gene | Integrated DNA Technologies (IDT) | N/A |
| miniGq chimera | Integrated DNA Technologies (IDT) | N/A |
| Gβ1 subunit | Integrated DNA Technologies (IDT) | N/A |
| Gγ2 subunit | Integrated DNA Technologies (IDT) | N/A |
| P2C2 Fab | Integrated DNA Technologies (IDT) | N/A |
| scFv16 | Integrated DNA Technologies (IDT) | N/A |
| pFastBac dual expression vector | Invitrogen | Cat#10712024 |
| pFastBac 1 expression vector | Invitrogen | Cat#10359016 |
| Software and Algorithms | ||
| SerialEM | (Mastronarde, 2005) | https://bio3d.colorado.edu/SerialEM/ |
| cryoSPARC | (Punjani et al., 2017) | https://cryosparc.com/ |
| COOT | (Emsley et al., 2010) | www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Chimera X | (Pettersen et al., 2021) | https://www.rbvi.ucsf.edu/chimerax/ |
| Phenix | (Liebschner et al., 2019) | https://www.phenix-online.org |
| PyMOL | Schrödinger | https://www.pymol.org/ |
| Prism v9.0 | GraphPad Software Inc. | N/A |
| Adobe Illustrator CC | Adobe | www.adobe.com |
| Other | ||
| Poly-Prep Chromatography Columns | Bio-Rad | Cat#731-1550 |
| PD MiniTrap G-25 columns | GE Healthcare | Cat#28-9180-07 |
| 100 kDa molecular weight cut-off Vivaspin 20 concentrator |
Sartorius Stedim | Cat#VS2042 |
| 100 kDa molecular weight cut-off Vivaspin 500 centrifuge concentrator |
Sartorius Stedim | Cat#VS0142 |
| Superdex 200 Increase 10/300 column | GE healthcare | Cat#289909944 |
| 96-well black plates | Greiner Bio-one GmbH | Cat#655090 |
| 96-well white plates | Greiner Bio-one GmbH | Cat#655098 |
| Amicon Ultra - 4 10K filter unit | Merck Millipore | Cat#UFC801024 |
| Amicon Ultra - 4 30K filter unit | Merck Millipore | Cat#UFC803024 |
| Quantifoil, Au300-R1.2/1.3 | ELECTRON MICROSCOPY SCIENCES | Cat#Q350AR13A |
| Quantifoil holey carbon (R1.2/1.3) | ELECTRON MICROSCOPY SCIENCES | Cat#Q250AR1.3 |
Highlights:
LSD-bound HTR2B with transducer-free, G protein-coupled and β-arrestin-1 structures
LSD-induced interactions between β-arrestin-1 and HTR2B revealed
TM6 of HTR2B has a larger outward movement for the coupling of β-arrestin-1
LSD-induced transducer coupling requires conformational changes of ICL2 and ICL3
ACKNOWLEDGMENTS
We thank Elizabeth Montabana at the Stanford cEMc facility for support with data collection. M.A.D. was supported by a Walter Benjamin Fellowship from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 471315262. C.-M.S. is supported by a Long-Term Fellowship (LT000916/2018-L) from the Human Frontier Science Program (HFSP). R.O.D., G.S. and B.L.R are supported by Defense Advanced Research Projects Agency (DARPA) under HR0011–20–2–0029. N.J.K. and R.H. are supported by the Defense Advanced Research Projects Agency (DARPA) under the Cooperative Agreements HR0011–19–2–0020 and HR0011–20–2–0029. The views, opinions, and/or findings contained in this material are those of the authors and should not be interpreted as representing the official views, policies, or endorsement of the Department of Defense or the US Government. An award of computer time was provided by the INCITE program. This research used resources of the Oak Ridge Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC05–00OR22725. B.L.R. and others in the Roth lab were also supported by the Michael Hooker Distinguished Professorship and NIH grants R37DA045657 and RO1MH112205. This work was also supported by NIH grant R01GM127359 (R.O.D.).
Footnotes
DECLARATION OF INTERESTS
The Krogan Laboratory has received research support from Vir Biotechnology, F. Hoffmann-La Roche, and Rezo Therapeutics. Nevan Krogan has financially compensated consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics, Interline Therapeutics, Rezo Therapeutics, GEn1E Lifesciences, Inc. and Twist Bioscience Corp. He is on the Board of Directors of Rezo Therapeutics and is a shareholder in Tenaya Therapeutics, Maze Therapeutics, Rezo Therapeutics, and Interline Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- BALLESTEROS JA & WEINSTEIN H 1995. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: SEALFON SC (ed.) Methods in Neurosciences. Academic Press. [Google Scholar]
- BONDE MM, HANSEN JT, SANNI SJ, HAUNSO S, GAMMELTOFT S, LYNGSO C & HANSEN JL 2010. Biased signaling of the angiotensin II type 1 receptor can be mediated through distinct mechanisms. PLoS One, 5, e14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN VB, ARENDALL WB 3RD, HEADD JJ, KEEDY DA, IMMORMINO RM, KAPRAL GJ, MURRAY LW, RICHARDSON JS & RICHARDSON DC 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASE DA, H. M. A., BELFON K, BEN-SHALOM IY, BROZELL SR, CERUTTI DS, CHEATHAM TE III, CISNEROS GA, CRUZEIRO VWD, DARDEN TA, DUKE RE, GIAMBASU G, GILSON MK, GOHLKE H, GOETZ AW, HARRIS R, IZADI S, IZMAILOV SA, JIN C, KASAVAJHALA K, KAYMAK MC, KING E, KOVALENKO A, KURTZMAN T, LEE TS, LEGRAND S, LI P, LIN C, LIU J, LUCHKO T, LUO R, MACHADO M, MAN V, MANATHUNGA M, MERZ KM, MIAO Y, MIKHAILOVSKII O, MONARD G, NGUYEN H, O’HEARN KA, ONUFRIEV A, PAN F, PANTANO S, QI R, RAHNAMOUN A, ROE DR, ROITBERG A, SAGUI C, SCHOTT-VERDUGO S, SHEN J, SIMMERLING CL, SKRYNNIKOV NR, SMITH J, SWAILS J, WALKER RC, WANG J, WEI H, WOLF RM, WU X, XUE Y, YORK DM, ZHAO S, AND KOLLMAN PA (2021), AMBER 2021, UNIVERSITY OF CALIFORNIA, SAN FRANCISCO. 2021. Amber. University of California, San Francisco. [Google Scholar]
- DWIVEDI-AGNIHOTRI H, CHATURVEDI M, BAIDYA M, STEPNIEWSKI TM, PANDEY S, MAHARANA J, SRIVASTAVA A, CAENGPRASATH N, HANYALOGLU AC, SELENT J & SHUKLA AK 2020. Distinct phosphorylation sites in a prototypical GPCR differently orchestrate beta-arrestin interaction, trafficking, and signaling. Sci Adv, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EICHEL K, JULLIE D, BARSI-RHYNE B, LATORRACA NR, MASUREEL M, SIBARITA JB, DROR RO & VON ZASTROW M 2018. Catalytic activation of beta-arrestin by GPCRs. Nature, 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMSLEY P, LOHKAMP B, SCOTT WG & COWTAN K 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLOCK T, RAVARANI CNJ, SUN D, VENKATAKRISHNAN AJ, KAYIKCI M, TATE CG, VEPRINTSEV DB & BABU MM 2015. Universal allosteric mechanism for Galpha activation by GPCRs. Nature, 524, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GELBER EI, KROEZE WK, WILLINS DL, GRAY JA, SINAR CA, HYDE EG, GUREVICH V, BENOVIC J & ROTH BL 1999. Structure and function of the third intracellular loop of the 5-hydroxytryptamine2A receptor: the third intracellular loop is alpha-helical and binds purified arrestins. J Neurochem, 72, 2206–14. [DOI] [PubMed] [Google Scholar]
- GHANOUNI P, SCHAMBYE H, SEIFERT R, LEE TW, RASMUSSEN SG, GETHER U & KOBILKA BK 2000. The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J Biol Chem, 275, 3121–7. [DOI] [PubMed] [Google Scholar]
- GUREVICH VV & GUREVICH EV 2006. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther, 110, 465–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HE QT, XIAO P, HUANG SM, JIA YL, ZHU ZL, LIN JY, YANG F, TAO XN, ZHAO RJ, GAO FY, NIU XG, XIAO KH, WANG J, JIN C, SUN JP & YU X 2021. Structural studies of phosphorylation-dependent interactions between the V2R receptor and arrestin-2. Nat Commun, 12, 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFER A 1965. D-Lysergic Acid Diethylamide (Lsd): A Review of Its Present Status. Clin Pharmacol Ther, 6, 183–255. [DOI] [PubMed] [Google Scholar]
- HOFMANN A 1979. How LSD originated. J Psychedelic Drugs, 11, 53–60. [DOI] [PubMed] [Google Scholar]
- HOPKINS CW, LE GRAND S, WALKER RC & ROITBERG AE 2015. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J Chem Theory Comput, 11, 1864–74. [DOI] [PubMed] [Google Scholar]
- HUANG J, RAUSCHER S, NAWROCKI G, RAN T, FEIG M, DE GROOT BL, GRUBMULLER H & MACKERELL AD JR. 2017. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG W, MASUREEL M, QU Q, JANETZKO J, INOUE A, KATO HE, ROBERTSON MJ, NGUYEN KC, GLENN JS, SKINIOTIS G & KOBILKA BK 2020. Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature, 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUMPHREY W, DALKE A & SCHULTEN K 1996. VMD: visual molecular dynamics. J Mol Graph, 14, 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
- HUNTER JD 2007. Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering, 9, 90–95. [Google Scholar]
- ISHCHENKO A, WACKER D, KAPOOR M, ZHANG A, HAN GW, BASU S, PATEL N, MESSERSCHMIDT M, WEIERSTALL U, LIU W, KATRITCH V, ROTH BL, STEVENS RC & CHEREZOV V 2017. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc Natl Acad Sci U S A, 114, 8223–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG Y, ZHOU XE, GAO X, HE Y, LIU W, ISHCHENKO A, BARTY A, WHITE TA, YEFANOV O, HAN GW, XU Q, DE WAAL PW, KE J, TAN MH, ZHANG C, MOELLER A, WEST GM, PASCAL BD, VAN EPS N, CARO LN, VISHNIVETSKIY SA, LEE RJ, SUINO-POWELL KM, GU X, PAL K, MA J, ZHI X, BOUTET S, WILLIAMS GJ, MESSERSCHMIDT M, GATI C, ZATSEPIN NA, WANG D, JAMES D, BASU S, ROY-CHOWDHURY S, CONRAD CE, COE J, LIU H, LISOVA S, KUPITZ C, GROTJOHANN I, FROMME R, JIANG Y, TAN M, YANG H, LI J, WANG M, ZHENG Z, LI D, HOWE N, ZHAO Y, STANDFUSS J, DIEDERICHS K, DONG Y, POTTER CS, CARRAGHER B, CAFFREY M, JIANG H, CHAPMAN HN, SPENCE JC, FROMME P, WEIERSTALL U, ERNST OP, KATRITCH V, GUREVICH VV, GRIFFIN PR, HUBBELL WL, STEVENS RC, CHEREZOV V, MELCHER K & XU HE 2015. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature, 523, 561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENAKIN T 2011. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther, 336, 296–302. [DOI] [PubMed] [Google Scholar]
- KIM K, CHE T, PANOVA O, DIBERTO JF, LYU J, KRUMM BE, WACKER D, ROBERTSON MJ, SEVEN AB, NICHOLS DE, SHOICHET BK, SKINIOTIS G & ROTH BL 2020. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell, 182, 1574–1588 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAUDA JB, VENABLE RM, FREITES JA, O’CONNOR JW, TOBIAS DJ, MONDRAGON-RAMIREZ C, VOROBYOV I, MACKERELL AD JR. & PASTOR RW 2010. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B, 114, 7830–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRISHNA KUMAR K, SHALEV-BENAMI M, ROBERTSON MJ, HU H, BANISTER SD, HOLLINGSWORTH SA, LATORRACA NR, KATO HE, HILGER D, MAEDA S, WEIS WI, FARRENS DL, DROR RO, MALHOTRA SV, KOBILKA BK & SKINIOTIS G 2019. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell, 176, 448–458 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KROEZE WK, SASSANO MF, HUANG XP, LANSU K, MCCORVY JD, GIGUERE PM, SCIAKY N & ROTH BL 2015. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol, 22, 362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRUMM B & ROTH BL 2018. Activation mechanisms for a universal signalling protein. Nature, 557, 318–319. [DOI] [PubMed] [Google Scholar]
- KUHN H, HALL SW & WILDEN U 1984. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett, 176, 473–8. [DOI] [PubMed] [Google Scholar]
- KUYPERS KP, NG L, ERRITZOE D, KNUDSEN GM, NICHOLS CD, NICHOLS DE, PANI L, SOULA A & NUTT D 2019. Microdosing psychedelics: More questions than answers? An overview and suggestions for future research. J Psychopharmacol, 33, 1039–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUYPERS KPC 2020. The therapeutic potential of microdosing psychedelics in depression. Ther Adv Psychopharmacol, 10, 2045125320950567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LATORRACA NR, MASUREEL M, HOLLINGSWORTH SA, HEYDENREICH FM, SUOMIVUORI CM, BRINTON C, TOWNSHEND RJL, BOUVIER M, KOBILKA BK & DROR RO 2020. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell, 183, 1813–1825 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE Y, WARNE T, NEHME R, PANDEY S, DWIVEDI-AGNIHOTRI H, CHATURVEDI M, EDWARDS PC, GARCIA-NAFRIA J, LESLIE AGW, SHUKLA AK & TATE CG 2020. Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature, 583, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEBSCHNER D, AFONINE PV, BAKER ML, BUNKOCZI G, CHEN VB, CROLL TI, HINTZE B, HUNG LW, JAIN S, MCCOY AJ, MORIARTY NW, OEFFNER RD, POON BK, PRISANT MG, READ RJ, RICHARDSON JS, RICHARDSON DC, SAMMITO MD, SOBOLEV OV, STOCKWELL DH, TERWILLIGER TC, URZHUMTSEV AG, VIDEAU LL, WILLIAMS CJ & ADAMS PD 2019. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol, 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU JJ, HORST R, KATRITCH V, STEVENS RC & WUTHRICH K 2012. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science, 335, 1106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOHSE MJ, BENOVIC JL, CODINA J, CARON MG & LEFKOWITZ RJ 1990. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science, 248, 1547–50. [DOI] [PubMed] [Google Scholar]
- LOMIZE MA, LOMIZE AL, POGOZHEVA ID & MOSBERG HI 2006. OPM: orientations of proteins in membranes database. Bioinformatics, 22, 623–5. [DOI] [PubMed] [Google Scholar]
- MANGLIK A, KIM TH, MASUREEL M, ALTENBACH C, YANG Z, HILGER D, LERCH MT, KOBILKA TS, THIAN FS, HUBBELL WL, PROSSER RS & KOBILKA BK 2015. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell, 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASTRONARDE DN 2005. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol, 152, 36–51. [DOI] [PubMed] [Google Scholar]
- MAYER D, DAMBERGER FF, SAMARASIMHAREDDY M, FELDMUELLER M, VUCKOVIC Z, FLOCK T, BAUER B, MUTT E, ZOSEL F, ALLAIN FHT, STANDFUSS J, SCHERTLER GFX, DEUPI X, SOMMER ME, HUREVICH M, FRIEDLER A & VEPRINTSEV DB 2019. Distinct G protein-coupled receptor phosphorylation motifs modulate arrestin affinity and activation and global conformation. Nat Commun, 10, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCLURE-BEGLEY TD & ROTH BL 2022. The promises and perils of psychedelic pharmacology for psychiatry. Nature Reviews Drug Discovery. [DOI] [PubMed] [Google Scholar]
- MCCORVY JD, WACKER D, WANG S, AGEGNEHU B, LIU J, LANSU K, TRIBO AR, OLSEN RHJ, CHE T, JIN J & ROTH BL 2018. Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol, 25, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLS DE 2016. Psychedelics. Pharmacol Rev, 68, 264–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOBLES KN, XIAO K, AHN S, SHUKLA AK, LAM CM, RAJAGOPAL S, STRACHAN RT, HUANG TY, BRESSLER EA, HARA MR, SHENOY SK, GYGI SP & LEFKOWITZ RJ 2011. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal, 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYGAARD R, ZOU Y, DROR RO, MILDORF TJ, ARLOW DH, MANGLIK A, PAN AC, LIU CW, FUNG JJ, BOKOCH MP, THIAN FS, KOBILKA TS, SHAW DE, MUELLER L, PROSSER RS & KOBILKA BK 2013. The dynamic process of beta(2)-adrenergic receptor activation. Cell, 152, 532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANDY-SZEKERES G, MUNK C, TSONKOV TM, MORDALSKI S, HARPSOE K, HAUSER AS, BOJARSKI AJ & GLORIAM DE 2018. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res, 46, D440–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASSIE T, HALPERN JH, STICHTENOTH DO, EMRICH HM & HINTZEN A 2008. The pharmacology of lysergic acid diethylamide: a review. CNS Neurosci Ther, 14, 295–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEREZ-RIVEROL Y, CSORDAS A, BAI J, BERNAL-LLINARES M, HEWAPATHIRANA S, KUNDU DJ, INUGANTI A, GRISS J, MAYER G, EISENACHER M, PEREZ E, USZKOREIT J, PFEUFFER J, SACHSENBERG T, YILMAZ S, TIWARY S, COX J, AUDAIN E, WALZER M, JARNUCZAK AF, TERNENT T, BRAZMA A & VIZCAINO JA 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETTERSEN EF, GODDARD TD, HUANG CC, COUCH GS, GREENBLATT DM, MENG EC & FERRIN TE 2004. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem, 25, 1605–12. [DOI] [PubMed] [Google Scholar]
- PETTERSEN EF, GODDARD TD, HUANG CC, MENG EC, COUCH GS, CROLL TI, MORRIS JH & FERRIN TE 2021. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci, 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUNJANI A, RUBINSTEIN JL, FLEET DJ & BRUBAKER MA 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods, 14, 290–296. [DOI] [PubMed] [Google Scholar]
- RAHMEH R, DAMIAN M, COTTET M, ORCEL H, MENDRE C, DURROUX T, SHARMA KS, DURAND G, PUCCI B, TRINQUET E, ZWIER JM, DEUPI X, BRON P, BANERES JL, MOUILLAC B & GRANIER S 2012. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A, 109, 6733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANGANATHAN A, DROR RO & CARLSSON J 2014. Insights into the role of Asp79(2.50) in beta2 adrenergic receptor activation from molecular dynamics simulations. Biochemistry, 53, 7283–96. [DOI] [PubMed] [Google Scholar]
- RM B 2017. Dabble. Zenodo. [Google Scholar]
- ROE DR & CHEATHAM TE 3RD 2013. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput, 9, 3084–95. [DOI] [PubMed] [Google Scholar]
- ROTH BL 2007. Drugs and valvular heart disease. N Engl J Med, 356, 6–9. [DOI] [PubMed] [Google Scholar]
- ROTH BL, BANER K, WESTKAEMPER R, SIEBERT D, RICE KC, STEINBERG S, ERNSBERGER P & ROTHMAN RB 2002. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A, 99, 11934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALOMON-FERRER R, GOTZ AW, POOLE D, LE GRAND S & WALKER RC 2013. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput, 9, 3878–88. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-GARCIA R, GOMEZ-BLANCO J, CUERVO A, CARAZO JM, SORZANO COS & VARGAS J 2021. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun Biol, 4, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHUKLA AK, MANGLIK A, KRUSE AC, XIAO K, REIS RI, TSENG WC, STAUS DP, HILGER D, UYSAL S, HUANG LY, PADUCH M, TRIPATHI-SHUKLA P, KOIDE A, KOIDE S, WEIS WI, KOSSIAKOFF AA, KOBILKA BK & LEFKOWITZ RJ 2013. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature, 497, 137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH JS, LEFKOWITZ RJ & RAJAGOPAL S 2018. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov, 17, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAUS DP, HU H, ROBERTSON MJ, KLEINHENZ ALW, WINGLER LM, CAPEL WD, LATORRACA NR, LEFKOWITZ RJ & SKINIOTIS G 2020. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature, 579, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUOMIVUORI CM, LATORRACA NR, WINGLER LM, EISMANN S, KING MC, KLEINHENZ ALW, SKIBA MA, STAUS DP, KRUSE AC, LEFKOWITZ RJ & DROR RO 2020. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science, 367, 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZCZEPEK M, BEYRIERE F, HOFMANN KP, ELGETI M, KAZMIN R, ROSE A, BARTL FJ, VON STETTEN D, HECK M, SOMMER ME, HILDEBRAND PW & SCHEERER P 2014. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun, 5, 4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMSEN ARB, PLOUFFE B, CAHILL TJ 3RD, SHUKLA AK, TARRASCH JT, DOSEY AM, KAHSAI AW, STRACHAN RT, PANI B, MAHONEY JP, HUANG L, BRETON B, HEYDENREICH FM, SUNAHARA RK, SKINIOTIS G, BOUVIER M & LEFKOWITZ RJ 2016. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell, 166, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- URBAN JD, CLARKE WP, VON ZASTROW M, NICHOLS DE, KOBILKA B, WEINSTEIN H, JAVITCH JA, ROTH BL, CHRISTOPOULOS A, SEXTON PM, MILLER KJ, SPEDDING M & MAILMAN RB 2007. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther, 320, 1–13. [DOI] [PubMed] [Google Scholar]
- VIOLIN JD, CROMBIE AL, SOERGEL DG & LARK MW 2014. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci, 35, 308–16. [DOI] [PubMed] [Google Scholar]
- WACKER D, WANG C, KATRITCH V, HAN GW, HUANG XP, VARDY E, MCCORVY JD, JIANG Y, CHU M, SIU FY, LIU W, XU HE, CHEREZOV V, ROTH BL & STEVENS RC 2013. Structural features for functional selectivity at serotonin receptors. Science, 340, 615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WACKER D, WANG S, MCCORVY JD, BETZ RM, VENKATAKRISHNAN AJ, LEVIT A, LANSU K, SCHOOLS ZL, CHE T, NICHOLS DE, SHOICHET BK, DROR RO & ROTH BL 2017. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell, 168, 377–389 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEDEGAERTNER PB, WILSON PT & BOURNE HR 1995. Lipid modifications of trimeric G proteins. J Biol Chem, 270, 503–6. [DOI] [PubMed] [Google Scholar]
- WINGLER LM, SKIBA MA, MCMAHON C, STAUS DP, KLEINHENZ ALW, SUOMIVUORI CM, LATORRACA NR, DROR RO, LEFKOWITZ RJ & KRUSE AC 2020. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science, 367, 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG F, YU X, LIU C, QU CX, GONG Z, LIU HD, LI FH, WANG HM, HE DF, YI F, SONG C, TIAN CL, XIAO KH, WANG JY & SUN JP 2015. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun, 6, 8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Z, YANG F, ZHANG D, LIU Z, LIN A, LIU C, XIAO P, YU X & SUN JP 2017. Phosphorylation of G Protein-Coupled Receptors: From the Barcode Hypothesis to the Flute Model. Mol Pharmacol, 92, 201–210. [DOI] [PubMed] [Google Scholar]
- YIN W, LI Z, JIN M, YIN YL, DE WAAL PW, PAL K, YIN Y, GAO X, HE Y, GAO J, WANG X, ZHANG Y, ZHOU H, MELCHER K, JIANG Y, CONG Y, EDWARD ZHOU X, YU X & ERIC XU H 2019. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res, 29, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG L & HERMANS J 1996. Hydrophilicity of cavities in proteins. Proteins, 24, 433–8. [DOI] [PubMed] [Google Scholar]
- ZHOU XE, HE Y, DE WAAL PW, GAO X, KANG Y, VAN EPS N, YIN Y, PAL K, GOSWAMI D, WHITE TA, BARTY A, LATORRACA NR, CHAPMAN HN, HUBBELL WL, DROR RO, STEVENS RC, CHEREZOV V, GUREVICH VV, GRIFFIN PR, ERNST OP, MELCHER K & XU HE 2017. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell, 170, 457–469 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed in this study are included in this article and the Supplementary Information. The cryo-EM density maps and corresponding coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB), respectively, under the following accession codes: EMD-25403 and 7SRS (HTR2B/β-arrestin-1), EMD-25402 and 7SRR (HTR2B-LSD/miniGq) and EMD-25401 and 7SRQ (HTR2B-LSD). The mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD030752.