

Abstract
Despite decades of research and an enormity of resultant data, cancer remains a significant public health problem. New tools and fresh perspectives are needed to obtain fundamental insights, to develop better prognostic and predictive tools, and to identify improved therapeutic interventions. With increasingly common genome-scale data, one suite of algorithms and concepts with potential to shed light on cancer biology is phylogenetics, a scientific discipline used in diverse fields. From grouping subsets of cancer samples to tracing subclonal evolution during cancer progression and metastasis, the use of phylogenetics is a powerful systems biology approach. Well-developed phylogenetic applications provide fast, robust approaches to analyze high-dimensional, heterogeneous cancer data sets.
Keywords: Tumor heterogeneity, Cancer stratification, Clonal evolution, Tumor trees, Cancer types
1. Introduction
Cancer results from a breakdown in multicellular cooperation [1], evolving changes in DNA sequence, gene expression patterns, and/or epigenetic modifications that permit unchecked growth. These molecular changes induce phenotypes that can increase the ability of a cell to compete, survive and reproduce, and ultimately lead to cancer. Advantageous phenotypes include 1) self-sufficiency in growth signals, 2) insensitivity to anti-growth signals, 3) evasion of apoptosis, 4) limitless replicative potential, 5) sustained angiogenesis, 6) ability to invade and metastasize to surrounding tissue and distant organs, 7) deregulated cellular energetics, and 8) avoidance of immune destruction [2,3]. In many cases, these hallmarks are the consequences of mutations that result in a cell with increased fitness compared to its healthy counterparts, followed by selective pressures that increase the prevalence of that cell lineage. Continued rounds of mutation and selection putatively lead to more extreme phenotypes in comparison to normal tissue, and thereby more aggressive metastatic disease.
From the initial transforming event to dissemination, seeding, and eventual metastatic colonization, cancer progression represents a process of selection over time. Nowell first drew this parallel between the selective forces acting on cancer cells within the body and those acting on individuals within populations in nature [36]. Nowell proposed that the heterogeneity observed in tumors is due to an increase in genetic instability as cancer progresses [36]. Indeed, evidence of increased genetic instability over time has been recently shown in the progression of Barrett’s esophagus to esophageal adenocarcinoma in a longitudinal study of patients for over 20 years [37]. This increased genetic instability enhances the genetic diversity of the cancer cell population, and presumably the phenotypic diversity as well, which is acted upon by selective forces within the tumor, such as immune surveillance, hypoxia, glucose deprivation, and the production of reactive oxygen species, to produce sub-clones capable of thriving despite the barriers to progression [38]. These concepts of increasing heterogeneity coupled with selection in the context of cancer progression have been borne out by studies using both first generation and next-generation sequencing technologies [39]. For example, analysis of breast cancer primary and matched metastases found that just over half of the coding mutations identified in the metastases (19/32) were not detected in the primary tumor [40]. Of the mutations common to the primary tumor and metastases, 6/13 were found in only 1–13% of cells in the primary tumor [40]. Similarly, sequencing of pancreatic cancer primary tumors and metastases revealed that different metastases are seeded from unique clones [41]. These authors also concluded that metastatic clones may have seeded tertiary subclones [41], though the physical history of metastatic departures cannot be inferred from sequence data without a complete sampling of clonal lineages within the primary tumor [42]. In the largest multi-region sampling paper published to date, sampling of 40 patients with primary tumors and 3–8 matched metastases demonstrated diverse patterns of molecular genetic divergence along the time course of cancer progression [28]. These and other studies clearly demonstrate that cancer progression, from indolent neoplasia to aggressive and metastatic disease, is a process in which cells change in a spatio-temporal manner while under selective forces.
Given that cancer progression is governed by selective forces, tools developed to elucidate evolutionary relationships should generally be appropriate for use in the analysis of cancer. One of the most well-developed and successful evolutionary approaches is phylogenetics. Originally designed to model and infer evolutionary relationships among organisms, this suite of algorithms, concepts, and tools has been usefully applied in a wide array of diverse fields, even fields in which the data have no true evolutionary context [43–47]. Below, we briefly review phylogenetic concepts and methods and discuss the possibilities for the application of phylogenetics to analysis of cancer data sets in the following three capacities: 1) as a suite of classification algorithms that could be applied to assign specimens as coming from either healthy individuals, patients with localized disease, or those with metastasis; 2) as a means to deconstruct the complex heterogeneity within tumors; and 3) as a natural method to determine the branching evolution of cancer cells within individuals during cancer progression.
2. Phylogenetics: revealing relationships between states
The field of phylogenetic systematics was born from a need to sort and classify organisms in such a way as to capture their relationships by descent. Phylogenetics utilizes a data matrix of input characteristics from a group of organisms (Fig. 1A) to produce a graphical “tree” (Fig. 1B) where the branching pattern, or tree-topology, represents bifurcations between individuals, species, or higher taxa, depending on the scope of the taxonomic question of interest. A phylogeny, or an evolutionary tree, provides a basic structure to statistically analyze the evolutionary relationships (differences and similarities) among distantly-related taxa (species or larger groups of inclusively-related species). The most recent common ancestor (MRCA) of a group is the node furthest from the root that contains all members of the group as descendants. Pairs of taxa that share a more recent common ancestor are more closely related than those whose MRCA occurs more deeply in the tree (Fig. 1B). When numerous taxon divergences are represented along a lineage, it becomes possible to chart the accumulation of traits or features that have resulted from evolution over time. Tree topologies can be rooted or unrooted. In a rooted tree, some extrinsic information is used to root the tree. This information is typically in the form of an assumed outgroup. Outgroups typically represent distantly-related taxa that provide information on the ancestral condition (state) of a character prior to its transformation to a more derived condition. In principle, this use of outgroups enables the researcher to establish the directionality of change for a set of characters [48], though it must be cautioned that outgroups often have undergone significant evolutionary change themselves and are not always suitable proxies for the ancestor. Unlike rooted trees, unrooted trees reveal the relatedness of taxa within the nodes of the tree without assuming a relationship of a group of taxa to an ancestral state. Phylogenetic algorithms are widely used in the study of the evolutionary dynamics of molecular sequences themselves, using each homologous position in a sequence as an individual character [49–51]. Moreover, phylogenetic methods can be applied to numerous types of data, including morphological characteristics and/or other data that can be converted into discrete character states, and even can be applied to quantitative characters under appropriate models of evolutionary change.
Fig. 1.
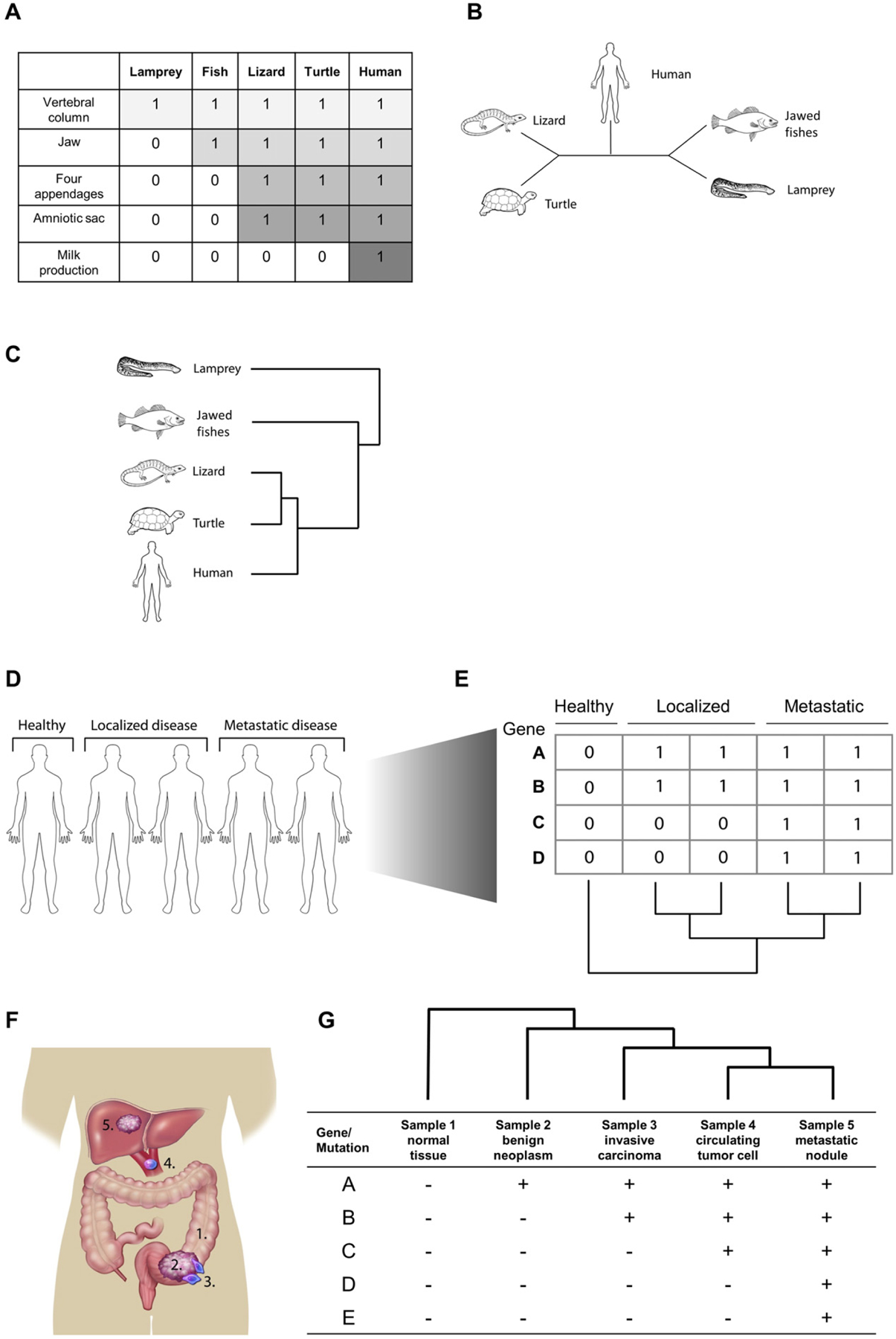
Phylogenetics reveals evolutionary relationships between states. A. Characteristics from various species under study can be transformed into a binary character state matrix. A species that is known to possess the ancestral state of the given characters (e.g. here, the lamprey) can be included as an “outgroup” as a means by which to polarize the resulting tree. B. An unrooted most parsimonious tree obtained by choosing the topology requiring the fewest number of character changes. C. The unrooted tree is converted to a rooted tree by assuming that jawed vertebrates share a more recent common ancestor than the most recent common ancestor (MRCA) of the entire group. C. As a cancer research tool, phylogenetic analyses can be used strictly as a clustering algorithm to segregate individual patients by their progression status. D. Samples are collected from individual patients, a matrix of characters is constructed using gene expression, mutation status, or some other information, and a phylogenetic tree is generated. E. In a more direct application of phylogenetic methods, they can be used to analyze phenotypic/genotypic heterogeneity within a patient or disease location. In this example, samples are collected at different sites to construct a matrix and tree of progression F. Depending on the question being asked, samples can be collected longitudinally or from neighboring areas of a tissue a single site (e.g. primary tumor and metastatic nodules) to reconstruct the evolutionary history of the disease progression.
3. Phylogenetic analysis of cancer data
The multiple and diverse paths of progression to cancer are forged by genetic mutations and alterations to epigenetics, gene expression, and protein signaling. Because these changes tend to accumulate over time in diversifying somatic lineages, phylogenetic analysis provides a natural tool set for evaluating the branching history of cancer onset and progression. The development of these tools has been considered an emerging field of inquiry, termed herein as PhyloOncology or Cancer Phylogenetics, which represents diverse applications of phylogenetic algorithms to the analysis of cancer data. We highlight below three general types of analyses in which phylogenetics has provided insight in the understanding of cancer biology: 1) classification of cancer specimens (Fig. 1C and D); 2) analyses of intratumoral heterogeneity (Fig. 1E and F), and 3) tracking clonal evolution and progression (Fig. 1E and F).
3.1. Applying phylogenetic methods to classify gene expression profiles
The application of phylogenetics methods to the analysis of gene expression profiles from individual tumors may not be its most natural usage, but does provide an alternative to other approaches for the classification of microarray or transcriptomic analyses, such as hierarchical clustering (Fig. 1C). In this utilization of phylogenetics, genes can be coded as the ‘characters’, expression levels can be coded as discrete ‘character states’, and individual samples can be the ‘taxa’ (Fig. 1C and D). In one such discretization, gene expression changes can be converted into discrete character states based on whether they are upregulated (1), downregulated (−1), or effectively unchanged (0). Application of a phylogenetically-based algorithm then produces one or more trees based on the similarities and differences in gene-expression profiles, putatively grouping (or classifying) cancer or disease tissues relative to normal tissue expression profiles (Fig. 1D). In traditional phylogenetics, a distantly-related taxon is used as an outgroup; however, in the analysis of cancer vs. normal tissue, the “outgroup” comprises either a mixture of normal tissues from representative individuals or, ideally, normal samples from the same individual as each tumor sample. A number of studies using maximum parsimony [52–54] and distance [55,56] algorithms on gene expression data have suggested that the methodology classifies tumors into monophyletic groupings compared to ‘normal’ tissue controls [57]. These analyses suggest that phylogenetic algorithms or algorithms developed from phylogenetic algorithms could serve as clinically-relevant tools for diagnosis, prognosis, and/or prediction of clinical outcomes.
3.2. Limitations of phylogenetics for analyzing gene expression data
The use of phylogenetic algorithms to classify individual tumors by their gene expression profiles presents some conceptual as well as technical challenges. For example, the extent to which gene expression profiles can be appropriately modeled within the discrete taxonomic character matrices typically used by most phylogenetic algorithms remains to be more fully explored. In addition, any reduction of a continuous character to discrete states, though sometimes useful, represents a loss of information. In general, the application of phylogenetics for classification of individuals is susceptible to criticisms that unlike phylogenetic algorithms applied as ‘classifiers’, other non-phylogenetic algorithms have been specifically developed for their particular application.
It is also important to note that these applications of phylogenetics algorithms do not necessarily assume that true evolutionary relationships exist between samples. Rather, the phylogenetic algorithm can be applied as a clustering algorithm to reveal relationships, evolutionary or otherwise, between samples. For example, when applied to gene expression data, phylogenetics algorithms would likely cluster samples together that have similar responses to microenvironmental cues, such as hypoxia or upregulation of epithelial plasticity pathways. While it remains to be determined whether this application of phylogenetics will add meaningfully to our understanding of cancer, it is worth considering that phylogenetics has been applied as a clustering method across diverse fields, including geology [47], astrophysics [58–60], comparative linguistics [61], and other disciplines for which no underlying biological evolutionary relationships exist between comparators. To further develop phylogenetics as a useful general tool for analysis of gene expression or other data across individuals, a rigorous comparison of phylogenetics algorithms with other clustering methods needs to be performed, as has been performed in a limited capacity on a relatively small dataset [52]. As a contrasting example, methods for quality assessment and control, normalization, quantification, and statistical analysis of microarray data are mature and highly standardized–and the limitations have therefore been well documented for nearly a decade [62]. Application of such a large-scale technical validation effort would set clear standards for claims of appropriate algorithms to provide superior or significantly complementary results. Of course, claims that these tools could be diagnostic, prognostic, or predictive will need especially conscientious validation. For example, classifiers identified in these studies have not been tested for prediction of pre-metastatic indications in blinded clinical data. Moreover, these classifications of tumors have not been overlaid with any clinical outcomes (e.g. recurrence-free survival, overall survival). These analyses must be done before phylogenetic methods can truly be validated as a clinically-useful analysis suite for classification of samples.
3.3. Tracking intratumoral heterogeneity
Enhanced breadth and depth of sequencing in recent years have revealed extensive inter-patient heterogeneity [63–66]. These technological advances have also demonstrated remarkable intratumoral heterogeneity, with many subclones arising and diversifying from a set of early tumor-initiating events, or “trunk” of the tree [66–70]. For example, phylogenetic analyses have been applied to data from fluorescence in situ hybridization (FISH) probes used to detect copy number or gene fusion abnormalities at PAX5, CDKN2A, RUNX1, and ETV6 genes (used as characters) in 200 individual cells from the bulk leukemic blast population (used as the individual taxa) per patient in a total of 30 acute lymphoblastic leukemia cases [71]. Leukemic cells from two of these patients were injected in immunodeficient mice, and showed striking changes in frequencies of phylogenetic subclones determined by cells’ FISH probe patterns between the original leukemia and after primary and secondary transplantations [71]. These subclone frequency data and inferred phylogenies provided evidence of complex subclonal architecture, and supported the hypothesis that individual subclones carrying distinct configurations of abnormalities can exhibit differential competitive potency, differential biological functions, and differential therapy resistance within an individual patient. A recent study built on these findings using novel microfluidics technology for single cell separation and typing of a limited number of gene fusions, copy number abnormalities, and nucleotide variants [72]. Similarly, in an analysis of B-cell chronic lymphocytic leukemia, an unrooted most-parsimonious-tree analysis was carried out on ultra-deep pyrosequencing of the immunoglobulin locus in a total of 22 patients [73]. Phylogenetic analysis revealed multiple co-existing subclonal cell populations detected within individuals.
Work by [74] used whole genome single-cell sequencing of 100 breast cancer cells to construct distance-based phylogenies. This work revealed multiple independent clonal expansions within a single breast tumor [74]. Similarly, work by [69] used phylogenetic reconstruction of multi-site sequencing from a renal cell carcinoma primary tumor and several metastases to reveal extensive heterogeneity, with only 30–40% mutations in common across sites within a single tumor [69]. Distance-based phylogenetic reconstructions of multi-site biopsies from a single ovarian cancer patient also revealed a branching pattern of accumulating mutations from a shared TP53 mutant clone, with diverse, unique mutations from each biopsy site [75]. A recent study even claims to support a hypothesis that no two cells share an absolutely identical genome in some breast cancers [76]. These results underscore the importance of developing better tools to understand intratumoral heterogeneity as personalized therapies become more common.
3.4. Limitations to analyses of intratumoral heterogeneity
Analysis of intratumoral heterogeneity faces several challenges. With regard to tumor sample collection, tumor biopsies to be used for analysis are comprised of heterogeneous mixtures of cancerous and non-cancerous cells. To exclude contamination based on this heterogeneity, researchers can take advantage of improved sampling methods, such as laser-capture microdissection [77–79] and flow cytometry [80] to separate cancer from non-cancer cells. Advances in technology have also enabled the analysis of increasingly smaller quantities of cells, thereby paving the way for the use of fine needle aspirates, circulating tumor cells, single cells, and cell-free biomolecules in the blood of cancer patients [63,81]. Lower costs of sequencing have also enabled the higher depth of reads necessary to detect cancer-specific signals and rare mutations, as well as facilitating increased frequency of sampling to capture a greater depth of intra-tumoral heterogeneity [69]. Importantly, when taking multiple bulk samples from within a tumor, researchers have attributed heterogeneity in mutation content between bulk samples to mutation rate differences; however, simulations have shown that such heterogeneity can be spurious and can be generated under neutral mutation and spatial growth models [82]. Further advances in single-cell sampling and sequencing as well as improvements in deconvolution algorithms will be needed to address the issues associated with sequencing from bulk samples within a tumor.
In addition to these technical challenges and limitations, it is worthwhile to consider carefully the phylogenetic methodologies applied to the inference of the evolutionary history of cancer molecular evolution. For example, Navin et al. used a distance-based method to analyze whole-genome copy number data from 100 single cells isolated from two primary breast tumors and their corresponding liver metastases [83]. While these analyses are interesting, the use of distance methods may not be the most powerful phylogenetic approach for analysis of the data. Distance methods reduce complex state-based data to summary statistics, which leaves out of the analysis some information on higher-order combinations of character states.
3.5. Tracking clonal evolution and progression
In the study of clonal evolution, phylogenetic algorithms are applied to samples from a single tumor, a single site, or an individual. If multiple samples from the same tumor are taken, each sample can be treated as an individual species or taxon [28]. Ideally, clones within the bulk tumor population should be differentiated; to do so, subclonal deconvolution methods can be applied to decompose the bulk population into subclones that can then be treated as individual species or taxa [51, 85–88]. Data characters can be any characteristics of the sample or of the inferred subclone, e.g. point mutation presence/absence, nucleotide state, codon state, or chromosomal abnormalities, such as translocations, duplications, or deletions of large chromosomal segments. Epigenetic changes in DNA methylation states can also change over time and can be analyzed phylogenetically [89–94], although there is yet no agreed-upon model for the evolution of epigenetic state changes. Each distinct character type has its own mechanism of inheritance—its own rate of evolutionary change—and therefore can be modeled uniquely (e.g. for mutations of DNA nucleotides, these range from simple (e.g. Jukes-Cantor, [95]) to increasingly complex (e.g. Kimura 2-Parameter [96], TrNef [97], Kimura3-Parameter [98], F81 [99], or HKY/F84 [100, 101]).
In one of the earliest demonstrations of the utility of phylogenetics for analysis of cancer data, the authors analyzed a set of chromosomal break points in 135 ovarian adenocarcinoma patients using distance-based methods and compared these results to six other multivariate analyses [102]. Interestingly, the multiple analysis methods came to similar conclusions regarding the relative timing of specific breaks and the segregation of tumors into different sub-classes [102]. Distance-based phylogenetic algorithms were also applied to the classification of an array comparative hybridization dataset from renal cancer samples, demonstrating the strengths of the phylogenetic analyses for the identification of co-occurrences of pairs of events and early events in tumor progression [103]. These early analyses provided support for the potential of applications of phylogenetics to the study of cancer datasets. A parsimony analysis using loss of heterozygosity (LOH) at 29 microsatellite markers on chromosome 9 of multiple biopsies from tumors in an individual bladder tracked initiation and development of heterogeneous tumors and revealed the progression and evolution of different locations in each tumor. This phylogenetic analysis [104], along with additional probabilistic analyses [105], supported a hypothesis of monoclonal origin in these multifocal bladder cancers, as opposed to multi- or poly-clonal origins. Phylogenetics analyses can also incorporate cell-extrinsic microenvironmental influences on clonal evolution, for example, by estimating rates of change before and after treatment [70].
In other cancers as well, deep sequencing followed by phylogenetic analysis has been used to analyze changes in subclonal architecture during treatment and at relapse, or in primary tumors, invasive sites, and metastases. Phylogenetic analysis of next-generation sequence technologies was performed on a large analysis of matched normal, primary, and metastatic samples from 40 patients, in which whole exome sequencing data were analyzed by parsimony, maximum likelihood, and Bayesian inference methods [28]. In this investigation, the authors traced the evolution of metastases from primary tumor tissue. The phylogenetic analyses revealed that the timing of the first genetic divergence of metastatic lineages often arose prior to initial diagnosis (Fig. 2). In addition, the results demonstrated that the application of well-established phylogenetic approaches to the estimation of chronograms can reveal the temporal orders of driver mutations, which could allow clinicians to detect and treat potential metastases with actionable therapies earlier in the disease course [28]. These analyses are consistent with earlier phylogenetics-based analyses of gene expression data from prostate cancer, in which benign prostate cancer samples were grouped with metastatic samples, suggesting that the gene expression differences observed in metastatic samples can arise early in the progression to metastatic disease [54]. (See Fig. 2.)
Fig. 2.
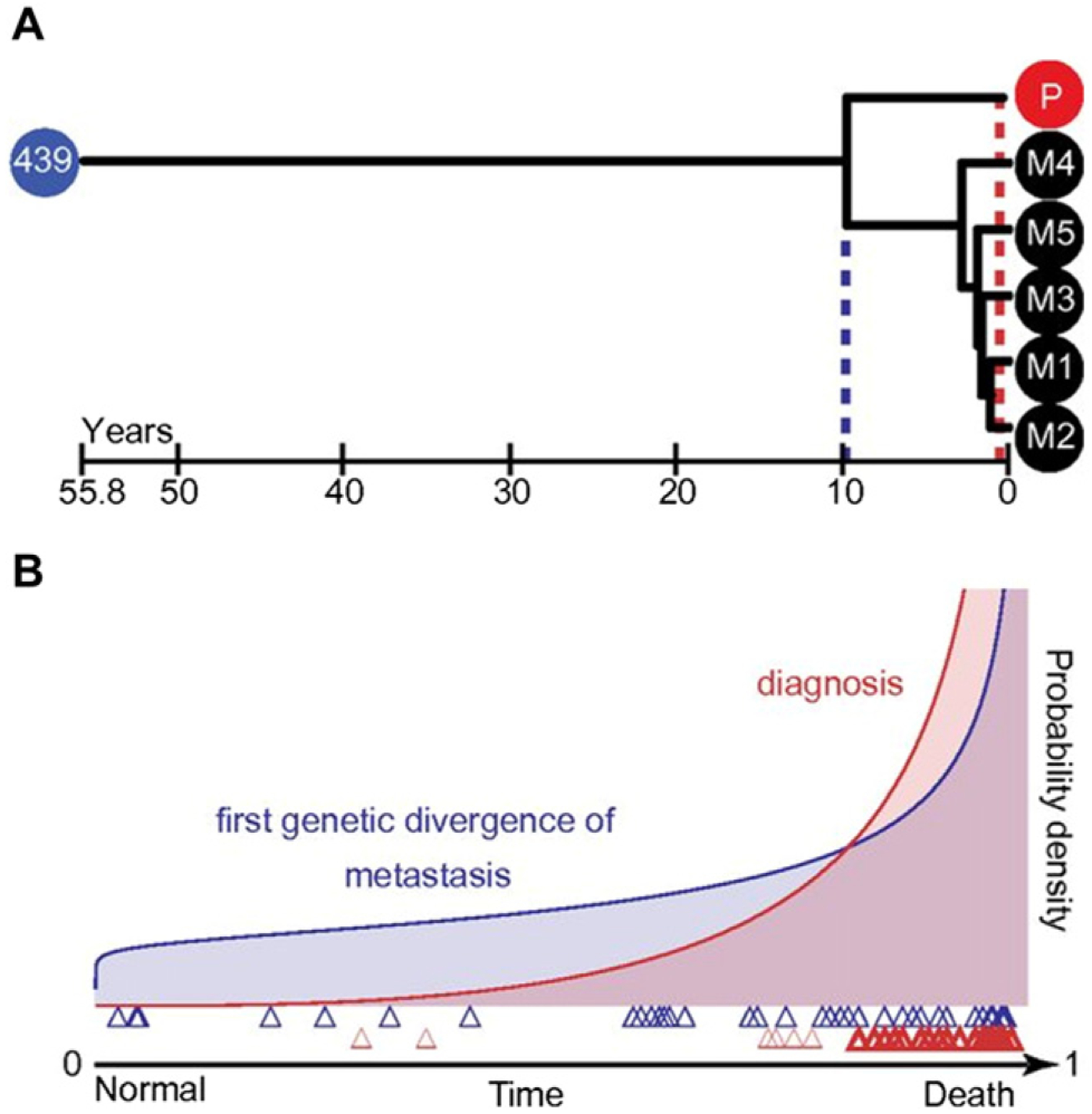
Reconstructing the chronology of metastatic lineages using phylogenetics. Timings of the first genetic divergence from normal tissue sequence (blue circle), of the first genetic divergence of metastases (blue dashes) and of diagnosis (red dashes) during tumor progression. A. A patient with renal clear cell carcinoma exemplifies late metastasis in which diagnosis of the primary tumor and metastases occurred after the first genetic divergence of metastasis. B. Probability density for the occurrence of the first genetic divergence of metastases and for the time of diagnosis. The x axis is scaled from 0 (the first genetic divergence of primary tumor tissue from normal tissue) to 1 (death). In the set of 40 lethal cancers analyzed in the study, the first genetic divergences of metastatic lineages (blue triangles) are distributed so as to often occur earlier than diagnosis time (red triangles). Figure reproduced from [28]. Early and multiple origins of metastatic lineages within primary tumors. 113:8, 2140–2145. doi:10.1073/pnas.1525677113. Copyright Proceedings of the National Academy of Sciences.
3.6. Limitations of using phylogenetics to elucidate cancer progression
Deep sequencing of tumor samples has revealed a vast diversity of cancer sub-populations, not only between individuals, but within a single tumor nodule [66–69]. Similarly, investigations have revealed that tumor-propagating cells often exist as rare subsets of cells within a tumor population [115]. Consistent with these analyses, gene expression signatures indicating good and poor prognosis have been observed in different areas of a single tumor [69]. In these instances, ‘omics’ analyses from single biopsies would be likely to wholly miss these key cells; even analyses from whole tumor specimens would likely not detect very rare cells; the signal from the bulk of the tumor would wash out any signal from them [42].
While identifying the rare subsets of tumor cells that drive resistance and progression from an entire tumor is, at present, an extremely costly and time-consuming proposition from an ‘omics’ perspective, innovative solutions have begun to arise that confront these challenges. For instance, reliable phylogenetic inference among subclones depends on knowledge of the ‘phase’ among mutation variants. Phasing refers to determining whether two or more mutations exist on the same or different haploid chromosome copies. Phase information can in principle be obtained experimentally, by evaluating the co-occurrence of mutation variants in single cells. For example, complete phase information was experimentally determined in an acute lymphoblastic leukemia study, where FISH was used to detect the state configuration of presence/absence of copy number changes at five loci and to quantify the frequency of those state configurations in the population [71]. Data of this type do not need subclonal deconvolution.
Most commonly, however, phase is inferred computationally from mutant allele frequencies from multiple individual samples per tumor obtained over space or time (e.g. at different time points during treatment and relapse). Deconvolution methods can help to infer subclones, mutational state configurations, and phylogenetic relationships/distances among subclones that are most likely given the observed mutation allele frequencies. Unfortunately, there is no guarantee that somatic variant frequency data will provide sufficient information to deconvolute phase, especially for somatic variants of low frequency. Current deconvolution methods differ significantly in how they accomplish this task. Consequently, different deconvolution approaches to phasing often yield different results. Moreover, clinical “tumor” samples are often mixtures of tumor tissue and normal tissue, thus driving tumor subclones to even lower (and harder-to-differentiate) frequencies. Despite these limitations, deconvolution has the advantage that it supplies an explicit approach to address the well-understood ‘problem of heterogeneity’. By analyzing individual cells or deconvoluted subclones within a population, these methods—at least in principle—portray the relationships of clonal mutations that are observed during progression or metastasis. To the degree that they are successful in phasing, therefore, they provide advantageous insight into the genotype-phenotype relationship of clonal evolution of tumor cell populations. Powerful approaches simultaneously use copy number and single nucleotide alterations to infer phylogenies and deconvolute cancer subpopulations from bulk samples [116].
In addition to computational and theoretical advances, assays of cells and molecules of tumoral origin that are transiting the blood of cancer patients can be devised to identify known, rare “driver” or resistance-conferring sequences. Numerous platforms and methodologies have been developed that enrich and detect cells and cell-free biomolecules in the circulation of patients with a variety of cancer types [83]. This technology has led to the conception of a “liquid biopsy”, in which researchers can track prognostic and/or predictive cells and/or cell-free biomolecules longitudinally during disease progression. The ability to assay for cells and other biomarkers via assays of the circulatory system over time brings significant clinical advantages over the use of single-time-point biopsies and whole tumor analysis [117]. In addition, the liquid biopsy strategy enables researchers to identify gene expression pathways that are critical to invasion, dissemination, and metastasis [118–121]. Indeed, future investigations combining circulating tumor cell/biomolecule enrichment with ‘omics-based phylogenetics analysis could prove to be a powerful approach to study disease progression and metastasis.
While it is important to understand the benefits and shortcomings of particular methodologies, it is equally important to carefully evaluate the assumptions that they make about the data structure. For example, maximum parsimony makes assumptions about the rate at which characters change in different regions of the tree, and violation of these assumptions can lead to long-branch attraction. Maximum likelihood and Bayesian methods require explicit models of molecular evolution to explain the data. Although these methods tend to be robust to violations of their assumptions, strong model mis-specifications can lead them to infer the incorrect tree.
4. Summary/conclusions
Analysis of high-throughput ‘omics’ data, such as deep sequencing of DNA mutations, gene expression, as well as increasingly-sensitive proteomics, metabolomics, and epigenomics data, has shed light on the vast heterogeneity of cancer, both within and between individual patients as well as during disease progression and in response to treatment. It has become clear from these global analyses that cancer onset and progression is a multi-step, branching process in which the genetic mutations, metabolites, epigenetics, and gene expression change over time—an evolutionary process that is appropriately analyzed by the long-standing tools of phylogenetics. In this evolutionary process, subsets of cells within the primary tumor that are, at one point, a rare breed might become selected for during metastatic dissemination and colonization. The characteristic progression over time exhibited by cancer makes it an ideal system for the application of phylogenetics, a suite of analytical approaches developed specifically to study evolutionary history. Not only are these strategies appropriate for the analysis of molecular evolutionary divergence of cellular lineages, but they might have additional utility as classifiers that can compete on an even footing with other methodologies for clustering samples. Phylogenetics, and an evolutionary perspective in general [122,123], should be an additional instrument in the toolkit of the cancer biologist, providing both essential conceptual paradigms and new computational strategies for understanding cancer.
Acknowledgments
JAS acknowledges support from the Duke Cancer Institute, The Duke University Genitourinary Oncology Laboratory, and the Duke University Department of Orthopaedics. JPT acknowledges support from Gilead Sciences and from Notsew Orm Sands Foundation. The authors would like to thank Dr. Shyamal Pedadda (National Institute of Environmental Health and Safety) for his helpful discussions. The conception of this manuscript was the result of a meeting of the PhyloOncology Working Group, which was sponsored by the National Evolutionary Synthesis Center (NESCent, NSF #EF-0905606) and the Triangle Center for Evolutionary Medicine (TriCEM).
Footnotes
Transparency document
The Transparency document associated with this article can be found, in the online version.
References
- [1].Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS, Cancer across the tree of life: cooperation and cheating in multicellularity, Philos. Trans. R. Soc. B Biol. Sci 370 (1673) (2015) 10.1098/rstb.2014.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hanahan D, Weinberg RA, The hallmarks of cancer, Cell 100 (1) (2000) 57–70 (Epub 2000/01/27. doi: S0092–8674(00)81683–9 [pii].). [DOI] [PubMed] [Google Scholar]
- [3].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (5) (2011) 646–674, 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [28].Zhao ZM, Zhao B, Bai Y, Iamarino A, Gaffney SG, Schlessinger J, Lifton RP, Rimm DL, Townsend JP, Early and multiple origins of metastatic lineages within primary tumors, Proc. Natl. Acad. Sci. U. S. A 113 (8) (2016) 2140–2145, 10.1073/pnas.1525677113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nowell PC, The clonal evolution of tumor cell populations, Science 194 (4260) (1976) 23–28. [DOI] [PubMed] [Google Scholar]
- [37].Li X, Galipeau PC, Paulson TG, Sanchez CA, Arnaudo J, Liu K, Sather CL, Kostadinov RL, Odze RD, Kuhner MK, Maley CC, Self SG, Vaughan TL, Blount PL, Reid BJ, Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus, Cancer Prev. Res. (Phila.) 7 (1) (2014) 114–127, 10.1158/1940-6207.CAPR-13-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cahill DP, Kinzler KW, Vogelstein B, Lengauer C, Genetic instability and Darwinian selection in tumours, Trends Cell Biol. 9 (12) (1999) M57–M60. [PubMed] [Google Scholar]
- [39].Brosnan JA, Iacobuzio-Donahue CA, A new branch on the tree: next-generation sequencing in the study of cancer evolution, Semin. Cell Dev. Biol 23 (2) (2012) 237–242, 10.1016/j.semcdb.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, Severson T, Taylor GA, Teschendorff AE, Tse K, Turashvili G, Varhol R, Warren RL, Watson P, Zhao Y, Caldas C, Huntsman D, Hirst M, Marra MA, Aparicio S, Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution, Nature 461 (7265) (2009) 809–813, 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- [41].Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Griffin CA, Burton J, Swerdlow H, Quail MA, Stratton MR, Iacobuzio-Donahue C, Futreal PA, The patterns and dynamics of genomic instability in metastatic pancreatic cancer, Nature 467 (7319) (2010) 1109–1113, 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hong WS, Shpak M, Townsend JP, Inferring the origin of metastases from cancer phylogenies, Cancer Res. 75 (19) (2015) 4021–4025, 10.1158/0008-5472.CAN-15-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Young GC, Application of cladistics to terrane history—parsimony analysis of qualitative geological data, J. SE Asian Earth Sci 11 (3) (1995) 167–176, 10.1016/0743-9547(95)00011-G. [DOI] [Google Scholar]
- [44].Fraix-Burnet D, Thuillard M, Chattopadhyay AK, Multivariate Approaches to Classification in Extragalactic Astronomy, arXivorg, 2015. 10.3389/fspas.2015.00003 (arXiv:1508.06756). [DOI] [Google Scholar]
- [45].Platnick NIaDC H, Cladistic methods in textual, linguistic, and phylogenetic analysis, Syst. Zool 26 (4) (1977) 380–385, 10.2307/2412794. [DOI] [Google Scholar]
- [46].AlGeddawy T, A DSM cladistics model for product family architecture design, Procedia CIRP 21 (2014) 87–92, 10.1016/j.procir.2014.03.122. [DOI] [Google Scholar]
- [47].Echeverry A, Silva-Romo G, Morrone JJ, Tectonostratigraphic terrane relationships: a glimpse into the Caribbean under a cladistic approach, Palaeogeogr. Palaeoclimatol. Palaeoecol 353–355 (2012) 87–92, 10.1016/j.palaeo.2012.07.007. [DOI] [Google Scholar]
- [48].Graham SW, Olmstead RG, Barrett SC, Rooting phylogenetic trees with distant outgroups: a case study from the commelinoid monocots, Mol. Biol. Evol 19 (10) (2002) 1769–1781. [DOI] [PubMed] [Google Scholar]
- [49].Abu-Asab MS, Laassri M, Amri H, Algorithmic assessment of vaccine-induced selective pressure and its implications on future vaccine candidates, Adv. Bioinforma 2010 (2010) 178069, 10.1155/2010/178069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Whelan S, Lio P, Goldman N, Molecular phylogenetics: state-of-the-art methods for looking into the past, Trends Genet. 17 (5) (2001) 262–272. [DOI] [PubMed] [Google Scholar]
- [51].Clement M, Posada D, Crandall KA, TCS: a computer program to estimate gene genealogies, Mol. Ecol 9 (10) (2000) 1657–1659. [DOI] [PubMed] [Google Scholar]
- [52].Abu-Asab M, Chaouchi M, Amri H, Evolutionary medicine: a meaningful connection between omics, disease, and treatment, Proteomics Clin. Appl 2 (2) (2008) 122–134, 10.1002/prca.200780047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Abu-Asab MS, Abu-Asab N, Loffredo CA, Clarke R, Amri H, Identifying early events of gene expression in breast cancer with systems biology phylogenetics, Cytogenet. Genome Res 139 (3) (2013) 206–214, 10.1159/000348433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Abu-Asab MS, Chaouchi M, Alesci S, Galli S, Laassri M, Cheema AK, Atouf F, VanMeter J, Amri H, Biomarkers in the age of omics: time for a systems biology approach, OMICS 15 (3) (2011) 105–112, 10.1089/omi.2010.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Desper R, Khan J, Schaffer AA, Tumor classification using phylogenetic methods on expression data, J. Theor. Biol 228 (4) (2004) 477–496, 10.1016/j.jtbi.2004.02.021. [DOI] [PubMed] [Google Scholar]
- [56].Riester M, Stephan-Otto Attolini C, Downey RJ, Singer S, Michor F, A differentiation-based phylogeny of cancer subtypes, PLoS Comput. Biol 6 (5) (2010) e1000777, 10.1371/journal.pcbi.1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Abu-Asab MS, Chaouchi M, Amri H, Phylogenetic modeling of heterogeneous gene-expression microarray data from cancerous specimens, OMICS 12 (3) (2008) 183–199, 10.1089/omi.2008.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fraix-Burnet D, Thuillard M, Chattopadhyay AK, Multivariate Approaches to Classification in Extragalactic Astronomy, arXiv preprint arXiv, 2015. 150806756. [Google Scholar]
- [59].Fraix-Burnet D, Chattopadhyay T, Chattopadhyay AK, Davoust E, Thuillard M, A six-parameter space to describe galaxy diversification, Astron. Astrophys 545 (2012) A80. [Google Scholar]
- [60].Barret D, Casoli F, Contini T, Lagache G, Lecavelier A, Determining the Evolutionary History of Galaxies by Astrocladistics: Some Results on Close Galaxies(arXiv preprint astroph/0610190) 2006.
- [61].Longobardi G, Ghirotto S, Guardiano C, Tassi F, Benazzo A, Ceolin A, Barbujani G, Across language families: genome diversity mirrors linguistic variation within Europe, Am. J. Phys. Anthropol 157 (4) (2015) 630–640, 10.1002/ajpa.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Consortium M, Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Amur S, Bao W, Barbacioru CC, Lucas AB, Bertholet V, Boysen C, Bromley B, Brown D, Brunner A, Canales R, Cao XM, Cebula TA, Chen JJ, Cheng J, Chu TM, Chudin E, Corson J, Corton JC, Croner LJ, Davies C, Davison TS, Delenstarr G, Deng X, Dorris D, Eklund AC, Fan XH, Fang H, Fulmer-Smentek S, Fuscoe JC, Gallagher K, Ge W, Guo L, Guo X, Hager J, Haje PK, Han J, Han T, Harbottle HC, Harris SC, Hatchwell E, Hauser CA, Hester S, Hong H, Hurban P, Jackson SA, Ji H, Knight CR, Kuo WP, LeClerc JE, Levy S, Li QZ, Liu C, Liu Y, Lombardi MJ, Ma Y, Magnuson SR, Maqsodi B, McDaniel T, Mei N, Myklebost O, Ning B, Novoradovskaya N, Orr MS, Osborn TW, Papallo A, Patterson TA, Perkins RG, Peters EH, Peterson R, Philips KL, Pine PS, Pusztai L, Qian F, Ren H, Rosen M, Rosenzweig BA, Samaha RR, Schena M, Schroth GP, Shchegrova S, Smith DD, Staedtler F, Su Z, Sun H, Szallasi Z, Tezak Z, Thierry-Mieg D, Thompson KL, Tikhonova I, Turpaz Y, Vallanat B, Van C, Walker SJ, Wang SJ, Wang Y, Wolfinger R, Wong A, Wu J, Xiao C, Xie Q, Xu J, Yang W, Zhang L, Zhong S, Zong Y, Slikker W Jr., The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements, Nat. Biotechnol 24 (9) (2006) 1151–1161, 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].De Luca F, Rotunno G, Salvianti F, Galardi F, Pestrin M, Gabellini S, Simi L, Mancini I, Vannucchi AM, Pazzagli M, Di Leo A, Pinzani P, Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer, Oncotarget (2016) 10.18632/oncotarget.8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Szalat R, Munshi NC, Genomic heterogeneity in multiple myeloma, Curr. Opin. Genet. Dev 30 (2015) 56–65, 10.1016/j.gde.2015.03.008. [DOI] [PubMed] [Google Scholar]
- [65].Jhunjhunwala S, Jiang Z, Stawiski EW, Gnad F, Liu J, Mayba O, Du P, Diao J, Johnson S, Wong KF, Gao Z, Li Y, Wu TD, Kapadia SB, Modrusan Z, French DM, Luk JM, Seshagiri S, Zhang Z, Diverse modes of genomic alteration in hepatocellular carcinoma, Genome Biol. 15 (8) (2014) 436, 10.1186/s13059-014-0436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, Vizan P, Di Croce L, Puente DA, Lopez-Guerra M, Moros A, Roue G, Aymerich M, Villamor N, Colomo L, Martinez A, Valera A, Martin-Subero JI, Amador V, Hernandez L, Rozman M, Enjuanes A, Forcada P, Muntanola A, Hartmann EM, Calasanz MJ, Rosenwald A, Ott G, Hernandez-Rivas JM, Klapper W, Siebert R, Wiestner A, Wilson WH, Colomer D, Lopez-Guillermo A, Lopez-Otin C, Puente XS, Campo E, Landscape of somatic mutations and clonal evolution in mantle cell lymphoma, Proc. Natl. Acad. Sci. U. S. A 110 (45) (2013) 18250–18255, 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kogita A, Yoshioka Y, Sakai K, Togashi Y, Sogabe S, Nakai T, Okuno K, Nishio K, Inter-and intra-tumor profiling of multi-regional colon cancer and metastasis, Biochem. Biophys. Res. Commun 458 (1) (2015) 52–56, 10.1016/j.bbrc.2015.01.064. [DOI] [PubMed] [Google Scholar]
- [68].Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, Fenwick K, Kozarewa I, Gonzalez D, Lord CJ, Ashworth A, Davies FE, Morgan GJ, Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma, Blood 120 (5) (2012) 1077–1086, 10.1182/blood-2012-03-412981. [DOI] [PubMed] [Google Scholar]
- [69].Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C, Intratumor heterogeneity and branched evolution revealed by multiregion sequencing, N. Engl. J. Med 366 (10) (2012) 883–892, 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kostadinov RL, Kuhner MK, Li X, Sanchez CA, Galipeau PC, Paulson TG, Sather CL, Srivastava A, Odze RD, Blount PL, Vaughan TL, Reid BJ, Maley CC, NSAIDs modulate clonal evolution in Barrett’s esophagus, PLoS Genet. 9 (6) (2013) e1003553, 10.1371/journal.pgen.1003553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, Kearney L, Enver T, Greaves M, Genetic variegation of clonal architecture and propagating cells in leukaemia, Nature 469 (7330) (2011) 356–361, 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- [72].Potter NE, Ermini L, Papaemmanuil E, Cazzaniga G, Vijayaraghavan G, Titley I, Ford A, Campbell P, Kearney L, Greaves M, Single-cell mutational profiling and clonal phylogeny in cancer, Genome Res. 23 (12) (2013) 2115–2125, 10.1101/gr.159913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, Stratton MR, Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing, Proc. Natl. Acad. Sci. U. S. A 105 (35) (2008) 13081–13086, 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M, Tumour evolution inferred by single-cell sequencing, Nature 472 (7341) (2011) 90–94, 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lee JY, Yoon JK, Kim B, Kim S, Kim MA, Lim H, Bang D, Song YS, Tumor evolution and intratumor heterogeneity of an epithelial ovarian cancer investigated using next-generation sequencing, BMC Cancer 15 (2015) 85, 10.1186/s12885-015-1077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, Michor F, Meric-Bernstam F, Navin NE, Clonal evolution in breast cancer revealed by single nucleus genome sequencing, Nature 512 (7513) (2014) 155–160, 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP, Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer, Cancer Res. 66 (5) (2006) 2815–2825, 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- [78].De Marchi T, Braakman RB, Stingl C, van Duijn MM, Smid M, Foekens JA, Luider TM, Martens JW, Umar A, The advantage of laser-capture microdissection over whole tissue analysis in proteomic profiling studies, Proteomics (2016) 10.1002/pmic.201600004. [DOI] [PubMed] [Google Scholar]
- [79].Jensen DH, Dabelsteen E, Specht L, Fiehn AM, Therkildsen MH, Jonson L, Vikesaa J, Nielsen FC, von Buchwald C, Molecular profiling of tumour budding implicates TGFbeta-mediated epithelial-mesenchymal transition as a therapeutic target in oral squamous cell carcinoma, J. Pathol 236 (4) (2015) 505–516, 10.1002/path.4550. [DOI] [PubMed] [Google Scholar]
- [80].Boyd ZS, Raja R, Johnson S, Eberhard DA, Lackner MR, A tumor sorting protocol that enables enrichment of pancreatic adenocarcinoma cells and facilitation of genetic analyses, J. Mol. Diagn 11 (4) (2009) 290–297, 10.2353/jmoldx.2009.080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang X, Marjani SL, Hu Z, Weissman SM, Pan X, Wu S, Single-cell sequencing for precise cancer research: progress and prospects, Cancer Res. 76 (6) (2016) 1305–1312, 10.1158/0008-5472.CAN-15-1907. [DOI] [PubMed] [Google Scholar]
- [82].Kostadinov R, Maley CC, Kuhner MK, Bulk genotyping of biopsies can create spurious evidence for hetereogeneity in mutation content, PLoS Comput. Biol 12 (4) (2016) e1004413, 10.1371/journal.pcbi.1004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ferreira MM, Ramani VC, Jeffrey SS, Circulating tumor cell technologies, Mol. Oncol 10 (3) (2016) 374–394, 10.1016/j.molonc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Schwartz R, Shackney SE, Applying unmixing to gene expression data for tumor phylogeny inference, BMC Bioinforma. 11 (2010) 42, 10.1186/1471-2105-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Strino F, Parisi F, Micsinai M, Kluger Y, TrAp: a tree approach for fingerprinting subclonal tumor composition, Nucleic Acids Res. 41 (17) (2013) e165, 10.1093/nar/gkt641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, Vij R, Tomasson MH, Graubert TA, Walter MJ, Ellis MJ, Schierding W, DiPersio JF, Ley TJ, Mardis ER, Wilson RK, Ding L, SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution, PLoS Comput. Biol 10 (8) (2014) e1003665, 10.1371/journal.pcbi.1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Qiao Y, Quinlan AR, Jazaeri AA, Verhaak RG, Wheeler DA, Marth GT, SubcloneSeeker: a computational framework for reconstructing tumor clone structure for cancer variant interpretation and prioritization, Genome Biol. 15 (8) (2014) 443, 10.1186/s13059-014-0443-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Skinner MK, Gurerrero-Bosagna C, Haque MM, Nilsson EE, Koop JA, Knutie SA, Clayton DH, Epigenetics and the evolution of Darwin’s finches, Genome Biol. Evol 6 (8) (2014) 1972–1989, 10.1093/gbe/evu158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Alonso C, Perez R, Bazaga P, Herrera CM, Global DNA cytosine methylation as an evolving trait: phylogenetic signal and correlated evolution with genome size in angiosperms, Front. Genet 6 (4) (2015) 10.3389/fgene.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Siegmund KD, Marjoram P, Woo YJ, Tavare S, Shibata D, Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers, Proc. Natl. Acad. Sci. U. S. A 106 (12) (2009) 4828–4833, 10.1073/pnas.0810276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJ, Smirnov IV, Reis GF, Phillips JJ, Barnes MJ, Idbaih A, Alentorn A, Kloezeman JJ, Lamfers ML, Bollen AW, Taylor BS, Molinaro AM, Olshen AB, Chang SM, Song JS, Costello JF, DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors, Cancer Cell 28 (3) (2015) 307–317, 10.1016/j.ccell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nicolas P, Kim KM, Shibata D, Tavare S, The stem cell population of the human colon crypt: analysis via methylation patterns, PLoS Comput. Biol 3 (3) (2007) e28, 10.1371/journal.pcbi.0030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Graham TA, Humphries A, Sanders T, Rodriguez-Justo M, Tadrous PJ, Preston SL, Novelli MR, Leedham SJ, McDonald SA, Wright NA, Use of methylation patterns to determine expansion of stem cell clones in human colon tissue, Gastroenterology 140 (4) (2011) 10.1053/j.gastro.2010.12.036. [DOI] [PubMed] [Google Scholar]
- [95].JTaC CR, Evolution of Protein Molecules, Academic Press, New York, 1969. (21–132 pp.). [Google Scholar]
- [96].Kimura M, A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences, J. Mol. Evol 16 (2) (1980) 111–120. [DOI] [PubMed] [Google Scholar]
- [97].Tamura K, Nei M, Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees, Mol. Biol. Evol 10 (3) (1993) 512–526. [DOI] [PubMed] [Google Scholar]
- [98].Kimura M, Estimation of evolutionary distances between homologous nucleotide sequences, Proc. Natl. Acad. Sci. U. S. A 78 (1) (1981) 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Felsenstein J, Evolutionary trees from DNA sequences: a maximum likelihood approach, J. Mol. Evol 17 (6) (1981) 368–376. [DOI] [PubMed] [Google Scholar]
- [100].Kishino H, Hasegawa M, Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea, J. Mol. Evol 29 (2) (1989) 170–179. [DOI] [PubMed] [Google Scholar]
- [101].Felsenstein J, Churchill GA, A hidden Markov model approach to variation among sites in rate of evolution, Mol. Biol. Evol 13 (1) (1996) 93–104. [DOI] [PubMed] [Google Scholar]
- [102].Simon R, Desper R, Papadimitriou CH, Peng A, Alberts DS, Taetle R, Trent JM, Schaffer AA, Chromosome abnormalities in ovarian adenocarcinoma: III. Using breakpoint data to infer and test mathematical models for oncogenesis, Genes Chromosom. Cancer 28 (1) (2000) 106–120. [PubMed] [Google Scholar]
- [103].Desper R, Jiang F, Kallioniemi OP, Moch H, Papadimitriou CH, Schaffer AA, Distance-based reconstruction of tree models for oncogenesis, J. Comput. Biol 7 (6) (2000) 789–803, 10.1089/10665270050514936. [DOI] [PubMed] [Google Scholar]
- [104].Louhelainen J, Wijkstrom H, Hemminki K, Initiation-development modelling of allelic losses on chromosome 9 in multifocal bladder cancer, Eur. J. Cancer 36 (11) (2000) 1441–1451. [DOI] [PubMed] [Google Scholar]
- [105].Louhelainen J, Wijkstrom H, Hemminki K, Allelic losses demonstrate monoclonality of multifocal bladder tumors, Int. J. Cancer 87 (4) (2000) 522–527. [PubMed] [Google Scholar]
- [115].Wei Q, Tang YJ, Voisin V, Sato S, Hirata M, Whetstone H, Han I, Ailles L, Bader GD, Wunder J, Alman BA, Identification of CD146 as a marker enriched for tumor-propagating capacity reveals targetable pathways in primary human sarcoma, Oncotarget 6 (37) (2015) 40283–40294, 10.18632/oncotarget.5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Jiang Y, Qiu Y, Minn AJ, Zhang NR, Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing, Proc. Natl. Acad. Sci. U. S. A 113 (37) (2016) E5528–E5537, 10.1073/pnas.1522203113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Gupta S, Li J, Kemeny G, Bitting RL, Beaver J, Somarelli J, Ware KE, Gregory S, Armstrong AJ, Whole genomic copy number alterations in circulating tumor cells from men with abiraterone or enzalutamide resistant metastatic castration-resistant prostate cancer, Clin. Cancer Res (2016) 10.1158/1078-0432.CCR-16-1211. [DOI] [PubMed] [Google Scholar]
- [118].Alonso-Alconada L, Muinelo-Romay L, Madissoo K, Diaz-Lopez A, Krakstad C, Trovik J, Wik E, Hapangama D, Coenegrachts L, Cano A, Gil-Moreno A, Chiva L, Cueva J, Vieito M, Ortega E, Mariscal J, Colas E, Castellvi J, Cusido M, Dolcet X, Nijman HW, Bosse T, Green JA, Romano A, Reventos J, Lopez-Lopez R, Salvesen HB, Amant F, Matias-Guiu X, Moreno-Bueno G, Abal M, Consortium E, Molecular profiling of circulating tumor cells links plasticity to the metastatic process in endometrial cancer, Mol. Cancer 13 (2014) 223, 10.1186/1476-4598-13-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Markou A, Zavridou M, Sourvinou I, Yousef G, Kounelis S, Malamos N, Georgoulias V, Lianidou E, Direct comparison of metastasis-related miRNAs expression levels in circulating tumor cells, corresponding plasma, and primary tumors of breast cancer patients, Clin. Chem (2016) 10.1373/clinchem.2015.253716. [DOI] [PubMed] [Google Scholar]
- [120].Scholch S, Garcia SA, Iwata N, Niemietz T, Betzler AM, Nanduri LK, Bork U, Kahlert C, Thepkaysone ML, Swiersy A, Buchler MW, Reissfelder C, Weitz J, Rahbari NN, Circulating tumor cells exhibit stem cell characteristics in an orthotopic mouse model of colorectal cancer, Oncotarget (2016) 10.18632/oncotarget.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Cho WJ, Oliveira DS, Najy AJ, Mainetti LE, Aoun HD, Cher ML, Heath E, Kim HR, Bonfil RD, Gene expression analysis of bone metastasis and circulating tumor cells from metastatic castrate-resistant prostate cancer patients, J. Transl. Med 14 (1) (2016) 72, 10.1186/s12967-016-0829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Merlo LM, Pepper JW, Reid BJ, Maley CC, Cancer as an evolutionary and ecological process, Nat. Rev. Cancer 6 (12) (2006) 924–935, 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- [123].Thomas F, Fisher D, Fort P, Marie JP, Daoust S, Roche B, Grunau C, Cosseau C, Mitta G, Baghdiguian S, Rousset F, Lassus P, Assenat E, Gregoire D, Misse D, Lorz A, Billy F, Vainchenker W, Delhommeau F, Koscielny S, Itzykson R, Tang R, Fava F, Ballesta A, Lepoutre T, Krasinska L, Dulic V, Raynaud P, Blache P, Quittau-Prevostel C, Vignal E, Trauchessec H, Perthame B, Clairambault J, Volpert V, Solary E, Hibner U, Hochberg ME, Applying ecological and evolutionary theory to cancer: a long and winding road, Evol. Appl 6 (1) (2013) 1–10, 10.1111/eva.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]