

Abstract
Reversing reversible deactivation radical polymerization (RDRP) to regenerate the original monomer is an attractive prospect for both fundamental research and industry. However, current depolymerization strategies are often applied to highly heat-tolerant polymers with a specific end-group and can only be performed in a specific solvent. Herein, we depolymerize a variety of poly(methyl methacrylate) materials made by reversible addition–fragmentation chain-transfer (RAFT) polymerization and terminated by various end groups (dithiobenzoate, trithiocarbonate, and pyrazole carbodithioate). The effect of the nature of the solvent on the depolymerization conversion was also investigated, and key solvents such as dioxane, xylene, toluene, and dimethylformamide were shown to facilitate efficient depolymerization reactions. Notably, our approach could selectively regenerate pure heat-sensitive monomers (e.g., tert-butyl methacrylate and glycidyl methacrylate) in the absence of previously reported side reactions. This work pushes the boundaries of reversing RAFT polymerization and considerably expands the chemical toolbox for recovering starting materials under relatively mild conditions.
Reversible deactivation radical polymerization (RDRP), also sometimes referred to as controlled radical polymerization, is an advanced form of radical polymerization that enables access to extremely high control over the molecular weight, end-group fidelity, dispersity, and architecture of the resulting polymer.1−3 A key distinguishing feature of RDRP is the presence of a well-defined chemical moiety at the chain terminus, often referred to as the end-group, enabling the synthesis of block copolymers for use in applications such as self-assembly in bulk and solution.4−9 One of the most widely used RDRP techniques is reversible addition–fragmentation chain-transfer (RAFT) polymerization, which operates through a degenerative chain-transfer process.10−13 Through an effective regulation of the equilibrium between active and dormant species, polymers with high livingness can be synthesized (i.e., the overwhelming majority of chains have a chain-transfer agent as the end group).14
Although the field of RDRP has generated a large body of literature on controlling the polymerization of vinyl monomers, publications on reversing the process (i.e., depolymerization) and regenerating the monomer are scarce despite the fact that depolymerization has been classified by IUPAC as one of the top 10 emerging technologies in chemistry.15 As such, several groups have initiated important lines of research toward developing new depolymerization strategies. For instance, Haddleton and co-workers reported an aqueous depolymerization of bromine-terminated water-soluble polymers in the presence of a metal catalyst and CO2.16 In another report, Ouchi and co-workers presented the depolymerization of Cl-capped poly(methyl methacrylate) in toluene using a ruthenium catalyst, yielding relatively modest depolymerization conversions (e.g., <24%).17 More recently, Matyjaszewski and co-workers demonstrated the depolymerization of a Cl-terminated bottlebrush polymer (i.e., poly(poly(dimethylsiloxane) methacrylate) in a trichlorobenzene/hexadecane solvent mixture by utilizing a highly active copper catalyst (CuCl2/tris(2-pyridylmethyl)amine) at 170 °C.18,19 The same group subsequently employed a similar catalytic system to depolymerize poly(n-butyl methacrylate) in trichlorobenzene, also at 170 °C.19,20 In the RAFT arena, Gramlich and co-workers reported the depolymerization of bulky bottlebrush polymethacrylates (poly(oligo(dimethylsiloxane) methacrylate) and poly(oligo(ethylene glycol) methyl ether methacrylate)) with a trithiocarbonate end group regenerating up to 35% of the respective monomers.21 More recently, our group reported the depolymerization of bulky and nonbulky polymethacrylates synthesized by RAFT polymerization, yielding high depolymerization conversions, albeit under specific conditions (e.g., a single RAFT end-group, largely limited to a single solvent, thermally robust pendent groups, etc.).22 Overall, current depolymerization strategies have been applied to a specific polymer/solvent/end-group system and have not been utilized to regenerate the monomer from the respective heat-sensitive polymers (i.e., polymers with thermally unstable side chains), thus limiting their scope and potential applications.
In this work, we first explored the compatibility of our previously developed RAFT-based depolymerization methodology with various end groups. The ability to depolymerize polymers formed through different RAFT agents is important for expanding the scope of our approach, as a different RAFT agent may be desirable depending on the intended application. For example, trithiocarbonates are preferably employed over dithiobenzoates for the synthesis of polymethacrylates in aqueous media (and subsequently for bioapplications), as they exhibit lower toxicity and enhanced stability toward hydrolysis.23 Other end groups and mixtures thereof are also utilized for the synthesis of polymethacrylates with tunable dispersities.24,25
To investigate the possibility of depolymerization from a range of RAFT end groups, we initially polymerized methyl methacrylate (MMA) at 70 °C with 2-cyano-2-propyl dithiobenzoate (DTB), 2-cyano 2-propyl dodecyltrithiocarbonate (TTC), and 2-cyanobutan-2-yl 4-chloro-3,5-dimethyl-1H-pyrazole-1-carbodithioate (pyrazoleCD) as the RAFT agents (Figures S1–S6). All three PMMAs were purified by precipitation in methanol to remove unreacted monomer, yielding pure PMMAs with comparable degrees of polymerization (DP) of 72–75 (Figures S2, S4, and S6). Deoxygenated solutions of PMMA (5 mM with respect to MMA repeat units) were prepared in 1,4-dioxane and subsequently heated at 120 °C. Reactions were periodically sampled and analyzed by 1H NMR to determine the extent of the depolymerization by monitoring the gradual appearance of the characteristic MMA vinylidene proton signals. Within 15 min, a clear generation of MMA accompanied by an expected decrease in the backbone signals was observed for all cases, indicating that depolymerization of polymers synthesized via different RAFT agents is possible (Figure 1). In particular, polymers synthesized by pyrazoleCD exhibited relatively slow depolymerization kinetics yielding 52% of conversion at 8 h. In contrast, PMMAs synthesized by either DTB or TTC could reach higher depolymerization conversions regenerating 85% and 72% of monomer respectively at 8 h. The difference in depolymerization behavior can be attributed to multiple factors including the varying strength of the C–S bond between the terminal MMA unit and the CTA and the different initial dispersities of the three macroCTAs. It is also worth noting that pyrazoleCD-PMMA exhibited high end-group fidelity (94–97%) and thus the difference in the depolymerization behavior can largely be attributed to the end group itself (Figure S7). Nevertheless, this data clearly illustrates that the depolymerization of nonbulky polymers synthesized by a range of RAFT agents was successful, thus expanding the scope of the depolymerizable materials.
Figure 1.

1H NMR spectra of the depolymerization reaction of PMMA with various end groups: (a) dithiobenzoate, (b) trithiocarbonate, and (c) pyrazole carbodithioate. Reactions were run in 1,4-dioxane at 5 mM and 120 °C.
Next, we explored the possibility of the depolymerization to commence in different solvents. Having a range of compatible solvents is critical for the depolymerization of various polymers as it allows dissolution of polymethacrylates of varying solubility (e.g., poly(stearyl methacrylate) has poor solubility in dioxane). As such, a range of solvents were tested including dimethylformamide (DMF), dimethyl sulfoxide (DMSO), benzene, toluene, xylenes (mixture of isomers), 1,2-dichlorobenzene (DCB), methyl ethyl ketone (MEK), and 2-methyl tetrahydrofuran (MeTHF). Importantly, all solvents were capable of facilitating the depolymerization of DTB-PMMA, with xylene reaching the highest depolymerization conversion (i.e., 60%), while DMF, a commonly employed polymerization solvent, led to 45% monomer regeneration (Figure 2a). It is noted that xylene and DMF presented a lower extent of depolymerization when compared with dioxane (i.e., 85% depolymerization). The observed solvent dependence was investigated by comparing the relative end-group fidelity during the depolymerization in these three different solvents. To do so, three depolymerization reactions were conducted in xylene, DMF and dioxane and were stopped at comparable conversions (i.e., 26–30%). The obtained polymers were then analyzed by both RI and UV size-exclusion chromatography (SEC); the results of which are shown in Figure 2b,c. In dioxane, a 23% reduction in the UV signal of the polymer was observed, very close to the 26% reduction of the RI signal, thus suggesting minimal loss of end-group. In contrast, in xylene and DMF, a much higher UV signal reduction was observed (44% and 63%, respectively), clearly evidencing a significant loss in the end-group fidelity at an early stage of the reaction. Thus, the lower depolymerization conversions in xylene and DMF compared with dioxane can largely be attributed to faster decomposition of the end group and side reactions, leading to a faster loss of end group. The same trend could be seen in depolymerization reactions in DCB wherein a greater reduction in the UV signal was observed (Figure S8). We speculate that end-group loss can occur through various mechanisms including Chugaev-like elimination of PMMA from the dithiobenzoate,26 disproportionation between two PMMA radicals, hydrolysis27 by trace amounts of water, and aminolysis (in the case of DMF) by trace amounts of amine.28 We confirmed that the remaining polymer did not exhibit a UV signal after the cessation of the depolymerization (Figure S9). Other solvents also resulted in appreciable depolymerization conversions including toluene (48% of depolymerization), DMSO (32%), MEK (30%), MeTHF (30%), and benzene (26%) while the lowest conversion was observed for DCB (10%). For pyrazoleCD-PMMA, the same solvent trend was observed, and the extent of depolymerization increased in the order DCB < DMSO < xylenes < dioxane (Figure S10). We thus concluded that the best solvents to conduct depolymerization of polymers synthesized by RAFT polymerization are dioxane and xylene.
Figure 2.
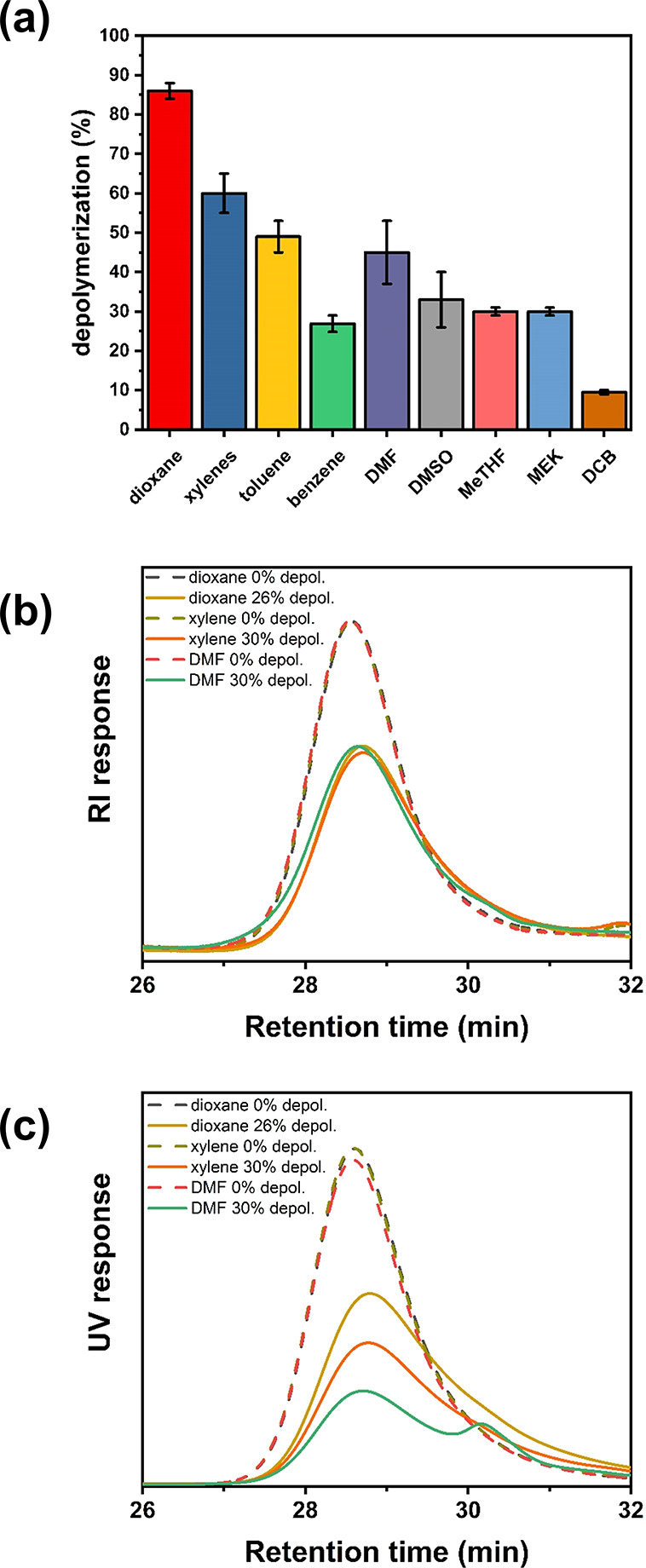
(a) Final depolymerization conversion of PMMA-DTB in various solvents after 8 h when subjected to 5 mM and 120 °C. Reactions were run at least three times in each solvent, and the error bars indicate the full range of values. (b) RI and (c) UV SEC traces of PMMA before and after depolymerization in dioxane, xylene, and dimethylformamide.
We were also interested in exploring the effect of the molecular weight on the depolymerization reaction. To investigate this, we synthesized a range of DTB-PMMAs with DPs of 29, 75, 92, 152, and 260 and subjected them to our optimized depolymerization conditions. For DP = 29 and DP = 75, comparable final depolymerization conversions were obtained (85–87%). However, for higher DPs, a gradual decrease in conversion was observed, with DP = 260 regenerating only 52% of monomer (Figure 3). It is noted that for all these experiments the repeat unit concentration was kept constant at 5 mM, meaning that the higher DPs contain a lower CTA concentration. The decrease in conversion for the higher DPs was attributed to the faster loss of the end group (Figure S11), as in the case of the various solvents.
Figure 3.
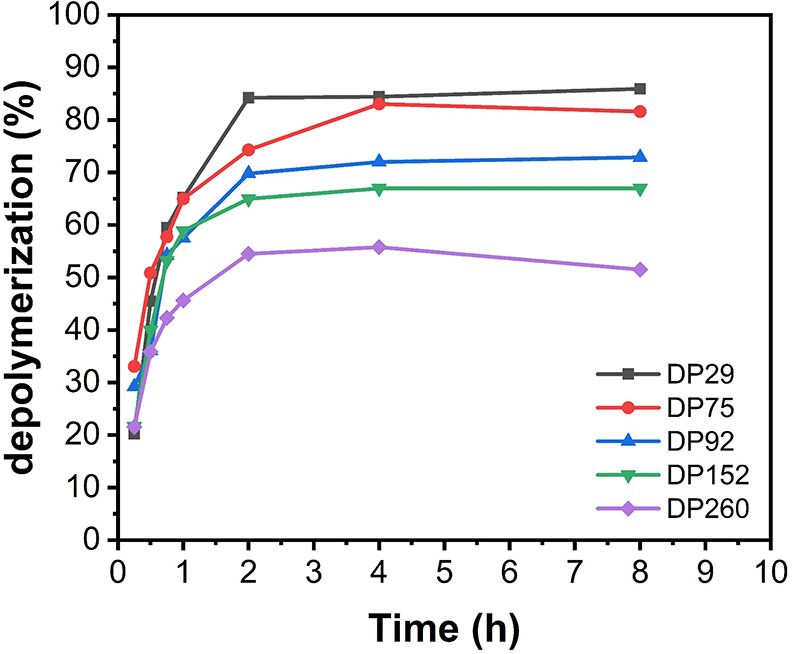
Depolymerization kinetics of PMMA with various degrees of polymerization. Reactions were run in dioxane at 5 mM and 120 °C.
Finally, we took advantage of the relatively low temperature of our reactions (compared with pyrolytic temperatures above 400 °C) to depolymerize polymethacrylates with heat-sensitive functional groups. Poly(tert-butyl methacrylate) (PtBMA) is a prime example of a polymer with heat-sensitive groups as the six β-hydrogens in the methacrylic ester makes the tert-butyl group highly susceptible to thermolytic de-esterification via elimination at temperatures around 200 °C.29−32 Thus, it is not possible to meaningfully regenerate pure tBMA monomer via pyrolysis due to simultaneous side reactions yielding methacrylic acid and methacrylic anhydride (after dehydration of two methacrylic acid units). We hypothesized that our mild reaction conditions would be optimal to both trigger depolymerization and selectively regenerate tBMA as we operate at sufficiently high temperatures for efficient depolymerization but well below the temperature for de-esterification. In order to test this theory, we synthesized and purified PtBMA-DTB (Mn = 7900; Đ = 1.13, Figures S12 and S13) and subsequently subjected it to 5 mM and 120 °C conditions in dioxane. 1H NMR analysis of the crude reaction mixture showed the appearance of characteristic vinylidene protons corresponding to tBMA and the absence of any methacrylic acid and methacrylic anhydride, with a total depolymerization conversion of 85% (Figure 4a; separate 1H NMR for tBMA, methacrylic acid, and methacrylic anhydride are also shown for comparison). To the best of our knowledge, this is the first demonstration of the selective regeneration of tBMA via thermal depolymerization. To investigate other heat-sensitive polymers, we also explored the depolymerization of poly(glycidyl methacrylate) (PGMA)-DTB (Mn = 13600; Đ = 1.20, Figures S14 and S15). PGMA is a widely used functional polymethacrylate, as the pendent ring-strained oxirane group makes it an easy target for “click” reactions with a nucleophile. Although PGMA is quite stable under ambient conditions, elevated temperatures can cause ring-opening and even cross-linking in the absence of a strong nucleophile.33−35 To examine whether it is possible to regenerate GMA without triggering ring-opening reactions in parallel, PGMA-DTB was subjected to 5 mM/120 °C conditions in dioxane and analyzed with 1H NMR. Similar to the depolymerization of PtBMA-DTB, no side products (i.e., ring-opened version of the monomer) were identified, and the product consisted entirely of GMA (Figure 4b; separate 1H NMR for GMA and the ring-opened counterpart are shown for comparison) with a depolymerization conversion of 84%, confirming that our low-temperature methodology is suitable, if not necessary, for the regeneration of heat-sensitive monomers.
Figure 4.

Depolymerization of heat-sensitive polymers (a) poly(tert-butyl methacrylate) and (b) poly(glycidyl methacrylate) at 5 mM concentration in dioxane and 120 °C.
In summary, we have reported the catalyst-free depolymerization of RAFT polymethacrylates consisting of various end groups such as trithiocarbonate and pyrazole carbodithioate. Our method also proved to be compatible with a range of solvents, with dioxane, xylene, toluene, and DMF giving the highest depolymerization conversions. In addition, the depolymerization of polymers consisting of different molecular weights was presented, and the observed differences in the final conversions were attributed to the loss of end-group fidelity. Last but not least, we demonstrated that this low-temperature methodology has a key advantage over pyrolysis in that heat-sensitive polymers (e.g., poly(tert-butyl methacrylate)) can also be depolymerized back into their original monomers in the absence of any side reactions.
Acknowledgments
A.A. gratefully acknowledges ETH Zurich for financial support. N.P.T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076). H.S.W. acknowledges the award of the Swiss Government Excellence Scholarship (ESKAS No. 2020.0324). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (DEPO: Grant Agreement No. 949219).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmacrolett.2c00506.
General information, experimental procedures, 1H NMR spectra, and SEC traces (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Parkatzidis K.; Wang H. S.; Truong N. P.; Anastasaki A. Recent developments and future challenges in controlled radical polymerization: A 2020 update. Chem. 2020, 6 (7), 1575–1588. 10.1016/j.chempr.2020.06.014. [DOI] [Google Scholar]
- Corrigan N.; Jung K.; Moad G.; Hawker C. J.; Matyjaszewski K.; Boyer C. Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications. Prog. Polym. Sci. 2020, 111, 101311. 10.1016/j.progpolymsci.2020.101311. [DOI] [Google Scholar]
- Gentekos D. T.; Sifri R. J.; Fors B. P. Controlling polymer properties through the shape of the molecular-weight distribution. Nat. Rev. Mater. 2019, 4 (12), 761–774. 10.1038/s41578-019-0138-8. [DOI] [Google Scholar]
- De Neve J.; Haven J. J.; Maes L.; Junkers T. Sequence-definition from controlled polymerization: the next generation of materials. Polym. Chem. 2018, 9 (38), 4692–4705. 10.1039/C8PY01190G. [DOI] [Google Scholar]
- Gody G.; Maschmeyer T.; Zetterlund P. B.; Perrier S. Rapid and quantitative one-pot synthesis of sequence-controlled polymers by radical polymerization. Nat. Commun. 2013, 4 (1), 2505. 10.1038/ncomms3505. [DOI] [PubMed] [Google Scholar]
- Dadashi-Silab S.; Atilla Tasdelen M.; Yagci Y. Photoinitiated atom transfer radical polymerization: Current status and future perspectives. J. Polym. Sci., Part A: Polym. Chem. 2014, 52 (20), 2878–2888. 10.1002/pola.27327. [DOI] [Google Scholar]
- Bates C. M.; Maher M. J.; Janes D. W.; Ellison C. J.; Willson C. G. Block Copolymer Lithography. Macromolecules 2014, 47 (1), 2–12. 10.1021/ma401762n. [DOI] [PubMed] [Google Scholar]
- Figg C. A.; Simula A.; Gebre K. A.; Tucker B. S.; Haddleton D. M.; Sumerlin B. S. Polymerization-induced thermal self-assembly (PITSA). Chem. Sci. 2015, 6 (2), 1230–1236. 10.1039/C4SC03334E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Farias-Mancilla B.; Kulai I.; Hoeppener S.; Lonetti B.; Prévost S.; Ulbrich J.; Destarac M.; Colombani O.; Schubert U. S.; Guerrero-Sanchez C.; Harrisson S. Effect of Hydrophilic Monomer Distribution on Self-Assembly of a pH-Responsive Copolymer: Spheres, Worms and Vesicles from a Single Copolymer Composition. Angew. Chem., Int. Ed. 2021, 60 (9), 4925–4930. 10.1002/anie.202010501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58 (6), 379–410. 10.1071/CH05072. [DOI] [Google Scholar]
- Barner-Kowollik C.Handbook of RAFT Polymerization; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Xu J.; Jung K.; Atme A.; Shanmugam S.; Boyer C. A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and Its Oxygen Tolerance. J. Am. Chem. Soc. 2014, 136 (14), 5508–5519. 10.1021/ja501745g. [DOI] [PubMed] [Google Scholar]
- Allegrezza M. L.; Konkolewicz D. PET-RAFT Polymerization: Mechanistic Perspectives for Future Materials. ACS Macro Lett. 2021, 10 (4), 433–446. 10.1021/acsmacrolett.1c00046. [DOI] [PubMed] [Google Scholar]
- Truong N. P.; Jones G. R.; Bradford K. G. E.; Konkolewicz D.; Anastasaki A. A comparison of RAFT and ATRP methods for controlled radical polymerization. Nat. Rev. Chem. 2021, 5, 859–869. 10.1038/s41570-021-00328-8. [DOI] [PubMed] [Google Scholar]
- IUPAC Top Ten Emerging Technologies in Chemistry. https://iupac.org/what-we-do/top-ten/.
- Lloyd D. J.; Nikolaou V.; Collins J.; Waldron C.; Anastasaki A.; Bassett S. P.; Howdle S. M.; Blanazs A.; Wilson P.; Kempe K.; Haddleton D. M. Controlled aqueous polymerization of acrylamides and acrylates and “in situ” depolymerization in the presence of dissolved CO2. Chem. Commun. 2016, 52 (39), 6533–6536. 10.1039/C6CC03027K. [DOI] [PubMed] [Google Scholar]
- Sano Y.; Konishi T.; Sawamoto M.; Ouchi M. Controlled radical depolymerization of chlorine-capped PMMA via reversible activation of the terminal group by ruthenium catalyst. Eur. Polym. J. 2019, 120, 109181. 10.1016/j.eurpolymj.2019.08.008. [DOI] [Google Scholar]
- Martinez M. R.; Dadashi-Silab S.; Lorandi F.; Zhao Y.; Matyjaszewski K. Depolymerization of P(PDMS11MA) Bottlebrushes via Atom Transfer Radical Polymerization with Activator Regeneration. Macromolecules 2021, 54 (12), 5526–5538. 10.1021/acs.macromol.1c00415. [DOI] [Google Scholar]
- Martinez M. R.; Matyjaszewski K. Degradable and Recyclable Polymers by Reversible Deactivation Radical Polymerization. CCS Chem. 2022, 4 (7), 2176–2211. 10.31635/ccschem.022.202201987. [DOI] [Google Scholar]
- Martinez M. R.; De Luca Bossa F.; Olszewski M.; Matyjaszewski K. Copper(II) Chloride/Tris(2-pyridylmethyl)amine-Catalyzed Depolymerization of Poly(n-butyl methacrylate). Macromolecules 2022, 55 (1), 78–87. 10.1021/acs.macromol.1c02246. [DOI] [Google Scholar]
- Flanders M. J.; Gramlich W. M. Reversible-addition fragmentation chain transfer (RAFT) mediated depolymerization of brush polymers. Polym. Chem. 2018, 9 (17), 2328–2335. 10.1039/C8PY00446C. [DOI] [Google Scholar]
- Wang H. S.; Truong N. P.; Pei Z.; Coote M. L.; Anastasaki A. Reversing RAFT Polymerization: Near-Quantitative Monomer Generation Via a Catalyst-Free Depolymerization Approach. J. Am. Chem. Soc. 2022, 144 (10), 4678–4684. 10.1021/jacs.2c00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-W.; Bays E.; Tao L.; Alconcel S. N. S.; Maynard H. D. Differences in cytotoxicity of poly(PEGA)s synthesized by reversible addition–fragmentation chain transfer polymerization. Chem. Commun. 2009, 24, 3580–3582. 10.1039/b904456f. [DOI] [PubMed] [Google Scholar]
- Whitfield R.; Parkatzidis K.; Truong N. P.; Junkers T.; Anastasaki A. Tailoring polymer dispersity by RAFT polymerization: A versatile approach. Chem. 2020, 6 (6), 1340–1352. 10.1016/j.chempr.2020.04.020. [DOI] [Google Scholar]
- Benaglia M.; Chiefari J.; Chong Y. K.; Moad G.; Rizzardo E.; Thang S. H. Universal (Switchable) RAFT Agents. J. Am. Chem. Soc. 2009, 131 (20), 6914–6915. 10.1021/ja901955n. [DOI] [PubMed] [Google Scholar]
- Chong B.; Moad G.; Rizzardo E.; Skidmore M.; Thang S. H. Thermolysis of RAFT-Synthesized Poly(Methyl Methacrylate). Aust. J. Chem. 2006, 59 (10), 755–762. 10.1071/CH06229. [DOI] [Google Scholar]
- Thomas D. B.; Convertine A. J.; Hester R. D.; Lowe A. B.; McCormick C. L. Hydrolytic Susceptibility of Dithioester Chain Transfer Agents and Implications in Aqueous RAFT Polymerizations. Macromolecules 2004, 37 (5), 1735–1741. 10.1021/ma035572t. [DOI] [Google Scholar]
- Xu J.; He J.; Fan D.; Wang X.; Yang Y. Aminolysis of Polymers with Thiocarbonylthio Termini Prepared by RAFT Polymerization: The Difference between Polystyrene and Polymethacrylates. Macromolecules 2006, 39 (25), 8616–8624. 10.1021/ma061961m. [DOI] [Google Scholar]
- Grant D. H.; Grassie N. The thermal decomposition of poly(t-butyl methacrylate). Polymer 1960, 1, 445–455. 10.1016/0032-3861(60)90060-4. [DOI] [Google Scholar]
- Novaković K.; Katsikas L.; Popović I. G. The thermal degradation of poly (iso-butyl methacrylate) and poly (sec-butyl methacrylate). J. Serb. Chem. Soc. 2000, 65 (12), 867–875. 10.2298/JSC0012867N. [DOI] [Google Scholar]
- Wang H. S.; Oh S.; Choi J.; Jang W.; Kim K. H.; Arellano C. L.; Huh J.; Bang J.; Im S. G. High-Fidelity, Sub-5 nm Patterns from High-χ Block Copolymer Films with Vapor-Deposited Ultrathin, Cross-Linked Surface-Modification Layers. Macromol. Rapid Commun. 2020, 41 (4), 1900514. 10.1002/marc.201900514. [DOI] [PubMed] [Google Scholar]
- Grassie N. Recent work on the thermal degradation of acrylate and methacrylate homopolymers and copolymers. Pure Appl. Chem. 1972, 30, 119–134. 10.1351/pac197230010119. [DOI] [Google Scholar]
- Jones M.-C.; Tewari P.; Blei C.; Hales K.; Pochan D. J.; Leroux J.-C. Self-Assembled Nanocages for Hydrophilic Guest Molecules. J. Am. Chem. Soc. 2006, 128 (45), 14599–14605. 10.1021/ja065462c. [DOI] [PubMed] [Google Scholar]
- Muzammil E. M.; Khan A.; Stuparu M. C. Post-polymerization modification reactions of poly(glycidyl methacrylate)s. RSC. Adv. 2017, 7 (88), 55874–55884. 10.1039/C7RA11093F. [DOI] [Google Scholar]
- Han E.; Gopalan P. Cross-Linked Random Copolymer Mats As Ultrathin Nonpreferential Layers for Block Copolymer Self-Assembly. Langmuir 2010, 26 (2), 1311–1315. 10.1021/la902483m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.