

Conspectus

Nearly a century after its first description, configurationally stable axial chirality remains a rare feature in marketed drugs. In the development of the KRASG12C inhibitor sotorasib (LUMAKRAS/LUMYKRAS), an axially chiral biaryl moiety proved a critical structural element in engaging a “cryptic” protein binding pocket and enhancing inhibitor potency. Restricted rotation about this axis of chirality gave rise to configurationally stable atropisomers that demonstrated a 10-fold difference in potency. The decision to develop sotorasib as a single-atropisomer drug gave rise to a range of analytical and synthetic challenges, whose resolution we review here.
Assessing the configurational stability of differentially substituted biaryl units in early inhibitor candidates represented the first challenge to be overcome, as differing atropisomer stability profiles called for differing development strategies (e.g., as rapidly equilibrating rotamers vs as single atropisomers). We relied on a range of NMR, HPLC, and computational methods to assess atropisomer stability. Here, we describe the various variable-temperature NMR, time-course NMR, and chiral HPLC approaches used to assess the configurational stability of axially chiral bonds displaying a range of rotational barriers.
As optimal engagement of the “cryptic” pocket of KRASG12C was ultimately achieved with a configurationally stable atropisomeric linkage, the second challenge to be overcome entailed preparing the preferred (M)-atropisomer of sotorasib on industrial scale. This synthetic challenge centered on the large-scale synthesis of an atropisomerically pure building block comprising the central azaquinazolinone and pyridine rings of sotorasib. We examined a range of strategies to prepare this compound as a single atropisomer: asymmetric catalysis, chiral chromatographic purification, and classical resolution. Although chiral liquid and simulated moving bed chromatography provided expedient access to initial multikilo supplies of this key intermediate, a classical resolution process was ultimately developed that proved significantly more efficient on metric-ton scale. To avoid discarding half of the material from this resolution, this process was subsequently refined to enable thermal recycling of the undesired atropisomer, providing an even more efficient commercial process that proved both robust and green.
While the preparation of sotorasib as a single atropisomer significantly increased both the analytical and synthetic complexity of its development, the axially chiral biaryl linkage that gave rise to the atropisomerism of sotorasib proved a key design element in optimizing sotorasib’s binding to KRASG12C. It is hoped that this review will help in outlining the range of analytical techniques and synthetic strategies that can be brought to bear in addressing the challenges posed by such axially chiral compounds and that this account may provide helpful guidelines for future efforts aimed at the development of such single atropisomer, axially chiral pharmaceutical agents.
Key References
Lanman B. A.; Allen J. R.; Allen J. G.; Amegadzie A. K.; Ashton K. S.; Booker S. K.; Chen J. J.; Chen N.; Frohn M. J.; Goodman G.; Kopecky D. J.; Liu L.; Lopez P.; Low J. D.; Ma V.; Minatti A. E.; Nguyen T. T.; Nishimura N.; Pickrell A. J.; Reed A. B.; Shin Y.; Siegmund A. C.; Tamayo N. A.; Tegley C. M.; Walton M. C.; Wang H. L.; Wurz R. P.; Xue M.; Yang K. C.; Achanta P.; Bartberger M. D.; Canon J.; Hollis L. S.; McCarter J. D.; Mohr C.; Rex K.; Saiki A. Y.; San Miguel T.; Volak L. P.; Wang K. H.; Whittington D. A.; Zech S. G.; Lipford J. R.; Cee V. J.. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65.1Details the structure-based design of sotorasib (AMG 510), motivation for introducing an axially chiral biaryl linkage, and efforts to address the configurational instability of early biaryl atropisomers. Additionally describes the biopharmaceutical optimization and pharmacological characterization of sotorasib.
Parsons A. T.; Caille S.; Caporini M. A.; Griffin D. J.; Lovette M. A.; Powazinik IV W.; St-Pierre G.. Axial chirality in the sotorasib drug substance, Part 1: Development of a classical resolution to prepare an atropisomerically-pure sotorasib intermediate. Org. Process Res. Dev. 2022, 26, 2629–2635.2Reports the discovery of (+)-2,3-dibenzoyl-d-tartaric acid [(+)-DBTA] as a resolving agent that enables the classical resolution of a key atropisomeric intermediate in the synthesis of sotorasib. Development of a commercial process that provides >2000:1 selectivity on >500 kg scale is described.
Beaver M. G.; Brown D. B.; Campbell K.; Fang Y.-Q.; Ford D. D.; Mardirossian N.; Nagy K. D.; Rotheli A. R.; Sheeran J. W.; Telmesani R.; Parsons A. T.. Axial chirality in the sotorasib drug substance, Part 2: Leveraging a high-temperature thermal racemization to recycle the classical resolution waste stream. Org. Process Res. Dev. 2022, 26, 2636–2645.3Describes the development of a continuous-flow racemization process to recycle the off-enantiomer of an atropisomeric sotorasib intermediate obtained from the classical resolution. Additionally reports the demonstration of this process on kilogram scale.
1. Ligand Design: Axial Chirality as a Structural Tool
Although the topic of atropisomerism4 in the pharmaceutical sciences has received growing attention over the past decade,5−9 the presence of configurationally stable, axially chiral stereochemical elements in marketed drugs remains a rarity a century after the first report of axial atropisomerism.10 To date, only four United States Food and Drug Administration (FDA)-approved drugs incorporate such a stereochemical feature: the natural products vancomycin and colchicine, whose axial chirality is enforced as a result of conventional stereocenters in rings bridging their biaryl motifs;8,11 lesinurad, whose configurational stability was only recognized postlaunch (through separation of the racemic marketed product);12 and sotorasib (LUMAKRAS/LUMYKRAS), which represents the first FDA-approved therapy to be manufactured and marketed as a configurationally stable, atropisomerically pure compound (Figure 1).1 In this work, we describe the factors that led to the decision to incorporate an axially chiral linkage in the structure of sotorasib as well as the analytical and synthetic techniques used to address the consequences of that decision.
Figure 1.
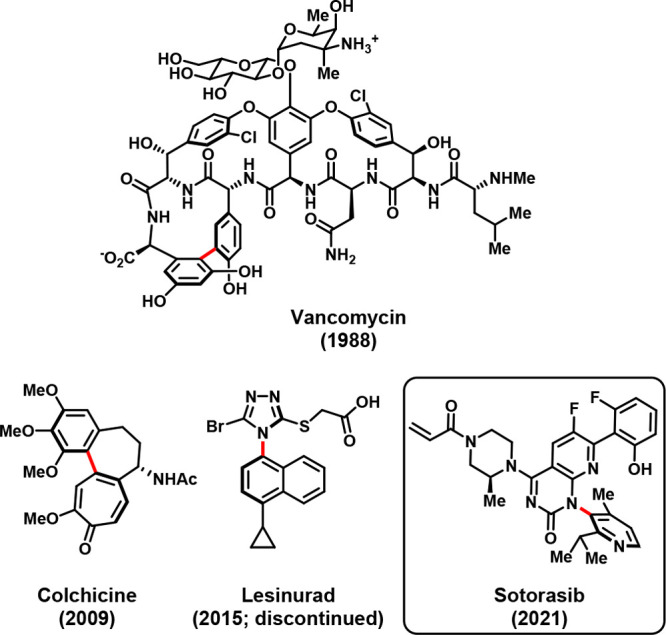
FDA-approved drugs containing axially chiral atropisomeric bonds.
The decision to incorporate an axially chiral linkage in sotorasib arose from a specific structural need: to provide a structural motif capable of accessing a “cryptic” pocket13 on the surface of the target protein, KRASG12C, that had been discovered through our earlier screening efforts.14 As shown in Figure 2, the biaryl linkage15 of quinazolinone lead 2 provided a synthetically accessible, readily diversified structural element capable of positioning a substituent in the H95/Y96/Q99 cryptic pocket (magenta), thereby mimicking the role played by the tetrahydroisoquinoline amide of indole lead 1 (which similarly accessed this cryptic pocket via an atropisomeric linkage (red bond)—albeit a nonconfigurationally stable one).16
Figure 2.

Axial chirality as a structural element in accessing a novel pocket in the design of sotorasib. An axis of chirality (red bond) in indole lead 1 facilitated occupancy of the H95/Y96/Q99 “cryptic” pocket (magenta) of KRASG12C by a tetrahydroisoquinoline substituent (green), enhancing inhibit or potency. The metabolic instability of compound 1 prevented its further development; however, analogous use of an axially chiral linkage in quinazolinone lead 2 enabled similar occupancy of the H95/Y96/Q99 pocket while affording KRASG12C inhibitors with significantly enhanced metabolic stability. Optimization of scaffold 2 led to the invention of sotorasib. Data from ref (1) and ref (14).
The decision to retain the atropisomeric linkage of quinazolinone 2 in the final structure of sotorasib was not taken lightly. Axial chirality differs from more conventional point chirality in that the racemization of atropisomers does not depend on bond-breaking processes, but only upon simple rotational interconversion. Therefore, the energetic barriers to atropisomer isomerization are typically significantly lower than those of point stereocenter racemization processes, posing considerable added analytical and synthetic challenges for the development of axially chiral compounds.6,17
In the following sections, we survey the range of analytical techniques we used to characterize the configurational stability of the diverse atropisomeric analogs prepared in the optimization of sotorasib as well as the various synthetic strategies employed in accessing atropisomerically pure, axially chiral compounds efficiently, economically, and on commercial scale.18 As such efforts were central to the registration of the first atropisomerically pure, axially chiral pharmaceutical agent, it is hoped that this overview may provide helpful guidelines for future efforts seeking to bring single-atropisomer compounds to market.
2. Conformational Analysis: Interconversion Barriers and Conformational Stability
Efforts to optimize quinazolinone scaffold 2 initially focused on modification of the ortho substituent of the biaryl moiety (green ball, Figure 2) to optimally engage the cryptic pocket of KRASG12C as well as modification of the monocyclic arene ring. Both modifications contributed to changes in the configurational stability of the axially chiral biaryl system (Figure 3).
Figure 3.
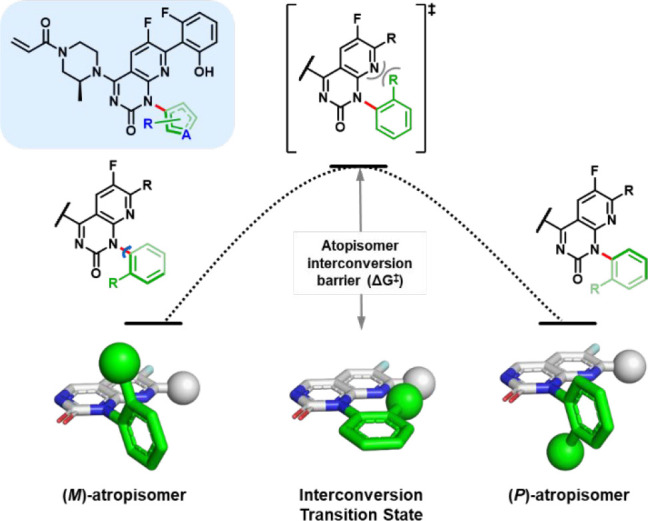
Interconversion of biaryl atropisomers. Rotation about the axially chiral biaryl bond (red) allows for (M)- and (P)-atropisomer interconversion. Intramolecular interactions (e.g., steric, electronic) in the interconversion transition state lead to varying atropisomer interconversion barriers (ΔG⧧), whose magnitudes depend on the identity and substitution pattern of the rings of the biaryl system.
The configurational stability of an atropisomeric linkage, such as that present in quinazolinone 2, can have significant implications for the way in which an atropisomeric compound is developed for pharmaceutical use (e.g., as a mixture or single atropisomer; Table 1), as has been expertly reviewed.5,6 Conformational stability is typically assessed by estimation of the free-energy of activation for atropisomer interconversion (ΔG⧧) (Figure 3). During research efforts toward the discovery of sotorasib, we sought to optimize the biaryl substituent of scaffold 2 (e.g., ring-size and substitution pattern) to maximize productive interactions with the H95/Y96/Q99 pocket of KRASG12C while avoiding the generation of analogs with low-to-moderate atropisomeric stability (ΔG⧧ of 20–30 kcal/mol),1 as the configurational instability of such compounds was expected to pose significant regulatory challenges.19 To assess atropisomer configurational stability, we relied on a range of analytical techniques (see Table 2), each of which proved useful for characterizing conformational dynamics occurring on differing time scales and at differing interconversion energies.
Table 1. Favored Development Strategy As a Function of Atropisomer Interconversion Barrier (ΔG⧧)a.
| Interconversion barrier (ΔG⧧; kcal/mol) | Isomerization time (1% conversion; 23 °C) | Development strategy |
|---|---|---|
| <20 (‘Class 1’) | <1 s | Rapidly equilibrating mixtureb |
| 20–30 (‘Class 2’) | 1 s to 9 months | Case dependent; disfavored for developmentc |
| >30 (‘Class 3’) | >9 months | Single atropisomerd |
Interconversion rapid versus pharmacological and manufacturing processes.
Interconversion on the same time scale as pharmacological and manufacturing processes.
Interconversion slow versus pharmacological and manufacturing processes.
Table 2. Choosing an Experimental Technique for Configurational Stability Assessment.
| Technique | Favored by | Disfavored when | Compound requirement |
|---|---|---|---|
| VT-NMR: Line-shape analysis, EXSY, or kinetic analyses | Fast interconversion (t1/2 < 1 s,26 e.g., analysis of nonchromatographically separable isomer mixtures), ΔG‡ < 23 kcal/mol27 | Atropisomers are enantiomers or chemically unstable at elevated temperature | ∼3 mg of atropisomer mixture (or atropisomerically enriched material) |
| Time-course NMR | Medium-to-slow interconversion within the accessible temperature rage (t1/2 > 5 min23), ΔG‡ = 22–33 kcal/mol28 | Atropisomers are enantiomers or unstable, chiral separation is not feasible | ∼3 mg of atropisomerically enriched material |
| Chiral Chromatography (e.g., HPLC, SFC) | Slow interconversion (t1/2 > 20 min23), ΔG‡ > 24 kcal/mol | Atropisomers are rapidly converting, or chiral separation is not feasible | <1 mg of atropisomerically enriched material |
NMR techniques proved one of the most versatile means of detecting and analyzing atropisomer mixtures. In the case of quinazolinone analogs such as 2, the presence of a second stereocenter in a molecule (i.e., the (S)-methyl piperazine) rendered the two (M)- and (P)-atropisomers21 diastereomeric. Diastereotopic NMR resonances arising from the two atropisomers therefore provided a first indication of the presence of restricted rotation arising from the axial chirality in compound 2.22
In the case of scaffold 2, variable-temperature NMR (VT-NMR) studies proved a highly useful tool in assessing the activation energy barrier (ΔG⧧) for atropisomer interconversion. For compounds with low interconversion barriers (ΔG⧧), spectral line broadening (and/or merging) at elevated temperatures allowed for the direct calculation of ΔG⧧, ΔH⧧, and ΔS⧧ from the exchange rate constants (k) or half-lives (t1/2) of the interconversion processes. For compounds with slower exchange rate constants (i.e., higher interconversion energy barriers), a range of additional NMR techniques also proved useful:23
When interconversion rates were short on an NMR time-scale (i.e., t1/2 < 1 s), line-shape analysis could be used to determine interconversion rate constants (k) by simulating exchange processes using dynamic NMR models.24,25
In cases where an intermediate exchange regime29 was observed (i.e., k ≈ frequency difference (Δν0) of monitored nuclei), rate constants (k) could be calculated from Δν0 of the diastereotopic nuclei at the coalescence temperature (Tc) of the interconverting atropisomers.30
In the slow-to-intermediate exchange regime29 (k < Δν0), 2D exchange spectroscopy (EXSY) could be used to calculate k with increased sensitivity by measuring the intensity of chemical-exchange cross peaks as a function of mixing time.31,32
For systems with higher rotational barriers, kinetic analysis (i.e., time-course NMR) could be used to monitor the isomerization of purified atropisomers as a function of time and temperature, deriving the exchange rate (k) and reaction half-life (t1/2) from first-order reaction kinetics.33
Atropisomer configurational stability analysis also took place against a background of other rotational and conformationally dynamic processes in the sotorasib scaffold, all of which additionally contributed to the generation of diastereotopic 1H NMR resonances on the NMR-time scale (Figure 4). Conformationally dynamic features of the sotorasib scaffold included:
-
A.
The axially chiral biaryl linkage (source of atropisomerism in most sotorasib analogs)
-
B.
A second axially chiral bond (not typically a source of configurationally stable rotamers due to low interconversion barriers at ambient temperature)
-
C.
A conformationally flexible piperazine ring/acrylamide “warhead”, giving rise to E-/Z-acrylamide isomers, piperazine conformers, and rotational isomers about the piperazine/quinazolinone bond (not typically a source of configurationally stable isomers at ambient temperature)
-
D.
Proton exchange between the phenolic hydroxyl group and basic sites on the molecule/solvent.
Figure 4.
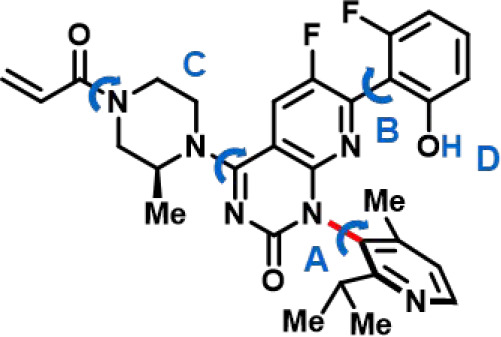
Conformationally/chemically dynamic features of the sotorasib scaffold (A–D), each demonstrating distinct activation energy barriers and NMR exchange kinetics. Restricted rotation about bond A (red) gives rise to configurationally stable (M)- and (P)-atropisomers.
As an illustration of the use of VT-NMR line-shape analysis in determining the interconversion energy barriers for several of these dynamic processes, below, we describe two processes within the sotorasib scaffold (bond A and B rotation) that have sufficiently different activation energy barriers to allow for their concurrent analysis using simple two-site exchange models (Figure 5A,B).
Figure 5.

VT-NMR analysis: (A) Structure of compound (M)-3 and temperature dependence of the proton-decoupled 19F NMR spectrum, monitoring fluorophenyl ring rotation from slow- to fast-exchange regimes. (B) Structure of compound 4 (mixture of atropisomers) and temperature dependence of 1H NMR spectra of the azaquinazolinone H5 proton (blue), monitoring (M)- and (P)-atropisomer interconversion. Peak separations in the slow-exchange limit are indicated by Δv0. Both samples were dissolved at ∼5 mg/mL in DSMO-d6. NMR measurements were performed on a Bruker Avance III 500 MHz spectrometer with a BBFO probe.
In Figure 5A, proton-decoupled 19F VT-NMR was used to determine the barrier for fluorophenyl ring rotamer interconversion in compound (M)-3.34,35 At 300 K, the 19F NMR spectrum of (M)-3 consists of two signals with similar integrals, reflecting two rotational isomers that are in slow exchange on the NMR time scale. This two-site exchange can be described by a first-order reaction approaching equilibrium:23
| 1 |
As the forward and reverse rates of interconversion between the (M)- and (P)-fluorophenol rotamers are very similar (k+1 ≈ k–1), nearly equal concentrations of the two rotamers are observed at equilibrium (k+1/k–1 = [M]eq/[P]eq ≈ 1), as is reflected by the similar integrals for the two 19F signals.
At elevated temperatures, the 19F NMR signals for (M)-3 gradually broaden (reflecting increasingly rapid rotation about bond B), ultimately reaching coalescence at ∼327 K.36 At the coalescence temperature (Tc), the exchange rate (k) between the two rotamers can be calculated from the frequency difference Δv0 between these 19F peaks in the limit of slow exchange:23,30
| 2 |
The activation free energy for this rotational exchange process (ΔG⧧) can then be calculated from the Eyring equation:23,37,38
| 3 |
By this methodology, given a frequency difference (Δv0) of 97 ± 2 Hz in the absence of exchange, the exchange rate at Tc = 327 ± 2 K can be calculated as k = 215 ± 4 s–1, corresponding to a ΔG⧧ of 15.7 ± 0.1 kcal mol–1.
Figure 5B illustrates a similar VT-NMR analysis of atropisomer interconversion rates, this time using the diastereotopic 1H NMR resonances of the quinazolinone H5 hydrogen atom to monitor the interconversion of biaryl (“bond A”) atropisomers. This figure additionally illustrates the complex NMR spectra that can result from the concomitant contributions of multiple rotationally mobile bonds. At ambient temperature, H5 shows not only two distinct 1H NMR resonances arising from the two “bond A” atropisomers, but also significant broadening/splitting of these resonances resulting from the presence of two “bond B” (fluorophenyl ring) atropisomers (cf.Figure 4).
At temperatures >340 K, fluorophenyl rotation rates enter the fast-exchange regime, resulting in the sharpening of the H5 resonances into two narrow peaks of similar intensity, corresponding to the two “bond A” atropisomers. With further increases in temperature, these two peaks broaden and begin to coalesce, with a Tc ≈ 380 ± 2 K. Given a peak separation of Δv0 = 27 ± 1 Hz for the “bond A” atropisomers (i.e., at 343 K), this results in a calculated exchange rate of k = 60 ± 2 s–1 at Tc and a ΔG⧧ = 19.3 ± 0.1 kcal mol–1.
As outlined by LaPlante (Table 1),6 the low interconversion barriers for both of the atropisomer interconversion processes depicted in Figure 5 make these processes of little concern from a development perspective: atropisomer conversion about “bond A” of compound 4 and “bond B” of (M)-3 is rapid on a pharmacological and manufacturing time scale, and so no effort was made to separately isolate and characterize these rotational isomers. Unfortunately, neither of these compounds ultimately proved suitable for further development, and optimization efforts continued with more highly conformationally restricted biaryl atropisomers.1
A key limitation of VT-NMR methods is the requirement that rapid atropisomer interconversion rates be obtained at temperatures compatible with NMR analysis. To assess KRAS inhibitors possessing higher atropisomer interconversion barriers (i.e., up to 32–33 kcal/mol), we turned to time-course NMR experiments, where we monitored interconversion dynamics over extended periods at elevated temperatures.
Figure 6 illustrates the use of time-course NMR studies to assess the atropisomer (“bond A”) interconversion barrier of a more highly conformationally restricted inhibitor, compound 5. In this experiment, an (M)-atropisomer-enriched sample of 5 was incubated at 325 K for ∼19 h, and the diastereotopic quinazolinone F6 19F NMR resonances (Figure 6C; corresponding to the two “bond A” atropisomers) were integrated at successive time-points to provide a time-course profile of evolving (M)- and (P)-5 concentrations (Figure 6B).
Figure 6.

Time-course NMR analysis: (A) Structure of compound 5 (mixture of atropisomers). Fluorine atom (F6, magenta) was used to monitor (M)- and (P)-atropisomer interconversion (note: F6 shows a splitting of 5JF–F = 8.6 Hz resulting from the proximal fluorophenol ring). (B) (M)- and (P)-atropisomer interconversion kinetics (T = 325 K). Atropisomer ratios (colored data points) were obtained by integration, normalizing the sum of the integrals to 100%. Solid lines show a fit to first-order kinetics with a half-life (t1/2) of 334 min. (C) Proton-decoupled 19F NMR spectra (Bruker AVIII-500, ∼5 mg/mL in DMSO-d6) of F6 collected at 325 K (time points as indicated).
For a two-site exchange process (eq 1), the rate of (M)-atropisomer change over time is given by
| 4 |
where k+1 and k–1 are the rate constants for forward and reverse reaction, respectively.33 With the initial concentration of [M] = [M]0 and [P] = 0, this time dependence can be expressed as
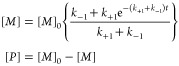 |
5 |
Fitting the (M)- and (P)-fractions in Figure 6B to eq 5 revealed rate constants of k+1 ≈ k–1 = 3.46 ± 0.05 × 10–5 s–1 and a corresponding half-life of t1/2 = 334 ± 5 min, which (using eq 3) equated to an activation energy of ΔG⧧ = 25.7 ± 0.1 kcal mol–1 at 325 ± 1 K.
This represented a significantly higher barrier to atropisomer interconversion than for the previously described compounds, and one expected to pose a significant development challenge, as atropisomer interconversion of compound 5 would be expected to occur on the same time-scale as that of pharmacological and manufacturing processes. Accordingly, although compounds such as quinazolinone 5 proved potent KRASG12C inhibitors, further optimization was pursued to identify similarly potent inhibitors with atropisomer interconversion barriers <20 kcal/mol or >30 kcal/mol (cf. Table 1 guidelines).
Such optimization efforts, more fully described in ref (1), ultimately led to the identification of sotorasib (Figure 1). Using similar time-course NMR studies, the atropisomer interconversion barrier for sotorasib was determined to be >33.5 kcal mol–1, corresponding to an atropisomer interconversion half-life of >1000 h at 373 K. Slow decomposition of sotorasib in DMSO at T > 380 K prevented more precise determination of the interconversion barrier; however, experiments with synthetic precursors (see below) have helped to more precisely define the interconversion barrier for the pyridine/quinazolinone system of sotorasib.
As previously noted, direct NMR analysis of atropisomer mixtures relies on the presence of a second stereocenter in the analyte molecule to render the atropisomers diastereotopic. Although enantiomeric atropisomers may be studied spectroscopically by using chemical shift reagents or anisotropic media,39,40 a potentially more straightforward approach employs chiral chromatographic analysis. In the case of sotorasib, the synthetic precursor rac-6 (Figure 7) possessed a single chiral axis (red) and no additional stereocenters. Restricted rotation about this axis resulted in the formation of two enantiomeric atropisomers (M)- and (P)-6, which could not be distinguished using standard NMR approaches.
Figure 7.

Chiral chromatographic analysis: (A) Thermal racemization of (P)-6. (B) HPLC chromatograms illustrating the racemization (P)-6 as a function of time. (C) Plot of (P)-6 enantiomeric excess (derived from HPLC peak integration) as a function of time, following incubation at 280 °C in diphenyl ether.
To assess the atropisomer interconversion barrier of compound 6, a chiral high-performance liquid chromatography (HPLC) method was developed to separate (M)- and (P)-6, enabling time-course studies of the thermally induced racemization of (P)-6.41 In such studies, a solution of (P)-6 in diphenyl ether was superheated to 280 °C (boiling point = 258 °C) and samples were collected over ∼2 h to determine the degree of racemization at each time point (Figure 7B,C). Fitting the exponential decay of (P)-6 concentrations over time allowed for the calculation of the racemization rate constant, k (2.21 × 10–4 s–1), and corresponding interconversion free energy, ΔG⧧ (42.3 kcal/mol), resulting in a calculated racemization half-life of >10 billion years (at 25 °C), confirming the exceptional configurational stability of this biaryl linkage.
Although the prior discussion focused on experimental methods to establish atropisomer interconversion barriers, computational (quantum-mechanical) methods of estimating such barriers have also become quite reliable6,20,42 and can serve as a convenient method for assessing rotational restriction about an axially chiral bond [and for predicting the most appropriate experimental approach to use in assessing atropisomer interconversion barriers (cf. Table 2)]. Figure 8 illustrates the application of density functional theory (DFT) calculations at the B3LYP/6-31+G(d,p) level of theory to estimate the interconversion barrier of rac-6, which is predicted to be 40.7 kcal/mol, in good correspondence with the experimentally determined barrier of 42.3 kcal/mol.
Figure 8.
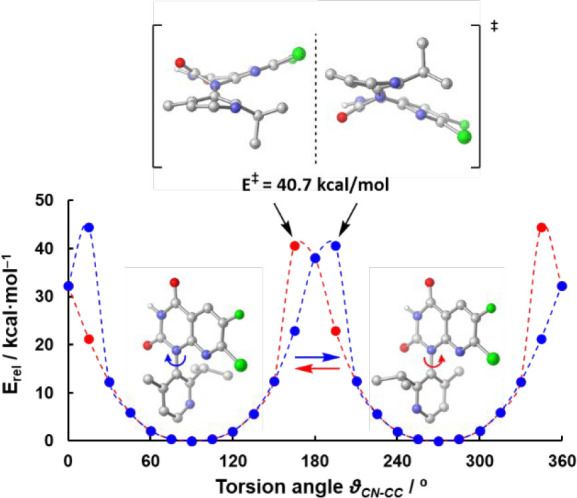
Computational analysis: DFT analysis (B3LYP/6-31+G(d,p)) of the torsional energy profile of the atropisomeric axis of compound 6.
3. Synthetic Considerations: Preparing Single Atropisomers on Scale
Having identified the atropisomeric biaryl bond of sotorasib as a key structural element in the design of potent KRASG12C inhibitors and established the exceptional configurational stability of the specific biaryl system present in sotorasib through careful analytical work, our efforts subsequently turned to the practical question of how best to prepare the (M)-atropisomer of sotorasib on industrial scale. During discovery efforts, the (M)-atropisomer was isolated by chiral chromatographic separation of the two atropisomers obtained upon completion of the synthesis.1 Given the low throughput, significant solvent waste, and poor scalability of such a chromatographic separation, finding ways to install the (M)-atropisomeric axis without resorting to late-stage chiral separation became a key focus at the outset of process development efforts.
The earliest atropisomeric intermediate in the synthesis of sotorasib, rac-6, was formed in just two steps from raw materials nicotinamide 7 and aniline 8, proceeding through nonisolated intermediate 9 (Figure 9).43 Several strategies were envisioned for accessing atropisomerically pure (M)-6 while retaining 7 and 8 as raw materials: (1) asymmetric ring closure of intermediate 9, (2) chiral chromatographic separation of rac-6, and (3) classical resolution of rac-6.
Figure 9.

Preparation of rac-6 and approaches to prepare (M)-6.
Asymmetric synthesis was initially envisioned as being the most efficient means of accessing (M)-6, and initial efforts toward this goal focused on the identification of suitable catalysts for the asymmetric ring closure of intermediate 9, given established supply chains for starting materials 7 and 8, which promised to expedite future scale-up. Although multiple approaches44−46 were pursued—including asymmetric intramolecular cross-coupling, various organocatalytic methods (hydrogen bonding, chiral Brønsted acids, and nucleophilic catalysis), and stochiometric methods (chiral bases)—ultimately, none of these approaches met the required yield or selectivity requirements for the program.47
Given an aggressive timeline to deliver kilogram quantities of (M)-6 to support early development efforts, it was determined that chromatographic separation of rac-6 would be a pragmatic, phase-appropriate approach to provide material for Investigational New Drug-enabling toxicology studies and first-in-human clinical trials. Indeed, the first delivery of sotorasib for clinical studies relied on traditional chiral chromatography and delivered ∼2 kg of sotorasib drug substance. For a subsequent 10 kg delivery of drug substance, simulated moving bed chromatography was utilized for chiral separation, which reduced both processing times and solvent use when compared to traditional chromatographic separation.
While preparative chromatography was key to establishing material supplies for early clinical development, it was not considered a preferred option for commercialization. In addition to the previously mentioned shortcomings (i.e., solvent waste, low throughput, etc.), such an approach also complicated supply chains and increased cycle time through the need to enlist third parties for chromatographic separation. This prompted the team to pursue the development of a classical resolution48 of rac-6, which would both reduce cycle times and allow for implementation in standard processing equipment, eliminating the need for specialist third-party partners.
In developing a classical resolution of rac-6, the team leveraged high-throughput experimentation (HTE) to identify a suitable resolving agent and solvent system to enable the selective crystallization of the (M)-6 isomer.41 Two rounds of screening experiments were performed, both focusing on readily available chiral acid resolving agents and a variety of solvent systems. While an initial screen of >250 conditions failed to produce promising leads, a follow-up screen of >100 conditions—primarily focused on the use of orthogonal solvent systems—resulted in several conditions that produced complexes between 6 and several chiral acids (Table 3).
Table 3. Chiral Acids That Form Complexes with Compound 6.
| Chiral acida | Solvent | M/P ratio |
|---|---|---|
| (1S)-(−)-camphanic acid | EtOAc | 50:50 |
| (1S)-(−)-camphanic acid | 2-MeTHF/heptane (1:1) | 50:50 |
| d-(+)-malic acid | EtOAc | 50:50 |
| (+)-2,3-dibenzoyl-d-tartaric acid | 2-MeTHF/heptane (1:1) | 81:19(10:90)b |
1 equiv.
M/P ratio in the supernatant. Adapted with permission from ref (2). Copyright 2022 The American Chemical Society.
Although (1S)-(−)-camphanic acid and d-(+)-malic acid produced crystalline complexes with 6 as determined by X-ray powder diffraction, neither acid provided any resolution when the resulting complexes were analyzed by chiral HPLC. (+)-2,3-Dibenzoyl-d-tartaric acid (DBTA) in a mixture of 2-methyltetrahydrofuran (2-MeTHF) and heptane, however, fortuitously generated a diastereomeric complex with (M)-6 that provided significant enantioenrichment (M/P ratio of 81:19; 62% de). Interestingly, analysis by single crystal X-ray diffraction revealed that the resolved (M)-6/DBTA complex was not a salt but rather a cocrystalline 2-MeTHF solvate with a 2:1 ratio of (M)-6 to DBTA.49
Having achieved proof-of-principle, the process development team subsequently sought to optimize these conditions to provide a commercially viable process. Through optimization of the solvent ratio and by increasing the resolving agent stoichiometry, the team was able to both optimize (M)-6 recovery and maximize process robustness, ultimately delivering a process that consistently provided (M)-6 with an M/P ratio of >2000:1 (>99.9% de) in 42% yield (theoretical maximum yield = 50%) on >500 kg scale per batch (Figure 10).41,50−52
Figure 10.
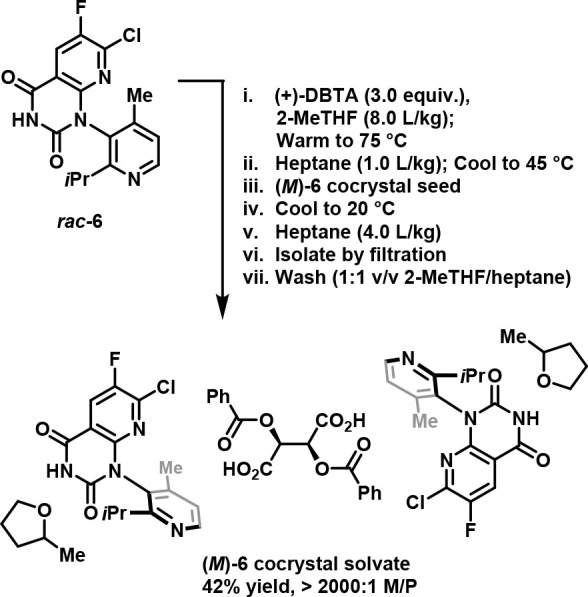
Commercial process to prepare (M)-6 cocrystal solvate. Adapted with permission from ref (2). Copyright 2022 The American Chemical Society.
The development of a classical resolution to deliver atropisomerically pure (M)-6 was not only crucial in supporting accelerated commercialization timelines (by ensuring clinical and commercial supplies of sotorasib drug substance), but also significantly improved the overall “greenness” of the manufacturing process by substantially decreasing organic solvent use and reducing processing cycle times (Table 4).
Table 4. Process Efficiency Comparison (Chromatography vs Classical Resolution).
| Metric | Chromatographya | Resolutionb |
|---|---|---|
| PMIc | 942 | 113 (↓88%) |
| Solvent used | 817 L | 118 L (↓86%) |
| Yield (2-step)e | 39% | 39% |
| Processing time | 36 days | 14 days (↓61%) |
Data based on a clinical chromatography process not optimized for commercial production.
Includes solvent contributions from free-basing/crystallizing free (M)-6.
Process mass intensity relative to sotorasib drug substance.
Total solvent used per kg of (M)-6 produced.
Yield after crystallization (chromatography) or free-basing/crystallization (resolution).
Despite these significant improvements in the efficient preparation of (M)-6, classical resolutions nonetheless have inherent inefficiencies, such as the need to discard the undesired stereoisomer as waste. To circumvent this shortcoming, we envisioned a process wherein the recovered undesired atropisomer (P)-6 could be recycled to serve as an alternative source of rac-6 through high temperature racemization,53−55 substantially improving the process greenness metrics for this resolution (Scheme 1).56
Scheme 1. (P)-6 Recycling via Thermal Racemization.
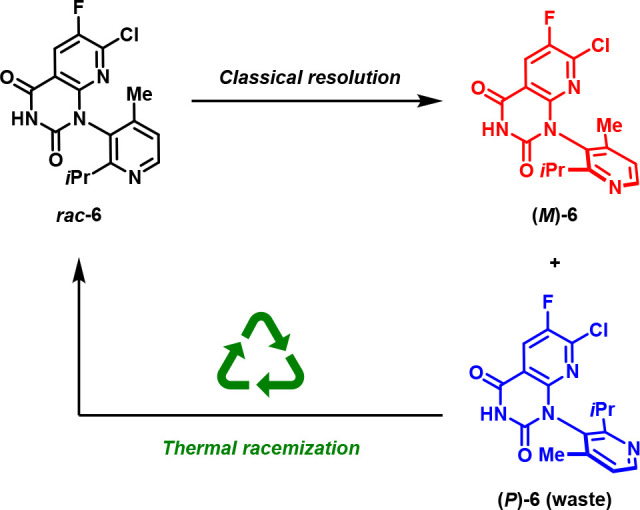
As noted above, the atropisomer interconversion barrier for (P)-6 was known to be quite substantial (42.3 kcal/mol), and accordingly, it was expected that temperatures approaching 300 °C or higher would likely be required to achieve reasonable racemization rates. Initial attempts to racemize (P)-6 (>99.5% ee) in a range of polar solvents led to significant decomposition; however, high-temperature racemization in nonpolar solvents was found to proceed with minimal decomposition (Table 5). Heating a solution of (P)-6 in anisole at 315 °C in a plug flow reactor resulted in near complete racemization in only 20 min (mean residence time) while maintaining high overall purity. This process was subsequently executed on kilogram scale, affording >1.4 kg of racemized 6 in 77% isolated yield with a P/M ratio of 50.5:49.5 following direct crystallization from the reaction mixture.
Table 5. Initial Screening Experiments to Racemize (P)-6.
| Entry | Solvent | Conditionsa | %eeb | LC area% |
|---|---|---|---|---|
| 1 | NMP | 200 °C (2 h) | 99.1 | 94.4 |
| 2 | NMP | 200 °C (8 h) | 96.3 | 80.3 |
| 3 | DMI | 250 °C (4 h) | 61.1 | 59.5 |
| 4 | sulfolane | 250 °C (4 h) | 75.5 | 86.3 |
| 5 | sulfolane | 280 °C (4 h) | 50.9 | 76.1 |
| 6 | diphenyl ether | 250 °C (4 h) | 69.1 | 97.5 |
| 7 | 1-octadecenec | 250 °C (4 h) | 74.2 | 98.5 |
| 8 | anisoled | 315 °C (0.3 h) | 2.0 | 99.1 |
Experiments performed at 100 mg scale.
Chiral purity at t0 = 99.6% ee.
Slurry-to-slurry transformation.
Experiment was performed in a plug-flow reactor. Adapted with permission from ref (3). Copyright 2022 The American Chemical Society.
Implementing this procedure on a production scale required a means of recovering (P)-6 from the waste stream of the (+)-DBTA-mediated resolution. This was achieved by treating the 2-MeTHF/heptane waste stream with potassium carbonate to extract the resulting potassium DBTA salt into the aqueous phase, followed by crystallization of (P)-6, which was isolated in 90% yield with a P/M ratio of 88:12 (76% ee). Acidification of the aqueous extract allowed for recovery of (+)-DBTA (93% yield, crystallized) and subsequent reuse in the resolution without a reduction in efficiency. A depiction of the resolution/recycling processes is shown in Scheme 2.56
Scheme 2. Classical Resolution of (M)-6 and Recovery/Recycling of (P)-6 and (+)-DBTA.

Successful implementation of this resolution process allowed for the preparation of multimetric ton quantities of (M)-6 while avoiding the drawbacks initially posed by the requirement for large-scale chiral chromatography. Through optimization of the kilo-scale (P)-6/(+)-DBTA recovery and recycling process, we were additionally able to further enhance the efficiency of the recovery process, providing a greener approach to rac-6 that reduced its process mass intensity (PMI) contribution to the drug substance manufacturing process from 336 to 141 (58% reduction).
As this brief case history illustrates, while large-scale chiral chromatography played a key role in the delivery of atropisomerically pure sotorasib for early development activities, chromatographic approaches to atropisomer separation posed a significant hurdle to efficient drug synthesis. Classical resolution ultimately proved a highly selective, scalable, and efficient approach to the preparation of atropisomerically pure drug substance, particularly when coupled with process refinements that allowed for recycling of the undesired atropisomer.
4. Concluding Remarks
An axially chiral biaryl linkage played a central architectural role in the design and optimization of the KRASG12C inhibitor sotorasib (LUMAKRAS/LUMYKRAS), providing stereodefined access to a novel cryptic pocket on the surface of the KRAS protein. Restricted rotation about this axially chiral linkage gave rise to configurationally stable atropisomers, which demonstrated a 10-fold difference in potency. The decision to develop sotorasib as a wholly synthetic, axially chiral single-atropisomer drug presented several analytical and synthetic challenges to be overcome, particularly as sotorasib has now become the first such molecule to receive marketing authorization.
In this account, we provide an overview of the analytical techniques (NMR- and HPLC-based) that were used to assess the configurational stability of the atropisomeric intermediates prepared during the optimization of sotorasib, along with descriptive guidelines for the selection of appropriate techniques to characterize the configurational stability of axially chiral compounds with differing degrees of rotational restriction.
We additionally survey some of the practical synthetic considerations that impacted our strategy in supporting the early clinical development of sotorasib as well as its later commercial synthesis. Whereas large-scale chromatography provided efficient access to early supplies of atropisomerically pure sotorasib, commercial synthesis ultimately relied upon classical resolution to provide the operational simplicity and robustness required for synthesis on the metric ton scale. Further process refinements, allowing for recycling of the undesired isomer from the classical resolution, ultimately led to dramatic efficiency improvements in this resolution process, delivering a commercial process that was both robust and green.
While the incorporation of an atropisomerically pure axis of chirality in sotorasib significantly increased the synthetic and analytical complexity of its development, this review illustrates the useful structural role that axial chirality can play in the design of efficient small molecule inhibitors, the range of analytical techniques that can be brought to bear in analyzing their configurational stability, and the synthetic strategies that can be used to enable the practical commercial-scale synthesis of atropisomerically pure pharmaceutical agents. It is hoped that this account may provide helpful guidelines for future efforts aimed at the development of such single atropisomer, axially chiral compounds.
Acknowledgments
We thank Steve Hollis for performing initial VT-NMR experiments, Mike Bartberger, Narbe Mardirossian, and Andreas Rotheli for computational simulation of atropisomer interconversion barriers, Bill Powazinik for experimental support in development of the classical resolution, and Matthew Beaver and Snapdragon Chemistry for their work in developing the off-enantiomer recycling processes. The authors thank Amgen, Inc., for financial support of this research.
Glossary
Abbreviations
- DBTA
2,3-dibenzoyl-d-tartaric acid
- DFT
density functional theory
- EtOAc
ethyl acetate
- EXSY
exchange spectroscopy
- FDA
(US) Food and Drug Administration
- HPLC
high-performance liquid chromatography
- HTE
high-throughput experimentation
- KRAS
Kirsten rat sarcoma virus oncogene
- 2-MeTHF
2-methyltetrahydrofuran
- NMR
nuclear magnetic resonance
- PMI
process mass intensity
- VT-NMR
variable-temperature NMR
Biographies
Brian A. Lanman is a Director of Research in the Medicinal Chemistry department at Amgen, Inc. Brian received his A.B. in Chemistry from Harvard University (1998), conducting undergraduate research on the total synthesis of Taxol with Yoshito Kishi. He subsequently received A.M. (2000) and Ph.D. (2004) degrees from Harvard for research as an NSF fellow on the solid-supported synthesis of tetrahydroisoquinoline antitumor antibiotics in the laboratories of Andrew Myers. In 2004, he joined Larry Overman’s group at UC Irvine as an NIH postdoctoral fellow, developing methods to access the architecturally complex bis-guanidine natural product palau’amine. Brian joined Amgen’s Medicinal Chemistry department in 2006, where he has since led chemistry and discovery research efforts in the inflammation, oncology, and cardiovascular therapeutic areas. Brian led the medicinal chemistry team that discovered LUMAKRAS/LUMYKRAS (sotorasib), Amgen’s first-in-class KRASG12C inhibitor.
Andrew T. Parsons is a Director of Process Development in the Pivotal and Commercial Synthetics group at Amgen, Inc. Andrew received his A.B. in Chemistry from Bowdoin College (2005), conducting undergraduate research with Richard D. Broene. He subsequently received a Ph.D. from the University of North Carolina at Chapel Hill (2010) for research on catalytic annulations of strained cycloalkanes to synthesize five- and six-membered heterocycles in the laboratories of Jeffrey S. Johnson. In 2010, he joined Stephen L. Buchwald’s group at MIT as an NIH postdoctoral fellow where he developed new catalytic trifluoromethylation reactions. After spending a year in the Process Development group at Bristol-Myers Squibb, Andrew joined Amgen’s Process Development group in 2013, where he has since supported the development efforts for numerous clinical and commercial assets. Most recently, Andrew led the process chemistry team that developed and commercialized the drug substance manufacturing process for LUMAKRAS/LUMYKRAS (sotorasib).
Stephan G. Zech is a Principal Scientist in the Lead Discovery and Characterization group at Amgen, Inc. Stephan received a Ph.D. (1998) in Biophysical Chemistry from Technical University in Berlin, Germany, conducting EPR studies on membrane proteins in the group of Wolfgang Lubitz. As a fellow of the Alexander-von-Humboldt foundation, he joined Prof. Ann McDermott at Columbia University in New York (2001) for postdoctoral studies in protein structure determination by solid-state NMR. In 2004, he joined the biopharmaceutical industry and has contributed to the discovery and development of multiple marketed oncology drugs including ponatinib, brigatinib, mobocertinib, and sotorasib.
Author Contributions
CRediT: Brian A Lanman conceptualization (lead), data curation (equal), formal analysis (equal), writing-original draft (equal), writing-review & editing (lead); Andrew T Parsons conceptualization (supporting), data curation (equal), formal analysis (equal), writing-original draft (equal), writing-review & editing (supporting); Stephan G Zech conceptualization (supporting), data curation (equal), formal analysis (equal), writing-original draft (equal), writing-review & editing (supporting).
The authors declare the following competing financial interest(s): B.A.L., A.T.P., and S.G.Z. are employees and stockholders of Amgen, Inc.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Atropisomers: Synthesis, Analysis, and Applications”.
References
- Lanman B. A.; Allen J. R.; Allen J. G.; Amegadzie A. K.; Ashton K. S.; Booker S. K.; Chen J. J.; Chen N.; Frohn M. J.; Goodman G.; Kopecky D. J.; Liu L.; Lopez P.; Low J. D.; Ma V.; Minatti A. E.; Nguyen T. T.; Nishimura N.; Pickrell A. J.; Reed A. B.; Shin Y.; Siegmund A. C.; Tamayo N. A.; Tegley C. M.; Walton M. C.; Wang H. L.; Wurz R. P.; Xue M.; Yang K. C.; Achanta P.; Bartberger M. D.; Canon J.; Hollis L. S.; McCarter J. D.; Mohr C.; Rex K.; Saiki A. Y.; San Miguel T.; Volak L. P.; Wang K. H.; Whittington D. A.; Zech S. G.; Lipford J. R.; Cee V. J. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- Parsons A. T.; Caille S.; Caporini M. A.; Griffin D. J.; Lovette M. A.; Powazinik IV W.; St-Pierre G. Axial chirality in the sotorasib drug substance, Part 1: Development of a classical resolution to prepare an atropisomerically-pure sotorasib intermediate. Org. Process Res. Dev. 2022, 26, 2629–2635. 10.1021/acs.oprd.2c00176. [DOI] [Google Scholar]
- Beaver M. G.; Brown D. B.; Campbell K.; Fang Y.-Q.; Ford D. D.; Mardirossian N.; Nagy K. D.; Rotheli A. R.; Sheeran J. W.; Telmesani R.; Parsons A. T. Axial chirality in the sotorasib drug substance, Part 2: Leveraging a high-temperature thermal racemization to recycle the classical resolution waste stream. Org. Process Res. Dev. 2022, 26, 2636–2645. 10.1021/acs.oprd.2c00177. [DOI] [Google Scholar]
- Atropisomers are stereoisomers that arise due to restricted rotation about a single bond (which leads to the formation of separately isolable, nonsuperimposable rotational isomers).
- Clayden J.; Moran W. J.; Edwards P. J.; LaPlante S. R. The challenge of atropisomerism in drug discovery. Angew. Chem. 2009, 48, 6398–6401. 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]
- Laplante S. R.; Fader L. D.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P.; Edwards P. J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54, 7005–7022. 10.1021/jm200584g. [DOI] [PubMed] [Google Scholar]
- Glunz P. W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett. 2018, 28, 53–60. 10.1016/j.bmcl.2017.11.050. [DOI] [PubMed] [Google Scholar]
- Toenjes S. T.; Gustafson J. L. Atropisomerism in medicinal chemistry: challenges and opportunities. Future Med. Chem. 2018, 10, 409–422. 10.4155/fmc-2017-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zask A.; Murphy J.; Ellestad G. A. Biological stereoselectivity of atropisomeric natural products and drugs. Chirality 2013, 25, 265–274. 10.1002/chir.22145. [DOI] [PubMed] [Google Scholar]
- Christie G. H.; Kenner J. LXXI.—The molecular configurations of polynuclear aromatic compounds. Part I. The resolution of γ-6:6′-dinitro- and 4:6:4′:6′-tetranitro-diphenic acids into optically active components. J. Chem. Soc., Trans. 1922, 121, 614–620. 10.1039/CT9222100614. [DOI] [Google Scholar]
- Evans D. A.; Dinsmore C. J.; Ratz A. M.; Evrard D. A.; Barrow J. C. Synthesis and Conformational Properties of the M(4–6)(5–7) Bicyclic Tetrapeptide Common to the Vancomycin Antibiotics. J. Am. Chem. Soc. 1997, 119, 3417–3418. 10.1021/ja964419u. [DOI] [Google Scholar]
- Wang J.; Zeng W.; Li S.; Shen L.; Gu Z.; Zhang Y.; Li J.; Chen S.; Jia X. Discovery and Assessment of Atropisomers of (±)-Lesinurad. ACS Med. Chem. Lett. 2017, 8, 299–303. 10.1021/acsmedchemlett.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajda S.; Beglov D.; Wakefield A. E.; Egbert M.; Whitty A. Cryptic binding sites on proteins: definition, detection, and druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. 10.1016/j.cbpa.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y.; Jeong J. W.; Wurz R. P.; Achanta P.; Arvedson T.; Bartberger M. D.; Campuzano I. D. G.; Fucini R.; Hansen S. K.; Ingersoll J.; Iwig J. S.; Lipford J. R.; Ma V.; Kopecky D. J.; McCarter J.; San Miguel T.; Mohr C.; Sabet S.; Saiki A. Y.; Sawayama A.; Sethofer S.; Tegley C. M.; Volak L. P.; Yang K.; Lanman B. A.; Erlanson D. A.; Cee V. J. Discovery of N-(1-Acryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as Covalent Inhibitors of KRAS(G12C). ACS Med. Chem. Lett. 2019, 10, 1302–1308. 10.1021/acsmedchemlett.9b00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ‘biaryl’ linkage of quinazoline 2 (and sotorasib) can also be viewed as an N-aryl-substituted amide linkage, a related structural motif known to give rise to axial chirality due to nonplanarity between the amide and arene pi system. In this account, we emphasize the ‘biaryl’ character of this axially chiral linkage, however, given that this amide moiety is part of a larger quinazolinone ring system, similar approaches are used in analyzing N-aryl amide and nonamide biaryl configurational stability questions, and this axially chiral substituted quinazolinone feature evolved from nonamide precursors (see ref (1)).
- Stereodefined access to the H95/Y96/Q99 cryptic pocket was essential for inhibitor potency. In early analogs, the (M)-atropisomer, which positioned an ortho substituent in the cryptic pocket, was 10-fold more potent than the (P)-atropisomer, which did not similarly position a substituent in this pocket.
- Mancinelli M.; Bencivenni G.; Pecorari D.; Mazzanti A. Stereochemistry and Recent Applications of Axially Chiral Organic Molecules. Eur. J. Org. Chem. 2020, 2020, 4070–4086. 10.1002/ejoc.201901918. [DOI] [Google Scholar]
- Nikitidis G.; Carlsson A.-C. C.; Karlsson S.; Campbell A. D.; Cook C.; Dai K.; Emtenäs H.; Jonson A. C.; Leek H.; Malmgren M.; Moravčík Š.; Pithani S.; Tatton M. R.; Zhao H.; Öhlén K. Synthetic and Chromatographic Challenges and Strategies for Multigram Manufacture of KRASG12C Inhibitors. Org. Process Res. Dev. 2022, 26, 710–729. 10.1021/acs.oprd.1c00179. [DOI] [Google Scholar]
- Both chemical manufacturing and control (CMC)-related (isomerization during manufacturing and distribution) and pharmacology/toxicology-related (isomerization in vivo).
- LaPlante S. R.; Edwards P. J.; Fader L. D.; Jakalian A.; Hucke O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem. 2011, 6, 505–513. 10.1002/cmdc.201000485. [DOI] [PubMed] [Google Scholar]
- Two nomenclatures for the description of axial chirality have been adopted by the International Union of Pure and Applied Chemistry (IUPAC): ‘R/S’ (sometimes written as Ra/Sa to denote axial chirality) and ‘M/P’ nomenclature. In the case of axial chirality, ‘R’ = ‘M’ and ‘S’ = ‘P’, allowing for straightforward translation between the two systems. In this manuscript, we have adopted the ‘M/P’ convention, given its intuitive application to the ‘minus’ (anticlockwise) and ‘plus’ (clockwise) disposition of Cahn–Ingold–Prelog priority substituents about an axially chiral bond. For authoritative treatment of both nomenclatures, see:; Favre H. A.; Powell W. H.. Specification of Configuration and Conformation. In Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013; Royal Society of Chemistry: London, 2013; pp 1156–1292. [Google Scholar]
- Were it not for the piperazine stereocenter in compound 2, anisotropic media or chiral derivatization reagents would have been required to generate distinct NMR signals from the enantiotopic nuclei.
- Claridge T. D. W.High-resolution NMR techniques in organic chemistry; Elsevier: Amsterdam, 2016. [Google Scholar]
- Stephenson D. S.; Binsch G. Iterative computer analysis of complex exchange-broadened NMR bandshapes. J. Magn. Reson. 1978, 32, 145–152. [Google Scholar]
- Szalay Z.; Rohonczy J. Monte Carlo simulation of DNMR spectra of coupled spin systems. J. Magn. Reson. 2008, 191, 56–65. 10.1016/j.jmr.2007.12.002. [DOI] [PubMed] [Google Scholar]
- At the temperature at which the experiment is conducted.
- VT-NMR allows for the analysis of a significant range of dynamic processes in small molecules, as temperatures between 260 and 420 K are readily accessible using standard NMR equipment, allowing for the assessment of atropisomer interconversion processes with every very low interconversion barriers (provided a suitable NMR solvent is employed).
- 33 kcal/mol represents a practical upper limit to the interconversion barriers accessible using this approach, given the probe operating temperatures accessible using standard NMR equipment.
- Kleckner I. R.; Foster M. P. An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 2011, 1814, 942–968. 10.1016/j.bbapap.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparro F. P.; Kolodny N. H. NMR determination of the rotational barrier in N, N-dimethylacetamide. A physical chemistry experiment. J. Chem. Educ. 1977, 54, 258. 10.1021/ed054p258. [DOI] [Google Scholar]
- Perrin C. L.; Dwyer T. J. Application of two-dimensional NMR to kinetics of chemical exchange. Chem. Rev. 1990, 90, 935–967. 10.1021/cr00104a002. [DOI] [Google Scholar]
- This approach could also be used when an exchange process did not cause significant line broadening in the 1D NMR spectra (k ∼ 1 s–1).
- Atkins P.; de Paula J.. Physical Chemistry: Thermodynamics, Structure, and Change, W. H. Freeman: New York, 2014. [Google Scholar]
- 19F NMR offers several advantages over 1H detection: low-background spectra free of solvent and water signals, increased separation of 19F signals due to a wider chemical shift range relative to 1H NMR spectra, and high sensitivity and narrow line widths (comparable to 1H detection). Since fluorine atoms are usually sparsely distributed in pharmaceutical compounds, the absence of J-couplings can also further simplify analysis.
- Sun M.; Chen W.; Zhang T.; Liu Z.; Wei J.; Xi N. 19F NMR spectroscopy as a tool to detect rotations in fluorine substituted phenyl compounds. Tetrahedron 2020, 76, 131679. 10.1016/j.tet.2020.131679. [DOI] [Google Scholar]
- Above 330 K, a single line is observed that narrows with increasing temperature, as predicted by a two-site exchange model.
- Where kB is the Boltzmann constant, ℏ is Planck’s constant, and R is the gas constant.
- Measuring the exchange rate at various temperatures T also allows for separate determination of enthalpy (ΔH⧧) and entropy (ΔS⧧):Rotzler J.; Gsellinger H.; Bihlmeier A.; Gantenbein M.; Vonlanthen D.; Häussinger D.; Klopper W.; Mayor M. J. O. Atropisomerization of di-para-substituted propyl-bridged biphenyl cyclophanes. Org. Biomol. Chem. 2013, 11, 110–118. 10.1039/C2OB26243F. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Navarro-Vazquez A.; Gil R. R.; Griesinger C.; Martin G. E.; Williamson R. T. Application of anisotropic NMR parameters to the confirmation of molecular structure. Nat. Protoc. 2019, 14, 217–247. 10.1038/s41596-018-0091-9. [DOI] [PubMed] [Google Scholar]
- Casy A. F. Chiral discrimination by NMR spectroscopy. Trac-Trend. Anal. Chem. 1993, 12, 185–189. 10.1016/0165-9936(93)87021-O. [DOI] [Google Scholar]
- Parsons A. T.; Cochran B. M.; Powazinik W. IV; Caporini M. A.. Improved Synthesis of Key Intermediate of KRAS G12C Inhibitor Compound WO 2020102730A1, May 22, 2020.
- Kobayashi T.; Ishiwari F.; Fukushima T.; Hanaya K.; Sugai T.; Higashibayashi S. Analysis of Interconversion between Atropisomers of Chiral Substituted 9,9’-Bicarbazole. Eur. J. Org. Chem. 2021, 2021, 449–451. 10.1002/ejoc.202001385. [DOI] [Google Scholar]
- Sotorasib is then prepared in four chemical transformations from (M)-6.
- Cheng J. K.; Xiang S. H.; Li S.; Ye L.; Tan B. Recent Advances in Catalytic Asymmetric Construction of Atropisomers. Chem. Rev. 2021, 121, 4805–4902. 10.1021/acs.chemrev.0c01306. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Salamanca P.; Fernandez R.; Hornillos V.; Lassaletta J. M. Asymmetric Synthesis of Axially Chiral C-N Atropisomers. Chemistry 2022, 28, e202104442 10.1002/chem.202104442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski S. R.; Carrasquillo-Flores R.; Lora Gonzalez F.; Ramirez A.; Casey M.; Soumeillant M.; Razler T. M.; Mack B. Adventures in Atropisomerism: Development of a Robust, Diastereoselective, Lithium-Catalyzed Atropisomer-Forming Active Pharmaceutical Ingredient Step. Org. Process Res. Dev. 2018, 22, 1426–1431. 10.1021/acs.oprd.8b00246. [DOI] [Google Scholar]
- It warrants mention that the selectivity requirement for such an asymmetric synthesis is quite stringent: We targeted an enantiomer ratio (M:P) of at least 99.7:0.3. Although atroposelective cyclizations delivering lower enantiomer ratios might subsequently be upgraded via resolution to provide an improved yield of (M)-6, the development of the (P)-6 recycling process described in ref (3) reduced the need to pursue such approaches.
- Douglas J. J.; Tatton M. R.; de Bruin D.; Buttar D.; Cook C.; Dai K.; Ferrer C.; Leslie K.; Morrison J.; Munday R.; Ronson T. O.; Zhao H. Exploration of a Nitromethane-Carbonylation Strategy during Route Design of an Atropisomeric KRASG12C Inhibitor. J. Org. Chem. 2022, 87, 2075–2086. 10.1021/acs.joc.1c01736. [DOI] [PubMed] [Google Scholar]
- Grothe E.; Meekes H.; Vlieg E.; ter Horst J. H.; de Gelder R. Solvates, Salts, and Cocrystals: A Proposal for a Feasible Classification System. Cryst. Growth Des. 2016, 16, 3237–3243. 10.1021/acs.cgd.6b00200. [DOI] [Google Scholar]
- While the theoretical amount of DBTA required is 0.5 equiv based on the 2:1 ratio of (M)-6:DBTA, a superstoichiometric amount of DBTA improved robustness by increasing the rate of cocrystal formation and reducing its thermodynamic solubility in the resolution medium.
- Nehm S. J.; Rodriguez-Spong B.; Rodriguez-Hornedo N. Phase Solubility Diagrams of Cocrystals Are Explained by Solubility Product and Solution Complexation. Cryst. Growth Des. 2006, 6, 592–600. 10.1021/cg0503346. [DOI] [Google Scholar]
- Rodriguez-Horned N.; Nehm S. J.; Jayasankar A.: Cocrystals: Design, Properties and Formation Mechanisms. In Encyclopedia of Pharmaceutical Technology, 3rd ed.; CRC Press: Boca Raton, FL, 2007; pp 615–635. [Google Scholar]
- Parker J. S.; Smith N. A.; Welham M. J.; Moss W. O. A New Approach to the Rapid Parallel Development of Four Neurokinin Antagonists. Part 5. Preparation of ZM374979 Cyanoacid and Selective Crystallisation of ZM374979 Atropisomers. Org. Process Res. Dev. 2004, 8, 45–50. 10.1021/op030041q. [DOI] [Google Scholar]
- Liu Y.; Prashad M.; Shieh W.-C. A Scalable Synthesis of an Atropisomeric Drug Substance via Buchwald-Hartwig Amination and Bruylants Reactions. Org. Process Res. Dev. 2014, 18, 239–245. 10.1021/op400250s. [DOI] [Google Scholar]
- Achmatowicz M. M.; Chen C.-y.; Snead D. R.. Simultaneous Processing of Antagonistic Chemical Events (SPACE): An Atroposelective Dynamic Kinetic Resolution of MRTX1719. ChemRxiv, June 20, 2022, ver. 2. 10.26434/chemrxiv-2022-pwtdx-v2 (accessed September 28, 2022). [DOI]
- Beaver M. G.; Corbett M. T.; Fang Y.-Q.; Ford D. D.; Parsons A. T.; St-Pierre G.; Telmesani R.. Process for Racemizing and Isolating Atropisomers of 7-Chloro-6-fluoro-l-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-D]pyrimidine-2,4(1H,3H)-dione. WO2022076623A1, April 14, 2022.