

Abstract
Background:
Substantial evidence from recent research suggests an influential and underappreciated force in Alzheimer’s disease (AD) pathogenesis: the pathological signals originate from outside the brain. Pathogenic bacteria produce amyloid-like proteins “curli” that form biofilms and show functional similarities to human amyloid-β (Aβ). These proteins may contribute to neurological disease progression via signaling cascade from the gut to the brain.
Objective:
We propose that curli causes neuroendocrine activation from the gut to brain that promotes central Aβ pathology.
Methods:
PGP9.5 and TLR2 levels in response to curli in the lumen of Tg2576 AD mice were analyzed by immunohistochemical and qRT-PCR analysis. Western blot and human 3D in vitro enteroids culture systems were also used. 16S rRNA gene sequencing was used to investigate bacterial dysbiosis.
Results:
We found significant increase in bacterial-amyloid curli with elevated TLR2 at the mRNA level in the pre- and symptomatic Tg-AD gut compared to littermate WT controls. This data associates with increased gram-positive bacterial colonization in the ileum of the symptomatic AD mice. We found fundamental evidence for vagus nerve activation in response to bacterial curli. Neuroendocrine marker PGP9.5 was significantly elevated in the gut epithelium of symptomatic AD mice, and this was colocalized with increased TLR2 expression. Enteroids, 3D-human ileal mini-gut monolayer in vitro model system also revealed increase levels of TLR2 upon stimulation with purified bacterial curli fibrils.
Conclusion:
These findings reveal the importance of pathological changes within the gut-vagus-brain signaling in response to luminal bacterial amyloid that might play a vital role in central Aβ pathogenesis seen in the AD brain.
Keywords: Alzheimer’s disease, amyloid-β, bacterial amyloid, curli, dysbiosis, enteroendocrine cell, gut barrier, gut-brain axis, neuroendocrine, PGP9.5, TLR2, vagus nerve
INTRODUCTION
Our understanding of the “gut-brain axis” (GBA) and the mechanisms by which peripheral events can affect processes in the central nervous system (CNS) remains relatively unexplored [1]. Recent studies have provided robust evidence that gut impairment is connected to the progression of neurodegenerative diseases including Alzheimer’s disease (AD) [1]. A hallmark of aging is the presence of chronic low-grade systemic inflammation (i.e., “inflammaging”) [2]. We propose that a potential source of this inflammation come from the gut mucosa [1]. Age-related alterations in the gut microbiota composition have been documented, with decreased bacterial diversity and stability [3, 4]. This leads to gut barrier breakdown, a further increase in pro-inflammatory cytokines with the presence of bacteria-derived products into host and subsequent blood-brain barrier impairment and neuroinflammation [5, 6]. Others and our previous studies show that aging itself is associated with mild gut impairment and central inflammation, and this can be worsened in young or reversed in aged mice by modifying the microbiota [3, 4]. However, vagus communication within the gut-brain axis is still poorly understood. It establishes one of the major extrinsic connections between the brain and the gastrointestinal tract and sends information about the state of the inner organs to the brain via afferent fibers [7]. There is preliminary evidence that vagus nerve stimulation is a promising add-on treatment for gastrointestinal disorders [7] but is not fully understood and also it lacks information in terms of other disease types. Currently, all treatments that exist for AD patients are initiated after the emergence of clinical symptoms, when significant pathology is already present in the brain. Once the disease is symptomatic, the pathology is so severe that any treatment is likely to be futile. Investigation at pre-symptomatic stage, well before the disease has progressed in the brain, is crucial to understand the initiation and progression of the disease.
It is clear that late-onset AD is collectively modified by number of genetic factors that govern diverse cellular and molecular pathways, including many genes involved in the immune responses [8]. The role played by the immune system in AD pathogenesis is prominent but is by no means limited to the brain. Copious evidence from clinical and experimental research suggests an influential, yet largely underappreciated, force in AD pathogenesis: immune signals originating outside the brain. In the gut, there is a single layer of epithelial cells separating the lumen from the underlying tissue. Within the epithelial layer lies highly specialized cells called enterochromaffin cells, a subset of enteroendocrine cells, that are dispersed and can be excited electrically. These enterochromaffin cells (ECs) sense ingested nutrients and microbial products from the lumen [9]. The mechanism we propose is that part of intestinal bacteria can form biofilms by producing amyloid-like proteins that behaves similar to mammalian Aβ [10–12]. Gut inflammation in reaction to bacterial amyloid can alter gut integrity and allow the translocation of antigens [13–16] including bacterial biofilm associated amyloids “curli” into the gut mucosa [10–12]. This may trigger a specific immune response just not only in the systemic pathway but also in the vagal pathway that contributes to the central Aβ pathology [16] with age.
Therefore, we hypothesize that the gut physiology in response to its microbiota composition might play a vital role in AD progression. This is possible by toll-like receptor (TLR) activation in the gut. TLR2 activation has been recently shown to increase amyloid aggregation [17] and inhibition of TLR2 attenuates neuronal Aβ levels in AD mice [17, 18]. We propose that microbial amyloid-like proteins may cause TLR associated inflammatory cascade that promotes central Aβ pathology [19] via gut-brain axis interaction. This interaction might be stronger via afferent vagus signals from the gut to the brain. In support of our hypothesis, we found significantly increased luminal curli (bacterial-amyloid), associated with elevated gut TLR2 mRNA levels in the pre-symptomatic transgenic (Tg) 6-month-old and symptomatic Tg≥15-month-old AD mice compared to littermate wild-type (WT) controls. In association with TLR2 elevation, we found increased gram-positive bacterial colonization in the ileum of the symptomatic AD mice compared to WT controls and the bacterial colonies appeared morphologically different with increased filamentous shape compared to cocci shape found in the lumen of aged WT mice gut. In addition, we found increased curli expression in pre-symptomatic Tg 6-month-old compared to WT littermate control mice gut. It is possible that TLR2 activation is due to increased translocation of bacterial products like curli (bacterial amyloids) for a prolonged period of time and that causes gut dysfunction. We found fundamental evidence for vagus nerve activation in response to bacterial curli. This was investigated by studying the expression levels of PGP9.5 (neuroendocrine marker) within the ileum. The data obtained clearly shows that TLR2 activation caused significant elevation of PGP9.5 expression within the ECs of AD mice ileum compared to WT mice. TLR2 + PGP9.5 + signals were also elevated in the sub-mucosa where vagus nerve fibers are enriched [20]. In addition, enteroids 3D human ileal mini-gut monolayer in vitro model also revealed increase levels of TLR2 upon stimulation with purified bacterial curli fibrils, adding supportive evidence to the in vivo study. Overall, our study reveals the first set of evidence on the importance of immune changes within the gut vagal pathway in response to luminal bacterial amyloid that might play a vital role in central Aβ pathogenesis seen in the AD brain (Fig. 1).
Fig. 1.

Schematic representation of the gut neuroepithelial circuit activation (the neuropod cells) in response to bacterial curli breach in the presence of central Aβ pathology observed in AD mice. TLR2, Toll-Like Receptor 2; Aβ, amyloid-beta. (Designed using Bio Render).
METHODS
Mice
The Tg2576 transgenic mouse model of AD was used in this study. These mice overexpress the 695-amino acid isoform of human Alzheimer amyloid precursor protein (APP695) containing a double mutation of Lys670Asn, Met671Leu (i.e., Swedish mutation) under the control of the hamster prion protein promoter [21], resulting in elevated levels of Aβ aggregates, extensive cerebral amyloid pathology, and cognitive deficits [22]. These animals develop parenchymal plaques beginning at around 9–10 months of age with some vascular amyloid which eventually becomes widespread in the cortical and limbic structures by around 15 to 16 months of age [23].
The study was conducted under protocols approved by the Center for Laboratory Animal Medicine and Care (CLAMC)/Institutional Animal Care and Use Committee (IACUC) at the University of Texas McGovern Medical School. We formed the breeding pairs with young (2 months old) Tg/WT males (129S6.Cg-Tg(APPSWE)2576Kha N20+?) mice (model 1349) and young WT/WT (129S6.Cg-Tg(APPSWE)2576Kha N20+?) females mice, that were purchased from Taconic. Prior to breeding, all the animals purchased were housed for one month to normalize their microbiota and adapt to the new environment. The breeding pairs were used to generate F1 generation followed by genotyping to confirm the presence of inherited mutation. Tg2576 and WT littermate mice obtained from F1 generation were used in the study. All the experiments shown here were performed with animals obtained from F1 generation. The pups were weaned 21 days after birth and both sexes were used in the study. Mice were housed up to 5 per cage (individually ventilated, changed weekly or bi-weekly under HEPA-filtered workstations) in standard facilities with a 12-h light/dark schedule (lights on at 7AM) in a temperature- (21.7–22.8 C) and humidity-controlled (40–60 RH) controlled vivarium, with ad libitum access to food (LabDiet 5053 and 5058, pelleted, irradiated at manufacturer, stored at room temperature for up to 6 months) and water (filtered tap water, pH 6–8, not acidified nor chlorinated). The animal used in the study were pre-symptomatic Tg mice that were 6 months of age and symptomatic Tg mice that were15 to 16 months of age with their respective age-matched littermate controls.
Immunohistochemistry (IHC) and microscopy
IHC was performed as previously described in [1]. Formalin-fixed, paraffin-embedded 5 μm-thick intestinal sections were used for the following staining protocols. For lectin staining, terminal mucin glycans were examined using a panel of FITC-conjugated lectins: Ulex europaeus agglutinin-1 (UEA-1) for terminal fucose. De-paraffinized sections were rinse in PBS-Tween and block with PBS containing 10% FBS for 1 h at room temperature (RT). Sections were then stained with labeled lectin (10 μg/ml) for 1 h at RT. Sections were washed with ice-cold 1X PBS and counterstained with DAPI for 10 min at RT and mounting using fluoroshield (F6182–20 ml, Sigma).
For all other gut staining protocols, after dewaxing and rehydration steps, a heat-inducing antigen retrieval procedure using 1 mM EDTA buffer at pH 8.0 was performed, with subsequent washing in PBS. After a blocking step, sections were incubated with primary antibodies (Table 1) overnight followed by incubation with fluorescent secondary or HRP immunoglobulins (1 : 1000, multiple routinely used vendors) for 60 min.
Table 1.
The information of primary antibodies
| Primary antibody | Host | Dilution ratio | Mono/Polyclonal | Source | Catalog number |
|---|---|---|---|---|---|
| CsgA | Rabbit | 1 : 10000 | Monoclonal | Obtained from Dr. Chapman lab | |
| Muc2 | Rabbit | 1 : 100 | Polyclonal | Novus Biologicals | NBP1-31231 |
| CD282 (TLR2) | Rat | 1 : 100 | Monoclonal | Invitrogen | 14-9021-82 |
| TLR2 | Rabbit | 1 : 100 | Monoclonal | Invitrogen | MA5-32787 |
| PGP9.5 | Rabbit | 1 : 200 | Polyclonal | GeneTex | GTX109637 |
Immunoreactions were developed with diaminobenzidine (DAB) diluted in DAB Substrate Buffer (Peroxidase chromogen or Substrate solution from N-Histofine DAB-2V, Tokyo, Japan). The sections were counterstained with hematoxylin for IHC or DAPI (ThermoFischer) for f-IHC prior to visualization. Images were obtained on a Keyence BZ-X810 all in one confocal microscope.
Brain sections fixed in tissue freezing media were cut with 12 μm thickness. Thioflavin S (Millipore Sigma, Mfr. No. T1892–25G) staining for detection of Aβ plaques in the brain sections was performed by incubating the sections with 1% thioflavin-S in 0.01 mol/L PBS in the dark for 8 min at 22°C, followed by washing in 70% ethanol for 3 min.
CsgA expression was quantified by our newly established scoring protocol. Briefly, Curli expression per 10 μm of lumen was quantified using scores from the 20X images obtained. Five images per section and 3 slides per animal were used. In total n of 6 animals per group for symptomatic timepoints and 3 animals per group for pre-symptomatic timepoints were used. The scores were performed as, score: 4 (dark brown IHC stain with disrupted mucus barrier), score: 3.5 (dark brown IHC stain with intact mucus barrier), score: 3 (medium brown IHC stain with disrupted mucus barrier), score: 2.5 (medium brown stain with intact mucus barrier), score: 2 (light brown IHC stain with reduced intact mucus barrier), score: 1 (light brown IHC stain with intact mucus barrier) and score: 0 (little to no brown stain).
Fluorescent in situ hybridization (FISH)
Formalin-fixed paraffin-embedded intestinal tissue sections (5 μm) were initially treated with lysis buffer for 1 h at 37°C and then hybridized at 51°C with a 20 bp bacteria-specific probe (EUB 338: GCT-GCCTCCCGTAGGAGT) to visualize and quantify the proximity of luminal bacterial colonies and the corresponding antigen load on the epithelium. Following the overnight hybridization, the intestinal sections were counter-stained with 4,6-diamidino-2-phenylindole (DAPI, Dihydrochloride, Cat #: D1306 by Invitrogen) for visualization of cell nuclei. Images were obtained and analyzed using a Keyence BZ-X810 all in one fluorescence microscope.
Curli bacterial amyloid protein purification
CsgA protein from E. coli was purified as described previously [24]. Briefly, E. coli CsgA was overexpressed in NEB 3016 cells. Induction performed at OD 0.9 with 0.5 mM IPTG for 1 h and cells were pelleted down. Pellet was resuspended in 25 mL 8 M guanidine chloride in 50 mM KPi buffer pH 7.4 and incubated at room temperature for 1 h with shaking. Following this, the cell lysate was centrifuged at 10000 g for 20 min at 4°C. The supernatant was then sonicated with a probe sonicator at level 3 for three 20-s bursts. Then 800 μL of Sigma Ni-NTA resins were added and incubated at room temperature for 1 h with continuous slow shaking. The supernatant was then loaded on to 5 ml polypropylene columns and washed with 10 mL of 50 mM KPi buffer pH 7.4, followed by 3 mL of wash buffer (12.5 mM Imidazole in 50 mM KPi buffer pH 7.4) and finally CsgA protein was eluted out with 3 mL of elution buffer (125 mM Imidazole in 50 mM KPi buffer pH 7.4. Fraction 1 was 500 μL and fraction 2 was 1000 μL (only two fractions were collected). Fraction 2 contained CsgA. This fraction was then first buffer exchanged to 50 mM KPi buffer pH 7.4 and then passed through a 30 kD cut off column and the filtrate was collected (CsgA). The CsgA protein concentration was estimated using BCA assay prior to use (this protein purification is performed in Chapman lab).
Human ileal enteroids (HIE) cultures
HIE cultures were generated from crypts isolated from the ileal tissues of adult patients undergoing bariatric surgery as previously described [25]. These established cultures were obtained at Baylor College of Medicine through the Texas Medical Center Digestive Diseases Center Gastrointestinal Experimental Model Systems (GEMS) Core. Three-dimensional HIE cultures were prepared from the tissue samples and maintained in culture as described previously [25]. For these studies, ileal HIEs from patient IL103 were used. Complete medium without growth factors (CMGF−) and CMGF+ were prepared as previously described [25]. Briefly, CMGF− consisted of advanced Dulbecco’s modified Eagle medium (DMEM)/F-12 medium supplemented with 100 U/mL penicillin-streptomycin, 10 mmol/L HEPES buffer, and 1× GlutaMAX (all Invitrogen, Carlsbad, CA). CMGF+consisted of CMGF−medium with 50% (vol/vol) Wnt3A-conditioned medium, 20% (vol/vol) R-spondin–conditioned medium, 10% (vol/vol) Noggin-conditioned medium, 1× B-27 supplement (Invitrogen), 1× N-2 supplement (Invitrogen), 1 mmol/L N-acetylcysteine (Sigma-Aldrich, St. Louis, MO), 50 ng/mL mouse epidermal growth factor (Invitrogen), 10 mmol/L nicotinamide (Sigma-Aldrich), 10 nmol/L Leu-Gastrin I (Sigma-Aldrich), 500 nmol/L A-83-01 (Tocris Bioscience, Bristol, UK), and 10 nmol/L SB202190 (Sigma-Aldrich).
Differentiation medium consisted of the same components as CMGF + without Wnt3A-conditioned medium, R-spondin–conditioned medium, SB202190, and nicotinamide, and only 5% (vol/vol) Noggin-conditioned medium. hW-CMGF+, for creating and maintaining lentivirus-transduced HIEs, consisted of CMGF+mixed with an additional 50% (vol/vol) Wnt3a-conditioned medium. All HIEs were passaged in phenol red-free, growth factor–reduced Matrigel (Corning, NY).
HIE monolayer preparation
HIE monolayers were prepared from 3D cultures and seeded into flat 96-well plates. In brief, 96-wells (Corning) were pretreated with Matrigel diluted in 1×PBS (1 : 40) and incubated at 37°C. 3D HIEs were lifted from Matrigel and washed with an ice-cold solution of 0.5 mmol/L EDTA in 1×PBS and dissociated with 0.05% trypsin/0.5 mmol/L EDTA for 4 min at 37°C. Trypsin was inactivated with CMGF− +10% fetal bovine serum and the cell solution was pipetted vigorously and filtered using a 40-μm nylon cell strainer (Corning) to dissociate into single cells. Then cells were centrifuged for 5 min at 400×g, resuspended with CMGF+ and 10 μmol/L Y-27632 Rock inhibitor, and plated into prepared wells. After 48 h in CMGF+ and 10 μmol/L Y-27632 Rock inhibitor, the medium was changed to differentiation media with the addition of 10 μmol/L Y-27632 Rock inhibitor and indicated concentrations of doxycycline (Thermo Fisher Scientific, Waltham, MA). Differentiation medium with Y-27632 and doxycycline was changed every day for 4–5 days to differentiate cells.
Bacterial curli incubation with HIEs
HIE monolayers were differentiated for 4 to 5 days. Upon differentiation, the monolayers were treated with bacterial curli at three different concentrations. HPLC purified bacterial curli of 40 μM was used. We used saline control, 0.025 nM, 0.05 nM, and 0.5 nM curli concentrations to treat bacterial enteroids.
The enteroids were incubated for 12 h at 37°C. After incubation, supernatants were removed and enteroids were washed in 1x Pbs. Enteroids were treated with RIPA lysis buffer for western blot protein analysis. These were reproduced five times. Lysed enteroids were stored at −80°C until analyzed.
mRNA gene expression
To quantify relative mRNA expression levels of TLR2, TLR4, and GAPDH mRNA was extracted from intestinal mucosa samples using the miRNeasy® mini kit (QIAGEN). 1.0 μg of RNA was reverse-transcribed to single-stranded cDNA using the RevertAid H minus First Strand cDNA Synthesis Kit (Thermo Fischer, USA). Reverse transcriptase real-time (RT) PCR was performed using the Quant Studio 3 Real-Time PCR system (Applied Biosystems, USA). The RT-PCR reaction mix (adjusted with H2O to a total volume of 20 μl) contained 2 μl template DNA, 10 μl Sybr green master mix (Thermo Fischer, USA), Relative mRNA target gene expression levels (Ratio = [(Etarget) dCPtarget(control−sample)] / [(Eref.) dCPref.(control−sample)]) were normalized to the house keeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and used as a reference. Subsequently, intestinal mucosal gene of interest of the WT littermate control group at 16 months (Yg-WT) was set to 1.0 and used as the calibrator to identify the relative mRNA fold difference between the WT littermate controls and Tg2576 mice at 6 and 15 months. Similar methodology was used for mRNA extraction from enteroids treated with bacterial curli and saline treated control group.
Immunoblotting
Cell pellets were collected from enteroids monolayers and solubilized in RIPA lysis buffer containing protease and phosphatases inhibitor (Roche), followed by homogenization by sonication. After centrifugation at 5000 g for 10 min, the supernatant was collected as the protein lysate. Protein concentrations were determined using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Twenty-five micrograms of protein from each sample were resolved onto 4 to 15% SDS-PAGE gradient gels (Bio-Rad). Following electrophoresis, proteins were transferred to nitrocellulose membrane. After blocking with 5% non-fat milk in TBST, blots were probed with 1 : 500 dilution of primary antibody (TLR2, cat# MA5–32787, Invitrogen) overnight at 4°C, followed by a 1 : 2000 dilution of anti-rabbit HRP secondary antibody (Cat# 7074, Cell Signaling Technology) for 1 h at room temperature. SuperSignalTM West Femto Maximum Sensitivity Substrate (Thermo Fisher) was applied and the blot was imaged on a chemiluminescent imaging system. The blot was then stripped and probed with HRP-conjugated mouse anti-beta actin diluted 1 : 3000 (Cell Signaling Technology) before substrate addition and imaging. Beta actin was used as the loading control. Image J is used to analyze the band intensity.
Statistics
Data were tested for normal distribution using the Kolmogorov–Smirnov test. Normally distributed data are presented as means with standard error while the medians with their range are given for non-normally distributed data. Significance of differences between Yg-WT (age-matched littermate controls for pre-symptomatic Tg2576), Yg-Tg (pre-symptomatic, 6-month-old Tg2576), Ag-WT (age-matched littermate controls for symptomatic Tg2576), and Ag-Tg (symptomatic,≥15-month-old Tg2576) mice were analyzed using the One-way of variance test for normally distributed data (or) the Kruskal-Wallis test for non-normally distributed data, followed by either Bonferroni /Tukey or Dunn’s comparison post-hoc tests. Differences between Yg-WT and Yg-Tg group was tested using students t-test followed by the Mann-Whitney test for non-normally distributed data. All the experiments are performed blinded. Differences between the groups were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001. SPSS 16.0 (IBM, USA) for Windows 7 was used for data analysis. Prism 5.0 software (Graph Pad Software, Inc., La Jolla, CA, USA) for Windows, was used for data presentation and also for data analysis. All investigators were blinded to genotype.
RESULTS
Dysbiosis was significantly found in the small intestinal microbial colonization of symptomatic AD mice compared to WT littermate controls
First and foremost, we confirmed the Aβ pathology in symptomatic AD mice brain showing the presence of Aβ plaques in the cortex, hippocampus, and thalamus regions (Fig. 2A). The composition of gut microbiota can be influenced by intestinal epithelial dysfunction associated immune activation [1]. We have previously reported the presence of large intestinal gut dysfunction in pre-symptomatic and symptomatic Tg mice compared to their respective WT littermate controls [1]. Similarly, we performed 16S rRNA sequencing to examine compositional differences in the gut microbiota of Tg2576 mice at 15 to 16 months of age with the littermate controls. We examined the bacterial diversity of gut microbiota in the small intestinal (SI) luminal content (ileal) samples. After visualization and analysis of alpha-diversity, or within-sample diversity, we observed no differences in OTUs between the symptomatic AD compared to WT littermate controls groups (p = 0.24, Fig. 2B). Upon visualization of beta-diversity, or between-samples diversity, with Unweighted UniFrac distances by principal coordinate analysis (PCoA), a significant clustering effect (p = 0.003) emerged along the PC1 axis (23.9% variation explained) at the symptomatic timepoint in Tg2576, when compared to age-matched WT littermates (Fig. 2C). Examination of 16S data at the family level showed a significant increase in Lactobacillaceae abundance in the symptomatic Tg2576 mice when compared to age-matched WT littermates. Our 16S data show that significant bacterial compositional differences termed as dysbiosis (imbalanced in beneficial versus detrimental bacterial composition), exist in the SI of symptomatic Tg2576 mice gut microbiota when compared to WT littermate controls and that shift denoted significant increase in gram positives within the SI lumen.
Fig. 2.

Compositional differences in gut microbiota by 16S rRNA sequencing of small intestinal luminal content. A) Presence of Aβ plaques (green signal) in the symptomatic Tg2576 AD mice brain compared to age-matched WT controls using thioflavin-S staining. Magnification-2X. B) Visualization of alpha diversity shows changed with-in the sample diversity. They show no significant changes with-in the bacterial population of animals from the same groups. C) Beta-diversity, or between-samples diversity, with Unweighted UniFrac distances by principal coordinate analysis (PCoA) shows a clustering effect by strain between Tg2576 and WT littermate controls at symptomatic timepoint of 15 months, within the small intestine. n = 5–7 per group. p-values were calculated using ALTIMA sequencing software platform.
Bacterial amyloid “Curli” breach observed in symptomatic AD mice gut when compared to littermate WT controls
Dysbiotic bacteria within the gut lumen may form biofilms by producing bacterial amyloid-like proteins known as curli [10–12]. To investigate if dysbiosis found in Tg2576 AD mice show increased curli burden in the gut lumen, we used immunohistochemistry technique against bacterial curli biofilm in the cecal gut tissue and found significant increase in curli abundance in both pre-symptomatic (6-months of age) and symptomatic AD mice (≥15 months of age) compared to age-matched WT littermate controls. We found significant increase in bacterial curli at both pre- and symptomatic timepoints showing the presence of biofilm bloom within the gut earlier than Aβ pathology occurs in the brain which persists into symptomatic age (Fig. 3A (i)-(iv), B). In addition, ileum also showed increased curli abundance in symptomatic-AD mice compared to WT littermate control. Bacterial curli was proportionately increased in aged WT-littermate controls (Fig. 3C) compared to young WT controls. It is known that aging induces dysbiosis [3, 4], and we found increased curli expression in aged gut compared to young gut in WT littermate controls (Fig. 3A (i), 3A (iii)). Together with curli analysis, bacterial biofilm using FISH quantification in the SI tissue revealed that bacterial colonies appeared morphologically different with filamentous shape in the ileum of symptomatic AD mice compared to cocci shaped found in WT littermate controls and gut bacterial breach was proportionately increased in AD mice as previously observed [1] (Fig. 3D).
Fig. 3.

Bacterial amyloid curli protein expression by IHC in the gut lumen. A-C) Bacterial amyloid Curli protein (CsgA) staining within the large intestinal (A) and small intestinal (C) gut lumen (bacterial mass), showing a significant increase expression in pre-symptomatic and symptomatic Tg2576 AD mice when compared to age-matched WT controls (B) quantification of curli expression. A(i) and A(ii) are from pre-symptomatic timepoints of 6 months of age; A(iii), A(iv), C (i), and C(ii) are from symptomatic time-points of 15 months of age. n = 3,6 per group. D) Bacterial breach through the mucosal barrier onto the intestinal epithelium detected by FISH in the small intestine of Tg2576 mice and their WT littermate controls at 15 months. Red - bacterial biofilm. Blue - host epithelial nuclei. n = 6 per group. A, C, D) Magnification 20x. n = 3, 6 per group. Data are expressed as mean ± s.d., as well as individual values, and are obtained from > three independent experiments at various times. *p < 0.05; **p < 0.01; ***p < 0.001. p-values were calculated using One-Way analysis of variance with Bonferroni correction.
Significant increase in the gut mRNA and protein TLR2 levels of AD mice and this was associated with increased amyloid plaque formation in the brain
TLRs are the first line of defense in recognizing pathogens and activate innate immune responses. They are expressed in the gut epithelial cells, lamina mucosa especially dendritic cells, intraepithelial lymphocytes, and vagus nerve cells within the submucosa. TLR2 is activated by enrichment of materials from gram-positive and TLR4 is activated due to gram-negative bacteria [26]. We set to understand the host-microbiota link due to bacterial dysbiosis and its associated increase in bacterial amyloid-curli seen in symptomatic AD mice compared to WT littermate controls. TLRs sensing might play a major role in immune activation against bacterial curli in these animals. We found significant increase in the mRNA levels of TLR2 (Fig. 4A) and TLR4 (Supplementary Figure 1) in the ileum of symptomatic AD mice compared to the WT littermate controls. Interestingly this difference was strikingly elevated in the SI where vagus nerve and immune activation is dominant. In addition, TLR2 activation was significantly stronger than TLR4 activation which provides us the clue that the pathogenic molecule activates TLR2 associated pathway. Importantly, we found TLR2 activation was ≥5-fold elevated in the ileum of the pre-symptomatic AD mice at 6 months of age compared to littermate WT control (Fig. 4C). At 6 months of age the Tg2576 animals show no pathology in the brain [1]. In addition, we found 2-fold increase in TLR2 mRNA levels of symptomatic AD mice compared to WT-littermate controls. Immunohistochemical protein analysis (Fig. 4B) also supported the mRNA data that TLR2 was found elevated in the gut sub-mucosa and in the intestinal epithelium of AD mice compared to WT mice.
Fig. 4.
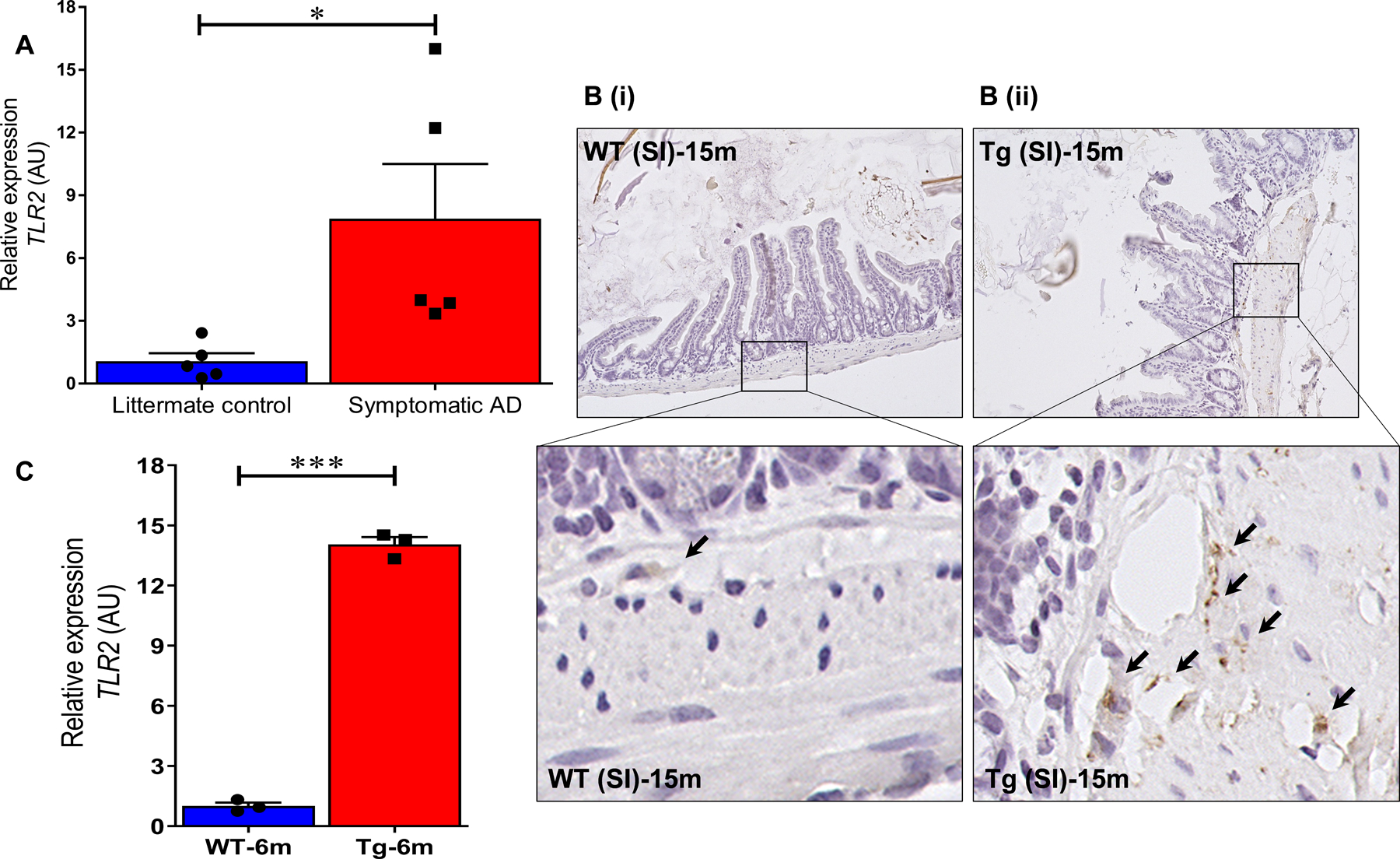
TLR2 is elevated in the gut tissue of AD mice. A) TLR2 mRNA expression is significantly increased in symptomatic Tg2576 measured by qRT-PCR. B) Immunohistochemical staining of small intestine shows a significant increase in TLR2 expression in Tg2576 mice (B(ii)) when compared to WT littermate controls (B(i)) at 15 months. Brown-TLR2, blue-nuclei. C) The transcriptomic analysis targeting TLR2 in the small intestine of Tg2576 mice at pre-symptomatic timepoint of 6 months of age shows significant increased TLR2 to the WT controls. n = 3 or 5 per group. B) Magnification 20x. n = 6 per group. A, C) Data are expressed as mean ± s.d., as well as individual values, and are obtained from three independent experiments at various times. *p < 0.05; **p < 0.01; ***p < 0.001. p-values were calculated using students t-test.
Increased PGP9.5 neuroendocrine marker and TLR2 activation seen in the ileal epithelial enterochromaffin cells of the symptomatic AD mice
Because we were interested in both the systemic and vagus nerve role in the gut-brain axis (GBA) communication of the central Aβ pathogenesis and to understand the TLR2 activation associated signaling mechanism within the GBA, we performed fluorescence-IHC on the ileal gut tissues of AD and control WT mice. Intriguing changes were recorded at the TLR2 protein levels. We additionally co-stained these gut sections with PGP9.5 (stains neuroendocrine cells). We found increased vagal afferent activation within the ileal tissue observed in symptomatic AD mice compared to littermate controls. Interestingly, PGP9.5 neuroendocrine marker was colocalized with TLR2 and this was present in ECs of the ileal epithelium (Fig. 5). TLR2 expression was also found sparsely in age-matched WT littermate controls but was significantly lower than AD mice. However, the number of TLR2+ PGP9.5+ cells were significantly increased in AD mice compared to the control mice (Fig. 5m). Additionally, these TLR2+ PGP9.5+ cells were also strongly expressed at the sub-mucosa that defines vagus nerve activation from the lumen to the mucosa (Figs. 4B, 5k, 5l) and potentially to the CNS.
Fig. 5.

TLR2 colocalizes with neuroendocrine marker PGP9.5 in the enterochromaffin cells within the epithelium and sub-mucosa of AD mice ileum. Immunofluorescent microscopy image of small intestinal villus showing TLR2 (green color) in ECs (subset of enteroendocrine cells) co-localized with PGP 9.5 neuroendocrine marker (red color) of vagus nervous system. Expression of TLR2 and PGP 9.5 in Symptomatic Tg2576 (e-h) are higher than WT (a-d) in the small intestine. The merged cells with overlapping expression are highlighted in the enlarged view, WT ileum epithelium (i) and sub-mucosa (j), whereas Tg AD mice ileum epithelium (k) and sub-mucosa (l). Images are taken at 60x magnification and exposures are set using staining of WT mouse tissues. All scale bars indicate 20 μm. (m) The percentage of TLR2+ & PGP9.5+ cells per villi in small intestine of symptomatic Tg2576 is higher than WT-littermate controls. Data are represented the mean ± s.d. of 5 mice in each group. TLR2 positive cells were calculated per villi for 15 villi per animal tissue. The TLR2 expression was also associated with PGP9.5 expression (low in WT animals). *p < 0.05; **p < 0.01; ***p < 0.001. p-values were calculated using students t-test. LP, lamina propria; SM, sub-mucosa; L, lumen.
Central TLR2 levels increased with Aβ plaque formation in the symptomatic AD mouse brain
Since we found increased TLR2 signals together with afferent vagal activation at the sub-mucosa in the AD mouse gut (Figs. 4 and 5), we set out to understand the TLR2 activation in the brain. Interestingly, we found significantly elevated levels of TLR2 activation in the cortex of AD mouse brain (Fig. 6) by immunohistochemical analysis and this was associated with significant presence of Aβ plaque deposits in the brain (Fig. 2A). This was observed significantly in the cortex and hippocampus regions of the AD brain compared to age-matched WT littermate control mice.
Fig. 6.

Expression of TLR2 is markedly increased in symptomatic Tg2576-mice brain (Cortex). A) Immunofluorescence staining of TLR2 in the brain indicates significant increase of TLR2 expression in the cortex of Tg2576 AD mice (Tg) compared to the wild-type littermate controls (WT). Images are taken at 2x and 60x magnification and exposures are set using staining of WT mouse tissues. Images are representative of at least 3 independent experiments, each including 5 mice of each group. Scale bars indicate 20 μm. B) Fluorescence intensities of TLR2 expression in cortex sections from Tg and WT mice. Data are expressed as fluorescence intensity in arbitrary unit (a.u.) and represent the mean ± s.d. of 5 mice. *p < 0.05; **p < 0.01; ***p < 0.001. p-values were calculated using students t-test.
Elevated TLR2 levels after bacterial curli treatment in the human ileal enteroids
To validate if bacterial curli activates TLR2 specific responses in the gut epithelium containing enterochromaffin cells, we used an in vivo specialized method of gut 3D enteroids cultures from human ileum. This was then prepared as monolayer. These enteroids monolayer where then treated with saline control and purified bacterial curli at varying concentrations (high-0.5 nM, medium-0.05 nM, and low-0.025 nM). We found increased TLR2 protein levels after 12 h of incubation with bacterial curli (Fig. 7A). Enteroids showed 2-fold increased levels of TLR2 with as low as 0.025 nM of curli protein incubation for 12-hours (Fig. 7B).
Fig. 7.

Bacterial Curli accelerate TLR2 protein expression in human ileal enteroids (HIE). A, B) Immunoblotting analysis of TLR2 expression in HIE. The assay is performed in triplicate and analyzed by NIH ImageJ Software. Samples are compared to β-actin as a housekeeping protein. Data are presented as the mean ± s.d. *p < 0.05; **p < 0.01; ***p < 0.001. p-values were calculated using One-Way analysis of variance with Bonferroni correction.
DISCUSSION
A growing body of evidence suggests that intestinal pathobiontic bacteria produce amyloid-like proteins that forms biofilms [10–12]. These proteins may contribute to neurological disease progression from the gut to the brain [13–16]. Amyloids are a class of protein with unique self-aggregation properties [27], and their aberrant accumulation can lead to cellular dysfunction associated with neurodegenerative diseases [27, 28]. While genetic and environmental factors can influence amyloid formation, molecular triggers are not well defined. Growing evidence suggests that non-identical amyloid proteins may accelerate reciprocal amyloid aggregation in a prion-like fashion [29]. While humans encode ∼30 amyloidogenic proteins, the gut microbiome also produces functional amyloids to build the biofilm matrix [30] that lines the intestinal mucus layer. Gut inflammation can alter gut integrity, gut bacterial composition as well and allow the translocation of antigens including biofilm associated amyloids into the gut mucosa [10–12]. Bacterial amyloid may trigger those local inflammation [31]. Bacterial amyloid translocation [13–15] may trigger [31] and accelerate amyloid aggregation in the brain [32–35]. Viral, bacterial, and fungal infections might be causative factors for the inflammatory responses found in AD [36]. We have already documented in our previous study that gut dysfunction occurs prior to Aβ accumulation in the brain using the same Tg2576 AD mouse model [1]. Most of the previous analysis was from large intestine, however, to understand gut-brain axis communication, the SI is of utmost importance due to its primary role in vagal activation [37]. Similar to the large intestinal gut dysbiosis found earlier [1], in our current study using 16S rRNA gene sequencing, data show significant bacterial compositional differences termed as dysbiosis (imbalance in normal gut microbiota compared to WT controls), found in the SI of symptomatic Tg2576 mice (15 to 16 months of age) gut microbiota when compared to WT littermate controls, and that shift denoted significant increase in gram positives within the SI lumen. We also found increased proportion of family Lactobacillaceae that again relates to our previous observation [1]. We sort to understand the main role of increase in Lactobacillaceae family. We will in our future studies understand the strain type of Lactobacillaceae bloom in these AD gut. However, since we found significant dysbiosis occur in AD mice SI, we were interested to look at immune activation from the gut. Therefore, we explored the gut integrity by looking at bacterial biofilm using FISH and found that the bacterial type differs in AD mice compared to WT controls. They were found more of rod shape and filamentous in the AD mice gut compared to cocci shaped bacteria found in WT mice. As a consequence, we will focus our future analysis based on these morphological changes. Most importantly, our control animals were age matched WT littermates and they live in the same cage. Since mice are coprophagic, the bacterial cross seeding might be an existing problem. Therefore, our future experiments will have age-matched non-littermate WT controls in addition. We mainly focused this study in SI because, microbial diversity in the SI is of utmost importance due to its specialized nature of containing ECs that are known to activate not just the systemic but primarily vagus nerve signaling pathways [37].
Loss of gut barrier integrity may cause pathogenic bacterial amyloid [10–12] translocation [13–15] and peripheral inflammation [38–40], and may influence toll-like receptor 2 induced innate immune responses [31]. We hypothesize that gut TLR2 activation may accelerate central Aβ pathology [19]. Interestingly, we found increased levels of bacterial amyloid-curli in the Tg AD mouse large intestine at pre-symptomatic timepoint before the Aβ pathology was found in the brain [1] compared to WT controls. However, we observed bacterial-curli presence in both aged WT and symptomatic AD mice large intestine. Similarly, we also found curli proteins in SI gut tissue of both WT and symptomatic AD mice. The increased bacterial amyloid-curli biofilm in WT animals’ gut lumen might be due to age-associated changes in the gut microbiota that are known to have detrimental effects [3, 4]. However, gut barrier integrity is impaired significantly in AD mice than in WT mice with age. Therefore, curli translocation and TLR2 activation would be profound in aged AD mice. We found TLR2 activation was 4- to 5-fold increase in pre-symptomatic AD mice compared to WT controls in the ileum. In addition, symptomatic aged AD mice also showed significantly increased TLR2 compared to WT controls but was not as high as pre-symptomatic timepoint. The increased levels of gut TLR2 was associated with loss of gut barrier integrity [1] and luminal bacterialcurli biofilm abundance found in pre-symptomatic AD mice compared to young WT control mice. Interestingly, we found TLR2 activation to be significantly stronger at the sub-mucosa regions of the ileal tissue in the symptomatic AD mice analyzed with IHC. It is previously known that vagal nerve plexus is present abundantly in the sub-mucosal regions and lamina propria within the gut tissue [20] and they extend the nodes to the epithelium and directly communicate with gut epithelial ECs [9]. ECs serve as bidirectional signal transducers between the gut lumen and nervous system [41, 42]. Vagal, afferent innervation of ECs provides a direct pathway for enterochromaffin-cell signaling to neuronal circuits [42, 43]. Since we found increased protein levels of TLR2 at the sub-mucosa, the data guided us to look at the intestinal epithelium more closely. PGP9.5 is a neuroendocrine/enteroendocrine marker that is known to be activated during vagus nerve stimulation [44]. Using fluorescence IHC, we found that ileal gut ECs significantly express TLR2 and this expression was co-localized with PGP9.5 (neuronal endocrine marker) [9, 45].
Dysbiotic pathogenic bacteria in the gut lumen form biofilms and they might produce amyloid-like proteins [10–12]. Specifically, Salmonella derived bacterial amyloid curli fibrils contribute to Nos2 expression in animal models by stimulating TLR2 signaling [31, 46]. Bacterial amyloids can activate TLR 2/9 receptors [31] and exacerbate memory deficits [10–15]. TLR2 activation also worsens white matter damage in AD [19]. TLR2 activation has been shown to increase amyloid aggregation and inhibition of TLR2 attenuates neuronal Aβ levels in AD mice [17, 18]. It is possible that TLR2 activation is due to increased translocation of bacterial products like curli (bacterial amyloids) [31] due to gut dysfunction in AD [1]. On that note, we also found increased TLR2 levels in the symptomatic AD brain compared to age-matched WT controls. We believe that TLR2 activation mainly progress from the gut in response to bacterial amyloid-curli. We have shown that gut TLR2 elevation colocalizes with neuroendocrine marker within the ECs. Our data provided the first evidences that gut TLR2 activation causes afferent vagus nerve signaling in response to bacterial-curli that travel to the brain in an AD mouse model. More research on this line of thought is necessary and we are doing further experiments to understand this pathway more in detail. However, to verify that bacterial curli activates TLR2 within the gut epithelium, we used 3D enteroids culture of human ileum which also has intact ECs apart from other specialized epithelial cells and where treated with purified bacterial curli protein fibrils. This experiment revealed that bacterial-curli induces TLR2 activation.
In conclusion, our data clearly shows that bacterial amyloid burden is present in AD mouse gut even before Aβ pathology occurs in the brain. Even though WT aged mice show curli presence, an intact gut barrier is significantly reduced in AD mice gut [1] which might cause increased curli translocation in AD mice. Bacterial curli activates TLR2 specific signals within the epithelium and in the sub-mucosa of the gut. Importantly, TLR2 activation co-localized with significantly elevated neuroendocrine marker PGP9.5 within the gut of AD mice that provides evidence for vagus nerve activation in response to bacterial curli [44]. We also found increased levels of TLR2 activation within the brain of AD mice as seen by others [17, 18]. Therefore, it provides preliminary evidence that TLR2 activation from the gut to brain in response to bacterial curli might play a major role in central Aβ pathology of AD. However, we need more evidence to prove whether this signaling pathway is clinically important in order to treat the pre-symptomatic AD patients before pathology is seen in the brain when treatments are likely to be ineffective. Our future studies will primarily focus on unraveling the gutvagus-brain connection with clinical implications in AD.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIA 1R01AG070934–01 to B.P.G). The Gastrointestinal Experimental Model Systems Core, a part of Silvio O Conte Digestive Diseases Research Core Center was supported by the NIH-NIDDK grant.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0106r2).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220106.
REFERENCES
- [1].Honarpisheh P, Reynolds CR, Blasco Conesa MP, Moruno Manchon JF, Putluri N, Bhattacharjee MB, Urayama A, McCullough LD, Ganesh BP (2020) Dysregulated gut homeostasis observed prior to the accumulation of the brain amyloid-beta in Tg2576 mice. Int J Mol Sci 21, 1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chung HY, Kim DH, Lee EK, Chung KW, Chung S, Lee B, Seo AY, Chung JH, Jung YS, Im E, Lee J, Kim ND, Choi YJ, Im DS, Yu BP (2019) Redefining chronic inflammation in aging and age-related diseases: Proposal of the senoinflammation concept. Aging Dis 10, 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ritzel RM, Lai Y-J, Crapser JD, Patel AR, Schrecengost A, Grenier JM, Mancini NS, Patrizz A, Jellison ER, Morales-Scheihing D, Venna VR, Kofler JK, Liu F, Verma R, McCullough LD (2018) Aging alters the immunological response to ischemic stroke. Acta Neuropathol 136, 89–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spychala MS, Honarpisheh P, McCullough LD (2017) Sex differences in neuroinflammation and neuroprotection in ischemic stroke. J Neurosci Res 95, 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].DeJong EN, Surette MG, Bowdish DME (2020) The gut microbiota and unhealthy aging: Disentangling cause from consequence. Cell Host Microbe 28, 180–189. [DOI] [PubMed] [Google Scholar]
- [6].Erickson MA, Banks WA (2019) Age-associated changes in the immune system and blood(−)brain barrier functions. Int J Mol Sci 20, 1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Breit S, Kupferberg A, Rogler G, Hasler G (2018) Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front Psychiatry 9, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cao W, Zheng H (2018) Peripheral immune system in aging and Alzheimer’s disease. Mol Neurodegener 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kaelberer MM, Buchanan KL, Klein ME, Barth BB, Montoya MM, Shen X, Bohorquez DV (2018) A gut-brain neural circuit for nutrient sensory transduction. Science 361, eaat5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Romero D, Aguilar C, Losick R, Kolter R (2010) Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad SciUSA 107, 2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Erskine E, MacPhee CE, Stanley-Wall NR (2018) Functional amyloid and other protein fibers in the biofilm matrix. J Mol Biol 430, 3642–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tursi SA, Tukel C (2018) Curli-containing enteric biofilms inside and out: Matrix composition, immune recognition, and disease implications. Microbiol Mol Biol Rev 82, e00028–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet MF, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, Mazmanian SK (2016) Gut microbiota regulate motor deficits and neuroin-flammation in a model of Parkinson’s disease. Cell 167, 1469–1480 e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, Mazmanian SK, Volpicelli-Daley LA, Gradinaru V (2020) Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 23, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, Shastri GG, Thron T, Needham BD, Horvath I, Debelius JW, Janssen S, Knight R, Wittung-Stafshede P, Gradinaru V, Chapman M, Mazmanian SK (2020) A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. eLife 9, e53111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, Garaci E, Grassi C, Palamara AT (2012) Infectious agents and neurodegeneration. Mol Neurobiol 46, 614–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, Menger MD, Fassbender K (2012) TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J Immunol 188, 1098–1107. [DOI] [PubMed] [Google Scholar]
- [18].McDonald CL, Hennessy E, Rubio-Araiz A, Keogh B, McCormack W, McGuirk P, Reilly M, Lynch MA (2016) Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav Immun 58, 191–200. [DOI] [PubMed] [Google Scholar]
- [19].Zhou C, Sun X, Hu Y, Song J, Dong S, Kong D, Wang Y, Hua X, Han J, Zhou Y, Jin G, Yang X, Shi H, Zhang Z, Hua F (2019) Genomic deletion of TLR2 induces aggravated white matter damage and deteriorated neurobehavioral functions in mouse models of Alzheimer’s disease. Aging 11, 7257–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Browning KN, Verheijden S, Boeckxstaens GE (2017) The vagus nerve in appetite regulation, mood, and intestinal inflammation. Gastroenterology 152, 730–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Scott MR, Kohler R, Foster D, Prusiner SB (1992) Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci 1, 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- [23].Elder GA, Gama Sosa MA, De Gasperi R (2010) Transgenic mouse models of Alzheimer’s disease. Mt Sinai J Med 77, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhou Y, Smith DR, Hufnagel DA, Chapman MR (2013) Experimental manipulation of the microbial functional amyloid called curli. Methods Mol Biol 966, 53–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saxena K, Blutt SE, Ettayebi K, Zeng XL, Broughman JR, Crawford SE, Karandikar UC, Sastri NP, Conner ME, Opekun AR, Graham DY, Qureshi W, Sherman V, Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Donowitz M, Estes MK (2016) Human intestinal enteroids: A new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol 90, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S (1999) Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451. [DOI] [PubMed] [Google Scholar]
- [27].Soto C, Pritzkow S (2018) Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci 21, 1332–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Soto C (2012) Transmissible proteins: Expanding the prion heresy. Cell 149, 968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Visanji NP, Lang AE, Kovacs GG (2019) Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl Neurodegener 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Taglialegna A, Navarro S, Ventura S, Garnett JA, Matthews S, Penades JR, Lasa I, Valle J (2016) Staphylococcal Bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathogens 12, e1005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tukel C, Wilson RP, Nishimori JH, Pezeshki M, Chromy BA, Baumler AJ (2009) Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe 6, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Evans ML, Gichana E, Zhou Y, Chapman MR (2018) Bacterial amyloids. Methods Mol Biol 1779, 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Friedland RP, McMillan JD, Kurlawala Z (2020) What are the molecular mechanisms by which functional bacterial amyloids influence amyloid beta deposition and neuroinflammation in neurodegenerative disorders? Int J Mol Sci 21, 1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Spaulding CN, Dodson KW, Chapman MR, Hultgren SJ (2015) Fueling the fire with fibers: Bacterial amyloids promote inflammatory disorders. Cell Host Microbe 18, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhou Y, Blanco LP, Smith DR, Chapman MR (2012) Bacterial amyloids. Methods Mol Biol 849, 303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sochocka M, Zwolinska K, Leszek J (2017) The infectious etiology of Alzheimer’s disease. Curr Neuropharmacol 15, 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, Tirosh I, Beyaz S, Dionne D, Zhang M, Raychowdhury R, Garrett WS, Rozenblatt-Rosen O, Shi HN, Yilmaz O, Xavier RJ, Regev A (2017) A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lobionda S, Sittipo P, Kwon HY, Lee YK (2019) The role of gut microbiota in intestinal inflammation with respect to diet and extrinsic stressors. Microorganisms 7, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ganesh BP, Nelson JW, Eskew JR, Ganesan A, Ajami NJ, Petrosino JF, Bryan RM, Jr., Durgan DJ (2018) Prebiotics, probiotics, and acetate supplementation prevent hypertension in a model of obstructive sleep apnea. Hypertension 72, 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ganesh BP, Klopfleisch R, Loh G, Blaut M (2013) Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PloS One 8, e74963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bohorquez DV, Samsa LA, Roholt A, Medicetty S, Chandra R, Liddle RA (2014) An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PloS One 9, e89881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Latorre R, Sternini C, De Giorgio R, Greenwood-Van Meerveld B (2016) Enteroendocrine cells: A review of their role in brain-gut communication. Neurogastroenterol Motil 28, 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rhee SH, Pothoulakis C, Mayer EA (2009) Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol 6, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Booth LC, Yao ST, Korsak A, Farmer DGS, Hood SG, McCormick D, Boesley Q, Connelly AA, McDougall SJ, Korim WS, Guild SJ, Mastitskaya S, Le P, Teschemacher AG, Kasparov S, Ackland GL, Malpas SC, McAllen RM, Allen AM, May CN, Gourine AV (2021) Selective optogenetic stimulation of efferent fibers in the vagus nerve of a large mammal. Brain Stimul 14, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].West PW, Canning BJ, Merlo-Pich E, Woodcock AA, Smith JA (2015) Morphologic characterization of nerves in wholemount airway biopsies. Am J Respir Crit Care Med 192, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tursi SA, Lee EY, Medeiros NJ, Lee MH, Nicastro LK, Buttaro B, Gallucci S, Wilson RP, Wong GCL, Tukel C (2017) Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathogens 13, e1006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.