

Abstract
The ability to associate temporally segregated information and assign positive or negative valence to environmental cues is paramount for survival. Studies have shown that different basolateral amygdala (BLA) projections are potentiated following reward or punishment learning1–7. However, we do not yet understand how valence specific information is routed to the BLA neurons with the appropriate downstream projections. Nor do we understand how to reconcile the subsecond timescales of synaptic plasticity8–11 with the longer timescales separating the predictive cues from their outcomes. Here, we demonstrate that neurotensin (NT) neurons in the paraventricular nucleus of the thalamus (PVT) projecting to the BLA (PVT-BLA:NT) mediate valence assignment by exerting concentration-dependent modulation in BLA during associative learning. We found that optogenetic activation of the PVT-BLA:NT projection promotes reward learning, while PVT-BLA projection-specific Nt gene knockout augments punishment learning. Using genetically encoded calcium and NT sensors, we further revealed that both calcium dynamics within the PVT-BLA:NT projection and NT concentrations in the BLA are enhanced after reward learning and reduced after punishment learning. Finally, we showed that CRISPR-mediated knockout of the Nt gene in the PVT-BLA pathway blunts BLA neural dynamics and attenuates the preference to active behavioral strategies to reward and punishment predictive cues. Taken together, we have identified NT as a neuropeptide that signals valence in the BLA, and showed that NT is a critical neuromodulator that orchestrates positive and negative valence assignment in amygdala neurons by extending valence-specific plasticity to behaviorally-relevant timescales.
During associative learning, the basolateral complex of the amygdala (BLA) forms associations with positive or negative outcomes, as supported by evidence from neural recordings2,3,12,13, synaptic changes1,14,15, and optogenetic manipulations1,5,16,17. Positive and negative associative learning is mediated by synaptic plasticity onto divergent paths1,18–24, yet how the correct path is chosen and the correct valence is assigned on behaviorally-relevant timescales remains a mystery. How does the brain solve the “valence assignment problem”?
Specifically, how are predictive stimuli linked to the rewards or punishments that follow tens of seconds later when the temporal dynamics of synaptic plasticity operate on a sub-second timescale? Neuromodulatory signals are believed to produce slow acting modulations and extend spike timing-dependent plasticity (STDP) at functionally-relevant timescales25,26. Thus, we speculated that a neuromodulator or neuropeptidergic signal that might selectively gate plasticity to positive or negative valence encoding neurons could be the solution to the valence assignment problem.
In the amygdala, dopamine has been shown to gate synaptic plasticity, but codes for “absolute value” in that dopamine receptor 1 (D1) signaling is necessary for both reward and punishment learning and does not differentiate between positive and negative valence27–29, and therefore does not signal valence. To facilitate valence assignment, a GPCR-signal would need to be transmitted differentially on positive and negative valence encoding neurons.
Neurotensin (NT) is a 13 amino acid peptide implicated in both reward and punishment processing30,31, and its signaling in the BLA has been shown to affect long-term potentiation (LTP) and fear learning32,33. We previously identified that the expression of NT receptor type 1 gene (Ntsr1) is enriched BLA neurons that project to the centromedial nucleus (CeM; BLA-CeM) of the central amygdala (CeA) compared to the nucleus accumbens (NAc; BLA-NAc)1, two BLA populations that have been implicated in associative learning of both positive and negative valence1–3,14,15, though each population includes further genetic and functional heterogeneity4–7. Using fluorescent mRNA in situ hybridization, we further confirmed this differential receptor expression among these two BLA projection populations (Figure E1a–f). Together, the evidence suggested that NT was well-positioned to play an essential role in guiding valence assignment in the BLA.
Figure 5: Computational analysis reveals BLA population dynamics, encoding of valence, and the behavioral preference in action selection.
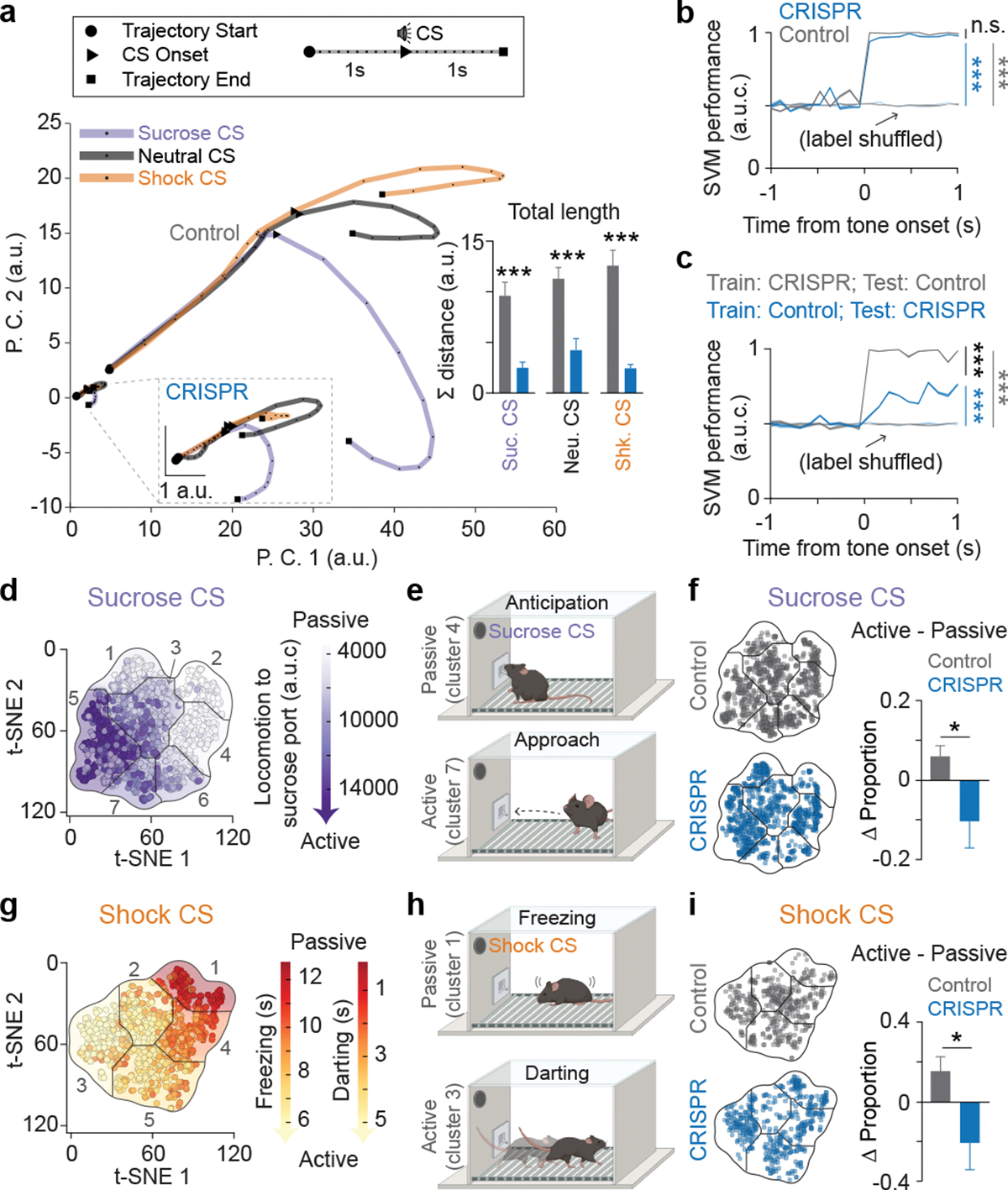
(a) Principal component analysis (PCA) was applied on the pooled raw firing rates of all neurons to reward, neutral, and shock CS, and the first 2 components were used for plotting the neural trajectories. Inset: neural trajectories of the control and CRISPR group in the same PCA subspace reveals group differences in both trajectory length and distance between CSs (Two-tailed unpaired t-test, N=12 and 14 for control and CRISPR group using a leave-one-out approach, respectively. See Methods for details. ***P<0.0001 for sucrose and shock CSs and ***P=0.0002 for neutral CS). (b) A support vector machine (SVM) was used to decode trial types (sucrose or shock trial) using the first 10 PC scores of the averaged neural activity of each trial. The SVM decoder performed equally well when trained and tested on the same groups (trained and tested on the control data or on the CRISPR data), while the decoder performance remained at the baseline level when the model was trained and tested on the shuffled trial labels (5-fold validations for all groups. Control vs. CRISPR: P=0.2122; Control vs. Control Shuffled: ***P<0.0001; CRISPR vs. CRISPR Shuffled: ***P<0.0001). (c) SVM decoder performance significantly dropped when the model was trained on the control data and tested on the CRISPR data, compared to trained on the CRISPR data and tested on the control data (5-fold validations for all groups. Trained on control vs. trained on CRISPR: ***P<0.0001; trained on control vs. Shuffled: ***P<0.0001; trained on CRISPR vs. Shuffled: ***P<0.0001). One-way ANOVA with Holm-Sidak’s multiple comparisons test for b and c. (d, g) Mice exhibited distinct behavioral strategies that are distributed across the total behavioral space for sucrose and shock trials. Passive or active strategy in sucrose trials were described by the low or high changes in distance moved toward port after the sucrose CS onsets, and in shock trials were described by high freezing and low darting or low freezing and high darting in response to shock CS, respectively. (e, h) Illustrations of behavior in passive and active strategies during both sucrose and shock trials. (f, i) Control mice showed more active trials in responses to both sucrose and shock CSs compared to CRISPR-treated mice (Two-tailed unpaired t-test, *P=0.0325 for sucrose trials and *P=0.0251 for shock trials; N=15). Error bars and solid shaded regions around the mean indicate s.e.m.
NT inputs to the BLA co-release glutamate
To explore the role of NT in the BLA during valence assignment, we first located NT neural populations that project to the BLA, by injecting retrograde tracer into the BLA in mice that express a fluorescent reporter, tdTomato, in all NT neurons (Figure 1a, b; E1g). We observed that the medial geniculate nucleus (MGN), ventral subiculum and ventral CA1 of the hippocampus (vHPC), and the paraventricular nucleus of the thalamus (PVT) had NT neurons (tdTomato expressing) projecting to the BLA (labeled with the retrograde tracer Figure 1c).
Figure 1: Identification of functional NT sources to the BLA in associative learning.
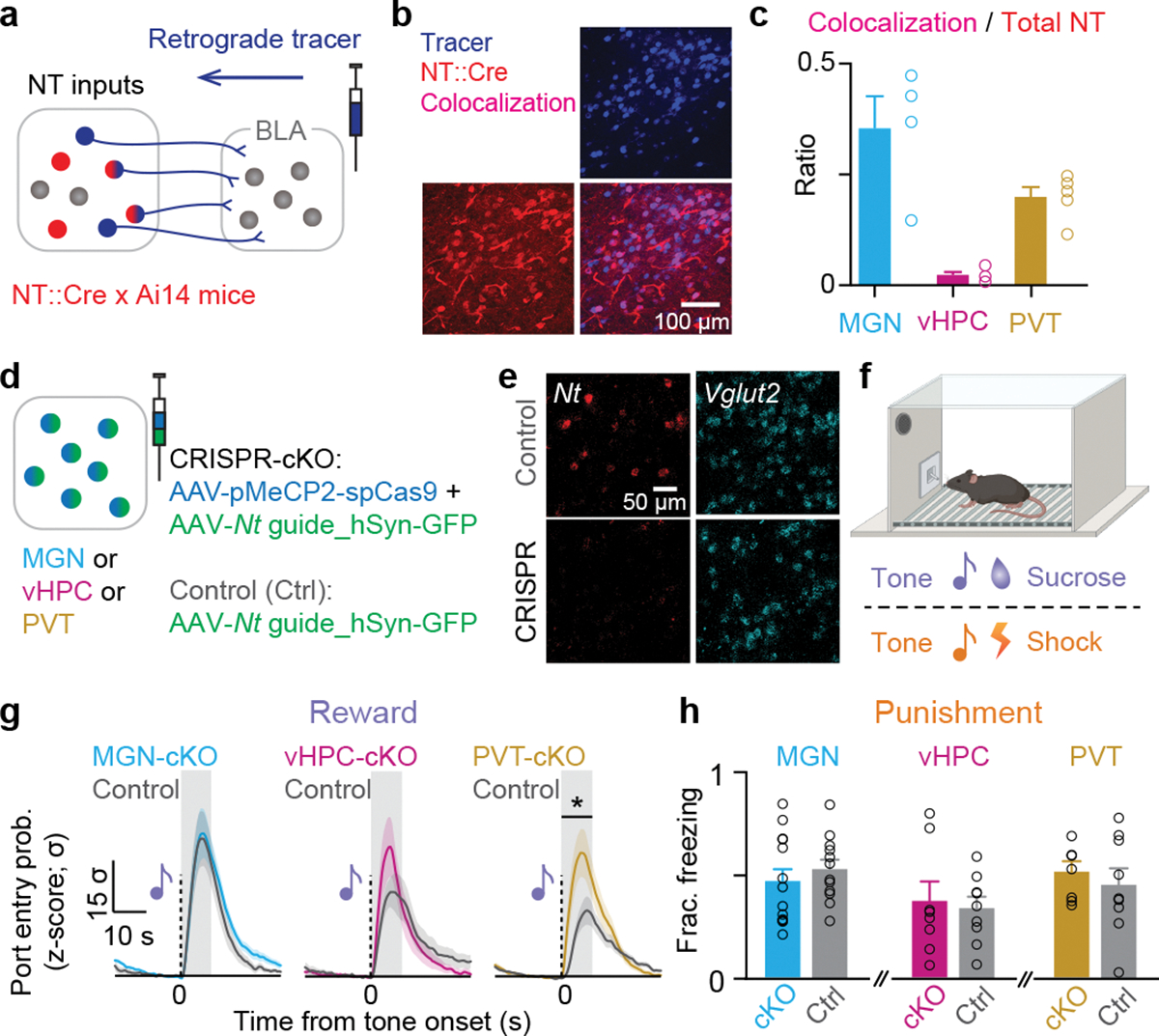
(a) Schematic of viral injection. (b) Representative confocal image showing the overlap of fluorescence from the retrograde tracer depicting BLA projectors and the fluorophore tdTomato depicting NT populations. (c) Quantification of overlap between NT expressing and BLA projecting neurons as a fraction of the NT population. (d) Schematic of viral-mediated CRISPR-Cas9 Nt gene knockout in each NT source. (e) Confocal images from a representative brain slice showing fluorescence from in situ hybridization of Nt mRNA and Vglut2 mRNA from the control and experimental groups. (f) Experimental design to test the acquisition of reward and punishment learning upon depletion of Nt gene. (g) Mice with PVT:Nt CRISPR-cKO showed a significantly higher port entry probability in last session of tone-sucrose conditioning, while MGN:Nt and vHPC:Nt CRISPR-cKO did not show significant changes compared to their cage-mate controls (P=0.9195, P=0.3976, and *P=0.0148, respectively for MGN, vHPC, and PVT. For CRISPR and control, N=18 and 17, N=11 and 9, and N=15 and 13, respectively for MGN, vHPC, and PVT). (h) CRISPR-cKO in none of the regions affected tone-shock acquisition in second session of tone-shock conditioning (P=0.4122, P=0.7818, and P=0.5751, respectively for MGN, vHPC, and PVT. For CRISPR and control, N=14 and 12, N=8 and 9, and N=8 and 9, respectively for MGN, vHPC, and PVT). Two-tailed unpaired t-test was used for all statistical tests. N denote number of mice in each group. Error bars and solid shaded regions around the mean indicate s.e.m. Shaded regions in panel g indicate the analysis windows used for statistical tests.
To confirm that NT cell populations in the MGN, vHPC, and PVT (represented as MGN:NT, vHPC:NT, and PVT:NT, respectively) indeed send axons to the BLA, we expressed channelrhodopsin-2 (ChR2) with a different fluorophore (ChR2-eYFP or ChR2-mCherry) in each population and imaged their axonal fields in the BLA (Figure E1h). Their axonal fields were topographically separable (Figure E1h–j).
Furthermore, we confirmed that axons from all three NT populations form functional synapses in the BLA by stimulating ChR2-expressing axon terminals in the BLA while recording synaptic currents from principal BLA neurons using patch-clamp electrophysiology (Figure E2a). Stimulating axon terminals evoked both excitatory and inhibitory post-synaptic currents (EPSCs and IPSCs; Figure E2b, c). Using ChR2 assisted circuit mapping34, we determined that the EPSCs evoked in BLA neurons by stimulating ChR2-expressing axon terminals from all three NT populations were monosynaptic and can be blocked by antagonizing the AMPA and NMDA glutamate receptors (Figure E2d), whereas the IPSCs were not (Figure E2e). Altogether, these data suggest that the BLA receives monosynaptic inputs from three NT populations, all of which co-release glutamate in the BLA.
Global Nt gene knockout in the PVT promotes reward learning
To explore the functional role of NT sources, we selectively disrupted NT from each of the upstream sources while preserving the glutamatergic machinery by using the clustered regularly interspaced short palindromic repeats (CRISPR)-associated endonuclease Cas935,36 along with guide RNAs targeted to exons 1 and 3 of the Nt gene. The Cas9 protein induces a double-strand break in the gene targeted by the guide RNAs, leading to the formation of loss-of-function insertion-deletion (indel) mutations by non-homologous end joining37,38, allowing site-specific targeted mutation, in vivo.
We verified that our guide RNAs (guide RNA 1 and guide RNA 2) had high editing efficiency (~90 %) in vivo (Figure E3a, c), and that the most frequent edit was a frame-shifting single-nucleotide insertion into the target gene (Figure E3b, d). Furthermore, guide RNA 1 mutated the start codon of the Nt gene, therefore possibly even preventing translation of the Nt gene. Finally, we validated that our viral system targeted cells with high efficiency and reduced Nt mRNA levels in the injected region without affecting the Vglut mRNA levels (Figure E3e, f).
Using the CRISPR-Cas9 system, we then knocked out Nt gene expression in the MGN:NT, the vHPC:NT, or the PVT:NT populations of separate animals before exposing the animals to tone-sucrose and tone-shock associations (Figure 1d–f; E4). We found that the CRISPR-Cas9 mediated conditional knockout (CRISPR-cKO) of Nt gene in the PVT, but not the MGN nor the vHPC, enhanced tone-sucrose association (Figure 1g), but not tone-shock association (Figure 1h), implicating the PVT:NT population in reward learning.
PVT-BLA:NT neurons are a functionally-distinct PVT population and bidirectionally modulate both reward and punishment learning
As the global CRISPR-cKO of Nt gene affected all PVT:NT neurons providing potential direct and indirect effects in the BLA, it was unclear how PVT:NT inputs to the BLA contribute to associative learning. Given the functional heterogeneity of the PVT, which contains a mix of reward and aversion encoding neurons39–43, we speculated that the PVT-BLA:NT projection could be a functionally distinct PVT population.
To confirm this, we selectively knocked out the Nt gene in the PVT-BLA projection by injecting a retrogradely-transported virus encoding the guide RNAs into the BLA and another virus encoding Cas9 into the PVT, and examined the effects of the PVT-BLA:Nt CRISPR-cKO on reward and punishment learning (Figure 2a–b). We validated that our retrograde CRISPR-Cas9 approach had high efficiency targeting NT in PVT-BLA projecting neurons but not glutamate (Figure E5a–g). As speculated, we found that the PVT-BLA:Nt CRISPR-cKO impaired tone-sucrose association and enhanced tone-shock association relative to the scrambled guide RNA controls (Figure 2c, d), which are different from the behavioral effects of the PVT:Nt global CRISPR-cKO. Consistently, we found that optogenetic activation of PVT:NT axon terminals in the BLA of NT::Cre mice during the presentation of the tones enhanced tone-sucrose association and impaired tone-shock association, relative to mCherry controls (Figure 2e–h; E5h–k).
Figure 2: Causal manipulations show PVT-BLA:NT bidirectionally modulate both reward and punishment learning.
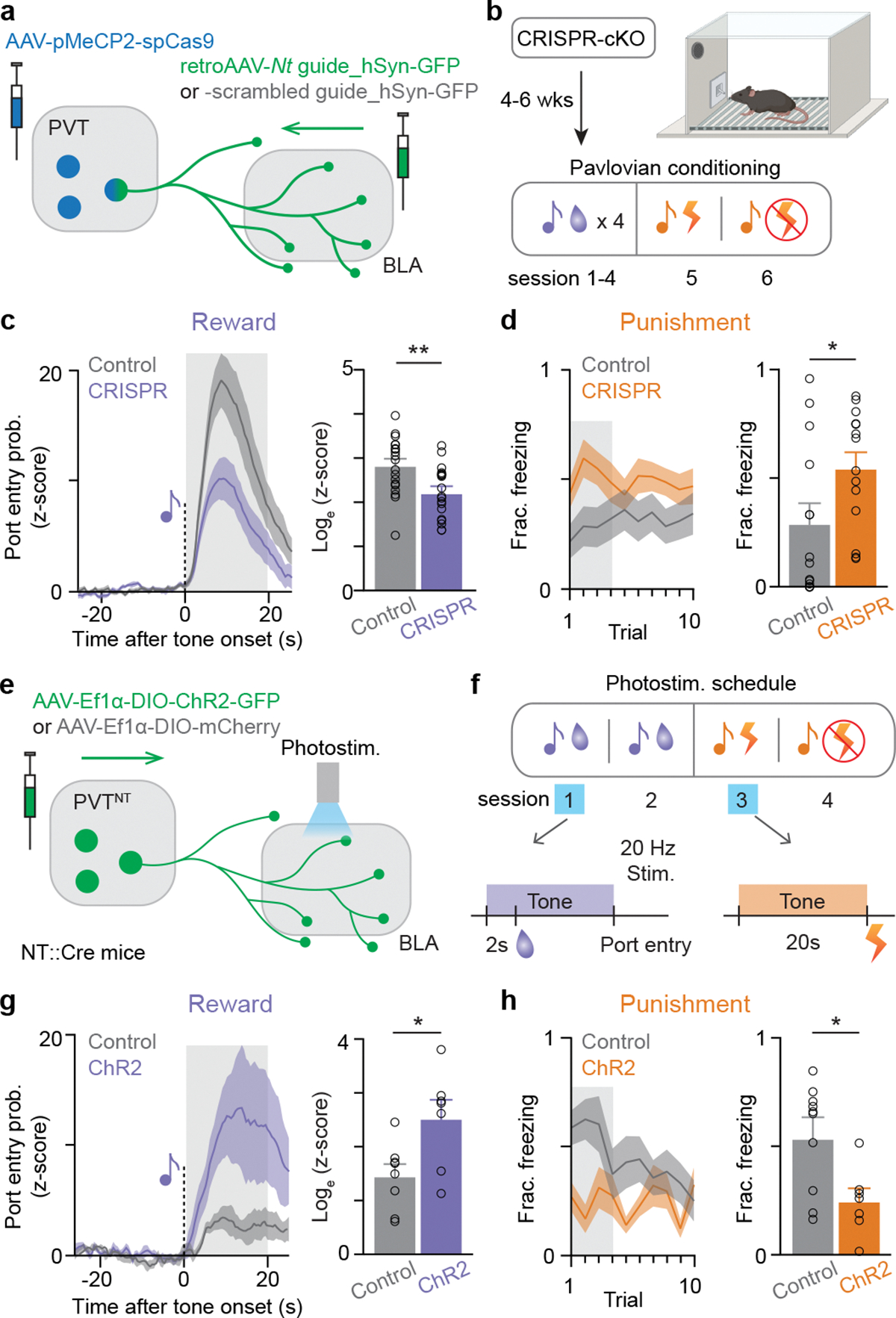
(a) Schematics of viral injection for CRISPR-mediated Nt gene cKO in the PVT-BLA projection. (b) Behavioral procedure of reward and punishment learning. Following the tone-shock conditioning session, an additional session of tone-shock testing was added in the next day, in which shocks were omitted. (c) The CRISPR group showed significantly reduced port entry probability in response to the sucrose-predictive CS (**P=0.0052, N=17 and 19 for CRISPR and control, respectively). (d) The CRISPR group also showed significantly higher proportion of time spent freezing during the duration of shock-predictive CS in the tone-shock testing session (*P=0.0396, N=14 and 14 for CRISPR and control, respectively). (e) Schematics of viral injection for optogenetic activation of PVT:NT terminals in the BLA. (f) Photostimulation protocol took place during the presentation of the tones at the first day of tone-sucrose and tone-shock association. (g) The ChR2 group showed significantly enhanced port entry probability in response to sucrose-predictive CS (*P=0.018, N=7 and 8 for ChR2 and control, respectively). (h) The ChR2 group showed significantly reduced freezing in the tone-shock testing session during the presentation of shock-predictive CS compared to the control group (*P=0.0279, N=7 and 8 for CRISPR and control, respectively). Two-tailed unpaired t-test was used for all statistical tests. Shaded regions indicate the analysis windows used for the sub-panels in c, d, g, and h. N denotes number of mice in each group, respectively. Error bars and solid shaded regions around the mean indicate s.e.m.
To further explore the possibility that PVT-BLA:NT neurons are a functionally distinct subpopulation of PVT:NT, we monitored calcium activity of the two populations using fiber photometry. Specifically, we injected a Cre-dependent genetically-encoded calcium indicator GCaMP6m in the PVT of NT::Cre mice and recorded PVT:NT somatic or PVT-BLA:NT axon terminal calcium activity in separate animals. Mice were trained to associate one auditory tone with sucrose and another with airpuff in a head-fixed preparation (Figure 3a; E6a). The acquisition of tone-sucrose and tone-airpuff associations was assessed by anticipatory lick responses and anticipatory eyelid closure upon tone onset, respectively (Figure 3b–c; E6e–g). We found that behaviorally-defined acquisition of either task selectively changed PVT-BLA:NT axon terminal calcium responses (Figure 3d–f; E7a), but not PVT:NT somatic calcium responses (Figure E6b–d). Specifically, we found that PVT-BLA:NT terminal calcium responses to outcomes (sucrose or airpuff delivery) increased after the acquisition of a positive association (tone-sucrose) and decreased after the acquisition of a negative association (tone-airpuff; Figure 3d–f), while somatic calcium responses of the NT three sources including PVT:NT had no learning-induced changes (Figure E6b–d).
Figure 3: Genetically-encoded fluorescent sensors reveal enhanced and suppressed NT dynamics in the BLA after reward and punishment learning, respectively.
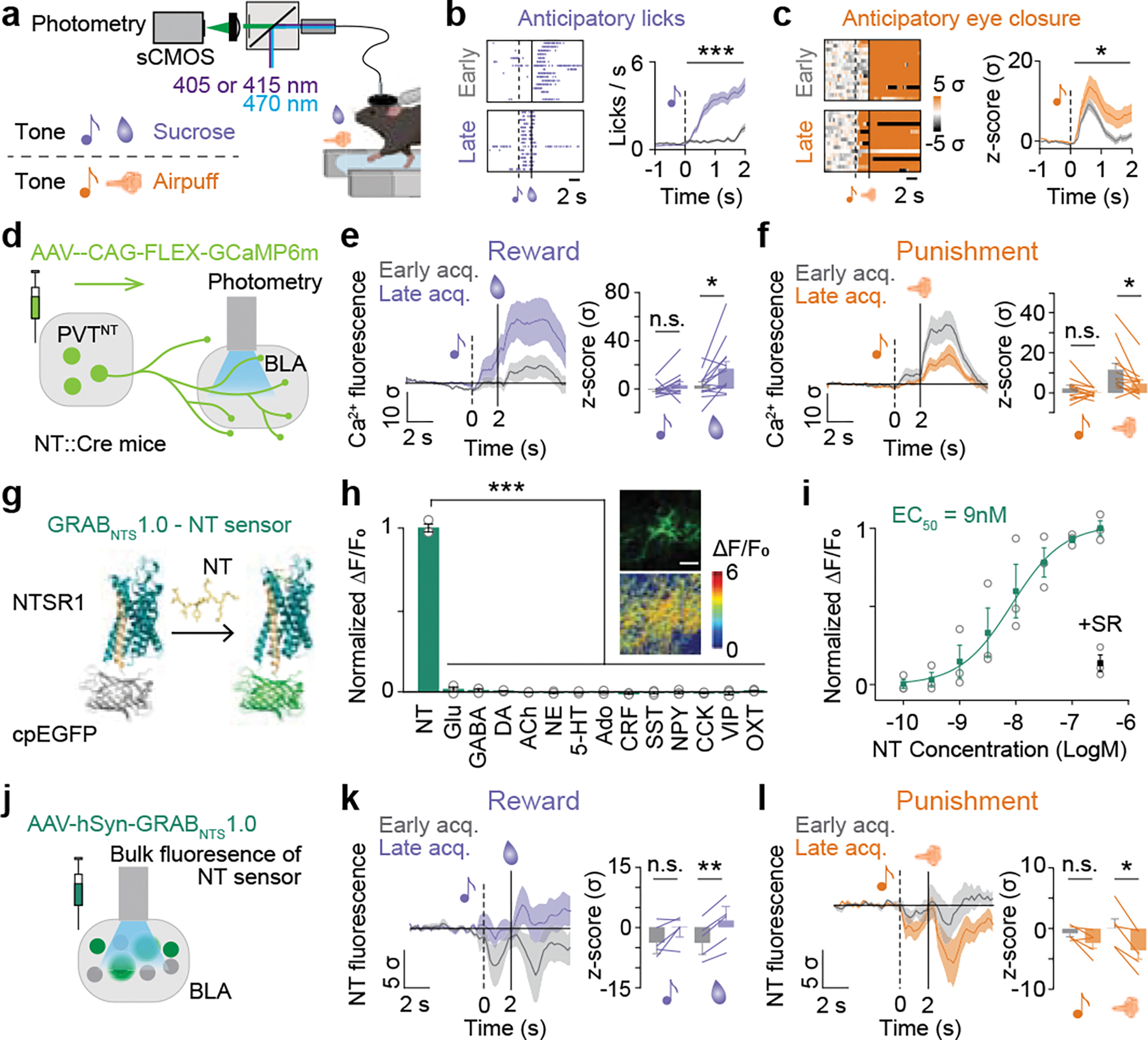
(a) Schematics of fiber photometry recording and behavioral paradigm of head-fixed associative learning. (b) Representative plots of licks (Left) and averaged anticipatory licks to the sucrose tone for the first 16 trials of first and last tone-sucrose sessions (Right; Two-tailed paired t-test; ***P<0.0001, N=13). (c) Representative plots of normalized eye closure index (Left) and averaged anticipatory eye closure to the airpuff tone for the first 16 trials of first and last tone-airpuff conditioning sessions (Right; Two-tailed paired t-test; *P=0.03, N=12). (d) Schematic of viral injection of a genetically-encoded calcium indicator and fiber implantation. (e) Calcium responses of the PVT-BLA:NT axon terminals to sucrose were significantly enhanced by reward conditioning (Two-tailed paired t-test, CS: P=0.1663; US: *P=0.0125; N=13). (f) Responses to airpuff were significantly suppressed by punishment conditioning. (Two-tailed paired t-test, CS: P=0.169; US: *P=0.0145; N=12). (g) Schematic drawing of the GRABNTS1.0 sensor. The third intracellular loop of the human NTSR1 was replaced with cpEGFP, thus the binding of neurotensin induces a conformational change that increases the intensity of cpEGFP fluorescence. (h) Normalized ΔF/F0 in sensor-expressing neurons following the application of NT alone, glutamate (Glu), gamma-aminobutyric acid (GABA), dopamine (DA), acetylcholine (ACh), norepinephrine (NE), serotonin (5-HT), adenosine (Ado), corticotrophin releasing factor (CRF), somatostatin (SST), neuropeptide Y (NPY), cholecystokinin (CCK), vasoactive intestinal peptide (VIP) and oxytocin (OXT; One-way ANOVA with Dunnett’s multiple comparisons test; ***P<0.0001). Inset: representative images of sensor expression (top) and responses to 100 nM NT (down) in cultured neurons. Scale bar represents to 100 μm. (i) Normalized dose-response curve for GRABNTS1.0 expressing neurons in response to NT, and the black dot indicates subsequent addition of 1 μM NTSR1 (+SR) antagonist at the 300 nM NT condition. (j) Schematic of viral injection of a genetically-encoded NT sensor and fiber implantation. (k) BLA NT sensor response to sucrose was enhanced by reward conditioning (Two-tailed paired t-test, CS: P=0.1483; US: **P=0.001; N=5). (l) BLA NT sensor response to airpuff was reduced by punishment conditioning (Two-tailed paired t-test, CS: P=0.0994; US: *P=0.0162; N=5). Analysis windows for the CSs and the USs were 0–2s and 2–7s from CS onsets, respectively. N denotes number of mice in each group. Error bars and solid shaded regions around the mean indicate s.e.m.
Genetically-encoded fluorescent NT sensor reveals distinct changes in BLA NT dynamics after reward and punishment learning
Since the calcium activity would reflect any neural activity, including glutamate release, it was still unclear how NT concentration in the BLA was changed by associative learning. To selectively monitor BLA NT dynamics, we developed a genetically-encoded fluorescent NT sensor GRABNTS1.0, by replacing the third intracellular loop of the human NTSR1 with a circularly permutated EGFP (cpEGFP; Figure 3g), and validated the sensor to show high sensitivity and specificity for NT (Figure 3h, i; E7b–j). We then expressed the GRABNTS1.0 in BLA neurons and quantified the dynamics of NT release in vivo during associative learning using fiber photometry (Figure 3j; E7k, l).
Initially, we observed transient decreases in NT sensor responses to both sucrose and airpuff during early acquisition trials, suggesting a pause in basal NT innervation to the BLA. After learning, we observed an increased NT sensor response to sucrose, and a decreased NT sensor response to airpuff (Figure 3k, l), consistent with the learning-induced changes in the PVT-BLA:NT terminal calcium activity. These data suggest that PVT-BLA:NT neurons differentially respond to positive and negative valence across learning and modulate BLA NT concentration in a valence-specific manner – with increased NT after reward learning and decreased NT after punishment learning. We speculate that when salient events are uncertain, NT concentrations drop, but when a rewarding or aversive context is detected, this is signaled to the BLA with increased or further decreased concentrations of NT to gate plasticity, respectively.
NT inputs from the PVT amplify BLA representation of learned valences
As we observed distinct NT dynamics across reward and punishment learning, we next investigated whether NT dynamics were necessary for encoding valence assignment in BLA neurons. We performed in vivo electrophysiological recordings in the BLA in mice with and without the PVT-BLA:Nt CRISPR-cKO (Figure 4a; E8a–c). We recorded a total of 685 BLA neurons from 30 mice while they were performing a discrimination task where three types of trials with distinct auditory tones predicting sucrose, shock, or no outcome (Sucrose, Shock, and Neutral CSs) were randomly presented at a 2:1:1 ratio (Figure 4b). At the end of each session, we optogenetically photoidentified BLA-NAc and BLA-CeA neurons based on previously reported photoresponse latencies2,3 (Figure 4c).
Figure 4: The CRISPR-cKO of Nt gene in the PVT to BLA projection attenuates BLA responses to learned valences.
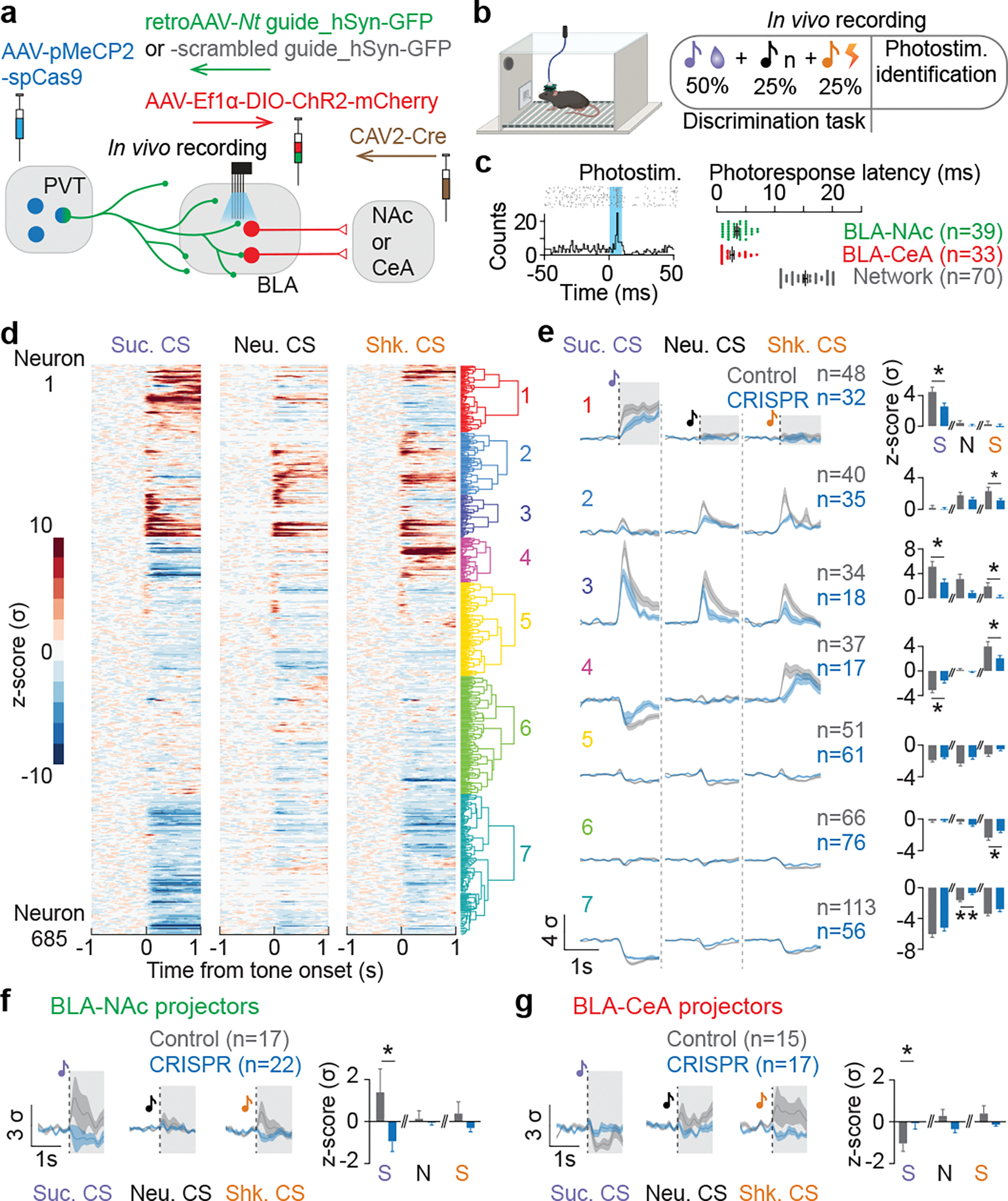
(a) Schematics of in vivo electrophysiological recordings in the BLA with BLA-NAc or BLA-CeA phototagging in mice with or without Nt gene inactivation in BLA-projecting PVT neurons. (b) Behavioral paradigm of in vivo recordings during a discrimination task and a photoidentification session. (c) An example raster histogram of a photo-responsive BLA neuron (left), and photoresponse latencies of identified BLA-NAc, BLA-CeA, and in-network neurons (right). (d) Seven different clusters were identified using hierarchical clustering based on neural responses to sucrose, neutral, and shock CSs. (e) Comparing to the control group, the CRISPR group showed reduced neural responses to sucrose CS and shock CS, while responses to the neutral CS across clusters were largely indifferent between the control and CRISPR group (For sucrose, neutral, and shock CS, cluster 1: *P=0.03, P=0.32, P=0.52; cluster 2: P=0.31, P=0.14, *P=0.02; cluster 3: *P=0.04, P=0.051, *P=0.04; cluster 4: *P=0.03, P=0.43, *P=0.03; cluster 5: P=0.21, P=0.07, P=0.06; cluster 6: P=0.936, P=0.457, *P=0.027; cluster 7: P=0.23, **P=0.004, P=0.16; two-tailed unpaired t-test). (f, g) BLA-NAc and BLA-CeA population in the CRISPR group showed significantly reduced excitation and inhibition to the sucrose CS compared to the control group, respectively (BLA-NAc: *P=0.0465; BLA-CeA: *P=0.0481; two-tailed unpaired t-test). The shaded regions in e indicate the time window used for analysis of each CS in the adjacent panels for all clusters. Black dotted lines on the mean traces in e-g indicate the onset of CSs. n denotes number of neurons in each group. Error bars and solid shaded regions around the mean indicate s.e.m.
We first noticed that the CRISPR group show reduced basal firing (Figure E8d), and that the CRISPR group contains smaller proportions of neurons that were significantly excited or inhibited by CSs compared to the control group receiving scrambled control guides (Figure E8f), while the magnitude of responses among these responsive neurons did not show significant differences compared to the control group (Figure E8g). It suggests that the CRISPR-cKO might selectively affect certain BLA ensembles. Indeed, we clustered BLA neurons into seven functional groups based on their responses across all task events using unsupervised hierarchical clustering (Figure 4d, E8h) and found that the CRISPR-cKO decreased the proportions of CS-responsive neurons and attenuated the magnitude of neural responses to Sucrose and Shock CSs within each task-relevant cluster, compared to the control group (Figure 4e, E8i). Among photoidentified BLA-NAc and BLA-CeA neurons, we replicated our previous reports in control mice showing that BLA-NAc and BLA-CeA neurons are predominantly excited by Sucrose CS and Shock CS, respectively2,3, and found that the CRISPR-cKO abolished the excitatory and inhibitory responses to the Sucrose CS in BLA-NAc and BLA-CeA neurons, respectively (Figure 4f, g, E8j).
To further compare the BLA neural dynamics at a population level, we pooled all recorded neurons and performed principal component analysis (PCA) to reduce the dimensionality and enable visualization of the entire ensemble dynamics across time in trial-averaged neural trajectories. Separating the neural trajectories by trial type and group in the activity space revealed that trajectories in the CRISPR group showed significant decreases in total trajectory length and trajectory distance between sucrose and shock trials compared to the controls (Figure 5a), indicating reduced BLA neural dynamics across trial types in the PVT-BLA:Nt CRISPR-cKO mice.
The population dynamics of BLA ensembles – reflected in the normalized population plots and the neural trajectory visualizations – suggest that the NT dynamics are critical for amplifying existing glutamatergic signals, enabling the appropriate ensemble formation and maximally discriminable representations of positive and negative valence broadly in BLA neurons.
NT inputs from the PVT to the BLA mediates passive-active behavioral strategies
At baseline, we found that mice in the control group exhibited a bias towards active behavioral strategies within both reward and punishment contexts. Behaviorally, we noticed that mice that received CRISPR-mediated KO of Nt in PVT-BLA showed significantly enhanced baseline port entries and reduced port entry specificity during sucrose trials, compared to the control group (Figure E8e), suggesting alterations in the behavioral strategies in responses to the predictive cues.
To test this hypothesis, we first examined BLA encoding of valence using machine learning analysis. We used a support vector machine (SVM) classifier trained on the principal components from all neurons recorded on a trial-by-trial basis to determine whether we could decode trial type (sucrose or shock) from neural activity to the predictive cues before reward or punishment was delivered. We found that BLA neurons from both control and CRISPR groups could decode the trial type compared to label-shuffled trials, when the SVM classifier was trained and tested within-group (Figure 5b). However, when the classifier was trained and tested on the opposite groups, we found that decoding performance significantly dropped (Figure 5c). Specifically, this asymmetry suggests that a classifier trained on the blunted neural dynamics used by CRISPR-treated mice was still able to decode the dynamics of control mice, but that a classifier trained on the coding rules of control animals was not able to decode the muted signals from CRISPR-treated mice, indicating an alteration in the valence encoding principles of BLA neurons induced by the Nt cKO.
By performing an unsupervised trial-by-trial clustering of behavioral responses to Sucrose and Shock CSs based on automatic pose estimation44,45 (Figure 5d, g; E8k), we further separated the behavior into several clusters that represent to passive and active behavioral strategies (Figure 5e, h; E8l–n, p–r). We found that control mice preferred active behavioral strategies during both sucrose and shock trials, including sucrose port approaching behaviors and active avoidance responses, while the PVT-BLA:Nt CRISPR-cKO abolished this behavioral preference to active strategies (Figure 5f, i), consistent with increased baseline sucrose anticipation and freezing behaviors (Figure E8e). Finally, we observed predominant BLA neural responses to CSs during the active behavioral trials, which were diminished by the PVT-BLA:Nt CRISPR-cKO (Figure E8o, s). Together these results suggest that the PVT-BLA:Nt CRISPR-cKO altered valence-coding principles in the BLA and impaired cue-driven active behaviors.
The temporal dynamics of NT are critical for valence assignment
Given the rich temporal dynamics we observed in vivo of NT and its effects on BLA neural encoding, we compared readouts from different temporal scales and found that manipulations with different temporal scales could dramatically affect behavioral outcomes, but were all internally consistent across our findings.
On a sub-second timescale, we found that NT rapidly facilitated glutamatergic transmission onto BLA-NAc neurons and suppressed it onto BLA-CeM neurons in an NTSR1-dependent manner (Figure E9a–g), consistent with enhanced reward learning and suppressed punishment learning by optogenetic activation of the PVT-BLA:NT projection.
At the timescale of minutes, we found that pre-incubation of NT attenuated the amount of plasticity induced onto BLA-NAc, but not BLA-CeM neurons when using a STDP LTP induction protocol46 (Figure E9h–l). This is consistent with pharmacological inhibition of NTSR1, in which NTSR1 antagonist delivered in the BLA 15 minutes before the conditioning session enhanced reward learning but had no effects on punishment learning (Figure E10a–f).
On a timescale spanning weeks, we found that CRISPR-cKO of Ntsr1 gene in the BLA suppressed reward learning (Figure E10g–p), consistent with the effects of PVT-BLA Nt CRISPR-cKO on reward learning.
Conclusion
In summary, our findings demonstrate that NT inputs from PVT NT neurons to the BLA provide a critical neuromodulatory signal modulating valence assignment in the BLA, which is distinct from other neuromodulators, such as dopamine27–29. With the applications of NT-specific signal monitoring and manipulation in combination of projection-specific neural recordings, our study highlights that PVT-BLA:NT neurons facilitate reward and punishment learning in a dose-dependent manner. Our findings further reveal that NT guides valence assignment in the BLA by amplifying the encoding of both positive and negative valences, and promotes the selection of active behavioral strategies. Together, our findings complement and extend the growing evidence supporting a role for the PVT in valence-associated behaviors39–43 and action selection47,48.
Although NT clearly exerts a valence-specific effect given each of the causal manipulations made (Figure 2 and 3), we found that temporal dynamics are critically involved in NT function and NT action across short, medium and long time scales resulted in distinct behavioral outcomes, suggesting that temporal dynamics could be a common mechanistic principle by which NT or even neuropeptides in general modulate the patterning of long-term synaptic changes in the brain, thereby regulating a wide variety of behaviors. Future work enabled by the development of peptide-specific sensors and in vivo CRISPR-Cas9 gene manipulation could further interrogate the temporal dynamics by which neuropeptides can influence neural ensembles or circuit nodes in motivated behaviors.
To conclude, we have identified a specific neuropeptide, NT, which can mediate neural dynamics and behavior in a valence-specific manner via dose-dependent NT signaling. These empirical data demonstrate how a neuromodulatory gain circuit motif can work to direct signaling downstream to enable synaptic plasticity on behaviorally relevant timescales during valence assignment49. This work represents a circuit-based approach to drug discovery and implicates NT and its receptors as potential targets for therapeutic interventions of psychiatric diseases affecting emotional valence such as depression, anxiety, and addiction.
MATERIALS AND METHODS
Animals and Stereotaxic Surgery
Adult wild-type C57BL/6J mice (186 mice), NT::Cre mice (178 mice; Stock #017525), and NT::Cre mice crossed with the Ai14 reporter line (7 mice; Stock #007914) were all at least 2 months (Jackson Laboratory) before being used for experiments. Mice were housed with randomly assigned littermates in cages of 3–4, maintained under a 12-hour reverse light/dark cycle, and given access to food and water ad libitum except where indicated in experimental proceedings. Experiments were conducted during the light-off light cycle phase. For all experiments involving cranial implants or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-mediated gene CRISPR-cKO, only male mice were used. For all other experiments, both male and female mice were used. Following surgery, mice were maintained on a reverse 12 hours light/dark cycle with ad libitum food and water, upwards of 10 days to allow Fast Blue or retrobead migration, or upwards of 6 weeks to allow viral expression. All procedures of handling were in accordance with the guidelines from NIH, and with approval of the Salk and MIT Institutional Animal Care and Use Committee. All surgeries were conducted under aseptic conditions using a digital small animal stereotaxic instrument (David Kopf Instruments, Tujunga, CA). Mice were anaesthetized with isoflurane (5 % for induction, 1.5–2.0 % after) in the stereotaxic frame for the entire session and their body temperature was maintained with a heating pad.
For pharmacological experiments (Figure E10a–f), bilateral cannulae (22GA, PlasticsOne, Roanoke, VA) were implanted above the basolateral amygdala (BLA, −1.6 mm anteroposterior -AP-; ±3.4 mm mediolateral -ML-; −4.4 mm dorsoventral -DV-) bilaterally for infusion of Neurotensin receptor 1 (NTSR1) antagonist SR48692. Implanted cannulae were anchored to the skull with a layer of adhesive cement (C&B Metabond; Parkell, Edgewood, NY) followed by dental cement (Ortho-Jet; Lainactivatng, Wheeling, IL). Behavioral experiments were conducted after at least 2 weeks of recovery.
For retrograde tracers viral injections, we used glass micropipettes (1–5 μl; Drummond Scientific, Broomall, PA) pulled with a puller (Narishige PC-10, East Meadow, NY) or a beveled 34 gauge microinjection needle (Hamilton) mounted on 10 μL microsyringe (Hamilton Microliter 701, Hamilton co., Reno, NV). We delivered retrograde tracers at the rate of 2 nL/s and virus at a rate of 1 to 2 nL/s, using a microsyringe pump (UMP3; WPI, Sarasota, FL) and controller (Micro4; WPI, Sarasota, FL). After completion of the injection, the pipette or needle was left for an additional 10 minutes to allow diffusion of the tracer or the virus at the injection site and then slowly withdrawn and the skin incision was sutured. After surgery, the animal’s body temperature was maintained using a heat lamp or pad until it fully recovered from anesthesia.
In order to label BLA neurons projecting to the nucleus accumbens (NAc), 120 nL of red or green retrobeads (Lumafluor Inc.) were injected into the NAc at stereotaxic coordinates from bregma: +1.42 mm AP, ±0.86 mm ML and −4.7 mm DV. In order to label BLA neurons projecting to the medial part of the central amygdala (CeM), 75 nL of retrobeads (different color from NAc injection) were injected in the contralateral CeM (−0.80 mm AP, ±2.36 mm ML and −5.10 mm DV; Figure E1a–f, E9).
To label neurotensin (NT) neurons projecting to the BLA (Figure 1a–c), Fast Blue tracer (Polysciences, Inc.) was injected bilaterally into the BLA (−1.6 mm AP, ±3.35 mm ML and −4.9 mm DV) of NT::Cre x Ai14 mice, which express the fluorescent reporter tdTomato in the presence of Cre. To label the axons in the BLA from the medial geniculate nucleus (MGN), ventral hippocampus (vHPC) and the paraventricular nucleus of the thalamus (PVT), either an adeno associated virus serotype 5 (AAV5) carrying the fluorophore mCherry in a double floxed open inverted reading frame expressing under the promoter EF1α (AAV5-Ef1α-DIO-mCherry), or a similar virus expressing the enhanced yellow fluorescent protein tagged to Channelrhodopsin-2 (AAV5-Ef1α-DIO-ChR2-eYFP) was injected in the MGN (−3.00 mm AP, ±1.7 mm ML and −3.5 mm DV), the vHPC (−3.9 mm AP, ±3.34 mm ML and −3.85 mm DV) and/or the PVT (−0.4 mm AP, ±1.47 mm ML and −3.5 mm DV, 20° angle) and the virus(es) were allowed to express for 8 weeks in NT::Cre mice. For the patch-clamp electrophysiology experiments, AAV5-Ef1α-DIO-ChR2-eYFP was allowed to express in the MGN, the vHPC or the PVT for upwards of 8 weeks in NT::Cre mice (Figure E1h–j, E2).
For the somatic calcium photometry experiments, an AAV5 virus carrying the Cre-inducible calcium indicator GCaMP6m expressing under the CAG promoter (AAV5-CAG-FLEX-GCaMP6m) was injected into the MGN, the vHPC and the PVT (500 nL in each region) of NT::Cre mice. In order to deliver light to the BLA and record calcium signals, a 400 μm optic fiber (0.50 numerical aperture, NA) glued to a 1.25 mm ferrule was implanted in the MGN, vHPC and the PVT for multisite photometry experiments (Figure E6).
For PVT-BLA:NT calcium photometry experiments, the same GCaMP6m virus was injected in the PVT and an optical fiber was implanted in the BLA (Figure 3a–f, E7a). For the NT sensor photometry experiments, an AAV9 virus carrying NT fluorescent sensor (AAV9-hSyn-GRABNTS1.0) was injected and a 200 μm optic fiber was implanted into the BLA (300 nL) of wild type mice (Figure 3g–l, E7b–l).
For head-fixed surgeries, an aluminum bar (2 × 0.2 × 0.2 cm) was placed on the skull (head bar) ~1.5 mm anterior to Bregma. One layer of adhesive cement (C&B metabond; Parkell, Edgewood, NY) followed by cranioplastic cement (Dental cement; Stoelting, Wood Dale, IL) was used to secure the fiber ferrules and the head bar to the skull and 20 min after, the incision was sutured. Behavioral experiments were conducted at least 8 weeks after surgery.
For the photostimulation of NT terminals from PVT to the BLA (Figure 2e–h, E5h–k), Cre-dependent channelrhodopsin-2 (AAV5-Ef1α-DIO-ChR2-eYFP) or control (AAV5-Ef1α-DIO-mCherry) was injected into the PVT of NT::Cre mice (500 nL), and 200 μm optical fibers were implanted bilaterally above the BLA at the same time. Behavioral experiments were conducted after 4–7 weeks of surgery.
For the experiments involving non-pathway specific CRISPR knockdown of the Nt gene (Figure 1d–h, E3, E4), an adeno associated virus serotype 9 (AAV9) carrying the DNA for CRISPR associated protein 9 (Cas9) under the methyl CpG (Cytosine Phosphate Guanine) binding protein 2 (MeCP2) promoter (AAV-Cas9) and an AAV9 carrying DNA to express Nt guide RNA and the green fluorescent protein (GFP) tagged to a Klarsicht, ANC-1, Syne Homology (KASH) domain for targeting the GFP to the nuclear membrane expressed under the U6 promoter (AAV-NT-guide) were prepared. Guides targeting different regions of the Nt gene (Guide 1 and Guide 2) were packaged in separate viruses. To inactivate NT, the AAV-Cas9 virus was mixed with the virus carrying Nt guide 1, Nt guide 2 in a 50:50 ratio, or the three viruses were mixed in a 2:1:1 (Cas9:Guide1:Guide2) ratio and injected either into the MGN, the vHPC or the PVT. The corresponding control group in the pavlovian association experiments received an injection of a 50:50 mixture of viruses carrying guide 1 and guide 2 without Cas9. Behavioral experiments were conducted 4–7 weeks after surgery.
For the experiments involving extracellular electrophysiological recordings and PVT-BLA CRISPR-cKO of the Nt gene (Figure 2a–d, 4, 5, E8), the same AAV virus carrying Cas9 (AAV9-pMeCP2-Cas9) was injected into the PVT (300 nL). A cocktail of Cre-dependent channelrhodopsin-2 (AAV5-Ef1α-DIO-ChR2-mCherry) and two retrogradely-transported AAV carrying two guide RNAs of Nt gene (AAV2-retro-sgRNA_hSyn-GFP-KASH-bGH) or a single virus carrying the negative control guide RNA was injected into the BLA (150 nL, mixture of a 1:1:1 ratio for Nt guides and 1:2 for control guide). A retrogradely-transported virus carrying Cre-recombinase (Cav2-Cre) was injected into the CeA or the NAc in the ipsilateral hemisphere of the BLA injection (100 nL). Mice were allowed to recover for 4–6 weeks before an optrode was implanted in the BLA. During the optrode implantation, a customized optrode was slowly lowered into the BLA over the course of 10 minutes, and the ground wire was inserted into the cortex by 1–2mm. Once the optrode reached 500μm above the desired depth for the BLA, Metabond was applied on the surface of the skull before continually lowering the optrode for 500μm. Finally, dental cement was applied to secure and cover the optrode. Behavioral and recordings experiments were conducted 2 weeks after the optrode implantation.
For ex vivo validation of PVT-BLA CRISPR -cKO of the Nt gene experiments (Figure E5a–g) a cocktail of Cre-dependent channelrhodopsin-2 (AAV5-Ef1α-DIO-ChR2-mCherry) and the same AAV virus carrying Cas9 (AAV9-pMeCP2-Cas9) was injected in the PVT and the Nt guides virus were injected in the BLA of NT::Cre mice.
For the Ntsr1 gene CRISPR-cKO experiments (Figure E10g–p), a cocktail of retrograde AAV carrying Ntsr1 guide 1 and guide 2 or carrying negative control guide was injected bilaterally into the NAc, the CeA, or the BLA (100 nL, 1:1 ratio for Ntsr1 guides) and AAV-Cas9 was bilaterally injected into the BLA. For the BLA global Ntsr1 CRISPR-cKO group, the equal numbers of experimental and control mice received the viral injections before or after 4 sessions of tone-sucrose association. Behavioral experiments were conducted 4–8 weeks after surgery.
In vivo pharmacology
A 26GA stainless steel internal canulae (PlasticsOne) projecting 0.5 mm beyond the tip of the guide cannula were connected to a syringe pump (Harvard Apparatus, MA). NTSR1 antagonist SR 48692 (Sigma) was diluted (20 nM) in saline solution containing 2% dimethylsulfoxide (DMSO) and its vehicle solution (Veh) was used for control injections. NTSR1 antagonist was infused into the BLA in a volume of 0.3 μl at a rate of 0.1μl per minute. The internal canulae were withdrawn 5 min after the end of infusion and mice underwent either tone-sucrose or -shock conditioning 15–20 min following drug infusions (Figure E10a–f).
Freely moving behavior - Pavlovian sucrose association
Behavioral assay.
After overnight food restriction, mice were conditioned in sound-proof boxes (MedAssociates, St Albans, VT). Each box contained a modular test cage assembled with a sucrose delivery port, a speaker and a house light placed under the sucrose port. The CS consisted of a pure tone cue, which ended 400 ms after a port entry beam break by the mouse was detected. For every CS predictive of sucrose, about 15 μL of a 30 % sucrose solution was delivered into the port 2 s after CS onset (motor for delivering the sucrose started 1 s after CS onset). This only occurred if the mouse had entered the port after the onset of the previous CS, to prevent sucrose accumulation. Mice received ad libitum food for 4 hours between sucrose association sessions.
For the NTSR1 antagonist experiment (Figure E10a–f), the CS consisted of either a 10 kHz or 2 kHz, 80 dB pure tone. The tone lasted until mice made a port entry, or a total of 20 s. The inter-trial interval (ITI) of the CS presentations was chosen randomly from a list at runtime and was in a range of 100 to 180 s for the first 20 CS presentations and 60 to 165 s for subsequent CS presentations. The conditioning session was terminated after 120 sucrose deliveries and lasted for about 4 hours. This protocol was repeated for two days.
For the Ntsr1 gene CRISPR-cKO experiments (Figure E10g–p), the CS consisted of either a 2 kHz or 20 kHz, 60 dB pure tone. The tone lasted until mice made a port entry, or a total of 20 s. The inter-trial interval (ITI) of the CS presentations was chosen randomly from a list at runtime and was in a range of 90 to 120 s. The conditioning session was terminated after 120 sucrose deliveries and lasted for ~3 hours. This protocol was repeated for four days. For the BLA global Ntsr1 CRISPR-cKO post-conditioning group, an additional tone-sucrose session was given to both experimental and control group 6 weeks after surgery. No significant difference in port entry probability was observed in the control groups between pre- and post-conditioning procedures.
For the non-pathway specific Nt gene CRISPR-cKO experiment (Figure 1f–h), the CS consisted of either a 2 kHz or a 20 kHz, 60 dB pure tone. If the mouse did not enter the port, the tone lasted for 10 s. The inter-trial interval (ITI) of the CS presentations was chosen randomly from a list at runtime and was in a range of 60 to 100 s for the first 20 CS presentations and 30 to 58 s for subsequent CS presentations. Mice also experienced either a 20 kHz or 2 kHz pure tone that was not predictive of sucrose (50% of all the tones, data not shown). The conditioning session was terminated after ~125 sucrose deliveries and lasted for about 3 hours. This protocol was repeated for 4 days. Tone frequencies were counter-balanced between sucrose-predictive and neutral CS for all groups of mice. Results shown in the analysis were from the last day of the conditioning.
For the photostimulation of PVT-BLA:NT experiments (Figure 2e–h), the CS consisted of either a 2 kHz or 20 kHz, 60 dB pure tone. The tone lasted until mice made a port entry, or a total of 10 s. The inter-trial interval (ITI) of the CS presentations was chosen randomly from a list at runtime and was in a range of 100 to 140 s for the first 20 CS presentations and 60 to 150 s for subsequent CS presentations. Laser stimulation (473mm, 20 Hz pulse, 5–10 mW) was turned on 500ms before the onset of the CS and lasted until 500ms after the offset of the CS. The conditioning session was terminated after 120 sucrose deliveries and lasted for about 4 hours. This protocol was repeated for two days. Results shown in the analysis were from the last day of the conditioning.
Data analysis.
Sucrose reward port entry probability were computed across all trials within the session with 500 ms bins for 25s before and after the onsets of the CS. The CS PSTHs were z-transformed using the histogram values in a 25 s baseline period prior to the onset of the CS.
Freely moving behavior - Pavlovian shock association
The mice were conditioned using behavioral hardware boxes (MedAssociates, St Albans, VT) placed in custom made sound attenuating chambers. Each box contained a modular test cage with an electric floor grid and a speaker. Videos of the mice were acquired during all sessions. To allow freezing quantification to the tone an infrared LED was toggled for the period of the tone for synchronizing the video to the tones. A period of acclimation lasting 3 minutes preceded the presentation of the first tone.
Tone-shock acquisition:
For the NTSR1 antagonist and Ntsr1 CRISPR-cKO experiments (Figure E10g–p), mice were exposed to one session of tone-foot shock conditioning. The CS consisted of either a 2 kHz or 10 kHz, 60–80 dB pure tone lasting 20 seconds. The US consisted of a scrambled 0.4 mA foot shock lasting 2 s. Cue presentations were separated by 70 to 130 seconds.
For the photostimulation of PVT-BLA:NT experiments (Figure 2e–h), mice were exposed to the same conditioning protocol as the NTSR1 antagonist experiment. Laser stimulation (473mm, 20 Hz pulse, 5–10 mWatt) was turned on 500ms before the onset of the CS and lasted until 500ms after the offset of the CS during the conditioning day.
For the non-pathway specific Nt gene CRISPR-cKO experiment (Figure 1f–h), mice were exposed to one session of tone-shock conditioning. The CS consisted of either a 1 kHz or 10 kHz, 60 dB pure tone lasting 10 seconds. The US consisted of a scrambled 0.7 mA foot shock lasting 0.1 s. Cue presentations were separated by 150 to 210 seconds. These animals also experienced a different pure tone (15 kHz or 1 kHz) that was not paired with a foot-shock (data not shown).
Tone-shock recall:
Following conditioning, mice were returned to their home cages. On the next day, mice were put through the same protocol as the conditioning session, but the shocks and photostimulation were omitted.
Measurement of fear behavior.
Percentage time freezing during CS presentation was quantified using custom written software in MATLAB. For each of the trials, a segment of the video containing 30 s before and 30 s after the onset of the tone was extracted frame by frame and exported into MATLAB. Mice were tracked using an algorithm that performed background subtraction using a median filter, frame-by-frame Gaussian smoothing and thresholding. Distances in the video were calibrated using the physical length of the shock grid. Mice were considered to be freezing at a velocity of less than 0.3 cm/s. We first verified the freeze detection algorithm with a small number of files. For these video files, every single trial was scored by 2 human observers. They were blind to the experimental condition. The sequence of trials was also scrambled (meaning, the software might ask to score the 6th pairing of fear conditioning first, followed by the second pairing, etc). The correlation between human scores and auto scores was above 90% (human score 1 correlated with human score 2, and each human score correlated with auto score).
After freeze detection, every single file was then verified by a human observer (still blind to the experimental manipulation). In cases where the algorithm was not able to accurately detect the outline of the mouse, or the algorithm incorrectly segmented a glare as a mouse, the freezing detection algorithm would be erroneous. In rare cases, when the mouse made small movements with its head while sitting in one place, the algorithm would sometimes label these frames as freezing. In these cases, the human observer’s scoring replaced the auto score for the whole trial.
Freely moving behavior - Discrimination task
For the CRISPR-cKO experiments (Figure 2a–d) and photostimulation experiments (Figure 2e–h), the time mice spent freezing during the CS presentation was manually scored by two individuals separately and averaged for the final score. The final freezing scores were spot-checked by an experienced scorer.
For the PVT-BLA Nt gene CRISPR-cKO experiments (Figure 4, 5, E8), mice were trained in three different training blocks before the discrimination task. First, mice were trained in a similar tone-sucrose association paradigm as described earlier, in which mice received 120 pairs of tone-sucrose association. 15 μl of 30% sucrose was delivered into the port 2 s after the onset of either a 2 kHz or 20 kHz, 60 dB pure tone. The tone lasted for 20 s unless mice made a port entry after the CS onset. The session consisted of 120 sucrose trials. This protocol was repeated for four days. On the fifth day, mice received an additional 9 kHz, 60dB tone which was followed by nothing (neutral trial). The tone lasted for 20 s. The session still consisted of 120 trials and sucrose and neutral trials were randomly selected at 50% probability. This protocol was repeated for two days.
Following this, mice received one day of tone-shock conditioning and one day of tone-shock recall task. During the tone-shock conditioning, mice experienced a distinct tone (20 kHz or 2 kHz, 60dB), which lasted for 20 s. For the first 20 trials, the tone was followed by nothing. Starting from 21 trials, a 2s 0.4mA scrambled footshock was delivered at the offset of the tone. This session consisted of 30 trials. On the following day, mice were put through the same protocol, but the shocks were omitted.
Once mice completed tone-shock recall task, mice experienced the three-cued discrimination task, in which sucrose, neutral, and shock trials was randomly selected at 50%, 25%, and 25% probability, respectively. All the parameters remained the same as previous training blocks, except that footshocks were changed from 2s of 0.4mA to 100ms of 0.6mA. The session consisted of 120 trials, and the protocol was repeated for 4–5 days
Freely moving behavior - Open Field Test
The open field chamber was made of transparent plastic (50 × 50 cm) and divided into a central field (center, 25 × 25 cm) and an outer field (border). Individual mice were placed in the center of the open field and allowed to recover from handling 1–5 minutes before the start of the session. The open field test consisted of a 10-minute session. Behavioral tests were recorded by a video camera. The EthoVision XT video tracking system (Noldus, Wageningen, Netherlands) was used to track mouse location, velocity, and movement of head, body, and tail. All measurements displayed are relative to the mouse body (Figure E5, E10).
For the photostimulation of PVT-BLA:NT pathway experiments (Figure E5), the open field test consisted of a 15-minute session. 473mm photostimulation was on for minute 5 through 10 (5–10 mW, 20Hz, 5ms pulses).
Head-fixed behavior
Tone-sucrose association experimental setup.
Mice learned to acquire the association between a tone and sucrose over 5–7 sessions of tone-sucrose association in a head-fixed preparation. Mice were food restricted overnight prior to each session of sucrose association. The CS predictive of sucrose was a pure tone of either 1 kHz or 8 kHz, and mice were presented with about 60 CS-US pairings for every session. The CS duration was 4 s. The US was a drop of 30% sucrose solution (~4 μl) and was delivered 2 s after the onset of the CS. Mice were also presented with a CS that was not predictive of a sucrose reward (~50% of all CS; data not shown). Tone frequencies were counter-balanced across animals. All the hardware (speaker, solenoid, and beam break) interfaced with the same NIDAQ used to control the multi-site photometry hardware (Figure 3, E6).
Measurement of tone-sucrose association.
Acquisition of the association was based on anticipatory lick responses of the mouse. The raw analog signal from the infra-red beam break was digitized at 40 kHz using the same hardware clock as the one used to control the speaker and solenoid valves. Lick onsets and offsets were detected by thresholding the trace, which was performed using the following algorithm implemented in MATLAB. The lick trace was concatenated across all trials and sessions, downsampled to 1 kHz, corrected for drifts on the hectoseconds scale using a median filter and thresholded using a principle used for spike detection in in vivo electrophysiology50. Significant anticipatory licking was determined by comparing the number of licks from the CS to the US to the number of licks in the 2 s period prior to CS onset using a Wilcoxon rank sum test. Only animals with significant anticipatory licking behavior were considered for further analysis. Five out of 7 animals acquired the task.
Tone-air puff association experimental setup.
Mice learned to acquire the association between a tone and an air puff over 2 sessions of tone-air puff conditioning in a head-fixed preparation. The CS predictive of air puff was either white noise, or 100 ms long 4500 kHz pips repeated every 500 ms. Mice were presented with about 30 CS-US pairings for every session. The CS duration was 4 s. The US was a puff of pressurized air (20 psi) presented 2 s after the onset of the CS for a duration of 100 ms. Mice were also presented with a CS that was not predictive of air puff punishment (~50% of all CS; data not shown). Tone frequencies were counter-balanced across animals. All the hardware (speaker, solenoid, and beam break) interfaced with the same NIDAQ used to control the multi-site photometry hardware. Videos of the mouse performing the task were collected using an IR camera for measuring behavior (Figure 3, E6).
Measurement of tone-air puff association.
Acquisition of the association was based on monitoring the eye of the mouse. An eye blink trace was extracted from the video and aligned to the onset of the CS for each animal. Regions of interest (ROI) of the eye were drawn around the CS and US LEDs, and the average pixel intensities in the ROI were thresholded to identify the CS and US onset frames. An ROI encompassing the eye of the mouse was drawn, and each frame was smoothed using a Gaussian filter. The average intensity of all the pixels within the eye ROI was used as a metric for the size of the eye. To control for background changes in intensity, the reference ROI time course was regressed from the eye ROI time course. The area under the eye blink trace from the onset of the CS to the onset of the US (for every CS-US pairing) was compared using an unpaired t-test. Only animals that showed a significant difference between the first 15 CS-US presentations on the first session and all the CS-US presentations in the second session were considered to have acquired the association between the tone and the airpuff and were used for further analysis.
CRISPR-mediated gene cKO
The CRISPR-associated endonuclease Cas9 from Streptococcus pyogenes (SpCas9) was used to generate indels in the neurotensin gene (NC_000076). Neurotensin (Nt) and Neurotensin receptor 1 (Ntsr1) -specific single guide RNAs were designed on Benchling (https://benchling.com/) using the GRCM38 (Mus musculus) reference genome. Based on the computed specificity and efficiency scores, we cloned guides 1 and 2 (GAAGGCTGAGAGAAGATGAG and GCTAAAACCATCCAGAACAA for Nt guides, and GGAGCTGTTGAGGTGCATGG and AGGATGGAGATGATCAGCAT for Ntsr1 guides) to target exon 1 of the reverse strand and exon 3 of the forward strand of the neurotensin coding region, and exon 1 and exon 2 of the neurotensin receptor 1 coding region. Non-targeting negative control guide was designed with a scrambled sequencing with no known targets in the genome (GGAAGAGCGAGCTCTTCT). Each gRNA was cloned into vector PX552 (pAAV-U6-sgRNA_hSyn-GFP-KASH-bGH; Addgene # 60958). The two AAV vectors targeting the same gene were pooled with equal titer prior to the stereotactic injection.
Virus construction and packaging:
For non-pathway specific CRISPR-mediated Nt gene CRISPR-cKO (Figure 1,E3,E4), high-titer AAV9 virions encoding AAV-SpCas9 (PX551: pAAV-pMecp2-SpCas9-spA; Addgene plasmid # 60957) and pAAV-U6sgRNA_hSyn-GFP-KASH-bGH vectors encoding neurotensin-specific sgRNAs were produced as described previously51. Briefly, HEK293T cells were transfected with AAV9 serotype plasmids in equal ratios, transgene plasmid and pDF6 helper plasmid using polyethyleneimine. 72 h after transfection, cells were harvested and high-titer AAV9 virus was purified by iodixanol gradient ultracentrifugation. The titer of AAV vectors was determined by real-time quantitative PCR (qPCR) using probes and primers specific for the hSyn1 promoter sequence (Integrated DNA Technologies). We obtained titers of 1.7 × 1013, 3.9 × 1013 and 5.7 × 1013 vector genomes/ml for the Cas9, guide1 and guide2 viruses respectively. For PVT-BLA specific Nt gene CRISPR-cKO, Nt-specific sgRNAs were packaged into AAV2-retro (titers of 1.55 × 1014 and 1.77 × 1014 for guide 1 and guide 2, respectively). For Ntsr1 gene CRISPR-cKO, Ntsr1-specific sgRNAs were packaged into AAV2-retro (titers of 1.5 × 1014 and 1.35 × 1014 for guide 1 and guide 2, respectively). Negative control guide was packaged into AAV2-retro (titers of 1.35 × 1014).
In vivo validation of DNA mutation.
Fresh-frozen brains were sliced using a cryostat at 150 μm thickness, and tissue around the injection site was punched out using a 1 mm tissue punch (Cat#57401, Stoelting, Illinois, USA). Nuclei were extracted using nuclear isolation protocol described in52. GFP-KASH expressing nuclei were isolated using fluorescence activated cell sorting (FACS; MoFlo Astrios EQ sorter, Beckman-Coulter). Genomic DNA was then extracted and the parts of the DNA targeted by each guide were amplified using the following primer sequences for extracting DNA around the target of Nt guide 1 - forward AGCTCCTTCAGTGTCTGAGTG and reverse GGAGTTGTGAGATGCAGTTGAGC (product length 150) and Nt guide 2 – forward CATGATGACGACCTTGTTGGC and reverse TGGGTTCTGATACCTCCCAGT (product length 139). After second round PCR with barcoded primers, the extracted DNA was prepared for Illumina sequencing using the MiSeq Reagent Nano Kit, v2 (300 cycles) Cat: MS-103–1001MiSeq reagent kit (Cat# MS-102–2002, Illumina, San Diego, CA). Resulting data was analyzed using the website outknocker.org (v2 beta).
Design and characterization of the GRABNTS1.0 sensor
The GRABNTS1.0 sensor design.
The cDNA encoding human NTSR1 receptor was amplified from the human GPCR cDNA library with primers (Tsingke) and the third intracellular loop (ICL3) was replaced with ICL3 from GRABNE and assembled into mammalian expression pDisplay vector (Invitrogen) using Gibson assembly method53. The insertion sites on NTSR1, the amino acid composition between NTSR1 and ICL3 and the critical sties on cpGFP were systematically screened to obtain GRABNTS1.0. Plasmids of NTS sensor candidates were transformed, amplified and mini-preped. The NTS sensor was cloned into the pAAV vector using a human synapsin promoter (hSyn) for AAV virus packing. GRABNTS1.0 was then expressed in HEK293T cells and cultured rat primary neurons for further characterization of its response, sensitivity, specificity, kinetics and spectra (Figure 3g–i, E7b–l).
Cell cultures.
HEK293T cells and primary neurons were cultured at 37°C with 5% CO2 in incubators. HEK293T cells and HTLA cells were cultured in DMEM (Biological Industries) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Gibco). Rat primary neurons were dissected from 0-day (P0) pups and cortical neurons were dissociated from the brains in 0.25% Trypsin-EDTA (Gibco) and plated on glass coverslips coated with poly-D-lysin (Sigma-Aldrich). Neurobasal medium (Gibco) supplemented with 2% B-27 (Gibco), 1% GlutaMAX (Gibco) and 1% penicillin-streptomycin (Gibco) were added to culture the primary neurons.
Expression of GRABNTS1.0 sensor in cultured cells.
HEK293T cells were plated into CellCarrier-96Ultra microplates (PerkinElmer) and grown to 60–80% confluence for transfection. A mixture containing 0.3 μg DNA and 0.3 μg PEI was added to each well and transfected for 4–6 h, then cultured medium was changed to remove DNA and PEI. Cells were imaged 34–36 h after transfection. AAV9 virus with a tilter around 5×10^13 v.g./mL were added to cultured neurons at DIV6–8 for transfection. Culture medium was half changed 48 h after AAV virus transfection. Cultured neurons were imaged at DIV11–14.
Imaging of GRABNTS1.0 sensor in cultured cells.
HEK293T cells expressing GRABNTS1.0 sensors and other sensor candidates were imaged using Opera Phenix high content screening system (PerkinElmer). HEK293T cells were bathed in Tyrode’s solution containing (in mM): 180 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES and 10 glucose (pH 7.35–7.45). To image the GRABNTS1.0 sensor, a 488-nm laser and a 525/50-nm emission filter was used.
GRABNTS1.0 expressing HEK293T cells and cultured neurons were imaged using a Ti-E A1 inverted confocal microscope (Nikon). The microscope was equipped with a 20x/0.75 NA objective and a 10x/0.5 NA objective. A 488-nm laser and a 525/50-nm emission filter was used to collect GRABNTS1.0 sensor signals. During imaging, the cells were bathed with Tyrode’s solution in the imaging chamber.
Spectra measuring of GRABNTS1.0 sensor in cultured cells.
HEK293T cells expressing GRABNTS1.0 sensors were plated into a well of 384-well microplate and measured by Tecan Safire 2 Plate Reader. Cells were bathed with Tyrode’s solution with or without 1 μM NT (Tocris). Excitation spectrum was scanned using excitation wavelength from 300-nm to 540-nm with a step size of 5-nm, and emission signal was collected at 560/20-nm. Emission spectrum was scanned using excitation wavelength as 455-nm and collected emission wavelength from 495-nm to 800-nm with a step size of 5-nm.
Fiber photometry
Data acquisition.
For the photometry experiments (Figure 3d–f, E6, E7), the hardware setup for acquisition of bulk fluorescence from multiple sites is adapted from54. The setup consisted of wide-field microscope for imaging fluorescence emission from multiple fibers, while exciting the sample with two wavelengths of light (405 and 470 nm). The excitation path consisted of a 405 and 470 nm LED (Thorlabs M405FP1 and M470F3) which were collimated (Thorlab F671SMA-405) and coupled to 405 nm and 469 nm excitation filters (Thorlabs FB400–10 and MF469–35), respectively. Unlike 470 nm excitation, 405 nm excitation produces calcium-independent GCaMP and NT sensor fluorescence, and thus can be used to assess movement- and autofluorescence-related noise. Light from these two excitation sources were combined into one path via a series of dichroic mirrors and were coupled to a 20x air objective (Nikon CFI Plan Apo Lambda). A fiber optic patch cord (Doric) containing 4 fibers bundled into a single ferrule (400 μM diameter, 0.48 NA for each fiber) was positioned at the working distance of the objective. Emission resulting from the 405 or 470 nm excitation was passed through a 525 nm emission filter (Thorlabs MF525–39) and focused through a tube lens (Thorlabs AC254–100-A-ML) onto the face of CMOS camera (Hamamatsu ORCA-Flash V2). The system was controlled through custom MATLAB scripts made available by Kim et al. (2016)54 and averaged fluorescence within each fiber was quantified for online data visualization (frames were also saved for later analysis). For the BLA NT sensor photometry experiments (Figure 3j–l), the Neurophotometrics system was used (Neurophotometrics V2), where 415 nm LED was used for the reference channel. Frames were captured at 40 Hz and each LED was modulated at 20 Hz in an alternating fashion, resulting in a 20 Hz sample rate in the control and signal channels. LED and camera timing as well as recording of timestamps from behavioral equipment was achieved using a data acquisition board (National Instruments NI BNC-2110). Prior to the start of each session, the entire system was shielded from outside light using blackout cloth.
LED power was calibrated to emit 470 nm light in a range of 100–160 μW for GCaMP activation and 400 nm light in a range of 15–50 μW (measured at the end of the patch cable) through each ferrule, which then interfaced with a ferrule implanted in the mouse (carrying fibers of efficiencies between 75 and 95%). LEDs were turned on and data was collected continuously for Figure 3j–l, while LEDs were turned on and data was collected starting 10 s before the start of each CS until 10 s after the onset of each CS for the rest of all relevant experiments.
Data analysis.
Both the calcium signal (responses to 470 nm excitation) and reference signals (responses to 405 or 415 nm excitation) were down-sampled to 5Hz and filtered to reject high frequency noise (found more in the reference channel) using a median filter with a span of 200 ms. Data from the reference channel was then regressed from data in the 470 nm channel. Regression coefficients were computed using data averaged across trials in a session in a time window of −5 s to 0 s from the start of the CS to minimize any possible regression artifacts introduced by calcium transients recorded in the 470 nm channel evoked by the sensory stimuli or the animal’s response. Residuals from the regression were z-transformed using data a baseline window of −5 s to 0 s relative to the start of the CS.
In vivo electrophysiology
Recordings and phototagging with ChR2
A customized optrode consisted of a bundle of 32-micron Formvar-insulated nichrome wires (California Fine Wire, Stablohm 675) and a 200-micron optical fiber (Thorlabs) attached to an omnetics connector (Omnetics, NPD-36-VV-GS) was implanted into the BLA 1–2 weeks before behavioral sessions and recordings. Neural signals were recorded via the Open Ephys acquisition board (Open Ephys) at 30000 Hz with high-pass and low-pass filter cutoffs of 300 Hz and 6 kHz, respectively.
In vivo electrophysiological recordings in the BLA were attempted for every session throughout all training blocks and the discrimination task. Only the recordings from the last day of the discrimination task are shown in the analysis. Following completion of the task, a phototagging session was conducted after a “fake” phototagging session in which the patch cord was disconnected from the optrode and aimed to detect any electrical noise generated by the system during laser delivery. In these two phototagging sessions, pseudorandomly dispersed stimulations of 1 s constant 473 nm laser, 10 s of 1 Hz (5 or 10 ms pulses), 2 s of 10 Hz (5 ms pulses) and 100 ms of 100 Hz (5 ms pulses) were delivered, with at least 5 iterations of each.
Analysis for in vivo recordings
Recording files were converted to PLX format using customized MATLAB scripts and Offline sorter (Plexon, Dallas, TX, USA) were used to sort single units. Neural responses to cues and light stimulation were visualized through peristimulus time histograms (PSTH) and rasters for every unit using NeuroExplorer. Data from Plexon and Neuroexplorer data files were then imported into MATLAB and analyzed using customized scripts. Units with a baseline firing rate less than 0.3 Hz were excluded. PSTHs were computed with 20 ms bins for 1s before and after the onsets of sucrose, neutral, and shock CS, and with 1 ms bins for 50 ms before and after for responses evoked by light stimulation. The CS PSTHs were z-transformed using the histogram values in a 1 s baseline period prior to the onset of the CSs and 50 ms baseline for the photoidentification. A z-score of ±1.98 was used to test significance of neural responses. A unit was called photo-responsive if it showed (1) significant excitations within 6 ms after the stimulation onset based on our previous study 2 , (2) insignificant responses during 7–50ms after stimulation onset with a moving average of 6 ms, (3) insignificant responses during the “fake” phototagging session.
Functional hierarchical clustering algorithm for trial-averaged data
Prior to clustering, the data were pre-processed as follows: The PSTH was computed using 20 ms bin widths. Z-scores were calculated using the mean and standard deviation during the baseline period (−1 sec to cue onset) individually for each neuron. Finally, the data were smoothed along the time dimension using a Gaussian-weighted moving average with a smoothing factor of 0.85. Data from each experiment protocol (Sucrose CS, Neutral CS, Shock CS) were concatenated to calculate universal clusters, allowing for comparisons between and within R, N, and S protocols. A hierarchical cluster tree was generated using Ward’s method, which uses inner squared distance to determine hierarchy using correlation for the distance metric. A cutoff threshold was used to determine clusters; the value selected was 35% of the maximum value of the linkage distance. Heatmaps plotted for each region are the smoothed z-score input data; clusters for each region are color-coded based on the original cluster tree (Figure 4d, E8h).
Neural trajectory analysis
Principal component analysis (PCA) was conducted to evaluate the population-level firing rate dynamics. PCA is a commonly used dimensionality reduction tool that maximizes the variance in the data55. A single global PCA was done on a matrix containing all the data for all groups such that we could compare neural trajectories across groups56. Neurons from the control group were randomly selected to make the total number in CRISPR group. This matrix had neurons in rows, and columns contained the time-varying mean firing rates computed using 20 ms bins over the interval −1 to 1 seconds post task-relevant event. The neural trajectories in PCA space for each task-relevant event were created per group by projecting the time-varying mean firing rates of the neurons onto the top PCA eigenvectors (by multiplying the firing rates with the PCA coefficients). For each trajectory, the length was calculated as the sum of euclidean distances between adjacent 20ms bins. Distance between trajectories was calculated as the euclidean distance between the two trajectories bin-by-bin. To allow for statistical comparisons the neural trajectory metrics were calculated using the leave one out (LOO) method, leaving out all the neurons from a single mouse in every iteration. Importantly in every iteration the same coefficients were used, since the PCA was done once prior to this step, but the neurons included varied. For visualization purposes we plotted the first 2 PC subspaces. For quantification of trajectory lengths and distance between trajectories, a total of number of PCs that explain 95% of variance were used (Figure 5a).
Support Vector Machine classifier
To test if trial types (sucrose or shock trial) in the discrimination task could be decoded from single trial BLA population activity, we used a support vector machine (SVM) with a linear kernel. To obtain single trial BLA population activity we used the coefficients obtained for each neuron in the global PCA and created a single trial neural trajectory using the firing rate for that trial. We trained the SVM using the first 10 PCs per trial as features and the trial type as the label. We did a 5-fold cross validation, briefly, the data were split into five parts and in each iteration the training consisted of a different 80% subset of the data and the testing was done with the remaining 20% of the data. Since the number of sucrose trials is twice than the number of shock trials in the task, we randomly subsampled the sucrose trials to match the number of shock trials 50 times for each iteration. Thus, we generated area under the curve (AUC) scores from plotting the binary classification output in a receiver operating characteristic (ROC) curve, as a function of time. For control, we repeated the same process with the training data labels shuffled in order to see the performance of a chance model (Figure 5b, c).
Unsupervised trial-based classification of behaviors
We performed unsupervised classification of mice behaviors during the cue discrimination task for sucrose and shock trials. Each mouse was video recorded (15 frames per second) performing the discrimination task, and videos were subsequently labeled using automated pose estimation algorithms DeepLabCut44. The head, electrode, right ear, left ear, and tail of mice were labeled. Following video labeling, we extracted eight unique measurements to describe general mouse movement during each sucrose and shock trial. These measurements included the following: velocity (pixels/s), distance to port (pixels), direction (radians), and difference in mouse direction relative to the reward port (degrees), acceleration (pixels/s2), change in distance to port (pixels/s), change in direction (radians/s), and change in mouse direction relative to reward port (degrees/s). Each of the eight measurements were extracted for 300 frames (20 seconds) following the sucrose and shock cue, respectively.
After obtaining 300 frame time series for each of the eight measurements for all sucrose and shock trials, we performed time-series feature extraction. Using the Python library tsfresh (version 0.16.0), we extracted hundreds of features for each 300 frame time series for each measurement across sucrose and shock trials, respectively. These features consisted of, but were not limited to, the following: min-max values, number of peaks in the time series, forward and reverse autocorrelations at various lags, summary statistics such as mean, median, and variance, and many more. Using tsfresh (https://tsfresh.readthedocs.io/), we extracted ~6000 features to describe the behavior for each trial, with sucrose and shock trials being processed separately.
Following feature extraction, we performed dimensionality reduction and unsupervised clustering in line with the method proposed by Berman et al., 201445. Notably, we applied the analysis on sucrose and shock trials separately. Briefly, we performed principal components analysis (PCA) to reduce the dimensionality of our feature matrix from ~6000 features to a multidimensional set of principal components. Following linear dimensionality reduction with PCA, we non-linearly embedded the multidimensional principal components into two-dimensional space using t-distributed stochastic neighbor embedding (t-SNE). After t-SNE, we computed a probability density function (PDF) over the 2-D t-SNE output using seaborn.kdeplot (https://seaborn.pydata.org/), which aims to approximate the underlying probability density function by binning and counting observations, to obtain a probability map and identify regions of dense points. The PDF output was then rasterized, and local maxima were identified and selected as markers for watershed segmentation. Following watershed segmentation, we obtained seven clusters for sucrose trials and five clusters for shock trials (Figure 5d–i, E8k–s).
Ex vivo electrophysiology
Brain tissue preparation.
Mice were deeply anesthetized with 90 mg/kg pentobarbital and perfused transcardially, with 10 mL of modified artificial cerebrospinal fluid (ACSF, at ~4°C) containing (in mM): 75 sucrose, 87 NaCl, 2.5 KCl, 1.3 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3 and 5 ascorbic acid. The brain was then extracted and glued (Roti coll 1; Carh Roth GmbH, Karlsruhe, Germany) on the platform of a semiautomatic vibrating blade microtome (VT1200; Leica, Buffalo Grove, IL). The platform was then placed in the slicing chamber containing modified ACSF at 4°C. Coronal sections of 300 μm containing the NAc, CeM, MGN, PVT, vHPC and/or BLA were collected in a holding chamber filled with ACSF saturated with 95% O2 and 5% CO2, containing (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.0 MgCl2, 2.4 CaCl2, 26.0 NaHCO3, 10 glucose. Recordings started 1 h after slicing and the temperature was maintained at approximately 31°C in the holding chamber. Temperature was ~31°C during recordings assessing input synapses onto BLA-NAc and BLA-CeM neurons (Figure E9), and ~25°C for experiments assaying the MGN, vHPC and PVT inputs to the BLA (Figure E2, E5).
All injection sites were checked and imaged with a camera (Hamatsu, Bridgewater, NJ) attached to the microscope (BX51; Olympus, Center Valley, PA). The slice images were reported on the mouse brain atlas57.
Whole-cell patch-clamp recording.
Recordings were made from visually identified neurons containing retrobeads for recordings in Figure E9. Patched cells were filled with Alexa Fluor (AF) 350 and biocytin, visualized and superimposed with retrobeads fluorescence to confirm whether the patched cell was retro bead positive. Recordings in Figure E2, and E5 were made from putative projection neurons in an area of the BLA showing fluorescence from axon terminals.
Brain slices containing the BLA were then placed in the recording chamber perfused with ACSF. A bipolar stimulation electrode (~80 μm spacing between tips) was placed in the amygdala-striatal transition zone containing internal capsule fibers. Electric stimulation intensity used was comprised between 0.01 and 0.2 mA. To test the effect of NT on LTP induction we used a spike timing dependent plasticity (STDP) protocol where the presynaptic inputs (internal capsule) were stimulated 7 ms prior to post-synaptic depolarization (1nA, 5 ms) 10 times at 30 Hz. This was repeated 15 times every 10 seconds.
Voltage-clamp recordings were made using glass microelectrodes (4–9 MΩ) shaped with a horizontal puller (P-1000, Sutter, CA) and filled with a solution containing (in mM): 120 cesium methansulphonate, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 tetraethylammonium chloride, 2.5 MgATP, 0.25 NaGTP, 8 biocytin and 2 Alexa Fluor 350 (pH 7.25–7.4, 280–290 milliosmol). ChR2 was activated using 5 ms pulses from an LED light source (470 nm, Thorlabs, Newton, NJ) and intensity adjusted to generate 100 – 300 pA baseline currents at −70 mV. Cells were clamped at −70 mV for EPSC measurement and 0 mV for IPSC measurement. After baseline measurements at −70 mV and 0 mV, TTX and 4-AP were added to block polysynaptic action potential propogation. Measurements were repeated at 0 mV and −70 mV. NBQX and AP5 were added to block AMPA and NMDA glutamate receptors.
Current-clamp recordings to characterize electrophysiological properties of BLA-NAc and BLA-CeM projectors we made using similar glass microelectrodes (4–9 MΩ) filled with a solution containing (in mM): 125 potassium gluconate, 20 HEPES, 10 NaCl, 3 MgATP, 8 biocytin and 2 Alexa Fluor 350 (pH 7.25–7.4; 208–290 milliosmol). The cells were first voltage clamped at −70mV to determine optimal intensity for the electric stimulation of the internal capsule.
Recorded signals were amplified using Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Analog signals were digitized at 10 kHz using a Digidata 1440 and pClamp 10 software (Molecular Devices, Sunnyvale, CA). ACSF and drugs were applied to the slice via a peristaltic pump (Minipuls3; Gilson, Middleton, WI) at 3 mL/min.
Drug concentrations.
Neurotensin Fragment 8–13 acetate salt (Sigma-Aldrich, St. Louis, MO) was applied at a concentration of 2, 10, 100, or 500 nM.
For experiments using the Neurotensin receptor-1 antagonist SR 48692 (Sigma-Aldrich, St. Louis, MO) dissolved in DMSO, slices were pre-incubated in ACSF containing 150nM of SR 48692.
Tetrodotoxin (TTX ; Tocris, Minneapolis, MN) was applied at a concentration of 1 μM. 4-Aminopyridine (4-AP; Sigma-Aldrich, St. Louis, MO) was applied at a concentration of 100–400 μM.
Glutamate receptor selectively activated by N-methyl-D-aspartate (NMDA-R) antagonist D-(−)-2amino-5-phosphonopentanoate was applied at a concentration of 50 mM (AP5; R&D systems, Minneapolis, MN).
Glutamate receptor selectively activated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA-R) receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione was applied at a concentration of 20 mM (NBQX; Tocis, Minneapolis, MN).
Histology.
The location of all recorded neurons was checked after the recording. Co-localization of AF 350 and retrobeads was confirmed at the end of the recording and double-checked with confocal microscopy for the cells that were recovered with the streptavidin staining (for example, see Figure E2 and E5). For each experiment, the slices containing viral injections or a recorded BLA neuron were fixed overnight at 4°C in 4 % PFA, and then kept in PBS. Slices containing patched neurons were incubated for 2 hours in streptavidin-AF405 (2 mg/ml, dilution 1:500, Biotum, Hayward, CA), mounted on microscope slides with PVA-DABCO and imaged under the confocal microscope.
Data analysis.
Data from the LTP experiment was analyzed using Clampfit (Molecular Devices) and Excel. EPSC amplitude and latencies were estimated by custom MATLAB code (R2016b; Mathworks, Natick, MA).
General Histology
Preparation of fixed-sections.
After the pharmacology, anatomy and photometry experiments, all mice were anesthetized with pentobarbital sodium, and transcardially perfused with ice-cold saline followed by ice-cold 4% paraformaldehyde (PFA) in PBS (pH 7.3). Extracted brains were fixed in 4% PFA overnight and then equilibrated in 30% sucrose in PBS. 40–60 μm thick coronal sections were sliced using a sliding microtome (HM430; Thermo Fisher Scientific, Waltham, MA) and stored in PBS at 4°C until processed for histology. Sections were then incubated with a DNA specific fluorescent probe (DAPI: 4’,6-Diamidino-2-Phenylindole (1:50,000)) for 30 minutes and lastly washed 4 times with PBS-1X followed by mounting on microscope slides with PVA-DABCO.
Confocal microscopy
Confocal fluorescence images were acquired on an Olympus FV1000 confocal laser scanning microscope using a 20x/0.75NA objective or a 40x/1.30NA oil immersion objective for imaging streptavidin-CF405 stained neurons. Serial Z-stack images were acquired using the FluoView software (Olympus, Center Valley, PA) to confirm viral injections and fiber placements. The tip of the fiber was determined by the ~50 μm thick gliosis generated by the fiber. Neurons recovered from the Streptavidin staining were imaged covering the whole dendritic arborization contained in the slice.
For identifying NT-expressing BLA projectors, every third section from the entire brain was imaged. Structures with overlap between NT-expressing and BLA-projecting neurons were identified visually. The extent of overlap was quantified by imaging just these structures in multiple brains.
The number of cells were quantified with the Imaris software (Bitplane Inc., South Windsor, CT). Quantification of fluorescence for comparing axon terminal fields from NT populations was performed by thresholding the fluorescence images as described in2, and using the percentage of pixels above threshold (referred as fluorescent pixels in the manuscript) as a metric to compare between samples. Images were reported on the mouse brain atlas57.
In-situ hybridization
Preparation of fresh sections.
All mice were anesthetized with 5% isoflurane, their brains immediately extracted and covered by powdered dry ice for ~5 minutes, and stored at −80°C. These brains were sectioned using a cryostat (CM3050 S; Leica, Buffalo Grove, IL) at −16°C. Sections were 16 μm thick and they were placed on a glass slide, heated from the bottom to encourage adhesion of the slice to the slide, and stored at −80°C until further processing.
In-situ hybridization using RNAscope.
Fluorescence in-situ hybridization was performed using the ACDBio V2 RNAScope kit (Advanced Cell Diagnostics, Newark, CA). The following products were used: RNAscope Multiplex Fluorescent Reagent Kit V2 (Catalog #323110), Fluorescent Multiplex Detection Reagents (#323110), probes for Nts (#420441), Slc17a6 (#319171-C2), Cas9 (#475781-C1), Ntsr1 (422411-C2), and Ntsr2 (452311-C3), and the Perkin Elmer TSA Plus Fluorescence Palette Kit (NEL760001KT).
The following steps were performed to label mRNA while preventing tissue degradation: fresh frozen slices were fixed in 4% paraformaldehyde for 1 hour at 4°C. Slices were dehydrated in an ethanol series and incubated in hydrogen peroxide for 8 minutes. No protease treatment was used in order to prevent tissue degradation. Slides were incubated in desired probe combinations for 2 hours at 40°C. Signal amplification molecules were hybridized to the target probes in sequential steps as per the V2 RNAscope protocol. For fluorescent labelling of each probe, slides were incubated in channel-specific HRP for 10 minutes, followed by incubation with Tyramide Signal Amplification fluorophores (PerkinElmer, Waltham, MA) for 20 minutes, and then incubation in HRP-blocker for 10 minutes. Nt and Ntsr1 were labelled in far red (TSA Plus Cyanine 5) and Cas9 was labelled in red (TSA Plus Cyanine 3). Between all steps, slides were washed 2 times in 1x RNAscope Wash Buffer for 1–2 minutes. Slides were then incubated in ACDBio DAPI for 10 minutes, washed, dried for 20 minutes, coverslipped with PVA-DABCO, (Sigma-Aldrich, St. Louis, MO) and left to dry overnight before imaging.
Image Analysis:
Automated cell identification and analysis of mRNA fluorescent labelling was done using CellProfiler using an optimized version of the Colocalization pipeline as published on the CellProfiler website (https://cellprofiler.org/examples/). Brightness and contrast of confocal images were moderately adjusted using ImageJ, with consistent thresholding levels used for each probe-fluorophore combination. Cell outlines were identified based on DAPI labelling and individual mRNA puncta (2–10 pixels in diameter) colocalized within each cell were quantified. Cells with >6 puncta of Nt or Ntsr1 mRNA were considered to be expressing cells (Figure E5e, E5f). The quantity of Nt or Ntsr1-expressing cells was then quantified in the PVT and BLAusing custom-written MATLAB script (Mathworks, Natick, MA; Figure E10h, E10j).
Statistical analysis
Statistical analyses were performed using commercial software (GraphPad Prism 8.0; GraphPad Software, Inc, La Jolla, CA). Within subject comparisons were made using paired comparisons. Single-variable differences were detected with two-tailed (unless specified otherwise) paired or unpaired (as noted) Student t-test. Reported effect sizes are different between means when using unpaired comparisons and mean of differences when using paired comparisons. For all results, significance threshold was placed at P = 0.05 (represented by P<0.05 = *, P<0.01 = **, P<0.001 = ***). All data are shown ± standard error to the mean (s.e.m.). To assess learning during head-fixed reward task, we used a one-sided Wilcoxon ranksum test (MATLAB) and set the threshold for learning at P < 0.05.
Additional statistical information:
1g: unpaired t-test
The MGN: t33=0.1019, P= 0.9195, effect size= 0.8488 ± 8.331, CI95= −16.10 to 17.80.
The vHPC: t18=0.8666, P=0.3976, effect size= 6.356 ± 7.335, CI95= −9.054 to 21.77.
The PVT: t26=2.611, *P=0.0148, effect size= 20.52 ± 7.860, CI95= 4.363 to 36.67.
1h: unpaired t-test
The MGN: t24=0.8346, P=0.4122, effect size=−0.06289 ± 0.07535, CI95=−0.2184 to 0.09263.
The vHPC: t15=0.2820, P=0.7818, effect size=0.02992 ± 0.1061, CI95=−0.1962 to 0.2561.
The PVT: t14=0.5740, P=0.5751, effect size=0.05790 ± 0.1009, CI95=−0.1585 to 0.2743.
2c: Unpaired t-test. t34=2.988, **P=0.0052, effect size=−0.6385 ± 0.2137, CI95=−1.073 to −0.2042.
2d: Unpaired t-test. t26=2.167, *P=0.0396, effect size=0.2567 ± 0.1185, CI95=0.01324 to 0.5002.
2g: Unpaired t-test. t13=2.706, *P=0.018, effect size=1.068 ± 0.3948, CI95=0.2156 to 1.921.
2f: Unpaired t-test. t13=2.474, *P=0.0279, effect size=−0.2915 ± 0.1178, CI95=−0.546 to −0.03698.
3b: Paired t-test. t12=6.497, ***P<0.0001, effect size= 1.82 ± 0.2802, CI95= 1.210 to 2.431.
3c: Paired t-test. t11=2.48, *P=0.0306, effect size= 4.311 ± 1.738, CI95= 0.4852 to 8.136.
3e: Paired t-test. CS: t12=1.474, P=0.1663, effect size= 3.887 ± 2.637, CI95= −1.860 to 9.633.
US: t12=2.935, *P=0.0125, effect size= 14.12 ± 4.81, CI95= 3.635 to 24.60.
3f: Paired t-test. CS: t11=1.472, P=0.169, effect size= −2.116 ± 1.437, CI95= −5.280 to 1.047.
US: t11=2.896, *P=0.0145, effect size= −7.08 ± 2.444, CI95= −12.46 to −1.700.
3h: One-way ANOVA with Dunnett’s multiple comparisons test: F (13, 28) = 1059, ***P<0.0001.
NT vs. Glu: q28=84.87, ***P<0.0001, effect size= 0.9866 ± 0.01163, CI95= 0.9519 to 1.021.
NT vs. GABA: q28=85.34, ***P<0.0001, effect size= 0.9921 ± 0.01163, CI95= 0.9574 to 1.027.
NT vs. DA: q28=85.62, ***P<0.0001, effect size= 0.9954 ± 0.01163, CI95= 0.9607 to 1.030.
NT vs. ACh: q28=86.28, ***P<0.0001, effect size= 1.003 ± 0.01163, CI95= 0.9683 to 1.038.
NT vs. NE: q28=86.42, ***P<0.0001, effect size= 1.005 ± 0.01163, CI95= 0.9699 to 1.039.
NT vs. 5-HT: q28=86.26, ***P<0.0001, effect size= 1.003 ± 0.01163, CI95= 0.9681 to 1.038.
NT vs. Ado: q28=86.13, ***P<0.0001, effect size= 1.001 ± 0.01163, CI95= 0.9666 to 1.036.
NT vs. CRF: q28=86.90, ***P<0.0001, effect size= 1.010 ± 0.01163, CI95= 0.9755 to 1.045.
NT vs. SST: q28=86.36, ***P<0.0001, effect size= 1.004 ± 0.01163, CI95= 0.9693 to 1.039.
NT vs. NPY: q28=86.28, ***P<0.0001, effect size= 1.003 ± 0.01163, CI95= 0.9683 to 1.038.
NT vs. CCK: q28=86.56, ***P<0.0001, effect size= 1.006 ± 0.01163, CI95= 0.9716 to 1.041.
NT vs. VIP: q28=86.57, ***P<0.0001, effect size= 1.006 ± 0.01163, CI95= 0.9716 to 1.041.
NT vs. OXT: q28=85.52, ***P<0.0001, effect size= 0.9943 ± 0.01163, CI95= 0.9595 to 1.029.
3k: Paired t-test. CS: t4=1.788, P=0.1483, effect size= 2.754 ± 1.54, CI95= −1.523 to 7.031.
US: t4=8.709, *P=0.001, effect size= 4.14 ± 0.4759, CI95= 2.744 to 5.538.
3l: Paired t-test. CS: t4=2.137, P=0.0994, effect size= −1.598 ± 0.7475, CI95= −2.585 to 0.3570.
US: t4=3.996, *P=0.0162, effect size= −3.657 ± 0.9152, CI95= −6.083 to −0.9583.
4e: Unpaired t-test, for sucrose, neutral, and shock CS:
Cluster #1: t78=2.203, *P=0.0303, effect size=1.881 ± 0.8537, t78=0.99, P=0.3252, effect size=0.4098 ± 0.4098, t78=0.6334, P=0.5283, effect size=0.3699 ± 0.5841;
Cluster #2: t73=1.008, P=0.3166, effect size=0.2745 ± 2745, t73=1.474, P=0.1444, effect size=0.4075 ± 0.4075, t73=2.365, *P=0.02, effect size=0.5161 ± 0.5161;
Cluster #3: t50=2.071, *P=0.0435, effect size=2.552 ± 1.232, t50=1.996, P=0.0513, effect size=2.251 ± 1.127, t50=2.076, *P=0.043, effect size=1.748 ± 0.8419;
Cluster #4: t52=2.219, *P=0.031, effect size=−1.564 ± 0.7046, t52=0.7807, P=0.4385, effect size=0.2421 ± 0.3101, t52=2.225, *P=0.03, effect size=2.65 ± 1.191;
Cluster #5: t111=1.243, P=0.216, effect size=−0.1798 ± 0.1146, t111=1.836, P=0.069, effect size=−0.3948 ± 0.2151, t111=1.89, P=0.061, effect size=−0.3178 ± 0.1681;
Cluster #6: t140=0.08, P=0.936, effect size=− 0.009792± 0.1222, t140=0.7447, P=0.457, effect size=0.1128 ± 0.1514, t140=2.228, *P=0.0276, effect size=−0.429 ± 0.1928;
Cluster #7: t167=1.206, P=0.229, effect size=−0.4117 ± 0.3413, t167=2.924, **P=0.003, effect size=−0.4571 ± 0.1563, t167=1.394, P=0.165, effect size=−0.3006 ± 0.2157.
4f: Unpaired t-test, for sucrose CS: t37=2.06, *P=0.0465, effect size=−2.317 ± 1.125, CI95=−4.596 to −0.0376, for neutral CS: t37=0.4676, P=0.6428, effect size=−0.168 ± 0.3594, CI95=−0.8962 to 0.5601, for shock CS: t37=1.397, P=0.1707, effect size=−0.7888 ± 0.5646, CI95=−1.933 to 0.3552.
4g: Unpaired t-test, for sucrose CS: t30=2.06, *P=0.0481, effect size=1.026 ± 0.4978, CI95=0.0009 to 2.042, for neutral CS: t30=1.758, P=0.089, effect size=−0.9119 ± 0.5189, CI95=−1.972 to 0.1477, for shock CS: t30=1.542, P=0.1336, effect size=−1.798 ± 1.166, CI95=−4.179 to 0.5837.
5a: Trajectory length was calculated for 12 iterations for the control group and 14 iterations for the CRISPR group using LOO approach (see Methods). Two-tailed unpaired t-test.
Sucrose CS: t11= 4.786, ***P<0.0001, effect size= −7.124 ± 1.376, CI95= −9.963 to −4.284.
Neutral CS: t11= 1.224, ***P=0.0002, effect size= −7.053 ± 1.607, CI95= −10.37 to −3.736.
Shock CS: t11= 14.99, ***P<0.0001, effect size= −10.13 ± 1.464, CI95= −13.15 to −7.113.
5b: One-way ANOVA with Holm-Sidak’s multiple comparisons test. F(3, 16)= 668.6, ***P<0.0001.
Control vs. CRISPR: t16=1.592, P=0.1309, effect size= 0.02392 ± 0.01502.
Control vs. Control-Shuffled: t16= 33.45, ***P<0.0001, effect size= 0.5025± 0.01502.
CRISPR vs. CRISPR-Shuffled: t16= 29.78, ***P<0.0001, effect size= 0.5025± 0.01502.
5c: One-way ANOVA with Holm-Sidak’s multiple comparisons test. F(3, 16)= 34196, ***P<0.0001.
trained with CRISPR and tested with Control vs. trained with Control and tested with CRISPR: t16= 181.0, ***P<0.0001, effect size=−0.3114 ± 0.001721.
trained with CRISPR and tested with Control vs. Shuffled: t16= 96.57, ***P<0.0001, effect size=0.1662 ± 0.001721.
trained with Control and tested with CRISPR vs. Shuffled: t16= 277.1, ***P<0.0001, effect size=0.4769 ± 0.001721.
5f: Two-tailed unpaired t-test. t28= 2.25, *P=0.0325, effect size= −0.1644 ± 0.07308, CI95= −0.3141 to −0.01474.
5i: Two-tailed unpaired t-test. t28= 2.366, *P=0.0251, effect size= −0.3644 ± 0.1540, CI95= −0.6799 to −0.04895.
E8d: Two-tailed unpaired t-test, non-projectors: t611=3.132, **P=0.0018, effect size=−1.296 ± 0.4138, CI95=−2.108 to −0.4834; BLA-NAc: t37=0.4665, P=0.6436, effect size= −1.165 ± 2.498, CI95= −6.226 to 3.896; BLA-CeA: t30=2.429, *P=0.021, effect size= −3.784 ± 1.558, CI95= −6.961 to −0.6071.
E8e: Two-tailed unpaired t-test, sucrose trials: raw probability baseline, t33=2.37, *P=0.012, effect size=0.1396 ± 0.05889, CI95=0.01974 to 0.2594; raw probability 10–25s post sucrose, t33=3.797, ***P=0.0006, effect size=0.2368 ± 0.06238, CI95=0.1099 to 0.3638; z-score, **P=0.0038, effect size=−0.6448 ± 0.2078, CI95=−1.067 to −0.2225. Shock trial: raw probability, t33=0.1401, P=0.8894, effect size=−0.0049± 0.0358, CI95=−0.0771 to 0.06722; z-score, t33=2.495, *P=0.0178, effect size=1.241 ± 0.4976, CI95=0.229 to 2.254.
E8j: Two-tailed unpaired t-test, BLA-NAc: t37=2.043, *P=0.0483, effect size=−1.611 ± 0.7888, CI95=−3.209 to −0.01291. BLA-CeA: t30=2.46, *P=0.0199, effect size=2.668 ± 1.084, CI95=0.453 to 4.882.
E8o: Two-way ANOVA with Holm-Sidak’s multiple comparisons test. Non-phototagged: Interaction: F(1, 1284)=4.083, *P=0.0435, “State”: F(1, 1284)=6.238, *P=0.0126, Treatment: F(1, 1284)=0.7362, P=0.3910. Control vs. CRISPR: “Passive”, t1284=0.7418, P=0.4523, effect size=−0.3049 ± 0.4056; “Active”, t1284=2.27, *P=0.046, effect size= 0.7551 ± 0.3327. BLA-NAc: Interaction: F(1, 434)=10.96, **P=0.001, “State”: F(1, 434)=3.264, P=0.0715, Treatment: F(1, 434)=0.0003, P=0.9849. Control vs. CRISPR: “Passive”, t434=2.287, *P=0.033, effect size=−0.5420 ± 0.237; “Active”, t434=2.4, *P=0.033, effect size= 0.5358 ± 0.2233. BLA-CeA: Interaction: F(1, 744)=2.405, P=0.1213, “State”: F(1, 744)=0.614, P=0.4335, Treatment: F(1, 744)=0.8808, P=0.3483. Control vs. CRISPR: “Passive”, t744=1.402, P=0.2969, effect size=−0.1867 ± 0.1332; “Active”, t744=0.6663, P=0.5054, effect size= 0.0459 ± 0.0689. The number of trials used for analysis were matched between passive and active trials.
E8s: Two-way ANOVA with Holm-Sidak’s multiple comparisons test. Non-phototagged: Interaction: F(1, 780)=6.198, *P=0.013, “State”: F(1, 780)=0.628, P=0.4282, Treatment: F(1, 780)=0.1508, P=0.6978. Control vs. CRISPR: “Passive”, t780=1.368, P=0.1716, effect size=−0.6263 ± 0.4577; “Active”, t780=2.245, *P=0.049, effect size= 0.8579 ± 0.3821. BLA-NAc: Interaction: F(1, 268)=7.422, **P=0.0069, “State”: F(1, 268)=0.737, P=0.3934, Treatment: F(1, 268)=0.062, P=0.8024. Control vs. CRISPR: “Passive”, t268=1.583, P=0.1145, effect size=−0.5154 ± 0.3255; “Active”, t268=2.383, *P=0.0354, effect size= 0.6197 ± 0.2601. BLA-CeA: Interaction: F(1, 396)=3.516, P=0.0615, “State”: F(1, 396)=6.734, **P=0.0098, Treatment: F(1, 396)=2.648, P=0.1045. Control vs. CRISPR: “Passive”, t396=2.28, *P=0.045, effect size=0.4653 ± 0.2041; “Active”, t396=0.1935, P=0.8467, effect size= −0.0392 ± 0.1701. The number of trials used for analysis were matched between passive and active trials.
Sample size
Sample sizes were not predetermined and based on similar studies in the literature 1,2. Sample size is reported in the legends and methods.
Data exclusions
Animals were excluded based on histological verification. For the pharmacology experiments involving bilateral cannula injections, all animals with one or both cannula placements not targeting the BLA were excluded from analysis by an experimenter blind to the experimental manipulation. For tone-sucrose association behavior, behavioral outliers were excluded after using Grubbs test (with a P value threshold of 0.05). A total of 4 mice out of 91 mice used in Figure 1g, h were excluded. Data from each individual mouse (with the excluded outliers identified) is available upon request. Grubbs test was also used to discard outliers from the NGS data in Figure E3a and c. Data from one sample was discarded across all 19 samples. Data from each sample (with the outlier identified) is available upon request. In multisite photometry experiments, mice that did not acquire the task were excluded. Two out of 7 mice were excluded based on these criteria for tone-air puff association, and two out of 7 mice were excluded for tone-sucrose association. Data from each individual mouse (with the non-learners identified) is available upon request. In in situ validation of CRISPR-cKO of Nt gene, 6 out of 19 mice from control groups were excluded from the plot due a power outage of our freezer which made these tissues undesirable for in situ hybridization. In situ validation of Vglut2 mRNA was only done on a group of randomly selected mice. Freezing videos recorded during the test session were lost for 5 control and 3 CRISPR mice, therefore, they were excluded from the analysis. For the in vivo electrophysiological recording, 4 control mice and 1 CRISPR mice were excluded due to the optrode placement. One of 6 mice was excluded from the NT sensor in vivo recording due to its fiber placement. One neurons from BLA-CeA group was excluded for the analysis except the basal firing due to the loss of its signal. Other than fiber placement, one of 13 mice was excluded from the terminal photometry experiment due to the poor quality of its anticipatory eye closure video recording.
Reproducibility
Experiments included in Figure 1g and h were repeated twice across two different institutes (MIT and Salk). Experiments included in Figure 2c and d were repeated four times by the same investigator. Experiments included in Figure 3h were repeated 3 times means using 3 coverslips of neuron cultures, each coverslip was analyzed with 30–40 ROIs and the averaged response of 30–40 ROIs are plotted as a single circle, the bars represent average of 3 coverslips. Inset: repeated 3 times means using 3 coverslips of neuron cultures, inset showing one representative coverslip. Experiments included in Figure 3i were repeated 3 times means using 3 coverslips of neuron cultures, each coverslip was analyzed with 30–40 ROIs and the averaged response of 30–40 ROIs are plotted as a single open circle, the solid rectangles represent average of 3 coverslips. Experiments included in Figure E7b were repeated 4 times using 4 batches of HEK293T cells, each time cells were tested in 3 individual wells, each well contains 10 image fields. Experiments included in Figure E7c were repeated 3 times means using 3 coverslips of transfected HEK293T cells and 30–40 cells from each coverslip are analyzed; the lines with shading indicate average and s.e.m. of 30–40 ROIs from a single representative coverslip. Experiments included in Figure E7d were repeated 3 times means using 3 coverslips of transfected HEK293T cells and 30–40 cells from each coverslip are analyzed; the response of each cell are plotted as a single circle and the bars represent average of all cells from 3 coverslips. Experiments included in Figure E7f were repeated 3 times; means measured 3 times using one bath of HEK293T cells stably expressing NT1.0 sensor. The spectrum is plotted using data from 1 representative measurement. Experiments included in Figure E9 were repeated by 4 different investigators across 56 animals with 1–3 cells per animal. Results involving freezing scoring and histological quantifications were independently evaluated by at least two investigators.
All other experiments included in the manuscript were repeated with multiple cohorts by multiple investigators. However, data evaluation was not performed independently across each cohort.
Randomization
Mice in each cage were randomly divided into experimental and control groups, with 2 experimental and 2 control mice in cage of 4, or 2 experimental and 1 control, or 1 experimental and 2 control mice in a cage of 3. All the conditioning stimuli were counter-balanced across mice. For experiments involving multiple conditioning boxes, approximately equal number of mice belonging to the experimental and the control groups were conditioned in each box. Same number of mice from experimental and control groups were tested together at a time. For experiments other than those involving mice, samples were randomly allocated into experimental groups.
Blinding
During behavioral testing investigators were not always blind to the group affiliation (experimental vs control) given familiarity with the subjects. However, for histology, optogenetic experiments, and ex vivo electrophysiological recordings, the experimenters were blinded to the group assignment of the animals (experimental vs control). During electrophysiological data processing and analysis experimenters were blinded to the group affiliation until the point that all data was processed such that group comparisons could be made.
Data availability
All the raw and/or processed data shown in this manuscript are available upon request. Sequences of custom AAV9 viruses and guide RNAs used for CRISPR-based CRISPR-cKO experiments are available upon request. All mouse illustrations included in the main figures were created with BioRender.com.
Code availability
All the custom code used to process data shown in this manuscript is available upon request.
Extended Data
Extended Data Figure 1: In situ hybridization of Ntsr1 and Ntsr2 mRNA, validation of the NT::Cre x Ai14 mouse line, identification of NT source to the BLA, and their terminal fields of NT axons in the BLA.
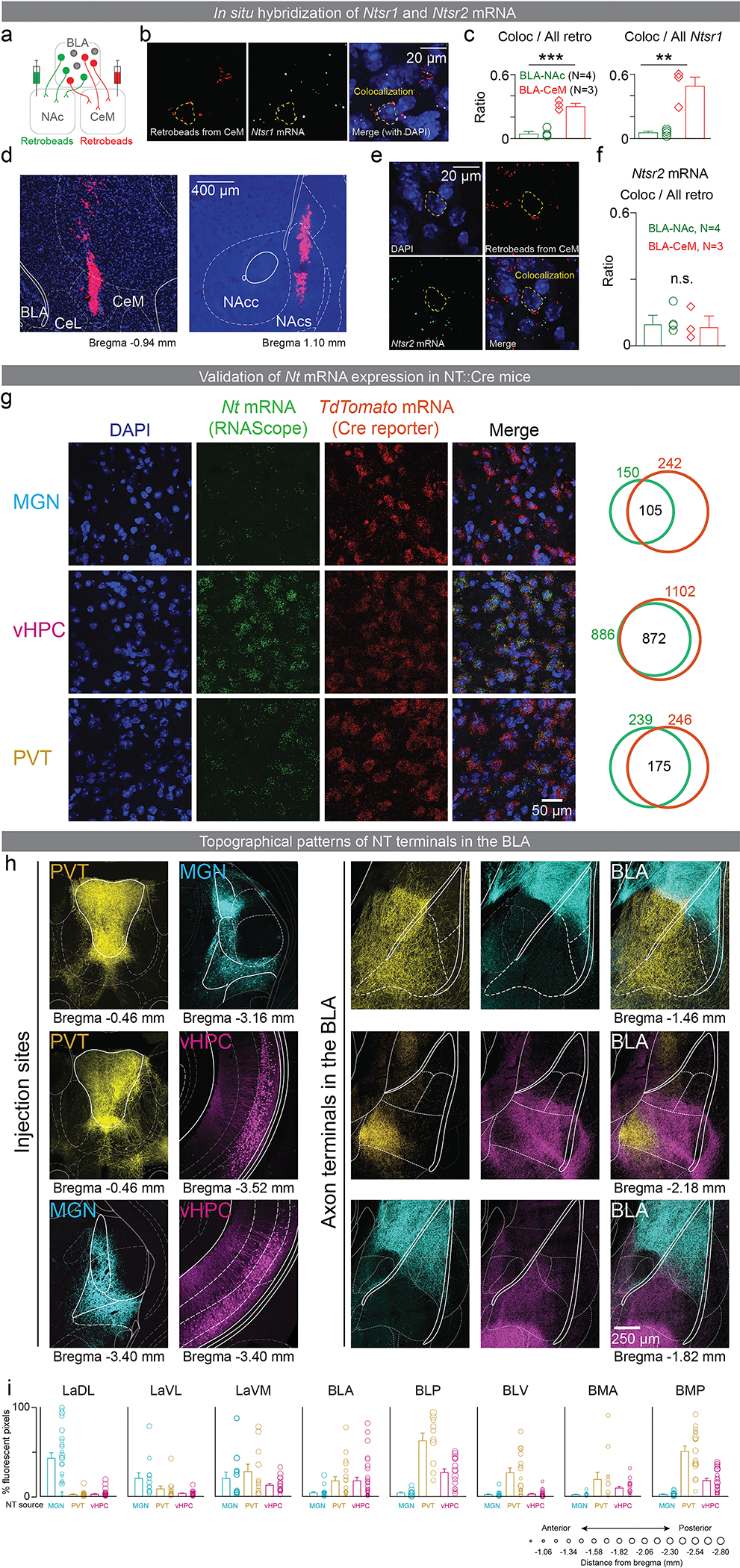
a-c: Ntsr1 mRNA was predominantly expressed in BLA-CeM neurons compared to BLA-NAc neurons. (a) Schematics of labeling BLA-NAc and BLA-CeM neurons with retrobeads. (b) Representative confocal images of retrobeads from the CeM (left panel), fluorescence in situ hybridization of Ntsr1 mRNA (middle panel) in the BLA, and the merged image (right panel). The white arrow indicates a retrobead-positive neuron that is colocalized with Ntsr1 mRNA staining. The dotted yellow line delineates a cell co-expressing retrobeads and Ntsr1 mRNA. (c) BLA-CeM neurons showed significant higher proportions of colocalization with Ntsr1 mRNA staining-positive neurons compared to the BLA-NAc neurons (Unpaired t-test, for coloc to tracer: t5=7.901, ***P=0.0005, effect size=0.2574 ± 0.0325, CI95=0.1737 to 0.3412, for coloc to Ntsr1, t5=6.383, **P=0.0014, effect size=0.4375 ± 0.06854, CI95=0.2613 to 0.6137).
d-f: Ntsr2 mRNA is equally distributed among the BLA-NAc and the BLA-CeM neurons. (d) Representative images of injection sites of retrobeads in the CeM (left panel) and the NAc shell (right panel). (e) Pseudocolored confocal images of DAPI, retrobeads from the CeM, and RNAscope staining of Ntsr2 mRNA. The dotted yellow line delineates a cell co-expressing retrobeads and Ntsr2 mRNA. (f) The proportions of colocalized neurons (retrobeads and Ntsr2 mRNA puncta) do not show significant difference between the BLA-CeA and the BLA-NAc neurons (unpaired t-test, two-tailed, t5=0.3729, P=0.7245, effect size= −0.01523 ± 0.04803, CI95= −0.1202 to 0.08974).
g: To confirm the tdTomato expressing neurons in the Ai14 reporter line when crossed with the NT::Cre line we used in-situ hybridization (RNAScope) to co-label Nt mRNA and tdTomato mRNA. We found a large amount of overlap between cells co-expressing Nt mRNA and tdTomato mRNA. Numbers in the Venn diagrams indicate the total number of cells counted in each category.
h-i: MGN:NT, vHPC:NT and PVT:NT populations send axons to largely topographically segregated sub-regions of the BLA. (h) Pseudocolored confocal images showing cell bodies expressing fluorophores under the control of Cre in two of the three NT populations containing BLA projectors (left two columns), and their corresponding axon terminals in the BLA (right three columns). The anterior-posterior direction from bregma is indicated below (in mm). (i) Fluorescence from the PVT:NT, vHPC:NT and MGN:NT axon terminals was quantified in the following sub-divisions of the amygdala (as defined in the mouse brain atlas57) – lateral amygdaloid nucleus, dorsolateral part (LaDL), lateral amygdaloid nucleus, ventrolateral part (LaVL), lateral amygdaloid nucleus, ventromedial part (LaVM), basolateral amygdaloid nucleus, anterior part (BLA), basolateral amygdaloid nucleus, posterior part (BLP), basolateral amygdaloid nucleus, ventral part (BLV), basomedial amygdaloid nucleus, anterior part (BMA), basomedial amygdaloid nucleus, posterior part (BMP). These regions are labeled across 6 coronal sections in (h). Each circle in (j) shows data from an image obtained from one coronal section from a single mouse. The size of the circle represents the anterior-posterior coordinate relative to bregma, and the mapping is shown in the legend at the bottom.
N denotes number of mice in each group. Error bars and solid shaded regions around the mean indicate s.e.m.
Extended Data Figure 2: MGN:NT, vHPC:NT and PVT:NT populations evokes both EPSCs and IPSCs in BLA neurons, but IPSCs are not monosynaptic.
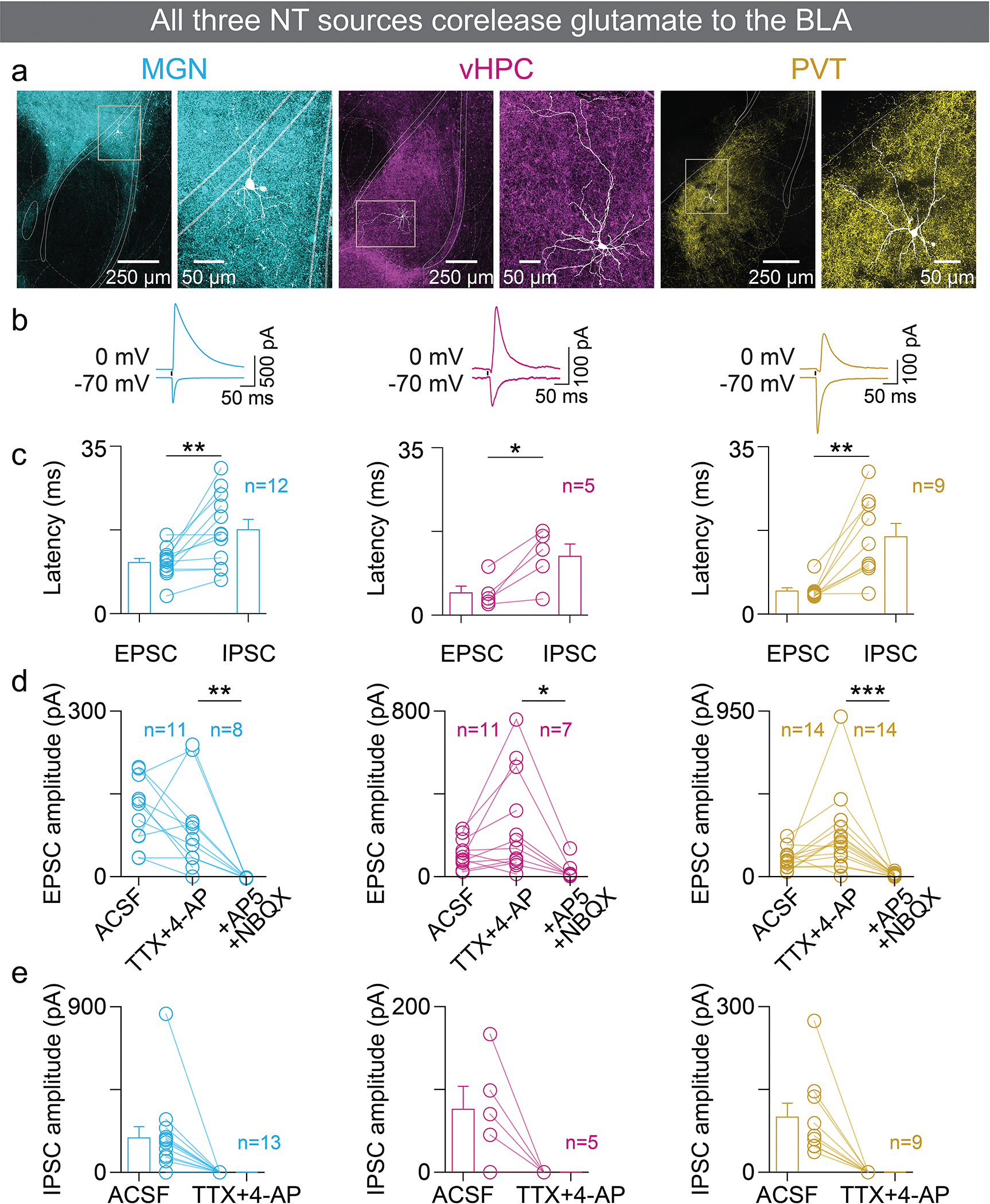
a: Representative images of BLA neurons at lower (left) and higher (right) magnifications used to assay synaptic currents from axon terminals coming from the MGN, the vHPC or the PVT.
b: Representative traces of excitatory and inhibitory post-synaptic currents (EPSCs and IPSCs) recorded from putative principal neurons in the BLA upon stimulating ChR2-expressing axon terminals from NT populations in the MGN (left), the vHPC (middle) and the PVT (right).
c: The EPSCs had faster onset times than the IPSCs upon stimulation of NT population axon terminals from the MGN (Two-tailed paired t-test, t11=3.587, **P=0.0043), the vHPC (Two-tailed paired t-test, t4=3.730, *P=0.0203) and the PVT (Two-tailed paired t-test, t8=4.354, **P=0.0024).
d: Light-evoked EPSCs were monosynaptic as they were still evoked upon stimulation of axon terminals from NT populations of all three regions after the application of TTX+4-AP. Monosynaptic EPSCs were significantly reduced by the application of glutamate blockers for AMPA and NMDA receptors, AP5 and NBQX respectively (One-way ANOVA with Holm-Sidak’s multiple comparisons test. The MGN: F(2, 27)=9.613, TTX+4-AP vs. ACSF: t27=1.209, P=0.2372, effect size=−31.28 ± 28.87; TTX+4-AP vs. AP5+NBQX: t27=3.19, **P=0.0072, effect size=89.95 ± 28.2; The PVT: F(2,39)=10.67, TTX+4-AP vs. ACSF: t39=2.786, **P=0.008, effect size=141.9 ± 50.93; TTX+4-AP vs. AP5+NBQX: t39=4.585, ***P<0.0001, effect size=233.5 ± 50.93; The vHPC: F(2, 26)=4.304; TTX+4-AP vs. ACSF: t26=2.013, P=0.0545, effect size=141.9 ± 70.48; TTX+4-AP vs. AP5+NBQX: t26=28.03, *P=0.0188, effect size=224 ± 79.91.
e: IPSCs were abolished upon application of TTX+4-AP confirming that they are not monosynaptic. There was no detectable positive deflection in the current after application of TTX+4-AP from any cell.
n denotes number of neurons in each group. Error bars around the mean indicate s.e.m.
Extended Data Figure 3: Detection of insertion-deletion mutations introduced by the CRISPR-Cas9 system, and Nt mRNA depletion.
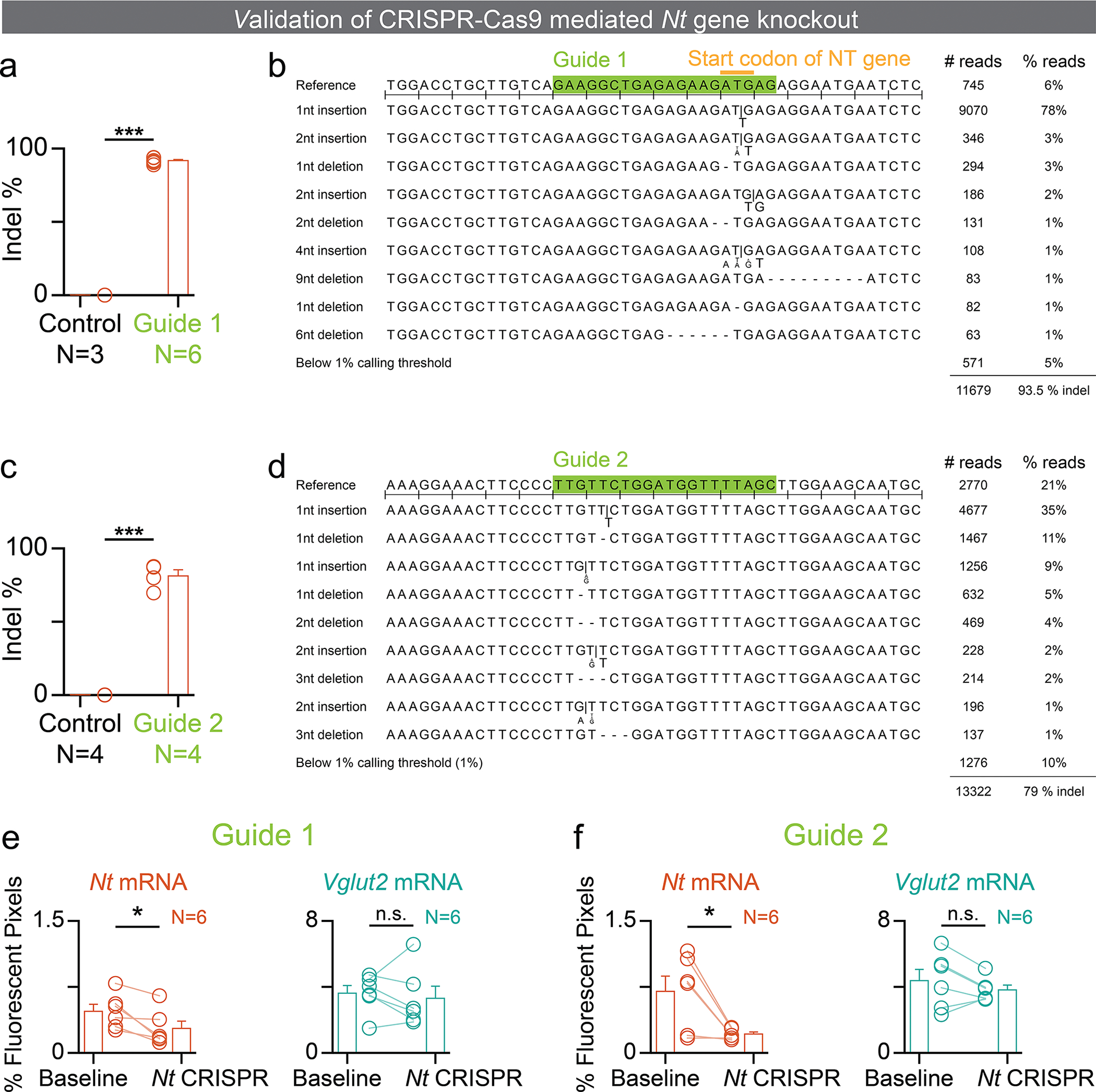
a-d: To compute the percentage of insertion-deletion (indel) mutations introduced into the Nt gene by the CRISPR-Cas9 system in vivo, we harvested tissue from viral-injection sites (AAV-Cas9 + AAV-Nt guide 1 or AAV-Cas9 + AAV-Nt guide 2 in the MGN), sorted the nuclei using fluorescence, extracted the genomic DNA, used PCR to amplify the parts of the DNA targeted by each guide and used next-generation sequencing (NGS) to sequence the DNA. For the control group, tissue was harvested from the same brain region, but from either the hemisphere contralateral to the viral injection, or the same brain region in an un-injected mouse. The percentage of indel mutations were quantified using the website outknocker.org (a, c). For the Guide 1 and Guide 2 groups in (a) and (c) respectively, only one tissue sample was derived from each injected mouse. For the control groups, the 4 and 5 samples in (a) and (c) were derived from 2 and 3 mice, respectively. There were significantly higher number of indel mutations in the Cas9+Guide 1 (a; unpaired t-test, two-tailed, t8=85.99, ***P<0.0001, effect size= 90.82 ± 1.056, CI95= 88.38 to 93.25) and Cas9+Guide 2 (c; unpaired t-test, two-tailed, t7=21.20, ***P<0.0001, effect size= 80.27 ± 3.787, CI95= 71.32 to 89.23) injected tissue, compared with mutations in the control tissue. (b,d)The percentage of reads from a representative mouse in each of the Guide 1-injected (b) and Guide 2-injected (d) groups are shown.
N denotes number of tissue samples in each group. Error bars around the mean indicate s.e.m.
e-f: Quantification of Nt mRNA and Vglut2 mRNA using fluorescence in-situ hybridization (RNAScope), where both guides were co-introduced into the brain. Data shown here are from separate sets of mice that received an AAV-Cas9 injection along with an injection of either AAV-Nt guide 1 (e) or AAV-Nt guide 2 (f). Using Cas9 in combination with guide 1 depleted Nt mRNA levels (e, left; paired t-test, t5=3.251, *P=0.0227, effect size= 0.1950, CI95= 0.04078 to 0.3492), while Vglut2 mRNA levels were not affected (e, right; paired t-test, t5=0.5491, P=0.6065, effect size=0.3050, CI95= −1.123 to 1.733). Using Cas9 in combination with guide 2 depleted Nt mRNA levels (f, left; paired t-test, t5=3.193, *P=0.0242, effect size= 0.4850, CI95= 0.09443 to 0.8756), while Vglut2 mRNA levels were not affected (f, right; paired t-test, t5=1.280, P=0.2568, effect size=0.5617, CI95= −0.5666 to 1.690).
N denotes number of mice in each group. Error bars around the mean indicate s.e.m.
Extended Data Figure 4: In vivo validation of CRISPR-Cas9 mediated Nt gene cKO.
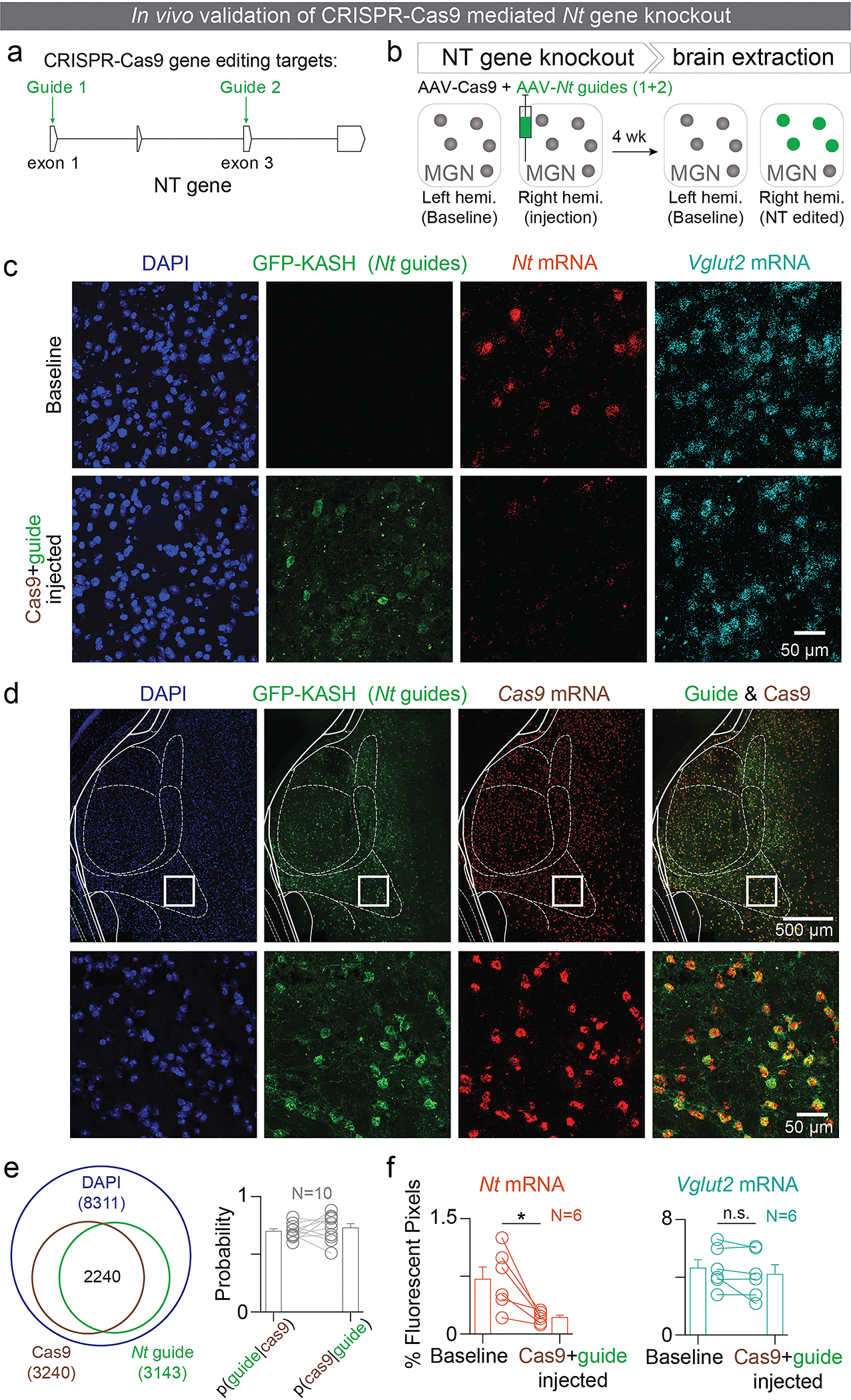
a: The CRISPR-associated endonuclease Cas9 was used to generate indels in the Nt gene for depleting NT levels in specific NT populations. NT-specific single guide RNAs were designed to target exon 1 and 3 of the NT coding region.
b: Experimental design to validate the efficacy of the CRISPR-Cas9 system in depleting Nt mRNA levels. Four weeks after steretotaxic injection of AAV9 viral vectors encoding Cas9 and both the guides into the right MGN, brains were extracted and mRNA levels of Nt and Vglut2 transcripts were assessed using fluorescence in-situ hybridization (ISH; RNAScope).
c: Confocal images from a representative brain slice showing fluorescence from the nuclear stain DAPI, GFP in the Nt guides, ISH of Nt mRNA and Vglut2 mRNA from the control (top) and Cas9+guide injected hemispheres (bottom).
d: Representative images showing overlap between the GFP-KASH and Cas9 mRNA expressing neurons. We used a multi-AAV viral system to deliver the different components of the CRISPR-Cas9 gene editing system (Cas9 and guides) to neurons in vivo. Since gene editing requires the Cas9 protein and one or more guides to be present in the same cell, we quantified the amount of overlap between cells expressing the Cas9 and the guide components. The images shown here are from a representative mouse. The bottom row is a magnified version of the area indicated by the solid white square in the top row.
e: Quantification of penetrance of the AAV vectors carrying Cas9 and the guides. Both the Cas9 protein and guide RNAs targeting the Nt gene need to be present in a cell for the Cas9 protein to edit the Nt gene.
f: Percentage of fluorescent pixels was used as a metric to quantify Nt and Vglut2 mRNA levels. Nt mRNA levels were significantly reduced in the Cas9+guide injected hemisphere, compared to the non-injected hemisphere (paired t-test, t5=3.315, *P=0.0211, effect size=0.4983, CI95= 0.1118 to 0.8849; left panel), while there was no detectable difference in the levels of Vglut2 mRNA between the two hemispheres (paired t-test, t5=1.442, P=0.2089, effect size=0.4283, CI95= −0.3355 to 1.192; right panel).
N denotes number of mice in each group. Error bars around the mean indicate s.e.m.
Extended Data Figure 5: Validations of the PVT-BLA projection-specific CRISPR-Cas9 mediated Nt gene cKO and supplementary data for PVT-BLA:NT photostimulation.
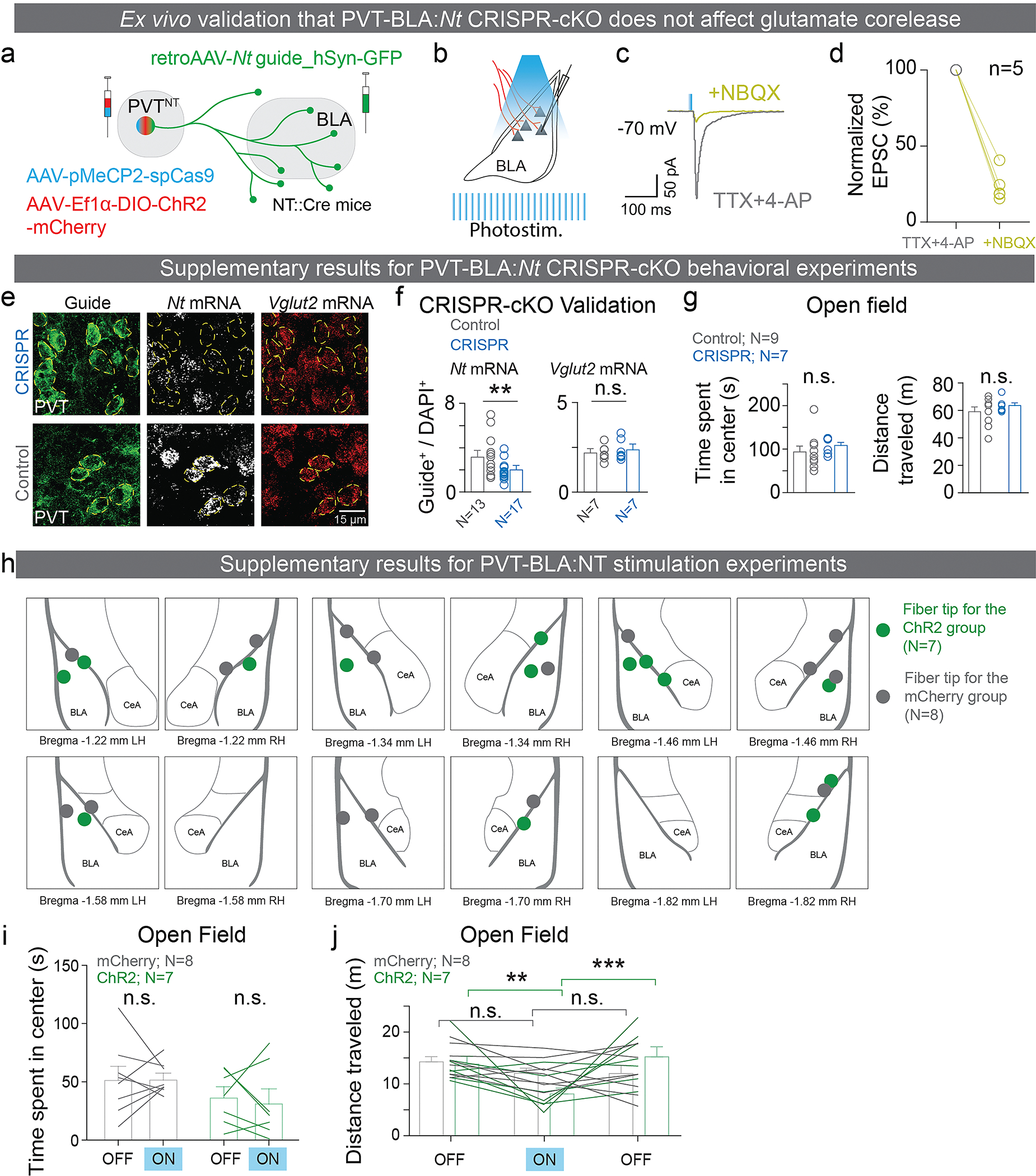
a-d: PVT-BLA:Nt CRISPR-cKO does not affect glutamate release onto BLA neurons. (a) Schematics of the experiment. (b-d) In mice with CRISPR-cKO of Nt in the PVT-BLA projection, glutamatergic transmission was still observed (5 cells recorded from 4 separate mice; Wilcoxon test, *P = 0.0156).
e, f: PVT-BLA:Nt CRISPR-cKO in situ validation. (e) Representative confocal images of fluorescent in situ hybridization in the PVT of control and CRISPR mice. Dotted lines outline the guide positive cells. (f) Mice receiving Nt guides injections (CRISPR group) showed significantly reduced Nt mRNA levels in the guide-positive cells compared to mice receiving control guide injections, while Vglut2 mRNA levels were unaffected (Unpaired t-test, Nt mRNA: t28=2.788, **P=0.009, effect size=−1.257 ± 0.4508, CI95=−2.18 to −0.3335. Vglut2 mRNA: t12=0.724, P=0.483, effect size=0.1755 ± 0.2425, CI95=−0.3528 to 0.7039). The mRNA level was calculated as the ratio of average number of mRNA puncta in guide-positive neurons to DAPI-positive neurons.
g: In the PVT-BLA:Nt CRISPR-cKO experiments, a subset of control and CRISPR mice were further tested with the open field test (OFT). There were no significant differences between the control and CRISPR groups in time spent in the center and distance traveled during open field test (unpaired t-test, two-tailed, OFT: t14=0.9669, P=0.35, effect size= 16.57 ± 17.14, CI95= −20.19 to 53.33, distance traveled: t14=1.048, P=0.3123, effect size= 446.1 ± 425.6, CI95= −466.7 to 1359).
h: Diagrams of fiber placements for PVT-BLA:NT photostimulation experiments. Placements are indicated on outlines in the coronal plane, and the numbers below indicate the anterior-posterior distance from bregma.
i: Photostimulation of the PVT-BLA:NT pathway does not affect time spent freezing in response to the shock-predictive CS during tone-shock acquisition (unpaired t-test, two-tailed, t13=0.6903, P=0.5022, effect size= 0.05650 ± 0.08184, CI95= −0.1203 to 0.2333).
j, k: After mice were tested in sucrose and shock association tasks, they received a single session of the OFT, consisting of three 5-minute blocks in a laser OFF-ON-OFF manner. (j) Photostimulation of the PVT-BLA:NT pathway did not affect the amount of time mice spent in the center (paired t-test, two-tailed, mCherry group, t7=0.032, P=0.9753, effect size=0.3253 ± 10.14, CI95=−23.66 to 24.31; ChR2 group, t6=0.4472, P=0.6704, effect size=−5191 ± 11.61, CI95=−33.59 to 23.21). (k) The ChR2 group showed a significant reduction in distance traveled during the OFT when the laser was ON (Two-way ANOVA repeated measurement, stimulation OFF vs ON: F(2,26)=7.1, **P=0.0035, ChR2 vs. mCherry: F(1,13)=0.0786, P=0.786, and interaction: F(2,26)=4.769, *P=0.0172, Holm-Sidak’s multiple comparisons test, mCherry: 1st OFF vs. ON, P=0.4207, 2nd OFF vs. ON, P=0.9779, 1st OFF vs. 2nd OFF, P=0.4207; ChR2:1st OFF vs. ON, **P=0.0043, 2nd OFF vs. ON, ***P=0.0008, 1st OFF vs. 2nd ON, P=0.4335).
n and N denote number of neurons and mice in each group, respectively. Error bars and solid shaded regions around the mean indicate s.e.m.
Extended Data Figure 6: Multisite photometry recordings of NT populations in the MGN, the PVT, and the vHPC.
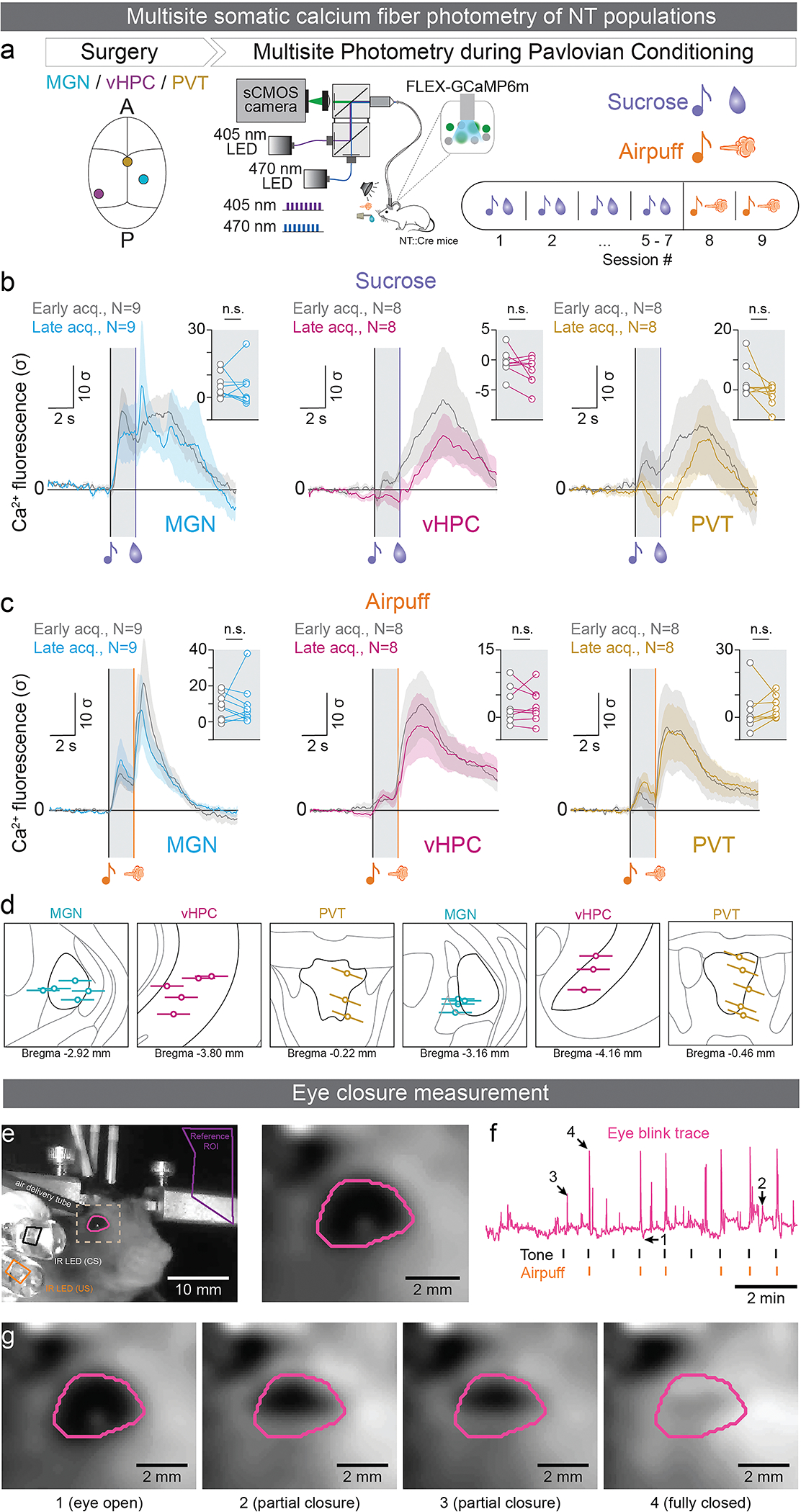
a: Experimental design for assaying neural activity from the MGN, the vHPC and the PVT:NT populations during reward and punishment associations. A virus expressing the genetically encoded calcium indicator GCaMP6m under the control of Cre was injected into the MGN, the vHPC and the PVT of NT::Cre mice. A sketch of the top-down view of the mouse skull depicting placement positions for viral injections and optic fibers is shown on the left. Upon viral incubation for at least 8 weeks, mice learned to associate a tone with sucrose reward over 5–7 sessions, and a different tone with airpuff over two sessions (right) in a head-fixed preparation while calcium activity from NT populations was being recorded using a multi-site photometry system depicted in the middle.
b-c: Population averages of z-scored Ca2+ fluorescence responses during early and late acquisition of anticipatory lick behavior (b) or anticipatory eye blink behavior (c) from the NT populations in the MGN (left), the vHPC (middle) and the PVT (right). Insets compare area under the curve during early and late acquisition from the onset of the CS to the onset of the US (light gray shaded region) during tone-sucrose association (b; paired t-tests, two-tailed, t8=0.3214, P=0.7561, effect size= −0.7986 ± 2.484, CI95= −6.528 to 4.931 for MGN, t7=1.615, P=0.1504, effect size= −1.444 ± 0.894, CI95= −3.558 to 0.6703 for vHPC, t7=1.76, P=0.1218, effect size= −4.543 ± 2.581, CI95= −10.65 to 1.56 for PVT), or tone-airpuff association (c; paired t-tests, two-tailed, t8=0.2102, P=0.8388, effect size=0.7795 ± 3.709, CI95= −7.774 to 9.333 for MGN; t7=0.1973, P=0.8492, effect size= −0.1977 ± 1.002, CI95= −2.567 to 2.172 for vHPC; and t7=0.5676, P=0.588, effect size= 1.456 ± 2.564, CI95= −4.608 to 7.519 for PVT).
d: Representation of fiber placements in all mice. Placements are indicated on outlines in the coronal plane, and the numbers below indicate distance from bregma.
e-g: Mice were head-fixed and pressurized air (20 psi) was delivered through a tube directly onto their eye for 0.1 s. The video frames were synchronized to the behavior data using two infra-red (IR) light emitting diodes (LEDs), one to indicate the CS tone, and the other to indicate the open state of the solenoid valve gating the air delivery. (e) Regions of interest (ROI) were drawn around the CS and US LEDs, and the average pixel intensities in the ROI were thresholded to identify the CS and US onset frames. An ROI encompassing the eye of the mouse was drawn (pink), and each frame was smoothed using a Gaussian filter. (f) The average intensity of all the pixels within the eye ROI was used as a metric for the size of the eye. (g) To control for background changes in intensity, the reference ROI time course was regressed from the eye ROI time course. A post-Gaussian smoothed frame of the region around the eye (dotted line) is shown on the right. Distances were calibrated using the size of the right screw that holds the head bar.
N denotes number of mice in each group. Solid shaded regions around the mean indicate s.e.m.
Extended Data Figure 7: Validations of NT fluorescent sensor.
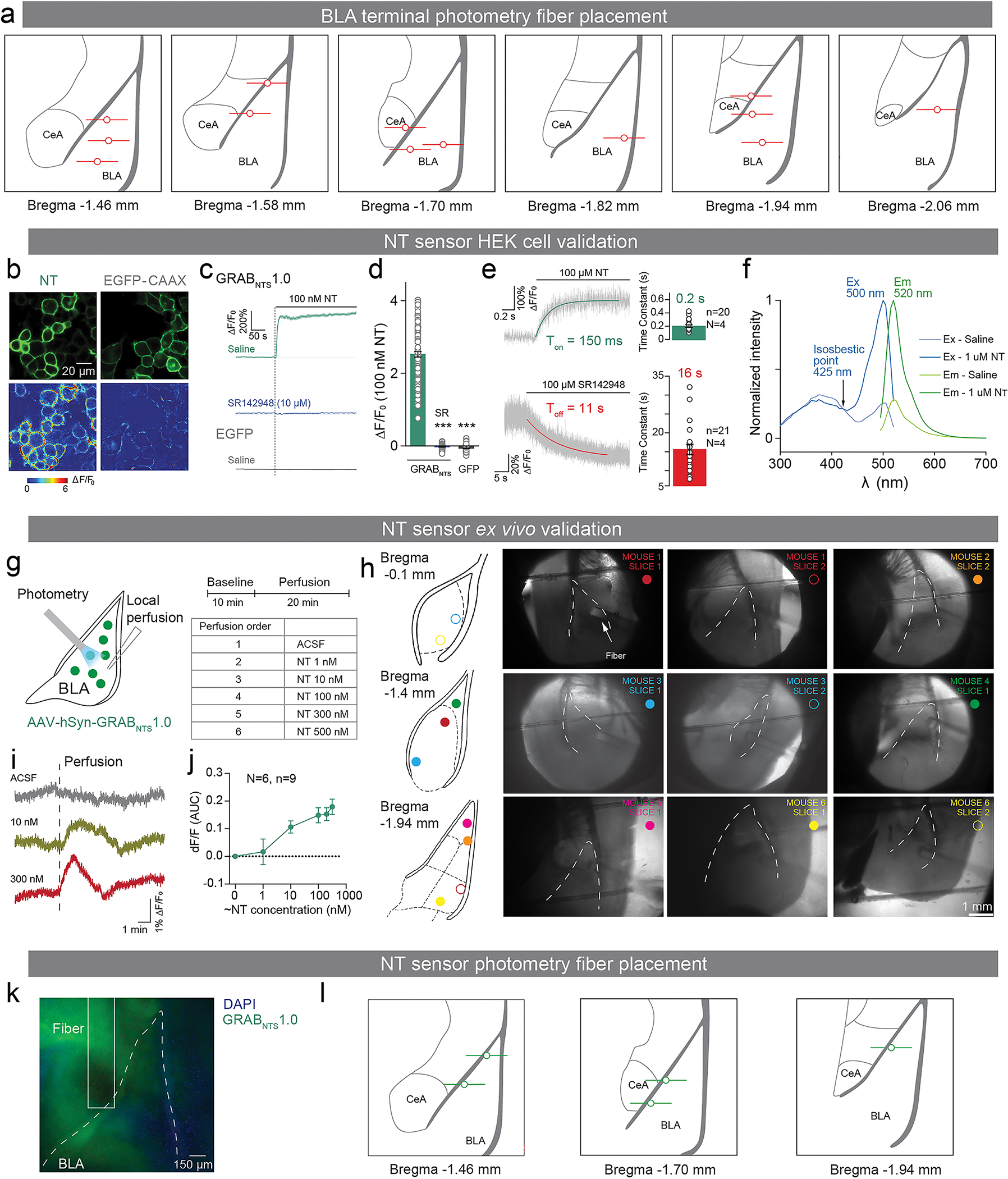
a: Illustrations of fiber placements for the PVT-BLA NT calcium photometry experiment.
b-f: NT sensor validation in HEK cells. (b) Representative images of sensor expression (top) and response to 100 nM NT (bottom) of the GRABNTS1.0 sensor and EGFP-CAAX in HEK293T cells. (Scale, 20 μm). (c, d) Time courses (left) and group summary of peak response ΔF/F0 (right) of GRABNTS1.0 sensor and EGFP-CAAX towards 100 nM NT; SR: SR142948 NTSR1 antagonist; n = 101, 102 and 115 cells from 3 cultures for Saline, SR and GFP respectively. (Unpaired t-test with Welch’s correction was performed. Saline vs. SR: t101.2=34.18, ***P<0.0001, effect size= −2.550 ± 0.07461, CI95= −2.698 to −2.402. Saline vs. GFP: t102=34.56, ***P<0.0001, effect size= −2.583 ± 0.07474, CI95= −2.731 to −2.435.) (e) Representative traces showing the response to NT (top) and subsequent addition of SR (bottom). The traces were the average of 3 different regions of interest (ROIs) on the scanning line. Each trace was fitted with a single-exponential function to determine ton and toff, and their group summary. n = 20 and 21 cells from 4 cultures for ton and toff. (f) Excitation and emission spectra for the GRABNTS1.0 sensor in the absence and the presence of NT.
g-j: NT sensor validation ex vivo on BLA slices. (g) After mice performed reward and punishment learning task, we sliced the BLA and recorded fluorescent responses to different concentrations of NT perfusions using fiber photometry by placing the fiber on the top of the BLA. (h) Illustrations and microphotographs of fiber placements and BLA slices. (i) Example traces of NT fluorescence in responses to single perfusions of ACSF, 10 nM NT, and 300 nM NT. (j) Normalized dose-response curve for GRABNTS1.0 in response to different concentration of NT perfusions (9 slices from 5 separate mice).
k-l: Representative images and illustrations of fiber placements for the NT sensor photometry experiment.
n and N denote number of neurons or slices and mice in each group, respectively. Error bars and shaded regions around the mean indicate s.e.m.
Extended Data Figure 8: supplementary analysis for in vivo electrophysiology.
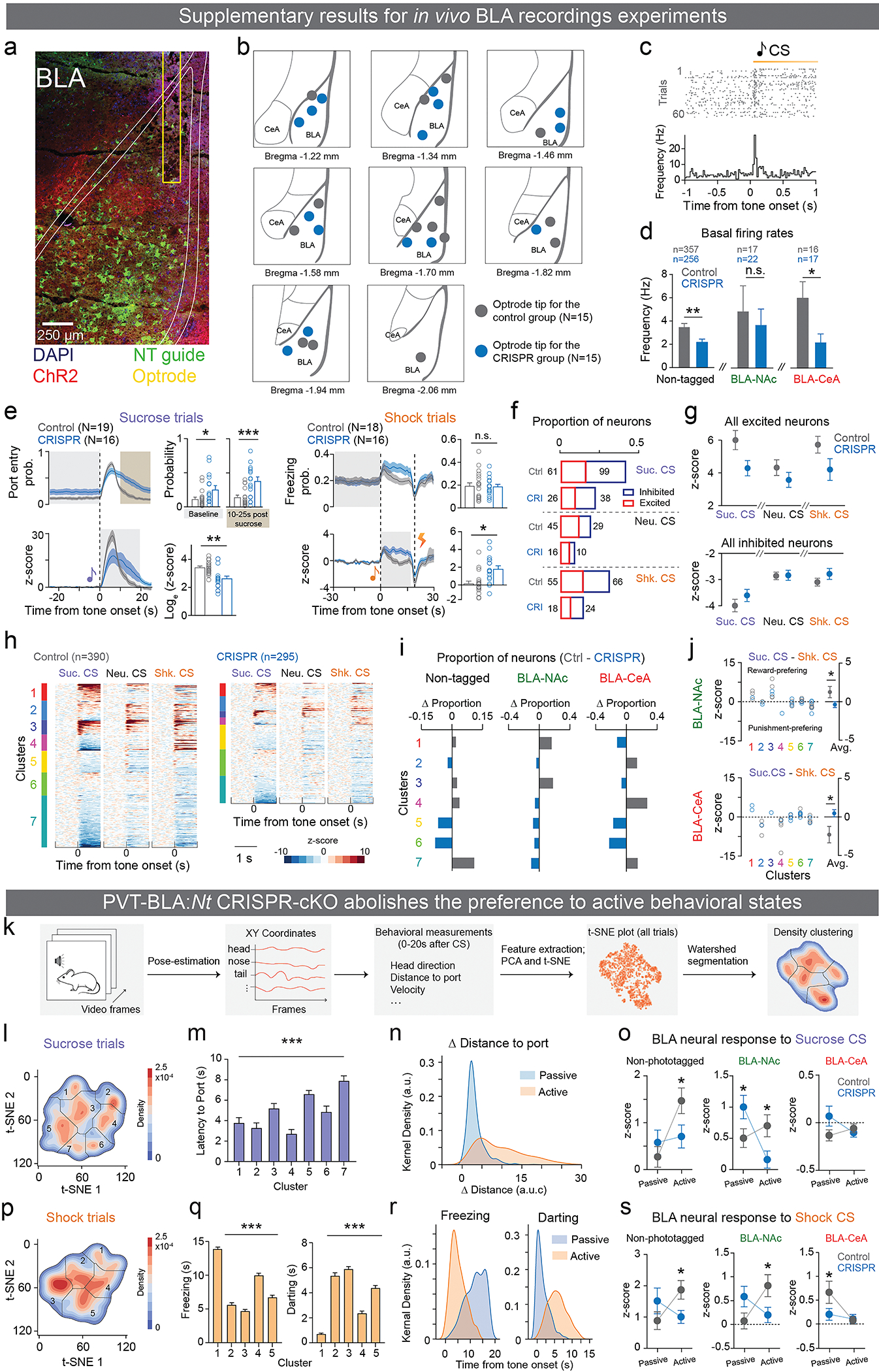
a-c: (a) A representative confocal image of a recording site in the BLA. (b) A summary of recording sites in the BLA from all mice. (c) A raster plot and histogram of a representative BLA neuron in response to sucrose-predictive cues.
d: Both non-tagged BLA neurons and the BLA-CeA neurons showed significantly lower basal firing rates (in Hz) in the CRISPR group compared to the control group (Two-tailed unpaired t-test, non-projectors: **P=0.0018; BLA-NAc: P=0.6436; BLA-CeA: *P=0.021).
e: The CRISPR group showed significantly reduced port entry probability in sucrose trials, while enhanced freezing to CS during shock trials (Two-tailed unpaired t-test, sucrose trials: raw probability baseline, *P=0.012; raw probability 10–25s post sucrose, ***P=0.0006, shock trials: raw probability, P=0.8894, z-score, *P=0.0178).
f, g: CRISPR mice showed overall reduced proportions, but not responsive amplitudes of excited and inhibited neurons to all three cues relative to the control group. Numbers in f indicate the total number of neurons that were excited and inhibited included in g.
h: Functional clustering plotted separately for neurons in the control and CRISPR groups.
i: Differences in proportions in neurons clustered in each functional cluster between the control and CRISPR groups. The proportion of BLA-NAc neurons in cluster 1 and 3 which preferentially encode positive valence was less in the CRISPR group compared to the control group. The proportion of BLA-CeA neurons in clusters 2 and 4 which preferentially encode negative valence was less in the CRISPR group compared to the control group.
j: The positive valence encoding pattern observed in BLA-NAc neurons was abolished in the CRISPR group. The negative valence encoding pattern observed in BLA-CeA neurons was abolished in the CRISPR group (Two-tailed unpaired t-test, BLA-NAc: *P=0.0483; BLA-CeA: *P=0.0199).
k-s: Unsupervised clustering of behavioral states. (k) First-order features were extracted from pose coordinates obtained from the videos. For each of these features, second-order features were extracted which were decomposed using PCA. Following PCA, t-distributed stochastic neighbor embedding (t-SNE) was performed on the top 30 principal components. A probability density function was computed over the 2-D t-SNE output and finally a watershed algorithm was used to form clusters. Each point in the t-SNE output plot represents a single trial. (l, p) Images depict the probability density function (PDF) estimate computed separately on t-SNE output for sucrose and shock trials, respectively. (m, q) The average latency to port in each sucrose trial cluster (m; One-way ANOVA, F6=20.924, ***P<0.0001), and the average time spent freezing to CS (q, left) and average darting time (q, right) in each shock trial cluster (One-way ANOVA, freezing: F4=212.917, ***P<0.0001, darting: F4=150.806, ***P<0.0001). (n, r) Kernel density of changes of difference in distance to port in sucrose trials (n), and kernel density of time spent freezing (r, left panel) and time spent darting (r, right panel) in shock trials. (o, s) Control and CRISPR group showed vastly different neural responses to both sucrose and shock CSs in passive and active trials. In responses to sucrose CS, non-phototagged BLA neurons in the CRISRP group showed significantly smaller response in active trials, while the BLA-NAc neurons showed bigger response in passive trials and smaller response in active trials compared to the control group. In response to shock CS, non-phototagged BLA neurons in the CRISRP group also showed significantly smaller response in active trials, while the BLA-NAc neurons showed smaller response in active trials and the BLA-CeA neurons showed smaller response during passive trials compared to the control group (Two-way ANOVA with Holm-Sidak’s multiple comparisons test. Sucrose CS: non-phototagged: P=0.4523 for passive, *P=0.0462 for active, BLA-NAc: *P=0.0334 for both passive and active, BLA-CeA: P=0.2969 for passive, P=0.5054 for active. Shock CS: non-phototagged: P=0.1716 for passive, *P=0.0495 for active, BLA-NAc: P=0.1145 for passive, *P=0.0354 for active, BLA-CeA: *P=0.0458 for passive, P=0.8467 for active).
See Methods for detailed statistical values. n and N denote number of neurons and mice in each group, respectively. Error bars and shaded regions around the mean indicate s.e.m.
Extended Data Figure 9: Acute and pre-incubation of NT applications produce distinct synaptic effects on BLA neurons.
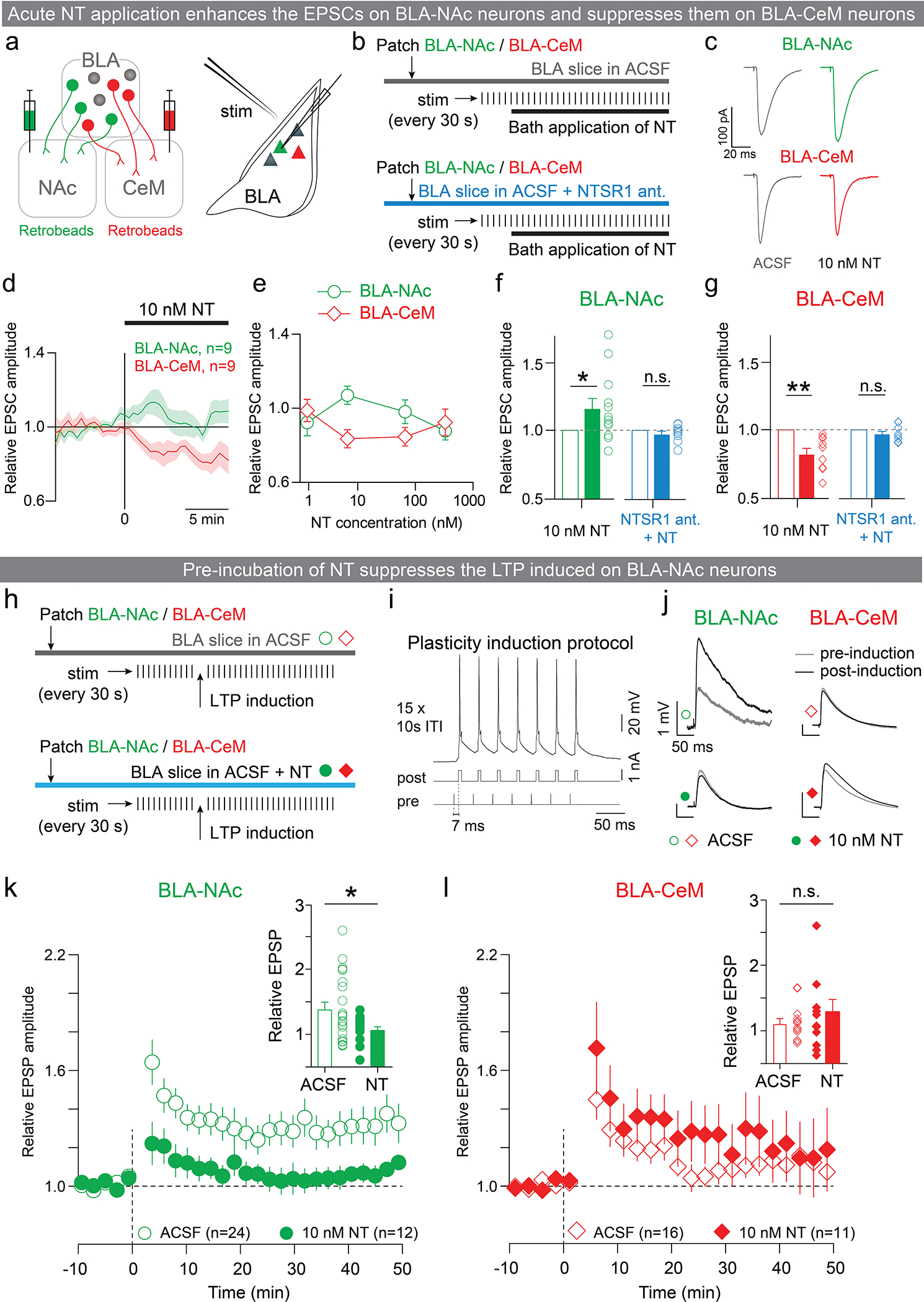
a-g: Phasic NT application enhanced EPSCs on BLA-NAc neurons and suppressed it on BLA-CeM neurons. (a) Experimental design for recording from BLA-NAc or BLA-CeM neurons ex vivo. About two weeks after injecting retrobeads into the NAc and CeM, BLA-NAc or BLA-CeM neurons were targeted in acute slices using fluorescence from the retrobeads. EPSCs evoked by internal capsule stimulation were assayed from these neurons. (b) EPSCs were assayed every 30 s during voltage clamp recordings. Once the EPSC amplitude stabilized, NT was applied to the bath. EPSC modulation of NT was assessed by the ratio of amplitudes of EPSCs after and before the bath application of NT. (c) Average EPSCs from representative BLA-NAc and BLA-CeM neurons before (gray; left column) and after (right column) the bath application of NT. (d) Average relative EPSC amplitude time course for BLA-NAc and BLA-CeM populations for a concentration of 10 nM NT in the bath. (e) Relative EPSC amplitudes of BLA-NAc and BLA-CeM neurons for different concentrations of NT (2, 10, 100 and 500 nM). (f-g) At a concentration of 10 nM, NT significantly enhanced EPSCs in BLA-NAc neurons and suppressed EPSCs in BLA-CeM neurons (Wilcoxon signed-rank test, NT: **P=0.0039, W=−45, n=9, sum of positive and negative ranks=0 and −45. NTSR1: P=0.0781, W=−26, n=8, sum of positive and negative ranks=5 and −31). Differential EPSC modulation by NT was abolished when the slices were bathed in NTSR1 antagonist (Wilcoxon signed-rank test, NT: *P=0.0425, W=52, n=12, sum of positive and negative ranks=65 and −13; NTSR1: P=0.4961, W=−13, n=9, sum of positive and negative ranks=16 and −29).
h-l: Pre-incubation of NT suppressed LTP induced on BLA-NAc neurons. (h) Schematics of LTP induction protocol. (i) Spike timing dependent plasticity induction protocol: each pulse in a 10 pulse, 30 Hz stimulus train delivered through a bipolar stimulating electrode to the internal capsule was followed after 7 ms by a 5 ms, 1 nA current pulse delivered directly to the recorded cell in the BLA through the recording electrode. This protocol was repeated 15 times with a 10 second inter-trial interval, and reliably induced spiking in recorded neurons. (j) EPSP traces from representative BLA-NAc and BLA-CeM neurons before (gray traces) and after (black traces) LTP induction. (k-l) STDP-induced LTP in the presence of NT (10 nM) 10–50 minutes after induction was reduced in BLA-NAc neurons, but unchanged in BLA-CeM neurons compared with the ACSF control (unpaired t-test, k, Unpaired t-test, t31=2.177, *P=0.0367, effect size=−0.3355 ± 0.1541, CI95=−0.649 to −0.022. l, Unpaired t-test, t25=1.193, P=0.2442, effect size=0.1795 ± 0.1505, CI95=−0.1305 to 0.4896).
n denotes number of neurons in each group. Error bars and shaded regions around the mean indicate s.e.m.
Extended Data Figure 10: Temporal and permanent blockades of NTSR1 signal in the BLA produces the different behavioral effects.
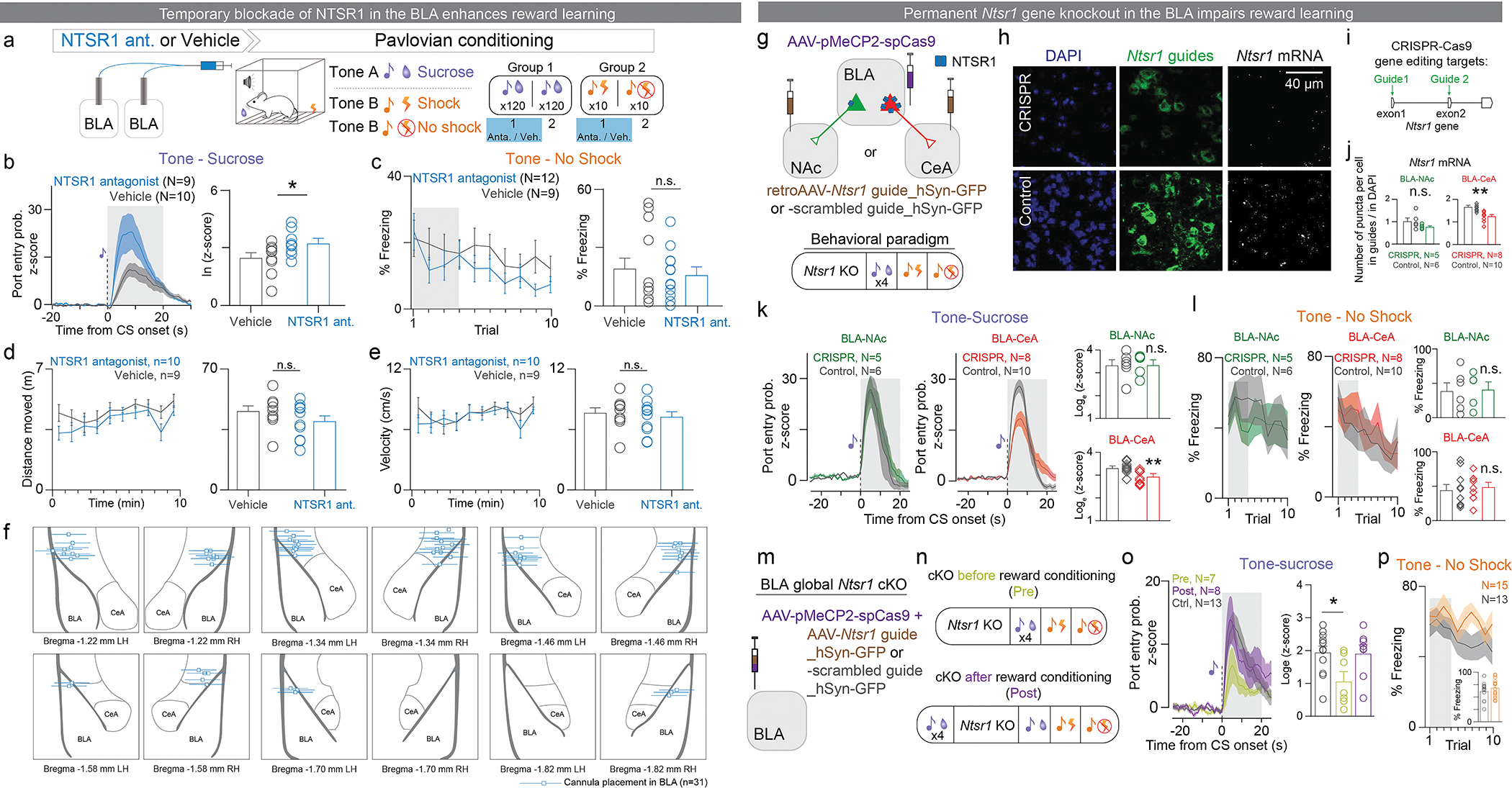
a-f: Temporal inhibition of NTSR1 in the BLA enhanced tone-sucrose association. (a) Experimental design to test the role of NTSR1 in the BLA during reward and punishment learning. Mice received a bilateral intracranial injection locally into the BLA of either NTSR1 antagonist (SR48692) or vehicle 15–20 minutes before the first session of Pavlovian sucrose or shock conditioning. Mice were reward conditioned by pairing a tone with a sucrose reward 120 times per session over two sessions, and punishment conditioned by pairing a different tone with a footshock 10 times in one session. (b) Normalized population average for the entire 120 pairings of session 2 (Shaded region indicates the time window when the peak was selected; Unpaired t-test, t16=2.128, *P=0.049, effect size = 0.7431 ± 0.3491, CI95=0.0029 to 1.483). (c) Population data quantifying percentage of freezing during the tone (Unpaired t-test, t19=1.061, P=0.3021, effect size=7.785 ± 7.339, CI95=−7.577 to 23.15). (d) Distance moved in a 10-minute OFT by mice receiving NTSR1 antagonist or vehicle bilaterally into the BLA (left). Total distance travelled in 10 minutes did not differ between mice treated with intra-BLA NTSR1 antagonist or vehicle (right; unpaired t-test, two-tailed, t17=1.212, P=0.2420, effect size= 5.569 ± 4.594, CI95= −4.124 to 15.26). (e) Velocity in the open field (left). The average rate of movement in the OFT did not differ between mice treated with intra-BLA NTSR1 antagonist vs. vehicle (right; unpaired t-test, two-tailed, t17=0.5012, P=0.6227, effect size= 0.3852 ± 0.7686, CI95= −1.237 to 2.007). (f) Placement of bilateral cannula tips above the BLA for delivery of NTSR1 antagonist in mice tested in sucrose and shock learning paradigms.
g-p: Permanent Ntsr1 gene CRISPR-cKO in the BLA impaired tone-sucrose association. (g) Schematic of viral injections. Retrograde AAV encoding Ntsr1 guide RNAs or the negative control guide were injected bilaterally in the NAc or the CeA and AAV5 or AAV9 encoding Cas9 was injected bilaterally in the BLA. (h) Representative confocal images of DAPI, Ntsr1 guide positive cells, and NRSR1 mRNA in situ hybridization. (i) Ntsr1 guide RNAs were designed to target exon1 and exon2 of the Ntsr1 gene. (j) Ntsr1 mRNA levels in BLA-CeA neurons is significantly reduced in the CRISPR group compared to the control group (Unpaired t-test, two-tailed, t16=4.503, ***P=0.0004, effect size= −0.3844 ± 0.0853, CI95= −0.5654 to −0.2035). There is no difference in Ntsr1 mRNA levels in BLA-NAc neurons, possibly due to the small amount of basal Ntsr1 signal in BLA-NAc neurons (Unpaired t-test, two-tailed, t9=1.629, P=0.1377, effect size= −0.2457 ± 0.1508, CI95= −0.5869 to 0.00945). mRNA levels were calculated as the ratio of the average number of mRNA puncta in guide-positive neurons to DAPI-positive neurons. (k) Ntsr1 CRISPR cKO in BLA-CeA neurons, but not in BLA-NAc neurons reduced port entry probability in response to sucrose-predictive cues (Unpaired t-test, two-tailed, BLA-CeA, t16=3.577, **P=0.0025, effect size= −0.4462 ± 0.1248, CI95= −0.7107 to −0.1818, BLA-NAc, t9=0.03604, P=0.972, effect size= 0.01156 ± 0.3208, CI95= −0.7141 to 0.7373). (l) Ntsr1 cKO in either BLA-CeA or BLA-NAc neurons affected tone-shock recall test (Unpaired t-test, two-tailed, BLA-NAc, t9=0.1234, P= 0.9045, effect size= 0.02031 ± 0.1646, CI95= −0.3520 to 0.3927; BLA-CeA, t16=0.4075, P=0.689, effect size= 0.04435 ± 0.1088, CI95= −0.1864 to 0.2751). (m) Schematic of viral injections of global BLA Ntsr1 CRISPR-cKO. (n) The BLA global Ntsr1 CRISPR-cKO was performed before or after the acquisition of tone-sucrose association in separate groups. (o) The BLA global Ntsr1 CRISPR-cKO prior to the acquisition impaired subsequent tone-sucrose association, while the CRISPR-cKO post the acquisition did not affect the memory recall (One-way ANOVA, F(2,25)= 4.324, *P= 0.024. Holm-Sidak’s multiple comparisons test, Control vs. Pre: *P= 0.019; Control vs. Post: P= 0.99). (q) The CRISPR-cKO globally in BLA neurons did not affect tone-shock association (Unpaired t-test, two-tailed, t26=0.871, P=0.3917, effect size= 0.05526 ± 0.06345, CI95= −0.07516 to 0.1857).
N denotes number of mice in each group. Error bars and solid shaded regions around the mean indicate s.e.m.
Supplementary Material
ACKNOWLEDGMENTS
We thank Craig Wildes for technical assistance. We thank the entire Tye Laboratory for helpful discussion. We thank Jesse Grey and Suzuko Yorozu for the RNA-seq experiment in our 2015 Nature paper that led to the discovery of NT modulation in the BLA; Amy Guo for preparing NTSR1 plasmids; Gillian Matthews for discussions on the analysis of RNAscope images; Sebastien Hausmann for DeepLabCut troubleshooting; Calvin Lee, Jessica Du, Kiana Miyamoto, Shuqi Chen, Margaux Silvestre, Meghan Cum, Faith Aloboudi, and Natalie Giles for the helps with data collection; Shan Shao for the helps with MATLAB scripts; Jolene Diedrich and Antonio Pinto in the mass spectrometry core at Salk for their efforts to quantify NT in the microdialysis samples; Jacque Ip, Vincent Pham, and the Sur Lab at MIT for generously sharing their cryostat.
K.M.T. is the Wylie Vale chair at Salk Institute for Biological Studies, HHMI Investigator, a New York Stem Cell Foundation - Robertson Investigator and McKnight Scholar and this work was supported by funding from the JPB Foundation, the PIIF, PNDRF, JFDP, Alfred P Sloan Foundation, New York Stem Cell Foundation, Klingenstein Foundation, McKnight Foundation, Clayton Foundation, Kavli Foundation, Dolby Family Fund, R01-MH102441 (NIMH), R37-MH102441 (NIMH), the NIH Director’s New Innovator Award DP2-DK102256 (NIDDK), Pioneer Award DP1-AT009925 (NCCIH). H.L. was supported by the K99/R00 NIH Pathway to Independence Award (K99 DA055111). P.N. was supported by Singleton, Leventhal and Whitaker fellowships, and A. Beyeler was supported by a fellowship from the Swiss National Science Foundation and NARSAD. N.H-I. was supported by Grant-in-Aid for Scientific Research on Innovative Areas [15K21744, 17H06043] from MEXT and the Uehara Memorial Foundation. R.W. was supported by a NARSAD Young Investigator Award (Brain and Behavior Research Foundation). J.M.O. was supported by a Brain Initiative F32 from NIMH (F32 MH115446–01). V.d.l.F. was supported by a Fulbright fellowship.
Footnotes
COMPETING INTEREST STATEMENT
No competing Interests.
REFERENCES
- 1.Namburi P et al. A circuit mechanism for differentiating positive and negative associations. Nature 520, 675–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beyeler A et al. Divergent Routing of Positive and Negative Information from the Amygdala during Memory Retrieval. Neuron (2016) doi: 10.1016/j.neuron.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyeler A et al. Organization of Valence-Encoding and Projection-Defined Neurons in the Basolateral Amygdala. Cell Rep. 22, 905–918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen C-J et al. Cannabinoid CB1 receptors in the amygdalar cholecystokinin glutamatergic afferents to nucleus accumbens modulate depressive-like behavior. Nat. Med. 25, 337–349 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Kim J, Pignatelli M, Xu S, Itohara S & Tonegawa S Antagonistic negative and positive neurons of the basolateral amygdala. Nat. Neurosci. 19, 1636–1646 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X et al. Genetically identified amygdala-striatal circuits for valence-specific behaviors. Nat. Neurosci. 24, 1586–1600 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corder G et al. An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clugnet MC & LeDoux JE Synaptic plasticity in fear conditioning circuits: induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate body. J. Neurosci. Off. J. Soc. Neurosci. 10, 2818–2824 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang YY & Kandel ER Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21, 169–178 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Dan Y & Poo M Spike Timing-Dependent Plasticity of Neural Circuits. Neuron 44, 23–30 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Bittner KC, Milstein AD, Grienberger C, Romani S & Magee JC Behavioral time scale synaptic plasticity underlies CA1 place fields. Science 357, 1033–1036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuster JM & Uyeda AA Reactivity of limbic neurons of the monkey to appetitive and aversive signals. Electroencephalogr. Clin. Neurophysiol. 30, 281–293 (1971). [DOI] [PubMed] [Google Scholar]
- 13.Paton JJ, Belova MA, Morrison SE & Salzman CD The primate amygdala represents the positive and negative value of visual stimuli during learning. Nature 439, 865–870 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Britt JP et al. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76, 790–803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gd S et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature vol. 475 https://pubmed.ncbi.nlm.nih.gov/21716290/ (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gore F et al. Neural Representations of Unconditioned Stimuli in Basolateral Amygdala Mediate Innate and Learned Responses. Cell 162, 134–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Redondo RL et al. Bidirectional switch of the valence associated with a hippocampal contextual memory engram. Nature 513, 426–430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clem RL & Huganir RL Calcium-Permeable AMPA Receptor Dynamics Mediate Fear Memory Erasure. Science 330, 1108–1112 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janak PH & Tye KM From circuits to behaviour in the amygdala. Nature 517, 284–292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKernan MG & Shinnick-Gallagher P Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Nabavi S et al. Engineering a memory with LTD and LTP. Nature 511, 348–352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogan MT, Stäubli UV & LeDoux JE Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390, 604–607 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Rumpel S, LeDoux J, Zador A & Malinow R Postsynaptic receptor trafficking underlying a form of associative learning. Science 308, 83–88 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Tye KM, Stuber GD, De Ridder B, Bonci A & Janak PH Rapid strengthening of thalamo-amygdala synapses mediates cue–reward learning. Nature 453, 1253–1257 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brzosko Z, Mierau SB & Paulsen O Neuromodulation of Spike-Timing-Dependent Plasticity: Past, Present, and Future. Neuron 103, 563–581 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Cassenaer S & Laurent G Conditional modulation of spike-timing-dependent plasticity for olfactory learning. Nature 482, 47–52 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Lutas A et al. State-specific gating of salient cues by midbrain dopaminergic input to basal amygdala. Nat. Neurosci. 22, 1820–1833 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bissière S, Humeau Y & Luthi A Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat. Neurosci. 6, 587–592 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Tye KM et al. Methylphenidate facilitates learning-induced amygdala plasticity. Nat. Neurosci. 13, 475–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steele FF, Whitehouse SC, Aday JS & Prus AJ Neurotensin NTS1 and NTS2 receptor agonists produce anxiolytic-like effects in the 22-kHz ultrasonic vocalization model in rats. Brain Res. 1658, 31–35 (2017). [DOI] [PubMed] [Google Scholar]
- 31.László K, Tóth K, Kertes E, Péczely L & Lénárd L The role of neurotensin in positive reinforcement in the rat central nucleus of amygdala. Behav. Brain Res. 208, 430–435 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Amano T et al. Heightened amygdala long-term potentiation in neurotensin receptor type-1 knockout mice. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol 33, 3135–3145 (2008). [DOI] [PubMed] [Google Scholar]
- 33.McCullough KM et al. Molecular characterization of Thy1 expressing fear-inhibiting neurons within the basolateral amygdala. Nat. Commun. 7, 13149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petreanu L, Mao T, Sternson SM & Svoboda K The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deltcheva E et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livneh Y et al. Homeostatic circuits selectively gate food cue responses in insular cortex. Nature 546, 611–616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otis JM et al. Paraventricular Thalamus Projection Neurons Integrate Cortical and Hypothalamic Signals for Cue-Reward Processing. Neuron 103, 423–431.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Penzo MA et al. The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Y, Wienecke CFR, Nachtrab G & Chen X A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 530, 219–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Do-Monte FH, Minier-Toribio A, Quiñones-Laracuente K, Medina-Colón EM & Quirk GJ Thalamic Regulation of Sucrose Seeking during Unexpected Reward Omission. Neuron 94, 388–400.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathis A et al. DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat. Neurosci. 21, 1281–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Berman GJ, Choi DM, Bialek W & Shaevitz JW Mapping the stereotyped behaviour of freely moving fruit flies. J. R. Soc. Interface 11, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weisskopf MG, Bauer EP & LeDoux JE L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J. Neurosci. Off. J. Soc. Neurosci. 19, 10512–10519 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flagel SB et al. A food predictive cue must be attributed with incentive salience for it to induce c-fos mRNA expression in cortico-striatal-thalamic brain regions. Neuroscience 196, 80–96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma J et al. Divergent projections of the paraventricular nucleus of the thalamus mediate the selection of passive and active defensive behaviors. Nat. Neurosci. (2021) doi: 10.1038/s41593-021-00912-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tye KM Neural Circuit Motifs in Valence Processing. Neuron 100, 436–452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quiroga RQ, Nadasdy Z & Ben-Shaul Y Unsupervised spike detection and sorting with wavelets and superparamagnetic clustering. Neural Comput. 16, 1661–1687 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Choudhury SR et al. In Vivo Selection Yields AAV-B1 Capsid for Central Nervous System and Muscle Gene Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 24, 1247–1257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enzymatic assembly of DNA molecules up to several hundred kilobases | Nature Methods. https://www.nature.com/articles/nmeth.1318. [DOI] [PubMed]
- 54.Kim CK et al. Simultaneous fast measurement of circuit dynamics at multiple sites across the mammalian brain. Nat. Methods 13, 325–328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cunningham JP & Yu BM Dimensionality reduction for large-scale neural recordings. Nat. Neurosci. 17, 1500–1509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linsenbardt DN, Timme NM & Lapish CC Encoding of the Intent to Drink Alcohol by the Prefrontal Cortex Is Blunted in Rats with a Family History of Excessive Drinking. eNeuro 6, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paxinos G & Franklin KBJ The Mouse Brain in Stereotaxic Coordinates. (Gulf Professional Publishing, 2004). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw and/or processed data shown in this manuscript are available upon request. Sequences of custom AAV9 viruses and guide RNAs used for CRISPR-based CRISPR-cKO experiments are available upon request. All mouse illustrations included in the main figures were created with BioRender.com.