

This case series reports data for 3 patients from 2 unrelated families with congenital stationary night blindness to describe an unreported phenotype and the associated gene defect.
Key Points
Question
What are the detailed clinical picture and underlying gene defect in patients with a peculiar form of congenital stationary night blindness (CSNB)?
Findings
In this case series, we report on 3 patients from 2 families with CSNB, anterior segment abnormalities (ciliary body hypoplasia and lens luxation), and all-bipolar cell dysfunction. We identified 2 genetic variants in VSX2, a major gene important for ocular development and bipolar cell fate, in those families with CSNB.
Meaning
This genetic defect of an inherited ocular disease has both functional and anatomical eye defects.
Abstract
Importance
Congenital stationary night blindness (CSNB) is an inherited stationary retinal disorder that is clinically and genetically heterogeneous. To date, the genetic association between some cases with CSNB and an unusual complex clinical picture is unclear.
Objective
To describe an unreported CSNB phenotype and the associated gene defect in 3 patients from 2 unrelated families.
Design, Setting, and Participants
This retrospective case series was conducted in 2021 and 2022 at a national referral center for rare ocular diseases. Data for 3 patients from a cohort of 140 genetically unsolved CSNB cases were analyzed clinically and genetically.
Exposures
Complete ocular examination including full-field electroretinography and multimodal fundus imaging (spectral-domain optical coherence tomography, color, infrared reflectance, and short-wavelength autofluorescence photographs) were performed. The gene defect was identified by exome sequencing and confirmed by Sanger sequencing and co-segregation analysis in 1 family. Screening was performed for genetically unsolved CSNB cases for VSX2 variants by direct Sanger sequencing.
Main Outcomes and Measures
Ocular and molecular biology findings.
Results
The series included 3 patients whose clinical investigations occurred at ages in the early 30s, younger than 12 years, and in the mid 40s. They had nystagmus, low stable visual acuity, and myopia from birth and experienced night blindness. Two older patients had bilateral lens luxation and underwent lens extraction. Full-field electroretinography revealed an electronegative Schubert-Bornschein appearance, combining characteristics of incomplete and complete CSNB, affecting the function of rod and cone ON- and OFF-bipolar cells. Exome sequencing and co-segregation analysis in a consanguineous family with 2 affected members identified a homozygous variant in VSX2. Subsequently, screening of the CSNB cohort identified another unrelated patient harboring a distinct VSX2 variant.
Conclusions and Relevance
This case series revealed a peculiar pan-bipolar cell retinopathy with lens luxation associated with variants in VSX2. Clinicians should be aware of this association and VSX2 added to CSNB diagnostic gene panels.
Introduction
Congenital stationary night blindness (CSNB) with largely normal fundus appearance is a vast group of genetically and clinically heterogeneous congenital retinal disorders. The most common form manifests with infantile nystagmus, reduced visual acuity, variable degree of myopia, poor visual behavior in dim lighting, and/or photophobia.1 The diagnosis is ascertained by full-field electroretinography (ffERG) displaying characteristic waveform changes.
Under dark adaptation (DA), the stimulation with a dim flash (DA 0.01 ERG, dark-adapted 0.01 cd/m2) generates a positive-going b-wave reflecting the rod ON-bipolar cell depolarization.2 In the same dark-adapted conditions, the stimulations with brighter flashes (DA 3.0 ERG, DA 10.0 ERG) produce a negative a-wave, reflecting the hyperpolarization of photoreceptors, mainly dominated by rods in this adaptation state,3,4 followed by a positive b-wave, for which the ascending limb is dominated by ON-bipolar cell depolarization.5 Under light-adapted conditions, the stimulations with bright flashes (LA 3.0 ERG, light adapted 3.0 cd/m2) lead to a negative a-wave generated by the hyperpolarization of cones and then of OFF-bipolar cells. The second deflection is a positive b-wave, for which the ascending limb is dominated by cone ON-bipolar cell depolarization and descending limb by OFF-bipolar cell hyperpolarization.6,7 Long-duration stimulations allow the separation of responses generated by cone ON- and OFF-bipolar pathway.6,8
Most of the patients with CSNB have a distinct Schubert-Bornschein electronegative waveform (b/a amplitude ratio <1) under dark-adapted conditions,9 in relation to a signaling defect from photoreceptors to adjacent bipolar cells. Two subtypes of Schubert-Bornschein CSNB can be distinguished on the basis of additional ffERG features: incomplete (icCSNB) and complete (cCSNB).10 In icCSNB, there is a preserved but reduced and delayed b-wave to the DA 0.01 ERG.10 Light-adapted responses are markedly reduced and delayed, reflecting a cone-to-cone ON- and OFF-bipolar cell transmission defect, which can be documented using long-duration stimulations.10,11 Patients have variable degrees of night blindness, strabismus, and nystagmus; a wide range of refractive errors12; and a normal fundus, apart from myopic changes. Photophobia may be the main concern for some patients who also experience difficulties in dimly lit environments. Pathogenic variants in CACNA1F (OMIM 300110)13,14 and CABP4 (OMIM 608965)15 lead to icCSNB. Variants were also identified in CACNA2D4 (OMIM 608171)16 in patients initially diagnosed with icCSNB but later associated with a cone dystrophy.17 Proteins encoded by these genes are localized at the presynaptic membrane of both rods and cones. They play a role in glutamate release into the synaptic cleft. Thus, icCSNB represents a photoreceptor to ON- and OFF-bipolar cell transmission defect.1 Light sensitivity present in patients with icCSNB may be explained by the worse dysfunction in cone bipolar circuitry, unlike in other forms of CSNB.12 In cCSNB, the b-wave is typically absent on the DA 0.01 ERG. The a-wave has typically a square shape with a sharply arising b-wave and a reduced b/a amplitude ratio at the LA 3.0 ERG,10 with absence of ON-bipolar responses but preserved OFF responses to long-duration stimulations.18 Patients with cCSNB present consistently with high myopia, nystagmus, night blindness, and low visual acuity.12 Variants in NYX (OMIM 300278),13,19 GRM6 (OMIM 604096),20,21 TRPM1 (OMIM 603576),22,23,24 GPR179 (OMIM 614515),25 and LRIT3 (OMIM 615004)26 lead to cCSNB. Genes listed above code for proteins localized mainly in the dendritic tips of the ON-bipolar cells. They are important for glutamate-induced signaling. Thus, cCSNB represents a photoreceptor to ON-bipolar cell transmission defect.1
Other rare forms of CSNB with a different ERG and clinical phenotype have been associated with SAG (OMIM 181031), GNAT1 (OMIM 139330), RHO (OMIM 180380), PDE6B (OMIM 180072), GRK1 (OMIM 180381), and SLC24A1 (OMIM 603617).1 The goal of this study was to report an unusual CSNB phenotype and associated gene defects in 3 patients from 2 unrelated Turkish families.
Methods
Research procedures adhered to the tenets of the Declaration of Helsinki and were approved by the local ethics committee (CPP, Ile de France V, project 06693, N°EUDRACT 2006-A00347-44, December 11, 2006, and CPP, Ile de France II). Prior to testing, written informed consent was obtained from 2 adult participants and the parents of a participant who was younger than 18 years. No compensation or incentive was offered to patients to participate in the study. Relevant reporting guidelines were followed.
Clinical Studies
The cases were clinically investigated at the national reference center for rare ocular diseases REFERET of the Centre Hospitalier National d’Ophtalmologie (CHNO) des Quinze-Vingts as previously described.27 Patient 2 was also assessed at the national reference center for rare ocular diseases OPHTARA of Necker-Enfants Malades Hospital as an infant (<6 months old).
Genetic Analysis
Blood samples from cases and all available family members were collected for genetic research, and genomic DNA was extracted as previously reported.28 These DNA samples were collected within the NeuroSensCol DNA bank for research in neuroscience.
Direct Sanger sequencing was performed for patient 1 for all exons and exon-intron boundaries of the genes CACNA1F, NYX, TRPM1, LRIT3, GRM6, GPR179, CABP4, and CACNA2D4 and for some other genes associated with inner retinal dysfunctions: RIMS2, GNB3, and GNB5. (The amplification and sequencing conditions can be obtained on request.)
Patient 2 was analyzed on a next-generation sequencing (NGS) molecular diagnostic panel, including CSNB- and pediatric retinal degeneration–associated genes (eTable 1 in the Supplement). Subsequently, exome sequencing was performed in the patient and both parents. Genomic DNA libraries were generated from DNA sheared with a Covaris S2 Ultrasonicator via SureSelectXT Library Prep Kit (Agilent). Regions of interest were captured with the SureSelect All Exon V5 kit (Agilent) and sequenced on an Illumina HiSeq2500 HT system (Illumina). Data analysis was performed with a homemade pipeline (POLYWEB)29 created by the Imagine Institute Bioinformatics core facilities of Paris University. Considering that pathogenic variants are uncommon, we searched for recessive variants absent in the dbSNP, 1000 Genomes, EVS, ExAC, gnomAD, and all in-house databases and for variants with minor allele frequency up to 0.01. We searched in priority for homozygous consensus splice-site changes, nonsense variants, insertions, and deletions in coding regions. After this stringent filtering, variants in 4 candidate genes (ITIH2, PRDM10, VSX2, and BCR) were identified. All variants were classified using Human Genome Variation Society nomenclature,30 and their pathogenicity was assessed in accordance with the American College of Medical Genetics and Genomics (ACMG) recommendations.31 Validation of putative pathogenic variants and a co-segregation study were performed by direct Sanger sequencing of exons of interest in all available family members.
In a second step, a cohort of 140 unsolved CSNB cases was screened for VSX2 variants. One additional patient (patient 3) was identified. He previously had negative results on an NGS panel32 including all known-to-date CSNB-associated genes.
Results
Patients 1, 2, and 3 were clinically investigated at ages in the early 30s, younger than 12 years, and in the mid 40s, respectively. All 3 patients had infantile nystagmus, low visual acuity, myopia, and night blindness from birth, but the adult patients did not report changes in their symptoms or vision since childhood. Clinical data are summarized in the Table.
Table. Clinical Characteristics of Patients.
| Patient No., age at first assessment | BCVA, Snellen | Refraction | Axial length, mm | Color vision (15 hue Lanthony) | Visual field | ffERG | Anterior segment | Fundus | SW-FAF | OCT |
|---|---|---|---|---|---|---|---|---|---|---|
| Family with c.595C>T, p.(Arg199Cys) | ||||||||||
| Patient 1, early 30s | 20/200 OD, 20/200 OS; 20/63 OD, 20/63 OS after cataract surgery | −15.0 OD, −15.0 OS | 28.76 OD, 28.23 OS | Normal | GP: superior flattening at all target sizes; SP: diffuse reduction of retinal sensitivity; MD: −6.6 dB OD, −10.2 dB OS, FT: 25 dB OU | DA 0.01: undetectable; DA 3.0, DA 10.0: electronegative Schubert-Bornschein configuration; LA 3.0, LA 3.0 flicker: severely | Superior lens subluxation OU; cataract OU; phaco- and iridodonesis | Oval pale tilted discs, peripapillary chorioretinal atrophy, vascular narrowing, increased visibility of choroidal vasculature; white glistening crystals, atrophic patches, and retinal tears in peripheral retina | Normal | NA |
| Patient 2, <12 y | 20/200 OU | −10.50 (−1.50) 175° OD; −8.75 (−1.50) 155° OS, cycloplegic | 24.77 OD, 24.43 OS | NA | NA | DA 0.01: undetectable; DA 3.0, DA 10.0: electronegative Schubert-Bornschein configuration; LA 3.0, LA 3.0 flicker: reduced and delayed | Normal | Oval pale tilted discs, peripapillary chorioretinal atrophy, vascular narrowing, increased visibility of choroidal vasculature; white without pressure in peripheral retina | Normal | Preserved outer retina; thinned inner retinal layers |
| Family with c.698C>T, p.(Pro233Leu) | ||||||||||
| Patient 3, mid 40s | 20/50 OD, 20/100 OS | +2.0 OD, +3.50 (−0.50) 90° OS after cataract surgery; −2.75 (−3.0) 0° OD, −3.0 (−3.5) 25° OS before surgery | 22.3 OD, 21.94 OS | NA | SP: diffuse reduction of retinal sensitivity | DA 0.01: undetectable; DA 3.0, DA 10.0: electronegative Schubert-Bornschein configuration; LA 3.0, LA 3.0 flicker: reduced | Pseudophakic OU; UBM: hypoplastic ciliary body; surgery for lens subluxation and cataracts in both eyes in early 40s | Normal discs, mild vascular narrowing, vessels crossing fovea, increased visibility of choroidal vasculature, triangular zone of chorioretinal atrophy pointing macula inferiorly | Triangular hypoautofluorescent lesion with hyperautofluorescent edges | Grade 3 foveal hypoplasia; disappearance of outer reflective layers corresponding to atrophic fundus lesion |
Abbreviations: BCVA, best-corrected visual acuity; DA, dark adapted; ffERG, full-field electroretinography; FT, foveal threshold; GP, Goldman perimetry; LA, light adapted; MD, mean deficit; NA, not available; OCT, optical coherence tomography; SP, automated static perimetry; SW-FAF, short-wavelength fundus autofluorescence; UBM, ultrasound biomicroscopy.
Their ffERG recordings (Figure 1) under dark-adapted conditions showed no detectable responses to a dim 0.01 cd/m2 flash (DA 0.01 ERG), a normal a-wave, but a severely reduced b-wave, leading to an electronegative waveform to bright flashes (DA 3.0 and DA 10.0 ERG). These alterations resembled ON-bipolar dysfunction in cCSNB. However, light-adapted responses to a 3 cd/m2 single flash and to a 30 Hz, 3 cd/m2 flicker (LA 3.0 ERG and LA 3.0 flicker) were reduced and delayed, unlike patients with cCSNB but resembling functional alterations in icCSNB.1
Figure 1. Full-Field Electroretinography According to the Standard by the International Society for Clinical Electrophysiology of Vision.
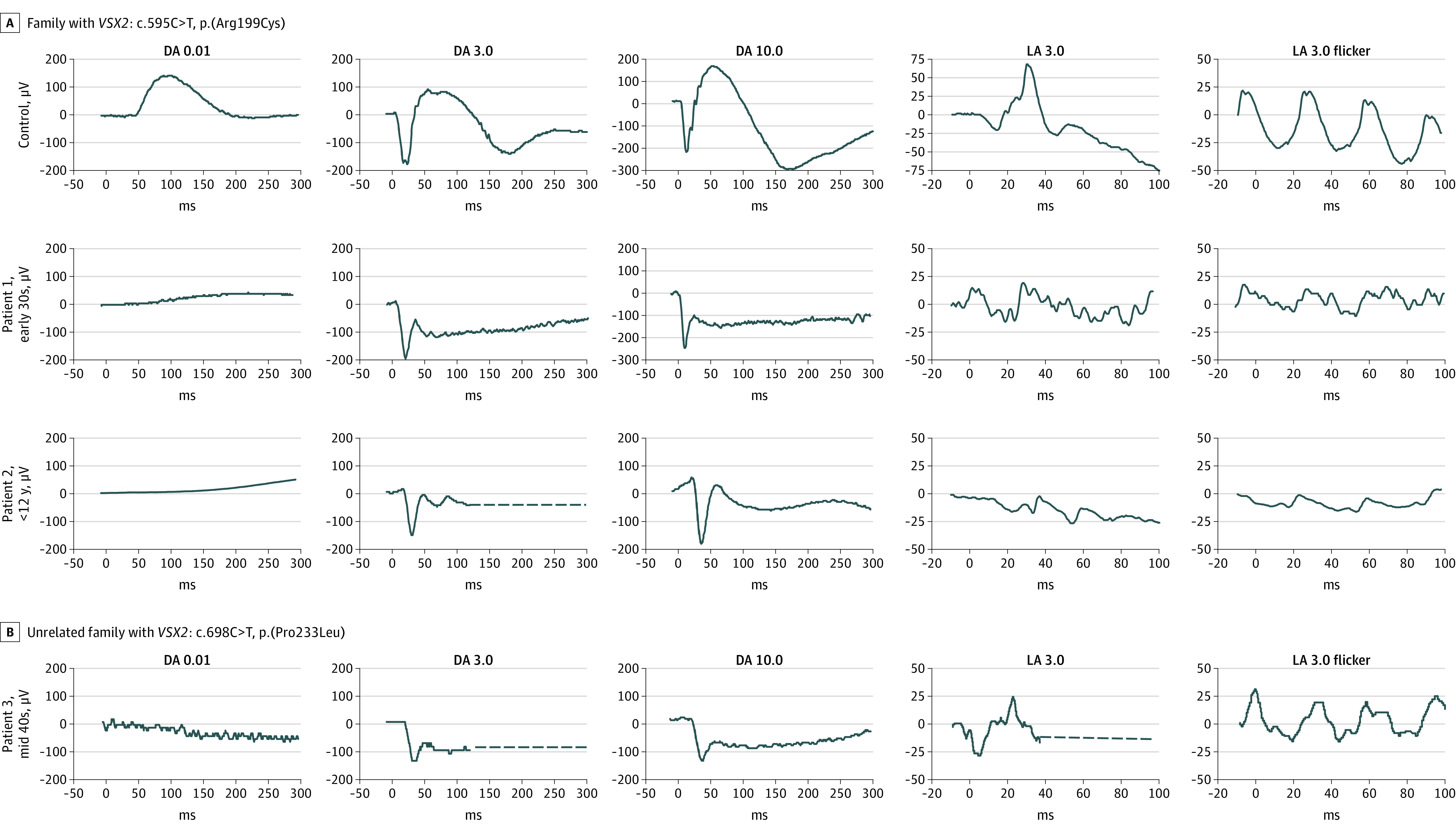
Undetectable responses to a dim flash under dark-adapted condition (DA 0.01). Electronegative waveform (b/a amplitude ratio <1) to bright flashes under dark-adapted conditions (DA 3.0 and DA 10.0). Severely reduced light-adapted responses (LA 3.0 and LA 3.0 flicker).
The female patient 1 was referred to our clinic in her early 30s. Best-corrected visual acuity was 20/200 OU. Color vision was normal. Goldmann visual field was flattened in the superior part on all target sizes. Static visual field found a diffuse reduction of retinal sensitivity and reduction of foveal threshold. Both lenses were subluxated superiorly and showed cataract. Ophthalmoscopic evaluation revealed typical myopic changes, including oval tilted optic discs surrounded with peripapillary chorioretinal atrophy, thin retinas with increased visibility of choroidal vasculature, and narrowed retinal vessels (Figure 2A). Numerous glistening white crystals and atrophic chorioretinal patches were visible in the peripheral retina (Figure 2A). Infrared reflectance (eFigure 1B in the Supplement) and short-wavelength fundus autofluorescence (eFigure 1C in the Supplement) were unremarkable. Spectral-domain optical coherence tomography (SD-OCT) was not performed at the first visit. The patient underwent intracapsular cataract extraction without intraocular lens implantation. Best-corrected visual acuity improved to 20/63 OU.
Figure 2. Retinal Findings in Patients 1 and 2 From the Same Family.

A, Myopic fundus abnormalities with oval tilted optic discs, temporal pallor and peripapillary chorioretinal atrophy, narrowed retinal vessels, and thinned retina with increased choroidal visibility. Peripheral snowflake degeneration and patches of chorioretinal atrophy. B, Myopic fundus abnormalities with oval titled optic disc, temporal pallor, narrowed retinal vessels, retinal thinning. C, Spectral-domain optical coherence tomography horizontal scans passing through fovea. Preserved outer retina with inner retinal thinning.
The male patient 2, a nephew of patient 1, was referred to the clinic as a young child. Convergent squint and infantile nystagmus syndrome with an initial intermittent and transient upbeat component were present during infancy. Slitlamp examination results were unremarkable. Fundus examination revealed similar myopic changes as reported in the aunt (Figure 2B). In the peripheral retina, there was 360° white without pressure. Preserved outer retina and thinned inner retinal layers were shown on SD-OCT (Figure 2C). Multimodal retinal imaging was difficult due to nystagmus, with unremarkable infrared reflectance and short-wavelength fundus autofluorescence (eFigure 1E in the Supplement).
The male patient 3, from an unrelated family, was referred to the clinic when in his middle 40s. He had infantile nystagmus and poor vision since birth. In his early 40s, he underwent intracapsular lens extraction for bilateral subluxated cataract. Ultrasound biomicroscopy revealed hypoplastic ciliary body and posterior iris bowing (eFigure 2 in the Supplement). Ophthalmoscopic examination revealed triangular atrophic chorioretinal lesions inferior to the macula and white without pressure in the peripheral retina (Figure 3A). Atrophic chorioretinal lesions were hypoautofluorescent with hyperautofluorescent edges on short-wavelength fundus autofluorescence (Figure 3B) and were bright on infrared reflectance (Figure 3C) with outer retinal disruption on SD-OCT. Grade 3 foveal hypoplasia33 as well as a thin epimacular membrane could be seen on SD-OCT (Figure 3D).
Figure 3. Retinal Findings in Patient 3 From an Unrelated Family.

A, Normal optic disc, narrowed retinal vessels, and a triangular zone of chorioretinal atrophy inferior to the macula. B, Zone of extinguished autofluorescence with hyperautofluorescent edges inferior to the macula. C, Increased reflectance inferior to the macula. Vessels crossing fovea. Arrow indicates the direction of the spectral-domain optical coherence tomography (SD-OCT) scan. D, Grade 3 foveal hypoplasia. Epiretinal membrane. Loss of outer reflective layers in scan passing through the zone of chorioretinal atrophy.
Genetic Assessment
Patients 1 and 2 were members of a family with North Turkish ancestry and were born from consanguineous unions (Figure 4A). Patient 3 was also of Turkish ancestry (Figure 4B). He did not report parental consanguinity.
Figure 4. Pedigrees for the 2 Unrelated Families and the Schematic VSX2 Gene and Protein Structure.
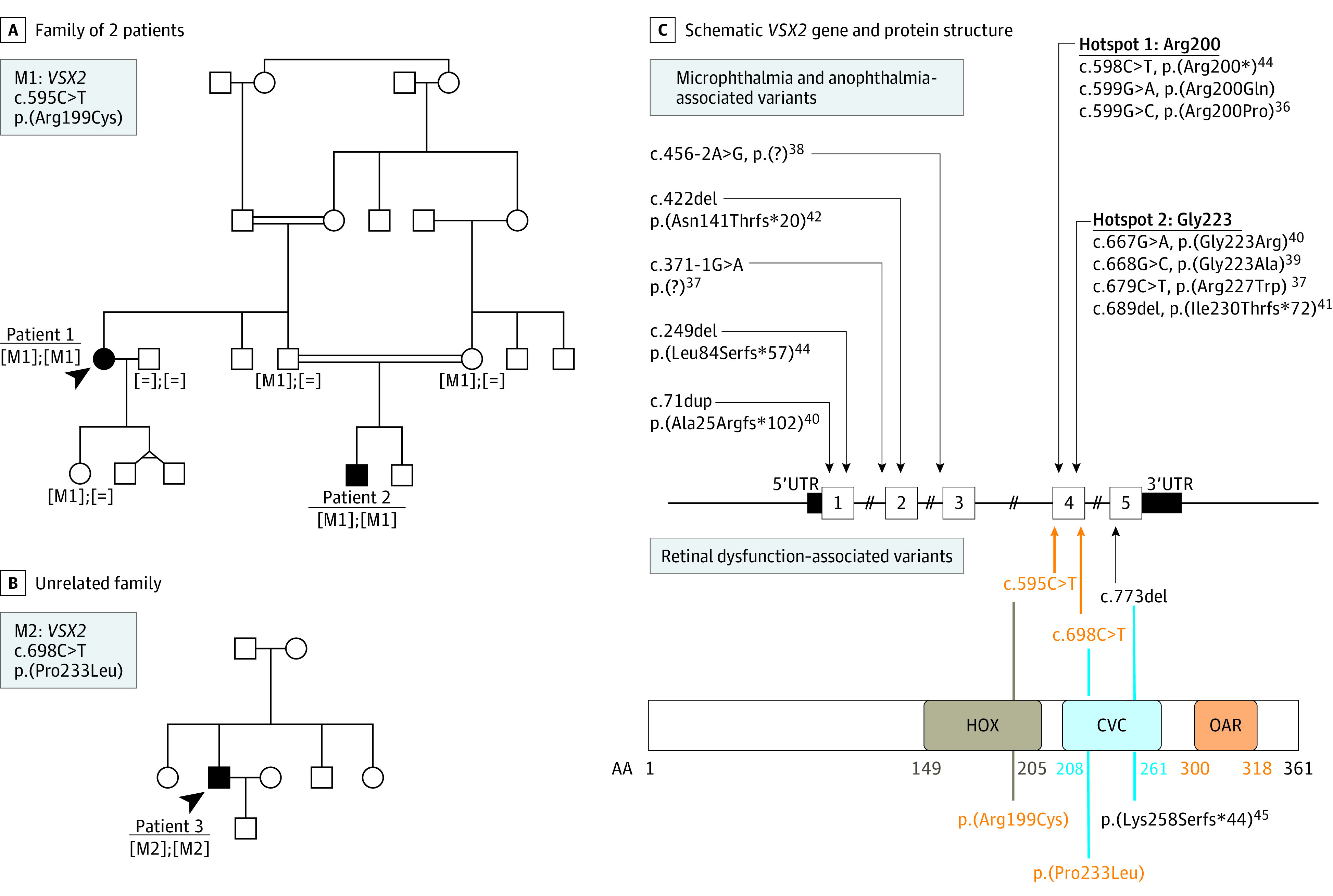
A, The c.595C>T variant in VSX2 co-segregated with disease in this consanguineous Turkish family. B, The c.698C>T variant was identified in 1 affected member; other family members were unavailable for co-segregation analyses. C, Microphthalmia- and anophthalmia-associated variants are listed above and retinal dysfunction associated variants below the schematic gene structure. In addition, the consequences on protein levels are depicted and the schematic protein structure given. Variants identified by us are highlighted in orange. The different domains of VSX2 are indicated as follows: HOX, homeobox domain; CVC, CHX10, VSX1, and CEH10 domains; OAR, OTP, aristaless, and RAX domains. Amino acid residue positions of each domain are depicted in brown, light blue, and orange, respectively.
Sanger sequencing and targeted NGS revealed the absence of disease-causing variants in patients 1 and 2, respectively. Exome sequencing performed in patient 2 and his parents revealed homozygous putative disease-causing variants in the genes ITIH2 (OMIM 146640), PRDM10 (OMIM 618319), BCR (OMIM 151410), and VSX2 (OMIM 142993), consistent with autosomal-recessive inheritance (eTable 2 in the Supplement). However, only the missense variant in VSX2 (NM_182894.3): c.595C>T, p.(Arg199Cys) co-segregated with the disease (Figure 4C). This variant is rare in population databases (minor allele frequency 8 × 10−6 in gnomAD; not present in the ExAC database) is predicted to be damaging by 25 of 40 prediction algorithms available at VarSome.34 Thus, this variant is considered likely pathogenic by ACMG standards (with evidence classified as PM1, PM2, PP1-M, and PP3).31 VSX2 Sanger sequencing in patient 3 revealed another missense homozygous variant: c.698C>T, p.(Pro233Leu) (Figure 4C). This variant is absent in population databases and predicted damaging by all the prediction algorithms available at VarSome.34 It is considered a variant of unknown significance by ACMG standards (PM2, PM1, PP3). Unfortunately, other members of the family were not available for co-segregation analyses.
Discussion
The clinical picture of the patients presented herein that included infantile nystagmus, low stable best-corrected visual acuity, and myopia was suggestive of CSNB. The upbeat component of nystagmus, present in patient 2, has also been reported as an early and transient feature of infantile nystagmus in CSNB.35 Regarding functional alteration, the ffERG revealed an electronegative dark-adapted response in keeping with a Schubert-Bornschein type of CSNB. However, both dark- and light-adapted response alterations were different than in cCSNB or icCSNB.10 On one hand, dark-adapted responses were in line with cCSNB with absent b-wave, and on the other hand, severely reduced light-adapted responses were suggestive for icCSNB. These ffERG alterations would reflect generalized both rod and cone post-phototransduction disorder. Because these patients had absent rod and cone ON- and OFF-bipolar cell responses, in contrast to cCSNB and icCSNB, we propose to call this finding a pan-bipolar cell dysfunction.
In general, VSX2 (visual system homeobox 2, formerly described as HOX10/CHX10) recessive gene defects have been reported in association with isolated microphthalmia/anophthalmia (OMIM 610093),36 microphthalmia with coloboma (OMIM 610092),37 microphthalmia with cataracts, and iris abnormalities38,39,40,41,42 (Figure 4). The adult patients presented lens subluxations and cataracts. None of the patients assessed in this study had microphthalmia. One report mentions lens ectopia without microphthalmia in a patient with a c.456-6C>G, r.(?) change in VSX2, but detailed data are lacking.43
These patients presented a peculiar retinal dysfunction, between cCSNB and icCSNB, in keeping with generalized rod and cone post-phototransduction disorder. Only few data on retinal dysfunction in both heterozygous and homozygous pathogenic variants in VSX2 are available44,45 as discussed in the eAppendix in the Supplement.
VSX2 is a 5-exon gene (Figure 4) located on chromosome 14q24.346 with a conserved sequence across vertebrates.36 It is abundantly expressed in retinal progenitor cells during embryonic and fetal eye development.47,48 This abundant expression is transient: in adult vertebrate retina, the protein is found only in the nuclei of bipolar cells.36,47,49,50,51 According to available retinal gene expression databases, VSX2 is present in all types of bipolar cells, and thus it is considered as a pan-bipolar cell marker.52,53,54,55 VSX2 encodes a 361–amino-acid residue protein (Figure 4) with transcription factor activity. VSX2 is composed of several domains: residues 149 to 205 constitute the homeobox domain (HOX); residues 208 to 261, the CVC domain (standing for CHX10, VSX1 and CEH10); and residues 300 to 318, the OAR domain (OTP, aristaless, and RAX). HOX or homeodomain has a helix-turn-helix structure binding directly regulatory DNA sequences of target genes.56 Disease-causing truncating or missense VSX2 variants were identified all over the gene (Figure 4). VSX2 presence defines the bipolar cell fate of retinal precursor cells.57 Several retinal targets of VSX2 have been identified. VSX2 acts as a transcriptional repressor of RHO, cone opsin locus control region, and SAG (S-antigen visual arrestin), thus suppressing photoreceptor gene expression in bipolar cell precursors.58,59
HOX is a common peptide found in transcription factors.60 Within the HOX peptide, residue 50 and its surrounding amino acids are important for DNA binding.61 Residue 50 binds the DNA backbone, while the surrounding residues bind to neighbor DNA bases. These important residues would correspond to residue 200 and its surrounding amino acids in VSX2 that would explain the pathogenic mechanism associated with Arg200 missense variants leading to severe ocular malformations (microphthalmia and anophthalmia).36,62 Computational structure-based evaluation of the p.(Arg199Cys) variant predicts a modification of VSX2 binding to a neighbor base of targeted DNA sequence.63 We hypothesize that Arg199 residue is probably less crucial for protein function as a regulator of ocular morphogenesis but is still required for correct bipolar cell differentiation and function. Indeed, in the 2 patients from 1 family, we are reporting that the p.(Arg199Cys) substitution leads to the CSNB phenotype without microphthalmia. The CVC domain and OAR domain, both less commonly found in the structure of transcription factors and found in proteins involved in neural tube and ocular development, are supposed to be involved in the specificity of DNA recognition, protein-protein interactions, and stability.64,65,66 The p.(Pro233Leu) variant found in patient 3 is in the CVC domain. The pathogenic variant described by Khan et al,45 c.773delA, p.(Lys258Serfs*44), is also located in the CVC domain and leads to an ocular phenotype close to the phenotype of the patients presented herein. Missense variants in the CVC domain could change the pattern of gene interactions because of impairment of specific DNA-binding activity and lead to severe ocular phenotypes in mice67 and humans.39,40,41 It is unclear why mutations in this region lead either to micropthalmia/anophthalmia or the CSNB-like phenotype. The 3-dimensional structures of different VSX2 mutations were similar (data not shown). Different variants may influence gene interactions differently, leading to the variability of the VSX2-associated phenotypes.
A naturally occurring mouse model carrying a nonsense variant in VSX2, p.Tyr176* (in the HOX domain), is known by the term ocular retardation phenotype (Or). Or mice are blind with microphthalmia, congenital cataracts, and an underdeveloped iris, retina, and optic nerve.68,69,70 They have abnormal retinal development with reduced retinal progenitor proliferations and a complete absence of bipolar cells.70 Dependent on the genetic background of the mice, the ocular phenotype could be milder. In modified Or mice71 with only mild microphthalmia and better visual behavior, there was evidence of underdeveloped ciliary body.71 Lenses became cataractous by 6 months of age. The retina extended less in the periphery and did not reach the ciliary body root. Bipolar cells were present, but their number was severely reduced compared with wild-type mice. Bipolar cell connections were abnormal: their dendrites extended into the outer nuclear layer, whereas the axons were not found in the inner plexiform layer. ERG recordings of modified Or mice showed an electronegative response to a bright flash (DA 5.0) in dark-adapted animals, resembling the ffERG alteration of the patients presented herein. We hypothesize that the underdeveloped ciliary body might be a cause for lens subluxation. Peripheral retinal underdevelopment might be a cause of peripheral retinal alterations in our patients. The depletion of the bipolar cell population and abnormal bipolar cell wiring might be linked to a peculiar ffERG waveform, which combines features of incomplete and complete CSNB. Future studies need to be performed to better understand the different phenotypes associated with VSX2 variants.
Limitations
This study is limited to only 2 families, making it difficult to determine the frequency or clinical relevance of this genetic defect. Functional in vitro or in vivo studies are needed to validate the pathogenic mechanism of the VSX2 variants leading to different eye phenotypes.
Conclusions
While only identified in 3 patients of 2 distinct families, the clinical phenotype of patients harboring missense variants in VSX2 is in accordance with retinal VSX2 expression/localization data and resembles the phenotype reported in a Vsx2 knockout animal model. The peculiar pan-bipolar cell CSNB phenotype might be due to the global lack or dysfunction of bipolar cells.
eFigure 1. Multimodal retinal imaging in patients 1 and 2 from the same family
eFigure 2. Ultrasound biomicroscopy in patient 3
eTable 1. The molecular diagnosis NGS panel including 164 genes associated with pediatric inherited retinal diseases
eTable 2. Homozygous variants found in ES for autosomal recessive model
eAppendix. Discussion of VSX2 variants identified herein in respect to the literature
eReferences
References
- 1.Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res. 2015;45:58-110. doi: 10.1016/j.preteyeres.2014.09.001 [DOI] [PubMed] [Google Scholar]
- 2.McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130(1):1-12. doi: 10.1007/s10633-014-9473-7 [DOI] [PubMed] [Google Scholar]
- 3.Penn RD, Hagins WA. Signal transmission along retinal rods and the origin of the electroretinographic a-wave. Nature. 1969;223(5202):201-204. doi: 10.1038/223201a0 [DOI] [PubMed] [Google Scholar]
- 4.Baylor DA, Nunn BJ, Schnapf JL. The photocurrent, noise and spectral sensitivity of rods of the monkey Macaca fascicularis. J Physiol. 1984;357:575-607. doi: 10.1113/jphysiol.1984.sp015518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hood DC, Birch DG. Beta wave of the scotopic (rod) electroretinogram as a measure of the activity of human on-bipolar cells. J Opt Soc Am A Opt Image Sci Vis. 1996;13(3):623-633. doi: 10.1364/JOSAA.13.000623 [DOI] [PubMed] [Google Scholar]
- 6.Sieving PA, Murayama K, Naarendorp F. Push-pull model of the primate photopic electroretinogram: a role for hyperpolarizing neurons in shaping the b-wave. Vis Neurosci. 1994;11(3):519-532. doi: 10.1017/S0952523800002431 [DOI] [PubMed] [Google Scholar]
- 7.Ueno S, Kondo M, Niwa Y, Terasaki H, Miyake Y. Luminance dependence of neural components that underlies the primate photopic electroretinogram. Invest Ophthalmol Vis Sci. 2004;45(3):1033-1040. doi: 10.1167/iovs.03-0657 [DOI] [PubMed] [Google Scholar]
- 8.Audo I, Robson AG, Holder GE, Moore AT. The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv Ophthalmol. 2008;53(1):16-40. doi: 10.1016/j.survophthal.2007.10.010 [DOI] [PubMed] [Google Scholar]
- 9.Schubert G, Bornschein H. Beitrag zur Analyse des menschlichen Elektroretinogramms [Analysis of the human electroretinogram]. Ophthalmologica. 1952;123(6):396-413. doi: 10.1159/000301211 [DOI] [PubMed] [Google Scholar]
- 10.Miyake Y, Yagasaki K, Horiguchi M, Kawase Y, Kanda T. Congenital stationary night blindness with negative electroretinogram: a new classification. Arch Ophthalmol. 1986;104(7):1013-1020. doi: 10.1001/archopht.1986.01050190071042 [DOI] [PubMed] [Google Scholar]
- 11.Langrová H, Gamer D, Friedburg C, Besch D, Zrenner E, Apfelstedt-Sylla E. Abnormalities of the long flash ERG in congenital stationary night blindness of the Schubert-Bornschein type. Vision Res. 2002;42(11):1475-1483. doi: 10.1016/S0042-6989(02)00068-8 [DOI] [PubMed] [Google Scholar]
- 12.Bijveld MMC, Florijn RJ, Bergen AAB, et al. Genotype and phenotype of 101 Dutch patients with congenital stationary night blindness. Ophthalmology. 2013;120(10):2072-2081. doi: 10.1016/j.ophtha.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 13.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19(3):264-267. doi: 10.1038/947 [DOI] [PubMed] [Google Scholar]
- 14.Strom TM, Nyakatura G, Apfelstedt-Sylla E, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19(3):260-263. doi: 10.1038/940 [DOI] [PubMed] [Google Scholar]
- 15.Zeitz C, Kloeckener-Gruissem B, Forster U, et al. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006;79(4):657-667. doi: 10.1086/508067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wycisk KA, Budde B, Feil S, et al. Structural and functional abnormalities of retinal ribbon synapses due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci. 2006;47(8):3523-3530. doi: 10.1167/iovs.06-0271 [DOI] [PubMed] [Google Scholar]
- 17.Wycisk KA, Zeitz C, Feil S, et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet. 2006;79(5):973-977. doi: 10.1086/508944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quigley M, Roy MS, Barsoum-Homsy M, Chevrette L, Jacob JL, Milot J. On- and off-responses in the photopic electroretinogram in complete-type congenital stationary night blindness. Doc Ophthalmol. 1996-1997;92(3):159-165. doi: 10.1007/BF02583287 [DOI] [PubMed] [Google Scholar]
- 19.Pusch CM, Zeitz C, Brandau O, et al. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet. 2000;26(3):324-327. doi: 10.1038/81627 [DOI] [PubMed] [Google Scholar]
- 20.Dryja TP, McGee TL, Berson EL, et al. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci U S A. 2005;102(13):4884-4889. doi: 10.1073/pnas.0501233102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeitz C, van Genderen M, Neidhardt J, et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci. 2005;46(11):4328-4335. doi: 10.1167/iovs.05-0526 [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Sergouniotis PI, Michaelides M, et al. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet. 2009;85(5):711-719. doi: 10.1016/j.ajhg.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Audo I, Kohl S, Leroy BP, et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2009;85(5):720-729. doi: 10.1016/j.ajhg.2009.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Genderen MM, Bijveld MMC, Claassen YB, et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009;85(5):730-736. doi: 10.1016/j.ajhg.2009.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Audo I, Bujakowska K, Orhan E, et al. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2012;90(2):321-330. doi: 10.1016/j.ajhg.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeitz C, Jacobson SG, Hamel CP, et al. ; Congenital Stationary Night Blindness Consortium . Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2013;92(1):67-75. doi: 10.1016/j.ajhg.2012.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Audo I, Friedrich A, Mohand-Saïd S, et al. An unusual retinal phenotype associated with a novel mutation in RHO. Arch Ophthalmol. 2010;128(8):1036-1045. doi: 10.1001/archophthalmol.2010.162 [DOI] [PubMed] [Google Scholar]
- 28.Audo I, Lancelot ME, Mohand-Saïd S, et al. Novel C2orf71 mutations account for ∼1% of cases in a large French arRP cohort. Hum Mutat. 2011;32(4):E2091-E2103. doi: 10.1002/humu.21460 [DOI] [PubMed] [Google Scholar]
- 29.Gerber S, Alzayady KJ, Burglen L, et al. Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome. Am J Hum Genet. 2016;98(5):971-980. doi: 10.1016/j.ajhg.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Human Genome Variation Society . Updated March 1, 2021. http://www.hgvs.org/
- 31.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Audo I, Bujakowska KM, Léveillard T, et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 2012;7:8. doi: 10.1186/1750-1172-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas MG, Kumar A, Mohammad S, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011;118(8):1653-1660. doi: 10.1016/j.ophtha.2011.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–1980. doi: 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simonsz HJ, Florijn RJ, van Minderhout HM, Bergen AA, Kamermans M. Nightblindness-associated transient tonic downgaze (NATTD) in infant boys with chin-up head posture. Strabismus. 2009;17(4):158-164. doi: 10.3109/09273970903396893 [DOI] [PubMed] [Google Scholar]
- 36.Ferda Percin E, Ploder LA, Yu JJ, et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000;25(4):397-401. doi: 10.1038/78071 [DOI] [PubMed] [Google Scholar]
- 37.Bar-Yosef U, Abuelaish I, Harel T, Hendler N, Ofir R, Birk OS. CHX10 mutations cause non-syndromic microphthalmia/anophthalmia in Arab and Jewish kindreds. Hum Genet. 2004;115(4):302-309. doi: 10.1007/s00439-004-1154-2 [DOI] [PubMed] [Google Scholar]
- 38.Burkitt Wright EM, Perveen R, Bowers N, et al. VSX2 in microphthalmia: a novel splice site mutation producing a severe microphthalmia phenotype. Br J Ophthalmol. 2010;94(3):386-388. doi: 10.1136/bjo.2009.159996 [DOI] [PubMed] [Google Scholar]
- 39.Reis LM, Khan A, Kariminejad A, Ebadi F, Tyler RC, Semina EV. VSX2 mutations in autosomal recessive microphthalmia. Mol Vis. 2011;17:2527-2532. [PMC free article] [PubMed] [Google Scholar]
- 40.Chassaing N, Causse A, Vigouroux A, et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin Genet. 2014;86(4):326-334. doi: 10.1111/cge.12275 [DOI] [PubMed] [Google Scholar]
- 41.Monies D, Abouelhoda M, Assoum M, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104(6):1182-1201. doi: 10.1016/j.ajhg.2019.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakobsson C. Compound heterozygous VSX2 mutation causing bilateral anophthalmia in a consanguineous Egyptian family. J Clin Exp Ophthalmol. 2015;06(03). doi: 10.4172/2155-9570.1000441 [DOI] [Google Scholar]
- 43.Maddirevula S, Kuwahara H, Ewida N, et al. Analysis of transcript-deleterious variants in Mendelian disorders: implications for RNA-based diagnostics. Genome Biol. 2020;21(1):145. doi: 10.1186/s13059-020-02053-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iseri SU, Wyatt AW, Nürnberg G, et al. Use of genome-wide SNP homozygosity mapping in small pedigrees to identify new mutations in VSX2 causing recessive microphthalmia and a semidominant inner retinal dystrophy. Hum Genet. 2010;128(1):51-60. doi: 10.1007/s00439-010-0823-6 [DOI] [PubMed] [Google Scholar]
- 45.Khan AO, Aldahmesh MA, Noor J, Salem A, Alkuraya FS. Lens subluxation and retinal dysfunction in a girl with homozygous VSX2 mutation. Ophthalmic Genet. 2015;36(1):8-13. doi: 10.3109/13816810.2013.827217 [DOI] [PubMed] [Google Scholar]
- 46.De Chen J, Ploder L, Collins L, et al. Chromosomal sublocalization and cellular expression of the retinal homeobox gene HOX10 [abstract]. Am J Hum Genet. 1990;47(A102). [Google Scholar]
- 47.Liu IS, Chen JD, Ploder L, et al. Developmental expression of a novel murine homeobox gene (Chx10): evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron. 1994;13(2):377-393. doi: 10.1016/0896-6273(94)90354-9 [DOI] [PubMed] [Google Scholar]
- 48.Belecky-Adams T, Tomarev S, Li HS, et al. Pax-6, Prox 1, and Chx10 homeobox gene expression correlates with phenotypic fate of retinal precursor cells. Invest Ophthalmol Vis Sci. 1997;38(7):1293-1303. [PubMed] [Google Scholar]
- 49.Passini MA, Levine EM, Canger AK, Raymond PA, Schechter N. Vsx-1 and Vsx-2: differential expression of two paired-like homeobox genes during zebrafish and goldfish retinogenesis. J Comp Neurol. 1997;388(3):495-505. doi: [DOI] [PubMed] [Google Scholar]
- 50.Chen CM, Cepko CL. Expression of Chx10 and Chx10-1 in the developing chicken retina. Mech Dev. 2000;90(2):293-297. doi: 10.1016/S0925-4773(99)00251-8 [DOI] [PubMed] [Google Scholar]
- 51.Vitorino M, Jusuf PR, Maurus D, Kimura Y, Higashijima S, Harris WA. Vsx2 in the zebrafish retina: restricted lineages through derepression. Neural Dev. 2009;4:14. doi: 10.1186/1749-8104-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shekhar K, Lapan SW, Whitney IE, et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166(5):1308-1323.e30. doi: 10.1016/j.cell.2016.07.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics: tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 54.Siegert S, Cabuy E, Scherf BG, et al. Transcriptional code and disease map for adult retinal cell types. Nat Neurosci. 2012;15(3):487-495,S1-2. doi: 10.1038/nn.3032 [DOI] [PubMed] [Google Scholar]
- 55.Clark BS, Stein-O’Brien GL, Shiau F, et al. Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron. 2019;102(6):1111-1126.e5. doi: 10.1016/j.neuron.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scott MP, Tamkun JW, Hartzell GW III. The structure and function of the homeodomain. Biochim Biophys Acta. 1989;989(1):25-48. doi: 10.1016/S0167-4838(00)00120-5 [DOI] [PubMed] [Google Scholar]
- 57.Livne-Bar I, Pacal M, Cheung MC, et al. Chx10 is required to block photoreceptor differentiation but is dispensable for progenitor proliferation in the postnatal retina. Proc Natl Acad Sci U S A. 2006;103(13):4988-4993. doi: 10.1073/pnas.0600083103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dorval KM, Bobechko BP, Fujieda H, Chen S, Zack DJ, Bremner R. CHX10 targets a subset of photoreceptor genes. J Biol Chem. 2006;281(2):744-751. doi: 10.1074/jbc.M509470200 [DOI] [PubMed] [Google Scholar]
- 59.West ER, Cepko CL. Development and diversification of bipolar interneurons in the mammalian retina. Dev Biol. 2022;481:30-42. doi: 10.1016/j.ydbio.2021.09.005 [DOI] [PubMed] [Google Scholar]
- 60.Bürglin TR, Affolter M. Homeodomain proteins: an update. Chromosoma. 2016;125(3):497-521. doi: 10.1007/s00412-015-0543-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson DS, Guenther B, Desplan C, Kuriyan J. High resolution crystal structure of a paired (Pax) class cooperative homeodomain dimer on DNA. Cell. 1995;82(5):709-719. doi: 10.1016/0092-8674(95)90468-9 [DOI] [PubMed] [Google Scholar]
- 62.Faiyaz-Ul-Haque M, Zaidi SHE, Al-Mureikhi MS, Peltekova I, Tsui LC, Teebi AS. Mutations in the CHX10 gene in non-syndromic microphthalmia/anophthalmia patients from Qatar. Clin Genet. 2007;72(2):164-166. doi: 10.1111/j.1399-0004.2007.00846.x [DOI] [PubMed] [Google Scholar]
- 63.Barrera LA, Vedenko A, Kurland JV, et al. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science. 2016;351(6280):1450-1454. doi: 10.1126/science.aad2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohtoshi A, Justice MJ, Behringer RR. Isolation and characterization of Vsx1, a novel mouse CVC paired-like homeobox gene expressed during embryogenesis and in the retina. Biochem Biophys Res Commun. 2001;286(1):133-140. doi: 10.1006/bbrc.2001.5372 [DOI] [PubMed] [Google Scholar]
- 65.Knauer SK, Carra G, Stauber RH. Nuclear export is evolutionarily conserved in CVC paired-like homeobox proteins and influences protein stability, transcriptional activation, and extracellular secretion. Mol Cell Biol. 2005;25(7):2573-2582. doi: 10.1128/MCB.25.7.2573-2582.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furukawa T, Kozak CA, Cepko CL. rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc Natl Acad Sci U S A. 1997;94(7):3088-3093. doi: 10.1073/pnas.94.7.3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zou C, Levine EM. Vsx2 controls eye organogenesis and retinal progenitor identity via homeodomain and non-homeodomain residues required for high affinity DNA binding. PLoS Genet. 2012;8(9):e1002924. doi: 10.1371/journal.pgen.1002924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Truslove GM. A gene causing ocular retardation in the mouse. J Embryol Exp Morphol. 1962;10:652-660. doi: 10.1242/dev.10.4.652 [DOI] [PubMed] [Google Scholar]
- 69.Robb RM, Silver J, Sullivan RT. Ocular retardation (or) in the mouse. Invest Ophthalmol Vis Sci. 1978;17(5):468-473. [PubMed] [Google Scholar]
- 70.Burmeister M, Novak J, Liang MY, et al. Ocular retardation mouse caused by Chx10 homeobox null allele: impaired retinal progenitor proliferation and bipolar cell differentiation. Nat Genet. 1996;12(4):376-384. doi: 10.1038/ng0496-376 [DOI] [PubMed] [Google Scholar]
- 71.Bone-Larson C, Basu S, Radel JD, et al. Partial rescue of the ocular retardation phenotype by genetic modifiers. J Neurobiol. 2000;42(2):232-247. doi: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. Multimodal retinal imaging in patients 1 and 2 from the same family
eFigure 2. Ultrasound biomicroscopy in patient 3
eTable 1. The molecular diagnosis NGS panel including 164 genes associated with pediatric inherited retinal diseases
eTable 2. Homozygous variants found in ES for autosomal recessive model
eAppendix. Discussion of VSX2 variants identified herein in respect to the literature
eReferences