

Abstract
Müller glia (MG) in mammalian retinas are incapable of regenerating neurons after damage, whereas the MG in lower vertebrates regenerate functional neurons. Identification of cell signaling pathways and gene regulatory networks that regulate MG-mediated regeneration is key to harnessing the regenerative potential of MG. Here we study how NFkB-signaling influences glial responses to damage and reprogramming of MG into neurons in the rodent retina. We find activation of NFkB and dynamic expression of NFkB-associated genes in MG after damage, however damage-induced NFkB activation is inhibited by microglia ablation. Knockout of NFkB in MG suppressed the accumulation of immune cells after damage. Inhibition of NFkB following NMDA-damage significantly enhanced the reprogramming of Ascl1-overexpressing MG into neuron-like cells. scRNA-seq of retinal glia following inhibition of NFkB reveals coordination with signaling via TGFβ2 and suppression of NFI and Id transcription factors. Inhibition of Smad3 signal transducer or Id transcription factors increased numbers of neuron-like cells produced by Ascl1-overexpressing MG. We conclude that NFkB is a key signaling hub that is activated in MG after damage, mediates the accumulation of immune cells, and suppresses the neurogenic potential of MG.
Keywords: Retina, Müller glia, microglia, neuroinflammation, regeneration
Introduction:
The phenomenon of retinal regeneration has been studied for many decades. Within the last 2 decades Müller glia (MG) have been identified as the primary cellular source of retinal regeneration (Fausett & Goldman, 2006; Fischer & Reh, 2001; Lenkowski & Raymond, 2014; Thummel et al., 2008). In normal healthy retinas MG are the primary support cells providing structural, metabolic, and synaptic support (Reichenbach & Bringmann, 2013). However, after damage MG can be stimulated to become activated, de-differentiate, proliferate as progenitor-like cells and produce progeny that differentiate into functional neurons that restore vision (Bernardos et al., 2007; Fausett et al., 2008; Fausett & Goldman, 2006; Fischer & Reh, 2001; Ooto et al., 2004). However, the neurogenic capacity of MG varies widely across species (Gallina et al., 2014; Lahne et al., 2020). In lower vertebrate species, such as zebrafish, MG have an extraordinary capacity to become proliferating progenitors that regenerate functional neurons (Fischer & Reh, 2001; Goldman, 2014; Lenkowski & Raymond, 2014). In birds, MG are capable of forming numerous proliferating progenitor-like cells with limited neurogenic potential (Fischer & Reh, 2001). By contrast, MG in the mammalian retina lack a significant regenerative response and, instead, rapidly activate a gliotic and quiescence-restoring programs (Bringmann et al., 2009).
Comparative epigenomic and transcriptomic analysis of MG revealed that following damage, zebrafish and chick MG transition to a reactive/activated state prior to reprogramming into progenitor cells whereas mammalian MG enter a reactive/activated state prior to restoring quiescence (Hoang et al., 2020). NFkB has been implicated as a master regulator of inflammation (Hayden & Ghosh, 2004), and may be a key difference between species wherein components of this pathway are prevalently expressed in mammalian MG but to a lesser degree in MG chick (Palazzo et al., 2020) and fish retinas (Hoang et al., 2020). Thus, we hypothesize that NFkB signaling may be part of the regulatory networks that drive reactive MG to restore quiescence and suppress gene modules that promote the de-differentiation and the formation of neurogenic progenitors. We have recently reported that NFkB signaling in the chick retina suppresses the formation of Müller glia derived progenitor cells (MGPCs) and this process depends on signals from reactive microglia (Palazzo et al., 2020). NFkB signaling is activated in the retina following different types of neuronal damage (Lebrun-Julien et al., 2009; Sakamoto et al., 2017; Yang et al., 2020) or chronic degeneration (Brambilla et al., 2012; Zeng et al., 2008). NFkB promotes inflammation and exacerbates cell death in the mammalian retina (Lebrun-Julien et al., 2009; Sakamoto et al., 2017). However, nothing is known about how this pathway influences the ability of MG to reprogram into progenitor cells and generate neurons in the mammalian retina.
Although mammalian MG do not spontaneously undergo reprogramming into neurogenic progenitor-like cells in response to retinal damage, overexpression of Ascl1 in combination with damage stimulates the reprogramming of MG into functional neurons (Jorstad et al., 2017). It was recently reported that Jak/Stat signaling (Jorstad et al., 2020) and signals produced by reactive microglia suppress neuronal regeneration from Ascl1-overexpressing MG (Todd et al., 2020). The purpose of this study was to investigate how NFkB influences glial responses to retinal damage and Ascl1-mediated reprogramming of MG. We find that NFkB is rapidly activated in MG and that is dependent on signals from microglia. Further, we find that NFkB signaling interferes with Ascl1-mediated neuronal regeneration by promoting expression of pro-glial transcription factors that drive MG to restore quiescence.
Materials and Methods:
Animals:
The use of animals in these experiments was in accordance with the guidelines established by the National Institutes of Health and the IACUC committee at the Ohio State University. Mice were kept on a cycle of 12 hours light, 12 hours dark (lights on at 8:00 AM). NFkB-eGFP reporter mice, which have eGFP-expression driven by a chimeric promoter containing three HIV-NFkB cis elements (Magness et al., 2004) and Ikkbfl/fl mice, with insertion of Cre recombinase binding sites (LoxP) into the intronic regions flanking exon 3 of the wild type Ikkb gene (Li et al., 2003) were kindly provided Dr. Denis Guttridge’s laboratory at The Ohio State University. We crossed Ikkbfl/fl mice onto Rlbp1-CreERT;R26-stop-flox-CAG-tdTomato mice (provided by Dr. Ed Levine; Vanderbilt University), wherein Cre-mediated recombination occurs in a tamoxifen-dependent manner specifically in MG under the control of the retinaldehyde binding protein 1 (Rlbp1) promoter, herein referred to as (Rlbp1-CreERT:Ikkbfl/fl). The use of Ascl1 over-expressing mice (Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP) was as previously described (Ueki et al., 2015).
Injections:
Mice were anesthetized via inhalation of 2.5% isoflurane in oxygen and intraocular injections performed as described previously (Todd et al., 2015). For all experiments, the vitreous chamber of right eyes of mice were injected with the experimental compound and the contra-lateral left eyes were injected with a control vehicle. Compounds used in these studies included N-methyl-D-aspartate (NMDA; Sigma; 1.5 ul at concentration of 100mM in PBS), 15-Deoxy-delta12,14-prostaglandin J2 (PGJ2; Alfa Aesar; 5.0 ug/dose in DMSO), TSA (Sigma; 1.0 ug/dose in DMSO), Sis3 (Sigma; 2.5 ug/dose in DMSO), AGX51 (Fischer; 10.0 ug/dose in DMSO). Intraperitoneal injections of tamoxifen (Sigma; 1.5 mg/100 μl corn oil per dose) were applied for 4 consecutive days to induce ER-Cre activity.
Single Cell RNA-sequencing of retinas
Retinas were obtained from adult (age ≥ postnatal day 60) mouse retinas at various times after NMDA treatment. Retinas were dissociated in papain/DNase (Worthington Biochemical) for 15 minutes at 37°C then triturated to form single cell suspensions. Ovomucoid (Worthington Biochemical) was added to halt the proteolysis and samples were centrifuged at 300xG for 10 minutes at 4°C and resuspended in a 1:1:10 solution of DNase:ovomucoid:Neurobasal media (Worthington:Worthington:Gibco). Cell suspensions were incubated with primary antibodies to CD11b and CD45 (table 1) at a dilution of 1:1000 in DNase:ovomucoid:Neurobasal (1:1:10) for 15 minutes. Cells were sedimented and rinsed in DNase:ovomucoid:Neurobasal (1:1:10) twice for 10 minutes. The cell suspension was passed through a 40 μm nylon filter. FACs was performed on a BD FACS ARIAIII Cell Sorter for GFP+ MG or TdTomato+ MG and CD45+/Cd11b+ immune cells (Flow Cytometry Shared Resource core at OSU).
Table 1:
Antibody table
| Chicken anti-GFP | 1:1000 | Cat#: Ab13970 | Abcam |
| Goat anti-Sox2 | 1:1000 | Cat#: KOY0418121 | R&D Systems |
| Rabbit anti-Iba1 | 1:300 | Cat#: 019–19741 | Wako Pure Chemical Industries |
| Rat anti-CD45 | 1:300 | Cat#: MCA1031G | Bio-Rad |
| Rabbit anti-GFAP | 1:1000 | Cat#: Z0334 | DakoCytomation |
| Rabbit anti-Psd95 | 1:500 | Cat#: ab18258 | Abcam |
| Mouse anti-CtBP2 | 1:500 | Cat#: 612044 | BD Transduction Laboratories |
| Goat anti-Otx2 | 1:1000 | Cat#: BAF1979 | R&D Systems |
| Rabbit anti-Secretogogin | 1:500 | Cat#: 14037T | Cell Signaling Technology |
| Rabbit anti-Lhx4 | 1:200 | Cat#: 11183–1-AP | Proteintech |
| Rabbit anti-ATF3 | 1:200 | Cat#: NBP1–85816 | Novus Biologicals |
| Rat anti-Cd45 (PE-Cy7) | 1:1000 | Cat#: 552848 | BD biosciences |
| Rat anti-Cd11b (APC) | 1:1000 | Cat#: 17-0112-82 | Thermo Fischer |
Following FACS, cell suspensions were assessed for viability (Countess II; Invitrogen) and cell-density diluted to 700 cell/μl. Each single cell cDNA library was prepared for a target of 10,000 cells per sample. Cells and 10X Genomics Chromium Single Cell 3’ V3 reagents were loaded onto chips to capture cells with gel beads in emulsion (GEMs) using 10X Chromium Controller. Library preparation was according to manufacturer’s protocols. Sequencing was conducted using an S4 flowcell and NovaSeq 6000 (Novogene) with the following parameters: Read 1 i7 Index i5 Index Read 2 28 cycles 8 cycles 0 cycles 91 cycles. Fasta sequence files were de-multiplexed, aligned and annotated to the mm10 genome using 10X Genomics Cell Ranger software.
Using Seurat toolkits (Butler et al., 2018; Satija et al., 2015), Uniform Manifold Approximation and Projection (UMAP) for dimensional reduction plots were generated from aggregates of multiple scRNA-seq libraries. Seurat was used to construct gene lists for differentially expressed genes (DEGs), violin/scatter plots and dot plots. Significance of difference in violin/scatter plots was determined using a Wilcoxon Rank Sum or Poisson test with Bonferroni correction. Monocle was used to construct unguided pseudotime trajectories and scatter plotters for MG and MGPCs across pseudotime (Qiu, Hill, et al., 2017; Qiu, Mao, et al., 2017; Trapnell et al., 2012). SingleCellSignalR was used to assess potential ligand-receptor interactions between cells within scRNA-seq datasets (Cabello-Aguilar et al., 2020). Lists of DEGs were uploaded to ShinyGo v0.66 (http://bioinformatics.sdstate.edu/go/) and AmiGO 2 (http://amigo.geneontology.org/amigo) to perform Gene Ontology (GO) enrichment analyses.
Genes that were used to identify different types of retinal cells included the following: (1) Müller glia: Glul, Nes, Vim, Scl1a3, Rlbp1, (2) microglia: C1qa, C1qb, Csf1r, Apoe, Aif1 (3) ganglion cells: Thy1, Pou4f2, Rbpms2, Nefl, Nefm, (4) amacrine cells: Gad67, Calb2, Tfap2a, (5) horizontal cells: Prox1, Calb2, (6) bipolar cells: Vsx1, Otx2, Grik1, Gabra1, and (7) cone photoreceptors: Gnat2, Opn1lw, and (8) rod photoreceptors: Rho, Nr2e3, Arr1.
Quantitative RT-PCR:
Total RNA was isolated from whole retinas using Qiagen RNeasy Mini Kits. cDNA was synthesized using SuperScript® IV Reverse Transcriptase (Invitrogen) with oligo dT primers according to the manufacturer’s protocol. qRT-PCR reactions were performed in triplicate using GoTaq® qPCR Master Mix (Promega) on a StepOne™ Real-Time PCR System (Thermo Fisher). Primers included: Id1-F 5’-TACGACATGAACGGCTGCTACTCA-3’; Id1-R 5’-TTACATGCTGCAGGATCTCCACCT-3’; Id3-F 5’-GCTCACTCCGGAACTTGTGA-3’; Id3-R 5’-CTCTCGACACCCCATTCTCG-3’; Rela-F 5’-CTTCCTCAGCCATGGTACCTCT-3’; Rela-R 5’-CAAGTCTTCATCAGCATCAAACTG-3’; Nfkb1-F 5’-GAAATTCCTGATCCAGACAAAAAC-3’; Nfkb1-R 5’-ATCACTTCAATGGCCTCTGTGTAG-3’; Nfkbia-F 5’-CCTGACCTGGTTTCGCTCTT-3’; Nfkbia-R 5’-AGGTAAGCTGGTAGGGGGAG-3’; Ikbkb-F 5’-CGTTCTGCAGCAAGGAGAGA-3’; Ikbkb-R 5’-GTCAACGGTCACGGTGTACT-3’; Tgfb2-F 5’-TCCCCTCCGAAAATGCCATC-3’; Tgfb2-R 5’-ACTCTGCCTTCACCAGATTCG-3’. Relative expression was quantified via ΔΔCt and normalized to Gapdh. Statistical significance was determined via a Student’s t-test.
Fixation, sectioning and immunocytochemistry
Tissues were fixed in 4% PFA, sectioned, and immunolabeled as described previously (Fischer et al., 1998, p. 199). Working dilutions and sources of antibodies used in this study are listed in table 1. None of the observed labeling was due to non-specific labeling of secondary antibodies or autofluorescence because sections labeled with secondary antibodies alone were devoid of fluorescence. Secondary antibodies included donkey-anti-goat-Alexa488/568, goat-anti-rabbit-Alexa488/568/647, goat-anti-mouse-Alexa488/568/647, goat anti-rat-Alexa488 (Life Technologies) diluted to 1:1000 in PBS plus 0.01% Triton X-100, and incubated for 1 hour.
Labeling for EdU:
Regular drinking water was removed 24hr prior to NMDA injections and replaced with water containing 5-ethynyl-2′-deoxyuridine (EdU; Sigma; 50 mg/100 mL dH2O) and EdU water was replaced every third day. Mice were maintained on EdU water until 4th day after TSA treatment.
Immunolabeled tissue sections were fixed in 4% formaldehyde in PBS for 5 minutes at room temperature, washed for 10 minutes with PBS, permeabilized with 0.01% Triton X-100 in PBS for 1 minute at room temperature, and washed in PBS for 10 minutes. Sections were incubated for 30 minutes at room temperature in 2M Tris, 50 mM CuSO4, Alexa Fluor 568 or 647 Azide (Thermo Fisher Scientific), and 0.5M ascorbic acid in dH2O. Sections were washed with PBS and further processed for immunofluorescence as required.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL):
To identify dying cells that contained fragmented DNA the TUNEL assay was used. We used an In Situ Cell Death Kit (TMR red or Fluorescein; Sigma-Aldrich), as per the manufacturer’s instructions.
Photography, immunofluorescence measurements, and statistics:
Wide-field photomicroscopy was performed using a Leica DM5000B microscope equipped with epifluorescence and Leica DC500 digital camera or Zeiss AxioImager M2 equipped with epifluorescence and Zeiss AxioCam MRc. Confocal images were obtained using a Leica SP8 imaging system at Department of Neuroscience Imaging Facility at The Ohio State University. Images were optimized for color, brightness and contrast, multiple channels overlaid and figures constructed by using Adobe Photoshop. Cell counts were performed on representative images. To avoid the possibility of region-specific differences within the retina, cell counts were consistently made from the same region of retina for each data set.
Similar to previous reports (Fischer et al., 2009; Ghai et al., 2009), immunofluorescence was quantified by using Image J (NIH). Identical illumination, microscope, and camera settings were used to obtain images for quantification. Retinal areas were sampled from 5.4 MP digital images. Measurements of fluorescence of Id1 within the nuclei of MG were made by selecting the total area of pixel values ≥70 for tdTomato immunofluorescence (in the red channel) and copying the fluorescence of Id1 within this area in the green channel. This copied channel data was pasted into a separate file for quantification or onto 70% grayscale background for figures. Measurements were made for regions containing pixels with intensity values greater than set threshold (0 = black and 255 = saturated). The total area was calculated for regions with pixel intensities >threshold. The intensity sum was calculated as the total of pixel values for all pixels within thresholded regions.
Where significance of difference was determined between two treatment groups accounting for inter-individual variability (means of treated-control values) we performed a two-tailed, paired t-test. Where significance of difference was determined between two treatment groups, we performed a two-tailed, unpaired t-test. A Levene’s test was performed to assess homogeneity of variance prior to performing a t-test or ANOVA. For data where homogeneity of variance was not found, a Kruskal-Wallis test was performed. Where evaluating significance in difference between multiple groups we performed ANOVA followed by Tukey’s test. GraphPad Prism 6 was used for statistical analyses and generation of histograms and bar graphs.
Results:
scRNA-seq analysis of NFkB-related genes in damaged mouse retina.
We first assessed the patterns of expression of NFkB-related factors across different cell types in normal and damaged mouse retinas. We analyzed single cell RNA sequencing (scRNA-seq) libraries from control and NMDA-damaged WT retinas(Hoang et al., 2020). Uniform Manifold Approximation and Projection (UMAP) plots revealed discrete clusters of different retinal cell types (Fig. 1a). Cell clusters were labeled based on gene expression patterns described in the Methods. Neuronal cells from control and damaged retinas were clustered together regardless of time after NMDA-treatment (Fig. 1a). By contrast, resting MG, including cells from 48 and 72 hrs after NMDA, and activated MG from 3, 6, 12, and 24 hrs after NMDA were spatially separated in UMAP plots (Fig.1a,b). Pseudotime analysis generated a trajectory of cells with resting MG (control MG and some MG from 48 to 72 hr after treatment) to the left, MG from 3 to 6 hr after treatment to the far right, and MG from 12 to 24 hr bridging the middle (Supplemental Fig. S1a–d).
Figure 1: scRNA-seq analysis of NFkB-related genes in damaged mouse retina.
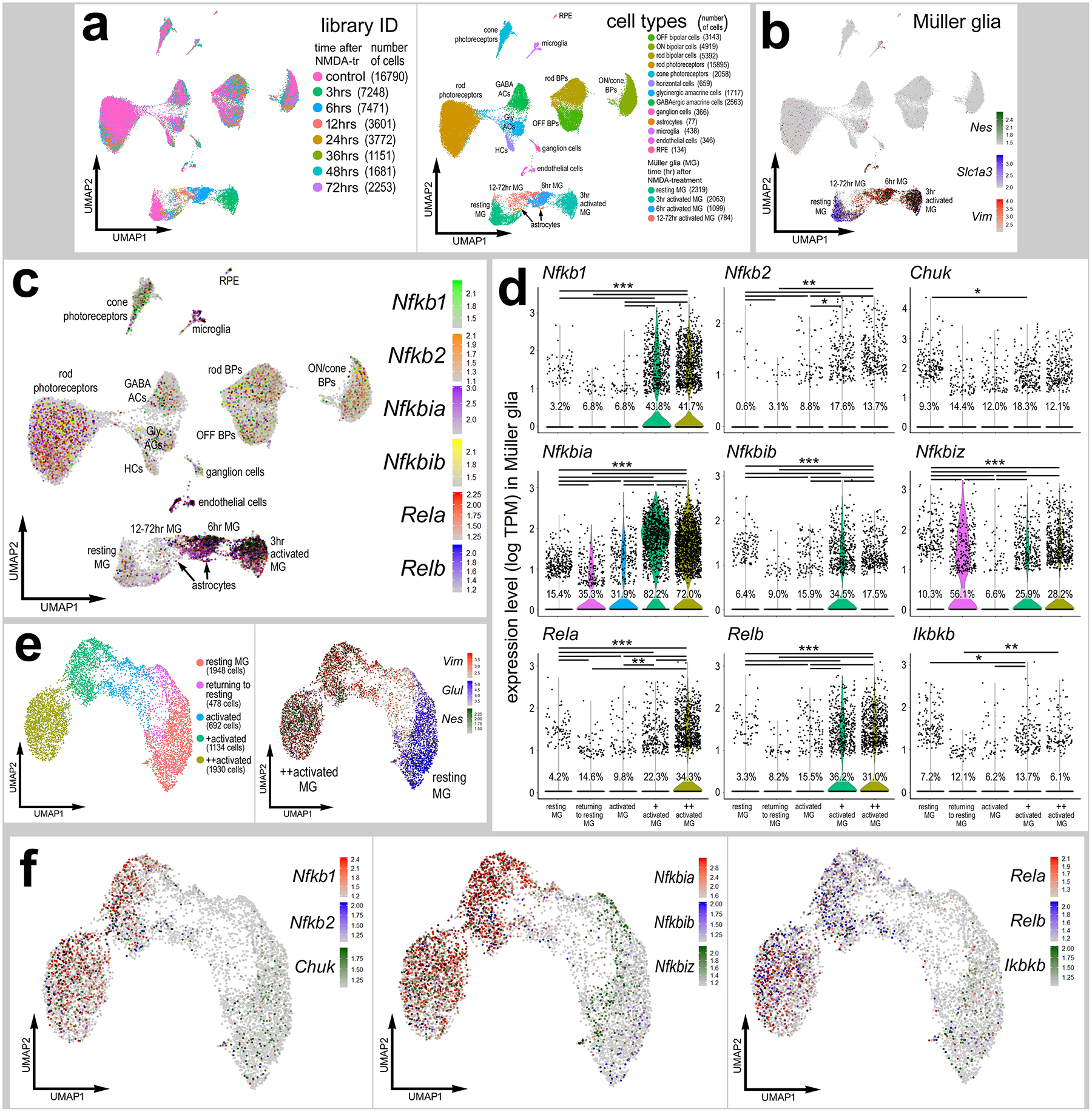
UMAPs of aggregated scRNA-seq libraries prepared from whole wild type control retinas and retinas 3hr, 6hr, 12hr, 24hr, 36hr, 48hr, and 72hr after NMDA damage; numbers of cells per library or per cluster in parentheses (a). Clusters of different types of retinal cells were identified based on collective expression of different cell-distinguishing markers as described in the Materials and Methods. Resting MG and reactive MG identified by expression of Slc1a3 or Nes and Vim, respectively (b). UMAP plots illustrate expression of Nfkb1, Nfkb2, Nfkbia, Nfkbib, Nfkbiz, Rela, Relb, Ikbkb, and Chuk (c). Violin/scatter plots illustrate expression levels of these factors within MG (d). Significance of difference (**P<0.01, ***P<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction. UMAP plots of isolated and re-embedded MG with activation states identified by expression of Glul, Vim, and Nes (e). UMAP plots of MG illustrate expression of Nfkb1, Nfkb2, Chuk, Nfkbia, Nfkbib, Nfkbiz, Rela, Relb, Ikbkb (f).
We examined changes in expression levels of NFkB-related genes including transcription factors Nfkb1 (p105/50), Nfkb2 (p100/52), Rela (p65), and Relb, inhibitor of kappa-B (IkB) components Nfkbia (IkB-a), Nfkbib (IkB-b), Nfkbiz (IkB-z), and inhibitor of kappa-B kinase (IKK) component Chuk (IKK-alpha). Although some NFkB-related genes were detected in different types of retinal neurons, most NFkB-related genes were primarily expressed in endothelial cells, microglia, astrocytes, and MG (Fig. 1c). In microglia, for example, Nfkb1, Nfkb2 and Nfkbia/b/z were significantly upregulated shortly after NMDA-treatment (Supplemental Fig. S2). We next isolated the MG and identified significant increases in expression levels of 9 different NFkB-related genes in activated MG at 3 and 6hrs after NMDA treatment compared to levels seen in resting and undamaged MG (Fig. 1d–f). Additionally, we examined the expression patterns of Nfkb1, Rela, Nfkbia, and Ikbkb by using qRT-PCR. Consistent with scRNA-seq data, we found significant increases in the expression of the NFkB signaling components after damage (Supplemental Fig. S1e). These data suggest that genes involved in NFkB signaling are rapidly and highly upregulated in MG after retinal damage.
NFkB activation in response to retinal damage is dependent on microglia.
To validate findings from scRNA-seq analyses, we identified patterns of NFkB activation in situ in the retina. We injected NMDA into the vitreous chamber of transgenic mice wherein eGFP is under the transcriptional control of NFkB cis regulatory elements (NFkB-eGFP) (Magness et al., 2004). We observed significant increases in numbers of GFP+/Sox2+ MG at 24, 48, and 72hrs after damage (Fig. 2a–b). Numbers of reporter-positive MG remained elevated at 120hrs after NMDA-treatment, but this may have resulted from a combination of continued NFkB activation and perdurance of eGFP. At 2 weeks after treatment there were few reporter-positive MG (not shown). NFkB reporter was observed primarily in MG but was not detected in astrocytes or microglia, despite scRNA-seq evidence for expression of some NFkB-related genes in these cell types (Fig. 1c–d; Supplemental Fig. S2). The absence of NFkB-reporter in microglia may be due to the specific NFkB cis-regulatory elements used in this transgenic line or may represent limited NFkB signaling activity without widespread expression of all components of this pathway.
Figure 2: NFkB activation in response to retinal damage is dependent on microglia.
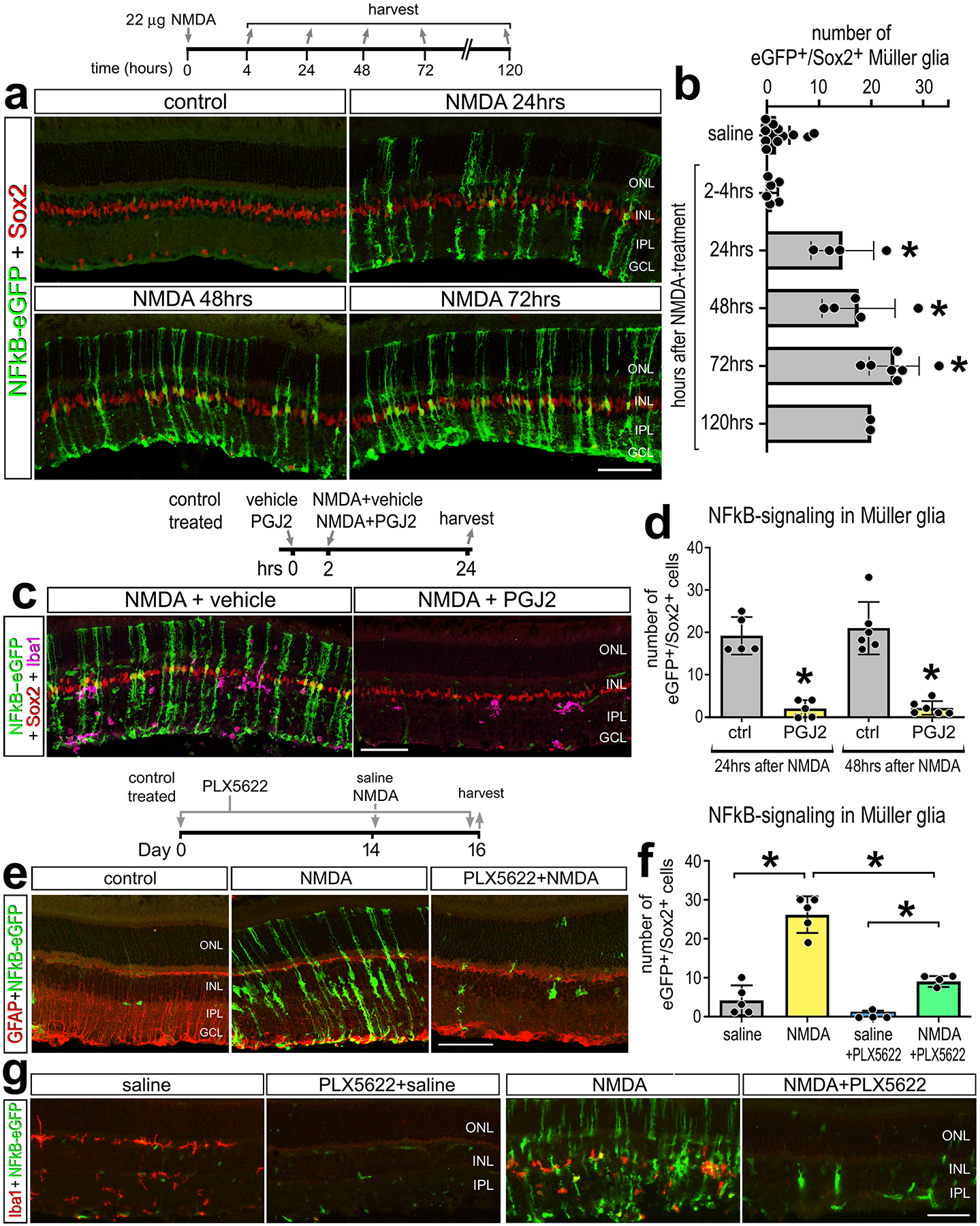
Representative images of undamaged retinas and retinas 24hr, 48hr, or 72hr after NMDA damage from NFkB-eGFP reporter mice. Retinal sections were immunolabeled for GFP (green) and Sox2 (red) (a). The histogram in (b) illustrated the mean (±SD and individual data points) numbers of Sox2+/GFP+ cells per field of view. Eyes were injected with vehicle control (left eye) or PGJ2 (NFkB inhibitor; right eye) 2h prior to NMDA treatment. Retinal sections were labeled for GFP (green), Sox2 (red) and Iba1 (magenta) (c). The histogram in (d) illustrates mean (±SD and individual data points) numbers of Sox2+/GFP+ cells per field of view. Mice were fed a control diet or PLX5622-diet for 2 weeks, eyes were injected with saline or NMDA, and retinas harvest 2 days later (e-g). Retinal sections were immuno-labeled for GFP (green, e,g), GFAP (red, e), or Iba1 (red, g). Significance of difference (*p<0.05, **p<0.001) was determined by using one way ANOVA and Tukey’s post-hoc test. The calibration bars in panels a, c, e and g represent 50 μm. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
We next injected a small molecule inhibitor, 15-deoxy-delta-12,14-prostaglandin J2 (PGJ2), into NMDA damaged eyes to test the efficacy of this NFkB inhibitor. PGJ2 has been previously shown to effectively inhibit NFkB signaling in different cell lines and chick retina (Palazzo et al., 2020; Straus et al., 2000). PGJ2 has also been shown to act at PI3K/Akt upstream of NFkB (REFS) and act at PPAR-gamma in an astrocyte cell line (REFS). However, we did not detect PPAR-gamma in the scRNA-seq libraries (not shown). We observed a significant decrease in numbers of GFP+/Sox2+ MG in damaged retinas treated with PGJ2 compared to controls (Fig. 2c–d). NFkB can be activated by various proinflammatory cytokines (Hayden & Ghosh, 2004, 2014), including IL1α, IL1β and TNF which are rapidly upregulated by retinal microglia after damage (Todd et al., 2019). Thus, we examined how elimination of microglia influenced damage-induced activation of the NFkB-reporter in the retina. Microglia were ablated using PLX5622, a Csf1r antagonist that ablates CNS microglia within 2 weeks of treatment (Dharmarajan et al., 2017; Elmore et al., 2014). Following 2 weeks of PLX5622 administration, we injected NMDA and observed a large significant decrease in numbers of GFP+/Sox2+ MG compared to numbers seen in control damaged retinas with microglia present (Fig. 2e–f). The efficacy of the PLX5622 induced ablation of microglia was confirmed by immunolabeling labeling for Iba1 (Fig. 2g). Taken together, these data indicate that NFkB signaling is robustly activated in MG after damage and this activation can be blocked by small molecule inhibitors or ablation of microglia, implying that reactive microglia secrete factors that selectively activate NFkB signaling in MG.
Conditional knockout of NFkB signaling in MG impairs immune cell responses in damaged retinas
We next examined how genetic knockout of NFkB signaling in MG influences cell survival and the responses of immune cells. Upon NFkB activation, the IKK complex is activated and phosphorylates IkBa/IkBb, thereby releasing NFkB transcription factors and allowing them to translocate into the nucleus to regulate transcription of target genes(Zhang et al., 2017). Conditional knockout of Ikkb blocks signaling through the canonical NFkB pathway due to the loss of IKK-mediated phosphorylation and degradation of IkBa/IkBb, thereby leading to maintained sequestration of NFkB transcription factors in the cytoplasm(Li et al., 2003; Zhang et al., 2017). To permit tamoxifen-inducible knockout of Ikkb specifically in MG, mice with floxed alleles of Ikkb (Ikkbfl/fl) (Li et al., 2003) were crossed with Rlbp1-CreERT mice (Rlbp1-CreERT:Ikkbfl/fl), where expression of Cre is only expressed by MG in the retina (Vazquez-Chona et al., 2009). We administered 4 consecutive daily doses of tamoxifen prior to NMDA-treatment. There was a significant reduction in the total number of Iba1+ microglia/macrophages in Rlbp1-CreERT:Ikkbfl/fl retinas compared to Rlbp1-CreERT controls at 48hrs after NMDA damage (Fig. 3a–b). In addition to activation of resident microglia, reactive peripheral immune cells migrate into the retina after damage or disease (Mitchell et al., 2018; White et al., 2017; Yu et al., 2020). Resident microglia and macrophages are CD45(lo), whereas peripheral monocyte derived macrophages are CD45(hi) expressing cells(O’Koren et al., 2016; Sedgwick et al., 1991). We identified CD45+/Iba1− putative recruited monocyte-derived macrophages within the retina following NMDA-treatment; these cells likely originated from outside of the retina (Fig. 3a). We found a significant reduction in the total numbers of CD45+/Iba1− immune cells in damaged retinas with Ikkb-cKO MG compared to controls (Fig. 3b).
Figure 3: Conditional knockout of NFkB in Müller glia impairs immune cell responses after damage.
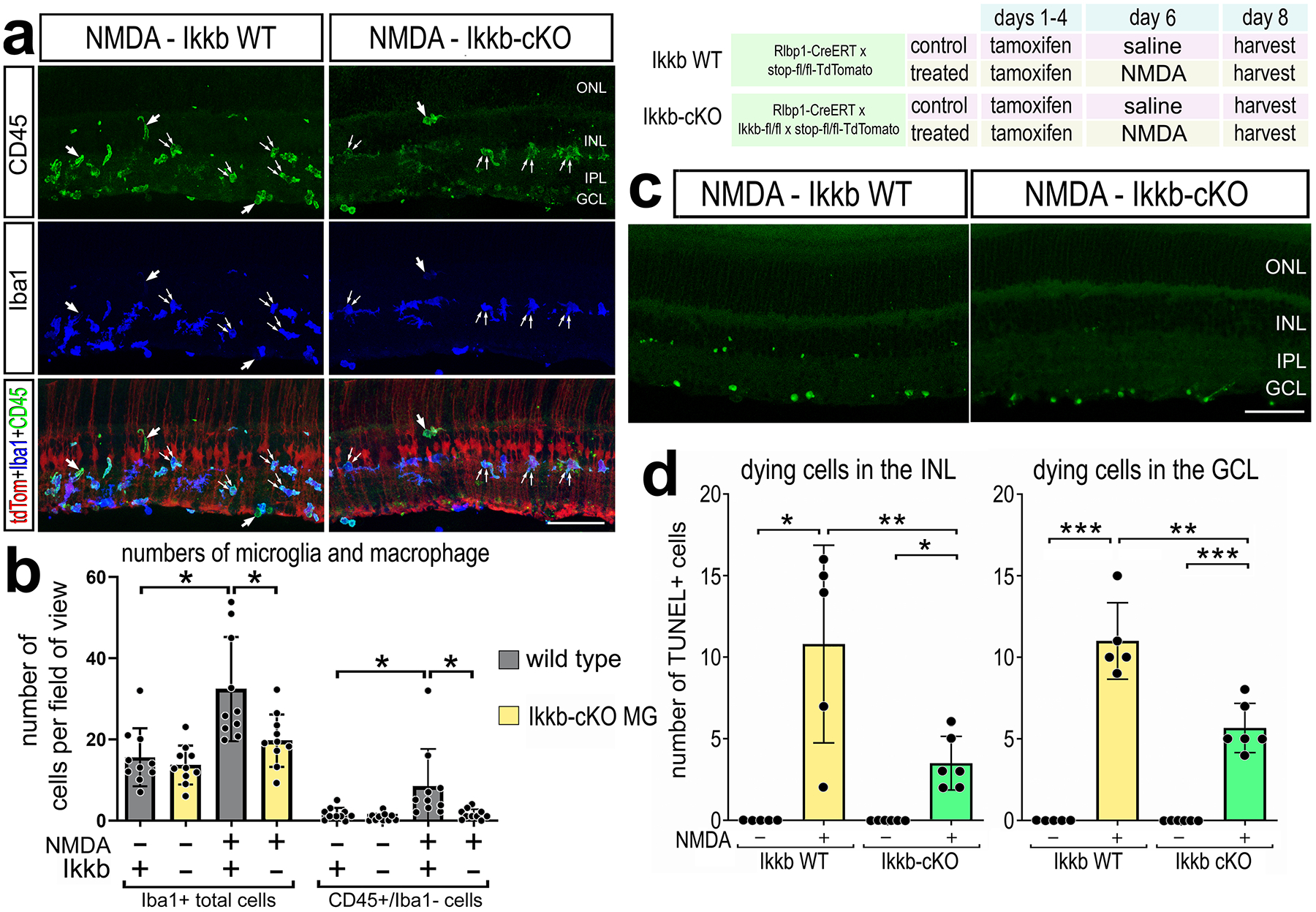
Rlbp1-creERT and Rlbp1-creERT:Ikkbfl/fl mice were injected IP with tamoxifen 1x daily for 4 consecutive days. Left eyes were injected with saline and right eyes were injected with NMDA on D6, and retinas were harvested on D8. (a) Retinal sections were labeled for CD45 (green), Iba1 (blue), and TdTomato (red). Single arrows in a represent CD45+/Iba1− cells; double arrows indicate CD45+/Iba1+ cells. The histogram illustrates the mean number (±SD and individual data points) of Iba1+ or CD45+/Iba1− cells and significance of difference (*p<0.05) was determined by a Kruskal-Wallis test (b). TUNEL assays were performed on retinal sections to identify dying cells. Histogram illustrate the mean number (±SD and individual data points) of TUNEL+ cells in the INL and GCL and significance of difference (*p<0.05, **p<0.01) was determined by paired t-test (d). The calibration bars in panels a,c represent 50 μm. Abbreviations: ONL – outer nuclear layer, INL – inner nuclear layer, IPL – inner plexiform layer, GCL – ganglion cell layer.
Additionally, we assayed for changes in cell death considering previous studies indicating that inhibition of NFkB is neuroprotective damaged mammalian (Lebrun-Julien et al., 2009; Sakamoto et al., 2017) and avian retinas (Palazzo et al., 2020). We found a significant decrease in numbers of TUNEL positive cells in both the INL and GCL in Ikkb-cKO retinas compared to numbers seen in Rlbp1-CreERT retinas at 48hr after NMDA treatment (Fig. 3c–d). Taken together these data indicate that NFkB signaling in MG influences both neuronal survival and recruitment of peripheral immune cells into damaged retina.
scRNA-seq analyses of glial cells from retinas with Ikkb-cKO Müller glia.
To further investigate the effects of disrupting NFkB signaling in MG we conducted scRNA-seq analysis to probe transcriptomic changes in retinal glia. Rlbp1-CreERT (control) mice and Rlbp1-CreERT:Ikkbfl/fl (Ikkb-cKO) mice received 4 consecutive daily IP doses of tamoxifen. Three days later both control and Ikkb-cKO mice received intravitreal injections of NMDA. Retinas were harvested for scRNA-seq 8 hours after NMDA treatment. Distinct types of retinal neurons and glia were identified based on expression of cell-distinguishing markers (Supplemental Fig. S3). UMAP ordering of MG revealed clusters of resting and activated MG of mixed library origin (Fig. 4a,b). We identified differentially expressed genes (DEGs) between control MG and Ikkb-cKO MG, 603 DEGs were down-regulated, whereas there were 242 upregulated DEGs in Ikkb-cKO MG. Gene Ontology (GO) enrichment analysis of DEGs between control and Ikkb-cKO MG revealed significant downregulation of gene modules associated with the regulation of immune cell responses, inflammatory cytokine signaling, and signaling pathways known to influence NFkB-, Akt- and MAPK-signaling (Fig. 4c). Additionally, chemokine signaling and leukocyte chemotaxis pathways were downregulated in Ikkb-cKO MG. Thus, we probed scRNA-seq libraries for Ccl2 which is known to be involved in recruiting peripheral monocyte-derived cells into the retina(Feng et al., 2017; Karlen et al., 2018; Rangasamy et al., 2014; Rutar et al., 2011). We find Ccl2 is expressed in endothelial cells, microglia, and is significantly upregulated in MG at 3, 6 and 12hrs after NMDA-treatment in wild type retinas (Supplemental Fig. 4d–e). We found a significant decrease in Ccl2 expression levels in Ikkb-cKO MG compared to levels seen in WT MG (Fig. 4d–e).
Figure 4: scRNA-seq analyses of glial cells from retinas with Ikkb-cKO Müller glia.
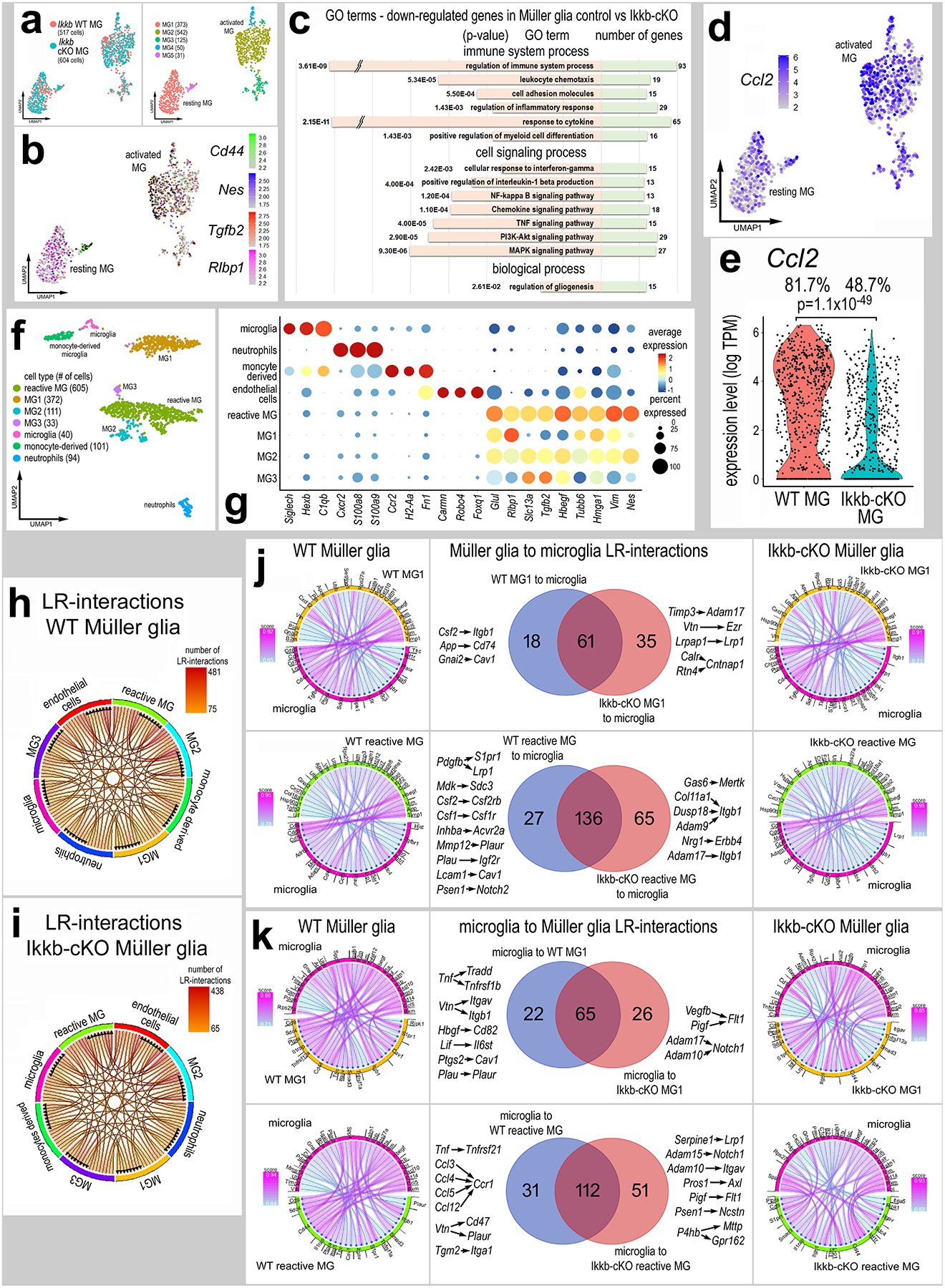
UMAP plots of isolated and re-embedded MG from scRNA-seq libraries of Rlbp1-creERT (Ikkb-WT) and Rlbp1-creERT:Ikkbfl/fl (Ikkb-cKO) retinas 8hr after NMDA (a). Resting and reactive MG were identified by expression of Rlbp1 or Cd44, Nes, and Tgfb2 (b). GO analysis on the significant (adjusted p-value ≤ 0.05) DEGs between Ikkb-WT and Ikkb-cKO MG (c). UMAP plot (d) and violin plot (e) of MG clusters illustrate expression levels of Ccl2. UMAP plots of aggregated MG and immune cells (f). Dot plot distinct patterns of gene expression of various clusters and illustrates changes in resting and reactivity genes in MG clusters 1–3 (g). Chord diagrams illustrate potential autocrine and paracrine ligand receptor (LR-) interactions generated from SingleCellSignalR for Ikkb-WT control and Ikkb-cKO libraries (h-i). Chord plots show the 40 most significant LR-interactions from MG1 to microglia or from reactive MG to microglia from Ikkb-WT control and Ikkb-cKO libraries (j). Chord plots show significant LR interactions from microglia to MG1 or to reactive MG from Ikkb-WT control or Ikkb-cKO libraries (k).
Since Ikkb-cKO in MG resulted in diminished recruitment of reactive microglia/macrophage (Fig. 3a–b), we bioinformatically combined MG and immune cells and ordered these cells in UMAP plots (Fig. 4f). The UMAP-clustered cells had distinct patterns of gene expression identifying clusters of microglia, neutrophils, monocyte-derived cells, endothelial cells, and 4 clusters of MG (Fig. 4f–g). There was one cluster of highly reactive MG, characterized based on the expression of reactive glial genes (Tgfb2, Vim, Hmgb1, Nes), while a separate cluster, MG1, expressed lower levels of genes associated with reactivity (Fig. 4g). We probed for changes in cell signaling networks and putative ligand-receptor (LR) interactions between immune cells and MG (Fig. 4h–i) using SingleCellSignalR, a package that uses a curated LR database and regularized score to identify significant LR interactions between cells (Cabello-Aguilar et al., 2020). We focused our analyses on MG, microglia, and monocytes because there is significant evidence indicating signaling among these cells in the retina(Wang et al., 2011; Wang & Wong, 2014). We identified 114 significant LR-interactions from MG1 signaling to microglia, of which 18 were specific to only control MG, and 35 were specific to Ikkb-cKO MG (Fig. 4j). We identified 228 significant LR-interactions between reactive MG to microglia, with 27 specific to control MG and 65 specific to Ikkb-cKO MG (Fig. 4j). LR-interactions specific to control MG-microglia include Csf1/2-Csf1r/Csf2rb, which is involved in promoting macrophage differentiation and survival(Elmore et al., 2014; Patel & Player, 2009). Additionally, LR-interactions that promote inflammation were identified in control libraries with WT MG (App-Cd74, Inhba-Acvr2a, Mmp12-Plaur, Mdk-Sdc3), and not in libraries with Ikkb-cKO MG (Fig. 4j). This indicates that knocking out NFkB signaling in MG impairs microglia/macrophage support pathways
We next assessed the significant LR-interactions from microglia to MG upon knockout of Ikkb in MG. We identified 117 significant interactions between microglia and MG, with 22 specific to libraries with WT MG and 26 specific to libraries with Ikkb-cKO MG (Fig. 4k). We identified 194 significant LR-interactions between microglia and reactive MG, with 31 specific to libraries with WT MG and 51 specific to libraries with Ikkb-cKO MG (Fig. 4k). LR-interactions enriched in Ikkb-cKO libraries include factors involved in modulating angiogenesis (Vegfb/Pigf-Flt1) as well as A Disintegrin and Metalloproteinase Domain (ADAM) -containing proteins to Notch1 and Itgav. LR-interactions specific to control libraries are enriched for TNF to Tradd/Tnfrsf1b/Tnfrsf21, vitronectin to integrins/CD47/Plaur, and C-C motif chemokine ligands to Ccr1 (Fig. 4k). In retinas with Ikkb-cKO MG we see losses of putative LR-interactions between MG and microglia involving pro-inflammatory signaling, including Ccl’s to Ccr’s, Cxcl’s to Cxcr’s, and Tnf and Il1b to different receptors. These LR-interactions likely underlie a diminished inflammatory glial environment that results from Ikkb-cKO in MG. Taken together, these data indicate that NFkB signaling in MG is an essential signaling hub that mediates cross-talk between MG and microglia/macrophages and is involved in mediating glial reactivity and immune cell recruitment in response to damage.
Inhibition of NFkB promotes Ascl1-mediated neuron regeneration.
It has recently been shown that microglia provide signals to suppress the ability of MG to reprogram into neuronal cells following forced over expression of Ascl1 in the mammalian retina (Todd et al., 2020). Given our findings indicating that the ablation of microglia prevents the activation of NFkB in MG (Fig. 2e–f), we hypothesized that actived NFkB in MG may suppress neurogenic reprogramming. To test this hypothesis we applied NFkB inhibitor (PGJ2), which potently suppresses NFkB activity (Fig. 2c–d), to the retinas of Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice (Jorstad et al., 2017). To induce neuron regeneration from MG treatment with NMDA, to induce neuronal damage, and HDAC inhibitor trichostatin A (TSA) is required (Jorstad et al., 2017). The paradigm of Ascl1+NMDA+TSA (hereafter referred to as ANT treatment) selectively drives expression of Ascl1 and GFP-reporter (to lineage trace) in MG to produce bipolar-like neurons and a few amacrine-like neurons that integrate into local circuitry and respond to light (Jorstad et al., 2017). These regenerated neurons are formed primarily through transdifferentiation, but a subset of Ascl1-overexpressing MG proliferate prior to giving rise to new neurons (Jorstad et al., 2020). We first validated the ANT paradigm to confirm efficacy and baseline numbers for neuronal regeneration. Consistent with previous results (Jorstad et al., 2017, 2020; Todd et al., 2020), we found that treatment of retinas with NMDA and TSA significantly increased the percentage of GFP+/Otx2+ cells (bipolar-like cells) produced by Ascl1-overexpressing MG (Supplemental Fig. S5c–d). Conversely, we found that treatment of retinas with NMDA and TSA significantly decreased the percentage of GFP+/Sox2+ cells (cells maintaining glial phenotype) in Ascl1-overexpressing MG (Supplemental Fig. S5a–b).
We next tested whether inhibition of NFkB increased the formation of neurons derived from Ascl1-overexpressing MG. Ascl1 expression in MG was activated by IP delivery of 4 consecutive daily doses of tamoxifen in adult mice (P90-P140). This was followed by intravitreal injection of NMDA or NMDA+PGJ2 on D8 and injection of TSA+vehicle or TSA+PGJ2 on D10 (Fig. 5). Eyes were harvested and retinas were processed for immunolabeling 2 weeks after the final injection. We found a significant increase in the proportion of Ascl1-overexpressing GFP+ cells that co-express Otx2, Scgn, or Lhx4, but not HuC/D (Supplemental Fig. S9a,b) in retinas treated with NFkB inhibitor (Fig. 5a–g), suggesting increased formation of bipolar-like neurons. Conversely, we found a significant decrease in the proportion of GFP+ cells that co-express Sox2+ (Fig. 5h–i), suggesting fewer Ascl1-MG remaining as glia. Additionally, GFP+ MG-derived neurons had morphologies distinct from mature MG, with spherical rather than fusiform cell bodies and processes retracted from the outer limiting membrane (Fig. 5c,e,g). Cells with neuronal morphology may have formed synapses with photoreceptors and inner retinal neurons indicated by apposition of synaptic markers CtBP2 with Psd95 on GFP+ processes in the OPL and IPL (Fig. 5j,k).
Figure 5: Inhibition of NFkB promotes Ascl1-mediated neuron regeneration.
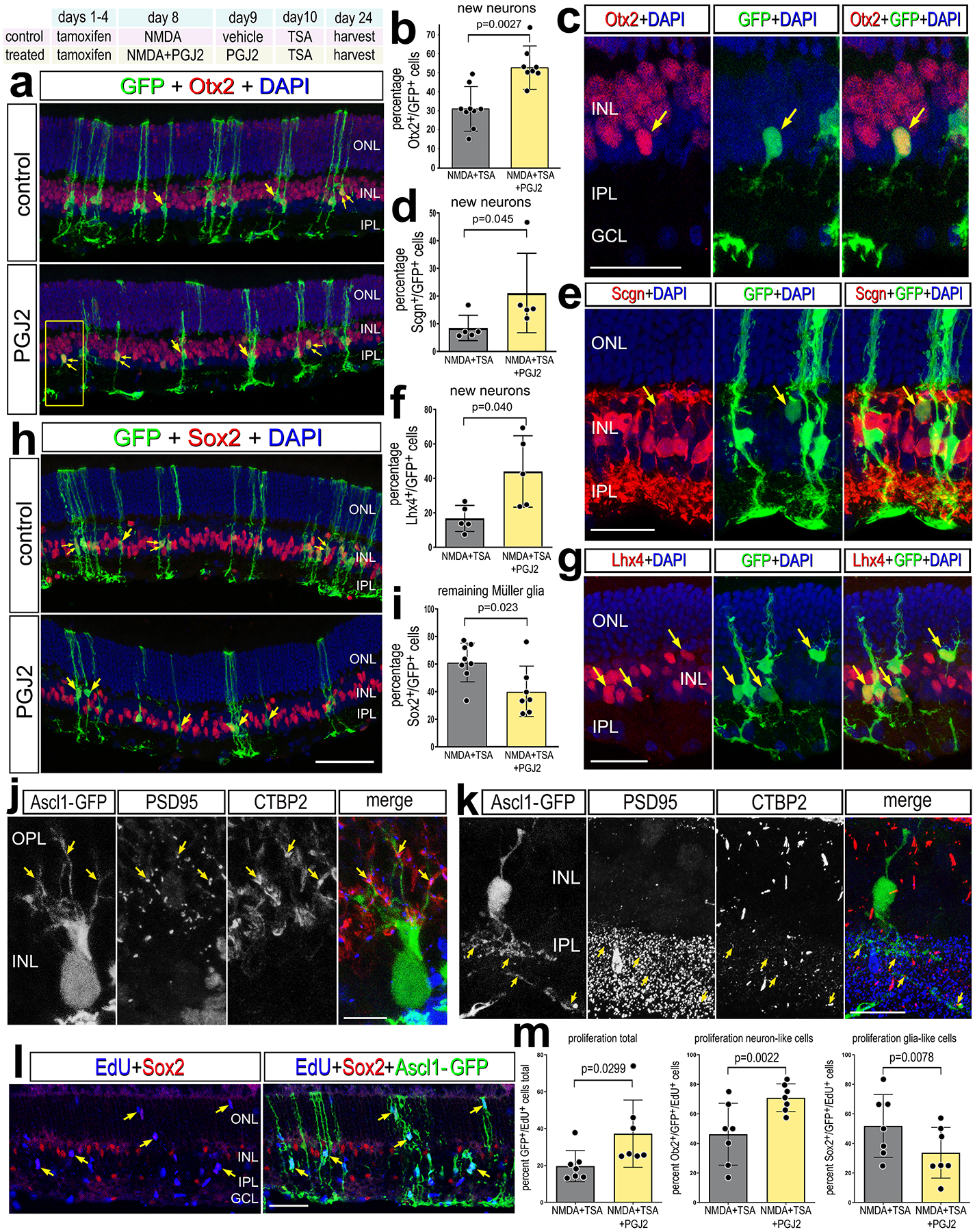
Experimental paradigm outlined at top. Tamoxifen was administered IP 1x daily for 4 consecutive days to Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. NMDA was injected intravitreally in left (control) eyes and NMDA+ PGJ2 in right (treated) eyes on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested 2 weeks after TSA injection. Retinal sections were labeled for GFP (green), DAPI (blue), and Otx2 (red; a,c), Scgn (red; e), Lhx4 (red; g), or Sox2 (red; h). Arrows in a,c,e,h and g indicate cells double-labeled for GFP and Otx2, Scgn, Lhx4 or Sox2, and small double-arrows indicate GFP alone. Histogram illustrate the mean percentage (±SD and individual data points) of GFP+ cells that are Otx2+ (b), SCGN+ (d), Lhx4+ (f), or Sox2+ (i). Significance of difference (p-values shown) was determined by using a paired t-test. Retinal sections were labeled for GFP, PSD95, and CTBP2 to identify apposing pre- and post-synaptic markers in the OPL (j) and IPL (k). (l-m) EdU was administered in the drinking water 24h prior to damage and sustained until harvesting retinas 4 days after TSA treatment. Retinal sections were labeled for GFP (green), Sox2 (red), EdU (blue) (I). Single arrows in l indicate GFP+/EdU+ cells. The histogram in (m) illustrates the mean percentage (±SD and individual data points) of GFP+ cells that are EdU+, and the percent of EdU+/GFP+ cells that are Otx2+ or Sox2+(m). The calibration bars represent 50 μm. Abbreviations: ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
ANT-treatment is known to stimulate some Ascl1-overexpressing MG to re-enter the cell cycle before generating neuron-like cells (Jorstad et al., 2020). Thus, we probed for proliferation in ANT+PGJ2 treated retinas to determine whether inhibition of NFkB promoted the proliferation of Ascl1-overexpressing MG. To identify proliferating cells we applied EdU in the drinking water prior to damage and sustained the EdU exposure until harvesting retinas at 4 days after TSA treatment. We found a significant increase in the proportion of EdU+/GFP + cells in ANT+PGJ2 treated retinas compared to ANT alone (Fig. 5l–m). This finding suggests that inhibition of NFkB promotes proliferation of Ascl1-overexpressing MG. Some of these EdU+/GFP + cells were positive for Sox2, while some EdU+/GFP + were negative for Sox2 (Supplemental Fig. S6a–d). There was a significant increase in the proportion of EdU+/Ascl1-GFP + MG that were positive for Otx2 in ANT+PGJ2 treated retinas compared to controls, and a significant decrease in the proportion of EdU+/Ascl1-GFP + MG that were positive for Sox2 in ANT+PGJ2 treated eyes compared to controls (Fig. 5l–m, Supplemental Fig. S6a–d). This suggests that inhibition of NFkB stimulates proliferation of cells with bias toward acquiring neuronal phenotype.
Additionally, we probed for changes in cell death since previous studies have shown that inhibition of NFkB is protective against excitotoxic retinal damage (Lebrun-Julien et al., 2009; Sakamoto et al., 2017), and increased levels of damage from the drug treatment could result in increased proliferation. However, we found a significant decrease in the number of TUNEL-positive cells at 24hrs after NMDA-treatment in ANT retinas treated with PGJ2 compared to ANT controls (Supplemental Fig. S6e–f). Similarly, we found a significant decrease in the total number of Iba1+ microglia in ANT+PGJ2 retinas compared to ANT controls (Supplemental Fig. S6g–h). Taken together, these data indicate the inhibition of NFkB is neuroprotective, diminishes immune cell presence, and promotes reprogramming of Ascl1-overexpressing MG into bipolar-like neurons.
Inhibition of NFkB and changes in transcriptomic profiles
We next sought to identify gene regulatory networks (GRNs) that are changed in MG upon inhibition of NFkB. We performed the ANT±PGJ2 regeneration paradigm on both Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice and on Rlbp1-CreERT mice as controls. We performed FACs to enrich for GFP+ Ascl1-overexpressing MG, or TdTomato+ control MG, as well as CD45+/Cd11b+ microglia and macrophages. We generated scRNA-seq libraries for sorted glial cells from the following transgenic lines and treatment conditions: (1) Rlbp1-CreERT+NMDA+TSA, (2) Rlbp1-CreERT+NMDA+TSA+PGJ2, (3) ANT, and (4) ANT+PGJ2 (Fig. 6a). We identified distinct clusters of cells that were identified as microglia, monocyte derived macrophages, neutrophils, dendritic cells and MG, as well as some photoreceptor cells from contamination (Fig. 6b, Supplemental Fig. S7).
Figure 6: Inhibition of NFkB signaling and changes in transcriptomic profiles.
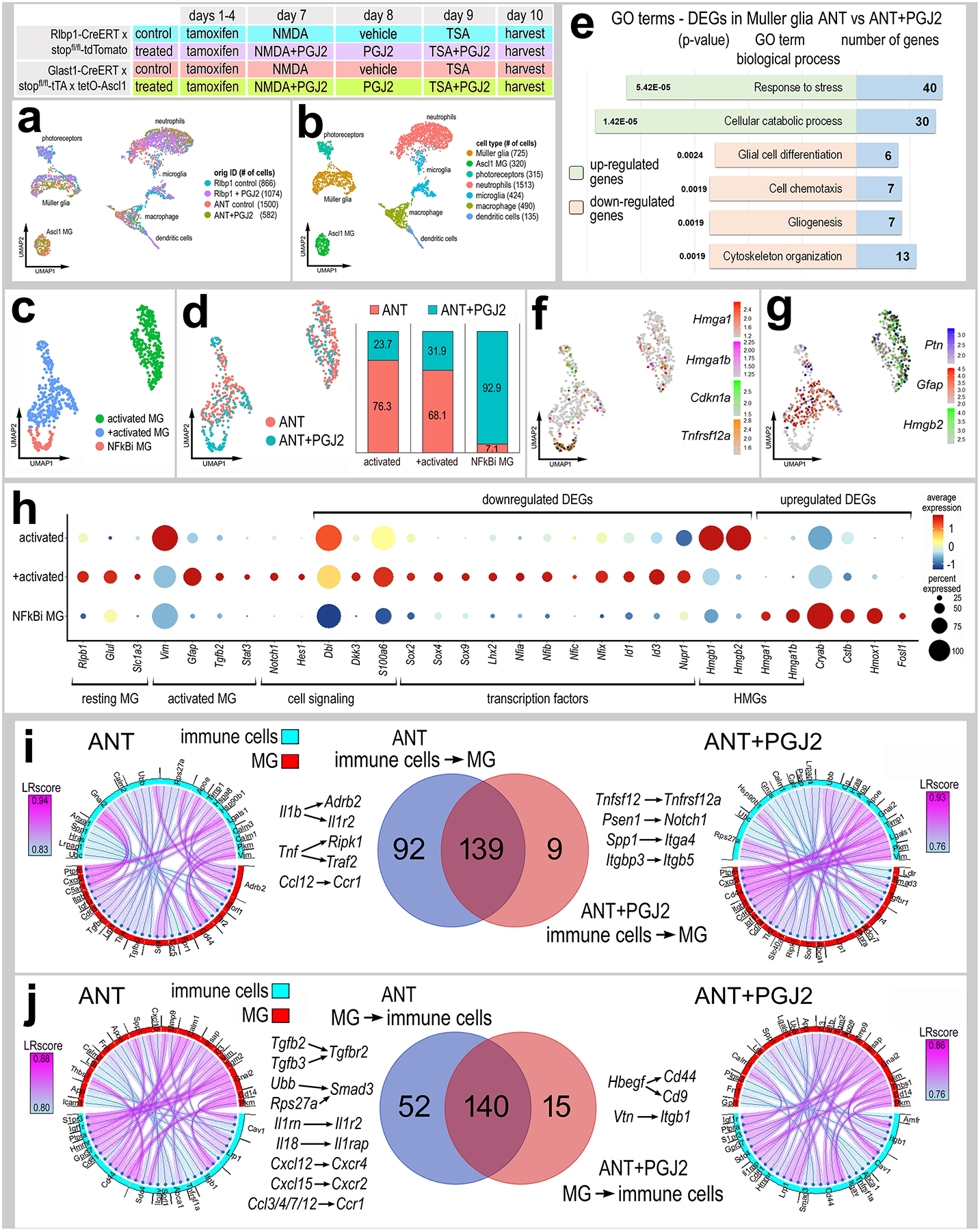
Experimental paradigm outlined at top. scRNA-seq libraries were prepared from Rlbp1-CreERT and Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. Adult mice were treated with tamoxifen IP 1x daily for 4 consecutive days. NMDA was intravitreally injected into left (control) eyes and NMDA+PGJ2 in right (treated eyes) on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retina were harvested 24h after TSA treatment. Cells were FACS enriched for either GFP+ and CD45+/Cd11b+ (from Ascl1 over-expressing mice) or TdTomato+ cells and CD45+/Cd11b+ cells (from Rlbp1-CreERT mice). UMAP plots illustrate aggregated scRNA-seq libraries for sorted MG and immune cells (a). Clusters of different types of cells were identified (b). UMAP plots of isolated and re-embedded MG from scRNA-seq libraries of ANT (Ascl1-NMDA-TSA) and ANT+PGJ2 treated retinas (c-d). Stacked bar histograms represent occupancy of each cluster by treatment (ANT or ANT+ PGJ2) (d). GO analysis of significant (adjusted P value ≤.05) DEGs in MG from ANT and ANT+PGJ2 libraries (e). UMAP plots of MG clusters illustrate patterns of expression of Hmga1, Hmga1b, Cdkn1a, Tnfsf12a (f) or Ptn, Gfap, Hmgb2 (g). Changes in resting- and reactivity-associated genes in MG clusters 1–3 were illustrated in dot plot, with dot size representing percentage of cells expressing and dot color representing level of expression (h). Ligand receptor (LR-) interactions were assessed by using SingleCellSignalR. Chord plots illustrate the 40 most significant LR-interactions from immune cells to MG (i) or from MG to immune cells (j) from ANT (control) and ANT+PGJ2 (treated) conditions. The Venn diagrams illustrate numbers of common and differential LR-interactions between ANT (control) and ANT+PGJ2 (treated) conditions.
To probe for changes in gene expression specifically in MG we isolated and re-normalized MG from all 4 libraries. UMAP plots revealed 2 clusters of Ascl1-overexpressing MG containing cells from both ANT (control) and ANT+PGJ2 (treated) libraries (Ascl1 MG1 and Ascl1 MG2); the cells were distinguished by differential expression of genes for resting glia (Rlbp1, Id1, Id3 and Nfix) or activated glia (Ptn, Hmgb1 and Hmgb2) (Supplemental Fig. S7d–e). We also identified a cluster of cells containing both Rlbp1-CreERT control (NMDA) and Rlbp1-CreERT treated (NMDA+PGJ2) cells. This cluster of cells maintained high expression of resting glial genes such as Rlbp1, Slc1a3, Dkk3 and Glul (Supplemental Fig. S7d–e). Finally, we identified one cluster of cells consisting of only NFkB-inhibitor (NFkBi) treated cells (Rlbp1-CreERT treated (NMDA+PGJ2) and ANT+PGJ2) (Supplemental Fig. S7d). This cluster of cells had a significant reduction in expression of pro-glial genes including Lhx2, NFia/b/x, Hes1 and Id1/3 (Supplemental Fig. S7e) (Clark et al., 2019; Hoang et al., 2020; Melo et al., 2016). Additionally, the NFkBi cluster had decreased expression of genes associated with activation of glial reactivity including Stat3, Notch2, and Tgfb2 (Supplemental Fig. S7e).
We next isolated the MG from ANT (control) and ANT+PGJ2 (treated) libraries to probe for differences in Ascl1-overexpressing MG resulting from NFkB inhibition (Fig. 6c–d). We performed GO analysis on significant up- or downregulated DEGs between control and treated libraries. GO enrichment analysis revealed upregulated groups of genes associated with cell stress and catabolism in Ascl1-MG treated with NFkB inhibitor (Fig. 6e). GO enrichment analysis revealed downregulated groups of genes associated with glial differentiation and development, cytoskeleton organization, and chemotaxis in Ascl1-MG treated with NFkB inhibitor (Fig. 6e). UMAP plots of MG from control and treated libraries revealed one cluster of exclusively ANT+PGJ2 cells (NFkBi MG cluster), and two clusters with mixed origin identity (Fig. 6c–d). The NFkBi MG cluster showed enriched expression of Hmga1, Hmga1b, Cdkn1a, and Tnfrsf12a (Fig. 6f). Hmga1 is a high mobility group AT-hook 1 gene, which is an important factor driving the transition from reactive MG to progenitor state in zebrafish (Hoang et al., 2020). Hmga1 expression is induced in reactive MG in the zebrafish retina after damage, and knockdown of Hmga1 decreased MG proliferation and resulted in increased MG reactivity (Hoang et al., 2020). Thus, the increased Hmga1 expression is a promising candidate underlying the increase in transition of reactive glia to a progenitor state in the mammalian retina following inhibition of NFkB.
The NFkBi MG cluster had a significant reduction in expression of reactivity genes (Gfap, Ptn, Tgfb2, Hmgb1/2, Sox9) (Fig. 6g–h). Additionally, there was a significant reduction in expression of pro-glial factors that suppress neurogenesis, including Nfia/b/x (Clark et al., 2019; Hoang et al., 2020), Id3 and Hes1 (Bai et al., 2007) (Fig. 6h). Taken together, these data indicate that inhibition of NFkB decreases expression of genes associated with glial reactivity and diminishes pro-glial/anti-neuronal transcriptional networks.
We used SingleCellSignalR (Cabello-Aguilar et al., 2020) to identify putative Ligand-Receptor (LR) interactions among DEGs in immune cells (microglia, macrophages, neutrophils) and MG in ANT control and ANT+PGJ2 conditions (Fig. 6i–j). Significant LR-interactions from immune cells to MG included Il1b-IL1r2, TNF-Traf, and Ccl12-Ccr1 in ANT control conditions but are absent in ANT+PGJ2 conditions (Fig. 6i). Comparatively, Tnfsf12-tnfrsf12a, Spp1-Itga4, and Itgbp3-Itgb5 were enriched in ANT+PGJ2 libraries (Fig. 6i). Similarly, significant LR-interactions from MG to immune cells included Tgfb2/3-Tgfbr2, Ubb-Smad3, Il18-Il1r2, Cxcl12/15-Cxcr4/2, Ccl3/4/7/12-Ccr1 in ANT control conditions, but are absent in ANT+PGJ2 treated conditions (Fig. 6j). Comparatively, Hbegf-Cd44/Cd9 and Vtn-Itgb1 LR-interactions were significantly enriched in ANT+PGJ2 conditions (Fig. 6j). These findings suggest that NFkB signaling in MG facilitates pro-inflammatory cascades, recruitment of peripheral immune cells into the retina, and activation of pro-glial signals and transcriptional networks coordinated through communication with immune cells.
Lastly, we isolated immune cells from ANT (control) and ANT+PGJ2 (treated) libraries to probe for differences in gene expression resulting from NFkB inhibition. We identified clusters of microglia, macrophages, dendritic cells, neutrophils, and a mixed population of putative monocyte-derived cells in both ANT and ANT+PGJ2 libraries based on expression of Cxcr2, Ly6g, Ccr2, Fn1, C1qa, Hexb, Siglecf, and Ccr7 (Supplemental Fig. S8a–c). We identified DEGs in immune cells between ANT and ANT+PGJ2 scRNA-seq libraries (Supplemental Fig. S8d–e) and found a significant downregulation of genes related to inflammatory signaling pathways in ANT+PGJ2 treated retinas, including Il1b/Il1rn, Spp1, Tnf and interferon-related genes (Ifi47, Irf7) (Supplemental Fig. S8e). These data indicate that NFkB inhibition diminishes the inflammatory profile of immune cells in ANT retinas.
Inhibition of TGFβ/Smad3 promotes neurogenesis.
TGFβ/Smad signaling may be a point of divergence regarding the responses of MG to damage across species (Bringmann et al., 2009; Reichenbach & Bringmann, 2013; Robel et al., 2011). In the fish retina, some reports find that TGFβ/Smad3 signaling is activated in MG in response to light damage (Lenkowski et al., 2013) or chemical induced damage (Tappeiner et al., 2016); others find TGFβ Smad signaling to be active in quiescent glia and repressed following injury (Lee et al., 2020). Regardless of different patterns of TGFβ/Smad signaling, these studies indicate that TGFβ/Smad repress MG proliferation and neuronal regeneration (Lee et al., 2020; Lenkowski et al., 2013; Tappeiner et al., 2016). TGFβ 1/2 promotes reactivity/gliosis in MG in the mouse retina (Conedera, Quintela Pousa, et al., 2021), and TGFβ/Smad3 signaling suppresses the formation and proliferation of MG-derived progenitor cells in the chick retina (Todd et al., 2017). We found a significant reduction in expression of Tgfb2 in MG upon inhibition of NFkB with ANT+PGJ2 treatment (Fig. 6h) or IKKb-cKO in MG (Fig. 7h). Thus, we tested whether inhibition of TGFβ/Smad signaling influenced Ascl1-mediated reprogramming of MG. First, we analyzed expression patterns of TGFβ ligands and Smad signal transducers in scRNA-seq libraries of WT NMDA damaged mouse retina (Fig. 7a–d). We found that Tgfb2 is highly expressed in activated MG and in MG that are returning to a resting state (Fig. 7e–f). We validated expression of Tgfb2 via qRT-PCR and confirmed increased expression of Tgfb2 in the retina 4h after NMDA damage (Supplemental Fig. 1e). Tgfb3 is not widely expressed (Fig. 7c,e). Additionally, Tgfb1/2 are expressed by microglia, amacrine cells, endothelial cells, rod bipolar cells and ON/cone bipolar cells (Fig. 7c,g). Levels of Tgfb1 in microglia and Tgfb2 in glycinergic amacrine cells significantly changed after damage (Fig. 7g). Smads1-5 were expressed by scattered cells in nearly all types of neurons and glia (Fig. 7d).
Figure 7: Inhibition of TGFβ/Smad3 promotes neurogenesis.
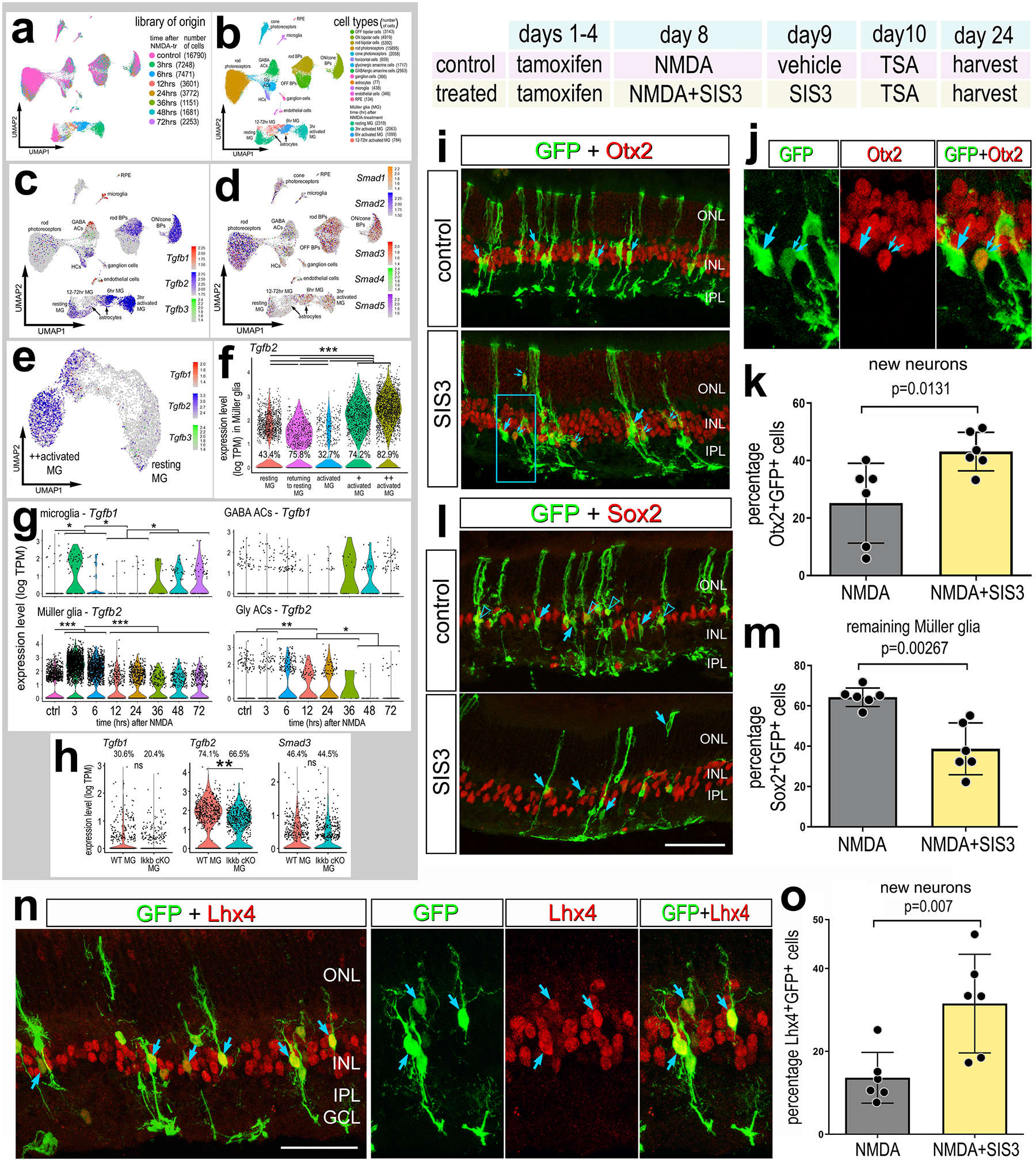
Aggregated scRNA-seq libraries were prepared from wild type control retinas and retinas 3, 6,12, 24, 36, 48 and 72hr after NMDA damage, and numbers of cells per library or per cluster in parentheses (see Fig.1 for legend). UMAP plots illustrate expression patterns of Tgfb1, Tgfb2, Tgfb3 (c) or Smad1, Smad2, Smad3, Smad4, Smad5 (d). UMAP plots of isolated and re-embedded MG illustrate patterns expression of Tgfb1, Tgfb2, and Tgfb3 across a gradient of resting and activated MG from different times after NMDA treatment (e). Violin plots illustrate expression levels and percentage of expressing for Tgfb2 in MG (f), and Tgfb1 and Tgfb2 in microglia, GABAergic amacrine cells, glycinergic amacrine cells and MG (g). Violin plots in (h) illustrated expression levels of Tgfb1/2 and Smad3 from Ikkb-WT control MG and Ikkb-cKO MG (f). Significance of difference (**p<0.01, ***p<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction (f-h). Paradigm: tamoxifen was administered IP 1x daily for 4 consecutive days in Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice, intravitreal injection of NMDA±SIS3 on D8, TSA ± SIS3 on D9, and retinas were harvested 2 weeks after the last injection (i-o). Retinal sections were labeled for GFP (green), Otx2 (red; i-j), Sox2 (red; l), or Lhx4 (red; n). Histograms illustrate the percentage (±SD and individual data points) of GFP+ cells that are Otx2+ (k), Sox2+ (m), or Lhx4+ (o). Significance of difference (p-values shown) was determined by using a paired t-test (m,o) or Kruskal-Wallis test (k). Arrows in i-j indicate GFP+ cells negative for Otx2, small double-arrows in i-j indicate GFP+/Otx2+ cells. Arrows in l indicate GFP+ cells negative for Sox2, and hollow arrow-heads indicate GFP+/Sox2+ cells. Arrows in n indicate GFP+/Lhx4+ double-labeled cells. Calibration bars represent 50 μm. Abbreviations: ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Next, we applied SIS3, a small molecule known to selectively inhibit Smad3 (Conedera, Pousa, et al., 2021; Jinnin et al., 2006; Todd et al., 2017), to the ANT regeneration paradigm. Previous studies have shown that treatment with SIS3 increases the formation of proliferating MGPCs in damaged chick retinas (Todd et al., 2017). The ANT+SIS3 treatment induced a significant increase in the proportion of GFP+/Otx2+ (Fig. 7i–k) and GFP+/Lhx4+ MG-derived neurons (Fig. 7n–o), but not GFP+/HuC/D + cells (Supplemental Fig. S9a,c), suggesting increased differentiation of bipolar-like cells, not amacrine-like cells. By contrast, ANT+SIS3 treatment induced a significant decrease in the proportion of GFP+/Sox2+ MG that remained as glia (Fig. 7l–m). Taken together, these data indicate that NFkB coordinates with TGFβ2/Smad3 signaling to suppress the neurogenic potential of MG.
Inhibition of Id transcription factors promotes neurogenesis.
Our findings suggest that levels of Id1 and Id3 are significantly reduced in Ascl1-overexpressing MG by NFkB inhibition (Fig. 6h). Ids are pro-glial transcriptional regulators that can bind to and prevent pro-neural bHLH transcription factors from binding to DNA (Benezra et al., 1990), and repress neurogenesis (Bai et al., 2007; Cai et al., 2000). Ascl1-overexpressing MG that remain as glia and don’t differentiate into neurons express higher levels of Id1 and Id3 (Jorstad et al., 2020). Analysis of WT scRNA-seq databases indicates expression of Id1, Id2 and Id3 primarily in MG, and Id1/3 and Id2 were also expressed by endothelial cells and microglia, respectively (Fig. 8a–d). Id4 was detected predominantly in GABAergic amacrine cells (Fig. 8a–d). We validated expression of Id1/3 via qRT-PCR and found significant increases in expression at 4hr after NMDA damage (Supplemental Fig.1e). We find levels of Id1, Id2, and Id3 were significantly reduced in Ascl1-overexpressing MG upon inhibition of NFkB and in Ikkb-cKO MG (Fig. 8e–g). Consistent with these findings, we found a significant decrease in levels Id1 immunofluoresence in Ikkb-cKO MG relative to control retinas at 48h after damage (Fig. 8h–j).
Figure 8: Inhibition of ID transcription factors promotes neurogenesis.
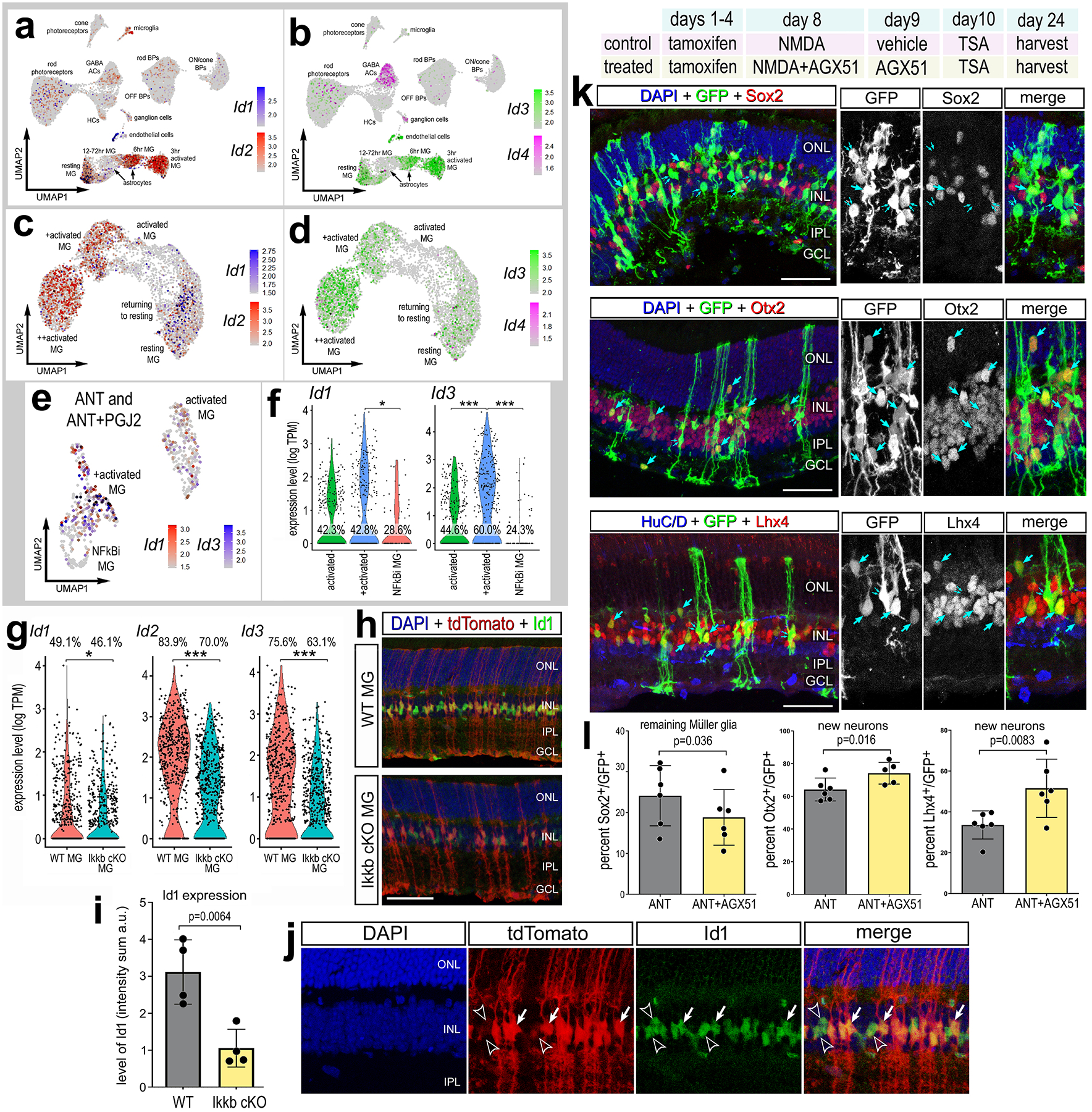
Aggregated scRNA-seq libraries were prepared from wild type control retinas and retinas 3, 6,12, 24, 36, 48 and 72hr after NMDA damage. UMAP plots illustrate expression of Id1, Id2 (a) or Id3, Id4 (b). UMAP plots of isolated and re-embedded WT MG illustrate expression of Id1, Id2 (c) or Id3, Id4 (d). UMAP and violin plots of isolated and re-embedded ANT±PGJ2 MG show expression of Id1 and Id3 (e-f) or from Ikkb-WT control and Ikkb-cKO libraries (g). Significance of difference (*p<0.05, **p<0.01, ***p<0.001) was determined by using a Poisson test with Bonferoni correction. Rlbp1-creERT and Rlbp1-creERT:Ikkbfl/fl mice were injected IP with tamoxifen daily for 4 consecutive days. Left eyes were injected with saline and right eyes were injected with NMDA on D6, and retinas were harvested on D8. Retinal sections were labeled for Id1 (green), DAPI (blue), and TdTomato (red) (h,j). Solid arrows in j represent Id1 expression in TdTomato+; hollow arrows represent Id1 in tdTomato− cells. Histogram illustrates the mean (±SD and individual data points) pixel intensity above threshold for Id1 immunofluorescence within MG nuclei (i). Experimental paradigm for k-l outlined: tamoxifen administered IP 1x daily for 4 consecutive days in Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice, NMDA ± AGX51 on D8, vehicle ± AGX51 on D9, TSA ± AGX51 on D10, and retinas were harvested 3 weeks after TSA. Representative images of treated eyes, retinal sections were labeled for GFP (green), Sox2 (red), Otx2 (red), or Lhx4 (red) (k). Single arrows in k indicate co-expression of GFP with each marker of interest (Sox2, Otx2, Lhx4), double arrows indicate GFP+ cells not co-expressing each marker. Histograms illustrate the percentage (±SD and individual data points) of GFP+ cells that are Sox2+, Otx2+, or Lhx4+ (l). Significance of difference (p-values shown) was determined by paired t-test (i,l). Calibration bars in images represent 50 μm. Abbreviations: ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Thus, we next investigated whether inhibition of Ids was sufficient to promote increased neurogenesis from Ascl1-overexpressing MG. We applied a small molecule Id inhibitor, AGX51, to the ANT paradigm. AGX51 is a pan Id inhibitor and has been shown to phenocopy genetic deletion Id1/Id3 in the mouse retina (Wojnarowicz et al., 2019). We found a significant increase in the proportion of GFP+/Otx2+ and GFP+/Lhx4+ MG-derived neurons in ANT retinas treated with Id inhibitor (Fig. 8k–l). We found a corresponding decrease in the proportion of GFP+/Sox2+ MG in response to Id inhibitor (Fig. 8k–l). However, there was no change in the proportion of GFP+ cells that co-expressed HuC/D (Supplemental Fig. S9a,d). Taken together, these data indicate that NFkB signaling is involved in promoting expression of Ids, which in turn may repress the formation of bipolar-like cells from Ascl1-overexpressing MG.
Discussion:
The process of MG-mediated retinal regeneration is contingent upon a balance between MG activation, reactive gliosis, de-differentiation and neurogenesis. MG reactivity is necessary prior to proceeding to neurogenic reprogramming in lower vertebrates (Hoang et al., 2020; Thomas et al., 2016). However, in mammalian MG, reactivity and quiescence are tightly regulated by different networks of genes (Hoang et al., 2020). Our findings suggest that NFkB drives MG reactivity and pro-glial/quiescence programs, thereby blocking transition toward a neurogenic progenitor-like state, and this occurs rapidly within 2 days after damage. By comparison, the differentiation of neuron-like cells from Ascl1-overexpressing MG is a slow process that takes 2–3 weeks after retinal damage in adult rodents (Jorstad et al., 2017). Our findings indicate that there is activation of NFkB-signaling within 48hrs of damage, and inhibition of NFkB during this early time increases numbers of neuron-like cells that differentiate over the following 2 weeks. Similarly, inhibition of TGFβ signaling or Id transcription factors during the first 2 days after damage is sufficient to increase numbers of neuron-like cells derived from Ascl1-overexpressing MG. These findings indicate that shortly after acute retinal injury, MG activate a program involving NFkB signaling, TGFβ signaling, and Id transcription factors that suppress neurogenic potential, and likely act to restore and maintain glial phenotype. This is consistent with our findings in the chick retina that activation of NFkB signaling suppresses the formation of MGPCs and promotes differentiation of progeny into glial cells (Palazzo et al., 2020). However, we cannot eliminate the possibility of off-target effects of the Tgfβ or Id inhibitors used in this study. A further limitation of this study is the use of only a single inhibitor to each target. Further work is required to thoroughly investigate and validate the roles of Id transcription factors and TGFβ signaling in the reprogramming of MG.
The temporal patterns of NFkB signaling activation (Fig. 2) lag behind the detection of NMDA-induced upregulation of genes encoding various signaling components of the NFkB pathway (Fig.1). The scRNA-seq data indicates increased transcription of NFkB signaling components, and thus suggests increased availability for NFkB signaling. However, this is different from the time-course of the NFkB-GFP reporter, which represents activation at the transcriptional end of the signaling cascade, where eGFP is driven by NFkB transcription factor binding. Thus, it is expected that upregulation of mRNA for components of the signaling pathway may not temporally correlate with reporter expression that is downstream of activation of cell signaling and, thereafter, transcriptional activation and reporter expression.
NFkB signaling is a well-established mediator of inflammation following damage or stress, in addition to regulating genes involved in cell survival, apoptosis, proliferation and differentiation (Hayden & Ghosh, 2014). NFkB signaling is activated by a variety of cytokines, primarily members of the interleukin (IL) and TNF ligand/receptor families (Osborn et al., 1989). NMDA-induced excitotoxic damage in the retina is known to activate NFkB in MG (Lebrun-Julien et al., 2009). Here we establish an essential role of microglia in activating NFkB within MG in response to damage. Microglia are the resident immune cells of the retina and are a primary source of pro-inflammatory cytokines. In response to retinal injury, microglia acquire an activated phenotype, associated with inflammation, and migrate to the site of injury (Aguzzi et al., 2013). There is evidence showing bi-directional communication between microglia and MG that regulates inflammatory responses after retinal injury (Wang et al., 2011). Previous studies have shown that ablation of microglia results in increased neuronal death following NMDA damage in mouse retinas (Todd et al., 2019). In addition, reactive microglia appear to be the only source of Il1a, Il1b and Tnf in damaged retinas (Todd et al., 2019), and these pro-inflammatory cytokines likely activate NFkB signaling (Osborn et al., 1989). Considering these reports with our current findings that NFkB activation in MG is lost in the absence of microglia, we propose that elimination of microglia eliminates the source of the cytokines that activate NFkB in MG. Our data establishes NFkB signaling as a major hub of communication between microglia and MG that is activated by microglia-derived cytokines following neuronal injury.
Our data indicate that activation of NFkB in MG induces an inflammatory response in MG that involves activation of chemokine pathways involved in recruiting peripheral immune cells into the retina. Signaling through Ccl2 and Ccr2 is essential for infiltration of monocytes into the retina (Rangasamy et al., 2014; Robbie et al., 2016). We observed a significant reduction in expression of Ccl2 in MG when NFkB signaling had been conditionally knocked-out. This reduction of Ccl2 in MG may be responsible for the diminished accumulation of reactive microglia and recruitment of peripheral immune cells to retinas where NFkB signaling was disrupted in MG. Consistent with prior studies suggesting that inhibition of NFkB in the retina is neuroprotective (Lebrun-Julien et al., 2009; Sakamoto et al., 2017), we found that Ikkb-cKO in MG resulted in neuroprotection. These findings are consistent with observations that factors, such as CNTF or FGF2, that activate cell signaling primarily in MG provide potent neuroprotection (Fischer et al., 2004, 2009), implicating MG as key mediators of cell survival in the retina. We cannot eliminate the possibility that the diminished numbers of dying neurons in retinas with Ikkb-cKO MG influenced the decreased infiltration of immune cells. Further studies are required to determine the mechanisms by which NFkB signaling regulates recruitment of peripheral immune cells into damaged retinas. Additionally, inhibition of NFkB in astrocytes has been shown to protect against axon degeneration in a model of experimental autoimmune encephalomyelitis (Brambilla et al., 2012). NFkB signaling has been shown to promote the inflammatory microenvironment of the brain and is largely mediated by interactions between astrocytes and microglia (reviewed by (Dresselhaus & Meffert, 2019)). Unfortunately, our scRNA-seq libraries from mouse retinas did not contain large enough numbers of astrocytes to permit a detailed analysis.
Reactive microglia and macrophages are known to influence the reprogramming response of MG after damage. In the zebrafish, inflammatory signals are necessary for MG reprogramming and ablation of microglia in the retina prior to injury impairs the process of neuronal regeneration (Conedera et al., 2019; White et al., 2017). In the chick retina, the ablation of microglia in the retina prevents the formation of proliferating MGPCs (Fischer et al., 2014), but this can be rescued by delivery of a pro-inflammatory cytokine (TNFSF15) or by pharmacological activation of NFkB-signaling (Palazzo et al., 2020). However, in mammalian retinas, microglia interfere with MG-mediated neuronal regeneration; ablation of microglia promotes Ascl1-mediated neurogenesis and treatment with TNF decreases Ascl1-mediated MG reprogramming (Todd et al., 2020). These findings are consistent with our results indicating that activation of NFkB in MG is mediated by reactive microglia, and inhibition of NFkB promotes the formation of MG-derived neurons following overexpression of Ascl1.
Following retinal damage, zebrafish MG activate gene networks that drive reactivity prior to de-differentiation and entering the cell cycle as a progenitor cell (Hoang et al., 2020). Similarly in chick retinas, MG upregulate reactivity genes and proceed to de-differentiate and become proliferating progenitor cells or re-enter quiescence (Hoang et al., 2020). However, MG in damaged mouse retinas upregulate gene networks associated with reactivity and networks that rapidly restore quiescence (Hoang et al., 2020).The networks driving restoration of quiescence in mammalian MG include Nfia/b/x, Hes5, and Ids, and Nfia/b/x knockout is sufficient to promote neuronal reprogramming of MG in damaged mouse retinas (Hoang et al., 2020). We find that inhibition of NFkB in Ascl1-overexpressing MG decreases expression of genes (Nfia/b/x, Hes5, Ids) that promote pro-glial and quiescence networks. We propose that decreased levels of genes associated with pro-glial and quiescence networks underlie the increased neurogenesis following NFkB inhibition. In addition, inhibition of NFkB decreases expression of genes associated with reactivity networks involving Tgfb2, Stat3, and Notch1/Hes. This is consistent with a recent study demonstrating that the inhibition of Jak/Stat-signaling results in increased neurogenesis from Ascl1-overexpressing MG (Jorstad et al., 2020).
The increased neurogenesis from Ascl1-overexpressing MG treated with NFkB inhibitor may be mediated by HMG factors including Hmga1 and Hmga1b. Hmga1 is a non-histone chromatin modifying protein (Lund et al., 1983) and is highly expressed in malignant cancers and stem cells during embryogenesis (Chiappetta et al., 1996; Sumter et al., 2016). Hmga1 is expressed during embryonic development, but is undetectable in most adult tissues, except for cells residing in adult stem cell niches where this factor plays an important role in promoting stemness (Parisi et al., 2020). Further, Hmga1 enhances reprogramming of somatic cells into induced pluripotent stem cells when combined with Sox2, Oct4, c-Myc and Klf4 (Shah et al., 2012). Hmga1 expression is induced in reactive MG in the zebrafish retina after damage and is required for the transition from reactivity to a progenitor-like state; knockdown of Hmga1 decreased proliferation of MGPCs and increased MG reactivity (Hoang et al., 2020). We find a significant increase in expression of Hmga1 and Hmga1b following NFkB inhibition. Thus, we propose that Hmga1 and Hmga1b may enhance the transition from a reactive MG state to progenitor-like state in the mammalian retina.
TGFβ/Smad3 may be another important signaling hub that coordinates with NFkB to suppress the reprogramming of mammalian MG. We find that inhibition of NFkB or cKO of Ikkb results in diminished levels of Tgfb2, and inhibition of Smad3 significantly increased in the formation of bipolar-like neurons derived from Ascl1-overexpressing MG, similar to inhibition of NFkB. TGFβ/Smad3 signaling has been shown to promote inflammation, scar formation, and fibrosis (Ashcroft et al., 1999). In the retina, TGFβ signaling through TGFβR2 drives increased expression of GFAP in MG, a symptom of glial reactivity (Hisatomi et al., 2002), and TGFβ1/2 promotes a gliotic gene networks in the mammalian retina after damage (Conedera, Quintela Pousa, et al., 2021). We find a significant reduction in Gfap following inhibition of NFkB, and this may be caused indirectly by diminished TGFβ signaling. However, there are conflicting reports regarding the influence of TGFβ/Smad-signaling on MG reprogramming and retinal regeneration in the zebrafish. Lee and colleagues (2020) reported that resting MG accumulate pSmad3 and express tgfb3, activated MG rapidly downregulate pSmad3 and tgfb3, and TGFβ/Smad3-signaling suppresses the proliferation of MGPCs in zebrafish retinas (Lee et al., 2020). Similarly, Tappenier and colleagues (2016) reported that TGFβ/Smad-signaling suppresses the proliferation of MGPCs, although the patterns of expression of tgfb3 and pSmad3 were opposite to those reported by Lee and colleagues (Tappeiner et al., 2016). Lenkowski and colleagues (2013) found that genetic deletion of tgif1, a repressor of TGFβ signaling, inhibits photoreceptor regeneration in the fish retina (Lenkowski et al., 2013). Similarly in the chick retina, TGFβ/Smad3-signaling suppresses MGPC proliferation (Todd et al., 2017). Collectively, these findings are consistent with our current findings that inhibition of TGFβ/Smad3-signaling enhances neurogenesis from Ascl1-overexpressing MG in damaged mouse retinas, and that Tgfb2/Smad-signaling may act downstream of NFkB to suppress neuronal reprogramming of MG. However, these findings are opposite to those reported by Sharma and colleagues (Sharma et al., 2020) and Conedera and colleague (Conedera, Quintela Pousa, et al., 2021), wherein TGFβ1/3 and Smad-signaling promote the proliferation of MGPCs and retinal regeneration in the zebrafish. Our data indicate that TGFβ signaling may coordinate with NFkB-signaling to promote inflammation/reactive gene networks that suppress reprogramming of MG in the mammalian retina.
Previous studies have shown that Ascl1-overexpressing MG that fail to differentiate into neurons maintain higher expression levels of Ids compared to those cells that acquire neuron-like phenotype (Jorstad et al., 2020). Ids can bind to pro-neural bHLH transcription factors to prevent expression of neuronal genes (Benezra et al., 1990), while promoting expression of the pro-glial bHLH transcription factor Hes1 to prevent neurogenesis from neural stem cells (Bai et al., 2007). Unlike MG in fish and chicks, mammalian MG rapidly undergo a transition to a reactive state after damage to restore quiescence (Hoang et al., 2020), and expression of Ids may be, in part, responsible for this inability to proceed to neurogenic reprogramming. We find that inhibition of NFkB diminishes expression of Id1/3, which may allow for increased neurogenic reprogramming by reducing the inhibition pro-neural gene networks. It has been shown that overexpression of Id1 can drive degradation of Ascl1(Viñals et al., 2004), thus MG that maintain Id expression may fail to differentiation into neurons because of Id-mediated degradation of Ascl1. Consistent with these observations, we found that inhibition of NFkB decreased expression of Ids and increased neuronal differentiation with Ascl1 overexpression.
Various inflammatory pathways converge on Id family members. For example, TGFβ can induce expression of Id1 (Stankic et al., 2013), and IL-1b and IL6 can induce expression of Id2(Hwang et al., 2018). We find a significant reduction in expression of both Il1b and Tgfb2 upon inhibition of NFkB signaling, which may be responsible for the reduction of Id expression. Collectively our findings suggest that Id- and TGFβ signaling are downstream of NFkB-signaling and collectively promote pro-inflammatory gene networks that converge on pro-glial transcription factors, including NFIs and Ids. Inhibition of NFkB alleviates pro-inflammatory signaling and diminishes expression of pro-glial transcription factors, thereby allowing MG to transition to neurogenic reprogramming rather than restoring quiescence and maintenance of glial phenotype.
Conclusions
Our findings indicate that NFkB is an important signaling hub that is activated in MG in response to damage. The activation of NFkB in MG relies on signals derived from reactive microglia and regulates the recruitment of peripheral immune cells into damaged retinas. Importantly, our findings indicate that NFkB-signaling suppresses the capacity of MG to de-differentiate and reprogram into neurons with over-expression of Ascl1, and TGFβ/Smad-signaling and Id factors are downstream of NFkB. NFkB-signaling coordinates pro-glial and pro-inflammation gene networks that represses MG reprogramming, promotes reactivity and drives a return to quiescence.
Supplementary Material
Supplemental Fig. S1: Expression of NFKB factors across pseudotime in damaged Müller glia. Pseudotime trajectory plots of MG from control and 3, 6, 12, 24, 36, 48 and 72h after NMDA damage separate into 3 distinct clusters (a). Resting glia are identified by expression of Glul, activated glia are identified by expression of Scg2 and Srxn1. The pseudotime trajectory plots (b) demonstrate Nfkbia, Nfkbib, Nfkbiz enriched in activated glia. Pseudotime reduction plots (c) and violin plots (d) illustrate change in expression levels of Nfkb1, Nfkb2, Nfkbia, Nfkbib, Nfkbiz, Ikbkb, Rela, and Relb. (e) qRT-PCR was used to measure changes in expression (fold-change) of Rela, Nfkb1, Nfkbia, Ikbkb, Tgfb2, Id1 and Id3 in whole retinas at 4hrs after NMDA treatment. Significance of difference (*p<0.05, **p<0.01, ***p<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction (d) or t-test (e).
Supplemental Fig. S2: NFkB factors expressed in microglia. UMAP plots of isolated and re-embedded microglia from WT control and NMDA 3, 6, 12, 24, 36, 48 and 72h scRNA-Seq libraries show 4 distinct clusters (a,b). Clusters were identified based on expression of Apoe, Aif1, Cyba, Klf2, and Ids (c). Violin plots illustrate levels of expression and percentage of expressing cells for Nfkb1, Nfkb2, Chuk, Nfkbia, Nfkbib, Nfkbiz, Rela, Relb, Ikbkb in different UMAP clusters of microglia (d). Significance of difference (**p<0.01, ***p<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction.
Supplemental Fig. S3: scRNA-Seq for whole retina Ikkb-cKO. UMAP plots for whole retina scRNA-seq libraries of Rlbp1-CreERT (control) and Rlbp1-CreERT:Ikkbfl/fl (treated) mice at 8h after NMDA damage (a). Distinct cell clusters of UMAP ordered cells (b) were characterized based on expression of markers such as Rlbp1, Slc1a3 and Glul (c) and DEGs identified from heatmap plots (d).
Supplemental Fig. S4: Expression patterns Ccl2 in WT scRNA-Seq libraries from control and damaged retinas. Aggregated scRNA-seq libraries were prepared from WT control retinas and retinas 3, 6,12, 24, 36, 48 and 72h after NMDA damage (a). Clusters were identified based on expression of cell-distinguishing markers, as described in the Methods (b). Resting MG and reactive MG identified by expression of Slc1a3 or Nes and Vim, respectively (c). UMAP plots illustrate patterns of expression of Ccl2 across retinal cell types (d). Violin plots illustrate expression levels Ccl2 in MG (e). Significance of difference (**p<0.001, **p<1×10–10, ***p<1×10–20) was determined by using a Wilcoxon rank sum with Bonferroni correction.
Supplemental Fig. S5: Validation of ANT regeneration paradigm. Experimental paradigm outlined at top. Retinal sections were labeled for GFP (green), DAPI (blue), Sox2 (red; a), or Otx2 (red; c). Arrows indicate double-labeled cells. Histograms illustrate the mean percentage (±SD and individual data points) of GFP+/Sox2+ (b) or GFP+/Otx2+ (d) cells. Significance of difference (p-values shown) was determined by using a paired t-test.
Supplemental Fig. S6: Inhibition of NFkB signaling promotes proliferation and neuroprotection following ANT treatment. Experimental paradigm outlined at top. Tamoxifen was administered IP 1x daily for 4 consecutive days in Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice, NMDA was injected intravitreally in left (control) eyes or NMDA+PGJ2 in right (treated) eyes on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested 4 days after TSA. EdU was added to the drinking water and administered starting on D7 and sustained until harvesting retinas on D14. Retinal sections were labeled for GFP (green), Otx2 (red;a-b), Sox2 (red;c-d), and EdU (blue). Arrows in a and b indicate GFP+/EdU+/Otx2+ cells, and arrows in c and d indicate GFP+/EdU+/Sox2+ cells. TUNEL assays were performed on ANT or ANT+PGJ2 treated retinas at 24h after NMDA (e). Retinal sections were labeled for GFP (green), Iba1 (red), and Draq5 (blue) (g). Histograms illustrate the mean (±SD and individual data points) number of TUNEL+ cells (f) or Iba1+ cells (h). Significance of difference (**p<0.01) was determined by using a Kruskal-wallis test (f), or paired t-test (h).
Supplemental Fig. S7: scRNA-Seq libraries from Ascl1 overexpressing mice. scRNA seq libraries were prepared from Rlbp1-CreERT and Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. Tamoxifen was injected IP 1x daily for 4 consecutive days. NMDA was injected intravitreally into left (control) eyes and NMDA+PGJ2 in right (treated eyes) on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested for scRNA-seq library prep 24h after TSA treatment. Samples were enriched via FACS for either GFP+ cells and CD45+/Cd11b+ cells (from Ascl1 mice) or TdTomato+ cells and CD45+/Cd11b+ cells (from Rlbp1-CreERT mice). Clusters of different types of retinal cells were identified based on expression patterns of different cell-distinguishing markers (a). (b-c) UMAP plots of aggregated scRNA-seq libraries for MG and immune cells. (d) UMAP plots of isolated and re-embedded MG from Rlbp1-CreERT control (NMDA) and treated (NMDA+PGJ2) and Ascl1 control (NMDA) and treated (NMDA+PGJ2). Stacked bar graphs illustrate UMAP cluster occupancy by library origin identity (d). The dot plot in (e) illustrates changes in expression of resting and reactivity genes, cell signaling factors, and transcription factors. Dot size represents percent of cells expressing and dot color representing expression levels.
Supplemental Fig. 8: Decreased expression of genes associated with inflammatory signaling in immune cells from ANT+PGJ2 scRNA-seq libraries. scRNA seq libraries were prepared from Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. Tamoxifen was injected IP 1x daily for 4 consecutive days. NMDA was injected intravitreally into left (control) eyes and NMDA+PGJ2 in right (treated eyes) on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested for scRNA-seq library prep 24h after TSA treatment. Samples were enriched via FACS for GFP+ cells and CD45+/Cd11b+ cells. UMAP plots of immune cells from ANT and ANT+PGJ2 scRNA-seq libraries (a-b). Types of immune cells were identified based on expression of cell type-specific markers (c). Heatmap of differentially expressed genes (adj_p<0.05) (d). Dot Plot illustrates the percent of expressing cells (dot size) and genes that were upregulated (red) or downregulated (blue) in ANT and ANT+PGJ2 scRNA-seq libraries (e).
Supplemental Fig. S9: Ascl1-overexpressing MG fail to differentiate into amacrine cells. Experimental paradigm outlined at top. Tamoxifen was administered IP 1x daily for 4 consecutive days to Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. NMDA was injected intravitreally in left (control) eyes and NMDA+ PGJ2, or SIS3, or AGX51 in right (treated) eyes on D8, vehicle ± inhibitor on D9, TSA ± inhibitor on D10, and retinas were harvested 2 weeks after TSA injection. Retinal sections were labeled for GFP (green), DAPI (blue), and HuC/D (red) (a). Histograms illustrate mean percentage (±SD and individual data points) of GFP+ cells that are HuC/D+ (b-d). Significance of difference (p-values shown) was determined by using a paired t-test. Arrows in indicate GFP+ cells negative for HuC/D. Calibration bar in panel a represents 50 μm. Abbreviations: ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Acknowledgements:
This work was supported by RO1 EY022030-08 (AJF), UO1 EY027267-04 (SB, AJF), and T32 NS105864 (IP). We thank Dr. Ed Levine for providing the Rlbp1-CreERT mice and Dr. Dennis Guttridge for providing the NFkB-eGFP reporter mice and Ikkbfl/fl mice. We thank Heithem El-Hodiri, Lisa Kelly and Olivia Taylor for comments and discussions that contributed to the final form of the paper.
Footnotes
Conflict of Interest: None.
Data availability:
gene-cell matrices for scRNA-seq data are deposited at GitHub https://github.com/jiewwwang/Single-cell-retinal-regeneration
https://github.com/fischerlab3140/
Some of the scRNA-seq data can be queried at https://proteinpaint.stjude.org/F/2019.retina.scRNA.html.
References:
- Aguzzi A, Barres BA, & Bennett ML (2013). Microglia: Scapegoat, saboteur, or something else? Science, 339(6116), 156–161. 10.1126/science.1227901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, & Roberts AB (1999). Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nature Cell Biology, 1(5), 260–266. 10.1038/12971 [DOI] [PubMed] [Google Scholar]
- Bai G, Sheng N, Xie Z, Bian W, Yokota Y, Benezra R, Kageyama R, Guillemot F, & Jing N (2007). Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Developmental Cell, 13(2), 283–297. 10.1016/j.devcel.2007.05.014 [DOI] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, & Weintraub H (1990). The protein Id: A negative regulator of helix-loop-helix DNA binding proteins. Cell, 61(1), 49–59. 10.1016/0092-8674(90)90214-y [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, & Raymond PA (2007). Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. J Neurosci, 27(26), 7028–7040. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17596452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, & Shestopalov VI (2012). Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. Journal of Neuroinflammation, 9, 213. 10.1186/1742-2094-9-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, & Reichenbach A (2009). Cellular signaling and factors involved in Muller cell gliosis: Neuroprotective and detrimental effects. Prog Retin Eye Res, 28(6), 423–451. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19660572 [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, & Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology, 36(5), 411–420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello-Aguilar S, Alame M, Kon-Sun-Tack F, Fau C, Lacroix M, & Colinge J (2020). SingleCellSignalR: Inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Research, 48(10), e55. 10.1093/nar/gkaa183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Morrow EM, & Cepko CL (2000). Misexpression of basic helix-loop-helix genes in the murine cerebral cortex affects cell fate choices and neuronal survival. Development (Cambridge, England), 127(14), 3021–3030. [DOI] [PubMed] [Google Scholar]
- Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, Merciai BM, Fidanza V, Giancotti V, Santoro M, Simeone A, & Fusco A (1996). High level expression of the HMGI (Y) gene during embryonic development. Oncogene, 13(11), 2439–2446. [PubMed] [Google Scholar]
- Clark BS, Stein-O’Brien GL, Shiau F, Cannon GH, Davis-Marcisak E, Sherman T, Santiago CP, Hoang TV, Rajaii F, James-Esposito RE, Gronostajski RM, Fertig EJ, Goff LA, & Blackshaw S (2019). Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron, 102(6), 1111–1126.e5. 10.1016/j.neuron.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conedera FM, Pousa AMQ, Mercader N, Tschopp M, & Enzmann V (2019). Retinal microglia signaling affects Müller cell behavior in the zebrafish following laser injury induction. Glia, 67(6), 1150–1166. 10.1002/glia.23601 [DOI] [PubMed] [Google Scholar]
- Conedera FM, Pousa AMQ, Mercader N, Tschopp M, & Enzmann V (2021). The TGFβ/Notch axis facilitates Müller cell-to-epithelial transition to ultimately form a chronic glial scar. Molecular Neurodegeneration, 16(1), 69. 10.1186/s13024-021-00482-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conedera FM, Quintela Pousa AM, Presby DM, Mercader N, Enzmann V, & Tschopp M (2021). Diverse Signaling by TGFβ Isoforms in Response to Focal Injury is Associated with Either Retinal Regeneration or Reactive Gliosis. Cellular and Molecular Neurobiology, 41(1), 43–62. 10.1007/s10571-020-00830-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmarajan S, Fisk DL, Sorenson CM, Sheibani N, & Belecky-Adams TL (2017). Microglia activation is essential for BMP7-mediated retinal reactive gliosis. J Neuroinflammation, 14(1), 76. 10.1186/s12974-017-0855-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresselhaus EC, & Meffert MK (2019). Cellular Specificity of NF-κB Function in the Nervous System. Frontiers in Immunology, 10, 1043. 10.3389/fimmu.2019.01043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, & Green KN (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82(2), 380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, & Goldman D (2006). A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J Neurosci, 26(23), 6303–6313. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16763038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, Gumerson JD, & Goldman D (2008). The proneural basic helix-loop-helix gene ascl1a is required for retina regeneration. J Neurosci, 28(5), 1109–1117. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18234889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Wang X, Liu T, Zhang M, Xu G, & Ni Y (2017). Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Molecular Vision, 23, 765–777. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5669614/ [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, & Reh TA (2001). Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci, 4(3), 247–252. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11224540 [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Schmidt M, Omar G, & Reh TA (2004). BMP4 and CNTF are neuroprotective and suppress damage-induced proliferation of Muller glia in the retina. Mol Cell Neurosci, 27(4), 531–542. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15555930 [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Ritchey ER, & Sherwood P (2009). Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia, 57(14), 1538–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Seltner RLP, Poon J, & Stell WK (1998). Immunocytochemical characterization of quisqualic acid- and N-methyl-D-aspartate-induced excitotoxicity in the retina of chicks. Journal of Comparative Neurology, 393(1), 1–15. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Gallina D, Scott MA, & Todd L (2014). Reactive microglia and macrophage facilitate the formation of Muller glia-derived retinal progenitors. Glia, 62(10), 1608–1628. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=24916856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina D, Todd L, & Fischer AJ (2014). A comparative analysis of Muller glia-mediated regeneration in the vertebrate retina. Exp Eye Res, 123, 121–130. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23851023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, & Fischer AJ (2009). Serotonin released from amacrine neurons is scavenged and degraded in bipolar neurons in the retina. J Neurochem, 111(1), 1–14. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19619137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D (2014). Muller glial cell reprogramming and retina regeneration. Nat Rev Neurosci, 15(7), 431–442. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=24894585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, & Ghosh S (2004). Signaling to NF-kappaB. Genes Dev, 18(18), 2195–2224. 10.1101/gad.1228704 [DOI] [PubMed] [Google Scholar]
- Hayden MS, & Ghosh S (2014). Regulation of NF-κB by TNF family cytokines. Seminars in Immunology, 26(3), 253–266. 10.1016/j.smim.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisatomi T, Sakamoto T, Yamanaka I, Sassa Y, Kubota T, Ueno H, Ohnishi Y, & Ishibashi T (2002). Photocoagulation-Induced Retinal Gliosis Is Inhibited by Systemically Expressed Soluble TGF-β Receptor Type II via Adenovirus Mediated Gene Transfer. Laboratory Investigation, 82(7), 863–870. 10.1097/01.LAB.0000018829.49754.DD [DOI] [PubMed] [Google Scholar]
- Hoang T, Wang J, Boyd P, Wang F, Santiago C, Jiang L, Yoo S, Lahne M, Todd LJ, Jia M, Saez C, Keuthan C, Palazzo I, Squires N, Campbell WA, Rajaii F, Parayil T, Trinh V, Kim DW, … Blackshaw S (2020). Gene regulatory networks controlling vertebrate retinal regeneration. Science, 370(6519). 10.1126/science.abb8598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S-M, Sharma G, Verma R, Byun S, Rudra D, & Im S-H (2018). Inflammation-induced Id2 promotes plasticity in regulatory T cells. Nature Communications, 9(1), 4736. 10.1038/s41467-018-07254-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, & Tamaki K (2006). Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Molecular Pharmacology, 69(2), 597–607. 10.1124/mol.105.017483 [DOI] [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, Wohl SG, VandenBosch LS, Yoshimatsu T, Wong RO, Rieke F, & Reh TA (2017). Stimulation of functional neuronal regeneration from Muller glia in adult mice. Nature. 10.1038/nature23283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Todd L, Finkbeiner C, Nakamura P, Radulovich N, Hooper MJ, Chitsazan A, Wilkerson BA, Rieke F, & Reh TA (2020). STAT Signaling Modifies Ascl1 Chromatin Binding and Limits Neural Regeneration from Muller Glia in Adult Mouse Retina. Cell Reports, 30(7), 2195–2208.e5. 10.1016/j.celrep.2020.01.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlen SJ, Miller EB, Wang X, Levine ES, Zawadzki RJ, & Burns ME (2018). Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. Journal of Neuroinflammation, 15(1), 1–16. 10.1186/s12974-018-1365-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Nagashima M, Hyde DR, & Hitchcock PF (2020). Reprogramming Müller Glia to Regenerate Retinal Neurons. Annual Review of Vision Science, 6, 171–193. 10.1146/annurev-vision-121219-081808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Julien F, Duplan L, Pernet V, Osswald I, Sapieha P, Bourgeois P, Dickson K, Bowie D, Barker PA, & Di Polo A (2009). Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J Neurosci, 29(17), 5536–5545. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19403821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-S, Wan J, & Goldman D (2020). Tgfb3 collaborates with PP2A and notch signaling pathways to inhibit retina regeneration. ELife, 9, e55137. 10.7554/eLife.55137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski JR, Qin Z, Sifuentes CJ, Thummel R, Soto CM, Moens CB, & Raymond PA (2013). Retinal regeneration in adult zebrafish requires regulation of TGFbeta signaling. Glia, 61(10), 1687–1697. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23918319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski JR, & Raymond PA (2014). Muller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Prog Retin Eye Res, in press(40), 94–123. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=24412518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZW, Omori SA, Labuda T, Karin M, & Rickert RC (2003). IKK beta is required for peripheral B cell survival and proliferation. J Immunol, 170(9), 4630–4637. https://www.ncbi.nlm.nih.gov/pubmed/12707341 [DOI] [PubMed] [Google Scholar]
- Lund T, Holtlund J, Fredriksen M, & Laland SG (1983). On the presence of two new high mobility group-like proteins in HeLa S3 cells. FEBS Letters, 152(2), 163–167. 10.1016/0014-5793(83)80370-6 [DOI] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, & Jobin C (2004). In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol, 173(3), 1561–1570. https://www.ncbi.nlm.nih.gov/pubmed/15265883 [DOI] [PubMed] [Google Scholar]
- Melo J. de, Zibetti C, Clark BS, Hwang W, Miranda-Angulo AL, Qian J, & Blackshaw S (2016). Lhx2 Is an Essential Factor for Retinal Gliogenesis and Notch Signaling. Journal of Neuroscience, 36(8), 2391–2405. 10.1523/JNEUROSCI.3145-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DM, Lovel AG, & Stenkamp DL (2018). Dynamic changes in microglial and macrophage characteristics during degeneration and regeneration of the zebrafish retina. Journal of Neuroinflammation, 15, 163. 10.1186/s12974-018-1185-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Koren EG, Mathew R, & Saban DR (2016). Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Scientific Reports, 6, 20636. 10.1038/srep20636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooto S, Akagi T, Kageyama R, Akita J, Mandai M, Honda Y, & Takahashi M (2004). Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc Natl Acad Sci U S A, 101(37), 13654–13659. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15353594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn L, Kunkel S, & Nabel GJ (1989). Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proceedings of the National Academy of Sciences of the United States of America, 86(7), 2336–2340. 10.1073/pnas.86.7.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo I, Deistler K, Hoang TV, Blackshaw S, & Fischer AJ (2020). NF-κB signaling regulates the formation of proliferating Müller glia-derived progenitor cells in the avian retina. Development. 10.1242/dev.183418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi S, Piscitelli S, Passaro F, & Russo T (2020). HMGA Proteins in Stemness and Differentiation of Embryonic and Adult Stem Cells. International Journal of Molecular Sciences, 21(1), 362. 10.3390/ijms21010362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, & Player MR (2009). Colony-stimulating factor-1 receptor inhibitors for the treatment of cancer and inflammatory disease. Current Topics in Medicinal Chemistry, 9(7), 599–610. 10.2174/156802609789007327 [DOI] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma Y-A, & Trapnell C (2017). Single-cell mRNA quantification and differential analysis with Census. Nature Methods, 14(3), 309–315. 10.1038/nmeth.4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, & Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nature Methods, 14(10), 979–982. 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy S, McGuire PG, Franco Nitta C, Monickaraj F, Oruganti SR, & Das A (2014). Chemokine Mediated Monocyte Trafficking into the Retina: Role of Inflammation in Alteration of the Blood-Retinal Barrier in Diabetic Retinopathy. PLoS ONE, 9(10), e108508. 10.1371/journal.pone.0108508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach A, & Bringmann A (2013). New functions of Muller cells. Glia, 61(5), 651–678. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23440929 [DOI] [PubMed] [Google Scholar]
- Robbie SJ, Georgiadis A, Barker SE, Duran Y, Smith AJ, Ali RR, Luhmann UFO, & Bainbridge JW (2016). Enhanced Ccl2-Ccr2 signaling drives more severe choroidal neovascularization with aging. Neurobiology of Aging, 40, 110–119. 10.1016/j.neurobiolaging.2015.12.019 [DOI] [PubMed] [Google Scholar]
- Robel S, Berninger B, & Götz M (2011). The stem cell potential of glia: Lessons from reactive gliosis. Nature Reviews Neuroscience, 12(2), 88–104. 10.1038/nrn2978 [DOI] [PubMed] [Google Scholar]
- Rutar M, Natoli R, Valter K, & Provis JM (2011). Early Focal Expression of the Chemokine Ccl2 by Müller Cells during Exposure to Damage-Inducing Bright Continuous Light. Investigative Ophthalmology & Visual Science, 52(5), 2379–2388. 10.1167/iovs.10-6010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Okuwaki T, Ushikubo H, Mori A, Nakahara T, & Ishii K (2017). Activation inhibitors of nuclear factor kappa B protect neurons against the NMDA-induced damage in the rat retina. Journal of Pharmacological Sciences, 135(2), 72–80. 10.1016/j.jphs.2017.09.031 [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, & Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol, 33(5), 495–502. 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, & ter Meulen V (1991). Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proceedings of the National Academy of Sciences of the United States of America, 88(16), 7438–7442. 10.1073/pnas.88.16.7438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SN, Kerr C, Cope L, Zambidis E, Liu C, Hillion J, Belton A, Huso DL, & Resar LMS (2012). HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks. PloS One, 7(11), e48533. 10.1371/journal.pone.0048533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Gupta S, Chaudhary M, Mitra S, Chawla B, Khursheed MA, Saran NK, & Ramachandran R (2020). Biphasic Role of Tgf-β Signaling during Müller Glia Reprogramming and Retinal Regeneration in Zebrafish. IScience, 23(2), 100817. 10.1016/j.isci.2019.100817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massagué J, & Benezra R (2013). TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Reports, 5(5), 1228–1242. 10.1016/j.celrep.2013.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, & Glass CK (2000). 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proceedings of the National Academy of Sciences of the United States of America, 97(9), 4844–4849. 10.1073/pnas.97.9.4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumter TF, Xian L, Huso T, Koo M, Chang Y-T, Almasri TN, Chia L, Inglis C, Reid D, & Resar LMS (2016). The High Mobility Group A1 (HMGA1) Transcriptome in Cancer and Development. Current Molecular Medicine, 16(4), 353–393. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5408585/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappeiner C, Maurer E, Sallin P, Bise T, Enzmann V, & Tschopp M (2016). Inhibition of the TGFβ Pathway Enhances Retinal Regeneration in Adult Zebrafish. PLoS ONE, 11(11), e0167073. 10.1371/journal.pone.0167073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JL, Ranski AH, Morgan GW, & Thummel R (2016). Reactive gliosis in the adult zebrafish retina. Experimental Eye Research, 143, 98–109. 10.1016/j.exer.2015.09.017 [DOI] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Enright JM, Nelson CM, Montgomery JE, & Hyde DR (2008). Characterization of Müller glia and neuronal progenitors during adult zebrafish retinal regeneration. Experimental Eye Research, 87(5), 433–444. 10.1016/j.exer.2008.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Finkbeiner C, Wong CK, Hooper MJ, & Reh TA (2020). Microglia Suppress Ascl1-Induced Retinal Regeneration in Mice. Cell Reports, 33(11), 108507. 10.1016/j.celrep.2020.108507 [DOI] [PubMed] [Google Scholar]
- Todd L, Palazzo I, Squires N, Mendonca N, & Fischer AJ (2017). BMP- and TGFbeta-signaling regulate the formation of Muller glia-derived progenitor cells in the avian retina. Glia. 10.1002/glia.23185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Palazzo I, Suarez L, Liu X, Volkov L, Hoang TV, Campbell WA, Blackshaw S, Quan N, & Fischer AJ (2019). Reactive microglia and IL1β/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. Journal of Neuroinflammation, 16(1), 118. 10.1186/s12974-019-1505-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Volkov LI, Zelinka C, Squires N, & Fischer AJ (2015). Heparin-binding EGF-like growth factor (HB-EGF) stimulates the proliferation of Muller glia-derived progenitor cells in avian and murine retinas. Mol Cell Neurosci, 69, 54–64. 10.1016/j.mcn.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, & Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc, 7(3), 562–578. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22383036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y, Wilken MS, Cox KE, Chipman L, Jorstad N, Sternhagen K, Simic M, Ullom K, Nakafuku M, & Reh TA (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proceedings of the National Academy of Sciences of the United States of America, 112(44), 13717–13722. 10.1073/pnas.1510595112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Chona FR, Clark AM, & Levine EM (2009). Rlbp1 promoter drives robust Muller glial GFP expression in transgenic mice. Invest Ophthalmol Vis Sci, 50(8), 3996–4003. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19324864 [DOI] [PubMed] [Google Scholar]
- Viñals F, Reiriz J, Ambrosio S, Bartrons R, Rosa JL, & Ventura F (2004). BMP-2 decreases Mash1 stability by increasing Id1 expression. The EMBO Journal, 23(17), 3527–3537. 10.1038/sj.emboj.7600360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ma W, Zhao L, Fariss RN, & Wong WT (2011). Adaptive Muller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation, 8, 173. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22152278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, & Wong WT (2014). Microglia-Muller cell interactions in the retina. Adv Exp Med Biol, 801, 333–338. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=24664715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DT, Sengupta S, Saxena MT, Xu Q, Hanes J, Ding D, Ji H, & Mumm JS (2017). Immunomodulation-accelerated neuronal regeneration following selective rod photoreceptor cell ablation in the zebrafish retina. Proceedings of the National Academy of Sciences of the United States of America, 114(18), E3719–E3728. 10.1073/pnas.1617721114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnarowicz PM, Lima e Silva R, Ohnaka M, Lee SB, Chin Y, Kulukian A, Chang S-H, Desai B, Garcia Escolano M, Shah R, Garcia-Cao M, Xu S, Kadam R, Goldgur Y, Miller MA, Ouerfelli O, Yang G, Arakawa T, Albanese SK, … Benezra R (2019). A Small-Molecule Pan-Id Antagonist Inhibits Pathologic Ocular Neovascularization. Cell Reports, 29(1), 62–75.e7. 10.1016/j.celrep.2019.08.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zeng Q, Barış M, & Tezel G (2020). Transgenic inhibition of astroglial NF-κB restrains the neuroinflammatory and neurodegenerative outcomes of experimental mouse glaucoma. Journal of Neuroinflammation, 17(1), 1–18. 10.1186/s12974-020-01930-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Roubeix C, Sennlaub F, & Saban DR (2020). Perspective: Microglia and Monocyte Distinct Roles in Degenerative Diseases of the Retina. Trends in Neurosciences, 43(6), 433–449. 10.1016/j.tins.2020.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Tso MOM, Lai S, & Lai H (2008). Activation of nuclear factor-κB during retinal degeneration in rd Mice. Molecular Vision, 14, 1075–1080. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2426720/ [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Lenardo MJ, & Baltimore D (2017). 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell, 168(1–2), 37–57. 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: Expression of NFKB factors across pseudotime in damaged Müller glia. Pseudotime trajectory plots of MG from control and 3, 6, 12, 24, 36, 48 and 72h after NMDA damage separate into 3 distinct clusters (a). Resting glia are identified by expression of Glul, activated glia are identified by expression of Scg2 and Srxn1. The pseudotime trajectory plots (b) demonstrate Nfkbia, Nfkbib, Nfkbiz enriched in activated glia. Pseudotime reduction plots (c) and violin plots (d) illustrate change in expression levels of Nfkb1, Nfkb2, Nfkbia, Nfkbib, Nfkbiz, Ikbkb, Rela, and Relb. (e) qRT-PCR was used to measure changes in expression (fold-change) of Rela, Nfkb1, Nfkbia, Ikbkb, Tgfb2, Id1 and Id3 in whole retinas at 4hrs after NMDA treatment. Significance of difference (*p<0.05, **p<0.01, ***p<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction (d) or t-test (e).
Supplemental Fig. S2: NFkB factors expressed in microglia. UMAP plots of isolated and re-embedded microglia from WT control and NMDA 3, 6, 12, 24, 36, 48 and 72h scRNA-Seq libraries show 4 distinct clusters (a,b). Clusters were identified based on expression of Apoe, Aif1, Cyba, Klf2, and Ids (c). Violin plots illustrate levels of expression and percentage of expressing cells for Nfkb1, Nfkb2, Chuk, Nfkbia, Nfkbib, Nfkbiz, Rela, Relb, Ikbkb in different UMAP clusters of microglia (d). Significance of difference (**p<0.01, ***p<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction.
Supplemental Fig. S3: scRNA-Seq for whole retina Ikkb-cKO. UMAP plots for whole retina scRNA-seq libraries of Rlbp1-CreERT (control) and Rlbp1-CreERT:Ikkbfl/fl (treated) mice at 8h after NMDA damage (a). Distinct cell clusters of UMAP ordered cells (b) were characterized based on expression of markers such as Rlbp1, Slc1a3 and Glul (c) and DEGs identified from heatmap plots (d).
Supplemental Fig. S4: Expression patterns Ccl2 in WT scRNA-Seq libraries from control and damaged retinas. Aggregated scRNA-seq libraries were prepared from WT control retinas and retinas 3, 6,12, 24, 36, 48 and 72h after NMDA damage (a). Clusters were identified based on expression of cell-distinguishing markers, as described in the Methods (b). Resting MG and reactive MG identified by expression of Slc1a3 or Nes and Vim, respectively (c). UMAP plots illustrate patterns of expression of Ccl2 across retinal cell types (d). Violin plots illustrate expression levels Ccl2 in MG (e). Significance of difference (**p<0.001, **p<1×10–10, ***p<1×10–20) was determined by using a Wilcoxon rank sum with Bonferroni correction.
Supplemental Fig. S5: Validation of ANT regeneration paradigm. Experimental paradigm outlined at top. Retinal sections were labeled for GFP (green), DAPI (blue), Sox2 (red; a), or Otx2 (red; c). Arrows indicate double-labeled cells. Histograms illustrate the mean percentage (±SD and individual data points) of GFP+/Sox2+ (b) or GFP+/Otx2+ (d) cells. Significance of difference (p-values shown) was determined by using a paired t-test.
Supplemental Fig. S6: Inhibition of NFkB signaling promotes proliferation and neuroprotection following ANT treatment. Experimental paradigm outlined at top. Tamoxifen was administered IP 1x daily for 4 consecutive days in Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice, NMDA was injected intravitreally in left (control) eyes or NMDA+PGJ2 in right (treated) eyes on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested 4 days after TSA. EdU was added to the drinking water and administered starting on D7 and sustained until harvesting retinas on D14. Retinal sections were labeled for GFP (green), Otx2 (red;a-b), Sox2 (red;c-d), and EdU (blue). Arrows in a and b indicate GFP+/EdU+/Otx2+ cells, and arrows in c and d indicate GFP+/EdU+/Sox2+ cells. TUNEL assays were performed on ANT or ANT+PGJ2 treated retinas at 24h after NMDA (e). Retinal sections were labeled for GFP (green), Iba1 (red), and Draq5 (blue) (g). Histograms illustrate the mean (±SD and individual data points) number of TUNEL+ cells (f) or Iba1+ cells (h). Significance of difference (**p<0.01) was determined by using a Kruskal-wallis test (f), or paired t-test (h).
Supplemental Fig. S7: scRNA-Seq libraries from Ascl1 overexpressing mice. scRNA seq libraries were prepared from Rlbp1-CreERT and Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. Tamoxifen was injected IP 1x daily for 4 consecutive days. NMDA was injected intravitreally into left (control) eyes and NMDA+PGJ2 in right (treated eyes) on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested for scRNA-seq library prep 24h after TSA treatment. Samples were enriched via FACS for either GFP+ cells and CD45+/Cd11b+ cells (from Ascl1 mice) or TdTomato+ cells and CD45+/Cd11b+ cells (from Rlbp1-CreERT mice). Clusters of different types of retinal cells were identified based on expression patterns of different cell-distinguishing markers (a). (b-c) UMAP plots of aggregated scRNA-seq libraries for MG and immune cells. (d) UMAP plots of isolated and re-embedded MG from Rlbp1-CreERT control (NMDA) and treated (NMDA+PGJ2) and Ascl1 control (NMDA) and treated (NMDA+PGJ2). Stacked bar graphs illustrate UMAP cluster occupancy by library origin identity (d). The dot plot in (e) illustrates changes in expression of resting and reactivity genes, cell signaling factors, and transcription factors. Dot size represents percent of cells expressing and dot color representing expression levels.
Supplemental Fig. 8: Decreased expression of genes associated with inflammatory signaling in immune cells from ANT+PGJ2 scRNA-seq libraries. scRNA seq libraries were prepared from Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. Tamoxifen was injected IP 1x daily for 4 consecutive days. NMDA was injected intravitreally into left (control) eyes and NMDA+PGJ2 in right (treated eyes) on D8, vehicle ± PGJ2 on D9, TSA ± PGJ2 on D10, and retinas were harvested for scRNA-seq library prep 24h after TSA treatment. Samples were enriched via FACS for GFP+ cells and CD45+/Cd11b+ cells. UMAP plots of immune cells from ANT and ANT+PGJ2 scRNA-seq libraries (a-b). Types of immune cells were identified based on expression of cell type-specific markers (c). Heatmap of differentially expressed genes (adj_p<0.05) (d). Dot Plot illustrates the percent of expressing cells (dot size) and genes that were upregulated (red) or downregulated (blue) in ANT and ANT+PGJ2 scRNA-seq libraries (e).
Supplemental Fig. S9: Ascl1-overexpressing MG fail to differentiate into amacrine cells. Experimental paradigm outlined at top. Tamoxifen was administered IP 1x daily for 4 consecutive days to Glast-CreER:LNL-tTA:tetO-mAscl1-ires-GFP mice. NMDA was injected intravitreally in left (control) eyes and NMDA+ PGJ2, or SIS3, or AGX51 in right (treated) eyes on D8, vehicle ± inhibitor on D9, TSA ± inhibitor on D10, and retinas were harvested 2 weeks after TSA injection. Retinal sections were labeled for GFP (green), DAPI (blue), and HuC/D (red) (a). Histograms illustrate mean percentage (±SD and individual data points) of GFP+ cells that are HuC/D+ (b-d). Significance of difference (p-values shown) was determined by using a paired t-test. Arrows in indicate GFP+ cells negative for HuC/D. Calibration bar in panel a represents 50 μm. Abbreviations: ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Data Availability Statement
gene-cell matrices for scRNA-seq data are deposited at GitHub https://github.com/jiewwwang/Single-cell-retinal-regeneration
https://github.com/fischerlab3140/
Some of the scRNA-seq data can be queried at https://proteinpaint.stjude.org/F/2019.retina.scRNA.html.