

Abstract
The devastating human pathogenMycobacterium tuberculosis (Mtb) is able to parasitize phagosomal compartments within alveolar macrophage cells due, in part, to the activity of its cell-surface lipids. Prominent among these is 1-tuberculosinyl-adenosine (1-TbAd), a derivative of the diterpenoid tuberculosinyl (halima-5,13-dienyl) diphosphate produced by the class II diterpene cyclase encoded by Rv3377c, termed here MtHPS. Given the demonstrated ability of 1-TbAd to act as a virulence factor for Mtb and the necessity for Rv3377c for its production, there is significant interest in MtHPS activity. Class II diterpene cyclases catalyze a general acid–base-mediated carbocation cascade reaction initiated by protonation of the terminal alkene in the general diterpenoid precursor (E,E,E)-geranylgeranyl diphosphate and terminated by deprotonation of the final cyclized (and sometimes also rearranged) intermediate. Here, structure-guided mutagenesis was applied to characterize the various residues contributing to activation of the enzymatic acid, as well as identify the enzymatic base in MtHPS. Particularly given the ability of conservative substitution for the enzymatic base (Y479F) to generate an alternative product (labda-7,13-dienyl diphosphate) via deprotonation of an earlier unrearranged intermediate, further mutational analysis was carried out to introduce potential alternative catalytic bases. The results were combined with mechanistic molecular modeling to elucidate how these mutations affect the catalytic activity of this important enzyme. This not only provided detailed structure–function insight into MtHPS but also further emphasized the inert nature of the active site of MtHPS and class II diterpene cyclases more generally.
Keywords: biosynthesis, terpene synthases, diterpene cyclases, diterpenoids, enzymology, natural products, tuberculosis, carbocation cascade
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of the human disease tuberculosis, which is a leading cause of death globally.1 This devastating pathogen is known to parasitize the endocytic compartments in which the bacterium is taken up by alveolar macrophage cells, blocking maturation of these into phagosomes and, hence, enabling Mtb persistence in human hosts.2 The ability of Mtb to block phagosomal maturation is due, at least in part, to the effects exerted by its unusual cell wall lipids.3 Prominent among these is the unique diterpene nucleoside 1-tuberculosinyl-adenosine (1-TbAd),4 which not only blocks acidification of endocytic compartments but also modulates their morphology.5
Production of 1-TbAd depends on Mtb genes Rv3377c and Rv3378c, encoding a class II diterpene cyclase6 and diterpenyl-adenosine transferase,7 respectively. The importance of this was first shown from a genetic screening targeted at the early stages of the Mtb infection process, specifically the ability of the bacterium to block acidification of the endocytic compartment, which identified roles for Rv3377c and Rv3378c.8 The ability of the class II diterpene cyclase encoded by Rv3377c to react with the general diterpenoid precursor (E,E,E)-geranylgeranyl diphosphate (GGPP, 1) to produce halima-5,13-dienyl diphosphate (HPP, 2), also termed tuberculosinyl diphosphate, was then quickly elucidated.6 While initial studies suggested that the enzyme encoded by Rv3378c might act as a phosphatase,9−11 with matching products found in Mtb cultures,12,13 it was later found that this primarily serves as an adenosine transferase that produces 1-TbAd, which was found to be produced in substantial quantities by Mtb and was actually first identified via a lipidomics screening.7 Indeed, 1-TbAd can serve as a biomarker for Mtb infection.14,15
Rv3377c and Rv3378c are part of a small biosynthetic operon that is present in only Mtb and Mycobacterium bovis.6,8 However, M. bovis is unable to produce 1-TbAd due to an inactivating frame-shift mutation in the gene (Mb3411c) encoding the halimadienyl diphosphate synthase (HPS), leaving production of 1-TbAd specific to the human pathogen Mtb.16 The operon also contains genes encoding enzymes acting in the upstream isoprenoid precursor supply pathway and for the subsequent more specific production of 1, but these are redundant, as other genes found in Mtb, and the Mycobacterium genus more broadly encodes enzymes with equivalent activity.17 Indeed, expression of just Rv3377c and Rv3378c is sufficient to impart production of 1-TbAd in other Mycobacterium species.7,18 This highlights the importance of the two encoded enzymes in tuberculosis, with the Rv3377c-encoded MtHPS catalyzing a particularly interesting reaction involving rearrangement following initial bicyclization (Figure 1A).19
Figure 1.

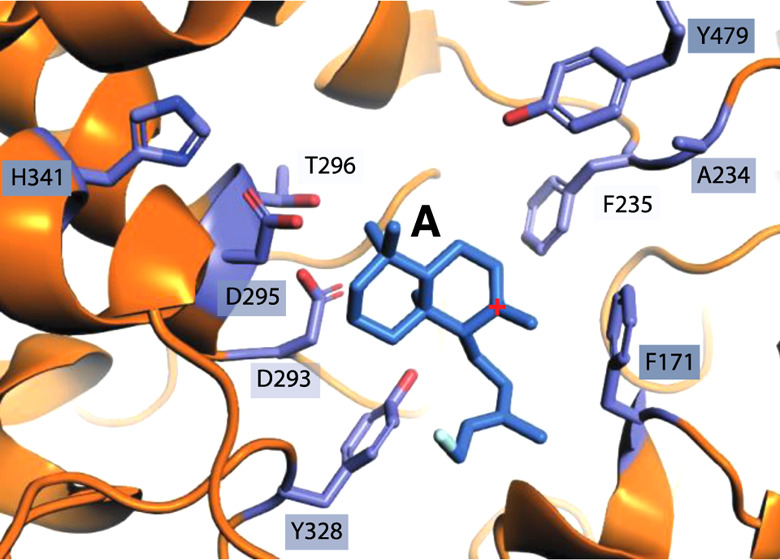
(A) Scheme for bicyclization and rearrangement of GGPP (1) to HPP (2) catalyzed by MtHPS. (B) TerDockin-based depiction of intermediate A (with diphosphate removed for clarity) docked into the MtHPS active site, with residues targeted for mutagenesis indicated (only side chains shown).20
As a class II diterpene, cyclase MtHPS utilizes a general acid–base mechanism.21 The catalytic acid is the middle aspartic acid from a highly conserved DxDD motif,22 which characterizes this enzymatic family as well as the ancestral squalene-hopene cyclases.23 The ability of this aspartic acid to protonate the terminal alkene of 1 to initiate the carbocationic bicyclization reaction is promoted not only by the flanking aspartates, which hydrogen-bond each other and ensure a nearby negative charge to impose the protonated state,24 but also by the activating hydrogen bond to another residue, typically conserved as a histidine in bacteria and asparagine in plants,23,25 as this helps establish the more reactive anti-orientation.26 By contrast, the catalytic base is not broadly conserved as it must be proximal to the final carbocation, whose position relative to the conserved DxDD motif may vary depending on the product outcome, including bicycle configuration (stereochemistry) and/or rearrangement.27 Nevertheless, it has been possible to identify the catalytic base in certain cases, most notably in the family members producing ent-copalyl diphosphate for gibberellin phytohormone biosynthesis in not only plants but also bacteria, where a conserved dyad composed of a histidine and an asparagine cooperatively serves this function.28,29 Notably, these residues seem to be important for shaping the product outcome in this enzymatic family more broadly.30−35
In the case of MtHPS, it has been noted that the catalytic acid motif contains a threonine in place of the last aspartate (i.e., DxDT rather than DxDD), with replacement by aspartate suggested to inactivate the enzyme.6,19 Kinetic analysis of MtHPS demonstrated inhibition by the divalent magnesium ion cofactor at concentrations above 0.1 mM.16,19 Intriguingly, magnesium levels are reduced to approximately this concentration during phagosomal maturation and such depletion of this cation is sufficient to induce MtHPS-dependent diterpenoid accumulation in Mtb cell cultures.13 In addition, building on previous work with plant class II diterpene cyclases,36 mimics of the carbocation initially formed by protonation of 1 (15-aza-dihydrogeranylgeranyl diphosphate and its thiolo analogue) were found to be potent inhibitors of MtHPS,16 as were the dephosphorylated analogue and even simpler plant growth inhibitor Amo-1618.19 More recently, a crystal structure has been reported for MtHPS along with mechanistic modeling of the catalyzed reaction using the TerDockin approach (Figure 1B).20 Here, structure-guided mutagenesis was applied to characterize the various residues contributing to activation of the enzymatic acid, as well as identify the enzymatic base in MtHPS, with the results combined with further mechanistic molecular modeling to elucidate the catalytic mechanism utilized by this important enzyme and further emphasize the inert nature of the active site of not only MtHPS but also class II diterpene cyclases more generally.
Methods
General
All reagents were purchased from Fisher Scientific unless otherwise noted.
Recombinant Constructs
For construction of recombinant expression vectors, MtHPS was cloned into pDEST14 using the Invitrogen Gateway System. All mutants were generated via site-directed mutagenesis performed using overlapping primers and whole plasmid PCR amplification verified by complete gene sequencing. The mutant enzymatic function was assessed through cotransformation with the compatible, previously described pGI vector (this contains the GGPP synthase and isopentenyl diphosphate isomerase from the gibberellin biosynthetic operon found in Erwinia tracheiphila).28
Assessment of CPP stereochemistry employed stereoselective class I DTSs recombined into pDEST14 expression vectors. An alternative system was employed to enable the necessary coexpression. In particular, the GGPP synthase from Abies grandis and MtHPS were amplified by the PCR designed with overlapping regions of homology to the amplified, linear expression vector. This expression vector was originally based on pDEST14 but altered to replace the antibiotic resistance gene with that for spectinomycin and the origin of replication with CloDF13, both originating from the pCDFDuet vector. These three linear fragments were then hybridized together and amplified by PCR into a single circularized plasmid, the nicks of which were sealed by endogenous ligases upon transformation into Escherichia coli. The resulting vector provided efficient flux of the class II product to the class I DTS expressed from the compatible pDEST14 expression vector.
Metabolic Engineering
Relevant expression constructs were transformed into the BL21-Star strain of E. coli, and the recombinant strains were used for functional screening via metabolic engineering. Recombinant cultures were inoculated into 45 mL of liquid TB media with appropriate antibiotics in 250 mL Erlenmeyer flasks. These cultures were first shaken at 37 °C and 180 rpm until an OD600 of 0.4–0.6 was reached and then phosphate buffer (pH 7.0) was added to 100 mM, pyruvate to 40 mM, and MgCl2 to 1 mM final concentrations were added. The temperature was decreased to 16 °C for 0.5 h, and the cultures were induced with 1 mM IPTG. After 72 h growth at 16 °C, enzymatic products were extracted from the metabolically engineered cultures via the addition of 1:1 hexanes and gentle shaking. The organic solvent was separated and concentrated under N2 and analyzed by GC–MS.
Diterpene Product Analysis by GC–MS
GC–MS analyses were carried out using an Agilent 8890 GC System with a 5977B mass spectrometer in electron ionization (70 eV) mode using an Agilent HP-5MS column (Agilent, 19091S-433) with a 1 mL/min helium flow rate. Samples were introduced (1 μL) in a splitless mode by a 7650A automatic liquid sampler by injection at 250 °C. The following temperature program is used: initial column oven temperature set to 50 °C, maintained for 3 min, followed by a ramp rate of 15 °C/min to 300 °C, where the temperature was held for 3 min. Mass spectra were recorded by mass-to-charge ratio (m/z) values in the range from 90 to 600, with detection beginning 13 min after sample injection for completion of the run.
Docking Simulations
Mechanistically relevant intermediates A and B (Scheme 1) had been previously optimized by our group using the mPW1PW91//6-31+G(d,p) level of theory.20 In line with the previous paper, both truncated (isoprenyl diphosphate group substituted by a methyl group) and nontruncated (built by adding the relevant chain substituents from A) intermediates A and B were docked. The TerDockin method37−40 was employed to dock intermediates A and B into the MtHPS double-mutant Y328F/Y479F (PDB 6VPT) protein structure, while maintaining the Asp295—carbocation intermediate constraint used previously (representing the initial protonation between the DxDD motif and GGPP).20 Each simulation generated 10,000 poses, which were then filtered based on constraint satisfaction, total energy after Tukey’s trimean assessment (see the Supporting Information; keeping all of the poses one standard deviation below the mean), and interface energy (keeping 10% of the lowest energy population). Based on results obtained with truncated intermediate B (see the Supporting Information), a model including a water molecule as the potential base was examined. The water molecule was included as part of the substrate, and its acidic C–H bond was elongated to 1.5 Å to mimic a breaking bond. The water molecule was coordinated with Asp293 during docking, which was carried out as above.
Results and Discussion
While some mutational analysis of MtHPS has been previously reported,6,19 that work utilized a 6xHis-tagged construct to carry out in vitro assays for kinetic analysis. To focus on altered product outcome, the work reported here utilizes untagged constructs expressed in E. coli engineered to produce substrate 1. The resulting cultures are extracted and product outcomes are analyzed by GC–MS, with detection of the dephosphorylated derivatives produced by endogenous phosphatases such as geranylgeraniol (1′) from 1 and halima-5,13-dien-15-ol (2′) from 2 (Figure 2A). As previously reported,41,42 cultures expressing wild-type MtHPS only yield 2, with no observation of 1, demonstrating robust activity (Figure 2B). Given mutational changes can affect protein folding and/or stability as well as catalytic activity, only effects on the metabolic flux (i.e., the relative yield from 1) are measured here.
Figure 2.

Investigation of catalytic acid. (A) Scheme for dephosphorylation to the detected alcohols. (B) Extracted ion (m/z = 275) chromatograms from GC–MS analysis of extracts from E. coli engineered to produce 1 and express the wild-type (WT) or indicated mutant of MtHPS. (C) TerDockin-based depiction of activating hydrogen bonds (dotted yellow lines) formed between D293 and T296, as well as H341 and D295, which acts as the general acid.20
To verify the importance of the DxDT motif for MtHPS activity, alanine was substituted for the middle aspartic acid of the motif (D295A), which was found to almost entirely abrogate the production of 2, with essentially only 1 observed. Particularly given that the equivalent position in squalene-hopene cyclases has been shown to impact the product outcome,43 the highly unusual presence of threonine in place of the last aspartate (T296) was then probed by site-directed mutagenesis. Specifically, this threonine was substituted with three other amino acids, i.e., alanine, glycine, and aspartate. Although none of the mutants led to an altered product outcome, there was some effect on the yield (Figure 2B). Somewhat surprisingly, the T296A mutant still mediated significant amounts of metabolic flux, producing 2 with only very small amounts of 1 observed, while less metabolic flux was observed with the T296G mutant, albeit with more 2 observed than 1. By contrast, changing this threonine back to the presumably ancestral aspartate led to even lower metabolic flux, with more 1 than 2 accumulating with the T296D mutant. While this contrasts with the complete loss of activity reported for the T296D mutant in the context of in vitro assays,19 it seems likely that there is a substantial decrease in catalytic efficiency by replacement of this threonine with aspartate. This suggests that MtHPS has adapted to use this threonine to recreate the catalytic acid group formed by the DxDD motif in other class II diterpene cyclases. In particular, the hydrogen bond observed between T296 and D293 (Figure 2C) positions the latter, presumably negatively charged aspartate to activate D295 (i.e., as an aspartic acid).
To probe the importance of the interaction between the middle aspartic acid (D295) and the conserved activating histidine (H341), H341 was substituted with alanine, as well as asparagine. Given the presence of asparagine at the corresponding position in plant class II diterpene cyclases, it is perhaps not surprising that the H341N mutant still mediated a significant amount of metabolic flux, producing 2 with only very small amounts of 1 observed (Figure 2B). It seems somewhat counterintuitive that the H341A mutant also exhibits substantial metabolic flux, with still only relatively small amounts of 1 observed. However, it should be noted that in the MtHPS crystal structure D295 and H341 do not directly interact, only sharing hydrogen bonding to a common water molecule, although the reported modeling does restore the direct interaction seen in other class II diterpene cyclase structures (even in the absence of the substrate or intermediate analogues).20 Regardless, these results are largely consistent with the previous investigation of plant class II diterpene cyclases.24 Accordingly, despite the observed conservation of this interaction, it seems to be of somewhat limited importance.
From the MtHPS crystal structure and derived mechanistic modeling, it has been suggested that the catalytic base is an active site tyrosine (Y479) in close proximity to the hydrogen removed from carbon-6 (C6) of the final intermediate halima-13-en-5-yl+ diphosphate (D) to form 2 (Figure 3).20 This hypothesis was examined by site-directed mutagenesis, specifically replacement with phenylalanine (Y479F). Notably, the resulting MtHPS:Y479F exhibits very efficient metabolic flux but to an alternative product, labda-8,13-dienyl diphosphate (3). Given the derivation of 2 from rearrangement of the initially bicyclized intermediate A, which exhibits normal stereochemistry,19 it is assumed here that all alternative products such as 3 are derived from this same configuration of the reactant (and is shown in one case, see below). However, 3 is produced by removal of the hydrogen found at C9 in the initially formed bicyclic carbocation intermediate labda-13-en-8-yl+ diphosphate (A), rather than the rearranged D that is deprotonated to yield 2. Based on the precedent set by previous studies of class II diterpene cyclases where mutational alteration of the catalytic base leads to such alternative product outcomes,27−33 the Y479F mutation enables using an alternative/secondary base that produces 3 in MtHPS.
Figure 3.

Y479 acts as the catalytic base. (A) TerDockin-based depiction of the proximity of Y479 hydroxyl to hydrogen on C6 of final intermediate D removed to produce 2.20 (B) Extracted ion (m/z = 275) chromatogram from GC–MS analysis of the extract from E. coli engineered to produce 1 and express MtHPS:Y479F. (C) Structure of alternative product 3, labda-8,13-dienyl diphosphate (PP).
Initial examination of the MtHPS active site suggested that another tyrosine (Y328) might serve as this secondary base in the Y479F mutant. However, rather than blocking production of 3, the Y328F/Y479F double mutant selectively produced 3 and exhibited very efficient metabolic flux (Figure 4A). Indeed, the Y328F single mutant also produced significant amounts of 3, as well as 2, and mediated efficient metabolic flux. A closer examination of the recently reported modeling indicates that the Y328 hydroxyl group, while in reasonably close proximity to C9 (4.4 Å), is on the opposite side of the hydrogen substituent that is removed (Figure 4B). Accordingly, it seems that removal of this hydroxyl group with the Y328F mutant enables increased access to the actual secondary base.
Figure 4.

Y328 does not serve as the secondary catalytic base. (A) Extracted ion (m/z = 275) chromatograms from GC–MS analysis of the extract from E. coli engineered to produce 1 and express indicated mutants of MtHPS, leading to alternative products. (B) TerDockin-based depiction of the distance from Y328 to the hydrogen on C9 of intermediate A (diphosphate not depicted for clarity) removed to produce 3.20
Given the lack of any other obvious candidates to serve this function, the TerDockin approach was applied to the Y328F/Y479F double mutant to identify the relevant catalytic base. Intriguingly, the results suggest that a tryptophan (W380) shields the C9 hydrogen in A, ruling out direct deprotonation of this intermediate to generate 3. Accordingly, the possibility that this proton was removed following its migration in a 1,2-hydride shift to C8, which forms the labda-13-en-9-yl+ diphosphate intermediate (B), was examined. While initial studies suggested the possibility of a water molecule serving as the secondary base, this was not correctly oriented for deprotonation and was based on a truncated model of B (Figure S1). Further work with a full model for B indicated a distinct orientation where the C8 hydrogen is only 3.2 Å away and appropriately oriented toward the backbone carbonyl of I474 (Figure 5). Thus, it seems at least possible that this carbonyl acts as the secondary base that deprotonates C8 in B to generate 3. This is consistent with their relative pKa’s (approximately −7 for a protonated carbonyl but less than −10 for carbocations such as B), and a recent report that a backbone carbonyl serves the same function in a class I (sesqui)terpene synthase; i.e., as a catalytic base for carbocation deprotonation.44 Such deprotonation seems to be restricted in wild-type MtHPS due to the steric constraints on reactant orientation imposed by the hydroxyl groups of both Y328 and Y479, and removal via phenylalanine substitution then enables the reactive orientation shown in Figure 5, which is otherwise energetically unfavorable, explaining the results observed here.
Figure 5.
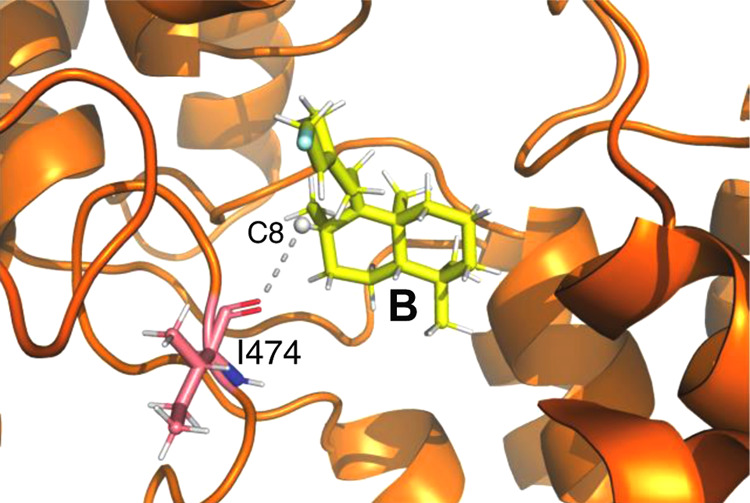
Use of the I457 backbone carbonyl as a secondary base for production of 3 via deprotonation at C8 in B (with diphosphate replaced by single blue atom for clarity) indicated by TerDockin.
Previous reports have highlighted the importance of the residues corresponding to the histidine–asparagine catalytic base dyad in the ent-CPP synthases (CPSs) from gibberellin phytohormone biosynthesis to product outcome, even in those from bacteria.28−35 Accordingly, the corresponding residues in MtHPS (F171 and A234, as determined by structure-based alignment to the only other structurally defined bacterial class II diterpene cyclase25 and previous sequence alignment28) were investigated here by mutational analysis. Although substitution of histidine for F171, as found in the CPSs, led to decreased metabolic flux (as evidenced by the observation of 1), this MtHPS:F171H mutant also produces significant amounts of copalyl diphosphate (CPP, 4), along with small amounts of 2 (Figure 6A,B). Coexpression of this with stereoselective class I diterpene synthases verified that 4 exhibits the expected normal stereochemistry (Figure S2) and must be derived from deprotontation of the methyl (C17) adjacent to the carbocation in A (note that 4 can also be labeled labda-8(17),13-dienyl diphosphate). Indeed, application of the TerDockin modeling approach suggested that the introduced histidine in F171H has its δN positioned for such deprotonation, sitting only 3.2 Å away from C17 in A (Figure 6C). Somewhat surprisingly, while substitution of F171 with aspartate essentially eliminates metabolic flux, some metabolic flux is observed with the MtHPS:F171A mutant, which forms small amounts of 2 and 3, along with trace amounts of labda-7,13-dienyl diphosphate (5), as well as an unknown product (Figure 6A, B, and D). On the other hand, substitution of A234 with asparagine, as also found in the CPSs, only reduces the metabolic flux without significantly altering the product outcome, yielding largely 2 (with only trace amounts of 3). Given that the following residue is conserved as a valine in the CPSs and this has shown to be important for the preceding asparagine to exert an effect on the production outcome,31 a double A234N/F235V mutant was constructed. Although small amounts of the same unknown product observed with the F171A mutant were found with MtHPS:A234N/F235V, this still predominantly produces 2, with trace amounts of 3 and 5, as well as exhibits some reduction in metabolic flux (Figure 6A). Introduction of all three residues from the CPSs led to an almost complete loss of metabolic flux, as only trace amounts of 2 were observed with the F171H/A234N/F235V triple mutant (Figure 6A). Perhaps more surprisingly, while combining replacement of the native base for production of 2 and introduction of the base for production of 4 was hypothesized to lead to more efficient production of 4, the MtHPS:F171H/Y479F double mutant instead produces a mixture of significant amounts of 3, as found with the Y479F mutation alone, and slightly larger amounts of 4.
Figure 6.
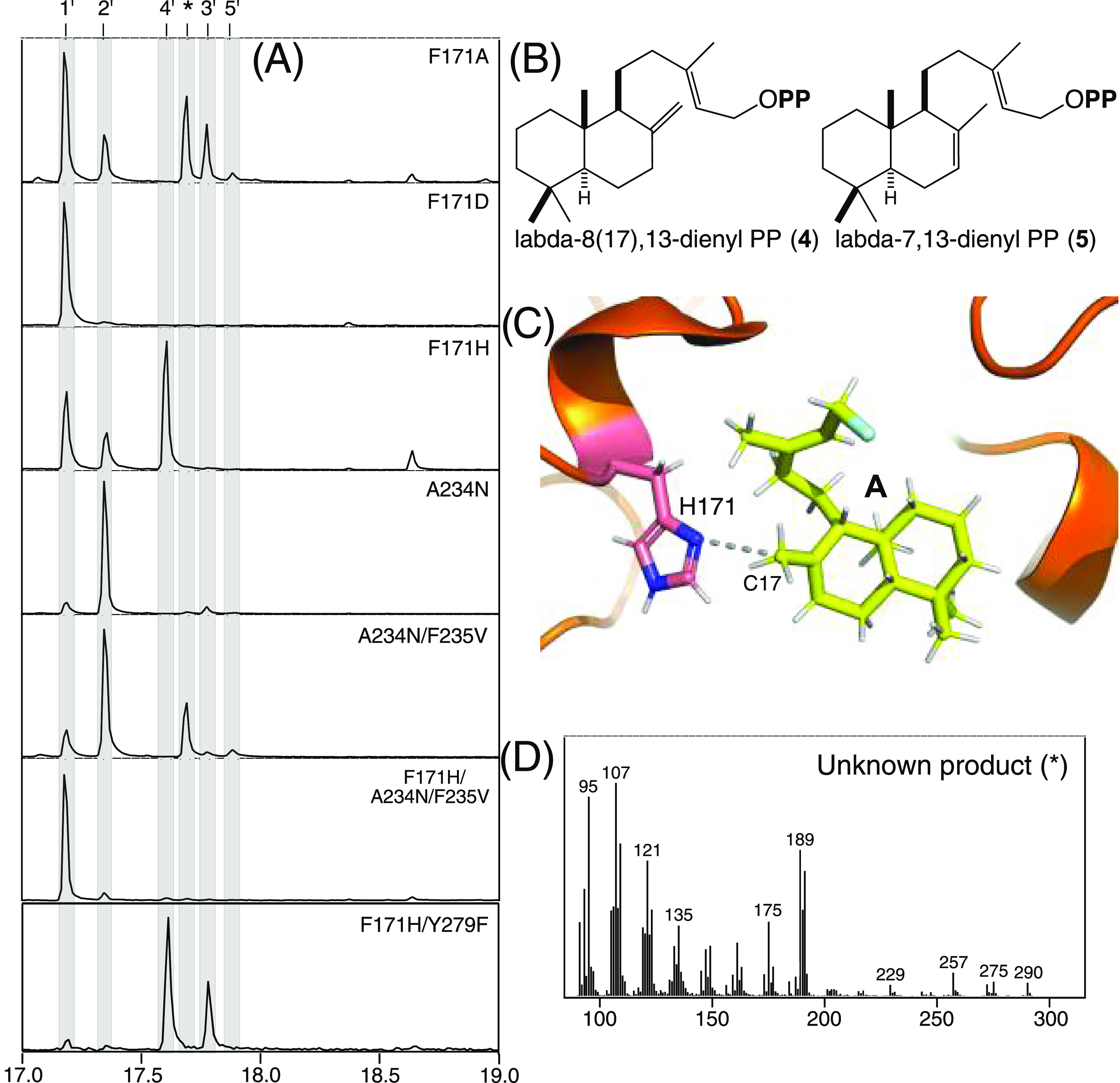
Substitution of histidine for F171 enables production of CPP (4). (A) Extracted ion (m/z = 275) chromatograms from GC–MS analysis of extracts from E. coli engineered to produce 1 and expressing indicated mutants of MtHPS. (B) Structures of alternative products 4, labda-8(17),13-dienyl diphosphate (PP), and 5, labda-7,13-dienyl PP (*, unknown product). (C) Depiction of the proximity of H171 (from MtHPS:F171H) to C17 of intermediate A (with diphospahte replaced by single blue atom for clarity), which is deprotonated to produce 4, indicated by TerDockin analysis. (D) Mass spectra of the unknown product (*).
These results further emphasize the importance of base positioning within the active site for class II diterpene synthases, especially upon comparison to previously reported studies. For example, mutational analysis of the CPS from Arabidopsis thaliana (AtCPS) demonstrated that substitution of another aromatic residue (tyrosine or phenylalanine) for the histidine of the catalytic base dyad led to complete rearrangement of the initially bicyclized ent-A, with removal of the originally added proton from C3 (presumably by the same “middle” aspartate of the DxDD catalytic acid motif), to generate ent-kolav-3,13-dienyl diphosphate.30 Given the results here suggest that a backbone carbonyl might serve as a catalytic base, this highlights the inert nature of the AtCPS active site. Indeed, the tryptophan noted above as shielding the C9 hydrogen is part of a (D/E)KW motif highly conserved in class II diterpene cyclases, and many other widely conserved aromatic residues line their active sites. These have been hypothesized to stabilize various carbocationic intermediates, and in certain cases, substitution for these residues has been shown to affect the activity, including the product outcome.27−35
Given that the highly exothermic bicyclization of 1 to A traps the reactant, and the precedent set by the facile conversion of AtCPS to production of ent-kolav-3,13-dienyl diphosphate,30 it might be asked why MtHPS:Y479F does not exhibit similar production of fully rearranged kolav-3,13-dienyl diphosphate. However, it has been shown that the last 1,2-methyl migration (from C4 to C5 in the final halima-13-en-5-yl+ diphosphate intermediate (D) deprotonated to yield 2) has a much higher energetic barrier than the preceding 1,2-shifts (15 versus <8 kcal/mol; as calculated for the enantiomeric reaction30). This in combination with the contrasting effects of the Y328F versus F171H mutations on product outcome in the context of the Y479F mutant prompts speculation that the reactant may be shifting between the more energetically accessible, upstream series of tertiary carbocation intermediates until one is properly oriented relative to a general base for deprotonation.
Conclusions
Here, based on the recently reported crystal structure for the class II diterpene cyclase associated with tuberculosis virulence, MtHPS,20 investigation of the underlying catalytic acid–base mechanism was carried out. While MtHPS contains a threonine in place of the last aspartate of the DxDD motif prototypically defining this family of diterpene cyclases, the results reported here indicate that MtHPS has adapted to retain the key interactions required for the middle residue to act as the catalytic acid. In addition, experimental evidence is provided supporting the previously proposed function of Y479 as the catalytic base. More interestingly, it was possible to introduce a new catalytic base as well as provide some evidence for an unanticipated secondary base in the form of a peptide backbone carbonyl. These results further emphasize the otherwise inert nature of the active site in not only MtHPS but also class II diterpene cyclases more generally, which must then isolate their reactants from not only polar side chains but also the peptide backbone, as accomplished at least in part through an array of conserved aromatic, as well as less conserved aliphatic, residues whose side chains line these cavities. Indeed, it can be speculated that this enables reactant equilibration via 1,2-migrations between a series of tertiary carbocationic states. Regardless, the results reported here support the applicability of the TerDockin approach to class II diterpene cyclases and provide insight into the enzymatic structure–function relationships underlying the activity of MtHPS, which may have some implications for treatment of tuberculosis.
Acknowledgments
This work was supported by a grant from the NIH to R.J.P. (GM131885).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00023.
Figure S1, depiction of TerDockin results for truncated B in MtHPS:Y328F/Y479F; Figure S2, coupled assay demonstrating stereochemistry of 4; Figure S3, Tukey’s trimean plot for TerDockin total energy distribution with B; and intermediate B xyz coordinates (PDF)
Author Contributions
⊥ C.L. and K.R. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Chakaya J.; Khan M.; Ntoumi F.; Aklillu E.; Fatima R.; Mwaba P.; Kapata N.; Mfinanga S.; Hasnain S. E.; Katoto P.; Bulabula A. N. H.; Sam-Agudu N. A.; Nachega J. B.; Tiberi S.; McHugh T. D.; Abubakar I.; Zumla A. Global Tuberculosis Report 2020 - Reflections on the Global TB burden, treatment and prevention efforts. Int. J. Infect. Dis. 2021, 113, S7–S12. 10.1016/j.ijid.2021.02.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D. G.; Barry C. E. III.; Flynn J. L. Tuberculosis: what we don’t know can, and does, hurt us. Science 2010, 328, 852–856. 10.1126/science.1184784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D. G. Who puts the tubercle in tuberculosis?. Nat. Rev. Microbiol. 2007, 5, 39–47. 10.1038/nrmicro1538. [DOI] [PubMed] [Google Scholar]
- Brodin P.; Hoffmann E. T(oo)bAd. Nat. Chem. Biol. 2019, 15, 849–850. 10.1038/s41589-019-0347-x. [DOI] [PubMed] [Google Scholar]
- Buter J.; Cheng T. Y.; Ghanem M.; Grootemaat A. E.; Raman S.; Feng X.; Plantijn A. R.; Ennis T.; Wang J.; Cotton R. N.; Layre E.; Ramnarine A. K.; Mayfield J. A.; Young D. C.; Jezek Martinot A.; Siddiqi N.; Wakabayashi S.; Botella H.; Calderon R.; Murray M.; Ehrt S.; Snider B. B.; Reed M. B.; Oldfield E.; Tan S.; Rubin E. J.; Behr M. A.; van der Wel N. N.; Minnaard A. J.; Moody D. B. Mycobacterium tuberculosis releases an antacid that remodels phagosomes. Nat. Chem. Biol. 2019, 15, 889–899. 10.1038/s41589-019-0336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano C.; Okamura T.; Sato T.; Dairi T.; Hoshino T. Mycobacterium tuberculosis H37Rv3377c encodes the diterpene cyclase for producing the halimane skeleton. Chem. Commun. 2005, 2005, 1016–1018. 10.1039/b415346d. [DOI] [PubMed] [Google Scholar]
- Layre E.; Lee H. J.; Young D. C.; Martinot A. J.; Buter J.; Minnaard A. J.; Annand J. W.; Fortune S. M.; Snider B. B.; Matsunaga I.; Rubin E. J.; Alber T.; Moody D. B. Molecular profiling of Mycobacterium tuberculosis identifies tuberculosinyl nucleoside products of the virulence-associated enzyme Rv3378c. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 2978–2983. 10.1073/pnas.1315883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K.; Swenson D. L.; Alonso S.; Anderson J.; Wang C.; Russell D. G. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13642–13647. 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano C.; Ootsuka T.; Takayama K.; Mitsui T.; Sato T.; Hoshino T. Characterization of the Rv3378c gene product, a new diterpene synthase for producing tuberculosinol and (13R, S)-isotuberculosinol (nosyberkol), from the Mycobacterium tuberculosis H37Rv genome. Biosci., Biotechnol., Biochem. 2011, 75, 75–81. 10.1271/bbb.100570. [DOI] [PubMed] [Google Scholar]
- Mann F. M.; Xu M.; Chen X.; Fulton D. B.; Russell D. G.; Peters R. J. Edaxadiene: a new bioactive diterpene from Mycobacterium tuberculosis. J. Am. Chem. Soc. 2009, 131, 17526–17527. 10.1021/ja9019287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugel N.; Mann F. M.; Hillwig M. L.; Peters R. J.; Snider B. B. Synthesis of (+/-)-nosyberkol (isotuberculosinol, revised structure of edaxadiene) and (+/-)-tuberculosinol. Org. Lett. 2010, 12, 2626–2629. 10.1021/ol100832h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prach L.; Kirby J.; Keasling J. D.; Alber T. Diterpene production in Mycobacterium tuberculosis. FEBS J. 2010, 277, 3588–3595. 10.1111/j.1742-4658.2010.07767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann F. M.; VanderVen B. C.; Peters R. J. Magnesium depletion triggers production of an immune modulating diterpenoid in Mycobacterium tuberculosis. Mol. Microbiol. 2011, 79, 1594–1601. 10.1111/j.1365-2958.2011.07545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. C.; Layre E.; Pan S. J.; Tapley A.; Adamson J.; Seshadri C.; Wu Z.; Buter J.; Minnaard A. J.; Coscolla M.; Gagneux S.; Copin R.; Ernst J. D.; Bishai W. R.; Snider B. B.; Moody D. B. In vivo biosynthesis of terpene nucleosides provides unique chemical markers of Mycobacterium tuberculosis infection. Chem. Biol. 2015, 22, 516–526. 10.1016/j.chembiol.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S. J.; Tapley A.; Adamson J.; Little T.; Urbanowski M.; Cohen K.; Pym A.; Almeida D.; Dorasamy A.; Layre E.; Young D. C.; Singh R.; Patel V. B.; Wallengren K.; Ndung’u T.; Wilson D.; Moody D. B.; Bishai W. Biomarkers for Tuberculosis Based on Secreted, Species-Specific, Bacterial Small Molecules. J. Infect. Dis. 2015, 212, 1827–1834. 10.1093/infdis/jiv312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann F. M.; Prisic S.; Hu H.; Xu M.; Coates R. M.; Peters R. J. Characterization and inhibition of a class II diterpene cyclase from Mycobacterium tuberculosis: implications for tuberculosis. J. Biol. Chem. 2009, 284, 23574–23579. 10.1074/jbc.M109.023788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann F. M.; Xu M.; Davenport E. K.; Peters R. J. Functional characterization and evolution of the isotuberculosinol operon in Mycobacterium tuberculosis and related mycobacteria. Front. Microbiol. 2012, 3, 368 10.3389/fmicb.2012.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem M.; Dube J. Y.; Wang J.; McIntosh F.; Houle D.; Domenech P.; Reed M. B.; Raman S.; Buter J.; Minnaard A. J.; Moody D. B.; Behr M. A. Heterologous Production of 1-Tuberculosinyladenosine in Mycobacterium kansasii Models Pathoevolution towards the Transcellular Lifestyle of Mycobacterium tuberculosis. mBio 2020, 11, e02645 10.1128/mBio.02645-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano C.; Hoshino T. Characterization of the Rv3377c gene product, a type-B diterpene cyclase, from the Mycobacterium tuberculosis H37 genome. Chembiochem 2009, 10, 2060–2071. 10.1002/cbic.200900248. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Prach L. M.; O’Brien T. E.; DiMaio F.; Prigozhin D. M.; Corn J. E.; Alber T.; Siegel J. B.; Tantillo D. J. Crystal Structure and Mechanistic Molecular Modeling Studies of Mycobacterium tuberculosis Diterpene Cyclase Rv3377c. Biochemistry 2020, 59, 4507–4515. 10.1021/acs.biochem.0c00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters R. J. Two rings in them all: the labdane-related diterpenoids. Nat. Prod. Rep. 2010, 27, 1521–1530. 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisic S.; Xu J.; Coates R. M.; Peters R. J. Probing the role of the DXDD motif in Class II diterpene cyclases. Chembiochem 2007, 8, 869–874. 10.1002/cbic.200700045. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Honzatko R. B.; Peters R. J. Terpenoid synthase structures: a so far incomplete view of complex catalysis. Nat. Prod. Rep. 2012, 29, 1153–1175. 10.1039/c2np20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köksal M.; Potter K.; Peters R. J.; Christianson D. W. 1.55A-resolution structure of ent-copalyl diphosphate synthase and exploration of general acid function by site-directed mutagenesis. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 184–190. 10.1016/j.bbagen.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf J. D.; Dong L. B.; Cao H.; Hatzos-Skintges C.; Osipiuk J.; Endres M.; Chang C. Y.; Ma M.; Babnigg G.; Joachimiak A.; Phillips G. N. Jr; Shen B. Structure of the ent-Copalyl Diphosphate Synthase PtmT2 from Streptomyces platensis CB00739, a Bacterial Type II Diterpene Synthase. J. Am. Chem. Soc. 2016, 138, 10905–10915. 10.1021/jacs.6b04317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köksal M.; Hu H.; Coates R. M.; Peters R. J.; Christianson D. W. Structure and mechanism of the diterpene cyclase ent-copalyl diphosphate synthase. Nat. Chem. Biol. 2011, 7, 431–433. 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter K. C.; Jia M.; Hong Y. J.; Tantillo D.; Peters R. J. Product Rearrangement from Altering a Single Residue in the Rice syn-Copalyl Diphosphate Synthase. Org. Lett. 2016, 18, 1060–1063. 10.1021/acs.orglett.6b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke C.; Potter K. C.; Schulte S.; Peters R. J. Conserved bases for the initial cyclase in gibberellin biosynthesis: from bacteria to plants. Biochem. J. 2019, 476, 2607–2621. 10.1042/BCJ20190479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter K.; Criswell J.; Zi J.; Stubbs A.; Peters R. J. Novel product chemistry from mechanistic analysis of ent-copalyl diphosphate synthases from plant hormone biosynthesis. Angew. Chem., Int. Ed. 2014, 53, 7198–7202. 10.1002/anie.201402911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter K. C.; Zi J.; Hong Y. J.; Schulte S.; Malchow B.; Tantillo D. J.; Peters R. J. Blocking Deprotonation with Retention of Aromaticity in a Plant ent-Copalyl Diphosphate Synthase Leads to Product Rearrangement. Angew. Chem., Int. Ed. 2016, 55, 634–638. 10.1002/anie.201509060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte S.; Potter K.; Lemke C.; Peters R. J. Catalytic bases and stereo-control in Lamiaceae class II diterpene cyclases. Biochemistry 2018, 57, 3473–3479. 10.1021/acs.biochem.8b00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafu S.; Potter K. C.; Hillwig M. L.; Schulte S.; Criswell J.; Peters R. J. Efficient heterocyclisation by (di)terpene synthases. Chem. Commun. 2015, 51, 13485–13487. 10.1039/C5CC05754J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell J.; Potter K.; Shephard F.; Beale M. H.; Peters R. J. A single residue change leads to a hydroxylated product from the class II diterpene cyclization catalyzed by abietadiene synthase. Org. Lett. 2012, 14, 5828–5831. 10.1021/ol3026022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelot K. A.; Mitchell R.; Kwon M.; Hagelthorn D. M.; Wardman J. F.; Chiang A.; Bohlmann J.; Ro D. K.; Zerbe P. Biosynthesis of the psychotropic plant diterpene salvinorin A: Discovery and characterization of the Salvia divinorum clerodienyl diphosphate synthase. Plant J. 2017, 89, 885–897. 10.1111/tpj.13427. [DOI] [PubMed] [Google Scholar]
- Hansen N. L.; Nissen J. N.; Hamberger B. Two residues determine the product profile of the class II diterpene synthases TPS14 and TPS21 of Tripterygium wilfordii. Phytochemistry 2017, 138, 52–56. 10.1016/j.phytochem.2017.02.022. [DOI] [PubMed] [Google Scholar]
- Peters R. J.; Ravn M. M.; Coates R. M.; Croteau R. B. Bifunctional abietadiene synthase: Free diffusive transfer of the (+)-copalyl diphosphate intermediate between two distinct active sites. J. Am. Chem. Soc. 2001, 123, 8974–8978. 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]
- Jia M.; O’Brien T. E.; Zhang Y.; Siegel J. B.; Tantillo D. J.; Peters R. J. Changing Face: A Key Residue for the Addition of Water by Sclareol Synthase. ACS Catal. 2018, 8, 3133–3137. 10.1021/acscatal.8b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien T. E.; Bertolani S. J.; Tantillo D. J.; Siegel J. B. Mechanisticallly informed predictions of binding modes for carbocation intermediates of a sesquiterpene synthase reaction. Chem. Sci. 2016, 7, 4009–4015. 10.1039/C6SC00635C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien T. E.; Bertolani S. J.; Zhang Y.; Siegel J. B.; Tantillo D. J. Predicting Productive Binding Modes for Substrates and Carbocation Intermediates in Terpene Synthases-Bornyl Diphosphate Synthase as a Representative Case. ACS Catal. 2018, 8, 3322–3330. 10.1021/acscatal.8b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; Zhang Y.; Siegel J. B.; Tantillo D. J.; Peters R. J. Switching on a Nontraditional Enzymatic Base - Deprotonation by Serine in the ent-Kaurene Synthase from Bradyrhizobium japonicum. ACS Catal. 2019, 9, 8867–8871. 10.1021/acscatal.9b02783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; Mishra S. K.; Tufts S.; Jernigan R. L.; Peters R. J. Combinatorial biosynthesis and the basis for substrate promiscuity in class I diterpene synthases. Metab. Eng. 2019, 55, 44–58. 10.1016/j.ymben.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M.; Potter K. C.; Peters R. J. Extreme promiscuity of a bacterial and a plant diterpene synthase enables combinatorial biosynthesis. Metab. Eng. 2016, 37, 24–34. 10.1016/j.ymben.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.; Hoshino T. Functional analysis of the DXDDTA motif in squalene-hopene cyclase by site-directed mutagenesis experiments: Initiation site of the polycyclization reaction and stabilization site of the carbocation intermediate of the initially cyclized A-ring. Biosci., Biotechnol., Biochem. 1999, 63, 2189–2198. 10.1271/bbb.63.2189. [DOI] [PubMed] [Google Scholar]
- Wang Y.-H.; Xu H.; Zou J.; Chen X.-B.; Zhuang Y.-Q.; Liu W.-L.; Celik E.; Chen G.-D.; Hu D.; Gao H.; Wu R.; Sun P.-H.; Dickschat J. S. Catalytic role of carbonyl oxygens and water in selinadiene synthase. Nat. Catal. 2022, 5, 128–135. 10.1038/s41929-022-00735-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.