

Abstract
Rhinovirus (RV), which is associated with acute exacerbations, also causes persistent lung inflammation in patients with chronic obstructive pulmonary disease (COPD), but the underlying mechanisms are not well-known. Recently, we demonstrated that RV causes persistent lung inflammation with accumulation of a subset of macrophages (CD11b+/CD11c+), and CD8+ T cells, and progression of emphysema. In the present study, we examined the mechanisms underlying the RV-induced persistent inflammation and progression of emphysema in mice with COPD phenotype. Our results demonstrate that at 14 days post-RV infection, in addition to sustained increase in CCL3, CXCL-10 and IFN-γ expression as previously observed, levels of interleukin-33 (IL-33), a ligand for ST2 receptor, and matrix metalloproteinase (MMP)12 are also elevated in mice with COPD phenotype, but not in normal mice. Further, MMP12 was primarily expressed in CD11b+/CD11c+ macrophages. Neutralization of ST2, reduced the expression of CXCL-10 and IFN-γ and attenuated accumulation of CD11b+/CD11c+ macrophages, neutrophils and CD8+ T cells in COPD mice. Neutralization of IFN-γ, or ST2 attenuated MMP12 expression and prevented progression of emphysema in these mice. Taken together, our results indicate that RV may stimulate expression of CXCL-10 and IFN-γ via activation of ST2/IL-33 signaling axis, which in turn promote accumulation of CD11b+/CD11c+ macrophages and CD8+ T cells. Furthermore, RV-induced IFN-γ stimulates MMP12 expression particularly in CD11b+/CD11c+ macrophages, which may degrade alveolar walls thus leading to progression of emphysema in these mice. In conclusion, our data suggest an important role for ST2/IL-33 signaling axis in RV-induced pathological changes in COPD mice.
Introduction
Acute exacerbations often lead to accelerated loss of lung function in subjects with chronic obstructive pulmonary disease (COPD). Rhinovirus (RV) is associated with one-third to half of all the viral-related COPD exacerbations. Additionally, RV infection is relatively common in patients with frequent exacerbations and associated with severe respiratory symptoms [1]. Experimental infection with RV caused lower airway obstruction, systemic, and airway inflammation that was more severe and prolonged in COPD than in healthy non-smokers [2,3]. Although experimentally infected COPD patients showed slightly increased viral load in the lower airways up to 12 days post infection, there was no difference in the kinetics of viral clearance between COPD and normal persons [2]. Interestingly, RV-infected COPD, but not normal persons also showed increase in neutrophils and lymphocytes even after the virus was cleared indicating an abnormal host response to RV infection. The mechanisms underlying these abnormal host responses that lead to accumulation of inflammatory cells and their contribution to progression of lung disease are not well-understood.
Our recent studies demonstrate that RV infection is associated with sustained lung inflammation up to 14 days in a mouse model of COPD [4,5]. This was associated with accumulation of macrophages, particularly CD11b+/CD11c+ inflammatory macrophages and CD8+ T cells. The sustained inflammation was not due to persistence of virus, because, although mice with COPD phenotype show slightly higher viral load than normal mice during initial stages of infection, both normal mice and mice with COPD phenotype clear the virus by 7 days. The persistent lung inflammation in RV-infected mice with COPD phenotype was associated with sustained up-regulation of CCL3, CXCL-10, and IFN-γ in the absence of virus indicating abnormal host responses
Levels of CXCL-10 and IFN-γ are elevated in COPD patients and further increases during viral exacerbations [6,7]. CXCL-10 is a potent chemoattractant for T cells, inflammatory CXCR3+ macrophages and neutrophils [8,9]. CXCL-10 also activates CD8+ T cells during viral infection and stimulates type 1 inflammation including the expression of IFN-γ [10]. IFN-γ plays an essential role in killing virus-infected cells. However, exaggerated levels of IFN-γ can further increase expression of CXCL-10, CXCL-9, thus enhancing recruitment and accumulation of T cells, neutrophils and macrophages. IFN-γ also promotes survival of recruited macrophages and stimulates expression of matrix metalloproteinases (MMP)12 in macrophages [11–13], and MMP12 has been thought to contribute to development of emphysema in COPD [14,15].
Previously, we demonstrated that RV stimulates CXCL-10 in vivo, primarily by activating interleukin-33 (IL-33)/ST2 signaling axis [16]. In addition, IL-33/ST2 also plays a major role in RV-stimulated CXCL-10 in vitro in human bronchial epithelial cells and peripheral blood-derived macrophages [16]. In the lung, bronchial epithelium constitutively expresses IL-33 and is significantly increased in airway progenitor cells of COPD patients [17,18]. IL-33 is released from the epithelial cells in response to stress or infection to alert the immune system, but exaggerated expression and release of IL-33 may result in inflammation. Accordingly, respiratory viruses including RV have been demonstrated to induce type 2 inflammation via exaggerated IL-33/ST2 signaling axis in an allergic environment [19,20]. On the other hand, in cigarette smoke exposed mice, IL-33/ST2 signaling stimulated by influenza virus enhances type 1 inflammation [18]. Since RV stimulates CXCL-10 primarily via IL-33/ST2 signaling axis, we hypothesized that IL-33/ST2 signaling pathway may contribute to continued recruitment and accumulation of inflammatory cells, primarily CD8+ T cells, CD11b+/CD11c+ inflammatory macrophages and neutrophils via CXCL-10 leading to sustained lung inflammation and progression of emphysema in mice with COPD phenotype.
CCL3 is a chemoattractant for monocytes and lung macrophages [21], and therefore, CCL3 can also potentially contribute to lung inflammation by enhancing recruitment of these cells following RV infection in mice with COPD phenotype. Therefore in the present study, we assessed the role of CCL3 and ST2/IL-33 signaling axis in the accumulation of inflammatory cells in RV-infected mouse model of COPD. We also delineated one of the mechanisms underlying sustained expression of CXCL-10 and IFN-γ and determined the contribution of IFN- γ to MMP12 expression and progression of emphysema in these mice.
Methods and materials
Mice
Mild COPD-like lung disease was induced in C57BL/6 mice (both males and females) by exposing to combination of cigarette smoke and heat-killed non-typeable Hemophilus influenzae as described previously [4,5]. We refer to these mice as mice with COPD phenotype or COPD mice. Mice neither exposed to cigarette smoke nor treated with heat-killed non-typeable H. influenzae were used as controls for mice with COPD phenotype and we refer to these as mice as “normal mice”. All the experiments were approved by the Animal Care and User Committee of the University of Michigan, Ann Arbor and Temple University, Philadelphia. Majority of the experiments were conducted at the Temple University.
Rhinovirus and infection
Stocks of RV1B were prepared by infecting H1HeLa cells with RV1B and subjecting HeLa cell supernatants to ultrafiltration as described previously [22,29]. Similarly, concentrated and purified cell supernatants from uninfected HeLa cells were used as sham controls. COPD and normal mice infected with RV or an equal volume of sham by intranasal route as described previously [22] and killed at 14 days post-infection. Some COPD mice were treated with 100 μl of endotoxin free PBS containing neutralizing antibody to CCL3 (2 μg/ml), ST2 (1 μg/ml) or IFN-γ (1 μg/ml), or similar concentration of isotype IgG control (all from R&D Systems, Minneapolis, MN) every other day starting from day 4 post-infection by intraperitoneal route as previously described [16] and killed on day 14.
To determine the efficacy of neutralizing antibody to CCL3 or ST2, normal mice were administered with either recombinant mouse CCL3 (0.5 μg) or IL-33 (10 ng) (both purchased from PeproTech, Rocky Hill, NJ) by intranasal route and immediately treated with neutralizing antibody to CCL3 or ST2 receptor, respectively. Mice were killed 6-h later and bronchoalveolar lavage (BAL) performed. Cytospins were prepared from the BAL, stained with DiffQuick and number of macrophages counted for CCL3-treated mice and number of macrophages and T cells were counted for IL-33-treated mice. Protein levels of CXCL-10 in BAL fluid were also determined in IL-33-treated mice. Isotype IgG-treated mice served as controls.
RNA isolation and qPCR
Total RNA isolated from the lungs was used for cDNA synthesis and probe-based qPCR. Primetime probe based assays for β-actin, CXCL-10, ST2, IFN-γ, MMP3, MMP9, and MMP12 were purchased from Integrated DNA Technologies (Coralville, IA).
ELISA
Supernatants of BAL fluid were subjected to ELISA to quantify IFN-γ, CXCL-10, IL-33 (all from R & D systems, Minneapolis, MN), and MMP12 (Abeam, Cambridge, MA).
Gelatin zymography
Gelatin zymogaphy was performed on supernatants of BAL fluid to determine MMP activity as previously described [22]. Briefly, after relevant treatment, BAL was performed, centrifuged and equal amount of supernatant was subjected to electrophoresis on gelatin impregnated polyacrylamide gels. Gels were washed with 1% Triton X-100, developed in Tris buffer containing 10 mM calcium chloride and 5 μM zinc chloride and stained with 0.5% Coomassie blue.
Histology and morphometry
Lungs were inflation fixed at a constant pressure of 30 cm. H2O for 30 min and embedded in paraffin. Five micron thick sagittal sections were taken at 5-mm intervals through the length of the lungs, stained with hematoxylin and eosin (H&E) and the diameter of the alveolar spaces measured in random fields using NIH image J analysis software to assess alveolar chord length [23].
Flow cytometry
Flow cytometry was performed on lung digests as previously described [5,24]. Briefly, lungs were perfused with cold PBS via right ventricle, minced and digested in collagenase IV (5 mg/ml) and DNase I. Red blood cells were lysed, then single cell suspensions were incubated with Zombie UV™ (BioLegend, San Diego, CA) to label dead cells and stained with fluorescence-labeled antibodies against surface markers of leukocytes, such as CD45, CD11c, CD11b, F4/80, Ly6G. Appropriate isotype-matched controls and fluorescence minus one (FMO) were used in all experiments. All antibodies were purchased from BioLegend. Cells were fixed and analyzed in BD LSR II Flow cytometer (BD Biosciences) and data were analyzed using FlowJO version 10 (Tree Star, Ashland, OR). For sorting macrophages, CD45+/F480+/CD11c+ or CD45+/F480+/CD11b+ positive cells were gated by CD11b+ or CD11c+ expression using Aria II flow cytometer (BD Biosciences) equipped with three lasers and DiVa software.
Statistical analysis
Data were expressed as mean ± S.D. or median with range of data. Data were analyzed by using SigmaStat statistical software (Systat Software, San Jose, CA). One way ANOVA with Bonferroni post hoc test, ANOVA on ranks with Kruskal–Wallis H-test or unpaired t-test was performed as appropriate to compare groups and a P-value ≤ 0.05 was considered significant.
Results
Neutralization of CCL3 does not attenuate accumulation of CD11b+/CD11c+ macrophages in RV-infected mice with COPD phenotype
Recently, we showed that RV-infected mice with COPD phenotype show sustained increase in CD11b+/CD11c+ macrophages at 14 days post-infection and this was accompanied by increase in CCL3 [5]. Since, CCL3 is a chemoattractant for macrophages [21], we assessed the contribution of CCL3 in RV-induced accumulation of CD11b+/CD11c+ macrophages in the lungs of COPD mice by treating with neutralizing antibody to CCL3 or isotype control. Figure 1A shows the gating strategy for quantification of CD11b+/CD11c+ cells. As observed previously, irrespective of infection, normal mice did not show significant increase in CD11b+/CD11c+ macrophage population (Figure 1B). In contrast, mice with COPD phenotype showed significant increase in CD11b+/CD11c+ macrophages compared with normal mice, which further increased following RV-infection. Treatment with CCL3 antibody did not reduce CD11b+/CD11c+ macrophage population in either sham or RV-infected mice with COPD phenotype (Figure 1C), indicating that CCL3 may not contribute to RV-induced accumulation of CD11b+/CD11c+ macrophage in these mice. Normal mice treated with recombinant CCL3 by intranasal route showed significant increase in the number of total cells and macrophages in the lungs, and this was inhibited by CCL3 antibody (Supplementary Figure S1) confirming the neutralizing capacity of CCL3 antibody used in these studies.
Figure 1. Blocking CCL3 does not affect RV-induced accumulation of CD11b+/CD11c+ macrophages in mice with COPD phenotype.

Normal mice and mice with COPD phenotype were infected with sham or RV and after 14 days mice were killed and lungs were harvested. Single cell suspensions isolated from lung digests were stained with antibodies to CD45, F/480, CD11c and CD11b to detect subtypes of macrophages. (A) Gating strategy to detect different subtypes of macrophages. (B) Quantification of subtypes of macrophages at 14 days post-infection in normal and COPD mice. (C) Sham or RV-infected COPD mice were treated every other day with neutralization antibody to CCL3 or isotype control starting at 4 days post-infection until the mice were harvested and CD11b+ and CD11c+ macrophage population was quantified. Data in B and C show median with range calculated from three independent experiment with two mice per group to a total of sixmice per group(*P≤0.05, different from respective sham; §P≤0.05, different from normal sham; ‡P≤0.05, different from normal RV; ANOVA on Ranks with Kruskal–Wallis H-test).
RV-stimulated CXCL-10 via ST2/IL-33 pathway contributes to accumulation of CD11b+/CD11c+ macrophages and T cells in COPD mice
CXCL-10 is a chemoattractant for both T cells and macrophages. As previously observed, we found that compared with sham-, RV-infected mice with COPD phenotype show significant increase in CXCL-10 protein levels (Figure 2A). Since, commercially available neutralizing antibody to CXCL-10 was not efficient in the neutralization of CXCL-10 in vivo, we targeted the IL-33/ST2 signaling, which plays an essential role in RV-induced acute CXCL-10 expression in normal mice [16]. First, we determined whether expression of ST2 receptor and IL-33 is altered in the lungs of RV-infected normal and COPD mice at 14 days post-RV infection. There was no difference in the expression of either ST2 receptor or IL-33 between sham and RV-infected normal mice (Figure 2B–D). On the other hand, mice with COPD phenotype showed increased expression of both ST2 and IL-33 compared with normal mice without viral infection. Following RV infection, IL-33, but not ST2 expression increased further at both mRNA and protein levels in COPD mice. As observed earlier, IFN-γ protein was also increased in COPD mice following RV infection (Figure 2E) [5]. Since only COPD, but not normal mice showed sustained expression of CXCL-10 and IFN-γ (Figure 2A,E), and accumulation of T cells, neutrophils and macrophages at 14 days post-RV infection [5], we focused on mice with COPD phenotype in the subsequent neutralization experiments.
Figure 2. Neutralization of ST2 receptor inhibits persistent expression of CXCL-10 and IFN-γ RV-infected mice with COPD phenotype.
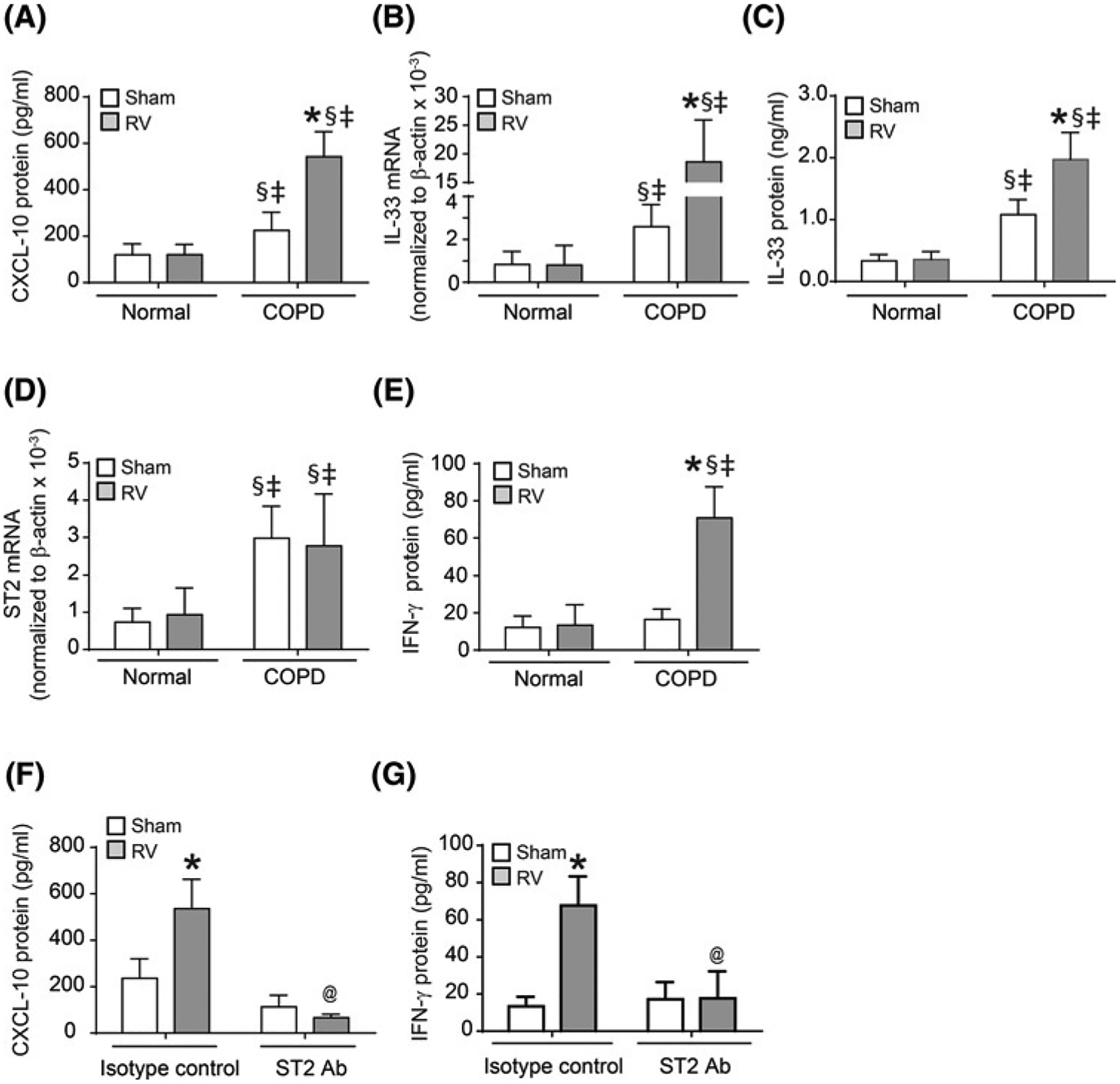
Normal mice and mice with COPD phenotype were infected with sham or RV, and BAL was performed at 14 days post-infection. (A, C and E). Protein levels of CXCL-10, IL-33, and IFN-γ in the BAL fluid were determined by ELISA. (B and D). Total RNA was isolated from the lungs and mRNA expression of ST2 and IL-33 was determined by qPCR. (F and G) Mice with COPD phenotype were infected with RV or sham and treated with neutralizing antibody to ST2 or isotype IgG control starting from 4 days post-infection every other day and protein levels of CXCL-10 and IFN-γ in BAL fluid was determined at 14 days post-infection. Data represent mean ± S.D. calculated from three independent experiments with two to three animals per group to a total of six to eight mice per group (*P≤0.05, different from respective sham; §P≤0.05, different from normal sham; ‡P≤0.05, different from normal RV; @P≤0.05, different from RV-infected COPD mice treated with isotype control; ANOVA with Tukey post hoc test).
Sham- or RV-infected mice with COPD phenotype were treated with neutralizing antibody to ST2 or isotype IgG and examined for the expression of CXCL-10 and IFN-γ, and accumulation of T cells, neutrophils and CD11b+/CD11c+ macrophages to assess the contribution of ST2/IL-33 signaling axis to these processes. Treatment with ST2 neutralizing antibody, but not isotype IgG control inhibited the protein expression of both CXCL-10 and IFN-γ in RV-infected COPD mice (Figure 2F,G). In contrast, ST2 neutralizing antibody had no effect on RV-induced expression of CCL3 or IL-17A, which are also persistently enhanced by RV (data not shown). We then performed flow cytometry on the lung digests to quantify T cells and CD11b+/CD11c+ macrophage populations. Figure 3A,B respectively demonstrates the gating strategy for T cells and representative histograms for CD4+ and CD8+ cells for each treatment groups. Quantification of each T cell population indicated that RV increases CD8+ T-cell population in mice with COPD phenotype and treatment with ST2 neutralizing antibody significantly inhibits this process (Figure 3C,D). Additionally, neutralizing antibody to ST2 also significantly reduced CD11b+/CD11c+ macrophage population in RV-infected mice with COPD phenotype (Figure 3E,F). These results indicate that ST2/IL-33 signaling axis may be involved in recruitment of both T cells and CD11b+/CD11c+ macrophages in RV-infected mice with COPD phenotype.
Figure 3. Accumulation of CD8+ cells and CD11b+/CD11c+ macrophages In RV-lnfected mice with COPD phenotype is abrogated by neutralizing antibody to ST2.

Mice with COPD phenotype were infected with RV or sham and treated with neutralizing antibody to ST2 or isotype IgG control as described under Figure 2. Mice were killed at 14 days post-infection and CD4+ and CD8+ cells, and macrophages were analyzed by flow cytometry using total lung digests. (A) Gating strategy for detecting CD4+ and CD8+ T cells. (B and E) Representative histograms of CD4+ and CD8+ cells, and CD11b+/CD11c+ macrophages, respectively in sham and RV infected COPD mice treated with isotype IgG control or ST2 antibody. (C, D and F) Quantification of CD4+ T cells, CD8+ T cells and CD11b+/CD11c+ macrophages, respectively. Data represent median and range calculated from three independent experiments carried out using two or three mice per group with a total of six to eight mice per group (*P≤0.05, different from respective sham; @P≤0.05, different from RV-infected COPD mice treated with isotype control; ANOVA on Ranks with Kruskal–Wallis H-test).
Previously, we have demonstrated that RV-infected COPD mice also show increase in the number of neutrophils [5]. Since CXCL-10 also contributes to recruitment of neutrophils in the lungs [9], we examined whether blocking ST2/IL-33 signaling affects neutrophil recruitment in RV-infected mice with COPD phenotype. Flow cytometry analysis confirmed the previous finding that RV enhances neutrophil accumulation in COPD, but not in normal mice (Figure 4A,B). Treatment with ST2 receptor antibody reduced neutrophil population by 48% in RV-infected mice with COPD mice (Figure 4C,D) indicating that ST2/IL-33 signaling axis may partially contribute to RV-induced neutrophil recruitment in these mice.
Figure 4. Neutralization of ST2 partially Inhibits RV-Induced accumulation of neutrophils In COPD mice.
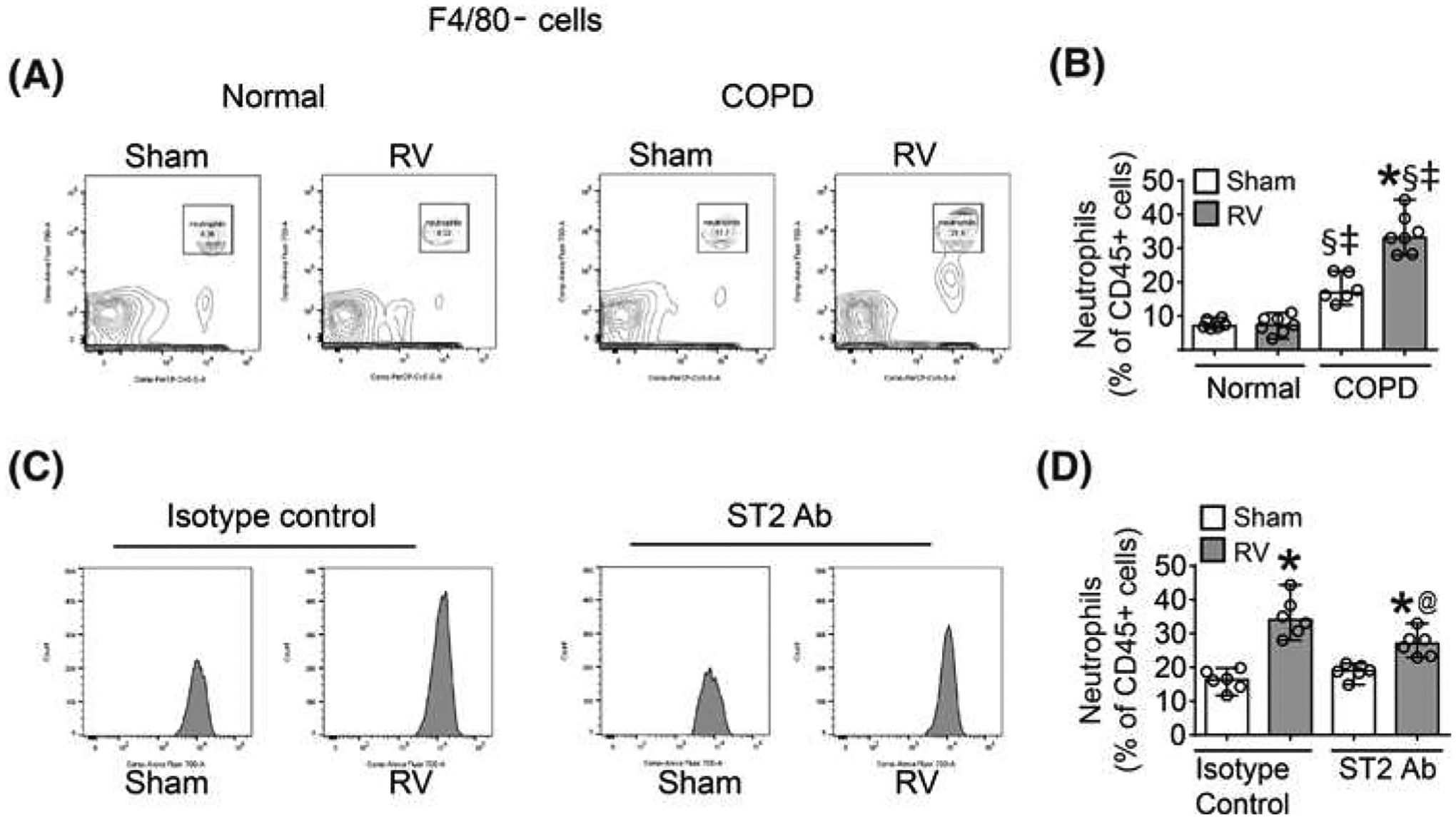
Mice with COPD phenotype were infected with RV or sham and treated with neutralizing antibody to ST2 or isotype IgG control as described under Figure 2. Mice were killed at 14 days post-infection and neutrophil population was determined by flow cytometry using total lung digests. (A) CD45+F4/80- cells were further gated on Ly6G and CD11B to detect neutrophils. (C) Representative histograms of neutrophils in sham and RV-infected COPD mice treated with isotype IgG control or ST2 antibody. (A and D) Quantification of neutrophils. Data represent median and range calculated from three independent experiments carried out using two mice per group with a total of six mice per group (*P≤0.05, different from respective sham; @P≤0.05, different from RV-infected COPD mice treated with isotype control; §P≤0.05, different from normal sham; ‡P≤0.05, different from normal RV, ANOVA on Ranks with Kruskal–Wallis H-test).
To confirm the efficacy of ST2 neutralizing antibody, we treated normal mice with 10 ng of recombinant IL-33 by intranasal route and then treated with either neutralizing antibody to ST2 antibody or isotype IgG and examined for CXCL-10 expression and the recruitment of T cells and macrophages. As anticipated, neutralization of ST2 receptor completely blocked IL-33-induced CXCL-10 expression (Supplementary Figure S2A). Similarly, neutralization of ST2 receptor also inhibited IL-33-stimulated recruitment of T cells and macrophages into the lungs (Supplementary Figure S2B–D). These results indicate that ST2/IL-33 signaling may contribute to recruitment of both T cells and macrophages and recruitment of neutrophils partially.
Treatment with IFN-γ or ST2 neutralizing antibody abrogates RV-induced MMP12 expression in CD11b+/CD11c+ macrophages
MMPs, particularly MMP9 and MMP12 contribute to development of emphysema and increased secretion of mucin glycoproteins in COPD. Further, IFN-γ activates lung macrophages and promotes expression of MMP12 [12]. Therefore, we examined MMP9 and MMP12 mRNA expression in the whole lungs by qPCR in mice with COPD phenotype. Compared with sham, RV-infected mice showed significant increase in the expression of MMP12, but not MMP9 mRNA (Figure 5A,B). Therefore, we only determined the protein level and the activity of MMP12 in BAL fluid and RV-infected mice showed increase in both protein and activity levels of MMP12 (Figure 5C,D). RV-infected mice treated with neutralizing antibody to IFN-γ showed reduction in MMP12 expression and activity compared with isotype IgG-treated mice. Additionally, neutralizing antibody to ST2 also inhibited both expression and activity of MMP12. Neutralizing antibody to either IFN-γ or ST2 receptor did not affect either MMP12 mRNA or protein levels in sham-infected animals and was similar to isotype IgG-treated mice (data not shown).
Figure 5. MMP12 expression In RV-lnfected mice displaying COPD phenotype was Inhibited by neutralizing antibody to IFN-γ and ST2.

Normal mice or mice with COPD phenotype were infected with sham or RV. Some RV-infected COPD mice were treated with neutralizing antibody to IFN-γ or ST2 or with isotype IgG control. All mice were killed at 14 days post-infection. (A and B) Total RNA from the lungs was isolated and subjected to qPCR to determine the expression of MMP9 and MMP12, respectively. (C and D) From identically treated mice, BAL fluid was collected, centrifuged and the supernatant subjected to ELISA or zymography, respectively. Data in (A–C), represent mean ± S.D. calculated from three independent experiments with two to three animals per group to a total of six to eight mice per group (*P≤0.05, different from respective sham; unpaired t-test, @P≤0.05, different from RV-infected COPD mice treated with isotype control; unpaired t-test). (D) Gelatin zymogram from three representative animals from each group. (E and F) Lung cells from RV-infected animals either treated with neutralization antibody to IFN-γ or isotype IgG control were flow sorted to isolate subsets of macrophages, RNA was isolated from sorted cells and subjected to qPCR. Data represent mean ± S.D. from four independent experiments and in each experiment, cells were pooled from three mice (*P≤0.05, different from CD11b+ cells; unpaired t-test, @P≤0.05, different from RV-infected COPD mice treated with isotype control; unpaired t-test).
We then examined whether reduction in MMP12 by IFN-γ neutralization is due to reduction in accumulation of CD11b+/CD11c+ macrophages directly or via IL-33. We quantified IL-33 expression by qPCR and CD11b+/CD11c+ macrophages following treatment with neutralizing antibody to IFN-γ by flow cytometry. There was no difference in either the expression of IL-33 (data not shown) or population of CD11b+/CD11c+ macrophages between control IgG and IFN-γ antibody treated RY-infected COPD mice (Supplementary Figure 3S). These results indicate that IFN-γ is not required for RV-induced CD11b+/CD11c+ macrophage accumulation, but maybe required for MMP12 expression in macrophages.
To identify the subpopulation of macrophages that express MMP12 in RV-infected COPD mice, we sorted the macrophages based on CD11c and CD11b and determined the expression of MMP12 mRNA. Compared with CD11c+/CD11b− and CD11b+/CD11c− macrophages, CD11b+/CD11c+ subpopulation showed higher expression of MMP12 mRNA (Figure 5E). Expression of MMP12 was considerably reduced in RV-infected mice treated with neutralizing antibody to IFN-γ (Figure 5F). We were not able to obtain sufficient numbers of CD11b+/CD11c+ macrophages from mice treated with neutralizing antibody to ST2 to assess MMP12 expression.
Treatment with neutralization antibody to ST2 or IFN-γ attenuates RV-induced progression of emphysema in COPD mice
Previously, we reported that RV causes progression of emphysema in mice with COPD phenotype [5]. Since MMP12 has been thought to induce emphysema, we examined whether neutralization of ST2 or IFN-γ blocks RV-induced emphysema in these mice by histology and quantified the changes by measuring alveolar chord length. As previously observed [5], mice with COPD phenotype showed enlargement of air space an indication of emphysematous change (Figure 6A,B). RV infection further increased emphysematous changes in COPD mice (Figure 6C). We also observed enhanced mononuclear cell infiltration in alveoli (inflammation) in RV-infected COPD mice (Figure 6D). Compared to RV-infected COPD mice treated with isotype IgG control, similarly-infected animals treated with neutralizing antibody to either IFN-γ or ST2 showed less emphysematous changes and inflammation (Figure 6E–G). Measurement of alveolar chord length confirmed the visual observations of emphysematous changes (Figure 6H,I).
Figure 6. Neutralization of either ST2 or IFN-γ Inhibits progression of emphysema In RV-lnfected mice with COPD phenotype.

(A) H&E-stained lung section from normal mice infected RV and killed at 14 days post-nfection. (B to D). Mice with COPD phenotype infected with sham or RV and assessed for lung histology at 14 days post-infection, respectively. D represents an enlarged Image marked in rectangle in C. Images are representative of three independent experiments. (E-G). H&E-stained lung sections from RV-infected COPD mice treated with isotype control or neutralizing antibody to IFN-γ or ST2 and killed at 14 days post-Infection. Asterisks in panels B,C, E, F and G represents emphysematous change. (H and I). Alveolar chord length was assessed from sham or RV-Infected normal mice or mice with COPD phenotype and RV-infected COPD mice treated with neutralization antibody to IFN-γ or ST2 or isotype IgG control. Data represent mean ± S.D. from three independent experiments with two to three mice per group to a total of six to eight mice per group (*P≤0.05, different from respective sham; §P≤0.05, different from normal sham; *P≤0.05, different from normal RV; @P≤0.05, different from RV-infected COPD mice treated with isotype control; ANOVA with Tukey post hoc test).
Taken together these results indicate a major role for IL-33/ST2 signaling in sustained lung inflammation in mice with COPD phenotype infected with RV. IFN-γ expressed by recruited CD8+ T cells may promote expression of MMP12 by CD11b+CD11c+ macrophages leading to progression of emphysema. In addition, sustained expression of IFN-γ can also stimulate expression of CXCL-10 in CD8+ T cells thus further enhancing recruitment of T cells and CD11b+CD11c+ macrophages. This vicious inflammatory loop induced by RV may lead to progression of lung disease in COPD mice.
Discussion
The present study highlights one of the mechanisms by which RV causes persistent lung inflammation and progression of emphysematous changes in mice with mild COPD phenotype. We demonstrate that RV-stimulated IL-33 may activate ST2 receptor signaling and contribute to accumulation of both CD11b+/CD11c+ macrophages and CD8+ T cells via expression of CXCL-10 in the lungs of mice with COPD phenotype. Further, we demonstrate that compared with alveolar or interstitial macrophages, intermediate CD11b+/CD11c+ macrophages express more MMP12 and blocking ST2 or IFN-γ inhibits RV-induced MMP12 expression and emphysematous changes.
Lung macrophages play an important role in acute inflammation and also in resolving the inflammation [25,26]. In COPD, however, there are more macrophages in the lung and the number of macrophages correlates with disease severity [27–29]. Interestingly, latent adenoviral infection was associated with increase in lung macrophage number and enhanced emphysema in COPD patients indicating that viral infections may promote development and/or progression of lung disease [30]. Consistent with this finding, chronic cigarette smoke along with influenza viral infection or treatment with double stranded RNA, a viral RNA mimic, was shown to exaggerate accumulation of macrophages and accelerate development of emphysematous changes in mice [31]. Further, exudate (CD11b+/CD11c+) macrophages were shown to express MMP12 and contribute to influenza virus-induced pulmonary pathology [32,33]. In the present study, we observed accumulation of CD11b+/CD11c+ macrophages in the lungs of RV-infected mice with COPD phenotype and these cells expressed higher levels of MMP12 compared with CD11b−/CD11c+ and CD11b+/CD11c− macrophages. This was associated with enhanced MMP12 activity and emphysematous changes in the lungs of these mice. Further, neutralization of virus-induced IFN-γ abrogated MMP12 expression in lungs as well as in CD11b+/CD11c+ macrophages. This was not surprising, because IFN-γ has been previously demonstrated to promote MMP12 expression in macrophages [11–13]. Since MMP12 plays an important role in the development of emphysema clinically [14,15], it is conceivable that RV-induced MMP12 in CD11b+/CD11c+ macrophages may contribute to progression of emphysema in mice with COPD phenotype.
Accumulation of lung macrophages during post-viral infection may occur as a result of continued recruitment or increased survival of infiltrated macrophages. CCL chemokines play a major role in the recruitment of macrophages. We found that, although RV-induces persistent expression of CCL3, a potent chemoattractant for macrophages [21], CCL3 did not play a role in the recruitment CD11b+/CD11c+ macrophages in RV-infected COPD mice. Triggering receptor expressed on myeloid cells-2 (TREM2) is another molecule that was shown to promote survival, thus accumulation of macrophages in mouse lungs following Sendai virus infection [34]. However, TREM2 levels were not changed in RV-infected COPD mice (data not shown) ruling out its contribution in the accumulation of CD11b+/CD11c+ macrophages in these mice. Interestingly, a subset of peripheral blood monocytes and recruited monocytes in inflamed tissue express CXCR3, a receptor for CXCL-10 [8,35]. We demonstrate that CXCL-10 protein levels are significantly elevated in RV-infected mice with COPD phenotype. Therefore, it is conceivable that CXCL-10 may contribute to recruitment of CD11b+/CD11c+ macrophages into the lungs. Since commercially available neutralizing antibody to CXCL-10 fails to block CXCL-10 induced inflammation in vivo and neutralizing antibody to CXCR3 is not available commercially, it was not possible to obtain direct evidence in support of our notion.
Previously, we demonstrated that RV stimulates acute expression of CXCL-10 primarily via ST2/IL-33 signaling axis in normal mice [16]. ST2 is IL-1-like receptor and signals via MyD88 to activate API and NF-kB [36]. We have also demonstrated that binding of NF-B to CXCL-10 promoter further enhances DNA accessibility around the TATA box region in the CXCL-10 promoter, thus increasing the transcription of CXCL-10 [16]. Depletion of IRAK-1, a kinase required for ST2 receptor signaling or inhibition of ST2 abrogated RV-induced CXCL-10 expression presumably due to reduced NF-B activation. In the present study, we demonstrate that ST2/IL-33 signaling axis is also responsible for sustained expression of CXCL-10 in RV-infected COPD mice. This was not due to enhanced expression of ST2 receptor but was due to significantly elevated levels of IL-33, a ligand for ST2, in RV-infected COPD mice. Blocking ST2 receptor signaling completely abolished CXCL-10 expression, but not CCL3 or IL-17A indicating the role of IL-33/ST2 signaling axis in RV-induced CXCL-10. Blocking ST2 receptor completely abrogated the recruitment and accumulation of CD11b+/CD11c+ macrophages. Further blocking ST2 receptor also completely inhibited accumulation of RV-induced CD8+ T cells, but neutrophil accumulation was only reduced partially. These results indicate an important role for IL-33/ST2 signaling in the recruitment of CD11b+/CD11c+ macrophages and CD8+ T cells probably via CXCL-10, but not for neutrophils. Since IL-17A also contributes to neutrophil recruitment, and IL-17A is increased in RV-infected COPD mice, it is possible that IL-17A may also play a role in RV-induced neutrophil recruitment in these mice [37]. However, further experiments with CXCR3 and IL-17R knockout mice are necessary to confirm these findings and are beyond the scope of this study.
CXCL-10 regulates migration of activated CD8+ T cells during virus infection [10]. CD8+ T cells, which play an important role in antiviral immunity are increased in patients with severe COPD and express elevated levels of IFN-γ in response to influenza virus infection [38]. IFN-γ can in turn stimulate production of CXCL-10 from activated CD8+ T cells, which can further increase recruitment of both T cells and macrophages, thus creating vicious inflammatory loop. In addition, both IFN-γ and CXCL-10 may also induce or enhance emphysema by increasing the expression of MMP12. Accordingly, mice over expressing IFN-γ shows constitutive expression of MMP12 in the lungs and spontaneous development of emphysema [39]. On the other hand, CXCL-10 has been shown to stimulate MMP12 expression in lung macrophages following exposure to cigarette smoke in mice [40]. Finally, lung macrophages from COPD subjects show heightened expression of MMP12 compared with control subjects [41]. Since we observed increase in both CXCL-10 and IFN-γ, and inhibition of MMP12 expression in CD11b+/CD11c+ by neutralizing antibody to IFN-γ or ST2, we think that RV-induced IFN-γ together with CXCL-10 via IL-33/ST2 signaling may stimulate MMP12 particularly in exudate CD11b+/CD11c+ macrophages.
In summary, we show that RV-induced CXCL-10 via ST2/IL-33 signaling activation causes prolonged infiltration of primarily CD11b+/CD11c+ macrophages and CD8+ T cells in the lungs leading to sustained lung inflammation in mice with COPD phenotype. Second, RV-induced IFN-γ and CXCL-10 may enhance MMP12 expression in CD11b+/CD11c+ macrophages thus leading to progression of emphysema in these mice (Figure 7).
Figure 7. Illustration showing RV-induced sustained lung inflammation and progression of emphysema in mice with COPD phenotype.
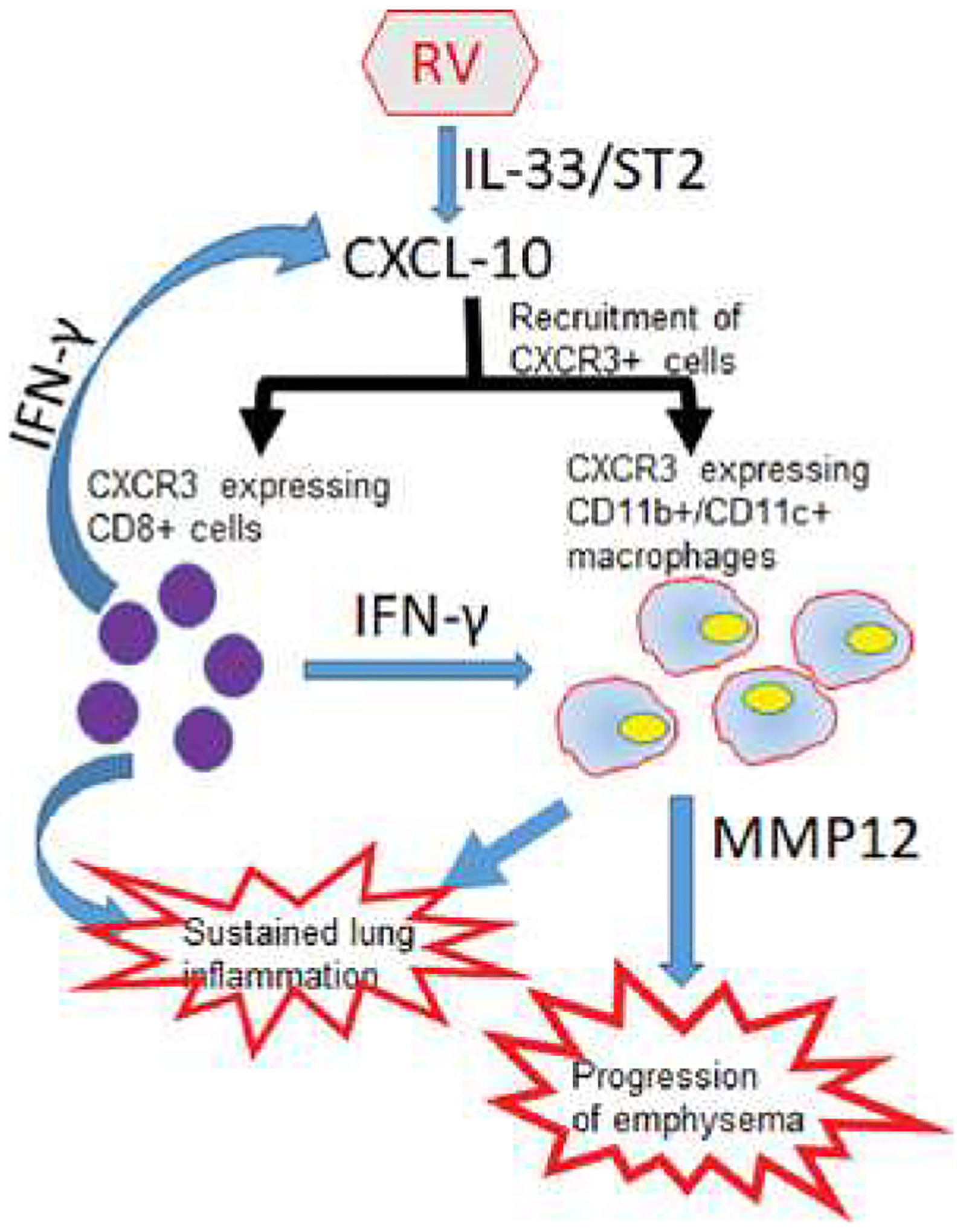
RV caused sustained activation of IL-33/ST2 signaling axis stimulates expression of CXCL-10, which recruits primarily CXCR3+ CD8+ T cells and CD11b+/CD11c+ macrophages leading to sustained lung inflammation. IFN-γ produced by recruited CD8+ T cells activates CD11b+/CD11c+ macrophages and induces expression of MMP12, a tissue-degrading enzyme, leading to the progression of emphysema
Supplementary Material
Clinical perspectives.
Rhinovirus is associated with severe exacerbations and sometimes leads to progression of lung disease in COPD, but the underlying mechanisms are poorly understood.
The present study demonstrates that, rhinovirus induced sustained lung inflammation and progression of emphysema in a mouse of model COPD is dependent on of ST2/IL-33 signaling axis.
Rhinovirus also induces IFN-γ via ST2/IL-33 signaling, which stimulate recruited macrophages to express MMP12, which can cause emphysema.
Acknowledgments
The authors thank Nicole Owuor and Francis Mele for their assistance in measuring alveolar chord length.
Funding
This work was supported by NIH (grant number AT007620 to U.S.).
Abbreviations
- BAL
bronchoalveolar lavage
- COPD
chronic obstructive pulmonary disease
- H&E
hematoxylin and eosin
- IFN
interferon
- IL-33
interleukin-33
- MMP
matrix metalloproteinase
- RV
rhinovirus
- TREM2
triggering receptor expressed on myeloid cells-2
Footnotes
Competing interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.George SN, Garcha DS, Mackay AJ, Patel AR, Singh R, Sapsford RJ et al. (2014) Human rhinovirus infection during naturally occurring COPD exacerbations. Eur. Respir. J 44,87–96, 10.1183/09031936.00223113 [DOI] [PubMed] [Google Scholar]
- 2.Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T et al. (2011) Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am. J. Respir. Crit. Care Med 183,734–742, 10.1164/rccm.201006-08330C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mallia P, Message SD, Contoli M, Gray K, Telcian A, Laza-Stanca V et al. (2014) Lymphocyte subsets in experimental rhinovirus infection in chronic obstructive pulmonary disease. Respir Med 108,78–85, 10.1016/j.rmed.2013.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganesan S, Comstock AT, Kinker B, Mancuso P, Beck JM and Sajjan US (2014) Combined exposure to cigarette smoke and nontypeable Haemophilus influenzae drives development of a COPD phenotype in mice. Respir. Res 15,11, 10.1186/1465-9921-15-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farazuddin M, Mishra R, Jing Y, Srivastava V, Comstock AT and Sajjan US (2018) Quercetin prevents rhinovims-induced progression of lung disease in mice with COPD phenotype. PLoS ONE 13, e0199612, 10.1371/journal.pone.0199612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P et al. (2011) Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am. J. Respir. Crit. Care Med 184,662–671, 10.1164/rccm.201104-05970C [DOI] [PubMed] [Google Scholar]
- 7.Singh M, Lee SH, Porter P, Xu C, Ohno A, Atmar RL et al. (2010) Human rhinovirus proteinase 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol 125,1369–1378 e1362, 10.1016/j.jaci.2010.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Tang PC, Qin L, Gayed PM, Li W, Skokos EA et al. (2010) CXCR3-dependent accumulation and activation of perivascular macrophages is necessary for homeostatic arterial remodeling to hemodynamic stresses. J. Exp. Med 207,1951–1966, 10.1084/jem.20100098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H et al. (2013) CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit Care Med 187,65–77, 10.1164/rccm.201203-05080C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen JE, de Lemos C, Moos T, Christensen JP and Thomsen AR (2006) CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J. Immunol 176,4235–4243, 10.4049/jimmunol.176.7.4235 [DOI] [PubMed] [Google Scholar]
- 11.McDermott AJ, Falkowski NR, McDonald RA, Frank CR, Pandit CR, Young VB et al. (2017) Role of interferon-gamma and inflammatory monocytes in driving colonic inflammation during acute Clostridium difficile infection in mice. Immunology 150,468–477, 10.1111/imm.12700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geraghty P, Greene CM, O’Mahony M, O’Neill SJ, Taggart CC and McElvaney NG (2007) Secretory leucocyte protease inhibitor inhibits interferon-gamma-induced cathepsin S expression. J. Biol. Chem 282,33389–33395, 10.1074/jbc.M706884200 [DOI] [PubMed] [Google Scholar]
- 13.Xaus J, Cardo M, Valledor AF, Soler C, Lloberas J and Celada A (1999) Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity 11,103–113, 10.1016/S1074-7613(00)80085-0 [DOI] [PubMed] [Google Scholar]
- 14.Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J et al. (2009) MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med 361, 2599–2608, 10.1056/NEJMoa0904006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace AM, Sandford AJ, English JC, Burkett KM, Li H, Finley RJ et al. (2008) Matrix metalloproteinase expression by human alveolar macrophages in relation to emphysema. COPD 5,13–23, 10.1080/15412550701817789 [DOI] [PubMed] [Google Scholar]
- 16.Ganesan S, Pham D, Jing Y, Farazuddin M, Hudy MH, Unger B et al. (2016) TLR2 activation limits rhinovirus-stimulated CXCL-10 by attenuating IRAK-1-dependent IL-33 receptor signaling in human bronchial epithelial cells. J. Immunol 197,2409–2420, 10.4049/jimmunol.1502702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X et al. (2013) Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Invest 123, 3967–3982, 10.1172/JCI65570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N et al. (2015) Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 42,566–579, 10.1016/j.immuni.2015.02.011 [DOI] [PubMed] [Google Scholar]
- 19.Werder RB, Zhang V, Lynch JP, Snape N, Upham JW, Spann K et al. (2018) Chronic IL-33 expression predisposes to virus-induced asthma exacerbations by increasing type 2 inflammation and dampening antiviral immunity. J. Allergy Clin. Immunol 141,1607–1619 e1609, 10.1016/j.jaci.2017.07.051 [DOI] [PubMed] [Google Scholar]
- 20.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J et al. (2014) IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med 190,1373–1382, 10.1164/rccm.201406-10390C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Opalek JM, Ali NA, Lobb JM, Hunter MG and Marsh CB (2007) Alveolar macrophages lack CCR2 expression and do not migrate to CCL2. J. Inflamm. (Lond.)A, 19, 10.1186/1476-9255-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesan S, Faris AN, Comstock AT, Chattoraj SS, Chattoraj A, Burgess JR et al. (2010) Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir. Res 11,131, 10.1186/1465-9921-11-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sajjan U, Ganesan S, Comstock AT, Shim J, Wang Q, Nagarkar DR et al. (2009) Elastase- and LPS-exposed mice display altered responses to rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol 297, L931–944, 10.1152/ajplung.00150.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong JY, Bentley JK, Chung Y, Lei J, Steenrod JM, Chen Q et al. (2014) Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness through IL-25 and type 2 innate lymphoid cells. J. Allergy Clin. Immunol 134,429–439, 10.1016/j.jaci.2014.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wynn TA, Chawla A and Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496,445–455, 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon S (2007) The macrophage: past, present and future. Bur. J. Immunol 37, S9–S17, 10.1002/eji.200737638 [DOI] [PubMed] [Google Scholar]
- 27.Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P et al. (1998) Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit Care Med 158,1277–1285, 10.1164/ajrocm.158.4.9802078 [DOI] [PubMed] [Google Scholar]
- 28.Bazzan E, Turato G, Tine M, Radu CM, Balestro E, Rigobello C et al. (2017) Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res 18,40, 10.1186/s12931-017-0522-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L et al. (2004) The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med 350,2645–2653, 10.1056/NEJMoa032158 [DOI] [PubMed] [Google Scholar]
- 30.Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC et al. (2001) Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am. J. Respir. Crit Care Med 164,469–473, 10.1164/ajrccm.164.3.2007149 [DOI] [PubMed] [Google Scholar]
- 31.Kang MJ, Lee CG, Lee JY, Dela Cruz CS, Chen ZJ, Enelow R et al. (2008) Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J. Clin. Invest 118,2771–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin KL, Suzuki Y, Nakano H, Ramsburg E and Gunn MD (2008) CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol 180,2562–2572, 10.4049/jimmunol.180.4.2562 [DOI] [PubMed] [Google Scholar]
- 33.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP and Hibbs ML (2012) Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J. Immunol 189,946–955, 10.4049/jimmunol.1200660 [DOI] [PubMed] [Google Scholar]
- 34.Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC et al. (2015) TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med 212,681–697, 10.1084/jem.20141732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janatpour MJ, Hudak S, Sathe M, Sedgwick JD and McEvoy LM (2001) Tumor necrosis factor-dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. J. Exp. Med 194,1375–1384, 10.1084/jem.194.9.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brint EK, Fitzgerald KA, Smith P, Coyle AJ, Gutierrez-Ramos JC, Fallon PG et al. (2002) Characterization of signaling pathways activated by the interleukin 1 (IL-1) receptor homologue T1/ST2. A role for Jun N-terminal kinase in IL-4 induction. J. Biol. Chem 277,49205–49211, 10.1074/jbc.M209685200 [DOI] [PubMed] [Google Scholar]
- 37.Chang Y, Al-Alwan L, Audusseau S, Chouiali F, Carlevaro-Fita J, Iwakura Y et al. (2014) Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol 306, L132–L143, 10.1152/ajplung.00111.2013 [DOI] [PubMed] [Google Scholar]
- 38.McKendry RT, Spalluto CM, Burke H, Nicholas B, Cellura D, Al-Shamkhani A et al. (2016) Dysregulation of antiviral function of CD8(+) T cells in the chronic obstructive pulmonary disease lung. Role of the PD-1-PD-L1 axis. Am. J. Respir. Crit. Care Med 193,642–651, 10.1164/rccm.201504-07820C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr. et al. (2000) Interferon gamma induction of pulmonary emphysema in the adult murine lung. J. Exp. Med 192,1587–1600, 10.1084/jem.192.11.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA and Shapiro SD (2007) CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J. Immunol 178,8090–8096, 10.4049/jimmunol.178.12.8090 [DOI] [PubMed] [Google Scholar]
- 41.Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD et al. (2005) Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm. Res 54,31–36, 10.1007/s00011-004-1319-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.