

Abstract
Mobile Genetic Elements (MGEs) are integrated in bacterial genomes and key elements that drive prokaryote genome evolution. Among them are Integrative and Conjugative Elements (ICEs) and Integrative Mobilizable Elements (IMEs) which are important for bacterial fitness since they frequently carry genes participating in important bacterial adaptation phenotypes such as antibiotic resistance, virulence or specialized metabolic pathways. Although ICEs and IMEs are widespread, they are as yet almost never annotated in public bacterial genomes. To address the need of dedicated strategies for the annotation of these elements, we developed ICEscreen, a tool that introduces two new features to detect ICEs and IMEs in Firmicute genomes. First, ICEscreen uses an efficient strategy to detect Signature Proteins of ICEs and IMEs based on a database dedicated to Firmicutes and composed of manually curated proteins and Hidden Markov Models (HMM) profiles. Second, ICEscreen includes a new original algorithm that detects composite structures of ICEs and IMEs that are frequent in genomes of Firmicutes but are currently not resolved by any other tool. We benchmarked ICEscreen on experimentally supported elements and on a public dataset of 246 manually annotated elements including the genomes of 40 Firmicutes and demonstrate its efficiency to detect ICEs and IMEs.
INTRODUCTION
Horizontal gene transfer constitutes a major evolutionary force among bacterial genomes (1,2). This can be achieved through Mobile Genetic Elements (MGEs) that can transfer from one cell to another and carry fitness genes that may increase the adaptability or resilience of their host to the environment. Among them, transposons and conjugative plasmids are well known to be involved in the spread of antibiotic resistance in both Gram-negative and Gram-positive bacteria, which is a major public health concern (3). Recently, numerous studies have been focusing on the contribution of Integrative and Conjugative Elements (ICEs) to antibiotic resistance dissemination. ICEs are mobile elements integrated in bacterial genomes, which encode their own excision, conjugative transfer and integration (4,5). The contribution of Integrative and Mobilizable Elements (IMEs) to antibiotic resistance has also begun to emerge. IMEs are mobilizable elements that encode all the functions necessary for their excision and integration but, unlike ICEs, are not autonomous for their conjugative transfer. In other words, IMEs use for their transfer the conjugation machinery of another conjugative element (conjugative plasmid or ICE) located in the same cell. At the time of writing, the transfer mechanism of ICEs and especially IMEs in Firmicutes are not fully understood but are probably similar to the one of conjugative and mobilizable plasmids (5,6).
ICEs and IMEs exhibit a broad range of sizes (10–700 kb for ICEs and 2–50 kb for IMEs) and are organized into functional modules that contain all the genes and the sequences involved in the same biological function (5,6). Module exchanges and acquisition/deletion are the main mechanisms driving ICE and IME expansion and evolution. ICEs carry four main types of modules: maintenance, conjugation, regulation and fitness (5,6). Each ICE possesses one maintenance module, one conjugation module, one regulation module as well as one or multiple fitness modules. Within IMEs, the conjugation module is replaced by a mobilization module. The recombination module of ICEs and IMEs encodes the enzymes and carries the sequences required for the integration and excision of the mobile element. Three types of recombination modules have been observed: modules encoding a tyrosine integrase, modules encoding one to three serine integrases and modules encoding a DDE transposase (5,6). In Firmicutes, studies have focused mainly on Streptococci and results indicate that the conjugation module ensures the processing and the translocation of the single strand DNA from the donor cell to the recipient cell. The DNA transfer is initiated by the relaxase which nicks the origin of transfer (oriT) of the ICE and covalently attaches to the 5′ end of the cut strand (7). The transfer of the single strand DNA-relaxase molecule is carried out by a coupling protein which is thought to initiate passage through the conjugation pore (8,9). Known coupling proteins in Firmicutes belong to two unrelated superfamilies, VirD4 and TcpA (8,9). The conjugation pore is a multiprotein complex related to the type IV secretion system (T4SS). It ensures the transport of the DNA-relaxase complex through the cell membranes and walls of both the donor and the recipient bacteria. This transport requires energy supplied by the T4SS ATPases, including the coupling protein and VirB4 (7,10). Based on their conjugation modules, seven families of ICEs have been identified in Streptococci, belonging to three superfamilies, namely Tn916, Tn5252 and TnGBS1 (11). The IME mobilization module contains an oriT and may also carry some genes involved in the conjugative transfer. Almost all known IMEs of Firmicutes encode a relaxase and many putative IMEs from Streptococcus encode a coupling protein (12). However, none of them encodes T4SS proteins. For instance, they are all deprived of the VirB4 ATPase, the most conserved protein of conjugative T4SSs (13). Although few studies describe the diversity of IMEs, analyses performed in streptococcal genomes suggest that their diversity is even larger than that of ICEs. A classification of these IMEs has been proposed based on their relaxase, resulting in nine distinct superfamilies (12). The regulatory modules of ICEs and IMEs are still poorly described and often contain proteins related to those of prophages. The adaptation modules of ICEs and IMEs are highly variable and contain genes that are not involved in the life cycle of the element but can confer different traits to the host organism. Those traits can provide a selective advantage to the host such as resistance to antibiotics or heavy metals, virulence and symbiosis (6,14).
Several studies point to the abundance and diversity of ICEs and IMEs in bacterial genomes. However, only one exhaustive search of conjugation modules was performed on a very large array of genomes (1124 archaeal and bacterial genomes) (15). It found 335 putative chromosomal conjugative modules that probably belong to ICEs and 402 chromosomal relaxase genes lacking neighboring T4SS genes that can be interpreted as putative IMEs. This study was based on HMM profiles deduced from known relaxases, coupling proteins and some T4SS proteins, all derived mainly from conjugation modules of proteobacterial plasmids. However, the focus on proteobacteria might have led to an underestimation of elements in other phyla. Subsequently, other phyla-specific studies were carried out to search for ICEs and IMEs. Studies of actinobacterial genomes (16) or of streptococcal ones (11,12,17) confirmed the great prevalence and diversity of these elements.
There have been only two initiatives in recent times to develop dedicated bioinformatic strategies for detecting ICEs and IMEs. A first initiative was published by the team of Prof. Ou of Shanghai university in 2012 who posed a dedicated resource named ICEberg that gathers ICEs and IMEs described in literature using text-mining methods (18). In its latest version (ICEberg2), it provides an expanded content of elements together with a new tool dedicated to ICE and IME annotation named ICEfinder (19). ICEfinder uses several tools to detect proteins encoded by the integration and transfer modules and attempts to delineate some of the detected and well-known elements by looking for direct repetitions corresponding to the 3′ ends of tRNA genes targeted by various ICEs and IMEs. The second initiative was led by the team of E. Rocha at the Pasteur Institute, France, who developed Conjscan (20), a module of the MacSyFinder software (21) that identifies conjugative modules on plasmids and chromosomes by looking for the genes encoding their protein components and then checking that the composition and genetic organization of the system is consistent with that expected from a conjugative system. A complementary method based on comparative genomics is provided to delineate elements by synteny when at least four closely related genomes of the same species are available (22).
However, designing an automatic tool to identify and annotate ICEs and IMEs in bacterial genomes remains a challenge: although the bioinformatic approaches mentioned above are relevant, they seem to be poorly adapted to Firmicutes. Firstly, these approaches are mostly based on data from proteobacteria. However, Firmicutes are assumed to use specific conjugation and mobilization systems, most of them as yet uncharacterized and probably very distant from the ones of the well-studied enterobacteria (for reviews, see (6,8,23)). Secondly, none of these approaches takes into account the existence of composite elements: within Firmicutes, ICEs, IMEs and the defective elements that derive from them are very often grouped together leading to complex genomic islands. They can form composite structures including tandems (accretions, (5)) of up to four elements. They can also form Matryoshkas where elements are inserted (nested) within others; up to three elements were found to be integrated within another (5,6,24). Several levels of nested elements may also exist.
As previously stated, knowledge on ICEs and IMEs from Firmicutes remains scarce. Our recent studies of ICEs and IMEs of Streptococci have led to the identification of numerous ICEs and even more IMEs and raise questions about the prevalence and diversity of these elements more broadly in Firmicutes. This is why we set out to develop a new tool that identifies regions of co-localized Signature Proteins (SPs) and implements a dedicated algorithm able to resolve the composite structures of ICEs and IMEs particularly in Firmicutes. In particular, we sought to address two needs of the microbiologist community working on MGE of Firmicute genomes: (i) being able to detect already characterized but also putative new ICE and IME using a SP based approach that does not rely on other variable parts of conjugative elements such as fitness genes (ii) helping to decipher complex composite structure according to their ICE and IME content.
In this article we present the result of our work, a new tool named ICEscreen. We describe the strategy implemented in ICEscreen and demonstrate its value for the scientific community through a benchmark study based both on the detection of 11 published and experimentally supported elements as well as on a dataset named FirmiData (25). The latter includes 246 ICEs and IMEs manually annotated in 40 Firmicute genomes, among which 104 are located in complex composite structures.
MATERIALS AND METHODS
ICEscreen ICE and IME detection rely on the presence of SP CDS grouped on the genomes and that were previously demonstrated to be a valuable clue to the presence of an integrative element (11,12).
Construction of the ICEscreen Signature Protein database
Four types of SPs were used in order to detect ICEs and IMEs: (i) integrases which are required for the excision and integration of the element, (ii) relaxases which are proteins catalyzing a site-specific nick of DNA, (iii) the coupling proteins which initiate the transfer of the element through the conjugation pore and (iv) VirB4 which plays an essential role in the functionality of the conjugation pore. VirB4 and the coupling proteins are the most conserved conjugation proteins and therefore the easiest to detect even in elements with a distant conjugation module.
The ICEscreen SP database was built in two distinct parts: (i) a protein database of curated SPs of ICEs and IMEs identified in Firmicutes, mainly in Streptococci (11,12) and (ii) an HMM profile bank designed to detect distant homologs of SPs in all other non-Streptococcus genera of Firmicutes.
The BlastP dataset of reference SPs was enriched with a method similar to the original protocol (11,12). The metadata associated with SP families and superfamilies were manually curated and standardized.
The HMM profile bank was built, composed of two kinds of HMM profiles: (i) publicly available profiles from either the PFAM (26), the TXSScan (20) or the MOBscan (27) resources and (ii) newly designed HMM profiles that characterize new domains of relaxases found in IMEs of Firmicutes and previously published (11,12,28,29). To create the new HMM profiles of relaxases, one or more reference sequences were selected from the ICEscreen protein database of SPs and/or from the NCBI GenBank public database. The functional domain architecture was identified using CD-Search (30) and SPARCLE (31). Coding DNA Sequences (CDSs) from GenBank with a similar functional domain architecture were retrieved. Redundant sequences were removed by a clustering at 95% identity over 100% of the length using CD-HIT (32–34). To further decrease the number of seed sequences if needed, a second clustering at 40% identity over 100% of the length was performed. A multiple alignment was constructed with MAFFT v7.407 using the FFTNS1 algorithm (35). Manual curation of the multiple alignment was carried out to remove too divergent proteins by using a phylogenetic tree constructed with SeaView 4.2 (36). The Neighbor-Joining method BioNJ (37) was used with the following parameters: gaps excluded, Poisson distance and bootstrap of 100 iterations. When a set of relaxases was still too distant, the multiple alignment was further divided into smaller but coherent sets of alignments to subsequently create HMM profiles. When the functional domain was found at the N-terminal position (38), Clustal Omega v1.2.4 with default parameters (39) was used instead of MAFFT to align subsets of sequences and minimize gaps in the N-terminal part of the alignment. To prune away the ends of the multiple alignments carrying little information on conservation, BMGE (40) was used with a BLOSUM 30 substitution matrix. The HMM profile was then built with the HMMbuild tool from the HMMER 3.2.1 suite (41) with default parameters.
Implementation and distribution of the ICEscreen workflow
ICEscreen was implemented using the Python 3 language and Bash scripts. The pipeline was managed with Snakemake (42). Two external tools were required for the detection of SPs by homology: BlastP version 2.9.0 (43) for the identification of close homologs and HMMscan from the HMMER3 suite version 3.3.2 (41) for the detection of remote homologs. BlastP results were filtered out to remove false positive results using four types of filters detailed in Supplementary Table S1(a). Filters used to remove false positive HMMscan results are detailed in Supplementary Table S1(b). Specific additional filters were designed to remove known SP false positive results and are described in Supplementary Table S1(c). A Conda package for ICEscreen was developed encapsulating all the dependencies and automating the installation process. A wrapper for ICEscreen was integrated into the main Galaxy ToolShed (44) in order to make the tool more accessible to researchers and easier to integrate into workflows.
Benchmarking
ICEscreen was compared with ICEfinder (19) and CONJscan version 1.0.5 which is a module of MacSyFinder (20,21). ICEfinder was used with default parameters. As CONJscan is a generic tool to search for conjugative T4SS in all bacteria and archaea, only the subsets FA (corresponding to the Tn916 superfamily) and FATA (corresponding to the Tn5252 superfamily) of conjugation modules known to be related to ICEs and IMEs in Firmicutes were analyzed for this benchmark. FA and FATA systems (8) were searched using the CONJ model to detect conjugation modules of ICEs (relaxase + VirB4 + coupling protein). Mobilizable modules of IMEs were searched using a modified version of the MOB model to match our definition of IMEs (mandatory relaxase, accessory coupling protein and absence of VirB4). As the exact delineation of the elements is not implemented in ICEscreen, we compared the three tools solely on their ability to (i) accurately detect the SPs they have been programmed to detect and (ii) gather the SPs accordingly to highlight the correct ICE and IME structures (SP composition and ICE or IME type correctly assigned).
Benchmark on experimentally supported elements from Firmicutes
First, a benchmarking was performed on a set of 11 elements from Firmicute genomes collected from publications and with experimental support (see ‘Genomes sheet’ of Supplementary Table S2). We took into account elements whose delineation was reliable and carried by assembled and well annotated genomes (RefSeq genomes) and not included in the FirmiData dataset (25) (see next sub-section). An element of the reference dataset was considered correctly detected by a tool when (i) at least two of its SPs were identified and grouped together and (ii) the type of the detected element was also correct (identical to the one mentioned in the publication). An element of the reference dataset was considered partially detected by a tool if less than two SPs were correctly detected. This situation often leads to an error in element type assignment (e.g. IME detected as ICE). An element of the reference dataset was considered not detected if none of its SPs was detected.
Benchmark on FirmiData
The FirmiData dataset (25) was used as a reference to evaluate and compare the performances of the three tools using a diverse set of manually annotated ICEs and IMEs, including complete, partial and nested structures as well as elements in accretion. These elements were annotated in 25 Streptococcus and 15 other Firmicute public complete genomes issued from RefSeq using data from the literature and a semi-automated procedure that was described in (11) for ICEs and in (12) for IMEs. Regarding SPs, FirmiData includes a total of 137 tyrosine Integrases, 113 Serine Integrases, 12 DDE transposase integrases, 250 relaxases, 143 coupling proteins, and 98 VirB4. The number of manually curated structures annotated in FimiData includes: 98 ICEs and 148 IMEs with 42.3% of these elements being found in complex structures (nested or in accretion). We used three levels of result evaluation:
An element of the reference dataset was considered detected by a tool when at least two of its SPs were identified and grouped together. Consequently, for an IME including two SPs in the reference dataset, both SPs had to be detected and grouped.
The type of the detected element was considered correct if it was consistent with the type attributed in the reference dataset. Thus, an ICE was considered correctly typed if a tool had characterized it as ICE, partial ICE or conjugative module. In the same way, an IME had to be characterized as either IME, mobilizable element or mobilizable module.
We also evaluated the ability of the tools to detect the correct number of ICE and IMEs included in composite elements.
Case studies: three examples of ICEs and IMEs detection in three Firmicute genomes
We selected three genomes of FirmiData to illustrate differences in ICE/IME prediction compared to the FirmiData manual annotation: Streptococcus gallolyticus subsp.gallolyticus BAA-2069, Roseburia hominis A2-183 and Clostridioides difficile R20291. ICEfinder, CONJscan and ICEscreen results were obtained as described as described above. Comparisons and graphical representations of ICE and IME contents in the FirmiData reference dataset and in the results produced by the three tools were performed using customized R scripts.
RESULTS
The ICEscreen workflow
ICEscreen takes as input annotated genomes with predicted coding genes in GenBank format. It generates three output result files: (i) one summary file including general information about the ICEscreen run (like parameters, number of detected SPs and number of detected elements), (ii) a second file listing the detected SPs in the query genome(s) and (iii) a third one listing the features of the detected ICEs and IMEs in the query genome(s). Output files including ICE and IME SP annotation are provided in gff and GenBank format. The ICEscreen workflow is described in Figure 1 and consists of four steps: (A) detection of SPs, (B) search of possible transfer modules by creation, extension and fusion of anchors, (C) assignation of the integrases to the anchors, and (D) classification of the ICEs and IMEs elements based on their content in SPs.
Figure 1.

The four main steps of the ICEscreen workflow. The input query genome(s) must be annotated genomes in GenBank format. Step A: The first step consists in detecting three types of Signature Proteins (SPs), which are relaxases, coupling proteins and VirB4. The resulting ordered list of SPs per genome is then segmented into groups of SPs where the distance between two subsequent SPs is less than 100. Step B: The second step consists in searching for all potential conjugation modules by ‘creating, extending and fusing’ anchors. An anchor is created when a relaxase, a coupling protein or a VirB4 is detected. Then, the anchor is extended left and right to detect group of SPs (excluding Integrases) corresponding to putative elements. When all potential anchors have been created, the algorithm evaluates the possibility of combining anchors in order to resolve the cases of complex structures (for instance nested elements). Step C: The integrase is then searched on both sides of each anchor. Step D: The next step consists in typing each element according to its SP content and size as described in the two panels on the right.
Step A: detection of Signature Proteins and segmentation
ICEscreen first searches for homologs of SPs with BlastP using the ICEscreen protein database. This includes SPs that were chosen for their diversity and that are encoded by demonstrated or predicted ICEs or IMEs from Firmicutes. Currently, the ICEscreen protein database comprises 1022 non-redundant SPs, including 317 relaxases, 231 coupling proteins, 140 VirB4 and 334 integrases (239 tyrosine integrases, 73 serine recombinases and 22 DDE transposases). According to the classification proposed by Ambroset et al. (11) and Coluzzi et al. (12), a family is assigned to relaxases, coupling proteins, and VirB4 when it shares more than 40% identity with proteins of the database encoded by ICEs. In order to identify genome regions including closely located genes encoding VirB4, CPs and relaxases and to limit the number of combinations for the grouping of these SPs into structures, a preliminary step is implemented that defines segments as lists of SPs where the distance between two subsequent SPs is less than 100 CDSs. This cutoff is based on elements from Firmicutes where the most distant SP encoding genes are less than 50 CDSs apart and takes into account a safety margin.
ICEscreen also searches for distant proteins with HMMscan using the ICEscreen HMM profile bank. The ICEscreen HMM profile bank contains 22 profiles, including 15 for relaxases, 3 for coupling proteins, 1 for VirB4 and 3 for integrases (see Supplementary Table S3 in supplementary material). Twelve HMM profiles have been incorporated from reliable resources: three integrase profiles from PFAM (26), three coupling protein profiles and five relaxase profiles from TXSScan (20) and one relaxase profile from MOBfamDB (27). Seven HMM profiles have been newly created: one profile for the relaxase MOBL of ICEs and six for new IME relaxases described in (12) (one profile for PF01719-like relaxases, two profiles for PHA00330-like relaxases and three profiles for PF02407-like relaxases). The domain content of SPs determined by HMM analysis has been used to classify the SPs in superfamilies as previously proposed (11,12). The steps described in the following are carried out with each segment.
Step B: creation, extension and fusion of anchors
For each segment of the query genome(s), the second step consists in detecting all possible anchors that correspond to putative complete or partial conjugation modules. To do so, the list of SPs arranged by genomic location is scanned from left to right and an anchor is created when one of the conjugation module's SPs (relaxase, coupling protein or VirB4) is found. The sequence of SPs continues to be scanned from left to right to extend the current anchor. Anchor extension is stopped when one of the following situations is encountered: (i) two successive genes encoding SPs are separated by >100 CDSs (because SPs of an anchor cannot be on distinct segments), (ii) two adjacent genes encoding VirB4 or two adjacent genes encoding coupling proteins are found (in both cases, a new anchor is created starting at the second VirB4 or coupling protein-encoding gene), (iii) two relaxase genes separated by more than one CDS are found (in this case a new anchor is created starting at the second relaxase gene), (iv) integrase genes are found, as they are dealt with at a later stage. SPs issued from BlastP hits of the same family of ICEs (families described in (11)) are grouped together into an anchor while those coming from different superfamilies of elements are separated into different anchors. Unassigned SPs can be added to any anchor if they are issued from the same superfamily of elements: for example, an unassigned relaxase belonging to the MobP superfamily, that is typical of ICEs belonging to the Tn5252 superfamily, can be assigned to an anchor including CP and VirB4 related to Tn1549 family, as the Tn1549 family belongs to the Tn5252 superfamily. Once an anchor has been created and possibly extended from left to right, the algorithm attempts to extend it from right to left in a similar way. ICEs and IMEs are not oriented on the genome, so the algorithm is independent of the choice of the initial scanning direction. Some SPs may be attributed to two different anchors at this stage. Anchor creation and extension are repeated to consider all the SPs of each segment. Finally, a last procedure consists in merging distant compatible anchors to find nested structures. The merging is exhaustive as all combinations of merging of anchors are tested. When multiple valid solutions are possible, priority is given to the merging of the nearest anchors. The algorithm is recursive and can detect multiple levels of nesting when several ICEs/IMEs are inserted into an element. The conditions for merging anchors are identical to the conditions for extending an anchor (see step B). The merging of distantly located compatible anchors can sometimes help resolve SPs previously attributed to two different anchors.
Step C: assignment of integrases to anchors
The third step of the workflow consists in assigning integrases to anchors located within the segment, using dedicated rules and an iterative algorithm. Any integrase, regardless of its family or superfamily, can be associated with any anchor. The number of integrases associated with an anchor is based on the integrase types. An anchor can be associated with (i) one or two identically oriented adjacent genes of tyrosine integrase (ii) one DDE transposase gene (iii) one, two or three identically oriented serine integrase(s) encoding genes and separated by up to one CDS.
The iterative algorithm works as follows. In a first step, integrase genes located directly upstream or downstream of anchors are assigned. For anchors including VirB4, integrases must also be facing outwards. This step generally permits to unambiguously assign a subset of integrases to anchors. For unassigned integrases, a second step consists in considering more distantly located integrase genes (up to three anchors away from the nearest one and possibly coming from nested elements) and iteratively assigning integrases to anchors that have not yet been assigned integrases. Once they have been assigned to an anchor, they are masked. This iterative process is performed until all possible integrase assignments have been explored.
At the end of this step, all integrases will be either assigned or not assigned to anchors. Unassigned integrases can be integrases with an ambiguous assignment (e.g. assigned to two distinct transfer modules) or integrases not associated with an anchor. Unassigned integrases are kept in the ICEscreen output files and are annotated as either ‘to be manually verified’ or ‘unassigned integrases’.
Step D: classification of elements into complete or partial ICEs and IMEs
The last step of ICEscreen consists in characterizing the elements found in the previous step according to their SPs and integrase composition and number of CDSs. We defined six different categories of elements:
complete ICE composed of one relaxase, one coupling protein, one VirB4 and of one or several integrases;
complete IME composed of either a) one relaxase and one or several integrases or b) of one relaxase, one coupling protein and one or several integrases. In IMEs, the maximum number of CDSs separating the most distantly located SPs is 10;
conjugation module composed of one relaxase, one coupling protein and one VirB4;
mobilizable element composed of one relaxase and one coupling protein and with the maximum number of CDS separating the most distant SPs being 10;
partial ICE defined as any structure of SPs that contains at least one VirB4 and that is not a complete ICE;
other partial elements defined as any structure that contains at least two SPs and that does not fall into any of the above categories. This category may also include degraded elements including pseudogenes of SPs.
Finally, when possible, a family and/or superfamily is assigned to the element based on the nature of its SP. This assignment takes into account the nature of the relaxase (38,45,46) and the nature of the mating pore proteins (47). Superfamilies of ICEs and IMEs are assigned using the known three ICE superfamilies (Tn916, Tn5252 and TnGBS1) and nine IME superfamilies in Streptococci (MOBT, MOBQ, MOBV, MOBP, MOBC, PF01719, PF01719-PF00910, PF02407 and PHA00330). Superfamilies of ICEs are assigned based on the relaxase and coupling protein families as defined in Ambroset et al., 2016 (11). Superfamilies of IMEs are assigned based solely on the relaxase family as defined in (12).
Benchmarking results
Benchmark on experimentally supported elements from Firmicutes
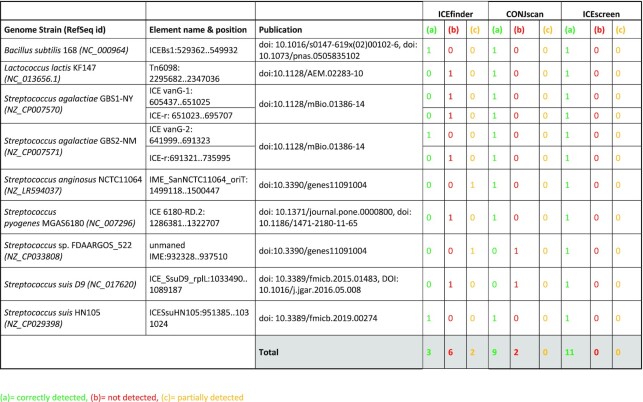
The ICEscreen tool was first evaluated using nine publicly available RefSeq genomes carrying 11 published elements (nine ICEs and two IMEs) with experimental support demonstrating their transfer and/or excision. Table 1 summarizes the performances of ICEfinder, CONJscan and ICEscreen on this first dataset. Detailed results obtained by each tool are provided in Supplementary Table S2. ICEfinder exhibited the poorest performance by detecting only three of the 11 elements correctly and two partially with either a wrong assignment (e.g.: ICE instead of IME) or a wrong SP composition. Six elements were not at all detected by ICEfinder: Tn6098 of Lactococcus lactis KF147, ICE vanG-1 and ICE-r of Streptococcus agalactiae GBS1-NY, ICE-r of S. agalactiae GBS2-NY, ICE 6180-RD.2 of S. pyogenes MGAS6180 and ICE_SsuD9_rplL of S. suis D9. CONJscan exhibited rather good performances by correctly detecting nine out the 11 elements. The two undetected elements corresponded to the two IMEs of the dataset (IME_SanNCTC11064_oriT of Streptococcus anginosus NCTC11064 and IME_SanNCTC11064_oriT of Streptococcus sp. FDAARGOS_522). ICEscreen correctly detected all the 11 elements of this dataset. Overall, ICEfinder did not achieve good results whilst CONJscan and ICEscreen performed well in the detection of elements whose excision and/or transfer has been demonstrated or is based on strong biological evidence.
Table 1.
Results of ICEfinder, CONJscan and ICEscreen on nine Refseq genomes including 11 elements with experimental support. (a) column (green) indicates correctly detected elements: at least two SP of the element are identified and grouped together and the type of the element is identical to the one mentioned in the publication. (b) column (red) indicates undetected elements and corresponds to elements with no SP detected. (c) column (orange) indicates partially detected elements and corresponds to elements with only one SP detected and possibly an error in element type assignment (e.g.: IME detected as ICE)
|
Benchmark on FirmiData
To evaluate more extensively the performance of ICEscreen, we used FirmiData, a public dataset of 40 genomes of Firmicutes that were chosen to illustrate the diversity of ICEs and IMEs as well as of their various organization into composite elements.
We first compared the ratio of FirmiData's SPs successfully detected by ICEscreen, CONJscan and ICEfinder (see Figure 2). Results are classified by SP type (Relaxase, Coupling Protein, VirB4 and Integrase). ICEscreen outperformed the two other tools and detected between 85.6% and 98% of each type of SP. CONJscan performed well and detected 73.1% of the coupling proteins, 75.4% of the relaxases and 82.7% of the VirB4. It is important to note that CONJscan does not search for integrases, and therefore this criterion cannot be used as a basis for comparison. ICEfinder had the lowest performance, detecting only 30% of the coupling proteins, 40.6% of integrases, 42.1% of the relaxases and 36.5% of the VirB4 proteins.
Figure 2.

Percentage of CDSs corresponding to FirmiData Signature Proteins that are detected by ICEscreen, CONJscan and ICEfinder. Each type of Signature Protein is indicated by a distinct color. CONJscan does not search for integrases. Some SPs may be fragmented due to insertion of IMEs, ICEs or mobile introns (for example composite structures composed of nested elements). The total number of CDSs corresponding to each SP type is reported at the left of the barplots.
We then used the elements annotated in the FirmiData reference dataset to compare the results of ICEscreen, CONJscan and ICEfinder. In FirmiData, 98 ICEs and 148 IMEs were annotated. Among them, 55 ICEs and 87 IMEs are isolated whilst 43 ICEs and 61 IMEs are in composite structures. We compared the number of correct FirmiData elements found by ICEscreen, CONJscan and ICEfinder taking into account whether they are isolated or in composite structures (see Figure 3). ICEscreen detected all the ICEs, whether isolated (47 perfectly and 8 with one SP missing and/or wrongly added) or in ICE/ICE composite structures (15 perfectly and 2 with one SP missing and/or wrongly added) (top A and top B panels). For ICEs in ICE/IME composite structures ICEscreen also performs well with 13 ICEs perfectly detected, 10 with one SP missing and/or wrongly added and 3 ICEs combined with other elements (bottom B panel, solid colour). ICEscreen results were also excellent for isolated IMEs with 72 out of 87 IMEs perfectly detected, 13 IMEs with either missing and/or wrongly added SPs or wrong element type associated and only 2 IMEs detected with an incorrect structure (bottom A panel). ICEscreen performance decreased slightly for IMEs in composite structures but was still quite good, with 48 out of 61 IME perfectly detected and only one missed IME (bottom B panel, hashed colour). CONJscan performed rather well for isolated ICEs (33 out of 55 perfectly detected), but results became fairly poor for ICEs in ICE/ICE composite structures (1 out of 17) and in ICE/IME composite structures (4 out of 26 perfectly detected). CONJscan, which was not designed to identify IMEs, generally missed a large number of IMEs whether isolated or in composite structures (62.8% IMEs were missed and the ones detected were generally not correct, either in terms of SP composition, element typing or structure resolution). ICEfinder performance was generally poor on the FirmiData genomes but slightly better for isolated IMEs (37 out of 87 detected, including 17 perfectly detected, nine with either additional and/or missing SPs or element typing error, 11 wrongly combined with other elements, bottom A panel). In summary, ICEscreen outperformed both CONJscan and ICEfinder due to its specific algorithm facilitating composite structure resolution and its SP databases adapted to Firmicute ICEs and IMEs composition.
Figure 3.

Number of elements reported by ICEscreen, CONJscan and ICEfinder compared to the ICEs and IMEs of the FirmiData reference. Elements of FirmiData that were detected by a tool and share exactly the same SP composition (all SPs detected and co-localized without additional SPs) and a similar element type as the reference are represented in green. Wrongly characterized elements sharing strictly the same SPs are represented in blue. Elements of FirmiData that have been detected by a tool but for which SPs are missing and/or wrongly added are also represented in blue. Elements of the reference that were detected by a tool but combined with other elements are represented in orange. Elements of the reference that were not detected by a tool are represented in black. The top A panel and the bottom A panel refer to isolated ICEs and IMEs, respectively. The panels on the right show the number of elements in composite structures (only composed of ICEs: top B panel; composed of ICEs & IMEs or IMEs only: bottom B panel). The number of ICEs and IMEs are represented in solid color and in hashed color, respectively.
Case studies: three examples of ICEs and IMEs detection in three Firmicute genomes
To examine the performance of ICEscreen in resolving composite structures, three case studies were carried out, presented in Figure 4. The first case study was performed on the complete chromosome of Streptococcus gallolyticus subs. gallolyticus BAA-2069 (Figure 4A) which includes 6 IMEs and 3 ICEs, the last three elements on the genome being in accretion. ICEscreen detected all ICEs and all IMEs, only missing two SPs of one IME. It also resolved the composite structure including one ICE and two IMEs. CONJscan detected the conjugation modules of the three ICEs but failed to detect any IME SPs (except one relaxase). ICEfinder only detected one IME correctly. It also identified two of the ICEs but added or missed some of their SPs. It also merged all three accreted elements into one ICE. This example illustrates that the detection of SPs is crucial to correctly detecting ICEs and IMEs and that falsely grouping SPs generates errors in the detection of elements.
Figure 4.

Three case studies of ICE and IME detection in Firmicute genomes. FirmiData reference annotation was compared to elements detected by ICEscreen, CONJscan and ICEfinder. The type of element is mentioned on the left hand-side of the figure. The coloured panels indicate is elements were annotated as accreted (orange) or nested (green) in the FirmiData reference set. The coloured lines indicate the type of element (blue: ICE, green: IME, red: conjugation module, orange: partial element). The coloured arrows indicate orientation and type of Signature Proteins (orange: relaxase, purple: coupling protein, green: VirB4, red: Integrase). Fragmented Signature Proteins are indicated with half-circles. The x-axis is an arbitrary sequential numbering of the genes that indicates the relative position of the Signature Protein genes. (A) ICE and IME detection in the Streptococcus gallolyticus subsp. gallolyticus BAA-2069 genome. (B) ICE and IME detection in a region of the Roseburia hominis A2-183 genome (positions 2 893 300–3 033 202). (C) ICE and IME detection in the Clostridioides difficile R20291 genome.
The second case study was carried out on a region of the Roseburia hominis A2-183 genome (positions 2 893 300–3 033 202) (Figure 4B). According to the FirmiData reference, this small region contains two IMEs and one ICE integrated in close but distinct sites. All three elements were correctly detected by ICEscreen. Interestingly, both CONJscan and ICEfinder correctly detected the SPs composing these 3 elements but failed in identifying the element structures by wrongly merging close SPs in a single ICE. Additionally, ICEfinder wrongly included three SPs in the ICE (one VirB4 and two coupling proteins). This example illustrates that CONJscan and ICEfinder, which were not designed to resolve composite elements, frequently erroneously merge close or adjacent elements. It also shows that the correct detection of SPs is mandatory but not sufficient to correctly detect ICEs and IMEs. This is why a dedicated algorithm like the one designed in ICEscreen is needed to correctly assign each SP to IMEs and ICEs, especially if they are close in the genome.
The third case study was performed on the complete chromosome of C. difficile R20291 (Figure 4C). According to the FirmiData reference annotation, this chromosome includes two isolated elements (one IME and one ICE) and a complex structure with three IMEs nested into one ICE. ICEscreen correctly detected the 2 ICEs and 3 out of the 4 IMEs. It only failed in assigning the correct integrase of the last IME. It also resolved the complex nested structure. CONJscan correctly detected most of the SPS but wrongly merged the elements included in the complex structure and considered them as one single ICE. ICEfinder missed several SPs of the IME/ICE nested element, failed to detect the SPs of the isolated IME and wrongly assigned a VirB4 to the isolated ICE.
These three cases illustrate the complexity of ICE and IME detection in Firmicute genomes. They also demonstrate ICEscreen's ability to find most ICEs and IMEs in FirmiData genomes and show that ICEscreen compares favourably with CONJscan and ICEfinder in correctly detecting and assigning SPs to ICEs and IMEs. These case studies also show the limitations of CONJscan and ICEfinder in resolving element structures, especially for adjacent and nested elements.
DISCUSSION
In this article, we present ICEscreen, a new tool to detect ICEs and IMEs in chromosomes of Firmicutes and compare its performance with two existing tools: ICEfinder which is associated with the ICEberg2 database (18,19), and CONJscan which is a module of MacSyFinder that was designed to detect conjugative T4SS (20,21). Compared to CONJscan and ICEfinder, ICEscreen brings several new important features: (i) it relies on an ad hoc new SP database composed of proteins and HMM profiles that reflect the state of the art of known Firmicute ICE and IME SPs, (ii) it combines the results of two tools (Blast and HMMscan) and specific filters to efficiently detect these SPs in Firmicute genomes while limiting false positives due to the lack of specificity of certain SPs, (iii) it implements a new algorithm capable of resolving regions carrying several elements (referred to as composite structures), (iv) it provides much valuable information, such as the precise structure and SP composition of elements, the family of Relaxase and coupling proteins (where possible) and other isolated SPs that may indicate the presence of new and as yet unknown element and (v) it is based on an automated approach and is packaged to provide a simple and valuable resource to the microbiologist community interested in ICE/IME detection and annotation in any species of Firmicutes. We evaluate ICEscreen performances using a list of 11 published elements (nine ICEs and two IMEs) with experimental support of their transfer and/or excision and on the FirmiData public dataset including 98 ICEs and 148 IMEs annotated in 40 genomes of Firmicutes (25). We also present three case studies illustrating typical composite structures in Firmicute genomes. Taking together, this dataset includes 109 ICEs and 150 IMEs that represent the full diversity of elements known to exist in Firmicutes, whether they have been experimentally demonstrated, are predicted members of well-known families or are members of predicted non-canonical families.
Overall, our results highlight ICEscreen good performances for the detection of Firmicute ICEs and IMEs. ICEscreen detects all the 109 ICEs and 149 out of the 150 IMEs of our two evaluation sets, whether isolated or in composite structures. ICEscreen performance decreases slightly for ICEs and IMEs in composite structures but was still very good. On this dataset, CONJscan performs quite well in correctly detecting conjugation modules of ICEs but does not neither detect detects IMEs nor correctly resolves the structures of most composite elements. ICEfinder has the weakest performance on our benchmark by missing an important number of SPs and adding other false positives to elements, especially in composite structures including nested or accreted ICEs and IMEs. Composite structures mixing ICEs and IMEs are particularly frequent in Firmicutes. Thanks to its dedicated algorithm, ICEscreen detects all elements and solves most intricate composite structures while the two other existing tools miss some of them and generally merge some elements together. This is due to the fact that the existence of composite elements is not taken into account in the current versions of neither CONJscan nor ICEfinder.
It is important to mention that ICEscreen, is still imperfect, as many factors render the detection of some ICEs and IMEs elusive: (i) ICEScreen cannot detect IMEs that do not encode any relaxase. However, many known IMEs of Gammaproteobacteria are devoid of relaxase (6) and the 40 genomes composing the FirmiData data set carry many integrative elements that could be IMEs devoid of relaxases (data not shown); (ii) knowledge of the different families of SPs of ICEs and IMEs is still highly partial, which could prevent detection of some yet unknown elements, (iii) ICEscreen algorithm does not permit detection of too decayed elements, i.e. IMEs that have lost one SP gene, or ICEs that have lost VirB4 and two other SP genes. Such elements are frequent in bacterial genomes (5), (iv) correct attribution of the integrases to the elements is very challenging, since integrases are not specific to ICEs and IMEs and may belong to other mobile elements such as prophages or satellite prophages (5). Moreover, many exchanges of integrase genes between ICEs and IMEs of Streptococci have been highlighted (12), (v) various ICEs or IMEs from Firmicutes carry genes of SPs that are fragmented by the insertion of one or multiple other mobile elements (such as ICEs, IMEs or type II introns), generating difficulties in detecting complex structures of ICEs and IMEs. This is for example the case of the genome of Clostridioides difficile R20291 (Figure 4C) which carries three IMEs, two of which (Tn6104 and Tn6105) disrupt the coupling protein gene of an ICE and (v) some composite structures cannot be solved because they contain many (decayed or complete) elements and may also contain other types of MGEs (such as the Lachnoclostridium phocaeense Marseille-P3177 genome included in the FirmiData set (25)). These cases remain difficult to completely resolve even by ICEscreen.
ICEscreen performs well for Firmicute ICEs and IMEs (functional or slightly decayed) detection but does not yet carry out a precise delimitation of the boundaries of the elements at the gene or nucleotide level; the latter is facilitated by ICEscreen but still needs to be carried out manually. The precise delimitation of the elements would be an asset to characterize elements and automatically study their fitness genes but also to improve the resolution of complex structures. Delimiting the elements is a complex problem. This difficulty is due (i) to the great diversity of integrases and of their specificity of integration and (ii) to the composite structures. Several methods could possibly handle this challenge. A first one is based on the detection of direct repeats flanking the elements. ICEfinder uses this method to delimit the ICEs integrated in the 3' end of the tRNAs (19). This is not fully satisfying, because (i) various Firmicute elements, such as Tn916, TnGBS1 or TnGBS2, integrate with a low specificity (48,49); (ii) many elements, that do integrate in a site-specific manner, target not 3′ end of tRNA genes, but different sites (3′end, 5′ end, internal sites) of various genes encoding proteins (11,12). A second possible method to delimitate elements is based on comparative genomics. Cury et al (50) uses this method to delineate ICEs. This method may be challenging because it requires closely related genomes from the same species, including fully assembled genomes, some having the element and others that do not. At least in some cases, it is difficult to find genomes deprived of element inserted in a specific site (for an example, see (51)). Moreover, the presence of composite structures and/or degraded elements impede the delineation analysis. A third method would be to use the sequence of empirically demonstrated elements to delineate related elements in genomes. This method is useful for identifying the presence of widespread elements such as Tn916-related ICEs in Firmicutes. However, it only allows the delineation of elements closely related to the known ones. To our opinion, no single method can adequately delimit all the elements. Therefore, a strategy for selecting the optimal method, or the best combination of methods, should be defined for each element, depending on the integrase that it encodes. For the next version of ICEscreen we plan to include an additional module of automatic or semi-automatic delineation of the elements. ICEscreen's ability to solve composite structures will be valuable.
As a perspective for this work, we also plan to extend ICEscreen's scope to address the detection of ICEs and IMEs from other bacterial phyla (Proteobacteria, Actinobacteria, Bacteroidetes). This implies two main tasks: (i) the extension of our SP database to include representative families specific of these phyla and (ii) the addition of new detection rules to the ICEscreen algorithm to take into account the structures observed in non-Firmicute genomes. Together with the ICE/IME delineation module, these will be the main new features of the next version of ICEscreen software.
DATA AVAILABILITY
The ICEscreen software is freely and publicly available on the gitlab repository of the MathNum division of INRAE at https://forgemia.inra.fr/ices_imes_analysis/icescreen. It is available to install from source, Conda (https://anaconda.org/search?q=icescreen), and the Galaxy tool shed (https://toolshed.g2.bx.psu.edu/). The documentation of the tool is available at https://icescreen.migale.inrae.fr. ICEscreen is a free software under the Affero GPLV3 licence.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the INRAE MIGALE bioinformatics facility (MIGALE, INRAE, 2020. Migale bioinformatics Facility, doi: 10.15454/1.5572390655343293E12) for providing help and computing resources. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Notes
Present address: Julie Lao, Infection, Antimicrobiens, Modélisation, Évolution (IAME), UMR 1137 Inserm, EVRest team Faculté de Médecine Site Xavier Bichat 16 rue Henri Huchard, F-75877 Paris Cedex 18, France.
Present address: Charles Coluzzi, Institut Pasteur, Université de Paris, CNRS, UMR3525, Microbial Evolutionary Genomics, Paris, F-75015 Paris, France.
Contributor Information
Julie Lao, Université Paris-Saclay, INRAE, MaIAGE, F-78350 Jouy-en-Josas, France; Université de Lorraine, INRAE, DynAMic, F-54000 Nancy, France.
Thomas Lacroix, Université Paris-Saclay, INRAE, MaIAGE, F-78350 Jouy-en-Josas, France.
Gérard Guédon, Université de Lorraine, INRAE, DynAMic, F-54000 Nancy, France.
Charles Coluzzi, Université Paris-Saclay, INRAE, MaIAGE, F-78350 Jouy-en-Josas, France; Université de Lorraine, INRAE, DynAMic, F-54000 Nancy, France.
Sophie Payot, Université de Lorraine, INRAE, DynAMic, F-54000 Nancy, France.
Nathalie Leblond-Bourget, Université de Lorraine, INRAE, DynAMic, F-54000 Nancy, France.
Hélène Chiapello, Université Paris-Saclay, INRAE, MaIAGE, F-78350 Jouy-en-Josas, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Institut National de Recherche pour l’Agriculture, l’alimentation et l’Environnement (INRAE); Région Grand-Est and the European Union through the Regional Operational Program of the European Regional Development Fund (ERDF).
Conflict of interest statement. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1. Frost L.S., Leplae R., Summers A.O., Toussaint A.. Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 2005; 3:722–732. [DOI] [PubMed] [Google Scholar]
- 2. Treangen T.J., Rocha E.P.C.. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011; 7:e1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Partridge S.R., Kwong S.M., Firth N., Jensen S.O.. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018; 31:e00088-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burrus V., Pavlovic G., Decaris B., Guédon G.. Conjugative transposons: the tip of the iceberg. Mol. Microbiol. 2002; 46:601–610. [DOI] [PubMed] [Google Scholar]
- 5. Bellanger X., Payot S., Leblond-Bourget N., Guédon G.. Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol. Rev. 2014; 38:720–760. [DOI] [PubMed] [Google Scholar]
- 6. Guédon G., Libante V., Coluzzi C., Payot S., Leblond-Bourget N.. The obscure world of integrative and mobilizable elements, highly widespread elements that pirate bacterial conjugative systems. Genes. 2017; 8:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alvarez-Martinez C.E., Christie P.J.. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. MMBR. 2009; 73:775–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guglielmini J., de la Cruz F., Rocha E.P.C.. Evolution of conjugation and type IV secretion systems. Mol. Biol. Evol. 2013; 30:315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ilangovan A., Connery S., Waksman G.. Structural biology of the Gram-negative bacterial conjugation systems. Trends Microbiol. 2015; 23:301–310. [DOI] [PubMed] [Google Scholar]
- 10. Redzej A., Ukleja M., Connery S., Trokter M., Felisberto-Rodrigues C., Cryar A., Thalassinos K., Hayward R.D., Orlova E.V., Waksman G.. Structure of a vird4 coupling protein bound to a VirB type IV secretion machinery. EMBO J. 2017; 36:3080–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ambroset C., Coluzzi C., Guédon G., Devignes M.-D., Loux V., Lacroix T., Payot S., Leblond-Bourget N.. New insights into the classification and integration specificity of streptococcus integrative conjugative elements through extensive genome exploration. Front. Microbiol. 2016; 6:1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coluzzi C., Guédon G., Devignes M.-D., Ambroset C., Loux V., Lacroix T., Payot S., Leblond-Bourget N.. A glimpse into the world of integrative and mobilizable elements in streptococci reveals an unexpected diversity and novel families of mobilization proteins. Front. Microbiol. 2017; 8:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramsay J.P., Firth N.. Diverse mobilization strategies facilitate transfer of non-conjugative mobile genetic elements. Curr. Opin. Microbiol. 2017; 38:1–9. [DOI] [PubMed] [Google Scholar]
- 14. Botelho J., Schulenburg H.. The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 2021; 29:8–18. [DOI] [PubMed] [Google Scholar]
- 15. Guglielmini J., Quintais L., Garcillán-Barcia M.P., de la Cruz F., Rocha E.P.C.. The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet. 2011; 7:e1002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghinet M.G., Bordeleau E., Beaudin J., Brzezinski R., Roy S., Burrus V.. Uncovering the prevalence and diversity of integrating conjugative elements in actinobacteria. PLoS One. 2011; 6:e27846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lao J., Guédon G., Lacroix T., Charron-Bourgoin F., Libante V., Loux V., Chiapello H., Payot S., Leblond-Bourget N.. Abundance, diversity and role of ICEs and IMEs in the adaptation of streptococcus salivarius to the environment. Genes. 2020; 11:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bi D., Xu Z., Harrison E.M., Tai C., Wei Y., He X., Jia S., Deng Z., Rajakumar K., Ou H.-Y.. ICEberg: a web-based resource for integrative and conjugative elements found in bacteria. Nucleic Acids Res. 2012; 40:D621–D626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu M., Li X., Xie Y., Bi D., Sun J., Li J., Tai C., Deng Z., Ou H.-Y.. ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019; 47:D660–D665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abby S.S., Cury J., Guglielmini J., Néron B., Touchon M., Rocha E.P.C.. Identification of protein secretion systems in bacterial genomes. Sci. Rep. 2016; 6:23080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abby S.S., Néron B., Ménager H., Touchon M., Rocha E.P.C.. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLOS ONE. 2014; 9:e110726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cury J., Abby S.S., Doppelt-Azeroual O., Néron B., Rocha E.P.C.. de la Cruz F. Identifying conjugative plasmids and integrative conjugative elements with CONJscan. Horizontal Gene Transfer: Methods and Protocols, Methods in Molecular Biology. 2020; NY: Springer US; 265–283. [DOI] [PubMed] [Google Scholar]
- 23. Goessweiner-Mohr N., Arends K., Keller W., Grohmann E.. Conjugation in gram-positive bacteria. Microbiol. Spectr. 2014; 2:PLAS–0004–2013. [DOI] [PubMed] [Google Scholar]
- 24. Brouwer M.S.M., Warburton P.J., Roberts A.P., Mullany P., Allan E.. Genetic organisation, mobility and predicted functions of genes on integrated, mobile genetic elements in sequenced strains of clostridium difficile. PLoS One. 2011; 6:e23014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guédon G., Lao J., Payot S., Lacroix T., Chiapello H., Leblond-Bourget N.. FirmiData: a set of 40 genomes of firmicutes with a curated annotation of ICEs and IMEs. BMC Res. Notes. 2022; 15:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A.et al.. The pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcillán-Barcia M.P., Redondo-Salvo S., Vielva L., de laCruz F.. de la Cruz F. MOBscan: automated annotation of MOB relaxases. Horizontal Gene Transfer: Methods and Protocols, Methods in Molecular Biology. 2020; NY: Springer US; 295–308. [DOI] [PubMed] [Google Scholar]
- 28. Lee C.A., Thomas J., Grossman A.D.. The bacillus subtilis conjugative transposon ICEBs1 mobilizes plasmids lacking dedicated mobilization functions. J. Bacteriol. 2012; 194:3165–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramachandran G., Miguel-Arribas A., Abia D., Singh P.K., Crespo I., Gago-Córdoba C., Hao J.A., Luque-Ortega J.R., Alfonso C., Wu L.J.et al.. Discovery of a new family of relaxases in firmicutes bacteria. PLoS Genet. 2017; 13:e1006586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marchler-Bauer A., Bryant S.H.. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004; 32:W327–W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C.J., Lu S., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R.et al.. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017; 45:D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinforma. Oxf. Engl. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Y., Niu B., Gao Y., Fu L., Li W.. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinforma. Oxf. Engl. 2010; 26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinforma. Oxf. Engl. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 35. Katoh K., Misawa K., Kuma K., Miyata T.. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gouy M., Guindon S., Gascuel O.. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010; 27:221–224. [DOI] [PubMed] [Google Scholar]
- 37. Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997; 14:685–695. [DOI] [PubMed] [Google Scholar]
- 38. Garcillán-Barcia M.P., Francia M.V., de La Cruz F.. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 2009; 33:657–687. [DOI] [PubMed] [Google Scholar]
- 39. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J.et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Criscuolo A., Gribaldo S.. BMGE (Block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010; 10:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Köster J., Rahmann S.. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 43. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blankenberg D., Von Kuster G., Bouvier E., Baker D., Afgan E., Stoler N.the Galaxy Team the Galaxy Team Taylor J., Nekrutenko A. Dissemination of scientific software with galaxy toolshed. Genome Biol. 2014; 15:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Francia M.V., Varsaki A., Garcillán-Barcia M.P., Latorre A., Drainas C., Cruz F.dela. A classification scheme for mobilization regions of bacterial plasmids. FEMS Microbiol. Rev. 2004; 28:79–100. [DOI] [PubMed] [Google Scholar]
- 46. Garcillán-Barcia M.P., Alvarado A., de la Cruz F.. Identification of bacterial plasmids based on mobility and plasmid population biology. FEMS Microbiol. Rev. 2011; 35:936–956. [DOI] [PubMed] [Google Scholar]
- 47. Smillie C., Garcillán-Barcia M.P., Francia M.V., Rocha E.P.C., Cruz F.dela. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 2010; 74:434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brochet M., Da Cunha V., Couvé E., Rusniok C., Trieu-Cuot P., Glaser P.. Atypical association of DDE transposition with conjugation specifies a new family of mobile elements. Mol. Microbiol. 2009; 71:948–959. [DOI] [PubMed] [Google Scholar]
- 49. Cookson A.L., Noel S., Hussein H., Perry R., Sang C., Moon C.D., Leahy S.C., Altermann E., Kelly W.J., Attwood G.T.. Transposition of Tn916 in the four replicons of the butyrivibrio proteoclasticus B316(T) genome. FEMS Microbiol. Lett. 2011; 316:144–151. [DOI] [PubMed] [Google Scholar]
- 50. Cury J., Touchon M., Rocha E.P.C.. Integrative and conjugative elements and their hosts: composition, distribution and organization. Nucleic Acids Res. 2017; 45:8943–8956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Puymège A., Bertin S., Guédon G., Payot S.. Analysis of streptococcus agalactiae pan-genome for prevalence, diversity and functionality of integrative and conjugative or mobilizable elements integrated in the tRNALys CTT gene. Mol. Genet. Genomics. 2015; 290:1727–1740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ICEscreen software is freely and publicly available on the gitlab repository of the MathNum division of INRAE at https://forgemia.inra.fr/ices_imes_analysis/icescreen. It is available to install from source, Conda (https://anaconda.org/search?q=icescreen), and the Galaxy tool shed (https://toolshed.g2.bx.psu.edu/). The documentation of the tool is available at https://icescreen.migale.inrae.fr. ICEscreen is a free software under the Affero GPLV3 licence.