

Abstract
Epstein–Barr virus (EBV) is a widely prevalent human herpes virus infecting over 95% of all adults and is associated with a variety of B-cell cancers and induction of multiple sclerosis. EBV accomplishes this in part by expression of coding and noncoding RNAs and alteration of the host cell transcriptome. To better understand the structures which are forming in the viral and host transcriptomes of infected cells, the RNA structure probing technique Structure-seq2 was applied to the BJAB-B1 cell line (an EBV infected B-cell lymphoma). This resulted in reactivity profiles and secondary structural analyses for over 10000 human mRNAs and lncRNAs, along with 19 lytic and latent EBV transcripts. We report in-depth structural analyses for the human MYC mRNA and the human lncRNA CYTOR. Additionally, we provide a new model for the EBV noncoding RNA EBER2 and provide the first reported model for the EBV tandem terminal repeat RNA. In-depth thermodynamic and structural analyses were carried out with the motif discovery tool ScanFold and RNAfold prediction tool; subsequent covariation analyses were performed on resulting models finding various levels of support. ScanFold results for all analyzed transcripts are made available for viewing and download on the user-friendly RNAStructuromeDB.
INTRODUCTION
Epstein–Barr virus (EBV) is a widely prevalent human herpes virus infecting over 95% of all adults. Most people become infected with the virus at young ages and show minimal to mild cold-like symptoms; for those that become infected later in life, infection presents as mononucleosis where the symptoms can be more severe and long lasting. After EBV’s initial infection and lytic cycle, it goes into a latency phase and primarily resides in a subset of B-cell lymphocytes. There are several latency profiles in which EBV can exist (0, I, II or III) and each one is distinguished by a unique transcription profile of latent genes (1).
It is not entirely clear how latent expression of EBV transcripts and proteins affects regular B-cell function and canonical regulation. However, in certain forms of B-cell cancers (e.g. Burkitt's lymphoma, Hodgkin's lymphoma, and nasopharyngeal carcinoma), EBV is found to be lytically active and highly associated with cancerous cells, indicating an interplay between the virus and the dysregulation of cellular biology that stimulates cancerous growth. Furthermore, EBV infection is sufficient to immortalize B-cells in culture and EBV has been recently linked to the induction of multiple sclerosis (2). There is a clear oncogenic and disease relevant phenotype associated with EBV and a deeper understanding of the interplay between host and virus is needed.
EBV can influence and alter the regulation of B-cells at many levels: transcription, post-transcription, translation, or post-translation (3,4). There are examples of EBV ncRNAs which exert control over the cell by altering transcription pathways, modulating host RNA levels, depletion of host trans-regulatory machinery, and more (5–7). The most highly expressed RNAs by latent EBV are the non-coding Epstein–Barr encoded RNAs (EBER1 and EBER2), which promote a pro-tumorigenic environment and can bind to several host proteins (e.g. PAX5, several hnRNPs and La antigen) to promote infection (5,8,9). Notably, both EBERs are structured ncRNAs, with EBER1 being significantly more structured than EBER2 (8). RNA structure is important to EBER function (5,8,9) however, secondary structure models for EBERs vary (particularly the less stable EBER2) (6,10,11). Structure modeling of EBERs has relied on chemical and enzymatic probing data from cell lysates and limited comparative analysis (6,10,11). To date, no in cellulo probing of EBER structure has been used to inform models. Additional noncoding transcripts are also expressed in latency. The stable intronic sequence (sis)RNAs 1 and 2 were described as arising from the EBNA-LP locus of EBV (12). While sisRNA function is an ongoing area of study, sisRNA-1 is highly conserved in sequence/structure and is highly expressed in latency III, suggesting function. A number of host regulatory proteins can bind sisRNA-2 (13) and this RNA appears to be necessary for cell transformation after infection (14). Aside from these RNAs, there are other latent transcripts expressed which have potential to modulate canonical cellular activity (e.g. LMPs, EBNAs and BART RNAs).
To discover additional functional RNAs or structural motifs present in EBV and other herpes viruses, our lab previously applied the motif discovery tool ScanFold (15,16) to all human herpes virus genomes—cataloging a myriad of potentially functional and significantly stable regions within each virus (17). Here, ScanFold was able to home in on a functional motif in the 3′UTR of BFRF1 which exhibited activity in a dual luciferase assay, indicating roles in post-transcriptional control. ScanFold excels at identifying regions of significant thermodynamic stability (using a thermodynamic z-score), which indicates a non-random sequence order (i.e. an evolved sequence) with high propensity for structure/function. However, while ScanFold can identify these regions, it only predicts local RNA structure (typically base pairs within 120 nucleotides or less) and the resulting structural models are purely computational. Therefore, these results can be enhanced by the incorporation of additional, experimentally derived structural data.
Unlike proteins, RNA structure is less amenable to atomic (or near-atomic) structure determination methods (e.g. NMR, X-ray crystallography and cryo-EM). While RNA molecules can include regions of rigid, static structures, much of an RNA is loosely structured and highly dynamic making it unsuitable (in many cases) for analyses using the high-resolution tools of structural biology (18). The folding of RNA is, however, hierarchical: the formation of secondary structure (i.e. base pairing) occurs first and accounts for a majority of the overall thermodynamic energy of structure formation (19). The ensuing formation of tertiary structure is largely constrained by the presence of secondary structure and, because of this, knowledge of the 2D structural landscape is highly informative for RNAs. This landscape can be predicted with limited accuracy (∼70% correct for RNAs < ∼700 nt (20)) using experimentally derived thermodynamic parameters. A number of approaches have been developed to improve predicted 2D models which typically limit the scope of predictions and incorporate complementary data. For example, ScanFold limits the size of 2D structures to a small scanning window size and informs models based on recurring base pairs with high propensity for ordered stability (15,16). Other methods incorporate phylogenetic comparative data into predictions to identify base pairing with evolutionary support: e.g. RNAalifold (21) and Multilign (22,23). All 2D prediction methods can be greatly improved by the incorporation of experimental structure probing data, where RNAs are exposed to structure-sensitive reagents to collect information on their 2D conformations (18). The collection of such data has been vastly improved through the use of high-throughput sequencing (24): e.g. as implemented by the RNA structure probing technique Structure-seq2 (25).
Structure-seq2 (25) utilizes dimethyl sulfate (DMS), a small, cell-permeable molecule, to modify unpaired or loosely structured adenosine (A) and cytidine (C) bases. DMS methylates the N1 and N3 position of A and C bases respectively, which present on the Watson–Crick base pairing face of each. Therefore, highly structured, base paired nucleotides are not accessible for reaction with DMS, making DMS a direct probe of base pairing. Here, the locations of DMS modifications are detected via accumulation of reverse transcriptase (RT) stops, where modified bases induce the RT enzyme to terminate transcription one nucleotide downstream from the modified base. When coupled to high throughput sequencing, whole transcriptomes can be probed for secondary structure in a single experiment. Reactivity profiles, normalized per transcript and calculated from the raw RT-stop counts at each nucleotide, represent a probability of individual nucleotides being paired or unpaired and are most informative when coupled with an RNA folding algorithm (24): e.g. as a soft constraint, where the reactivity is converted to a pseudo-energy which alters the overall predicted minimum free energy (MFE) of structure proportionate to the reactivity—with highly reactive nucleotides being less likely to be base paired due to their energy penalty contribution to the predicted free energy.
To better understand structure-function relationships of EBV transcripts and human transcripts present during latent viral infection, we have applied the Structure-seq2 method to the BJAB-B1 cell line—a B-cell lymphoma artificially infected with EBV that expresses a latency III gene expression program (the most transcriptionally active type of EBV latent infection). The resulting sequencing data were analyzed with the bioinformatics package StructureFold2 (26) to generate reactivity profiles for >10000 human and viral transcripts. For mRNAs and lncRNAs, reactivity profiles were coupled with ScanFold as pseudo-energies to yield experimentally informed structural models with a focus on significantly stable (i.e. potentially functional) local motifs. For shorter transcripts, reactivity profiles were used alongside RNAfold to generate global 2D models. High value motifs and models were further assessed using the cm-builder pipeline (27), which utilizes INFERNAL (28) and R-scape (29,30) to identify and align homologous sequences and to detect evidence of significant sequence covariation (correlated evolution between sites paired in our model structures). Additionally, we focus on several high value transcripts to showcase how researchers can use data from this study to generate models of longer-range interactions, identify functional motifs, and build structure-function hypotheses for future studies (e.g. functional assays, small-molecule targeting, stability assays, etc.). Resulting models and processed reactivity data are made available on the RNAStructuromeDB (31) to facilitate their usage by researchers interested in studying RNAs significant to EBV infection, B-cell biology, and disease: e.g. cancer and autoimmune diseases associated with EBV-mediated deregulation of B-cells.
MATERIALS AND METHODS
BJABJ-B1 cell culturing
BJAB-B1 cells were grown in suspension at 37°C and 5% CO2 using RPMI media supplemented with 2 mM l-glutamine, 1% penicillin–streptomycin, 10 mM HEPES, 1 mM sodium pyruvate (Life Technologies), and 10% FBS (Atlanta Biologicals). Cells were used in experiments when between 5–30 passages, were passed at v/v ratios of 1:20–1:4 and were regularly tested for mycoplasma contamination.
DMS probing of BJAB-B1 cells
BJAB-B1 cells were grown to a volume of ∼80 ml to ensure enough cells for adequate total RNA isolation yields. Cells were counted, centrifuged at 150 × g for 3 min, then normalized to 5000000 cells/ml in DPBS. Then, following recommended safety protocols (32), cells were treated with DMS (2% v/v) for 2 min at room temperature prior to neutralization with DTT (powder at a 5 times molar ratio to DMS). Neutralized cells were centrifuged at 4°C and 200 × g for 2 min, supernatant was removed, and the cell pellet was dissolved in TRIzol (Invitrogen). DMS probing of cells (DMS+) was completed in triplicate on separate days. Additionally, three DMS negative control samples (DMS−) were processed identically but without the addition of DMS.
RNA isolation and quality control
Total RNA for all DMS+/− samples was extracted via standard TRIzol protocol and two rounds of standard RNA ethanol precipitation. Quality of total RNA was assessed using both a ThermoFisher Scientific NanoDrop One and an Agilent Bioanalyzer 2100. All samples had a RNA Integrity Number (RIN) of 8.5–9.9, where a 10 would indicate the highest quality total RNA with no degradation and a 1 would indicate completely degraded RNA.
cDNA library generation and quality control
The cDNA library preparation protocol outlined by Structure-seq2 (25), was followed closely. We will briefly detail each step as it applied to the samples in this paper and any minor deviations which were taken.
Each DMS+ and DMS− sample had ∼400 ug of total RNA used for polyA selection using the Poly(A)Purist MAG Kit (ThermoFisher) and reactions were cleaned up using RNA ethanol precipitation per the manufacturer protocol. Following this, each sample underwent DNase I treatment (NEB) and reactions were purified with the RNA Clean and Concentrator kit (Zymo). PolyA selected RNA quality was assessed via the Agilent Bioanalyzer 2100. All samples showed minimal rRNA contamination (3.4–8.5%) and electropherogram traces characteristic of mRNA.
Reverse transcriptase (RT) reactions were performed using SuperScript III (ThermoFisher), biotinylated dCTPs (Trilink), random hexamer primers fused with a partial Illumina sequencing adapter (Supplemental Table S1) following the reaction conditions detailed in the Structure-seq2 method. After RT was complete, cDNA product was purified using hydrophilic magnetic streptavidin beads (NEB) and the RNA Clean and Concentrator kit (Zymo), as described in the ‘Streptavidin Version’ of Structure-seq2.
Ligation of partial sequencing adapter to the 3′ end of cDNA products was accomplished using a hairpin adapter (Supplemental Table S1), T4 DNA Ligase (NEB), and reaction conditions detailed in the Structure-seq2 method. Ligated product was purified from the reaction via the same streptavidin bead process used after RT reactions.
Ligated cDNA samples were then PCR amplified using full TruSeq adapter primers (Supplemental Table S1), Q5 polymerase (NEB), and reaction conditions previously detailed (25). Subsequently, PCR amplified cDNA libraries were purified, and size-selected between ∼200–800 nucleotides via PAGE using a 10% acrylamide, 8.3 M urea gel.
Resulting cDNA libraries were analyzed on the Agilent Bioanalyzer 2100 to confirm size selection. Additionally, libraries were analyzed via qPCR using primers targeting the ends of the sequencing adapters (Supplemental Table S1), and serial dilutions showed a concentration dependent shift in ΔΔCt values. The Bioanalyzer and qPCR results confirm the size of the cDNA libraries and the presence of proper adapter sequences.
Sequencing of cDNA libraries
The three DMS+ and three DMS− cDNA libraries were single-end sequenced on a single lane of a Illumina HiSeq3000 using standard indexing and sequencing primers with 150 cycles. This was completed at the Iowa State University DNA Facility. This sequencing generated FASTQ files used in downstream analyses.
Acquisition of FASTA sequences for sequence read mapping
The FASTA file used for mapping FASTQ file reference reads to human mRNA transcripts was downloaded from NCBI on 4 August 2020 and was filtered to include the longest isoform transcript of each protein coding gene. The FASTA file used for mapping FASTQ file reference reads to human lncRNAs was obtained from Ensembl database on 18 March 2021. The EBV sequence NC_009334.1 was downloaded from NCBI on 30 October 2020 and used for mapping and analysis of EBV transcripts. The 18s ribosome sequence NT_167214.1 was downloaded from NCBI on 9 April 2021.
StructureFold2 processing of sequencing data
FASTQ files were processed via the StructureFold2 (SF2) bioinformatic pipeline using default parameters as described previously (26).
Briefly, FASTQ files were initially analyzed with the program Fastqc (version 0.11.5). Identified adapter sequences and overrepresented sequences were trimmed using the SF2 script fastq_trimmer.py which utilizes Cutadapt (version 1.13) to trim and filter reads which are <30 Phred. Utilizing Bowtie2 (version 2.3.4.1) the mRNA, lncRNA, ribosome and EBV FASTA files had corresponding mapping indexes created and the SF2 script fastq_mapper.py was used to map trimmed FASTQ files to each reference index. Resulting SAM files were then filtered using the SF2 script sam_filter.py with default settings. Next, filtered SAM files were converted to reverse transcriptase stop count (RTSC) files via the SF2 script sam_to_rtsc.py. The three DMS+ and three DMS− RTSC files were combined into two respective RTSC files via rtsc_combine.py. Coverage and overlap files (useful for reactivity generation) were generated for the combined DMS+ RTSC file using the rtsc_coverage.py script utilizing the default parameters and the ‘-ol’ flag. Finally, the combined DMS+ and DMS− RTSC files were used to generate a REACT file via the rtsc_to_react.py script with default parameters and the ‘-restrict’ flag which limited reactivity data generation to transcripts present on the previously generated overlap list.
Statistical analysis of reactivity datasets
SAM files generated for each DMS+ and DMS− sample via the fastq_mapper.py script were analyzed using the program Samtools (version 1.10). SAM files for each mapping condition (mRNAs, lncRNAs, EBV transcripts, and 18S rRNA) were converted to BAM files then sorted, indexed, and merged into a DMS+ BAM file and a DMS− BAM file. The Samtools stats module was then executed on all corresponding DMS+ and DMS− BAM files.
Additionally, the scripts rtsc_coverage.py and react_statistics.py from the SF2 package were used to obtain details of the reactivity datasets. Here, a coverage and average transcript reactivity were calculated. Coverage in this case is defined as the number of observed RT-stops per transcripts dived by the total number of potential reactive nucleotides (A and C bases). Using this definition, a coverage of 1 indicates that there are an equal number of RT-stops as there are total A and C bases.
18S rRNA optimization of pseudo-energy parameters
In this study, reactivities are transformed to pseudo-energies following the Deigan method (33) which is written as Equation 1:
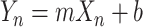 |
(1) |
Here, Yn is the pseudo-energy of a given nucleotide at position n, Xn is the normalized reactivity at nucleotide position n, and m and b are constant fitting parameters.
To determine the appropriate value of m and b to use with ScanFold, fitting optimizations using a crystal structure reference model of the human 18S rRNA were performed. A DMS reactivity file for the 18S rRNA was obtained by analysis of our FASTQ files with the 18S rRNA sequence via SF2 (FASTA acquisition and SF2 processing is described above). ScanFold was run on the 18S rRNA sequence using a 120-nucleotide sized window, a 1-nucleotide step size, and 100 randomizations. Additionally, it used the 18S rRNA react file as a pseudo-energy constraint varying the m and b parameters from 0.2 to 3.0 and −0.2 to −3.0, respectively, at 0.2 intervals for each parameter. This resulted in 225 unique parameter conditions. The resulting No Filter, −1 z-score, −2 z-score, and −1 z-score global refold files for each ScanFold model for each parameter condition (in CT file format) were compared to the reference structure model of the 18S rRNA using the script ct_sensitivity_ppv_120.py (see Data Availability for script access). In this process, the base pairs from the experimental and reference CT files are cross referenced and any identical (i, j) pairing is counted as consistent and any unique base pairs are counted as conflicting. Additionally, this process compares the local base pairs (120 nucleotide base pair span or less) as this is what ScanFold is optimized to model and the ScanFold results analyzed below focus on these local −1 and −2 z-score motifs. Using this definition, the sensitivity and positive predictive value (PPV) formulas can be seen in equation 2 and 3, respectively:
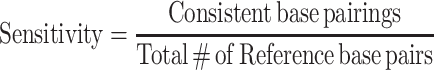 |
(2) |
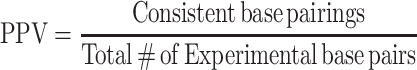 |
(3) |
The output for this analysis was used to identify an appropriate slope (m) and intercept (b) parameter for pseudo-energy incorporation with ScanFold (Supplemental Table S2).
Incorporation of reactivity values with ScanFold
A description of the ScanFold process can be found in a methods paper (15) and in several applications of ScanFold to human and viral targets (16,17,34,35). Briefly, ScanFold can be broken down into two major modules: ScanFold-Scan and ScanFold-Fold. During ScanFold-Scan, a small 120 nucleotide analysis window scans at 1 nucleotide intervals from the 5′ to the 3′ end of the transcript. In each analysis window, several thermodynamic metrics are calculated including a minimum free energy (MFE) (i.e. Gibbs free energy or ΔG), ΔG z-score, ensemble diversity (ED), and a metric indicating the fraction of random sequences which were more stable than the original sequence during z-score determination (termed the P-value). ScanFold-Fold then takes the data from all overlapping analysis windows and builds a consensus model of significantly stable, local 2D structure by assessing which modelled base pairs from all overlapping windows contribute the most to significant thermodynamic stability (i.e. which base pairs yield the lowest ΔG z-scores). The result of this process is windowed scanning data along the entirety of a transcript and a local 2D structure base pair consensus model, with an emphasis on significantly ordered/stable structures. While ScanFold does not predict the global structure of these transcripts, it was previously shown that ScanFold can accurately identify and model regions of well-structured and functional RNAs, meaning the ScanFold identified structures are of high value for potential functional assays and therapeutic targeting (35,36).
Each human mRNA, human lncRNA and EBV transcript which had SF2 generated reactivity profiles had the associated reactivity profile and transcript sequence analyzed with ScanFold. Reactivity files which were output from SF2 were parsed and reformatted for input with ScanFold using the script fasta_react_parser.py (see Data Availability for script access). This script also parsed the input FASTA files used for mapping and pulled out individual sequences to individual FASTA files. The following ScanFold command was used for analyses:
$ python /path/to/ScanFold.py <Input.fasta> –out_name <Input> –react <Input.react> –name <Input> -m 0.6 -b -1.0 –global_refold
Here, the ‘–out_name’ flag denotes a name for the output directory, ‘–react’ points to the react file, ‘–name’ specifies the name to use in file headers (e.g. CTs and WIGs), ‘-m’ and ‘-b’ denote the slope and intercept values used for pseudo-energy calculations, and ‘–global_refold’ enables global refolding of transcripts with the DMS informed −1 and −2 z-score DBN files as constraints.
DMS informed ScanFold models compared to in silico ScanFold models
DMS informed ScanFold models for over 6000 human mRNA sequences were compared to their corresponding in silico (i.e. purely computational) ScanFold models to determine the effect of incorporating probing constraints. This was accomplished by calculating the PPV and sensitivity for each model via comparison of all predicted base pairs, −1 z-score base pairs, and −2 z-score base pairs present in CT files produced by ScanFold (Supplemental File 1). This process utilized the script ct_sensitivity_ppv.py and has been previously described (34).
In addition to comparing the resulting structural models produced by ScanFold, the effect on the per nucleotide z-score values (found in the average per nucleotide z-score WIG file produced by ScanFold) were assessed between DMS informed and in silico ScanFold models. Here, a Pearson correlation assessment was conducted on a per transcript basis comparing the DMS informed and in silico per nucleotide z-score values (Supplemental File 1).
Incorporation of reactivity values with RNAfold
RNAfold (37,38) was used in several capacities throughout this study. RNAfold was used as the folding algorithm in ScanFold which calculates thermodynamic metrics (most importantly the MFE) of each WT and randomized ScanFold analysis window. Additionally, RNAfold was used to model several transcripts with reactivity profiles as pseudo-energy constraints. When used in this capacity, the Deigan model of reactivity incorporation was used along with the default slope and intercept values of RNAfold. In some cases, the probability of structure (determined from the partition function and indicates the likelihood of a given nucleotide being paired or unpaired) were extracted from RNAfold analyses of individual targets.
Covariation analysis of structured motifs
For certain high-value structure motifs and transcripts which underwent more targeted analyses, the cm-builder pipeline (27) was used to assess covariation. The cm-builder pipeline automates the use of INFERNAL (28) and R-scape (29,30) to make sequence and structure homology alignments and then assess primary and secondary structure to identify statistically significant events of structure preserving covariation. Initial alignment databases for analyzed sequences were generated using BLAST and either the Refseq database or the NT collections database.
To run cm-builder, the following command line was used:
$ perl /path/to/cm-builder -s <input.fasta> -m <input.dbn> -d <alignment_database.fasta> -c 24 -k
Here, ‘-s’ denotes the fasta file, ‘-m’ is the motif DBN being analyzed, ‘-d’ is the alignment database fasta, ‘-c’ denotes how many cpu nodes to utilize, and ‘-k’ specifies to keep the final Stockholm alignment. During the analyses INFERNAL (version 1.1.2) was used to make the covariance model (CM) files. CM files were then passed to R-scape (version 2.0.0) and Stockholm files, power files, and summary PDFs were generated. All results from cm-builder analyses can be found in Supplemental File 2.
Visualizations of modeled RNA
For visualization of data and generation of figure elements, the programs IGV (39) and VARNA (40) were used, and individual elements were then combined and finalized in Adobe Illustrator. For generation of figure elements, select ScanFold output files (see the ScanFold methods paper for details of file outputs (15)) were uploaded onto the IGV webserver and an SVG image was extracted. The DBN tracks of select motifs were loaded onto VARNA and SVG files were downloaded accordingly.
Transcriptome metrics analyses
The resulting ScanFold data for all human mRNAs, lncRNAs and EBV transcripts were globally analyzed, and several metrics were calculated per transcript including the average of all MFE windows (ΔG) (resulting from all ScanFold-Scan analysis windows), the average of ΔG z-score windows, the number and percent of windows below −1 and −2, the transcript length, and the number of ScanFold-Fold extracted motifs. This was accomplished using the python script transcriptome_metrics.py (see Data Availability). Additional analyses of the data were completed in excel and data are accessible in Supplemental File 3.
Regional ΔG z-score analyses
The average per-nucleotide z-score is a metric calculated for each nucleotide in a transcript based on all the ScanFold-Scan analysis windows the nucleotide appears in. This metric is reported per transcript in the ScanFold ‘Zavg_metrics’ WIG file (which is viewable on IGV) and present in each available download from RNAStructuromeDB (more details in Data Availability). For each analyzed human mRNA this data was parsed to the 5′UTR, the coding sequence (CDS), and the 3′UTR as found in the Gencode database. Next, the average per-nucleotide z-score data was parsed to each region of each transcript and the average value for the region was calculated using the python script regional_zavg.py (see Data Availability). Resulting data are discussed in the Results and viewable in Supplemental File 3.
Differential gene expression analyses
Nine gene expression datasets were obtained from The Human Protein Atlas on 1 February 2022 (41,42). These datasets contain genes that exhibit tissue specific expression, genes that exhibit tissue enriched expression in at least one analyzed tissue, housekeeping genes (HKGs), and genes of transcription factors (TFs). There are 10992 genes that exhibit tissue specific expression, 8839 HKGs and 1490 TF genes. Within the list of genes exhibiting tissue specific expression, there are subsets including 3107 tissue enriched genes (at least four-fold higher mRNA level in a particular tissue compared to any other tissue), 1691 group enriched genes (at least 4-fold higher average mRNA level in a group of 2–5 tissues compared to any other tissue), and enhanced genes (at least four-fold higher mRNA level in a particular tissue compared to the average level in all other tissues). Additionally, we found subsets of specificity-based genes using their tissue distribution. These subsets contain detection in a single tissue, some tissue (more than one but less than one third of tissues), many tissues (at least one third of tissues), and all tissues (HKGs).
The data generated from the transcriptome metrics analyses and the regional ΔG z-score analyses were cross referenced to each of the differential gene expression datasets to analyze trends of certain metrics across the differential datasets. These analyses were accomplished via the python scripts differential_expression_metrics.py and zavg_regional_diff_exp.py (see Data Availability). Output from these analyses can be found in Supplemental File 4.
Receiver operator characteristic analyses
The proposed alternative secondary structure for EBER2 (see Results) along with the EBER2 reference model (10) were compared to the DMS reactivity profile generated in the study using a receiver operator characteristic (ROC) analysis. The ROC analysis strategy was the same as we have previously reported (34). Briefly, the CT data files for each EBER2 secondary structure was cross referenced to the DMS reactivity profile generated in this study. The reactivity data had a threshold sequentially set from lowest to highest reactivity values at 1% intervals (i.e. 1, 2, 3… 100%) and any associated nucleotide below the threshold was defined as being paired. Each reactivity threshold was then referenced to the EBER2 secondary structure CT files and a true positive rate (TPR), and a false positive rate (FPR) were calculated. The TPR and FPR can be plotted for each reactivity threshold, generating an ROC curve. If a secondary structure model fits or agrees with the reactivity profile, a larger area under the curve (AUC) will be observed and a model that is more ‘random’ compared to the data will have a lower AUC and an ROC curve that roughly follows a 45-degree line as the TPR and FPR are expected to increase at the same rate for a random model.
Thermodynamic analyses of miRNA binding
To assess miRNA binding potential to CYTOR, the ΔΔG method presented by Kertesz et al. (43) was used. Here, the ΔΔG is a measure of the energy gained by miRNA binding minus the energy needed to break the existing structures and is represented in Equation 4:
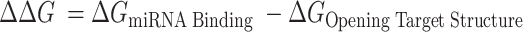 |
(4) |
For the binding of miR-4767 to CYTOR, the region of CYTOR from nucleotides 31–119 were assessed as this was the major structure present around the binding site. For the binding of miR-138, the region of CYTOR assessed was from nucleotides 129–588. This second region is larger as base pairs which were disrupted from miRNA binding were participating in long range interactions. The thermodynamics of RNA binding were assessed via RNAduplex (38) and the energy needed to break base pairs and stems involved in miRNA binding were assessed in RNAFold (37,38).
RESULTS
Transcriptome-wide analyses
Sequencing and read mapping overview
After execution of the RNA structure probing method Structure-seq2 on BJAB-B1 cells, a total of ∼218 million reads for DMS+ cDNA libraries and ∼132 million reads for DMS− cDNA libraries were acquired. The resulting reads were mapped via StructureFold2 to several reference sequences and genomes which included hg38 mRNAs and lncRNAs, the EBV genome (NC_0093341.1) and associated transcripts, and the human 18S ribosomal RNA (NT_167214.1) sequence. DMS reactivity profiles were generated for 6588 human mRNAs, 3427 human lncRNAs, and 19 individual EBV transcripts (along with a reactivity profile for the entire EBV genome sequence). Additionally, reactivity data were acquired for the human 18S rRNA sequence as a control for the DMS probing and data processing steps.
Across the 10015 human transcripts and 19 EBV transcripts that had detectible DMS reactivity, the read coverage, read depth, RTSC coverage and reactivity averages were calculated for each class of transcripts (mRNA, lncRNA, and EBV transcripts) (Supplemental File 5). The EBV transcripts had a higher average read coverage and depth per transcript (96.8 and 14539 reads, respectively) when compared to the mRNA and lncRNAs. These trends can be recapitulated using a different metric, RTSC coverage, that assesses the coverage of structure probing information per transcript. RTSC coverage differs from read coverage, as it indicates the number of RT stop events per transcript, divided by the total number of potentially reactive bases (see Materials and Methods for more details). The trends in RTSC coverage can be seen in Table 1 and follow the same pattern as read coverage, where EBV transcripts have the highest RTSC coverage, followed by mRNAs, then lncRNAs. EBV transcripts have the lowest maximal RTSC coverage and lncRNAs have the highest RTSC coverage, followed by mRNAs (Table 1). The average reactivity for each transcript of each RNA class was analyzed (Supplemental File 5) and the EBV transcripts had the highest average transcript reactivity, followed by mRNAs, then lncRNAs (Table 1).
Table 1.
Reactivity and sequencing statistics for multiple classes of transcripts
| Transcript class | RTSC coverage average (Min/Max) | Reactivity average (Min/Max) | Read coverage average (Min/Max) | Read depth average (Min/Max) |
|---|---|---|---|---|
| Human mRNAs | 12.16 (1/1362) | 0.203 (0.119/0.359) | 81.2 (0.45/100) | 512.43 (0.01/165048) |
| Human lncRNAs | 7.51 (1/4411) | 0.171 (0.112/0.34) | 45.82 (1.97/100) | 286.79 (0.02/201838) |
| EBV Transcripts | 106.07 (0.02/837) | 0.251 (0.174/0.29) | 96.82 (73.95/100) | 14539.97 (5.14/147319) |
ScanFold transcriptome metrics
The acquired DMS reactivity data were incorporated as pseudo-energies into ScanFold analyses of large transcripts. Use of these soft constraints informs and restricts the folding landscape during secondary structural predictions, leading to experimentally consistent structure models that may be biologically and/or functionally relevant. The DMS informed ScanFold results for human transcripts predict 3.225 million base pairs to be significantly stable (z-scores < −1), which is reduced to 0.879 million base pairs when limited to nucleotides with z-scores <−2. The percent of nucleotides per transcript (both mRNA and lncRNAs) predicted to contain unusually stable structure ranged from 0 to ∼62% with an average of ∼20% for <−1 z-score nucleotides and 0 to ∼52% with an average of ∼6% for <−2 z-score nucleotides (Table 2, Supplemental File 6).
Table 2.
Human mRNA and lncRNA base pair (BP) partitioned z-score (ZS) statistics
| Avg. # of −1 ZS BPs per transcript | Avg. # of −2 ZS BPs per transcript | Average transcript length | Average % of transcript length with −1 ZS BPs | Average % of transcript length with −2 ZS BPs | |
|---|---|---|---|---|---|
| Total | 322.03 | 87.77 | 3153.44 | 21 | 5.7 |
| mRNAs | 366.92 | 96.56 | 3704.7 | 19 | 4.9 |
| lncRNAs | 235.74 | 70.87 | 2093.72 | 23 | 7.3 |
The ScanFold generated data present in the analyzed human mRNAs and lncRNAs had the average windowed ΔG z-score, windowed MFE, and number of significantly stable motifs for each transcript calculated. The mRNA and lncRNA averaged data were compared as seen in Figure 1. The lncRNAs in this dataset had a lower median ΔG z-score (−0.70) compared to mRNAs (−0.51) and lncRNAs also contained the transcripts with the overall lowest average ΔG z-score (Figure 1A). Additionally, the median MFE of mRNAs (−78.98 kcal/mol) was slightly lower than the lncRNAs (−78.19 kcal/mol) and the mRNAs had the lowest overall average MFE transcripts (Figure 1B). The lncRNAs contained the transcripts with highest number of significantly stable structures (Figure 1C), but both the mRNA and lncRNA class of RNAs contained transcripts which were enriched for significantly stable structures (i.e. ScanFold-Fold extracted structures). Transcriptome metrics results for all mRNA and lncRNA transcripts can be found in Supplemental File 3.
Figure 1.

Transcriptome wide metric analyses of human mRNAs and lncRNAs which had DMS reactivity profiles and were analyzed by ScanFold. (A) Violin plots showing transcript average (from all ScanFold-Scan analysis windows of a given transcript) ΔG z-score for mRNAs and lncRNAs. (B) Violin plots showing transcript average (from all ScanFold-Scan analysis windows of a given transcript) MFE for mRNAs and lncRNAs. (C) Violin plots of the distribution of mRNA and lncRNA number of significantly stable motifs (i.e. −2 z-score structures).
Differential gene expression
Within an mRNA, the 3′UTR is known to contain regulatory elements and act as a hub of post-transcriptional control (44). The average length of the 3′UTR in organisms is associated with increasing organismal complexity due to the increased ability for regulatory control of the associated transcript (45). With this in mind, we hypothesized that mRNAs of genes, which undergo more selective, or tissue specific expression likely experience increased regulatory control to modulate their expression. RNA regions of significantly low ΔG z-score have thermodynamic stability that is far lower than randomized sequences of identical nucleotide composition. Significantly low z-score sequences are thus presumed to have a non-random sequence order that has been directed and preserved by evolution for a function (46). Therefore, it would be expected that more regulatory elements (i.e. lower ΔG z-scores and higher number of significantly stable motifs) would occur in mRNA that exhibit restricted expression.
To examine this, nine gene lists from the Protein Atlas, which contain the name of genes expressed in all cell types (i.e. housekeeping genes (HKGs)), genes expressed in a variable number of tissues, down to genes expressed in just single tissues, were obtained. These gene lists were cross referenced to the human mRNA transcripts analyzed by ScanFold in this study. For each transcript in each identified group, the average MFE, z-score and number of significantly stable motifs were calculated and parsed to each gene group (see Materials and Methods) and results can be seen in Table 3. Consistent with our hypothesis, the Group Enriched Genes, which are defined as having >4-fold higher expression in only 2–5 tissues, had the lowest average MFE, z-score, and the largest average number of motifs per transcript, whereas the Detected in All (HKGs) group had the highest ΔG z-score and lowest average number of motifs per transcript.
Table 3.
Analysis of ScanFold metrics and trends when cross referenced to lists of differentially expressed genes
| Full length mRNAs transcripts | Regional per-NT z-score for mRNAs | ||||||
|---|---|---|---|---|---|---|---|
| Gene groups (# of transcripts in group) | Avg. MFE | Avg. windowed ZS | Avg. # of Motifs | 5ʹUTR | CDS | 3ʹUTR | Description of group |
| Detected in all (HKGs) (694) | −78.88 | −0.47 | 7.2 | −0.586 | −0.854 | −1.044 | Genes that have detectable levels (nTPM ≥ 1 or transcription frequency ≥ 1) of transcribed mRNA molecules in all tissues. |
| Detected in many (247) | −81.76 | −0.56 | 9.45 | −0.707 | −0.877 | −1.136 | Genes that have detectable levels (nTPM ≥ 1 or transcription frequency ≥ 1) of transcribed mRNA molecules in at least one third but not all tissues. |
| Detected in some (113) | −81.49 | −0.59 | 10.89 | −0.741 | −0.907 | −1.107 | Genes that have detectable levels (nTPM ≥ 1 or transcription frequency ≥ 1) of transcribed mRNA molecules in more than one but less than one third of tissues. |
| Detected in single (25) | −77.69 | −0.57 | 8.48 | −0.613 | −1.003 | −1.491 | Genes that have detectable levels (nTPM ≥ 1 or transcription frequency ≥ 1) of transcribed mRNA molecules in a single tissue. |
| enhanced genes (307) | −81.04 | −0.52 | 7.93 | −0.609 | −0.887 | −1.06 | Genes that display at least four-fold higher mRNA level in a particular tissue compared to any other tissue. |
| group enriched genes (56) | −82.46 | −0.63 | 12.5 | −0.659 | −0.912 | −1.174 | Genes that display at least four-fold higher average mRNA level in a group of 2–5 tissues compared to any other tissue. |
| reg TFs (69) | -81.81 | -0.52 | 11.61 | -0.686 | -0.877 | -1.105 | Genes of transcription factors that are regulatory proteins known to bind to consensus DNA sequences and activate transcription. |
| tissue enriched expression (457) | −81.07 | −0.54 | 8.37 | −0.656 | −0.895 | −1.105 | Genes that display elevated expression in at least one of the analyzed tissues. |
| tissue enriched genes (94) | −80.35 | −0.55 | 7.36 | −0.81 | −0.91 | −1.215 | Genes that display at least four-fold higher mRNA level in a particular tissue compared to the average level in all other tissues. |
To further characterize the ΔG z-score trends present in the mRNAs of these groups, the average per nucleotide z-score in the 5′UTR, CDS and 3′UTR was analyzed for each transcript. Across all groups, the 5′UTRs had the highest average per-nucleotide z-scores, while the CDS averages were consistently between the 5′UTR and 3′UTR averages, and the 3′UTR averages were consistently the lowest, showing a bias for ordered structure in the 3′UTR for all separate gene groups (Table 3). The Tissue Enriched Genes showed the lowest average per-nucleotide z-score in the 5′UTR, while the Detected in Single transcript group had the lowest CDS and 3′UTR average per-nucleotide z-score. The Detected in All (HKGs) group had the highest average per-nucleotide z-score across all mRNA regions.
DMS informed ScanFold versus in silico ScanFold models
To assess the effects of DMS probing constraints on ScanFold model generation, over 6000 human mRNA sequences had their corresponding DMS−informed and in silico (i.e. purely computational) ScanFold models compared using the PPV and sensitivity metrics, where the DMS informed model was considered the reference model and the in silico model was considered the predicted model. Here, PPV indicates the fraction of base pairs from the in silico model that are consistent with the DMS model and the sensitivity measures the fraction of DMS informed base pairs which are consistent with the in silico model (Equations 2 and 3 in Materials and Methods). This allows for assessment of how accurately in silico ScanFold is modelling structure by comparison to DMS informed structures (via PPV) and how well in silico ScanFold performs at finding low z-score structures by comparison to DMS informed structures (sensitivity).
This analysis used all modelled base pairs, base pairs below a −1 z-score threshold, and base pairs below a −2 z-score threshold found in ScanFold outputs and compared how many consistent pairs were present between the DMS informed and in silico models (Figure 2A). The median PPV for the unfiltered base pairs, −1 z-score base pairs, and −2 z-score base pairs were 0.60, 0.66 and 0.76, respectively. The median sensitivity for the unfiltered base pairs, −1 z-score base pairs and −2 z-score base pairs was 0.57, 0.62 and 0.18, respectively (Figure 2A). The close similarities between the PPV and sensitivity between the unfiltered base pairs and the −1 z-score base pairs is due in part to both the DMS informed models and the in silico models having similar number of average base pairs per transcript. However, in the −2 z-score analyses, the DMS informed models had almost 4 times the amount of average base pairs per transcript compared to the in silico models (Supplemental File 1).
Figure 2.

Box plots comparing structures and per-nucleotide z-scores of in silico and DMS informed ScanFold models. (A) Box plots showing the results of PPV and sensitivity analyses on unfiltered base pairs (BPs), –1 z-score BPs and –2 z-score BPs resulting from in silico and DMS informed ScanFold models. (B) Box plot showing the distribution of correlation values which compared the per-nucleotide z-score values of individual transcripts for in silico and DMS informed ScanFold models.
To assess how much the z-score trend was affected by the incorporation of DMS reactivity values, the average per nucleotide z-score values for both the DMS informed and in silico ScanFold models for individual human mRNA transcripts were used in a Pearson correlation assessment. This revealed a median correlation of 0.74 with upper and lower quartiles of 0.51 and 0.97, respectively (Figure 2B). Additionally, the average z-score per transcript between DMS informed and in silico models had a median difference of only −0.046.
Global EBV data analyses
There were 19 EBV transcripts which had enough coverage to generate DMS reactivity profiles (Table 4). Of the 19 transcripts, 18 had their reactivity profiles analyzed with ScanFold; the 81 nt sisRNA-1 transcript had probing coverage, however, it was too short for comparative ScanFold analysis. Interestingly, the transcripts probed in BJAB-B1 cells contain both lytic and latent transcripts (the expression phase for each transcript is indicated in Table 4). Additionally, DMS reactivity profiles were generated for the whole genome in both the forward and reverse orientation. While detailed analyses for two of the EBV transcripts is provided below (EBER2 and the tandem terminal repeat (TR) RNA), here we provide an overview of the reactivity and ScanFold metrics for all EBV transcripts with DMS coverage.
Table 4.
Reactivity overview for EBV transcripts
| EBV-transcripts | Max reactivity | Average reactivity | Std. deviation | Gini | Latent/ Lytic |
|---|---|---|---|---|---|
| BFLF1 | 1.79 | 0.26 | 0.37 | 0.70 | Lytic |
| BFLF2 | 1.46 | 0.26 | 0.36 | 0.69 | Lytic |
| BFRF1 | 1.84 | 0.25 | 0.38 | 0.73 | Lytic |
| BFRF1A | 1.82 | 0.20 | 0.37 | 0.79 | Lytic |
| BFRF2 | 2.03 | 0.25 | 0.37 | 0.72 | Lytic |
| BHRF1 | 1.58 | 0.34 | 0.36 | 0.56 | Lytic |
| BNLF2a | 1.75 | 0.25 | 0.38 | 0.71 | Lytic |
| BNLF2b | 1.31 | 0.28 | 0.36 | 0.66 | Lytic |
| DR-Left | 1.67 | 0.27 | 0.36 | 0.68 | Latent |
| DR-Right | 1.71 | 0.25 | 0.36 | 0.71 | Latent |
| EBER1 | 1.94 | 0.27 | 0.38 | 0.66 | Latent |
| EBER2 | 1.37 | 0.25 | 0.35 | 0.69 | Latent |
| EBNA-LP | 1.56 | 0.29 | 0.37 | 0.65 | Latent |
| FGAM-synthase | 2.20 | 0.22 | 0.37 | 0.76 | Lytic |
| LMP1 | 1.88 | 0.25 | 0.37 | 0.70 | Latent |
| LMP2A | 2.01 | 0.25 | 0.37 | 0.71 | Latent |
| LMP-2B | 1.80 | 0.32 | 0.36 | 0.59 | Latent |
| sisRNA1 | 1.12 | 0.23 | 0.37 | 0.74 | Latent |
| sisRNA2 | 1.83 | 0.25 | 0.37 | 0.71 | Latent |
| TR-Region | 1.46 | 0.28 | 0.36 | 0.66 | Latent |
| Genome-FWD | 3.87 | 0.13 | 0.42 | 0.92 | |
| Genome-REV | 4.54 | 0.13 | 0.47 | 0.93 |
The reactivity coverage for each EBV transcript ranged from 1 (the minimum coverage needed to generate a reactivity profile) to 837, observed for the EBER1 transcript. The high coverage is due to the high abundance of EBER1 in BJAB-B1 cells. The average reactivity for each transcript was calculated using the SF2 package with BHRF1 having the highest average reactivity (0.345) followed closely by LMP-2B (0.321). The transcript with the lowest average reactivity was BFRF1A, a transcript encoding a protein necessary for the processing of DNA concatemers during viral replication and packaging (47) (Table 4).
The same transcriptome metric analysis that was applied above to ScanFold analyzed human mRNAs and lncRNAs was also applied to the EBV transcripts; resulting data are in Table 5. Average windowed MFE values ranged from −106.5 kcal/mol for the TR RNA to −68.02 kcal/mol for LMP1. Two repeat regions within the EBV genome, the direct repeat left (DRL) and direct repeat right (DRR), had the lowest average windowed ΔG z-score in the set at −1.70 and −1.66, respectively. These were followed closely by EBER1 with an average windowed z-score at −1.58. The lytic transcript BHRF1 had the highest average windowed z-score at 0.62 and there were four transcripts from the set whose average windowed z-score were positive (Table 5).
Table 5.
ScanFold metrics for EBV transcripts
| EBV-Transcripts | Avg. window ΔG | Avg. windowed z-score | # of windows | # of ZS windows <= –1 | % of ZS windows <= –1 | # of ZS windows <= –2 | % of ZS windows <= –2 | Sequence length | # of Motifs |
|---|---|---|---|---|---|---|---|---|---|
| BFLF1 | −79.94 | −0.74 | 1459 | 556 | 38.1 | 136 | 9.3 | 1578 | 5 |
| BFLF2 | −76.08 | −0.74 | 838 | 307 | 36.6 | 80 | 9.5 | 957 | 1 |
| BFRF1 | −84.47 | 0.24 | 892 | 124 | 13.9 | 5 | 0.6 | 1011 | 0 |
| BFRF1A | −77.04 | 0.14 | 289 | 7 | 2.4 | 0 | 0.0 | 408 | 0 |
| BFRF2 | −91.10 | −0.22 | 1657 | 376 | 22.7 | 116 | 7.0 | 1776 | 2 |
| BHRF1 | −69.31 | 0.62 | 457 | 38 | 8.3 | 3 | 0.7 | 576 | 0 |
| BNLF2a | −91.53 | −0.22 | 64 | 5 | 7.8 | 0 | 0.0 | 183 | 0 |
| BNLF2b | −88.19 | 0.11 | 178 | 37 | 20.8 | 9 | 5.1 | 297 | 0 |
| DR-Left | −89.17 | −1.70 | 925 | 578 | 62.5 | 399 | 43.1 | 1044 | 10 |
| DR-Right | −90.60 | −1.66 | 926 | 598 | 64.6 | 381 | 41.1 | 1045 | 11 |
| EBER1 | −92.11 | −1.58 | 48 | 48 | 100.0 | 7 | 14.6 | 167 | 0 |
| EBER2 | −89.27 | −0.51 | 54 | 18 | 33.3 | 0 | 0.0 | 173 | 0 |
| EBNA-LP | −96.15 | −0.25 | 76 | 1 | 1.3 | 0 | 0.0 | 195 | 0 |
| FGAM-synthase | −93.31 | −0.30 | 3838 | 1030 | 26.8 | 327 | 8.5 | 3957 | 8 |
| LMP1 | −68.02 | −0.17 | 997 | 303 | 30.4 | 53 | 5.3 | 1116 | 0 |
| LMP2A | −85.45 | −0.48 | 6740 | 2084 | 30.9 | 760 | 11.3 | 6859 | 11 |
| LMP-2B | −75.72 | −0.62 | 1018 | 356 | 35.0 | 125 | 12.3 | 1137 | 4 |
| sisRNA2 | −92.16 | −1.04 | 2672 | 1295 | 48.5 | 592 | 22.2 | 2791 | 11 |
| TR-Region | −106.48 | −0.93 | 1539 | 726 | 47.2 | 351 | 22.8 | 1658 | 7 |
| FWD-Genome | −90.43 | −0.53 | 172 645 | 55 559 | 32.2 | 19 580 | 11.3 | 172 764 | – |
| REV-Genome | −89.57 | −0.50 | 172 645 | 53 525 | 31.0 | 18 742 | 10.9 | 172 764 | – |
In cellulo models
Epstein─Barr virus encoded RNAs (EBERs)
Using the in cellulo DMS reactivity, pseudo-energy constrained MFE models for both EBER1 and 2 were generated using RNAfold. The DMS informed model for EBER1 is highly similar to the previously established model of Glickman et al. (10) (Supplemental Figure S1), hereafter referred to as the EBER1 reference model. High reactivity sites occurred throughout the transcript model with only a few sites having strong reactivity values embedded in more structured areas (e.g. nucleotides position A18 and A115). In contrast, while some consistencies in 2D structure between our EBER2 model and the Glickman et al. (10) EBER2 reference model are observed, there are notable structural rearrangements between the two models (Figure 3). First off, the Basal Stems of both structures are identical, and Stem 1 is highly similar with subtle differences in the size of the terminal hairpin loop. The Stem 1 structure does contain two additional base pairs (A40-U48 and C41-G47) in the hairpin loop region in our model compared to the reference model which has a larger loop in this region and the reference model has two additional base pairs at the base of Stem 1 (G21-C63 and G22-C64) whereas, in our model, these Gs are in a looped-out region and the Cs are involved in base pairing of Stem 2.
Figure 3.

Secondary structure models of EBER2. DMS reactivity data is overlaid on the models as red shaded nucleotides with the DMS reactivity scale ranging from 0.0 (white) to 1.0 (dark red). The nucleotides on each model are numbered at every 20-nucleotide interval. Covarying base pairs, as identified by R-scape, are highlighted with blue boxes. (A) Our proposed EBER2 model generated from RNAfold using DMS reactivity data as pseudo-energies. (B) The previously established reference model of EBER2.
The region of greatest difference between our proposed EBER2 model (in cellulo based) and the EBER2 reference model (in vitro based) is the rearrangement of the reference model Stem 2 into Stems 2–4 of our model with little structural similarity. Notably though, the looped-out region from nucleotides 106–124 in the reference model retain nucleotides 110–122 in a looped-out region. This region was previously shown to be loosely structured and available for binding oligonucleotides used in pulldowns of EBER2 and is accessible to single strand specific RNases (8,10).
A few positions in our model also contain DMS reactivity in structured regions. Notably, positions A108 and A125 showed strong reactivity values and are in stems flanked by G–C pairings. To assess how well both EBER2 models agree with or fit the in cellulo generated DMS reactivity data, we performed a receiver-operator characteristic (ROC) analysis using the in cellulo DMS reactivity profile and the corresponding CT files for both 2D structure models in Figure 3 (see Materials and Methods). Briefly, reactivity values are constrained at regular, increasing thresholds from lowest to highest reactivity. When constrained, the corresponding nucleotide is defined as being paired and we cross reference this position to the 2D models of EBER2 to gain a true positive rate (TPR) and false positive rate (FPR). The TPR and FPR can then be plotted at each interval to form an ROC curve, where a larger area under the curve (AUC) indicates a greater initial increase in TPR and a better fit to the data. As the reactivity values here are experimentally derived from an in cellulo system, they represent a probabilistic likelihood of specific nucleotides being paired or unpaired and cross referencing these with several potential models can be informative, regardless of how the model was generated. Unsurprisingly, the DMS informed model had a larger AUC, 0.718, when compared to the DMS reactivity profile and the reference EBER2 model had an AUC of 0.612.
To further assess the quality of the proposed and reference EBER models, we performed a covariation analysis using the cm-builder pipeline. The proposed EBER2 model in Figure 3A had a total of six base pairs identified as having statistically significant covariation (i.e. E-value < 0.05) with all base pairs having a power over 0.1. Here, the power metric is a measure of the amount of sequence variation observed in the alignment and the depth of the alignment. Covariation analysis on the EBER2 reference model identified no statistically significant covarying base pair (Figure 3B). Similarly, the EBER1 reference model had no statistically significant covarying base pairs identified. However, our proposed model of EBER1, which differs by only three base pairs from the reference model, had five base pairs identified as having statistically significant covariation, all with power above 0.1 (Supplemental Figure S1).
Tandem terminal repeat RNA of Epstein–Barr virus
The EBV genome contains a region of tandem terminal repeats which can contain several copies up to ∼20 (48). During lytic reactivation, the genome is linearized with the terminal repeats flanking the ends of the genome. When the virus enters latency, the genome is circularized at the terminal repeats through a homologous recombination process which is only partially understood (49). Interestingly, the terminal repeats are transcribed during latency and participate in an interaction with EBER2 that is mediated by base pairing. EBER2 also interacts with the transcription factor PAX5, and this complex will then bind the TR RNA (Figure 4). The overall result of the interactions between EBER2, TR, and PAX5 facilitates the recruitment of this complex to the EBV genome for transcriptional control—a novel regulatory mechanism (8).
Figure 4.

Secondary structural models of mono and di-terminal repeat RNA units of the EBV type II genome. (A) A DMS informed RNAfold 2D structure model of the EBV TR RNA mono-segment. The DMS reactivity data used is overlaid on the model as red shaded nucleotides with the DMS reactivity scale ranging from 0.0 (white) to 1.0 (dark red). The model has nucleotide positions labelled at every 20-nucleotide interval. The site which binds EBER2 is highlighted in purple. Sites of R-scape identified covariation are highlighted in green and blue. (B) The same TR RNA mono-segment as modeled in panel A. Here, structure probabilities (as determined by RNAfold) are overlaid on the model and ScanFold predicted base pairs with ΔG z-scores < –1 are highlighted with green base pair lines. (C) A DMS informed, RNAfold 2D structural model of the EBV TR RNA di-segment. Here, the first segment is highlighted in green, and the second segment is highlighted in red to help differentiate their positions. The EBER2 binding sites are highlighted in purple. This model has nucleotide positions labelled at every 40-nucleotide interval.
To model how the TR RNA might fold on its own and in the presence of multiple copies we generated a DMS informed model of a single (i.e. mono-) segment of the TR RNA and a double (i.e. di-) segment of the RNA using RNAfold (Figure 4). The mono- and di-segments of the TR RNA were also analyzed with ScanFold to identify regions and nucleotides which have significant thermodynamic stability. The TR RNA mono-segment (Figure 4A and B) forms several long stems and multi-branched regions where ∼64% of nucleotides are involved in local or long-range base pairs. Overall, moderate to highly reactive nucleotides are modeled in single-stranded regions, loops, bulges, or in base pairs flanked by unpaired regions or non-canonical base pairs. Several helical regions contain highly reactive nucleotides, notably at positions 14, 75 and 227 in Figure 4A. Additionally, the TR RNA mono-segment was assessed for covariation and 8 statistically significant covarying base pairs were identified in a cluster between nucleotides 50–130 (Figure 4A). Of these 8 covarying base pairs, 4 had power above 0.1 and 4 were <0.1.
To further interrogate the thermodynamic landscape, we overlaid the structure probabilities (as determined by RNAfold from the partition function) onto the 2D structural model of the TR RNA mono-segment (Figure 4B). Most of the probabilities are moderate in value, an indication of potentially dynamic nature of the TR RNA structure. The region known to bind EBER2 (nucleotides 177–200) has an average probability of 0.504. The region of consistent highest probability is from nucleotides 296–373 (average probability of 0.894) and the region from nucleotides 114–137 has the lowest probability (average probability of 0.126). To assess how the 2D structural landscape may differ between a TR RNA mono-segment and a TR RNA di-segment, a DMS informed 2D structural model of the di-segment using RNAfold was generated (Figure 4C). Interestingly, almost all the structure predicted for the mono-segment was preserved in the di-segment, except for two regions. The region of the mono-segment which had the lowest probabilities (nucleotides 114–137) unfolds, and forms a long-range, continuous 18 nucleotide stem with the corresponding region of the second segment (Figure 4C). Additionally, in the mono-segment, the 5′ and 3′ ends of the RNA came together to form a six base pair stem. In the di-segment, the 5′ end of the first segment forms a homologous six base pair stem with the 3′ end of the second segment. Similarly, the 5′ end of the second segment forms this stem with the 3′ end of the first segment.
The oncogenic MYC mRNA
The resulting ScanFold data for the 10015 human mRNA and lncRNA transcripts are available for download from the RNAStructuromeDB. Here, we show how researchers interested in using the DMS informed ScanFold data for the transcripts in this dataset can assess the thermodynamic and structural landscape of an RNA of interest. This will lower the barrier of entry for those interested in studying these transcripts as DMS informed structural models and thermodynamic analyses are already complete and available for visualization or download. Researchers can visualize local structural motifs (using genomic viewing software such as IGV (39) and VARNA (40)) and home in on regions of significant thermodynamic stability, an indication of potential functionality. Identified regions of significant thermodynamic stability and their local structure models can serve as starting points for functional assay design (e.g. dual luciferase assays, half-life experiments, etc.), targets for therapeutic targeting, or targets for more intensive structural studies: such as identifying significantly stable motifs for structural biology.
Here, we focus on the MYC mRNA (ENST00000621592.7) as an example of how to view and use DMS informed ScanFold data. MYC encodes a transcription factor which is a master regulator of several cell cycle control pathways including apoptosis and cell proliferation. MYC is considered an oncogene and its dysregulation and overexpression is a major contributor to over 60 percent of all cancers, including EBV-associated cancers (50). There is a need to understand the regulation of the MYC mRNA and how native structure contributes to regulatory processes. Aside from understanding structure-function mechanisms within the MYC mRNA, accurate structure models can also be used for small molecule or antisense oligonucleotide (ASO) design. This is particularly significant, as attempts to drug the MYC protein have been unsuccessful (50).
Data files from ScanFold output (description of files in Table 1 of (15)) can be loaded into the IGV software (or viewed on the RNAStructuromeDB) and an example of the MYC MFE and z-score analysis tracks are in Figure 5A. The z-score data from overlapping windows are used to deduce the base pairs which most contribute to significant thermodynamic stability, and these are modeled by the base pair track (Figure 5A). From here, regions which have base pairs with ΔG z-scores <−2 are identified and are pulled out as extracted structures (i.e. motifs). In this study, each extracted structure from the MYC mRNA underwent covariation analyses via the cm-builder pipeline. A total of 5 out of the 13 extracted structures (extracted structures 1, 6, 10, 11 and 13; Figure 5A) showed evidence of at least one statistically significant covarying base pair (Supplemental File 2).
Figure 5.

DMS informed ScanFold analysis of the MYC mRNA. (A) An IGV visualization of ScanFold data for the MYC mRNA. At the very top is a positional marker with every 500 base pairs marked. Below this is the MFE track and the ΔG z-score track from the ScanFold-Scan analysis. Next is the base pair track resulting from ScanFold-Fold, followed by a heatmap of DMS reactivity data used in analysis. Further down is a track showing the locations of ScanFold extracted structures and finally, a cartoon representation of the MYC mRNA where the 5′ and 3′ UTRs are represented by thinner grey lines and the coding sequence is depicted with a larger black box. Regions of additional focus are highlighted with opaque blue boxes. (B) Extracted structure 1 with a downstream hairpin. The start and end of the structure are labelled with the transcript nucleotide position. The DMS reactivity data for the region is overlaid on the model as red shaded nucleotides with the DMS reactivity scale ranging from 0.0 (white) to 1.0 (dark red). Base pairs with ΔG z-scores <–1 and <–2 are colored green and blue respectively. Covarying pairs, as identified by R-scape, are highlighted with dark green and blue opaque boxes. (C) Extracted structure 3–4 with a downstream hairpin. The start and end of the structure are labelled with the transcript nucleotide position. The DMS reactivity data for the region is overlaid on the model. Base pairs with ΔG z-scores <–1 and <–2 are colored green and blue respectively. (D) Extracted structure 5–8. The start and end of the structure are labelled with the transcript nucleotide position. The DMS reactivity data for the region is overlaid on the model as red shaded nucleotides. Base pairs with ΔG z-scores <–1 and <–2 are colored green and blue respectively. A covarying pair, as identified by R-scape, is highlighted with a dark green, opaque box.
The region of MYC which contains extracted structure 1 along with an adjacent hairpin which contains −1 ΔG z-score base pairs is depicted in Figure 5B. This region resides in the 5′UTR as the 5′ most motif and when analyzed, both hairpins show evidence of covariation. The region depicted in Figure 5C represents a 3′UTR region which we (35) and others (51) have previously tested in dual luciferase assay reporter systems and the DMS informed −2 z-score structure presented here matches our previous in silico predicted structure of the region (35). Despite deep phylogenetic conservation, this region showed no evidence of statistically significant covariation. There is evidence of functionality, however, as this region acts as a hub for protein and micro-RNA binding and has been previously shown to significantly affect the translation efficiency of reporter transcripts through miRNA targeted repression. Both mutations to the miRNA seed sequence and mutations to adjacent structure which reinforces structural stability ablated the translational repression associated with this region (35,51). Finally, the region encompassing MYC extracted structures 5–8, shown in Figure 5D, is the second cluster of low z-score structures in the 3′UTR after the region depicted in Figure 5C. To our knowledge, this second clustered region has not been previously described or tested. Covariation analysis with cm-builder highlighted one significantly covarying base pair at C2073-G2084.
CYTOR lncRNA
In this study, reactivity profiles for 3427 human lncRNAs were generated and these transcripts were analyzed with ScanFold. Individual thermodynamic and structure results for these lncRNAs can be downloaded from the RNAStructuromeDB. These results contain FASTA and react files for a given sequence and can be used to complete further analyses, such as global MFE modelling with the program RNAfold. The lncRNA cytoskeleton regulator RNA (CYTOR) provides an example of how to use the generated ScanFold data and how to further process these data into informative models.
In a study of lncRNAs dysregulated in EBV infected lymphoblastoid cell lines (LCLs), CYTOR was identified as significantly upregulated (52). The function of CYTOR has been linked to differentiation, stress response and cytoskeletal maintenance, while its overexpression promotes oncogenic and disease states (53). CYTOR is involved in the progression of a myriad of cancers including breast cancer, gastric cancer, colorectal cancer, hepatocellular carcinoma and more (53). Its over expression can drive tumor growth while reduced expression is associated with tumor shrinkage and increased life expectancy of cancer patients. While there is still much to learn about the mechanisms of CYTOR’s oncogenicity, it is known to bind several RBPs (e.g. NCL and SAM68 (54), β-catenin (55) and EZH2 (56)) and acts as a miRNA sponge (e.g. miR-4767 (57) and miR-138 (58)) altering the miRNA and transcriptome landscape of associated cells.
Figure 6A shows the DMS reactivity data overlaid on the nucleotides of CYTOR (ENST00000646636.1, 1020 nucleotides long). This model has several motifs and regions that have statistically significant covariation (Figure 6A; data in Supplemental File 2). The region of highest enrichment for covarying base pairs occurs from nucleotides 327–575 where 9 covarying base pairs are observed ranging in power from 0.00 to 0.59. The structural probabilities, as determined by RNAfold, are overlaid on the CYTOR model (Figure 6B). There are several regions which show clustered high probability nucleotides, notably the regions encompassed by nucleotides 131–144:576–589 (average probability of 0.96), 164–275 (average probability of 0.97), 338–484 (average probability of 0.94), 590–656 (average probability of 0.97), 671–693 (average probability of 0.95) and 809–818 and 999–1008 (average probability of 0.99). The region which is enriched in covarying base pairs has an average probability of 0.55, indicating that the region may be loosely structured or conformationally dynamic.
Figure 6.

A DMS informed RNAfold model of the lncRNA CYTOR with multiple data overlaid on the model. (A) The DMS reactivity data which was used to inform the model is overlaid on the nucleotides and ranges from 0.0 (white) to 1.0 (red). Sites of R-scape identified covariation are also indicated with green and blue base pair shading. Two known miRNA sites within CYTOR are highlighted as light green and purple annotations (B) Nucleotide probabilities, derived from RNAfold, are overlaid on the model. (C) The per-nucleotide z-score, derived from ScanFold, are overlaid on the model. (D) Nucleotides which were identically structured between the DMS informed RNAfold model and the DMS informed ScanFold model are highlighted in green.
From the ScanFold-Scan analysis, CYTOR had an average windowed ΔG z-score value of −0.278 with the lowest window being −3.55 and the highest being 1.83. The average windowed MFE values were −79.97 kcal/mol with a lowest and highest window value of −118.71 and 0.0 kcal/mol respectively. From the 5′ to 3′ end, the z-score and MFE showed no trend or biases across the transcript. The ScanFold-Fold analysis of the data generated per nucleotide ΔG z-scores (which are overlaid on the RNAfold generated model in Figure 6C) and there were no identified consensus base pairs with a z-score <−2. However, several −1 z-score motifs were identified throughout the transcript, and these were used as constraints to generate a global refolded model of CYTOR (using RNAfold) which locked in the −1 z-score structures and allowed for longer range interactions to form around them (Supplemental Figure S2). The −1 z-score constrained and refolded model of CYTOR was then compared to the DMS constrained RNAfold model and nucleotides that were similar structured between the two models are highlighted in Figure 6D. Notably, the low z-score regions correspond well with some regions of high structure probability, indicating that these regions are well structured and potentially functional motifs within the larger transcript.
Additionally, to assess the binding potential of the miRNAs which target CYTOR (Figure 6A), we used the ΔΔG method described by Kertesz et al. (43) which accounts for the energy gained by miRNA binding and the energy required to break existing structures (more details in the Materials and Methods). Here, a positive ΔΔG would indicate that miRNA binding is thermodynamically unfavorable as input energy would be required for binding, whereas a negative ΔΔG indicates that miRNA binding is thermodynamically spontaneous. The region where miR-4767 binds to CYTOR had a ΔΔG of −17.38 (kcal/mol). The region of miR-138 binding to CYTOR had a ΔΔG of + 1.6 (kcal/mol) and occurred in a region with more structure and long-range interactions.
DISCUSSION
Insights from in silico and DMS informed ScanFold comparisons
The comparison of in silico ScanFold to DMS informed ScanFold models allowed for an in-depth assessment about how well in silico ScanFold performs in the absence of probing data and how the incorporation of probing data is influencing model generation. The PPV of in silico ScanFold increases as the z-score cutoff becomes more stringent, from unfiltered base pairs to a −2 z-score threshold. When compared to DMS informed models, in silico ScanFold models −2 z-score structures with great consistency (Figure 2A). This is an observation we previously reported in our analyses of SARS-CoV-2 where the −2 z-scores structures agreed better (than higher z-score structures) with a myriad of structure probing reactivity datasets and reactivity informed structural models. While the PPV was considerably higher for −2 z-score base pairs in this study, the sensitivity was much lower compared to the −1 z-score pairs. This suggests that while in silico ScanFold is modeling −2 z-score base pairs with high confidence, it is missing many −2 z-score pairs which are present in the DMS informed ScanFold models. Data indicate that DMS informed ScanFold models contain nearly four times as many −2 z-score base pairs (on average) per transcript, compared to the corresponding in silico models (Supplemental File 1). Presumably, the incorporation of DMS reactivity data disallow the formation of higher (i.e. less negative) z-score structures with more spurious base pairs during the ScanFold-Scan step of the program. When the ScanFold-Fold module then parses the DMS informed scanning data, more −2 z-score pairs are identified because of less competition from more poorly predicted structures. This finding highlights the value of considering experimental probing data not only in secondary structure modeling, but also in functional motif discovery—something not previously appreciated.
Notably, the per nucleotide z-score correlation analyses revealed a high global correlation between DMS informed and in silico ScanFold z-score data and minimal changes in the average z-score per transcript (Figure 2B, Supplemental File 1). This is an indication that the z-score metric is less affected by the incorporation of DMS reactivity values across a transcript and what is affected is the identification of significantly stable base pairs. Taken together, these observations yield several key findings. In the absence of RNA structure probing reactivity data, the z-score metric of in silico ScanFold can home in on regions of well-ordered, stable structures. The −2 z-score structures modeled by in silico ScanFold have high agreement with DMS informed models making ScanFold a fast and powerful tool to identify well-structured and presumably functional regions within mRNAs. When it comes to identifying potentially functional and/or therapeutic regions in pathogens or disease related human RNAs, −2 z-score structures, as identified by in silico ScanFold, provide high confidence model predictions and a valuable starting point for additional studies. There is, however, great potential utility in combining experimental probing data with ScanFold. While in silico −2 z-score models are robust, they are limited to the best-predicted structures, whereas the experimental data, while validating those well-predicted regions, also expands the list of potentially functional structure.
Highly regulated human genes have greater propensity for ordered RNA structure
In the DMS informed ScanFold analyses of human mRNAs and lncRNAs, we found that mRNAs had a slightly more negative average window MFE when compared to lncRNAs, while lncRNAs had a slightly lower average ΔG z-score. The lower ΔG z-score transcripts and regions can be interpreted as having an increased potential for structure-related functionality and thus their biases for sequence-ordered thermodynamic stability. This indicates that lncRNAs have similar, if not more potential for regulatory function as mRNAs have. Furthermore, the transcriptome-wide analysis of ScanFold metrics resulting from BJAB-B1 mRNAs show that transcripts which have higher likelihoods of being regulated (i.e. tissue specific transcripts) have lower ΔG z-scores, per-nucleotide z-scores and a higher number of ScanFold extracted structures than transcripts which are expected to be less regulated (i.e. HKGs). These results support the hypothesis that these transcripts are under increased regulatory pressures and gives evidence for ScanFold extracted structures having potential regulatory roles. Additionally, a global decrease in z-score values from the 5′ to 3′ end of transcripts was observed for all gene groups. An indication that the 3′UTRs are enriched with low z-score structure, supporting previous observation that 3′UTRs are hubs for transcript regulation (44).
For example, our detailed analysis of MYC, which requires exquisite levels or posttranscriptional control for normal cellular development and function, revealed thirteen −2 z-score structures. One of these structures was in the 5′UTR, one in the CDS, and eleven structures were observed in four distinct clusters in the 3′UTR. The 5′UTR motif may represent a minimal structural motif needed to impart some form of translational regulation, or stability regulation to the transcript and this motif merits further testing in a functional reporter assay (59,60). The first cluster of −2 z-score structure in the 3′UTR has been experimentally validated by our lab and others to exhibit translational repression of the transcript via miRNA and RBP binding (35,51). Disruption of the miRNA binding sites or alteration of structure in the region can ablate miRNA targeting. Interestingly, covariation analysis of this region did not show evidence for statistically significant covarying base pairs. However, downstream structure in the 3′UTR did contain regions of statistically significant covariation. These remaining clusters of −2 z-score structure in the 3′UTR, to our knowledge, remain unstudied for their functional impact on MYC regulation, but due to their low z-score structure and evidence of covariation, merit further analyses.
Differing strategies provide novel structural insights into the human lncRNA CYTOR
Our focused analysis of the lncRNA CYTOR was stimulated by the high degree of induction of this transcript due to EBV infection. Even with DMS reactivity incorporation, the CYTOR lncRNA is on the upper end of sequence length (1020 nucleotides) for accurate and robust global MFE model generation and some regions may be more poorly modeled than others. Therefore, modeling of 2D structure relied on several orthogonal approaches to try and identify regions of robust, well-formed structure and an attempt to identify potential functional regions within the longer transcript. Two 2D structural models of CYTOR were generated, the first using the associated DMS reactivity profile and RNAfold and another model was made using the DMS reactivity profile and ScanFold to initially identify local structure motifs followed by a global refold where ScanFold identified motifs were used as initial constraints. The regions that are similar between the two CYTOR models (Figure 6D) are of interest as both model generation strategies (the DMS constrained RNAfold global fold model and the DMS informed ScanFold constrained refolded model) converged on similar structures. As ScanFold focuses on accurate prediction of local structures, initially identifying local structures and then constraining these regions to be paired in a global refold helps to limit the global folding landscape to allow for more robust long-range interaction modelling. While it is likely that some regions of the CYTOR transcript are conformationally dynamic and loosely structured, the regions of similarity between the models may represent well-structured and biologically relevant structures (as both models were DMS informed). These may be regions which assist in structure-function based modes of regulation and these regions could also serve as starting points for potential structure-specific targeted therapeutics.
Several of the high structure probability regions within CYTOR (Figure 6B), overlap well with both the low z-score regions (Figure 6C) and the regions which were structurally similar between models (Figure 6D). It is possible that these well-structured regions could be serving as hubs for trans-acting factors or helping to direct positions of binding motifs in 3D space. Furthermore, the regions of low probability (Figure 6B) are likely loosely structured and more dynamic which could allow for binding of miRNAs, other lncRNAs, genomic sequences, or allow for room to accommodate larger RNP complexes in adjacent structured regions. Two known miRNA binders of CYTOR bind in regions of higher probability (Figure 6A and B). While the binding of miR-4767 to CYTOR is predicted to be highly favorable (ΔΔG of −17.38 (kcal/mol)), the binding of miR-138 is predicted to be slightly unfavorable (ΔΔG of + 1.6 (kcal/mol)). However, the presence of unknown trans-acting factors around miR-138 may influence local stability, allowing for miRNA targeting. Additionally, the nucleotide probabilities (Figure 6B) are moderate to low in this region, implying a potential for conformational dynamics which may allow for increased propensity of miRNA binding. Structural modulation around the site of miRNA targeting is something we have previously observed within the MYC mRNA (35) and could be a mechanism present in CYTOR to finely tune the regulation of expression. As more information becomes available on known binders and resulting functions of CYTOR, these structural maps can be used to help infer potential structure-function mechanisms of action.
Analyses of EBV RNAs provides novel structural insights
There were 19 EBV transcripts present in BJAB-B1 cells that had enough read and RTSC coverage to generate DMS reactivity profiles; notably, both lytic and latent transcripts were probed (Table 4). While the BJAB-B1 cell line is considered to primarily expresses a latency III program, periodically the virus will become lytically active in a fraction of the cultured cells allowing the collection of structural information on not just latent transcripts, but some lytic transcripts as well (1).
The lytic transcript BHRF1, which is a viral homologue of cellular B-cell lymphoma 2 (BCL-2), is known to have anti-apoptotic function in the cell, help to accelerate MYC-induced lymphoma development and is considered a therapeutic target of lytic EBV infection (61). BHRF1 had the highest average reactivity and the highest average windowed ΔG z-score (0.34 and 0.62, respectively). These two metrics taken together indicate that the BHRF1 transcript is less structured than the other EBV transcripts as the increased reactivity values indicate greater nucleotide accessibility and the high, positive ΔG z-score indicate the transcript is lacking in significantly stable structure and may even have propensity for being significantly unstable. This could potentially allow for rapid translation of the mRNA into protein as there is little structure present to inhibit the ribosome and this could lead to a faster inhibition of apoptosis. Additionally, the apparent unstructured nature of BHRF1 suggest that, in addition to trying to therapeutically target the BHRF1 protein, the mRNA transcript may be amenable to sequence specific ASO targeting.
The EBV regions of lowest ΔG z-score were the two direct repeat regions (DRL and DRR), followed closely by EBER1. It is somewhat expected that the direct repeat regions had lower ΔG z-scores as the z-score metric excels at finding regions of non-random sequence which folds into stable structure and this is very characteristic of repeat regions themselves (which often form complementary hairpin structures). The finding that the ΔG z-score of EBER1 is −1.58 is interesting as it indicates the short ncRNA is highly organized to be very thermodynamically stable. In contrast, EBER2 has a ΔG z-score of −0.51 and the overall structure appears to be less significantly ordered and there are several competing structural models for this RNA. It may be that the conformational dynamics of EBER2 are greater than that of EBER1, perhaps to allow for binding of multiple trans-acting factors (e.g. PAX5, TR RNA, La antigen, etc.). Regarding potential conformational dynamics, there were several nucleotides that were highly reactive but are modelled to be embedded in Watson–Crick helices. Some of these nucleotides are adjacent to the large looped-out region present in both models. A certain amount of helical breathing or structural rearrangement may be occurring depending on the presence or absence of potential trans-regulatory interactors. It should be noted that the reference EBER2 model was generated from in vitro probing experiments conducted on Raji cell lysate (10), while our DMS reactivity data comes from in cellulo BJAB-B1 probing. These highly different probing environments affect the presence and abundance of the trans-acting factors that bind EBER2, which can affect its conformational equilibrium. Future work on the EBER targets could include comprehensive probing analyses across a variety of cell types to help clarify the level of conformational dynamics and contexts in which they are occurring.
The EBER2 reference model had good agreement with its corresponding chemical and enzymatic probing data, with only a few nucleotides and reactivities being inconsistent. As noted by the authors, most of these were at the ends of helices or in weaker base pairs (A–U or G–U pairs) and a certain degree of ‘helical breathing’ may explain some of the inconsistencies. From the Glickman et al. study, there were two highly reactive cobra venom RNase (which targets and cuts double stranded RNA) reactivity sites at position C99 and C100 (Figure 2 of (10)) ,which were inconsistent with the reference model. However, our proposed model has these nucleotides in base pairs at the end of Stem 3, an indication that the previously generated in vitro data is also consistent with our model.
Notably, both our alternative models proposed for EBER1 and EBER2 had 5 and 6 co-varying base pairs, respectively, and the reference models had no covarying base pairs (Supplemental Figure S1 and Figure 3). This is somewhat surprising as 5 of the EBER2 co-varying base pairs appear in a region which is structurally identical between our proposed model and the reference model. Additionally, our proposed EBER1 model differs by only three base pairs from the reference model and has 4 covarying base pairs identified in regions of identical structural context. These results can be explained in part by understanding that the initial alignments with which covariation is determined is based on both a sequence and secondary structure alignment (via INFERNAL) and even small structural differences can lead to varying alignments. In the case of EBER1, analysis of the Stockholm alignments showed our proposed model and the reference model had 45 aligned sequences in common with 14 and 6 sequences being unique to our proposed model and the reference model, respectively. This analysis exemplifies that identification of significantly co-varying base pairs can lend additional evolutionary support to models, but it can also be a mercurial technique and the absence of co-variation does not indicate an absence of function.
The TR RNA is an emerging transcript of interest within EBV. While the tandem terminal repeats have been known to assist in circularization, linearization, and sorting of EBV genomes in the genomic context, the transcription of the tandem terminal repeats into TR RNA is a more recent observation and preliminarily it is known to play roles in regulating transcription of the EBV genome by forming a complex with PAX5 and EBER2. As the region of the TR RNA which binds the genomic TR is still unknown, it may be a region of low probability which is capable of breaking cis-structure to form the trans-interaction. In our proposed model of the TR RNA, the region known to bind to EBER2 (Figure 4A) is in a region of low probability (Figure 4B) and the adjacent nucleotides (nucleotides ∼400–420) which form long-range interactions with the EBER2 binding site are presumably left unpaired; it is possible that this region is available for interaction with genomic sequence. Future studies could be conducted which mutate this region to assess if there is a loss of binding of TR RNA to the genomic sequence.
To compare the folding of a single or dimeric TR RNA molecule, we folded the mono- and di-segments of the TR RNA (Figure 4C). Surprisingly, almost all the structure present in the mono-segment was preserved in the di-segment, except for the 5′ and 3′ ends of the transcript and the region of lowest structure probability (the stem encompassed by nucleotides 114–131), which formed a completely complementary, long-range intersegmental interaction with the corresponding nucleotides in the second segment. This indicates that the segments have potential for forming larger, multi-segment structures, which may be stabilized by this interaction. If the TR RNAs are forming inter-segmental structure, disruption of this region, may disrupt complex formation and function. More work is needed to parse out the interactions of TR RNAs.
CONCLUSION
In this study, we report the application Structure-seq2 to the BJAB-B1 cell line (an EBV infected B-cell lymphoma). DMS modified RNAs underwent transcriptome-wide library preparation and sequencing. Bioinformatic analysis of resulting sequencing reads (using StructureFold2) generated DMS reactivity profiles for over 10000 human mRNAs and lncRNAs, along with 19 latent and lytic EBV RNAs. Using the DMS reactivity profiles from in cellulo probing, we provide an alternative model of the highly abundant EBV ncRNA, EBER2. Additionally, we provide the first structural model of the EBV tandem terminal repeat RNA (in both a mono- and di-segment configurations), which interacts with EBER2 to drive lytic reactivation—indicating unique and common structural features of the mono- and di-segment sequences, as well as insights into interactions with EBER2.
ScanFold was used to analyze 6588 human mRNAs and 3427 human lncRNAs which had a sufficient coverage to generate a DMS reactivity profile. These DMS informed ScanFold results are available for individual viewing and download from the RNAStructuromeDB. These results provided high resolution thermodynamic analyses of each transcript and contain robust, biologically relevant 2D local structural models with an emphasis on regions of significant thermodynamic stability and propensity for function. As DMS probing, RNA sequencing/analysis, and reactivity modelling of RNAs is highly technical and time consuming, the availability of the structure models generated in this study should significantly lower the barrier of entry for researchers interested in studying any of these transcripts. We show how to use the resulting ScanFold data to further investigate transcripts using the MYC mRNA and the CYTOR lncRNA as examples: here, we provide the first experimentally informed model of CYTOR secondary structure.
As our discovery of novel RNA sequences (both within mRNAs and lncRNAs) continues to grow, so does our need for accurate and robust structural analyses of these transcripts. Deducing the structure–function mechanism of RNAs is of vital importance as is identifying regions for potential therapeutic targeting and the data generated in this study is a crucial step towards these goals.
DATA AVAILABILITY
Processed data and analyses generated in this study can be found in the Supplemental Data section. ScanFold generated data along with input reactivity profiles for human mRNAs and lncRNAs can be viewed and downloaded from the RNAStructuromeDB. To access this data, the following link can be used: https://structurome.bb.iastate.edu/transcript-search.
To search the database, enter the ENST identifier for a transcript into the search bar and by clicking the ‘Experimentally informed transcripts’ toggle button, human mRNAs and lncRNAs from this study can be accessed. By clicking the ‘View Results’ button, an IGV window will be generated displaying the ScanFold data for the transcript of interest. Additionally, the ‘Download Results’ button will allow for download of a zipped directory containing all ScanFold output for the associated ENST identifier, including the reactivity file used
EBV transcripts and genomes analyzed by ScanFold with DMS pseudo-energy incorporation can be downloaded from the RNAStructuromeDB at the following link: https://structurome.bb.iastate.edu/download/DMS−informed-scanfold-analysis-ebv-bjab-b1.
Additionally, custom python scripts used in this study and mentioned above can be found on Github in the two following repositories: https://github.com/moss-lab/Transcriptome_Scriptshttps://github.com/moss-lab/SARS-CoV-2.
FASTQ files and resulting REACT files from this study have been uploaded to the NCBI GEO database (Accession number GSE210478).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge Dr. Phil Bevilacqua and his lab for their advice and assistance in answering our technical questions as we implemented both the Structure-seq2 protocol and analyses with the StructureFold2 package. We acknowledge Dr. Michael Baker and the ISU DNA facility for assisting with quality control, troubleshooting, and sequencing of our cDNA libraries. Finally, we acknowledge the ISU Research IT department, particularly Nick Booher, for computational support, management of the high-performance computing system, and data hosting.
Contributor Information
Collin A O’Leary, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Van S Tompkins, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Warren B Rouse, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Gijong Nam, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Walter N Moss, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB online.
FUNDING
National Institute of General Medical Sciences [R01GM133810 to W.N.M. and F31CA257090 to W.B.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Munz C. Latency and lytic replication in epstein-barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019; 17:691–700. [DOI] [PubMed] [Google Scholar]
- 2. Bjornevik K., Cortese M., Healy B.C., Kuhle J., Mina M.J., Leng Y., Elledge S.J., Niebuhr D.W., Scher A.I., Munger K.L.et al.. Longitudinal analysis reveals high prevalence of epstein-barr virus associated with multiple sclerosis. Science. 2022; 375:296–301. [DOI] [PubMed] [Google Scholar]
- 3. Saha A., Robertson E.S.. Mechanisms of B-cell oncogenesis induced by epstein-barr virus. J. Virol. 2019; 93:e00238-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Healy J.A., Dave S.S.. The role of EBV in the pathogenesis of diffuse large b cell lymphoma. Curr. Top. Microbiol. Immunol. 2015; 390:315–337. [DOI] [PubMed] [Google Scholar]
- 5. Moss W.N., Lee N., Pimienta G., Steitz J.A.. RNA families in epstein-barr virus. RNA Biol. 2014; 11:10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iwakiri D. Epstein-Barr virus-encoded RNAs: key molecules in viral pathogenesis. Cancers (Basel). 2014; 6:1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Falco G., Antonicelli G., Onnis A., Lazzi S., Bellan C., Leoncini L.. Role of EBV in microRNA dysregulation in burkitt lymphoma. Semin. Cancer Biol. 2009; 19:401–406. [DOI] [PubMed] [Google Scholar]
- 8. Lee N., Moss W.N., Yario T.A., Steitz J.A.. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell. 2015; 160:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee N., Yario T.A., Gao J.S., Steitz J.A.. EBV noncoding RNA EBER2 interacts with host RNA-binding proteins to regulate viral gene expression. Proc. Natl. Acad. Sci. U. S. A. 2016; 113:3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glickman J.N., Howe J.G., Steitz J.A.. Structural analyses of EBER1 and EBER2 ribonucleoprotein particles present in epstein-barr virus-infected cells. J. Virol. 1988; 62:902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosa M.D., Gottlieb E., Lerner M.R., Steitz J.A.. Striking similarities are exhibited by two small epstein-barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol. Cell. Biol. 1981; 1:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moss W.N., Steitz J.A.. Genome-wide analyses of epstein-barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genomics. 2013; 14:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tompkins V.S., Valverde D.P., Moss W.N.. Human regulatory proteins associate with non-coding RNAs from the EBV IR1 region. BMC Res Notes. 2018; 11:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bridges R., Correia S., Wegner F., Venturini C., Palser A., White R.E., Kellam P., Breuer J., Farrell P.J.. Essential role of inverted repeat in epstein-barr virus IR-1 in b cell transformation; geographical variation of the viral genome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019; 374:20180299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrews R.J., Baber L., Moss W.N.. Mapping the RNA structural landscape of viral genomes. Methods. 2020; 183:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andrews R.J., Roche J., Moss W.N.. ScanFold: an approach for genome-wide discovery of local RNA structural elements-applications to zika virus and HIV. PeerJ. 2018; 6:e6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrews R.J., O’Leary C.A., Moss W.N.. A survey of RNA secondary structural propensity encoded within human herpesvirus genomes: global comparisons and local motifs. PeerJ. 2020; 8:e9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mailler E., Paillart J.C., Marquet R., Smyth R.P., Vivet-Boudou V.. The evolution of RNA structural probing methods: from gels to next-generation sequencing. Wiley Interdiscip. Rev. RNA. 2019; 10:e1518. [DOI] [PubMed] [Google Scholar]
- 19. Herschlag D., Bonilla S., Bisaria N.. The story of RNA folding, as told in epochs. Cold Spring Harb. Perspect. Biol. 2018; 10:a032433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mathews D.H., Disney M.D., Childs J.L., Schroeder S.J., Zuker M., Turner D.H.. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernhart S.H., Hofacker I.L., Will S., Gruber A.R., Stadler P.F.. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008; 9:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fu Y., Xu Z.Z., Lu Z.J., Zhao S., Mathews D.H.. Discovery of novel ncRNA sequences in multiple genome alignments on the basis of conserved and stable secondary structures. PLoS One. 2015; 10:e0130200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Z., Mathews D.H.. Multilign: an algorithm to predict secondary structures conserved in multiple RNA sequences. Bioinformatics. 2011; 27:626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strobel E.J., Yu A.M., Lucks J.B.. High-throughput determination of RNA structures. Nat. Rev. Genet. 2018; 19:615–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ritchey L.E., Su Z., Tang Y., Tack D.C., Assmann S.M., Bevilacqua P.C.. Structure-seq2: sensitive and accurate genome-wide profiling of RNA structure in vivo. Nucleic Acids Res. 2017; 45:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tack D.C., Tang Y., Ritchey L.E., Assmann S.M., Bevilacqua P.C.. StructureFold2: bringing chemical probing data into the computational fold of RNA structural analysis. Methods. 2018; 143:12–15. [DOI] [PubMed] [Google Scholar]
- 27. Manfredonia I., Nithin C., Ponce-Salvatierra A., Ghosh P., Wirecki T.K., Marinus T., Ogando N.S., Snijder E.J., van Hemert M.J., Bujnicki J.M.et al.. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 2020; 48:12436–12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rivas E., Clements J., Eddy S.R.. Estimating the power of sequence covariation for detecting conserved RNA structure. Bioinformatics. 2020; 36:3072–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rivas E., Clements J., Eddy S.R.. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods. 2017; 14:45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andrews R.J., Baber L., Moss W.N.. RNAStructuromeDB: a genome-wide database for RNA structural inference. Sci. Rep. 2017; 7:17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ritchey L.E., Su Z., Assmann S.M., Bevilacqua P.C.. In vivo genome-wide RNA structure probing with Structure-seq. Methods Mol. Biol. 2019; 1933:305–341. [DOI] [PubMed] [Google Scholar]
- 33. Busan S., Weeks K.M.. Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with shapemapper 2. 2018; 24:143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrews R.J., O’Leary C.A., Tompkins V.S., Peterson J.M., Haniff H.S., Williams C., Disney M.D., Moss W.N.. A map of the SARS-CoV-2 RNA structurome. NAR Genom. Bioinform. 2021; 3:lqab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Leary C.A., Andrews R.J., Tompkins V.S., Chen J.L., Childs-Disney J.L., Disney M.D., Moss W.N.. RNA structural analysis of the MYC mRNA reveals conserved motifs that affect gene expression. PLoS One. 2019; 14:e0213758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haniff H.S., Tong Y., Liu X., Chen J.L., Suresh B.M., Andrews R.J., Peterson J.M., O’Leary C.A., Benhamou R.I., Moss W.N.et al.. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent. Sci. 2020; 6:1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gruber A.R., Bernhart S.H., Lorenz R.. The ViennaRNA web services. Methods Mol. Biol. 2015; 1269:307–326. [DOI] [PubMed] [Google Scholar]
- 38. Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Darty K., Denise A., Ponty Y.. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 42. Fagerberg L., Hallstrom B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K.et al.. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014; 13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E.. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007; 39:1278–1284. [DOI] [PubMed] [Google Scholar]
- 44. Mayr C. Regulation by 3'-Untranslated regions. Annu. Rev. Genet. 2017; 51:171–194. [DOI] [PubMed] [Google Scholar]
- 45. Mayya V.K., Duchaine T.F.. Ciphers and executioners: how 3'-Untranslated regions determine the fate of messenger RNAs. Front. Genet. 2019; 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clote P., Ferre F., Kranakis E., Krizanc D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005; 11:578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pavlova S., Feederle R., Gartner K., Fuchs W., Granzow H., Delecluse H.J.. An epstein-barr virus mutant produces immunogenic defective particles devoid of viral DNA. J. Virol. 2013; 87:2011–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moody C.A., Scott R.S., Su T., Sixbey J.W.. Length of epstein-barr virus termini as a determinant of epithelial cell clonal emergence. J. Virol. 2003; 77:8555–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laux G., Perricaudet M., Farrell P.J.. A spliced epstein-barr virus gene expressed in immortalized lymphocytes is created by circularization of the linear viral genome. EMBO J. 1988; 7:769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lourenco C., Resetca D., Redel C., Lin P., MacDonald A.S., Ciaccio R., Kenney T.M.G., Wei Y., Andrews D.W., Sunnerhagen M.et al.. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer. 2021; 21:579–591. [DOI] [PubMed] [Google Scholar]
- 51. Kong Y.W., Cannell I.G., de Moor C.H., Hill K., Garside P.G., Hamilton T.L., Meijer H.A., Dobbyn H.C., Stoneley M., Spriggs K.A.et al.. The mechanism of micro-RNA-mediated translation repression is determined by the promoter of the target gene. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8866–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang C., Li D., Zhang L., Jiang S., Liang J., Narita Y., Hou I., Zhong Q., Zheng Z., Xiao H.et al.. RNA sequencing analyses of gene expression during epstein-barr virus infection of primary b lymphocytes. J. Virol. 2019; 93: 10.1128/JVI.00226-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ou C.H., He X., Liu Y., Zhang X. lncRNA cytoskeleton regulator RNA (CYTOR): diverse functions in metabolism, inflammation and tumorigenesis, and potential applications in precision oncology. Genes Dis. 2021; 10.1016/j.gendis.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang X., Yu H., Sun W., Kong J., Zhang L., Tang J., Wang J., Xu E., Lai M., Zhang H.. The long non-coding RNA CYTOR drives colorectal cancer progression by interacting with NCL and sam68. Mol. Cancer. 2018; 17:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yue B., Liu C., Sun H., Liu M., Song C., Cui R., Qiu S., Zhong M.. A positive feed-forward loop between LncRNA-CYTOR and Wnt/beta-Catenin signaling promotes metastasis of colon cancer. Mol. Ther. 2018; 26:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen W.M., Huang M.D., Sun D.P., Kong R., Xu T.P., Xia R., Zhang E.B., Shu Y.Q.. Long intergenic non-coding RNA 00152 promotes tumor cell cycle progression by binding to EZH2 and repressing p15 and p21 in gastric cancer. Oncotarget. 2016; 7:9773–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Teng W., Qiu C., He Z., Wang G., Xue Y., Hui X.. Linc00152 suppresses apoptosis and promotes migration by sponging miR-4767 in vascular endothelial cells. Oncotarget. 2017; 8:85014–85023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cai Q., Wang Z., Wang S., Weng M., Zhou D., Li C., Wang J., Chen E., Quan Z.. Long non-coding RNA LINC00152 promotes gallbladder cancer metastasis and epithelial-mesenchymal transition by regulating HIF-1alpha via miR-138. Open Biol. 2017; 7:160247. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59. Haizel S.A., Bhardwaj U., Gonzalez R.L., Mitra S., Goss D.J. 2020. 5'-UTR recruitment of the translation initiation factor eIF4GI or DAP5 drives cap-independent translation of a subset of human mRNAs. J. Biol. Chem. 295:11693–11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leppek K., Das R., Barna M.. Author correction: functional 5' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018; 19:673. [DOI] [PubMed] [Google Scholar]
- 61. Fitzsimmons L., Cartlidge R., Chang C., Sejic N., Galbraith L.C.A., Suraweera C.D., Croom-Carter D., Dewson G., Tierney R.J., Bell A.I.et al.. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020; 27:1554–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data and analyses generated in this study can be found in the Supplemental Data section. ScanFold generated data along with input reactivity profiles for human mRNAs and lncRNAs can be viewed and downloaded from the RNAStructuromeDB. To access this data, the following link can be used: https://structurome.bb.iastate.edu/transcript-search.
To search the database, enter the ENST identifier for a transcript into the search bar and by clicking the ‘Experimentally informed transcripts’ toggle button, human mRNAs and lncRNAs from this study can be accessed. By clicking the ‘View Results’ button, an IGV window will be generated displaying the ScanFold data for the transcript of interest. Additionally, the ‘Download Results’ button will allow for download of a zipped directory containing all ScanFold output for the associated ENST identifier, including the reactivity file used
EBV transcripts and genomes analyzed by ScanFold with DMS pseudo-energy incorporation can be downloaded from the RNAStructuromeDB at the following link: https://structurome.bb.iastate.edu/download/DMS−informed-scanfold-analysis-ebv-bjab-b1.
Additionally, custom python scripts used in this study and mentioned above can be found on Github in the two following repositories: https://github.com/moss-lab/Transcriptome_Scriptshttps://github.com/moss-lab/SARS-CoV-2.
FASTQ files and resulting REACT files from this study have been uploaded to the NCBI GEO database (Accession number GSE210478).