

Abstract
Background
Dry eye disease (DED), arising from various etiologic factors, leads to tear film instability, ocular surface damage, and neurosensory changes. DED causes symptoms such as ocular dryness, burning, itching, pain, and visual impairment. Given their well‐established anti‐inflammatory effects, topical steroid preparations have been widely used as a short‐term treatment option for DED. Because of potential risks of ocular hypertension, cataracts, and infections associated with the long‐term use of topical steroids, published trials comparing the efficacy and safety of topical steroids (versus placebo) have mostly been of short duration (three to eight weeks).
Objectives
To evaluate the effectiveness and safety of topical corticosteroids compared with no treatment, placebo, other steroidal or non‐steroidal therapies, or a combination of therapies for DED.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL, which contains the Cochrane Eyes and Vision Trials Register; 2021, Issue 8); Ovid MEDLINE; Ovid Embase; Latin American and Caribbean Health Sciences database (LILACS); ClinicalTrials.gov; and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP), without restriction on language or year of publication. The date of the last search was 20 August 2021.
Selection criteria
We included randomized controlled trials (RCTs) in which topical corticosteroids, alone or in combination with tobramycin, were compared with no treatment, artificial tears (AT), vehicles, AT plus tobramycin, or cyclosporine A (CsA).
Data collection and analysis
We applied standard Cochrane methodology.
Main results
We identified 22 RCTs conducted in the USA, Italy, Spain, China, South Korea, and India. These RCTs reported outcome data from a total of 4169 participants with DED.
Study characteristics and risk of bias
All trials recruited adults aged 18 years or older, except one trial that enrolled children and adolescents aged between 3 and 14 years. Half of these trials involved predominantly female participants (median 79%, interquartile range [IQR] 76% to 80%). On average, each trial enrolled 86 participants (IQR 40 to 158). The treatment duration of topical steroids ranged between one week and three months; trial duration lasted between one week and six months. Eight trials were sponsored exclusively by industry, and four trials were co‐sponsored by industry and institutional or governmental funds. We assessed the risk of bias of both subjective and objective outcomes using RoB 2, finding nearly half of the trials to be at high risk of bias associated with selective outcome reporting.
Findings
Of the 22 trials, 16 evaluated effects of topical steroids, alone or in combination with tobramycin, as compared with lubricants (AT, vehicle), AT plus tobramycin, or no treatment. Corticosteroids probably have a small to moderate effect on improving patient‐reported symptoms by 0.29 standardized mean difference (SMD) (95% confidence interval [CI] 0.16 to 0.42) as compared with lubricants (moderate certainty evidence). Topical steroids also likely have a small to moderate effect on lowering corneal staining scores by 0.4 SMDs (95% CI 0.18 to 0.62) (moderate certainty evidence). However, steroids may increase tear film break‐up time (TBUT) slightly (mean difference [MD] 0.70 s, 95% CI 0.06 to 1.34; low certainty evidence) but not tear osmolarity (MD 1.60 mOsm/kg, 95% CI −10.47 to 13.67; very low certainty evidence).
Six trials examined topical steroids, either alone or in combination with CsA, against CsA alone. Low certainty evidence indicates that steroid‐based interventions may have a small to moderate effect on improving participants' symptoms (SMD −0.33, 95% CI −0.51 to −0.15), but little to no effect on corneal staining scores (SMD 0.05, 95% CI −0.25 to 0.35) as compared with CsA. The effect of topical steroids compared to CsA alone on TBUT (MD 0.37 s, 95% CI −0.13 to 0.87) or tear osmolarity (MD 5.80 mOsm/kg, 95% CI −0.94 to 12.54; loteprednol etabonate alone) is uncertain because the certainty of the evidence is low or very low. None of the included trials reported on quality of life scores.
Adverse effects
The evidence for adverse ocular effects of topical corticosteroids is very uncertain. Topical corticosteroids may increase participants' risk of intraocular pressure (IOP) elevation (risk ratio [RR] 5.96, 95% CI 1.30 to 27.38) as compared with lubricants. However, when compared with CsA, steroids alone or combined with CsA may decrease or increase IOP elevation (RR 1.45, 95% CI 0.25 to 8.33). It is also uncertain whether topical steroids may increase risk of cataract formation when compared with lubricants (RR 0.34, 95% CI 0.01 to 8.22), given the short‐term use and study duration (four weeks or less) to observe longer‐term adverse effects.
Authors' conclusions
Overall, the evidence for the specified review outcomes was of moderate to very low certainty, mostly due to high risk of bias associated with selective results reporting. For dry eye patients whose symptoms require anti‐inflammatory control, topical corticosteroids probably provide small to moderate degrees of symptom relief beyond lubricants, and may provide small to moderate degrees of symptom relief beyond CsA. However, the current evidence is less certain about the effects of steroids on improved tear film quality or quantity. The available evidence is also very uncertain regarding the adverse effects of topical corticosteroids on IOP elevation or cataract formation or progression. Future trials should generate high certainty evidence to inform physicians and patients of the optimal treatment strategies with topical corticosteroids in terms of regimen (types, formulations, dosages), duration, and its time‐dependent adverse profile.
Keywords: Adolescent; Adult; Child; Child, Preschool; Female; Humans; Male; Adrenal Cortex Hormones; Adrenal Cortex Hormones/adverse effects; Cataract; Cataract/drug therapy; Cyclosporine; Cyclosporine/adverse effects; Dry Eye Syndromes; Dry Eye Syndromes/drug therapy; Glucocorticoids; Glucocorticoids/adverse effects; Loteprednol Etabonate; Lubricant Eye Drops; Randomized Controlled Trials as Topic; Tobramycin
Plain language summary
What are the benefits and harms of topical corticosteroids for treating dry eye?
What is dry eye?
Dry eye is a common condition that occurs when a person's tears cannot lubricate their eyes sufficiently. Tears can be inadequate and unstable for many reasons. For example, dry eye may occur when tear production is reduced or when the tear quality is poor. This tear instability leads to inflammation and damage of the eye's surface. Dry eye is uncomfortable. People with dry eye often feel stinging or burning and sometimes experience blurred vision.
How is it treated?
Many treatment options are available for dry eye. For dry eye caused by the relative lack of the water layer in tears, treatments may include artificial tears, tear stimulants, serum eye drops, and punctal plugs. For dry eye caused by the blocked secretion of the lipid layer in tears, treatment options may include topical antibiotics, warm compresses, and anti‐inflammatory agents, such as corticosteroids and cyclosporine A. Corticosteroids eye drops aim to reduce the inflammatory process and provide symptom relief with short‐term use. High eye pressure and cataract formation are common concerns with longer‐term use of corticosteroids.
What did we want to find out?
We evaluated whether corticosteroids eye drops, alone or in combination with other medications, can improve dry eye symptoms or test results used to diagnose or monitor dry eye. We also examined whether corticosteroids eye drops cause any unwanted effects on the eyes.
What we did
We conducted a systematic review. We searched for studies that compared corticosteroids eye drops with lubricating controls, other active treatment, or no treatment. We summarized these study findings and rated the evidence based on numbers of study participants and methods used in the studies.
What we found
We identified 22 clinical trials that enrolled a total of 4169 participants with dry eye. Most trials involved adults with a mean age between 50 and 67 years, except for one trial that exclusively involved children aged 3 to 14 years. Treatment duration ranged between 7 days and 3 months. When compared with lubricants, such as artificial tears, or with cyclosporine A, corticosteroids eye drops were probably effective in improving patient‐reported symptoms and clinical tests, such as corneal staining. Clinicians may use corneal staining as a test for cornea damage. However, corticosteroids eye drops may result in little to no difference in tear quality or quantity. At the same time, it is uncertain whether steroid use may increase or decrease the chance of increased eye pressure, new cataract formation, or worsening of an existing cataract.
What are the limitations of the evidence?
More than half of the included trials had flawed study methods or did not report their results fully. These deficiencies led to concerns about the study findings and decreased our confidence in the evidence generated in this systematic review.
How up‐to‐date is this evidence?
The evidence is up‐to‐date as of August 2021.
Summary of findings
Summary of findings 1. Steroids compared with lubricants.
| Steroid treatment compared with lubricant (artificial tears alone or with tobramycin, vehicle, or no treatment) for dry eye | ||||||
|
Patient or population: people with dry eye Setting: eye clinics or medical centers Intervention: steroid alone (clobetasone, difluprednate, loteprednol etabonate, fluorometholone, corticosteroid) or in combination with tobramycin Comparison: artificial tears (including hyaluronate, PVP, Soothe Emollient), vehicle, no treatment, or artificial tears with tobramycin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) |
Relative effect (95% CI) |
No. of participants (studies) |
Certainty of the evidence (GRADE) |
Comments | |
|
Assumed risk Artificial tears |
Corresponding risk Steroid intervention |
|||||
|
Change in patient‐reported symptom scores (lower is favored) |
Change in symptom scores in the steroid groups was on average 0.29 SMD (95% CI 0.16 to 0.42) lower than in the artificial tears groups. |
SMD −0.29 (95% CI −0.42 to −0.16) |
3654 (15) | ⊕⊕⊕⊝ Moderate1 | As suggested in Cohen 1988, 0.2 SMD represents a small difference, and 0.5 a moderate difference. | |
| Change in patient‐reported quality of life scores | No studies measured this outcome. | ‐ | ‐ | ‐ | ||
|
Change in TBUT (seconds) (longer is favored) |
Change in TBUT in the steroid groups was on average 0.70 (95% CI 0.06 to 1.34) longer than in the artificial tears groups. |
MD 0.70 (95% CI 0.06 to 1.34) |
587 (7) | ⊕⊕⊝⊝ Low1,2 | MID 5 s (Wolffsohn 2017) | |
|
Change in fluorescein corneal staining scores (lower is favored) |
Change in fluorescein corneal staining scores in the steroid groups was on average 0.40 SMD (95% CI 0.18 to 0.62) lower than in the artificial tears groups. |
SMD −0.40 (95% CI −0.62 to −0.18) |
3583 (15) | ⊕⊕⊕⊝ Moderate1 | As suggested in Cohen 1988, 0.2 SMD represents a small difference, and 0.5 a moderate difference. | |
|
Change in tear osmolarity (mOsm/kg) (lower is favored) |
336.9 (SD 22.23) | 338.5 (SD 15.81) |
MD 1.60 (95% CI −10.47 to 13.67) |
40 (1) | ⊕⊝⊝⊝ Very low3,4 | MID 5 mOsm/L (Wolffsohn 2017) |
|
Adverse effect: incident elevated IOP (follow‐up 14 days to 2 months) |
9 incidents per 10,000 participants |
54 incidents (95% CI 12 to 246) per 10,000 participants |
RR 5.96 (95% CI 1.30 to 27.38) |
2264 (8) | ⊕⊝⊝⊝ Very low3,4 | |
|
Adverse effect: new cataract formation (follow‐up 14 days to 4 weeks) |
16 incidents per 10,000 participants |
5 incidents (95% CI 0.2 to 132) per 10,000 participants |
RR 0.34 (95% CI 0.01 to 8.22) |
1205 (3) | ⊕⊝⊝⊝ Very low3,4 | |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and the associated 95% CI). CI, confidence interval; IOP, intraocular pressure; MD, mean difference; MID, minimally important difference; mOsm, milliosmoles; PVP, polyvinylpyrrolidone; RR, risk ratio; SD, standard deviations; SMD, standardized mean difference; TBUT, tear film break‐up time | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded for risk of bias (−1). 2 Downgraded for unexplained heterogeneity (−1). 3 Downgraded for imprecision (−1). 4 Downgraded for high risk of bias (−2).
Summary of findings 2. Steroids compared with cyclosporine A.
| Steroid alone or in combination treatment compared with cyclosporine A for dry eye | ||||||
|
Patient or population: people with dry eye Setting: eye clinics or medical centers Intervention: steroid alone (fluorometholone, loteprednol etabonate, methylprednisolone) or in combination with cyclosporine A Comparison: cyclosporine A | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) |
Relative effect (95% CI) |
No. of participants (studies) |
Certainty of the evidence (GRADE) |
Comments | |
|
Assumed risk Cyclosporine A |
Corresponding risk Steroid intervention |
|||||
|
Change in patient‐reported symptom scores (lower is favored) |
Change in symptom scores in the steroid groups was on average 0.33 SMD (0.15 to 0.51) lower than in the cyclosporine A groups. |
SMD −0.33 (95% CI −0.51 to −0.15) |
465 (6) | ⊕⊕⊝⊝ Low1,2 | As suggested in Cohen 1988, 0.2 SMD represents a small difference, and 0.5 a moderate difference. | |
| Change in patient‐reported quality of life scores | No studies measured this outcome. | ‐ | ‐ | ‐ | ||
|
Change in TBUT (seconds) (longer is favored) |
Change in TBUT in the steroid groups was on average 0.37 longer (0.13 shorter to 0.87 longer) than in the cyclosporine A groups. |
MD 0.37 (95% CI −0.13 to 0.87) |
353 (5) | ⊕⊕⊝⊝ Low1,2 | MID 5 s (Wolffsohn 2017) | |
|
Change in fluorescein corneal staining scores (lower is favored) |
Change in fluorescein corneal staining scores in the steroid groups was on average 0.05 SMD higher (0.25 lower to 0.35 higher) than in the cyclosporine A groups. |
SMD 0.05 (95% CI −0.25 to 0.35) |
465 (6) | ⊕⊕⊝⊝ Low1,2 | As suggested in Cohen 1988, 0.2 SMD represents a small difference, and 0.5 a moderate difference. | |
|
Change in tear osmolarity (mOsm/kg) (lower is favored) |
1.50 lower (SD 17.33) |
LE alone: 4.30 higher (2.44 lower to 11.04 higher) |
MD 5.80 (95% CI −0.94 to 12.54) |
69 (1) | ⊕⊝⊝⊝ Very low2,3 | MID 5 mOsm/L (Wolffsohn 2017) |
|
LE + CsA: 2.20 higher (6.00 lower to 10.4 higher) |
MD 2.20 (95% CI −6.00 to 10.4) |
66 (1) | ||||
|
Adverse effect: incident elevated IOP (follow‐up 8 weeks to 6 months) |
12 incidents per 1000 participants |
17 incidents (3 to 100) per 1000 participants |
RR 1.45 (95% CI 0.25 to 8.33) |
331 (4) | ⊕⊝⊝⊝ Very low2,3 | The duration of steroid use ranged from 3 weeks to 3 months. |
|
Adverse effect: new cataract formation |
No studies measured this outcome. | ‐ | ‐ | ‐ | ||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and the associated 95% CI). CI, confidence interval; CsA, cyclosporine A; IOP, intraocular pressure; LE, loteprednol etabonate; MD, mean difference; MID, minimally important difference; mOsm, milliosmoles; RR, risk ratio; SD, standard deviations; SMD, standardized mean difference; TBUT, tear film break‐up time | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded for risk of bias (−1). 2 Downgraded for imprecision (−1). 3 Downgraded for high risk of bias (−2).
Background
Description of the condition
Dry eye disease (DED), arising from various etiologic factors, leads to tear film instability, ocular surface damage, and neurosensory changes (Bron 2017). DED causes symptoms such as ocular dryness, burning, itching, pain, and visual impairment (Messmer 2015). There was a lack of consensus in disease definition before 2017, and prevalence estimates of symptomatic DED varied widely between 5% and 50% (Stapleton 2017). In a recent cross‐sectional survey on 16 selected towns in Palestine's northern West Bank, Shanti and colleagues reported that 64% of the study population fulfilled the diagnostic criteria for DED (Shanti 2020). Yu and colleagues estimated the average annual healthcare cost for a patient with DED in the USA to be USD 783, and the overall cost of DED to the healthcare system to be USD 3840 million (Yu 2011). It is estimated that patients with DED in the UK spent USD 1.10 million (2003/2004 prices) seeking ophthalmologic care, with nearly 50% of the cost attributable to prescription drugs (Nichols 2016). Although older age and female sex are consistent risk factors for DED, the pathophysiological mechanisms underlying these correlations remain unclear (Nelson 2017). Besides environmental predispositions (low humidity, high temperature, windy conditions) (Bron 2017), other well‐characterized risk factors include prolonged screen time, contact lens wearing, androgen deficiency, medication use, and surgical and cosmetic procedures (Gomes 2017; Stapleton 2017).
To guide clinical management, DED has been categorized historically into aqueous‐deficient (due to tear insufficiency) and evaporative (due to increased tear evaporation) subtypes (Messmer 2015). Sjögren syndrome is a major underlying contributor to aqueous‐deficient dry eye. Meibomian gland diseases, including meibomian gland dysfunction (MGD) and ocular surface‐related causes, can lead to evaporative dry eye (Bron 2017).
Differentiating between aqueous‐deficient dry eye, evaporative dry eye, and a mixed mechanism comprised of both subtypes is crucial for guiding treatment plans (Bron 2017; Jones 2017). For aqueous‐deficient dry eye, treatment options comprise tear supplements, tear stimulants, and, in more severe cases, punctal plugs to preserve tears. Recent systematic reviews have demonstrated the safety and efficacy of artificial tears (Pucker 2016), but not of punctal plugs, Ervin 2017, or autologous serum eye drops (Pan 2017). For evaporative dry eye, cause‐specific therapies are available for various meibomian gland diseases, such as lid hygiene and topical antibiotics for anterior blepharitis (Jones 2017); warm compresses for meibomian gland dysfunction (Jones 2017); and anti‐inflammatory agents, such as topical corticosteroids (steroids) (Jones 2017), cyclosporine A (De Paiva 2019), and rebamipide for ocular surface inflammation (Holland 2019; Kojima 2020).
Description of the intervention
Given their well‐established anti‐inflammatory effects, topical steroid preparations have been widely used as a short‐term treatment option for DED. Several trials have shown one‐month use of topical steroid drops to improve symptoms and clinical signs (Avunduk 2003; Lee 2006; Pflugfelder 2004). The rapid onset of therapeutic effects of topical steroids could make them a useful pre‐treatment (or induction) choice before initiating long‐term cyclosporin (non‐steroidal) treatment (Byun 2012; Sheppard 2014).
How the intervention might work
Accumulating evidence has demonstrated the presence of pro‐inflammatory cytokines and T helper cells in the ocular surface regardless of DED etiologies, suggesting that ocular inflammation is a key factor in DED pathophysiology (Bron 2017). Topical steroids have been shown to exert anti‐inflammatory actions on multiple targets associated with DED symptoms and signs, including decreasing expression of cytokines, maintaining the integrity of corneal epithelium (De Paiva 2006a; De Paiva 2006b), and restoring tear production in animal models (Lekhanont 2007). In humans, topical steroids have been show to reduce pro‐inflammatory cytokines in tears (Lekhanont 2007).
Why it is important to do this review
Based on 2013 Medicare data, medications for DED ranked the second highest total costs generated by eye care (Newman‐Casey 2018). In England, DED was reportedly a major contributor to prescription costs by general practitioners in the National Health Service (Stephenson 2016). Within the ophthalmic medication group of ocular inflammation medications, prednisolone acetate was the most commonly prescribed ocular anti‐inflammatory drug by volume and cost (Newman‐Casey 2018). Despite widespread use of topical steroids clinically, significant debates about their role in DED remain. Because of potential risks of ocular hypertension, cataracts, and infections associated with the long‐term use of topical steroids, published trials comparing the efficacy and safety of topical steroids (versus placebo) in individuals with DED have mostly been of short duration (three to eight weeks) (Jones 2017). Heterogeneity in outcome measures, patient populations, and follow‐up durations, albeit short, suggests that the evidence supporting the routine use of topical steroids for DED may not be robust. In large surveys that we conducted, clinicians treating patients with dry eye prioritized the effectiveness of topical anti‐inflammatory treatments (such as corticosteroids) as the most important unanswered question (Saldanha 2017), and patients prioritized it as the third‐most important question (Saldanha 2018).
A systematic review that critically appraises the currently available data on the effects of topical steroids will provide clinicians, patients, and policymakers with robust and updated research evidence for treating DED. Along with previously published Cochrane Reviews on other local treatments for DED (De Paiva 2019; Downie 2017; Ervin 2017; Pan 2017; Pucker 2016), the current review will inform physicians of the risk‐benefit trade‐offs for prescribing topical steroids, even for short‐term use. The findings of the current review may also highlight evidence gaps and suggest potential directions for future research to address patient‐important clinical outcomes.
Objectives
To evaluate the effectiveness and safety of topical corticosteroids compared with no treatment, placebo, other steroidal or non‐steroidal therapies, or a combination of therapies for DED.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) only. We excluded within‐person studies, where eyes were randomly allocated to the intervention and comparator, because we were mostly interested in outcomes at the individual rather than the eye level.
Types of participants
We included RCTs that enrolled participants with clinically diagnosed DED regardless of etiology, or participants who reported dry eye symptoms regardless of severity. We excluded trials of patients with DED secondary to medications or medical procedures, because patients with iatrogenic DED likely shared a distinct profile of risk factors from those with primary DED.
Types of interventions
We included trials comparing topical steroids with no treatment, placebo, artificial tears, other steroidal or non‐steroidal therapy, or a combination of therapies. We planned to include trials that examined the following topical steroidal preparations.
Betamethasone
Clobetasone butyrate
Dexamethasone
Difluprednate
Fluorometholone
Loteprednol etabonate
Prednisolone
We did not require a minimum treatment frequency or duration as an eligibility criterion.
Types of outcome measures
Critical outcomes
Improvement in patient‐reported symptoms, quantified by patient questionnaires, such as the Ocular Surface Disease Index or other validated questionnaires (Schiffman 2000).
Improvement in patient‐reported general or vision‐related quality of life, measured by patient questionnaires such as the Dry Eye‐Related Quality of Life Score (DEQS).
Change in visual function, quantified as differences in reading speed using tests such as the short‐duration out‐loud reading test (Legge 1989), the 30‐minute sustained silent reading test, and the International Reading Speed Texts (IReST).
Change in tear film stability (tear film break‐up time [TBUT]).
We planned to collect outcomes at key time points that were short term (1 to 3 months), intermediate term (3 to < 6 months), or long term (≥ 6 months). All included trials had followed participants no longer than three months. For eligible studies that reported outcomes at multiple time points, we extracted outcome data, change scores, or post‐treatment measures, reported at the longest follow‐up time point.
Important outcomes
Change in ocular surface staining (Rose Bengal score/Van Bijsterveld score, fluorescein dye, or Lissamine green dye).
Proportion of participants who showed a decrease in tear osmolarity from baseline, or mean change in tear osmolarity (mOsm/kg).
Change in aqueous tear production (Schirmer test score or Jones basal secretion test).
We collected these important outcomes at the same time points as for critical outcomes.
Adverse events
We collected the proportion of participants with any ocular complication, elevated intraocular pressure (≥ 21 mmHg), new cataract formation, or delayed or impaired wound healing. Because few trials reported these specific ocular adverse events separately, we also collected total numbers of ocular and total (ocular plus systemic) adverse events documented for each comparison group as reported by the included studies. We extracted data on adverse events reported at the longest time point provided in each included RCT.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) (which contains the Cochrane Eyes and Vision Trials Register) (Issue 8, 2021), Ovid MEDLINE, Ovid MEDLINE E‐pub Ahead of Print, Ovid MEDLINE In‐Process and Other Non‐Indexed Citations, Ovid MEDLINE Daily (January 1946 to 20 August 2021), Embase (January 1947 to 20 August 2021), PubMed (1946 to 20 August 2021), Latin American and Caribbean Health Sciences Literature database (LILACS) (1982 to 20 August 2021), ClinicalTrials.gov (www.clinicaltrials.gov), and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp/search/en). We did not use any date or language restrictions in the electronic search for trials. Our last date of search was 20 August 2021.
See: Appendices for details of search strategies for CENTRAL (Appendix 1), MEDLINE (Appendix 2), Embase (Appendix 3), PubMed (Appendix 4), LILACS (Appendix 5), ClinicalTrials.gov (Appendix 6), and the WHO ICTRP (Appendix 7).
Searching other resources
We manually searched the reference lists of included studies, review articles, and guidelines for additional eligible trials, but did not identify any. We did not handsearch conference proceedings or journals, as these are included in CENTRAL.
Data collection and analysis
Selection of studies
The Information Specialist provided separate search results from the electronic databases and the trial registries. We then applied the web‐based review management software Covidence to automatically identify and remove duplicate references among the imported citations (Covidence). Two review authors worked in pairs to independently screen the titles and abstracts resulting from the searches using Covidence. Based on the eligibility criteria, each review author classified each citation as 'relevant (yes),' 'maybe relevant,' or 'not relevant (no)' for subsequent full‐text review. We then retrieved the full‐text articles for the records classified as 'relevant' or 'maybe relevant.' Two review authors worked in pairs to independently assess the full‐text records for eligibility as described in Criteria for considering studies for this review. Any disagreements were resolved by discussion.
We also contacted the investigators of potentially eligible studies to request additional information to determine the eligibility of studies as needed. If the study authors did not respond within two weeks, we used the information available from publications and trial registries to determine eligibility whenever feasible. We listed all excluded studies with the reasons for their exclusion in Characteristics of excluded studies. Regarding eligible studies identified on trials registers, we included any such studies in the review irrespective of whether we could identify or access published or unpublished data. In particular, we classified eligible trials as 'awaiting classification' if the trials were completed but no study results were publicly available, or 'ongoing' if the trials were not yet completed. Any discrepancies were resolved by discussion within the review author team.
Data extraction and management
One review author (SL) extracted data using an online structured form developed by Cochrane Eyes and Vision (Covidence), and a second review author (TR) independently verified the data entered into Covidence. We contacted trial investigators or sponsors for missing data. If the trial investigators or sponsors did not respond within two weeks, we extracted the relevant data available to us from trials registers or clinical study reports and other regulatory documents. We imported adjudicated data into Review Manager Web (RevMan Web 2022), which was again verified by a second review author for accuracy.
We extracted the following information from each included study: trial setting, countries where participants were recruited, sample size, study duration, and other trial‐level characteristics; participants' composition of age, sex, or major medical comorbidities; outcome data and adverse events. We collected continuous variables as mean, standard deviation or the associated 95% confidence intervals (95% CI), and dichotomous variables as number of participants for which the outcome was measured. In some studies, numerical data were only available in figures, from which we applied a free, web‐based software to extract outcome data for meta‐analysis (WebPlotDigitizer), as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Li 2021). For multi‐arm studies, we only collected data relevant to our intervention and comparator groups. If two groups contained relevant data, we combined the groups using the calculator within Review Manager Web, or included each group in relevant meta‐analyses separately in order to avoid double counting the comparator group.
Assessment of risk of bias in included studies
Two review authors (SL and TR) independently assessed risk of bias using Cochrane's RoB 2 tool for two critical outcomes (Higgins 2021a). As prespecified in the protocol, we chose to apply the RoB 2 tool to patient‐reported symptom scores and corneal fluorescein staining scores, as these two outcomes were the most frequently reported outcomes in the included trials. Any disagreements on the risk of bias assessments were resolved via discussion within the review author team.
We specifically considered and reported on the following domains.
Bias arising from the randomization process
Bias introduced by deviations from intended interventions
Bias due to missing outcome data
Bias in outcome measurement
Bias in selective reporting of outcome data
We judged each domain for each study as low risk of bias, high risk of bias, or some concerns as guided by signaling questions in each domain. Overall, we assessed each trial as having:
'low risk of bias' if all domains were judged to be at low risk;
'some concerns' if one or more domains were judged to be with some concerns, and none were at high risk;
'high risk of bias' if one or more domains were considered as at high risk, or if multiple domains were judged to be with some concerns such that we had low confidence in the validity of the reported findings (Higgins 2021a).
Measures of treatment effect
We calculated mean differences (MD) with 95% CI for continuous outcomes, and risk ratios (RR) with 95% CI for dichotomous outcomes. Where possible, we checked for the skewness of continuous data (Altman 1996). We used the standardized mean difference (SMD) for patient‐reported symptom scores and corneal staining scores because not every trial utilized the same symptom questionnaire or staining scoring system. Interpretation of treatment effects expressed in units of SMD, such as patient‐reported symptoms scores and fluorescein corneal staining scores, may follow the rule of thumb as suggested by Cohen, which considers an SMD of 0.2 a small effect, 0.5 a moderate effect, and 0.8 a large effect (Cohen 1988).
Unit of analysis issues
In trials where individuals were randomly allocated to treatment, but only one eye per person was included in the trial, we documented how the eye was selected or how the investigators decided from which eye to report data. If participants were randomly allocated to treatment, and both eyes were included but reported separately, we chose to collect and analyze outcome data for the right eye, rather than analyzing data of both eyes as planned in the protocol (Differences between protocol and review). We excluded studies that allocated different eyes to different treatments, as there might be cross‐over effects due to systemic absorption or biased reporting of patient‐reported symptom scores for each eye.
Dealing with missing data
We planned to use imputed data if computed by the trial investigators using an appropriate method; we did not plan to impute missing data ourselves. We contacted the trial investigators and requested clarification or missing information when the trial publication did not include outcome data for all randomized participants. If we did not hear back from the trial investigators within two weeks, we proceeded by conducting a complete‐case analysis, assuming that the data were missing completely at random (Bhaskaran 2014). We assessed whether this assumption was reasonable by collecting data from each included trial on the number of participants excluded or lost to follow‐up and the reasons for loss to follow‐up by treatment group, if reported.
Assessment of heterogeneity
We examined the overall characteristics of the studies, in particular the types of participants, types of interventions, and study design, to assess the extent to which the studies were sufficiently similar to permit a meaningful meta‐analysis for a given outcome. We considered the size and direction of intervention effects and took into account the amount of heterogeneity as quantified by the I² statistic (Higgins 2002). As suggested in Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2021), we used the following thresholds to interpret I² values:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
We assessed selective outcome reporting for each included trial as guided by relevant signaling questions in the RoB 2 tool (Higgins 2021a). In the protocol development stage, we planned to evaluate potential risk of bias arising from non‐reporting (missing evidence) for the critical outcomes using the Risk of Bias due to Missing Evidence tool (ROB‐ME, Page 2021). However, because the tool is still in its preliminary version, we decided not to perform this assessment for the current version of the review. We will assess the tool's availability in future updates of this review.
Data synthesis
In addition to qualitative synthesis of the included trials, we combined data using a random‐effects model as default if there were three or more trials reporting on the same outcome. When we judged the evidence as having considerable clinical, methodological, or statistical heterogeneity, we did not combine the data in a meta‐analysis but instead described the data qualitatively.
Subgroup analysis and investigation of heterogeneity
We planned that when there were sufficient trials (> 10), we would conduct subgroup analysis on critical outcomes by sex and etiology of dry eye (Sjögren syndrome, non‐Sjögren syndrome, meibomian gland dysfunction), separately. Because few studies reported on TBUT, we performed post hoc subgroup analysis on fluorescein staining results by etiology of dry eye (Differences between protocol and review). To explore potential sources of heterogeneity, we also performed additional post hoc subgroup analyses on patient‐reported symptoms and fluorescein staining scores by scoring system, source of trial funding, and intervention regimen (Differences between protocol and review).
Sensitivity analysis
We performed sensitivity analyses for each critical outcome by excluding trials at high risk of bias for that particular outcome and by excluding industry‐controlled studies.
Summary of findings and assessment of the certainty of the evidence
We prepared summary of findings tables presenting relative or absolute risks (Schünemann 2019). Two review authors independently graded the overall certainty of the evidence for each of the following outcomes using the GRADE approach (Schünemann 2013).
Improvement in patient‐reported symptom scores
Improvement in patient‐reported general or vision‐related quality of life scores
Improvement in TBUT
Improvement in ocular surface staining
Improvement in tear osmolarity
Ocular adverse event: incident elevated intraocular pressure (IOP) ≥ 21 mmHg
Ocular adverse event: new cataract formation
We graded the certainty of the evidence as 'high,' 'moderate,' 'low,' or 'very low' according to (1) risk of bias among the included trials; (2) indirectness of evidence; (3) unexplained heterogeneity or inconsistency of results; (4) low precision of results; and (5) risk of publication bias (Schünemann 2013). Any discrepancies between the two review authors were resolved by discussion.
Results
Description of studies
Results of the search
We searched the electronic databases in August 2021, identifying 5143 records (5127 studies) after removal of duplicates. After excluding 5073 irrelevant records, we screened 54 full‐text reports and included 22 studies in the review (Figure 1). We excluded 21 studies, with reasons for their exclusion reported in Characteristics of excluded studies, and assessed 4 studies as 'ongoing' (CTRI/2021/02/031182; ISRCTN16288419; NCT04734197; NCT04734210), and 7 studies as 'awaiting classification' (ChiCTR‐IPR‐15007196; Herman 2005; NCT00471419; NCT00560638; NCT01562795; NCT03418727; NTR2291).
1.
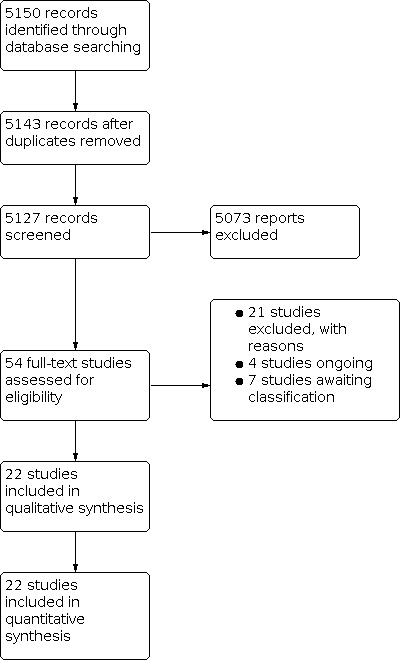
Study flow diagram.
Included studies
Types of studies
All 22 included RCTs had a parallel‐group design and compared topical corticosteroids alone or in combination versus lubricants or another pharmacological intervention in participants with dry eye. Eleven trials were conducted in the USA, nine in Asia (six in China, two in South Korea, one in India), and two in Europe (one each in Spain and Italy). The trials were published between 2003 and 2021. Twelve trials were registered on trial registries, but protocols were publicly available for only five trials (Akhlaq 2019; KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3)). Eleven trials provided power or sample size calculations for at least one study outcome.
Seven trials were multisite (KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); NCT01276223; Pflugfelder 2004; Sheppard 2014), while the rest were single‐site trials conducted in university‐affiliated medical centers. Eight trials had pharmaceutical sponsorship as the sole source of funding (36%) (Akhlaq 2019; Bausch 2013; KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); NCT01276223; Pflugfelder 2004). Another four trials reported dual funding sources from the industry and the affiliated institution or the government (18%) (Aragona 2013; Pinto‐Fraga 2016; Qazi 2015; Sheppard 2014). Five trials were supported by either the investigators' affiliated institution, Byun 2012, or government funding (Avunduk 2003; Chen 2020; Lee 2014; Wan 2012). Authors of another four trials did not disclose any financial support (Cao 2018; Li 2021; Luo 2013; Singla 2019). Lin and colleagues reported receiving no funding and disclosed no conflicts of interest (Lin 2015).
Eighteen trials (82%) randomized the interventions at the participant level and reported the outcomes as such. Four trials randomized participants to the interventions but reported findings exclusively at the eye level (Li 2021; Sheppard 2014; Singla 2019; Wan 2012). Most trials randomized participants into two groups, except for four trials that randomized participants into three or more groups (Avunduk 2003; Bausch 2013; Luo 2013; Qazi 2015). Corticosteroid treatment durations ranged between one week and three months, and follow‐up durations was generally short, ranging from one week to six months.
For details on the included trials, see Characteristics of included studies.
Types of participants
The 22 included trials reported data for a total of 4169 participants, excluding those randomized to treatments that were irrelevant to the current review. Two trials did not report numbers of participants initially randomized, overall, or for each comparator group, but only reported those included for outcomes analyzed (Avunduk 2003; Sheppard 2014). On average, each trial enrolled (or reported data for) a median number of 86 participants (interquartile range [IQR] 40 to 158). All trials but one recruited adult participants aged 18 years or older, with the majority of trials enrolling middle‐aged participants with mean ages ranging between 50 and 67 years. The exception was a trial that exclusively recruited children and adolescents aged between 3 and 14 years (Cao 2018).
In seven trials, both males and females were nearly equally represented, with females consisting of between 55% and 64% of trial populations (Avunduk 2003; Cao 2018; Lee 2014; Li 2021; Qazi 2015; Singla 2019; Wan 2012). However, in 11 trials, the investigators enrolled predominantly female participants (median 79%, IQR 76% to 80%; maximum 95%). Authors of another two trials did not report on sex distribution of the study participants (Akhlaq 2019; Lin 2015).
Five trials exclusively enrolled participants with Sjögren's, Aragona 2013; Lin 2015, or meibomian gland dysfunction (Lee 2014; Luo 2013; Qazi 2015); the other 17 trials did not report on the underlying etiologies of dry eye.
Types of interventions
Topical corticosteroids were used as stand‐alone or combination interventions in 18 and 4 trials, respectively. Treatment duration of the corticosteroid intervention ranged from one week to three months.
Of the 18 trials that evaluated corticosteroids alone, 12 used loteprednol etabonate (LE) 0.1% (Chen 2020), 0.25% (KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3)), or 0.5% (Akhlaq 2019; Bausch 2013; Lee 2014; Pflugfelder 2004; Qazi 2015; Wan 2012). Another trial also compared LE with hyaluronic acid (HA), but the authors did not report the drug concentration (Cao 2018). Six trials evaluated difluprednate 0.05% (Durezol) (NCT01276223), clobetasone butyrate (CB) 0.1% (Aragona 2013), or fluorometholone (FML) 0.1% (Li 2021; Lin 2015; Pinto‐Fraga 2016). In Avunduk 2003, the intervention group also received FML, but its concentration was not reported.
Four trials evaluated topical corticosteroids in combination with either cyclosporine A (CsA) 0.05%, Byun 2012; Sheppard 2014; Singla 2019, or tobramycin (Luo 2013), in comparison with CsA. Corticosteroids in these combination interventions were LE 0.5% (Sheppard 2014; Singla 2019), methylprednisolone (MP) 1% (Byun 2012), and dexamethasone (DEXA, unspecified concentration) (Luo 2013). In addition to one intervention group with LE 0.5% treatment alone, investigators in two of the four three‐arm trials also compared the combination effects of LE 0.5% plus CsA 0.05%, Bausch 2013, or LE 0.5% plus tobramycin (Qazi 2015). In four trials that combined topical corticosteroid with CsA, two trials evaluated the benefits of pre‐treatment with corticosteroid for two weeks before initiating CsA treatment (Bausch 2013; Sheppard 2014); the other two trials evaluated the effects of concurrent initiation of topical corticosteroid and CsA treatment (Byun 2012; Singla 2019).
Comparator interventions included no treatment (Lee 2014), lubricating solutions, emollients, or gels, or another active therapy. We did not identify any true placebo‐controlled trials when reviewing the literature, and we use 'lubricant' to reference all inert comparators that did not exhibit anti‐inflammatory effects. These lubricants consisted of artificial tears (AT) in various formulations (Avunduk 2003; Qazi 2015), emollients (Akhlaq 2019), HA (Cao 2018; Chen 2020; Li 2021), and vehicle (Aragona 2013; KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); NCT01276223; Pflugfelder 2004; Pinto‐Fraga 2016). We also considered a combination of AT and tobramycin as equivalent to a lubricating control when compared with DEXA plus tobramycin (Luo 2013).
Active therapies consisted of mostly topical CsA 0.5% (Bausch 2013; Byun 2012; Lin 2015; Sheppard 2014; Singla 2019; Wan 2012). In most active‐controlled trials, participants in both groups were allowed to apply lubricants of the same formulation to study eyes during the study period, except in one study where participants in the comparison group received only CsA 0.5% (Byun 2012).
Types of outcomes
We planned to evaluate the effects of topical corticosteroid on four critical outcomes and three important outcomes (Liu 2021). However, none of the included trials described patient‐reported quality of life or change in visual function, two of our prespecified critical outcomes.
All trial investigators randomized and reported at the individual level, except for three trials that reported at the eye level (Sheppard 2014; Singla 2019; Wan 2012); we included only data from the right eyes in data analysis from these three trials. Another two study teams prespecified how they selected study eyes for outcome reporting (right eyes or the worst eyes) (Chen 2020; Lee 2014). In two trials, the authors did not specify the unit of analysis for outcomes reported (Cao 2018; Luo 2013); post hoc Student t‐test results suggested that the unit of analysis might be eye, and the comparisons had accounted for within‐person correlation.
Critical outcomes
Change in patient‐reported symptom scores
Twenty‐one trials reported changes in patient‐reported symptoms; the remaining trial only reported changes in ocular surface signs (Cao 2018). Change scores were provided by 6 of the 21 trials (Bausch 2013; KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); NCT01276223; Pflugfelder 2004). These change scores were analyzed together with post‐treatment symptom scores from the other 15 trials, as suggested in Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b).
Thirteen trials used the Ocular Surface Disease Index (OSDI) (Akhlaq 2019; Bausch 2013; KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); Lee 2014; Li 2021; Lin 2015; Pinto‐Fraga 2016; Qazi 2015; Sheppard 2014; Singla 2019), in which participants were asked to rate different aspects of their symptom severity on a scale of 0 to 4; the total score ranged from 0 to 100 (Schiffman 2000).
Aragona 2013 and Pflugfelder 2004 applied visual analogue scales (VAS, 0 to 100 points) to document participants' symptoms: burning/stinging, itching, grittiness/scratchiness/foreign body sensation, photophobia/blurred vision, sticky eye, and dryness/tired eye sensation. In a separate trial (NCT01276223), the investigators also used VAS to record symptom severity and frequency, scored each from 0 to 100, and reported the composite score ranging up to 200.
Chen 2020 used the Standard Patient Evaluation of Eye Dryness (SPEED) Questionnaire, an 8‐item questionnaire with scores of 0 to 3 for each item (Ngo 2013), but the authors assessed only dryness, foreign body sensation, burning sensation, and eye irritation, with a maximum score of 12.
Avunduk 2003 used the Dry Eye Screening Questionnaire (DESQ), a 14‐item instrument (each with a 4‐point scale) developed for screening purposes (Oden 1998).
Three trials did not report on the specific questionnaires used for symptoms (Byun 2012; Luo 2013; Wan 2012).
Improvement in patient‐reported general or vision‐related quality of life
None of the included trials measured or reported this outcome.
Change in visual function
None of the included trials measured or reported this outcome.
Change in tear film break‐up time
Twelve trials reported on changes in tear break‐up time (TBUT). Ten trials measured changes in TBUT with fluorescein dye instilled (Aragona 2013; Byun 2012; Chen 2020; Lee 2014; Lin 2015; Luo 2013; Pinto‐Fraga 2016; Qazi 2015; Singla 2019; Wan 2012). In two trials, the investigators applied a novel, non‐invasive technique for measuring TBUT (non‐invasive keratograph break‐up time [NIKBUT]) alone ( Cao 2018) or along with the routine fluorescein dye instillation (Li 2021).
Important outcomes
Change in ocular surface staining
Twenty trials reported on changes in corneal staining, and eight trials reported on changes in conjunctival staining. We chose to report corneal staining data to assess corticosteroid effects on changes in ocular surface staining. Eleven of the 20 trials that implemented corneal staining exam followed the National Eye Institute (NEI) scoring scheme (Akhlaq 2019; Aragona 2013; Avunduk 2003; Bausch 2013; Byun 2012; KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); Sheppard 2014; Singla 2019), and reported an overall score ranging from 0 to 15 (Lemp 1995). Lin 2015 used a modified scoring system based on a previously published grading system (Macri 2000; Qiu 2011).
Pinto‐Fraga 2016 applied two scoring schemes to grade the corneal staining image (Bron 2003; Jones 2002); we chose to extract and include staining scores based on the Oxford system (Jones 2002).
Change in tear osmolarity
Only two trials reported on tear osmolarity (Bausch 2013; Pinto‐Fraga 2016). Bausch 2013 reported on changes in tear osmolarity of the worse eye between baseline and 12 weeks of intervention. Pinto‐Fraga 2016 reported on changes in tear osmolarity from baseline to 21 days after treatment with FML 0.1% or the vehicle, right before participants were to be exposed to adverse controlled environment (Pinto‐Fraga 2016).
Change in aqueous tear production
Nine trials used the Schirmer's test, without anesthesia (Byun 2012; Chen 2020; Li 2021; Lin 2015; Luo 2013; Pinto‐Fraga 2016; Sheppard 2014; Singla 2019; Wan 2012), while one trial used it with anesthesia (Qazi 2015). One trial did not report numeric results, only stating that "no significant change in the mean Schirmer test score was observed in either group" (Lin 2015).
Adverse effects
We prespecified four adverse effects of interest: ocular complications, elevated IOP, new cataract formation, and delayed or impaired wound healing (Liu 2021). Five trials reported on 'any' systemic or ocular adverse events, as well as specific ocular adverse events, such as elevated IOP (KPI‐121 (Phase 2); KPI‐121 (STRIDE1); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); NCT01276223). Six other trials reported specifically on IOP elevation, Bausch 2013; Byun 2012; Lin 2015; Pflugfelder 2004; Sheppard 2014, or new cataracts (KPI‐121 (STRIDE3); Pflugfelder 2004). Only two trials reported on delayed or impaired wound healing (Pinto‐Fraga 2016; Sheppard 2014).
Excluded studies
After screening 54 full‐text reports or trial registry records, we excluded 21 studies, with reasons for their exclusion provided in Characteristics of excluded studies. Among these 21 studies, 16 had an ineligible study design; two enrolled ineligible patient populations; two examined ineligible interventions; and one was terminated before participant recruitment.
Ongoing studies and studies awaiting classification
Four trials, registered between February 2020 and May 2021 on trial registries (CTRI/2021/02/031182; ISRCTN16288419; NCT04734197; NCT04734210), are still recruiting participants. We assessed a further six trials as awaiting classification because, to our knowledge, no published results are available despite their being completed (ChiCTR‐IPR‐15007196; NCT00471419; NCT00560638; NCT01562795; NCT03418727; NTR2291). We contacted these trial investigators (or the sponsoring companies) at least twice, but have not received any response. We also assessed Herman 2005 as awaiting classification because the findings were available only in a conference abstract, with no details about the sample size of the comparison group; the first author has not yet responded to our inquiries.
Risk of bias in included studies
We applied the RoB 2 tool to assess two critical outcomes: patient‐reported symptoms (Figure 2) and corneal fluorescein staining (Figure 3). Twenty‐one trials (95% of the 22 included trials) reported either outcome. Domain‐specific judgements and the supporting statements for each study are provided in Characteristics of included studies. For both outcomes, we judged two trials to be at low risk of bias across all the domains assessed (KPI‐121 (Phase 2); KPI‐121 (STRIDE1)); we also judged a third trial to have a low risk of bias for the objective outcome, but not for patient‐reported symptoms, due to concerns regarding selective reporting of trial results (KPI‐121 (STRIDE2)). We judged the other trials as having some concerns or high risk of bias.
2.

Risk of bias summary: review authors' judgements about each risk of bias domain for each included trial that reported patient‐reported symptom scores.
3.

Risk of bias summary: review authors' judgements about each risk of bias domain for each included trial reporting corneal fluorescein staining results.
Domain 1: Bias arising from the randomization process
Change in patient‐reported symptom scores, change in corneal fluorescein staining scores
For both outcomes, seven of the 21 trials adequately described the process of random number generation and whether the allocation was concealed before assigning participants (33%), and were thus considered to be at low risk of bias. In the other 14 trials (67%), study authors simply mentioned randomization, but did not provide sufficient details to permit a risk of bias assessment, therefore we categorized these trials as having some concerns for this domain.
Domain 2: Bias arising from deviations from intended interventions
Change in patient‐reported symptom scores, change in corneal fluorescein staining scores
We judged Singla 2019 to have high risk of bias because it was unclear whether the authors applied intention‐to‐treat analysis to estimate the effect of assignment. Six trials did not report whether trial participants or assessors were masked, therefore we had some concerns about potential biases associated with deviating from the intended interventions. We also had some concerns in one of the three‐arm trials, Qazi 2015, regarding how participants were masked because of inconsistent reporting in the trial report and on ClinicalTrials.gov. We assessed the remaining 13 trials (62%) and 12 trials (57%) as at low risk of bias for patient‐reported symptom scores and change in corneal fluorescein staining scores, respectively.
Domain 3: Bias due to missing outcome data
Change in patient‐reported symptom scores, change in corneal fluorescein staining scores
In 18 of the 21 trials (86%) that reported symptoms or corneal staining scores, study authors reported data for both outcomes for all or nearly all participants initially randomized to the intervention and comparator treatments. For example, Sheppard and colleagues initially randomized 116 participants into two comparison groups and reported data for both outcomes in 112 individuals (Sheppard 2014). The authors did not specify the numbers of participants randomized or lost to follow‐up in each group, but we judged that the bias associated with missing data would be minimal, and therefore assessed the study as at low risk of bias, similar to the other 17 trials.
In contrast, we assessed two trials with small or moderate sample sizes as having some concerns associated with missing outcome data, because the authors reported that four participants in total were withdrawn from the study (Avunduk 2003, N = 19), or an unknown number of participants who did not complete the study visits were excluded from data analysis (Singla 2019). We also judged one additional trial to have some concerns for this domain because of inconsistent reporting between the full‐text publication and the conference abstract (Byun 2012).
Domain 4: Bias in outcome measurement
Change in patient‐reported symptom scores
We judged nine trials (43%) that were open‐label (Lin 2015), unmasked to participants (Avunduk 2003; Lee 2014), or unclear about the masking status of participants (Byun 2012; Chen 2020; Li 2021; Luo 2013; Singla 2019; Wan 2012), as at high risk of bias in measuring this outcome. In comparison, we considered the other 13 trials as at low risk of bias because participants were masked to the treatment received.
Change in corneal fluorescein staining scores
The experience of clinical authors of this review suggests that grading of the corneal staining images could be subjective and might be influenced by the knowledge of the intervention. As such, we judged one open‐label trial, Lin 2015, and nine trials that did not provide masking information about the examiners or assessors who performed or graded the corneal staining images, Aragona 2013; Byun 2012; Cao 2018; Chen 2020; Li 2021; Luo 2013; Pflugfelder 2004; Singla 2019; Wan 2012, as having high risk of bias in measuring or assessing this outcome. Although there was no statistical evidence that the outcome assessment was differentially influenced by the open‐label design in Bausch 2013, we still considered this study as having some concerns related to risk of bias in measuring this outcome. We judged the other six trials as at low risk of bias for this domain.
Domain 5: Bias in selective reporting of outcome data
Change in patient‐reported symptom scores
In seven trials, the investigators analyzed and reported this outcome as prespecified in the study protocol or as one of the outcome variables considered in sample size calculations (Aragona 2013; KPI‐121 (STRIDE1); NCT01276223; Pflugfelder 2004; Pinto‐Fraga 2016), or in a standard manner as described in the methods section (KPI‐121 (Phase 2); Luo 2013); we judged these trials as at low risk of bias for selective outcome reporting.
We considered five trials as having some concerns due to inconsistent reporting of outcome metrics with what was specified in the study protocol, the analytic plan, or the trial registry records (Akhlaq 2019; KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); Lin 2015; Qazi 2015). We had some concerns with another five trials because no prespecified analytic plans were available for assessment (Bausch 2013; Byun 2012; Chen 2020; Singla 2019; Wan 2012).
We judged four trials as at high risk of bias due to apparent deviations from the analytic plan (Avunduk 2003; Lee 2014; Li 2021; Sheppard 2014).
Change in corneal fluorescein staining scores
Two trials reported this outcome as planned and were thus judged to be at low risk of bias for this domain (Lin 2015; Pinto‐Fraga 2016). We judged two trials as having some concerns related to risk bias because of incomplete reporting (Avunduk 2003; Pflugfelder 2004), along with another six trials that did not provide access to protocols for evaluation (Akhlaq 2019; Aragona 2013; Bausch 2013; Byun 2012; Chen 2020; Qazi 2015).
We considered three trials as at high risk of bias because of deviations from their planned analysis (Lee 2014; Li 2021; Sheppard 2014). We also considered Cao 2018 and Luo 2013 to have high risk of bias for selective outcome reporting because of incomplete reporting.
Overall assessment of bias
Change in patient‐reported symptom scores
For this subjective outcome, we judged 10 trials (48%) as at high risk of bias, mostly because of potential risks associated with biased measurement or selective reporting (Figure 2). We considered another nine trials (43%) to have some concerns associated with the process of allocation concealment or selective outcome reporting. Overall, selective outcome reporting was a major source of bias for trials that had reported this subjective outcome.
Change in corneal fluorescein staining scores
For this outcome, we judged 10 trials (48%) as having a high risk of bias, potentially due to biased measurement or selective reporting of results (Figure 3). Singla 2019 was the only trial that we judged to be at high risk of potential deviations from the intended intervention. We deemed another eight trials (38%) as having some concerns in the randomization process or outcome reporting. In summary, corneal staining scores reported by the included trials also shared a similar risk of bias profile to that of the subjective outcome, with selective outcome reporting being the most common source of potential bias.
Effects of interventions
Sixteen of the 22 included trials (73%) evaluated the effectiveness and safety of topical corticosteroid, alone or in combination therapy, with a lubricant‐like control treatment, such as AT, HA, vehicle, or no treatment (Table 1). We also included Luo 2013, which compared DEXA plus tobramycin with AT plus tobramycin, in Comparison 1. In Comparison 2, we included six trials that compared corticosteroid with CsA (Table 2).
Comparison 1: topical corticosteroids versus lubricants
Critical outcomes
Change in patient‐reported symptom scores
A total of 15 trials comparing topical corticosteroid alone or in combination with tobramycin measured patient‐reported symptoms (Analysis 1.1). One three‐arm study contributed data of two interventions, LE 0.5% alone and LE 0.5% plus tobramycin, separately, to this comparison (Qazi 2015).
1.1. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 1: Patient‐reported symptom scores—Qazi: LE alone
When including LE data from the three‐arm trial, Qazi 2015, topical corticosteroids alone or with tobramycin probably improve patient‐reported symptom by reducing 0.29 standardized mean difference (SMD) (95% confidence interval [CI] 0.16 to 0.42 ; n = 3654) relative to lubricants (I2 = 62%, P < 0.001) (Figure 4). Based on estimates obtained from the subgroup that applied the OSDI scale (9 trials, n = 3122), steroids may reduce 4.0 out of 100 points (95% CI 1.6 to 6.4) on the OSDI scale when compared with lubricants (Analysis 1.2). Results were similar when the analysis included the LE + tobramycin data from Qazi 2015 (Analysis 1.3; Analysis 1.4) or excluded a trial that had reported at eye level (Li 2021). A planned subgroup analysis by etiology found no statistical support for differential impacts of steroids on symptoms in study participants with Sjögren syndrome, MGD, or mixed etiologies (P = 0.32) (Analysis 1.5; Figure 5).
4.

Forest plot of comparison 1: Steroids versus lubricants, outcome: 1.1 Patient‐reported symptom scores. The analysis included data of the LE group in Qazi 2015.
1.2. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 2: Patient‐reported symptom scores—by questionnaire, Qazi: LE alone
1.3. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 3: Patient‐reported symptom scores—Qazi: LE + tobramycin
1.4. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 4: Patient‐reported symptom scores—by questionnaire, Qazi: LE + tobramycin
1.5. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 5: Patient‐reported symptom scores—by etiology
5.

Forest plot of comparison 1: Steroids versus lubricants, outcome: 1.5 Patient‐reported symptom scores, with subgroup analysis by etiology of dry eye. The analysis included data of the LE group in Qazi 2015.
In contrast, we identified substantial subgroup differences in the estimated steroid effects by quality of the trials (P < 0.001, I2 = 93.6%) (Analysis 1.6) and source of trial sponsorship (P < 0.001, I2 = 89.8%) (Analysis 1.7). The combined effect of corticosteroids on symptoms was reduced by 34% when excluding trials that were judged to be at high risk of bias (SMD −0.17, 95% CI −0.27 to −0.07; 10 trials, n = 3243) (Analysis 1.6). Trial quality and source of funding were closely related with each other (Analysis 1.7). Additional analysis by steroid structure (Analysis 1.8) or treatment duration (Analysis 1.9) found no evidence for subgroup differences.
1.6. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 6: Patient‐reported symptom scores—by risk of bias
1.7. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 7: Patient‐reported symptom scores—by sponsorship
1.8. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 8: Patient‐reported symptom scores—by regimen and steroid type
1.9. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 9: Patient‐reported symptom scores—by steroid treatment duration
We judged the certainty of the evidence for this outcome to be moderate, downgrading for associated risk of bias in outcome measurement and selective reporting (−1).
Change in patient‐reported quality of life scores
None of the included trials measured this outcome.
Change in visual function
None of the included trials measured this outcome.
Change in tear film break‐up time
Investigators of seven trials measured TBUT using the conventional invasive technique (Aragona 2013; Cao 2018; Chen 2020; Lee 2014; Li 2021; Pinto‐Fraga 2016; Qazi 2015). The combined estimate from 587 participants suggested that steroids may slightly increase TBUT by 0.70 seconds (95% CI 0.06 to 1.34) as compared with lubricants (I2 = 79%, P < 0.001) (Analysis 1.10).
1.10. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 10: Tear film break‐up time—Qazin: LE alone
Estimates were similar when including the LE + tobramycin data of Qazi 2015 or the NIKBUT data reported by Li 2021 (Analysis 1.11). Post hoc subgroup analysis by structure of steroids suggested no differential effects by structural variants of topical steroids (P = 0.94, I2 = 0%) (Analysis 1.12).
1.11. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 11: Tear film break‐up time—Qazi: LE + tobramycin
1.12. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 12: Tear film break‐up time—by steroid type
We consider the certainty of the evidence as low for this outcome because of potential risk of bias in the randomization process (−1) and unexplained heterogeneity (−1).
Important outcomes
Change in fluorescein corneal staining scores
Of the 16 trials comparing corticosteroid, alone or with tobramycin, with lubricants, 15 trials measured corneal staining scores at one or more postintervention visits (Analysis 1.13). Based on data from 3583 participants, topical steroids probably improve corneal staining scores by 0.40 SMD (95% CI 0.18 to 0.62 ) as compared with lubricants (I2 = 87%, P < 0.001) (Figure 6). The estimated effect was reduced by 70% in a subgroup of trials that used the NEI grading scheme (mean difference [MD] −0.12, 95% CI −0.19 to −0.04) (Analysis 1.14).
1.13. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 13: Corneal fluorescein staining scores—Qazi: LE alone
6.

Forest plot of comparison 1: Steroids versus lubricants, outcome: 1.13 Corneal fluorescein staining scores. The analysis included data of the LE group in Qazi 2015.
1.14. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 14: Corneal fluorescein staining score—by grading system, Qazi: LE alone
Including the combined intervention data of Qazi 2015 (Analysis 1.15; Analysis 1.16), or excluding eye‐level data (Li 2021), did not alter the results. There were no significant subgroup differences in subgroup analysis by etiology (Figure 7), risk of bias assessment results (Analysis 1.18), source of sponsorship (Analysis 1.19), or treatment duration (Analysis 1.20).
1.15. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 15: Corneal fluorescein staining score—Qazi: LE + tobramycin
1.16. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 16: Corneal fluorescein staining score—by grading system, Qazi: LE + tobramycin
7.

Forest plot of comparison 1: Steroids versus lubricants, outcome: 1.17 Corneal fluorescein staining scores, with subgroup analysis by etiology of dry eye. The analysis included data of the LE group in Qazi 2015.
1.18. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 18: Corneal fluorescein staining score—by risk of bias
1.19. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 19: Corneal fluorescein staining score—by sponsorship
1.20. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 20: Corneal fluorescein staining score—by steroid treatment duration
Overall, we judged the certainty of the evidence for this outcome as moderate after considering the associated risk of bias in outcome measurement and selective reporting (−1).
Change in tear osmolarity
Pinto‐Fraga 2016 was the only trial to report this outcome. The single study estimate suggested that corticosteroids may decrease or increase tear osmolarity (MD 1.60, 95% CI −10.47 to 13.67 mOsm/kg) after 21‐day treatment but before the study participants (n = 40) were exposed to adverse controlled environment (ACE) exposure (Analysis 1.21). The certainty of the evidence for this outcome estimate was very low because of risk of bias (−2) and imprecision (−1).
1.21. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 21: Tear osmolarity
Change in Schirmer's test scores
Only five of the 16 trials reported results of Schirmer's test, with or without anesthesia, before and after 21 to 30 days of corticosteroid treatment (Analysis 1.22). Based on the combined estimate, the evidence was very uncertain regarding the effect of steroids on Schirmer's test results (MD 0.94 mm, 95% CI 0.39 to 1.49) when compared with lubricants (n = 425; I2 = 41%, P = 0.15). However, sensitivity analysis that included the LE + tobramycin data of Qazi 2015 showed that topical steroids might have little or no effect on tear production (MD 0.69, 95% CI −0.02 to 1.39). Whether LE exerted a greater effect on improving Schirmer's test than FML when compared with lubricants also varied by which intervention data (LE versus LE + tobramycin) of Qazi 2015 were being considered (Analysis 1.24). As such, we judged the certainty of the evidence for this outcome to be very low because of inconsistency (−1), risk of bias associated with the randomization process (−1), and imprecision due to the small sample size of this one trial (−1).
1.22. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 22: Schirmer's test—Qazi: LE alone
1.24. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 24: Schirmer test—by steroid type, Qazi: LE alone
Adverse effects
Proportion of participants with elevated IOP
Combining data reported by eight of the 16 trials on the occurrence of elevated IOP, the estimate suggested that steroids may increase the risk of elevated IOP by nearly five‐fold when compared with lubricants (risk ratio [RR] 5.96, 95% CI 1.30 to 27.38; n = 2264); however, the evidence was very uncertain. Results of subgroup analysis by steroid structure did not support differential risks by steroid type (P = 0.31, I2 = 3.1%) (Analysis 1.26). Because of varied definitions for IOP elevation across the included trials and non‐reporting of this important adverse effect, we considered the certainty of the evidence as very low due to high risk of bias associated with biased measurement and selective reporting (−2) as well as imprecision (−1).
1.26. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 26: Proportion of participants with increased IOP—by steroid type
Proportion of participants with new cataract formation
The majority of the included trials did not measure or report incidents of cataract formation. Only one of the three trials that monitored the occurrence of cataract formation documented one incident case at the end of the 14‐day trial period (Analysis 1.27). The single study estimated RR was 0.34 (95% CI 0.01 to 8.22; n = 1205), suggesting that the evidence was very uncertain for the effect of topical steroids on cataract formation. Besides imprecision (−1), concerns about undersurveillance and selective outcome reporting (−2) led to a judgement of the certainty of the evidence for this outcome as very low.
1.27. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 27: Proportion of participants with new cataract formation
Proportion of participants with any ocular complication
Thirteen trials (81%) reported proportions of participants with any systemic or ocular adverse events during the study period, but none of these trials provided separate estimates for systemic and ocular incidents. Investigators in four of the 13 trials stated that they had not noticed any complaints from the study participants during a trial period lasting between 21 day and 2 months (Aragona 2013; Avunduk 2003; Lee 2014; Pinto‐Fraga 2016). Based on published results on ClinicalTrials.gov (KPI‐121 (Phase 2); KPI‐121 (STRIDE2); KPI‐121 (STRIDE3); NCT01276223), the combined estimated RR was 3.03 (95% CI 0.82 to 11.13; n = 2167; I2 = 0%), suggesting that the evidence was very uncertain for differences in risk of serious adverse effects, either systemic or ocular, when comparing topical steroids with lubricants (Analysis 1.28). Overall, we judged the certainty of the evidence to be very low because of risk of bias associated with under‐reporting or selective reporting (−2) and imprecision (−1).
1.28. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 28: Proportion of participants with serious adverse events (systemic or ocular)
Comparison 2: topical corticosteroid versus cyclosporine A
Critical outcomes
Change in patient‐reported symptom scores
All six trials comparing corticosteroid with CsA reported this outcome (Analysis 2.1). One three‐arm trial, Bausch 2013, contributed data to two intervention groups, one with LE gel 0.5% alone and the other with LE gel 0.5% and topical CsA. The combined estimate suggested that corticosteroids, alone or in combination, may slightly improve patient‐reported symptoms by 0.33 SMD (95% CI 0.15 to 0.51) as compared with CsA alone (I2 = 14%; n = 465) (Figure 8). Results were similar when considering the combined intervention from Bausch 2013 (Analysis 2.2).
2.1. Analysis.

Comparison 2: Steroids versus CsA, Outcome 1: Patient‐reported symptom scores—Bausch: LE alone
8.

Forest plot of comparison 2: Steroids versus cyclosporine A, outcome: 2.1 Patient‐reported symptom scores. The analysis included data of the LE group in Bausch 2013.
2.2. Analysis.

Comparison 2: Steroids versus CsA, Outcome 2: Patient‐reported symptom scores—Bausch: LE + CsA
The small number of trials precluded the performance of planned subgroup analysis by etiology. We did not perform a sensitivity analysis that excluded trials at high risk of bias or those sponsored by industry for the same reason. Exploratory subgroup analysis by intervention regimen (Analysis 2.3) or duration of steroid treatment (Analysis 2.4) did not find significant subgroup differences. We considered the certainty of the evidence to be low, downgrading for risk of bias (−1) and imprecision (−1).
2.3. Analysis.

Comparison 2: Steroids versus CsA, Outcome 3: Patient‐reported symptom scores—by regimen
2.4. Analysis.

Comparison 2: Steroids versus CsA, Outcome 4: Patient‐reported symptom scores—steroid treatment duration
Change in patient‐reported quality of life scores
None of the included trials measured this outcome.
Change in visual function
None of the included trials measured this outcome.
Change in tear film break‐up time
Five trials reported this outcome, evaluating the effects of corticosteroid alone, Bausch 2013; Lin 2015; Wan 2012, or in combination with CsA, Bausch 2013; Byun 2012; Singla 2019, compared with CsA (Analysis 2.5). Using data from 353 participants, the combined estimate for differences in TBUT was 0.37 seconds longer (95% CI 0.13 shorter to 0.87 longer) in the steroid group than in the CsA group, suggesting little to no difference in TBUT (I2 = 72%, P = 0.007). Considering data for the combined intervention group from Bausch 2013 did not alter the conclusions (Analysis 2.6). Exploratory subgroup analysis by intervention regimen (Analysis 2.7) or duration of steroid use (Analysis 2.8) did not help in identifying sources of heterogeneity. We judged the certainty of the evidence for this outcome as low because of risk of bias (−1) and imprecision (−1).
2.5. Analysis.

Comparison 2: Steroids versus CsA, Outcome 5: Tear film break‐up time—Bausch: LE alone
2.6. Analysis.

Comparison 2: Steroids versus CsA, Outcome 6: Tear film break‐up time—Bausch: LE + CsA
2.7. Analysis.

Comparison 2: Steroids versus CsA, Outcome 7: Tear film break‐up time—by regimen
2.8. Analysis.

Comparison 2: Steroids versus CsA, Outcome 8: Tear film break‐up time—by steroid treatment duration
Important outcomes
Change in fluorescein corneal staining scores
Six trials reported corneal fluorescein staining according to the NEI grading system, Bausch 2013; Byun 2012; Sheppard 2014; Singla 2019, or an unidentified scoring system (Lin 2015; Wan 2012). The combined estimated SMD was 0.05 (95% CI −0.25 to 0.35; n = 465) (Figure 9), suggesting that corticosteroids, alone or with CsA, may not reduce corneal staining scores when compared with CsA alone (Analysis 2.9). The result was similar when including data for the combined intervention group from Bausch 2013 (Analysis 2.10).
9.

Forest plot of comparison 2: Steroids versus cyclosporine A, outcome: 2.9 Corneal fluorescein staining scores. The analysis included data of the LE group in Bausch 2013.
2.9. Analysis.

Comparison 2: Steroids versus CsA, Outcome 9: Corneal fluorescein staining scores—Bausch: LE alone
2.10. Analysis.

Comparison 2: Steroids versus CsA, Outcome 10: Corneal fluorescein staining score—Bausch: LE + CsA
Post hoc subgroup analysis by grading system (Analysis 2.11), intervention regimen (Analysis 2.12), or duration of steroid treatment (Analysis 2.13) did not identify any potential sources of between‐study heterogeneity. We judged the certainty of the evidence as low because of associated risk of bias (−1) and imprecision (−1).
2.11. Analysis.

Comparison 2: Steroids versus CsA, Outcome 11: Corneal fluorescein staining score—by grading system
2.12. Analysis.

Comparison 2: Steroids versus CsA, Outcome 12: Corneal fluorescein staining score—by regimen
2.13. Analysis.

Comparison 2: Steroids versus CsA, Outcome 13: Corneal fluorescein staining score—by steroid treatment duration
Change in tear osmolarity
Bausch 2013 was the only trial to document changes in tear osmolarity (mOsm/kg) before and after the intervention with LE gel 0.5% alone or LE gel 0.5% plus CsA (Analysis 2.14).In comparison with tear samples from participants receiving CsA alone, tear samples collected from participants had a 5.80 mOsm/kg (95% CI −0.94 to 12.54) or 2.20 mOsm/kg (95% CI −6.00 to 10.4) higher osmolarity than those receiving CsA alone, suggesting LE alone or LE + CsA may decrease or increase tear osmolarity. We judged the certainty of the evidence as very low due to imprecision (−1) and risk of bias in the randomization and allocation process as well as selective reporting (−2).
2.14. Analysis.

Comparison 2: Steroids versus CsA, Outcome 14: Tear osmolarity
Change in Schirmer's test scores
Of the four trials that reported this outcome, Wan 2012 was the only trial comparing steroid monotherapy with CsA alone (Analysis 2.15). The overall combined estimate was 1.19 mm longer (95% CI 0.40 shorter to 2.77 longer; n = 361) in Schirmer's test strip in the intervention group than in the control group, suggesting little to no effects of steroids on tear production when compared with CsA alone.
2.15. Analysis.

Comparison 2: Steroids versus CsA, Outcome 15: Schirmer's test
In our exploration of potential sources of between‐study heterogeneity, we noted that excluding Singla 2019 alone could reduce the I2 statistics from 90% (P < 0.001) to 0%. Differences in the intervention regimen may also have contributed to the identified heterogeneity (Analysis 2.15). However, we found no statistical evidence for or against the use of an LE‐ versus MP‐based regimen (Analysis 2.16). We downgraded the certainty of the evidence to very low because of risk of bias (−2) and imprecision (−1).
2.16. Analysis.

Comparison 2: Steroids versus CsA, Outcome 16: Schirmer's test—by steroid type
Adverse effects
Proportion of participants with elevated IOP
Four trials monitored IOP elevation as adverse events during the trial period (Byun 2012; Lin 2015; Sheppard 2014; Singla 2019); three trials reported no incidents of IOP elevation, and one trial documented five incidents by the end of the 60‐day intervention (Sheppard 2014). The best available estimate was 45% increased risk associated with the use of LE + CsA (RR 1.45, 95% CI 0.25 to 8.33; n = 331), suggesting that steroids may decrease or increase the risk of elevated IOP as compared with CsA alone (Analysis 2.17). We judged the certainty of the evidence to be very low, downgrading for risk of bias in outcome measurement and selective reporting (−2) and imprecision of the estimate (−1).
2.17. Analysis.

Comparison 2: Steroids versus CsA, Outcome 17: Proportion of participants with increased IOP
Proportion of participants with new cataract formation
None of the six trials in this comparison reported this outcome.
Proportion of participants with any ocular complication
Three of the six trials comparing steroids with CsA reported this outcome; two provided numeric results for three comparisons (Bausch 2013; Byun 2012), and one trial described that during the observation period, they did not notice any adverse reactions to the medications [author translation] (Wan 2012). Topical steroids, either alone (RR 0.61, 95% CI 0.11 to 3.43; 2 trials, n = 103) or in combination with CsA (RR 0.65, 95% CI 0.11 to 3.80; 2 trials, n = 110), may show no effect on risk of any ocular complication when compared with CsA alone (Analysis 2.18). However, we judged the certainty of the evidence to be very low because of risk of bias associated with under‐reporting or selective reporting (−2) and imprecision (−1).
2.18. Analysis.

Comparison 2: Steroids versus CsA, Outcome 18: Proportion of participants with any ocular complication
Discussion
Summary of main results
Based on data from 22 RCTs that compared topical corticosteroids, alone or in combination, with lubricants or CsA, topical corticosteroids probably slightly improve patient‐reported symptoms when compared with lubricants (moderate certainty evidence) and may slightly improve patient‐reported symptoms when compared with CsA (low certainty evidence). Topical steroids probably result in a slight reduction in corneal staining scores (moderate certainty evidence) when compared with lubricants, but may result in no difference when compared with CsA (low certainty evidence). In general, the evidence suggests that corticosteroids do not increase tear production as quantified by TBUT or Schirmer's test scores (low certainty evidence). The evidence is also very uncertain about the effect of steroids on tear osmolarity.
Topical corticosteroids may increase IOP when compared with lubricants but may not do so when compared with CsA ; the evidence for each comparison is very uncertain. Similarly, very low certainty evidence suggests that steroids may have little to no effect on cataract formation when compared with lubricants. No trials comparing corticosteroids with CsA reported cataract information as adverse events.
Overall completeness and applicability of evidence
Population representativeness
The study populations of the included trials were representative of the dry eye populations generally seen in the outpatient clinic. The included trials had enrolled mainly middle‐aged, female participants with non‐specific (or unidentified) etiologies of DED as described in the literature (Nelson 2017). Nevertheless, the small to moderate numbers of participants enrolled in most trials, and the lack of reporting of group‐specific outcome data, precluded subgroup analysis by age, sex, or etiology to explore differential benefits (or harms) of topical corticosteroids as compared with lubricants or CsA.
Pharmacologic interventions
The current review did not find evidence of reduced ocular adverse effects in participants receiving LE versus other topical steroids. Among the seven steroidal preparations listed in the protocol (Liu 2021), LE of varying concentrations was the most frequently used corticosteroid in the intervention group, particularly in industry‐funded trials. As a structural variant to a typical corticosteroid such as prednisolone, LE has been described as having a low risk profile in terms of IOP elevation or cataract formation (Comstock 2018). However, we did not find statistical evidence for such differential risks of IOP elevation in a subgroup analysis by structural types of topical steroids. Insufficient numbers of included trials limited our ability to evaluate risk for cataract formation (or progression) comparing steroids with lubricants or CsA.
Given the small sample sizes and the high risk of bias in outcome measurement and reporting in trials that examined the effects of steroid in combination with CsA, we also concluded that the current evidence was insufficient in quantity and quality to suggest additional benefits of topical steroid in combination with CsA over steroid alone, when compared with CsA.
Outcome measurement
Although the current review found statistically significant improvement in patient‐reported symptoms and corneal staining scores when comparing steroids with lubricants, the clinical meaning of the effect sizes was unclear. By convention, an SMD of 0.2 is considered as a small effect, 0.5 a moderate effect, and 0.8 a large effect (Cohen 1988), suggesting that steroid effects on symptoms and corneal staining scores are probably small to moderate at most. Considering the subgroup of trials that had reported symptoms on the OSDI scale, the estimated steroid effect was also smaller than the previously published minimally important difference (MID) in OSDI scores for mild or moderate DED (4.5 to 7.3) (Miller 2010). Results of subgroup analysis that excluded trials at high risk of bias further suggested that the small to moderate effects of steroids might already be overestimated.
For the two most commonly reported review outcomes, patient‐reported symptom scores and corneal fluorescein staining scores, the included trials reported the use of varied standardized or non‐standardized questionnaires or grading schemes. Even within trials that had applied OSDI or the NEI scheme, the degree of between‐study heterogeneity was substantial, suggesting that inconsistent application of the same scales, variations among assessors in grading or interpreting imaging results, and reporting of post‐treatment endpoint scores or change scores, were all potential sources of between‐study heterogeneity. As such, we advise caution in the interpretation of SMD‐based findings and have provided alternative benchmarks to aid the interpretation and assessment for applicability (Table 1; Table 2).
Ocular adverse events were generally under‐reported, likely due to the lack of a systematic surveillance system implemented during the trial period and the short durations of the trials. Despite being well‐recognized as potential adverse effects of topical corticosteroid, elevated IOP was documented in only half of the included trials; incident cataract formation was reported by even fewer trials. As such, the small absolute risks for elevated IOP in the control groups, for instance 9 incidents per 10,000 participants using lubricants (Table 1), or 12 per 1000 participants receiving CsA (Table 2), might be underestimated. The small numbers of participants enrolled per trial and the short follow‐up periods further contributed to the imprecision of both the absolute and relative risk estimates for these safety outcomes.
Certainty of the evidence
We downgraded the certainty of the evidence for the review outcomes because of substantial risk of bias associated with: the procedure of randomly allocating treatment; biased outcome measurement due to unclear masking status of the participants or the outcome assessors; and selective results reporting. Particularly for safety outcomes, the small number of trials that had reported ocular adverse events might indicate non‐reporting or under‐reporting of these events. The potential impact on the combined estimates for adverse effects of topical steroids would be underestimation of the risks of IOP elevation or cataract formation as compared with lubricants or CsA.
Potential biases in the review process
An Information Specialist assisted with an exhaustive search in multiple electronic databases and trial registries. We applied standard Cochrane methods to conduct this review and to avoid potential biases associated with literature search, critical appraisal, data extraction, analysis, and interpretation. We also performed an additional handsearch of the references of the included trials, and made repeated efforts to contact trial investigators or study authors to clarify details regarding study design (NCT00471419), sample sizes (Herman 2005), concentrations of the intervention steroids (NCT03418727), or to request trial results (ChiCTR‐IPR‐15007196; NCT00560638; NCT01562795; NCT03418727; NTR2291). These efforts were not always successful, and contributed to the imprecision of the combined estimates for some comparisons.
Agreements and disagreements with other studies or reviews
In searching for relevant reviews that had been published since 2017, we identified four review articles that examined the efficacy or effectiveness of topical corticosteroids for dry eye disease (Beckman 2020; Cutolo 2019), ocular complications of Sjögren syndrome (Shih 2017), or ocular inflammatory conditions in general (Comstock 2018); none of these were systematic reviews.
Authors of both Beckman 2020 and Cutolo 2019 included not only RCTs but also observational studies in their narrative reviews. These two reviews also included trials of paired‐eye design and those that enrolled post‐transplant or postcataract patients. In Beckman 2020, the authors cited study findings from two conference proceedings that were not accessible through indexed databases (Barabino 2011; Evans 2017). Findings of Beckman 2020 and Cutolo 2019 agreed with the current review in finding that topical corticosteroids were effective in improving dry eye symptoms and tear film parameters, such as TBUT and corneal staining scores, as compared with lubricants. Their conclusions were also concordant with our findings that steroid use before or concurrently with CsA was beneficial for relieving symptoms when compared with CsA alone or with artificial tears/vehicle. However, our review did not find evidence supporting the role of topical steroid in ameliorating ocular signs as stated in Beckman 2020 or Comstock 2018. Differences in eligibility criteria for study populations or study design of the original studies might have contributed to these discrepancies in findings.
In three of the four narrative reviews (Beckman 2020; Comstock 2018; Cutolo 2019), the authors also summarized evidence regarding adverse effects, concluding that topical corticosteroids, particularly LE, "provide certain safety" based on the few included trials that had reported on incident IOP elevation, though authors of Cutolo 2019 recognized that "the incidence of side effects" could vary by different (types or formulas of) topical corticosteroids. In our review, the small number of trials comparing steroid alone or in combination with CsA did not allow for reliable estimation of risks for elevated IOP. In contrast with prior qualitative reviews, our review provided quantitative evidence showing that steroid use was associated with a nearly five‐fold increased risk of IOP elevation even with 'short‐term' use (up to four weeks).
Authors' conclusions
Implications for practice.
Topical corticosteroids likely provide small to moderate improvement in symptoms as compared with artificial tears (moderate certainty evidence), and may provide small to moderate improvement in symptoms as compared with cyclosporine A (low certainty evidence).
Evidence supports the anti‐inflammatory effects of steroids on corneal staining scores over lubricants (moderate certainty evidence), but not over cyclosporine A (low certainty evidence).
Steroids may have little to no effect on tear quality or production as shown by tear film break‐up time (low certainty evidence) and Schirmer's test (low certainty evidence).
The evidence is very uncertain regarding the effects of steroids on tear osmolarity, risk of elevated intraocular pressure, or risk of cataract formation.
Implications for research.
Whether topical corticosteroids have a role in the step‐up (from lubricants or cyclosporine A) treatment strategies for dry eye patients will be better informed by future trials that:
recruit patients of identifiable etiologies of dry eye and then report etiology‐specific treatment effects;
perform head‐to‐head comparisons between different types of steroids, such as ester steroids versus ketone steroids;
measure and report quality of life or visual function‐related outcomes;
last a reasonable duration of time to allow for the detection of uncommon yet important adverse events such as intraocular pressure elevation, cataract formation or progression.
Lack of consistency in correlating signs and symptoms within the patient population of dry eye creates difficulty in determining the minimally important differences in patient‐reported outcomes for different subgroups of patients. Variations in the clinical presentation of patients further emphasize the importance of unbiased measurement and reporting of these core outcomes in the same trial. Investigators should employ at least double‐masking of participants and assessors (rather than examiners) to avoid potential risk of bias in documenting both subjective and objective study outcomes.
History
Protocol first published: Issue 9, 2021
Risk of bias
Risk of bias for analysis 1.1 Patient‐reported symptom scores—Qazi: LE alone.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Avunduk 2003 | Some concerns | Patients were randomized to three groups according to a computerized list generated by the LSU Eye Center biostatistician. Details about allocation concealment were not reported. At the beginning of the study (day 0), no significant difference was detected between groups in terms of any parameters studied. | Low risk of bias | This was a single‐masked clinical tiral. The examiner was masked as to the medication used by the patients. The patients were instructed to discuss their medications only with the study coordinator and not with the examiner. | Some concerns | Besides excluding 4 participants who did not complete the study, all other participants randomzied were analyzed for the study outcomes. It was unclear the numbers of participants initially randomzied to each comparison group. | High risk of bias | The study outcomes included patient‐reported symptom severity scores and 4 objective measurements that required equipments performed by one examiner who was masked to the medication used. Patient‐reported symptom severity could be influenced by patient's awareness of the intervention received. The assessment of symptom outcome might be consciously or unconsciously influenced by patient's knowledge of the intervention received. | High risk of bias | There were no trial protocol available for comparison. Based on the statistical plan described in the Methods section, the authors reported many other numertical results other than the intended analysis for testing the null hypothesis. Additionally, for all comparison results, only P‐values were reported in the publication though data points were provided in multiple graphs. | High risk of bias | There was no information about how allocation concealment was performed. The questionnaired used to collect this patient‐reported outcome was not previously validated; it was unclear whether subjects were masked to the treatment received. There were also high risks of selective reporting of the study outcomes. |
| Li 2021 | Some concerns | Participants were randomly assigned to the intervention or control group using a random number table. The process of allocation concealment was not reported. There were no age or sex differences between the group. | Some concerns | Whether participants, carers, or assesors were masked was not reported. There were two and four participants who failed to complete the study in the intervention and control group, separately. The drop‐outs might have less favored outcome due to poor responses or inadequate treatment. The deviation rates appeared statistically balanced between the two groups. Complete‐case analysis was perofmred to estimate the effect of assignment, which might not be appropriate but the impact on the results might not be substantial. | Low risk of bias | Data for this outcome were reported for 116 out of 122 participants (95.1%). | Some concerns | OSDI was used to document patient‐reported symptoms. If participants were not masked to the treatment received, the outcome measurement could have differed between groups. | High risk of bias | No pre‐specified analysis plan available for assessment. Data for this outcome were analyzed and reported in a conventional way at the eye level, whereas the unit of randomization was person. | High risk of bias | There were some concerns about the performance of allocation concealment, whether participants were masked, and potentially biased measurement, mainly due to the lack of information. There were also high risks of selective outcome reporting. |
| Aragona 2013 | Some concerns | "Patients were randomized into 2 groups according to a computerized list." The authors did not report details about allocation concealment. Distribution of age, sex, and symptoms or eye test results at baseline appeared comparable between the two groups (Table 1). | Low risk of bias | Whether participants were masked was unclear, but the authors described that "the corticosteroid and vehicle eyedrops were dispensed in coded bottles; " the bottles and their content were identical in their appearance." "The statistical analysis of the results was carried out in a masked method by an investigator not involved in the study" | Low risk of bias | Data for this outcome was available for all participants included and randomized in the study. | Low risk of bias | "Symptom evaluation was obtained by VAS (from 0 = symptom absent to 100 = analogue to the maximum discomfort ever felt) for 7 pre‐specified symptoms". The total score was obtained adding up the result of each symptom. Based on the sham eyedrops described, the participants were likely masked and the measurement of this outcome could not have differed between the two groups. | Low risk of bias | Primary efficacy variables were the global symptoms score and other test results of the right eyes. Data for this outcome was analyzed as planned. | Some concerns | There were some concerns about the process of allocation concealment. |
| Lee 2014 | Low risk of bias | A randomization sequence was created using EXCEL 2007 (Microsoft, Redmond, Washington, USA) with a 1:1 allocation using random block sizes of 2, 4, and 6, by an independent doctor. No significant differences in any parameters were found between groups before treatment. | Some concerns | The allocation sequence was concealed from the physician enrolling and assessing pa‐ tients in sequentially numbered, opaque, and sealed envelopes. After the content of the envelope was revealed, the physician and patients were aware of the allocation and the corresponding treatment. However, outcome assessors and data analysts were kept masked to the allocation. Four and six participants in the intervention and control group dropped out of the study. It was unclear whether these drop‐outs were associated with deviations from the intended treatment allocation. | Low risk of bias | Data for this outcome were available for 60 out of 70 participants randomzeid (85.7%). | High risk of bias | OSDI was used for patient‐reported symptom severity. Participants were aware of the treatment they received and might have reported their symptoms differentially. | High risk of bias | There was no pre‐specified analytic plan available for assessment. The sample size estimation was based on ANCOVA of repeatedly measured inflammatory cytokine levels. Results presented for this outcome were based on post‐hoc linear mixed models. Model based estimates for OSDI scores were reported, instead of the raw scores. Post‐hoc regression analysis was applied, instead of ANCOVA used for sample size estimation. | High risk of bias | There were indications for potential high risks of bias in measuring and reporting this particular outcome. |
| Luo 2013 | Some concerns | Subjects were randomly grouped into three intervention arms, but how treatment allocation was performed was not reported. No differences in age or sex distribution among the three groups were reported without presenting data for each group. Baseline values of the symptoms and clinical signs were reported and no substantial differences were shown. | Some concerns | Whether the paritcipants, carers, or examiners were masked during the study period was not reported. Participants' tolerability to the treatment was not reported. Data of this outcome were analyzed in a conventional before versus after approach. | Low risk of bias | Data of this outcome were available for all participants reported randomzied. | High risk of bias | Symptom measurement was based on self‐reporting by participants. Participants if unmasked could have answered the questionnaire differentially among the treatment groups. | Low risk of bias | No pre‐specified analysis plan was avvailable for assessment. Results were analyzed in conventional before versus after comparison. | High risk of bias | There were some concerns about allocation concealment and participant masking. As a result, there were also high risks of measuring bias, depending on the masking status of the participants. |
| Chen 2020 | Some concerns | Particpants were reportedly randomzied to receiving either HA (Group A) or loteprednol plus HA (Group B), but the details about the randomization procedure or the allocation process were not reported. No significant differences in age or sex distribution were reported. | Some concerns | Whether participants or carers and other study personnels were masked or not was not reported. Data for participants randomized were analyzed as they were assigned to. | Some concerns | Data for participants randomized were available for all reported though the number of participants initially randomized was not reported. | High risk of bias | SPEED questionnaire was used to document symptom frequency for four symptoms. Depending on the masking status of participants, this outcome was patent‐reported and might be influenced by the knowledge of the treatment received. | Some concerns | No pres‐pecified analysis plan or study protocol was available for assessment. Outcome data were reported as before and after measurements. It was unclear whether there were potentially multiple eligible measurements after the planned 1 month of treatment. | High risk of bias | Because of lack of information, there are some concerns or high risks of bias across multiple domains. |
| Akhlaq 2019 | Low risk of bias | The randomization will be in a 1:1 ratio using a permuted‐block design (which will be obtained using a computer‐based random code generator) into the two groups. Baseline characteristics of the study population were not reported in the conference abstract, except for age and the baseline ODSI scores. The later was statistically higher in the intervention than in the control group (53.53±29.7 vs. 34.46±20.33, post‐hoc P = 0.0255). | Low risk of bias | The randomization scheme and treatment assignment will be held in the pharmacy to ensure that the investigator and study team remain masked. Participants, investigators, image analysts, and the sponsor (GSK) will be masked to the treatment group. | Low risk of bias | Study results of 38 participants (67 eyes) were reported; it was unclear how many participants were randomized initially. | Low risk of bias | Patient‐reported symptom severity or frequency was quantified using OASI and SANDE I & II at each follow‐up visit. According to the study protocol, participants were planned to be masked so the measurement of this outcome could not have been differed. | Some concerns | No analytic plan for this particular outcome was available for evaluation. Symptom scores based on OSDI and SANDE were pre‐specified as secondary outcomes in the study protocol but only results of OSDI were reported. | Some concerns | There were some concerns about selective reporting of patient‐reported symptom scores, which were not as planned in the study protocol. |
| KPI‐121 (STRIDE1) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. Four and 3 participants in the KPI‐121 and vehicle group did not completed the trial. This outcome data were reported for 907 participants at Day 15. | Low risk of bias | Data for this outcome were available for nearly all participants randomized (907/915=99.1%). | Low risk of bias | Ocular discomfort severity was self‐reported by patients using a visual analog scale (0‐100) and patients were masked to which intervention they received. | Low risk of bias | The study protocol did not specify analytic methods but described the use of t‐test for sample size consideration. Only the absolute measures for this outcome were reported. Only one set of analyses were applied to the data for this outcome. | Low risk of bias | The study was deemed as having low risk of bias across multiple domains. |
| KPI‐121 (STRIDE3) | Low risk of bias | "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the Sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. Eleven participants did not complete the trial. Data for 885 participants were reported for this outcome. | Low risk of bias | Data for this outcome were available for nearly all participants randomized (885/901=98.2%). | Low risk of bias | Ocular discomfort severity was self‐reported by patients using a visual analog scale (0‐100) and patients were masked to which intervention they received. | Some concerns | The study protocol did not specify analytic methods but described the use of t‐test for sample size consideration. However, "Change From Baseline/Visit 2 (Day 1) in Ocular Discomfort Severity at Visit 4 (Day 15) Using 7 Day Mean" was defined as an efficacy outcome on clinicaltrials.gov but no corresponding analytic plans were described in the study protocol. Only the absolute measures for this outcome were reported. The secondary outcome "Change From Baseline/Visit 2 (Day 1) in Ocular Discomfort Severity at Visit 4 (Day 15) Using 7 Day Mean" could be data‐driven. | Some concerns | There were some concerns about selective outcome reporting due to discrepancies in outcome specification on clinicaltrials.gov and the study protocol. |
| KPI‐121 (Phase 2) | Low risk of bias | The protocol stated that "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | Data for this outcome were available for all participants randomized. | Low risk of bias | OSDI was used to record ocular discomfort by the patients themselves. Patients were maksed to which intervention they received. | Low risk of bias | The study protocol did not specify analytic methods for producing this outcome. Only the absolute measures for this outcome were reported. Only one set of analyses were applied to the data for this outcome. | Low risk of bias | The study was judged to be at low risk of bias across all domains. |
| KPI‐121 (STRIDE2) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | Data for this outcome were available for nearly all participants randomized (889/905=98.2%). | Low risk of bias | Ocular discomfort severity was self‐reported by patients using a visual analog scale (0‐100) and patients were masked to which intervention they received. | Some concerns | The study protocol did not specify analytic methods but described the use of t‐test for sample size consideration. However, patient‐reported dryness was defined as a secondary outcome on clinicaltrials.gov but not in the study protocol. Only the absolute measures for this outcome were reported. Only one set of analyses were applied to the data for this outcome. | Some concerns | There were some concerns about selective outcome reporting due to discrepancies specifying this particular outcome between clinicaltrials.gov and the study protocol published. |
| NCT01276223 | Some concerns | According to the trial registry information. However, details about treatment allocation were not reported. Participants in the Difluprednate group were 5.7 years younger (post‐hoc P = 0.004); otherwise sex distribution was not different between the two groups. | Low risk of bias | According to the trial registry, the study was double‐masked to the participants and the investigators. An ITT analysis that included all participants randomized was performed; missing data was based on imputation. | Low risk of bias | Data for this outcome were available for nearly all participants randomized; missing data for 7 participants who did not complete the study were imputed based on mixed model repeated measure approach. | Low risk of bias | A Visual Analog Scale (VAS) was used by the subject to assess ocular discomfort, both frequency and severity. Each scale was 100 millimeters (mm) in length. The Global Ocular Discomfort Score is a composite of the two VAS scores, ranging from 0 (very mildly) to 100 (very severely uncomfortable). The participant was masked to the intervention received, so it was unlikely that the assessment of this outcome could be differentially influenced. | Low risk of bias | Data for this outcome was analyzed according to the primary study outcome pre‐specified. | Some concerns | There were some concerns about the process of allocation concealment, which was not described at all by the authors. |
| Qazi 2015 | Some concerns | No details about randomization process, including allocation concealment, were reported in the abstract, full‐text publication, or on clinicaltrials.gov. No significant differences in any parameters were found among comparison groups. | Some concerns | Quadruple masking (Participant, Care Provider, Investigator, Outcomes Assessor) was reported on clinicaltrials.gov. However, it was unclear how masking of participants were achieved except that, "during each visit, all participants had a complete masked ophthalmic evaluation." | Low risk of bias | "Six patients were withdrawn or lost to follow‐up before the 4‐week visit. Three aditional subjects, 1 in each group, did not complete the 8‐week visit". Data for this outcome were available for 17 out of 20 participants randomzeid (85%). | Low risk of bias | OSDI was applied to measure patient‐reported symptom severity. Participants were planned to be masked. | Some concerns | The numerical results were reported by convention, probably not being selected on the basis of significance. However, the trial registry history showed that the original timeframe for reporting outcomes was "the count of inflammatory cells at 4 weeks and 8 weeks after taking the first dose of treatment", which was not reported in the abstract or the full text publication. | Some concerns | There were some concerns about the process of treatment allocation, masking, and selective reporting of the outcome. |
| Pflugfelder 2004 | Some concerns | Eligible patients received a study number and received double‐masked study medication (either loteprednol or placebo) according to a predetermined random allocation schedule. There were multiple baseline characteristics disproportionately distributed between the two intervention groups: female (P = 0.015), history of previous surgical procedures (P = 0.022), and history of punctal occlusion use (P = 0.033) among less than 20 variables considered. | Low risk of bias | Loteprednol etabonate was supplied in 7.5‐ml white, low‐density polyethylene dropper bottles with what caps for masking purposes. Placebo (the vehicle of LE) was supplied in identical bottles. The medication was double‐masked. All participants randomized to each intervention were analyzed according the intervention assigned. | Low risk of bias | In primary analysis, the authors reported mean changes in sign and symptoms (this outcome) for all patients randomzied. | Low risk of bias | Patient‐reported worst symptom in the worst eye was measured using a visual analogue scale of a 100mm line, with labels of Absent at the left end and Unbearable at the right end. Each patient was asked to mark the line at the point between these two extremes that represented the severity of the symptom; the distance from the left end of the line to the mark was the VAS (symptom) score in milimeters. Patients were masked by the medication received and thus likely to answer differently from reasons other than their perceived severity of the worst symptom. This was a patient‐reported outcome and patients were masked. | Low risk of bias | No trial protocol or analysis plan available for assessment but based on the power and sample size consideration, there might be a proper analytic plan pre‐specified before the trial. Results of eligible measures at different time points were also reported. Possible but not reported or discussed. | Some concerns | There were some concerns about the performance of the randomization process, evidenced by excessive imbalance between the two comparison groups. |
| Pinto‐Fraga 2016 | Some concerns | 'The generation of random allocation sequence and the statistical analyses were carried out by a PhD‐licensed statistician (I.F.), using the R statistical package version 3.1.1'. There were no significant differences in age or gender distribution between the 2 groups (P 1⁄4 0.37 and P 1⁄4 0.66, respectively). The authors did not describe the process of allocation concealment. | Low risk of bias | The original labels of these bottles were removed and bottles were relabeled with the study number and a coded lot number to avoid potential identification of the treatment by site personnel or patients. Treatment was not delivered or collected by any investigator who performed evaluations during the clinical trial, and none of the investigators were involved in the masking process described above. | Low risk of bias | One patient dropped out of the study due to work‐related issues and additional one participant was recruited to fill the gap, whose outcome data was also analyzed as treated. | Low risk of bias | Symptom scores were documented using OSDI and SANDE version I questionnaire by patients. Patients were unaware of their treatment received. Patients were unaware of their treatment medication and might not answer the questionnares about symptoms differentially. | Low risk of bias | Reporting of the outcome variables and analytic approaches was consistent between trial registry, the methods section and the result section of the report. SANDE version I and ODSI were used for symptom scores as primary and secondary outcomes, separately. Both the absoulte scores and changes in proportions of participants with symptom reduction were reported. | Some concerns | There were some concerns about |
Risk of bias for analysis 1.3 Patient‐reported symptom scores—Qazi: LE + tobramycin.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Avunduk 2003 | Some concerns | Patients were randomized to three groups according to a computerized list generated by the LSU Eye Center biostatistician. Details about allocation concealment were not reported. At the beginning of the study (day 0), no significant difference was detected between groups in terms of any parameters studied. | Low risk of bias | This was a single‐masked clinical tiral. The examiner was masked as to the medication used by the patients. The patients were instructed to discuss their medications only with the study coordinator and not with the examiner. | Some concerns | Besides excluding 4 participants who did not complete the study, all other participants randomzied were analyzed for the study outcomes. It was unclear the numbers of participants initially randomzied to each comparison group. | High risk of bias | The study outcomes included patient‐reported symptom severity scores and 4 objective measurements that required equipments performed by one examiner who was masked to the medication used. Patient‐reported symptom severity could be influenced by patient's awareness of the intervention received. The assessment of symptom outcome might be consciously or unconsciously influenced by patient's knowledge of the intervention received. | High risk of bias | There were no trial protocol available for comparison. Based on the statistical plan described in the Methods section, the authors reported many other numertical results other than the intended analysis for testing the null hypothesis. Additionally, for all comparison results, only P‐values were reported in the publication though data points were provided in multiple graphs. | High risk of bias | There was no information about how allocation concealment was performed. The questionnaired used to collect this patient‐reported outcome was not previously validated; it was unclear whether subjects were masked to the treatment received. There were also high risks of selective reporting of the study outcomes. |
| Li 2021 | Some concerns | Participants were randomly assigned to the intervention or control group using a random number table. The process of allocation concealment was not reported. There were no age or sex differences between the group. | Some concerns | Whether participants, carers, or assesors were masked was not reported. There were two and four participants who failed to complete the study in the intervention and control group, separately. The drop‐outs might have less favored outcome due to poor responses or inadequate treatment. The deviation rates appeared statistically balanced between the two groups. Complete‐case analysis was perofmred to estimate the effect of assignment, which might not be appropriate but the impact on the results might not be substantial. | Low risk of bias | Data for this outcome were reported for 116 out of 122 participants (95.1%). | Some concerns | OSDI was used to document patient‐reported symptoms. If participants were not masked to the treatment received, the outcome measurement could have differed between groups. | High risk of bias | No pre‐specified analysis plan available for assessment. Data for this outcome were analyzed and reported in a conventional way at the eye level, whereas the unit of randomization was person. | High risk of bias | There were some concerns about the performance of allocation concealment, whether participants were masked, and potentially biased measurement, mainly due to the lack of information. There were also high risks of selective outcome reporting. |
| Aragona 2013 | Some concerns | "Patients were randomized into 2 groups according to a computerized list." The authors did not report details about allocation concealment. Distribution of age, sex, and symptoms or eye test results at baseline appeared comparable between the two groups (Table 1). | Low risk of bias | Whether participants were masked was unclear, but the authors described that "the corticosteroid and vehicle eyedrops were dispensed in coded bottles; " the bottles and their content were identical in their appearance." "The statistical analysis of the results was carried out in a masked method by an investigator not involved in the study" | Low risk of bias | Data for this outcome was available for all participants included and randomized in the study. | Low risk of bias | "Symptom evaluation was obtained by VAS (from 0 = symptom absent to 100 = analogue to the maximum discomfort ever felt) for 7 pre‐specified symptoms". The total score was obtained adding up the result of each symptom. Based on the sham eyedrops described, the participants were likely masked and the measurement of this outcome could not have differed between the two groups. | Low risk of bias | Primary efficacy variables were the global symptoms score and other test results of the right eyes. Data for this outcome was analyzed as planned. | Some concerns | There were some concerns about the process of allocation concealment. |
| Lee 2014 | Low risk of bias | A randomization sequence was created using EXCEL 2007 (Microsoft, Redmond, Washington, USA) with a 1:1 allocation using random block sizes of 2, 4, and 6, by an independent doctor. No significant differences in any parameters were found between groups before treatment. | Some concerns | The allocation sequence was concealed from the physician enrolling and assessing pa‐ tients in sequentially numbered, opaque, and sealed envelopes. After the content of the envelope was revealed, the physician and patients were aware of the allocation and the corresponding treatment. However, outcome assessors and data analysts were kept masked to the allocation. Four and six participants in the intervention and control group dropped out of the study. It was unclear whether these drop‐outs were associated with deviations from the intended treatment allocation. | Low risk of bias | Data for this outcome were available for 60 out of 70 participants randomzeid (85.7%). | High risk of bias | OSDI was used for patient‐reported symptom severity. Participants were aware of the treatment they received and might have reported their symptoms differentially. | High risk of bias | There was no pre‐specified analytic plan available for assessment. The sample size estimation was based on ANCOVA of repeatedly measured inflammatory cytokine levels. Results presented for this outcome were based on post‐hoc linear mixed models. Model based estimates for OSDI scores were reported, instead of the raw scores. Post‐hoc regression analysis was applied, instead of ANCOVA used for sample size estimation. | High risk of bias | There were indications for potential high risks of bias in measuring and reporting this particular outcome. |
| Luo 2013 | Some concerns | Subjects were randomly grouped into three intervention arms, but how treatment allocation was performed was not reported. No differences in age or sex distribution among the three groups were reported without presenting data for each group. Baseline values of the symptoms and clinical signs were reported and no substantial differences were shown. | Some concerns | Whether the paritcipants, carers, or examiners were masked during the study period was not reported. Participants' tolerability to the treatment was not reported. Data of this outcome were analyzed in a conventional before versus after approach. | Low risk of bias | Data of this outcome were available for all participants reported randomzied. | High risk of bias | Symptom measurement was based on self‐reporting by participants. Participants if unmasked could have answered the questionnaire differentially among the treatment groups. | Low risk of bias | No pre‐specified analysis plan was avvailable for assessment. Results were analyzed in conventional before versus after comparison. | High risk of bias | There were some concerns about allocation concealment and participant masking. As a result, there were also high risks of measuring bias, depending on the masking status of the participants. |
| Chen 2020 | Some concerns | Particpants were reportedly randomzied to receiving either HA (Group A) or loteprednol plus HA (Group B), but the details about the randomization procedure or the allocation process were not reported. No significant differences in age or sex distribution were reported. | Some concerns | Whether participants or carers and other study personnels were masked or not was not reported. Data for participants randomized were analyzed as they were assigned to. | Some concerns | Data for participants randomized were available for all reported though the number of participants initially randomized was not reported. | High risk of bias | SPEED questionnaire was used to document symptom frequency for four symptoms. Depending on the masking status of participants, this outcome was patent‐reported and might be influenced by the knowledge of the treatment received. | Some concerns | No pres‐pecified analysis plan or study protocol was available for assessment. Outcome data were reported as before and after measurements. It was unclear whether there were potentially multiple eligible measurements after the planned 1 month of treatment. | High risk of bias | Because of lack of information, there are some concerns or high risks of bias across multiple domains. |
| Akhlaq 2019 | Low risk of bias | The randomization will be in a 1:1 ratio using a permuted‐block design (which will be obtained using a computer‐based random code generator) into the two groups. Baseline characteristics of the study population were not reported in the conference abstract, except for age and the baseline ODSI scores. The later was statistically higher in the intervention than in the control group (53.53±29.7 vs. 34.46±20.33, post‐hoc P = 0.0255). | Low risk of bias | The randomization scheme and treatment assignment will be held in the pharmacy to ensure that the investigator and study team remain masked. Participants, investigators, image analysts, and the sponsor (GSK) will be masked to the treatment group. | Low risk of bias | Study results of 38 participants (67 eyes) were reported; it was unclear how many participants were randomized initially. | Low risk of bias | Patient‐reported symptom severity or frequency was quantified using OASI and SANDE I & II at each follow‐up visit. According to the study protocol, participants were planned to be masked so the measurement of this outcome could not have been differed. | Some concerns | No analytic plan for this particular outcome was available for evaluation. Symptom scores based on OSDI and SANDE were pre‐specified as secondary outcomes in the study protocol but only results of OSDI were reported. | Some concerns | There were some concerns about selective reporting of patient‐reported symptom scores, which were not as planned in the study protocol. |
| KPI‐121 (STRIDE1) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. Four and 3 participants in the KPI‐121 and vehicle group did not completed the trial. This outcome data were reported for 907 participants at Day 15. | Low risk of bias | Data for this outcome were available for nearly all participants randomized (907/915=99.1%). | Low risk of bias | Ocular discomfort severity was self‐reported by patients using a visual analog scale (0‐100) and patients were masked to which intervention they received. | Low risk of bias | The study protocol did not specify analytic methods but described the use of t‐test for sample size consideration. Only the absolute measures for this outcome were reported. Only one set of analyses were applied to the data for this outcome. | Low risk of bias | The study was deemed as having low risk of bias across multiple domains. |
| KPI‐121 (STRIDE3) | Low risk of bias | "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the Sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. Eleven participants did not complete the trial. Data for 885 participants were reported for this outcome. | Low risk of bias | Data for this outcome were available for nearly all participants randomized (885/901=98.2%). | Low risk of bias | Ocular discomfort severity was self‐reported by patients using a visual analog scale (0‐100) and patients were masked to which intervention they received. | Some concerns | The study protocol did not specify analytic methods but described the use of t‐test for sample size consideration. However, "Change From Baseline/Visit 2 (Day 1) in Ocular Discomfort Severity at Visit 4 (Day 15) Using 7 Day Mean" was defined as an efficacy outcome on clinicaltrials.gov but no corresponding analytic plans were described in the study protocol. Only the absolute measures for this outcome were reported. The secondary outcome "Change From Baseline/Visit 2 (Day 1) in Ocular Discomfort Severity at Visit 4 (Day 15) Using 7 Day Mean" could be data‐driven. | Some concerns | There were some concerns about selective outcome reporting due to discrepancies in outcome specification on clinicaltrials.gov and the study protocol. |
| KPI‐121 (Phase 2) | Low risk of bias | The protocol stated that "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | Data for this outcome were available for all participants randomized. | Low risk of bias | OSDI was used to record ocular discomfort by the patients themselves. Patients were maksed to which intervention they received. | Low risk of bias | The study protocol did not specify analytic methods for producing this outcome. Only the absolute measures for this outcome were reported. Only one set of analyses were applied to the data for this outcome. | Low risk of bias | The study was judged to be at low risk of bias across all domains. |
| KPI‐121 (STRIDE2) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | Data for this outcome were available for nearly all participants randomized (889/905=98.2%). | Low risk of bias | Ocular discomfort severity was self‐reported by patients using a visual analog scale (0‐100) and patients were masked to which intervention they received. | Some concerns | The study protocol did not specify analytic methods but described the use of t‐test for sample size consideration. However, patient‐reported dryness was defined as a secondary outcome on clinicaltrials.gov but not in the study protocol. Only the absolute measures for this outcome were reported. Only one set of analyses were applied to the data for this outcome. | Some concerns | There were some concerns about selective outcome reporting due to discrepancies specifying this particular outcome between clinicaltrials.gov and the study protocol published. |
| NCT01276223 | Some concerns | According to the trial registry information. However, details about treatment allocation were not reported. Participants in the Difluprednate group were 5.7 years younger (post‐hoc P = 0.004); otherwise sex distribution was not different between the two groups. | Low risk of bias | According to the trial registry, the study was double‐masked to the participants and the investigators. An ITT analysis that included all participants randomized was performed; missing data was based on imputation. | Low risk of bias | Data for this outcome were available for nearly all participants randomized; missing data for 7 participants who did not complete the study were imputed based on mixed model repeated measure approach. | Low risk of bias | A Visual Analog Scale (VAS) was used by the subject to assess ocular discomfort, both frequency and severity. Each scale was 100 millimeters (mm) in length. The Global Ocular Discomfort Score is a composite of the two VAS scores, ranging from 0 (very mildly) to 100 (very severely uncomfortable). The participant was masked to the intervention received, so it was unlikely that the assessment of this outcome could be differentially influenced. | Low risk of bias | Data for this outcome was analyzed according to the primary study outcome pre‐specified. | Some concerns | There were some concerns about the process of allocation concealment, which was not described at all by the authors. |
| Pflugfelder 2004 | Some concerns | Eligible patients received a study number and received double‐masked study medication (either loteprednol or placebo) according to a predetermined random allocation schedule. There were multiple baseline characteristics disproportionately distributed between the two intervention groups: female (P = 0.015), history of previous surgical procedures (P = 0.022), and history of punctal occlusion use (P = 0.033) among less than 20 variables considered. | Low risk of bias | Loteprednol etabonate was supplied in 7.5‐ml white, low‐density polyethylene dropper bottles with what caps for masking purposes. Placebo (the vehicle of LE) was supplied in identical bottles. The medication was double‐masked. All participants randomized to each intervention were analyzed according the intervention assigned. | Low risk of bias | In primary analysis, the authors reported mean changes in sign and symptoms (this outcome) for all patients randomzied. | Low risk of bias | Patient‐reported worst symptom in the worst eye was measured using a visual analogue scale of a 100mm line, with labels of Absent at the left end and Unbearable at the right end. Each patient was asked to mark the line at the point between these two extremes that represented the severity of the symptom; the distance from the left end of the line to the mark was the VAS (symptom) score in milimeters. Patients were masked by the medication received and thus likely to answer differently from reasons other than their perceived severity of the worst symptom. This was a patient‐reported outcome and patients were masked. | Low risk of bias | No trial protocol or analysis plan available for assessment but based on the power and sample size consideration, there might be a proper analytic plan pre‐specified before the trial. Results of eligible measures at different time points were also reported. Possible but not reported or discussed. | Some concerns | There were some concerns about the performance of the randomization process, evidenced by excessive imbalance between the two comparison groups. |
| Pinto‐Fraga 2016 | Some concerns | 'The generation of random allocation sequence and the statistical analyses were carried out by a PhD‐licensed statistician (I.F.), using the R statistical package version 3.1.1'. There were no significant differences in age or gender distribution between the 2 groups (P 1⁄4 0.37 and P 1⁄4 0.66, respectively). The authors did not describe the process of allocation concealment. | Low risk of bias | The original labels of these bottles were removed and bottles were relabeled with the study number and a coded lot number to avoid potential identification of the treatment by site personnel or patients. Treatment was not delivered or collected by any investigator who performed evaluations during the clinical trial, and none of the investigators were involved in the masking process described above. | Low risk of bias | One patient dropped out of the study due to work‐related issues and additional one participant was recruited to fill the gap, whose outcome data was also analyzed as treated. | Low risk of bias | Symptom scores were documented using OSDI and SANDE version I questionnaire by patients. Patients were unaware of their treatment received. Patients were unaware of their treatment medication and might not answer the questionnares about symptoms differentially. | Low risk of bias | Reporting of the outcome variables and analytic approaches was consistent between trial registry, the methods section and the result section of the report. SANDE version I and ODSI were used for symptom scores as primary and secondary outcomes, separately. Both the absoulte scores and changes in proportions of participants with symptom reduction were reported. | Some concerns | There were some concerns about |
| Qazi 2015 | Some concerns | No details about randomization process, including allocation concealment, were reported in the abstract, full‐text publication, or on clinicaltrials.gov. No significant differences in any parameters were found among comparison groups. | Some concerns | Quadruple masking (Participant, Care Provider, Investigator, Outcomes Assessor) was reported on clinicaltrials.gov. However, it was unclear how masking of participants were achieved except that, "during each visit, all participants had a complete masked ophthalmic evaluation." | Low risk of bias | "Six patients were withdrawn or lost to follow‐up before the 4‐week visit. Three aditional subjects, 1 in each group, did not complete the 8‐week visit". Data for this outcome were available for 17 out of 20 participants randomzeid (85%). | Low risk of bias | OSDI was applied to measure patient‐reported symptom severity. Participants were planned to be masked. | Some concerns | The numerical results were reported by convention, probably not being selected on the basis of significance. However, the trial registry history showed that the original timeframe for reporting outcomes was the count of inflammatory cells at 4 weeks and 8 weeks after taking the first dose of treatment, which was not reported in the abstract or the full text publication. | Some concerns | There were some concerns about the process of treatment allocation, masking, and selective reporting of the outcome. |
Risk of bias for analysis 1.13 Corneal fluorescein staining scores—Qazi: LE alone.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Cao 2018 | Some concerns | Eligible participants were randomly assigned to either the combined treatment or the control tretment gorup based on a random number table. No details about treatment allocation were reported. No information about baseline characteristics of the two comparison groups was provided. | Some concerns | Whether the participants or the carers/investigators were masked was not reported. There were no descriptions about deviations or lost‐to‐followup. | Low risk of bias | Data for this outcome were reported for all participants reportedly randomized. | High risk of bias | Examination procedure was described as clinical routines but the grading system was a unique one, without citing a reference. It was unclear if the examiner or the assessor was masked to the intervention received by the participant. If not masked, assessment of the outcome could have differed between the two groups. | High risk of bias | No pre‐specified analysis plan was available for assessment. Results of this and other study outcomes were reported as mean and standard deviations yet post‐hoc t‐test for between group differences at each follow‐up timepoint did not produce the same t‐statistics as reported in the publication (P values interpretation the same). | High risk of bias | There were concerns about the process of allocation concealment and whether examiners/carers were masked. As a result, the examination or assessment of the staining result could have been influenced by the knowledge of receiving the intervention treatment or not. Analysis of this ourcome data was not as described in the article and could not be verified by post‐hoc analysis. |
| Pinto‐Fraga 2016 | Some concerns | 'The generation of random allocation sequence and the statistical analyses were carried out by a PhD‐licensed statistician (I.F.), using the R statistical package version 3.1.1'. There were no significant differences in age or gender distribution between the 2 groups (P 1⁄4 0.37 and P 1⁄4 0.66, respectively). The authors did not describe the process of allocation concealment. | Low risk of bias | The original labels of these bottles were removed and bottles were relabeled with the study number and a coded lot number to avoid potential identification of the treatment by site personnel or patients. Treatment was not delivered or collected by any investigator who performed evaluations during the clinical trial, and none of the investigators were involved in the masking process described above. | Low risk of bias | Outcome data for all participants randomized were reported except for one who dropped out of the study. | Low risk of bias | Examination and grading of corneal staining were performed according to the Oxford global scheme and a modified Centre for Contact Lens Research (University of Waterloo, Ca) staining score. Treatment was not delivered or collected by any investigator who perfomred evaluations during the clinical trial, and none of the investigators were involved in the masking process described (here). | Low risk of bias | Percentages of participants with 1‐grade or more progress in staining scores were reported in accordance to the analytic approach described in the methods section. Absolute values in staining scores at each visit were also presented in the report. | Some concerns | There were some concerns about allocation concealment, which was not described. |
| Li 2021 | Some concerns | Participants were randomly assigned to the intervention or control group using a random number table. The process of allocation concealment was not reported. There were no age or sex differences between the group. | Some concerns | Whether participants, carers, or assesors were masked was not reported. There were two and four participants who failed to complete the study in the intervention and control group, separately; these drop‐outs might have less favored outcome due to poor responses or inadequate treatment. The deviation rates appeared statistically balanced between the two groups. Complete‐case analysis was perofmred to estimate the effect of assignment, which might not be appropriate but the impact on the results might not be substantial. | Low risk of bias | Data for this outcome were reported for 116 out of 122 participants (95.1%). | High risk of bias | The grading or scoring system used to assess the exam results was not reported. If assessors were not masked to the treatment received, the outcome grading could have differed between groups. | High risk of bias | No pre‐specified analysis plan available for assessment. The scoring or grading system was not reported either. The study randomized participants but reported this outcome for eyes. | High risk of bias | There were some concerns about the randomization process, particularly the procedure of allocation concealment, and in outcome measurement. There were also high risks of bias associated with selective outcome reporting. |
| Aragona 2013 | Some concerns | "Patients were randomized into 2 groups according to a computerized list." The authors did not report details about allocation concealment. Distribution of age, sex, and symptoms or eye test results at baseline appeared comparable between the two groups (Table 1). | Low risk of bias | Whether participants were masked was unclear, but the authors described that "the corticosteroid and vehicle eyedrops were dispensed in coded bottles; " the bottles and their content were identical in their appearance." "The statistical analysis of the results was carried out in a masked method by an investigator not involved in the study." | Low risk of bias | Data for this outcome were available for all participants randomized. | High risk of bias | The investigators applied the NEI scoring system to the corneal staining exam, the assessment of which could be differed between the intervention groups depending on the masking status of the assesors. The trial was reported to be double‐masked, however, it was unclear whether the participants or the carers were masked in addition to the analysts. | Some concerns | There was no pre‐specified statistical plan available for assessment. Data for this outcome were produced in a conventional way for continuous variables. | High risk of bias | There were some concerns about the process of allocation concealment and selective reporting. Besides, there was a high risk of bias associated with ascertainment of the staining results because it was unclear whether the carers or assessors were masked or not. |
| Akhlaq 2019 | Low risk of bias | The randomization will be in a 1:1 ratio using a permuted‐block design (which will be obtained using a computer‐based random code generator) into the two groups. Baseline characteristics of the study population were not reported in the conference abstract, except for age and the baseline ODSI scores. The later was statistically higher in the intervention than in the control group (53.53±29.7 vs. 34.46±20.33, post‐hoc P = 0.0255). | Low risk of bias | 'The randomization scheme and treatment assignment will be held in the pharmacy to ensure that the investigator and study team remain masked'. 'Participants, investigators, image analysts, and the sponsor (GSK) will be masked to the treatment group'. | Low risk of bias | Study results of 38 participants (67 eyes) were reported; it was unclear how many participants were randomized initially. | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol (0‐3 for each of 5 zones). The scoring system was based on a previously published scheme (NEI scoring system). According to the study protocol, investigators and image analysts were planned to be masked. | Some concerns | No analytic plan for this particular outcome was available, except for the scale/questionnaire to be used (NEI grading scheme). | Some concerns | The only concerns arose from the unclear reporting about how this outcome was planned to be analyzed and reported. There were no study protocol or analytic plan for evaluation. |
| Lee 2014 | Low risk of bias | A randomization sequence was created using EXCEL 2007 (Microsoft, Redmond, Washington, USA) with a 1:1 allocation using random block sizes of 2, 4, and 6, by an independent doctor. No significant differences in any parameters were found between groups before treatment. | Some concerns | The allocation sequence was concealed from the physician enrolling and assessing pa‐ tients in sequentially numbered, opaque, and sealed envelopes. After the content of the envelope was revealed, the physician and patients were aware of the allocation and the corresponding treatment. However, outcome assessors and data analysts were kept masked to the allocation. Four and six participants in the intervention and control group dropped out of the study. It was unclear whether these drop‐outs were associated with deviations from the intended treatment allocation. | Low risk of bias | Data for this outcome were available for 60 out of 70 participants randomzeid (85.7%). | Low risk of bias | Details about how corneal staining scores were graded in one or more corneal zones were not provided. It was unclear how "corneal staining" was different from the total scores based on the Oxford grading scheme. Outcome assessors were masked. | High risk of bias | There was no pre‐specified analytic plan available for assessment. The sample size estimation was based on ANCOVA of repeatedly measured inflammatory cytokine levels. Results presented for this outcome (corneal staining scores) were based on post‐hoc linear mixed models. Model based estimates for Oxford staining scores were reported, instead of the raw scores. | High risk of bias | Main concerns arose from potentially high risk of selected reporting of this outcome results. Post‐hoc regression analysis was applied to analyze this outcome findings, instead of ANCOVA that was used for sample size estimation. |
| Avunduk 2003 | Some concerns | Patients were randomized to three groups according to a computerized list generated by the LSU Eye Center biostatistician. Details about allocation concealment were not reported. At the beginning of the study (day 0), no significant difference was detected between groups in terms of any parameters studied. | Low risk of bias | This was a single‐masked clinical tiral. The examiner was masked as to the medication used by the patients. The patients were instructed to discuss their medications only with the study coordinator and not with the examiner. No additional information was reported. After excluding 4 participants who did not complete the study, all other participants randomzied were analyzed for the study outcomes. | Some concerns | Data for this outcome were available for 19 participants (8 in ATSand 11 in FML+ATS group) as well as 9 in NSAID group; outcome data for the 4 participants who discontinued the study were not reported. It was unclear the numbers of participants initially randomzied to each comparison group. | Low risk of bias | Examination procedures and the scoring system used followed the NEI Dry Eye Workshop. The examiner was masked to the medication used by the patients. | Some concerns | Data for this outcome was analyzed according to the analysis plan described in the methods section. However, reporting of this outcome was inadequate, with only P‐values reported though the data points presented on graphs. | Some concerns | There were som concerns about allocation concealment and selective outcome reporting. |
| Chen 2020 | Some concerns | Particpants were reportedly randomzied to receiving either HA (Group A) or loteprednol plus HA (Group B), but the details about the randomization procedure or the allocation process were not reported. No significant differences in age or sex distribution were reported. | Some concerns | Whether participants or carers and other study personnels were masked or not was not reported. | Low risk of bias | Data for participants randomized were available for this outcome. | High risk of bias | The grading system was different from the NEI or Oxford scoring system. The scale was 0 to 12, which is a sum of 0 to 3 from each of four quandrants assessed. Depending on the masking status of the examiner or assessor, the measurement and the grading of the outcome could have differed. | Some concerns | No pres‐pecified analysis plan or study protocol was available for assessment. Outcome data were reported as before and after measurements. It was unclear whether there were potentially multiple eligible measurements after the planned 1 month of treatment. | High risk of bias | Particpants were reportedly randomzied to receiving either HA (Group A) or loteprednol plus HA (Group B), but the details about the processs of allocation treatment were not reported. No significant differences in age or sex distribution were reported. Whether participants or carers and other study personnels were masked or not was not reported. |
| KPI‐121 (Phase 2) | Low risk of bias | The protocol stated that "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | All participants randomized were reported. | Low risk of bias | Examination procedures were described as in routine practice. Results assessment by examiners or assessors could not have been influenced because subjects and the investigative staff were masked. | Low risk of bias | The study protocol did not specify which study outcome as the basis for sample size estimation. However, the study protocol did specify to compare mean corneal staining (socres) between the two groups as one of the secondary efficacy endpoints. | Low risk of bias | The trial was judged to be at low risk across multiple domains. |
| KPI‐121 (STRIDE3) | Low risk of bias | "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the Sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. | Low risk of bias | Eleven participants did not complete this trial. Data for 890 participants were reported for this outcome (98.8%). | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol in accordance to the NEI Dry Eye Workshop. Assessors were masked to the intervention received by each participant. | Some concerns | Sample size estimation was not based on this outcome and there were no corresponding analytic plans described on how to analyze data for this outcome. | Some concerns | There were some concerns about selective outcome reporting as this outcome was not pre‐specified in the study protocol. |
| KPI‐121 (STRIDE1) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. | Low risk of bias | Seven participants did not complete this study. Data for 907 participants were reported for this outcome (99.1%). | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol in accordance to the NEI Dry Eye Workshop Protocol/ Guideline Assessors were masked to the intervention received by each participant. | Low risk of bias | Mean change in this outcome was used in sample size estimation. Data for this outcome was reported in accordance with the specified analysis. | Low risk of bias | The current study was judged to be at low risk of bias across multiple domains. |
| KPI‐121 (STRIDE2) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | Ten participants did not complete the study. Data for his outcome were available for 896 participants (99.0%). | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol in accordance to the NEI Dry Eye Workshop. Assessors were masked to the intervention received by each participant. | Low risk of bias | Mean change in this outcome was used in sample size estimation. Data for this outcome was reported in accordance with the specified analysis. | Low risk of bias | The study was assessed as having low risk of bias across multiple domains. |
| Pflugfelder 2004 | Some concerns | Eligible patients received a study number and received double‐masked study medication (either loteprednol or placebo) according to a predetermined random allocation schedule. There were multiple baseline characteristics disproportionately distributed between the two intervention groups: female (P = 0.015), history of previous surgical procedures (P = 0.022), and history of punctal occlusion use (P = 0.033) among less than 20 variables considered. | Low risk of bias | Loteprednol etabonate was supplied in 7.5‐ml white, low‐density polyethylene dropper bottles with what caps for masking purposes. Placebo (the vehicle of LE) was supplied in identical bottles. The medication was double‐masked. All participants randomized to each intervention were analyzed according the intervention assigned. | Low risk of bias | Data for this outcome were available for all participants randomzied. | High risk of bias | Examination procedures and the scoring system (0‐4 for rose bengal, 0‐6 for fluoroscein) were described; both eyes of the study participants were examined based on the same protocol. The medication described as double‐masked to the participants yet it was unclear who the other party was also masked. | Some concerns | Data for this outcome was analyzed according to the metrics (a continuous measure based on the composite fluorescein staining score in the worst eye) considered for sample size estimation. However, mean percentage changes from baseline were also presented in graphs; this scale of quantifying changes was not pre‐specified. | Some concerns | There were some concerns about the performance of proper randomization and selective outcome reporting. |
| Qazi 2015 | Some concerns | No details about randomization process, including allocation concealment, were reported in the abstract, full‐text publication, or on clinicaltrials.gov. No significant differences in any parameters were found among comparison groups. | Some concerns | Quadruple masking (Participant, Care Provider, Investigator, Outcomes Assessor) was reported on clinicaltrials.gov. However, it was unclear how masking of participants were achieved except that, "during each visit, all participants had a complete masked ophthalmic evaluation." | Low risk of bias | "Six patients were withdrawn or lost to follow‐up before the 4‐week visit. Three aditional subjects, 1 in each group, did not complete the 8‐week visit". Data for this outcome were available for 17 out of 20 participants randomzeid (85%). | Low risk of bias | The scale for grading corneal staining was reported (0‐15) but details about how corneal staining scores were graded in one or more corneal zones were not provided. Outcome assessors were planned to be masked. | Some concerns | The numerical results were reported by convention, probably not being selected on the basis of significance. However, the trial registry history showed that the original timeframe for reporting outcomes was the count of inflammatory cells at 4 weeks and 8 weeks after taking the first dose of treatment, which was not reported in the conference abstract or the full publication. | Some concerns | Trial registry history showed changes in the original timepoints for different outcomes specified in an earlier version of the record, leading to a main concern about potential risk of selected outcome reporting. There were also some concerns about the process of treatment allocation, and masking. |
| Luo 2013 | Some concerns | Subjects were randomly grouped into three intervention arms, but how treatment allocation was performed was not reported. No differences in age or sex distribution among the three groups were reported without presenting data for each group. Baseline values of the symptoms and clinical signs were reported and no substantial differences were shown. | Low risk of bias | Whether the paritcipants, carers, or examiners were masked during the study period was not reported. Participants' tolerability to the treatment was not reported. Data of this outcome were analyzed in a conventional before versus after approach. | Low risk of bias | Data of this outcome were available for all participants reported randomzied. | High risk of bias | Assessment of this outcome measurement incldued both cornea and conjunctiva (9 regions) with each scale ranging from 0 to 3. Whether the examiners or the assessors were masked was not reported. Assessors, if unmasked, could have graded the exam results differently among the groups. | High risk of bias | No pre‐specified analysis plan was avvailable for assessment. Results were analyzed by conventional before‐versus‐after comparisons. Assessment incldued both cornea and conjunctiva but reporting only included corneal staining scores. | High risk of bias | There were high risks of bias associated with assessing this particular outcome (whether the assessors were masked was unclear) and selective reporting of the outcome. |
Risk of bias for analysis 1.15 Corneal fluorescein staining score—Qazi: LE + tobramycin.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Cao 2018 | Some concerns | Eligible participants were randomly assigned to either the combined treatment or the control tretment gorup based on a random number table. No details about treatment allocation were reported. No information about baseline characteristics of the two comparison groups was provided. | Some concerns | Whether the participants or the carers/investigators were masked was not reported. There were no descriptions about deviations or lost‐to‐followup. | Low risk of bias | Data for this outcome were reported for all participants reportedly randomized. | High risk of bias | Examination procedure was described as clinical routines but the grading system was a unique one, without citing a reference. It was unclear if the examiner or the assessor was masked to the intervention received by the participant. If not masked, assessment of the outcome could have differed between the two groups. | High risk of bias | No pre‐specified analysis plan was available for assessment. Results of this and other study outcomes were reported as mean and standard deviations yet post‐hoc t‐test for between group differences at each follow‐up timepoint did not produce the same t‐statistics as reported in the publication (P values interpretation the same). | High risk of bias | There were concerns about the process of allocation concealment and whether examiners/carers were masked. As a result, the examination or assessment of the staining result could have been influenced by the knowledge of receiving the intervention treatment or not. Analysis of this ourcome data was not as described in the article and could not be verified by post‐hoc analysis. |
| Pinto‐Fraga 2016 | Some concerns | 'The generation of random allocation sequence and the statistical analyses were carried out by a PhD‐licensed statistician (I.F.), using the R statistical package version 3.1.1'. There were no significant differences in age or gender distribution between the 2 groups (P 1⁄4 0.37 and P 1⁄4 0.66, respectively). The authors did not describe the process of allocation concealment. | Low risk of bias | The original labels of these bottles were removed and bottles were relabeled with the study number and a coded lot number to avoid potential identification of the treatment by site personnel or patients. Treatment was not delivered or collected by any investigator who performed evaluations during the clinical trial, and none of the investigators were involved in the masking process described above. | Low risk of bias | Outcome data for all participants randomized were reported except for one who dropped out of the study. | Low risk of bias | Examination and grading of corneal staining were performed according to the Oxford global scheme and a modified Centre for Contact Lens Research (University of Waterloo, Ca) staining score. Treatment was not delivered or collected by any investigator who perfomred evaluations during the clinical trial, and none of the investigators were involved in the masking process described (here). | Low risk of bias | Percentages of participants with 1‐grade or more progress in staining scores were reported in accordance to the analytic approach described in the methods section. Absolute values in staining scores at each visit were also presented in the report. | Some concerns | There were some concerns about allocation concealment, which was not described. |
| Li 2021 | Some concerns | Participants were randomly assigned to the intervention or control group using a random number table. The process of allocation concealment was not reported. There were no age or sex differences between the group. | Some concerns | Whether participants, carers, or assesors were masked was not reported. There were two and four participants who failed to complete the study in the intervention and control group, separately; these drop‐outs might have less favored outcome due to poor responses or inadequate treatment. The deviation rates appeared statistically balanced between the two groups. Complete‐case analysis was perofmred to estimate the effect of assignment, which might not be appropriate but the impact on the results might not be substantial. | Low risk of bias | Data for this outcome were reported for 116 out of 122 participants (95.1%). | High risk of bias | The grading or scoring system used to assess the exam results was not reported. If assessors were not masked to the treatment received, the outcome grading could have differed between groups. | High risk of bias | No pre‐specified analysis plan available for assessment. The scoring or grading system was not reported either. The study randomized participants but reported this outcome for eyes. | High risk of bias | There were some concerns about the randomization process, particularly the procedure of allocation concealment, and in outcome measurement. There were also high risks of bias associated with selective outcome reporting. |
| Aragona 2013 | Some concerns | "Patients were randomized into 2 groups according to a computerized list." The authors did not report details about allocation concealment. Distribution of age, sex, and symptoms or eye test results at baseline appeared comparable between the two groups (Table 1). | Low risk of bias | Whether participants were masked was unclear, but the authors described that "the corticosteroid and vehicle eyedrops were dispensed in coded bottles; " the bottles and their content were identical in their appearance." "The statistical analysis of the results was carried out in a masked method by an investigator not involved in the study." | Low risk of bias | Data for this outcome were available for all participants randomized. | High risk of bias | The investigators applied the NEI scoring system to the corneal staining exam, the assessment of which could be differed between the intervention groups depending on the masking status of the assesors. The trial was reported to be double‐masked, however, it was unclear whether the participants or the carers were masked in addition to the analysts. | Some concerns | There was no pre‐specified statistical plan available for assessment. Data for this outcome were produced in a conventional way for continuous variables. | High risk of bias | There were some concerns about the process of allocation concealment and selective reporting. Besides, there was a high risk of bias associated with ascertainment of the staining results because it was unclear whether the carers or assessors were masked or not. |
| Akhlaq 2019 | Low risk of bias | The randomization will be in a 1:1 ratio using a permuted‐block design (which will be obtained using a computer‐based random code generator) into the two groups. Baseline characteristics of the study population were not reported in the conference abstract, except for age and the baseline ODSI scores. The later was statistically higher in the intervention than in the control group (53.53±29.7 vs. 34.46±20.33, post‐hoc P = 0.0255). | Low risk of bias | 'The randomization scheme and treatment assignment will be held in the pharmacy to ensure that the investigator and study team remain masked'. 'Participants, investigators, image analysts, and the sponsor (GSK) will be masked to the treatment group'. | Low risk of bias | Study results of 38 participants (67 eyes) were reported; it was unclear how many participants were randomized initially. | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol (0‐3 for each of 5 zones). The scoring system was based on a previously published scheme (NEI scoring system). According to the study protocol, investigators and image analysts were planned to be masked. | Some concerns | No analytic plan for this particular outcome was available, except for the scale/questionnaire to be used (NEI grading scheme). | Some concerns | The only concerns arose from the unclear reporting about how this outcome was planned to be analyzed and reported. There were no study protocol or analytic plan for evaluation. |
| Lee 2014 | Low risk of bias | A randomization sequence was created using EXCEL 2007 (Microsoft, Redmond, Washington, USA) with a 1:1 allocation using random block sizes of 2, 4, and 6, by an independent doctor. No significant differences in any parameters were found between groups before treatment. | Some concerns | The allocation sequence was concealed from the physician enrolling and assessing pa‐ tients in sequentially numbered, opaque, and sealed envelopes. After the content of the envelope was revealed, the physician and patients were aware of the allocation and the corresponding treatment. However, outcome assessors and data analysts were kept masked to the allocation. Four and six participants in the intervention and control group dropped out of the study. It was unclear whether these drop‐outs were associated with deviations from the intended treatment allocation. | Low risk of bias | Data for this outcome were available for 60 out of 70 participants randomzeid (85.7%). | Low risk of bias | Details about how corneal staining scores were graded in one or more corneal zones were not provided. It was unclear how "corneal staining" was different from the total scores based on the Oxford grading scheme. Outcome assessors were masked. | High risk of bias | There was no pre‐specified analytic plan available for assessment. The sample size estimation was based on ANCOVA of repeatedly measured inflammatory cytokine levels. Results presented for this outcome (corneal staining scores) were based on post‐hoc linear mixed models. Model based estimates for Oxford staining scores were reported, instead of the raw scores. | High risk of bias | Main concerns arose from potentially high risk of selected reporting of this outcome results. Post‐hoc regression analysis was applied to analyze this outcome findings, instead of ANCOVA that was used for sample size estimation. |
| Avunduk 2003 | Some concerns | Patients were randomized to three groups according to a computerized list generated by the LSU Eye Center biostatistician. Details about allocation concealment were not reported. At the beginning of the study (day 0), no significant difference was detected between groups in terms of any parameters studied. | Low risk of bias | This was a single‐masked clinical tiral. The examiner was masked as to the medication used by the patients. The patients were instructed to discuss their medications only with the study coordinator and not with the examiner. No additional information was reported. After excluding 4 participants who did not complete the study, all other participants randomzied were analyzed for the study outcomes. | Some concerns | Data for this outcome were available for 19 participants (8 in ATSand 11 in FML+ATS group) as well as 9 in NSAID group; outcome data for the 4 participants who discontinued the study were not reported. It was unclear the numbers of participants initially randomzied to each comparison group. | Low risk of bias | Examination procedures and the scoring system used followed the NEI Dry Eye Workshop. The examiner was masked to the medication used by the patients. | Some concerns | Data for this outcome was analyzed according to the analysis plan described in the methods section. However, reporting of this outcome was inadequate, with only P‐values reported though the data points presented on graphs. | Some concerns | There were som concerns about allocation concealment and selective outcome reporting. |
| Chen 2020 | Some concerns | Particpants were reportedly randomzied to receiving either HA (Group A) or loteprednol plus HA (Group B), but the details about the randomization procedure or the allocation process were not reported. No significant differences in age or sex distribution were reported. | Some concerns | Whether participants or carers and other study personnels were masked or not was not reported. | Low risk of bias | Data for participants randomized were available for this outcome. | High risk of bias | The grading system was different from the NEI or Oxford scoring system. The scale was 0 to 12, which is a sum of 0 to 3 from each of four quandrants assessed. Depending on the masking status of the examiner or assessor, the measurement and the grading of the outcome could have differed. | Some concerns | No pres‐pecified analysis plan or study protocol was available for assessment. Outcome data were reported as before and after measurements. It was unclear whether there were potentially multiple eligible measurements after the planned 1 month of treatment. | High risk of bias | Particpants were reportedly randomzied to receiving either HA (Group A) or loteprednol plus HA (Group B), but the details about the processs of allocation treatment were not reported. No significant differences in age or sex distribution were reported. Whether participants or carers and other study personnels were masked or not was not reported. |
| KPI‐121 (Phase 2) | Low risk of bias | The protocol stated that "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | All participants randomized were reported. | Low risk of bias | Examination procedures were described as in routine practice. Results assessment by examiners or assessors could not have been influenced because subjects and the investigative staff were masked. | Low risk of bias | The study protocol did not specify which study outcome as the basis for sample size estimation. However, the study protocol did specify to compare mean corneal staining (socres) between the two groups as one of the secondary efficacy endpoints. | Low risk of bias | The trial was judged to be at low risk across multiple domains. |
| KPI‐121 (STRIDE3) | Low risk of bias | "The randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the Sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. | Low risk of bias | Eleven participants did not complete this trial. Data for 890 participants were reported for this outcome (98.8%). | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol in accordance to the NEI Dry Eye Workshop. Assessors were masked to the intervention received by each participant. | Some concerns | Sample size estimation was not based on this outcome and there were no corresponding analytic plans described on how to analyze data for this outcome. | Some concerns | There were some concerns about selective outcome reporting as this outcome was not pre‐specified in the study protocol. |
| KPI‐121 (STRIDE1) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator. | Low risk of bias | Seven participants did not complete this study. Data for 907 participants were reported for this outcome (99.1%). | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol in accordance to the NEI Dry Eye Workshop Protocol/ Guideline Assessors were masked to the intervention received by each participant. | Low risk of bias | Mean change in this outcome was used in sample size estimation. Data for this outcome was reported in accordance with the specified analysis. | Low risk of bias | The current study was judged to be at low risk of bias across multiple domains. |
| KPI‐121 (STRIDE2) | Low risk of bias | The study protocol stated that "the randomization schedule will be generated by the randomization statistician (who is not on the project team) or designee and maintained in a secure and limited‐access location separate from the study Investigator and members of the project team". There were no significant differences in baseline characteristics between groups as shown in the results section on clinicaltrials.gov. | Low risk of bias | To minimize bias, investigational product allocation (KPI‐121 0.25% ophthalmic suspension versus vehicle) will be randomized and masked to the sponsor, subjects, and the investigative staff with the exception of a dosing coordinator who will be responsible for dispensing investigational product to subjects and instructing subjects regarding dosing of the investigational product. | Low risk of bias | Ten participants did not complete the study. Data for his outcome were available for 896 participants (99.0%). | Low risk of bias | Examination procedure and rading of corneal staining results were described in the study protocol in accordance to the NEI Dry Eye Workshop. Assessors were masked to the intervention received by each participant. | Low risk of bias | Mean change in this outcome was used in sample size estimation. Data for this outcome was reported in accordance with the specified analysis. | Low risk of bias | The study was assessed as having low risk of bias across multiple domains. |
| Pflugfelder 2004 | Some concerns | Eligible patients received a study number and received double‐masked study medication (either loteprednol or placebo) according to a predetermined random allocation schedule. There were multiple baseline characteristics disproportionately distributed between the two intervention groups: female (P = 0.015), history of previous surgical procedures (P = 0.022), and history of punctal occlusion use (P = 0.033) among less than 20 variables considered. | Low risk of bias | Loteprednol etabonate was supplied in 7.5‐ml white, low‐density polyethylene dropper bottles with what caps for masking purposes. Placebo (the vehicle of LE) was supplied in identical bottles. The medication was double‐masked. All participants randomized to each intervention were analyzed according the intervention assigned. | Low risk of bias | Data for this outcome were available for all participants randomzied. | High risk of bias | Examination procedures and the scoring system (0‐4 for rose bengal, 0‐6 for fluoroscein) were described; both eyes of the study participants were examined based on the same protocol. The medication described as double‐masked to the participants yet it was unclear who the other party was also masked. | Some concerns | Data for this outcome was analyzed according to the metrics (a continuous measure based on the composite fluorescein staining score in the worst eye) considered for sample size estimation. However, mean percentage changes from baseline were also presented in graphs; this scale of quantifying changes was not pre‐specified. | Some concerns | There were some concerns about the performance of proper randomization and selective outcome reporting. |
| Qazi 2015 | Some concerns | No details about randomization process, including allocation concealment, were reported in the abstract, full‐text publication, or on clinicaltrials.gov. No significant differences in any parameters were found among comparison groups. | Some concerns | Quadruple masking (Participant, Care Provider, Investigator, Outcomes Assessor) was reported on clinicaltrials.gov. However, it was unclear how masking of participants were achieved except that, "during each visit, all participants had a complete masked ophthalmic evaluation." | Low risk of bias | "Six patients were withdrawn or lost to follow‐up before the 4‐week visit. Three aditional subjects, 1 in each group, did not complete the 8‐week visit". Data for this outcome were available for 17 out of 20 participants randomzeid (85%). | Low risk of bias | The scale for grading corneal staining was reported (0‐15) but details about how corneal staining scores were graded in one or more corneal zones were not provided. Outcome assessors were planned to be masked. | Some concerns | The numerical results were reported by convention, probably not being selected on the basis of significance. However, the trial registry history showed that the original timeframe for reporting outcomes was the count of inflammatory cells at 4 weeks and 8 weeks after taking the first dose of treatment, which was not reported in the conference abstract or the full publication. | Some concerns | Trial registry history showed changes in the original timepoints for different outcomes specified in an earlier version of the record, leading to a main concern about potential risk of selected outcome reporting. There were also some concerns about the process of treatment allocation, and masking. |
| Luo 2013 | Some concerns | Subjects were randomly grouped into three intervention arms, but how treatment allocation was performed was not reported. No differences in age or sex distribution among the three groups were reported without presenting data for each group. Baseline values of the symptoms and clinical signs were reported and no substantial differences were shown. | Low risk of bias | Whether the paritcipants, carers, or examiners were masked during the study period was not reported. Participants' tolerability to the treatment was not reported. Data of this outcome were analyzed in a conventional before versus after approach. | Low risk of bias | Data of this outcome were available for all participants reported randomzied. | High risk of bias | Assessment of this outcome measurement incldued both cornea and conjunctiva (9 regions) with each scale ranging from 0 to 3. Whether the examiners or the assessors were masked was not reported. Assessors, if unmasked, could have graded the exam results differently among the groups. | High risk of bias | No pre‐specified analysis plan was avvailable for assessment. Results were analyzed by conventional before‐versus‐after comparisons. Assessment incldued both cornea and conjunctiva but reporting only included corneal staining scores. | High risk of bias | There were high risks of bias associated with assessing this particular outcome (whether the assessors were masked was unclear) and selective reporting of the outcome. |
Risk of bias for analysis 2.1 Patient‐reported symptom scores—Bausch: LE alone.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Byun 2012 | Some concerns | Subjects were randomy divided into two groups. However, the process of treatment allocation was not reported. No substantial differences or excessive similarity was noted between the two groups. | Some concerns | Whether masking was performed during the study was not reported. However, deviations from the intended intervention, such as cease of treatmnet, was reported in two participants in one of the group because of adverse effects. Analysis only included those who completed the study, thereby potentially biasing the effect estimate. | Some concerns | Data for this outcome were reported for 21 participants in group 1 (21 randomzied; 26 reported in the conference abstract) and 20 in group 2 (23 randomzied; 20 reported in the conference abstract). | Some concerns | Measurement was based on patient‐reported symptom using a scoring system not previously reported in the literature (0‐4) for 6 symptoms. Depending on the masking status of the participants to the treatment received, measurement could have differed between groups. | Some concerns | No pre‐specified analysis plan was avvailable for assessment. Results repeated measure analysis were reported for multiple timepoints for this outcome to answer the trial question that whether the addition of steroid to CsA could accelerate the improvement of one or more study outcomes. | High risk of bias | The trial was judged to have some concerns across multiple domains assessed for risk of bias. |
| Singla 2019 | Some concerns | Participants were randomly divided into two groups of 70 each. Details about how the random number sequence was generated or the treatment allocation was not reported. Both the groups had comparable baseline parameters, and there was no statistically significant difference between the two groups; there were no differences in mean age or sex distribution either. | High risk of bias | Whether masking was performed was not reported. There were no information regarding numbers of participants intitially randomized or of those who did not complete the study. Participants who did not complete the follow up for 6 months were excluded from the analysis (and the reporting). Results reported were likely based on complete‐case analysis; the degree of appropriateness would depend on the attrition rate at the end of the 3‐month treatment. | Some concerns | Data for this outcome were available for all participants analyzed. It was unclear how many participants were initially enrolled and randomized but were lost to follow up; the authors excluded these from the analysis. | Some concerns | OSDI questionnaire was used to document participant symptoms. Depending on whether the participants were masked or not, the measurement of patient‐reported symptoms could have differed. | Some concerns | No pre‐specified analysis plan was provided or reported. Results of this outcome were reported and analyzed in a conventional way, thus unlikely to have been selected based on the results. | High risk of bias | The trial was judged to have a high risk of participants deviating from the intended intervention due to an unclear attrition rate. There were also some concerns in the randomization process (excessive similarity between groups), missing outcome, outcome measurement and its reporting. |
| Wan 2012 | Some concerns | Subjects were randomly assigned to the intervention or control treatment. However, the process of allocation was not reported. There were no substantial differences or excessive similarity between the two groups. | Low risk of bias | Whether the participant, examiner, or assessor were masked to the intervention received by the paticipant was not reported. Participants might receive additional treatment outside of the study treatment but the possibility was unlikely increased by the trial. Analysis of this outcome was appropriate. | Low risk of bias | Data for this outcome were available for all participants reportedly randomized. | High risk of bias | Patient‐reported symptoms were based on an unvalidated scale for 5 related symptoms (0‐9); no relevant citations of the questionnaire was reported. Depending on the masking status of the participants, outcomes could have differed between the two groups. | Some concerns | No pre‐specified analysis plan was available for assessment. The unit of randomzation was participant; baseline values of the study outcomes were reported at the person level. However, outcome data collected at follow‐up were reported at the eye level though each participant contributed both eyes. | High risk of bias | The study was judged to be at high risk of measurement bias due to the use of unvalidated questionnaire; it was unclear whether participants were masked or not. There were also some concerns about selective outcome reporting. |
| Sheppard 2014 | Low risk of bias | Eligible patients were randomized 1:1 using a computer‐ generated randomization procedure. The identity of the drops (LE or AT) was masked to the subjects and the investigators. To achieve masking, an independent observer used an opaque label to conceal the manufacturers label on the drug bottles and applied nondescriptive identification tags. | Low risk of bias | The identity of the drops (LE or AT) was masked to the subjects and the investigators. | Low risk of bias | Data for this outcome were available for 112 out of 116 participants randomzeid (96.6%). | Low risk of bias | OSDI questionnaire was used to document participant symptoms. The participants were masked to the intervention received. | High risk of bias | According to the sample size calculation section, a repeated measure analysis of variance was planned but post‐hoc comparisons at multiple time points were reported in addition to results of Friedman repeated measures test, which was appropriate for ordinal measures. | High risk of bias | The trial was judged to be at high risk for selective outcome reporting. |
| Lin 2015 | Some concerns | The participants were randomly assigned to the FML or CsA group (n=20 each) at a 1:1 ratio using the permuted block method. Procedure of treatment allocation was not described. No significant differences in the baseline age or sex distribution were observed between the two groups. | Some concerns | The trial was open‐labeled (clinicaltrials.gov). One and 4 patients in the FML and the CsA group were lost to the follow‐up. It was unclear whether such differential lost‐to‐followup was related to deviatioins from the intervention assigned or not. | Low risk of bias | Data for this outcome were available for 35 participants (35/40 = 87.5%). | High risk of bias | OSDI qusetionnaire was used to document patient‐reported symptoms. Because of the self‐reporting nature of the questionnaire, knowing the intervention received could have potentially differentaily influenced the measurement of this outcome. | Some concerns | This outcome was not included in the pre‐specified outcome in the trial registry on clinicaltrials.gov. Results of this outcome were reported for multiple timepoints during the study in a conventional way. | High risk of bias | The study was judged to have a high risk of bias in selective outcome reporting and some concerns in other domains assessed. |
| Bausch 2013 | Some concerns | Participants were randomized to the treatments but the details about how random number sequences were generated or whether allocation was concealed were not reported. No significant differences in age or sex distribution among the three intervention arms were noted. | Low risk of bias | Participants were reported to be masked. Carers and intervention allocators might be aware of participants' assigned intervention. One participant in the combined intervention group was unmasked to the treatment received, yet none were found to be deviated from the intended intervention. Data were analyzed according to the treatment received. | Low risk of bias | Data for this outcome at week 4 was reported for all but one participants randomized (101/102 = 99.0%). | Low risk of bias | OSDI was applied to document patient‐reported symptom severity. Participants were masked to the treatment they received, so measurement of the outcome could not have differed among the intervention groups. | Some concerns | No pre‐specified analysis plan or study protocol was available for assessment. Eligible outcome measures were reported for the study eye at pre‐specified timepoints of the current protocol on the website. Results of this outcome were reported in the conventional way. | Some concerns | There were some concerns about random number generation, the process of allocation concealment, and selective outcome reporting. |
Risk of bias for analysis 2.9 Corneal fluorescein staining scores—Bausch: LE alone.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Singla 2019 | Some concerns | Participants were randomly divided into two groups of 70 each. Details about how the random number sequence was generated or the treatment allocation was not reported. Both the groups had comparable baseline parameters, and there was no statistically significant difference between the two groups; there were no differences in mean age or sex distribution either. | High risk of bias | Whether masking was performed was not reported. There was no information regarding numbers of participants intitially randomized or of those who did not complete the study. Participants who did not complete the follow up for 6 months were excluded from the analysis (and the reporting). Results reported were likely based on complete‐case analysis; the degree of appropriateness would depend on the attrition rate at the end of the 3‐month treatment. | Some concerns | Data for this outcome were available for all participants analyzed. It was unclear how many participants were initially enrolled and randomized but were lost to follow up; the authors excluded these from the analysis. | High risk of bias | Examination and scoring system were according to the NEI grading scheme. Depending on whether the examiners or the assessors were masked or not, the measurement or the assessment could have differed. | Some concerns | No pre‐specified analysis plan was provided or reported. Results of this outcome were reported and analyzed in a conventional way, thus unlikely to have been selected based on the results. | High risk of bias | The study was judged to be at high risk of deviation from the randomization process (excessive similarity between groups). There were also some concerns about biased measurement of the outcome and selective outcome reporting due to lack of information about masking and pre‐specified outcomes. |
| Byun 2012 | Some concerns | Subjects were randomy divided into two groups. However, the process of treatment allocation was not reported. No substantial differences or excessive similarity was noted between the two groups. | Some concerns | Whether masking was performed during the study was not reported. However, deviations from the intended intervention, such as cease of treatmnet, was reported in two participants in one of the group because of adverse effects. Analysis only included those who completed the study, thereby potentially biasing the effect estimate. | Some concerns | Data for this outcome were reported for 21 participants in group 1 (21 randomzied; 26 reported in the conference abstract) and 20 in group 2 (23 randomzied; 20 reported in the conference abstract). | High risk of bias | Measurement and grading were based on NEI workshop grid system. Depending on the masking status of the examiners or assessors to the treatment received, assessment could have differed between groups. | Some concerns | No pre‐specified analysis plan was avvailable for assessment. Results repeated measure analysis were reported for multiple timepoints for this outcome to answer the trial question that whether the addition of steroid to CsA could accelerate the improvement of one or more study outcomes. | High risk of bias | There were some concerns about risks of bias across all domains assessed. |
| Wan 2012 | Some concerns | Subjects were randomly assigned to the intervention or control treatment. However, the process of allocation was not reported. There were no substantial differences or excessive similarity between the two groups. | Low risk of bias | Whether the participant, examiner, or assessor were masked to the intervention received by the paticipant was not reported. Participants might receive additional treatment outside of the study treatment but the possibility was unlikely increased by the trial. Analysis of this outcome was appropriate. | Low risk of bias | Data for this outcome were available for all participants reportedly randomized. | High risk of bias | Examination procedures followed clinical routine though the scoring system was different from any previously‐validated scoring system. Depending on the masking status of the examiners or the assessors, outcomes could have differed between the two groups. | Some concerns | No pre‐specified analysis plan was available for assessment. The unit of randomzation was participant; baseline values of the study outcomes were reported at the person level. However, outcome data collected at follow‐up were reported at the eye level though each participant contributed both eyes. | High risk of bias | There were some concerns about allocation concealment, biased measurement due to unclear masking status of the assessors, and selective outcome reporting. |
| Sheppard 2014 | Low risk of bias | Eligible patients were randomized 1:1 using a computer‐ generated randomization procedure. The identity of the drops (LE or AT) was masked to the subjects and the investigators. To achieve masking, an independent observer used an opaque label to conceal the manufacturers label on the drug bottles and applied nondescriptive identification tags. | Low risk of bias | The identity of the drops (LE or AT) was masked to the subjects and the investigators. | Low risk of bias | Data for this outcome were available for 112 out of 116 participants randomzeid (96.6%). | Low risk of bias | Examination procedure was in accordance with routine clinical practice though the scoring system was applied to two regions of the corneal surface (central and inferior), rather than the standard 5 regions. The investigators were masked to the intervention received by the participants. | High risk of bias | According to the sample size calculation section, a repeated measure analysis of variance was planned but post‐hoc comparisons at multiple time points were reported in addition to results of Friedman repeated measures test, which was appropriate for ordinal measures. | High risk of bias | There were high risks of selective outcome reporting based on the sample size calculation reported. |
| Bausch 2013 | Some concerns | Participants were randomized to the treatments but the details about how random number sequences were generated or whether allocation was concealed were not reported. No significant differences in age or sex distribution among the three intervention arms were noted. | Low risk of bias | Participants were reported to be masked. Carers and intervention allocators might be aware of participants' assigned intervention. One participant in the combined intervention group was unmasked to the treatment received, yet none were found to be deviated from the intended intervention. Data were analyzed according to the treatment received. | Low risk of bias | Data for this outcome was reported for all but one participant randomized (101/102 = 99.0%). | Some concerns | Examination procedures and the grading system were reported in detail on clinicaltrials.gov. Outcome examiners and assesors were not masked to the treatment received by each participant, which might not influence the measurement but might influence the grading of the staining results. Based on the data reported, it did not appear that assessment of the outcome was biasedly influenced. | Some concerns | No pre‐specified analysis plan or study protocol was available for assessment. Eligible outcome measures were reported for the study eye at pre‐specified timepoints of the current protocol on the website. Results of eligible analyses were reported according to pre‐specified sets of data (the study eye and the average of both eyes). | Some concerns | There were some concerns about potential risk of bias across multiple domains, except for the one associated with missing outcome data. |
| Lin 2015 | Some concerns | The participants were randomly assigned to the FML or CsA group (n=20 each) at a 1:1 ratio using the permuted block method. Procedure of treatment allocation was not described. No significant differences in the baseline age or sex distribution were observed between the two groups. | Some concerns | The trial was open‐labeled (clinicaltrials.gov). One and 4 patients in the FML and the CsA group were lost to the follow‐up. It was unclear whether such differential lost‐to‐followup was related to deviatioins from the intervention assigned or not. | Low risk of bias | Data for this outcome were available for 35 participants (35/40 = 87.5%). | High risk of bias | Fluorescein staining scores were measured on 0‐3 point scale, in each quarter of corneal zone (0‐3). Grading of the corneal staining results was according to previously published literature. Because of the examiners and the assesors were unmasked to the intervention received by each participant, knowing the intervention received could have potentially differentaily influenced the measurement of this outcome. | Low risk of bias | This outcome was included in the pre‐specified outcomes in the trial registry on clinicaltrials.gov. Results of this outcome were reported for multiple timepoints during the study in a conventional way. | High risk of bias | There were some concerns about the process of allocation concealment, unmasking, and therefore bias associated with outcome measurement. |
Acknowledgements
We thank Lori Rosman, Information Specialist for Cochrane Eyes and Vision (CEV), who created and executed the electronic search strategies.
We also thank Genie Han and Renee Wilson, Assistant Managing Editors for CEV@US; and Anupa Shah, Managing Editor for CEV, for their support and guidance in the preparation of this review.
We would also like to thank the two anonymous reviewers for their comments for the review protocol; and Vishal Jhanji (University of Pittsburgh) and Tracy Doll (Pacific University) for the review manuscript.
Lastly, we thank Dr Ming‐Hsiung Shih for his assistance in independently reviewing and verifying data collected from full‐text reports that were published in Chinese.
This review was managed by CEV@US and was signed off for publication by Dr Gianni Virgili.
Appendices
Appendix 1. CENTRAL search strategy
#1 MeSH descriptor: [Dry Eye Syndromes] explode all trees #2 (dry near/2 eye*) #3 (ocular near/2 dry*) #4 MeSH descriptor: [Tears] explode all trees #5 tear* #6 MeSH descriptor: [Xerophthalmia] explode all trees #7 xerophthalmi* #8 MeSH descriptor: [Vitamin A Deficiency] explode all trees #9 ("vitamin A" near/3 deficien*) #10 ("avitaminosis a" or (retinol near/1 deficien*) or "hypovitaminosis A") #11 MeSH descriptor: [Keratoconjunctivitis Sicca] explode all trees #12 (Keratoconjunctiv* or "kerato conjunctivitis") #13 MeSH descriptor: [Sjogren's Syndrome] explode all trees #14 ((Sjogren* or Sjoegren*) near/2 (syndrom* or disease*)) #15 (sicca next/1 syndrom*) #16 MeSH descriptor: [Stevens‐Johnson Syndrome] explode all trees #17 (Steven* and Johnson and (syndrom* or disease*)) #18 MeSH descriptor: [Pemphigoid, Benign Mucous Membrane] explode all trees #19 (Benign and Muco* and Pemphigoid*) #20 (Cicatricial near/2 Pemphigoid*) #21 blepharoconjunctiviti* #22 MeSH descriptor: [Meibomian Glands] explode all trees #23 (meibomian or tarsal) #24 MeSH descriptor: [Lacrimal Apparatus Diseases] explode all trees #25 (lacrima* or epiphora) #26 {OR #1‐#25} #27 MeSH descriptor: [Adrenal Cortex Hormones] explode all trees #28 corticosteroid* OR glucocorticoid* OR mineralocorticoid* OR "adrenal cortex hormone" OR "adrenal cortex hormones" OR "adrenal cortical hormone" OR "adrenal cortical hormones" OR "adrenocortical hormone" OR "adrenocortical hormones" OR adrenocorticosteroid* OR corticoid* OR steroid* #29 MeSH descriptor: [Betamethasone] explode all trees #30 Betamethasone* OR adbeon OR becasone OR beprogel OR "beta methason" OR "beta methasone" OR "beta‐phos/ac" OR betacortril OR betadexamethasone OR betametasone OR betamethasolone OR betamethason OR betamethasonum OR betamethazone OR betason OR betnasol OR betnelan OR "betnesol v" OR "betnovate a" OR betsolan OR betsolon OR betsopart OR celestan OR celestene OR celeston OR celestona OR celestone OR cellestoderm OR cidoten OR dermobet OR diprolen OR flubenisolone OR methasone OR "nsc 39470" OR nsc39470 OR ophtamesone OR "rg 833" OR rg833 OR rinderon OR "sch 4831" OR sch4831 OR walacort OR "378‐44‐9" #31 "clobetasone butyrate" OR "cci 5537" OR cci5537 OR "clobetasone 17 butyrate" OR emovate OR eumovate OR "gr 2 1214" OR "gr 2‐1214" OR "gr 21214" OR "gr2‐1214" OR kindavate OR "sn 203" OR sn203 OR trimovate OR "25122‐57‐0" #32 MeSH descriptor: [Dexamethasone] explode all trees #33 Dexamethasone* OR adrecort OR adrenocot OR "aeroseb dex" OR "aeroseb‐d" OR aflucoson OR aflucosone OR alfalyl OR anaflogistico OR aphtasolon OR arcodexan OR arcodexane OR artrosone OR auxiron OR azium OR bidexol OR "bisu ds" OR calonat OR cebedex OR cetadexon OR colofoam OR corsona OR corsone OR cortastat OR cortidex OR cortidexason OR cortidrona OR cortidrone OR cortisumman OR "dacortina fuerte" OR "dacortine fuerte" OR dalalone OR danasone OR "de‐sone la" OR decacortin OR decadeltosona OR decadeltosone OR decaderm OR decadion OR decadran OR decadron OR decadronal OR decadrone OR decaesadril OR decagel OR decaject OR decalix OR decameth OR decamethasone OR decasone OR decaspray OR decasterolone OR decdan OR decilone OR decofluor OR dectancyl OR dekacort OR delladec OR deltafluoren OR deltafluorene OR dergramin OR deronil OR desacort OR desacortone OR desadrene OR desalark OR desameton OR desametone OR desigdron OR "dexa cortisyl" OR "dexa dabrosan" OR "dexa korti" OR "dexa scherosan" OR "dexa scherozon" OR "dexa scherozone" OR "dexa‐p" OR "dexacen 4" OR dexachel OR dexacort OR dexacortal OR dexacorten OR dexacortin OR dexacortisyl OR dexadabroson OR dexadecadrol OR dexadrol OR dexagel OR dexagen OR dexahelvacort OR dexakorti OR dexalien OR dexalocal OR dexame OR dexamecortin OR dexameson OR dexamesone OR dexametason OR dexametasone OR dexameth OR dexamethason OR dexamethazon OR dexamethazone OR dexamethonium OR dexamonozon OR dexan OR dexane OR dexano OR dexapot OR dexascheroson OR dexascherozon OR dexascherozone OR dexason OR dexasone OR dexinoral OR dexionil OR dexmethsone OR dexona OR dexone OR dexpak OR dextelan OR dextenza OR dextrasone OR dexycu OR dezone OR dibasona OR doxamethasone OR esacortene OR "ex s1" OR exadion OR exadione OR firmalone OR "fluormethyl prednisolone" OR fluormethylprednisolon OR fluormethylprednisolone OR fluormone OR fluorocort OR fluorodelta OR fluoromethylprednisolone OR fortecortin OR gammacorten OR gammacortene OR grosodexon OR grosodexone OR hemady OR hexadecadiol OR hexadecadrol OR hexadiol OR hexadrol OR isnacort OR "isopto dex" OR "isopto maxidex" OR "isopto‐dex" OR "isopto‐maxidex" OR isoptodex OR isoptomaxidex OR "lokalison f" OR loverine OR luxazone OR marvidione OR maxidex OR mediamethasone OR megacortin OR mephameson OR mephamesone OR metasolon OR metasolone OR "methazon ion" OR "methazone ion" OR methazonion OR methazonione OR methylfluorprednisolone OR "metisone lafi" OR mexasone OR millicorten OR millicortenol OR "mk 125" OR mk125 OR mymethasone OR neoforderx OR neofordex OR nisomethasona OR novocort OR "nsc 34521" OR nsc34521 OR "oftan‐dexa" OR opticorten OR opticortinol OR oradexan OR oradexon OR oradexone OR orgadrone OR ozurdex OR pidexon OR policort OR posurdex OR "predni f tablinen" OR "predni‐f" OR "prednisolone f" OR prodexona OR prodexone OR sanamethasone OR santenson OR santeson OR sawasone OR solurex OR spoloven OR sterasone OR thilodexine OR triamcimetil OR vexamet OR visumetazone OR visumethazone OR "50‐02‐2" #34 difluprednate* OR "cm 9155" OR cm9155 OR durezol OR epitopic OR myser OR "w 6309" OR w6309 OR "warner 6309" OR ENV905 OR "23674‐86‐4" #35 MeSH descriptor: [Fluorometholone] explode all trees #36 Fluorometholone* OR cortilet OR cortisdin OR delmeson OR delmesone OR efflumidex OR eflone OR flosef OR fluaton OR flucon OR fluforte liquifilm OR flulon OR flumelon OR flumetholon OR flumetholone OR flumex OR flumexo OR fluometholone OR fluoph OR "fluoro ophtal" OR "fluor‐op" OR fluorlon OR fluormetholon OR fluoromethalone OR fluoropos OR fml OR fuluson OR isopto flucon OR loticort OR methasite OR oxylone OR "426‐13‐1" #37 MeSH descriptor: [Loteprednol Etabonate] explode all trees #38 Loteprednol* OR alrex OR "cddd 5604" OR cddd5604 OR CEHOAC OR "Chloromethyl 17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate" OR "17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate, Chloromethyl" OR "Chloromethyl 17 ethoxycarbonyloxy 11 hydroxy 3 oxoandrosta 1,4 diene 17 carboxylate" OR eysuvis OR "hgp 1" OR hgp1 OR inveltys OR "kpi 121" OR kpi121 OR "le‐mpp" OR lotemax OR loterex OR loterox OR lotesoft OR "p 5604" OR p5604 OR "82034‐46‐6" #39 MeSH descriptor: [Prednisolone] explode all trees #40 Prednisolone* OR adelcort OR antisolon OR antisolone OR aprednislon OR aprednislone OR benisolon OR benisolone OR berisolon OR berisolone OR caberdelta OR capsoid OR "co hydeltra" OR codelcortone OR compresolon OR cortadeltona OR cortadeltone OR cortalone OR cortelinter OR cortisolone OR cotolone OR dacortin OR dacrotin OR decaprednil OR "decortin h" OR decortril OR "dehydro cortex" OR "dehydro hydrocortisone" OR "dehydro hydrocortisone" OR dehydrocortex OR dehydrocortisol OR dehydrocortisole OR dehydrohydrocortison OR dehydrohydrocortisone OR delcortol OR "delta 1 17 hydroxycorticosterone 21 acetate" OR "delta 1 hydrocortisone" OR "delta cortef" OR "delta cortril" OR "delta ef cortelan" OR "delta f" OR "delta hycortol" OR "delta hydrocortisone" OR "delta hydrocortisone" OR "delta ophticor" OR "delta stab" OR "delta‐cortef" OR "delta1 dehydrocortisol" OR "delta1 dehydrohydrocortisone" OR "delta1 hydrocortisone" OR deltacortef OR deltacortenolo OR deltacortil OR deltacortoil OR deltacortril OR deltaderm OR deltaglycortril OR deltahycortol OR deltahydrocortison OR deltahydrocortisone OR deltaophticor OR deltasolone OR deltastab OR deltidrosol OR deltisilone OR deltisolon OR deltisolone OR deltolasson OR deltolassone OR deltosona OR deltosone OR "depo‐predate" OR dermosolon OR dhasolone OR DiAdresonF OR "di adreson f" OR "di adresone f" OR "di‐adreson‐f" OR "diadreson f" OR "diadresone f" OR dicortol OR domucortone OR encortelon OR encortelone OR encortolon OR equisolon OR "fernisolone‐p" OR glistelone OR hefasolon OR "hostacortin h" OR hydeltra OR hydeltrone OR hydrelta OR hydrocortancyl OR hydrocortidelt OR hydrodeltalone OR hydrodeltisone OR hydroretrocortin OR hydroretrocortine OR inflanefran OR insolone OR "keteocort h" OR "key‐pred" OR lenisolone OR leocortol OR liquipred OR "lygal kopftinktur n" OR mediasolone OR meprisolon OR meprisolone OR metacortalon OR metacortalone OR metacortandralon OR metacortandralone OR metacortelone OR "meti derm" OR meticortelone OR metiderm OR morlone OR mydrapred OR "neo delta" OR nisolon OR nisolone OR "nsc 9120" OR nsc9120 OR opredsone OR panafcortelone OR panafcortolone OR panafort OR paracortol OR phlogex OR "pre cortisyl" OR preconin OR precortalon OR precortancyl OR precortisyl OR "pred‐ject‐50" OR "predacort 50" OR "predaject‐50" OR "predalone 50" OR predartrina OR predartrine OR predate OR "predate‐50" OR predeltilone OR predisole OR predisyr OR "predne dome" OR prednecort OR prednedome OR prednelan OR "predni coelin" OR "predni h tablinen" OR "predni‐helvacort" OR prednicoelin OR prednicort OR prednicortelone OR "prednifor drops" OR predniment OR predniretard OR prednis OR prednisil OR prednisolon OR prednisolona OR prednivet OR prednorsolon OR prednorsolone OR predonine OR predorgasolona OR predorgasolone OR "pregna 1, 4 diene 11beta, 17alpha, 21 triol 3, 20 dione" OR prelon OR prelone OR prenilone OR prenin OR prenolone OR preventan OR prezolon OR rubycort OR scherisolon OR scherisolona OR serilone OR solondo OR solone OR solupren OR soluprene OR spiricort OR spolotane OR sterane OR sterolone OR supercortisol OR supercortizol OR taracortelone OR walesolone OR wysolone OR "50‐24‐8" #41 {OR #27‐#40} #42 #26 AND #41 in Trials
Appendix 2. MEDLINE (Ovid) search strategy
1 Randomized Controlled Trial.pt. 2 Controlled Clinical Trial.pt. 3 (randomized or randomised).ab,ti. 4 placebo.ab,ti. 5 drug therapy.fs. 6 randomly.ab,ti. 7 trial.ab,ti. 8 groups.ab,ti. 9 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 10 exp animals/ not humans.sh. 11 9 not 10 12 exp dry eye syndromes/ 13 (dry adj2 eye*).tw. 14 (ocular adj2 dry*).tw. 15 exp tears/ 16 tear*.tw. 17 exp xerophthalmia/ 18 xerophthalmi*.tw. 19 exp vitamin A deficiency/ 20 (vitamin A adj3 deficien*).tw. 21 (avitaminosis a or retinol deficien* or hypovitaminosis A).tw. 22 exp keratoconjunctivitis sicca/ 23 (Keratoconjunctiv* or kerato conjunctivitis).tw. 24 exp Keratoconjunctivitis/ 25 limit 24 to yr="1966 ‐ 1985" 26 exp Sjogren's syndrome/ 27 ((Sjogren* or Sjoegren*) adj2 (syndrom* or disease*)).tw. 28 sicca syndrom*.tw. 29 exp Stevens Johnson syndrome/ 30 (Steven* and Johnson and (syndrom* or disease*)).tw. 31 exp Pemphigoid, Benign Mucous Membrane/ 32 Benign Muco* Pemphigoid*.tw. 33 (Cicatricial adj2 Pemphigoid*).tw. 34 blepharoconjunctiviti$.tw. 35 exp meibomian glands/ 36 (meibomian or tarsal).tw. 37 exp lacrimal apparatus diseases/ 38 (lacrima* or epiphora).tw. 39 or/12‐23,25‐38 40 exp Adrenal Cortex Hormones/ 41 (corticosteroid* or glucocorticoid* or mineralocorticoid* or "adrenal cortex hormone" or "adrenal cortex hormones" or "adrenal cortical hormone" or "adrenal cortical hormones" or "adrenocortical hormone" or "adrenocortical hormones" or adrenocorticosteroid* or corticoid* or steroid*).tw. 42 exp Betamethasone/ 43 (Betamethasone* or adbeon or becasone or beprogel or "beta methason" or "beta methasone" or "beta‐phos/ac" or betacortril or betadexamethasone or betametasone or betamethasolone or betamethason or betamethasonum or betamethazone or betason or betnasol or betnelan or "betnesol v" or "betnovate a" or betsolan or betsolon or betsopart or celestan or celestene or celeston or celestona or celestone or cellestoderm or cidoten or dermobet or diprolen or flubenisolone or methasone or "nsc 39470" or nsc39470 or ophtamesone or "rg 833" or rg833 or rinderon or "sch 4831" or sch4831 or walacort or "378‐44‐9").tw,rn. 44 ("clobetasone butyrate" or "cci 5537" or cci5537 or "clobetasone 17 butyrate" or emovate or eumovate or "gr 2 1214" or "gr 2‐1214" or "gr 21214" or "gr2‐1214" or kindavate or "sn 203" or sn203 or trimovate or "25122‐57‐0").tw,rn. 45 exp Dexamethasone/ 46 (Dexamethasone* or adrecort or adrenocot or "aeroseb dex" or "aeroseb‐d" or aflucoson or aflucosone or alfalyl or anaflogistico or aphtasolon or arcodexan or arcodexane or artrosone or auxiron or azium or bidexol or "bisu ds" or calonat or cebedex or cetadexon or colofoam or corsona or corsone or cortastat or cortidex or cortidexason or cortidrona or cortidrone or cortisumman or "dacortina fuerte" or "dacortine fuerte" or dalalone or danasone or "de‐sone la" or decacortin or decadeltosona or decadeltosone or decaderm or decadion or decadran or decadron or decadronal or decadrone or decaesadril or decagel or decaject or decalix or decameth or decamethasone or decasone or decaspray or decasterolone or decdan or decilone or decofluor or dectancyl or dekacort or delladec or deltafluoren or deltafluorene or dergramin or deronil or desacort or desacortone or desadrene or desalark or desameton or desametone or desigdron or "dexa cortisyl" or "dexa dabrosan" or "dexa korti" or "dexa scherosan" or "dexa scherozon" or "dexa scherozone" or "dexa‐p" or "dexacen 4" or dexachel or dexacort or dexacortal or dexacorten or dexacortin or dexacortisyl or dexadabroson or dexadecadrol or dexadrol or dexagel or dexagen or dexahelvacort or dexakorti or dexalien or dexalocal or dexame or dexamecortin or dexameson or dexamesone or dexametason or dexametasone or dexameth or dexamethason or dexamethazon or dexamethazone or dexamethonium or dexamonozon or dexan or dexane or dexano or dexapot or dexascheroson or dexascherozon or dexascherozone or dexason or dexasone or dexinoral or dexionil or dexmethsone or dexona or dexone or dexpak or dextelan or dextenza or dextrasone or dexycu or dezone or dibasona or doxamethasone or esacortene or "ex s1" or exadion or exadione or firmalone or "fluormethyl prednisolone" or fluormethylprednisolon or fluormethylprednisolone or fluormone or fluorocort or fluorodelta or fluoromethylprednisolone or fortecortin or gammacorten or gammacortene or grosodexon or grosodexone or hemady or hexadecadiol or hexadecadrol or hexadiol or hexadrol or isnacort or "isopto dex" or "isopto maxidex" or "isopto‐dex" or "isopto‐maxidex" or isoptodex or isoptomaxidex or "lokalison f" or loverine or luxazone or marvidione or maxidex or mediamethasone or megacortin or mephameson or mephamesone or metasolon or metasolone or "methazon ion" or "methazone ion" or methazonion or methazonione or methylfluorprednisolone or "metisone lafi" or mexasone or millicorten or millicortenol or "mk 125" or mk125 or mymethasone or neoforderx or neofordex or nisomethasona or novocort or "nsc 34521" or nsc34521 or "oftan‐dexa" or opticorten or opticortinol or oradexan or oradexon or oradexone or orgadrone or ozurdex or pidexon or policort or posurdex or "predni f tablinen" or "predni‐f" or "prednisolone f" or prodexona or prodexone or sanamethasone or santenson or santeson or sawasone or solurex or spoloven or sterasone or thilodexine or triamcimetil or vexamet or visumetazone or visumethazone or "50‐02‐2").tw,rn. 47 (difluprednate* or "cm 9155" or cm9155 or durezol or epitopic or myser or "w 6309" or w6309 or "warner 6309" or ENV905 or "23674‐86‐4").tw,rn. 48 exp Fluorometholone/ 49 (Fluorometholone* or cortilet or cortisdin or delmeson or delmesone or efflumidex or eflone or flosef or fluaton or flucon or fluforte liquifilm or flulon or flumelon or flumetholon or flumetholone or flumex or flumexo or fluometholone or fluoph or "fluoro ophtal" or "fluor‐op" or fluorlon or fluormetholon or fluoromethalone or fluoropos or fml or fuluson or isopto flucon or loticort or methasite or oxylone or "426‐13‐1").tw,rn. 50 exp Loteprednol Etabonate/ 51 (Loteprednol* or alrex or "cddd 5604" or cddd5604 or CEHOAC or "Chloromethyl 17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate" or "17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate, Chloromethyl" or "Chloromethyl 17 ethoxycarbonyloxy 11 hydroxy 3 oxoandrosta 1,4 diene 17 carboxylate" or eysuvis or "hgp 1" or hgp1 or inveltys or "kpi 121" or kpi121 or "le‐mpp" or lotemax or loterex or loterox or lotesoft or "p 5604" or p5604 or "82034‐46‐6").tw,rn. 52 exp Prednisolone/ 53 (Prednisolone* or adelcort or antisolon or antisolone or aprednislon or aprednislone or benisolon or benisolone or berisolon or berisolone or caberdelta or capsoid or "co hydeltra" or codelcortone or compresolon or cortadeltona or cortadeltone or cortalone or cortelinter or cortisolone or cotolone or dacortin or dacrotin or decaprednil or "decortin h" or decortril or "dehydro cortex" or "dehydro hydrocortisone" or "dehydro hydrocortisone" or dehydrocortex or dehydrocortisol or dehydrocortisole or dehydrohydrocortison or dehydrohydrocortisone or delcortol or "delta 1 17 hydroxycorticosterone 21 acetate" or "delta 1 hydrocortisone" or "delta cortef" or "delta cortril" or "delta ef cortelan" or "delta f" or "delta hycortol" or "delta hydrocortisone" or "delta hydrocortisone" or "delta ophticor" or "delta stab" or "delta‐cortef" or "delta1 dehydrocortisol" or "delta1 dehydrohydrocortisone" or "delta1 hydrocortisone" or deltacortef or deltacortenolo or deltacortil or deltacortoil or deltacortril or deltaderm or deltaglycortril or deltahycortol or deltahydrocortison or deltahydrocortisone or deltaophticor or deltasolone or deltastab or deltidrosol or deltisilone or deltisolon or deltisolone or deltolasson or deltolassone or deltosona or deltosone or "depo‐predate" or dermosolon or dhasolone or DiAdresonF or "di adreson f" or "di adresone f" or "di‐adreson‐f" or "diadreson f" or "diadresone f" or dicortol or domucortone or encortelon or encortelone or encortolon or equisolon or "fernisolone‐p" or glistelone or hefasolon or "hostacortin h" or hydeltra or hydeltrone or hydrelta or hydrocortancyl or hydrocortidelt or hydrodeltalone or hydrodeltisone or hydroretrocortin or hydroretrocortine or inflanefran or insolone or "keteocort h" or "key‐pred" or lenisolone or leocortol or liquipred or "lygal kopftinktur n" or mediasolone or meprisolon or meprisolone or metacortalon or metacortalone or metacortandralon or metacortandralone or metacortelone or "meti derm" or meticortelone or metiderm or morlone or mydrapred or "neo delta" or nisolon or nisolone or "nsc 9120" or nsc9120 or opredsone or panafcortelone or panafcortolone or panafort or paracortol or phlogex or "pre cortisyl" or preconin or precortalon or precortancyl or precortisyl or "pred‐ject‐50" or "predacort 50" or "predaject‐50" or "predalone 50" or predartrina or predartrine or predate or "predate‐50" or predeltilone or predisole or predisyr or "predne dome" or prednecort or prednedome or prednelan or "predni coelin" or "predni h tablinen" or "predni‐helvacort" or prednicoelin or prednicort or prednicortelone or "prednifor drops" or predniment or predniretard or prednis or prednisil or prednisolon or prednisolona or prednivet or prednorsolon or prednorsolone or predonine or predorgasolona or predorgasolone or "pregna 1, 4 diene 11beta, 17alpha, 21 triol 3, 20 dione" or prelon or prelone or prenilone or prenin or prenolone or preventan or prezolon or rubycort or scherisolon or scherisolona or serilone or solondo or solone or solupren or soluprene or spiricort or spolotane or sterane or sterolone or supercortisol or supercortizol or taracortelone or walesolone or wysolone or "50‐24‐8").tw,rn. 54 or/40‐53 55 39 and 54 56 11 and 55
The search filter for trials at the beginning of the MEDLINE strategy is from the published paper by Glanville et al (Glanville 2006).
Appendix 3. Embase.com search strategy
#1 'randomized controlled trial'/exp #2 'randomization'/exp #3 'double blind procedure'/exp #4 'single blind procedure'/exp #5 random*:ab,ti #6 #1 OR #2 OR #3 OR #4 OR #5 #7 'animal'/exp OR 'animal experiment'/exp #8 'human'/exp #9 #7 AND #8 #10 #7 NOT #9 #11 #6 NOT #10 #12 'clinical trial'/exp #13 (clin* NEAR/3 trial*):ab,ti #14 ((singl* OR doubl* OR trebl* OR tripl*) NEAR/3 (blind* OR mask*)):ab,ti #15 'placebo'/exp #16 placebo*:ab,ti #17 random*:ab,ti #18 'experimental design'/exp #19 'crossover procedure'/exp #20 'control group'/exp #21 'latin square design'/exp #22 #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 #23 #22 NOT #10 #24 #23 NOT #11 #25 'comparative study'/exp #26 'evaluation'/exp #27 'prospective study'/`exp #28 control*:ab,ti OR prospectiv*:ab,ti OR volunteer*:ab,ti #29 #25 OR #26 OR #27 OR #28 #30 #29 NOT #10 #31 #30 NOT (#11 OR #23) #32 #11 OR #24 OR #31 #33 'dry eye'/exp #34 (dry NEAR/2 eye*):ab,ti,kw #35 (ocular NEAR/2 dry*):ab,ti,kw #36 'lacrimal fluid'/exp #37 tear*:ab,ti,kw #38 'xerophthalmia'/exp #39 xerophthalmi*:ab,ti,kw #40 'retinol deficiency'/exp #41 ('vitamin a' NEAR/3 deficien*):ab,ti,kw #42 'avitaminosis a':ab,ti,kw OR (retinol NEAR/1 deficien*):ab,ti,kw OR 'hypovitaminosis a':ab,ti,kw #43 'keratoconjunctivitis sicca'/exp #44 keratoconjunctiv*:ab,ti,kw OR 'kerato conjunctivitis':ab,ti,kw #45 'sjoegren syndrome'/exp #46 ((sjogren* OR sjoegren*) NEAR/2 (syndrom* OR disease*)):ab,ti,kw #47 (sicca NEXT/1 syndrom*):ab,ti,kw #48 'stevens johnson syndrome'/exp #49 steven*:ab,ti,kw AND johnson:ab,ti,kw AND (syndrom*:ab,ti,kw OR disease*:ab,ti,kw) #50 'mucous membrane pemphigoid'/exp #51 benign AND muco* AND pemphigoid*:ab,ti,kw #52 (cicatricial NEAR/2 pemphigoid*):ab,ti,kw #53 blepharoconjunctiviti*:ab,ti,kw #54 'meibomian gland'/exp #55 meibomian:ab,ti,kw OR tarsal:ab,ti,kw #56 'lacrimal gland disease'/exp #57 lacrima*:ab,ti,kw OR epiphora:ab,ti,kw #58 #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 OR #41 OR #42 OR #43 OR #44 OR #45 OR #46 OR #47 OR #48 OR #49 OR #50 OR #51 OR #52 OR #53 OR #54 OR #55 OR #56 OR #57 #59 'corticosteroid'/exp #60 corticosteroid*:ab,ti,kw,tn OR glucocorticoid*:ab,ti,kw,tn OR mineralocorticoid*:ab,ti,kw,tn OR 'adrenal cortex hormone':ab,ti,kw,tn OR 'adrenal cortex hormones':ab,ti,kw,tn OR 'adrenal cortical hormone':ab,ti,kw,tn OR 'adrenal cortical hormones':ab,ti,kw,tn OR 'adrenocortical hormone':ab,ti,kw,tn OR 'adrenocortical hormones':ab,ti,kw,tn OR adrenocorticosteroid*:ab,ti,kw,tn OR corticoid*:ab,ti,kw,tn OR steroid*:ab,ti,kw,tn #61 'betamethasone'/exp #62 betamethasone*:ab,ti,kw,tn,rn OR adbeon:ab,ti,kw,tn,rn OR becasone:ab,ti,kw,tn,rn OR beprogel:ab,ti,kw,tn,rn OR 'beta methason':ab,ti,kw,tn,rn OR 'beta methasone':ab,ti,kw,tn,rn OR 'beta‐phos/ac':ab,ti,kw,tn,rn OR betacortril:ab,ti,kw,tn,rn OR betadexamethasone:ab,ti,kw,tn,rn OR betametasone:ab,ti,kw,tn,rn OR betamethasolone:ab,ti,kw,tn,rn OR betamethason:ab,ti,kw,tn,rn OR betamethasonum:ab,ti,kw,tn,rn OR betamethazone:ab,ti,kw,tn,rn OR betason:ab,ti,kw,tn,rn OR betnasol:ab,ti,kw,tn,rn OR betnelan:ab,ti,kw,tn,rn OR 'betnesol v':ab,ti,kw,tn,rn OR 'betnovate a':ab,ti,kw,tn,rn OR betsolan:ab,ti,kw,tn,rn OR betsolon:ab,ti,kw,tn,rn OR betsopart:ab,ti,kw,tn,rn OR celestan:ab,ti,kw,tn,rn OR celestene:ab,ti,kw,tn,rn OR celeston:ab,ti,kw,tn,rn OR celestona:ab,ti,kw,tn,rn OR celestone:ab,ti,kw,tn,rn OR cellestoderm:ab,ti,kw,tn,rn OR cidoten:ab,ti,kw,tn,rn OR dermobet:ab,ti,kw,tn,rn OR diprolen:ab,ti,kw,tn,rn OR flubenisolone:ab,ti,kw,tn,rn OR methasone:ab,ti,kw,tn,rn OR 'nsc 39470':ab,ti,kw,tn,rn OR nsc39470:ab,ti,kw,tn,rn OR ophtamesone:ab,ti,kw,tn,rn OR 'rg 833':ab,ti,kw,tn,rn OR rg833:ab,ti,kw,tn,rn OR rinderon:ab,ti,kw,tn,rn OR 'sch 4831':ab,ti,kw,tn,rn OR sch4831:ab,ti,kw,tn,rn OR walacort:ab,ti,kw,tn,rn OR '378‐44‐9':ab,ti,kw,tn,rn #63 'clobetasone butyrate'/exp #64 'clobetasone butyrate':ab,ti,kw,tn,rn OR 'cci 5537':ab,ti,kw,tn,rn OR cci5537:ab,ti,kw,tn,rn OR 'clobetasone 17 butyrate':ab,ti,kw,tn,rn OR emovate:ab,ti,kw,tn,rn OR eumovate:ab,ti,kw,tn,rn OR 'gr 2 1214':ab,ti,kw,tn,rn OR 'gr 2‐1214':ab,ti,kw,tn,rn OR 'gr 21214':ab,ti,kw,tn,rn OR 'gr2‐1214':ab,ti,kw,tn,rn OR kindavate:ab,ti,kw,tn,rn OR 'sn 203':ab,ti,kw,tn,rn OR sn203:ab,ti,kw,tn,rn OR trimovate:ab,ti,kw,tn,rn OR '25122‐57‐0':ab,ti,kw,tn,rn #65 'dexamethasone'/exp #66 dexamethasone*:ab,ti,kw,tn,rn OR adrecort:ab,ti,kw,tn,rn OR adrenocot:ab,ti,kw,tn,rn OR 'aeroseb dex':ab,ti,kw,tn,rn OR 'aeroseb‐d':ab,ti,kw,tn,rn OR aflucoson:ab,ti,kw,tn,rn OR aflucosone:ab,ti,kw,tn,rn OR alfalyl:ab,ti,kw,tn,rn OR anaflogistico:ab,ti,kw,tn,rn OR aphtasolon:ab,ti,kw,tn,rn OR arcodexan:ab,ti,kw,tn,rn OR arcodexane:ab,ti,kw,tn,rn OR artrosone:ab,ti,kw,tn,rn OR auxiron:ab,ti,kw,tn,rn OR azium:ab,ti,kw,tn,rn OR bidexol:ab,ti,kw,tn,rn OR 'bisu ds':ab,ti,kw,tn,rn OR calonat:ab,ti,kw,tn,rn OR cebedex:ab,ti,kw,tn,rn OR cetadexon:ab,ti,kw,tn,rn OR colofoam:ab,ti,kw,tn,rn OR corsona:ab,ti,kw,tn,rn OR corsone:ab,ti,kw,tn,rn OR cortastat:ab,ti,kw,tn,rn OR cortidex:ab,ti,kw,tn,rn OR cortidexason:ab,ti,kw,tn,rn OR cortidrona:ab,ti,kw,tn,rn OR cortidrone:ab,ti,kw,tn,rn OR cortisumman:ab,ti,kw,tn,rn OR 'dacortina fuerte':ab,ti,kw,tn,rn OR 'dacortine fuerte':ab,ti,kw,tn,rn OR dalalone:ab,ti,kw,tn,rn OR danasone:ab,ti,kw,tn,rn OR 'de‐sone la':ab,ti,kw,tn,rn OR decacortin:ab,ti,kw,tn,rn OR decadeltosona:ab,ti,kw,tn,rn OR decadeltosone:ab,ti,kw,tn,rn OR decaderm:ab,ti,kw,tn,rn OR decadion:ab,ti,kw,tn,rn OR decadran:ab,ti,kw,tn,rn OR decadron:ab,ti,kw,tn,rn OR decadronal:ab,ti,kw,tn,rn OR decadrone:ab,ti,kw,tn,rn OR decaesadril:ab,ti,kw,tn,rn OR decagel:ab,ti,kw,tn,rn OR decaject:ab,ti,kw,tn,rn OR decalix:ab,ti,kw,tn,rn OR decameth:ab,ti,kw,tn,rn OR decamethasone:ab,ti,kw,tn,rn OR decasone:ab,ti,kw,tn,rn OR decaspray:ab,ti,kw,tn,rn OR decasterolone:ab,ti,kw,tn,rn OR decdan:ab,ti,kw,tn,rn OR decilone:ab,ti,kw,tn,rn OR decofluor:ab,ti,kw,tn,rn OR dectancyl:ab,ti,kw,tn,rn OR dekacort:ab,ti,kw,tn,rn OR delladec:ab,ti,kw,tn,rn OR deltafluoren:ab,ti,kw,tn,rn OR deltafluorene:ab,ti,kw,tn,rn OR dergramin:ab,ti,kw,tn,rn OR deronil:ab,ti,kw,tn,rn OR desacort:ab,ti,kw,tn,rn OR desacortone:ab,ti,kw,tn,rn OR desadrene:ab,ti,kw,tn,rn OR desalark:ab,ti,kw,tn,rn OR desameton:ab,ti,kw,tn,rn OR desametone:ab,ti,kw,tn,rn OR desigdron:ab,ti,kw,tn,rn OR 'dexa cortisyl':ab,ti,kw,tn,rn OR 'dexa dabrosan':ab,ti,kw,tn,rn OR 'dexa korti':ab,ti,kw,tn,rn OR 'dexa scherosan':ab,ti,kw,tn,rn OR 'dexa scherozon':ab,ti,kw,tn,rn OR 'dexa scherozone':ab,ti,kw,tn,rn OR 'dexa‐p':ab,ti,kw,tn,rn OR 'dexacen 4':ab,ti,kw,tn,rn OR dexachel:ab,ti,kw,tn,rn OR dexacort:ab,ti,kw,tn,rn OR dexacortal:ab,ti,kw,tn,rn OR dexacorten:ab,ti,kw,tn,rn OR dexacortin:ab,ti,kw,tn,rn OR dexacortisyl:ab,ti,kw,tn,rn OR dexadabroson:ab,ti,kw,tn,rn OR dexadecadrol:ab,ti,kw,tn,rn OR dexadrol:ab,ti,kw,tn,rn OR dexagel:ab,ti,kw,tn,rn OR dexagen:ab,ti,kw,tn,rn OR dexahelvacort:ab,ti,kw,tn,rn OR dexakorti:ab,ti,kw,tn,rn OR dexalien:ab,ti,kw,tn,rn OR dexalocal:ab,ti,kw,tn,rn OR dexame:ab,ti,kw,tn,rn OR dexamecortin:ab,ti,kw,tn,rn OR dexameson:ab,ti,kw,tn,rn OR dexamesone:ab,ti,kw,tn,rn OR dexametason:ab,ti,kw,tn,rn OR dexametasone:ab,ti,kw,tn,rn OR dexameth:ab,ti,kw,tn,rn OR dexamethason:ab,ti,kw,tn,rn OR dexamethazon:ab,ti,kw,tn,rn OR dexamethazone:ab,ti,kw,tn,rn OR dexamethonium:ab,ti,kw,tn,rn OR dexamonozon:ab,ti,kw,tn,rn OR dexan:ab,ti,kw,tn,rn OR dexane:ab,ti,kw,tn,rn OR dexano:ab,ti,kw,tn,rn OR dexapot:ab,ti,kw,tn,rn OR dexascheroson:ab,ti,kw,tn,rn OR dexascherozon:ab,ti,kw,tn,rn OR dexascherozone:ab,ti,kw,tn,rn OR dexason:ab,ti,kw,tn,rn OR dexasone:ab,ti,kw,tn,rn OR dexinoral:ab,ti,kw,tn,rn OR dexionil:ab,ti,kw,tn,rn OR dexmethsone:ab,ti,kw,tn,rn OR dexona:ab,ti,kw,tn,rn OR dexone:ab,ti,kw,tn,rn OR dexpak:ab,ti,kw,tn,rn OR dextelan:ab,ti,kw,tn,rn OR dextenza:ab,ti,kw,tn,rn OR dextrasone:ab,ti,kw,tn,rn OR dexycu:ab,ti,kw,tn,rn OR dezone:ab,ti,kw,tn,rn OR dibasona:ab,ti,kw,tn,rn OR doxamethasone:ab,ti,kw,tn,rn OR esacortene:ab,ti,kw,tn,rn OR 'ex s1':ab,ti,kw,tn,rn OR exadion:ab,ti,kw,tn,rn OR exadione:ab,ti,kw,tn,rn OR firmalone:ab,ti,kw,tn,rn OR 'fluormethyl prednisolone':ab,ti,kw,tn,rn OR fluormethylprednisolon:ab,ti,kw,tn,rn OR fluormethylprednisolone:ab,ti,kw,tn,rn OR fluormone:ab,ti,kw,tn,rn OR fluorocort:ab,ti,kw,tn,rn OR fluorodelta:ab,ti,kw,tn,rn OR fluoromethylprednisolone:ab,ti,kw,tn,rn OR fortecortin:ab,ti,kw,tn,rn OR gammacorten:ab,ti,kw,tn,rn OR gammacortene:ab,ti,kw,tn,rn OR grosodexon:ab,ti,kw,tn,rn OR grosodexone:ab,ti,kw,tn,rn OR hemady:ab,ti,kw,tn,rn OR hexadecadiol:ab,ti,kw,tn,rn OR hexadecadrol:ab,ti,kw,tn,rn OR hexadiol:ab,ti,kw,tn,rn OR hexadrol:ab,ti,kw,tn,rn OR isnacort:ab,ti,kw,tn,rn OR 'isopto dex':ab,ti,kw,tn,rn OR 'isopto maxidex':ab,ti,kw,tn,rn OR 'isopto‐dex':ab,ti,kw,tn,rn OR 'isopto‐maxidex':ab,ti,kw,tn,rn OR isoptodex:ab,ti,kw,tn,rn OR isoptomaxidex:ab,ti,kw,tn,rn OR 'lokalison f':ab,ti,kw,tn,rn OR loverine:ab,ti,kw,tn,rn OR luxazone:ab,ti,kw,tn,rn OR marvidione:ab,ti,kw,tn,rn OR maxidex:ab,ti,kw,tn,rn OR mediamethasone:ab,ti,kw,tn,rn OR megacortin:ab,ti,kw,tn,rn OR mephameson:ab,ti,kw,tn,rn OR mephamesone:ab,ti,kw,tn,rn OR metasolon:ab,ti,kw,tn,rn OR metasolone:ab,ti,kw,tn,rn OR 'methazon ion':ab,ti,kw,tn,rn OR 'methazone ion':ab,ti,kw,tn,rn OR methazonion:ab,ti,kw,tn,rn OR methazonione:ab,ti,kw,tn,rn OR methylfluorprednisolone:ab,ti,kw,tn,rn OR 'metisone lafi':ab,ti,kw,tn,rn OR mexasone:ab,ti,kw,tn,rn OR millicorten:ab,ti,kw,tn,rn OR millicortenol:ab,ti,kw,tn,rn OR 'mk 125':ab,ti,kw,tn,rn OR mk125:ab,ti,kw,tn,rn OR mymethasone:ab,ti,kw,tn,rn OR neoforderx:ab,ti,kw,tn,rn OR neofordex:ab,ti,kw,tn,rn OR nisomethasona:ab,ti,kw,tn,rn OR novocort:ab,ti,kw,tn,rn OR 'nsc 34521':ab,ti,kw,tn,rn OR nsc34521:ab,ti,kw,tn,rn OR 'oftan‐dexa':ab,ti,kw,tn,rn OR opticorten:ab,ti,kw,tn,rn OR opticortinol:ab,ti,kw,tn,rn OR oradexan:ab,ti,kw,tn,rn OR oradexon:ab,ti,kw,tn,rn OR oradexone:ab,ti,kw,tn,rn OR orgadrone:ab,ti,kw,tn,rn OR ozurdex:ab,ti,kw,tn,rn OR pidexon:ab,ti,kw,tn,rn OR policort:ab,ti,kw,tn,rn OR posurdex:ab,ti,kw,tn,rn OR 'predni f tablinen':ab,ti,kw,tn,rn OR 'predni‐f':ab,ti,kw,tn,rn OR 'prednisolone f':ab,ti,kw,tn,rn OR prodexona:ab,ti,kw,tn,rn OR prodexone:ab,ti,kw,tn,rn OR sanamethasone:ab,ti,kw,tn,rn OR santenson:ab,ti,kw,tn,rn OR santeson:ab,ti,kw,tn,rn OR sawasone:ab,ti,kw,tn,rn OR solurex:ab,ti,kw,tn,rn OR spoloven:ab,ti,kw,tn,rn OR sterasone:ab,ti,kw,tn,rn OR thilodexine:ab,ti,kw,tn,rn OR triamcimetil:ab,ti,kw,tn,rn OR vexamet:ab,ti,kw,tn,rn OR visumetazone:ab,ti,kw,tn,rn OR visumethazone:ab,ti,kw,tn,rn OR '50‐02‐2':ab,ti,kw,tn,rn #67 'difluprednate'/exp #68 difluprednate*:ab,ti,kw,tn,rn OR 'cm 9155':ab,ti,kw,tn,rn OR cm9155:ab,ti,kw,tn,rn OR durezol:ab,ti,kw,tn,rn OR epitopic:ab,ti,kw,tn,rn OR myser:ab,ti,kw,tn,rn OR 'w 6309':ab,ti,kw,tn,rn OR w6309:ab,ti,kw,tn,rn OR 'warner 6309':ab,ti,kw,tn,rn OR env905:ab,ti,kw,tn,rn OR '23674‐86‐4':ab,ti,kw,tn,rn #69 'fluorometholone'/exp #70 ((fluorometholone*:ab,ti,kw,tn,rn OR cortilet:ab,ti,kw,tn,rn OR cortisdin:ab,ti,kw,tn,rn OR delmeson:ab,ti,kw,tn,rn OR delmesone:ab,ti,kw,tn,rn OR efflumidex:ab,ti,kw,tn,rn OR eflone:ab,ti,kw,tn,rn OR flosef:ab,ti,kw,tn,rn OR fluaton:ab,ti,kw,tn,rn OR flucon:ab,ti,kw,tn,rn OR fluforte:ab,ti,kw,tn,rn) AND liquifilm:ab,ti,kw,tn,rn OR flulon:ab,ti,kw,tn,rn OR flumelon:ab,ti,kw,tn,rn OR flumetholon:ab,ti,kw,tn,rn OR flumetholone:ab,ti,kw,tn,rn OR flumex:ab,ti,kw,tn,rn OR flumexo:ab,ti,kw,tn,rn OR fluometholone:ab,ti,kw,tn,rn OR fluoph:ab,ti,kw,tn,rn OR 'fluoro ophtal':ab,ti,kw,tn,rn OR 'fluor‐op':ab,ti,kw,tn,rn OR fluorlon:ab,ti,kw,tn,rn OR fluormetholon:ab,ti,kw,tn,rn OR fluoromethalone:ab,ti,kw,tn,rn OR fluoropos:ab,ti,kw,tn,rn OR fml:ab,ti,kw,tn,rn OR fuluson:ab,ti,kw,tn,rn OR isopto:ab,ti,kw,tn,rn) AND flucon:ab,ti,kw,tn,rn OR loticort:ab,ti,kw,tn,rn OR methasite:ab,ti,kw,tn,rn OR oxylone:ab,ti,kw,tn,rn OR '426‐13‐1':ab,ti,kw,tn,rn #71 'loteprednol etabonate'/exp #72 loteprednol*:ab,ti,kw,tn,rn OR alrex:ab,ti,kw,tn,rn OR 'cddd 5604':ab,ti,kw,tn,rn OR cddd5604:ab,ti,kw,tn,rn OR cehoac:ab,ti,kw,tn,rn OR 'chloromethyl 17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate':ab,ti,kw,tn,rn OR '17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate, chloromethyl':ab,ti,kw,tn,rn OR 'chloromethyl 17 ethoxycarbonyloxy 11 hydroxy 3 oxoandrosta 1,4 diene 17 carboxylate':ab,ti,kw,tn,rn OR eysuvis:ab,ti,kw,tn,rn OR 'hgp 1':ab,ti,kw,tn,rn OR hgp1:ab,ti,kw,tn,rn OR inveltys:ab,ti,kw,tn,rn OR 'kpi 121':ab,ti,kw,tn,rn OR kpi121:ab,ti,kw,tn,rn OR 'le‐mpp':ab,ti,kw,tn,rn OR lotemax:ab,ti,kw,tn,rn OR loterex:ab,ti,kw,tn,rn OR loterox:ab,ti,kw,tn,rn OR lotesoft:ab,ti,kw,tn,rn OR 'p 5604':ab,ti,kw,tn,rn OR p5604:ab,ti,kw,tn,rn OR '82034‐46‐6':ab,ti,kw,tn,rn #73 'prednisolone'/exp #74 prednisolone*:ab,ti,kw,tn,rn OR adelcort:ab,ti,kw,tn,rn OR antisolon:ab,ti,kw,tn,rn OR antisolone:ab,ti,kw,tn,rn OR aprednislon:ab,ti,kw,tn,rn OR aprednislone:ab,ti,kw,tn,rn OR benisolon:ab,ti,kw,tn,rn OR benisolone:ab,ti,kw,tn,rn OR berisolon:ab,ti,kw,tn,rn OR berisolone:ab,ti,kw,tn,rn OR caberdelta:ab,ti,kw,tn,rn OR capsoid:ab,ti,kw,tn,rn OR 'co hydeltra':ab,ti,kw,tn,rn OR codelcortone:ab,ti,kw,tn,rn OR compresolon:ab,ti,kw,tn,rn OR cortadeltona:ab,ti,kw,tn,rn OR cortadeltone:ab,ti,kw,tn,rn OR cortalone:ab,ti,kw,tn,rn OR cortelinter:ab,ti,kw,tn,rn OR cortisolone:ab,ti,kw,tn,rn OR cotolone:ab,ti,kw,tn,rn OR dacortin:ab,ti,kw,tn,rn OR dacrotin:ab,ti,kw,tn,rn OR decaprednil:ab,ti,kw,tn,rn OR 'decortin h':ab,ti,kw,tn,rn OR decortril:ab,ti,kw,tn,rn OR 'dehydro cortex':ab,ti,kw,tn,rn OR 'dehydro hydrocortisone':ab,ti,kw,tn,rn OR dehydrocortex:ab,ti,kw,tn,rn OR dehydrocortisol:ab,ti,kw,tn,rn OR dehydrocortisole:ab,ti,kw,tn,rn OR dehydrohydrocortison:ab,ti,kw,tn,rn OR dehydrohydrocortisone:ab,ti,kw,tn,rn OR delcortol:ab,ti,kw,tn,rn OR 'delta 1 17 hydroxycorticosterone 21 acetate':ab,ti,kw,tn,rn OR 'delta 1 hydrocortisone':ab,ti,kw,tn,rn OR 'delta cortef':ab,ti,kw,tn,rn OR 'delta cortril':ab,ti,kw,tn,rn OR 'delta ef cortelan':ab,ti,kw,tn,rn OR 'delta f':ab,ti,kw,tn,rn OR 'delta hycortol':ab,ti,kw,tn,rn OR 'delta hydrocortisone':ab,ti,kw,tn,rn OR 'delta ophticor':ab,ti,kw,tn,rn OR 'delta stab':ab,ti,kw,tn,rn OR 'delta‐cortef':ab,ti,kw,tn,rn OR 'delta1 dehydrocortisol':ab,ti,kw,tn,rn OR 'delta1 dehydrohydrocortisone':ab,ti,kw,tn,rn OR 'delta1 hydrocortisone':ab,ti,kw,tn,rn OR deltacortef:ab,ti,kw,tn,rn OR deltacortenolo:ab,ti,kw,tn,rn OR deltacortil:ab,ti,kw,tn,rn OR deltacortoil:ab,ti,kw,tn,rn OR deltacortril:ab,ti,kw,tn,rn OR deltaderm:ab,ti,kw,tn,rn OR deltaglycortril:ab,ti,kw,tn,rn OR deltahycortol:ab,ti,kw,tn,rn OR deltahydrocortison:ab,ti,kw,tn,rn OR deltahydrocortisone:ab,ti,kw,tn,rn OR deltaophticor:ab,ti,kw,tn,rn OR deltasolone:ab,ti,kw,tn,rn OR deltastab:ab,ti,kw,tn,rn OR deltidrosol:ab,ti,kw,tn,rn OR deltisilone:ab,ti,kw,tn,rn OR deltisolon:ab,ti,kw,tn,rn OR deltisolone:ab,ti,kw,tn,rn OR deltolasson:ab,ti,kw,tn,rn OR deltolassone:ab,ti,kw,tn,rn OR deltosona:ab,ti,kw,tn,rn OR deltosone:ab,ti,kw,tn,rn OR 'depo‐predate':ab,ti,kw,tn,rn OR dermosolon:ab,ti,kw,tn,rn OR dhasolone:ab,ti,kw,tn,rn OR diadresonf:ab,ti,kw,tn,rn OR 'di adreson f':ab,ti,kw,tn,rn OR 'di adresone f':ab,ti,kw,tn,rn OR 'di‐adreson‐f':ab,ti,kw,tn,rn OR 'diadreson f':ab,ti,kw,tn,rn OR 'diadresone f':ab,ti,kw,tn,rn OR dicortol:ab,ti,kw,tn,rn OR domucortone:ab,ti,kw,tn,rn OR encortelon:ab,ti,kw,tn,rn OR encortelone:ab,ti,kw,tn,rn OR encortolon:ab,ti,kw,tn,rn OR equisolon:ab,ti,kw,tn,rn OR 'fernisolone‐p':ab,ti,kw,tn,rn OR glistelone:ab,ti,kw,tn,rn OR hefasolon:ab,ti,kw,tn,rn OR 'hostacortin h':ab,ti,kw,tn,rn OR hydeltra:ab,ti,kw,tn,rn OR hydeltrone:ab,ti,kw,tn,rn OR hydrelta:ab,ti,kw,tn,rn OR hydrocortancyl:ab,ti,kw,tn,rn OR hydrocortidelt:ab,ti,kw,tn,rn OR hydrodeltalone:ab,ti,kw,tn,rn OR hydrodeltisone:ab,ti,kw,tn,rn OR hydroretrocortin:ab,ti,kw,tn,rn OR hydroretrocortine:ab,ti,kw,tn,rn OR inflanefran:ab,ti,kw,tn,rn OR insolone:ab,ti,kw,tn,rn OR 'keteocort h':ab,ti,kw,tn,rn OR 'key‐pred':ab,ti,kw,tn,rn OR lenisolone:ab,ti,kw,tn,rn OR leocortol:ab,ti,kw,tn,rn OR liquipred:ab,ti,kw,tn,rn OR 'lygal kopftinktur n':ab,ti,kw,tn,rn OR mediasolone:ab,ti,kw,tn,rn OR meprisolon:ab,ti,kw,tn,rn OR meprisolone:ab,ti,kw,tn,rn OR metacortalon:ab,ti,kw,tn,rn OR metacortalone:ab,ti,kw,tn,rn OR metacortandralon:ab,ti,kw,tn,rn OR metacortandralone:ab,ti,kw,tn,rn OR metacortelone:ab,ti,kw,tn,rn OR 'meti derm':ab,ti,kw,tn,rn OR meticortelone:ab,ti,kw,tn,rn OR metiderm:ab,ti,kw,tn,rn OR morlone:ab,ti,kw,tn,rn OR mydrapred:ab,ti,kw,tn,rn OR 'neo delta':ab,ti,kw,tn,rn OR nisolon:ab,ti,kw,tn,rn OR nisolone:ab,ti,kw,tn,rn OR 'nsc 9120':ab,ti,kw,tn,rn OR nsc9120:ab,ti,kw,tn,rn OR opredsone:ab,ti,kw,tn,rn OR panafcortelone:ab,ti,kw,tn,rn OR panafcortolone:ab,ti,kw,tn,rn OR panafort:ab,ti,kw,tn,rn OR paracortol:ab,ti,kw,tn,rn OR phlogex:ab,ti,kw,tn,rn OR 'pre cortisyl':ab,ti,kw,tn,rn OR preconin:ab,ti,kw,tn,rn OR precortalon:ab,ti,kw,tn,rn OR precortancyl:ab,ti,kw,tn,rn OR precortisyl:ab,ti,kw,tn,rn OR 'pred‐ject‐50':ab,ti,kw,tn,rn OR 'predacort 50':ab,ti,kw,tn,rn OR 'predaject‐50':ab,ti,kw,tn,rn OR 'predalone 50':ab,ti,kw,tn,rn OR predartrina:ab,ti,kw,tn,rn OR predartrine:ab,ti,kw,tn,rn OR predate:ab,ti,kw,tn,rn OR 'predate‐50':ab,ti,kw,tn,rn OR predeltilone:ab,ti,kw,tn,rn OR predisole:ab,ti,kw,tn,rn OR predisyr:ab,ti,kw,tn,rn OR 'predne dome':ab,ti,kw,tn,rn OR prednecort:ab,ti,kw,tn,rn OR prednedome:ab,ti,kw,tn,rn OR prednelan:ab,ti,kw,tn,rn OR 'predni coelin':ab,ti,kw,tn,rn OR 'predni h tablinen':ab,ti,kw,tn,rn OR 'predni‐helvacort':ab,ti,kw,tn,rn OR prednicoelin:ab,ti,kw,tn,rn OR prednicort:ab,ti,kw,tn,rn OR prednicortelone:ab,ti,kw,tn,rn OR 'prednifor drops':ab,ti,kw,tn,rn OR predniment:ab,ti,kw,tn,rn OR predniretard:ab,ti,kw,tn,rn OR prednis:ab,ti,kw,tn,rn OR prednisil:ab,ti,kw,tn,rn OR prednisolon:ab,ti,kw,tn,rn OR prednisolona:ab,ti,kw,tn,rn OR prednivet:ab,ti,kw,tn,rn OR prednorsolon:ab,ti,kw,tn,rn OR prednorsolone:ab,ti,kw,tn,rn OR predonine:ab,ti,kw,tn,rn OR predorgasolona:ab,ti,kw,tn,rn OR predorgasolone:ab,ti,kw,tn,rn OR 'pregna 1, 4 diene 11beta, 17alpha, 21 triol 3, 20 dione':ab,ti,kw,tn,rn OR prelon:ab,ti,kw,tn,rn OR prelone:ab,ti,kw,tn,rn OR prenilone:ab,ti,kw,tn,rn OR prenin:ab,ti,kw,tn,rn OR prenolone:ab,ti,kw,tn,rn OR preventan:ab,ti,kw,tn,rn OR prezolon:ab,ti,kw,tn,rn OR rubycort:ab,ti,kw,tn,rn OR scherisolon:ab,ti,kw,tn,rn OR scherisolona:ab,ti,kw,tn,rn OR serilone:ab,ti,kw,tn,rn OR solondo:ab,ti,kw,tn,rn OR solone:ab,ti,kw,tn,rn OR solupren:ab,ti,kw,tn,rn OR soluprene:ab,ti,kw,tn,rn OR spiricort:ab,ti,kw,tn,rn OR spolotane:ab,ti,kw,tn,rn OR sterane:ab,ti,kw,tn,rn OR sterolone:ab,ti,kw,tn,rn OR supercortisol:ab,ti,kw,tn,rn OR supercortizol:ab,ti,kw,tn,rn OR taracortelone:ab,ti,kw,tn,rn OR walesolone:ab,ti,kw,tn,rn OR wysolone:ab,ti,kw,tn,rn OR '50‐24‐8':ab,ti,kw,tn,rn #75 #59 OR #60 OR #61 OR #62 OR #63 OR #64 OR #65 OR #66 OR #67 OR #68 OR #69 OR #70 OR #71 OR #72 OR #73 OR #74 #76 #58 AND #75 #77 #32 AND #76
Appendix 4. PubMed search strategy
#1 ((randomized controlled trial[pt]) OR (controlled clinical trial[pt]) OR (randomised[tiab] OR randomized[tiab]) OR (placebo[tiab]) OR (drug therapy[sh]) OR (randomly[tiab]) OR (trial[tiab]) OR (groups[tiab])) NOT (animals[mh] NOT humans[mh]) #2 dry[tw] AND (eye[tw] OR eyes[tw] OR eyelid*[tw]) NOT Medline[sb] #3 (ocular[tw] AND dry[tw]) NOT Medline[sb] #4 tear*[tw] NOT Medline[sb] #5 xerophthalmi*[tw] NOT Medline[sb] #6 ("vitamin A"[tw] AND deficien*[tw]) NOT Medline[sb] #7 ("avitaminosis a"[tw] OR retinol deficien*[tw] OR "hypovitaminosis A"[tw]) NOT Medline[sb] #8 (Keratoconjunctiv*[tw] OR "kerato conjunctivitis"[tw]) NOT Medline[sb] #9 ((Sjogren*[tw] OR Sjoegren*[tw]) AND (syndrom*[tw] OR disease[tw] OR diseases[tw])) NOT Medline[sb] #10 sicca syndrom*[tw] NOT Medline[sb] #11 (Steven*[tw] AND Johnson[tw] AND (syndrom*[tw] OR disease[tw] OR diseases[tw])) NOT Medline[sb] #12 (Cicatricial [tw] AND Pemphigoid*[tw]) NOT Medline[sb] #13 Blepharoconjunctiviti*[tw] NOT Medline[sb] #14 (meibomian[tw] OR tarsal[tw]) NOT Medline[sb] #15 (lacrima*[tw] OR epiphora[tw]) NOT Medline[sb] #16 #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 #17 corticosteroid*[tw] OR glucocorticoid*[tw] OR mineralocorticoid*[tw] OR "adrenal cortex hormone"[tw] OR "adrenal cortex hormones"[tw] OR "adrenal cortical hormone"[tw] OR "adrenal cortical hormones"[tw] OR "adrenocortical hormone"[tw] OR "adrenocortical hormones"[tw] OR adrenocorticosteroid*[tw] OR corticoid*[tw] OR steroid*[tw] #18 Betamethasone*[tw] OR adbeon[tw] OR becasone[tw] OR beprogel[tw] OR "beta methason"[tw] OR "beta methasone"[tw] OR "beta‐phos/ac"[tw] OR betacortril[tw] OR betadexamethasone[tw] OR betamethasone[tw] OR betamethasolone[tw] OR betamethasone[tw] OR betamethasonum[tw] OR betamethasone[tw] OR betason[tw] OR betnasol[tw] OR betnelan[tw] OR "betnesol v"[tw] OR "betnovate a"[tw] OR betsolan[tw] OR betsolon[tw] OR betsopart[tw] OR celestan[tw] OR celestene[tw] OR celeston[tw] OR celestona[tw] OR celestone[tw] OR cellestoderm[tw] OR cidoten[tw] OR dermobet[tw] OR diprolen[tw] OR flubenisolone[tw] OR methasone[tw] OR "nsc 39470"[tw] OR nsc39470[tw] OR ophtamesone[tw] OR "rg 833"[tw] OR rg833[tw] OR rinderon[tw] OR "sch 4831"[tw] OR sch4831[tw] OR walacort[tw] OR "378‐44‐9"[tw] #19 "clobetasone butyrate"[tw] OR "cci 5537"[tw] OR cci5537[tw] OR "clobetasone 17 butyrate"[tw] OR emovate[tw] OR eumovate[tw] OR "gr 2 1214"[tw] OR "gr 2‐1214"[tw] OR "gr 21214"[tw] OR "gr2‐1214"[tw] OR kindavate[tw] OR "sn 203"[tw] OR sn203[tw] OR trimovate[tw] OR "25122‐57‐0"[tw] #20 Dexamethasone*[tw] OR adrecort[tw] OR adrenocot[tw] OR "aeroseb dex"[tw] OR "aeroseb‐d"[tw] OR aflucoson[tw] OR aflucosone[tw] OR alfalyl[tw] OR anaflogistico[tw] OR aphtasolon[tw] OR arcodexan[tw] OR arcodexane[tw] OR artrosone[tw] OR auxiron[tw] OR azium[tw] OR bidexol[tw] OR "bisu ds"[tw] OR calonat[tw] OR cebedex[tw] OR cetadexon[tw] OR colofoam[tw] OR corsona[tw] OR corsone[tw] OR cortastat[tw] OR cortidex[tw] OR cortidexason[tw] OR cortidrona[tw] OR cortidrone[tw] OR cortisumman[tw] OR "dacortina fuerte"[tw] OR "dacortine fuerte"[tw] OR dalalone[tw] OR danasone[tw] OR "de‐sone la"[tw] OR decacortin[tw] OR decadeltosona[tw] OR decadeltosone[tw] OR decaderm[tw] OR decadion[tw] OR decadran[tw] OR decadron[tw] OR decadronal[tw] OR decadrone[tw] OR decaesadril[tw] OR decagel[tw] OR decaject[tw] OR decalix[tw] OR decameth[tw] OR decamethasone[tw] OR decasone[tw] OR decaspray[tw] OR decasterolone[tw] OR decdan[tw] OR decilone[tw] OR decofluor[tw] OR dectancyl[tw] OR dekacort[tw] OR delladec[tw] OR deltafluoren[tw] OR deltafluorene[tw] OR dergramin[tw] OR deronil[tw] OR desacort[tw] OR desacortone[tw] OR desadrene[tw] OR desalark[tw] OR desameton[tw] OR desametone[tw] OR desigdron[tw] OR "dexa cortisyl"[tw] OR "dexa dabrosan"[tw] OR "dexa korti"[tw] OR "dexa scherosan"[tw] OR "dexa scherozon"[tw] OR "dexa scherozone"[tw] OR "dexa‐p"[tw] OR "dexacen 4"[tw] OR dexachel[tw] OR dexacort[tw] OR dexacortal[tw] OR dexacorten[tw] OR dexacortin[tw] OR dexacortisyl[tw] OR dexadabroson[tw] OR dexadecadrol[tw] OR dexadrol[tw] OR dexagel[tw] OR dexagen[tw] OR dexahelvacort[tw] OR dexakorti[tw] OR dexalien[tw] OR dexalocal[tw] OR dexame[tw] OR dexamecortin[tw] OR dexameson[tw] OR dexamesone[tw] OR dexametason[tw] OR dexametasone[tw] OR dexameth[tw] OR dexamethason[tw] OR dexamethazon[tw] OR dexamethazone[tw] OR dexamethonium[tw] OR dexamonozon[tw] OR dexan[tw] OR dexane[tw] OR dexano[tw] OR dexapot[tw] OR dexascheroson[tw] OR dexascherozon[tw] OR dexascherozone[tw] OR dexason[tw] OR dexasone[tw] OR dexinoral[tw] OR dexionil[tw] OR dexmethsone[tw] OR dexona[tw] OR dexone[tw] OR dexpak[tw] OR dextelan[tw] OR dextenza[tw] OR dextrasone[tw] OR dexycu[tw] OR dezone[tw] OR dibasona[tw] OR doxamethasone[tw] OR esacortene[tw] OR "ex s1"[tw] OR exadion[tw] OR exadione[tw] OR firmalone[tw] OR "fluormethyl prednisolone"[tw] OR fluormethylprednisolon[tw] OR fluormethylprednisolone[tw] OR fluormone[tw] OR fluorocort[tw] OR fluorodelta[tw] OR fluoromethylprednisolone[tw] OR fortecortin[tw] OR gammacorten[tw] OR gammacortene[tw] OR grosodexon[tw] OR grosodexone[tw] OR hemady[tw] OR hexadecadiol[tw] OR hexadecadrol[tw] OR hexadiol[tw] OR hexadrol[tw] OR isnacort[tw] OR "isopto dex"[tw] OR "isopto maxidex"[tw] OR "isopto‐dex"[tw] OR "isopto‐maxidex"[tw] OR isoptodex[tw] OR isoptomaxidex[tw] OR "lokalison f"[tw] OR loverine[tw] OR luxazone[tw] OR marvidione[tw] OR maxidex[tw] OR mediamethasone[tw] OR megacortin[tw] OR mephameson[tw] OR mephamesone[tw] OR metasolon[tw] OR metasolone[tw] OR "methazon ion"[tw] OR "methazone ion"[tw] OR methazonion[tw] OR methazonione[tw] OR methylfluorprednisolone[tw] OR "metisone lafi"[tw] OR mexasone[tw] OR millicorten[tw] OR millicortenol[tw] OR "mk 125"[tw] OR mk125[tw] OR mymethasone[tw] OR neoforderx[tw] OR neofordex[tw] OR nisomethasona[tw] OR novocort[tw] OR "nsc 34521"[tw] OR nsc34521[tw] OR "oftan‐dexa"[tw] OR opticorten[tw] OR opticortinol[tw] OR oradexan[tw] OR oradexon[tw] OR oradexone[tw] OR orgadrone[tw] OR ozurdex[tw] OR pidexon[tw] OR policort[tw] OR posurdex[tw] OR "predni f tablinen"[tw] OR "predni‐f"[tw] OR "prednisolone f"[tw] OR prodexona[tw] OR prodexone[tw] OR sanamethasone[tw] OR santenson[tw] OR santeson[tw] OR sawasone[tw] OR solurex[tw] OR spoloven[tw] OR sterasone[tw] OR thilodexine[tw] OR triamcimetil[tw] OR vexamet[tw] OR visumetazone[tw] OR visumethazone[tw] OR "50‐02‐2"[tw] #21 difluprednate*[tw] OR "cm 9155"[tw] OR cm9155[tw] OR durezol[tw] OR epitopic[tw] OR myser[tw] OR "w 6309"[tw] OR w6309[tw] OR "warner 6309"[tw] OR ENV905[tw] OR "23674‐86‐4"[tw] #22 Fluorometholone[tw]* OR cortilet[tw] OR cortisdin[tw] OR delmeson[tw] OR delmesone[tw] OR efflumidex[tw] OR eflone[tw] OR flosef[tw] OR fluaton[tw] OR flucon[tw] OR "fluforte liquifilm"[tw] OR flulon[tw] OR flumelon[tw] OR flumetholon[tw] OR flumetholone[tw] OR flumex[tw] OR flumexo[tw] OR fluometholone[tw] OR fluoph[tw] OR "fluoro ophtal"[tw] OR "fluor‐op"[tw] OR fluorlon[tw] OR fluormetholon[tw] OR fluoromethalone[tw] OR fluoropos[tw] OR fml[tw] OR fuluson[tw] OR "isopto flucon"[tw] OR loticort[tw] OR methasite[tw] OR oxylone[tw] OR "426‐13‐1"[tw] #23 Loteprednol*[tw] OR alrex[tw] OR "cddd 5604"[tw] OR cddd5604[tw] OR CEHOAC[tw] OR "Chloromethyl 17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate"[tw] OR "17‐ethoxycarbonyloxy‐11‐hydroxy‐3‐oxoandrosta‐1,4‐diene‐17‐carboxylate, Chloromethyl"[tw] OR "Chloromethyl 17 ethoxycarbonyloxy 11 hydroxy 3 oxoandrosta 1,4 diene 17 carboxylate"[tw] OR eysuvis[tw] OR "hgp 1"[tw] OR hgp1[tw] OR inveltys[tw] OR "kpi 121"[tw] OR kpi121[tw] OR "le‐mpp"[tw] OR lotemax[tw] OR loterex[tw] OR loterox[tw] OR lotesoft[tw] OR "p 5604"[tw] OR p5604[tw] OR "82034‐46‐6"[tw] #24 Prednisolone*[tw] OR adelcort[tw] OR antisolon[tw] OR antisolone[tw] OR aprednislon[tw] OR aprednislone[tw] OR benisolon[tw] OR benisolone[tw] OR berisolon[tw] OR berisolone[tw] OR caberdelta[tw] OR capsoid[tw] OR "co hydeltra"[tw] OR codelcortone[tw] OR compresolon[tw] OR cortadeltona[tw] OR cortadeltone[tw] OR cortalone[tw] OR cortelinter[tw] OR cortisolone[tw] OR cotolone[tw] OR dacortin[tw] OR dacrotin[tw] OR decaprednil[tw] OR "decortin h"[tw] OR decortril[tw] OR "dehydro cortex"[tw] OR "dehydro hydrocortisone"[tw] OR "dehydro hydrocortisone"[tw] OR dehydrocortex[tw] OR dehydrocortisol[tw] OR dehydrocortisole[tw] OR dehydrohydrocortison[tw] OR dehydrohydrocortisone[tw] OR delcortol[tw] OR "delta 1 17 hydroxycorticosterone 21 acetate"[tw] OR "delta 1 hydrocortisone"[tw] OR "delta cortef"[tw] OR "delta cortril"[tw] OR "delta ef cortelan"[tw] OR "delta f"[tw] OR "delta hycortol"[tw] OR "delta hydrocortisone"[tw] OR "delta hydrocortisone"[tw] OR "delta ophticor"[tw] OR "delta stab"[tw] OR "delta‐cortef"[tw] OR "delta1 dehydrocortisol"[tw] OR "delta1 dehydrohydrocortisone"[tw] OR "delta1 hydrocortisone"[tw] OR deltacortef[tw] OR deltacortenolo[tw] OR deltacortil[tw] OR deltacortoil[tw] OR deltacortril[tw] OR deltaderm[tw] OR deltaglycortril[tw] OR deltahycortol[tw] OR deltahydrocortison[tw] OR deltahydrocortisone[tw] OR deltaophticor[tw] OR deltasolone[tw] OR deltastab[tw] OR deltidrosol[tw] OR deltisilone[tw] OR deltisolon[tw] OR deltisolone[tw] OR deltolasson[tw] OR deltolassone[tw] OR deltosona[tw] OR deltosone[tw] OR "depo‐predate"[tw] OR dermosolon[tw] OR dhasolone[tw] OR DiAdresonF[tw] OR "di adreson f"[tw] OR "di adresone f"[tw] OR "di‐adreson‐f"[tw] OR "diadreson f"[tw] OR "diadresone f"[tw] OR dicortol[tw] OR domucortone[tw] OR encortelon[tw] OR encortelone[tw] OR encortolon[tw] OR equisolon[tw] OR "fernisolone‐p"[tw] OR glistelone[tw] OR hefasolon[tw] OR "hostacortin h"[tw] OR hydeltra[tw] OR hydeltrone[tw] OR hydrelta[tw] OR hydrocortancyl[tw] OR hydrocortidelt[tw] OR hydrodeltalone[tw] OR hydrodeltisone[tw] OR hydroretrocortin[tw] OR hydroretrocortine[tw] OR inflanefran[tw] OR insolone[tw] OR "keteocort h"[tw] OR "key‐pred"[tw] OR lenisolone[tw] OR leocortol[tw] OR liquipred[tw] OR "lygal kopftinktur n"[tw] OR mediasolone[tw] OR meprisolon[tw] OR meprisolone[tw] OR metacortalon[tw] OR metacortalone[tw] OR metacortandralon[tw] OR metacortandralone[tw] OR metacortelone[tw] OR "meti derm"[tw] OR meticortelone[tw] OR metiderm[tw] OR morlone[tw] OR mydrapred[tw] OR "neo delta"[tw] OR nisolon[tw] OR nisolone[tw] OR "nsc 9120"[tw] OR nsc9120[tw] OR opredsone[tw] OR panafcortelone[tw] OR panafcortolone[tw] OR panafort[tw] OR paracortol[tw] OR phlogex[tw] OR "pre cortisyl"[tw] OR preconin[tw] OR precortalon[tw] OR precortancyl[tw] OR precortisyl[tw] OR "pred‐ject‐50"[tw] OR "predacort 50"[tw] OR "predaject‐50"[tw] OR "predalone 50"[tw] OR predartrina[tw] OR predartrine[tw] OR predate[tw] OR "predate‐50"[tw] OR predeltilone[tw] OR predisole[tw] OR predisyr[tw] OR "predne dome"[tw] OR prednecort[tw] OR prednedome[tw] OR prednelan[tw] OR "predni coelin"[tw] OR "predni h tablinen"[tw] OR "predni‐helvacort"[tw] OR prednicoelin[tw] OR prednicort[tw] OR prednicortelone[tw] OR "prednifor drops"[tw] OR predniment[tw] OR predniretard[tw] OR prednis[tw] OR prednisil[tw] OR prednisolon[tw] OR prednisolona[tw] OR prednivet[tw] OR prednorsolon[tw] OR prednorsolone[tw] OR predonine[tw] OR predorgasolona[tw] OR predorgasolone[tw] OR "pregna 1, 4 diene 11beta, 17alpha, 21 triol 3, 20 dione"[tw] OR prelon[tw] OR prelone[tw] OR prenilone[tw] OR prenin[tw] OR prenolone[tw] OR preventan[tw] OR prezolon[tw] OR rubycort[tw] OR scherisolon[tw] OR scherisolona[tw] OR serilone[tw] OR solondo[tw] OR solone[tw] OR solupren[tw] OR soluprene[tw] OR spiricort[tw] OR spolotane[tw] OR sterane[tw] OR sterolone[tw] OR supercortisol[tw] OR supercortizol[tw] OR taracortelone[tw] OR walesolone[tw] OR wysolone[tw] OR "50‐24‐8"[tw] #25 #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 #26 #1 AND #16 AND #25
Appendix 5. LILACS search strategy
("Dry Eye" OR "Síndromes de Ojo Seco" OR "Síndromes do Olho Seco" OR MH:C11.496.260$ OR Tear$ OR Lágrimas OR MH:A12.200.882$ OR Xerophthalmia OR Xeroftalmia OR MH:C11.187.810$ OR MH:C11.496.260.892$ OR "Vitamin A Deficiency" OR "Deficiencia de Vitamina A" OR MH:C18.654.521.500.133.628$ OR MH:SP6.016.052.063.109$ OR "avitaminosis a" OR "retinol deficiency" OR "hypovitaminosis A" OR Keratoconjunctivitis OR " kerato conjunctivitis" OR Queratoconjuntivitis OR Ceratoconjuntivite OR MH:C11.187.183.394$ OR MH:C11.204.564.585$ OR MH:C11.496.260.394$ OR "Sjogren's Syndrome" OR "Síndrome de Sjögren" OR MH:C05.550.114.154.774$ OR MH:C05.799.114.774$ OR MH:C07.465.815.929.669$ OR MH:C11.496.260.719$ OR MH:C17.300.775.099.774$ OR MH:C20.111.199.774$ OR "sicca syndrome" OR "Stevens Johnson Syndrome" OR "Síndrome de Stevens Johnson" OR MH:C07.465.864.500$ OR MH:C17.800.229.400.683$ OR MH:C17.800.865.475.683$ OR "Pemphigoid Benign Mucous Membrane" OR "Penfigoide Benigno de la Membrana Mucosa" OR "Penfigoide Mucomembranoso Benigno" OR "Cicatricial Pemphigoid" OR MH:C11.187.482$ OR MH:C17.800.865.670$ OR blepharoconjunctiviti$ OR "Meibomian Glands" OR "Glándulas Tarsales" OR "Glândulas Tarsais" OR MH:A09.371.337.614 OR MH:A10.336.827.600 OR "Lacrimal Apparatus Diseases" OR "Enfermedades del Aparato Lagrimal" OR "Doenças do Aparelho Lacrimal" OR MH:C11.496$ OR lacrima$ or epiphora) AND (corticosteroid$ OR glucocorticoid$ OR mineralocorticoid$ OR "adrenal cortex hormone" OR "adrenal cortical hormone" OR "adrenocortical hormone" OR adrenocorticosteroid$ OR corticoid$ OR steroid$ OR MH:D06.472.040$ OR Betamethason$ OR Betametason$ OR MH:4.210.500.745.432.769.199$ OR MH:D04.210.500.908.093$ OR "Clobetasone butyrate" OR Dexamethason$ OR Dexametason$ OR MH:D04.210.500.745.432.769.344$ OR MH:D04.210.500.908.238$ OR Difluprednat$ OR Fluorometholon$ OR MH:D04.210.500.745.432.719.349$ OR MH:D04.210.500.908.431$ OR "Loteprednol etabonate" OR MH:D04.210.500.054.079.129.284$ OR "Etabonato de Loteprednol" OR Prednisolon$ OR MH:D04.210.500.745.432.769.795$)
Appendix 6. ClinicalTrials.gov search strategy
(dry eye OR Keratoconjunctivitis) AND (corticosteroid OR glucocorticoid OR mineralocorticoid OR adrenal cortex hormones OR adrenal cortical hormones OR adrenocortical hormones OR adrenocorticosteroid OR corticoid OR steroids OR betamethasone OR clobetasone butyrate OR dexamethasone OR difluprednate OR fluorometholone OR loteprednol etabonate OR prednisolone)
Appendix 7. WHO ICTRP search strategy
dry eye AND corticosteroid OR dry eye AND glucocorticoid OR dry eye AND mineralocorticoid OR dry eye AND adrenal cortex hormones OR dry eye AND adrenal cortical hormones OR dry eye AND adrenocortical hormones OR dry eye AND adrenocorticosteroid OR dry eye AND corticoid OR dry eye AND steroids OR dry eye AND betamethasone OR dry eye AND clobetasone butyrate OR dry eye AND dexamethasone OR dry eye AND difluprednate OR dry eye AND fluorometholone OR dry eye AND loteprednol etabonate OR dry eye AND prednisolone
Keratoconjunctivitis AND corticosteroid OR Keratoconjunctivitis AND glucocorticoid OR Keratoconjunctivitis AND mineralocorticoid OR Keratoconjunctivitis AND adrenal cortex hormones OR Keratoconjunctivitis AND adrenal cortical hormones OR Keratoconjunctivitis AND adrenocortical hormones OR Keratoconjunctivitis AND adrenocorticosteroid OR Keratoconjunctivitis AND corticoid OR Keratoconjunctivitis AND steroids OR Keratoconjunctivitis AND betamethasone OR Keratoconjunctivitis AND clobetasone butyrate OR Keratoconjunctivitis AND dexamethasone OR Keratoconjunctivitis AND difluprednate OR Keratoconjunctivitis AND fluorometholone OR Keratoconjunctivitis AND loteprednol etabonate OR Keratoconjunctivitis AND prednisolone
Data and analyses
Comparison 1. Steroids versus lubricants.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Patient‐reported symptom scores—Qazi: LE alone | 15 | 3770 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.42, ‐0.16] |
| 1.2 Patient‐reported symptom scores—by questionnaire, Qazi: LE alone | 15 | 3770 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.42, ‐0.16] |
| 1.2.1 OSDI | 9 | 3238 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.45, ‐0.13] |
| 1.2.2 Non‐OSDI | 6 | 532 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.56, ‐0.04] |
| 1.3 Patient‐reported symptom scores—Qazi: LE + tobramycin | 15 | 3770 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.41, ‐0.14] |
| 1.4 Patient‐reported symptom scores—by questionnaire, Qazi: LE + tobramycin | 15 | 3770 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.41, ‐0.14] |
| 1.4.1 OSDI | 9 | 3238 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.44, ‐0.10] |
| 1.4.2 Non‐OSDI | 6 | 532 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.56, ‐0.04] |
| 1.5 Patient‐reported symptom scores—by etiology | 15 | 3770 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.42, ‐0.16] |
| 1.5.1 Mixed patient populations | 11 | 3603 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.40, ‐0.12] |
| 1.5.2 Patients with Sjögren Syndrome | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.76 [‐1.40, ‐0.12] |
| 1.5.3 Patients with Meibomian Gland Dysfunction | 3 | 127 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐0.77, ‐0.06] |
| 1.6 Patient‐reported symptom scores—by risk of bias | 15 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.6.1 Low/some concerns RoB | 10 | 3263 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.28, ‐0.10] |
| 1.6.2 High RoB | 5 | 507 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.63 [‐0.84, ‐0.43] |
| 1.7 Patient‐reported symptom scores—by sponsorship | 15 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.7.1 Industry or affiliation & industry | 10 | 3300 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.27, ‐0.07] |
| 1.7.2 University or government | 3 | 245 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐0.75, ‐0.24] |
| 1.7.3 Not reported | 2 | 262 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐1.05, ‐0.50] |
| 1.8 Patient‐reported symptom scores—by regimen and steroid type | 15 | 3807 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.40, ‐0.14] |
| 1.8.1 (Ester) steroid alone | 9 | 3198 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.31, ‐0.13] |
| 1.8.2 (Ketone) steroid alone | 5 | 542 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐0.90, 0.02] |
| 1.8.3 (Ester) steroid plus antiseptic/antimicrobial | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | 0.39 [‐0.26, 1.04] |
| 1.8.4 (Ketone) steroid plus antiseptic/antimicrobial | 1 | 30 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐1.15, 0.30] |
| 1.9 Patient‐reported symptom scores—by steroid treatment duration | 15 | 3770 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.42, ‐0.16] |
| 1.9.1 Short‐term (< 28 days) | 5 | 2751 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.31, ‐0.10] |
| 1.9.2 Long‐term (28 to 42 days) | 9 | 959 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.60, ‐0.11] |
| 1.9.3 Long‐term > 42 days | 1 | 60 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐1.14, ‐0.10] |
| 1.10 Tear film break‐up time—Qazin: LE alone | 7 | 935 | Mean Difference (IV, Random, 95% CI) | 0.70 [0.23, 1.17] |
| 1.11 Tear film break‐up time—Qazi: LE + tobramycin | 7 | 935 | Mean Difference (IV, Random, 95% CI) | 0.78 [0.34, 1.21] |
| 1.12 Tear film break‐up time—by steroid type | 7 | 935 | Mean Difference (IV, Random, 95% CI) | 0.70 [0.23, 1.17] |
| 1.12.1 Loterprednol etabonate (LE): Ketone steroid | 4 | 391 | Mean Difference (IV, Random, 95% CI) | 0.85 [‐0.48, 2.17] |
| 1.12.2 Fluorometholone (FML)/Clobetasol (CB): Ester steroid | 3 | 544 | Mean Difference (IV, Random, 95% CI) | 0.52 [0.32, 0.71] |
| 1.13 Corneal fluorescein staining scores—Qazi: LE alone | 15 | 3699 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.62, ‐0.18] |
| 1.14 Corneal fluorescein staining score—by grading system, Qazi: LE alone | 15 | 3699 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.62, ‐0.18] |
| 1.14.1 NEI grading | 7 | 2940 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.22, ‐0.07] |
| 1.14.2 Other grading systems | 8 | 759 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐1.04, 0.01] |
| 1.15 Corneal fluorescein staining score—Qazi: LE + tobramycin | 15 | 3699 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.61, ‐0.17] |
| 1.16 Corneal fluorescein staining score—by grading system, Qazi: LE + tobramycin | 15 | 3699 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐0.61, ‐0.17] |
| 1.16.1 NEI grading | 7 | 2940 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.22, ‐0.07] |
| 1.16.2 Other grading systems | 8 | 759 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐1.01, 0.07] |
| 1.17 Corneal fluorescein staining scores—by etiology | 15 | 3699 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.62, ‐0.18] |
| 1.17.1 Mixed patient population | 11 | 3532 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐0.74, ‐0.25] |
| 1.17.2 Patients with Sjögren syndrome | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐1.24, 0.03] |
| 1.17.3 Patients with Meibomian Gland Dysfunction | 3 | 127 | Std. Mean Difference (IV, Random, 95% CI) | 0.17 [‐0.61, 0.96] |
| 1.18 Corneal fluorescein staining score—by risk of bias | 15 | 3736 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.58, ‐0.15] |
| 1.18.1 Low/some concerns RoB | 10 | 3120 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.34, ‐0.06] |
| 1.18.2 High RoB | 5 | 616 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.52 [‐1.21, 0.18] |
| 1.19 Corneal fluorescein staining score—by sponsorship | 15 | 3736 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.58, ‐0.15] |
| 1.19.1 Industry or affiliation & industry | 9 | 3101 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.34, ‐0.05] |
| 1.19.2 University or government | 3 | 245 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.60, ‐0.10] |
| 1.19.3 Not reported | 3 | 390 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.58 [‐1.87, 0.70] |
| 1.20 Corneal fluorescein staining score—by steroid treatment duration | 15 | 3736 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.58, ‐0.15] |
| 1.20.1 Short‐term (< 28 days) | 5 | 2763 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.38, 0.07] |
| 1.20.2 Short‐term (28 to 42 days) | 9 | 913 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.46 [‐0.84, ‐0.07] |
| 1.20.3 Duration ≥ 2 months | 1 | 60 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐0.93, 0.09] |
| 1.21 Tear osmolarity | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.22 Schirmer's test—Qazi: LE alone | 5 | 541 | Mean Difference (IV, Random, 95% CI) | 0.94 [0.39, 1.49] |
| 1.23 Schirmer's test—Qazi: LE + tobramycin | 5 | 541 | Mean Difference (IV, Random, 95% CI) | 0.69 [‐0.02, 1.39] |
| 1.24 Schirmer test—by steroid type, Qazi: LE alone | 5 | 541 | Mean Difference (IV, Random, 95% CI) | 0.94 [0.39, 1.49] |
| 1.24.1 Loteprednol etabonate (LE): Ketone steroid | 3 | 269 | Mean Difference (IV, Random, 95% CI) | 1.44 [0.88, 1.99] |
| 1.24.2 Fluorometholone (FML): Ester steroid | 2 | 272 | Mean Difference (IV, Random, 95% CI) | 0.61 [0.30, 0.92] |
| 1.25 Schirmer test—by steroid type, Qazi: LE + tobramycin | 5 | 541 | Mean Difference (IV, Random, 95% CI) | 0.69 [‐0.02, 1.39] |
| 1.25.1 Loteprednol etabonate (LE): Ketone steroid | 3 | 269 | Mean Difference (IV, Random, 95% CI) | 0.54 [‐1.21, 2.30] |
| 1.25.2 Fluorometholone (FML): Ester steroid | 2 | 272 | Mean Difference (IV, Random, 95% CI) | 0.61 [0.30, 0.92] |
| 1.26 Proportion of participants with increased IOP—by steroid type | 8 | 2264 | Risk Ratio (M‐H, Random, 95% CI) | 5.96 [1.30, 27.38] |
| 1.26.1 Ester steroid: LE | 4 | 1954 | Risk Ratio (M‐H, Random, 95% CI) | 3.59 [0.59, 21.97] |
| 1.26.2 Ketone steroids | 4 | 310 | Risk Ratio (M‐H, Random, 95% CI) | 20.42 [1.21, 344.00] |
| 1.27 Proportion of participants with new cataract formation | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.28 Proportion of participants with serious adverse events (systemic or ocular) | 4 | 2167 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.03 [0.82, 11.13] |
1.17. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 17: Corneal fluorescein staining scores—by etiology
1.23. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 23: Schirmer's test—Qazi: LE + tobramycin
1.25. Analysis.

Comparison 1: Steroids versus lubricants, Outcome 25: Schirmer test—by steroid type, Qazi: LE + tobramycin
Comparison 2. Steroids versus CsA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Patient‐reported symptom scores—Bausch: LE alone | 6 | 465 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.33 [‐0.51, ‐0.15] |
| 2.2 Patient‐reported symptom scores—Bausch: LE + CsA | 6 | 462 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐0.54, ‐0.17] |
| 2.3 Patient‐reported symptom scores—by regimen | 6 | 531 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.48, ‐0.12] |
| 2.3.1 Steroid alone vs. CsA 0.05% | 3 | 172 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.43, 0.17] |
| 2.3.2 Steroid run‐in before initiating CsA 0.05% | 2 | 178 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.50, 0.09] |
| 2.3.3 Steroid run‐in with CsA 0.05% | 2 | 181 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐0.86, ‐0.27] |
| 2.4 Patient‐reported symptom scores—steroid treatment duration | 6 | 530 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.26 [‐0.43, ‐0.08] |
| 2.4.1 High dose for 3‐4 weeks before tapering | 2 | 106 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.09 [‐0.47, 0.30] |
| 2.4.2 8 weeks | 4 | 355 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.37 [‐0.58, ‐0.16] |
| 2.4.3 12 weeks | 1 | 69 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.05 [‐0.42, 0.52] |
| 2.5 Tear film break‐up time—Bausch: LE alone | 5 | 353 | Mean Difference (IV, Random, 95% CI) | 0.37 [‐0.13, 0.87] |
| 2.6 Tear film break‐up time—Bausch: LE + CsA | 5 | 350 | Mean Difference (IV, Random, 95% CI) | 0.36 [‐0.15, 0.87] |
| 2.7 Tear film break‐up time—by regimen | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.7.1 Steroid alone | 3 | 172 | Mean Difference (IV, Random, 95% CI) | 0.28 [‐0.74, 1.30] |
| 2.7.2 Steroid + CsA | 3 | 247 | Mean Difference (IV, Random, 95% CI) | 0.46 [0.16, 0.76] |
| 2.8 Tear film break‐up time—by steroid treatment duration | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.8.1 High dose for 2‐4 weeks before tapering | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.86, 1.04] |
| 2.8.2 Fixed dose for 8 weeks | 2 | 103 | Mean Difference (IV, Random, 95% CI) | 0.32 [‐1.20, 1.84] |
| 2.8.3 Fixed dose for 3 months | 3 | 250 | Mean Difference (IV, Random, 95% CI) | 0.47 [0.17, 0.77] |
| 2.9 Corneal fluorescein staining scores—Bausch: LE alone | 6 | 465 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.25, 0.35] |
| 2.10 Corneal fluorescein staining score—Bausch: LE + CsA | 6 | 462 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.27, 0.29] |
| 2.11 Corneal fluorescein staining score—by grading system | 6 | 531 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.20, 0.31] |
| 2.11.1 NEI grading system | 4 | 428 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.31, 0.32] |
| 2.11.2 Other grading system | 2 | 103 | Std. Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.22, 0.62] |
| 2.12 Corneal fluorescein staining score—by regimen | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.12.1 Steroid alone vs. CsA 0.05% | 3 | 172 | Std. Mean Difference (IV, Random, 95% CI) | 0.25 [‐0.06, 0.55] |
| 2.12.2 Steroid run‐in before intitiating CsA 0.05% | 2 | 178 | Std. Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.14, 0.45] |
| 2.12.3 Steroid run‐in concurrently with CsA 0.05% | 2 | 181 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.33 [‐0.73, 0.07] |
| 2.13 Corneal fluorescein staining score—by steroid treatment duration | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.13.1 For 3‐4 weeks | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.32, 0.44] |
| 2.13.2 For 8 weeks | 4 | 355 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.39, 0.42] |
| 2.13.3 For 12 weeks | 1 | 69 | Std. Mean Difference (IV, Random, 95% CI) | 0.32 [‐0.15, 0.80] |
| 2.14 Tear osmolarity | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.14.1 LE gel 0.5% alone | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.14.2 LE gel 0.5% plus CsA | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.15 Schirmer's test | 4 | 361 | Mean Difference (IV, Random, 95% CI) | 1.19 [‐0.40, 2.77] |
| 2.15.1 Steroid alone | 1 | 68 | Mean Difference (IV, Random, 95% CI) | 0.12 [‐0.59, 0.83] |
| 2.15.2 Combination | 3 | 293 | Mean Difference (IV, Random, 95% CI) | 1.73 [0.32, 3.13] |
| 2.16 Schirmer's test—by steroid type | 4 | 361 | Mean Difference (IV, Random, 95% CI) | 1.19 [‐0.40, 2.77] |
| 2.16.1 Ketone steroid: MP + CsA | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐0.52, 2.12] |
| 2.16.2 Ester steroid: LE ± CsA | 3 | 320 | Mean Difference (IV, Random, 95% CI) | 1.32 [‐0.72, 3.36] |
| 2.17 Proportion of participants with increased IOP | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.18 Proportion of participants with any ocular complication | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.18.1 Steroid alone | 2 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.11, 3.43] |
| 2.18.2 Steroid plus CsA | 2 | 110 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.11, 3.80] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Akhlaq 2019.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2014/02 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2017/04 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant (both eyes) 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: double (participant, investigator) 05. Study visits and the corresponding time points: baseline, 2 weeks, and 6 weeks 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI; SANDE 07. Assessment for safety outcomes: the occurrence of adverse events: serious vs non‐serious; expected vs unexpected; changes to the use of concomitant medications; comprehensive eye examination including BCVA, measuring intraocular pressure, evaluation of the condition of conjunctiva, cornea, anterior chamber, iris/pupil, lens, vitreous, macula and optic nerve; conjunctival infections; if treatment with artificial tears is inadequate, or the participant develops a severe form of ocular surface disease and dry eye, for the safety and proper treatment of the participant, the investigator can unmask the participant's treatment assignment 08. Planned follow‐up duration: 6 weeks 09. Actual follow‐up duration: 6 weeks 10. Planned treatment duration (of the intervention steroid): 6 weeks 11. How missing data were handled: the study protocol specified an ITT analysis for any withdrawal from the study 12. Description of power and sample size calculation: "To calculate sample size, density of sub‐basal dendritic cells in the central cornea was used as an outcome measure. With α = 0.05 (Zα = 1.96), d = 52, and p = 44, a sample size of 22 was calculated for each group (44 for both groups). To compensate for potential loss to follow‐up, a total of 50 subjects will be enrolled in this study" |
|
| Participants |
Country: USA Setting: single‐site, university‐affiliated eye center Interventions
Age, mean/SD (range): 57.2/12.1 Female, n (%): NR Etiology, n (%): NR Participants (eyes) randomized: 18 (31 eyes) Participants (eyes) analyzed for primary study outcomes: 18 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): 55.4/15.6 Female, n (%): NR Etiology, n (%): NR Participants (eyes) randomized: 20 (36 eyes) Participants (eyes) analyzed for primary study outcomes: 20 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): 56.2/13.9 Female, n (%): NR Etiology, n (%): NR Participants (eyes) randomized: 38 (67 eyes) Participants (eyes) analyzed for primary study outcomes: 38 Participants (eyes) analyzed for safety outcomes: NR Inclusion criteria:
Exclusion criteria:
Baseline comparison: baseline characteristics of the study population were not reported, except the baseline OSDI scores, which suggested evident differences in patient‐reported symptom severity between groups (53.53 ± 29.7 vs 34.46 ± 20.33, post hoc P = 0.0255) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: at 6 weeks Primary outcomes of the study: IVCM for determination of:
Other outcomes of the study: corneal fluorescein staining using the NEI grading scheme; conjunctival Lissamine green staining using NEI grading scheme; TBUT, seconds; Schirmer's test with anesthesia, mm; IOP by measure of applanation tonometry, mmHg; OSDI questionnaire, total score of the OSDI questionnaire; SANDE questionnaire |
|
| Study Identification |
Sponsorship source: GlaxoSmithKline plc Ethics approval: a copy of the approved protocol and study‐related materials will be placed on a shared network drive accessible to authorized research staff involved in this study and on IRBnet.org Correspondence author's name: Pedram Hamrah; Institution: Tufts University, Boston, MA Additional information:
|
|
| Notes | ||
Aragona 2013.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): NR 02. Calendar time when the study completed follow‐up (YYYY/MM): NR 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: double masking 05. Study visits and the corresponding time points: baseline (T0), day 15 (T15), day 30 (T30), and day 45 (TFU) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: VAS (0 to 100) for each of the following symptoms: burning, itching, foreign body sensation, photophobia, sticky eye, blurred vision, and dryness. The total score was therefore obtained by adding up the results of each symptom. 07. Assessment for safety outcomes: IOP at each visit; fundus exam on day 30 08. Planned follow‐up duration: 45 days 09. Actual follow‐up duration: 45 days 10. Planned treatment duration (of the intervention steroid): 30 days 11. How missing data were handled: NR 12. Description of power and sample size calculation: "For the study population, it was considered an assumed efficacy of the study treatment of 50% and 5% of the control treatment for an α value of 0.05, using a 2‐tailed analysis, and a power of 90%, which gave a population of 38 patients in total (19 per arm) to achieve statistically significant results" |
|
| Participants |
Country: Italy Setting: single‐site, university‐affiliated eye center Interventions
Age, mean/SD (range): 58.8/6.7 Female, n (%): 19 (95%) Etiology, n (%): Sjögren syndrome, 20 (100%) Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 20 Participants (eyes) analyzed for safety outcomes: 20
Age, mean/SD (range): 60.1/5.1 Female, n (%): 19 (95%) Etiology, n (%): Sjögren syndrome, 20 (100%) Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 20 Participants (eyes) analyzed for safety outcomes: 20
Age, mean/SD (range): 59.5/5.9 Female, n (%): 38 (95%) Etiology, n (%): Sjögren syndrome, 20 (100%) Participants (eyes) randomized: 40 Participants (eyes) analyzed for primary study outcomes: 40 Participants (eyes) analyzed for safety outcomes: 40 Inclusion criteria: One or more dry eye–related symptoms including burning, itching, foreign body sensation, photophobia, sticky eye, blurred vision, and dryness, with a VAS value above 30 mm out of 100 mm for each symptom; TBUT ≤ 3 seconds; and corneal staining with a fluorescein score ≥ 2, on a scale from 0 (absent) to 3 (severe), in at least 2 out of 5 corneal areas as described by Lemp (NEI Workshop on clinical trials in dry eyes 1995). Exclusion criteria: Eye injury, infections, non–dry eye ocular inflammation, or trauma or surgery within the previous 6 months; concurrent treatment able to interfere with the interpretation of the study results (systemic corticosteroids, immunosuppressive therapy, parasympathetic agents); uncontrolled disease or significant illness; or pregnancy or lactation. Postmenopausal patients on hormonal replacement therapy were also excluded. Baseline comparison: no differences in age or sex distribution between groups |
|
| Interventions |
PVP (Wet‐Comod, Visufarma SpA, Rome, Italy); CB (Visucloben, Visufarma SpA) |
|
| Outcomes |
Time points of primary outcome data collected: at day 30 Primary outcomes of the study:
Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: the drugs used in the trial were provided by Visufarma SpA, Roma, Italy. The authors report no proprietary interest or financial support. Ethics approval: ethical approval was granted by the Ethics Committee of the University Hospital of Messina Correspondence author's name: Pasquale Aragona; Institution: Department of Surgical Specialties Section of Ophthalmology Ocular Surface Unit University Hospital of Messina, Italy Additional information:
|
|
| Notes | ||
Avunduk 2003.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): NR 02. Calendar time when the study completed follow‐up (YYYY/MM): NR 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: the examiner (AMA) was masked as to the medication used by the participants. The participants were instructed to discuss their medications only with the study co‐ordinator and not with the examiner. 05. Study visits and the corresponding time points: days 0, 15, and 30 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: "We also obtained symptom severity scores from patients who were instructed to grade their symptoms averaging the symptom severities for both eyes." The authors utilized a screening questionnaire for DEEP published by Oden NL and colleagues. 07. Assessment for safety outcomes: unclear—the authors did not report assessment for safety outcomes in the methods section. However, the authors mentioned in the results section that: "We did not observe any complication that could be linked to the study medications during the treatment period". 08. Planned follow‐up duration: 30 days 09. Actual follow‐up duration: 30 days 10. Planned treatment duration (of the intervention steroid): 30 days 11. How missing data were handled: complete‐case analysis on study outcomes. 12. Description on power and sample size calculation: power was calculated to detect an among‐group difference in change from baseline in symptom severity scores on day 30 |
|
| Participants |
Country: USA Setting: single‐site, university‐affiliated eye center Interventions:
Age, mean/SD (range): 57.6/12.4 Female, n (%): 7 (64%) Etiology, n (%): NR Participants (eyes) randomized: NR Participants (eyes) analyzed for primary study outcomes: 11 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): 51.2/12.4 Female, n (%): 5 (63%) Etiology, n (%): NR Participants (eyes) randomized: NR Participants (eyes) analyzed for primary study outcomes: 8 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): 57.2/12.1 Female, n (%): 12 (63%) Etiology, n (%): NR Participants (eyes) randomized: NR Participants (eyes) analyzed for primary study outcomes: 19 Participants (eyes) analyzed for safety outcomes: NR Note: data are not shown for a third group where participants were randomized to receive flurbiprofen and ATS 4 times a day for 30 days (N = 9) Inclusion criteria:
Exclusion criteria: Patients were excluded from the study if they:
Postmenopausal patients who were on hormonal replacement therapy were also excluded. Baseline comparison: at the beginning of the study (day 0), no significant difference was detected between groups in terms of any of the parameters studied |
|
| Interventions |
ATS (Refresh, Allergan Inc, Irvine, CA, USA); FML (Allergan Inc, Irvine, CA, USA), concentration unspecified Note: data not shown for a third group (N = 9, 5 females) treated with topical non‐steroidal anti‐inflammatory drug (NSAID) eyedrops, flurbiprofen (Ocufen, Allergan Inc, Irvine, CA, USA) 4 times a day plus ATS 4 to 8 times a day in both eyes |
|
| Outcomes |
Time points of primary outcome data collected: Primary outcomes of the study: Difference in change from baseline in symptom severity scores on day 30 (for considering power calculation) Other outcomes of the study:
|
|
| Study Identification |
Sponsorship Source: US Public Health Service Grant EY02377 (H.E.K.) from the National Eye Institute, National Institutes of Health, Bethesda, Maryland, and an unrestricted departmental grant from Research to Prevent Blindness Inc, New York, NY Ethics approval: informed consent was obtained from all participants, and the research was begun after obtaining approval from the Institutional Review Board of the Louisiana State University Health Sciences Center Correspondence author's name: Avni Murat Avunduk, MD; Institution: KTU (Karadeniz Technical University, Turkish: Karadeniz Teknik Üniversitesi or KTÜ), Konya, Turkey Additional information:
|
|
| Notes | ||
Bausch 2013.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2013/05 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2014/01 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: single (investigator) 05. Study visits and the corresponding time points: Visit 1 (14 days before randomization), Visit 2 (Day 0, randomization), Visit 3 (Week 4), Visit 4 (Week 12), Visit 5 (Week 13) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI is a 12‐item questionnaire developed to assess severity of DED. There are 3 question types: "Have you experienced any of following (light sensitivity, eye feel gritty, sore eyes, blurred vision, and poor vision) during last week?"(items 1 to 5); "Have problems with your eyes limited you in performing any of following (reading, driving at night, working with computer, and watching TV) during last week?" (items 6 to 9); and "Have your eyes felt uncomfortable in any of following situations (windy, low humidity, air conditioned) during the last week?" (items 10 to 12). Responses to each question were graded on a scale (that relates to the frequency of ocular surface disease effects) of 0 (none of the time) to 4 (all of the time). Total OSDI score was calculated using the following formula: OSDI = ([sum of scores for all questions answered] × 100)/([total number of questions answered] * 4). Total OSDI score ranged from 0 to 100, with higher scores representing greater disability. Participants scored their degree of comfort with their assigned study drug on a 4‐point scale (0 to 3 units) within 5 minutes after instillation of study drug. Comfort grade 0 indicated comfortable, discomfort absent; 1 indicated generally comfortable, mild discomfort; 2 indicated some discomfort but tolerable, moderate discomfort; 3 indicated severely uncomfortable or intolerable. The mean global ocular comfort grade was reported. 07. Assessment for safety outcomes: an AE was defined as any untoward medical occurrence in a participant who received study drug without regard to possibility of causal relationship. Serious AEs were defined as death, a life‐threatening AE, inpatient hospitalization or prolongation of existing hospitalization, persistent or significant disability or incapacity, a congenital anomaly or birth defect, or an important medical event that jeopardized the participant and required medical intervention to prevent 1 of the outcomes listed in this definition. 08. Planned follow‐up duration: 13 weeks 09. Actual follow‐up duration: 13 weeks 10. Planned treatment duration (of the intervention steroid): 12 weeks 11. How missing data were handled: imputation using mixed‐effect model for repeated measures (MMRM) method 12. Description of power and sample size calculation: NR |
|
| Participants |
Country: USA Setting: single medical center Interventions:
Age, mean/SE (range): 62.0/8.30 Female, n (%): 25 (76%) Etiology, n (%): NR Participants (eyes) randomized: 33 Participants (eyes) analyzed for primary study outcomes: 32 Participants (eyes) analyzed for safety outcomes: 33
Age, mean/SE (range): 64.3/9.1 Female, n (%): 30 (83%) Etiology, n (%): NR Participants (eyes) randomized: 33 Participants (eyes) analyzed for primary study outcomes: 32 Participants (eyes) analyzed for safety outcomes: 33
Age, mean/SE (range): 61.6/9.98 Female, n (%): 26 (79%) Etiology, n (%): NR Participants (eyes) randomized: 36 Participants (eyes) analyzed for primary study outcomes: 36 Participants (eyes) analyzed for safety outcomes: 36
Age, mean/SE (range): 62.7/9.1 Female, n (%): 81 (79%) Etiology, n (%): NR Participants (eyes) randomized: 102 Participants (eyes) analyzed for primary study outcomes: 101 Participants (eyes) analyzed for safety outcomes: 102 Inclusion criteria:
Exclusion criteria:
Baseline comparison: no differences in age or sex distribution among the three comparison groups |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: at week 4 Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship Source: Bausch & Lomb Inc Ethics approval: NR (on ClinicalTrials.gov) Correspondence author's name: Susan Harris; Institution: Bausch Health Americas Inc Additional information:
|
|
| Notes | ||
Byun 2012.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): NR 02. Calendar time when the study completed follow‐up (YYYY/MM): NR 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: NR 05. Study visits and the corresponding time points: baseline, month 1, 2, 3 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: patient‐reported symptom scores: according to a scoring system ranging from 0 (absent) to 4 (severe) in terms of 6 ocular surface symptoms: burning, itching, foreign body sensation, blurring, photophobia, and pain. A total score was obtained by adding the scores for each symptom, and this was considered in the evaluation of overall ocular discomfort. 07. Assessment for safety outcomes: instillation site disorders such as burning, stinging, conjunctival hyperemia, and bitter taste 08. Planned follow‐up duration: 3 months 09. Actual follow‐up duration: 3 months 10. Planned treatment duration (of the intervention steroid): 3 weeks before tapering off 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: Republic of Korea Setting: single‐site, university‐affiliated eye center Interventions:
Age, mean/SD (range): 51.2/14.0 Female, n (%): 18 (86%) Etiology, n (%): NR Participants (eyes) randomized: 21 Participants (eyes) analyzed for primary study outcomes: 21 Participants (eyes) analyzed for safety outcomes: 21
Age, mean/SD (range): 52.2/9.4 Female, n (%): 17 (74%) Etiology, n (%): NR Participants (eyes) randomized: 23 Participants (eyes) analyzed for primary study outcomes: 20 Participants (eyes) analyzed for safety outcomes: 23
Age, mean/SD (range): 51.7/11.7 Female, n (%): 35 (80%) Etiology, n (%): NR Participants (eyes) randomized: 44 Participants (eyes) analyzed for primary study outcomes: 41 Participants (eyes) analyzed for safety outcomes: 44 Inclusion criteria:
Exclusion criteria:
Baseline comparison: groups 1 and 2 were similar in terms of pretreatment (baseline) signs and symptoms (Table 1) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: at month 1, 2, 3 Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship Source: supported by the Dong‐A University Research Fund in 2007, and partially supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST No. 2009‐0066392) Ethics approval: all applicable institutional regulations concerning the ethical use of human volunteers were followed during this research (IRB no. 4‐2006‐0141) Correspondence author's name: Woo Chan Park; Institution: Department of Ophthalmology, Dong‐A University College of Medicine Additional information:
|
|
| Notes | ||
Cao 2018.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): NR 02. Calendar time when the study completed follow‐up (YYYY/MM): NR 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: NR 05. Study visits and the corresponding time points: baseline, week 2 and 6 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: NA 07. Assessment for safety outcomes: complications and ocular discomfort (irritation or sticky sensation) 08. Planned follow‐up duration: 6 weeks 09. Actual follow‐up duration: 6 weeks 10. Planned treatment duration (of the intervention steroid): 6 weeks 11. How missing data were handled: NA 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: China Setting: single medical center Intervention:
Age, mean/SD (range): NR Female, n (%): NR Etiology, n (%): NR Participants (eyes) randomized: 64 Participants (eyes) analyzed for primary study outcomes: 64 Participants (eyes) analyzed for safety outcomes: 64
Age, mean/SD (range): NR Female, n (%): NR Etiology, n (%): NR Participants (eyes) randomized: 64 Participants (eyes) analyzed for primary study outcomes: 64 Participants (eyes) analyzed for safety outcomes: 64
Age, mean/SD (range): 7.8/3.3 (3 to 14) Female, n (%): 70 (54.7%) Etiology, n (%): NR Participants (eyes) randomized: 128 Participants (eyes) analyzed for primary study outcomes: 128 Participants (eyes) analyzed for safety outcomes: 128 Inclusion criteria: Dry eye symptoms: frequent blinking, fatigue sensation, dryness, burning, itchiness, photophobia, redness, and at least 1 of the following signs: TBUT ≤ 5 s, or TBUT > 5 s but ≤ 10 s; corneal staining score > 3 points; tear meniscus height < 0.3 mm Exclusion criteria:
Baseline comparison: NR |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: week 2, 6 Primary outcomes of the study:
Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: NR Ethics approval: this study was approved by the institutional review board of the hospital Correspondence author's name: Xian‐Yong Cao; Institution: The Third Affiliated Hospital of Xinxiang Medical University, Henan, China Additional information:
|
|
| Notes | ||
Chen 2020.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2018/07 02. Calendar time when the study completed follow‐up (YYYY/MM): 2019/06 03. Unit of randomization (participant or eye): participant (the worst eye or the right eye) 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: NR 05. Study visits and the corresponding time points: before and after treatment 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: SPEED questionnaire: according to the frequency of symptoms, dry eye was divided into 4 grades (I: 0 score; II: 1 score; III: 2 scores; IV: 3 scores). The total score is calculated by adding the scores of the 4 symptoms (dryness, foreign body sensation, burning sensation, and eye irritation). The maximum is 12 scores. A total score < 10 points is the mild symptom group, and > 10 points is the severe symptom group. 07. Assessment for safety outcomes: NR 08. Planned follow‐up duration: NR 09. Actual follow‐up duration: NR 10. Planned treatment duration (of the intervention steroid): 1 month 11. How missing data were handled: NA 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: China Setting: single‐site, university‐affiliated eye center Interventions:
Age, mean/SD (range): 47.4/11.0 Female, n (%): 72 (87%) Etiology, n (%): NR Participants (eyes) randomized: 83 Participants (eyes) analyzed for primary study outcomes: 83 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): 51.1/11.8 Female, n (%): 68 (82%) Etiology, n (%): NR Participants (eyes) randomized: 83 Participants (eyes) analyzed for primary study outcomes: 83 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): 49.2/11.5 Female, n (%): 140 (84%) Etiology, n (%): NR Participants (eyes) randomized: 166 Participants (eyes) analyzed for primary study outcomes: 166 Participants (eyes) analyzed for safety outcomes: NR Inclusion criteria: dry eye patients and people who received simple optometry with glasses in the ophthalmic outpatient department Exclusion criteria: NR Baseline comparison: as shown in Table I, the differences of age and gender between the 3 groups were not significant (P > 0.05, Table I) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: before and after treatment Primary outcomes of the study:
Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: this study was supported by grants from the National Natural Science Foundation of China (NSFC no. 81970771), Huaxia Translation Medicine Funding (no. 2017‐A‐02), Xiamen Key Medical and Health Project (no.3502Z20191101), and Zhenjiang Science Technology Planning. Project (No. SH2019033) Ethics approval: the experimental protocol was reviewed and approved by the Ethics Committee of Xiamen University Medical College. All participants were informed of the purpose of this study and signed informed consent. Correspondence author's name: Nuo Dong, MD; Institution: Department of Ophthalmology, Affiliated People’s Hospital & Zhenjiang Kangfu Eye Hospital, Jiangsu University, Jiangsu, China Additional information:
|
|
| Notes | ||
KPI‐121 (Phase 2).
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2014/06 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2015/01 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: quadruple masking (participant, care provider, investigator, outcomes assessor) 05. Study visits and the corresponding time points: 6 visits: Visit 1 Screening (14 ± 1 days prior to Day 1), Visit 2 Randomization (Day 1), Visit 3 (Days 8 ± 1), Visit 4 (Day 15 ± 1), Visit 5 (Day 22 ± 1), Visit 6 (Day 29 ± 1) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: visual analog grading scale (OSDI) for ocular discomfort 07. Assessment for safety outcomes: adverse events, slit lamp biomicroscopy, IOP measurement, BCVA 08. Planned follow‐up duration: 29 days 09. Actual follow‐up duration: 29 days 10. Planned treatment duration (of the intervention steroid): 28 days 11. How missing data were handled: NA 12. Description on power and sample size calculation: descriptions about power and sample size calculation were partially blocked in the publicly available trial protocol |
|
| Participants |
Country: USA Setting: eye clinics at medical centers and private medical groups Interventions:
Age, mean/SD (range): 56.3/17.2 Female, n (%): 53 (73%) Etiology, n (%): NR Participants (eyes) randomized: 73 Participants (eyes) analyzed for primary study outcomes: 73 Participants (eyes) analyzed for safety outcomes: 72
Age, mean/SD (range): 54.9/12.3 Female, n (%): 61 (79%) Etiology, n (%): NR Participants (eyes) randomized: 77 Participants (eyes) analyzed for primary study outcomes: 77 Participants (eyes) analyzed for safety outcomes: 78
Age, mean/SD (range): 55.6/14.8 Female, n (%): 114 (76%) Etiology, n (%): NR Participants (eyes) randomized: 150 Participants (eyes) analyzed for primary study outcomes: 150 Participants (eyes) analyzed for safety outcomes: 150 Note: 1 participant was randomized to KPI‐121 0.25% but received vehicle. As a result, 72 participants are included in the KPI‐121 0.25% group, and 78 participants are included in the vehicle group in the safety analyses. Inclusion criteria:
*[blocked] were phrases or words in the study protocol that were blocked from viewing Exclusion criteria:
Baseline comparison: no significant differences in baseline characteristics between groups (source: ClinicalTrials.gov) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: Visit 6 (Day 29) Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship Source: Kala Pharmaceuticals Inc Ethics approval: IRB approval was required by the study protocol, but no specific institutional review boards were mentioned in the protocol or in the prescribing information Correspondence author's name: NR Additional information:
|
|
| Notes | 78% of study participants were white. | |
KPI‐121 (STRIDE1).
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2016/06 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2017/10 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: quadruple (participant, care provider, investigator, outcomes assessor) 05. Study visits and the corresponding time points: Visit 1 (14 ± 1 days before Visit 2) for screening Visit 2 (Day 1) for randomization Visit 3 (Day 8 ± 1) Visit 4 (Day 15 ± 1) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: participant‐rated assessment of ocular discomfort [blocked]* utilizing [blocked] VAS (0 to 100) 07. Assessment for safety outcomes: adverse events; slit lamp biomicroscopy; IOP measurement; BCVA 08. Planned follow‐up duration: 15 days 09. Actual follow‐up duration: 15 days 10. Planned treatment duration (of the intervention steroid): 14 days 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: descriptions about power and sample size calculation were partially blocked in the publicly available trial protocol |
|
| Participants |
Country: USA Setting: multicenter Interventions:
Age, mean/SD (range): 58.1/15.4 Female, n (%): 367 (80.0%) Etiology, n (%): NR Participants (eyes) randomized: 459 Participants (eyes) analyzed for primary study outcomes: 452/455 Participants (eyes) analyzed for safety outcomes: 459
Age, mean/SD (range): 58.3/14.7 Female, n (%): 359 (78.7%) Etiology, n (%): NR Participants (eyes) randomized: 456 Participants (eyes) analyzed for primary study outcomes: 451/452 Participants (eyes) analyzed for safety outcomes: 456
Age, mean/SD (range): 58.2/15.0 Female, n (%): 726 (79.3%) Etiology, n (%): NR Participants (eyes) randomized: 915 Participants (eyes) analyzed for primary study outcomes: 903/907 Participants (eyes) analyzed for safety outcomes: 915 Inclusion criteria:
*[blocked] were phrases or words in the study protocol that were blocked from viewing Exclusion criteria:
NOTE: oral corticosteroid use at a dose of prednisone > 11 mg/day or equivalent is excluded. (more criteria shown in the study protocol) Baseline comparison: no significant differences in baseline characteristics between groups (source: ClinicalTrials.gov) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: change from Visit 2 (Day 1) to Visit 4 (Day 15) Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship source: Kala Pharmaceuticals Inc Ethics approval: the study protocol states that: "This protocol and the informed consent form must be approved by an appropriately constituted and qualified IRB and the approvals made available to the sponsor or designee prior to the start of enrollment into the study based on these items" Correspondence author's name: NR Additional information:
|
|
| Notes | 79% of participants were white. | |
KPI‐121 (STRIDE2).
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2016/06 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2017/09 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: quadruple (participant, care provider, investigator, outcomes assessor) 05. Study visits and the corresponding time points: Visit 1 (14 ± 1 days before Visit 2) for screening Visit 2 (Day 1) for randomization Visit 3 (Day 8 ± 1) Visit 4 (Day 15 ± 1) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: participant‐rated assessment of ocular discomfort [blocked]* utilizing [blocked] VAS (0 to 100). *[blocked] were phrases or words in the study protocol that were blocked from viewing 07. Assessment for safety outcomes: adverse events; slit lamp biomicroscopy; IOP measurement; BCVA 08. Planned follow‐up duration: 15 days 09. Actual follow‐up duration: 15 days 10. Planned treatment duration (of the intervention steroid): 14 days 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: (same as in the study protocol for STRIDE 1) |
|
| Participants |
Country: USA Setting: multicenter Interventions:
Age, mean/SD (range): 59.1/14.5 Female, n (%): 332 (73.5%) Etiology, n (%): NR Participants (eyes) randomized: 452 Participants (eyes) analyzed for primary study outcomes: 445/446 Participants (eyes) analyzed for safety outcomes: 452
Age, mean/SD (range): 59.3/15.0 Female, n (%): 354 (78.1%) Etiology, n (%): NR Participants (eyes) randomized: 453 Participants (eyes) analyzed for primary study outcomes: 444/447 Participants (eyes) analyzed for safety outcomes: 453
Age, mean/SD (range): 59.2/14.7 Female, n (%): 686 (75.8%) Etiology, n (%): NR Participants (eyes) randomized: 905 Participants (eyes) analyzed for primary study outcomes: 889/893 Participants (eyes) analyzed for safety outcomes: 905 Inclusion criteria: (same study protocol for STRIDE 1) Exclusion criteria: (same study protocol for STRIDE 1) Baseline comparison: no significant differences in baseline characteristics between groups (source: ClinicalTrials.gov) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: baseline/visit 2 (day 1) and visit 4 (day 15) Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship source: Kala Pharmaceuticals Inc Ethics approval: the study protocol states that: "This protocol and the informed consent form must be approved by an appropriately constituted and qualified IRB and the approvals made available to the sponsor or designee prior to the start of enrollment into the study based on these items" Correspondence author's name: NR Additional information:
|
|
| Notes |
|
|
KPI‐121 (STRIDE3).
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2018/07 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2020/02 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: quadruple (participant, care provider, investigator, outcomes assessor) 05. Study visits and the corresponding time points: Visit 1 (14 ± 1 days before Visit 2) for screening Visit 2 (Day 1) for randomization Visit 3 (Day 8 ± 1) Visit 4 (Day 15 ± 1) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: participant‐rated assessment of ocular discomfort [blocked]* utilizing [blocked] VAS (0 to 100). *[blocked] were phrases or words in the study protocol that were blocked from viewing 07. Assessment for safety outcomes: adverse events; slit lamp biomicroscopy; IOP measurement; BCVA; dilated ophthalmoscopy; pregnancy screen (performed only at Visits 1 and 4) 08. Planned follow‐up duration: 15 days 09. Actual follow‐up duration: 15 days 10. Planned treatment duration (of the intervention steroid): 14 days 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: (same as in the study protocol for STRIDE 1 for ocular discomfort severity overall and in subgroup) |
|
| Participants |
Country: USA Setting: multicenter Interventions:
Age, mean/SD (range): 57.6/15.3 Female, n (%): 339 (75.8%) Etiology, n (%): NR Participants (eyes) randomized: 447 Participants (eyes) analyzed for primary study outcomes: 434 Participants (eyes) analyzed for safety outcomes: 449
Age, mean/SD (range): 57.3/15.5 Female, n (%): 330 (72.7%) Etiology, n (%): NR Participants (eyes) randomized: 454 Participants (eyes) analyzed for primary study outcomes: 451 Participants (eyes) analyzed for safety outcomes: 452
Age, mean/SD (range): 57.4/15.4 Female, n (%): 669 (74.3%) Etiology, n (%): NR Participants (eyes) randomized: 901 Participants (eyes) analyzed for primary study outcomes: 885 Participants (eyes) analyzed for safety outcomes: 901 Note: 2 participants were randomized to vehicle (and were included in the vehicle arm for efficacy analyses as part of the ITT population) but erroneously received KPI‐121 (and were included in the KPI‐121 treatment arm for safety analyses). Inclusion criteria: are willing and able to follow instructions and can be present for the required study visits for the duration of the study, including:
*[blocked] were phrases or words in the study protocol that were blocked from viewing Exclusion criteria:
(others same as in the study protocol for STRIDE 1) Baseline comparison: no significant differences in baseline characteristics between groups (source: ClinicalTrials.gov) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: baseline/visit 2 (day 1) and visit 4 (day 15) Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship source: Kala Pharmaceuticals Inc Ethics approval: the study protocol states that: "This protocol and the informed consent form must be approved by an appropriately constituted and qualified IRB and the approvals made available to the Sponsor or designee prior to the start of enrollment into the study based on these items" Correspondence author's name: NR Additional information:
|
|
| Notes | 76.5% of participants were white. | |
Lee 2014.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2012/08 02. Calendar time when the study completed follow‐up (YYYY/MM): 2013/03 03. Unit of randomization (participant or eye): participant (single eye, the study eye chosen was the one with a higher stage of MGD) 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: double‐masked (assessor, analyst); physicians and participants were aware of the treatment received 05. Study visits and the corresponding time points: baseline, 1 month, 2 months 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI 07. Assessment for safety outcomes: safety was assessed by monitoring any adverse events during the entire course of study 08. Planned follow‐up duration: 2 months 09. Actual follow‐up duration: 2 months 10. Planned treatment duration (of the intervention steroid): 2 months 11. How missing data were handled: complete‐case analysis; 4 participants (4 eyes) in group I and 6 participants (6 eyes) in group II were lost to follow‐up. Measurements of the remaining 60 eyes of 60 participants were used for statistical analysis. 12. Description on power and sample size calculation: the primary outcome measure of this study was inflammatory tear cytokine levels at 2 months after treatment |
|
| Participants |
Country: South Korea Setting: single university‐affiliated medical center Interventions:
Age, mean/SD (range): 66.8/10.1 (46 to 81) Female, n (%): 19 (56%) Etiology, n (%): MGD (100%) Participants (eyes) randomized: 34 Participants (eyes) analyzed for primary study outcomes: 30 Participants (eyes) analyzed for safety outcomes: 30
Age, mean/SD (range): 67.1/11.7 (44 to 81) Female, n (%): 20 (56%) Etiology, n (%): MGD (100%) Participants (eyes) randomized: 36 Participants (eyes) analyzed for primary study outcomes: 30 Participants (eyes) analyzed for safety outcomes: 30
Age, mean/SD (range): 66.9/10.9 Female, n (%): 39 (56%) Etiology, n (%): MGD (100%) Participants (eyes) randomized: 70 Participants (eyes) analyzed for primary study outcomes: 60 Participants (eyes) analyzed for safety outcomes: 60 Inclusion criteria:
Exclusion criteria:
Baseline comparison: no significant differences in any parameters were found between groups before treatment (Table 1) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: 2 months Primary outcomes of the study: inflammatory cytokine levels at 2 months after treatment Other outcomes of the study: data on both clinical tests and patient‐reported outcomes |
|
| Study Identification |
Sponsorship Source: Yonsei University Ethics approval: this randomized controlled trial was prospectively approved by the Institutional Review Board of Severance Hospital, Yonsei University College of Medicine (Seoul, South Korea) Correspondence author's name: Tae‐im Kim; Institution: Department of Ophthalmology, Yonsei University College of Medicine, Seoul, South Korea Additional information:
|
|
| Notes | Continuous outcomes were presented as least square mean (standard error), and P values are from linear mixed model with post hoc analysis considering the interaction effect between the 2 groups and the 3 time courses. Generalized estimating equations model for non‐continuous scale values: expressibility, n (%, proportion ≥ grade 1); ocular irritation symptom, n (%, proportion ≥ grade 2); MGD stage, n (%, proportion ≥ stage 3) | |
Li 2021.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2017/02 02. Calendar time when the study completed follow‐up (YYYY/MM): 2019/12 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: double (participant, investigator) 05. Study visits and the corresponding time points: baseline, week 2 and 4 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI 07. Assessment for safety outcomes: participants' tolerability to the treatment ; adverse symptoms 08. Planned follow‐up duration: 4 weeks 09. Actual follow‐up duration: 4 weeks 10. Planned treatment duration (of the intervention steroid): 4 weeks 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: China Setting: single medical center Interventions:
Age, mean/SD (range): 40.1/8.6 Female, n (%): 33 (55%) Etiology, n (%): NR Participants (eyes) randomized: 60 Participants (eyes) analyzed for primary study outcomes: 58 Participants (eyes) analyzed for safety outcomes: 58
Age, mean/SD (range): 39.7/9.1 Female, n (%): 36 (58%) Etiology, n (%): NR Participants (eyes) randomized: 62 Participants (eyes) analyzed for primary study outcomes: 58 Participants (eyes) analyzed for safety outcomes: 58
Age, mean/SD (range): 38.4/9.0 Female, n (%): 69 (57%) Etiology, n (%): NR Participants (eyes) randomized: 122 Participants (eyes) analyzed for primary study outcomes: 116 Participants (eyes) analyzed for safety outcomes: 116 Inclusion criteria:
Exclusion criteria: NR Baseline comparison: there were no statistical differences in pre‐treatment characteristics between groups |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: week 2 and 4 Primary outcomes of the study:
Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: NR Ethics approval: NR Correspondence author's name: Neng Li; Institution: Department of Ophthalmology, Hangzhou Hospital of Traditional Chinese Medicine, Zhejiang, China Additional information:
|
|
| Notes | ||
Lin 2015.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2013/01 02. Calendar time when the study completed follow‐up (YYYY/MM): 2013/09 03. Unit of randomization (participant or eye): participant (the right eye or the worst eye identified by the corneal staining score) 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: open‐label (ClinicalTrials.gov) 05. Study visits and the corresponding time points: 14 ± 2, 28 ± 2, and 56 ± 2 days after the first treatment 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI 07. Assessment for safety outcomes: safety was evaluated based on the incidence of adverse events in each treatment group 08. Planned follow‐up duration: 8 weeks 09. Actual follow‐up duration: 8 weeks 10. Planned treatment duration (of the intervention steroid): 56 ± 2 days 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: China Setting: single medical center Interventions:
Age, mean/SD (range): 50.4/10.9 Female, n (%): NR Etiology, n (%): Sjögren syndrome (100%) Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 19 Participants (eyes) analyzed for safety outcomes: 19
Age, mean/SD (range): 49.9/10.7 Female, n (%): NR Etiology, n (%): Sjögren syndrome (100%) Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 16 Participants (eyes) analyzed for safety outcomes: 16
Age, mean/SD (range): 50.2/10.6 Female, n (%): NR Etiology, n (%): Sjögren syndrome (100%) Participants (eyes) randomized: 40 Participants (eyes) analyzed for primary study outcomes: 35 Participants (eyes) analyzed for safety outcomes: 35 Inclusion criteria:
Exclusion criteria: Patients who had suffered an injury or infection to their eye, who had ocular inflammation unrelated to dry eye, who had undergone ophthalmological surgery within the previous 6 months, had another uncontrolled illness, or who were pregnant or lactating, were excluded from the study. Postmenopausal women receiving hormonal replacement therapy were also excluded. Baseline comparison: no significant differences in the baseline demographic and ocular characteristics were observed between the FML and the CsA groups (Table 1) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: week 2, 4, and 8 after the first treatment Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship source: the authors have no funding and conflicts of interest to disclose Ethics approval: "Our study was conducted in compliance with the Declaration of Helsinki for research involving human participants and was approved by the Ethics Committee of the EENT Hospital of Fudan University" Correspondence author's name: Lan Gong; Institution: Department of Ophthalmology, Eye, Ear, Nose, and Throat Hospital of Fudan University, Shanghai, China Additional information:
|
|
| Notes | ||
Luo 2013.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2011/01 02. Calendar time when the study completed follow‐up (YYYY/MM): 2011/06 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: NR 05. Study visits and the corresponding time points: baseline (before) and 1 week after treatment (after) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: dryness symptom questionnaire for dryness, foreign body sensation, burning, fatigue, photophobia, and stinging; the scale (0 to 5), 0 for no symptom, 1 for occasional symptom (< 3 times/week occurrence and relieved with rest), 2 for frequent occurrences (≥ 3 times/week), 3 for frequent symptoms affecting daily life (≥ 6 times/week, not relieved by rest), 4 for ≥ 10 times/week and not relieved by medications, 5 for persistent symptoms that severely affected daily life 07. Assessment for safety outcomes: NR 08. Planned follow‐up duration: 1 week 09. Actual follow‐up duration: 1 week 10. Planned treatment duration (of the intervention steroid): 1 week 11. How missing data were handled: NA 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: China Setting: single medical center Intervention:
Age, mean/SD (range): NR Female, n (%): NR Etiology, n (%): MGD (100%) Participants (eyes) randomized: 15 Participants (eyes) analyzed for primary study outcomes: 15 Participants (eyes) analyzed for safety outcomes: NR
Age, mean/SD (range): NR Female, n (%): NR Etiology, n (%): MGD (100%) Participants (eyes) randomized: 15 Participants (eyes) analyzed for primary study outcomes: 15 Participants (eyes) analyzed for safety outcomes: 15
Age, mean/SD (range): 35.6/6.7 (18 to 72), including participants in the tobramycin‐alone group Female, n (%): 27 (60%), including participants in the tobramycin‐alone group Etiology, n (%): MGD (100%) Participants (eyes) randomized: 30 Participants (eyes) analyzed for primary study outcomes: 30 Participants (eyes) analyzed for safety outcomes: NR Note: data were not shown for a third group treated with tobramycin 3 g/L with local physical therapy (N = 15). Inclusion criteria: patients with MGD, which was diagnosed based on symptoms, slit lamp exam, and instability of the tear film Exclusion criteria: NR Baseline comparison: there were no statistical differences in baseline characteristics (age or sex), symptoms, and clinical signs among the 3 groups (Table 1) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: week 1 Primary outcomes of the study: symptom; TBUT; Schirmer's I test; corneal fluorescein staining (lid margin) score Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: NR Ethics approval: NR Correspondence author's name: Cheng Lei; Institution: Department of Ophthalmology, General Hospital of Wuhan Steel and Iron Group Corp, Wuhan Hubei, China Additional information:
|
|
| Notes | ||
NCT01276223.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2011/02 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2012/01 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: double (participant, investigator) 05. Study visits and the corresponding time points: baseline, week 1, 2, 3, 4 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: VAS was used by the participant to assess ocular discomfort, both frequency and severity, at baseline (pre‐treatment) and weekly thereafter for 4 additional weeks. Each scale was 100 mm in length. The VAS score was calculated by measuring the length in millimeters from the start of the line to the intersection point of the vertical mark made by the participant. The Global Ocular Discomfort Score is a composite of the 2 VAS scores, ranging from 0 (very mildly) to 100 (very severely uncomfortable). 07. Assessment for safety outcomes: NR 08. Planned follow‐up duration: 4 weeks 09. Actual follow‐up duration: 4 weeks 10. Planned treatment duration (of the intervention steroid): 4 weeks 11. How missing data were handled: of the 722 participants enrolled, 433 did not qualify for run‐in and were exited without exposure to product. Of the 289 participants entering run‐in, 78 did not qualify for treatment. The 211 participants qualifying for treatment were randomized 1:1 to receive either difluprednate (Durezol) or vehicle. Mixed model repeated measure (MMRM) approach was used to handle missing data during the randomized treatment period; 4 and 3 participants in the difluprednate and vehicle groups did not complete the study. 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: USA Setting: participants were recruited from 25 investigative sites Interventions:
Age, mean/SD (range): 54.4/14.8 Female, n (%): 90 (84.1%) Etiology, n (%): NR Participants (eyes) randomized: 107 Participants (eyes) analyzed for primary study outcomes: 107 Participants (eyes) analyzed for safety outcomes: 107
Age, mean/SD (range): 60.1/13.8 Female, n (%): 85 (81.7%) Etiology, n (%): NR Participants (eyes) randomized: 104 Participants (eyes) analyzed for primary study outcomes: 104 Participants (eyes) analyzed for safety outcomes: 104
Age, mean/SD (range): 57.2/14.6 Female, n (%): 175 (82.9%) Etiology, n (%): NR Participants (eyes) randomized: 211 Participants (eyes) analyzed for primary study outcomes: 211 Participants (eyes) analyzed for safety outcomes: 211 Inclusion criteria:
Exclusion criteria:
Baseline comparison: participants in the difluprednate group were 5.7 years younger than those in the vehicle group (post hoc P = 0.004) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: at week 4 Primary outcomes of the study: mean change from baseline (Week 0) in VAS Global Ocular Discomfort Score over 4 weeks Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: Alcon Research Ethics approval: NR Correspondence author's name: NR Additional information:
|
|
| Notes | ||
Pflugfelder 2004.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): NR 02. Calendar time when the study completed follow‐up (YYYY/MM): NR 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: "Eligible patients received a study number and received double‐masked study medication (either loteprednol or placebo) according to a predetermined random allocation schedule" 05. Study visits and the corresponding time points: Visit 1 (day −14 to −7), Visit 2 (day 1 or baseline), Visit 3 (day 14 ± 3), Visit 4 (day 28 ± 3), Visit 5 (day 42 ± 3) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: "A VAS score for the worst symptom at visit 2 was chosen as the subjective variable. The patients were asked to grade the severity of the following symptoms: burning/stinging, itching, grittiness/scratchiness/foreign body sensation, dryness, stickiness, redness of the eye, and tired eye sensation. Symptoms were graded for each eye separately and for the conditions upon waking and during the day. The VAS—a 100‐mm line, marked at the left end“Absent” and at the right end “Unbearable”—was provided for this purpose. The patient was asked to mark the line at the point between these two extremes that represented the severity of the symptom; the distance from the left end of the line to the mark was the VAS score in millimeters." 07. Assessment for safety outcomes: "Safety was assessed by funduscopy, lens examination and biomicroscopy, tests of visual acuity and intraocular pressure, and monitoring adverse events and changes in symptoms. Adverse events were evaluated by the investigator as to severity (mild, moderate, severe) and relationship of the event to the study drug (probable, possible, unlikely, or unknown)." 08. Planned follow‐up duration: 42 ± 3 days 09. Actual follow‐up duration: 42 ± 3 days 10. Planned treatment duration (of the intervention steroid): 28 ± 3 days 11. How missing data were handled: NR 12. Description on power and sample size calculation: "Since this was a pilot study, the calculation of the sample size was based on the best available relevant publications. ... Thus, the planned sample size of 30 per group was expected to provide adequate sensitivity to differentiate between the loteprednol and the placebo groups with respect to both the subjective and objective outcome parameters" |
|
| Participants |
Country: USA Setting: multicenter pilot study Interventions:
Age, mean/SD (range): 57.6 Female, n (%): 20 (63%) Etiology, n (%): NR Participants (eyes) randomized: 32 Participants (eyes) analyzed for primary study outcomes: 32 Participants (eyes) analyzed for safety outcomes: 32
Age, mean/SD (range): 56.2 Female, n (%): 30 (88%) Etiology, n (%): NR Participants (eyes) randomized: 34 Participants (eyes) analyzed for primary study outcomes: 34 Participants (eyes) analyzed for safety outcomes: 34
Age, mean/SD (range): 56.9 Female, n (%): 50 (76%) Etiology, n (%): NR Participants (eyes) randomized: 66 Participants (eyes) analyzed for primary study outcomes: 66 Participants (eyes) analyzed for safety outcomes: 66 Inclusion criteria:
Exclusion criteria: Female patients were either postmenopausal or using a recognized, reliable method of contraception; pregnant or lactating females; patients with contraindications to the use of topical corticosteroid eye drops or their components; patients with illnesses that could interfere with the study; ocular infections or a history of herpes simplex infection were also excluded. Further exclusions were made for contact lens use, punctal occlusion within 3 months, systemic corticosteroid use in the last 6 months, topical corticosteroid use in the last 2 months, and concurrent use of ophthalmic medications for conditions other than keratoconjunctivitis sicca. Ophthalmic surgery in the past 6 months, participation in another clinical study, or use of experimental medication in the past 30 days also disqualified patients from participation. Baseline comparison: the majority of participants were female (75.7%), and there was a slightly larger preponderance of female participants in the vehicle‐treated group (88.2%) vs the loteprednol‐treated group (62.5%) (Table 3) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: 28 ± 3 days Primary outcomes of the study: changes in the following 2 variables at week 4 from baseline:
Other outcomes of the study: secondary analyses included the following, calculated as a change from Visit 2 to Visits 4 and 5, respectively:
|
|
| Study Identification |
Sponsorship source: Bausch & Lomb, Rochester, NY, USA Ethics approval: approval was obtained from an IRB for each study site before the study was initiated Correspondence author's name: Stephen P Bartels; Institution: Bausch & Lomb Additional information:
|
|
| Notes | 92.4% of the participants were white. | |
Pinto‐Fraga 2016.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2014/03 (ClinicalTrials.gov: 2014/02) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2014/11 (ClinicalTrials.gov: 2014/12) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: participants, treatment allocators, and outcome assessors (examiners) 05. Study visits and the corresponding time points: Day 0 (Visit 1), Day 21 (Visit 2 and Visit 3), Day 22 (Visit 4) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI, SANDE version I 07. Assessment for safety outcomes: the safety of both treatments was assessed by recording the nature, severity, and duration of all adverse events and their relationship to the study medication. 4 additional safety endpoints were included in the trial (Table 1): changes in BCVA, fundus evaluation and optic cup‐to‐disc ratio, anterior segment anomalies (especially corneal epithelial problems or signs of infection), and IOP. 08. Planned follow‐up duration: 22 days 09. Actual follow‐up duration: 22 days 10. Planned treatment duration (of the intervention steroid): 21 days 11. How missing data were handled: "because 1 patient dropped from the study because of work‐related issues, 1 more individual was recruited, and thus a total of 41 patients eventually were included in the study, but data from only 40 patients were analyzed" 12. Description on power and sample size calculation: the sample size was estimated to detect a 1‐point difference (Oxford scale) in the main efficacy variable (corneal fluorescein staining), at a significance level of 0.05, with a statistical power of 0.9, and with an estimate of a 10% loss of the sample size |
|
| Participants |
Country: Spain Setting: single medical center Interventions:
Age, mean (95% CI): 59.0 (55.2 to 62.7) Female, n (%): 17 (81%) Etiology, n (%): NR Participants (eyes) randomized: 21 Participants (eyes) analyzed for primary study outcomes: 21 Participants (eyes) analyzed for safety outcomes: 21
Age, mean (95% CI): 60.3 (56.0 to 64.7) Female, n (%): 17 (89%) Etiology, n (%): NR Participants (eyes) randomized: 19 Participants (eyes) analyzed for primary study outcomes: 19 Participants (eyes) analyzed for safety outcomes: 19
Age, mean (95% CI): 59.6 (58.9 to 60.3) Female, n (%): 34 (85%) Etiology, n (%): NR Participants (eyes) randomized: 40 Participants (eyes) analyzed for primary study outcomes: 40 Participants (eyes) analyzed for safety outcomes: 40 Inclusion criteria: Corneal fluorescein staining score of 1 or more (Oxford scale) in both eyes, a TBUT of 7 seconds or less in both eyes, unanesthetized Schirmer test results of 10 mm/5 min or less in both eyes, and an OSDI score of 12 points or more. Importantly, the patient had to express a worsening of DED‐related symptoms when exposed to adverse environmental conditions during their daily life and the use of artificial tears before beginning the study. Patients were accepted into the study if they were taking other topical or systemic treatment if it was begun at least 3 months before inclusion, and the dosage was to be maintained throughout the entire study. Finally, patients had to have a BCVA of 1.0 logarithm of the minimum angle of resolution or less in each eye. Exclusion criteria: Patients were excluded if they had a known sensitivity or intolerance to any of the treatments used in the study; a history of ocular infection or severe ocular inflammation (other than DED related) 6 months before inclusion in the study; any active ocular disease (different from DED); any uncontrolled severe systemic disease that may affect the eye (except Sjögren syndrome); any ocular surgery or trauma that could affect corneal sensitivity, normal tear distribution in the 6 previous months, any ocular or systemic surgery or procedure planned during the study duration that could affect outcomes, or a combination thereof; occlusion of the lacrimal puncta either surgically or with plugs within 3 months before study; contact lenses wear within 3 months or during the study; or use of any topical medication except for DED. Other exclusion criteria included initiation, discontinuation, or change of dosage of antihistaminic, cholinergic agents, beta‐blocking agents, antidepressants, or any systemic medication with possible effects over the tear film; history of glaucoma or IOP of more than 22 mmHg in any measurements 2 months before baseline; optic cup‐to‐disc ratio of more than 0.6 mm; pregnancy; lactation; or inadequate contraception. Also, patients were excluded if undergoing topical cyclosporine A eye drops within 3 months before study inclusion, topical corticosteroid eye drops within 1 month before inclusion, or both. Baseline comparison: for baseline clinical characteristics, there were no statistical differences (P = 0.06) between groups in corneal and conjunctival staining, hyperemia, TBUT, unanesthetized Schirmer test results, BCVA, tear osmolarity, or IOP (Table 4) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: between Visits 2 and 3, i.e. before and after 2 hours of desiccating stress in participants treated for 21 days Primary outcomes of the study:
Other outcomes of the study: All other evaluations were considered as secondary variables measuring efficacy, except those included as safety measures (Table 1); these included OSDI scores, BCVA (high and low contrast), tear osmolarity, treatment satisfaction, TBUT, Lissamine green conjunctival staining, biomicroscopy with slit lamp, Schirmer test (unanesthetized), IOP, fundus evaluation, and optic cup‐to‐disc ratio. These were evaluated between Visits 1 and 2, 2 and 3, and 3 and 4. |
|
| Study Identification |
Sponsorship source: the study was sponsored by and conducted at Instituto Universitario de Oftalmobiología Aplicada, University of Valladolid, Valladolid, Spain (in the text). According to the footnotes of the article, the study was supported by the Spanish Ministry of Economy and Competitiveness (grant no.: SAF2010‐15361); European Social Funds, Operative Program for Castilla y León, Castilla y León Council, Spain (grant no.: EDU/346/2013); and the Inflammation Research Program, Allergan Inc (Irvine, CA, USA) (contributed extra funding for environmental chamber use). Ethics approval: "This clinical trial was approved by the University Hospital Ethics Committee (Valladolid, Spain) and by the Spanish Regulatory Agency (Spanish Drugs and Health Products Administration; www.aemps.gob.es/en/home.htm) with EUDRA (European Union Drug Regulating Authorities) number 2013‐002183‐63" Correspondence author's name: Margarita Calonge, MD, PhD; Institution: Instituto Universitario de Oftalmobiología Aplicada, Universidad de Valladolid Additional information:
|
|
| Notes | ||
Qazi 2015.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2011/08 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2013/03; 2017/06 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: quadruple (participant, care provider, investigator, outcomes assessor) 05. Study visits and the corresponding time points: baseline, week 4 (after treatment), week 8 (4 weeks after treatment cessation) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI; SANDE 1. OSDI: a 12‐question survey used to measure the symptoms of dry eye disease 2. SANDE: the range of the SANDE frequency scale is 0 to 100, with 0 being the minimum level of frequency of dry eye symptoms, and 100 being the maximum level of frequency of dry eye symptoms; the range of the SANDE severity scale is 0 to 100, with 0 being the minimum level of severity of dry eye symptoms, and 100 being the maximum level of severity of dry eye symptoms 07. Assessment for safety outcomes: NR 08. Planned follow‐up duration: 8 weeks 09. Actual follow‐up duration: 8 weeks 10. Planned treatment duration (of the intervention steroid): 4 weeks 11. How missing data were handled: complete‐case analysis for outcome data 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: USA Setting: single‐site Interventions
Age, mean/SD (range): 52/12 Female, n (%): 9 (45%) Etiology, n (%): NR Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 17 Participants (eyes) analyzed for safety outcomes: 20
Age, mean/SD (range): 55/13 Female, n (%): 8 (47%) Etiology, n (%): NR Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 17 Participants (eyes) analyzed for safety outcomes: 20
Age, mean/SD (range): 57/12 Female, n (%): 15 (30%) Etiology, n (%): NR Participants (eyes) randomized: 20 Participants (eyes) analyzed for primary study outcomes: 20 Participants (eyes) analyzed for safety outcomes: 20
Age, mean/SD (range): 55/12 Female, n (%): 24 (53%) Etiology, n (%): MGD 54 (100%) Participants (eyes) randomized: 60 Participants (eyes) analyzed for primary study outcomes: 54 Participants (eyes) analyzed for safety outcomes: 60 Inclusion criteria:
Exclusion criteria:
Baseline comparison: no significant differences in baseline characteristics of participants among the 3 comparison groups (ClinicalTrials.gov) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: 4 weeks Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship source: Massachusetts Eye and Ear Infirmary (MEEI); Bausch & Lomb Inc (conference abstract); Bausch & Lomb Inc, National Institutes of Health K24‐ EY019098, Falk Medical Research Trust (full publication) Ethics approval: "The study protocol was approved by the Human Studies Committee of the Massachusetts Eye and Ear Infirmary (Boston, MA), and the research was conducted in accord with the requirements of the Health Insurance Portability and Accountability Act and the tenets of the Declaration of Helsinki." Correspondence author's name: Reza Dana; Institution: Massachusetts Eye and Ear Infirmary Additional information: 01. Trial registration no.: NCT01456780 02. Trial registration website: ClinicalTrials.gov 03. Financial disclosure or conflicts of interest statement from authors: Conference abstract: Yureeda Qazi, none; Ahmad Kheirkhah, none; Thomas Dohlman, none; Andrea Cruzat, none; Bernardo Cavalcanti, none; Clara Colon, none; Reza Dana, Bausch & Lomb (F), MEEI (P); Pedram Hamrah, MEEI (P) Full‐text publication: RD, personal fees: Eleven Biotherapeutics, Alcon Laboratories Inc, Allergan Inc, Bausch & Lomb Inc, Genentech, outside the submitted work, and has a US Patent Application pending |
|
| Notes | From the full‐text publication: "This study was partly presented at: the Annual Meeting of the Association for Research in Vision and Ophthalmology, May 4e8, 2014, Orlando, Florida. This manuscript is not an Annual Meeting paper or poster" | |
Sheppard 2014.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2006/11 (ClinicalTrials.gov) 02. Calendar time when the study completed follow‐up (YYYY/MM): 2007/09 (ClinicalTrials.gov) 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: double (participant, investigator) 05. Study visits and the corresponding time points: study visits occurred on day 1 (screening and baseline, Visit 1), 14 ± 2 days (Visit 2), 30 ± 3 days (Visit 3), and 60 ± 5 days (Visit 4) 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI: a global assessment of their perceptions about their dry eye condition and how it had impacted their vision and daily activities at Visits 2, 3, and 4 and also rated the tolerability of the topical CsA drops since the previous study visit at the third and fourth study visits 07. Assessment for safety outcomes: safety outcomes included ocular and systemic adverse events, IOP, and ophthalmoscopic examination findings 08. Planned follow‐up duration: 60 days 09. Actual follow‐up duration: 60 ± 5 days 10. Planned treatment duration (of the intervention steroid): 60 days 11. How missing data were handled: complete‐case analysis 12. Description on power and sample size calculation: the calculations were based on a power of 80% with an alpha of 0.05 (2‐tailed) and also used published data to estimate standard deviations for individual parameters of the study, which included OSDI, Lissamine staining (NEI scale), central fluorescein staining (NEI scale), and Schirmer test |
|
| Participants |
Country: USA Setting: multicenter Interventions:
Age, mean/SD (range): 59.6/12.1 (27 to 80) Female, n (%): 44 (77%) Etiology, n (%): NR Participants (eyes) randomized: NR Participants (eyes) analyzed for primary study outcomes: 57 Participants (eyes) analyzed for safety outcomes: 57
Age, mean/SD (range): 57.9/1038 (36 to 79) Female, n (%): 43 (78%) Etiology, n (%): NR Participants (eyes) randomized: NR Participants (eyes) analyzed for primary study outcomes: 55 Participants (eyes) analyzed for safety outcomes: 55
Age, mean/SD (range): 58.7/11.4 (24 to 80) Female, n (%): 87 (78%) Etiology, n (%): NR Participants (eyes) randomized: 116 Participants (eyes) analyzed for primary study outcomes: 112 Participants (eyes) analyzed for safety outcomes: 112 Inclusion criteria:
Exclusion criteria:
Baseline comparison: there were no statistically significant differences in baseline characteristics within and between treatment groups (Table 2) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: day 14, 30, and 60 Primary outcomes of the study: The primary efficacy outcomes included:
Other outcomes of the study: frequency of adjunctive tear use (> 6/d, 3 to 6/d, 1 to 2/d, none) |
|
| Study Identification |
Sponsorship source: Supported by an unrestricted research grant from Bausch & Lomb Inc, Rochester, NY, USA, and the Virginia Eye Foundation, Norfolk, VA, USA Ethics approval: NR Correspondence author's name: John D Sheppard; Institution: Virginia Eye Consultants, Norfolk, VA, USA Additional information:
|
|
| Notes |
|
|
Singla 2019.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): NR 02. Calendar time when the study completed follow‐up (YYYY/MM): NR 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: NR 05. Study visits and the corresponding time points: baseline, 2 weeks, 6 weeks, 3 months, and 6 months 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: OSDI questionnaire was used for grading the severity of dry eye. This questionnaire consists of 12 questions and is graded on a scale from 0 to 100. OSDI score of 16 to 30 was included for the diagnosis of moderate dry eye disease. 07. Assessment for safety outcomes: IOP 08. Planned follow‐up duration: 6 months 09. Actual follow‐up duration: 6 months 10. Planned treatment duration (of the intervention steroid): 3 months 11. How missing data were handled: NA 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: India Setting: single‐site Interventions
Age, mean (range): 44.4 (25 to 68) Female, n (%): 45 (64%) Etiology, n (%): NR Participants (eyes) randomized: 70 Participants (eyes) analyzed for primary study outcomes: 70 Participants (eyes) analyzed for safety outcomes: 70
Age, mean (range): 44.6 (23 to 69) Female, n (%): 45 (64%) Etiology, n (%): NR Participants (eyes) randomized: 70 Participants (eyes) analyzed for primary study outcomes: 70 Participants (eyes) analyzed for safety outcomes: 70
Age, mean/SE (range): 44.5/7.7 (23 to 69) Female, n (%): 90 (64.3%) Etiology, n (%): NR Participants (eyes) randomized: 140 Participants (eyes) analyzed for primary study outcomes: 140 Participants (eyes) analyzed for safety outcomes: 140 Inclusion criteria: Patients above 18 years of age, diagnosed with moderate dry eye, who were not wearing contact lenses for at least 1 month before the study and agree not to wear the same during the study period were included in the study Exclusion criteria:
Baseline comparison: both groups had comparable baseline parameters, and there was no statistically significant difference between groups in terms of OSDI scores, TBUT values, Schirmer’s test values, corneal fluorescein staining scores, and Lissamine green conjunctival staining scores, which may suggest excessive balance between the two groups |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: 2 weeks, 6 weeks, 3 months, and 6 months Primary outcomes of the study:
Other outcomes of the study: NR |
|
| Study Identification |
Sponsorship source: NR Ethics approval: approval of the Institute’s Ethics Committee was obtained before starting the study Correspondence author's name: Mukesh Joshi; Institution: Safdarjung Hospital, New Delhi, India Additional information:
|
|
| Notes | ||
Wan 2012.
| Study characteristics | ||
| Methods |
00. Study design: randomized controlled trial, parallel group 01. Calendar time when the study enrolled the first participant (YYYY/MM): 2009/03 02. Calendar time when the study completed follow‐up (YYYY/MM): 2010/09 03. Unit of randomization (participant or eye): participant 04. Masking of participants, treatment allocator, outcome assessor, or data analyzer: NR 05. Study visits and the corresponding time points: baseline, weeks 2, 4, 6, 8 06. Instruments and the scales used for documenting patient‐reported symptoms or quality of life: symptom scale (0 to 9) for dryness, foreign body sensation, burning, photophobia, and blurring 07. Assessment for safety outcomes: IOP and tolerability 08. Planned follow‐up duration: 8 weeks 09. Actual follow‐up duration: 8 weeks 10. Planned treatment duration (of the intervention steroid): 8 weeks 11. How missing data were handled: NA 12. Description on power and sample size calculation: NR |
|
| Participants |
Country: China Setting: single medical center Intervention:
Age, mean/SE (range): 36.4/4.5 Female, n (%): 11 (58%) Etiology, n (%): NR Participants (eyes) randomized: 19 Participants (eyes) analyzed for primary study outcomes: 19 Participants (eyes) analyzed for safety outcomes: 19
Age, mean/SE (range): 33.6/5.0 Female, n (%): 10 (67%) Etiology, n (%): NR Participants (eyes) randomized: 15 Participants (eyes) analyzed for primary study outcomes: 15 Participants (eyes) analyzed for safety outcomes: 15
Age, mean/SE (range): 35/5.0 (23 to 56) Female, n (%): 21 (62%) Etiology, n (%): NR Participants (eyes) randomized: 34 Participants (eyes) analyzed for primary study outcomes: 34 Participants (eyes) analyzed for safety outcomes: 34 Inclusion criteria:
Exclusion criteria:
Baseline comparison: no statistical differences in baseline characteristics of the study participants (Table 1) |
|
| Interventions |
|
|
| Outcomes |
Time points of primary outcome data collected: weeks 2, 4, 6, and 8 Primary outcomes of the study:
Other outcomes of the study:
|
|
| Study Identification |
Sponsorship source: National Natural Science Foundation (Grant no. 30973246), Provincial Science and Technology Research Grant (Grant no. 2009 A030200004), Provincial Medical Research and Development Fund (B2011106), Provincial Natural Science Foundation (S2011040004327) Ethics approval: ethics approval requirement or process not reported Correspondence author's name: Zungcze Wang; Institution: Chung Sang University Eye Center, Guangdong, China Additional information:
|
|
| Notes | ||
AE, adverse event; ATS, artificial tear substitute; BCVA, best‐corrected visual acuity; CAE, controlled adverse environment; CB, clobetasone butyrate; CI, confidence interval; CXCL10, C‐X‐C motif chemokine ligand 10; DE, dry eye; DED, dry eye disease; DEEP, Dry Eye Epidemiology Projects; ETDRS, Early Treatment of Diabetic Retinopathy Study; FML, fluorometholone; HLA‐DR, human leukocyte antigen–DR isotype; IL‐6, interleukin 6; IL‐8, interleukin 8; IOP, intraocular pressure; IRB, institutional review board; ITT, intention‐to‐treat; IVCM, in vivo confocal microscopy; LASIK, laser in situ keratomileusis; MGD, meibomian gland dysfunction; NA, not applicable; NCT, clinical trial registry provided by the US National Library of Medicine; NEI, National Eye Institute; NIKBUT, non‐invasive keratographic tear film break‐up time; no., number; NR, not reported; NSAIDs, non‐steroidal anti‐inflammatory drugs; OSDI, Ocular Surface Disease Index; OTC, over the counter; PVP, polyvinylpyrrolidone (tear substitute); SANDE, Symptom Assessment iN Dry Eye questionnaire; SD, standard deviation; SE, standard error; SPEED, Standard Patient Evaluation of Eye Dryness; SS, Sjögren syndrome; TBUT, tear film break‐up time; TFOS, Tear Film & Ocular Surface Society; TNF‐α, tumor necrosis factor‐alpha; VAS, visual analogue scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abud 2016 | Ineligible study population |
| Acord 2010 | Ineligible study design (cross‐over trial, conference poster) |
| Asbell 2011 | Ineligible study design (workshop report) |
| Boynton 2015 | Ineligible study population |
| ChiCTR‐IPQ‐15006773 | Ineligible study design (quasi‐randomization) |
| Edward 2014 | Trial withdrawn by sponsor (before participant enrollment) |
| EUCTR2006‐003391‐35‐NL | Ineligible study design (within‐person comparison) |
| EUCTR2019‐000747‐27‐IT | Ineligible intervention (within‐person comparison) |
| Gupta 2021 | Ineligible study design (review article) |
| ISRCTN13765551 | Ineligible study design (same intervention, preservative‐free versus preserved formula) |
| Jee 2014 | Ineligible study design (same intervention, preservative‐free versus preserved formula) |
| JPRN‐UMIN000025159 | Ineligible study design (non‐pharmacological intervention) |
| Kallab 2020 | Ineligible study design (comparing dosing schedules) |
| Korenfeld 2021 | Ineligible study design (secondary data analysis) |
| Lee 2006 | Ineligible study design (within‐person comparison) |
| NCT03907865 | Ineligible study design (comparing dosing schedules) |
| Rolando 2008 | Ineligible study design (comparing dosing schedules) |
| Ryu 2005 | Ineligible study design (within‐person comparison) |
| Shen 2015 | Ineligible intervention (FML combined with contact lens) |
| Sindhu 2015 | Ineligible study design (observational study) |
| Wong 2021 | Ineligible study design (review article) |
FML, fluorometholone
Characteristics of studies awaiting classification [ordered by study ID]
ChiCTR‐IPR‐15007196.
| Methods | Randomized parallel controlled trial |
| Participants |
Age limitation: minimum 18; maximum 70 Gender: both (sex) Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Target sample size: 40 in each group
|
| Outcomes |
Primary outcomes:
Secondary outcomes:
|
| Notes | Sources of financial support: Senju Pharmaceutical Ltd Date of first enrollment: 12 November 2015 Contacts: Zuguo Liu, Caihong Huang; Xiamen University, Fujian, China Contact efforts made: emailed Dr Liu twice but no responses received within 2 weeks Contact reasons: no trial results available |
Herman 2005.
| Methods | Randomized controlled trial, parallel group |
| Participants | Eligibility criteria not reported |
| Interventions |
Actual enrollment: 30 participants in the steroid group, unknown number of participants in the control group
|
| Outcomes |
|
| Notes | Sources of financial support: Ocular Research of Boston Inc and The Walter and Valerie Winchester Research Grant Presented as ARVO Annual meeting abstract May 2005 Contacts efforts made: emailed JP Herman twice; no response received within 2 weeks Contact reasons: the sample size of the control group was not reported in the conference abstract |
NCT00471419.
| Methods | Randomized parallel trial, double masking |
| Participants |
Sex: all eligible for study Inclusion criteria:
Exclusion criteria: under 18 years |
| Interventions | Estimated enrollment: 750 participants
|
| Outcomes |
Primary outcomes:
Secondary outcomes:
|
| Notes | Source of financial support: Alcon Research Study start date: July 2006 Actual study completion date: August 2007 Investigator(s): Michael Brubaker, PhD; Alcon Research Contact efforts made: emailed Alcon Research personnel 3 times; no responses received within 2 weeks Contact reasons: unclear comparator treatment; no published trial results available |
NCT00560638.
| Methods | Randomized controlled trial, parallel assignment, double masked (participant and investigator) |
| Participants |
Sex: all eligible for study Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Actual enrollment: 119 participants
|
| Outcomes |
Primary outcome measures:
Secondary outcome measures:
|
| Notes | Source of financial support: Bausch & Lomb Inc Study start date: November 2005 Actual study completion date: February 2006 Investigator(s): Gail Torkildsen MD; Ophthalmic Research Associates Inc Contact efforts made: emailed to medical research personnel twice; no responses received within 2 weeks Contact reasons: no trial results available |
NCT01562795.
| Methods | Randomized interventional trial, parallel group, open‐label |
| Participants |
Age: 18 to 70 years Sex: all eligible for study Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Actual enrollment: 48 participants
|
| Outcomes |
Primary outcome measures:
Secondary outcome measures:
|
| Notes | Sources of financial support: Wenzhou Medical University Study start date: March 2012 Actual study completion date: July 2012 Investigator(s): Wei Chen, MD PhD; Eye Hospital, Wenzhou Medical College, China Contact efforts made: emailed Dr Chen twice; no response received within 2 weeks Contact reasons: the sample size of each group was not reported; no trial results available |
NCT03418727.
| Methods | Randomized parallel trial, double masking |
| Participants |
Sex: all eligible for study Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Actual enrollment: 84 participants
|
| Outcomes |
Primary outcomes: tolerability [ Time Frame: Baseline ‐ Day 84 ] Participants will assess their tolerance to the administration of the study drug, utilizing a VAS. The VAS is a 100‐millimeter horizontal line with verbal descriptors at either end. The VAS ratings will be completed after administration of the study drug on day 1 (postdose), day 28, day 56, and day 84. Participants will place a single slash mark across the horizontal line between the end labeled "completely intolerable" (0 mm) and "easily tolerable" (100 mm). |
| Notes | Source of financial support: Ocugen Study start date: September 2017 Actual study completion date: March 2018 Investigator(s): Ocugen Contact efforts made: contacted Ocugen research personnel twice; no response received within 2 weeks Contact reasons: the specific type and concentration of interventional corticosteroid used was not reported; no trial results available |
NTR2291.
| Methods | Randomized, placebo‐controlled, interventional trial, parallel group |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
Target sample size: 20
|
| Outcomes |
Primary outcome:
Secondary outcomes:
|
| Notes | Source of financial support: University of Genoa; Bausch & Lomb IOM Start date: 1 January 2010 Stop date: 31 December 2010 Contact(s): Stefano Barabino, MD; Genoa, Italy Contact efforts made: emailed Dr Barabino twice; no responses received within 2 weeks Contact reasons: no trial results available |
BCVA, best‐corrected visual acuity; BUT, break‐up time; CAE, controlled adverse environment; DED, dry eye disease; ETDRS, Early Treatment of Diabetic Retinopathy Study; HLA‐DR, human leukocyte antigen–DR isotype; IOP, intraocular pressure; IRB, Institutional Review Board; LASIK, laser in situ keratomileusis; MGD, meibomian gland dysfunction; OSDI, Ocular Surface Disease Index; SANDE, Symptom Assessment iN Dry Eye questionnaire; SPEED, Standard Patient Evaluation of Eye Dryness; TBUT, tear film break‐up time; VAS, visual analogue scale
Characteristics of ongoing studies [ordered by study ID]
CTRI/2021/02/031182.
| Study name | Eye drops made of antibodies for dry eye disease patients: topical human immunoglobulin as adjunct therapy in dry eye disease |
| Methods |
|
| Participants |
Country: India Inclusion:
Exclusion:
Target sample size: 70 |
| Interventions |
Intervention: topical immunoglobulin
Control intervention: standard therapy for dry eye disease
|
| Outcomes |
Time point: outcomes will be measured at baseline and time intervals: 1 and 2 months Primary outcomes:
Secondary outcomes: Changes in meibomian gland and tear imaging |
| Starting date | 1 March 2021 (date of first enrollment planned) |
| Contact information | Livia Khan; Institution: AIIMS, New Delhi Prof DrM Vanathi; Institution: AIIMS, New Delhi |
| Notes | Status: not yet recruiting (as of 23 February 2021) |
ISRCTN16288419.
| Study name | Evaluation of the performance of new substitute tears in dry eye patients |
| Methods | Randomized interventional trial, double masked |
| Participants |
Country: Italy Inclusion:
Exclusion:
Target sample size: 40 |
| Interventions |
At the end of the first week, participants will keep using eyedrops 4 times a day for 6 months. |
| Outcomes |
Time point: outcomes will be measured at at day 7, 28, 56, 180 Primary outcomes: Symptom improvement measured by SANDE score Secondary outcomes:
|
| Starting date | 10 June 2020 (date of first enrollment) |
| Contact information | Stefano Barabino; Institution: University of Milan, Italy |
| Notes | Current status: ongoing (as of 17 August 2020) |
NCT04734197.
| Study name | A research study to see how well an eye drop, SURF‐100 (a mycophenolic acid/betamethasone sodium phosphate combination), works and what side effects there are in subjects with dry eye disease |
| Methods |
|
| Participants |
Country: USA Inclusion:
Exclusion:
Target sample size: 280 |
| Interventions |
|
| Outcomes |
Time point: outcomes will be measured at baseline and time intervals: day 84 Primary outcomes: UNC DEMS score [Time Frame: Day 84] A reduction of 10% in patient‐reported dry eye disease symptoms and reduction of impact of symptoms on daily life as defined by the UNC DEMS with SURF‐100 as compared to vehicle, Restasis, and Xiidra Secondary outcomes:
|
| Starting date | 1 January 2021 (date of actual study start) |
| Contact information | Kamran Hosseini; Institution: Surface Pharmaceuticals Inc |
| Notes | Status: recruiting (as of 18 May 2021) |
NCT04734210.
| Study name | A research study to see how well an eye drop, SURF‐200 (0.02% and 0.04% betamethasone sodium phosphate), works, what side effects there are, and to compare it with vehicle (placebo) in subjects diagnosed with dry eye disease and experiencing an episodic flare‐up |
| Methods |
|
| Participants |
Country: USA Inclusion:
Exclusion:
Target sample size: 120 |
| Interventions |
|
| Outcomes |
Time point: outcomes will be measured at baseline and time intervals: day 8 Primary outcomes: UNC DEMS score [Time Frame: Day 8] A minimum reduction of 1 unit in patient‐reported dry eye disease symptoms and reduction of impact of symptoms with SURF‐200 as compared to vehicle (as measured by the UNC DEMS) Secondary outcomes:
|
| Starting date | 7 January 2021 (date of actual study start) |
| Contact information | Kamran Hosseini; Institution: Surface Pharmaceuticals Inc |
| Notes | Status: recruiting (as of 27 July 2021) |
ATS, artificial tear substitute; BCVA, best‐corrected visual acuity; CAE, controlled adverse environment; CB, clobetasone butyrate; CsA, cyclosporine A; ETDRS, Early Treatment of Diabetic Retinopathy Study; FML, fluorometholone; HA, hyaluronic acid; HBV, hepatitis B virus; HCV, hepatitis C virus; IgG, immunoglobulin G; IOP, intraocular pressure; IVIG, intravenous immunoglobulin; LASIK, laser in situ keratomileusis; LE, loteprednol etabonate; MGD, meibomian gland dysfunction; MP, methylprednisolone; NA, not applicable; NCT, clinical trial registry provided by the US National Library of Medicine; NEI, National Eye Institute; no., number; NR, not reported; OSDI, Ocular Surface Disease Index; PVP, polyvinylpyrrolidone; SANDE, Symptom Assessment iN Dry Eye questionnaire; TBUT, tear film break‐up time; UCVA, uncorrected visual acuity; UNC DEMS, University of North Carolina Dry Eye Management Scale
Differences between protocol and review
We excluded trials that compared topical corticosteroid therapy, alone or in combination therapy, with the following comparator therapy: (1) intense pulsed light therapy; (2) steroid iontophoresis or inserts; (3) herbal medicines; these comparators were not specifically listed in the protocol.
For trials that randomized participants but reported outcome data for both eyes without specifying which eye was the study eye, we extracted and analyzed data for the right eye. We did not define this lateral preference in the protocol.
We did not perform planned subgroup analysis by sex because the included trials did not report sex‐specific symptom scores or corneal staining results.
We did not perform subgroup analysis on one of the critical outcomes, tear film break‐up time, by etiology as planned in the protocol because fewer than 10 studies reported this outcome for either comparison. Instead, we chose to perform subgroup analysis on one of the important outcomes, fluorescein corneal staining scores, by etiology.
For the subjective and objective outcomes that we chose for risk of bias assessment, we also performed post hoc subgroup analysis by questionnaire or scale, source of funding, and intervention regimen (steroid type, duration).
Contributions of authors
All review authors contributed to the conception and design of the study, participated in study selection, data extraction, and/or analysis, drafted portions of the review, commented on drafts critically regarding intellectual content, and approved the final version for publication.
Sources of support
Internal sources
No sources of support provided
External sources
-
National Eye Institute, National Institutes of Health, USA
Cochrane Eyes and Vision US Project, supported by grant UG1EY020522 (PI: Tianjing Li, MD, MHS, PhD)
-
Public Health Agency, UK
The HSC Research and Development (R&D) Division of the Public Health Agency funds the Cochrane Eyes and Vision editorial base at Queen's University Belfast.
-
Queen’s University Belfast, UK
The work of Gianni Virgili, Co‐ordinating Editor for Cochrane Eyes and Vision, is funded by the Centre for Public Health, Queen’s University of Belfast, Northern Ireland.
Declarations of interest
Su‐Hsun Liu: reports a grant from the National Eye Institute, National Institutes of Health, USA; payment to institution.
Ian J Saldanha: reports grants and contracts, Cochrane Eyes and Vision, from National Eye Institute, National Institutes of Health, USA; payment to institution. Travel reimbursement for making a talk on outcomes related to dry eye in 2018 from Johns Hopkins Wilmer Eye Institute; personal payment.
Alison G Abraham: declared that she has no conflicts of interest.
Thanitsara Rittiphairoj: declared that she has no conflicts of interest.
Scott Hauswirth: reports grants and contracts for paid investigator from TearSolutions and Sylentis (contract pending—study not yet underway as of 23 February 2021); paid to institution. Payments for presentations from Kala Pharmaceuticals, Dompe, Sun Pharmaceuticals, Takeda, and Avedro; personal payment. Support for INTREPID meeting (indirect) from Alcon/Novartis; personal payment. Stock shares in Oyster Point (paid for and owned individually, not as compensation), TearRestore (compensation for design and medical advisory work), and Horizon Pharma (paid for and owned individually, not as compensation); personal payment. Consulting fees for study design and analysis from Ocular Therapeutix (pending); personal payment. Advisory Board payments from Dompe, NuSight Medical, Kala Pharmaceuticals, Sun Pharmaceuticals, EyePoint Medical, EyeVance Pharma, Horizon Pharma, Avedro/Glaukos, Quidel, and Sight Sciences; personal payment. Writing assistance from Takeda (medical writer for review paper); personal payment. SH reports publishing opinions on topical immunomodulation in dry eye in the OSDocs Facebook group (moderator/co‐administrator role), and published 'When dry eye compromises corneal integrity' in Review of Optometry, Nov 2017 (contract pending—study not yet underway as of 23 February 2021). They report working as an Optometrist at the University of Colorado.
Darren Gregory: reports working as an Ophthalmologist at the University of Colorado Eye Center. Their clinical work focuses on the treatment of dry eyes, which sometimes involves the use of topical corticosteroid medications.
Cristos Ifantides: reports being an inventor with intellectual property assigned to their university. The design relates to dry eye, but does not relate to corticosteroid use for dry eye. It is a design for a new form of eyeglasses that can theoretically help with dry eye (patent application filed, University of Colorado), paid to institution. Ownership of stock in Pfizer. Their partner works for AbbVie, which makes medications related to dry eye. She does not work in the eye care space. CI reports working as a clinician at Denver Health and University of Colorado, where they are an attending physician.
Tianjing Li: reports a grant from the National Eye Institute, National Institutes of Health, USA; payment to institution.
New
References
References to studies included in this review
Akhlaq 2019 {published data only}
- Akhlaq A, Kheirkhah A, Aggarwal S, Cavalcanti B, Mueller R, Abbouda A, et al. Patients enrichment for increased dendritiform cells using in vivo confocal microscopy results in improved response to topical steroids in dry eye disease: results of the therapeutic response to antiinflammatory agents in the corneal epithelium (TRACE) study. Investigative Ophthalmology & Visual Science 2019;60:6753. [Google Scholar]
- Hamrah P, Akhlaq A, Ozmen MC, Kheirkhah A, Aggarwal S, Cavalcanti B, et al. Change in dendritiform cell density by in vivo confocal microscopy may be used as a surrogate biomarker for therapeutic response in ery eye disease patients enriched for presence of inflammation: results from the therapeutic response to anti-inflammatory. Investigative Ophthalmology & Visual Science 2019;60:5207. [Google Scholar]
- NCT02106377. Using in vivo confocal microscopy to assess cellular response and efficacy of steroid treatment in ery eye disease. clinicaltrials.gov/ct2/show/NCT02106377 (first received 18 April 2014).
- NCT02120079. The utility of IVCM to assess cellular response and efficacy of long-term topical steroid treatment in patients With DED. clinicaltrials.gov/show/NCT02120079 (first received 22 April 2014).
Aragona 2013 {published data only}
- Aragona P, Spinella R, Rania L, Postorino E, Sommario MS, Roszkowska AM, et al. Safety and efficacy of 0.1% clobetasone butyrate eyedrops in the treatment of dry eye in Sjögren syndrome. European Journal of Ophthalmology 2013;23(3):368‐76. [DOI] [PubMed] [Google Scholar]
Avunduk 2003 {published data only}
- Avunduk AM, Avunduk MC, Varnell ED, Kaufman HE. The comparison of efficacies of topical corticosteroids and nonsteroidal anti-inflammatory drops on dry eye patients: a clinical and immunocytochemical study. American Journal of Ophthalmology 2003;136(4):593‐602. [DOI] [PubMed] [Google Scholar]
- Chan CK, Lam DS. The comparison of efficacies of topical corticosteroids and nonsteroidal anti-inflammatory drops on dry eye patients: a clinical and immunocytochemical study. American Journal of Ophthalmology 2004;137(6):1157-8. [DOI] [PubMed] [Google Scholar]
Bausch 2013 {published data only}
- NCT01817582. Lotemax® Gel 0.5% and Restasis 0.05% in participants with mild or moderate keratoconjunctivitis sicca (ery eye disease). clinicaltrials.gov/show/NCT01817582 (first received 25 March 2013).
Byun 2012 {published data only}
- Byoun Y, Kim S, Kim TI, Kim E. Efficacy of combined treatment of cyclosporine 0.05% and 1% methylprednisolone on dry eye patients of Sjögren syndrome. Investigative Ophthalmology & Visual Science 2007;48:ARVO E‐Abstract 401. [Google Scholar]
- Byun YJ, Kim TI, Kwon SM, Seo KY, Kim SW, Kim EK, et al. Efficacy of combined 0.05% cyclosporine and 1% methylprednisolone treatment for chronic dry eye. Cornea 2012;31(5):509-13. [DOI] [PubMed] [Google Scholar]
Cao 2018 {published data only}
- Cao XY, He L, Li Y H, Li Y. Study on the effect of sodium hyaluronate combined with loteprednol eye drops on the treatment of dry eye in children. International Eye Science 2018;18(3):516‐9. [Google Scholar]
Chen 2020 {published data only}
- Chen W, Shi XL, He XH, Mao YH, Li C, Dong N. Loteprednol combined with sodium hyaluronate in the treatment of dry eye disease and its effect on tnf-α and cxcl10 in tears. Journal of Biological Regulators and Homeostatic Agents 2020;34(5):1825‐9. [DOI] [PubMed] [Google Scholar]
KPI‐121 (Phase 2) {published data only}
- NCT02188160. Safety and efficacy of KPI-121 in subjects with dry eye disease (Kauai). clinicaltrials.gov/show/NCT02188160 (first received 11 July 2014).
KPI‐121 (STRIDE1) {published data only}
- NCT02813265. Safety and efficacy of KPI-121 in subjects with dry eye disease. clinicaltrials.gov/show/NCT02813265 (first received 24 June 2016).
KPI‐121 (STRIDE2) {published data only}
- NCT02819284. Safety and efficacy of KPI-121 compared to placebo in subjects with dry eye disease. clinicaltrials.gov/show/NCT02819284 (first received 30 June 2016).
KPI‐121 (STRIDE3) {published data only}
- NCT03616899. Safety and efficacy of KPI-121 in subjects with DED. clinicaltrials.gov/show/NCT03616899 (first received 6 August 2018).
Lee 2014 {published data only}
- Lee H, Chung B, Kim KS, Seo KY, Choi BJ, Kim TI. Effects of topical loteprednol etabonate on tear cytokines and clinical outcomes in moderate and severe meibomian gland dysfunction: randomized clinical trial. American Journal of Ophthalmology 2014;158(6):1172‐83. e1. [DOI] [PubMed] [Google Scholar]
- NCT01692652. Changes of inflammatory cytokines in the tears of moderate and severe MGD treated with topical loteprednol etabonate. clinicaltrials.gov/show/NCT01692652 (first received 25 September 2012).
Li 2021 {published data only}
- Li N, Lai J. Effects of fluorometholone combined with sodium hyaluronate eye drops in the treatment of xerophthalmia and the influence on inflammatory factors in tears. International Eye Science 2021;21(3):509‐14. [Google Scholar]
Lin 2015 {published data only}
- Lin T, Gong L. Topical fluorometholone treatment for ocular dryness in patients with Sjögren syndrome: a randomized clinical trial in China. Medicine 2015;94(7):e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Luo 2013 {published data only}
- Luo XL, Lei C, Wang BL. Clinical study of corticosteriod for meibomian gland dysfunction. International Eye Science 2013;13(2):377-9. [Google Scholar]
NCT01276223 {published data only}
- NCT01276223. Evaluation of anti-inflammatory treatment in dry eye patients. clinicaltrials.gov/ct2/show/NCT01276223 (first received 12 January 2011).
Pflugfelder 2004 {published data only}
- Pflugfelder SC, Maskin SL, Anderson B, Chodosh J, Holland EJ, De Paiva CS, et al. A randomized, double-masked, placebo-controlled, multicenter comparison of loteprednol etabonate ophthalmic suspension, 0.5%, and placebo for treatment of keratoconjunctivitis sicca in patients with delayed tear clearance. American Journal of Ophthalmology 2004;138(3):444‐57. [DOI] [PubMed] [Google Scholar]
Pinto‐Fraga 2016 {published data only}
- Aapola U, Nattinen J, Jylha A, Pinto-Fraga J, Lpez-Miguel A, Gonzalez-Garcia MJ, et al. Tear fluid proteome reveals inflammation and immune response proteins as potential predictive biomarkers of the effects of desiccating stress and dry eye treatments. Investigative Ophthalmology & Visual Science 2016;57(12):397. [Google Scholar]
- Calonge M, Pinto-Fraga J, Enriquez-De-Salamanca A, Fernandez I, Gonzalez-Garcia MJ, Lopez-Miguel A, et al. Tear cytokine biomarkers in dry eye patients subjected to environmental stress and treated with topical 0.1% fluorometholone. Investigative Ophthalmology & Visual Science 2016;57(12):2861. [Google Scholar]
- Estevez J, Pesudovs K. Re: Pinto-Fraga et al.: topical fluorometholone protects the ocular surface of dry eye patients from desiccating stress: a randomized controlled clinical trial. Ophthalmology 2017;124(2):e14. [DOI] [PubMed] [Google Scholar]
- Nättinen J, Jylhä A, Aapola U, Enríquez-de-Salamanca A, Pinto-Fraga J, López-Miguel A, et al. Topical fluorometholone treatment and desiccating stress change inflammatory protein expression in tears. Ocular Surface 2018;16(1):84‐92. [DOI] [PubMed] [Google Scholar]
- NCT02051023. Efficacy and safety of fluorometholone (FML) in dry eye disease (keratoconjunctivitis sicca). clinicaltrials.gov/show/NCT02051023 (first received 31 January 2014).
- Pinto-Fraga J, Enríquez-de-Salamanca A, Calonge M, González-García MJ, López-Miguel A, López-de la Rosa A, et al. Severity, therapeutic, and activity tear biomarkers in dry eye disease: an analysis from a phase III clinical trial. Ocular Surface 2018;16(3):368‐76. [DOI] [PubMed] [Google Scholar]
- Pinto-Fraga J, López-Miguel A, González-García MJ, Fernández I, López-de-la-Rosa A, Enríquez-de-Salamanca A, et al. Topical fluorometholone protects the ocular surface of dry eye patients from desiccating stress: a randomized controlled clinical trial. Ophthalmology 2016;123(1):141‐53. [DOI] [PubMed] [Google Scholar]
Qazi 2015 {published data only}
- Efficacy of Zylet vs. Lotemax for the treatment of ocular surface inflammation/MGD/blepharitis. clinicaltrials.gov/show/NCT01456780 (first received 21 October 2011).
- Kheirkha A, Dohlman TH, Amparo F, Arnoldner MA, Jamali A, Hamrah P, et al. Effects of corneal nerve density on the response to treatment in dry eye disease. Ophthalmology 2015;122:662-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qazi Y, Kheirkhah A, Dohlman TH, Cruzat A, Cavalcanti B, Colon C, et al. Corneal dendritic cells as a surrogate biomarker of therapeutic efficacy in dry eye-associated corneal inflammation. Investigative Ophthalmology & Visual Science 2015;56(7):291. [Google Scholar]
Sheppard 2014 {published data only}
- Donnenfeld ED, Sheppard JD, Holland EJ, Slonim CH, Solomon R, Solomon KD, et al. Prospective, multicenter, randomized controlled study on the effect of loteprednol etabonate on initiating therapy with cyclosporin A. In: Annual Meeting of American Academy of Ophthalmology; 2007 Nov 10-13; New Orleans (LA). 2007.
- NCT00407043. Multicenter, randomized, controlled study of the effect of Lotemax on initiation of dry eye treatment with Restasis. clinicaltrials.gov/show/NCT00407043 (first received 4 December 2006).
- Sheppard JD, Donnenfeld ED, Holland EJ, Slonim CB, Solomon R, Solomon KD, et al. Effect of loteprednol etabonate 0.5% on initiation of dry eye treatment with topical cyclosporine 0.05%. Eye & Contact Lens 2014;40(5):289‐96. [DOI] [PubMed] [Google Scholar]
- Sheppard JD, Donnenfeld ED. Topical loteprednol 0.5% induction therapy improves topical cyclosporine emulsion tolerability in chronic dry eye disease. Investigative Ophthalmology & Vision Science 2008;49:ARVO E‐Abstract 99. [Google Scholar]
Singla 2019 {published data only}
- Singla S, Sarkar L, Joshi M. Comparison of topical cyclosporine alone and topical loteprednol with cyclosporine in moderate dry eye in Indian population: a prospective study. Taiwan Journal of Ophthalmology 2019;9(3):173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wan 2012 {published data only}
- Wan PX, Wang XR, Song YY, Li ZY, Duan HC, Zhang W, et al. Study on the treatment of dry eye with loteprednol etabonate. Zhonghua Yan Ke Za Zhi [Chinese Journal of Ophthalmology] 2012;48(2):142‐7. [PubMed] [Google Scholar]
References to studies excluded from this review
Abud 2016 {published data only}
- Abud TB, Amparo F, Saboo US, Di Zazzo A, Dohlman TH, Ciolino JB, et al. A clinical trial comparing the safety and efficacy of topical tacrolimus versus methylprednisolone in ocular graft-versus-host disease. Ophthalmology 2016;123(7):1449‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Acord 2010 {published data only}
- Acord C, Gonzales A, McKee AG. Loteprednol etabonate 0.2% in the possible treatment of dry eye syndrome. Optometry 2010;81:299‐300. [Google Scholar]
Asbell 2011 {published data only}
- Asbell PA, Stapleton FJ, Wickström K, Akpek EK, Aragona P, Dana R, et al. The international workshop on meibomian gland dysfunction: report of the clinical trials subcommittee. Investigative Ophthalmology & Visual Science 2011;52(4):2065-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boynton 2015 {published data only}
- Boynton GE, Raoof D, Niziol LM, Hussain M, Mian SI. Prospective randomized trial comparing efficacy of topical loteprednol etabonate 0.5% versus cyclosporine-a 0.05% for treatment of dry eye syndrome following hematopoietic stem cell transplantation. Cornea 2015;34(7):725‐32. [DOI] [PubMed] [Google Scholar]
ChiCTR‐IPQ‐15006773 {published data only}
- ChiCTR-IPQ-15006773. 0.1% tacrolimus (FK506) in the treatment of moderately severe dry eye clinical efficacy evaluation. trialsearch.who.int/Trial2.aspx?TrialID=ChiCTR-IPQ-15006773 (first received 27 March 2015).
Edward 2014 {published data only}
- NCT02028312. A phase IV, randomized, parallel group, investigator-masked evaluation of the effect of loteprednol etabonate ophthalmic gel 0.5% on the initiation of dry eye treatment with Restasis®. clinicaltrials.gov/show/NCT02028312 (first received 7 January 2014).
EUCTR2006‐003391‐35‐NL {published data only}
- EUCTR2006-003391-35-NL. Evaluation of the efficacy and safety of unpreserved dexamethasone phosphate 0.1% eye drops (T1910) versus placebo in patients with bilateral treated severe keratoconjunctivitis sicca due to Sjögrens' syndrome. trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2006-003391-35-NL (first received 7 May 2006).
EUCTR2019‐000747‐27‐IT {published data only}
- EUCTR2019-000747-27-IT. A clinical trial to evaluate the safety and efficacy of Pro-ocular™ topical gel in two different concentration, 0.5% and 1%, when administered in the forehead twice a day for 12 weeks in patients diagnosed with dry eye syndrome. trialsearch.who.int/Trial2.aspx?TrialID=EUCTR2019-000747-27-IT (first received 10 July 2020).
Gupta 2021 {published data only}
- Gupta PK, Venkateswaran N. The role of KPI-121 0.25% in the treatment of dry eye disease: penetrating the mucus barrier to treat periodic flares. Therapeutic Advances in Ophthalmology 2021;13:25158414211012797. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
ISRCTN13765551 {published data only}
- ISRCTN13765551. Comparison of the treatment effect of preservative-free vs preserved eye drops in patients with dry eye syndrome. trialsearch.who.int/Trial2.aspx?TrialID=ISRCTN13765551 (first received 2 May 2014).
Jee 2014 {published data only}
- Jee D, Park SH, Kim MS, Kim EC. Antioxidant and inflammatory cytokine in tears of patients with dry eye syndrome treated with preservative-free versus preserved eye drops. Investigative Ophthalmology & Vision Science 2014;55(8):5081‐9. [DOI] [PubMed] [Google Scholar]
JPRN‐UMIN000025159 {published data only}
- JPRN-UMIN000025159. Efficacy of expression treatment on o-MGD (obstructive meibomian gland dysfunction). trialsearch.who.int/Trial2.aspx?TrialID=JPRN-UMIN000025159 (first received 2 January 2017).
Kallab 2020 {published data only}
- Kallab M, Szegedi S, Hommer N, Stegmann H, Kaya S, Werkmeister RM, et al. Topical low dose preservative-free hydrocortisone reduces signs and symptoms in patients with chronic dry eye: a randomized clinical trial. Advances in Therapy 2020;37(1):329‐41. [DOI] [PubMed] [Google Scholar]
Korenfeld 2021 {published data only}
- Korenfeld M, Nichols KK, Goldberg D, Evans D, Sall K, Foulks G, et al. Safety of KPI-121 ophthalmic suspension 0.25% in patients with dry eye disease: a pooled analysis of 4 multicenter, randomized, vehicle-controlled studies. Cornea 2021;40(5):564-70. [DOI] [PubMed] [Google Scholar]
Lee 2006 {published data only}
- Lee HK, Ryu IH, Seo KY, Hong S, Kim HC, Kim EK. Topical 0.1% prednisolone lowers nerve growth factor expression in keratoconjunctivitis sicca patients. Ophthalmology 2006;113(2):198‐205. [DOI] [PubMed] [Google Scholar]
NCT03907865 {published data only}
- NCT03907865. Clinical efficacy of topical hydrocortisone 0.335% (Softacort®) in patients with chronic dry eye disease and associated ocular surface inflammation. clinicaltrials.gov/show/NCT03907865 (first received 9 April 2019).
Rolando 2008 {published data only}
- Rolando M, Solignani F, Valente C, Allavena F, Bertolotto M, Barabino S. Is there a role for a long term tapered small dose steroidal treatment for keratoconjunctivitis sicca? Investigative Ophthalmology & Vision Science 2008;49:ARVO E‐Abstract 97. [Google Scholar]
Ryu 2005 {published data only}
- Ryu I, Lee HK, Seo KR, Kim E. Change of nerve growth factor after 01% prednisolone instillation in dry eye syndrome patients and its correlation with clinical parameters. Investigative Ophthalmology & Vision Science 2005;46:ARVO E‐Abstract 2049. [Google Scholar]
Shen 2015 {published data only}
- Shen ZB, Li JL. Clinical effect of 1g/L fluorometholone drops combined with soft corneal contact lens for filamentary keratitis. International Eye Science 2015;15(9):1633‐5. [Google Scholar]
Sindhu 2015 {published data only}
- Sindhu S, Dutta S, Beg MA, Mittal SK, Gupta SD. Comparative evaluation of topical carboxymethyl cellulose either alone or in combination with topical corticosteroid in the treatment of dry eye in a tertiary-care teaching hospital. National Journal of Physiology, Pharmacy and Pharmacology 2015;5(3):207-11. [Google Scholar]
Wong 2021 {published data only}
- Wong S (Editor). Loteprednol 0.25% (Eysuvis) for dry eye disease. Medical Letter on Drugs and Therapeutics 2021;63(1624):75-7. [PubMed]
References to studies awaiting assessment
ChiCTR‐IPR‐15007196 {published data only (unpublished sought but not used)}
- ChiCTR-IPR-15007196. Clinical trial of sodium bromide hydrate eye drops and Pranoprofen eye drops for dry eye. trialsearch.who.int/Trial2.aspx?TrialID=ChiCTR-IPR-15007196 (first received 9 October 2015).
Herman 2005 {published data only (unpublished sought but not used)}
- Herman JP, Korb DR, Greiner JV, Scaffidi RC, Blackie CA. Treatment of lid wiper epitheliopathy with a metastable lipid emulsion or a corticosteroid. Investigative Ophthalmology & Vision Science 2005;46:ARVO E‐Abstract 2017. [Google Scholar]
NCT00471419 {published data only (unpublished sought but not used)}
- NCT00471419. Phase II study of AL-2178 (FID 109980) in the treatment of dry eye. clinicaltrials.gov/show/NCT00471419 (first received 7 May 2007).
NCT00560638 {published data only (unpublished sought but not used)}
- NCT00560638. Loteprednol etabonate ophthalmic suspension for the treatment of dry eye. clinicaltrials.gov/ct2/show/NCT00560638 (first received 19 November 2007).
NCT01562795 {published data only (unpublished sought but not used)}
- NCT01562795. Efficacy of nonsteroidal anti-inflammatory drugs in treatment of moderate and severe dry eye disease. clinicaltrials.gov/ct2/show/NCT01562795 (first received 23 March 2012).
NCT03418727 {published data only (unpublished sought but not used)}
- NCT03418727. Dry eye disease study with brimonidine. clinicaltrials.gov/show/NCT03418727 (first received 1 February 2018).
NTR2291 {published data only (unpublished sought but not used)}
- NTR2291. Ocular inflammation and dry eye. trialsearch.who.int/?trialid=NTR2291 (first received 15 April 2010).
References to ongoing studies
CTRI/2021/02/031182 {published data only (unpublished sought but not used)}
- CTRI/2021/02/031182. Eye drops made of antibodies for dry eye disease patients. trialsearch.who.int/Trial2.aspx?TrialID=CTRI/2021/02/031182 (first received 10 February 2021).
ISRCTN16288419 {published data only (unpublished sought but not used)}
- ISRCTN16288419. Evaluation of the performance of new substitute tears in dry eye patients. trialsearch.who.int/?TrialID=ISRCTN16288419 (first received 8 May 2020).
NCT04734197 {published data only}
- NCT04734197. A Research Study To See How Well an Eye Drop, SURF-100 (A Mycophenolic Acid/Betamethasone Sodium Phosphate Combination), Works and What Side Effects There Are in Subjects With Dry Eye Disease. clinicaltrials.gov/show/NCT04734197 (first received 2 February 2021).
NCT04734210 {published data only}
- NCT04734210. A research study to see how well an eye drop, SURF-200 (0.02% and 0.04% Betamethasone Sodium Phosphate), works, what side effects there are, and to compare it with vehicle (placebo) in subjects diagnosed with dry eye disease and experiencing an episodic flare-up. clinicaltrials.gov/show/NCT04734210 (first received 2 February 2021).
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Avunduk 2003
- Avunduk AM, Avunduk MC, Varnell ED, Kaufman HE. The comparison of efficacies of topical corticosteroids and nonsteroidal anti-inflammatory drops on dry eye patients: a clinical and immunocytochemical study. American Journal of Ophthalmology 2003;136(4):593-602. [DOI] [PubMed] [Google Scholar]
Barabino 2011
- Barabino S, Montaldo E, Corsi E, Valente C, Solignani F, Mingari MC, et al. The effect of tapered small dose steroidal treatment on symptoms, clinical signs, and ocular surface inflammation in patients with dry eye syndrome. Investigative Ophthalmology & Vision Science 2011;52:3826. [Google Scholar]
Beckman 2020
- Beckman K, Katz J, Majmudar P, Rostov A. Loteprednol etabonate for the treatment of dry eye disease. Journal of Ocular Pharmacology and Therapeutics 2020;36(7):497-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhaskaran 2014
- Bhaskaran K, Smeeth L. What is the difference between missing completely at random and missing at random? International Journal of Epidemiology 2014;43(4):1336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bron 2003
- Bron AJ, Evans VE, Smith JA. Grading of corneal and conjunctival staining in the context of other dry eye tests. Cornea 2003;22:640-50. [DOI] [PubMed] [Google Scholar]
Bron 2017
- Bron AJ, Paiva CS, Chauhan SK, Bonini S, Gabison EE, Jain S, et al. TFOS DEWS II pathophysiology report. Ocular Surface 2017;15(3):438-510. [DOI] [PubMed] [Google Scholar]
Byun 2012
- Byun YJ, Kim TI, Kwon SM, Seo KY, Kim SW, Kim EK, et al. Efficacy of combined 0.05% cyclosporine and 1% methylprednisolone treatment for chronic dry eye. Cornea 2012;31(5):509-13. [DOI] [PubMed] [Google Scholar]
Cohen 1988
- Cohen J. Statistical Power Analysis in the Behavioral Sciences. 2nd edition. Lawrence Erlbaum Associates Inc, 1988. [Google Scholar]
Comstock 2018
- Comstock TL, Sheppard JD. Loteprednol etabonate for inflammatory conditions of the anterior segment of the eye: twenty years of clinical experience with a retrometabolically designed corticosteroid. Expert Opinion on Pharmacotherapy 2018;19(4):337-53. [DOI] [PubMed] [Google Scholar]
Covidence [Computer program]
- Covidence. Version accessed 9 August 2022. Melbourne, Australia: Veritas Health Innovation. Available at covidence.org.
Cutolo 2019
- Cutolo CA, Barabino S, Bonzano C, Traverso CE. The use of topical corticosteroids for treatment of dry eye syndrome. Ocular Immunology and Inflammation 2019;27(2):266-75. [DOI] [PubMed] [Google Scholar]
Deeks 2021
- Deeks JJ, Higgins JPT, Altman DG. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
De Paiva 2006a
- De Paiva CS, Corrales RM, Villarreal AL, Farley WJ, Li DQ, Stern ME, et al. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Experimental Eye Research 2006;83(3):526-35. [DOI: 10.1016/j.exer.2006.02.004] [DOI] [PubMed] [Google Scholar]
De Paiva 2006b
- De Paiva SC, Corrales RM, Villarreal AL, Farley W, Li DQ, Stern ME, et al. Apical corneal barrier disruption in experimental murine dry eye is abrogated by methylprednisolone and doxycycline. Investigative Ophthalmology and Vision Science 2006;47(7):2847-56. [DOI: 10.1167/iovs.05-1281] [DOI] [PubMed] [Google Scholar]
De Paiva 2019
- De Paiva CS, Pflugfelder SC, Ng SM, Akpek EK. Topical cyclosporine A therapy for dry eye syndrome. Cochrane Database of Systematic Reviews 2019, Issue 9. Art. No: CD010051. [DOI: 10.1002/14651858.CD010051.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
DEQS
- Sakane Y, Yamaguchi M, Yokoi N, Uchino M, Dogru M, Oishi T, et al. Development and validation of the Dry Eye-Related Quality-of-Life Score questionnaire. JAMA Ophthalmology 2013;131(10):1331-8. [DOI: 10.1001/jamaophthalmol.2013.4503] [DOI] [PubMed] [Google Scholar]
Downie 2017
- Downie LE, Ng SM, Lindsley KB, Akpek EK. Omega-3 and omega-6 polyunsaturated fatty acids for dry eye disease. Cochrane Database of Systematic Reviews 2019, Issue 12. Art. No: CD011016. [DOI: 10.1002/14651858.CD011016.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ervin 2017
- Ervin AM, Law A, Pucker AD. Punctal occlusion for dry eye syndrome. Cochrane Database of Systematic Reviews 2017, Issue 6. Art. No: CD006775. [DOI: 10.1002/14651858.CD006775.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Evans 2017
- Evans DG, Sheppard JD, Williams JI. Loteprednol etabonate ophthalmic gel 0.5% for inflammation associated with dry eye disease: outcomes of a 12-week Phase 2 clinical study. In: Annual Meeting of the American Optometric Association; 2017 Sep 27-30; Washington, DC. 2017.
Gomes 2017
- Gomes JAP, Azar DT, Baudouin C, Efron N, Hirayama M, Horwath-Winter J, et al. TFOS DEWS II iatrogenic report. Ocular Surface 2017;15(3):511-38. [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Statistics in Medicine 2002;21(11):1539-58. [DOI] [PubMed] [Google Scholar]
Higgins 2021a
- Higgins JPT, Savović J, Page MJ, Elbers RG, Sterne JAC. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins J, Thomas J, Chandler J, Cumpston M, Li T, Page M, Welch V, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Higgins 2021b
- Higgins JPT, Li T, Deeks JJ. Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Holland 2019
- Holland EJ, Darvish M, Nichols KK, Jones L, Karpecki PM. Efficacy of topical ophthalmic drugs in the treatment of dry eye disease: a systematic literature review. Ocular Surface 2019;17(3):412-23. [DOI: 10.1016/j.jtos.2019.02.012] [DOI] [PubMed] [Google Scholar]
IReST
- Trauzettel-Klosinski S, Dietz K, IReST Study Group. Standardized assessment of reading performance: the New International Reading Speed Texts IReST. International Ophthalmology and Vision Science 2012;53:5452-61. [DOI: 10.1167/iovs.11-8284] [DOI] [PubMed] [Google Scholar]
Jones 2002
- Jones L, MacDougall N, Sorbara LG. Asymptomatic corneal staining associated with the use of balafilcon silicone-hydrogel contact lenses disinfected with a polyaminopropyl biguanide-preserved care regimen. Optomery and Visual Science 2002;79:753-61. [DOI] [PubMed] [Google Scholar]
Jones 2017
- Jones L, Downie LE, Korb D, Benitez-Del-Castillo JM, Dana R, Deng SX, et al. TFOS DEWS II Management and Therapy Report. Ocular Surface 2017;15(3):575-628. [DOI] [PubMed] [Google Scholar]
Kojima 2020
Lee 2006
- Lee HK, Ryu IH, Seo KY, Hong SW, Kim HC, Kim EK. Topical 0.1% prednisolone lowers nerve growth factor expression in keratoconjunctivitis sicca patients. Ophthalmology 2006;113:198e205. [DOI: 10.1016/j.ophtha.2005.09.033] [DOI] [PubMed] [Google Scholar]
Legge 1989
- Legge GE, Ross JA, Luebker A, Lamay J. Psychophysics of reading: VIII. The Minnesota low-vision reading test. Optometry and Vision Science 1989;66:843-53. [DOI] [PubMed] [Google Scholar]
Lekhanont 2007
- Lekhanont K, Leyngold IM, Suwan-Apichon O, Rangsin R, Chuck RS. Comparison of topical dry eye medications for the treatment of keratoconjunctivitis sicca in a botulinum toxin B-induced mouse model. Cornea 2007;26(1):84-9. [DOI: 10.1097/01.ico.0000240079.24583.a1] [DOI] [PubMed] [Google Scholar]
Lemp 1995
- Lemp MA. Report of the National Eye Institute/Industry Workshop on clinical trials in dry eyes. CLAO Journal 1995;21:211-32. [PubMed]
Li 2021
- Li T, Higgins JPT, Deeks JJ, editor(s). Chapter 5: Collecting data. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Macri 2000
- Macri A, Rolando M, Pflugfelder S. A standardized visual scale for evaluation of tear fluorescein clearance. Ophthalmology 2000;107:1338-43. [DOI] [PubMed] [Google Scholar]
Messmer 2015
- Messmer EM. The pathophysiology, diagnosis, and treatment of dry eye disease. Deutsches Ärzteblatt International 2015;112(5):71-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miller 2010
- Miller KL, Walt JG, Mink DR, Satram-Hoang S, Wilson SE, Perry HD, et al. Minimal clinically important difference for the ocular surface disease index. Archives of Ophthalmology 2010;128(1):94-101. [DOI] [PubMed] [Google Scholar]
Nelson 2017
- Nelson JD, Craig JP, Akpek EK, Azar DT, Belmonte C, Bron AJ, et al. TFOS DEWS II Introduction. Ocular Surface 2017;15(3):269-75. [DOI: 10.1016/j.jtos.2017.05.005] [DOI] [PubMed] [Google Scholar]
Newman‐Casey 2018
- Newman-Casey PA, Woodward MA, Niziol LM, Lee PP, De Lott LB. Brand medications and Medicare Part D: how eye care providers' prescribing patterns influence costs. Ophthalmology 2018;125(3):332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ngo 2013
- Ngo W, Situ P, Keir N, Korb D, Blackie C, Simpson T. Psychometric properties and validation of the Standard Patient Evaluation of Eye Dryness questionnaire. Cornea 2013;32(9):1204-10. [DOI] [PubMed] [Google Scholar]
Nichols 2016
- Nichols KK, Bacharach J, Holland E, Kislan T, Shettle L, Lunascek O, et al. Impact of dry eye disease on work productivity, and patients' satisfaction with over-the-counter dry eye treatments. Investigative Ophthalmology and Vision Science 2016;57(7):2975-82. [DOI: 10.1167/iovs.16-19419] [DOI] [PubMed] [Google Scholar]
Oden 1998
- Oden NL, Lilienfeld DE, Lemp MA, Nelson JD, Ederer F. Sensitivity and specificity of a screening questionnaire for dry eye. Advances in Experimental Medicine Biology 1998;438:807-20. [DOI] [PubMed] [Google Scholar]
Page 2021
- Page MJ, Higgins JPT, Sterne JAC. Chapter 13: Assessing risk of bias due to missing results in a synthesis. In: Higgins J, Thomas J, Chandler J, Cumpston M, Li T, Page M, Welch V, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2.
Pan 2017
- Pan Q, Angelina A, Marrone M, Stark WJ, Akpek EK. Autologous serum eye drops for dry eye. Cochrane Database of Systematic Reviews 2017, Issue 2. Art. No: CD009327. [DOI: 10.1002/14651858.CD009327.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pflugfelder 2004
- Pflugfelder SC, Maskin SL, Anderson B, Chodosh J, Holland EJ, Paiva CS, et al. A randomized, double-masked, placebo-controlled, multicenter comparison of loteprednol etabonate ophthalmic suspension, 0.5%, and placebo for treatment of keratoconjunctivitis sicca in patients with delayed tear clearance. American Journal of Ophthalmology 2004;138(3):444-57. [DOI: 10.1016/j.ajo.2004.04.052] [DOI] [PubMed] [Google Scholar]
Pucker 2016
- Pucker AD, Ng SM, Nichols JJ. Over the counter (OTC) artificial tear drops for dry eye syndrome. Cochrane Database of Systematic Reviews 2016, Issue 2. Art. No: CD009729. [DOI: 10.1002/14651858.CD009729.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Qiu 2011
- Qiu X, Gong L, Sun X, Jin H. Age-related variations of human tear meniscus and diagnosis of dry eye with Fourier-domain anterior segment optical coherence tomography. Cornea 2011;30:543-9. [DOI] [PubMed] [Google Scholar]
RevMan Web 2022 [Computer program]
- Review Manager Web (RevMan Web). Version 4.10.0. The Cochrane Collaboration, 2022. Available at revman.cochrane.org.
Saldanha 2017
- Saldanha IJ, Dickersin K, Hutfless ST, Akpek EK. Gaps in current knowledge and priorities for future research in dry eye. Cornea 2017;36(12):1584-91. [DOI: 10.1097/ICO.0000000000001350] [DOI] [PMC free article] [PubMed] [Google Scholar]
Saldanha 2018
- Saldanha IJ, Petris R, Han G, Dickersin K, Akpek EK. Research questions and outcomes prioritized by patients with dry eye. JAMA Ophthalmology 2018;136(10):1170-9. [DOI: 10.1001/jamaophthalmol.2018.3352] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schiffman 2000
- Schiffman RM, Christianson MD, Jacobsen G, Hirsch JD, Reis BL. Reliability and validity of the Ocular Surface Disease Index. Archives of Ophthalmology 2000;118(5):615-21. [DOI: 10.1001/archopht.118.5.615] [DOI] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
Schünemann 2019
- Schünemann HJ, Higgins JPT, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch V, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from training.cochrane.org/handbook/archive/v6.
Shanti 2020
- Shanti Y, Shehada R, Bakkar MM, Quaddumi J. Prevalence and associated risk factors of dry eye disease in 16 northern West bank towns in Palestine: a cross-sectional study. BMC Ophthalmology 2020;20(26):1-8. [DOI: 10.1186/s12886-019-1290-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sheppard 2014
- Sheppard JD, Torkildsen GL, Lonsdale JD, D'Ambrosio FA Jr, McLaurin EB, Eiferman RA, et al, OPUS-1 Study Group. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 Phase 3 Study. Clinical Trial 2014;121(2):475-83. [DOI: 10.1016/j.ophtha.2013.09.015] [DOI] [PubMed] [Google Scholar]
Shih 2017
- Shih KC, Lun CN, Jhanji V, Thong BY, Tong L. Systematic review of randomized controlled trials in the treatment of dry eye disease in Sjogren syndrome. Journal of Inflammation (London) 2017;14:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stapleton 2017
- Stapleton F, Alves M, Bunya VY, Jalbert I, Lekhanont K, Malet F, et al. TFOS DEWS II Epidemiology Report. Ocular Surface 2017;15(3):334-65. [DOI] [PubMed] [Google Scholar]
Stephenson 2016
- Stephenson L, Mistry V, Spink G, Morrison D, Thomas A, Barton S, et al. The management of dry eye. Drug and Therapeutics Bulletin 2016;54(1):9-12. [DOI] [PubMed] [Google Scholar]
WebPlotDigitizer [Computer program]
- WebPlotDigitizer. Version 4.5. Rohatgi A, accessed 22 February 2022. Available from: automeris.io/WebPlotDigitizer.
Wolffsohn 2017
- Wolffsohn JS, Arita R, Chalmers R, Djalilian A, Dogru M, Dumbleton K, et al. TFOS DEWS II Diagnostic Methodology report. Ocular Surface 2017;15:539-74. [DOI] [PubMed] [Google Scholar]
Yu 2011
- Yu J, Asche CV, Fairchild CJ. The economic burden of dry eye disease in the United States: a decision tree analysis. Cornea 2011;30(4):379-87. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Liu 2021
- Liu SH, Gregory D, Hauswirth S, Ifantides C, Abraham AG, Saldanha IJ, et al. Topical corticosteroids for dry eye. Cochrane Database of Systematic Reviews 2021, Issue 9. Art. No: CD015070. [DOI: 10.1002/14651858.CD015070] [DOI] [PMC free article] [PubMed] [Google Scholar]