

1. Introduction
Pathogenic microorganisms such as bacteria, viruses, fungi, and parasites have developed molecular strategies to breach host barriers. Following invasion, pathogens dynamically interact with surrounding immune cells, epithelial cells, endothelial cells, and the nervous system and actively attempt to evade host defenses. While host-pathogen interactions are a key area of research in infectious diseases, less is known about the role of the nervous system in sensing and defending against pathogens. To detect potentially damaging stimuli and respond to insults, a dense network of somatosensory neurons called nociceptors innervate tissues including the skin, meninges, joints, urinary tract, reproductive tract, and gut [21; 32; 51]. Upon activation by noxious stimuli, nociceptors transduce signals to the central nervous system (CNS) to mediate the sensation of pain [8]. Many microbial infections are accompanied by pain, but until recently the molecular mechanisms of pain during infection were not well understood. It was originally thought that pain during infection was mainly due to the release of inflammatory mediators such as prostaglandins or cytokines by immune cells that act on nociceptors to generate pain [32; 51]. Recent work has shown that pathogens can directly activate nociceptors during infection, thereby generating pain [22](Fig. 1). In this review, we describe molecular interactions that occur between microbes and nociceptors in pain and discuss how this knowledge could lead to new treatments for pain and infection.
Figure 1. Bacterial pathogens that directly activate nociceptors to produce pain during infection.
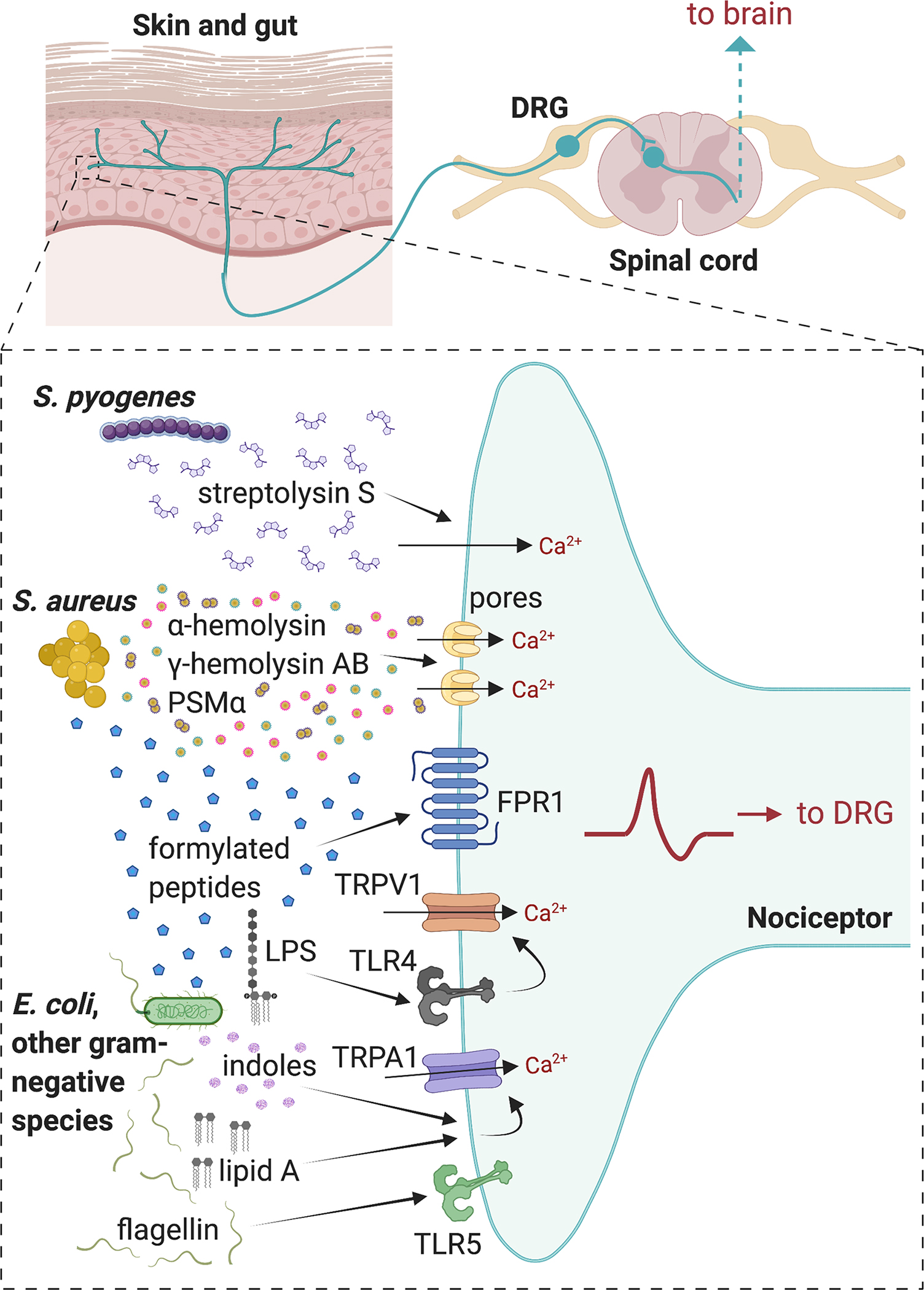
In skin and soft tissue infections, S. pyogenes produces the cytolytic toxin Streptolysin S (SLS) which activates TRPV1+ nociceptors to produce pain. S. aureus secretes pore-forming toxins α-hemolysin (Hla), HlgAB, and PSMα, which are able to induce cation influx into nociceptors, leading to action potential generation and pain during subcutaneous infections. Formylated peptides from S. aureus and E. coli bind to FPR1 on nociceptors to induce mechanical allodynia. LPS from gram-negative bacteria can bind to TLR4 which sensitizes TRPV1 channels in nociceptors during gram-negative pathogen infection. The lipid A moiety of LPS can also act via TRPA1 ion channels on nociceptors independent of TLR4. Flagellin from gram-negative bacteria can bind to TLR5 on A-fibers that mediate neuropathic pain. Image created with BioRender.com
Pain (dolor) was originally defined as one of four cardinal signs of inflammation by the Roman physician Celsius [67]. Nociceptors mediate pain and also control the magnitude of the other three inflammatory signs (redness, heat, swelling) via neurovascular and neuroimmune signaling [23; 53]. In this process termed “neurogenic inflammation”, calcium influx at peripheral nerve terminals of nociceptors leads to axonal reflexes and subsequent release of neuropeptides and neurotransmitters along neighboring branches of the nociceptive neuron in peripheral tissues such as the skin [23; 53; 67]. These neuronal mediators, such as calcitonin-gene related peptide (CGRP) or substance P (SP), then bind to neuropeptide receptors on vascular smooth muscle and endothelial cells to mediate vasodilation [23; 91]. In addition, growing work is showing that neuropeptides or neurotransmitters can act directly on their cognate receptors expressed by innate or adaptive immune cells, leading to changes in transcription, cytokine production, and immune phenotypic polarization [22; 78; 99]. In the context of host defense, this rapid neuroinflammatory response could mediate or regulate the outcome of pathogen infection.
Nociceptors are equipped with many molecular sensors, including but not limited to G protein-coupled receptors (GPCRs), cytokine receptors, thermosensitive and chemosensitive ion channels including transient receptor potential channels (e.g., TRPA1, TRPV1, TRPM2), P2X3 channels, and voltage-gated sodium channels (e.g., NaV1.7, NaV1.8, and NaV1.9) [8; 78]. Nociceptors also express receptors that can detect pathogenic components, which will be described in detail below. These functionally and molecularly heterogenous classes of molecular sensors at peripheral terminals tune nociceptors to detect a diversity of environmental threats [8; 99]. Upon sensing noxious or inflammatory stimuli, action potentials are generated at nociceptor peripheral terminals that are transduced to the dorsal horn of the spinal cord and relayed to the brain to be processed and perceived as pain [8; 32].
While the concept that pathogens can directly activate or sensitize nociceptors is relatively new, there are several reasons why the ability of nociceptors to detect pathogens could be advantageous to the host. The speed of neural reflexes (withing seconds) compared to immune responses (minutes to hours) ideally positions nociceptors to be the first responders to pathogens and fundamental contributors to set the immune stage of barrier tissues [26; 77]. Pain could induce sickness or avoidance behaviors to allow proper healing of wounds or infected areas. While the effect of pain blockade on infection outcome varies, in some cases analgesics can adversely affect the ability of the immune system to combat pathogens and are contraindicated for certain infections [3; 44].
Some pathogens may also exploit activating or silencing nociceptors for their advantage [21; 62; 76] (Fig. 2a). Induction of analgesia during some specific phases of their life cycles could facilitate pathogen spread from host to host [30; 62; 74; 76]. Neuroimmune signaling is in some cases highly immunosuppressive of antimicrobial immunity, and some pathogens actively induce nociception to facilitate their survival in tissues [76]. Therefore, understanding the complexities of host-pathogen interactions in pain could be important for developing ways to prevent or treat infection. Furthermore, some molecular toxins or mediators produced by pathogens that act on neurons and silence pain could be repurposed as novel and selective therapies to treat pain in non-infectious settings (Fig. 2b and c).
Figure 2. Bacterial pathogens and microbial toxins that silence pain.
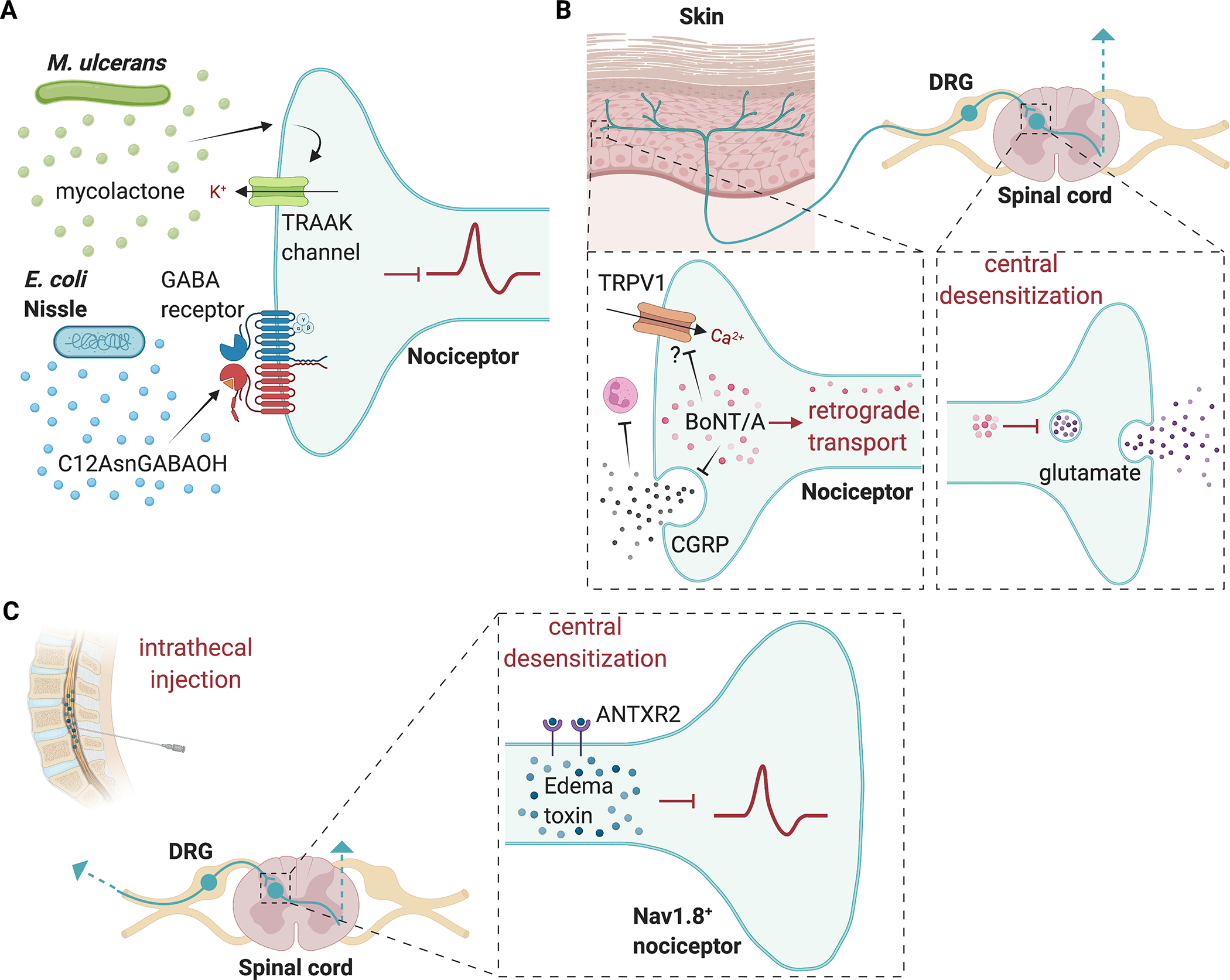
(A) M. ulcerans produces a mycolactone that induces potassium efflux through TRAAK family potassium channels; E. coli Nissle produces the lipopeptide C12AsmGABAOH which inhibits pain by acting on GABAB receptors. (B) BoNT/A is internalized into nerve terminals and through retrograde transport the toxin can reach central terminals where it blocks glutamate release. BoNT/A can also block release of CGRP from nerve terminals, which affects neutrophil function. BoNT/A may also interact with TRPV1 ion channels to alter nociceptor activity. (C) Nav1.8+ nociceptors express the high affinity anthrax toxin receptor ANTXR2. Intrathecal injection of anthrax edema toxin (composed of Protective antigen and edema factor) can silence pain. Edema toxin alters cAMP and intracellular signaling at nociceptor central terminals to suppress pain. Image created with BioRender.com
In this Review, we will discuss the recent studies that highlight how pathogens affect nociceptors and the perception of pain. The studies addressing pathogen-nociceptor interactions are also summarized in Table 1. We will focus on the known molecular mechanisms through which major classes of pathogens (bacterial, viral, fungal, parasites) communicate with sensory afferent neurons to induce pain behavior or silence it. These mechanisms will be dissected from the immune-mediated mechanisms of nociceptor sensitization. We also highlight how certain analgesic factors could be engineered to treat pain. Lastly, we discuss gaps in knowledge that require future investigation.
Table 1.
Microbial pathogens that produce pain or analgesia.
| Pathogen or microbial molecule | Type of pathogen | Pain phenotype/Behavioral outcome | Infection site(s) | Mechanism |
|---|---|---|---|---|
| Staphylococcus aureus | Gram-positive bacteria | Spontaneous nocifensive behaviors, mechanical and thermal hypersensitivity | Skin and soft tissues | Sensitization via α-hemolysin and N-formylated peptides [14; 22] |
| Streptococcus pyogenes | Gram-positive bacteria | Spontaneous nocifensive behaviors, mechanical and thermal hypersensitivity | Skin and soft tissues | Activation of TRPV1+ nociceptors via streptolysin S [74] |
| Mycobacterium tuberculosis | Gram-positive bacteria | Cough | Lungs and respiratory tract | Activation of TRPV1+ vagal neurons via MTb derived glycolipid sulfolipid [82] |
| Listeria monocytogenes | Gram-positive bacteria | Mechanical and thermal hypersensitivity | Gastrointestinal tract, Skin, Meninges | Unknown |
| Botulinum neurotoxins from Clostridium botulinum | Gram-positive bacteria | Analgesia | Neuromuscular junctions | Inhibition of neurotransmission in neurons via SNAP-25 cleavage [12] |
| Edema Toxin from Bacillus anthracis | Gram-positive bacteria | Analgesia | Skin, Lungs, Gut | Silencing of Nav 1.8+ DRG neurons via interaction between anthrax edema toxin and neuronal ANTXR2 [105] |
| LPS derived from E. coli, S. typhimurium, K. pneumoniae, S. marcescens, P. aeruginosa, R. sphaeroides | Gram-negative bacteria | Cutaneous and pelvic hypersensitivity, Trigeminal pain | Trigeminal nerve, Skin, Genitourinary tract | LPS activates TLR4 (which sensitizes TRPV1) or TRPA1 independently to drive pain [31; 47; 48; 67] |
| Escherichia coli | Gram-negative bacteria | Spontaneous nocifensive behaviors | Skin | Activation of TRPA1+ nociceptors via indole metabolites [25] |
| Pseudomonas aeruginosa | Gram-negative bacteria | Blink reflexes | Eye | Activation of trigeminal TRPV1+ nociceptors via pili, flagella, Type III secretion system [56] |
| Double-stranded viral RNA mimetic poly(I:C) | Synthetic mimetic of a double-stranded viral RNA | Mechanical and thermal hypersensitivity | Skin | Type I IFN receptor signaling through MNK-eIF4E pathway in Nav1.8+ nociceptors [7] |
| SARS-CoV-2 virus | Virus | Headache, myalgias, joint pain | Meninges, lungs, joints, DRGs | Unknown |
| Herpes simplex virus (HSV-1) | Virus | Mechanical hypersensitivity | Skin, DRG and trigeminal ganglia | Activation of peptidergic nociceptors by leukocytes via TNF/TNFR1; Kir4.1+ downregulation of satellite glial cells [40; 86] |
| Varicella zoster | Virus | Pain and itch | Skin, DRG and trigeminal ganglia | Unknown |
| Candida albicans | Fungus | Mechanical hypersensitivity, pelvic hypersensitivity | Skin, Gut, Genitourinary tract | Activation of TRPV1+/TRPA1+ nociceptors via zymosan or β-glucan/dectin-1 [26; 38]; Activation of nociceptors via keratinocyte-released ATP and P2X receptors [62] |
| Leishmania sp, | Parasite | Mechanical and thermal hypersensitivity | Skin | Production of NGF, IL-1β and TNFα, and spinal cord gliosis [16; 17; 49] |
| Trypanosoma cruzi | Parasite | Mechanical and thermal hypersensitivity | Skin | TNF-α and IL-1β induction, as well as gliosis and NF- κB activation [15] |
| Schistosoma mansoni | Parasites | Visceral pain | Gut | Activation of TRPV1+ nociceptors and crosstalk with mast cells via CGRP release [29] |
In this table, we list bacterial, fungal, viral, and parasitic pathogens or their products that have been linked to production or silencing of pain. For each pathogen, we describe the type of pathogen, the pain phenotype or behavioral outcome following infection, the tissue or site of infection, and known mechanisms of pain with accompanying references.
2. Bacterial pathogens and pain
2.1. Gram-positive bacteria
Bacterial infections are often anecdotally and empirically associated with pain (Fig. 1). Staphylococcus aureus is a leading cause of human infections, and a major cause of skin and soft tissue infections [22] and joint infections [94; 102]. In a mouse model of methicillin-resistant S. aureus (MRSA) subcutaneous infection with USA300 (current endemic strain of S. aureus), bacterial infections caused acute nociception, mechanical and thermal hyperalgesia [14; 22]. This hyperalgesia during S. aureus infection correlated with bacterial burden in tissues rather than tissue swelling or immune cell influx, suggesting that nociceptors may directly detect bacteria to produce pain [22]. It was found that S. aureus was able to directly activate nociceptor neurons through two secreted factors: the pore-forming toxin (PFT) α-hemolysin (Hla) and N-formylated peptides [22]. Hla binds to A disintegrin and metalloproteinase domain–containing protein 10 (ADAM10) on the surface of host cells, and then forms membrane pores that allows influx of cations such as calcium and sodium. In ADAM10+ nociceptors, this leads to firing of action potentials and pain production [22]. The second factor found to induce pain secreted by S. aureus included N-formylated peptides. It was found that the N-formylated peptides fMLF from Escherichia coli and fMIFL from S. aureus activates formyl peptide receptor 1 (FPR1), a G-protein coupled receptor, expressed on DRG neurons. N-formylated peptides acted only on neurons that responded to both capsaicin, a TRPV1 ligand, and allyl isothiocyanate, a TRPA1 ligand [22]. While Hla contributes to both mechanical and thermal pain hypersensitivity, N-formylated peptides only contributed to mechanical hypersensitivity. Of note, peptidoglycans and lipoteichoic acid (LTA) which are present in S. aureus cell walls did not produce nociceptor activation [22]. Following this first study, it was found that other S. aureus derived toxins can also induce nociceptor activation and pain [14]. In addition to Hla, S. aureus secretes toxins including phenol soluble modulins and the bicomponent leuckocidin HlgAB [14]. Hla, PSMs and HlgAB could all induce action potential firing in DRG neurons and produce acute nociceptive pain in vivo when injected into mice [14]. Furthermore, S. aureus lacking the agr quorum sensing system, which controls production of PFTs [72], was unable to induce either acute nociception or hyperalgesia during infection [14]. Utilizing this knowledge that pore-mediating toxins drive pain during infection [14; 22], it was found that the normally membrane-impermeable sodium channel inhibitor QX-314 was able to pass through bacterial PFTs to block action potential generation in DRG neurons and potentially produce analgesia in vivo during S. aureus infection [11; 14].
In the context of S. aureus infection, nociceptors also play a major role in regulating the host immune response. Nociceptor activation leads to release of neuropeptides from nociceptor peripheral terminals that modulate the activity of innate and adaptive immune cells [23; 78; 99]. Upon neuronal activation by S. aureus Hla, nociceptors were found to release the neuropeptide CGRP, which in turn acted directly on innate immune cells [22]. Neutrophils, monocytes, and macrophages, key myeloid cells that help fight against bacterial infections, express RAMP1 and CALCRL [22], which form the cognate receptor complex for CGRP [66]. It was found that CGRP inhibited S. aureus induced TNF-alpha secretion by macrophages [22]. CGRP signaling through RAMP1/CALCRL induces activation of inducible cAMP early repressor (ICER), which shuts down NF-κB signaling in macrophages and dendritic cells to suppress cytokine production [1; 22; 70]. In S. aureus infections in mice, ablation of Nav 1.8+ nociceptors increased recruitment of monocytes and neutrophils into the infection site, and increased hypertrophy and leukocyte recruitment in skin-draining lymph nodes [22]. These findings suggest that pain and neuroimmune signaling in the skin and lymph nodes directly regulates host responses against this major skin pathogen.
Streptococcus pyogenes is another major gram-positive bacterial pathogen that causes skin and soft tissue infections in humans. S. pyogenes is a leading cause of necrotizing fasciitis (flesh-eating disease), which is characterized by “pain out of proportion” at early stages of infection [18]. Pain and neuroimmune signaling was recently found to mediate S. pyogenes pathogenesis in mice [76]. S. pyogenes, like S. aureus, secretes PFTs which facilitate its spread in the host, namely the peptide toxin streptolysin S (SLS) and larger cholesterol dependent cytolysin streptolysin O (SLO). Using clinical isolates of S. pyogenes, it was demonstrated that significant acute nociception, mechanical and thermal hyperalgesia was produced during subcutaneous infection of mice [76]. S. pyogenes bacteria and culture supernatant were able to directly induce calcium influx in TRPV1+ DRG neurons and calcitonin gene-related peptide CGRP release [76]. Nociceptor activation and pain was dependent on bacterial expression of SLS. Specifically, isogenic mutant bacterial strains lacking the sagA gene (which encodes SLS), but not SLO, showed absence of the ability to evoke neuronal calcium influx in vitro and nocifensive pain, thermal or mechanical hyperalgesia in vivo during S. pyogenes infection [76]. Neuronal release of CGRP from nociceptor terminals directly inhibited bactericidal killing of S. pyogenes by neutrophils, potentially through decreased activity of the major antimicrobial enzyme myeloperoxidase [76]. Genetic ablation of TPRV1+ nociceptors or chemical ablation using the high affinity TRPV1 ligand resiniferatoxin led to significant improvement of and resolution of S. pyogenes necrotizing abscesses. This was associated with increased recruitment of neutrophils and decreased bacterial load during infection. Furthermore, local blockade of CGRP release using botulinum neurotoxin A (BoNT/A) injection or systemic inhibition of CGRP-RAMP1 signaling using the drug BIBN4096 improved the immune response. Therefore, blocking nociceptor activity or its signaling to immune cells increases antimicrobial defenses [76]. It is therefore possible that S. pyogenes may have evolved streptolysin S to induce host nociceptor activation for its advantage by activating a neuroimmune suppressive circuit.
Listeria monocytogenes is a gram-positive bacterium that is a major food-borne pathogen responsible for listeriosis, an infection with a high mortality rate [19]. Symptoms secondary to this condition depend on the anatomical site of infection and may include abdominal pain, bone or joint pain, and headache [19]. Recent work has shown a role for GPR37+ macrophages in mediating both protection against infection-induced sepsis and reduction of pain-like behaviors including mechanical and thermal hypersensitivity in L. monocytogenes infected mice [5]. Pharmacological treatment with the pro-resolving lipid neuroprotectin D1 and the antimalarial drug, artesunate, also ameliorates sepsis severity and pain-like behavior via GPR37 by inducing macrophage-driven host defense and resolution of the infection [5]. While it remains unclear whether L. monocytogenes causes pain through direct or indirect activation of nociceptors, it would be interesting to determine whether specific secreted factors such as N-formylated peptides [93] or other virulence factors directly sensitize neurons. Alternatively, proinflammatory macrophages could also release factors that drive pain [20].
2.2. Gram-negative bacteria
Gram-negative pathogens can infect many tissue sites, including the oral cavity, the urinary tract, and the gastrointestinal tract, and these infections are associated with pain [30; 68]. Gram-negative bacteria differ from gram-positive bacteria in their cell wall composition. Lipopolysaccharide (LPS) is a major component of gram-negative cell walls, which differs from gram-positive bacteria, which express peptidoglycans (PGN) and lipoteichoic acids (LTA) [98]. Depending on the stage of infection, gram-negative pathogens produce distinct forms of LPS [108]. LPS has been found to signal to neurons via multiple molecular mechanisms to drive inflammatory pain (Fig. 1). Toll-like receptor 4 (TLR4) is the major host receptor for bacterial LPS [98] and is expressed by peptidergic nociceptor neurons, as revealed by scRNA-Seq and dRNA-Seq [101; 107; 111]. TLR4 has been found to potentiate TRPV1 signaling in DRG neurons [56] and may be a key signaling receptor for inflammatory pain produced by LPS. LPS induces calcium influx and CGRP release in a PKA- and PKC-dependent manner through TLR4 on neurons from DRG, vagal ganglia, and trigeminal ganglia [31; 47; 48]. TLR4 was recently shown to be associated with TRPV1 via its cytoplasmic toll/interleukin 1 Receptor (TIR) domain, which could prevent activation induced desensitization of TRPV1[71].
TLR4 signaling also mediates pelvic pain in mice induced by uropathogenic E. coli (UPEC) [83]. UPEC is a leading cause of urinary tract infections and bladder cystitis, which are accompanied by significant pain. It was found that instillation of LPS purified from the UPEC strain NU14 into the bladders of mice was sufficient to induce pelvic pain in a manner dependent on the O-antigen, which was significantly decreased in TLR4-deficient animals [83]. By contrast, purified LPS from the asymptomatic bacteriuria E. coli strain 83972 did not cause pain in mice, suggesting distinctions between different types of LPS in pain production [83].
LPS may also signal to neurons through the large-pore cation channel TRPA1 independently of TLR4. LPS derived from several bacterial pathogens (E. coli, Salmonella typhimurium, Klebsiella pneumoniae, Serratia marcescens, Pseudomonas aeruginosa, Rhodobacter sphaeroides) directly activated DRG neurons [68]. This LPS-induced nociceptor activation required Lipid A, the membrane anchored moiety of LPS [68]. In this study, LPS induced mechanical hyperalgesia was abrogated in TRPA1 deficient mice but not in TLR4 deficient mice [68]. Mechanisms behind the dichotomy of results in the above studies are still unknown [31; 47; 48; 68], but these discrepancies could be due to different doses of LPS, in which TRPA1 mediates responses to low concentrations of LPS while TLR4 and TRPV1 activation might be responsible for shaping inflammation in response to LPS at high concentrations [31; 47; 48; 68].
Secreted metabolites from gram-negative bacteria can also act on host neurons to regulate physiological and pathological conditions [81]. Indole, a tryptophan-derived metabolite, can be produced by a variety of bacterial species including commensal E. coli strains and the pathogenic oral bacteria P. gingivalis and F. nucleatum [25; 81]. Injecting live E. coli into the footpads of mice was sufficient to produce nocifencive behaviors, which was significantly reduced when an isogenic E. coli strain lacking indole production (Δtryptophanase E. coli) was injected [25]. In addition to pain-like behaviors in mice, indole elicited calcium influx and evoked inward currents in TRPA1+ DRG neurons. Pain-like behaviors and neuronal responses induced by indole were pharmacologically blocked by the TRPA1 antagonist, HC-030031, and significantly abolished in TRPA1 knockout mice, indicating that E. coli-derived metabolites produces pain via TRPA1 activation [25].
Other Gram-negative bacterial components may also directly activate nociceptors and contribute to pain. Flagellin is a major component of bacterial flagella, which is utilized by multiple gram-negative pathogens including E. coli and Salmonella enterica for motility and survival in hosts [45]. A subset of Aβ DRG neurons were found to express toll-like receptor 5 (TLR5), which is the prototypic receptor for bacterial flagellin [104]. A recent study showed that TLR5+ DRG neurons were critical mediators of neuropathic pain [104]. Co-application of flagellin with the normally impermeable lidocaine derivative QX-314 allowed TLR5-dependent silencing of Aβ fibers, which potently inhibited neuropathic pain in mice [104]. While it is exciting that a bacterial ligand and analgesic can be utilized to silence pain, it is still unknown how TLR5 contributes to pain during infection. Indeed, the endogenous role of TLR5 in bacteria-induced pain and whether flagellin induces neuropathic-like pain has not been fully studied.
We note that much remains to be determined regarding the mechanisms of pain produced during infections by live bacteria. The studies we described above show that bacteria can produce factors that directly act on nociceptors to drive pain. However, pain during infection is likely to involve action of both microbial and inflammatory mediators. Immune cells release prostaglandins (e.g. PGE2), cytokines (e.g. TNF, IL1) and other factors that sensitize neurons peripherally and centrally [78]. Future studies are required to define the contributing factors that drive pain in each type of infection. The type of bacterial pathogen, its expression of surface-anchored components (e.g. flagellin, cell wall components), and secreted factors (e.g. toxins, enzymes), and how they are sensed by distinct host cells (e.g. neurons, immune cells) in vivo will determine the specific modalities and nature of pain produced. Targeted molecular and functional studies on both the host and pathogen side during live infections are required to elucidate these mechanisms.
3. Bacterial pathogens and analgesia
3.1. Microbes that silence pain
While many bacterial infections are painful, some infections are characterized by a complete lack of pain or are potentially analgesic in nature [21; 34; 62; 74] (Fig. 2A). Bacterial pathogens may silence pain for their selective advantage. For instance, Treponema pallidum, the causative agent of syphilis, produces painless lesions despite significant tissue inflammation and destruction [74], [89]. This bacterial pathogen is a spirochete that can also enter the brain and cause neurosyphilis [41; 74]. As a sexually transmitted pathogen, it may be beneficial for T. pallium to block pain. In contrast, Borrelia burgdorferi, which is also a spirochete and the causative agent of Lyme disease, causes arthritis and pain [58]. The mechanisms by which Treponema or Borrelia modulate pain during infection is unknown.
Another bacterial pathogen that can induce pain blockade during infection is Mycobacterium ulcerans [62]. M. ulcerans is the causal agent of Buruli ulcer, a chronic infectious disease characterized by the massive destruction of cutaneous tissue, leading to the development of large ulcerative and painless lesions [62]. This tissue destruction is caused by a unique lipid-like toxin called mycolactone, produced by M. ulcerans. Interestingly, mycolactone is cytotoxic at high doses, but, at lower doses, it modulates pain and immune responses, facilitating host colonization [62]. At these lower concentrations, rather than promoting nerve damage, mycolactone was found to signal trough type 2 angiotensin 2 (AT2R)/TRAAK channels to promote nociceptor hyperpolarization [62]. Mycolactone could potentially be developed as a therapeutic treatment as it was shown to be analgesic in mice [62]. This mechanism nevertheless raises controversy, as AT2R blockade, instead of activation, was previously reported to promote analgesia [27; 79]. Single-cell transcriptomic studies also reveal low or absence of expression of AT2R by nociceptors [86]. Therefore, additional studies are needed to clarify how M. ulcerans mycolactone blocks pain. A related pathogen that causes analgesic infections is Mycobacterium leprae, the causative agent of leprosy [34]. It is thought that in this case M. leprae induces significant destruction of nerves and Schwann cells, leading to lack of sensory transmission [34].
In addition to pathogens, some non-pathogenic bacteria may also have analgesic potential. This includes the gut probiotic Escherichia coli strain Nissle 1917 (EcN) [75]. It was found that EcN secretes a lipopeptide, C12AsnGABAOH, which can reduce visceral pain sensitivity in mice [75]. C12AsnGABAOH crosses the epithelial barrier to prevent capsaicin-induced visceral pain [75]. Furthermore, C12AsnGABAOH inhibits calcium influx in TRPV1+ nociceptors by signaling via the gamma-aminobutyric acid (GABAB receptor) [75]. Importantly, C12AsnGABAOH does not alter intestinal motility or modify the physiology of the intestinal epithelium, suggesting that it does not affect the enteric nervous system and is more specific for nociception [75]. This might have fewer side effects than prototypical analgesics such as morphine, which block gut motility. EcN is the active component of Mutaflor®, a commercial probiotic therapy. Therefore, repurposing this probiotic or its lipopeptide represents a promising candidate for analgesic therapy for acute or chronic abdominal pain.
3.2. Bacterial toxins and engineering to silence pain
3.2.1. Botulinum Neurotoxins (BoNTs)
Bacterial toxins are secreted proteins capable of multiple tasks. On the one hand, they facilitate bacterial pathogenesis. On the other hand, they have great potential for therapeutic applications. An example of utilizing bacterial toxins to treat disease are in the therapeutic use of botulinum neurotoxins (BoNTs) in pain (Fig. 2B) and other diseases [73]. Clostridium botulinum, Clostridium tetani and other related family members of bacteria have evolved the secretion of several major types of toxins including botulinum neurotoxins (BoNTs) and tetanus toxin (TenT) that target the nervous system [13; 73; 82]. These toxins are key mediators of bacterial virulence. By inhibiting neurotransmitter release at neuromuscular junctions, BoNTs causes the severe neuroparalytic disease that accompanies botulism and C. botulinum foodborne infections [13; 28; 82]. There are seven serologically distinct BoNT isoforms (denoted A-G), which exhibit significant amino acid sequence similarity [28; 82]. Different serotypes have distinct host receptor and target specificity. Among these seven types of botulinum toxin, BoNT/A has been the main serotype repurposed to treat human disease [73]. Because of its long-lasting activity and high efficiency, BoNT/A has been approved by the U.S. Food and Drug Administration for treating a variety of disorders [90], including pain. Clinical research demonstrated that after treatment with BoNT/A (formulated as BOTOX®), migraine patients reported significant alleviation of headache pain symptoms and reduced frequency and duration of migraines [35; 90]. Besides migraine, BoNT/A has also been clinically applied in the treatment of chronic neck pain, shoulder pain, back pain, and peripheral neuropathic pain [6; 35; 73; 90].
Structurally, BoNT/A consist of heavy-chain (HC) and light-chain (LC) moieties linked by a disulfide bridge. The heavy chain is responsible for binding the toxin to neuronal receptors and promoting essential light chain translocation across the endosomal membrane [12; 28]. The light chain is a zinc endopeptidase that cleaves synaptosome associated protein 25 (SNAP25), a component of the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex required for neuronal vesicle release [12; 28]. Once internalized within nerve terminals, the light chain of BoNT/A has the capacity to silence neurotransmission for several weeks to months, a property that may be due to deubiquitinating enzymes that decrease cytoplasmic degradation [12]. BoNT/E is another botulinum toxin that cleaves SNAP-25, albeit with shorter half-life than BoNT/A within the nerve terminals [12]. The analgesic effect of BoNT/A is thought to depend on retrograde delivery in nociceptors and blockade of the release of glutamate from central terminals [46]; in addition, BoNT/A blocks nociceptor release of the neuropeptides substance P [64] and calcitonin gene-related peptide (CGRP), which are important in driving pain perception [2; 76]. Other potential mechanisms of analgesia caused by BoNT/A involve functional and structural interactions between BoNT/A and the TRPV1 ion channel [37; 55]. BoNT/A also blocks peripheral release of neuropeptides which could drive neurogenic inflammation. As described above, BoNT/A can block CGRP release in S. pyogenes infection, leading to increased neutrophil recruitment and antibacterial function, thereby enhancing the peripheral immune response [76]. Therefore, BoNT/A has potential both in the treatment of pain and infection.
Beyond the use of native botulinum toxins in therapeutic applications to silence pain, these toxins or toxin fragments can be mutated or fused with exogenous protein domains to engineer novel proteins with translational potential [60]. Engineered BoNT/A fusion proteins have been generated as a platform to target nociceptors and central pain circuitry to silence pain [59; 60]. “Bitox” is an engineered toxin produced by “stapling” together the recombinant light chain/translocation domain and receptor-binding domain of BoNT/A [59; 60]. This version of BoNT/A lacked paralytic activity but was able to attenuate complete Freund’s adjuvant (CFA) inflammatory pain and spared-nerve injury (SNI) induced neuropathic pain [60]. Further work by the same group showed that engineered botulinum toxins can be used to selectively target and silence spinal neurons that mediate pain signaling [59]. The light chain/translocation domain of BoNT/A was “stapled” using SNARE proteins to either substance P (SP) or dermorphin (Derm). The fused proteins, SP-Botulin or Derm-Bot constructs were then injected intrathecally to target spinal neurons expressing the neurokinin receptor (NK1R) or mu opioid receptor (MOR), respectively. This caused cleavage of SNAP-25 in these target neurons and produced long-term amelioration of inflammatory and neuropathic pain [59], making engineered bacterial toxins a unique platform for relieving pain while avoiding widespread systemic effects. These exciting studies reveal the promising potential of both native botulinum toxin and engineered neurotoxins to block pain.
3.2.2. Anthrax toxins
Anthrax toxins are the key virulence factors of Bacillus anthracis, a gram-positive bacterial pathogen that can cause cutaneous infections of in humans and livestock, and severe or lethal pulmonary infections [95]. Recent work has shown that anthrax toxins could also act on nociceptors and have analgesic effects [105] (Fig. 2C). Anthrax toxins consist of three proteins: Protective antigen (PA), Edema Factor (EF) and Lethal Factor (LF). PA binds to its cognate receptors ANTXR1 and ANTXR2, with significantly higher affinity to the latter, and oligomerizes into a pore that translocate EF and LF into the cytoplasm [33; 106]. EF is an adenylate cyclase and LF is an enzyme that cleaves map kinase kinases [33]. The lethality of the toxins mainly occurs through the action of the toxins on the vasculature, liver, and immune systems [95]. It was found that the high affinity anthrax toxin receptor, ANTXR2, is expressed and enriched in Nav 1.8+ nociceptive neurons compared to Parvalbumin+ proprioceptive neurons, and that it was largely absent in other neuronal subtypes throughout the central nervous system [105]. Anthrax toxins induce signaling in both mouse DRG neurons and human iPSC-derived sensory neurons. Different combinations of anthrax toxins were injected intrathecally into mice to determine the effects on pain in vivo. Edema toxin (ET), which consists of PA+EF, was found to specifically induce analgesic effects in vivo in mice, showing blockade of both mechanical and thermal nociception at baseline, and mechanical allodynia following carrageenan induced inflammatory pain or spared nerve injury (SNI) induced neuropathic pain. ET was found to block neurotransmission at the central terminals of nociceptors. These pain blocking effects were not due to injury or death to sensory neurons, but rather likely due to changes in cAMP and intracellular signaling [105].
Anthrax toxins also hold potential to be engineered as a delivery system for transporting functional molecular cargo into nociceptors to silence pain [105]. The anthrax toxin system, with its two components, PA, which binds its receptor, and either EF or LF, which enters the cytoplasm, has been successful engineered to target tumor cells [4]. For example, it has been demonstrated that the N-terminus of LF retains its ability to bind PA and translocate enzymatic cargoes such as diphtheria toxin A (DTA) into tumor cells [4]. PA was also shown to be able to deliver LFn-DTA into DRG neurons and induce neuronal cell death [105]. Next, to silence pain, one idea was to utilize the light chain of BoNT/A as the molecular cargo for anthrax toxins, given its ability to cleave SNAP-25 and produce long-lasting pain blockade [12]. It was shown that PA+LFN-LC/A was able to deliver this cargo into DRG neurons to cleave SNAP-25, block CGRP release, and pain blockade in the spared nerve injury (SNI) model of neuropathic pain [105]. Future studies using the anthrax platform by engineering each specific component (PA, LF or EF) can increase or tune the receptor specificity of the system to distinct neuronal subtypes or to deliver distinct molecular cargoes with analgesic potential into neurons.
4. Viral pathogens and pain
Viral infections cause various clinical manifestations including pain, which can be one of the earliest signs of infection often occurring before fever starts [7]. Influenza viruses, Sars-COV-2, and other respiratory viral pathogens can induce headache, myalgias, and joint pain. Pain during viral infections could result from both direct action of viruses on neurons and indirect action by pain sensitization through inflammatory mediators [7; 21; 32; 88; 97; 100]. Recently, type 1 interferons which are key antiviral cytokines induced by viral infections, were found to cause pain and hyperalgesia by acting on IFN receptors expressed by nociceptors. Viral infection stimulates Type 1 IFN production (i.e. IFN-alpha, IFN-beta) by immune or epithelial cells, which then activate type I IFN receptor signaling through a MNK-eIF4E pathway in Nav1.8+ nociceptors to induce pain [7]. With regards to COVID19, viral effects on the nervous system, both acute and chronic, are a major area of current interest [87]. Some persistent neurological effects of SARS-CoV-2 associated with nociceptors have been described, including headache, chest, joint and nerve pain [87]. Angiotensin converting enzyme-2 (ACE2), the main receptor for the SARS-CoV-2 viral entry [85], is expressed by human DRG neurons [85]. However, it remains to be determined whether SARS-Cov-2 can directly infect neurons and if this leads to pain sensitivity changes.
Several types of herpesviruses, including herpes simplex viruses (HSV-1, HSV-2) and varicella zoster virus (VZV), exhibit neurotropism and preferentially infect nociceptor sensory neurons [39; 42; 80; 88] (Fig. 3A). In these viral infections, pain is a primary symptom of both acute infection and viral reactivation [39; 42; 88]. These neurotropic viruses enter the host by infecting mucocutaneous surfaces where they access sensory nerves from DRG and trigeminal ganglia via axonal transport [42; 88]. For VZV, infection is followed by established latency in nociceptors [42; 80; 103]. Stress or other triggers lead to reactivation of viral replication and spread of the virus to the skin innervated by these neurons, causing zoster, popularly known as “shingles”, which can be severely painful. Elderly patients are at high risk for subsequent VZV mediated neuronal cell death and persistent neuropathic pain, termed postherpetic neuralgia [42; 103].
Figure 3. Viruses and fungi that act on nociceptors to produce pain.
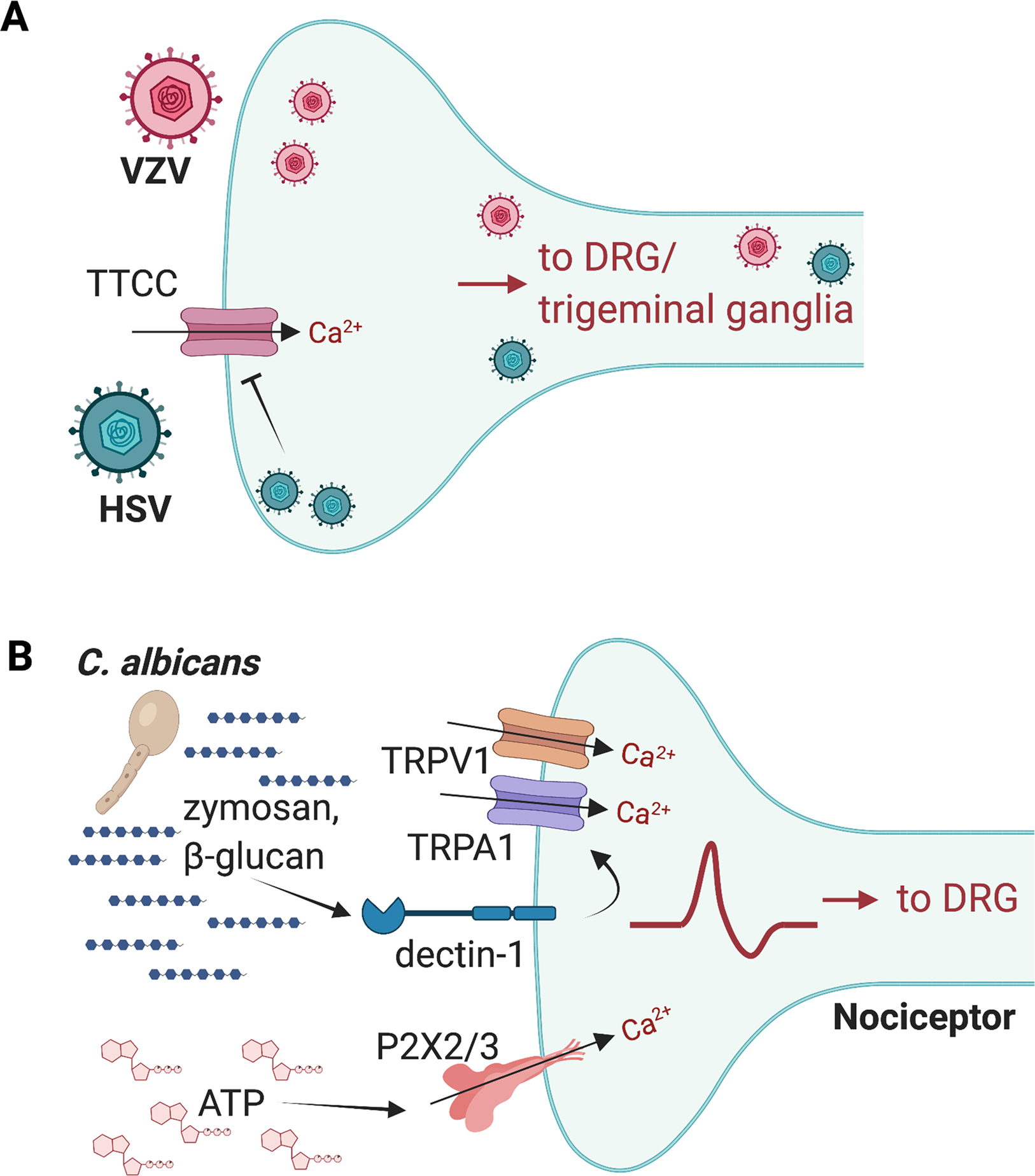
(A) Herpesviruses including HSV-1, HSV-2, and VZV infect skin or mucosal surface-innervating sensory nerve terminals and are retrogradely transported via axons to DRG and trigeminal ganglia. HSVs reduce the expression of T-type calcium channels in sensory neurons, which could potentiate pain. (B) C. albicans zymosan and β-glucans bind to the Dectin-1 receptor, which could act via TRPV1 and TRPA1 ion channels to induce neuronal activation. ATP released during C. albicans infection can also activate P2X2 and P2X3 receptors on nociceptors to amplify pain. Image created with BioRender.com
While it is likely that neuropathic damage induced by the virus is the cause of postherpetic neuralgia [39; 42], the underlying mechanisms are not yet fully determined. Some evidence suggests a role of neuron-satellite glia interactions in the pathogenesis of VZV infection [80], however, it remains unknown if this interaction contributes to the pain underlying VZV infection. As VZV cannot efficiently infect mice or other small animals, its mechanisms of pain are not as well understood, even though it is the major cause of postherpetic neuralgia.
HSV-1 and HSV-2 are viral pathogens capable of infecting both mouse and human DRG neurons [40; 88; 97; 103]. In humans, HSV-1 infections are clinical manifested as painful blisters or ulcers in or around the mouth, and HSV-2 induces similar painful infections in the genital area. In mice, HSV-1 infection can result in acute mechanical allodynia and hyperalgesia that persists over time [88; 97]. One mechanism by which this persistent pain occurs in mice during HSV-1 infection is via leukocyte infiltration into the DRG [88]. It was found that monocytes and neutrophils infiltrate the DRG, subsequently releasing the cytokine TNF-alpha, which in turn mediates the development of pain through downregulation of the inwardly rectifying K+ channel Kir4.1 in satellite glial cells which surround DRG neurons [88]. HSV-1 could also alter neuronal excitability that leads to pain, as it has been found to reduce the expression of T-type calcium channels and modulate electrical properties of a sensory neuron–like cell line [109]. Emerging evidence also suggests that Nav1.8+ nociceptors can regulate immune responses against HSV-1 [40]. In a skin infection model by HSV-1, nociceptor ablation led to significantly enhanced neutrophil recruitment and suppressed CD8+ T cell responses against the virus. This neural modulation of immunity may limit the severity of tissue damage and restore skin homeostasis [40]. Further investigations are required to determine the contribution of intrinsic changes in neurons due to viral infection and the extrinsic role of non-neuronal cells such as immune cells in pain induction during HSV-1 and HSV-2 infection and reactivation of these viruses.
It is important to note that other types of viral infections, including by arborviruses (i.e., Chikungunya virus and Dengue virus), are characteristically painful [110]. However, the underlying mechanism of pain for these and the majority of viral pathogens is unknown. Even beyond pain, other sensory neuron mediated protective reflexes including sneezing, vomiting, or cough are characteristic of viral infections, yet the neurological mechanisms driving these reflexes are largely unknown. Thus, deciphering how nociceptors are activated during viral infection and how they signal to the immune system is an area that warrants investigation.
5. Fungal Pathogens and Pain
Fungal opportunistic pathogens such as Candida albicans can cause painful infections in the skin, oral cavity, and genitourinary tract (Fig. 3B). Fungal pathogen-associated molecular patterns (PAMPs) that are known to induce host immune responses include cell wall components such as chitin, β-glucans, and mannoproteins [92; 96]. Injection of Zymosan or β-glucan (yeast cell wall components) induces inflammatory pain-like behaviors in mice, which have been found to be dependent on mouse expression of toll-like receptor 2 (TLR2), its downstream signaling component MyD88 [43], and on the receptor Dectin-1[36]. TLR2 and Dectin-1 were originally thought to detect zymosan or β-glucan and signal through innate immune cells to induce pain [36; 38; 43]. Another study suggests that nociceptors may also directly sense fungi, leading to pain sensitivity [26]. Zymosan or heat-killed C. albicans induced calcium influx and CGRP release in cultured DRG neurons in a manner dependent on Dectin-1 [26]. Injecting C. albicans into the hind paws of mice induced spontaneous nocifensive pain and mechanical hypersensitivity. Dectin-1 in neurons was thought to signal in a TRP channel–dependent mechanism; both dectin-1–deficient and TRPV1/TRPA1 double-deficient mice fail to develop mechanical allodynia after β-glucan treatment [26]. Further studies finds that C. albicans derived β-glucan can also stimulate Dectin-1 expressed by keratinocytes to release ATP that activates P2X2/P2X3 expressed by nociceptors to amplify pain [63]. C. albicans infections can also induce significant pain in the genital tract. In a mouse model of vulvodynia, repeated localized exposure of the vulva to C. albicans induces persistent mechanical allodynia and hyperinnervation characterized by increases in the density of peptidergic (CGRP+) and sympathetic fibers (VMAT2+) [38].
Nociceptor activation during fungal infection plays a key role in cutaneous immunity and host defenses [26; 50]. It was found that TRPV1+ nociceptors are activated by C. albicans and release the neuropeptide CGRP, which acts on CD301b+ dermal dendritic cells to produce the cytokine IL-23. In turn, IL-23 stimulates IL-17 production from γδ T cells and activates an effective immune response to the fungus in the skin [50]. Therefore, in the scenario of fungal infections, activation of pain could play a beneficial role in driving host defenses [50]. Optogenetic activation of cutaneous TRPV1+ neurons elicited a local response involving IL-17-secreting T helper cells (TH17 cells) without pathogen inoculation or tissue damage, and reduced susceptibility to subsequent infection with S. aureus or C. albicans, showing that this pathway could induce an anticipatory immune response to prepare the host for future pathogen exposure [26].
6. Parasitic Pathogens and Pain
Parasitic pathogens are highly diverse in nature, ranging from single cell protozoans to large multicellular helminths [24]. Most have complex life cycles that involve both an ability to feeding off, survive within, and transmit between multiple mammalian or insect hosts [24]. Compared to bacteria, viruses, and fungi, less is known about how parasites interact with the nervous system and the role of pain in infection. Certain types of parasite invasion of barriers can be painful and recent work is starting to investigate the mechanisms behind this pain. Leishmaniasis is a parasitic disease caused by the protozoan parasite from Leishmania genus. Murine models of Leishmania infection demonstrate that L. major or L. amazonensis induce persistent hyperalgesia during infection [17; 49]. In these studies, it was found that peripheral production of nerve growth factor (NGF) and the NF-κB-dependent cytokines IL-1β and TNFα mediated pain. Spinal cord astrocyte and microglia activation also occur in Leishmania infected mice in a manner dependent on these cytokines [16; 17].
Trypanosoma cruzi is an insect-born parasite that causes Chagas disease, a tropical infection with high mortality. In a mouse model of T. cruzi infection, mechanical and thermal hyperalgesia increased, and this correlated with induction of both TNF-a and IL-1b, as well as gliosis and NFκb activation [15]. Glial and NFκB inhibitors were able to diminish pain and reduce the production of the inflammatory mediators in T. cruzi infected mice [15].
Schistosomiasis is a parasitic infection caused by Schistosoma mansoni that can produce abdominal pain. Intestinal schistosomiasis in mice concurrently increased the number of CGRP+ fibers and mucosal mast cells, which were found closely associated with each other in the small intestine ileum [29]. Neonatal capsaicin treatment led to a 70% reduction in the number of mucosal mast cells induced by S. mansoni infection, suggesting that nociceptors may actively participate in mast cell expansion during parasite infection, suggesting a neuroimmune crosstalk [29].
While these studies have begun to reveal a role for pain in parasite infections, there is still a significant knowledge gap on the molecular and cellular mechanisms by which parasites could interact with neurons in the skin, gut, and other barrier sites to regulate pain or neuroimmune crosstalk.
7. Cough, itch, and other nociceptive reflexes
Beyond pain, infections are also associated with related but distinct protective sensory driven neural reflexes including coughing, sneezing, itching, or blinking. How sensory neurons are activated by pathogens to mediate these functions is less well studied, though the functional consequences could play major roles in infection pathogenesis. Coughing and sneezing may help the host expel harmful pathogens, but also concurrently facilitate the transmission of pathogens between hosts. Recent work has begun to identify the sensory neurons that mediate these important neural reflexes. A sneeze reflex circuit was found in mice that is driven by TRPV1+NMB+ trigeminal neurons [54]. Itch is driven by chemical or inflammatory pruritogens that activate skin-innervating DRG or trigeminal neurons, inducing spinal circuits that mediate a desire to scratch in mice [9]. Itch may remove harmful substances from the skin including pathogen-carrying insects, but also induce skin barrier damage and facilitate pathogen entry. Of note, very little is known about how pathogens cause itch and sneezing during infection, an area that requires significant future research.
The cough reflex is initiated by vagal sensory neurons that innervate the respiratory tract, and upon detection of noxious stimuli, vagal afferents send signals to the brainstem, which feeds back via motor signals to drive cough [32]. Mycobacterium tuberculosis (MTb) infection often manifests with a persistent cough, which could facilitate its transmission from host to host [30; 84]. Using a guinea pig model of MTb and cough, a recent study showed that MTb secretes sulfolipid-1 (SL-1) which is critical for driving the cough reflex [84]. SL-1 was also shown to directly induce calcium influx in mouse vagal and DRG TRPV1+ neurons. DRG neurons exposed to MTb, or its extract showed a rapid increase in intracellular calcium, and this was greatly diminished in isogenic mutant strains of MTb lacking genes required for the synthesis of the sulfolipid [84]. A few exciting questions remain from this study, including identification of the receptor for SL-1 on vagal sensory neurons, and to formally prove whether SL-1 is necessary for spread of MTb between animals via cough. However, it opens the possibility that pathogens may have evolved specific ways of inducing nociception or cough reflexes to facilitate pathogenesis and transmission.
The nociceptive blink reflex or eye blinking behavior is a protective trigeminofacial reflex aimed at facilitating eyelid closure as a response to threatening and potentially harmful stimuli [61]. Sensory neurons in the eye almost exclusively originate from the ophthalmic division of the trigeminal ganglion [10]. In a mouse model of P. aeruginosa induced keratitis, a rapid reduction in blink reflexes was observed, which is indicative of neuronal damage [57]. Consistent with the alterations in the blink reflexes, sub-basal nerve plexi in central and peripheral corneas were collapsed after P. aeruginosa infection, indicating the presence of neuronal damage and corneal neuropathy. Chemical ablation of TRPV1+ nociceptors decreased corneal bacterial burdens and improved myeloid immune cell trafficking in corneal tissues in mice infected with P. aeruginosa, indicating that sensory neurons play a role in disease progression. In vitro, trigeminal TRPV1+ neurons showed calcium influx in response to P. aeruginosa but less activation in response to strains of P. aeruginosa that lacked virulence factors such as pili, flagella, or type III secretion system. CGRP treatment of neutrophils inhibited bactericidal killing of P. aeruginosa [57]. Therefore, P. aeruginosa may exploit activation of nociceptors to regulate immune trafficking into the cornea and facilitate its survival during infection.
8. Conclusions and future directions
Pain is a hallmark of many types of infectious diseases, yet the mechanisms of pathogen-induced pain are only beginning to be understood. Given the vast diversity of bacterial, viral, fungal and parasitic organisms that mammals encounter, there is a growing appreciation of the intricate and context-dependent ways the host nervous system responds to infection. As reviewed here, nociceptors can sense and be activated by pathogen-derived products, and that activation triggers the release of mediators such as neuropeptides that impact host defense. However, these findings are only the tip of the iceberg in defining microbe-neuron-immune interactions. Many open questions remain in this burgeoning field of research. For pathogens that cause pain, what are the molecular factors being sensed by the nervous system that drives pain? What are the host receptors on neurons for these microbes and do pathogen receptors signal in the same way in a neuron as in immune cells?
Can these studies be translated into clinical application in humans? Comparison of human DRG neurons and their functional responses to microbes with rodent neurons is an important first step. There is also a growing interest in the use of human induced pluripotent stem cell (iPSC) derived sensory neurons to model and study nociceptor naïve and diseased states in vitro. Once iPSCs are generated and differentiated into peripheral sensory neurons, they offer eclectic potential for studying disease mechanisms and to search for individualized or population therapies [52; 69]. Detailed RNA sequencing has been performed on IPSC-derived nociceptors suggesting a DRG transcript ‘signature’ when compared to published human DRG RNA sequencing data, including the ion channel profiles [65]. iPSCs could also serve as a cellular platform to interrogate the translational value of targets and nociceptive mechanisms underlying pain during infectious diseases. For instance, the identification of receptors presents in neurons that sense microbes and then determining if microbial factors activate or silence human iPSC-derived neurons could be of interest. The genome editing of iPSCs also allows interrogation of causal genes involved in a specific neuronal response [52; 69].
Another growing area of future interest is how nociceptors regulate innate and adaptive immunity against pathogens. Pain has been shown to beneficial either for the host or for the pathogen, depending on the nature of the pathogen, anatomical site, and specific immune cell type involved. Some pathogens, such as S. pyogenes, hijack pain for its advantage by inducing nociceptor release of factors that polarize immune responses to suppress antibacterial killing [76]. By contrast, nociceptors mediate host defenses against C. albicans by activating dendritic cell and T cell cutaneous immunity [26]. Defining how microbes interact with neurons in pain and host defense also has important therapeutic implications. Chronic use of analgesics including opioids could also alter the ability of the host to respond to and defend against pathogens. It is also possible that with certain pathogens, targeting neuronal signaling could lead to novel approaches to enhance immunity and treat infection. Another exciting direction is identifying and defining the mechanisms by which microbes silence pain, which could lead to development of novel analgesics. As described above, bacterial toxins including BoNTs, and anthrax toxins act on nociceptors to silence neurotransmission and pain signaling. Engineering these bacterial toxins to not only target primary sensory afferents but central spinal neurons and deliver distinct molecular cargoes into neurons could lead to selective and potent analgesic approaches [59], [105]. Therefore, defining the molecular and cellular mechanisms by which pathogens interact with nociceptive neurons could lead to novel approaches to treat infectious diseases and chronic pain.
Acknowledgments
Work in the Chiu laboratory is supported by National institutes of Health (5R01DK127257, 5R01AI130019), Chan-Zuckerberg Initiative, Burroughs Wellcome Fund, Fairbairn Family Lyme Research Initiative, Drako Family Fund for Allergy Research, and Food Allergy Science Initiative.
Footnotes
Conflict of interest statement
I.M.C. serves on scientific advisory boards for GSK pharmaceuticals and Limm Therapeutics.
References
- [1].Altmayr F, Jusek G, Holzmann B. The neuropeptide calcitonin gene-related peptide causes repression of tumor necrosis factor-alpha transcription and suppression of ATF-2 promoter recruitment in Toll-like receptor-stimulated dendritic cells. J Biol Chem 2010;285(6):3525–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Antoniazzi C, Belinskaia M, Zurawski T, Kaza SK, Dolly JO, Lawrence GW. Botulinum Neurotoxin Chimeras Suppress Stimulation by Capsaicin of Rat Trigeminal Sensory Neurons In Vivo and In Vitro. Toxins (Basel) 2022;14(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Aronoff DM, Bloch KC. Assessing the relationship between the use of nonsteroidal antiinflammatory drugs and necrotizing fasciitis caused by group A streptococcus. Medicine (Baltimore) 2003;82(4):225–235. [DOI] [PubMed] [Google Scholar]
- [4].Bachran C, Leppla SH. Tumor Targeting and Drug Delivery by Anthrax Toxin. Toxins (Basel) 2016;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bang S, Donnelly CR, Luo X, Toro-Moreno M, Tao X, Wang Z, Chandra S, Bortsov AV, Derbyshire ER, Ji RR. Activation of GPR37 in macrophages confers protection against infection-induced sepsis and pain-like behaviour in mice. Nat Commun 2021;12(1):1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baron R, Binder A. Fighting neuropathic pain with botulinum toxin A. Lancet Neurol 2016;15(6):534–535. [DOI] [PubMed] [Google Scholar]
- [7].Barragan-Iglesias P, Franco-Enzastiga U, Jeevakumar V, Shiers S, Wangzhou A, Granados-Soto V, Campbell ZT, Dussor G, Price TJ. Type I Interferons Act Directly on Nociceptors to Produce Pain Sensitization: Implications for Viral Infection-Induced Pain. J Neurosci 2020;40(18):3517–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 2009;139(2):267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bautista DM, Wilson SR, Hoon MA. Why we scratch an itch: the molecules, cells and circuits of itch. Nat Neurosci 2014;17(2):175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Belmonte C, Aracil A, Acosta MC, Luna C, Gallar J. Nerves and sensations from the eye surface. Ocul Surf 2004;2(4):248–253. [DOI] [PubMed] [Google Scholar]
- [11].Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 2007;449(7162):607–610. [DOI] [PubMed] [Google Scholar]
- [12].Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, Sudhof TC, Jahn R, Niemann H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem 1994;269(3):1617–1620. [PubMed] [Google Scholar]
- [13].Binz T, Sikorra S, Mahrhold S. Clostridial neurotoxins: mechanism of SNARE cleavage and outlook on potential substrate specificity reengineering. Toxins (Basel) 2010;2(4):665–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, Roberson DP, Ma YC, Otto M, Woolf CJ, Torres VJ, Chiu IM. Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 2018;9(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Borghi SM, Fattori V, Carvalho TT, Tatakihara VLH, Zaninelli TH, Pinho-Ribeiro FA, Ferraz CR, Staurengo-Ferrari L, Casagrande R, Pavanelli WR, Cunha FQ, Cunha TM, Pinge-Filho P, Verri WA. Experimental Trypanosoma cruzi Infection Induces Pain in Mice Dependent on Early Spinal Cord Glial Cells and NFkappaB Activation and Cytokine Production. Front Immunol 2020;11:539086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Borghi SM, Fattori V, Pinho-Ribeiro FA, Domiciano TP, Miranda-Sapla MM, Zaninelli TH, Casagrande R, Pinge-Filho P, Pavanelli WR, Alves-Filho JC, Cunha FQ, Cunha TM, Verri WA Jr. Contribution of spinal cord glial cells to L. amazonensis experimental infection-induced pain in BALB/c mice. J Neuroinflammation 2019;16(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Borghi SM, Fattori V, Ruiz-Miyazawa KW, Miranda-Sapla MM, Casagrande R, Pinge-Filho P, Pavanelli WR, Verri WA Jr. Leishmania (L). amazonensis induces hyperalgesia in balb/c mice: Contribution of endogenous spinal cord TNFalpha and NFkappaB activation. Chem Biol Interact 2017;268:1–12. [DOI] [PubMed] [Google Scholar]
- [18].Borschitz T, Schlicht S, Siegel E, Hanke E, von Stebut E. Improvement of a Clinical Score for Necrotizing Fasciitis: ‘Pain Out of Proportion’ and High CRP Levels Aid the Diagnosis. PLoS One 2015;10(7):e0132775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Charlier C, Perrodeau E, Leclercq A, Cazenave B, Pilmis B, Henry B, Lopes A, Maury MM, Moura A, Goffinet F, Dieye HB, Thouvenot P, Ungeheuer MN, Tourdjman M, Goulet V, de Valk H, Lortholary O, Ravaud P, Lecuit M, group Ms. Clinical features and prognostic factors of listeriosis: the MONALISA national prospective cohort study. Lancet Infect Dis 2017;17(5):510–519. [DOI] [PubMed] [Google Scholar]
- [20].Chen O, Donnelly CR, Ji RR. Regulation of pain by neuro-immune interactions between macrophages and nociceptor sensory neurons. Curr Opin Neurobiol 2020;62:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chiu IM. Infection, Pain, and Itch. Neurosci Bull 2018;34(1):109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, Bubeck Wardenburg J, Hwang SW, Carroll MC, Woolf CJ. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013;501(7465):52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci 2012;15(8):1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chulanetra M, Chaicumpa W. Revisiting the Mechanisms of Immune Evasion Employed by Human Parasites. Front Cell Infect Microbiol 2021;11:702125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chung S, Kim H, Kim D, Lee JM, Lee CJ, Oh SB. Common bacterial metabolite indole directly activates nociceptive neuron through transient receptor potential ankyrin 1 channel. Pain 2021. [DOI] [PubMed] [Google Scholar]
- [26].Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y, Zhang S, Ho J, Davis BM, Albers KM, Kaplan DH. Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 2019;178(4):919–932 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Danser AH, Anand P. The angiotensin II type 2 receptor for pain control. Cell 2014;157(7):1504–1506. [DOI] [PubMed] [Google Scholar]
- [28].Darios F, Niranjan D, Ferrari E, Zhang F, Soloviev M, Rummel A, Bigalke H, Suckling J, Ushkaryov Y, Naumenko N, Shakirzyanova A, Giniatullin R, Maywood E, Hastings M, Binz T, Davletov B. SNARE tagging allows stepwise assembly of a multimodular medicinal toxin. Proc Natl Acad Sci U S A 2010;107(42):18197–18201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].De Jonge F, Van Nassauw L, Adriaensen D, Van Meir F, Miller HR, Van Marck E, Timmermans JP. Effect of intestinal inflammation on capsaicin-sensitive afferents in the ileum of Schistosoma mansoni-infected mice. Histochem Cell Biol 2003;119(6):477–484. [DOI] [PubMed] [Google Scholar]
- [30].Deng L, Chiu IM. Microbes and pain. PLoS Pathog 2021;17(4):e1009398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res 2011;90(6):759–764. [DOI] [PubMed] [Google Scholar]
- [32].Donnelly CR, Chen O, Ji RR. How Do Sensory Neurons Sense Danger Signals? Trends Neurosci 2020;43(10):822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998;280(5364):734–737. [DOI] [PubMed] [Google Scholar]
- [34].Ebenezer GJ, Scollard DM. Treatment and Evaluation Advances in Leprosy Neuropathy. Neurotherapeutics 2021;18(4):2337–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Escher CM, Paracka L, Dressler D, Kollewe K. Botulinum toxin in the management of chronic migraine: clinical evidence and experience. Ther Adv Neurol Disord 2017;10(2):127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Falsetta ML, Foster DC, Woeller CF, Pollock SJ, Bonham AD, Haidaris CG, Stodgell CJ, Phipps RP. Identification of novel mechanisms involved in generating localized vulvodynia pain. Am J Obstet Gynecol 2015;213(1):38 e31–38 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fan C, Chu X, Wang L, Shi H, Li T. Botulinum toxin type A reduces TRPV1 expression in the dorsal root ganglion in rats with adjuvant-arthritis pain. Toxicon 2017;133:116–122. [DOI] [PubMed] [Google Scholar]
- [38].Farmer MA, Taylor AM, Bailey AL, Tuttle AH, MacIntyre LC, Milagrosa ZE, Crissman HP, Bennett GJ, Ribeiro-da-Silva A, Binik YM, Mogil JS. Repeated vulvovaginal fungal infections cause persistent pain in a mouse model of vulvodynia. Sci Transl Med 2011;3(101):101ra191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fields HL, Rowbotham M, Baron R. Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiol Dis 1998;5(4):209–227. [DOI] [PubMed] [Google Scholar]
- [40].Filtjens J, Roger A, Quatrini L, Wieduwild E, Gouilly J, Hoeffel G, Rossignol R, Daher C, Debroas G, Henri S, Jones CM, Malissen B, Mackay LK, Moqrich A, Carbone FR, Ugolini S. Nociceptive sensory neurons promote CD8 T cell responses to HSV-1 infection. Nat Commun 2021;12(1):2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Garcia-Monco JC, Benach JL. A disconnect between the neurospirochetoses in humans and rodent models of disease. PLoS Pathog 2013;9(4):e1003288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gershon AA, Breuer J, Cohen JI, Cohrs RJ, Gershon MD, Gilden D, Grose C, Hambleton S, Kennedy PG, Oxman MN, Seward JF, Yamanishi K. Varicella zoster virus infection. Nat Rev Dis Primers 2015;1:15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Guerrero AT, Cunha TM, Verri WA Jr., Gazzinelli RT, Teixeira MM, Cunha FQ, Ferreira SH. Toll-like receptor 2/MyD88 signaling mediates zymosan-induced joint hypernociception in mice: participation of TNF-alpha, IL-1beta and CXCL1/KC. Eur J Pharmacol 2012;674(1):51–57. [DOI] [PubMed] [Google Scholar]
- [44].Hamilton SM, Bayer CR, Stevens DL, Bryant AE. Effects of selective and nonselective nonsteroidal anti-inflammatory drugs on antibiotic efficacy of experimental group A streptococcal myonecrosis. J Infect Dis 2014;209(9):1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001;410(6832):1099–1103. [DOI] [PubMed] [Google Scholar]
- [46].Hong B, Yao L, Ni L, Wang L, Hu X. Antinociceptive effect of botulinum toxin A involves alterations in AMPA receptor expression and glutamate release in spinal dorsal horn neurons. Neuroscience 2017;357:197–207. [DOI] [PubMed] [Google Scholar]
- [47].Hou L, Wang X. PKC and PKA, but not PKG mediate LPS-induced CGRP release and [Ca(2+)](i) elevation in DRG neurons of neonatal rats. J Neurosci Res 2001;66(4):592–600. [DOI] [PubMed] [Google Scholar]
- [48].Jia L, Lee S, Tierney JA, Elmquist JK, Burton MD, Gautron L. TLR4 Signaling Selectively and Directly Promotes CGRP Release from Vagal Afferents in the Mouse. eNeuro 2021;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kanaan SA, Saade NE, Karam M, Khansa H, Jabbur SJ, Jurjus AR. Hyperalgesia and upregulation of cytokines and nerve growth factor by cutaneous leishmaniasis in mice. Pain 2000;85(3):477–482. [DOI] [PubMed] [Google Scholar]
- [50].Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015;43(3):515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lagomarsino VN, Kostic AD, Chiu IM. Mechanisms of microbial-neuronal interactions in pain and nociception. Neurobiol Pain 2021;9:100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lampert A, Bennett DL, McDermott LA, Neureiter A, Eberhardt E, Winner B, Zenke M. Human sensory neurons derived from pluripotent stem cells for disease modelling and personalized medicine. Neurobiol Pain 2020;8:100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Levine JD, Khasar SG, Green PG. Neurogenic inflammation and arthritis. Ann N Y Acad Sci 2006;1069:155–167. [DOI] [PubMed] [Google Scholar]
- [54].Li F, Jiang H, Shen X, Yang W, Guo C, Wang Z, Xiao M, Cui L, Luo W, Kim BS, Chen Z, Huang AJW, Liu Q. Sneezing reflex is mediated by a peptidergic pathway from nose to brainstem. Cell 2021;184(14):3762–3773 e3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li X, Coffield JA. Structural and Functional Interactions between Transient Receptor Potential Vanilloid Subfamily 1 and Botulinum Neurotoxin Serotype A. PLoS One 2016;11(1):e0143024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Li Y, Adamek P, Zhang H, Tatsui CE, Rhines LD, Mrozkova P, Li Q, Kosturakis AK, Cassidy RM, Harrison DS, Cata JP, Sapire K, Zhang H, Kennamer-Chapman RM, Jawad AB, Ghetti A, Yan J, Palecek J, Dougherty PM. The Cancer Chemotherapeutic Paclitaxel Increases Human and Rodent Sensory Neuron Responses to TRPV1 by Activation of TLR4. J Neurosci 2015;35(39):13487–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lin T, Quellier D, Lamb J, Voisin T, Baral P, Bock F, Schonberg A, Mirchev R, Pier G, Chiu I, Gadjeva M. Pseudomonas aeruginosa-induced nociceptor activation increases susceptibility to infection. PLoS Pathog 2021;17(5):e1009557. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [58].Lochhead RB, Strle K, Arvikar SL, Weis JJ, Steere AC. Lyme arthritis: linking infection, inflammation and autoimmunity. Nat Rev Rheumatol 2021;17(8):449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Maiaru M, Leese C, Certo M, Echeverria-Altuna I, Mangione AS, Arsenault J, Davletov B, Hunt SP. Selective neuronal silencing using synthetic botulinum molecules alleviates chronic pain in mice. Sci Transl Med 2018;10(450). [DOI] [PubMed] [Google Scholar]
- [60].Mangione AS, Obara I, Maiaru M, Geranton SM, Tassorelli C, Ferrari E, Leese C, Davletov B, Hunt SP. Nonparalytic botulinum molecules for the control of pain. Pain 2016;157(5):1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Marin JC, Gantenbein AR, Paemeleire K, Kaube H, Goadsby PJ. Nociception-specific blink reflex: pharmacology in healthy volunteers. J Headache Pain 2015;16:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Marion E, Song OR, Christophe T, Babonneau J, Fenistein D, Eyer J, Letournel F, Henrion D, Clere N, Paille V, Guerineau NC, Saint Andre JP, Gersbach P, Altmann KH, Stinear TP, Comoglio Y, Sandoz G, Preisser L, Delneste Y, Yeramian E, Marsollier L, Brodin P. Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 2014;157(7):1565–1576. [DOI] [PubMed] [Google Scholar]
- [63].Maruyama K, Takayama Y, Sugisawa E, Yamanoi Y, Yokawa T, Kondo T, Ishibashi KI, Sahoo BR, Takemura N, Mori Y, Kanemaru H, Kumagai Y, Martino MM, Yoshioka Y, Nishijo H, Tanaka H, Sasaki A, Ohno N, Iwakura Y, Moriyama Y, Nomura M, Akira S, Tominaga M. The ATP Transporter VNUT Mediates Induction of Dectin-1-Triggered Candida Nociception. iScience 2018;6:306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Matak I, Tekus V, Bolcskei K, Lackovic Z, Helyes Z. Involvement of substance P in the antinociceptive effect of botulinum toxin type A: Evidence from knockout mice. Neuroscience 2017;358:137–145. [DOI] [PubMed] [Google Scholar]
- [65].McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, Clark AJ, Millar V, Peck LJ, Ebner D, Tracey I, Serra J, Bennett DL. Defining the Functional Role of NaV1.7 in Human Nociception. Neuron 2019;101(5):905–919 e908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998;393(6683):333–339. [DOI] [PubMed] [Google Scholar]
- [67].Inflammation Medzhitov R. 2010: new adventures of an old flame. Cell 2010;140(6):771–776. [DOI] [PubMed] [Google Scholar]
- [68].Meseguer V, Alpizar YA, Luis E, Tajada S, Denlinger B, Fajardo O, Manenschijn JA, Fernandez-Pena C, Talavera A, Kichko T, Navia B, Sanchez A, Senaris R, Reeh P, Perez-Garcia MT, Lopez-Lopez JR, Voets T, Belmonte C, Talavera K, Viana F. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun 2014;5:3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Middleton SJ, Barry AM, Comini M, Li Y, Ray PR, Shiers S, Themistocleous AC, Uhelski ML, Yang X, Dougherty PM, Price TJ, Bennett DL. Studying human nociceptors: from fundamentals to clinic. Brain 2021;144(5):1312–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Millet I, Phillips RJ, Sherwin RS, Ghosh S, Voll RE, Flavell RA, Vignery A, Rincon M. Inhibition of NF-kappaB activity and enhancement of apoptosis by the neuropeptide calcitonin gene-related peptide. J Biol Chem 2000;275(20):15114–15121. [DOI] [PubMed] [Google Scholar]
- [71].Min H, Cho WH, Lee H, Choi B, Kim YJ, Lee HK, Joo Y, Jung SJ, Choi SY, Lee S, Lee SJ. Association of TRPV1 and TLR4 through the TIR domain potentiates TRPV1 activity by blocking activation-induced desensitization. Mol Pain 2018;14:1744806918812636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet 2008;42:541–564. [DOI] [PubMed] [Google Scholar]
- [73].Patil S, Willett O, Thompkins T, Hermann R, Ramanathan S, Cornett EM, Fox CJ, Kaye AD. Botulinum Toxin: Pharmacology and Therapeutic Roles in Pain States. Curr Pain Headache Rep 2016;20(3):15. [DOI] [PubMed] [Google Scholar]
- [74].Peeling RW, Hook EW, 3rd. The pathogenesis of syphilis: the Great Mimicker, revisited. J Pathol 2006;208(2):224–232. [DOI] [PubMed] [Google Scholar]
- [75].Perez-Berezo T, Pujo J, Martin P, Le Faouder P, Galano JM, Guy A, Knauf C, Tabet JC, Tronnet S, Barreau F, Heuillet M, Dietrich G, Bertrand-Michel J, Durand T, Oswald E, Cenac N. Identification of an analgesic lipopeptide produced by the probiotic Escherichia coli strain Nissle 1917. Nat Commun 2017;8(1):1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Pinho-Ribeiro FA, Baddal B, Haarsma R, O’Seaghdha M, Yang NJ, Blake KJ, Portley M, Verri WA, Dale JB, Wessels MR, Chiu IM. Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 2018;173(5):1083–1097 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pinho-Ribeiro FA, Chiu IM. Nociceptor nerves set the stage for skin immunity. Cell Res 2019;29(11):877–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pinho-Ribeiro FA, Verri WA Jr., Chiu IM. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol 2017;38(1):5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pulakat L, Sumners C. Angiotensin Type 2 Receptors: Painful, or Not? Front Pharmacol 2020;11:571994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Reichelt M, Zerboni L, Arvin AM. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J Virol 2008;82(8):3971–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 2016;16(6):341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rossetto O, Pirazzini M, Montecucco C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol 2014;12(8):535–549. [DOI] [PubMed] [Google Scholar]
- [83].Rudick CN, Billips BK, Pavlov VI, Yaggie RE, Schaeffer AJ, Klumpp DJ. Host-pathogen interactions mediating pain of urinary tract infection. J Infect Dis 2010;201(8):1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ruhl CR, Pasko BL, Khan HS, Kindt LM, Stamm CE, Franco LH, Hsia CC, Zhou M, Davis CR, Qin T, Gautron L, Burton MD, Mejia GL, Naik DK, Dussor G, Price TJ, Shiloh MU. Mycobacterium tuberculosis Sulfolipid-1 Activates Nociceptive Neurons and Induces Cough. Cell 2020;181(2):293–305 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Samavati L, Uhal BD. ACE2, Much More Than Just a Receptor for SARS-COV-2. Front Cell Infect Microbiol 2020;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shepherd AJ, Mickle AD, Golden JP, Mack MR, Halabi CM, de Kloet AD, Samineni VK, Kim BS, Krause EG, Gereau RWt, Mohapatra DP. Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Proc Natl Acad Sci U S A 2018;115(34):E8057–E8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Shiers S, Ray PR, Wangzhou A, Sankaranarayanan I, Tatsui CE, Rhines LD, Li Y, Uhelski ML, Dougherty PM, Price TJ. ACE2 and SCARF expression in human dorsal root ganglion nociceptors: implications for SARS-CoV-2 virus neurological effects. Pain 2020;161(11):2494–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Silva JR, Lopes AH, Talbot J, Cecilio NT, Rossato MF, Silva RL, Souza GR, Silva CR, Lucas G, Fonseca BA, Arruda E, Alves-Filho JC, Cunha FQ, Cunha TM. Neuroimmune-Glia Interactions in the Sensory Ganglia Account for the Development of Acute Herpetic Neuralgia. J Neurosci 2017;37(27):6408–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Silver AC, Dunne DW, Zeiss CJ, Bockenstedt LK, Radolf JD, Salazar JC, Fikrig E. MyD88 deficiency markedly worsens tissue inflammation and bacterial clearance in mice infected with Treponema pallidum, the agent of syphilis. PLoS One 2013;8(8):e71388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Simpson DM, Hallett M, Ashman EJ, Comella CL, Green MW, Gronseth GS, Armstrong MJ, Gloss D, Potrebic S, Jankovic J, Karp BP, Naumann M, So YT, Yablon SA. Practice guideline update summary: Botulinum neurotoxin for the treatment of blepharospasm, cervical dystonia, adult spasticity, and headache: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016;86(19):1818–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sorkin LS, Eddinger KA, Woller SA, Yaksh TL. Origins of antidromic activity in sensory afferent fibers and neurogenic inflammation. Semin Immunopathol 2018;40(3):237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sorrell TC, Chen SC. Fungal-derived immune modulating molecules. Adv Exp Med Biol 2009;666:108–120. [DOI] [PubMed] [Google Scholar]
- [93].Southgate EL, He RL, Gao JL, Murphy PM, Nanamori M, Ye RD. Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J Immunol 2008;181(2):1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Staurengo-Ferrari L, Trevelin SC, Fattori V, Nascimento DC, de Lima KA, Pelayo JS, Figueiredo F, Casagrande R, Fukada SY, Teixeira MM, Cunha TM, Liew FY, Oliveira RD, Louzada-Junior P, Cunha FQ, Alves-Filho JC, Verri WA. Interleukin-33 Receptor (ST2) Deficiency Improves the Outcome of Staphylococcus aureus-Induced Septic Arthritis. Front Immunol 2018;9:962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sweeney DA, Hicks CW, Cui X, Li Y, Eichacker PQ. Anthrax infection. Am J Respir Crit Care Med 2011;184(12):1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Taghavi M, Khosravi A, Mortaz E, Nikaein D, Athari SS. Role of pathogen-associated molecular patterns (PAMPS) in immune responses to fungal infections. Eur J Pharmacol 2017;808:8–13. [DOI] [PubMed] [Google Scholar]
- [97].Takasaki I, Andoh T, Shiraki K, Kuraishi Y. Allodynia and hyperalgesia induced by herpes simplex virus type-1 infection in mice. Pain 2000;86(1–2):95–101. [DOI] [PubMed] [Google Scholar]
- [98].Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999;11(4):443–451. [DOI] [PubMed] [Google Scholar]
- [99].Talbot S, Foster SL, Woolf CJ. Neuroimmunity: Physiology and Pathology. Annu Rev Immunol 2016;34:421–447. [DOI] [PubMed] [Google Scholar]
- [100].Tan PH, Ji J, Yeh CC, Ji RR. Interferons in Pain and Infections: Emerging Roles in Neuro-Immune and Neuro-Glial Interactions. Front Immunol 2021;12:783725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015;18(1):145–153. [DOI] [PubMed] [Google Scholar]
- [102].Wang Y, Cheng LI, Helfer DR, Ashbaugh AG, Miller RJ, Tzomides AJ, Thompson JM, Ortines RV, Tsai AS, Liu H, Dillen CA, Archer NK, Cohen TS, Tkaczyk C, Stover CK, Sellman BR, Miller LS. Mouse model of hematogenous implant-related Staphylococcus aureus biofilm infection reveals therapeutic targets. Proc Natl Acad Sci U S A 2017;114(26):E5094–E5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wilson AC, Mohr I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 2012;20(12):604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Xu ZZ, Kim YH, Bang S, Zhang Y, Berta T, Wang F, Oh SB, Ji RR. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat Med 2015;21(11):1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yang NJ, Isensee J, Neel DV, Quadros AU, Zhang HB, Lauzadis J, Liu SM, Shiers S, Belu A, Palan S, Marlin S, Maignel J, Kennedy-Curran A, Tong VS, Moayeri M, Roderer P, Nitzsche A, Lu M, Pentelute BL, Brustle O, Tripathi V, Foster KA, Price TJ, Collier RJ, Leppla SH, Puopolo M, Bean BP, Cunha TM, Hucho T, Chiu IM. Anthrax toxins regulate pain signaling and can deliver molecular cargoes into ANTXR2(+) DRG sensory neurons. Nat Neurosci 2022;25(2):168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yang NJ, Neel DV, Deng L, Heyang M, Kennedy-Curran A, Tong VS, Park JM, Chiu IM. Nociceptive Sensory Neurons Mediate Inflammation Induced by Bacillus Anthracis Edema Toxin. Front Immunol 2021;12:642373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S. Molecular Architecture of the Mouse Nervous System. Cell 2018;174(4):999–1014 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zenk SF, Jantsch J, Hensel M. Role of Salmonella enterica lipopolysaccharide in activation of dendritic cell functions and bacterial containment. J Immunol 2009;183(4):2697–2707. [DOI] [PubMed] [Google Scholar]
- [109].Zhang Q, Hsia SC, Martin-Caraballo M. Regulation of T-type Ca(2+) channel expression by interleukin-6 in sensory-like ND7/23 cells post-herpes simplex virus (HSV-1) infection. J Neurochem 2019;151(2):238–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhang Y, Yan H, Li X, Zhou D, Zhong M, Yang J, Zhao B, Fan X, Fan J, Shu J, Lu M, Jin X, Zhang E, Yan H. A high-dose inoculum size results in persistent viral infection and arthritis in mice infected with chikungunya virus. PLoS Negl Trop Dis 2022;16(1):e0010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zheng Y, Liu P, Bai L, Trimmer JS, Bean BP, Ginty DD. Deep Sequencing of Somatosensory Neurons Reveals Molecular Determinants of Intrinsic Physiological Properties. Neuron 2019;103(4):598–616 e597. [DOI] [PMC free article] [PubMed] [Google Scholar]