

Abstract
BACKGROUND:
Traumatic brain injury (TBI) is an underrecognized public health threat. The constitutive activation of microglia after TBI has been linked to long-term neurocognitive deficits and the progression of neurodegenerative disease. Evolving evidence indicates a critical role for the gut-brain axis in this process. Specifically, TBI has been shown to induce the depletion of commensal gut bacteria. The resulting gut dysbiosis is associated with neuroinflammation and disease.
HYPOTHESIS:
We hypothesized that fecal microbiota transplantation would attenuate microglial activation and improve neuropathology after TBI.
METHODS:
C57Bl/6 mice were subjected to severe TBI (n=10) or sham-injury (n=10) via an open-head controlled cortical impact. The mice underwent fecal microbiota transplantation (FMT) or vehicle alone via oral gavage once weekly for four weeks post-injury. At 59 days post-TBI, mice underwent 3D, contrast-enhanced magnetic resonance imaging. Following imaging, mice were sacrificed, brains harvested at 60 DPI, and CD45+ cells isolated via florescence-activated cell sorting. cDNA libraries were prepared using the 10x Genomics Chromium Single Cell 3’ Reagent kit followed by sequencing on a HiSeq4000 instrument, and computational analysis was performed.
RESULTS:
Fecal microbiota transplantation resulted in a marked reduction of ventriculomegaly (p<0.002) and preservation of white matter connectivity at 59 days post-TBI (p<0.0001). Additionally, microglia from FMT-treated mice significantly reduced inflammatory gene expression and enriched pathways involving the heat shock response compared to mice treated with vehicle alone.
CONCLUSIONS:
We hypothesized that restoring gut microbial community structure via fecal microbiota transplantation would attenuate microglial activation and reduce neuropathology after TBI. Our data demonstrated significant preservation of cortical volume and white matter connectivity after an injury compared to mice treated with vehicle alone. This preservation of neuroanatomy after TBI was associated with a marked reduction in inflammatory gene expression within the microglia of FMT-treated mice. Microglia from FMT-treated mice enriched pathways in the heat shock response, which is known to play a neuroprotective role in TBI and other neurodegenerative disease processes.
Keywords: Traumatic Brain Injury, Fecal Microbiome Transplantation, Microbiome, Dysbiosis, Trauma, Controlled Cortical Impact, Behavior, Anxiety, Microglia, Transcriptome
Introduction
Traumatic brain injury (TBI) is an underrecognized public health threat, with over 10 million people afflicted globally annually (1, 2). Victims of TBI have a high rate of neurocognitive and neuropsychiatric morbidity due to the secondary ischemic, hypoxic, excitotoxic, and inflammatory processes that are hallmarks of TBI. In survivors, these mechanisms are linked to over 90,000 American disabilities each year, potentially progressing to chronic neurodegenerative disease (3). Consequently, TBI-related healthcare expenditures approach 80 billion dollars annually in the United States alone (4).
Traumatic brain injuries are heterogenous injury processes resulting in free radical generation, metabolic energy disturbance, neuronal excitotoxicity, and marked neuroinflammation. These processes have the potential to culminate in a gamut of behavioral, neurocognitive, and motor deficits (5, 6). Microglia, innate immune cells that are native to the brain under physiological conditions, play a critical role in the direction of the injury process (7). Likewise, emerging evidence has shown a strong relationship between the gut, the brain, and microglia via a bidirectional communication pathway known as the gut-brain axis. This bidirectionality is then capable of modulating the health of the other in either direction. This bidirectional influence between the gut and the brain has yet to be fully elucidated in TBI (8, 9). However, it is known that TBI results in increased gut-barrier permeability, systemic inflammation, and changes within the gut microbiome. These changes, in turn, can alter function within the brain (10–12). Indeed, preclinical evidence has demonstrated the modulatory effects of gut microbiota on the brain in “germ-free” models. In this model, the disease is studied in animals devoid of microorganisms, models in which microbial manipulation is accomplished with antibiotics, and models of fecal microbiota transplantation (13–16). These data suggest that the gut microbiota is mechanistically important for regulating several CNS processes, including TBI.
Our laboratory recently published work has shown a significant role of the gut-brain axis in the onset and progression of TBI-associated neurocognitive decline. Using our controlled cortical impact TBI model, we linked differences in TBI pathology and neurocognition to a divergent microbial community structure with both injury and age (17, 18). In follow-up work, we hypothesized that replacing a healthy microbiome through fecal microbiome transplantation (FMT) would attenuate neurocognitive deficits in TBI (Shock (19). We found that administering FMT in a clinically applicable post-injury model could restore gut microbial community structure. We also performed neurocognitive testing to assess the effect of FMT on TBI-mediated decreases in neurocognition. Remarkably, we found that FMT treatment also attenuated neurocognitive deficits after TBI in mice.
FMT is an accepted intervention in other disease processes. For example, FMT is now a widely accepted treatment for chronic Clostridium difficile colitis (20). Recent preclinical data has shown promising results, with FMT being used to combat neurologic deficits in stroke and spinal cord injury (21). Moreover, ongoing clinical trials utilizing FMT in human patients have shown promising early results in the autism spectrum of disorders, multiple sclerosis, Parkinson’s disease, epilepsy, Tourette syndrome, and diabetic neuropathy (22). Despite these early successes, the mechanisms underlying the beneficial effects of FMT have yet to be elucidated. Initial work from outside laboratories has suggested that FMT can function as an immunomodulator and confer protection against various disease processes linked to gut dysbiosis (23). Therefore, we hypothesized that fecal microbiota transplantation would attenuate microglial activation and improve neuropathology after TBI.
Methods
Experimental Design
A 2 × 2 experimental design was utilized with TBI and Sham injury groups treated with either fecal microbiota transfer or vehicle alone. Mice were randomly assigned to experimental groups. Experimental TBI or sham injury (n=20) was induced at 14 weeks old. An a priori power calculation was performed (Gpower 3.1), determining that five animals per experimental group were necessary to detect a 20% difference between groups (utilizing the conventional values of 0.05 and 0.2 for α and β). TBI was induced via an open head controlled cortical impact, as previously published (24). Two hours post-injury, sham injured and TBI mice underwent weekly oral gavage with either a slurry of healthy mouse stool or vehicle alone corresponding to their assigned group. Animal welfare, clinical applicability, and previous research were primary factors in the treatment regimen (25, 26). 3D, contrast-enhanced MRI was used to assess cortical volume loss and white matter connectivity at 59-days post-injury. Brains were then harvested during animal sacrifice for cell sorting and single-cell RNA sequencing (scRNA-seq) analysis. The duration of the study was 60 days. For more significant within- and between-groups comparison, all experimental investigations were performed, on the indicated days, in the same cohort of animals. At the start of the study animals were randomly assigned to the following 4 groups:
Sham (untreated, no injury)
TBI (Untreated, with injury)
FMT (Treated, No injury ((sham))
TBI-FMT (Treated, with injury)
Animals
C57BL/6 male mice (Mus musculus) (28–30 grams) were purchased from the Jackson Laboratory (Bar Harbor, Maine). Animals were delivered at 12 weeks of age and given a 2-week facility acclimation period before the start of the experiment. Bedding transfer and mixing were also performed upon arrival. Mice were housed and maintained in a pathogen-free barrier facility at the Northwestern University Center for Comparative Medicine. Standard chow (Harlan, Indianapolis, IN) and water were provided for ad libitum feeding. The animals were housed on a 12:12 light-dark cycle for the study duration. Mice were treated per the National Institutes of Health Guidelines for the Use of Laboratory Animals. All experimental protocols were approved by Northwestern University Institutional Animal Care and Use Committee.
Traumatic brain injury
Mice received anesthesia using an intraperitoneal injection of 125 mg/kg ketamine (Ketaset, Fort Dodge, IA) and 10 mg/kg xylazine (Anased, Shenandoah, IA). Following anesthesia, TBI mice received a 1-cm scalp incision to reveal the sagittal and coronal sutures of the skull. The injury site was marked 2 mm rostral to the coronal suture and 2 mm left of the sagittal suture. The brain’s impact area (5 mm diameter) was exposed via a craniectomy, leaving the dura mater intact. TBI mice were then stabilized within a stereotaxic operating frame. Impacts were delivered via a commercially available device (Impact One, Leica Biosystems, Des Planes IL), inducing a controlled cortical impact. A 3 mm diameter impacting rod was utilized. The rod impacted the brain at a velocity of 2.5 m/s, penetrating to a depth of 2 mm with a dwell time of 0.1 seconds. Sham mice underwent anesthesia and scalp incision alone. All scalp incisions were sealed with VetBond (3M) (Santa Cruz Animal Health, Dallas, TX) immediately following sham injury or TBI. Post-procedure analgesia with Buprenorphine SR (SR Veterinary Technologies, Windsor, CO) was administered to all animals via subcutaneous injection. Animals recovered in separate cages over a warming pad. Euthanasia of all mice was performed following AVMA guidelines. Mice were placed into a carbon dioxide chamber and euthanized by inhalation. The mice were then exsanguinated, followed by decapitation. Brains were harvested for analysis by single-cell RNA seq.
Fecal microbiota transfer
The stool was collected daily for two weeks from healthy, naïve, chow-fed donor mice (10-week-old, male), placed in 1mL cryogenic collection tubes, and flash-frozen in liquid nitrogen before −80C storage. The frozen stool was diluted (1g:1ml) in sterile room temperature water, homogenized, and mortared for each treatment through a 100μm mesh cell strainer to remove particulates. Mice were inoculated through oral gavage. The mice were given 200uL of stool slurry or sterile water vehicle control once weekly for four weeks. The first treatment was administered 2hrs post TBI.
Single-Cell Analysis
Tissue Processing for scRNA-seq
Samples were injected with 3mL of digestion buffer [2.5 mg/ml Liberase TL (Roche, Basel, Switzerland) and 1 mg/ml of DNase I in HBSS], morcellated, and placed into C-tubes (Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany) containing 1mL of the digestion buffer. C-tubes were placed on a MACS dissociator (Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany) and ran using the brain dissociation protocol. Samples were then incubated in a shaker at 200 rpm for 30 min at 37°C. Following incubation, the c-tubes were returned to the MACS dissociator per the manufacturer’s instructions. The heterogeneous tissue mixture was strained through a 40 μm nylon mesh strainer and washed with 100Ml of autoMACs Running Buffer (Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany) per brain. Microglia and infiltrating leukocytes were isolated using a 30/70 percoll gradient (Percoll Plus, GE Healthcare, Chicago, IL, USA). Microglia and infiltrating leukocytes were then collected from the interphase of the gradient and washed with HBSS. The cells were courted with a Countess automated cell counter (Invitrogen, Waltham, MA, USA). Cells were then stained with Live/Dead Aqua (Invitrogen, Waltham, MA, USA) viability dye, Fc-Block (BD Biosciences, San Jose, CA, USA), and fluorochrome-conjugated antibodies for CD45.1(A-20/BD Biosciences, San Jose, CA, USA) and CD11b (M1/70/BD Biosciences, San Jose, CA, USA). Cells were washed and prepared for sorting.
Fluorescent Activated Cell Sorting
Data were acquired, and microglia and infiltrating leukocytes were sorted on a BD FACSAria cell sorter (BD Biosciences, San Jose, CA, USA) 60-days post-TBI or sham surgery. Gates were established using “Fluorescence minus one” controls (Sup Fig. 1).
Library Preparation and Sequencing
Concentration and viability (>90%) were confirmed using K2 Cellometer (Nexcelom Bioscience LLC, Lawerence, MA, USA) with AO/PI reagent, and ~5,000–10,000 cells were loaded on 10x Genomics Chip A using Chromium Single Cell V3 Reagent Kit and Controller (10x Genomics, Pleasanton, CA, USA). Single-cell 3’ RNA-Seq libraries were prepared according to manufacturer protocol (10x Genomics, Pleasanton, CA, USA). Libraries were assessed for quality (TapeStation 4200, Agilent Technologies, Santa Carla, CA, USA) and then sequenced on HiSeq 4000 instrument (Illumina, San Diego, CA, USA), generating >25,000 read pairs/cell.
Preprocessing of scRNAseq data
Raw data were processed using the Cell Ranger, version 3.0 pipeline from 10x Genomics (Pleasanton, CA, USA). The reads were mapped to the mm10 mouse reference genome (Ensemble build 98). Individual sample expression matrices were loaded and read into R using the functions Read10x and CreateSeuratObject under the Seurat package version 4.0.3 from Satija Lab (https://satijalab.org/seurat/). The expression matrix for Sham, TBI, FMT sham, and FMT TBI were merged using the Merge function for assessing the quality metrics. As a result, cells from all groups expressing less than 1300 genes, less than 2800 unique molecular identifiers (UMIs), and more than 10% mitochondrial genes were excluded. After quality control, merged objects were explored for unwanted variation due to cell cycle phases and split using SplitObject. Doublets from each sample were removed using the Scrubblet package by Chris McGinnis(27). Normalization was performed using the SCTransform method to normalize, scale, select variable genes, and regress out mitochondrial mapping percentage (Seurat package version 4.0.3, Satija Lab). Correlation analysis (Supp Fig. 2) was conducted to test within-group variation as opposed to inter-group variation. Consequently, processing proceeded with no integration performed. Following PCA, 30 principal components were selected for unsupervised clustering of the cells. With unsupervised clustering, we annotated clusters using the SingleR package version 1.4.1 by Aaron Lun (28) and known markers for primary microglia and infiltrating immune cell types, retrieved from the Allen Brain Atlas, UCSC Cell Browser, PanglaoDB, Hammond, et al., Masuda et al., and Ochocka et al. (29–31). These markers were sufficient to define all major cell types. The R packages ggplot2 version 3.3.5 and Seurat version 4.0.3 were used to make the plots.
Analysis of scRNAseq data
The cluster of microglia was portioned using the Subset function in Seurat, followed by unsupervised clustering. Differential expression (DE) analysis was performed using the ”FindAllMarkers” function in Seurat version 4.0.3 with the Wilcoxon rank-sum test to identify differentially expressed genes of all the unsupervised clusters. Only genes expressed by at least 25% of cells and log2 fold change (FC) equal to and greater than 0.25 were included. Annotation of microglia clusters, i.e., microglia scoring (Fig. 2A and Fig. 2B), was performed based on their known functions and top expressed markers for each cluster (32). To compare inflammatory profiles between all four objects, we averaged the expression of SCT normalized data using the “AverageExpression” function from Seurat version 4.0.3, followed by comparing the expression of known and top-expressed inflammatory markers between all groups. A comparison of inflammatory markers between four groups was plotted using “DoHeatmap” from Seurat version 4.0.3. To identify sample-specific upregulated genes in microglia from FMT-treated TBI mice and TBI only mice, we firstly extracted genes highly upregulated in microglial cells, using the same parameters as in the DE analysis for microglia, from FMT TBI brain (significantly upregulated genes in FMT TBI compared to FMT sham) as well as genes highly upregulated in microglial cells from TBI brain (significantly upregulated genes in TBI compared to sham). Mitochondrial and ribosomal genes were filtered out. Subsequently, we compared those profiles in FMT TBI and TBI only microglia to find common or specific genes for each sample. Comparison of upregulated genes between FMT TBI and TBI only was plotted using ggplot2 packages with selected top-expressed genes labeled. Downstream gene ontology (GO) enrichment analysis was performed using Gorilla (33) with all genes within the cell type in the dataset as a background. The false discovery rate (FDR) was also calculated to correct for multiple testing. Enriched ontology terms for biological and functional processes were plotted using ggplot2 packages displaying FDR.q.values and enrichment scores.
Figure 2. FMT treatment prevents the expansion of the proportion of T cells after traumatic brain injury in mice.

Immune cells were isolated from mouse groups using Fluorescent Activated Cell Sorting and assessed via single-cell RNA sequencing for cell type. Immune cell proportions are shown here for all groups: Sham (untreated, no injury), TBI (Untreated, with injury), FMT (Treated, No injury ((sham)), TBI-FMT (Treated, with injury). A) UMAP plot demonstrating clustering obtained for each group (Sham, TBI, FMT, FMT-TBI) two biological replicates were combined. Cluster annotations: MG (microglia), T (T-cells) B (B-cells) NK (natural killer cells), NP (neutrophils), Mo_MΦ (monocytes/monocytes-derived macrophages), EC (epithelial cells). Number of total cells obtained from each group: Sham=3952, TBI=5570, FMT-Sham=3286, FMT-TBI=5832. B) Pie charts demonstrating the proportion of the identified cell types across samples. FMT mitigated the expansion in the proportion of T cells within the brain after TBI. C) Dim plots showing markers that identify each cell type. Average expression and percent expression are indicated by dot size.
Statistical Analyses
Data are presented as mean ± SEM. Group means were compared by analysis of variance (ANOVA) and Tukey post hoc analysis. Significance was set at α < 0.05. ((p ≤ 0.05(*), 0.001(**), & 0.0001(***)). All analyses were conducted using GraphPad Prism (GraphPad, La Jola, Ca, version 6).
Results
To determine whether fecal microbiome transfer would attenuate neurocognitive, anatomic, and pathologic outcomes after TBI, sham and TBI mice were administered FMT or vehicle control via weekly oral gavage. To maintain clinical relevance, treatment started 2 hours post-TBI. 3D contrast-enhanced magnetic resonance imaging and single-cell RNA sequencing analyses were performed 59 and 60-days post-injury (DPI), respectively.
MRI
We used ventricle size as a surrogate for cortical volume loss per our previously published work (Figure 1A) (34). At 59 DPI, 3D imaging revealed significant ventriculomegaly in vehicle-treated TBI mice. This ventriculomegaly was markedly attenuated in mice treated with post-injury FMT (p<0.002). Figure 1(a–b) shows a plot of average ventricle size for each group. Average ventricle size is consistent with an increase in relative preserved cortical volume in FMT treated TBI mice.
Figure 1. FMT attenuates cortical volume loss and preserves white matter connectivity after TBI.

A) Representative MRI scans of animals (60DPI) with injury cerebrospinal fluid (CSF) ventricles are denoted in yellow for all groups: Sham (untreated, no injury), TBI (Untreated, with injury), FMT (Treated, No injury ((sham)), TBI-FMT (Treated, with injury). (N=3) B) Representative longitudinal and transverse images reveal the pattern of whole-brain fractional anisotropy. C) Mean head size of animals 60 days post-TBI reveals a significant effect of injury (p<0.0001) and the interaction between FMT and injury (p<0.04). D) Fluid diffusion highlights enlargement of ventricles with TBI. E) Fractional anisotropy (connectivity) data was extracted from MRI. FMT attenuates TBI-induced ventriculomegaly, a well-described surrogate for cortical volume loss (p<0.002). In addition, TBI induced a decrease in fractional anisotropy (white matter connectivity) compared to sham injury (p<0.0001). FMT treatment attenuated this loss of connectivity compared to vehicle-treated TBI mice (p=0.04).
Additionally, fractional anisotropy maps were generated using the relative degree of diffusion, and we compared the modified fractional anisotropy (FA) above a threshold. This measurement is an accepted surrogate for overall white-matter connectivity, as our laboratory has previously published (34). Data revealed increased connectivity (FA) in FMT treated TBI mice versus vehicle-treated TBI mice (p=0.039) (Fig 1c–d). These anatomic data indicate a neuroprotective effect of FMT after TBI.
Single Cell RNA Sequencing Analyses
To characterize the role of immune cells in FMT treatment after TBI, both resident and infiltrating immune cells were sorted from mouse brains (n=8 total, two pooled mice per group) (14-weeks at the time of TBI). Animals were 22 weeks at the time of sacrifice. 14,247 cells from 8 mice were sequenced: 7,760 cells from young mice undergoing sham surgery (sham), 9,582 cells from young mice undergoing TBI (TBI), 4,919 cells from FMT treated mice undergoing sham surgery (FMT sham), and 9,221 cells from FMT treated mice undergoing TBI (FMT TBI). After QC, as described in the methods, we had 3,952 cells in the sham, 5,570 cells in the TBI, 3,286 cells in the FMT sham, and 5,832 cells in the FMT TBI. Arranging the data into a visual Uniform Manifold Approximation and Projection (UMAP), we observed the transcriptomic distribution of computed clusters in two dimensions (Fig. 2A). To characterize the cell identity of the computed clusters, we utilized the SingleR package, and the immune cell markers (Fig. 2B) curated from the literature to annotate individual cell identities. Most collected cells were identified as microglia (MG) and T cells (T) (Fig. 2B). Minor cell populations included B cells (B), natural killer cells (NK), monocytes (Mo), monocyte-derived macrophages (MΦ), neutrophils (NP), and epithelial cells (EC).
Overall, we identified a notable disparity between sham mice and TBI mice in their immune response post brain injury. Specifically, TBI mouse brains demonstrated an increased proportion of infiltrating immune cells, including T-cells, B-cells, and NK-cells, as compared to the brains of sham mice (T-cells: 24% vs. 11.0%.; B-cells: 7.0% vs. 5.0%; NK-cells: 4.5% vs. 4.0%) (Fig. 2B). While FMT treatment had little effect on the total number of immune cells that invaded the brain, there was a marked treatment effect in the proportion of T-cells after TBI (18.0% vs. 24%) (Fig. 2B). Additionally, the percentage of microglia demonstrated a differential response to injury between relevant groups. Post-injury, untreated mice had a lower proportion of microglia (MG: 75.0% vs. 59.5%) than sham-injured mice due to the increased proportion of infiltrating cells. In contrast, FMT-treated groups maintained their relative ratio of microglia post-injury (MG: 62% vs. 59.5%) while mitigating the increase in infiltrating T-cells compared to vehicle-treated TBI mice (18.0% vs. 24%). These results show a change in the overall immune cell composition after TBI with FMT treatment.
Following the annotation of resident and infiltrating immune cell populations within the brain, scRNA-seq data was sub-divided to analyze microglia according to function using top-expressed genes generated from the DE test as identifiers. According to the literature, we identified subclusters of microglia that expressed genes linked to specific functions. Microglia were grouped by gene expression according to function, revealing predominant microglia phenotypes within samples. The dominant phenotypes included microglia expressing Homeostatic (HMG), Disease-associated (DAM), Inflammatory, or Heat-shock (hspMG) related genes (Fig. 3A). These data reveal an increased proportion of DAM genes (Lpl, Cst7, & Apoe) in both TBI groups compared to sham (Fig. 3B). Differential expression analysis also demonstrated that FMT treatment increased the proportion of heat shock genes (Hsp1a, Hsp1b, & Hspb1) at the expense of inflammatory genes (Cd3, Ccl4, Il1a, TNF) in sham, a phenotype that was amplified post-TBI.
Figure 3. FMT administration alters microglial gene expression after sham and brain injury.
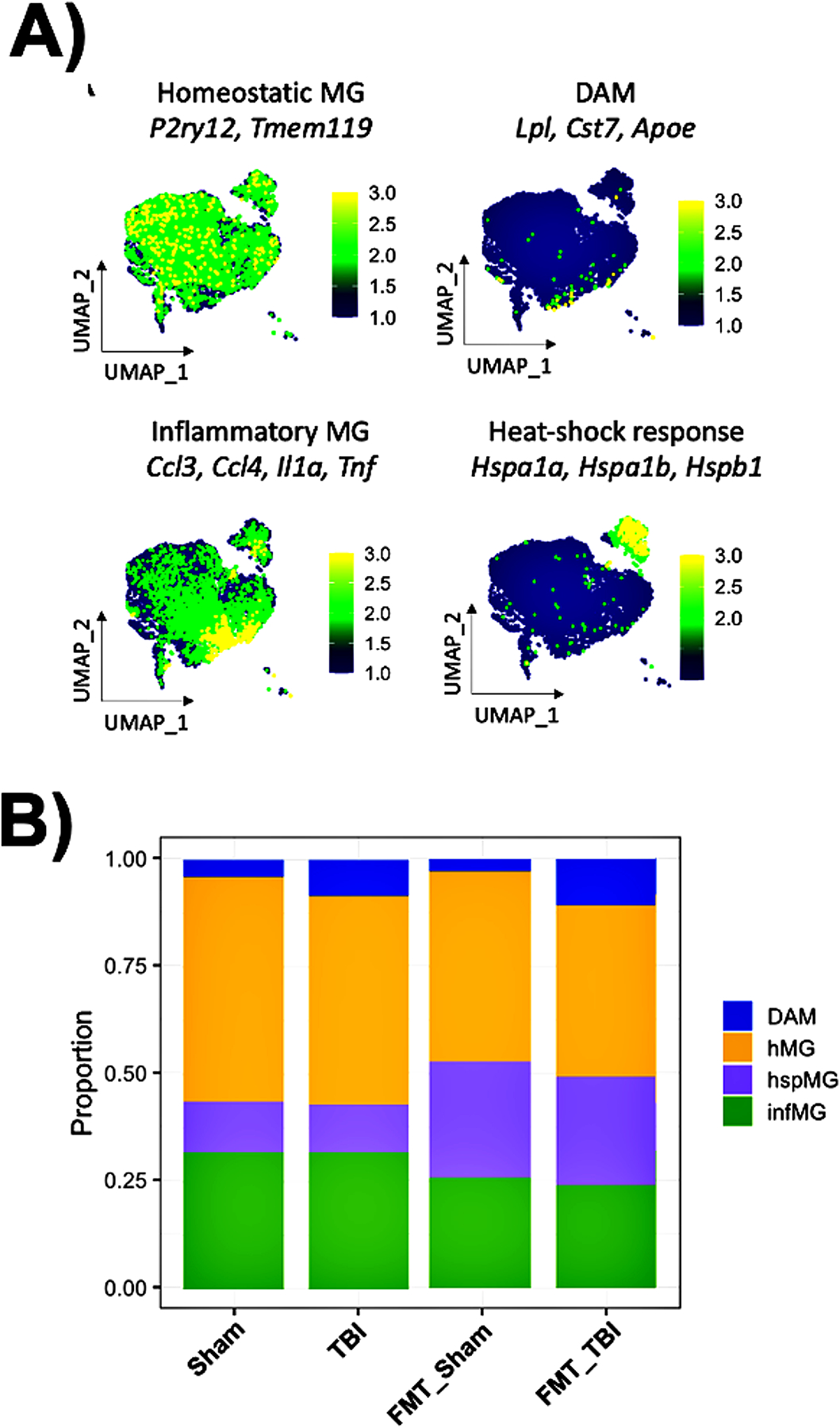
A) Microglia were grouped by gene expression according to function, revealing predominant microglia phenotypes within samples for all groups: Sham (untreated, no injury), TBI (Untreated, with injury), FMT (Treated, No injury ((sham)), TBI-FMT (Treated, with injury). The predominant phenotypes included microglia expressing 1. Homeostatic (HMG), 2. Disease-associated (DAM), 3. Inflammatory (INMG) and 4. Heat-shock (hspMG) related genes. B) Stacked bar charts demonstrating the proportion of DAM and hspMG microglia subclusters in injury and FMT, respectively. The microglia found within samples: FMT=2046, FMT_TBI=3435, Sham=2941, and TBI=3093.
Specific interrogation of inflammatory genes was conducted to address this work’s central hypothesis that FMT alters inflammation (Fig. 4). This subcluster presented data from highly expressed inflammation-linked gene signatures (Il1a, Il2b, Ccl3, Ccl4, Ccl2, Cd83, Ler3, Egr1, Tnf, Nfkbia, Il1b, & lnfgr1) within related microglia. Compared to sham, TBI mice demonstrated a notable increase in most inflammatory genes (Il2b, Ccl4, Ccl2, Egr1 Nfkbia, & Il1b). Ccl3, Cd83, & lnfgr1 decreased with injury in vehicle-treated animals, while IL1a & TNF remained unchanged compared to the sham group. Overall, untreated sham vs. TBI animals confirms an inflammatory microglial response within our mouse CCI model of TBI. Comparing FMT-treated sham mice to baseline, we saw an overall decrease in most inflammatory markers (Ccl3, Ccl4, Cd83, Ler3, Tnf). With FMT treatment alone, few inflammatory genes increased (Ccl2, Efr1, Nfrkbia, & Il1b) while the remaining genes remained unchanged. Finally, vehicle-treated TBI mice vs. the FMT-treated TBI group was the comparison that revealed the highest level of differential gene expression with nearly all predominant genes (Il1a, Il2b, Ccl3, Ccl4, Cd83, Ler3, Egr1, Tnf, Nfkbia, Il1b, & lnfgr1) showing FMT attenuated inflammatory gene expression with only Il1b displaying an increase due to FMT in TBI. Taken together these data indicate that FMT treatment alters the neuroinflammatory response with a marked decrease in the expression of several inflammatory genes not only at baseline but with an even greater downregulation after injury suggesting a significant anti-inflammatory effect of FMT.
Figure 4. Heatmap demonstrates a marked downregulation of inflammatory gene expression after FMT, further amplified after traumatic brain injury.

TBI results in a significant upregulation of inflammatory gene expression in microglia. Groups: Sham (untreated, no injury), TBI (Untreated, with injury), FMT (Treated, No injury ((sham)), TBI-FMT (Treated, with injury). Treatment with FMT markedly decreased inflammatory gene expression within the microglia of mice at baseline (sham-injury). This downregulation of inflammatory gene expression was further amplified after TBI.
Discussion
A growing body of evidence demonstrates the importance of the gut microbiome in the brain’s health, disease, and injury (35). After TBI, several physiologic and pathophysiologic processes within the brain-gut axis ensue resulting in a bidirectional inflammatory cascade between the gut and the brain (36). This inflammatory cascade leads to a breakdown of the gut mucosal barrier, and gut dysmotility, and generates dysbiosis within the gut microbial community. In turn, gut dysbiosis can propel the index brain injury via neural, humoral, and metabolic mechanisms that are only beginning to be discovered (35). This bidirectional feedback loop between the gut microbial community and the brain then has the potential to either propel and propagate the index injury or set up a microenvironment facilitating repair and regeneration (17–19, 36). Our laboratory recently published that restoring a pre-injury gut microbial community structure after TBI can mitigate or even ameliorate several cognitive, anatomic, and pathologic deficits seen after brain injury. These findings indicate that the gut microbial community may represent an untapped therapeutic tool after TBI (19). Our prior work showed that a severe TBI reduces alpha diversity, increases unfavorable microbial taxa, and decreases favorable microbial taxa. These changes were associated with marked deficits in learning, memory, and anxiety-like behaviors. However, we found that many of the TBI-induced neurocognitive deficits were preserved by restoring the gut microbial community structure to a pre-injury state via fecal microbiota transfer (FMT). Furthermore, we found that FMT decreased lesion size and maintained white matter connectivity between brain regions. Nonetheless, the mechanisms driving these findings remain opaque.
To begin unveiling the molecular mechanisms underlying the therapeutic effects of FMT, we hypothesized that FMT would attenuate neuroinflammation and limit the scope of neurological injury after TBI. We treated mice with FMT after TBI or sham injury to test this hypothesis. We then assessed brains for signs of attenuated post-injury neuroinflammation at an anatomic, cellular, and transcriptional level. Quantitative MRI of TBI mice in our current and prior studies shows a marked reduction of cortical volume loss and significant preservation of white matter connectivity in mice treated post-injury with FMT (Fig. 1)(19). In addition, neuropathologic analysis of these FMT-treated brains demonstrated increased levels of Iba-1 staining, indicating increased activation and/or many microglia in the FMT treated groups compared to vehicle-treated mice. Combined with the imaging analyses, we surmised that FMT might alter the inflammatory profile of microglia via the gut-brain axis, thereby mitigating secondary brain injury. To further interrogate this possibility, we isolated immune cells from the brain and performed single-cell RNA sequencing to determine the overall proportion of immune cells within the brain and the transcriptional signature of microglia in TBI mice treated with FMT or control.
After TBI, we noted a marked increase in infiltrating immune cell populations. We recorded a doubling in the proportion of T cells within the injured brain (Fig. 2). Indeed, T cells have been identified as a principal cell type generating secondary brain injury in stroke (37). T cell infiltration has also been noted in the neuroinflammatory response in various neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and Multiple Sclerosis(38). In addition, reports of microglia transcriptomics have also shown an upregulation of genes involved in APC-T cell interactions (39). In post-mortem TBI patients, T cells appear accumulated within the lesion area one-week post-TBI and persist there over time(40). However, FMT treatment in our model of TBI mitigated this increase in the overall proportion of T cells within the injured brain (Fig. 2). Given that microglia are the primary antigen-presenting cell (APC) in the brain and that recent studies have shown an upregulation of genes involved in APC-T-cell interactions, we reasoned that FMT-mediated alterations in microglial gene expression were responsible for this expansion in the proportion of T cells. Therefore, we focused on a deeper dive into microglial gene expression with and without FMT treatment.
Microglia are resident to the brain, activating when necessary to serve as the innate immune cells of the CNS. As such the microglial response to TBI is critical. Acutely, microglia phagocytose cellular debris and recruit other immune effector cells for repair and regeneration. However, microglia may also propel and propagate the initial insult via various secondary injury processes, including inflammatory, free-radical, and metabolic responses (24, 41). In our previously published work, we have reported on the peripheral inflammatory infiltrate into the injured brain and their interaction with microglia(5, 42). The TBI-mediated increase in peripheral immune cell infiltration into the injured brain after TBI is driven by a non-classical monocyte mediated neutrophil infiltration followed by a later lymphocyte infiltration and not due to a proliferation of the resident CNS immune cell population (5). To characterize the effect of this pathway in the context of the gut-brain axis, we employed single-cell RNA sequencing to identify injury and/or FMT-specific subpopulations of microglia (Fig. 3A). We identified four main subpopulations of microglia in this analysis corresponding to Homeostatic (HMG), Disease-associated microglia (DAM), Inflammatory, or Heat-shock (hspMG) related genes (Fig. 3A). HMG was identified in all four experimental groups, while DAMs were predominately identified in the TBI/vehicle-treated group. DAM is now well described in the neurodegenerative disease literature with a crucial role in the onset and development of Alzheimer’s disease (21).
Furthermore, DAM demonstrates an overall reduction in homeostatic gene expression with an upregulation in inflammatory genes—resulting in the constitutive activation of microglia over time (Fig 2B). However, in the current study, we identified no significant mitigation in the proportion of DAMs after FMT. This finding suggests that other microglial subtypes may be responsible for the neuroprotective effects. Indeed, we noted a marked increase in the proportion of heat shock protein-expressing microglia with a concomitant decrease in inflammatory microglia, offering some insight into our previously reported preservation of neurocognition and neuroanatomy (19).
ScRNA-seq analysis identified a subpopulation of microglia with significantly increased expression of heat-shock genes (hspMG) corresponding to FMT treatment. This effect was noted in both sham-injured and TBI mice after FMT treatment (Fig 3A–B). Heat shock proteins have a variety of roles, including a neuroprotective role against endotoxins and cellular injury in models of infection and stroke (43, 44). In particular, it has been shown that heat shock proteins activate the TLR-4 signaling pathway to reduce autoimmune-like responses in neurodegenerative diseases (44). More relevant to TBI, heat shock proteins have been shown to inhibit NF-kB mediated microglial activation resulting in neuroprotection after injury (43). In our model, the expression of heat shock proteins was increased in TBI mice after FMT and was also increased in sham-injured mice after FMT treatment. This suggests that FMT itself generates a cellular-protective phenotype and offers a putative mechanism behind the attenuated cortical volume loss, preserved white matter connectivity, and decreased neurocognitive deficits in FMT treated mice after TBI.
Most pertinent to the central hypothesis of the current study, we performed a deeper analysis of the relationship between FMT and microglia-associated inflammatory gene expression (Il1a, Il2b, Ccl3, Ccl4, Ccl2, Cd83, Ler3, Egr1, TNF, Nfkbia, Il1b, & lnfgr1) (Fig. 4). While scrutiny of individual genes could highlight specific components within the inflammatory process, in aggregate, the inflammatory signatures within microglia from FMT and vehicle-treated TBI mice supported our hypothesis— FMT treatment resulted in a markedly attenuated inflammatory transcriptional profile in FMT treated TBI mice as compared to control. Furthermore, FMT also resulted in downregulation of inflammatory gene expression in sham-injured mice, once again suggesting that FMT itself may be anti-inflammatory. This may be another one of the putative mechanisms by which FMT has shown clinical benefit in other infectious, inflammatory, and autoimmune disease processes such as Clostridium difficile colitis, multiple sclerosis, and Parkinson’s disease (20).
Our previous work demonstrated that TBI leads to the chronic activation of microglia, which, in turn, drives many of the neurodegeneration and cognitive sequelae of traumatic brain injury (34). The transcriptional analysis within the current study offers several putative mechanisms by which this occurs, including the expansion of T cells within the injured brain post-injury and the generation of inflammatory microglia subtypes. However, more importantly, these data provide novel insight into potentially protective mechanisms by which the age and activity of these injury-associated cell types can be attenuated, including both the heat-shock response and attenuation of inflammatory gene expression within microglial (24, 41, 42). To address limitations of this study, follow up studies are underway including sex-difference studies as well as metabolomic and serum-protein changes to assess whether FMT accomplishes this through short-chain fatty acid production, via the release of anti-inflammatory cytokines from gut bacteria, or by way of intermediary mechanisms. Despite limitations however, the current data begin to provide some insight into the protective mechanisms of FMT and suggest that FMT may represent a novel therapeutic in the treatment of traumatic brain injury.
Conclusion
Fecal Microbiota Transplant attenuated cortical volume loss, improved white matter connectivity, mitigated expansion of T cells, attenuated inflammatory gene expression, and increased heat shock protein gene expression within microglia after traumatic brain injury. These data highlight the potential of FMT as a novel therapeutic intervention for TBI. FMT is already an established, albeit non-traditional, therapy for other clinical disease entities; repurposing FMT for TBI treatment may represent a novel treatment paradigm for an injury process that is notorious for having few treatment options other than supportive care.
Supplementary Material
Supplement Figure 1 Flow cytometry gating strategy for microglia and infiltrating leukocytes. A forward scatter area (FSC-A) vs. forward scatter height (FSC-H) gating identifies singlets. Live cells can be isolated with the singlet gate based on Live/Dead fixable stain, which is further subdivided into Cd45+ Cd11b subpopulations.
Supplement Figure 2: Correlation heatmap of SCT normalized matrix counts across four samples. Groups Sham (untreated, no injury), TBI (Untreated, with injury), FMT (Treated, No injury ((sham)), TBI-FMT (Treated, with injury). All groups exhibit relatively similar within-group variation as opposed to inter-group.
Funding:
NIH Grant 1R01GM130662
NIH Grant 1R01GM130662 - S1
Footnotes
Conflict of Interest:
All authors declare no conflict(s) of interest.
- Ethical Approval and Consent to participate
This article does not contain any studies with human participants. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Animals were treated and cared for following the National Institutes of Health Guidelines for the Use of Laboratory Animals. The Northwestern University Institutional Animal Care and Use Committee approved the experimental protocol.
Consent for publication
All authors of the manuscript have read and agreed to its content and are accountable for all aspects of the accuracy and integrity of the manuscript per ICMJE criteria. This article is original, has not already been published in a journal, and is not currently under consideration by another journal.
Availability of supporting data
Data Availability
Reviewer link:
References
- 1.Nittayasoot N, Peterson AB, Thammawijaya P, Parker EM, Sathawornwiwat A, Boonthanapat N, Chantian T, Voradetwitaya L, Jiraphongsa C, Sagarasaeranee O, et al. : Evaluation of a hospital-based injury surveillance system for monitoring road traffic deaths in Phuket, Thailand. Traffic Inj Prev 20(4):365–371, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badhiwala JH, Wilson JR and Fehlings MG: Global burden of traumatic brain and spinal cord injury. Lancet Neurol 18(1):24–25, 2019. [DOI] [PubMed] [Google Scholar]
- 3.DeKosky ST and Asken BM: Injury Cascades in TBI- Related Neurodegeneration. Brain Inj 31(9):1177–82, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faul M and Coronado V: Chapter 1 - Epidemiology of traumatic brain injury. In Grafman J and Salazar AM (eds): Vol. 127. Elsevier, 2015, pp. 3–13. [DOI] [PubMed] [Google Scholar]
- 5.Makinde HM, Cuda CM, Just TB, Perlman HR and Schwulst SJ: Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J Immunol 199(10):3583–3591, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAllister TW: Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci 13(3):287–300, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, et al. : Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70(3):374–83, 2011. [DOI] [PubMed] [Google Scholar]
- 8.Romijn JA, Corssmit EP, Havekes LM and Pijl H: Gut-brain axis. Curr Opin Clin Nutr Metab Care 11(4):518–21, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Cryan JF and Dinan TG: Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13(10):701–12, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Ma EL, Smith AD, Desai N, Cheung L, Hanscom M, Stoica BA, Loane DJ, Shea-Donohue T and Faden AI: Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav Immun 66:56–69, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice MW, Pandya JD and Shear DA: Gut Microbiota as a Therapeutic Target to Ameliorate the Biochemical, Neuroanatomical, and Behavioral Effects of Traumatic Brain Injuries. Front Neurol 10:875, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundman MH, Chen NK, Subbian V and Chou YH: The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav Immun 66:31–44, 2017. [DOI] [PubMed] [Google Scholar]
- 13.Blaser M: Antibiotic overuse: Stop the killing of beneficial bacteria. Nature 476(7361):393–4, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Zhou L and Foster JA: Psychobiotics and the gut-brain axis: in the pursuit of happiness. Neuropsychiatr Dis Treat 11:715–23, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu MQ, Cao HL, Wang WQ, Wang S, Cao XC, Yan F and Wang BM: Fecal microbiota transplantation broadening its application beyond intestinal disorders. World J Gastroenterol 21(1):102–11, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J and Wesselingh S: From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry 21(6):738–48, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis B. T. t., Islam MBAR, Das P, Gilbert JA, Ho KJ and Schwulst SJ: Differential Fecal Microbiome Dysbiosis after Equivalent Traumatic Brain Injury in Aged Versus Young Adult Mice. Journal of experimental neurology 2(3):120–130, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Islam M, Davis B. T. t., Kando MJ, Mao Q, Procissi D, Weiss C and Schwulst SJ: Differential neuropathology and functional outcome after equivalent traumatic brain injury in aged versus young adult mice. Exp Neurol 341:113714, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis B. T. t., Chen Z, Islam M, Timken ME, Procissi D and Schwulst SJ: Fecal Microbiota Transfer Attenuates Gut Dysbiosis and Functional Deficits After Traumatic Brain Injury. Shock 57(6):251–259, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar V and Fischer M: Expert opinion on fecal microbiota transplantation for the treatment of Clostridioides difficile infection and beyond. Expert Opinion on Biological Therapy 20(1):73–81, 2020. [DOI] [PubMed] [Google Scholar]
- 21.Vendrik KEW, Ooijevaar RE, de Jong PRC, Laman JD, van Oosten BW, van Hilten JJ, Ducarmon QR, Keller JJ, Kuijper EJ and Contarino MF: Fecal Microbiota Transplantation in Neurological Disorders. Front Cell Infect Microbiol 10:98, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vendrik KEW, Ooijevaar RE, de Jong PRC, Laman JD, van Oosten BW, van Hilten JJ, Ducarmon QR, Keller JJ, Kuijper EJ and Contarino MF: Fecal Microbiota Transplantation in Neurological Disorders. Frontiers in cellular and infection microbiology 10:98–98, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan B, Lu X-J and Wu Q: Gut Microbiota and Acute Central Nervous System Injury: A New Target for Therapeutic Intervention. Frontiers in immunology 12:800796–800796, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makinde HM, Just TB, Gadhvi GT, Winter DR and Schwulst SJ: Microglia Adopt Longitudinal Transcriptional Changes After Traumatic Brain Injury. J Surg Res 246:113–122, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larkin GL, Claassen CA, Pelletier AJ and Camargo CA Jr.: National study of ambulance transports to United States emergency departments: importance of mental health problems. Prehosp Disaster Med 21(2):82–90, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Chen R, Xu Y, Wu P, Zhou H, Lasanajak Y, Fang Y, Tang L, Ye L, Li X, Cai Z, et al. : Transplantation of fecal microbiota rich in short chain fatty acids and butyric acid treat cerebral ischemic stroke by regulating gut microbiota. Pharmacol Res 148:104403, 2019. [DOI] [PubMed] [Google Scholar]
- 27.McGinnis CS, Murrow LM and Gartner ZJ: DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell systems 8(4):329–337.e4, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. : Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20(2):163–172, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. : Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50(1):253–271.e6, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones AR, Overly CC and Sunkin SM: The Allen Brain Atlas: 5 years and beyond. Nature reviews. Neuroscience 10(11):821–828, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Masuda T, Sankowski R, Staszewski O and Prinz M: Microglia Heterogeneity in the Single-Cell Era. Cell Rep 30(5):1271–1281, 2020. [DOI] [PubMed] [Google Scholar]
- 32.Popova G, Soliman SS, Kim CN, Keefe MG, Hennick KM, Jain S, Li T, Tejera D, Shin D, Chhun BB, et al. : Human microglia states are conserved across experimental models and regulate neural stem cell responses in chimeric organoids. Cell Stem Cell 28(12):2153–2166.e6, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eden E, Navon R, Steinfeld I, Lipson D and Yakhini Z: GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10:48, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makinde HM, Just TB, Cuda CM, Bertolino N, Procissi D and Schwulst SJ: Monocyte depletion attenuates the development of posttraumatic hydrocephalus and preserves white matter integrity after traumatic brain injury. PLoS One 13(11):e0202722, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanscom M, Loane DJ and Shea-Donohue T: Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J Clin Invest 131(12), 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rice MW, Pandya JD and Shear DA: Gut Microbiota as a Therapeutic Target to Ameliorate the Biochemical, Neuroanatomical, and Behavioral Effects of Traumatic Brain Injuries. Front Neurol 10, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loane DJ and Kumar A: Microglia in the TBI brain: The good, the bad, and the dysregulated. Experimental neurology 275 Pt 3(0 3):316–327, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.González H and Pacheco R: T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation 11:201, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ and Van Kooyk Y: Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front Immunol 8:1905, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dreßler J, Hanisch U, Kuhlisch E and Geiger KD: Neuronal and glial apoptosis in human traumatic brain injury. International Journal of Legal Medicine 121(5):365–375, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Makinde HM, Just TB, Cuda CM, Perlman H and Schwulst SJ: The Role of Microglia in the Etiology and Evolution of Chronic Traumatic Encephalopathy. Shock 48(3):276–283, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trahanas DM, Cuda CM, Perlman H and Schwulst SJ: Differential Activation of Infiltrating Monocyte-Derived Cells After Mild and Severe Traumatic Brain Injury. Shock 43(3):255–60, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheppard PW, Sun X, Khammash M and Giffard RG: Overexpression of heat shock protein 72 attenuates NF-κB activation using a combination of regulatory mechanisms in microglia. PLoS computational biology 10(2):e1003471–e1003471, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang R, Li Y, Hou X, Miao Z and Wang Y: Neuroprotective effect of heat shock protein 60 on matrine-suppressed microglial activation. Experimental and therapeutic medicine 14(2):1832–1836, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1 Flow cytometry gating strategy for microglia and infiltrating leukocytes. A forward scatter area (FSC-A) vs. forward scatter height (FSC-H) gating identifies singlets. Live cells can be isolated with the singlet gate based on Live/Dead fixable stain, which is further subdivided into Cd45+ Cd11b subpopulations.
Supplement Figure 2: Correlation heatmap of SCT normalized matrix counts across four samples. Groups Sham (untreated, no injury), TBI (Untreated, with injury), FMT (Treated, No injury ((sham)), TBI-FMT (Treated, with injury). All groups exhibit relatively similar within-group variation as opposed to inter-group.
Data Availability Statement
Reviewer link: