

Abstract
Objectives
Despite advances in antibody treatments and vaccines, COVID‐19 caused by SARS‐CoV‐2 infection remains a major health problem resulting in excessive morbidity and mortality and the emergence of new variants has reduced the effectiveness of current vaccines.
Methods
Here, as a proof‐of‐concept, we engineered primary CD8 T cells to express SARS‐CoV‐2 Spike protein‐specific CARs, using the extracellular region of ACE2 and demonstrated their highly specific and potent cytotoxicity towards Spike‐expressing target cells. To improve on this concept as a potential therapeutic, we developed a bispecific T cell engager combining ACE2 with an anti‐CD3 scFv (ACE2‐Bite) to target infected cells and the virus.
Results
As in CAR‐T cell approach, ACE2‐Bite endowed cytotoxic cells to selectively kill Spike‐expressing targets. Furthermore, ACE2‐Bite neutralized the pseudoviruses of SARS‐CoV, SARS‐CoV‐2 wild‐type, and variants including Delta and Omicron, as a decoy protein. Remarkably, ACE2‐Bite molecule showed a higher binding and neutralization affinity to Delta and Omicron variants compared to SARS‐CoV‐2 wild‐type Spike proteins.
Conclusion
In conclusion, these results suggest the potential of this approach as a variant‐proof, therapeutic strategy for future SARS‐CoV‐2 variants, employing both humoral and cellular arms of the adaptive immune response.
Keywords: ACE2‐Bite, CAR‐T cell, COVID‐19, Delta, Omicron, SARS‐CoV‐2
In this study, we utilised ACE2 to develop both a CAR molecule that was engineered to be expressed by T cells and as part of a bispecific (Bite) molecule (ACE2 and anti‐CD3), which in turn activated T cells to target and kill infected cells expressing the Spike protein. Further, ACE2‐Bite protein neutralied entry of viruses mediated by variants of Spike protein, including Omicron. Using a bispecific ACE2 molecule provides the basis for future development of a variant‐resistant treatment approach.
Introduction
Worldwide, coronavirus disease 2019 (COVID‐19) caused by SARS‐CoV‐2 infection has caused over 6 million deaths and a chronic debilitating condition called postacute COVID‐19 syndrome (PACS) in many millions more. An unprecedented effort by researchers around the world has resulted in the development of a spectrum of preventative and therapeutic approaches at an extraordinary speed. Vaccines focussed on virus Spike protein (such as messenger RNA vaccines, non‐replicating vector vaccines and virus‐like particle vaccines) are highly efficient in preventing infection. 1 Several therapeutic developments, such as synthetic neutralising antibodies, monoclonal antibodies to Spike protein 2 , 3 , 4 , 5 and immunomodulators like corticosteroids, 6 , 7 anti‐IL‐6, 8 anti‐IL‐1 9 and Interferon‐β‐1a agents 10 were shown to have a range of treatment efficacy from non‐effective to highly promising. Some of those treatments were repurposed to focus on blocking viral entry, while others were used to control the hyperinflammatory immune response during the disease. Beyond antibody therapies, specific SARS‐CoV‐2 immune modulators and cellular immunotherapies 11 have been developed; however, sustaining their effectiveness in the new variants has been challenging because of the antibody escape mutations. Implementing these approaches in the clinical settings of COVID‐19 is also a major challenge due to the requirements for ex vivo expansion of the therapeutic cells from the same donors. Therefore, it is of great interest to develop treatment approaches that remain effective against future SARS‐CoV‐2 variants and are practical to implement clinically or prophylactically.
SARS‐CoV‐2 uses its Spike protein to bind to the key host receptor angiotensin‐converting enzyme 2 (ACE2) on the target cell surface for cell entry, 12 which physiologically helps to modulate the activity of another protein called angiotensin II (ANG II) that increases the blood pressure. 13 Mutations in Spike protein in SARS‐CoV‐2 variants have resulted in higher affinity interactions with ACE2 14 and/or a better escaping mechanism from the immune system. 15 Following the cell entry, SARS‐CoV‐2 generates viral components by taking over the protein synthesis machinery of the host cell and displays Spike protein on the cell membrane. 16 Using ACE2 molecule to target these Spike‐expressing infected cells could be an effective strategy in preventative and therapeutic approaches to COVID‐19 in the future since the virus cannot avoid binding to ACE2 receptor, and in fact, variants are generally selected based on better interactions with ACE2 for cell entry.
Here, we used a synthetic biology approach to engineer primary human CD8 T cells to express Spike protein‐specific chimeric antigen receptors with ACE2 or anti‐Spike antibody (ACE2 CAR or anti‐Spike CAR) on the extracellular domain to target SARS‐CoV‐2‐infected cells. In addition, as a potential therapeutic, we also engineered a novel ACE2/anti‐CD3 bispecific T cell engager antibody (ACE2‐Bite) to target both SARS‐CoV‐2‐infected cells and neutralisation of the virus before cell entry. Bispecific T cell engager antibodies (Bites) are engineered chimeric molecules that are designed to bind to CD3 on T cells via an anti‐CD3 single‐chain variable fragment (ScFv) and to a target cell via a target‐specific molecule. Upon bridging the T cells with a target cell, Bite molecules in turn trigger T cell activation and target cell apoptosis. CAR‐T and bispecific T cell engager antibody approaches developed herein are inspired by targeting cancer cells, several of which have been approved for treatment. 17
In this study, we show that ACE2‐CAR and anti‐Spike CAR‐expressing CD8 T cells were activated and selectively killed different types of target cells expressing SARS‐CoV‐2 Spike protein on their surface. The ACE2‐Bite antibodies also led to T cell activation in the presence of Spike‐expressing cells and displayed cytotoxicity to these targets. In addition, ACE2‐Bite antibodies acted as a decoy receptor for pseudoviruses incorporated with Spike proteins of the Coronaviridae family including SARS‐CoV, SARS‐CoV‐2 wild‐type, Delta and Omicron variants and neutralised the latter two variants with significantly increased affinity. Taken together, these results suggest that the novel chimeric antigen receptors and bispecific antibodies may be used to redirect cytotoxic immune cells towards SARS‐CoV‐2‐infected host cells and to neutralise current and future variants of the virus.
Results
Development of SARS‐CoV‐2 specific synthetic CARs expressed in T cells
We first sought to develop a system to test whether cells that express SARS‐CoV‐2 Spike protein on their cell surface can be targeted and killed by T cells 18 (Figure 1a). In this experimental set‐up, we engineered effector human T cells to express CAR molecules that can recognise the Spike protein on the cell surface, by transfecting the cells with a plasmid containing a full‐length wild‐type Spike gene (Figure 1b). After 72 hours, the cells were stained with a recombinant ACE2‐Fc protein followed by a fluorescent anti‐Fc antibody to detect surface Spike protein expression and were compared with control cells transfected with a control plasmid. A total of 293 cells transfected with full‐length Spike gene plasmid displayed high cell surface Spike expression, demonstrating cell membrane localisation despite containing Endoplasmic Reticulum Retention Signal (ERRS) domain (Figure 1c). We then established a target 293 cell line that stably expressed Spike protein and Green Fluorescent Protein (GFP) as a reporter. Further, we deleted the ERRS domain to enhance the cell surface spike protein expression as previously shown. 19 , 20 At 72 hours after the transduction, engineered 293 cells showed concordant and high co‐expression of Spike and GFP in these cells (Figure 1d).
Figure 1.
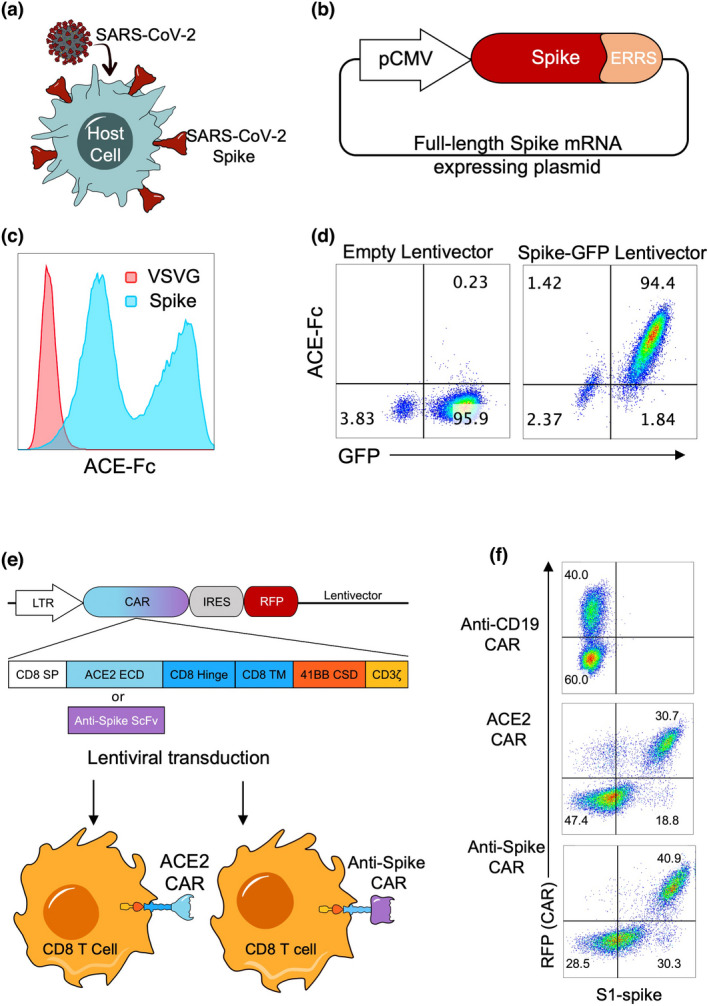
Engineering human primary CD8 T cells to express CAR molecules targeting SARS‐CoV‐2 Spike protein‐expressing cells. (a) Illustration of Spike protein localisation on the surface of SARS‐CoV‐2‐infected cells and (b) of full‐length SARS‐CoV‐2 Spike protein‐expressing plasmid including the Endoplasmic Reticulum Retention Signal (ERRS) of Spike protein on C terminal. (c) 293 cells transfected with full‐length Spike protein (blue histogram) or with VSVG as a negative control (red histogram) expressing vectors. The cells were stained with ACE2‐Fc and anti‐Fc‐APC secondary antibody, and flow cytometry data overlays are shown. (d) 293 cells transduced with a lentivirus encoding a truncated Spike protein gene without the ERRS domain and Green Fluorescent Protein (GFP) as a reporter. Transduced cells were stained with ACE2‐Fc and anti‐Fc‐APC secondary antibody, and representative flow cytometry data plots are shown. (e) Illustration of ACE2 CAR and anti‐SARS‐CoV‐2 Spike protein CAR constructs and their expression in CD8 T cells. A constitutive LTR promoter drives ACE2 or anti‐Spike CAR and RFP genes separated by an Internal Ribosomal Entry Site (IRES). CAR constructs consist of CD8 alpha signal peptide, ACE2 or single‐chain variable fragment of an anti‐Spike antibody, CD8 Hinge, CD8 transmembrane domain, 4‐1BB (CD137) co‐stimulatory domain and CD3ζ domain. Lentiviruses containing CARs were used to transduce primary CD8 T cells. (f) Expression of CAR constructs on CD8 T cells. Activated and transduced CD8 T cells were expanded for 10–12 days and stained with SARS‐CoV‐2 S1 protein fused to mouse Fc, and anti‐mouse Fc secondary antibody. Flow cytometry plots showing ACE2 or anti‐Spike surface expression versus RFP are shown. Anti‐CD19 CAR‐expressing CD8 T cells were used as control. The experiments were replicated three times with similar results.
Next, we designed lentivector constructs containing ACE2 CAR or anti‐Spike CAR cassettes followed by an Internal Ribosomal Entry Site (IRES) and Red Fluorescent Protein (RFP) and transduced human primary CD8 T cells (Figure 1e) as previously described. 21 ACE2 CAR and anti‐Spike CAR constructs composed of CD8 alpha signal peptide, ACE2 extracellular domain (ECD) or anti‐Spike ScFv, respectively, and intracellular 41BB co‐stimulatory domain (CSD) and CD3ζ (zeta) signalling domains (Figure 1e). An Anti‐CD19 CAR‐RFP lentiviral construct was also designed as another control. CD8 T cells were then activated and transduced with these lentiviruses encoding the CAR constructs and expanded in IL‐2 for 10–12 days. Surface expression of ACE2 and anti‐Spike was demonstrated on CAR‐engineered CD8 T cells, which also correlated with RFP reporter (Figure 1f).
Cytotoxicity assays with ACE2 CAR and anti‐Spike CAR‐expressing T cells
We then cocultured Spike+ target cell line and effector T cells expressing CARs and measured the cytotoxic activity of the T cells (Figure 2a). Briefly, after the ~2‐week proliferation of CAR‐T cells, the cells were cocultured for 72 h with Spike‐expressing target cells at different effector‐to‐target ratios. The CD8 T cells were then stained with anti‐CD25 to determine their activation. Target cells were identified via GFP, which was co‐expressed with Spike protein. Both ACE2 CAR and anti‐Spike CAR‐T cells became highly activated and killed the Spike+293 cells, whereas control anti‐CD19 CAR‐T cells were neither activated nor displayed any cytotoxicity (Figure 2b and c).
Figure 2.
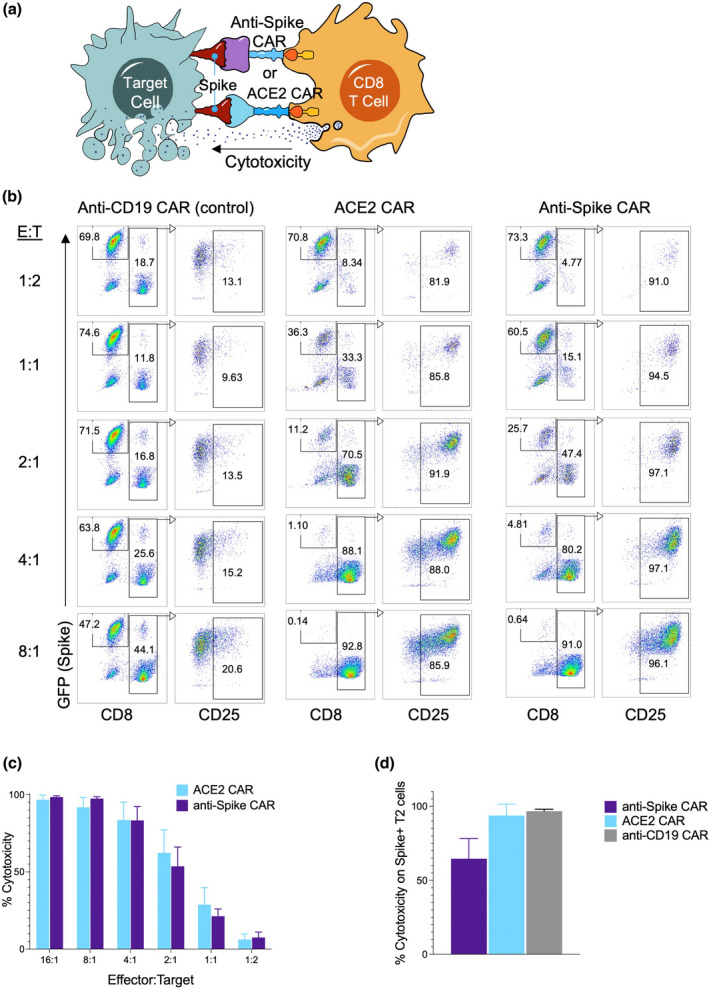
Cytotoxic activity of human primary CD8 T cells engineered to express ACE2 CAR or anti‐Spike CAR. (a) Cytotoxicity assay against Spike‐expressing target cells using ACE2 CAR or anti‐Spike CAR‐expressing CD8 T cells as effector cells. (b) CAR‐engineered T cells cytotoxicity assays with Spike‐expressing 293 target cells at different Effector:Target ratios. CD8 T cells transduced with anti‐CD19 CAR lentiviruses were used as control effector cells. Effector CD8 T cells were identified with CD8 staining, while target cells were gated based on GFP (Spike) expression. Activation of effector cells and CAR expression were determined with CD25 expression after gating on CD8 T cells 2 days after coculture. (c) Cytotoxicity of ACE2 CAR (blue) and anti‐Spike CAR (purple) T cells normalised to anti‐CD19 CAR‐T cells at different Effector:Target ratios and using Spike‐expressing 293 cells as the target. (d) CAR‐engineered T cells cytotoxicity assays with Spike‐expressing target B cell line (T2 cells) at 8:1 E:T ratio. Wild‐type CD8 T cells were used as a negative control and anti‐CD19 CAR‐expressing CD8 T cells were used as a positive control. Panels show representative experiments replicated three times with similar results.
We next tested whether ACE2 CAR and anti‐Spike CAR‐T cells can kill Spike‐expressing human B cell line, which can also be used as a positive control using anti‐CD19 CAR‐T cells. ACE2 CAR and anti‐Spike CAR‐T cells killed Spike‐expressing B cells as efficiently as 293 cells, indicating that different cell types infected with SARS‐CoV‐2 can be targeted using these novel CAR‐T cells (Figure 2d, Supplementary figure 1a). In addition, ACE2 CAR and anti‐Spike CAR‐T cells did not show cytotoxicity to GFP‐expressing, Spike‐negative control targets and were also not activated, showing a selective Spike protein‐mediated activation and killing (Supplementary figure 1b).
Development of bispecific antibodies to mobilise and activate T cells against SARS‐CoV‐2 Spike protein‐expressing target cells
Currently, the CAR‐T cell immunotherapy procedure requires a meticulous process of collecting cells from patients, engineering them in a good manufacturing process environment, re‐infusion and extensive clinical follow‐up of the patients. 22 As such, this may not be practical for the treatment of COVID‐19 patients. To overcome the hurdles of the CAR‐T cell approach, we engineered a similar approach using bispecific antibodies (Bites) as T cell activators, consisting of an anti‐CD3 scFv fused with the extracellular domain of ACE2 to redirect CD3 T cells to SARS‐CoV‐2‐infected cells (Figure 3a). The ACE2‐Bite cassette consisted of ACE2 signal peptide, ACE2 extracellular domain, a linker peptide, an anti‐CD3 antibody single‐chain variable fragment, a His‐Tag and a Hemagglutinin (HA) Tag (Figure 3b). ACE2‐Bite was produced by suspension 293 cells as described in Methods. The supernatant from these cells was then filtered to eliminate small molecules, which also resulted in a ~30‐fold concentration of ACE2‐Bite proteins. The supernatant of wild‐type suspension 293 cells was also collected and filtered/concentrated to be used as a control.
Figure 3.
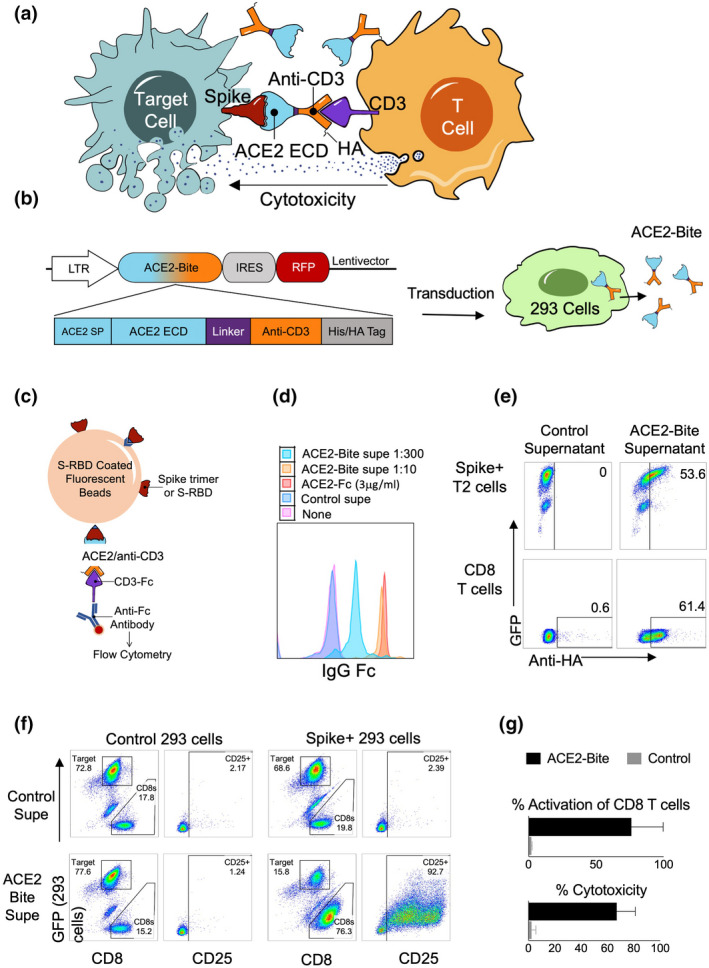
Functional ACE2/anti‐CD3 bispecific T cell engagers against SARS‐CoV‐2. (a). Mechanism of action of ACE2‐Bite. The extracellular domain (ECD) of ACE2 (blue) in ACE2‐Bite binds to Spike protein (red) expressed on the surface of SARS‐CoV‐2‐infected cells, and the anti‐CD3 fragment (orange) binds to CD3 molecule (purple) on T cells linking both cell types and inducing the activation of T cells, which subsequently results in apoptosis of infected target cells. ACE2‐Bite recombinant protein also contains a hemagglutinin (HA) tag at the C terminal. (b) ACE2‐Bite construct and protein production in 293 cells. A constitutive LTR promoter drives the expression of ACE2‐Bite and RFP genes separated by an Internal Ribosomal Entry Site (IRES). ACE2‐Bite cassette consists of ACE2 signal peptide (SP), ACE2 extracellular domain, a linker peptide, an anti‐CD3 antibody single‐chain variable fragment, a His‐Tag and a Hemagglutinin (HA) Tag. Lentiviruses expressing ACE2‐Bite were used to transduce suspension 293 cells that produce and secrete ACE2‐Bite protein in their culture supernatant. (c) Bead‐based ACE2‐Bite capture assay. Fluorescent beads coated with Spike protein trimer or Spike‐Receptor binding domain (S‐RBD) were used to capture ACE2‐Bite molecules, which were detected via a recombinant CD3‐Fc fusion protein and an anti‐Fc antibody then subsequently analysed by flow cytometry. ACE2‐Fc molecules were also detected with Spike trimer or S‐RBD coated beads and anti‐Fc antibody. (d) Detection of ACE2‐Bite concentrations. 1:10 and 1:300 dilutions were shown in orange and turquoise, respectively, and ACE2‐Fc (3 μg mL−1) (red) using bead‐based ACE2‐Bite capture assay. Wild‐type 293 cell supernatant (Control supe, Blue) and staining buffer (None, Pink) were used as negative controls. (e) Binding of ACE2‐Bite to Spike‐GFP‐expressing T2 cell line and primary human T cells. HA staining of Spike‐GFP‐expressing T2 cells (top panel) and CD8 T cells (bottom panel) when combined with ACE2‐Bite (right plot) or control (non‐transduced 293) (left plot) supernatants. (f) CD25 and GFP expressions show activation and cytotoxicity of resting CD8 T cells against Spike/GFP‐expressing or control (transduced with GFP‐expressing empty vector) 293 cells in the presence of ACE2‐Bite or control supernatant. (g) The bar graph demonstrating the results of the cytotoxicity assay of the experiment is represented in f. The experiments were replicated three times with similar results.
To test the correct folding of the recombinant ACE2‐Bite protein, we developed a fluorescent bead‐based ACE2‐Bite detection assay in which the fluorescent beads were coated with either SARS‐CoV‐2 Spike protein trimer or Spike‐Receptor Binding Domain (S‐RBD) and the ACE2‐Bite molecules captured by these beads were detected via a recombinant CD3‐Fc molecule, which was then stained with an anti‐Fc antibody (Figure 3c). A recombinant ACE2‐Fc molecule was used as a positive control since the ACE2 part could bind to Spike trimer or S‐RBD on the surface of beads and anti‐Fc antibody could recognise the Fc part of ACE2‐Fc. ACE2‐Bite detection assay with S‐RBD coated beads showed that secreted and concentrated ACE2‐Bite levels (1:10) were comparable to control ACE2‐Fc concentration (3 μg mL−1) (Figure 3d). We also confirmed that the ACE2‐Bite concentration protocol functioned as intended and increased the ACE2‐Bite concentration by an order of magnitude (Supplementary figure 2).
We then tested the ACE2‐Bite binding on human primary CD8 T and Spike‐expressing target cells. For this, ACE2‐Bite and wild‐type supernatants were added to primary human CD8 T cells and a B cell line (T2 cells), which was engineered to express Spike/GFP. The cells combined with ACE2‐Bite or control supernatants were then stained for HA Tag on their surface. Spike/GFP co‐expressing T2 cells and CD3 expressing primary human CD8 T cells combined with ACE2‐Bites were stained positive for HA Tag, suggesting Spike‐specific binding of ACE2 fragment and CD3‐specific binding of Anti‐CD3 fragment (Figure 3e).
We then performed a cytotoxicity assay to test the ability of ACE2‐Bites to trigger human T cell activation and effector function. Human primary CD8 T cells were cocultured with Spike‐expressing or control 293 cells in the presence of ACE2‐Bite or control supernatants. Two days later, cells were collected and stained for their CD8 and CD25 expression. GFP expressed by control and Spike lentivectors was used to identify the target cells. Indeed, resting human T cells became activated and were cytotoxic only in the presence of ACE2‐Bite supernatant and Spike‐expressing targets, suggesting Spike‐specific T cell activation functionality of ACE2‐Bites (Figure 3f and g).
Determining the function of ACE2‐Bite on Spike protein variants
We next determined the binding of ACE2‐Bite to a variety of Spike proteins with ACE2‐Bite/Spike affinity assays, first using a bead‐based antibody detection method we developed (Figure 3). In this assay, we labelled the beads with wild‐type and Omicron Spike protein trimers to determine the affinity of ACE2‐Bites to the natural trimeric Spike protein structure of these strains. Labelled beads were treated with ACE2‐Bite and control supernatants in threefold serial dilutions from 1 to 30 to generate titration curves. ACE2‐Bite treated beads were then stained with recombinant CD3‐Fc protein and anti‐Fc antibody and analysed via flow cytometry. The bead‐based assay using these Spike protein trimers revealed a significant increase in affinity of ACE2‐Bite molecule to Omicron Spike trimer compared with wild‐type (P < 0.0001) (Figure 4a and b).
Figure 4.
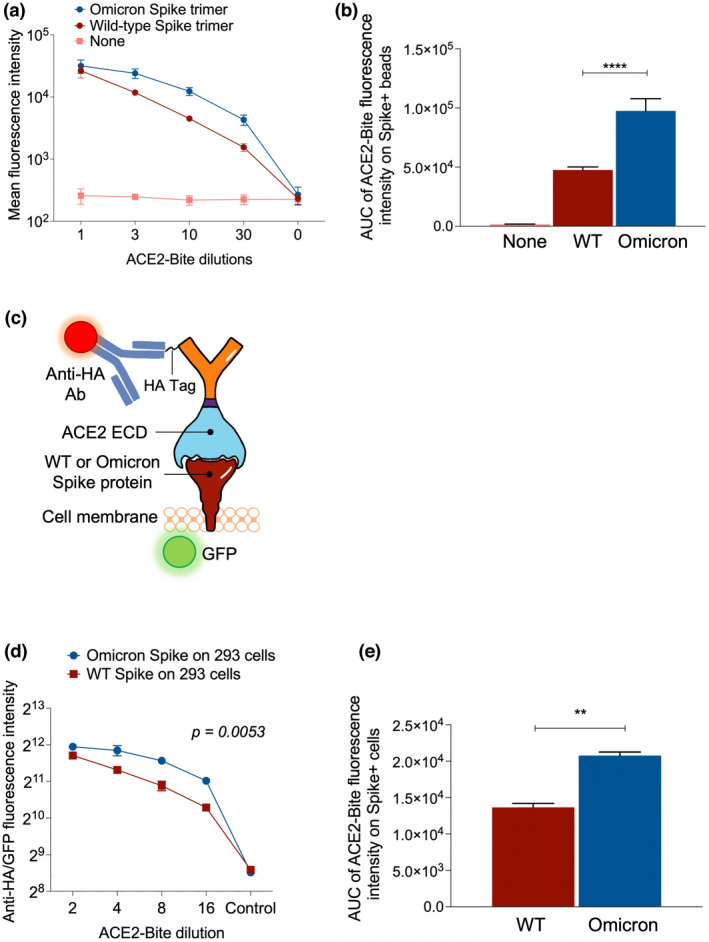
Binding of ACE2‐Bite to variant Spike proteins. (a) Bead‐based ACE2‐Bite/Spike binding assay. Beads were coated with full‐length SARS‐CoV‐2 wild‐type and omicron variant Spike protein trimers and treated with ACE2‐Bite and control media in threefold serial dilutions from 1 to 30. Treated beads were then stained with a CD3‐Fc recombinant protein and an anti‐Fc antibody. Geometric mean values of anti‐Fc antibody fluorescence were used to quantify the fluorescent intensity of samples. (b) Area under the curve (AUC) values of bead‐based ACE2‐Bite/Spike binding assay data from (a). Experiments were replicated three times. (c) ACE2‐Bite binding to spike protein (wild‐type or Omicron) expressed on the cell surface and its detection by immunostaining with an anti‐HA antibody. GFP was co‐expressed with Spike proteins as a reporter. (d) ACE2‐Bite binding assay on Spike‐expressing 293 cells. Geometric mean intensity of anti‐HA antibody staining was used to quantify the affinity of ACE2‐Bite molecules on Spike‐expressing cells. For each condition, gates with similar intensities of GFP were used as a Spike protein marker when assessing the fluorescence intensity of ACE2‐Bite‐stained cells to determine the quantitative value of ACE2‐Bite fluorescence per Spike protein. Fluorescence intensity was determined via geometric mean values. An unpaired t‐test was used to determine the statistical significance, and P‐values were corrected for multiple comparisons using the Holm–Sidak method. (e) Area under the curve (AUC) values of bead‐based ACE2‐Bite/Spike binding assay data from d. The experiments were replicated twice with similar results.
In addition to this bead‐based affinity assay, we also transduced 293 cells with GFP‐encoding lentivectors to express wild‐type and Omicron variant Spikes and determined the ACE2‐Bite affinity to these Spike variants on live cell surface (Figure 4c). Three days after the transduction, the cells were collected and co‐stained with ACE2‐Bite and anti‐HA antibody and analysed via flow cytometry. The ACE2‐Bite/Spike protein binding assay on living cells revealed a significantly higher affinity of ACE2‐Bite to Omicron variant Spike protein compared with wild‐type (P = 0.0053), confirming the result of the bead‐based affinity assay (Figure 4d and e). Taken together, these results suggest the ACE2‐Bite approach can be effective in all variants of SARS‐CoV‐2, in fact possibly with better neutralisation and T cell cytotoxicity, as the virus evolves into variants with higher affinity towards ACE2 protein.
Finally, we also tested the activity of ACE2‐Bite against target cells expressing Spike proteins with and without the ERRS to explore whether this domain would result in reduced Spike protein expression on the cell surface and in turn decrease ACE2‐Bite binding. For this assay, 293 cells were transfected with the plasmids expressing wild‐type Spike and ERRS‐deleted Spike proteins and combined with resting CD8 T cells in the presence and absence of ACE2‐Bite. After 72 h, the cells were collected, and T cell activation was determined by cell surface marker expressions of CD25 and CD69. We found that CD8 T cells were activated at very similar levels against the cells expressing Spike proteins with or without ERRS domains (Supplementary figure 2b).
ACE2‐Bite neutralises Spike‐pseudotyped lentiviruses
In addition to bridging infected cells to activate T cells, we reasoned that ACE2‐Bite might also effectively neutralise SARS‐CoV‐2 by binding to Spike proteins on the virus. To test this, we generated a set of lentiviruses pseudotyped with SARS‐CoV, SARS‐CoV‐2 wild‐type, Delta and Omicron Spike proteins and investigated ACE2‐Bite neutralisation on these viruses. Neutralisation assay was performed by pre‐culturing pseudotyped viruses with different dilutions of ACE2‐Bite supernatant and then adding to ACE2‐expressing 293 cells as previously described 23 (Figure 5a). We also incubated a recombinant ACE2‐Fc molecule at different concentrations with Spike‐pseudotyped lentivirus as a positive control. The infection levels were determined 3‐day postinfection based on the GFP expression of ACE2‐expressing 293 cells. As shown in the representative experiment, ACE2‐Fc and ACE2‐Bite molecules neutralised the Spike‐pseudotyped lentivirus (Supplementary figure 3). Importantly, the ACE2‐Bite molecule was able to neutralise all versions of Spike encoding lentiviruses with increased efficiency against Delta and Omicron variants compared with wild‐type SARS‐CoV‐2 pseudovirus (P = 0.016, 0.0008 and 0.016 for Delta 1:28, 1:14 and 1:7 dilutions, respectively; P = 0.0023 and 0.014 for Omicron 1:14 and 1:7 dilutions, respectively) (Figure 5b). This neutralisation assay suggests that novel ACE2‐Bite recombinant protein could also function as a decoy receptor for the virus and against all variants of the Spike protein. Taken together, these findings demonstrate the potential of ACE2‐Bite as a fool‐proof therapeutic approach for potential future emerging variants.
Figure 5.
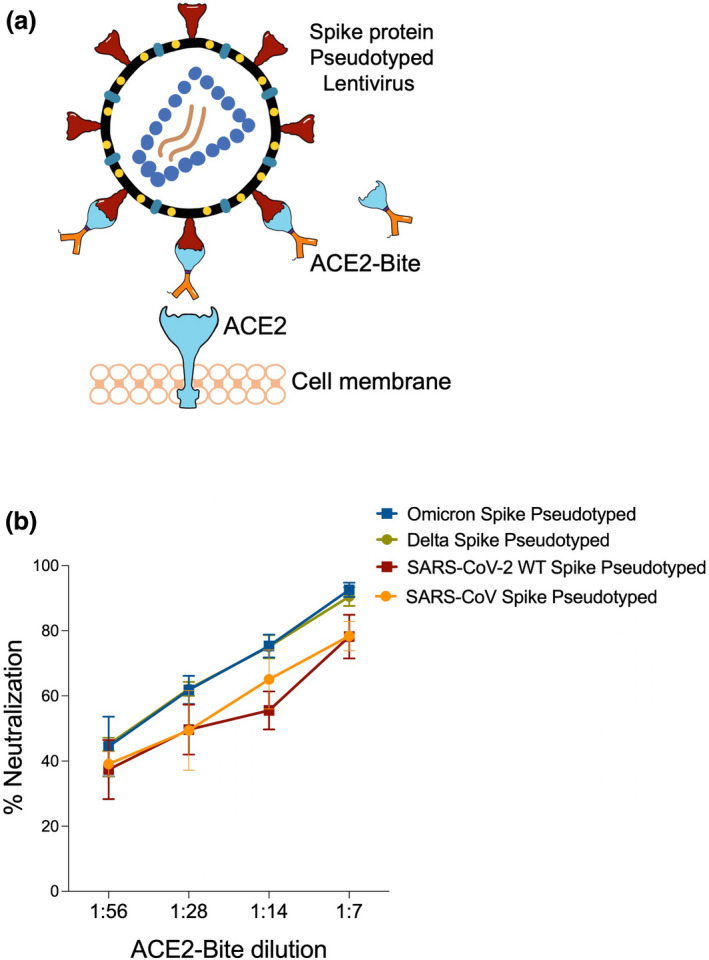
Binding of ACE2‐Bite to SARS‐CoV‐2 Spike protein variants on pseudotyped lentiviruses for virus neutralisation. (a) Virus neutralisation assay. ACE2‐Bite and Spike (SARS‐CoV, SARS‐CoV‐2, Delta and Omicron) pseudotyped lentiviruses are pre‐incubated and then added to ACE2‐overexpressing 293 cells. (b) Virus neutralisation data of the lentiviruses pseudotyped with different Spike proteins that pre‐incubated with ACE2‐Bite at different ACE2‐Bite:virus ratios then added to ACE2‐overexpressing 293s. The experiments were replicated twice with similar results.
Discussion
Despite advances in vaccine development, COVID‐19 is still a major cause of morbidity and mortality in the USA and throughout the world. The rapid evolution of the virus is also a major concern, suggesting the need to develop novel effective treatment strategies, as SARS‐CoV‐2 specific targeted therapeutic approaches such as monoclonal antibody therapies can lose their effectiveness as new escape variants emerge. To mitigate or override these potential problems, here we utilised synthetic biology approaches, to develop synthetic molecules that can bridge T cells with SARS‐CoV‐2‐infected cells, through recognition of cell surface expression of virus Spike protein and eliminate them through cytotoxic activity.
We first generated CD8 T cells expressing chimeric antigen receptors (CARs) specific to Spike protein with an anti‐Spike antibody or ACE2 surface domain on the extracellular region and tested their effectiveness against different cell types expressing Spike proteins. In this assay, engineered CAR‐T cells (anti‐Spike CARs and ACE2 CARs) became activated and killed the Spike‐expressing target cells selectively. Although cancer cells have predominantly been the focus of adaptive cellular immunotherapies, studies have suggested that autoimmune and infectious diseases could also be targeted via such approaches. 24 , 25 Several studies have employed or investigated the CAR‐T approach against infectious diseases, such as human immunodeficiency virus (HIV), 26 , 27 , 28 hepatitis B virus (HBV), 29 hepatitis C virus (HCV), 30 cytomegalovirus (CMV), 31 Epstein–Barr virus (EBV) 32 and Aspergillus fumigatus. 33 While it is not practical to apply this approach during acute COVID‐19 disease, given it takes about 2 weeks to generate CAR‐T cells, it is conceivable that this approach can be utilised as prophylaxis in high‐risk immune‐compromised patients, as CAR‐T cells can have a very long lifespan of many months to years. 34 , 35 It may also be possible to generate off‐the‐shelf Spike‐specific CAR‐T cells by removing genes in T cells that can cause allogeneic reaction or graft versus host disease 36 to be used in immune‐compromised or elderly individuals who are in the high‐risk category for mortality. However, it will also be important to develop and built‐in failsafe mechanisms for CAR‐T to minimise potential toxicities, and thus currently a proof‐of‐principle approach given functional alternatives such as Spike‐specific antibody cocktails and antiviral therapies. Yet, this may also be an important therapeutic for other deadly current or potential viral infections, including unexpected variants of SARS‐CoV‐2, for which vaccines or treatments may fail or are not yet available. 37 , 38
Because of current hurdles of Spike‐specific CAR‐T cells in clinical settings, we also developed a similar alternative potential treatment or prophylactic bispecific T cell engagers (Bites), with the potential to activate cytotoxic cells upon bridging with Spike protein expressed on the infected cell surface. We demonstrated that the ACE2‐Bite bispecific antibody‐receptor complex triggered effective CD8 T cell activation, which resulted in the selective killing of Spike+ target cells. Indeed, a B cell surface protein (CD19)‐specific Bite called Blinatumomab (CD19‐CD3 Bite) has already been approved to be used in B cell lymphoma patients in 2018. 39 Other studies also showed that Bites could be employed against Her2, 40 BCMA, 41 EpCAM, 42 EGFR, 43 CD20 44 and PDL1 45 expressing cancers; and diseased cells infected by CMV 46 and HIV. 47 Compared with current treatments (such as neutralising antibodies or antivirals), ACE2‐Bite approach may potentially be effective both at the early stages (as a neutraliser of the virus entry) and later stages of the infection when antibody immune defences are breached, and T cells become more critical in restricting the spread of the virus in vivo. Another major advantage of the ACE2‐Bite approach is using ACE2, the key host receptor of SARS‐CoV‐2, as the Spike protein recognising part of the bispecific antibody. As shown in Figure 4, this allowed us to target mutated Spike proteins from variants of concern with even better efficiency in contrast to conventional antibody approaches which lose their efficiency due to immune escape mutations. 15
In addition to recognising mutated Spike proteins, we found that the ACE2 part of the ACE2‐Bite functioned as a decoy receptor and neutralised the virus entry into cells. The neutralisation feature of the ACE2‐Bite molecule is promising for its use as preventive treatment, and it would conceivably have a synergistic effect with the cytotoxic effect by engaging T cells towards infected cells. Indeed, in line with our findings, a clinical study by Zoufaly et al. 48 found that infusion of soluble recombinant human ACE2 molecule in a 45‐year‐old COVID‐19 patient resulted in a dramatic decrease in viral copies in the patient plasma. Other studies also demonstrated the neutralisation capacity of soluble ACE2 molecule. 49 , 50 , 51 Considering the immunity of ACE2 to the Spike mutations, both as a CD3 T cell redirecting molecule and a decoy receptor, the efficacy of the ACE2‐Bite treatment is unlikely to be diminished by variants arising during the COVID‐19 pandemic or in possible future SARS pandemics. Conceivably, future variants with increased affinity to ACE2 would bind better to ACE2‐Bite, thus possibly further improving its efficacy.
One limitation of the ACE‐Bite approach described herein is that we do not know how well Spike expression levels we achieved in vitro would correspond to in vivo primary cells infected with SARS‐CoV‐2. We think these will be comparable, given findings by Ding et al., 52 which demonstrated the robust Spike protein expression on the surface of the cells infected with authentic SARS‐CoV‐2 variants. It is also important to note that the cells expressing Spike in vitro were also infected with lentiviruses and not transfections of plasmid DNA, which can artificially increase cell surface expression of the protein of interest. Ultimately, to what extent Spike expression on the cells during COVID‐19 in vivo will be sufficient to mediate T cell cytotoxicity will require in vivo animal studies. Another important limitation of the ACE2‐Bite approach is the partial dependence on the presence of effective cytotoxic T cells in patients. This may not be optimal for immunocompromised patients who may have T cell defects or in the elderly with higher exhausted T cell subsets. While we think ACE2‐Bite will still be effective in neutralising the virus entry, it may not be as efficient to activate cytotoxic T cells due to intrinsic defects of the patient‐specific immunity. Perhaps the off‐the‐shelf CAR‐T approach using ACE2‐CAR and anti‐Spike CARs would be more practical in such cases. Furthermore, it will be important to determine the efficiency of ACE2‐Bite within tissues such as lungs in vivo.
If these approaches are developed as therapeutics, it will also be important to consider the side effects of CAR‐T cells or ACE2‐Bite treatment, cytokine release syndrome and T cell exhaustion, similar to cancer‐focussed CAR‐T therapies. 53 Although, we think this will be less likely given that the number of infected cells would be expected to be much smaller than the tumour burden; thus overall, much fewer T cells would be stimulated during the infection. Another potential downside could be that the ACE2 in ACE2‐Bite may interact with its physiologic ligands and interfere with the renin‐angiotensin system, although, so far recombinant human ACE2 molecule has been tested in 89 patients with tolerable clinical outcomes. 54 , 55 In addition to that, while we did not find Angiotensin II to cause T cell activation through ACE2, this is something to be considered in the clinical setting or during in vivo testing. In that respect, using ACE2‐Bite for a short period of time rather than using ACE2 CAR‐T cells, which would last much longer could be more practical. Regardless, if these become a problem, it can be prevented by mutating the carboxypeptidase activity of ACE2.
In conclusion, engineered CD8 T cells expressing Spike protein‐specific chimeric antigen receptors and ACE2/anti‐CD3 bispecific T cell engagers developed in this study could be used to target SARS‐CoV‐2‐infected host cells and the virus itself and may be alternative future therapeutic strategies for COVID‐19.
Methods
ACE2 CAR construct
CAR constructs consisting of CD8 alpha signal peptide, the extracellular domain of ACE2 molecule or single‐chain variable fragment (scFv) of anti‐CD19 or anti‐Spike protein antibodies, CD8 hinge domain, CD8 transmembrane domain, 4‐1BB (CD137) intracellular domain and CD3ζ domain were designed with Snapgene and synthesised via Genscript. ACE2 extracellular domain, CD8a signal peptide, CD8 hinge, CD8 transmembrane domain, 4‐1BB intracellular domain and CD3ζ domain sequences were obtained from Ensembl Gene Browser and codon‐optimised with SnapGene by removing the restriction enzyme recognition sites that are necessary for subsequent molecular cloning steps while preserving the amino acid sequences. Anti‐CD19 and anti‐Spike scFv amino acid sequences were obtained from Addgene plasmids #79125 and #155364, respectively, reverse translated to DNA sequences and codon‐optimised with Snapgene 5.2.4. The constructs were then cloned into a lentiviral expression vector with a multiple cloning site separated from RFP reporter via an Internal Ribosomal Entry Site (IRES).
Spike protein constructs
Human codon‐optimised wild‐type and Omicron full‐length SARS‐CoV‐2 Spike protein sequences were synthesised by MolecularCloud (MC_0101081 and MC_0101272, respectively) and then cloned into pLP/VSVG plasmid from Thermo Fisher under CMV promoter after removing the VSVG sequence via EcoRI‐EcoRI restriction digestion. 5′‐ACGACGGAATTCATGTTCGTCTTCCTGGTCCTG‐3′ and 5′‐ACGACGGAATTCTTAACAGCAGGAGCCACAGC‐3′; and 5′‐ACGACGGAATTCATGTTCGTGTTCCTGGTGCT‐3′ and 5′‐ACGACGGAATTCTTAACAGCAACTGCCGCAG‐3′ primers were used to generate wild‐type and Omicron SARS‐CoV‐2 Spike protein sequences without the Endoplasmic Reticulum Retention Signal (ERRS, last 19 amino acids of Spike), 16 respectively. For stable wild‐type Spike protein overexpression, wild‐type and Omicron Spike protein sequences without ERRS domain were cloned into a lentivector with a GFP marker under LTR promoter. Human codon‐optimised Delta full‐length SARS‐CoV‐2 Spike protein plasmid was synthesised by Invivogen (plv‐spike‐v8).
VSVG and Spike protein pseudotyped lentivirus production
The lentiviruses pseudotyped with vesicular stomatitis virus G protein envelope were generated with HEK293T cells. Briefly, the lentivector plasmids containing the constructs were co‐transfected with vesicular stomatitis virus G protein, pLP1 and pLP2 plasmids into HEK293T cells at 80–90% confluency using Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol. In the case of Spike protein pseudotyped lentiviruses, a lentivector plasmid containing GFP reporter was co‐transfected with wild‐type or mutated SARS‐CoV‐2 Spike protein plasmids in the same manner. The transfection medium was replaced with RPMI 1640 with 10% FBS 6‐h post‐transfection. Viral supernatants were collected 24‐ to 48‐h post‐transfection and filtered through a 0.45‐μm syringe filter (Millipore) to remove cellular debris. A Lenti‐X concentrator (Takara Bio USA) was used according to the manufacturer's protocol to concentrate the virus 10–20×, and the resulting lentiviral stocks were aliquoted and stored at −80°C. To measure viral titres of VSVG pseudotyped lentiviruses, virus preparations were serially diluted on Jurkat cells and 3‐day postinfection, infected cells were measured using flow cytometry, and the number of cells transduced with 1 mL of virus supernatant was calculated as infectious units per millilitre. For spike protein pseudotyped lentiviruses, to measure viral titres, virus preparations were serially diluted on ACE2‐overexpressing 293 cells, which were stained for their ACE2 expressions and confirmed ~%100 positive. Seventy‐two hours after infection, GFP‐positive cells were counted using flow cytometry and the number of cells transduced with virus supernatant was calculated as infectious units per mL. Based on these titre values, primary T cells, 293 T cells and T2 cells were transduced with a multiplicity of infection (MOI) of 3–10.
ACE2‐Bite design and production
The ACE2‐Bite construct consists of ACE2 signal peptide, ACE2 extracellular domain, a linker peptide, an anti‐CD3 antibody single‐chain variable fragment, a His‐Tag, and a Hemagglutinin (HA) Tag was designed with Snapgene and synthesised via Genscript. ACE2 signal peptide and extracellular domain sequences were obtained from Ensembl Gene Browser (Transcript ID: ENST00000252519.8). Anti‐CD3 antibody single‐chain variable fragment, His‐Tag and Hemagglutinin (HA) Tag sequences were obtained from Addgene plasmid #85437. The ACE2‐Bite construct was cloned into an RFP marked lentivector under LTR promoter, and Expi293F™ suspension 293 cells from ThermoFisher were transduced with the ACE2‐Bite expressing VSVG pseudotyped lentiviruses with a multiplicity of infection of 5. The cells were then grown in Expi293™ Expression Medium in shaking flasks for 7 days until they reached maximum viable density. ACE2‐Bite containing supernatant was then collected and filtered/concentrated up to 30‐fold with 30 kDa MilliporeSigma™ Amicon™ Ultra Centrifugal Filter Units. Concentrated ACE2‐Bite and control supernatants were aliquoted and stored at 4°C.
Engineering CAR‐T cells and Spike‐expressing target cells
Healthy adult blood was obtained from AllCells. PBMCs were isolated using Ficoll‐paque plus (GE Health care). CD8 T cells were purified using Dynal CD8 Positive Isolation Kit (from Invitrogen). CD8 T cells were > 99% pure and assessed by flow cytometry staining with CD8‐Pacific Blue antibody (Biolegend). Total CD8 T cells were activated using anti‐CD3/CD28 coated beads (Invitrogen) at a 1:2 ratio (beads:cells) and infected with anti‐CD19 CAR, anti‐Spike CAR or ACE2 CAR VSVG pseudotyped lentiviral constructs at the multiplicity of infection (MOI) of 5–10. The cells were then expanded in complete RPMI 1640 medium supplemented with 10% Fetal Bovine Serum (FBS, Atlanta Biologicals), 1% penicillin/streptomycin (Corning Cellgro) and 20 ng mL−1 of IL‐2 and cultured at 37°C and 5% CO2 supplemented incubators. Respective viruses were added 24 h after the activation. Cells were expanded for 10–12 days, and cytotoxicity assays were performed following their expansion. To generate HEK‐293T cells that transiently expressed wild‐type and mutated spike protein (ATCC; mycoplasma‐free low passage stock), the cells were transfected with Spike protein‐expressing pLP plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol and stained for their spike protein expression 72 h after the transfection as described in Flow cytometry analysis. All engineered and wild‐type HEK‐293 and T2 cells were cultured in complete RPMI 1640 medium (RPMI 1640 supplemented with 10% FBS; Atlanta Biologicals, Lawrenceville, GA), 8% GlutaMAX (Life Technologies), 8% sodium pyruvate, 8% MEM vitamins, 8% MEM nonessential amino acid and 1% penicillin/streptomycin (all from Corning Cellgro). To generate T2s and 293s with stable Spike overexpression, wild‐type T2 and 293 cells were transduced with 3 MOI of Spike protein overexpressing VSVG lentivirus and proliferated. The infection levels were determined by GFP expression through Flow Cytometry analysis. For ACE2 overexpression in 293, a wild‐type ACE2 sequence was obtained from Ensembl Gene Browser (Transcript ID: ENST00000252519.8) and codon‐optimised with SnapGene by removing restriction enzyme recognition sites that are necessary for subsequent molecular cloning steps preserving the amino acid sequence, synthesised in GenScript and cloned into a lentiviral vector. VSVG pseudotyped lentiviruses of respective constructs were generated as mentioned above, and cells were transduced with this virus at an MOI of 3. Transduction levels were determined by ACE2 staining via Flow Cytometry 72 h after the infection. ACE2 staining is described in Flow cytometry analysis.
Flow cytometry analysis
Cells were resuspended in staining buffer (PBS + 2% FBS) and incubated with fluorochrome‐conjugated antibodies for 30 min at 4°C. CD8 T cells were identified with CD8‐Pacific Blue antibody (Biolegend). Activation of CD8 CAR‐T cells was determined with CD25 staining using the CD25‐APC antibody (Biolegend). CAR expressions of ACE2 CAR and anti‐Spike CAR and ACE2 expression of ACE2‐293 cells were determined with SARS‐CoV‐2 S1 protein, Mouse IgG2a Fc Tag (Acro Biosystems) incubation followed with APC Goat anti‐mouse IgG2a Fc Antibody (Invitrogen) staining and RFP expression. CAR expression of anti‐CD19 CAR was determined with Human CD19 (20–291) Protein, Fc Tag, low endotoxin (Super affinity) (Acro) followed by a secondary staining with APC conjugated anti‐human IgG Fc Antibody (Biolegend) and RFP expression. For cytotoxicity assay analysis, stably Spike protein‐expressing T2 and 293 cell lines were identified with GFP marker. For Spike protein flow cytometry analysis, the cells were stained with Biotinylated Human ACE2/ACEH Protein, Fc, Avitag (Acro Biosystems) and then stained with APC anti‐human IgG Fc Antibody clone HP6017 (Biolegend). Samples were acquired on a BD FACSymphony A5 analyser, and data were analysed using FlowJo (BD Biosciences).
Cytotoxicity assay
Following the expansion of engineered CAR‐T cells for 10–12 days, the cells were analysed for their RFP and CAR expressions. The effector‐to‐target cell ratio was calculated based on the number of CAR‐expressing cells. CAR‐expressing cells were titrated from 2:1 to 1:16 effector‐to‐target cell ratio at twofold dilutions, while the target cell number was constant. For ACE2‐Bite cytotoxicity assays, resting total CD8 T cells were combined with wild‐type Spike overexpressing 293 cells, empty vector transduced 293 cells, mutated Spike protein transfected 293 cells and wild‐type 293 cells in a 4:1 Effector/Target cell ratio, and ACE2‐Bite and control supernatant were added in 1:10 supernatant/cell medium ratio. Cytotoxicity assay conditions were analysed with Flow Cytometry at 72‐h postcoculture, and the cells were identified as described in Flow cytometry analysis.
ACE2‐Bite detection assay
Supernatants from ACE2‐Bite secreting and wild‐type suspension 293 cells were collected at several time points with different cell densities ranging from 3 to 7 million mL−1. ACE2‐Bite molecules taken from 3 million mL−1 cell culture supernatant were concentrated fivefold and 30‐fold by using 15 mL 30 kDa MilliporeSigma™ Amicon™ Ultra Centrifugal Filter Units. To capture the ACE2‐Bite or ACE2‐Fc molecules, The DevScreen SAv Bead kit (Essen BioScience, MI) was used. Biotinylated 2019‐nCoV (COVID‐19) spike protein RBD, His, Avitag, Biotinylated SARS‐CoV‐2 Spike Trimer, His, Avitag™ (B.1.1.529/Omicron) (MALS verified) and Biotinylated SARS‐CoV‐2 S protein, His, Avitag™, Super stable trimer (MALS verified) were coated to SAv Beads according to the manufacturer's instructions. Confirmation of successful bead conjugation was determined by staining with anti‐His‐Tag (Biolegend) and flow cytometry analysis. The conjugated beads were then used as capture beads in flow immunoassay where they were incubated with recombinant Human ACE2‐Fc (Acro Biosystems) or ACE2‐Bite supernatant samples for 1 h at room temperature. Supernatant samples were assayed at a 1:1 starting dilution and three additional tenfold serial dilutions. ACE2‐Fc was tested at a 30 μg mL−1 starting concentration and in additional five threefold serial dilutions. Detection reagent was prepared using Human CD3 epsilon Protein, Mouse IgG2a Fc Tag (Acro) and Phycoerythrin‐conjugated Goat anti‐Mouse IgG2a Cross‐Adsorbed Secondary Antibody (Fisher) for ACE2‐Bite and APC anti‐human IgG Fc Antibody clone HP6017 (Biolegend) for ACE2‐Fc were added to the wells and incubated for another hour at room temperature. Plates were then washed twice with PBS and analysed by flow cytometry using iQue Screener Plus (IntelliCyt, MI). Flow cytometry data were analysed using FlowJo (BD biosciences). DevScreen SAv Beads were gated using FSC‐H/SSC‐H, and singlet beads gate was created using FSC‐A/FSC‐H. Gates for different DevScreen SAv Beads were determined based on their fluorescence signature on the RL1‐H/RL2‐H plot (on iQue plus). The PE fluorescence median, directly associated with each single plex bead, was determined using BL2‐H (on iQue plus). Geometric means of PE fluorescence in different titrations were used to generate the titration curve, and the area under the curve was calculated using GraphPad Prism 9.0 software (GraphPad Software).
Spike‐pseudotyped virus neutralisation assay
Threefold serially diluted recombinant human ACE2‐Fc (Acro Biosystems) or twofold serially diluted ACE2‐Bite and control supernatants were incubated with GFP‐encoding SARS‐CoV‐2 Spike‐pseudotyped viruses with 0.2 multiplicity of infection (MOI) for 1 h at 37°C degrees. The mixtures were subsequently added to ACE2+ 293 cells, which were previously stained for their ACE2 expressions and confirmed ~%100 positive before neutralisation assays, for 72 h after which cells were collected, washed with FACS buffer (1×PBS + 2% FBS) and analysed by flow cytometry using a BD FACSymphony A5 analyser. Cells not expressing GFP were used to define the boundaries between non‐infected and infected cell populations. Per cent infection was normalised for samples derived from cells infected with SARS‐CoV‐2 pseudotyped virus in the absence of ACE2‐Fc or ACE2‐Bite.
Statistical analyses and reproducibility
All statistical analyses were performed, and graphs were prepared using GraphPad Prism V9 software. The number of repeats for each experiment is described in the figure captions.
Conflict of interest
MD and DU are inventors in a provisional patent application.
Author contributions
Mikail Dogan: Conceptualization; data curation; formal analysis; investigation; methodology; project administration; software; validation; visualization; writing – original draft. Lina Kozhaya: Conceptualization; data curation; formal analysis; investigation; methodology. Lindsey Placek: Methodology. Fatih Karabacak: Methodology. Mesut Yigit: Conceptualization; investigation; methodology. Derya Unutmaz: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; software; supervision; validation; visualization; writing – review and editing.
Supporting information
Supplementary figures 1–3
Acknowledgments
The research in this study was supported by the National Institute of Health (NIH) grant U19 AI142733‐01 (DU) and the Achelis and Bodman Foundation (DU). We thank Courtney Gunter and Sara Cassidy for critical reading and Mustafa Semih Elitok for advice on designing the anti‐Spike antibody ScFv protein.
References
- 1. Wu Q, Dudley MZ, Chen X et al. Evaluation of the safety profile of COVID‐19 vaccines: a rapid review. BMC Med 2021; 19: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baum A, Ajithdoss D, Copin R et al. REGN‐COV2 antibodies prevent and treat SARS‐CoV‐2 infection in rhesus macaques and hamsters. Science 2020; 370: 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gottlieb RL, Nirula A, Chen P et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID‐19: a randomized clinical trial. JAMA 2021; 325: 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marconi VC, Ramanan AV, de Bono S et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID‐19 (COV‐BARRIER): a randomised, double‐blind, parallel‐group, placebo‐controlled phase 3 trial. Lancet Respir Med 2021; 9: 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guimaraes PO, Quirk D, Furtado RH et al. Tofacitinib in patients hospitalized with Covid‐19 pneumonia. N Engl J Med 2021; 385: 406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gandhi RT, Lynch JB, Del Rio C. Mild or moderate Covid‐19. N Engl J Med 2020; 383: 1757–1766. [DOI] [PubMed] [Google Scholar]
- 7. RECOVERY Collaborative Group , Horby P, Lim WS et al. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med 2021; 384: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cellina M, Orsi M, Bombaci F, Sala M, Marino P, Oliva G. Favorable changes of CT findings in a patient with COVID‐19 pneumonia after treatment with tocilizumab. Diagn Interv Imaging 2020; 101: 323–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huet T, Beaussier H, Voisin O et al. Anakinra for severe forms of COVID‐19: a cohort study. Lancet Rheumatol 2020; 2: e393–e400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Monk PD, Marsden RJ, Tear VJ et al. Safety and efficacy of inhaled nebulised interferon beta‐1a (SNG001) for treatment of SARS‐CoV‐2 infection: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Respir Med 2021; 9: 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conway SR, Keller MD, Bollard CM. Cellular therapies for the treatment and prevention of SARS‐CoV‐2 infection. Blood 2022; 140: 208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoffmann M, Kleine‐Weber H, Schroeder S et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020; 181: 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fyhrquist F, Metsärinne K, Tikkanen I. Role of angiotensin II in blood pressure regulation and in the pathophysiology of cardiovascular disorders. J Hum Hypertens 1995; 9 Suppl 5: S19–S24. [PubMed] [Google Scholar]
- 14. Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA. Effects of common mutations in the SARS‐CoV‐2 spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife 2021; 10: e70658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harvey WT, Carabelli AM, Jackson B et al. SARS‐CoV‐2 variants, spike mutations and immune escape. Nat Rev Microbiol 2021; 19: 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ou X, Liu Y, Lei X et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun 2020; 11: 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmad A, Uddin S, Steinhoff M. CAR‐T cell therapies: an overview of clinical studies supporting their approved use against acute lymphoblastic leukemia and large B‐cell lymphomas. Int J Mol Sci 2020; 21: 3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cattin‐Ortola J, Welch LG, Maslen SL, Papa G, James LC, Munro S. Sequences in the cytoplasmic tail of SARS‐CoV‐2 spike facilitate expression at the cell surface and syncytia formation. Nat Commun 2021; 12: 5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dieterle ME, Haslwanter D, Bortz RH 3rd et al. A replication‐competent vesicular stomatitis virus for studies of SARS‐CoV‐2 spike‐mediated cell entry and its inhibition. Cell Host Microbe 2020; 28: 486–496.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H. The SARS‐CoV‐2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike‐based vaccine immunogens. Front Immunol 2020; 11: 576622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wan Q, Kozhaya L, Imberg K et al. Probing the effector and suppressive functions of human T cell subsets using antigen‐specific engineered T cell receptors. PLoS One 2013; 8: e56302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sterner RC, Sterner RM. CAR‐T cell therapy: current limitations and potential strategies. Blood Cancer J 2021; 11: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dogan M, Kozhaya L, Placek L et al. SARS‐CoV‐2 specific antibody and neutralization assays reveal the wide range of the humoral immune response to virus. Commun Biol 2021; 4: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol 2018; 18: 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wraith DC. The future of immunotherapy: a 20‐year perspective. Front Immunol 2017; 8: 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitsuyasu RT, Anton PA, Deeks SG et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene‐modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus‐infected subjects. Blood 2000; 96: 785–793. [PubMed] [Google Scholar]
- 27. Leibman RS, Richardson MW, Ellebrecht CT et al. Supraphysiologic control over HIV‐1 replication mediated by CD8 T cells expressing a re‐engineered CD4‐based chimeric antigen receptor. PLoS Pathog 2017; 13: e1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Savoldo B, Ramos CA, Liu E et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor‐modified T cells in lymphoma patients. J Clin Invest 2011; 121: 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kruse RL, Shum T, Tashiro H et al. HBsAg‐redirected T cells exhibit antiviral activity in HBV‐infected human liver chimeric mice. Cytotherapy 2018; 20: 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sautto GA, Wisskirchen K, Clementi N et al. Chimeric antigen receptor (CAR)‐engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 2016; 65: 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Full F, Lehner M, Thonn V et al. T cells engineered with a cytomegalovirus‐specific chimeric immunoreceptor. J Virol 2010; 84: 4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang X, Zhou Y, Li W et al. T cells expressing a LMP1‐specific chimeric antigen receptor mediate antitumor effects against LMP1‐positive nasopharyngeal carcinoma cells in vitro and in vivo . J Biomed Res 2014; 28: 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumaresan PR, Manuri PR, Albert ND et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci USA 2014; 111: 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melenhorst JJ, Chen GM, Wang M et al. Decade‐long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022; 602: 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalos M, Levine BL, Porter DL et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off‐the‐shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov 2020; 19: 185–199. [DOI] [PubMed] [Google Scholar]
- 37. Bailey JR, Barnes E, Cox AL. Approaches, progress, and challenges to hepatitis C vaccine development. Gastroenterology 2019; 156: 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnston MI, Fauci AS. An HIV vaccine–challenges and prospects. N Engl J Med 2008; 359: 888–890. [DOI] [PubMed] [Google Scholar]
- 39. Kantarjian H, Stein A, Gokbuget N et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 2017; 376: 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu S, Zhang J, Yan Y et al. A novel asymmetrical anti‐HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2‐positive tumor cells. J Exp Clin Cancer Res 2019; 38: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goldstein RL, Goyos A, Li CM et al. AMG 701 induces cytotoxicity of multiple myeloma cells and depletes plasma cells in cynomolgus monkeys. Blood Adv 2020; 4: 4180–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferrari F, Bellone S, Black J et al. Solitomab, an EpCAM/CD3 bispecific antibody construct (BiTE(R)), is highly active against primary uterine and ovarian carcinosarcoma cell lines in vitro . J Exp Clin Cancer Res 2015; 34: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lutterbuese R, Raum T, Kischel R et al. T cell‐engaging BiTE antibodies specific for EGFR potently eliminate KRAS‐ and BRAF‐mutated colorectal cancer cells. Proc Natl Acad Sci USA 2010; 107: 12605–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hosseini I, Gadkar K, Stefanich E et al. Mitigating the risk of cytokine release syndrome in a phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. NPJ Syst Biol Appl 2020; 6: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horn LA, Ciavattone NG, Atkinson R et al. CD3xPDL1 bi‐specific T cell engager (BiTE) simultaneously activates T cells and NKT cells, kills PDL1+ tumor cells, and extends the survival of tumor‐bearing humanized mice. Oncotarget 2017; 8: 57964–57980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brey CU, Proff J, Teufert N et al. A gB/CD3 bispecific BiTE antibody construct for targeting human cytomegalovirus‐infected cells. Sci Rep 2018; 8: 17453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sung JA, Pickeral J, Liu L et al. Dual‐affinity re‐targeting proteins direct T cell‐mediated cytolysis of latently HIV‐infected cells. J Clin Invest 2015; 125: 4077–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zoufaly A, Poglitsch M, Aberle JH et al. Human recombinant soluble ACE2 in severe COVID‐19. Lancet Respir Med 2020; 8: 1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Monteil V, Kwon H, Prado P et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell 2020; 181: 905–913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Glasgow A, Glasgow J, Limonta D et al. Engineered ACE2 receptor traps potently neutralize SARS‐CoV‐2. Proc Natl Acad Sci USA 2020; 117: 28046–28055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Case JB, Rothlauf PW, Chen RE et al. Neutralizing antibody and soluble ACE2 inhibition of a replication‐competent VSV‐SARS‐CoV‐2 and a clinical isolate of SARS‐CoV‐2. Cell Host Microbe 2020; 28: 475–485.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ding S, Adam D, Beaudoin‐Bussieres G et al. SARS‐CoV‐2 spike expression at the surface of infected primary human airway epithelial cells. Viruses 2021; 14: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Subklewe M. BiTEs better than CAR T cells. Blood Adv 2021; 5: 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haschke M, Schuster M, Poglitsch M et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin‐converting enzyme 2 in healthy human subjects. Clin Pharmacokinet 2013; 52: 783–792. [DOI] [PubMed] [Google Scholar]
- 55. Khan A, Benthin C, Zeno B et al. A pilot clinical trial of recombinant human angiotensin‐converting enzyme 2 in acute respiratory distress syndrome. Crit Care 2017; 21: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–3