

Abstract
The increasing prevalence of candidosis caused by Candida glabrata is related to its ability to acquire azole resistance. Although azole resistance mechanisms are well known, the mechanisms for azole import into fungal cells have remained obscure. In this work, we have characterized two hexose transporters in C. glabrata and further investigate their role as potential azole importers. Three azole susceptible C. glabrata clinical isolates were evolved towards azole resistance and the acquired resistance phenotype was found to be independent of CgPDR1 or CgERG11 mutations. Through whole-genome sequencing, CgHXT4/6/7 was found to be mutated in the three evolved strains, when compared to their susceptible parents. CgHxt4/6/7 and the 96% identical CgHxt6/7 were found to confer azole susceptibility and increase azole accumulation in C. glabrata cells, strikingly rescuing the susceptibility phenotype imposed by CgPDR1 deletion, while the identified loss-of-function mutation in CgHXT4/6/7, leads to increased azole resistance. In silico docking analysis shows that azoles display a strong predicted affinity for the glucose binding site of CgHxt4/6/7. Altogether, we hypothesize that hexose transporters, such as CgHxt4/6/7 and CgHxt6/7, may constitute a family of azole importers, involved in clinical drug resistance in fungal pathogens, and constituting promising targets for improved antifungal therapy.
Subject terms: Pathogenesis, Microbiology
Mutations in the hexose transporter, CgHXT4/6/7, contribute to increased antifungal (azole) resistance in the fungal pathogen, Candida glabrata, potentially by influencing azole accumulation.
Introduction
Candida species are one of the most common cause of fungemia in humans, responsible for >400,000 life-threatening infections yearly worldwide with an associated 46–75% mortality rate1. Clinical isolates resistant to all three classes of antifungals available have been found in hospitalized patients2. C. glabrata, particularly, is a prominent cause of invasive candidosis, best known for its ability to overcome azole antifungal therapy3–6.
Azoles act by inhibiting the 14α-demethylase Erg11 in the ergosterol biosynthesis pathway, causing the accumulation of a toxic sterol that permeabilizes the plasma membrane7. These drugs have been used for decades as the standard treatment against candidosis and are still the only oral treatment option. Instead of developing new drugs to overcome antifungal resistance, a process which is both very costly and time-consuming, therapeutic strategies can be improved by enhancing the efficacy of existing drugs, such as azoles. Therefore, it is worth identifying the genes and cellular pathways involved in the development of drug resistance8.
The main mechanism of acquired azole resistance is the up-regulation of ABC (ATP-binding Cassette) drug transporters, mainly CgCdr19–11, and, to a lower extent, of the major facilitator superfamily (MFS) drug:H + antiporters12–19, which catalyse the efflux of azoles preventing their intracellular accumulation. Generally, the constitutive up-regulation of the ABC efflux pumps is due to the emergence of gain-of-function (GOF) mutations in the CgPDR1 gene, encoding the key regulator of multidrug resistance in C. glabrata4, 20. In contrast to what is observed in C. albicans, several studies suggest that mutations in CgERG11 is not a relevant mechanism of acquired azole resistance in C. glabrata clinical isolates5, 9, 21. Although CgPDR1 GOF mutations are considered the major mechanism of clinical acquisition of azole resistance, several resistant clinical isolates do not to display any of the known mechanisms of resistance22, 23, suggesting the presence of yet unknown alternative paths to acquired azole resistance in C. glabrata.
In this study, prolonged exposure to posaconazole was used to induce in vitro C. glabrata azole susceptible clinical isolates to acquire azole resistance. The underlying molecular mechanisms were evaluated through whole-genome sequencing. Genes found to exhibit non-synonymous single-nucleotide polymorphisms (nsSNPs) in the resistant strains, when compared to their susceptible counterparts, were identified and evaluated for a possible role in azole resistance. The putative hexose transporter CgHXT4/6/7 (ORF CAGL0A02233g) stood out in the analysis, being hypothesized to work as an azole importer. Although little is known about how these drugs enter pathogenic fungal cells to exert their antifungal action24, it was demonstrated that azoles are imported by facilitated diffusion rather than passive diffusion or ATP-dependent import25. Moreover, fluconazole import differs among C. albicans resistant clinical isolates, which suggests that altered facilitated diffusion is possibly a previously uncharacterized mechanism of resistance to azole drugs25.
In this context, the role of CgHxt4/6/7 and of the 96% identical CgHxt6/7 (ORF CAGL0A02211g)26 as hexose transporters was confirmed, while their role as potential azole importers was further explored leading to the characterization of an alternative mechanism of acquired azole drug resistance in C. glabrata.
Results
In vitro evolution towards posaconazole resistance in C. glabrata clinical isolates
Four isolates, previously found to display azole susceptibility27, the CBS138 reference strain and three clinical isolates (040, 044 and OL152), were subjected to an in vitro directed evolution approach, to generate azole-resistant populations. After prolonged exposure to a therapeutic plasma concentration of the triazole posaconazole (1 mg L−1), the CBS138, 040 and OL152 isolates evolved towards posaconazole, fluconazole and clotrimazole resistance following 10 days of incubation, while the 044 isolate acquired resistance to clotrimazole, fluconazole, voriconazole and posaconazole after 25 days of incubation (Supplementary Table 1). Interestingly, MIC50 levels for voriconazole never reached resistance levels for CBS138, 040 and OL152 isolates (Supplementary Table 1). The evolved multiazole-resistant strains were denominated CBS138_Psc, 040_Psc, 044_Psc and OL152_Psc. The resistance pattern remained stable in all the evolved resistant isolates following sub-culture in drug-free medium for up to 30 days.
Acquisition of azole resistance can be independent of mutations in CgPDR1 or CgERG11
To understand whether the evolved resistance phenotype relied on the major mechanism of acquired azole resistance, the acquisition of Pdr1 GOF mutations, the transcription factor encoding gene CgPDR1 was sequenced in the four evolved resistant strains (CBS138_Psc, 040_Psc, 044_Psc and OL152_Psc). Remarkably, no sequence changes were detected in 040_Psc, 044_Psc or OL152_Psc and so the molecular basis underlying the development of azole resistance on these three evolved strains remained to be clarified. In the azole-resistant strain derived from the CBS138 strain, CBS138_Psc, a mutation in the CgPDR1 gene sequence was found. In this single case, indeed a non-synonymous mutation leading to a Trp297Leu substitution was observed, which corresponds to a previously characterized GOF mutation in CgPDR123. This mutation is likely underlying the observed resistance phenotype in the evolved strain, which was therefore excluded from further analysis. Furthermore, no changes in the sequence or expression (Supplementary Fig. 1) of the drug target, CgErg11, were found in the four evolved strains, reinforcing the assumption that alteration of the drug target is not a usual mechanism of acquired azole resistance in C. glabrata clinical isolates. Potential resistance-related mutations in the promoter regions of both CgPDR1 and CgERG11 were also not found (Supplementary Data 1).
Identification of nsSNPs acquired during posaconazole exposure
In the search for insights into the mechanism(s) underlying the development of azole resistance in the evolved strains 040_Psc, 044_Psc and OL152_Psc, their genomes were sequenced and compared to that of the corresponding susceptible parents 040, 044 and OL152, respectively (Supplementary Data 2; Supplementary Data 3). 40, 70 and 103 nsSNPs were registered for the 040_Psc, 044_Psc and OL152_Psc strains, respectively, when compared to the susceptible parents (Supplementary Fig. 2a). These nsSNPs were found to affect 19, 22 and 28 genes in 040_Psc, 044_Psc and OL152_Psc, respectively (Supplementary Fig. 2b), which were grouped based on their biological function (Supplementary Table 2). Most of the mutated genes were found to encode adhesins or adhesin-like proteins, specifically 70%, 92% and 70% of the mutated genes in the 040_Psc, 044_Psc and OL152_Psc strains, respectively. This is likely a direct consequence of adhesin encoding genes being mostly located in sub-telomeric regions and including tandem-repeat regions, which favours increased random mutation rates. Nonetheless, it is interesting to point out that at least the CgEPA3 adhesin has been implicated in azole resistance28.
Despite no common nsSNPs selected during prolonged posaconazole exposure among the 040_Psc, 044_Psc and OL152_Psc strains (Supplementary Fig. 2a) were registered, 5 genes were found to display non-synonymous mutations in the three resistant strains, when compared to the susceptible counterparts. These genes included 4 adhesin-like proteins, including CgEPA3, plus the putative hexose transporter CgHXT4/6/7 (Supplementary Fig. 2b), which was selected for further analysis.
CgHxt4/6/7 and CgHxt6/7 are plasma membrane hexose transporters
The subcellular localization of CgHxt4/6/7 and of the 96% identical CgHxt6/7 was inspected by fluorescence microscopy in both C. glabrata and S. cerevisiae cells. As expected, the CgHxt4/6/7_GFP and the CgHxt6/7_GFP fusion proteins were found to be predominantly localized to the cell periphery, which is consistent with its predicted plasma membrane localization and transporter function, in both C. glabrata and S. cerevisiae cells (Fig. 1a).
Fig. 1. CgHxt4/6/7 and CgHxt6/7 are plasma membrane hexose transporters.

a Fluorescence of exponential phase BY4741 S. cerevisiae cells harbouring the plasmids pGREG576_TEF_CgHXT4/6/7 (CgHxt4/6/7_GFP) or pGREG576_TEF_CgHXT6/7 (CgHxt6/7_GFP), and KUE100::URA- C. glabrata cells harbouring the plasmids pGREG576_PDC1_CgHXT4/6/7 (CgHxt4/6/7_GFP) or pGREG576_PDC1_CgHXT6/7 (CgHxt6/7_GFP), after 6 h of constitutive recombinant protein production. b Comparison of spot growth assays of the EBY.VW4000 (Schxt0) strain harbouring the pGREG567_TEF vector or the pGREG576_TEF_CgHXT4/6/7 or pGREG576_TEF_CgHXT6/7 expression plasmids in the presence of different indicated hexoses as carbon sources. The inocula were prepared as described in the Methods section. Cell suspensions used to prepare the spots were 1:5 (b) and 1:25 (c) dilutions of the cell suspensions used in (a). The displayed images are representative of at least three independent experiments.
The ability of the putative hexose transporters CgHxt4/6/7 and CgHxt6/7 to import the hexoses glucose, mannose, fructose and galactose into yeast cells was assessed using the hexose transporter-null S. cerevisiae strain (Schxt0), which grows normally on maltose as carbon source29. The Schxt0 strain is unable to grow on glucose, mannose or fructose as sole carbon sources and grows very poorly on galactose29. Results demonstrate that Schxt0 cells expressing either CgHxt4/6/7 or CgHxt6/7 were able to grow on glucose and mannose and slightly on fructose as carbon sources, while cells harbouring the cloning vector were not (Fig. 1b). Together, these results demonstrate that CgHxt4/6/7 and CgHxt6/7 are plasma membrane hexose transporters, which was expected considering the role of their S. cerevisiae orthologues ScHxt6 and ScHxt7, respectively30.
CgHXT4/6/7 and CgHXT6/7 promote azole drug susceptibility in C. glabrata, probably by facilitating azole entrance into fungal cells
Since the so far uncharacterized hexose transporter encoding gene CgHXT4/6/7 stood out in the genome-wide analysis of the evolved resistant strains 040_Psc, 044_Psc and OL152_Psc, the potential impact of this transporter in C. glabrata tolerance to azoles was further evaluated, along with its close homologue CgHXT6/7. By comparing the susceptibility of wild-type, single deletion strains (Δcghxt4/6/7 and Δcghxt6/7) and double deletion strain (Δcghxt4/6/7Δcghxt6/7), the absence of either HXT gene was found to increase C. glabrata resistance towards fluconazole and posaconazole (Fig. 2a). Accordingly, MIC50 values for fluconazole and posaconazole were found to be lower for the wild-type strain (16 and 0.5 mg L−1, respectively) when compared to the single deletion strains Δcghxt4/6/7 (32 and 1 mg L−1, respectively) or Δcghxt6/7 (32 and 1 mg L−1, respectively), and to the double deletion strain Δcghxt4/6/7Δcghxt6/7 (64 and 2 mg L−1, respectively).
Fig. 2. CgHxt4/6/7 and CgHxt6/7 promote azole susceptibility in C. glabrata.

a Comparison of the susceptibility to antifungal azole drugs, at the indicated concentrations, of the C. glabrata KEU100 wild-type strain or derived KUE100_Δcghxt4/6/7, KUE100_Δcghxt6/7 or KUE100_Δcghxt4/6/7Δcghxt6/7 mutant cells on YPD agar plates by spot assays. The inoculum was prepared as described in the Methods section. Cell suspensions used to prepare the spots correspond to 1:5 (b) and 1:25 (c) dilutions of the cell suspensions used in (a). The displayed images are representative of at least three independent experiments. b Time-course accumulation ratio of [3H]-Fluconazole in non-adapted KUE100 wild-type cells (black circles) or derived KUE100_Δcghxt4/6/7 mutant cells (grey squares) during cultivation in liquid YPD medium in the presence of unlabelled fluconazole. The accumulation ratio values are averages of, at least, n = 3 independent experiments. Error bars represent the corresponding standard deviations. Significance levels are attributed as follows: *p value < 0.05; **p value < 0.01; ***p value < 0.001.
The results obtained for either CgHxt4/6/7 or CgHxt6/7 are consistent, but moderate, likely because 10 other hexose transporters are predicted to be encoded by the C. glabrata genome, potentially displaying functional overlap. In fact, the double deletion strain Δcghxt4/6/7Δcghxt6/7 was found to display a fourfold increase in the MIC50 for both fluconazole and posaconazole, comparing to the wild-type. To assess the concept of functional overlap, the hexose transporter-null Schxt0 mutant was compared with the corresponding parental strain in terms of azole susceptibility. Hexose transporter-null Schxt0 cells are indeed less susceptible to the azoles tested (Fig. 3). Although this finding needs to be further assessed in C. glabrata, this result strongly supports the hypothesis that several of the HXT transporters might play a key role as azole importers, therefore contributing to azole susceptibility in fungal cells. Accordingly, the accumulation of [3H]-labelled fluconazole was evaluated in the single deletion strain ∆cghxt4/6/7 comparing to the wild-type. Under these conditions, cells lacking CgHXT4/6/7 accumulate significantly less fluconazole than the parental wild-type cells (Fig. 2b). Moreover, the same effect was observed when measuring the accumulation of the [3H]-labelled imidazole clotrimazole in cells lacking CgHXT4/6/7, comparing to wild-type cells (Supplementary Fig. 3), further supporting the hypothesis that CgHxt4/6/7 affects azole uptake in C. glabrata.
Fig. 3. HXT genes play a role in yeast susceptibility towards azole antifungals.
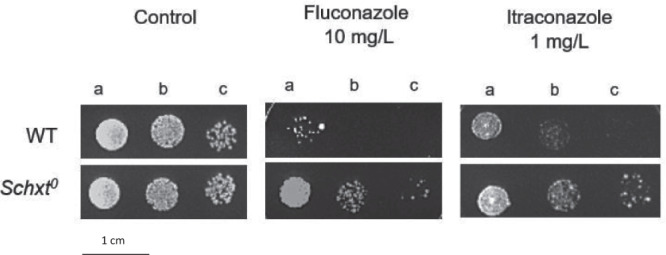
Comparison of the susceptibility to azole drugs, at the indicated concentrations, of the CEN.PK2-1C S. cerevisiae wild-type strain and the derived EBY.VW4000 (Schxt0) strain on YPM agar plates by spot assays. The inoculum was prepared as described in the Methods section. Cell suspensions used to prepare the spots correspond to 1:5 (b) and 1:25 (c) dilutions of the cell suspensions used in (a). The displayed images are representative of at least three independent experiments.
Deletion of CgHXT4/6/7 in the 040 susceptible clinical isolate promotes azole resistance
The relevance of CgHxt4/6/7 in the azole resistance phenotype developed by the 040 clinical isolate when exposed to posaconazole was further evaluated. Susceptibility testing demonstrates that the deletion of CgHXT4/6/7 in the 040 isolate decreases its susceptibility towards posaconazole and fluconazole (Fig. 4a). MIC50 values of fluconazole and posaconazole were found to be lower for the azole susceptible isolate 040 (16 and 0.125 mg L−1, respectively) when compared to the derived mutant strain 040_∆cghxt4/6/7 (32 and 2 mg L−1, respectively), while for the evolved 040_Psc resistant strain the values were even higher (≥64 and ≥8 mg L−1, respectively). Accordingly, the accumulation of [3H]-labelled fluconazole was shown to be significantly lower in the 040_∆cghxt4/6/7 strain when comparing to the susceptible parent isolate 040 (Fig. 4b). Although it is noteworthy that the overall accumulation of [3H]-labelled fluconazole appears to be higher in the 040 clinical isolate, when compared to the KUE100 lab strain, likely due to extensive genomic background differences, the impact of CgHXT4/6/7 expression in azole accumulation is clearly strain-independent. Additionally, the G361A missense mutation found in the CgHXT4/6/7 gene from the evolved 040_Psc strain, resulting in a Val121ILe amino acid residue substitution, was evaluated for its impact in the acquired azole-resistant phenotype (Fig. 4c). In that sense, the pGREG576_PDC1_mut_CgHXT4/6/7 plasmid was generated by site-directed mutagenesis. The introduction of the pGREG576_PDC1_mut_CgHXT4/6/7 plasmid in the single deletion mutant strain ∆cghxt4/6/7 did not affect the MIC50 values for fluconazole (MIC50 = 32mg L-1) and posaconazole (MIC50 = 1 mg L−1), while its complementation with the pGREG576_PDC1_CgHXT4/6/7 plasmid, expressing the wild-type CgHxt4/6/7 transporter, resulted in promoted susceptibility of the ∆cghxt4/6/7 strain, e.g. displaying lower MIC50 values for fluconazole (MIC50 = 16 mg L−1) and posaconazole (MIC50 = 0.5 mg L−1) (Fig. 4c).
Fig. 4. CgHxt4/6/7 promotes azole susceptibility in C. glabrata 040 clinical isolate by mediating azoles uptake.

a Comparison of the susceptibility to antifungal azole drugs, at the indicated concentrations, of the C. glabrata 040 clinical isolate, the derived mutant strain 040_Δcghxt4/6/7 and the evolved resistant strain 040_Psc on YPD agar plates by spot assays. The inoculum was prepared as described in the Methods section. Cell suspensions used to prepare the spots correspond to 1:5 (b) and 1:25 (c) dilutions of the cell suspensions used in (a). The displayed images are representative of at least three independent experiments. b Time-course accumulation ratio of [3H]-Fluconazole in non-adapted (black circles) 040 or derived (grey squares) 040_Δcghxt4/6/7 mutant cells during cultivation in liquid YPD medium in the presence of unlabelled fluconazole. The accumulation ratio values are averages of, at least, n = 3 independent experiments. Error bars represent the corresponding standard deviations. Significance levels are attributed as follows: *p value < 0.05; **p value < 0.01. c Comparison of the susceptibility to antifungal azole drugs, at the indicated concentrations, of the C. glabrata KUE100::URA-_Δcghxt4/6/7 mutant strain harbouring the pGREG576_PDC1_CgHXT4/6/7 or pGREG576_PDC1_mut_CgHXT6/7 on BM agar plates (without uracil, for plasmid maintenance) by spot assays. The inocula were prepared as described in the Methods section. Cell suspensions used to prepare the spots were 1:5 (b) and 1:25 (c) dilutions of the cell suspensions used in (a). The displayed images are representative of at least three independent experiments.
Altogether, these results strongly suggest that CgHXT4/6/7 plays a role in C. glabrata azole drug susceptibility and that the nsSNP selected in CgHXT4/6/7 of the 040 isolate during the in vitro evolution experiment impaired of the transporter function, contributing to the final resistant phenotype observed in the evolved strain 040_Psc.
Deletion of either CgHXT4/6/7 or CgHXT6/7 rescues the susceptibility phenotype imposed by the absence of the drug resistance regulator CgPDR1
CgPdr1 is the major regulator of azole resistance in C. glabrata, controlling the overexpression of azole drug efflux pumps. Consequently, cells lacking the CgPDR1 gene are highly susceptible to azoles. Surprisingly, the deletion of either CgHXT4/6/7 or CgHXT6/7 was found to rescue the azole susceptibility phenotype of CgPDR1 disrupted cells (Fig. 5a). Strikingly, MIC50 values for fluconazole and posaconazole were found to be similar for the wild-type strain (16 and 0.5 mg L−1, respectively) and for the double deletion strains ∆cgpdr1∆cghxt4/6/7 or ∆cgpdr1∆cghxt6/7 (16 and 0.5 mg L−1, respectively), while for the ∆cgpdr1 strain the MIC50 values were found to be 4–fold lower for fluconazole (4 mg L−1) and 32-fold lower for posaconazole (0.015625 mg L−1). Accordingly, growth curves obtained in the presence of inhibitory concentrations of posaconazole (4 mg L−1) demonstrate that the deletion of either CgHxt4/6/7 or CgHxt6/7 in the ∆cgpdr1 strain fully rescues its azole susceptibility phenotype (Fig. 5b).
Fig. 5. Deletion of either CgHXT4/6/7 or CgHXT6/7 in a ∆cgpdr1 background rescues the azole susceptible phenotype imposed by the absence of CgPdr1.

a Comparison of the susceptibility to antifungal azole drugs, at the indicated concentrations, of the KUE100 C. glabrata wild-type strain and the derived mutant strains KUE100_Δcgpdr1, KUE100_Δcgpdr1Δcghxt4/6/7, and KUE100_Δcgpdr1Δcghxt6/7 on YPD agar plates by spot assays. The inoculum was prepared as described in the “Methods” section. Cell suspensions used to prepare the spots correspond to 1:5 (b) and 1:25 (c) dilutions of the cell suspensions used in (a). The displayed images are representative of, at least, n = 3 independent experiments. b Comparison of the growth curves of the C. glabrata wild-type strain KUE100 and the derived mutant strains KUE100_Δcgpdr1, KUE100_Δcgpdr1Δcghxt4/6/7, and KUE100_Δcgpdr1Δcghxt6/7, in liquid YPD, in the absence or presence of 4 mg/L posaconazole, measured in terms of variation in OD600. The displayed growth curves are representative, at least, n = 3 independent experiments. Error bars represent the corresponding standard deviation. Significance levels are attributed as follows: ****p value <0.0001.
These results further support the hypothesis that these putative hexose transporters play a crucial role in C. glabrata azole drug susceptibility, probably by facilitating azole entrance into fungal cells. More importantly, this points out the critical relevance of these transporters in azole drug mode of action and in the consequent mechanism of drug resistance.
CgHxt4/6/7 is predicted to display high affinity for fluconazole binding
To strengthen our current hypothesis that CgHxt4/6/7 may act as an azole importer, the affinity of this hexose transporter to azoles was evaluated in silico. Since there is no available structure for CgHxt4/6/7, the CgHxt4/6/7 structure was modelled based on the 3D structure of the homologous E. coli transporter XylE (PDB ID 4GBZ) (Supplementary Fig. 4a). This was possible due to the sequence identity between both proteins, which is of 30% for all residues and 50% for the residues exposed to the transporting channel (Supplementary Fig. 4c). The crystal structure of E. coli XylE was solved with a resolution of 2.89 Å and features a D-glucose bound to its internal cavity (Supplementary Fig. 4b), providing information about the positioning of the ligands to be transported. Most of the structural differences between our model and the E. coli XylE crystal structure are found in the loops that connect the transmembrane helices (Fig. 6a). These loops are unstructured and exposed to the solvent regions that are far from the substrate binding regions, therefore not accountable for the quality of the model at the transmembrane region (Fig. 6a). As observed for the E. coli XylE (PDB ID 4GBZ) protein, our model is in an outward-facing, partially occluded conformation with a D-glucose molecule captured in the transmembrane channel. The ligand is trapped within the centre of the transmembrane domain, completely occluded from the intracellular side, yet solvent-accessible from the extracellular side through a channel that is too narrow (5.2 Å diameter) to allow the escape of the ligand31.
Fig. 6. Fluconazole displays a strong predicted affinity for the glucose binding site of CgHxt4/6/7.

a Overlap of the best DOPE score model and the template E. coli XylE (PDB ID 4GBZ) (pink). Loop regions that differ from the template are represented in green. b Docking of fluconazole on the modelled CgHxt4/6/7 structure, best pose. c Docking of D-glucose on the modelled CgHxt4/6/7 structure, best pose. Figures were prepared using PyMOL2.5.
Using the modelled structure of CgHxt4/6/7, its affinity to fluconazole was predicted through in silico docking, using AutoDock Vina, and compared to that of glucose, the natural substrate of this transporter. This study revealed that CgHxt4/6/7 is predicted to have strong affinity to glucose (−5.5 kcal/mol), as expected, and an even stronger predicted affinity to fluconazole (−7.3 kcal/mol) (Fig. 6b, c). Similar binding affinities to glucose and fluconazole were observed for the E. coli XylE transporter (Supplementary Fig. 5a, b). Although occupying the same region in the internal cavity, fluconazole and D-glucose revealed distinct patterns of interactions with the protein in our docking studies. D-glucose participates in more hydrogen bonds than fluconazole, whereas desolvation effects seem to play a major role in the binding affinity of the latter. Indeed, fluconazole shows a higher shape complementarity to the binding pocket compared to D-glucose (Fig. 6b, c). Taken together, our results suggest that Fluconazole is predicted to be a binder of CgHxt4/6/7. Despite these results, posaconazole docking failed to bind to the transporter, what would be expected given the size difference between the fluconazole and posaconazole. However, it is important to consider that this modelling was made from a partially occluded conformation, and, as such we can hypothesize that posaconazole may be accommodated and transported in an open conformation. In order to test this possibility, we decided to make a second modelling from the structure of Homo sapiens glucose transporter (PDB ID 4ZWC)32 that was obtained in an outward-open conformation (Supplementary Fig. 6). Indeed, docking studies showed that the open configuration allows binding of both posaconazole and fluconazole. Interestingly, both drugs bind in the same region in the outward-open structure (Supplementary Fig. 7). It is important to mention that posaconazole is a large molecule with a higher number of possible rotations, which makes it difficult to predict the precise mode of binding, beyond the base structure common to fluconazole, through molecular docking.
Discussion
In this study, two hexose transporters in C. glabrata were identified and characterized. More importantly, a possible role for hexose transporters as drug carriers is proposed in C. glabrata, shedding light into an important feature of azole drug mode of action: its uptake into fungal cells, possibly through hexose transporters. An in vitro directed evolution experiment was used to drive susceptible clinical isolates towards multi azole resistance, leading to the functional characterization of the CgHxt4/6/7 hexose transporter as a determinant of azole susceptibility in C. glabrata. The in vitro evolved resistant isolates 040_Psc, 044_Psc and OL152_Psc acquired multi azole resistance through previously unknown mechanisms. No CgPDR1 GOF mutations were found in these evolved strains, the main mechanism of acquired azole resistance in C. glabrata clinical isolates4, 20. Moreover, according to the general notion that alterations on the drug target are not a frequent mechanism of acquired azole resistance in C. glabrata clinical isolates5, 9, 21, we confirmed that no mutations arose in the CgERG11 gene during posaconazole exposure. Although there were no common nsSNPs among the three evolved resistant strains, CgHXT4/6/7 gene was found to be mutated in all three evolved resistant strains (Supplementary Fig. 2b). Our results show that, although CgPDR1 GOF mutations are a frequent event leading to acquisition of azole resistance, they are not the only mechanism. Whether the posaconazole-driven evolution, when compared to evolution driven by other azole drugs, or the genetic background of the used strains may underlie the results obtained in this study remains unclear and is certainly worthy of further scrutiny.
CgHxt4/6/7 was, so far, an uncharacterized putative hexose transporter in C. glabrata. Based on knowledge gathered for S. cerevisiae homologous transporters, CgHxt4/6/7 belongs to the big family of HXT genes, specifically to the high-affinity glucose transporter cluster33. With 96% amino acid sequence identity to CgHxt4/6/7, CgHxt6/7 was also characterized. According to their predicted function, CgHxt4/6/7 and CgHxt6/7 were found to be localized to the cell plasma membrane (Fig. 1a), and to be able to import hexoses (Fig. 1b).
Several studies have covered a panoply of drug resistance mechanisms that depend on drug efflux pumps belonging to the ABC and MFS. Nonetheless, despite some efforts25, 34, the study of drug uptake mechanisms has been, to some extent, overlooked in pathogenic fungi24. Besides being an important mechanism of drug effectiveness acknowledged in human parasites24, decreased accumulation of drugs have been associated with azole resistance in Candida clinical isolates25. Nonetheless, the players underlying drug uptake have remained elusive for at least 20 years. HXT genes belong to the MFS class of membrane proteins and were associated with the multidrug resistance phenomenon in S. cerevisiae in 1997, being implicated in cycloheximide, sulfomethuron methyl and 4-NQO (4-nitroquinoline-N-oxide) susceptibility35. In fact, CgHxt4/6/7 and CgHxt6/7 S. cerevisiae orthologues (ScHxt6 and ScHxt7, respectively) were previously shown to be major mediators of arsenic import in yeast36. Our study shows that at least some of S. cerevisiae HXT transporters also play a relevant role in yeast azole susceptibility, probably by being hijacked by these antifungals serving as channels for drug entry into fungal cells (Fig. 3). Consistently, CgHxt4/6/7 or CgHxt6/7 expression was found to confer azole susceptibility to C. glabrata cells (Fig. 2a). The accumulation of radiolabelled fluconazole was shown to be higher in wild-type C. glabrata cells when compared to cells devoid of CgHXT4/6/7 (Fig. 2b), suggesting that fluconazole can enter C. glabrata cells through facilitated diffusion, mediated by this transporter. Furthermore, in silico molecular docking results corroborate this hypothesis, as they predict that CgHxt4/6/7 has affinity to its natural substrate, glucose, but also affinity to fluconazole and posaconazole. Interestingly, the CgHxt4/6/7 related phenotypes were observed in the C. glabrata lab strain KUE100, but also in the 040 clinical isolate (Fig. 4a, b), suggesting that the role of this transporter is not strain dependent. Additionally, the Val121Ile mutation that arose in the 040 clinical isolate upon prolonged exposure to posaconazole was found to render this transporter inactive in terms of azole susceptibility, suggesting that similar point mutations may be used as clinically relevant mechanisms of evolution towards azole resistance (Fig. 4c).
In wild-type C. glabrata cells, the prime mechanism of azole drug resistance involves CgPdr13. Once this transcription factor acquires GOF mutations, induced by drug exposure, natural selection favours cells harbouring these mutations, ultimately leading to the disappearance of other subpopulations and to the development of fully azole-resistant cells20, 28. Therefore, CgPDR1 disruption in C. glabrata cells restores azole susceptibility4. Since there were no GOF mutations in CgPDR1 in the evolved resistant strains 040_Psc, 044_Psc and OL152_Psc, we assessed the impact of the proposed resistance mechanism, involving the reduced uptake of fluconazole, in a strain devoid of CgPDR1. While in wild-type cells the single deletion of CgHxt4/6/7 or CgHxt6/7 results only in a moderate increase in azole resistance, in cells devoid of CgPdr1 the single deletion of either HXT gene fully reverses the strong azole susceptibility phenotype imposed by the absence of CgPdr1 (Fig. 5a, b). This further supports the concept that azoles may enter C. glabrata cells through Hxt proteins and that the impairment of this pathway constitutes an important mechanism of resistance, that, when triggered, renders alternative azole resistance pathways, based on drug extrusion, almost irrelevant.
Overall, we show that hexose transporters affect azole accumulation in C. glabrata and play a role in the acquisition of resistance by clinical isolates as an alternative to the main mechanism of acquired azole resistance involving CgPDR1. Despite the extensive knowledge on the mechanisms of action/resistance to azoles, little was known about how these drugs enter fungal cells to exert their antifungal action. Although indirect effects of HXT transporters in azole susceptibility, eventually affecting energy availability or the expression of drug efflux pumps, cannot be fully discarded, our results show that CgHxt4/6/7 and CgHxt6/7 transporters not only play a role in the uptake of glucose, but also appear to mediate azole uptake into C. glabrata cells. Being two in a family of 11 HXT transporters in C. glabrata, it is likely that their role in azole uptake is just the tip of the iceberg. It will be important to assess how far other homologous hexose transporters may contribute to azole uptake and whether mutations acquired in additional hexose transporter encoding genes may contribute to azole resistance in fungal pathogens, in general.
Methods
Strains and growth media
The reference strain CBS138 and three Candida glabrata clinical isolates (named OL152, 040 and 044) obtained from patients admitted to Centro Hospitalar de S. João (CHSJ) in Porto, Portugal were used in this study27. Isolates 040 and 044 were recovered from blood cultures, while isolate OL152 was recovered from urine. These isolates were characterized by Vitek YBC identification cards (BioMérieux, Paris, France) and mass-spectrometry (Maldi TOF). Until testing, the strains were stored in Brain–Heart broth (Difco) with 5% glycerol at −70 °C. For each experiment, the strains were subcultured twice on Sabouraud agar (Difco) at 35 °C for 48 h.
Candida glabrata parental strain KUE10037 and derived KUE100_Δcgpdr138 were kindly provided by Hiroji Chibana, Medical Mycology Research Center at Chiba University, Japan. KUE100_Δcghxt4/6/7, KUE100_Δcghxt6/7, KUE100_Δcghxt4/6/7Δcghxt6/7, KUE100_Δcgpdr1Δcghxt4/6/7, KUE100_Δcgpdr1Δcghxt6/7, and KUE100::URA-, were all constructed in this study. Cells were batch-cultured at 30 °C, with orbital agitation (250 rpm) in Basal Medium (BM) or Yeast Extract-Peptone-Dextrose (YPD) medium. BM has the following composition (per litre): 1.7 g yeast nitrogen base without amino acids or NH4+ (Difco), 20 g glucose (Merck) and 2.65 g (NH4)2SO4 (Merck). YPD has the following composition (per liter): 20 g glucose (Merck), 20 g Peptone (Merck) and 10 g Yeast extract (Merck).
Saccharomyces cerevisiae parental strain BY4741 (MATa, ura3∆0, leu2∆0, his3∆1, met15∆0) was obtained from the Euroscarf collection. Wild-type yeast cells harbouring pGREG576 derived plasmids were batch-cultured at 30 °C, with orbital agitation (250 rpm) in BM supplemented with 20 mg L−1 methionine, 20 mg L−1 histidine and 60 mg L−1 leucine (all from Sigma).
S. cerevisiae parental strain CEN.PK2-1C39 and derived hexose transporter-null (Schxt0) strain EBY.VW400029 were kindly provided by Eckhard Boles, Institute of Molecular Biological Sciences, Goethe University, Frankfurt, Germany. Cells were batch-cultured at 30 °C, with orbital agitation (250 rpm) in Yeast Extract-Peptone-Maltose (YPM) medium containing (per liter): 20 g maltose (Merck), 20 g Peptone (Merck) and 10 g Yeast extract (Merck). CEN.PK2-1C and EBY.VW4000 cells harbouring pGREG576 derived plasmids were grown overnight in BM-Maltose medium containing (per liter): 1.7 g yeast nitrogen base without amino acids or NH4+ (Difco), 20 g maltose (Merck) and 2.65 g (NH4)2SO4 (Merck), supplemented with 20 mg L−1 tryptophan, 20 mg L−1 histidine and 60 mg L−1 leucine (all from Sigma).
Solid media contained, besides the above indicated ingredients, 20 g L−1 agar (Iberagar).
Gene expression analysis
The transcript level of the CgERG11 gene in the initial azole susceptible clinical isolates was compared to the evolved resistant strains using quantitative real-time PCR (qRT-PCR). Total-RNA samples were obtained from cell suspensions harvested upon reaching an OD600nm = 0.8 ± 0.08 in rich YPD medium. cDNA for real-time reverse transcription-PCR was synthesized from total-RNA samples by using the MultiScribeTM reverse transcriptase kit (Applied Biosystems) and the 7500 RT-PCR thermal cycler block (Applied Biosystems) according to the manufacturer’s instructions. The quantity of cDNA for subsequent reactions was kept at ca. 10 ng. The subsequent RT-PCR step was carried out using SYBR green reagents. Primers for the amplification of the CgERG11 and CgACT1 genes were designed using Primer Express software (Applied Biosystems) and are summarized in Supplementary Table 3. The RT-PCR was carried out using a thermal cycler block (7500 real-time PCR system, Applied Biosystems). Default parameters established by the manufacturer were used, and fluorescence was detected by the instrument and recorded in an amplification plot (7500 System SDS software, Applied Biosystems). The CgACT1 mRNA level was used as an internal control. The relative values obtained for the initial isolates exhibiting the lower gene expression level were set as 1 and the remaining values are presented relative to that control. To avoid false-positive signals, the absence of nonspecific amplification with the chosen primers was confirmed by the generation of a dissociation curve for each pair of primers.
Plasmids
For CgHXT4/6/7 or CgHXT6/7 gene cloning, the plasmid pGREG576 was obtained from the Drag&Drop collection40. For CgHXT4/6/7 or CgHXT6/7 gene disruption, the plasmid pYC44 (Addgene plasmid #63903) was used; for expression of the flippase enzyme, the plasmid pLS10 was used. For CgURA3 gene disruption, the plasmid pV138241 was obtained from Addgene (Addgene plasmid #111436).
Disruption of CgHXT4/6/7 (ORF CAGL0A02233g) or CgHXT6/7 (ORF CAGL0A02211g)
The wild-type strain KUE100, derived KUE100_Δcgpdr1 mutant, and 040 clinical isolate were transformed by electroporation with the deletion cassette (a nourseothricin cassette flanked by FRT sites and a ~500 bp region flanking the targeted gene). The deletion cassette was constructed by Gibson Assembly using the pYC44 plasmid, previously cut with XhoI and Not1, ~500 bp of the CgHXT4/6/7 promoter region and ~500 bp of the CgHXT4/6/7 terminator region, or ~500 bp of the CgHXT6/7 promoter region and ~500 bp of the CgHXT6/7 terminator region. Cells were plated on YPD agar medium supplemented with 200 mg L−1 nourseothricin. Transformants were checked for insertion of the deletion cassette by PCR using the control primers. Correct strains were subsequently transformed with plasmid pLS10 to induce expression of the flippase enzyme (300 mg L−1 hygromycin selection). Removal of the nourseothricin cassettes of the transformants was checked by PCR. The pLS10 plasmid was lost by growth on non-selective YPD medium and checked by replating on YPD supplemented with 300 mg L−1 hygromycin. All the primers used for the construction of the mutant strains are depicted in Supplementary Table 3.
Cloning of the CgHXT4/6/7 (ORF CAGL0A02233g) or CgHXT6/7 (ORF CAGL0A02211g)
The pGREG576 plasmid from the Drag&Drop collection was used to clone and express CgHXT4/6/7 or CgHXT6/7 genes, as described before14–17, 19. pGREG576 contains a galactose inducible promoter (GAL1), the yeast selectable marker URA3 and the GFP gene, encoding a Green Fluorescent Protein (GFPS65T), which allows monitoring of the expression and subcellular localization of the cloned fusion protein. CgHXT4/6/7 and CgHXT6/7 DNA was generated by PCR, using genomic DNA extracted from the sequenced CBS138 C. glabrata strain. The designed primers contain, besides a region with homology to the first ~25 and last ~25 nucleotides of the CgHXT4/6/7 or CgHXT6/7 coding region, nucleotide sequences with homology to the cloning site flanking regions of the pGREG576 vector. The amplified fragments were co-transformed into the parental S. cerevisiae strain BY4741 with the pGREG576 vector, previously cut with the restriction enzyme SalI, to obtain the pGREG576_CgHXT4/6/7 or pGREG576_CgHXT6/7 plasmids. Since the GAL1 promoter needs galactose for induction in S. cerevisiae, and only allows a slight expression of downstream genes in C. glabrata, new constructs were obtained. The GAL1 promoter was replaced by the pPDC1 C. glabrata constitutive promoter42, generating the pGREG576_PDC1_CgHXT4/6/7 or pGREG576_PDC1_CgHXT6/7 plasmids, or by the TEF S. cerevisiae constitutive promoter43, generating the pGREG576_TEF_CgHXT4/6/7 or pGREG576_TEF_CgHXT6/7 plasmids. The PDC1 promoter DNA was generated by PCR, using genomic DNA extracted from the sequenced CBS138 C. glabrata strain. The TEF promoter DNA was generated by PCR, using genomic DNA extracted from the sequenced BY4741 S. cerevisiae strain. The amplified fragments were co-transformed into the parental strain BY4741 with the pGREG576_CgHXT4/6/7 or pGREG576_CgHXT6/7 plasmid, previously cut with SacI and NotI restriction enzymes to remove the GAL1 promoter. The recombinant plasmids were obtained through homologous recombination in S. cerevisiae and verified by DNA sequencing. All primers used are present in Supplementary Table 3.
Site-directed mutagenesis
The CgHXT4/6/7 gene sequence in the recombinant pGREG576_PDC1_CgHXT4/6/7 plasmid was mutated by site-directed mutagenesis. The designed primers contain the mutation occurred in the 040_Psc resistant isolate during posaconazole exposure, resulting in the production of the mutated sequence by PCR amplification to obtain the pGREG576_PDC1_mut_CgHXT4/6/7 plasmid. The original template was then degraded by DpnI digestion. All primers used are present in Supplementary Table 3.
Disruption of the CgURA3 (ORF CAGL0I03080g)
The disruption of the C. glabrata URA3 gene, encoded by ORF CAGL0I03080g, was carried out in the KUE100 parental strain as described previously44, using the CRISPR-Cas9 system from Vyas et al.41 Briefly, a CgURA3 gRNA sequence selected from the resources made available by Vyas et al.41 was cloned in the pV1382 plasmid, previously linearized with the restriction enzyme BsmBI. The CgURA3 gRNA was obtained by oligonucleotide annealing and the product ligated into the previously linearized pV1382 plasmid to obtain the pV1382_CgURA3 vector. The construct was verified by DNA sequencing. The plasmid was transformed into C. glabrata cells which were then directly plated on 5-Fluoroorotic acid (5-FOA) to select for URA- cells. Sequential passages in nonselective medium (YPD) were performed to avoid detrimental effects of further Cas9 expression and CgURA3 loss of function was further confirmed by the inability to grow in medium without uracil. The introduction of pGREG576 derived plasmids in the edited strains was able to rescue the growth impairment in the absence of uracil. Sequencing of the CgURA3 gene from the selected candidates revealed the existence of frameshifts within the ORF, thus resulting in premature stop codons as it is expected from Non-Homologous End Joining (NHEJ) correction of double-strand breaks. All primers used are present in Supplementary Table 3.
In vitro induction of antifungal resistance in C. glabrata clinical isolates and CBS138 strain and assessment of its stability
Standard powders of fluconazole (Pfizer, Groton, CT), voriconazole (Pfizer, New York, NY), posaconazole (Schering-Plough, Kenilworth, NJ) and clotrimazole (Sigma, St. Louis, MO) were obtained from the respective manufacturers. A stock solution of fluconazole was prepared in distilled water, while voriconazole, posaconazole and clotrimazole were prepared with dimethyl sulfoxide (Sigma, St. Louis, MO). Antifungal drugs were diluted afterwards with Roswell Park Memorial Institute 1640 medium (RPMI 1640; Sigma, St. Louis, MO) buffered to pH 7.0 with 0.165 M morpholine propanesulfonic acid buffer (MOPS; Sigma) and stored at −70 °C until further use. The culture medium used for resistance induction assays was RPMI 1640 with 0.165 M MOPS, pH 7.0. A single, randomly selected, colony from each C. glabrata strain (040, 044, OL152 and CBS138) was incubated in 10 mL of RPMI 1640 overnight in a rotating drum at 150 rpm and 35 °C. An aliquot of this culture, containing 106 yeast cells, was transferred to different vials, each containing 10 mL of culture medium with or without posaconazole and incubated overnight as described above. The following day, aliquots from each culture containing 106 yeast cells were again transferred into fresh medium containing the same antifungal and re-incubated as described. Each day, for the 30 days of the assay, a 1 mL aliquot from each subculture was mixed with 0.5 mL of 50% glycerol and frozen at −70 °C for later testing. Incubation took place for 30 days with constant concentrations of posaconazole (1 mg L−1), corresponding to therapeutic plasma levels obtained during antifungal treatment45. To assess resistance stability, the evolved resistant isolates obtained were subcultured daily in the absence of the drug for 30 days. A single colony from each isolate was incubated in 10 mL drug-free RPMI 1640 at 35 °C and 150 rpm. In the following day, aliquots were transferred into fresh medium. At each subculture, a 1 mL aliquot of the suspension was mixed with 0.5 mL of 50% glycerol, and frozen at −70 °C, for further testing.
Genome sequencing and data analysis
The draft genome sequences of the three azole susceptible C. glabrata clinical isolates used in this study, 040, 044 and OL152, had been characterized previously, as described in Pais et al.46. The genome sequences of the selected in vitro evolved resistant strains 040_Psc, 044_Psc and OL152_Psc derived from 040, 044 and OL152 clinical isolates, respectively, were obtained and characterized in this study. Raw sequencing data and genome assemblies can be found accessing BioProject no. PRJNA525402 (initial isolates) and BioProject no. PRJNA694431 (evolved isolates).
Genome sequencing was performed by Illumina HiSeqX 150-bp, paired-end sequencing. Library preparation (Nextera XT) and sequencing were carried out by Admera Health, LLC. Each resistant isolate was confirmed to be the same as their correspondent initial isolate by Multi Locus Sequence Typing (MLST) using six loci (FKS, LEU2, NMT1, TRP1, UGP1, URA3)47 and each sequence typing (ST) was identified according to the Sequence Typing website48(https://pubmlst.org/organisms/candida-glabrata). Genome assembly and comparison with the C. glabrata reference genome was carried out as described previously46. Briefly, raw sequencing reads were trimmed, and duplicates removed to only keep high-quality reads. Reads were mapped against the C. glabrata reference genome yielding >97% of aligned reads for each isolate. Genomes were assembled using SPAdes49 and scaffolds <500 bp discarded to attain final draft genomes. Each assembly was aligned against the reference genome and found to align >97.6% in each case. The high sequencing depth was leveraged to performed Single Nucleotide Polymorphism (SNP) identification with increased sensitivity and sensibility across the genome. Briefly, reads were aligned against the reference genome and variant identification was performed using GATK50. Low-quality variants were subsequently filtered out with BCFtools51 as described previously46. For subsequent analysis, only variants occurring within gene coding sequences and resulting in missense or non-sense mutations in the encoded protein were considered.
Antifungal susceptibility assays
Clinical isolates
The Minimal Inhibitory Concentration (MIC50) of voriconazole, fluconazole, clotrimazole and posaconazole antifungal drugs was determined according to the M27-A3 protocol of the Clinical Laboratory Standards Institute52, for all the C. glabrata isolates under study. The MICs were determined after 24 h. Interpretative criteria for fluconazole were those of the CLSI document M6053: susceptible-dose dependent (S-DD)-MIC50≤32 mg L−1 and resistance (R)-MIC50≥64 mg L−1. Although susceptibility breakpoints have not yet been established for voriconazole, posaconazole or clotrimazole for C. glabrata, strains inhibited by ≤2m L−1, ≤1m L−1 and ≤2m L−1, respectively, were considered to be susceptible. Every 5 days of incubation, with or without antifungal, MIC50 values were redetermined for the four antifungals tested. The antifungal concentrations ranged from 0.03125 to 16 mg L−1 for posaconazole and voriconazole, and 0.125-64 mg L−1 for fluconazole and clotrimazole. C. glabrata reference strain CBS138 was used in each testing assay, as recommended.
Lab strains
C. glabrata cells susceptibility to toxic concentrations of the selected azoles was evaluated by spot assays. Cell suspensions used to inoculate agar plates were prepared with mid-exponential cells grown in YPD, YPM or BM until an OD600nm = 0.5 ± 0.05, and then diluted in sterile water to obtain suspensions with an OD600nm = 0.05 ± 0.005. These cell suspensions and subsequent 1:5 and 1:25 dilutions were applied as 4 µL spots onto the surface of appropriate solid media supplemented with adequate concentrations of chemical stressors. The drugs tested included the following compounds, used in the specified concentrations: fluconazole 10, 100 or 150 mg L−1, posaconazole 2 or 4 mg L−1 and Itraconazole 1 mg L−1 (all from Sigma).
Yeast cells used for susceptibility assays in liquid medium were grown overnight in YPD broth medium at 30 °C, 250 rpm. New cultures were made in fresh YPD medium, supplemented or not with 4 mg L−1 of posaconazole, with an initial OD600nm = 0.01 ± 0.001. Growth, taking place in Erlenmeyer flasks, at 30 °C, 250 rpm, was followed by measuring the optical density of the cell suspension at 600 nm. Drug concentrations used for spot and liquid susceptibility assays using YPD medium were higher than the standard range used for MIC determination since YPD is a richer medium, comparing to RPMI, and the initial cell concentration used was higher.
Posaconazole and fluconazole MIC50 values for each strain were determined in accordance with the CLSI guidelines by using 96-well plates containing RPMI 1640 2% glucose medium supplemented with the appropriate drug concentration. The antifungal concentrations ranged from 0.015625 to 8 mg L−1 for posaconazole and 0.125–64 mg L−1 for fluconazole.
CgHxt4/6/7 or CgHxt6/7 subcellular localization assessment
The subcellular localization of the CgHxt4/6/7 or CgHxt6/7 protein was determined based on the observation of KUE100::URA- C. glabrata cells and BY4741 S. cerevisiae cells transformed with pGREG576_PDC1_HXT4/6/7 or pGREG576_PDC1_HXT6/7, and pGREG576_TEF_CgHXT4/6/7 or pGREG576_TEF_CgHXT6/7 plasmids, respectively. These cells express either the CgHxt4/6/7_GFP or the CgHxt6/7_GFP fusion protein, whose localization may be determined using fluorescence microscopy. C. glabrata or S. cerevisiae cells were grown in BM without uracil, for plasmid maintenance, until mid-exponential phase, OD600nm = 0.5 ± 0.05. The distribution of fusion protein was assessed by fluorescence microscopy in a Zeiss Axioplan microscope (Carl Zeiss MicroImaging), using excitation and emission wavelength of 395 and 509 nm, respectively. Fluorescence images were captured using a cooled Zeiss Axiocam 503 colour (Carl Zeiss Microscopy).
Hexose utilization assays
The ability of CgHxt4/6/7 and CgHxt6/7 to import glucose, mannose, fructose or galactose into fungal cells was accessed in S. cerevisiae by using the hexose transporter-null EBY.VW4000 (Schxt0) strain, which grows normally on maltose, and barely on galactose as carbon sources. EBY.VW4000 cells harbouring pGREG576 empty vector or the pGREG576_TEF_CgHXT4/6/7 or pGREG576_TEF_CgHXT4/6/7 plasmids were grown overnight at 30 °C, with orbital agitation (250 rpm) in BM-Maltose, without uracil. Cell suspensions used to inoculate agar plates were mid-exponential cells grown until an OD600nm = 0.5 ± 0.05, and then suspended in sterile water to an OD600nm = 0.05 ± 0.005. These cell suspensions and subsequent 1:5 and 1:25 dilutions were applied as 4 µL spots onto the surface of solid BM media plates containing the indicated proportion of carbon sources, supplemented with 20 mg L−1 tryptophan, 20 mg L−1 histidine and 60 mg L−1 leucine.
[3H]-fluconazole and [3H]-clotrimazole accumulation assays
The intracellular accumulation of fluconazole or clotrimazole in the different C. glabrata strains was compared resorting to radiolabelled [3H]-fluconazole and [3H]-clotrimazole, respectively. The internal accumulation was determined by calculating the ratio between the value measured within yeast cells and in the external medium ([Intracellular]/[Extracellular]). The parental strains and the derived mutant strains were grown in YPD medium until mid-exponential phase and harvested by filtration. Cells were washed and resuspended in YPD medium, to obtain dense cell suspensions (OD600nm = 0.5 ± 0.1, equivalent to approximately 1.57 mg(dry weight) mL−1). Readily, 0.1 µM of [3H]-fluconazole (Moravek Inc.; 1.0 mCi mL−1) or [3H]-clotrimazole (American Radiolabelled Chemicals, St. Louis, MO;1 mCi mL−1) and 100 mg L−1 of unlabelled fluconazole or 30 mg L−1 of unlabelled clotrimazole, respectively, were added to the cell suspensions. The intracellular accumulation of labelled fluconazole was followed for a 15 min period by filtering 200 µL of cell suspension through pre-wetted glass microfiber filters (Whatman GF/C). The filters were washed with ice-cold TM buffer [0.1 M 2-(N-morpholino)ethanesulfonic acid (Sigma)/41 mM Tris (Sigma)], and the radioactivity measured in a PerkinElmer Tri-Carb 2810TR liquid scintillation analyser. Extracellular 3H-fluconazole was estimated by radioactivity assessment of 50 µL of the supernatant. To calculate the intracellular concentration of radiolabelled fluconazole, the internal cell volume (Vi) of the exponential cells, grown in the absence of drug and used for accumulation assays, was considered constant and equal to 2.5 µL mg (dry weight)−1.
Protein modelling and molecular docking
The homology modelling of the CgHxt4/6/7 membrane protein was performed using MODELLER version 9.2354. The crystal structure of E. coli XylE (PDB ID 4GBZ)31 was used as a template for the outward-facing, partially occluded conformation and the crystal structure of the H. sapiens glucose transporter (PDB ID 4ZWC)32 was used for the outward-open conformation. For each modulation, twenty independent models were generated. Subsequent analysis were carried on the model with the lowest DOPE score55.
The docking calculations were performed using AutoDock Vina56, with an exhaustiveness value of 50 and in a search box centralized at the ligand of the PDB ID 4GBZ and with sizes of 16, 16 and 14 Å. For each docking calculation, the docking pose with lowest energy was used in subsequent analysis. Figures of docking results were prepared using PyMOL2.557.
Statistics and reproducibility
All experiments represent the average of three or more independent experiments. Error bars represent the standard deviation. Statistical analysis was performed using one-way analysis of variance with Tukey’s correction. Significance levels are attributed as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by “Fundação para a Ciência e a Tecnologia” (FCT) (Contract PTDC/BII-BIO/28216/2017 and AEM PhD grants to M.G. and R.V.) and by the Research Council of the KU Leuven (grant # C14/17/063). Funding received by iBB-Institute for Bioengineering and Biosciences (UIDB/04565/2020) and by i4HB (LA/P/0140/2020) from FCT is also acknowledged. PVD was further supported by a grant from the FWO (Fund for Scientific Research, Flanders, grant # G0C0622N). We acknowledge Eckhard Boles, Institute of Molecular Biological Sciences, Goethe University, Frankfurt, Germany and Hiroji Chibana, Medical Mycology Research Center at Chiba University, Japan for kindly providing some of the strains used in this study, as indicated in the “Methods” section.
Author contributions
M.G. conducted most of the experiments. R.V., C.S.S. and C.M.S. conducted the protein modelling and molecular docking experiments. P.P. contributed to the genome sequencing data generation and analysis. A.S.-D., I.M.M., M.C., C.C. and J.B. contributed to in vitro evolution experiments. R.V., M.C., P.P. and M.V.E. contributed to genetic manipulation and strain construction. M.C.T., A.G.R. and P.V.D. designed the experiments and M.C.T. and A.G.R. coordinated the underlying project. The manuscript was written by M.G., A.G.R. and M.C.T. with contributions from all authors. All authors have given approval to the final version of the manuscript.
Peer review
Peer review information
Communications Biology thanks Yue Wang, Rajendra Prasad and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary handling editor: George Inglis.
Data availability
Raw sequencing data and genome assemblies can be found accessing NCBI BioProjects PRJNA525402 (initial isolates) and PRJNA694431 (evolved isolates). All other data are available from the corresponding author on reasonable request. Raw data underlying all graphs have been provided as Supplementary Data 4.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Acácio G. Rodrigues, Email: agr@med.up.pt
Miguel C. Teixeira, Email: mnpct@tecnico.ulisboa.pt
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-022-04087-0.
References
- 1.Brown GD, et al. Hidden killers: human fungal infections. Sci. Transl. Med. 2012;4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.Colombo AL, Júnior JNDA, Guinea J. Emerging multidrug-resistant Candida species. Curr. Opin. Infect. Dis. 2017;30:528–538. doi: 10.1097/QCO.0000000000000411. [DOI] [PubMed] [Google Scholar]
- 3.Tsai H-F, Krol AA, Sarti KE, Bennett JE. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 2006;50:1384–92. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vermitsky J-P, et al. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 2006;61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 5.Caudle KE, et al. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot. Cell. 2011;10:373–83. doi: 10.1128/EC.00073-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whaley, S. G. & Rogers, P. D. Azole resistance in Candida glabrata. Curr. Infect. Dis. Rep.18, 41 (2016). [DOI] [PubMed]
- 7.Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 2010;9:719–727. doi: 10.1038/nrd3074. [DOI] [PubMed] [Google Scholar]
- 8.Demuyser, L., Van Dyck, K., Timmermans, B. & Van Dijck, P. Inhibition of vesicular transport influences fungal susceptibility to fluconazole. Antimicrob. Agents Chemother. 63, e01998 (2019). [DOI] [PMC free article] [PubMed]
- 9.Sanguinetti M, et al. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob. Agents Chemother. 2005;49:668–79. doi: 10.1128/AAC.49.2.668-679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 1999;43:2753–65. doi: 10.1128/aac.43.11.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akins, R. A. An update on antifungal targets and mechanisms of resistance in Candida albicans. Med. Mycol. 43, 285–318 (2005). [DOI] [PubMed]
- 12.Costa C, et al. Clotrimazole drug resistance in Candida glabrata clinical isolates correlates with increased expression of the drug: H+ antiporters CgAqr1, CgTpo1_1, CgTpo3, and CgQdr2. Front. Microbiol. 2016;7:1–11. doi: 10.3389/fmicb.2016.00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costa, C., Dias, P. J., Sá-Correia, I. & Teixeira, M. C. MFS multidrug transporters in pathogenic fungi: Do they have real clinical impact? Front. Physiol.5, 197 (2014). [DOI] [PMC free article] [PubMed]
- 14.Costa C, et al. Candida glabrata drug:H+ antiporter CgQdr2 confers imidazole drug resistance, being activated by transcription factor CgPdr1. Antimicrob. Agents Chemother. 2013;57:3159–67. doi: 10.1128/AAC.00811-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pais P, et al. Membrane proteome-wide response to the antifungal drug clotrimazole in Candida glabrata: role of the transcription factor CgPdr1 and the Drug:H+ antiporters CgTpo1_1 and CgTpo1_2. Mol. Cell. Proteomics. 2016;15:57–72. doi: 10.1074/mcp.M114.045344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa C, et al. The dual role of Candida glabrata drug:H+ antiporter CgAqr1 (ORF CAGL0J09944g) in antifungal drug and acetic acid resistance. Front. Microbiol. 2013;4:170. doi: 10.3389/fmicb.2013.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa C, et al. Candida glabrata drug:H+ antiporter CgTpo3 (ORF CAGL0I10384G): Role in azole drug resistance and polyamine homeostasis. J. Antimicrob. Chemother. 2014;69:1767–1776. doi: 10.1093/jac/dku044. [DOI] [PubMed] [Google Scholar]
- 18.Cavalheiro M, Pais P, Galocha M, Teixeira M. Host-pathogen interactions mediated by MDR transporters in fungi: as pleiotropic as it gets! Genes. 2018;9:332. doi: 10.3390/genes9070332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pais P, et al. Membrane proteomics analysis of the candida glabrata response to 5-flucytosine: unveiling the role and regulation of the drug efflux transporters CgFlr1 and CgFlr2. Front. Microbiol. 2016;7:2045. doi: 10.3389/fmicb.2016.02045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrari S, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009;5:e1000268. doi: 10.1371/journal.ppat.1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vermitsky J-P, Edlind TD. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 2004;48:3773–3781. doi: 10.1128/AAC.48.10.3773-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben-Ami, R. et al. Heteroresistance to fluconazole is a continuously distributed phenotype among Candida glabrata clinical strains associated with in vivo persistence. MBio7, e00655-16 (2016). [DOI] [PMC free article] [PubMed]
- 23.Arastehfar, A. et al. Low level of antifungal resistance in iranian isolates of Candida glabrata recovered from blood samples in a multicenter study from 2015 to 2018 and potential prognostic values of genotyping and sequencing of PDR1. Antimicrob. Agents Chemother. 63, e02503-18 (2019). [DOI] [PMC free article] [PubMed]
- 24.Galocha M, Costa IV, Teixeira MC. Carrier-mediated drug uptake in fungal pathogens. Genes. 2020;11:1324. doi: 10.3390/genes11111324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mansfield BE, et al. Azole drugs are imported by facilitated diffusion in Candida albicans and other pathogenic fungi. PLoS Pathog. 2010;6:1–11. doi: 10.1371/journal.ppat.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy A, Dement AD, Cho KH, Kim JH. Assessing glucose uptake through the yeast hexose transporter 1 (Hxt1) PLoS ONE. 2015;10:1–14. doi: 10.1371/journal.pone.0121985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faria-Ramos I, et al. Species distribution and in vitro antifungal susceptibility profiles of yeast isolates from invasive infections during a Portuguese multicenter survey. Eur. J. Clin. Microbiol. Infect. Dis. 2014;33:2241–2247. doi: 10.1007/s10096-014-2194-8. [DOI] [PubMed] [Google Scholar]
- 28.Cavalheiro M, et al. A transcriptomics approach to unveiling the mechanisms of in vitro evolution towards fluconazole resistance of a Candida glabrata clinical isolate. Antimicrob. Agents Chemother. 2019;63:e00995-18. doi: 10.1128/AAC.00995-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boles, E. & Oreb, M. In Methods in Molecular Biology, Vol. 1713 123–135 (Humana Press Inc., 2018). [DOI] [PubMed]
- 30.Özcan S, Johnston M. Function and regulation of yeast hexose transporters. Microbiol. Mol. Biol. Rev. 1999;63:554–569. doi: 10.1128/mmbr.63.3.554-569.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun, L. et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature496, 361–366 (2012). [DOI] [PubMed]
- 32.Deng D, et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526:391–396. doi: 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- 33.Ng, T. S., Desa, M. N. M., Sandai, D., Chong, P. P. & Than, L. T. L. Phylogenetic and transcripts profiling of glucose sensing related genes in Candida glabrata. Jundishapur J. Microbiol. 8, e25177 (2015). [DOI] [PMC free article] [PubMed]
- 34.Esquivel BD, Smith AR, Zavrel M, White TC. Azole drug import into the pathogenic fungus Aspergillus fumigatus. Antimicrob. Agents Chemother. 2015;59:3390–3398. doi: 10.1128/AAC.05003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nourani A, Wesolowski-Louvel M, Delaveau T, Jacq C, Delahodde A. Multiple-drug-resistance phenomenon in the yeast Saccharomyces cerevisiae: involvement of two hexose transporters. Mol. Cell. Biol. 1997;17:5453–5460. doi: 10.1128/mcb.17.9.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, Boles E, Rosen BP. Arsenic trioxide uptake by hexose permeases in Saccharomyces cerevisiae. J. Biol. Chem. 2004;279:17312–17318. doi: 10.1074/jbc.M314006200. [DOI] [PubMed] [Google Scholar]
- 37.Ueno K, et al. Development of a highly efficient gene targeting system induced by transient repression of YKU80 expression in Candida glabrata. Eukaryot. Cell. 2007;6:1239–1247. doi: 10.1128/EC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galkina KV, Okamoto M, Chibana H, Knorre DA, Kajiwara S. Deletion of CDR1 reveals redox regulation of pleiotropic drug resistance in Candida glabrata. Biochimie. 2020;170:49–56. doi: 10.1016/j.biochi.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Entian KD, Kötter P. 25 Yeast genetic strain and plasmid collections. Methods Microbiol. 2007;36:629–666. [Google Scholar]
- 40.Jansen G, Wu C, Schade B, Thomas DY, Whiteway M. Drag&Drop cloning in yeast. Gene. 2005;344:43–51. doi: 10.1016/j.gene.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 41.Vyas, V. K. et al. New CRISPR mutagenesis strategies reveal variation in repair mechanisms among fungi. mSphere3, e00154-18 (2018). [DOI] [PMC free article] [PubMed]
- 42.Zordan RE, et al. Expression plasmids for use in Candida glabrata. G3 Genes Genomes Genet. 2013;3:1675–1686. doi: 10.1534/g3.113.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 44.P P, et al. A new regulator in the crossroads of oxidative stress resistance and virulence in Candida glabrata: the transcription factor CgTog1. Virulence. 2020;11:1522–1538. doi: 10.1080/21505594.2020.1839231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azanza, J. R., García-quetglas, E. & Sádaba, B. Farmacología de los azoles. Rev. Iberoamericana Micol.24, 223–227 (2007). [DOI] [PubMed]
- 46.Pais, P., Galocha, M., Miranda, I. M., Rodrigues, A. G. & Teixeira, M. C. Draft genome sequences of three clinical isolates of the pathogenic yeast Candida glabrata. Microbiol. Resour. Announc. 8, e00278-19 (2019). [DOI] [PMC free article] [PubMed]
- 47.AR D, C P, DW D, DR S, AJ F. Multilocus sequence typing of Candida glabrata reveals geographically enriched clades. J. Clin. Microbiol. 2003;41:5709–5717. doi: 10.1128/JCM.41.12.5709-5717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved] Wellcome Open Res. 2018;3:124. doi: 10.12688/wellcomeopenres.14826.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narasimhan V, et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32:1749–1751. doi: 10.1093/bioinformatics/btw044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.M27-A3. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard-third edition. www.clsi.org (2008).
- 53.M60Ed2. Performance standards for antifungal susceptibility testing of yeasts, 2nd edition. https://clsi.org/standards/products/microbiology/documents/m60/ (2020).
- 54.Šali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed]
- 55.Shen, M. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006). [DOI] [PMC free article] [PubMed]
- 56.Oleg, T. & Arthur J. O. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010). [DOI] [PMC free article] [PubMed]
- 57.The PyMOL Molecular Graphics System, Version 1.3r1 (Schrodinger, LLC, 2010).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Raw sequencing data and genome assemblies can be found accessing NCBI BioProjects PRJNA525402 (initial isolates) and PRJNA694431 (evolved isolates). All other data are available from the corresponding author on reasonable request. Raw data underlying all graphs have been provided as Supplementary Data 4.