

Abstract
Background
We sought to identify clinical and genetic predictors of temozolomide-related myelotoxicity among patients receiving therapy for glioblastoma.
Methods
Patients (n = 591) receiving therapy on NRG Oncology/RTOG 0825 were included in the analysis. Cases were patients with severe myelotoxicity (grade 3 and higher leukopenia, neutropenia, and/or thrombocytopenia); controls were patients without such toxicity. A risk-prediction model was built and cross-validated by logistic regression using only clinical variables and extended using polymorphisms associated with myelotoxicity.
Results
23% of patients developed myelotoxicity (n = 134). This toxicity was first reported during the concurrent phase of therapy for 56 patients; 30 stopped treatment due to toxicity. Among those who continued therapy (n = 26), 11 experienced myelotoxicity again. The final multivariable clinical factor model included treatment arm, gender, and anticonvulsant status and had low prediction accuracy (area under the curve [AUC] = 0.672). The final extended risk prediction model including four polymorphisms in MGMT had better prediction (AUC = 0.827). Receiving combination chemotherapy (OR, 1.82; 95% CI, 1.02–3.27) and being female (OR, 4.45; 95% CI, 2.45–8.08) significantly increased myelotoxicity risk. For each additional minor allele in the polymorphisms, the risk increased by 64% (OR, 1.64; 95% CI, 1.43–1.89).
Conclusions
Myelotoxicity during concurrent chemoradiation with temozolomide is an uncommon but serious event, often leading to treatment cessation. Successful prediction of toxicity may lead to more cost-effective individualized monitoring of at-risk subjects. The addition of genetic factors greatly enhanced our ability to predict toxicity among a group of similarly treated glioblastoma patients.
Keywords: genetic association, glioblastoma, myelotoxicity, risk prediction, temazolomide
Key Points.
Toxicity from chemotherapy can be a serious event leading to treatment cessation.
Genetic variants enhanced prediction of myelotoxicity among glioblastoma patients.
Individualized genetic monitoring of at-risk subjects could greatly improve outcomes.
Importance of the Study.
Based on results from other studies as well as our initial findings, we hypothesized that germline SNPs are logical predictors of chemotherapy toxicity and response, especially when examined in conjunction with clinical risk factors. For cytotoxic regimens like TMZ, variants from DNA repair pathways are likely shared with tumors, making prediction of both toxicity and response possible. We found that clinical variables alone were poor predictors of myelotoxicity in patients with GBM receiving TMZ; however, the addition of germline variants in the DNA repair gene MGMT increased the ability to predict toxicity among these patients. Such findings have the potential to impact the management of patients by predicting toxicity risk and better individualize initial temozolomide dosing and bloodwork monitoring schedules in patients with GBM receiving this drug.
Glioblastoma (GBM) is the most common primary malignant brain tumor in the United States and carries the worst survival for patients with this disease.1 Currently, temozolomide (TMZ) in conjunction with conventional fractionated external beam radiation therapy followed by adjuvant TMZ is the standard of care for GBM patients.2 Despite the widespread acceptance that TMZ is well tolerated, 16% of patients on NRG Oncology’s RTOG 0525, a phase III trial comparing conventional adjuvant TMZ with dose-intensive TMZ, experienced grade 3 or higher myelotoxicity during chemoradiation, and 23% of patients had similar levels of myelotoxicity during the standard dose adjuvant phase, typically during the first cycles of the treatment course.3 In addition to contributing to severe infections and bleeding events, grade 3 or 4 myelotoxicities often result in treatment delays or complete cessation of this life-prolonging therapy.4–6 The risk is underscored by reports of profound myelotoxicity, including aplastic anemia.7–10 For this reason, identifying patients at high risk of developing myelotoxicity would allow for a more suitable starting dose for this sensitive subpopulation. Conversely, it would also allow the remaining population to receive full-dose treatment from the initiation of therapy. Both scenarios potentially increase the therapeutic efficacy of this drug by shifting the therapeutic window and reducing toxicity, thus allowing for continuation of treatment and potentially impacting patient outcomes.
Genomic predictors of treatment-related toxicities have a long history.11,12 The seminal discovery of inherited Glucose-6-phosphate dehydrogenase (G6PD) deficiency leading to the profound development of hemolytic anemia when exposed to the quinines and other drugs such as dapsone provided clear evidence that genomic variability in genes that mediate drug metabolism can predict toxicity.13 In cancer treatment with irinotecan, there is a significant association between the UGT1A1 *28/*6 polymorphisms and increased toxicity. This association has demonstrated the clinical relevance of genome-based predictive markers for treatment-related toxicity.14 More recently, a series of polymorphisms that predict the development of thalidomide-associated peripheral neuropathy15,16 was detected in patients treated for multiple myeloma. We also recently reported that several clinical factors as well as specific concomitant medications could be incorporated into a formula to create a “risk score” that was highly correlated with the occurrence of significant myelotoxicity in patients treated with standard dose TMZ at recurrence.17 Interestingly, the toxicity risk score components varied markedly between male (higher body surface area [BSA], not currently using steroids, and currently using bowel medication) and female (no prior chemotherapy, higher creatinine levels, lower platelet levels, lower BSA, not currently using medication for gastroesophageal reflux disease, and currently using analgesics) patients. Furthermore, a small set of candidate polymorphisms also increased our ability to predict myelotoxicity in this group of GBM patients.
Taken together, these studies strongly support the identification of predictors of treatment toxicity to mitigate the adverse impact of therapy in select populations. For example, if a risk model is validated, patients with newly diagnosed GBM could be screened using a risk calculator for the likelihood of myelotoxicity. Patients at elevated risk of toxicity could then be started at a lower dose of TMZ (with escalation as tolerated) while the other patients could begin at full dose, thus increasing the likelihood that all patients are able to receive this proven life-extending treatment. We, therefore, conducted a study using a closely monitored patient population who received concurrent TMZ and radiation as a component of a large clinical trial, taking advantage of the trial-related mandatory systematic toxicity reporting and collection of peripheral blood.
Materials and Methods
Study Population
NRG Oncology’s RTOG 0825 was a phase III, randomized double-blind placebo-controlled trial comparing standard chemoradiotherapy using TMZ with or without the addition of bevacizumab (BEV) for patients with newly diagnosed GBM.18 Eligibility criteria included age ≥ 18 years, Karnofsky Performance Status (KPS) ≥ 70, and adequate hematologic, renal, and hepatic function. Patients with recent or ongoing cardiovascular problems were excluded as well as those with poorly controlled moderate to severe hypertension (baseline systolic blood pressure ≤ 160 mmHg, diastolic ≤ 90 mmHG). Eligibility requirements also included submission of tumor tissue for central pathology review and MGMT methylation analysis. A blood sample was requested, although not mandatory. Informed consent for future research was provided as part of the parent trial which was approved by appropriate institutional review boards; de-identified data/samples were analyzed as part of the current study. Approval for the analysis of the de-identified data used in this analysis was received from the University of Texas Health Sciences Center at Houston.
Detailed demographic and clinical characteristics were obtained, including age, height, weight, gender, race/ethnicity, concurrent medications, KPS, baseline laboratory studies, and tumor characteristics. Treatment-specific toxicities graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0 were also obtained.
Patients of all races/ethnicities were randomized (n = 621) to the clinical trial; however, 591 (95%) of the randomized subjects were non-Hispanic white and had complete clinical data. Therefore, we limited our analyses to non-Hispanic whites to minimize any confounding due to population structure in the genetic analyses. Blood samples were collected on 367 non-Hispanic white patients and were available for inclusion in the genetic analyses.
Processing of Blood Samples and Genotyping
Blood was collected as an amendment to the trial protocol. Germline DNA was extracted from blood using the QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany) according to manufacturer’s protocol. Germline DNA samples were genotyped using the HumanOmni2.5Exome beadchip (Illumina, San Diego, CA), according to manufacturer’s protocol.
Statistical Methods
We defined cases as patients with severe TMZ-related myelotoxicity using CTCAE version 3.0 for grade 3 and higher leukopenia, neutropenia, and/or thrombocytopenia, using the earliest event for each patient. Controls were defined as patients with grade 0–2 toxicity. The risk modeling was conducted in two steps: first, we built a model including only clinical factors; and second, we extended that model to include genetic polymorphisms. Clinical risk factors with a P value < 0.15 in the univariable logistic regression model were included for variable selection in the multivariable model. The final clinical risk model for toxicity was built using the backward selection method at P-value < .05. Where appropriate, a test for trend was used to assess significant trends in the association analysis. Next, we conducted a genome-wide association study (GWAS) of myelotoxicity. All of the samples had call rates ≥ 95%. Among available polymorphisms included on the beadchip (n = 2 583 651), we included in the analysis autosomal SNPs having call rates ≥ 95%, showing minimal departure from Hardy-Weinberg equilibrium (HWE; P > 10−5 in controls) and minor allele frequencies (MAF) ≥ 5% (n = 1 257 515). We dropped from further analysis all loci in strong linkage disequilibrium (LD) with another marker (D′ ≥ 0.99) to reduce redundant information. We used a standard GWAS nominal P-value ≤ 10−7 to select the most significant SNPs associated with myelotoxicity. We estimated the main effect of each genotype using the allele-based Chi-squared association test. Polymorphisms significantly associated with myelotoxicity in the main effects analyses were then included in the final multivariable regression models of clinical risk factors. We also conducted a joint effect analysis to test the hypothesized dose–response relationship between genotype and myelotoxicity, by summing the number of at-risk alleles (minor allele) identified from the main effect analysis. We used a 30-fold cross-validation of the entire process to check the performance of the final prediction model.19 We calculated the area under curve (AUC) for the receiver operating characteristic (ROC) curve as a way to assess the performance of the prediction models. We used PLINK v.1.9 to conduct the GWAS, and R v.3.2.3 (packages: genetics and haplo.stats) to perform all cross-validation and haplotype analyses. We conducted all other analyses using SAS v.9.4.
Data Availability
The data sets generated and/or analyzed during this study, as well as the computer code used to perform statistical analysis, are available from the corresponding authors on reasonable request. The datasets from the parent trial are available in dbGaP.
Results
Clinical Myelotoxicity Risk Model
Clinical characteristics and univariable logistic regression outcomes of the participants in this analysis (Figure 1) are listed in Table 1. There were 134 cases with myelotoxicity (23%) and 457 controls; median ages at diagnosis were 59 and 58 years, respectively. Overall, 61% of the subjects were male; however, there were more females among the cases (61%) compared to controls (33%). In addition, those receiving combination therapy (60%) were more likely to experience myelosuppression than those who received TMZ alone (40%).
Figure 1.
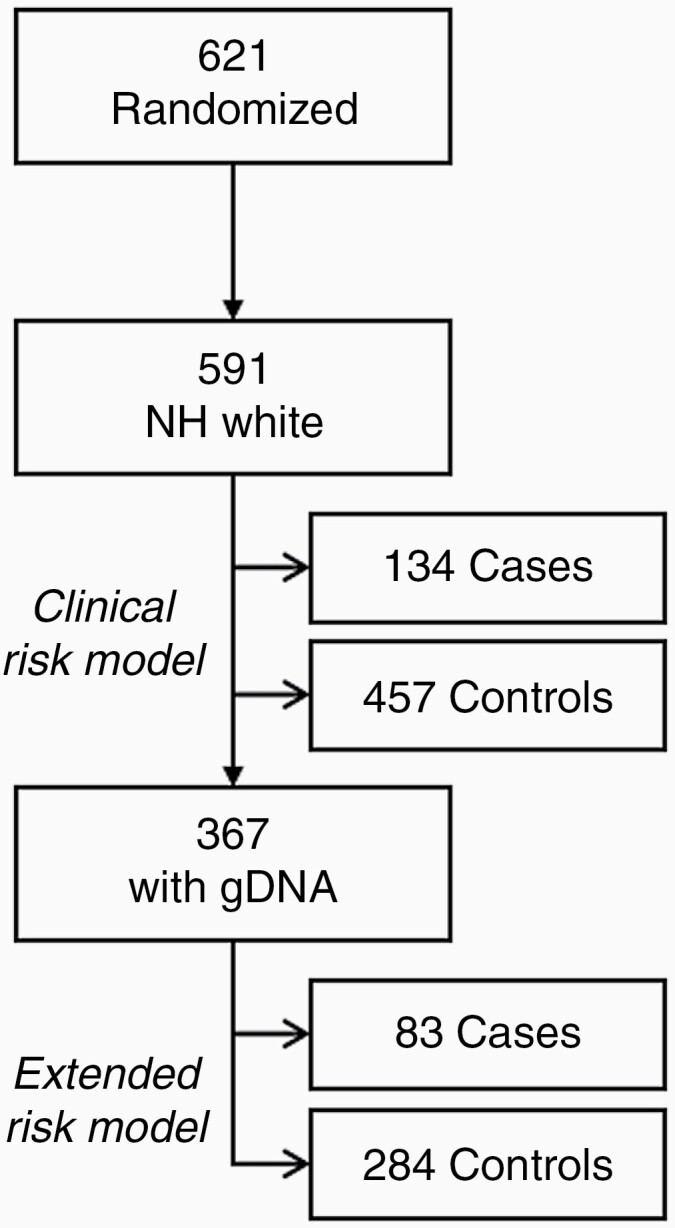
Flowchart of patients included in analysis.
Table 1.
Characteristics and Univariable Logistic Regression Outcomes of Myelotoxicity
| Categorical Characteristics | Cases (N = 134) | Controls (N = 457) | OR | 95% CI | P-value | ||
|---|---|---|---|---|---|---|---|
| No. | % | No. | % | ||||
| Treatment arm | .015 | ||||||
| TMZ+placebo | 54 | 40.3 | 239 | 52.3 | 1 | ||
| TMZ+BEV | 80 | 59.7 | 218 | 47.7 | 1.6 | 1.01–2.40 | |
| Gender | <.0001 | ||||||
| Male | 52 | 38.8 | 308 | 67.4 | 1 | ||
| Female | 82 | 61.2 | 149 | 32.6 | 3.26 | 2.19–4.86 | |
| KPS | .0356 | ||||||
| 70 | 23 | 17.2 | 52 | 11.4 | 2.45 | 1.20–4.99 | |
| 80 | 42 | 31.3 | 111 | 24.3 | 2.09 | 1.12–3.92 | |
| 90 | 52 | 38.8 | 200 | 43.7 | 1.44 | 0.79–2.62 | |
| 100 | 17 | 12.7 | 94 | 20.6 | 1 | ||
| Surgery | .365 | ||||||
| Subtotal | 55 | 41 | 157 | 34.4 | 1.33 | 0.49–3.68 | |
| Gross total | 76 | 56.7 | 288 | 63 | 1 | ||
| Other | 3 | 2.3 | 12 | 2.6 | 0.95 | 0.26–3.44 | |
| Tumor MGMT Methylation Status | .423 | ||||||
| Unmethylated | 90 | 68.7 | 321 | 72.3 | 1 | ||
| Methylated | 41 | 31.3 | 123 | 27.7 | 1.19 | 0.75–1.85 | |
| BMI | .4839 | ||||||
| Underweight & Normal | 39 | 29.1 | 116 | 25.4 | 1.36 | 0.81–2.29 | |
| Overweight | 60 | 44.8 | 197 | 43.1 | 1.24 | 0.77–1.98 | |
| Obese | 35 | 26.1 | 142 | 31.1 | 1 | ||
| Missing | 0 | 0 | 2 | 0.4 | |||
| Anticoagulant agents | .3368 | ||||||
| No | 128 | 95.5 | 426 | 93.2 | 1.55 | 0.63–3.80 | |
| Yes | 6 | 4.5 | 31 | 6.8 | 1 | ||
| Proarrhythmic potential agents | .2178 | ||||||
| No | 130 | 97 | 431 | 94.3 | 1.96 | 0.67–5.72 | |
| Yes | 4 | 3 | 26 | 5.7 | 1 | ||
| Herbal | .6509 | ||||||
| No | 128 | 95.5 | 432 | 94.5 | 1.24 | 0.50–3.08 | |
| Yes | 6 | 4.5 | 25 | 5.5 | 1 | ||
| Corticosteroids | .3701 | ||||||
| No | 32 | 23.9 | 127 | 27.8 | 1 | ||
| Yes | 102 | 76.1 | 330 | 72.2 | 1.23 | 0.79–1.92 | |
| Anticonvulsants | .0901 | ||||||
| No | 29 | 21.6 | 133 | 29.1 | 1 | ||
| Yes | 105 | 78.4 | 324 | 70.9 | 1.49 | 0.94–2.35 | |
| EIAEDs | .1646 | ||||||
| No | 117 | 87.3 | 374 | 81.8 | 1.49 | 0.85–2.62 | |
| Yes | 17 | 12.7 | 81 | 17.7 | 1 | ||
| Missing | 0 | 0 | 2 | 0.5 | |||
| NEIAEDs | .0472 | ||||||
| No | 77 | 57.5 | 304 | 66.5 | 1 | ||
| Yes | 57 | 42.5 | 151 | 33 | 1.49 | 1.01–2.21 | |
| Missing | 0 | 0 | 2 | 0.5 | |||
| Urine protein/creatinine ratio | .9768 | ||||||
| ≤0.5 | 115 | 85.8 | 393 | 86 | 1 | ||
| >0.5 | 18 | 13.4 | 61 | 13.3 | 1.01 | 0.57–1.78 | |
| Missing | 1 | 0.8 | 3 | 0.7 | |||
| Continuous Characteristics | Median | IQR | Median | IQR | |||
| Age, years | 59 | 51–64 | 58 | 51–65 | 1.00 | 0.98–1.02 | .9693 |
| Creatinine, mg/dL | 0.78 | 0.65–0.90 | 0.83 | 0.70–0.95 | 0.14 | 0.05–0.42 | .0005 |
| Platelet, 103/mm3 | 240.5 | 192–302 | 235 | 193.0–283.5 | 1.00 | 0.99–1.01 | .5535 |
| ANC, 102/mm3 | 60.4 | 35.6–90.0 | 63 | 41.0–90.4 | 1.00 | 0.99–1.01 | .6629 |
| Hemoglobin, g/dL | 13.4 | 12.3–14.2 | 13.5 | 12.5–14.4 | 0.94 | 0.82–1.09 | .4145 |
| WBC, 102/mm3 | 86 | 60–119 | 85 | 64–112 | 1.00 | 0.99–1.01 | .7684 |
| BUN, mg/dL | 17 | 13–22 | 17 | 14–21 | 0.99 | 0.96–1.03 | .7739 |
| Total Bilirubin, mg/dL | 0.4 | 0.3–0.6 | 0.5 | 0.3–0.6 | 0.84 | 0.42–1.71 | .6364 |
| SGOT, u/dL | 20 | 16–26 | 21 | 16–26 | 0.99 | 0.98–1.01 | .6985 |
| SGPT, u/dL | 31.5 | 23–47 | 36 | 25–50 | 0.99 | 0.98–1.01 | .2498 |
| PT, seconds | 11.3 | 10.2–12.7 | 11.1 | 10.2–12.8 | 0.97 | 0.91–1.04 | .3752 |
| PT/INR | 1 | 0.9–1.0 | 1 | 0.9–1.0 | 0.50 | 0.13–1.88 | .3017 |
| TMZ dose, 102 mg | 30.5 | 27.3–33.6 | 31.5 | 28.5–34.7 | 0.97 | 0.95–0.99 | .0439 |
| BSA | 1.87 | 1.72–2.08 | 2 | 1.84–2.18 | 0.15 | 0.06–0.34 | <.0001 |
Abbreviations: ANC, absolute neutrophil count; BEV, bevacizumab; BSA, body surface area; BUN, blood urea nitrogen; CI, confidence interval; EIAEDs, enzyme-inducing anti-epileptic drugs; INR, international normalized ratio; IQR, inter-quartile range; KPS, Karnofsky Performance Status; NEIAEDs, non-enzyme-inducing anti-epileptic drugs; OR, odds ratio; PT, prothrombin time; SGOT, serum glutamic oxaloacetic transaminase; SGPT, serum glutamic pyruvic transaminase; TMZ, temozolomide; WBC, white blood cell count.
Bold values represent factors with a univariable p value <0.15 and were considered for the multivariable model.
Of the 134 cases with myelotoxicity, 66 (49%) were first reported during the concurrent phase; an average of 40 days from the start of treatment (SD = 14 [range: 8–77]). Thirty-eight of these patients (57%) stopped treatment due to toxicity. Treatment termination due to any toxicity during the concurrent phase was more common among patients with myelotoxicity (60%) than those without myelotoxicity (20%) (P < .001). One patient continued to report myelotoxicity within 5 months of treatment termination, and one patient died within 1 month of treatment termination. Twenty-eight (42%) were able to continue to adjuvant treatment, 8 of which again experienced myelotoxicity; an average of 145 days from the start of treatment (SD = 91 [range: 91–364]) and an average of 111 days from the first event (SD = 91 [range: 61–333]). Sixty-seven (50%) of the remaining cases were first reported to have significant myelotoxicity during the adjuvant/follow-up phase; occurring an average of 194 days from the start of treatment (SD = 125 [range: 78–566]).
Before any adjustment, patients receiving both TMZ and BEV were 60% more likely to develop myelotoxicity compared to those patients who only received TMZ (OR, 1.6; 95% CI, 1.01–2.40; P value, .015). Females were 3.26 times more likely to have myelotoxicity compared to males (OR, 3.26; 95% CI, 2.19–4.86; P value, < .0001). Patients with impaired KPS had higher risk of myelotoxicity (KPS = 70: OR, 2.45; 95% CI, 1.2–4.99. KPS = 80: OR, 2.09; 95% CI, 1.12–3.92. KPS = 90: OR, 1.44; 95% CI, 0.79–2.62. Trend P value, .0356). Increased creatinine, BSA and TMZ dose were associated with a lower risk of myelotoxicity. The final multivariable clinical factor model included treatment arm, gender, and anticonvulsant status (Table 2). The AUC for the ROC curve was 0.672, which represented a low prediction accuracy for clinical factors alone. When limiting the dataset to the 367 patients with genotype data, the AUC for the ROC curve for the model was 0.68, which was not statistically different than the AUC in the full dataset (P = .77).
Table 2.
Multivariable Logistic Regression Model with Significant Clinical Risk Factors and Dose Effect of Top SNPs
| Characteristics | Cases | Controls | OR | 95% Wald CI | P-value | |||
|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | |||||
| Final Clinical Model (n = 591) | ||||||||
| Treatment arm | .0243 | |||||||
| TMZ+placebo | 54 | 40.3 | 239 | 52.3 | 1 | |||
| TMZ+BEV | 80 | 59.7 | 218 | 47.7 | 1.60 | 1.06 | 2.38 | |
| Gender | <.0001 | |||||||
| Male | 52 | 38.8 | 308 | 67.4 | 1 | |||
| Female | 82 | 61.2 | 149 | 32.6 | 2.98 | 2.00 | 4.45 | |
| Anticonvulsants | .0386 | |||||||
| No | 29 | 21.6 | 133 | 29.1 | 1 | |||
| Yes | 105 | 78.4 | 324 | 70.9 | 1.65 | 1.03 | 2.65 | |
| Final Clinical+SNP Model (n = 367) | ||||||||
| Treatment arm | .0441 | |||||||
| TMZ+placebo | 30 | 36.1 | 153 | 53.9 | 1 | |||
| TMZ+BEV | 53 | 63.9 | 131 | 46.1 | 1.82 | 1.02 | 3.27 | |
| Gender | <.0001 | |||||||
| Male | 31 | 37.4 | 194 | 68.3 | 1 | |||
| Female | 52 | 62.7 | 90 | 31.7 | 4.45 | 2.45 | 8.08 | |
| Anticonvulsants | .1442 | |||||||
| No | 20 | 24.1 | 88 | 31.1 | 1 | |||
| Yes | 63 | 75.9 | 196 | 69.0 | 1.64 | 0.85 | 3.18 | |
| Top SNPs dose effect, number at-risk alleles | Median = 6, IQR = 4–7 | Median = 3, IQR = 1–5 | 1.64 | 1.43 | 1.89 | <.0001 | ||
Abbreviations: BEV, bevacizumab; CI, confidence interval; IQR, inter-quartile range; OR, odds ratio; SNP, single nucleotide polymorphism; TMZ, temozolomide.
Genetic Main Effects on Myelotoxicity
Among 367 patients with genotype information, 5 SNPs reached genome-wide statistical significance (Table 3). Four of the five SNPs were in MGMT, and the MAF of each SNP ranged from 0.46 to 0.59 among cases and 0.25 to 0.36 among controls. The strongest association with myelotoxicity was observed for rs1008982 with unadjusted P value 1.36 × 10−10 (OR, 3.12; 95% CI, 2.19–4.46 in unadjusted model; OR, 4.55; 95% CI, 2.81–7.35 in adjusted model including significant clinical factors). The final significant SNP, rs1549102, is located in ANKS1B with unadjusted P value 3.62 × 10−7 (OR, 2.52; 95% CI, 1.75–3.62 in unadjusted model; OR, 2.50; 95% CI, 1.71–3.66 in adjusted model including significant clinical factors).
Table 3.
Top SNPs with P Values at Least 10−7 Levels Identified by Genotyping
| SNP | Gene | Location | Chr: Position | Allele | Minor Allele Freq. | Unadjusted P value | Unadjusted | Adjusteda | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | ORs | 95% CI | ORs | 95% CI | ||||||||
| rs1008982 | MGMT | Intron | 10: 131445731 | G/A | 0.560 | 0.290 | 1.36 × 10−10 | 3.12 | 2.19 | 4.46 | 4.55 | 2.81 | 7.35 |
| rs12266634 | MGMT | Intron | 10: 131412659 | G/C | 0.512 | 0.267 | 2.76 × 10−9 | 2.88 | 2.02 | 4.12 | 3.78 | 2.40 | 5.95 |
| rs1711667 | MGMT | Intron | 10: 131278410 | C/A | 0.590 | 0.357 | 7.52 × 10−8 | 2.60 | 1.82 | 3.70 | 3.47 | 2.23 | 5.37 |
| rs10466114 | MGMT | Intron | 10: 131367368 | A/G | 0.458 | 0.253 | 3.84 × 10−7 | 2.50 | 1.74 | 3.58 | 2.95 | 1.94 | 4.49 |
| rs1549102 | ANKS1B | Intron | 12: 99282746 | C/A | 0.663 | 0.438 | 3.62 × 10−7 | 2.52 | 1.75 | 3.62 | 2.50 | 1.71 | 3.66 |
Abbreviations: CI, confidence interval; OR, odds ratio.
aAdjusting for the selected clinical risk factors: treatment arm, gender, and anticonvulsants.
Multivariable Model of Myelotoxicity
We compared demographic and clinical characteristics and toxicity outcome incidence between participants with (n = 367) and without (n = 224) genotype data to ensure comparability between these groups. No significant differences between the two sets of patients were found for any variables (data not shown), including those in the final clinical model (Table 2).
Initially, we aimed to fit our original clinical model developed for those receiving TMZ at recurrence17 to this sample; however, that model did not adequately fit the current data from newly diagnosed patients (data not shown). Therefore, we initiated an agnostic model building process in the current dataset as outlined above. Table 2 summarizes the results of multivariable regression model including both selected clinical and genetic risk factors.
Receiving BEV in addition to TMZ increased myelotoxicity risk 80% (OR, 1.82; 95% CI, 1.02–3.27; adjusted P-value, .0441). Females were about 4.5 times more likely to develop toxicity (OR, 4.45; 95% CI, 2.45–8.08; adjusted P–value, < .0001). Taking anticonvulsants increased risk, but not statistically. However, because anticonvulsant use influenced the overall fit of the model (P-value for likelihood ratio test < .05), we included it in the final clinical model. For each additional at-risk (minor) allele in any of the top 5 SNPs, the risk increased by 64% (OR, 1.64; 95% CI, 1.43–1.89; adjusted P-value, < .0001). The AUC of the final model increased to 0.827 after including the SNP dose effect, indicating a dramatic improvement in prediction accuracy. Figure 2 shows the ROC curve for the cross-validated model; the AUC is 0.807.
Figure 2.

Receiver-operator characteristics (ROC) curve and area under the curve (AUC) metrics of the multivariable risk prediction model with both clinical and genetic factors conducting 30-fold cross-validation. The AUC of 0.807 indicates that the model provides excellent discrimination between those with and without toxicity.
Haplotype Block Structure and LD Analysis among SNPs from MGMT
Because four of the top five SNPs were in MGMT, we constructed haplotypes to better understand the contribution of variation across MGMT on toxicity. Haplotype block structures were defined using the LD confidence intervals method.20 The association between toxicity and each common haplotype (frequency > 0.05) was evaluated by logistic regression analysis adjusting for the selected clinical risk factors. Figure 3 shows a plot of the pairwise LD (D’) values for the 13 significant SNPs within MGMTat 10−7 level. Two blocks (both intronic) with high LD were identified: block 1, containing selected SNPs rs1711667 and rs10466114; block 2, containing selected SNPs rs12266634 and rs1008982. We examined the association between the common haplotypes in MGMT and toxicity (Table 4). Two risk haplotypes were identified in Block 1: CA (adjusted OR, 3.96; 95% CI, 2.45–6.40) and CG (adjusted OR, 2.98; 95% CI, 1.62–5.49). One risk haplotype was identified in Block 2: GG (adjusted OR, 4.98; 95% CI, 2.99–8.29). The global score test showed significant differences in the haplotype profile for both Blocks 1 and 2 (P < .0001). Finally, we explored the haplotype association between toxicity and the four selected SNPs globally using a block-free approach. Consistent with the genotype analysis, the most common haplotype AGCA was also the most favorable haplotype, containing zero risk alleles. When we used this favorable haplotype as the reference group, haplotype CAGG (containing all four risk alleles) showed statistically significant increased toxicity (adjusted OR, 6.40; 95% CI, 3.59–11.43).
Figure 3.

Linkage disequilibrium (LD) plot of MGMT SNPs identified at E-7 level and haplotype-block structure. The values indicate the LD relationship between each pair of SNPs; darker shading denotes a greater extent of LD between SNPs. The SNPs selected for inclusion in the haplotype analysis sufficiently capture the variation represented in this region of MGMT.
Table 4.
Association between Haplotypes in MGMT and Myelotoxicity
| Blocks | Haplotypea | Cases (N=83) | Controls (N=284) | Total % | ORb | 95% Wald CI | Global P-value | |||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | |||||||
| Block 1 | AG | 32 | 38.8 | 182 | 64.0 | 58.4 | 1.00 | <.0001 | ||
| CA | 36 | 43.6 | 70 | 24.8 | 29.1 | 3.96 | 2.45 | 6.40 | ||
| CG | 13 | 15.5 | 31 | 10.8 | 11.8 | 2.98 | 1.62 | 5.49 | ||
| Block 2 | CA | 34 | 40.7 | 197 | 69.5 | 63.0 | 1 | <.0001 | ||
| GG | 40 | 47.9 | 71 | 24.9 | 30.1 | 4.98 | 2.99 | 8.29 | ||
| Block free | AGCA | 30 | 36.0 | 178 | 62.8 | 56.7 | 1 | <.0001 | ||
| CAGG | 32 | 39.0 | 56 | 19.7 | 24.2 | 6.40 | 3.59 | 11.43 | ||
Note: Loci for Block 1: rs1711667 and rs10466114; Block 2: rs12266634 and rs1008982; block free: rs1711667, rs10466114, rs12266634, and rs1008982.
aHaplotypes with frequency less than 0.05 were excluded from the analysis.
bLogistic regression model adjusting for treatment arm, gender, and anticonvulsant use.
Discussion
The occurrence of myelotoxicity during concurrent chemoradiation with TMZ is an uncommon but serious event. In this study, as with others, occurrence was idiosyncratic, occurring early in treatment rather than after cumulative exposure. As expected,21 incidence was higher in those on combination therapy, but surprisingly dose and BSA did not predict occurrence. The significance of the early toxicity in this study is noteworthy. The majority of patients who experienced myelotoxicity stopped their treatment and several experienced myelotoxicity months afterwards. Patients who continued onto adjuvant therapy also were likely to subsequently experience significant myelotoxicity.
Gender was the only demographic factor associated with myelotoxicity in this study. Females were 4.5 times as likely to experience myelotoxicity than males. Gender as a predictor of toxicity was also shown in our earlier study of recurrent disease,17 as well as a smaller study of the concurrent phase of treatment.22 The propensity for females to experience more drug toxicity is increasingly being recognized. Overall, the degree to which gender relates to drug toxicity is not known, but it was reported that up to 7% of new drug applications that include gender analysis showed at least a 40% difference in pharmacokinetics between males and females.23 The underlying molecular mechanism is probably multifactorial, including increased drug exposure by hormone-dependent regulation of proteins in drug metabolism pathways and/or by direct action of sex hormones on the drug target.24
As noted by Church and colleagues,12 the burden of chemotherapy-associated toxicity is recognized but there are limited tools to increase the precision of prescribing chemotherapy to reduce risk. They coined the term “toxgnostics” as the systematic agnostic study of genetic predictors of such toxicity. Key elements framing these analyses were: analysis embedded within large, prospective, randomized, controlled clinical trials; phenotype clearly defined and clinically relevant; analysis using the maximum relevant genomic diversity; and comparison of the performance of individual variants to that of a combined risk score, which may outperform individual variants. We followed these key tenants in our study.
In this report, for every additional at-risk allele carried by a patient, the risk of myelotoxicity increased by 64%. Further, the inclusion of the SNP data in our risk prediction model resulted in a cross-validated AUC of 0.807. Interestingly, four of our five top SNPs are located in MGMT. In our study, there was no association between tumor MGMT promoter methylation status and myelotoxicity. This is not a surprising finding as the promoter methylation, to our knowledge, is restricted to tumor cells; therefore, reduction of MGMT enzyme activity in hematopoietic cells could be due to the polymorphisms rather than expression changes due to the methylation status of the promoter region of the gene. Furthermore, there were no associations between our top SNPs and MGMT promoter methylation status (all P-values > .226). However, two recent studies reported increased risk of myelotoxicity to be associated with female gender and with MGMT promoter methylation. Becker-Schiebe et al.25 identified the methylated MGMT promoter in the tumor tissue of 4/5 (80%) patients with grade 4 myelotoxicity compared to 19/64 (30%) among those without grade 4 toxicity. Lombardi et al.22 detected MGMT promoter methylation in peripheral blood DNA from 4/4 (100%) patients with grade 3–4 myelotoxicity and 0/9 (0%) patients without myelotoxicity. They also reported that the distribution of genotypes in 3 candidate SNPs in MGMT was not statistically different between patients with and without myelotoxicity; however, several SNPs in various drug metabolism genes were associated with myelotoxicity.
The differences between our study and previous ones are three-fold. First, our study is based on a large randomized, controlled clinical trial rather than institutional series of patients. Second, due to the multi-institutional nature of the parent trial, our study is much larger, providing the ability to build multivariable risk prediction models for toxicity. Third, our genetic analyses considered first genome-wide variation rather than candidate SNPs or pathways. One primary limitation to our study was the inability to validate our findings in an independent sample of patients. However, given the unique treatment combination provided under the parent trial, there was no existing comparable population. For this reason, we employed a highly robust cross-validation of our findings, as described in the methods above. This approach provided the opportunity to validate our results using a sophisticated statistical approach in the absence of an external validation population.
This unique collaborative research effort allows us to maximize power for the discovery of novel associations between clinical variables, germline polymorphisms, and myelotoxicity in this at-risk population. Our group was among the first to examine the effects of individual genomic variation on treatment toxicity and response among GBM patients. Our preliminary studies were limited by the retrospective nature of our single institutional repository and the application of our risk model for recurrent GBM. The current expansion of these efforts to perform analyses using the prospectively collected data and samples from RTOG 0825 has allowed for a more robust analysis using modern treatment paradigms. The findings of this report are limited in clinical utility, until further test development can be completed and integrated into clinical care. Use of this model for prediction of risk of toxicities has the potential to provide more cost-effective individualized monitoring of at-risk subjects and allow better individualized initial temozolomide dosing and bloodwork monitoring schedules in patients with GBM receiving this drug. In order to implement this as a clinical tool, development of a assay system to evaluate the pertinent polymorphisms would be necessary, as has been successfully done for other chemotherapy-associated toxities, such as to predict against fluoropyrimidine toxicity.26
Acknowledgments
We kindly thank Philip J Stella, MD for collecting/assembly data and reading and reviewing this manuscript. In addition to the work he did accruing patients under Michigan Cancer Research Consortium CCOP. Presented: 2015 Society for Neuro-Oncology Meeting, November 19-22, 2015.
Contributor Information
Michael E Scheurer, Baylor College of Medicine, Departments of Pediatrics and Medicine, Houston, Texas, USA.
Renke Zhou, Baylor College of Medicine, Departments of Pediatrics and Medicine, Houston, Texas, USA.
Mark R Gilbert, National Institutes of Health Clinical Center, Bethesda, MD, USA.
Melissa L Bondy, Baylor College of Medicine, Departments of Pediatrics and Medicine, Houston, Texas, USA.
Erik P Sulman, M D Anderson Cancer Center, Brain and Spine Center, Houston, TX, USA; Laura and Isaac Perlmutter Cancer Center at NYU Langone, New York, NY, USA.
Ying Yuan, M D Anderson Cancer Center, Brain and Spine Center, Houston, TX, USA.
Yanhong Liu, Baylor College of Medicine, Departments of Pediatrics and Medicine, Houston, Texas, USA.
Elizabeth Vera, National Institutes of Health Clinical Center, Bethesda, MD, USA; M D Anderson Cancer Center, Brain and Spine Center, Houston, TX, USA.
Merideth M Wendland, National Cancer Institute, Bethesda, MD, USA; Texas Oncology Cancer Center Sugar Land, Sugar Land, TX, USA.
Emad F Youssef, Arizona Oncology Services Foundation, Tucson, AZ, USA.
Volker W Stieber, Forsyth Regional Cancer Center, Winston-Salem, NC, USA.
Ritsuko R Komaki, M D Anderson Cancer Center, Brain and Spine Center, Houston, TX, USA.
John C Flickinger, UPMC-Shadyside Hospital, Pittsburgh, PA, USA.
Lawrence C Kenyon, Thomas Jefferson University Hospital, Philadelphia, PA, USA.
H Ian Robins, University of Wisconsin Hospital, Madison, WI, USA.
Grant K Hunter, Intermountain Medical Center, Murray, UT, USA.
Ian R Crocker, Emory University, Winship Cancer Institute, Atlanta, GA, USA.
Samuel T Chao, Cleveland Clinic Foundation, Cleveland, OH, USA.
Stephanie L Pugh, NRG Oncology Statistics and Data Management Center, Philadelphia, PA, USA.
Terri S Armstrong, National Institutes of Health Clinical Center, Bethesda, MD, USA.
Funding
This work was supported by National Cancer Institute (U10CA180868 (NRG Operations), U10CA180822 (NRG SDMC), UG1CA189867 (NCORP), U24CA196067 (NRG Biospecimen Bank)); National Institutes of Health (1R01NR013707, K07CA181480) by a research grants from the Voices Against Brain Cancer Foundation and Genentech.
Conflict of interest statement. None: Dr(s). Armstrong, Bondy, Crocker, Flickinger, Gilbert, Hunter, Kenyon, Komaki, Liu, Robins, Scheurer, Stella, Stieber, and Vera. Dr. Chao discloses honoraria from Varian, Zeiss, and Abbvie, and a consulting or advisory role from Novocure. Dr. Pugh discloses research funding from Millennium. Dr. Sulman discloses honoraria from Merck, consulting or advisory roles from Merck, AbbVie, and Novocure, research funding from AbbVie and Novocure, and travel, accommodations, or expenses from Merck, AbbVie, and Novocure. Dr. Wendland discloses employment from US Oncology. Dr. Youssef discloses stock or other ownership with Gammatile, and patents, royalties, or other intellectual property with Gammatile. Dr. Yuan discloses honoraria from Vertex Pharmaceuticals and Ono Pharmaceuticals, consulting or advisory roles from Akros Pharma Inc., and travel, accommodations, or expenses from Vertex Pharmaceuticals.
Authorship Statement. Conception and design- YY, EPS, MES, MRG, MLB, TSA. Collection and assembly of data- YY, EFY, MMW, EPS, MES, HIR, SLP, RRK, LCK, GKH, MRG, IRC, STC, TSA. Data analysis and interpretation- RZ, YY, EV, VWS, MES, SLP, YL, MRG, JCF, TSA. Drafting the work or revising it critically for important intellectual content- MES, RZ, MRG, MLB, EPS, YY, YL, EV, MMW, EFY, VWS, RRK, JCF, LCK, HIR, GKH, IRC, STC, SLP, TSA. Read and Approved the final version-MES, RZ, MRG, MLB, EPS, YY, YL, EV, MMW, EFY, VWS, RRK, JCF, LCK, HIR, GKH, IRC, STC, SLP, TSA.
References
- 1. Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015; 17(Suppl 4):iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 3. Gilbert MR, Wang M, Aldape KD, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J ClinOncol. 2013; 31(32):4085–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gerber DE, Grossman SA, Zeltzman M, Parisi MA, Kleinberg L. The impact of thrombocytopenia from temozolomide and radiation in newly diagnosed adults with high-grade gliomas. Neuro Oncol. 2007; 9(1):47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singhal N, Selva-Nayagam S, Brown MP. Prolonged and severe myelosuppression in two patients after low-dose temozolomide treatment- case study and review of literature. J Neurooncol. 2007; 85(2):229–230. [DOI] [PubMed] [Google Scholar]
- 6. Noronha V, Berliner N, Ballen KK, et al. Treatment-related myelodysplasia/AML in a patient with a history of breast cancer and an oligodendroglioma treated with temozolomide: case study and review of the literature. Neuro Oncol. 2006; 8(3):280–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Comez G, Sevinc A, Sever ON, et al. An unusual case of aplastic anemia caused by temozolomide. Case Rep Med. 2010; 2010:975039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oh J, Kutas GJ, Davey P, Morrison M, Perry JR. Aplastic anemia with concurrent temozolomide treatment in a patient with glioblastoma multiforme. Curr Oncol. 2010; 17(4):124–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jalali R, Singh P, Menon H, Gujral S. Unexpected case of aplastic anemia in a patient with glioblastoma multiforme treated with Temozolomide. J Neurooncol. 2007; 85(1):105–107. [DOI] [PubMed] [Google Scholar]
- 10. Morris EB, Kasow K, Reiss U, Ellison D, Broniscer A. Bone marrow transplantation for severe aplastic anemia secondary to temozolomide. J Neurooncol. 2009; 91(2):237–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang RS, Ratain MJ. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J Clin. 2009; 59(1):42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Church D, Kerr R, Domingo E, et al. “Toxgnostics”: an unmet need in cancer medicine. Nat Rev Cancer. 2014; 14(6):440–445. [DOI] [PubMed] [Google Scholar]
- 13. Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007; 447(7145):661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002; 2(1):43–47. [DOI] [PubMed] [Google Scholar]
- 15. Johnson DC, Corthals SL, Walker BA, et al. Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. J ClinOncol. 2011; 29(7):797–804. [DOI] [PubMed] [Google Scholar]
- 16. Cibeira MT, de Larrea CF, Navarro A, et al. Impact on response and survival of DNA repair single nucleotide polymorphisms in relapsed or refractory multiple myeloma patients treated with thalidomide. Leuk Res. 2011; 35(9):1178–1183. [DOI] [PubMed] [Google Scholar]
- 17. Armstrong TS, Cao Y, Scheurer ME, et al. Risk analysis of severe myelotoxicity with temozolomide: the effects of clinical and genetic factors. Neuro Oncol. 2009; 11(6):825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370(8):699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Molinaro AM, Simon R, Pfeiffer RM. Prediction error estimation: a comparison of resampling methods. Bioinformatics. 2005; 21(15):3301–3307. [DOI] [PubMed] [Google Scholar]
- 20. Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002; 296(5576):2225–2229. [DOI] [PubMed] [Google Scholar]
- 21. Kurtin S. Myeloid toxicity of cancer treatment. J Adv Pract Oncol. 2012; 3(4):209–224. [PMC free article] [PubMed] [Google Scholar]
- 22. Lombardi G, Rumiato E, Bertorelle R, et al. Clinical and genetic factors associated with severe hematological toxicity in glioblastoma patients during radiation plus temozolomide treatment: a prospective study. Am J Clin Oncol. 2015; 38(5):514–519. [DOI] [PubMed] [Google Scholar]
- 23. Anderson GD. Sex and racial differences in pharmacological response: where is the evidence? Pharmacogenetics, pharmacokinetics, and pharmacodynamics. J Womens Health (Larchmt). 2005; 14(1):19–29. [DOI] [PubMed] [Google Scholar]
- 24. Nicolson TJ, Mellor HR, Roberts RR. Gender differences in drug toxicity. Trends Pharmacol Sci. 2010; 31(3):108–114. [DOI] [PubMed] [Google Scholar]
- 25. Becker-Schiebe ME, Wetzel M, Wetzel F, Christansen H, Hoffmann W. Hematologic toxicity of temozolomide and radiation in glioblastoma patients - correlation with clinicopatholgoical factors. Clin Med J. 2015; 1:63–69. [Google Scholar]
- 26. Del Re M, Di Paolo A, van Schaik RH, et al. Dihydropyrimidine dehydrogenase polymorphisms and fluoropyrimidine toxicity: ready for routine clinical application within personalized medicine? EPMA J. 1(3):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets generated and/or analyzed during this study, as well as the computer code used to perform statistical analysis, are available from the corresponding authors on reasonable request. The datasets from the parent trial are available in dbGaP.