

Abstract
Small Extracellular Vesicles (sEVs) are 50–200 nm in diameter vesicles delimited by a lipid bilayer, formed within the endosomal network or derived from the plasma membrane. They are secreted in various biological fluids, including airway nasal mucus. The goal of this work was to understand the role of sEVs present in the mucus (mu‐sEVs) produced by human nasal epithelial cells (HNECs) in SARS‐CoV‐2 infection. We show that uninfected HNECs produce mu‐sEVs containing SARS‐CoV‐2 receptor ACE2 and activated protease TMPRSS2. mu‐sEVs cleave prefusion viral Spike proteins at the S1/S2 boundary, resulting in higher proportions of prefusion S proteins exposing their receptor binding domain in an ‘open’ conformation, thereby facilitating receptor binding at the cell surface. We show that the role of nasal mu‐sEVs is to complete prefusion Spike priming performed by intracellular furin during viral egress from infected cells. This effect is mediated by vesicular TMPRSS2 activity, rendering SARS‐CoV‐2 virions prone to entry into target cells using the ‘early’, TMPRSS2‐dependent pathway instead of the ‘late’, cathepsin‐dependent route. These results indicate that prefusion Spike priming by mu‐sEVs in the nasal cavity plays a role in viral tropism. They also show that nasal mucus does not protect from SARS‐CoV‐2 infection, but instead facilitates it.
Keywords: human nasal epithelial cells, SARS‐CoV‐2, small extracellular vesicles, Spike prefusion priming, TMPRSS2
1. INTRODUCTION
The emergence in December 2019 in Wuhan, Hubei province, China (Zhou et al., 2020; Zhu et al., 2020) and the rapid spread of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2) has been responsible for a worldwide pandemic that created a global sanitary, economic, political, and social crisis. As of July 20, 2022, coronavirus disease 2019 (COVID‐19) has been responsible for at least 569,898,918 cases and 6,392,173 deaths worldwide (https://www.worldometers.info/coronavirus/).
The most common clinical presentation of COVID‐19 includes fever, cough, shortness of breath and/or sputum production. COVID‐19 may lead to respiratory failure and acute respiratory distress syndrome (ARDS). Infected patients often report symptoms that suggest the involvement of organs outside of the respiratory tract, including signs of haematological, cardiovascular, renal, gastrointestinal, hepatobiliary, endocrinological, neurological, ophthalmological and/or dermatological involvement (Gupta et al., 2020). Loss of taste and smell has been considered as a frequent feature of COVID‐19 (Menni et al., 2020), as a result of smell receptor alteration, as previously observed with other respiratory viruses (Hopkins et al., 2020). Nevertheless, this symptom has become rarer with more recently emerged viral variants. Thus, although the main site of SARS‐CoV‐2 infection remains the epithelial mucosa of the upper respiratory tract and the lungs, infection of other organs and the systemic consequences of dysregulated immune host responses indicate that COVID‐19 is a systemic disease (Mehta et al., 2020; Synowiec et al., 2021).
SARS‐CoV‐2 entry into target cells has been well characterized. It is mediated by the viral Spike (S) protein, which belongs to the class I fusion protein family. The Spike protein ensures receptor binding and fusion of the viral lipid envelope with cellular membranes (Li, 2016; Perlman & Netland, 2009; Shang et al., 2020). The S protein is made of two subunits: (i) S1 which contains an N‐terminal domain (NTD), the receptor‐binding domain (RBD) and two subdomains (SD1 and SD2), and (ii) S2 which contains the fusion peptide (FP), a conserved motif principally composed of hydrophobic residues that triggers fusion after its insertion into host cell membranes, two heptad repeats (HR1 and HR2), the transmembrane domain (TM) and the cytoplasmic tail (CT) (Tang et al., 2020). SARS‐CoV‐2 S protein trimers bind the human angiotensin‐converting enzyme 2 (ACE2) receptor with high affinity (Hoffmann, Kleine‐Weber et al., 2020; Wang et al., 2020; Yan et al., 2020; Zhou et al., 2020). Several possible conformations of the RBD have been described within the structure of the prefusion SARS‐CoV‐2 S protein. In the ‘closed’ conformation, the RBD is in a ‘lying down’ position, that is, it caps the top of the S2 subunit and is not accessible to ACE2 (Benton et al., 2020). In the ‘open’ conformation, the RBD changes its orientation so that the ACE2 interacting surface becomes fully exposed at the top of the Spike monomer (Wrobel et al., 2020). In vitro experiments have shown that the vast majority of SARS‐CoV‐2 RBD trimers is fully closed (i.e., three RBDs in ‘closed’ conformation) or partially open (one RBD in ‘open’ and two in ‘closed’ conformations) (Ke et al., 2020; Walls et al., 2020; Wrapp et al., 2020).
Structural rearrangements of the S protein, from the closed pre‐fusion to the post‐fusion form, require proteolytic cleavages by host proteases that contribute to viral tropism (Cai et al., 2020; Fuentes‐Prior, 2021; Millet & Whittaker, 2015; Tang et al., 2020; White & Whittaker, 2016). The first cleavage separates the S1 subunit, responsible for receptor binding, from the S2 subunit that contains the fusion machinery, resulting in a non‐covalent association between S1 and transmembrane‐anchored S2. The second proteolytic cleavage occurs at the S2’ site within the S2 subunit, exposing the hydrophobic FP that can be inserted into the host cell membrane. This leads to the formation of coiled‐coil motifs by HR domains and, finally, to fusion of the viral and cellular membranes (Belouzard et al., 2012; Madu et al., 2009; Whittaker et al., 2021).
As described for SARS‐CoV‐1, SARS‐CoV‐2 can be triggered to fuse either at the plasma membrane (‘early pathway’) or at the endosome membrane (‘late pathway’), depending on the availability of host proteases in its target cells (Matsuyama et al., 2005). Type 2 transmembrane serine proteinase (TMPRSS2) has been shown to play a major role in SARS‐CoV‐2 entry, by cleaving the S protein at the S2’ site (R815) (Bestle et al., 2020; Hoffmann, Kleine‐Weber et al., 2020; Matsuyama et al., 2020). The SARS‐CoV‐2 Spike protein gene contains an insertion of 12 nucleotides at the S1/S2 boundary that creates a minimal proprotein convertase (PC) furin‐like cleavage site (PRRAR↓SV) (Coutard et al., 2020). Furin cleavage at the S1/S2 site has been demonstrated, as well as less efficient viral entry in the presence of PC inhibitors (Bestle et al., 2020; Hoffmann, Kleine‐Weber, & Pohlmann, 2020; Papa et al., 2021; Shang et al., 2020; Walls et al., 2020). The expression of furin in the trans‐Golgi could result in a pre‐activated state of the S protein (‘open’ RBD conformation of the prefusion S protein) in released virions (Shang et al., 2020; Wrobel et al., 2020).
Small Extracellular Vesicles (sEVs) are 50–200 nm in diameter vesicles delimited by a lipid bilayer, formed within the endosomal network or derived from the plasma membrane. They are secreted in various biological fluids, including airway nasal mucus (Akers et al., 2013; Thery et al., 2018; Wu et al., 2015). The molecular composition of the sEVs varies according to their cellular origin. They consist of selected cellular components located inside the vesicles or associated with their membrane. sEVs usually contain membrane proteins known to cluster into microdomains at the plasma or endosome membranes, mRNAs or non‐coding RNAs, and metabolites (Raposo & Stoorvogel, 2013; Zebrowska et al., 2019). sEVs are not only waste transporter. They are pivotal elements involved in intercellular communication, capable to transfer the aforementioned biomolecules from donor to recipient cells (Mathieu et al., 2019).
During the course of viral infections, viruses can hijack the sEV biogenesis system to favour viral particle secretion (Ramakrishnaiah et al., 2013). In addition, viruses can modify the sEV content in viral and host components to render healthy cells more susceptible to infection (Mack et al., 2000; Rozmyslowicz et al., 2003) or to modulate the immune response in favour of the pathogen or the host (Li et al., 2013; Schorey et al., 2015). Whether respiratory sEVs isolated from healthy cells contain ACE2 and TMPRSS2 and could therefore play a role during SARS‐CoV‐2 infection is unknown.
The goal of this work was to unravel the molecular composition of sEVs present in the mucus produced at the apical pole of human nasal epithelial cells (mu‐sEVs) and to understand the role of mu‐sEVs in priming the SARS‐CoV‐2 Spike protein and facilitating its entry through the ‘early pathway’, thereby favouring infection of TMPRSS2‐expressing cells.
2. RESULTS
2.1. Human nasal epithelial cells in primary culture can be infected with SARS‐CoV‐2 and their infection is reduced by mucus removal
We used an original model of uninfected primary human nasal epithelial cells (HNECs) which, unlike immortalized cell lines, recapitulate the most important characteristics of in vivo human respiratory epithelia (Bequignon et al., 2019; Coste et al., 2000; Muller et al., 2013; Papon et al., 2002). Such HNECs, isolated from nasal polyps (NP), constitute a stable differentiated epithelium containing three major cell types (e.g., basal, goblet and ciliated cells) after more than 21 days of culture at the air‐liquid interface (ALI) using a specific medium (Coste et al., 2000). When differentiated, HNECs exhibit morphological and functional characteristics that mimick the normal human nasal epithelium. Mucus, that constitutes the first barrier viruses encounter during infection, is notably produced by goblet cells at the apical pole of the epithelium (Harkema et al., 2006).
HNECs isolated from three different patients were infected at the apical pole of the epithelium with 20 μl of SARS‐CoV‐2 inoculum (∼2.04 × 104 TCID50/ml). As shown in Figure 1A, HNEC infection resulted in viral replication, as reflected by the production of intracellular SARS‐CoV‐2 RNA 72 h post‐infection. SARS‐CoV‐2 RNA production was reduced by 8.5‐fold when mucus produced by HNECs was removed (Figure 1A), suggesting that mucus contains components that favour viral infection. Mucus removal had no effect on HNEC vitality, as assessed by trans‐epithelial electrical resistance (TEER) (Figure S1).
FIGURE 1.
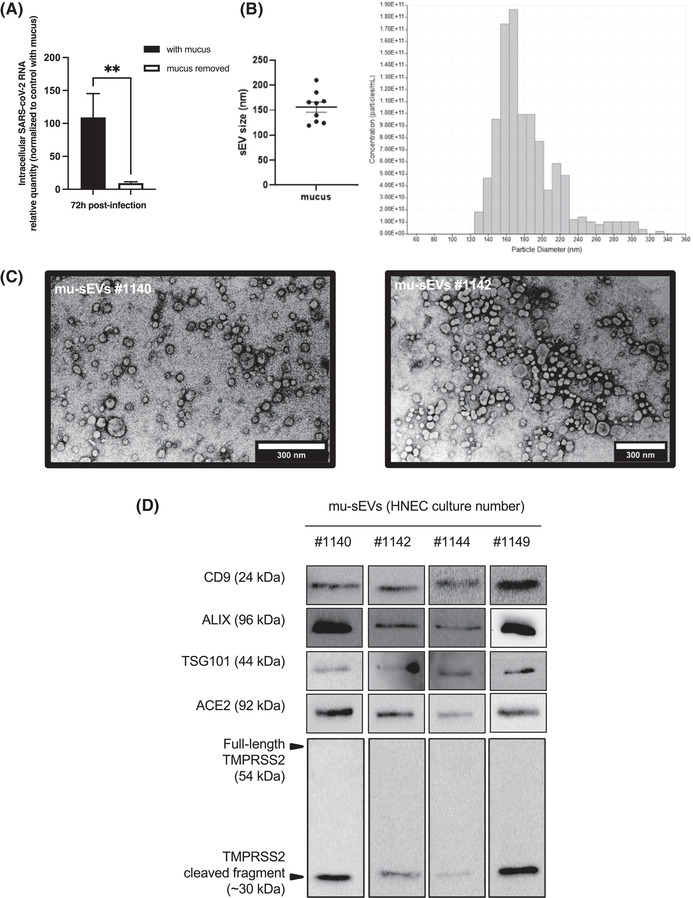
Effect of nasal mucus removal on SARS‐CoV‐2 infection of HNECs and characterization of mu‐sEVs. (A) Intracellular RNA was extracted 72 h post‐infection from HNECs isolated from three different patients, infected for 4 h with SARS‐CoV‐2 (20 μl of viral inoculum, ∼2.04 × 104 TCID50/ml) at the apical pole, in the presence or after removal of cell‐produced mucus. SARS‐CoV‐2 RNA was quantified by RT‐qPCR and the results were normalized to 18S rRNA, then to control with mucus ( = 100%). They are expressed as mean ± SEM. **p < 0.01. (B) sEVs isolated from ∼5 ml of mucus (mu‐sEVs) produced by HNECs from nine different patients analysed by tunable resistive pulse sensing (TRPS). Left panel: TRPS results from nine mu‐sEVs indicating a mean peak particle size of 155.9 ± 30.83 nm in diameter. Right panel: TRPS results obtained from one representative mucus. (C) mu‐sEVs isolated from two patients adsorbed on copper grid, dried at room temperature, stained with uranyl acetate 1% and imaged by transmission electron microscopy. Scale bar: 300 nm. (D) Western blot analysis of CD9, ALIX, TSG101, ACE2 and full‐length and cleaved TMPRSS2 proteins using the amount of 109 mu‐sEVs isolated from four different patients
2.2. Mucus produced by HNECs contains sEVs carrying host factors required for SARS‐CoV‐2 entry
The presence of extracellular vesicles produced by HNECs in the mucus was demonstrated by means of differential ultracentrifugation enrichment (Salem et al., 2020; Thery et al., 2006). Tunable resistive pulse sensing (TRPS) was used to estimate the amount of small (mu‐sEVs) and large extracellular vesicles (mu‐lEVs) produced by HNECs from 14 patients (Figure S2A). Mean concentrations of 3.3 ± 1.2 × 1010 mu‐sEVs and 9.6 ± 2.8 × 107 mu‐lEVs per ALI insert were observed, respectively. Because of the large excess of mu‐sEVs over mu‐lEVs, we focused our study on the characterization of sEVs and their potential effect on SARS‐CoV‐2 infection.
As shown in Figure 1B, TRPS of mu‐sEVs produced by HNECs from nine different patients revealed a mean peak particle size of approximately 155.9 ± 30.8 nm in diameter. Figure 1C shows the size and discoid morphology of mu‐sEVs isolated from mucus generated by HNECs from two patients, as assessed by transmission electron microscopy (TEM). Finally, Figure 1D shows western blot analysis of mu‐sEVs produced by HNECs from four different patients, demonstrating the presence of sEV membrane surface marker CD9 and packaged protein markers Tumour susceptibility gene 101 protein (TSG101) and ALG‐2‐interacting protein X (ALIX). Western blot analysis, performed on HNEC lysates, mu‐sEVs and EV‐deprived mucus (negative control) from two patients, showed the absence of EV markers in EV‐deprived mucus compared to mu‐sEVs and the absence of cytochrome C (mitochondrial marker) and calnexin (ER marker) in mu‐sEVs compared to HNEC lysates (Figure S2B).
Because the presence of ACE2 and TMPRSS2 at the apical pole of HNEC ciliated cells has been demonstrated (Ahn et al., 2021), we looked for the presence of these SARS‐CoV‐2 entry factors in mu‐sEVs. Western blot analysis demonstrated the presence of both full‐length ACE2 (∼100 kDa) and only the activated (cleaved) form of TMPRSS2 (∼30 kDa) in mu‐sEVs. These two SARS‐CoV‐2 entry factors were also detected in mu‐lEVs (Figure S2C). Together, these findings indicate that HNECs produce mucus that contains sEVs, which themselves contain both ACE2 and TMPRSS2, the key SARS‐CoV‐2 cellular entry factors.
2.3. mu‐sEVs facilitate SARS‐CoV‐2 infection of HNECs
To evaluate the effect of mu‐sEVs on SARS‐CoV‐2 infection, viral particles were incubated in the presence of increasing concentrations of mu‐sEVs or EV‐deprived mucus (negative control) generated by HNECs from two patients. Then, these viral particles were used to infect HNECs from a third, different patient after removal of recipient cell mucus. As shown in Figure 2A, a mu‐sEV concentration‐dependent augmentation of the amount of intracellular SARS‐CoV‐2 RNA was observed when mucus‐free HNECs were infected with viral particles preincubated with mu‐sEVs. The highest proviral effect was achieved with 109 mu‐sEVs, a concentration within the range of physiological HNEC production per ALI insert. No proviral effect was observed when viral particles were preincubated with EV‐deprived mucus (Figure 2A, hatched histogram).
FIGURE 2.
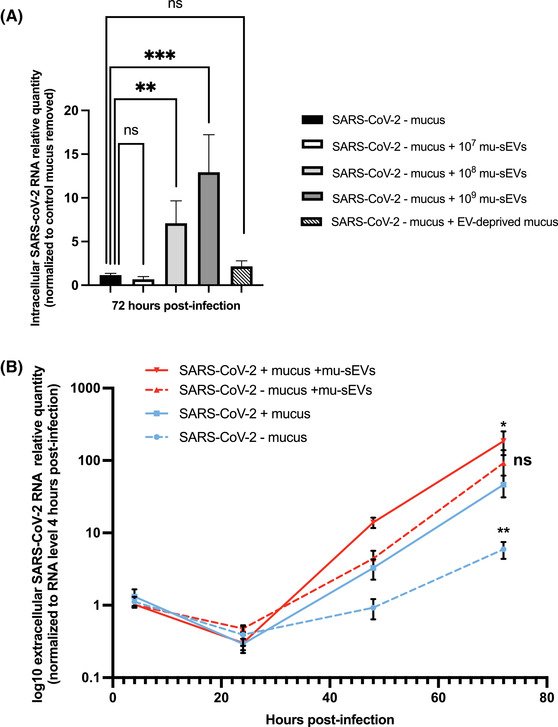
Effect of mu‐sEVs and mucus‐containing sEVs on HNEC infection by SARS‐CoV‐2. (A) Effect of preincubation of viral particles with different concentrations (107–109) of mu‐sEVs or with EV‐deprived mucus on SARS‐CoV‐2 infection (10 μl of viral inoculum, ∼2.04 × 105 TCID50/ml) at the apical pole of HNECs after removal of recipient cell mucus. Intracellular SARS‐CoV‐2 RNA was extracted 72 h post‐infection and quantified by RT‐qPCR. Results were normalized to 18S rRNA, then to mucus‐free control and viral particle preincubation with PBS alone (black bar). They are expressed as mean ± SEM of two independent experiments. **p < 0.01, ***p < 0.001. (B) Dynamics of SARS‐CoV‐2 RNA production at the apical pole of HNECs, as assessed by RT‐qPCR. Results were normalized to the viral RNA level 4 h post‐infection for each condition and expressed as log10 mean ± SEM of two independent experiments. *p < 0.05, **p < 0.01 (Mann‐Whitney U‐test versus extracellular SARS‐CoV‐2 RNA levels in the presence of recipient cell mucus 72 h post‐infection (blue line))
To investigate the effect of mu‐sEVs on virus production at the apical pole of HNECs, SARS‐CoV‐2 viral particles were preincubated with 109 mu‐sEVs generated by HNECS from two patients. These viral particles were used to infect HNECs from a third, different patient without or after removal of recipient cell mucus and virus production at the apical pole was monitored at different time points (Figure S3). The dynamics of SARS‐CoV‐2 RNA production after infection are shown in Figure 2B. The highest level of production was observed when HNECs with their mucus had been infected by viral particles preincubated with mu‐sEVs (Figure 2B, red line). Mucus removal only slightly and non‐significantly reduced viral production by cells infected with viruses preincubated with mu‐sEVs, to a level similar to that in cells that kept their mucus and were infected with viruses that had not been preincubated with mu‐sEVs (Figure 2B, dotted red line and blue line, respectively). Finally, viral production was significantly reduced when HNEC mucus had been removed and viruses had not been preincubated with mu‐sEVs (Figure 2B, dotted blue line). Together, the results indicate that both mucus‐containing sEVS and purified mu‐sEVS facilitate SARS‐CoV‐2 infection of HNECs.
2.4. TMPRSS2‐containing VCaP‐derived sEVs are a good model to study the mechanisms underlying the effect of mu‐sEVs on SARS‐CoV‐2 infection
Due to the limited amounts of mu‐sEVs produced by fully differentiated HNECs isolated from patients, we decided to use the in vitro model of sEVs isolated from immortalized cell lines to dissect the mechanisms underlying mu‐sEVs effects on SARS‐CoV‐2 infection. We selected the VCaP cell line, which has been previously demonstrated to express high levels of TMPRSS2 at both mRNA and protein levels (Li et al., 2021). VCaP enrichment was performed as previously reported (Ludwig et al., 2018) and VCaP‐sEVs produced in culture supernatants were characterized. Using TRPS technology, their mean peak particle size was approximately 154.6 ± 9.4 nm in diameter, comparable to that of mu‐sEVs (Figure S4A). Western blot analysis demonstrated the presence of both VCaP‐sEV markers (CD9, CD81, CD63, TSG101, ALIX) and SARS‐CoV‐2 entry factors (ACE2 and both full‐length and activated TMPRSS2) (Figure S4B).
Similar to what had been observed with mu‐sEVs, SARS‐CoV‐2 RNA production by HNECs was impaired by mucus removal, but it was restored when viral particles had been preincubated with VCaP‐sEVs. A significant increase in SARS‐CoV‐2 RNA production was observed when recipient cell mucus was not removed while viral particles had been preincubated with VCaP‐sEVs (Figure S3C). Based on these results, VCaP‐sEVs were subsequently used as a relevant model to unravel the mechanisms by which mu‐sEVs facilitate SARS‐CoV‐2 infection.
2.5. TMPRSS2‐expressing Calu‐3 cells are a good in vitro model to study the mechanisms underlying the effect of mu‐sEVs on SARS‐CoV‐2 infection
We tested different cellular models of SARS‐CoV‐2 infection, including Calu‐3 cells (in which the virus preferentially uses the early TMPRSS2‐dependent route of entry), VERO‐E6 and A549 cells (in which the virus preferentially uses the late, endosomal cathepsin‐dependent route of entry). Cells were infected at a final MOI of 0.1 and the intracellular amount of SARS‐CoV‐2 RNA was measured by RT‐qPCR 48 h post‐infection. Incubation of viral particles with VCaP‐sEVs prior to infection resulted in a significant, approximately 13‐fold increase in the intracellular amount of SARS‐CoV‐2 RNA in Calu‐3 cells, whereas a significant decrease was observed in infected A549 and VERO‐E6 cells (Figure 3A). This result indicates that VCaP‐sEVs can play a dual role in SARS‐CoV‐2 infection, depending on the virus preferential cellular route of entry. As shown in Figure 3B, the proviral effect of VCaP‐sEVs was confirmed with a SARS‐CoV‐2 Alpha variant strain in Calu‐3 cells. Thus, Calu‐3 cells, which express TMPRSS‐2 and thereby mimic natural infection, were used for further experiments.
FIGURE 3.
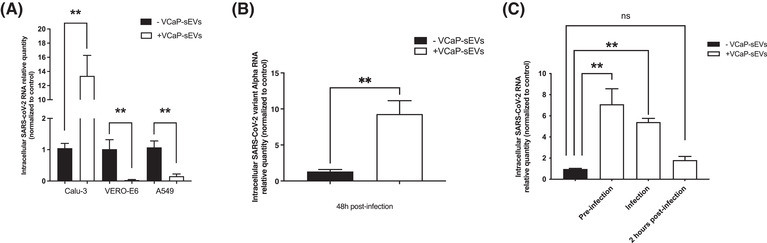
Proviral effect of VCaP‐sEVs on SARS‐CoV‐2 infection in TMPRSS2‐expressing cells. (A) Effect of VCaP‐sEV preincubation on SARS‐CoV‐2 infection of TMPRSS2‐expressing Calu‐3 cells, and TMPRSS2‐non‐expressing VERO‐E6 and A549 cells on intracellular viral RNA amounts assessed by RT‐qPCR 48 h post‐infection. Viral particles were incubated with 109 VCaP‐sEVS or PBS at a final MOI of 0.1. Results were normalized to 18S rRNA or GAPDH mRNA, then to control without VCaP‐sEVs incubation (black bars). Relative quantities are expressed as mean ± SEM of two independent experiments. (B) Effect of VCaP‐sEV preincubation on SARS‐CoV‐2 variant Alpha infection of Calu‐3 cells. (C) Time‐of‐addition experiment showing the effect of VCaP‐sEV incubation prior to infection, at the time of infection or 2 h post‐infection. **p < 0.01
2.6. sEVs favour SARS‐CoV‐2 entry into cells in the absence of fusion of their lipid envelopes
A time‐of‐addition experiment was performed to assess whether VCaP‐sEVs favour SARS‐CoV‐2 entry into Calu‐3 cells (Figure S5). Calu‐3 cells were infected with SARS‐CoV‐2 at an MOI of one in the absence of VCaP‐sEV exposure (control), 1 h after preincubation with VCaP‐sEVs, with VCaP‐sEVs added at the time of infection, and with VCaP‐sEVs added 2 h post‐infection (Figure S4). Total intracellular RNA extracted 24 h post‐infection was quantified by RT‐qPCR. As shown in Figure 3C, a significant increase in SARS‐CoV‐2 RNA quantity was detected when viral particles were preincubated with VCaP‐sEVs or when VCaP‐sEVs were added at the time of infection, but not when VCaP‐sEVs were added 2 h post‐infection. This result indicates that sEVs facilitate an early step of the SARS‐CoV‐2 life cycle, most likely viral entry.
To assess whether SARS‐CoV‐2 preincubation with VCaP‐sEVs leads to fusion of their viral and vesicular membranes, respectively, SARS‐CoV‐2 viral particles, VCaP‐sEVs and a mixture of both were immunoprecipitated overnight at 4°C by means of anti‐CD9 antibodies paired with magnetic beads (Figure S6A). Western blot analysis with anti‐SARS‐CoV‐2 nucleocapsid (N) protein and anti‐CD9 sEVs membrane marker showed the presence of CD9 in eluted fractions from both VCaP‐sEVs and VCaP‐sEVs/SARS‐CoV‐2, but not in those from SARS‐CoV‐2 alone fractions. Furthermore, SARS‐CoV‐2 N protein was detected in the supernatant of samples containing viral particles, but it was absent from eluted fractions from all samples (Figure S6B). These results indicate that preincubation of SARS‐CoV‐2 with VCaP‐sEVs does not lead to their fusion. Together, they suggest that VCap‐sEVs interact with SARS‐CoV‐2 particles without merging, and that this interaction facilitates viral entry into the TMPRSS2‐expressing cell line Calu‐3.
2.7. The integrity of surface sEV proteins is required for their proviral effect on SARS‐CoV‐2 infection
To assess whether surface protein integrity is required for VCaP‐sEVs to exert their proviral function, SARS‐CoV‐2 particles were incubated with intact or proteinase K (PK)‐treated VCaP‐sEVs. As shown by western blot analysis, PK treatment degraded vesicular ACE2, full‐length TMPRSS2, but not the ∼30 kDa activated TMPRSS2 fragment, probably because of its glycosylated status (Figure 4A). The luminal control ALIX protein remained detectable after PK treatment (Figure 4A). Similar results were obtained with PK‐treated mu‐sEVs generated by HNECs isolated from two distinct patients, using packaged sEV marker TSG101 (Figure 4B). Importantly, PK treatment of VCaP‐sEVs abolished their proviral effect in Calu‐3 cells after preincubation with SARS‐CoV‐2 (Figure 4C). Together, these results indicate that the integrity of surface‐accessible sEV proteins, including ACE2, is mandatory for the vesicles to exert their proviral effect in permissive cells.
FIGURE 4.
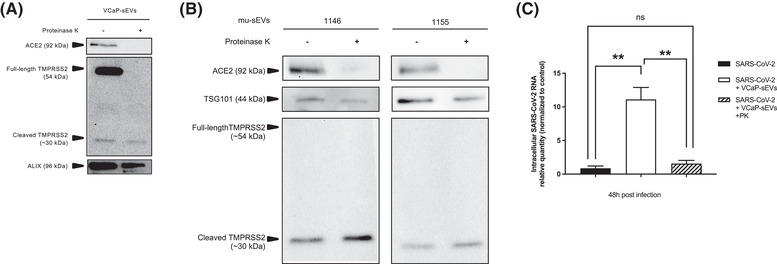
Role of sEV surface proteins in their proviral effect on SARS‐CoV‐2 infection. (A) Western blot analysis of ACE2, full‐length and cleaved TMPRSS2 and ALIX protein content in VCaP‐sEVs incubated in the absence or in the presence of proteinase K. (B) Western blot analysis of ACE2, full‐length and cleaved TMPRSS2 and TSG101 protein content in mu‐sEVs isolated from two patients, incubated in the absence or in the presence of proteinase K. (C) Effect of proteinase K (PK) treatment of VCaP‐sEVs on their proviral effect on SARS‐CoV‐2 infection of Calu‐3 cells. Intracellular viral RNA was quantified by RT‐qPCR 48 h post‐infection. Results were normalized to 18S rRNA, then to infection without VCaP‐sEV preincubation (black bar). Relative quantities are expressed as mean ± SEM of two independent experiments. **p < 0.01
2.8. Vesicular TMPRSS2 activates SARS‐CoV‐2 prefusion Spike protein but cellular TMPRSS2 remains required for cell entry
The capacity of VCaP‐sEVs to promote SARS‐CoV‐2 infection despite inhibition of cellular TMPRSS2 activity by camostat mesylate was explored. SARS‐CoV‐2 viral particles were incubated in the absence or presence of 109 VCaP‐sEVs prior to infection of Calu‐3 cells at an MOI of 0.1. Camostat mesylate (10 μM) was added or not at the time of infection and, when added, maintained until the end of the experiment. Intracellular SARS‐CoV‐2 RNA was quantified by RT‐qPCR 48 h post‐infection. As shown in Figure 5A, SARS‐CoV‐2 infection was significantly inhibited by camostat mesylate, regardless of prior preincubation with VCaP‐sEVs (∼193 fold‐decrease without preincubation, ∼333 fold‐decrease with VCaP‐sEV preincubation). This result suggests that cellular TMPRSS2 activity, which is required for the cleavage of cellular ACE2, remains necessary for viral entry in our experimental conditions.
FIGURE 5.
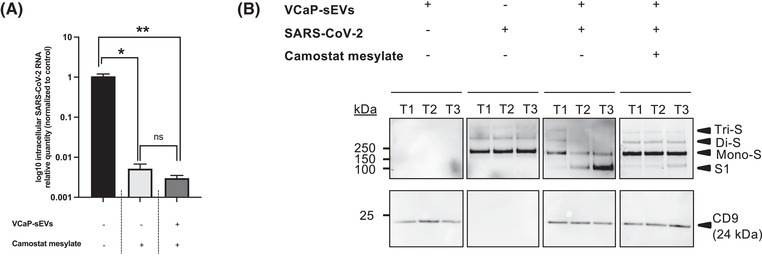
Role of TMPRSS2 in sEV proviral effect on SARS‐CoV‐2 infection of Calu‐3 cells. (A) Effect of the TMPRSS2 inhibitor camostat mesylate (10 μM) on Calu‐3 cell infection by SARS‐CoV‐2 viral particles preincubated or not with 109 VCaP‐sEVs. Intracellular SARS‐CoV‐2 RNA was extracted 48 h post‐infection and quantified by RT‐qPCR. Results were normalized to 18S rRNA, then to control (black bar). They are expressed as mean ± SEM of two independent experiments. *p < 0.05, **p < 0.01. (B) Time‐course analysis of the effect of VCaP‐sEVs on prefusion Spike protein priming and its inhibition by camostat mesylate. Western blot analyses of Spike (anti‐S1 antibodies) and CD9 proteins were performed on SARS‐CoV‐2 viral particles (10 μl of viral inoculum, ∼2.04 × 105 TCID50/ml), on VCaP‐sEVs (1010), and on mixtures of both, in the absence or presence of camostat mesylate (100 μM) at three time points: T1 (5 min à 37°C), T2 (1 h at 37°C) and T3 (overnight at 4°C). Tri‐S, Di‐S and Mono‐S: uncleaved prefusion Spike trimer, dimer, and monomer, respectively. S1: cleaved prefusion Spike fragment at the S1/S2 boundary
To better understand the role of vesicular TMPRSS2 activity during SARS‐CoV‐2 infection, western blot analysis of the viral Spike protein was performed under non‐reducing conditions. The dynamics of Spike processing were analysed at three time points, including T1 (5 min at 37°C), T2 (1 h at 37°C) and T3 (overnight at 4°C) post‐incubation with VCaP‐sEVs, in the absence or presence of camostat mesylate (100 μM) (Figure 5B). In the absence of VCaP‐sEV preincubation, uncleaved Spike prefusion trimers (>> 250 kDa, Tri‐S), dimers (> 250 kDa, Di‐S) and monomers (∼ 170 kDa, mono‐S) were detected at all time points. The absence of Spike cleavage at the S1/S2 boundary resulted from the fact that viral amplification had been performed in VERO‐E6 cells, generating acquisition of the R685H mutation that prevents intracellular furin cleavage. After preincubation with VCaP‐sEVs, SARS‐CoV‐2 Spike prefusion proteins were efficiently cleaved at the S1/S2 boundary, generating a fragment of ∼100 kDa, detectable with an anti‐S1 antibody at T2 and T3. This cleavage was inhibited by camostat mesylate, which restored the presence of uncleaved (Tri‐S, Di‐S and Mono‐S) Spike prefusion proteins at T2 and T3. This result indicates that the cleavage was TMPRSS2‐dependent. Thus, during preincubation of viral particles with VCaP‐sEVs, vesicular TMPRSS2 activates SARS‐CoV‐2 mutated (R685H) prefusion Spike by promoting its cleavage at the S1/S2 boundary. Nevertheless, ACE2 cleavage by cellular TMPRSS2 remains mandatory for viral entry.
2.9. mu‐sEVs activate both SARS‐CoV‐2 and SARS‐CoV‐2 variant Alpha Spike proteins
In order to confirm the results obtained with the VCaP‐sEV model on Spike protein processing, mu‐sEVs isolated from HNECs from two distinct patients (1010 and 5 × 1010 mu‐sEVs, respectively) were incubated with SARS‐CoV‐2 and SARS‐CoV‐2 variant Alpha viral particles. Variant Alpha was used because, unlike our initial SARS‐CoV‐2 virus, it was propagated in Calu‐3 cells and therefore contains the PRRAR furin‐like cleavage site. The dynamics of Spike processing were analysed by means of western blot under non‐reducing conditions at T1 (5 min at 37°C) and T2 (overnight at 4°C) (Figure 6). In the absence of mu‐sEVs, uncleaved SARS‐CoV‐2 Spike prefusion oligomers (Tri‐S, Di‐S and Mono‐S) were present at all time points, whereas in the case of SARS‐CoV‐2 variant Alpha, the uncleaved Spike prefusion monomer and the product of Spike prefusion cleavage at the S1/S2 boundary were predominant. Incubation of the original SARS‐CoV‐2 strain with mu‐sEVs induced Spike prefusion oligomer cleavage at the S1/S2 boundary, generating cleavage product intermediates and a fragment of ∼100 kDa detected with an anti‐S1 antibody (Figure 6A). Incubation of SARS‐CoV‐2 variant Alpha with mu‐sEVs induced cleavage of the uncleaved Spike prefusion monomer at the S1/S2 boundary (Figure 6B). For both SARS‐CoV‐2 viruses, mu‐sEV‐induced activation of the Spike prefusion proteins was observable at T1.
FIGURE 6.
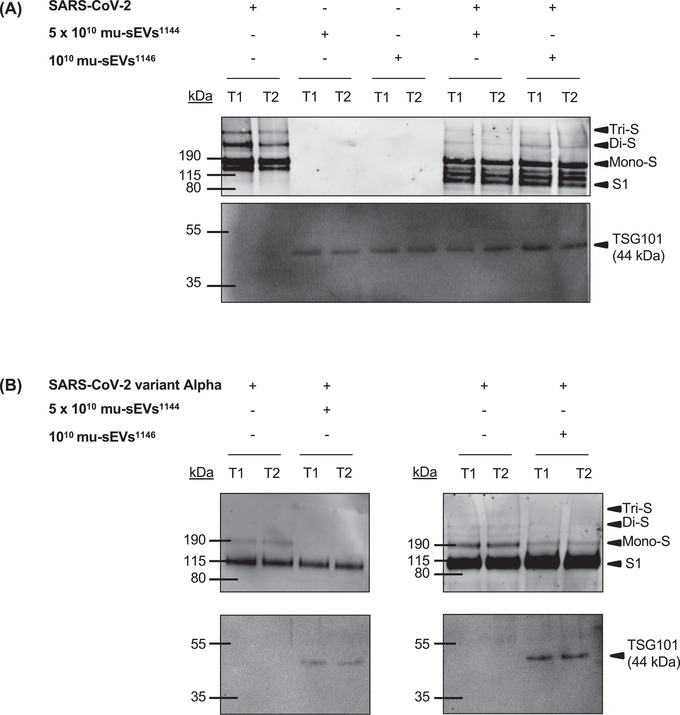
mu‐sEV priming of SARS‐CoV‐2 and SARS‐CoV‐2 variant Alpha prefusion Spike protein. Time‐course analysis of the effect of mu‐sEVs isolated from two patients (#1144 and #1146) on prefusion Spike protein priming from: (A) SARS‐CoV‐2, and (B) SARS‐CoV‐2 variant Alpha. Western blot analyses of Spike (anti‐S1 antibodies) and TSG101 were performed on SARS‐CoV‐2 viral particles (10 μl of viral inoculum, ∼2.04 × 105 TCID50/ml) or SARS‐CoV‐2 variant Alpha viral particles (10 μl of viral inoculum, ∼1 × 104 TCID50/ml), on mu‐sEVs from the two patients (1010 and 5 × 1010 mu‐sEVs, respectively), and on mixtures of both at two time points: T1 (5 min at 37°C) and T2 (overnight at 4°C). Tri‐S, Di‐S and Mono‐S: uncleaved prefusion Spike trimer, dimer, and monomer, respectively. S1: cleaved prefusion Spike fragment at the S1/S2 boundary
Together, these results demonstrate that, after short‐term incubation, mu‐sEVs activate Spike prefusion proteins from two different SARS‐CoV‐2 variants, including furin‐preactivated ones, by promoting cleavage at the S1/S2 boundary.
3. DISCUSSION
The first step of SARS‐CoV‐2 infection takes place in the upper airways, precisely in the nasal cavity, after airborne transmission by respiratory droplets. Airway mucus, which is principally produced by goblet cells, is theoretically the first barrier against infection. Under normal conditions, mucins and muco‐ciliary clearance both participate in protecting the respiratory system from harmful external agents, including pathogens.
In the present study, we evaluated the effect of mucus secreted by HNECs on their infection by SARS‐CoV‐2. In an ex vivo assay, we showed that removal of the airway mucus reduces SARS‐CoV‐2 infection, while mu‐sEVs isolated from different mucus exert a significant proviral effect. This effect is mu‐sEV concentration‐dependent, with a maximal effect obtained at mu‐sEV concentrations in the range of physiological HNEC production. Interestingly, a significant but weaker proviral effect was observed when SARS‐CoV‐2 particles were preincubated with physiological amounts of mu‐lEVs (Figure S7). Together, these results suggest that the mucus produced by HNECs contains EVs that favour SARS‐CoV‐2 infection at physiological concentrations. This proviral effect seems to be specific for sEVs contained in upper airway epithelial mucus; indeed, a recent study indicated that extracellular vesicles of salivary origin have no effect on SARS‐CoV‐2 infection (Conzelmann et al., 2020). The proviral effect of mu‐sEVs, that contain both ACE2 and TMPRSS2, does not appear to be related to the transfer of the ACE2 receptor to HNEC cell surface (Figure S8), but instead to the cleavage of viral Spike prefusion proteins mediated by vesicular TMPRSS2, as shown in our experiments using concentrations of mu‐sEVs comparable to those measured in HNEC‐generated mucus.
To understand the mechanisms explaining that mucus sEVs facilitate SARS‐CoV‐2 infection of respiratory cells, we used a 3‐component in vitro model highly relevant to the pathophysiological situation reflected by our experiments with HNECs. The model used: (i) two SARS‐CoV‐2 variant strains, including a variant derived from the original Wuhan strain and a SARS‐CoV‐2 variant Alpha strain, both originating from patients diagnosed in the laboratory of virology of our institution; (ii) sEVs produced by the TMPRSS2‐expressing VCaP cell line, the size of which is comparable to that of mu‐sEVs, that contain both ACE2 and full‐length and activated TMPRSS2 in their membranes (Deng et al., 2021) and exert a proviral effect on HNEC infection by SARS‐CoV‐2; and (iii) the TMPRSS2‐expressing, SARS‐CoV‐2 permissive lung carcinoma‐derived Calu‐3 cell line. This cell line is particularly relevant, because SARS‐CoV‐2 uses the ‘early’ TMPRSS2‐dependent entry pathway to infect it (Koch et al., 2021) and sEVs had the same proviral effect as mu‐sEVs in HNECs.
Other models were considered as less relevant because, in contrast with our observations with HNECs, mu‐sEVs and infectious viruses, sEV preincubation reduced infection and subsequent replication instead of increasing them. This was the case in our experiments when using A549 and VERO‐E6 cells, which express low levels of TMPRSS2 and therefore use the ‘late’ endosomal pathway for SARS‐CoV‐2 entry. Similarly, sEVs isolated from the kidney‐derived 293FT cell line overexpressing ACE2 and TMPRSS2 (293FT‐ACE2‐TMPRSS2) reduced infection of the colon‐derived Caco‐2 cell line with a lentivirus expressing the SARS‐CoV‐2 Spike protein (SARS‐CoV‐2‐S‐pseudotyped lentivirus) (Cocozza et al., 2020). One should therefore remain careful when suggesting the use of sEVs as therapeutic tools against SARS‐CoV‐2 infection, because our results strongly suggest the opposite (proviral) effect in real infected patients.
Our study indicates that the effect of sEVs on SARS‐CoV‐2 infection is to favour the TMPRSS2‐dependent entry pathway, at the expense of the ‘late’ endosomal entry route. Our time‐of‐addition experiments confirmed that sEVs favour entry into target cells. Interestingly, PK treatment of sEVs indicated that the integrity of surface‐accessible vesicular proteins (including ACE2) is mandatory for their proviral effect. PK treatment may also alter the effect of vesicular tetraspanins, that have been suggested to operate in cell membranes by bringing ACE2 and TMPRSS2 closer (Earnest et al., 2017; Earnest et al., 2015; Hantak et al., 2019). Our results suggest that SARS‐CoV‐2 Spike protein and vesicular ACE2 interact when they are incubated together. However, we did not observe any fusion of viral and sEV membranes in vitro, suggesting that the interaction between vesicular ACE2 and viral particles is transient and that, once released, viruses display activated prefusion Spike proteins.
We demonstrated that both VCaP‐sEV and mu‐sEV vesicular TMPRSS2 cleaves SARS‐CoV‐2 prefusion Spike at the S1/S2 boundary, with better efficiency of mu‐sEV vesicular TMPRSS2, detected as a cleaved (activated) fragment only. We also showed that, although incubation with vesicular TMPRSS2 induces SARS‐CoV‐2 Spike priming, cellular TMPRSS2 remains necessary for viral entry into Calu‐3 cells, probably to perform cleavage at the S2’ site which ultimately triggers fusion between viral and cellular membranes. Because the original SARS‐CoV‐2 strain used in our experiments had been propagated in VERO‐E6 cells, an R685H mutation that prevents furin cleavage at the S1/S2 boundary was present (Lamers et al., 2021), explaining the lack of S1 cleavage fragment when SARS‐CoV‐2 virions were incubated without sEVs. Using a SARS‐CoV‐2 Alpha variant strain propagated in Calu‐3 cells that contains the furin‐like cleavage site, we confirmed the facilitating effect of sEVs on infection and the ability of mu‐sEVs contained in airways mucus to induce Spike prefusion protein cleavage at the S1/S2 boundary.
In conclusion, our study shows that uninfected HNECs produce mucus that shelters sEVs containing both ACE2 and activated TMPRSS2. After binding SARS‐CoV‐2 viral particles, mu‐sEVs have the capacity to cleave wild‐type or R685H‐mutated prefusion Spike proteins at the S1/S2 boundary, resulting in higher proportions of prefusion S proteins exposing their RBD in the ‘open’ conformation, therefore capable to bind the receptor at the cell surface. Our observations confirm that S proteins containing the PRRAR polybasic cleavage site are not entirely primed after their release from producer cells (Koch et al., 2021; Wrobel et al., 2020). Thus, the role of mu‐sEVs produced by the nasal epithelium is to complete prefusion Spike priming performed by intracellular furin during viral egress from infected cells. This is mediated by vesicular TMPRSS2 activity, with the goal to render SARS‐CoV‐2 virions prone to entry into target cells using the ‘early’, TMPRSS2‐dependent pathway instead of the ‘late’, cathepsin‐dependent route. These results strongly suggest that prefusion Spike priming by mu‐sEVs produced in the nasal cavity plays a role in viral tropism. They also show that nasal mucus does not protect from SARS‐CoV‐2 infection, but instead facilitates it. It is now necessary to investigate the role of mu‐sEVs produced by HNECs on the priming of fusion proteins from other coronaviruses and respiratory enveloped viruses.
4. MATERIALS AND METHODS
4.1. Cell culture
VERO‐E6 cells (African green monkey kidney cells, ATCC CRL‐1856) and A549 cells (human tumorigenic lung epithelial cells, ATCC CCL‐185) were maintained in Dulbecco's modified Eagle medium (DMEM, ThermoFischer, Waltham, Massachusetts, USA) supplemented with 10% foetal bovine serum (FBS), 50 IU/ml penicillin, 100 μg/ml streptomycin, and 0.1 μg/ml amphotericin B (ThermoFischer Scientific) at 37°C with 5% CO2. Calu‐3 (ATCC, HTB‐55) and VCaP cells (vertebral‐cancer of the prostate cells, ATTC, Manassas, Virginia, USA) were cultured in the same medium supplemented with 1% non‐essential amino acids (NEAA, ThermoFischer Scientific) or 2 nM DHT (R&D Systems, Minneapolis, Minnesota, USA), respectively.
4.2. Preparation of human nasal epithelial cells
Primary HNECs were obtained from patients with chronic rhinosinusitis with nasal polyps undergoing ethmoidectomy, as previously described (Coste et al., 2000). All patients gave informed consent and the study was approved by the local ethics committee (Comité de Protection des Personnes IDF X 2016‐01‐01). All patients benefited from an earlier 24 h negative RT‐qPCR for SARS‐CoV‐2 RNA in nasopharyngeal swabs.
After surgical resection in the operating room, nasal polyps were immediately placed in DMEM/F‐12 supplemented with antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin, 2.5 g/ml amphotericin B, and 100 mg/ml gentamicin). For cellular dissociation, nasal polyps were washed with PBS (Phosphate Buffered Saline, Life Technologies, Carlsbad, California, USA) containing 5 nM dithiothreitol (Sigma Aldrich, Saint‐Louis, Missouri, USA) to eliminate mucus and blood. Enzymatic digestion was performed for 16 h at 4°C (0.1% [wt/vol] pronase [Sigma Aldrich] in culture medium). HNECs (106 cells) were then plated in inserts (12‐mm Costar Transwell, Sigma Aldrich) with 12‐mm‐diameter polycarbonate micropore membranes (pore size: 0.4 μm) coated with type IV collagen (Sigma Aldrich) and incubated at 37°C in 5% CO2. For the first 24 h, the cells were incubated with 1 ml of DMEM/F‐12‐antibiotics with 2% Ultroser G in the lower chamber and DMEM/F‐12‐antibiotics with 10% FCS in the insert. After 24 h, the medium at the surface was aspirated and cells were washed with PBS to eliminate non‐adherent cells. The culture medium in the insert (Pneumacult‐ALI, StemCell Technologies, Vancouver, Canada) was removed to place the cells at the air‐liquid interface (ALI). Cultures were maintained at 37°C with 5% CO2. The medium in the lower chamber was then changed every day. To assess cell viability, cells were observed daily with an optic microscope and trans‐epithelial electrical resistance (TEER) was measured (Epithelial Volt/Ohm (TEER) Meter 3, World Precision Instruments, Sarasota, Florida, USA). As already reported, HNECs reach a stable differentiated state with the detection of ciliated, goblet, and basal cells at day 21 of culture in the ALI medium (Papon et al., 2002).
4.3. Viruses
The SARS‐CoV‐2 strains used in this study were isolated from nasopharyngeal swabs from SARS‐CoV‐2‐infected patients seen at Henri Mondor University Hospital, Créteil, France. SARS‐CoV‐2 viral stock was propagated in VERO‐E6 cells, while SARS‐CoV‐2 variant Alpha viral stock was propagated in Calu‐3 cells, using modified culture media with 2% FBS. The viral stocks were aliquoted and stored at ‐80°C. Viral titers were measured by means of RT‐qPCR in VERO‐E6 cells and expressed as TCID50 per milliliter. VERO‐E6 cells were seeded in sextuplicate in 48‐well plates and incubated for 2 h with 10‐fold serial dilutions of viral stocks. The infectious inoculum was then replaced by the culture medium and cells were incubated for 48 h before intracellular viral RNA assessment by RT‐qPCR.
4.4. sEV preparation
Preparation of VCaP‐sEVs was adapted from a previous report (Ludwig et al., 2018). Briefly, ∼1.5 L of conditioned VcaP medium was collected and centrifuged twice at 2000 x g for 20 min. The supernatants were then supplemented with 10% PEG6000 and 75 mM NaCl. After overnight incubation at 4°C, sEVs were centrifuged at 2000 x g for 20 min at 4°C. The pellets were dissolved in NaCl 0.9% overnight and centrifuged at 16,500 x g for 20 min at 4°C. The supernatants were then collected and centrifuged at 100,000 x g for 90 min at 4°C. The pellets containing VCaP‐sEVs were rinsed one time with NaCl 0,9%, centrifuged at 100,000 x g for 90 min at 4°C and then resuspended in NaCl 0.9%.
m u‐sEVs were prepared by differential centrifugation using mucus produced by HNECs isolated from distinct patients. Briefly, mucus was recovered by washing the apical pole of HNECs cultured at the ALI every 48 h with 200 μl PBS per insert. After incubation for 5 min at room temperature, mucus‐containing PBS was collected and placed at 4°C for short‐term storage. The recovered biological samples (∼5 ml) were centrifuged at 2000 x g for 20 min at 4°C. 2K supernatants were then centrifuged at 16,500 x g for 20 min at 4°C. The 16,5K pellets obtained corresponded to mu‐lEVs. The 16,5K remaining supernatants were again ultracentrifuged at 100,000 x g for 90 min at 4°C. 100K supernatants, that correspond to EV‐deprived mucus, were collected. 100K pellets containing mu‐sEVs were rinsed with NaCl 0,9%, centrifuged at 100,000 x g for 90 min at 4°C and resuspended in NaCl 0.9%. All samples were stored at ‐20°C for short‐term or ‐80°C for long‐term conservation.
All relevant data from our experiments have been submitted to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV220307) (Van Deun et al., 2017).
4.5. Tunable resistive pulse sensing (TRPS)
The concentrations and sizes of EVs were measured by means of the TRPS technology, using the Gold qNano instrument (Izon Science, Medford, Massachusetts) and the Izon Control Suite software version V3.4.2.44 for analysis.
VCaP‐sEV‐containing samples were analysed using NP150 nanopores, stretched to 47.5 mm under 0.70 mV voltage and 7–10 mbar pressure. mu‐sEV‐containing samples were analysed using NP150 nanopores, stretched to 51 mm under 0.48 mV voltage and 5 mbar pressure. mu‐lEV‐containing samples were analysed using NP800 nanopores, stretched to 48 mm under 0.18 mV voltage and 7–10 mbar pressure. Calibrations were performed using CPC 200 or CPC 1000 carboxylated polystyrene beads. Each EV sample was processed with a minimum of 500 particle count in triplicate. Data were collected and analysed according to the manufacturer's instructions. The amount of vesicles per ALI insert was then estimated.
4.6. Morphological characterization of sEVs by transmission electron microscopy (TEM)
sEVs isolated from mucus secreted by HNECs from two donor patients were diluted 1:50 in PBS for analysis. Samples (3 μl) were deposited on copper grids, dried at room temperature and stained with uranyl acetate (1%). Morphology was investigated by transmission electron microscopy using a Tecnai F20 microscope (FEI Company, Hillsboro, Oregon, USA).
4.7. Proteinase K treatment
An amount of 109 mu‐sEVs or VCaP‐sEVs was incubated in the absence or in the presence of proteinase K (20 μg/ml) and 5 mM CaCl2 for 30 min at 37°C under gentle agitation. Proteinase K activity was inhibited by adding 5 mM phenylmethysulfonyl fluoride (PMSF) for 10 min at room temperature.
4.8. SARS‐CoV‐2 infection
SARS‐CoV‐2 infection of HNECs isolated from uninfected patients was performed at their apical pole with 20 μl of viral inoculum (∼2.04 × 104 TCID50/ml) for 4 h, in the presence or in the absence of mucus at 37°C with 5% CO2. To remove the mucus from the apical pole of HNECs, the wells were rinsed with 200 μl PBS two times 48 h prior to infection and two times at the time of infection. For Calu‐3, VERO‐E6 and A549 cell lines, 150,000, 75,000 and 75,000 cells, respectively, were seeded in 48‐well plates 24 h prior to SARS‐CoV‐2 infection. Cells were infected at a final MOI of 0.1 for 2 h in DMEM 2% FBS at 37°C with 5% CO2, with or without the addition of 10 μM of the protease inhibitor camostat mesylate (Merck, Darmstadt, Germany). When indicated, viral particles were incubated under gentle agitation overnight at 4°C in the absence or in the presence of 109 VCaP‐sEVs or mu‐sEVs. After infection, HNECs were rinsed at the apical pole with 200 μl PBS, while immortalized cell lines were washed and supplemented with fresh medium with or without the protease inhibitor. Supernatant and intracellular RNA were extracted at different time points post‐infection, as indicated.
4.9. Time‐of‐addition experiment
One hundred and fifty thousand Calu‐3 cells were seeded in 48‐well plates 24 h prior to SARS‐CoV‐2 infection at an MOI of 1. Culture medium or 109 VCaP‐sEVs were incubated with viral particles 1 h prior to infection, at the time of infection, or 2 h post‐infection. Cells were then washed and incubated with 109 VCaP‐sEVs for 24 h before viral RNA quantification was performed in cellular extracts.
4.10. Extraction and PCR amplification of supernatant and cellular viral RNAs
Extraction of SARS‐CoV‐2 RNA from the supernatant was performed by means of QIAmp Viral RNA Mini Kit (Qiagen, Hilden, Germany). Intracellular viral RNA was extracted using the RNeasy Mini Kit (Qiagen). Reverse transcription was performed using the High Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific). SARS‐CoV‐2 RNA was quantified with the TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, California, USA) using specific primers (forward primer 5′‐ACAGGTACGTTAATAGTTAATAGCGT‐3′ and reverse primer 3′‐ATATTGCAGCAGTACGCACACA‐5′) and qPCR probe (5′‐ACACTAGCCATCCTTACTGCGCTTCG[5′]Fam[3′]BHQ‐1‐3′). Quantitative PCR was performed with QuantStudio 5 Real‐Time PCR system (ThermoFisher Scientific). SARS‐CoV‐2 RNA levels were quantified according to the ΔΔCT method and normalized to GAPDH mRNA or 18S rRNA for intracellular samples. SARS‐CoV‐2 RNA relative quantities were plotted against infection without extracellular vesicles.
4.11. Western blot analysis
sEVs isolated from VCaP or from HNEC‐produced mucus (∼1010 sEVs) were incubated under gentle agitation in the absence or in the presence of 10 times‐concentrated SARS‐CoV‐2 or SARS‐CoV‐2 variant Alpha particles (∼2.04 × 105 TCID50/ml and ∼104 TCID50/ml, respectively) using Amicon Ultra‐4, PLTK, membrane Ultracel‐PL, 30 kD (Merck). Three incubation conditions were used: T1 (5 min at 37°C), T2 (1 h at 37°C) and T3 (overnight at 4°C). When indicated, incubations were performed in the presence of 100 μM camostat mesylate (Merck).
Samples were prepared in EZ buffer (Tris‐HCl 20 mM, NaCl 100 mM, EDTA 1 mM, NP40 0.5%, Glycerol 10%) before denaturation at 95°C and migration on NuPage 4–12%, Bis‐Tris gel (ThermoFisher Scientific). After transfer on a 0.45 μm nitrocellulose membrane (ThermoFisher Scientific), blocking was done in 5% bovine serum albumine (BSA, Euromedex, Souffelweyersheim, France) or 5% nonfat dry milk for 1 h. Membranes were then incubated overnight at 4°C with primary antibodies against the following proteins: SARS‐CoV‐2 nucleocapsid (#MA5‐29981, ThermoFisher Scientific), SARS‐CoV‐2 Spike S1 subunit (#PA5‐81795, ThermoFisher Scientific), TMPRSS2 (#109131, Abcam, Cambridge, UK), ACE2 (#15348, Abcam), TSG101 (#ab83, Abcam), CD9 (#92726, Abcam), ALIX (#117600, Abcam), CD81 (#79559, Abcam), CD63 (#59479, Abcam), Calnexin (#133615, Abcam), Cytochrome C (#133504, Abcam). Membranes were washed in TBS‐tween before incubation for 1 h with the corresponding HRP‐conjugated secondary antibody. Immune complexes were detected by chemiluminescence with ECL select western blotting detection reagent (GE Healthcare, Amersham, Buckinghamshire, UK) using ImageQuant LAS 4000 software.
4.12. VCaP‐sEV immunoprecipitation
VCaP‐sEVs (1010) were incubated overnight at 4°C with 100 μl of exosome‐human CD9 isolation beads (#10620D, Invitrogen), in the absence or in the presence of SARS‐CoV‐2 viral particles (∼2.04 × 104 TCID50/ml). All fractions were isolated using a magnetic separator and analysed by western blot using antibodies directed against CD9 or SARS‐CoV‐2 nucleocapsid.
4.13. SARS‐CoV‐2 Spike sequence analysis
SARS‐CoV‐2 RNA was extracted and reverse transcribed as described above. Amplification of the target sequence was performed using the PCR advantage kit (ThermoFisher Scientific) and the following primers: 1850‐Forward 5′‐GCACAGAAGTCCCTGTTGCT‐3′ and 2833‐Reverse 3′‐GTGCACTTGCTGTGGAAGAA‐5′. The BigDye Terminator kit (ThermoFisher Scientific) was used to prepare samples for Sanger sequencing using the following primers: 2293‐Reverse 3′‐GGTTTAATTGTGTACAAAAACTGCC‐5′ and 2691‐Reverse 3′‐TGGTATTTGTAATGCAGCACC‐5′. Sequences were analysed with Chromas Lite software.
4.14. Statistical analysis
The results are expressed as mean ± SEM of two or three replicate experiments, as indicated. Comparisons were performed by means of the Mann‐Whitney U‐test using Prism software.
AUTHOR CONTRIBUTIONS
François Berry: Conceptualization; Formal analysis; Investigation; Writing – original draft. Margot Morin‐Dewaele: Conceptualization; Formal analysis; Investigation; Writing – original draft. Amene Majidipur: Formal analysis; Investigation. Thibaud Jamet: Investigation; Resources. Sophie Bartier: Resources; Writing – review & editing. Eva Ignjatovic: Formal analysis; Investigation. Donatella Toniutti: Formal analysis; Investigation. Jeanne Gaspar Lopes: Formal analysis; Investigation. Pascale Soyeux‐Porte: Investigation; Resources. Pascale Maillé: Formal analysis; Investigation; Methodology. Carolina Saldana: Resources. Rozenn Brillet: Investigation; Resources. Nazim Ahnou: Investigation; Resources. Laurent Softic: Conceptualization; Investigation; Writing – review & editing. Benoit Couturaud: Formal analysis; Investigation; Methodology; Visualization. Hakim Ahmed‐Belkacem: Conceptualization; Formal analysis; Validation; Writing – review & editing. Slim Fourati: Formal analysis; Resources. Bruno Louis: Resources; Writing – review & editing. André Coste: Resources; Writing – review & editing. Alexandre De La Taille: Conceptualization; Resources. Damien Destouches: Conceptualization; Supervision; Validation; Writing – review & editing. Francis Vacherot: Conceptualization; Formal analysis; Funding acquisition; Resources; Supervision; Validation; Visualization; Writing – review & editing. Jean‐Michel Pawlotsky: Conceptualization; Supervision; Validation; Visualization; Writing – review & editing. Virginie Firlej: Conceptualization; Formal analysis; Funding acquisition; Resources; Supervision; Validation; Visualization; Writing – review & editing. Patrice Bruscella: Conceptualization; Formal analysis; Funding acquisition; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing.
CONFLICT OF INTEREST
The authors report no conflict of interest related to this study.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
This study was supported by an ‘UFR Santé‐UPEC’ cross‐teams COVID‐19 grant. The authors are grateful to the ‘Fondation pour la Recherche Médicale’ (FRM) for their financial support. We would also like to thank the technicians from the Department of Virology of Henri Mondor Hospital for their assistance with SARS‐CoV‐2 virus sampling and amplification.
Berry, F. , Morin‐Dewaele, M. , Majidipur, A. , Jamet, T. , Bartier, S. , Ignjatovic, E. , Toniutti, D. , Gaspar Lopes, J. , Soyeux‐Porte, P. , Maillé, P. , Saldana, C. , Brillet, R. , Ahnou, N. , Softic, L. , Couturaud, B. , Huet, É. , Ahmed‐Belkacem, A. , Fourati, S. , Louis, B. , … Bruscella, P. (2022). Proviral role of human respiratory epithelial cell‐derived small extracellular vesicles in SARS‐CoV‐2 infection. Journal of Extracellular Vesicles, 11, e12269. 10.1002/jev2.12269
François Berry and Margot Morin‐Dewaele participated equally.
Francis Vacherot, Jean‐Michel Pawlotsky, Virginie Firlej, and Patrice Bruscella participated equally.
REFERENCES
- Ahn, J. H. , Kim, J. , Hong, S. P. , Choi, S. Y. , Yang, M. J. , Ju, Y. S. , Kim, Y. T. , Kim, H. M. , Rahman, M. T. , Chung, M. K. , Hong, S. D. , Bae, H. , Lee, C. ‐S. , & Koh, G. Y. (2021). Nasal ciliated cells are primary targets for SARS‐CoV‐2 replication in the early stage of COVID‐19. Journal of Clinical Investigation, 131, e148517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akers, J. C. , Gonda, D. , Kim, R. , Carter, B. S. , & Chen, C. C. (2013). Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus‐like vesicles, and apoptotic bodies. Journal of Neuro‐Oncology, 113, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard, S. , Millet, J. K. , Licitra, B. N. , & Whittaker, G. R. (2012). Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses, 4, 1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton, D. J. , Wrobel, A. G. , Xu, P. , Roustan, C. , Martin, S. R. , Rosenthal, P. B. , Skehel, J. J. , & Gamblin, S. J. (2020). Receptor binding and priming of the spike protein of SARS‐CoV‐2 for membrane fusion. Nature, 588, 327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bequignon, E. , Dhommee, C. , Angely, C. , Thomas, L. , Bottier, M. , Escudier, E. , Isabey, D. , Coste, A. , Louis, B. , Papon, J. , & Gouilleux‐Gruart, V. (2019). FcRn‐dependent transcytosis of monoclonal antibody in human nasal epithelial cells in vitro: A prerequisite for a new delivery route for therapy? International Journal of Molecular Sciences, 20, 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestle, D. , Heindl, M. R. , Limburg, H. , Van Lam Van, T. , Pilgram, O. , Moulton, H. , Stein, D. A. , Hardes, K. , Eickmann, M. , Dolnik, O. , Rohde, C. , Klenk, H. ‐. D. , Garten, W. , Steinmetzer, T. , & Böttcher‐Friebertshäuser, E. (2020). TMPRSS2 and furin are both essential for proteolytic activation of SARS‐CoV‐2 in human airway cells. Life Science Alliance, 3, e202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. , Zhang, J. , Xiao, T. , Peng, H. , Sterling, S. M. , Walsh, R. M. Jr. , Rawson, S. , Rits‐Volloch, S. , & Chen, B. (2020). Distinct conformational states of SARS‐CoV‐2 spike protein. Science, 369, 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocozza, F. , Nevo, N. , Piovesana, E. , Lahaye, X. , Buchrieser, J. , Schwartz, O. , Manel, N. , Tkach, M. , Thery, C. , & Martin‐Jaular, L. (2020). Extracellular vesicles containing ACE2 efficiently prevent infection by SARS‐CoV‐2 Spike protein‐containing virus. Journal of Extracellular Vesicles, 10, e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann, C. , Gross, R. , Zou, M. , Kruger, F. , Gorgens, A. , Gustafsson, M. O. , El Andaloussi, S. , Munch, J. , & Muller, J. A. (2020). Salivary extracellular vesicles inhibit Zika virus but not SARS‐CoV‐2 infection. Journal of Extracellular Vesicles, 9, 1808281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste, A. , Brugel, L. , Maitre, B. , Boussat, S. , Papon, J. F. , Wingerstmann, L. , Peynegre, R. , & Escudier, E. (2000). Inflammatory cells as well as epithelial cells in nasal polyps express vascular endothelial growth factor. European Respiratory Journal, 15, 367–372. [DOI] [PubMed] [Google Scholar]
- Coutard, B. , Valle, C. , De Lamballerie, X. , Canard, B. , Seidah, N. G. , & Decroly, E. (2020). The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Research, 176, 104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Q. , Rasool, R. U. , Russell, R. M. , Natesan, R. , & Asangani, I. A. (2021). Targeting androgen regulation of TMPRSS2 and ACE2 as a therapeutic strategy to combat COVID‐19. iScience, 24, 102254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnest, J. T. , Hantak, M. P. , Li, K. , McCray, P. B. Jr. , Perlman, S. , & Gallagher, T. (2017). The tetraspanin CD9 facilitates MERS‐coronavirus entry by scaffolding host cell receptors and proteases. Plos Pathogens, 13, e1006546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnest, J. T. , Hantak, M. P. , Park, J. ‐. E. , & Gallagher, T. (2015). Coronavirus and influenza virus proteolytic priming takes place in tetraspanin‐enriched membrane microdomains. Journal of Virology, 89, 6093–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes‐Prior, P. (2021). Priming of SARS‐CoV‐2 S protein by several membrane‐bound serine proteinases could explain enhanced viral infectivity and systemic COVID‐19 infection. Journal of Biological Chemistry, 296, 100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A. , Madhavan, M. V. , Sehgal, K. , Nair, N. , Mahajan, S. , Sehrawat, T. S. , Bikdeli, B. , Ahluwalia, N. , Ausiello, J. C. , Wan, E. Y. , Freedberg, D. E. , Kirtane, A. J. , Parikh, S. A. , Maurer, M. S. , Nordvig, A. S. , Accili, D. , Bathon, J. M. , Mohan, S. , Bauer, K. A. , … Landry, D. W. (2020). Extrapulmonary manifestations of COVID‐19. Nature Medicine, 26, 1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantak, M. P. , Qing, E. , Earnest, J. T. , & Gallagher, T. (2019). Tetraspanins: Architects of viral entry and exit platforms. Journal of Virology, 93, e01429–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkema, J. R. , Carey, S. A. , & Wagner, J. G. (2006). The nose revisited: A brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicologic Pathology, 34, 252–269. [DOI] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , & Pohlmann, S. (2020a). A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Molecular Cell, 78, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , Schroeder, S. , Kruger, N. , Herrler, T. , Erichsen, S. , Schiergens, T. S. , Herrler, G. , Wu, N. H. , & Nitsche, A. (2020b). SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181, 271–280 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, C. , Surda, P. , Whitehead, E. , & Kumar, B. N. (2020). Early recovery following new onset anosmia during the COVID‐19 pandemic ‐ An observational cohort study. Journal of Otolaryngology Head & Neck Surgery, 49, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, Z. , Oton, J. , Qu, K. , Cortese, M. , Zila, V. , Mckeane, L. , Nakane, T. , Zivanov, J. , Neufeldt, C. J. , Cerikan, B. , Lu, J. M. , Peukes, J. , Xiong, X. , Kräusslich, H. ‐. G. , Scheres, S. H. W. , Bartenschlager, R. , & Briggs, J. A. G. (2020). Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature, 588, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, J. , Uckeley, Z. M. , Doldan, P. , Stanifer, M. , Boulant, S. , & Lozach, P. Y. (2021). TMPRSS2 expression dictates the entry route used by SARS‐CoV‐2 to infect host cells. EMBO Journal, 40, e107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers, M. M. , Mykytyn, A. Z. , Breugem, T. I. , Wang, Y. , Wu, D. C. , Riesebosch, S. , Van Den Doel, P. B. , Schipper, D. , Bestebroer, T. , Wu, N. C. , & Haagmans, B. L. (2021). Human airway cells prevent SARS‐CoV‐2 multibasic cleavage site cell culture adaptation. Elife, 10, e66815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. (2016). Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology, 3, 237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Han, M. , Dai, P. , Xu, W. , He, J. , Tao, X. , Wu, Y. , Tong, X. , Xia, X. , Guo, W. , Zhou, Y. , Li, Y. , Zhu, Y. , Zhang, X. , Liu, Z. , Aji, R. , Cai, X. , Li, Y. , Qu, D. , … Gao, D. (2021). Distinct mechanisms for TMPRSS2 expression explain organ‐specific inhibition of SARS‐CoV‐2 infection by enzalutamide. Nature Communication, 12, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Liu, K. , Liu, Y. , Xu, Y. , Zhang, F. , Yang, H. , Liu, J. , Pan, T. , Chen, J. , Wu, M. , Zhou, X. , & Yuan, Z. (2013). Exosomes mediate the cell‐to‐cell transmission of IFN‐alpha‐induced antiviral activity. Nature Immunology, 14, 793–803. [DOI] [PubMed] [Google Scholar]
- Ludwig, A. K. , De Miroschedji, K. , Doeppner, T. R. , Borger, V. , Ruesing, J. , Rebmann, V. , Durst, S. , Jansen, S. , Bremer, M. , & Behrmann, E. (2018). Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. Journal of Extracellular Vesicles, 7, 1528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, M. , Kleinschmidt, A. , Bruhl, H. , Klier, C. , Nelson, P. J. , Cihak, J. , Plachy, J. , Stangassinger, M. , Erfle, V. , & Schlondorff, D. (2000). Transfer of the chemokine receptor CCR5 between cells by membrane‐derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nature Medicine, 6, 769–775. [DOI] [PubMed] [Google Scholar]
- Madu, I. G. , Roth, S. L. , Belouzard, S. , & Whittaker, G. R. (2009). Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. Journal of Virology, 83, 7411–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, M. , Martin‐Jaular, L. , Lavieu, G. , & Thery, C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nature Cell Biology, 21, 9–17. [DOI] [PubMed] [Google Scholar]
- Matsuyama, S. , Nao, N. , Shirato, K. , Kawase, M. , Saito, S. , Takayama, I. , Nagata, N. , Sekizuka, T. , Katoh, H. , Kato, F. , Sakata, M. , Tahara, M. , Kutsuna, S. , Ohmagari, N. , Kuroda, M. , Suzuki, T. , Kageyama, T. , & Takeda, M. (2020). Enhanced isolation of SARS‐CoV‐2 by TMPRSS2‐expressing cells. Proceedings National Academy of Science United States of America, 117, 7001–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama, S. , Ujike, M. , Morikawa, S. , Tashiro, M. , & Taguchi, F. (2005). Protease‐mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proceedings National Academy of Science United States of America, 102, 12543–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, O. P. , Bhandari, P. , Raut, A. , Kacimi, S. E. O. , & Huy, N. T. (2020). Coronavirus Disease (COVID‐19): Comprehensive review of clinical presentation. Frontiers in Public Health, 8, 582932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menni, C. , Valdes, A. M. , Freidin, M. B. , Sudre, C. H. , Nguyen, L. H. , Drew, D. A. , Ganesh, S. , Varsavsky, T. , Cardoso, M. J. , El‐Sayed Moustafa, J. S. , Visconti, A. , Hysi, P. , Bowyer, R. C. E. , Mangino, M. , Falchi, M. , Wolf, J. , Ourselin, S. , Chan, A. T. , Steves, C. J. , & Spector, T. D. (2020). Real‐time tracking of self‐reported symptoms to predict potential COVID‐19. Nature Medicine, 26, 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet, J. K. , & Whittaker, G. R. (2015). Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Research, 202, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, L. , Brighton, L. E. , Carson, J. L. , Fischer, W. A. 2nd , & Jaspers, I. (2013). Culturing of human nasal epithelial cells at the air liquid interface. Journal of Visualized Experiments: JoVE, 50646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa, G. , Mallery, D. L. , Albecka, A. , Welch, L. G. , Cattin‐Ortola, J. , Luptak, J. , Paul, D. , McMahon, H. T. , Goodfellow, I. G. , Carter, A. , Munro, S. , & James, L. C. (2021). Furin cleavage of SARS‐CoV‐2 Spike promotes but is not essential for infection and cell‐cell fusion. Plos Pathogens, 17, e1009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papon, J. F. , Coste, A. , Gendron, M. C. , Cordonnier, C. , Wingerstmann, L. , Peynegre, R. , & Escudier, E. (2002). HLA‐DR and ICAM‐1 expression and modulation in epithelial cells from nasal polyps. Laryngoscope, 112, 2067–2075. [DOI] [PubMed] [Google Scholar]
- Perlman, S. , & Netland, J. (2009). Coronaviruses post‐SARS: Update on replication and pathogenesis. Nature Reviews Microbiology, 7, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnaiah, V. , Thumann, C. , Fofana, I. , Habersetzer, F. , Pan, Q. , De Ruiter, P. E. , Willemsen, R. , Demmers, J. A. A. , Stalin Raj, V. , Jenster, G. , Kwekkeboom, J. , Tilanus, H. W. , Haagmans, B. L. , Baumert, T. F. , & Van Der Laan, L. J. W. (2013). Exosome‐mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proceedings National Academy of Science United States of America, 110, 13109–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo, G. , & Stoorvogel, W. (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. Journal of Cell Biology, 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozmyslowicz, T. , Majka, M. , Kijowski, J. , Murphy, S. L. , Conover, D. O. , Poncz, M. , Ratajczak, J. , Gaulton, G. N. , & Ratajczak, M. Z. (2003). Platelet‐ and megakaryocyte‐derived microparticles transfer CXCR4 receptor to CXCR4‐null cells and make them susceptible to infection by X4‐HIV. Aids, 17, 33–42. [DOI] [PubMed] [Google Scholar]
- Salem, I. , Naranjo, N. M. , Singh, A. , DeRita, R. , Krishn, S. R. , Sirman, L. S. , Quaglia, F. , Duffy, A. , Bowler, N. , & Sayeed, A. (2020). Methods for extracellular vesicle isolation from cancer cells. Cancer Drug Resistance, 3, 371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorey, J. S. , Cheng, Y. , Singh, P. P. , & Smith, V. L. (2015). Exosomes and other extracellular vesicles in host‐pathogen interactions. EMBO Reports, 16, 24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Wan, Y. , Luo, C. , Ye, G. , Geng, Q. , Auerbach, A. , & Li, F. (2020). Cell entry mechanisms of SARS‐CoV‐2. Proceedings National Academy of Science United States of America, 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synowiec, A. , Szczepanski, A. , Barreto‐Duran, E. , Lie, L. K. , & Pyrc, K. (2021). Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2): A systemic infection. Clinical Microbiology Reviews, 34, e00133–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, T. , Bidon, M. , Jaimes, J. A. , Whittaker, G. R. , & Daniel, S. (2020). Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Research, 178, 104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery, C. , Amigorena, S. , Raposo, G. , & Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current Protocols in Cell Biology, 30: 3.22.1‐3.22.29. [DOI] [PubMed] [Google Scholar]
- Thery, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. ‐M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Agostinis, P. , Akay, O. , Anand, S. , Anckaert, J. , Martinez, Z. A. , Baetens, T. , Beghein, E. , & Bertier, L. (2017). EV‐TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods, 14, 228–232. [DOI] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y. ‐. J. , Tortorici, M. A. , Wall, A. , Mcguire, A. T. , & Veesler, D. (2020). Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell, 181, 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Zhang, Y. , Wu, L. , Niu, S. , Song, C. , Zhang, Z. , Lu, G. , Qiao, C. , Hu, Y. , Yuen, K. ‐Y. , Wang, Q. , Zhou, H. , Yan, J. , & Qi, J. (2020). Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell, 181, 894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, J. M. , & Whittaker, G. R. (2016). Fusion of enveloped viruses in endosomes. Traffic (Copenhagen, Denmark), 17, 593–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker, G. R. , Daniel, S. , & Millet, J. K. (2021). Coronavirus entry: How we arrived at SARS‐CoV‐2. Current Opinion in Virology, 47, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp, D. , Wang, N. , Corbett, K. S. , Goldsmith, J. A. , Hsieh, C. ‐. L. , Abiona, O. , Graham, B. S. , & Mclellan, J. S. (2020). Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel, A. G. , Benton, D. J. , Xu, P. , Roustan, C. , Martin, S. R. , Rosenthal, P. B. , Skehel, J. J. , & Gamblin, S. J. (2020). SARS‐CoV‐2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin‐cleavage effects. Nature Structural & Molecular Biology, 27, 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Yang, G. , Zhang, R. , Xu, G. , Zhang, L. , Wen, W. , Lu, J. , Liu, J. , & Yu, Y. (2015). Altered microRNA expression profiles of extracellular vesicles in nasal mucus from patients with allergic rhinitis. Allergy, Asthma & Immunology Research, 7, 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, R. , Zhang, Y. , Li, Y. , Xia, L. , Guo, Y. , & Zhou, Q. (2020). Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science, 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebrowska, A. , Skowronek, A. , Wojakowska, A. , Widlak, P. , & Pietrowska, M. (2019). Metabolome of exosomes: Focus on vesicles released by cancer cells and present in human body fluids. International Journal of Molecular Sciences, 20, 3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X. ‐L. , Wang, X. G. , Hu, B. , Zhang, L. , Zhang, W. , Si, H. ‐R. , Zhu, Y. , Li, B. , Huang, C. ‐L. , Chen, H. ‐D. , Chen, J. , Luo, Y. , Guo, H. , Jiang, R. ‐D. , Liu, M. ‐Q. , Chen, Y. , Shen, X. ‐R. , Wang, X. , … Shi, Z. ‐L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N. , Zhang, D. , Wang, W. , Li, X. , Yang, B. , Song, J. , Zhao, X. , Huang, B. , Shi, W. , Lu, R. , Niu, P. , Zhan, F. , Ma, X. , Wang, D. , Xu, W. , Wu, G. , Gao, G. F. , & Tan, W. (2020). A novel coronavirus from patients with pneumonia in China, 2019. New England Journal of Medicine, 382, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information