

Abstract
Endolysosomes are key regulators of iron metabolism and are central to iron trafficking and redox signaling. Iron homeostasis is linked to endolysosome acidity and inhibition of endolysosome acidity triggers iron dysregulation. Because of the physiological importance and pathological relevance of ferrous iron (Fe2+), we determined levels of Fe2+ specifically and quantitatively in endolysosomes as well as the effects of Fe2+ on endolysosome morphology, distribution patterns, and function. The fluorescence dye FeRhoNox-1 was specific for Fe2+ and localized to endolysosomes in U87MG astrocytoma cells and primary rat cortical neurons; in U87MG cells the endolysosome concentration of Fe2+ ([Fe2+]el) was 50.4 μM in control cells, 73.6 μM in ferric ammonium citrate (FAC) treated cells, and 12.4 μM in cells treated with the iron chelator deferoxamine (DFO). Under control conditions, in primary rat cortical neurons, [Fe2+]el was 32.7 μM. Endolysosomes containing the highest levels of Fe2+ were located perinuclearly. Treatment of cells with FAC resulted in endolysosomes that were less acidic, increased in numbers and sizes, and located further from the nucleus; opposite effects were observed for treatments with DFO. Thus, FeRhoNox-1 is a useful probe for the study of endolysosome Fe2+, and much more work is needed to understand better the physiological significance and pathological relevance of endolysosomes classified according to their heterogeneous iron content.
Keywords: Deferoxamine, Endolysosomes, FeRhoNox-1, ferric ammonium citrate, Golgi, iron
1 |. INTRODUCTION
Iron metabolism has increasingly become a focus of study in the field of cell biology and inter-organellar signaling because of its physiological importance and pathological relevance. Iron in endosomes and lysosomes (hereafter referred to as endolysosomes) is particularly important because endolysosomes have been termed “master regulators of iron metabolism” and this regulation is linked to endolysosome acidity (Rizzollo, More, Vangheluwe, & Agostinis, 2021; Weber et al., 2020). Indeed, endolysosome iron appears to be central to iron trafficking and redox signaling (Rizzollo et al., 2021; Weber et al., 2020). However, very little is known about actual levels of iron in endolysosomes and how changes in endolysosome iron content affects their functional and morphological features.
Iron is an essential regulator of numerous physiological processes including DNA and RNA synthesis, oxidative phosphorylation and ATP synthesis, immune responses, energy metabolism, mitochondrial homeostasis, and cell proliferation (Hill, 1985; Weber et al., 2020). Similarly, endolysosomes are regulators of many important physiological functions including plasma membrane repair, cell homeostasis, energy metabolism, nutrient-dependent signal transduction, immune responses, and are well-known signaling hubs that govern iron-related metabolic pathways (Perera & Zoncu, 2016; Settembre, Fraldi, Medina, & Ballabio, 2013).
A defining feature of endolysosomes is their acidic luminal pH (Appelmans, Wattiaux, & De Duve, 1955; Bainton, 1981; De Duve & Wattiaux, 1966), which is regulated mainly by vacuolar-ATPase (v-ATPase) that drives protons against their electrochemical gradients into the lumen of endolysosomes (Mindell, 2012; Perera & Zoncu, 2016). Endolysosome acidity is essential for the functionality of enzyme activity, and disruption of endolysosome pH is linked to a wide range of conditions including aging, cancer, and neurodegenerative diseases (Bergmann, Schütt, Holz, & Kopitz, 2004; Halcrow, Lynch, Geiger, & Ohm, 2021b; Klempner & Styrt, 1983; Koh, Kim, Hwang, Kim, & Park, 2019). De-acidification of endolysosomes triggers iron dysregulation as well as endolysosome and mitochondrial dysfunction (Abu-Remaileh et al., 2017; Halcrow et al., 2021a; Hughes et al., 2020; Weber et al., 2020). Endolysosomes are structurally heterogeneous and traffic dynamically in cells (Ballabio & Bonifacino, 2020; Cabukusta & Neefjes, 2018; Holtzman, 1989; Lie & Nixon, 2019), and morphological and functional changes to endolysosomes have been reported as very early events in the pathogenesis of neurodegenerative diseases (Colacurcio & Nixon, 2016).
Endolysosomes contain high levels of divalent transition metals including zinc, copper, and iron (Hui et al., 2015; Xu & Ren, 2015). For iron, endolysosomes accumulate extracellular ferric iron (Fe3+) bound to transferrin which enters cells by endocytosis where-upon it is reduced to unbound labile ferrous iron (Fe2+) via the Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3) enzyme (Fleming et al., 1998; Kaplan & Ward, 2013; Kurz, Eaton, & Brunk, 2011; Ohgami et al., 2005; Ohgami, Campagna, McDonald, & Fleming, 2006). Cellular processes, such as oxygen transfer depend on the capability of iron to cycle between Fe3+ and Fe2+. However, because of the reactive nature of Fe2+ via Fenton-like chemical reactions, disruption of iron homeostasis and excess Fe2+ leads to increased generation of reactive oxygen species (ROS) including hydroxyl radicals, damage to proteins, DNA and membranes, and when extreme enough cell death (Fenton, 1894).
Little is known about Fe2+ concentration ([Fe2+]el) in endolysosomes as well as how changes in [Fe2+]el affects endolysosome pH, morphology, and function. The fluorescence “turn-on” probe FeRhoNox-1 was found previously to have pronounced specificity for Fe2+ versus other biologically relevant divalent metal ions (Mukaide et al., 2014) and to label Fe2+ stores in acidic Golgi organelles in human hepatocellular carcinoma (HepG2) cells and in Michigan Cancer Foundation-7 (MCF-7) human breast carcinoma cells (Hirayama, Okuda, & Nagasawa, 2013). However, yet to be tested was the extent to which FeRhoNox-1 could be used to qualitatively and quantitatively label endolysosome stores of Fe2+. Here, using U87MG astrocytoma cells and primary rat neurons, we used FeRhoNox-1 to qualitatively and quantitatively measure [Fe2+]el and found that (1) FeRhoNox-1 specifically labeled stores of Fe2+, (2) FeRhoNox-1-positive stores of Fe2+ were localized to endolysosomes and not other acidic organelles (Golgi), (3) Fe2+ levels could be quantitated under control, iron-loaded and iron-chelated conditions, (4) endolysosomes contained widely varying levels of Fe2+ under control, iron-loaded and iron-chelated conditions, (5) endolysosomes containing higher levels of Fe2+ were more abundant near the nucleus and further away from plasma membranes, and (6) endolysosomes from iron-loaded cells were less acidic, were increased in numbers and sizes, and were further from the nucleus; opposite effects were observed for endolysosomes from iron-chelated cells. These findings increase our understanding of the heterogeneity of endolysosomes and the ability to determine [Fe2+]el will almost certainly increase the understanding of the physiological significance and pathological relevance of [Fe2+]el.
2 |. MATERIALS AND METHODS
The experiments performed in this manuscript were not pre-registered. No blinding procedures were performed. No statistical method was employed to pre-determine the sample size of the experiments. No randomization methods were used.
2.1 |. Materials
FeRhoNox-1, which is a rhodamine-based dye, was purchased from Goryo Chemical Company, Japan (cat. no. GC901). NBD_DFO, the specific indicator dye for Fe3+, was purchased from Squarix Biotechnology (cat. no. ME047.2). Deferoxamine (DFO) was purchased from Sigma-Aldrich (cat. no. D9533). Ferric ammonium citrate (FAC) was purchased from Fisher Scientific (cat. no. I72–500). Iron(II) chloride tetrahydrate (FeCl2) was purchased from Millipore Sigma (cat. no. 10386). Iron(III) chloride (FeCl3) was purchased from Millipore Sigma (cat. no. 103814). Methyl-β-cyclodextrin was purchased from Millipore Sigma (cat. no. C4555). Cytochalasin D was purchased from Millipore Sigma (cat. no. C8273). All other reagents were obtained from standard sources (Millipore Sigma and ThermoFisher). Citrate buffer was prepared as follows; 800 ml of distilled water, 24.2 g of sodium citrate dihydrate, 3.3 g of citric acid, the pH adjusted to 6.0 using an Accumet AB15 pH meter (Fisher Scientific, cat. no. 13–636-AB15), and then distilled water was added to reach a final volume of 1 L.
2.2 |. Cultures of U87MG astrocytoma cells
U87MG astrocytoma cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen, cat. no. 11995) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin (Invitrogen, cat. no. 15140122). Cells were grown at 37°C in a 5% CO2 environment to about 40% confluence (~15, 000 cells) in 35 mm2 plastic culture dishes coated with poly-d-lysine (Mattek, cat. no. P35GC-1.5–14-C). Cells were not used past their tenth passage. The cells used in this manuscript are not listed as a commonly misidentified cell line by the International Cell Line Authentication Committee and no authentication experiments were conducted.
2.3 |. Primary cultures of rat neurons
Primary neuronal cultures were prepared from cerebral cortices of male and female Holtzman E17 rat embryos as described previously (Nash et al., 2019). Pregnant dams (embryonic day 17) were sacrificed by asphyxiation with CO2. The fetuses were removed, decapitated, and meninges-free cerebral cortices were isolated, trypsinized, and plated onto 35-mm2 poly-D-lysine-coated glass-bottom tissue culture dishes (Mattek, cat. no. P35GC-1.5–14-C). Neurons were grown in Neurobasal™ medium (ThermoFisher, USA, A3582901) with L-glutamine, antibiotic/antimycotic, and B27 supplement, and were maintained at 37°C and 5% CO2 for 10–14 days at which time they were used for experimentation. Typically, the purity of the neuronal cultures was greater than 95% as determined by immunostaining with neuronal (mouse anti-NeuN or goat anti-MAP2 antibodies, Millipore, cat. no. AB15452 and MAB377) and astrocyte (mouse anti-GFAP antibody, Sigma, cat. no. G6171) markers. A total number of four pregnant dams and a total of 21 pups were used in this study to isolate neurons; the pups used were not separated by sex. Brains from the one mother were used for each separate culture.
2.4 |. Cell lysate preparation
For experiments with cell lysates, U87MG astrocytoma cells were first grown to 80% confluency in 100 mm2 cell culture dishes. Cells were trypsinized and scraped into centrifuge tubes containing RIPA lysis buffer (ThermoFisher, USA, cat. no. 89900) and protease phosphatase inhibitor (0.1 ml per 10 ml). Following 30 min on ice, lysate was centrifuged at 13 000 g for 5 min at 4°C and supernatant was collected into 1.5 ml tubes and stored at −20°C until taken for use.
2.5 |. FeRhoNox-1 specificity for Fe2+
To determine the specificity of FeRhoNox-1 for Fe2+, we used a cell lysate solution and our LSM800 laser scanning microscope system (Zeiss). FeRhoNox-1 was added to 96-well plates and 35 mm2 culture dishes at a final concentration of 10 μM, and FeCl2, FeCl3, MgCl2, CaCl2, and ZnCl2 were added at final concentrations ranging from 1 to 1000 μM in water. Following the addition of the reagents, plates were incubated at room temperature for 10 min and then analyzed at an excitation wavelength of 537 nm and an emission wavelength of 569 nm. We also tested for possible interference of FeCl2-induced FeRhoNox-1 fluorescence by other cations; FeCl2 (100 μM) was incubated with FeRhoNox-1 (10 μM) in the absence or presence of CaCl2 (10–1000 μM), ZnCl2 (1–20 μM), or MgCl2 (10–1000 μM). As before, following the addition of the reagents, plates were incubated at room temperature for 10 min and then analyzed at an excitation wavelength of 537 nm and an emission wavelength of 569 nm.
2.6 |. Identification of FeRhoNox-1-positive organelles in U87MG cells
Individual 35-mm2 poly-D-lysine-coated glass-bottom tissue culture dishes containing U87MG cells and primary neurons were incubated with FeRhoNox-1 (10 μM) for 10 min, washed three times with PBS, and imaged using our LSM800 laser scanning microscope system (Zeiss) at excitation and emission wavelengths of 537 and 569 nm, respectively. U87MG cells were incubated with the late endosome/lysosome marker LysoTracker Green DND-26 (10 μM, ThermoFisher, cat. no. L7526) for 10 min at 37°C. Fields were chosen at random and at least five images from every experimental condition were acquired by confocal microscopy (LSM800, Zeiss). Using Imaris 9.5 (Oxford Instruments) and ImageJ software, colocalization correlations for FeRhoNox-1 and LysoTracker on the merged images were determined using Pearson’s correlation coefficients, Mander’s and Li′s ICQ. Each fluorescence (wavelength) was quantified, and cellular background was excluded. The same methods were used for endosomes labeled with EEA1-GFP CellLight™ BacMam 2.0 (ThermoFisher, cat. no. C10586) and for lysosomes labeled with LAMP1-GFP CellLight™ BacMam 2.0 (ThermoFisher, cat. no. C10596); excitations were measured at 485 nm and emissions were measured at 520 nm.
2.7 |. Identification of Golgi-positive organelles
CellLight™ Golgi-GFP BacMam 2.0 (ThermoFisher, cat. no. C10592) was used to label Golgi in U87MG cells. Golgi-GFP was added to 35-mm2 poly-d-lysine-coated glass-bottom tissue culture dishes containing U87MG cells at a concentration of 1 × 108 CellLight® particles/ml, and after incubating cells overnight at 37°C in a 5% CO2 environment, cells were washed three times with PBS and then imaged using confocal microscopy (LSM800, Zeiss) at excitation and emission wavelengths of 488 and 510 nm, respectively. Using Imaris 9.5 (Oxford Instruments) and ImageJ software, colocalization correlations for FeRhoNox-1 and Golgi-GFP-positive organelles on the merged images were determined using Pearson’s correlation coefficients, Mander’s and Lis ICQ.
2.8 |. Endolysosome pH
As described previously (Hui, Chen, & Geiger, 2012a; Hui, Chen, Haughey, & Geiger, 2012b), endolysosome pH was measured using the ratiometric indicator-dye LysoSensor Yellow/Blue DND-160 (ThermoFisher, cat. no. L7545); a dual excitation dye that allows for the measurement of pH of acidic organelles independent of amount of dye up-taken by individual cells. U87MG cells were loaded with 10 μM DND-160 for 5 min at 37°C after which the dye-containing media was removed, and fresh media was added to the cells. Light emitted at 520 nm in response to excitation at 340 nm and 380 nm was measured every 30 s for a duration of 20 ms using a filter-based imaging system (Zeiss, Germany). The ratios of light excited (340/380 nm) versus light emitted (520 nm) were converted to pH units using a calibration curve established with a solution of 10 μM of the H+/Na+ ionophore monensin and 20 μM of the H+/K+ ionophore nigericin; both were dissolved in a buffer containing 20 mM 2-(N-morpholino) ethane sulfonic acid (MES), 110 mM KCl, and 20 mM NaCl, and the pH was adjusted between 3.0 and 7.0 with HCl and NaOH.
2.9 |. Statistical analyses
All data were presented as means ± standard deviation (SD). Statistical significance between two groups was analyzed with a two-tailed Student’s t-test, and statistical significance among multiple groups was analyzed with one-way ANOVA plus a Tukey post hoc test. p < 0.05 was accepted to be statistically significant. GraphPad Prism 9.2.0 software was used for statistical analyses. No exclusion criteria were pre-determined. The data were not assessed for normality and no test for outliers was conducted. No formal power calculations were performed to estimate the necessary sample size. The number of cells and animals used for this study was determined based on previous studies that were similar (Jiang et al., 2021; Uchida et al., 2019). Correlation coefficients according to Pearson’s, Mander’s, and Lis ICQ were performed using ImageJ (NIH) and Imaris 9.5 software (Oxford Instruments).
2.10 |. Ethical statement
All the procedures were approved by the local ethics committee at the University of North Dakota under the approved protocol IACUC2106–0004.
3 |. RESULTS
3.1 |. FeRhoNox-1 staining was specific for ferrous iron (Fe2+)
It was unclear from the literature the extent to which FeRhoNox-1 stained specifically for ferrous iron (Fe2+) and whether Fe2+-induced FeRhoNox-1 fluorescence was affected by other divalent cations. Accordingly, we determined first the specificity with which FeRhoNox-1-labeled Fe2+ in comparison to Fe3+. FeRhoNox-1 mean fluorescence intensity (MFI) in cell lysate was found to be highly specific for Fe2+ and not Fe3+ (Figure 1); the calculated EC50 for Fe2+ was 19.6 μM. Even at 200 μM, the highest concentration of FeCl3 and ferric ammonium citrate (FAC) tested, there were no significant increases in FeRhoNox-1 MFI observed (Figure 1). To demonstrate that FeRhoNox-1 can quantify decreasing levels of Fe2+ present as it oxidizes to Fe3+, we performed experiments in both cell lysate and citrate buffer in the presence of FeCl2 (100 μM) and FeRhoNox-1. The addition of aqueous potassium hydroxide (KOH) de-acidified the solutions and decreased FeRhoNox-1 MFI. In cell lysate, the KOH-induced decreases in FeRhoNox-1 MFI tended to subside over time and additional applications of KOH again decreased FeRhoNox-1 MFI. In citrate buffer, FeRhoNox-1 MFI remained decreased as Fe2+ is oxidized; Fe3+ is likely irreversibly complexed with citrate and other compounds, and subsequent additions of KOH decreased FeRhoNox-1 MFI (Figure S1).
FIGURE 1.

FeRhoNox-1 fluorescence intensity increased with increased concentrations of FeCl2: FeCl2 (closed circles), FeCl3 (open squares), and FAC (open triangles) were incubated at concentrations ranging from 0 to 200 μM with FeRhoNox-1 (10 μM) in cell lysate solution. Solutions were incubated for 10 min at room temperature and aliquots were added to 35 mm2 glass-bottom dishes; relative fluorescence units (RFU) were determined using a 63X oil objective lens and our LSM800 confocal microscope (Zeiss). The best fit line (R2) for FeCl2 was 0.99 and for FeCl3 and FAC was 0.33 and 0.37, respectively. The calculated EC50 for Fe2+ was 19.6 μM. Data represent the means ± SD of three experiments of independent preparations performed in three technical replicates (n = 9)
Next, we tested the ability of other divalent cations to increase FeRhoNox-1 fluorescence and for the divalent cations to affect Fe2+-induced increases in FeRhoNox-1 MFI. FeRhoNox-1 MFI was not affected by MgCl2 and CaCl2 at final concentrations ranging from 10 to 1000 μM nor by ZnCl2 at final concentrations ranging from 1 to 20 μM (Figure S2a). However, in testing for possible interference by other metals, we increased FeCl2 concentrations to 100 μM; levels that yield near-maximal MFI to enhance signal-to-noise ratios. High FeCl2 concentration-induced increases in FeRhoNox-1 MFI were not significantly affected by MgCl2 and CaCl2 at final concentrations ranging from 1 to 1000 μM nor by ZnCl2 at final concentrations ranging from 1 to 20 μM (Figure S2b).
To further verify the extent of FeRhoNox-1 specificity for Fe2+, we performed additional experiments at pH values ranging from 2 to 8. Results from these experiments showed that FeRhoNox-1 MFI for Fe2+ was not changed significantly between pH values of 4 and 7 (Figure S8); FeRhoNox-1 MFI decreased significantly starting at pH 7.5 (Figure S8). Similar results were observed with FeCl3 and FAC (Figure S3a,c,e). Under acidic conditions (pH ~ 2) in which Fe3+ is soluble (Kaplan & Ward, 2013), the FeRhoNox-1 MFI remained low with FAC and FeCl3 treatments but FeRhoNox-1 MFI was increased with FeCl2 treatment (Figure S3a,c,e). These results indicate that FeRhoNox-1 is specific for Fe2+ because soluble Fe3+ at low pH did not contribute to FeRhoNox-1 MFI. Additionally, to minimize potential error in FeRHoNox-1 MFI, we performed experiments where FeRhoNox-1 MFI was monitored for over 1 h (Figure S3b,d,f); FAC, FeCl2, and FeCl3 did not significantly affect FeRhoNox-1 MFI. Nevertheless, all experimental measurements were made 10 min after adding the dye.
3.2 |. FeRhoNox-1 stained Fe2+ in LysoTracker-positive endolysosomes
We next determined the extent to which FeRhoNox-1-positive stores of Fe2+ colocalized with late endosomes and lysosomes stained positively with LysoTracker. FeRhoNox-1, likely up-taken by passive diffusion across plasma membranes (Figure S4), robustly stained endolysosomes in U87MG cells (Figure 2) and in primary rat cortical neurons (Figure S9, S10). The late endosomes and lysosomes stained with LysoTracker exhibited staining patterns similar to those observed for FeRhoNox-1 in both U87MG cells (Figure 2a) and in primary cultures of rat neurons (Figure S9). When the images for FeRhoNox-1 and LysoTracker were merged, the staining patterns showed a high degree of colocalization in U87MG cells; the Pearson correlation coefficient was 0.84 ± 0.03, the Li′s ICQ was 0.37 ± 0.03, the Mander’s M1 (Red) was 0.70 ± 0.1, and the Mander’s M2 (Green) was 0.68 ± 0.05. To confirm further the localization of FeRhoNox-1-positive stores of Fe2+ in endolysosomes, we used Imaris software (version 9.5, Oxford Instruments) to observe individual endolysosomes from a horizontal view; there was a clear colocalization of LysoTracker and FeRhoNox-1 staining in individual endolysosomes (Figure 2b). In primary rat cerebral cortical neurons, similar colocalizations were observed between FeRhoNox-1 fluorescence and LysoTracker staining; 76.8 ± 0.5% of endolysosomes showed colocalization between FeRhoNox-1 and LAMP1-positive staining (Figure S9b).
FIGURE 2.

FeRhoNox-1-stained Fe2+ in LysoTracker-positive endolysosomes: FeRhoNox-1-positive stores (red) of Fe2+ (FeRhoNox-1), LysoTracker-positive (green) endolysosomes (LysoTracker), and DAPI (blue) were observed in U87MG cells. Cells were incubated with FeRhoNox-1 (10 μM), LysoTracker (10 μM), and DAPI (10 μM) for 10 min at 37°C. images were acquired by confocal microscopy (LSM800, Zeiss) using a 63X oil objective lens. (a) FeRhoNox-1 positive staining was highly colocalized with the LysoTracker-positive staining when the images were merged. The Pearson correlation coefficient for the colocalization determined from three independent experiments was 0.84 ± 0.03. (b) Individual endolysosomes from a horizontal viewpoint using Imaris imaging software (version 9.5, Oxford instruments) showed colocalization (merged) of LysoTracker and FeRhoNox-1 in a single endolysosome. Scale bars were 5 μm for (a) and 1 μm for (b). Images were taken from 10 independent cell preparations (n = 10) and cells were randomly selected from each culture dish
3.3 |. FeRhoNox-1 stained Fe2+ in EEA1-positive endosomes and LAMP1-positive lysosomes
To further differentiate the observed subcellular localization of FeRhoNox-1 positive staining in endolysosomes, we determined using U87MG cells the extent to which FeRhoNox-1 staining colocalized with early endosomes using EEA1 as a marker and late endosomes/lysosomes using LAMP1 as a marker. As illustrated, EEA1-positive endosomes (Figure 3a) and LAMP1-positive lysosomes (Figure 3b) were distributed similarly with FeRhoNox-1-positive stores of Fe2+. For EEA1-positive endosomes, FeRhoNox-1-positive staining showed a high degree of colocalization (Figure 3a); the Pearson correlation coefficient was 0.86 ± 0.07, the Li′s ICQ was 0.38 ± 0.06, the Mander’s M1 (Red) was 0.62 ± 0.1, and the Mander’s M2 (Green) was 0.55 ± 0.06. For LAMP1-positive late endosomes and lysosomes, FeRhoNox-1-positive staining showed a moderate degree of colocalization (Figure 3b); the Pearson correlation coefficient was 0.62 ± 0.05, the Li′s ICQ was 0.35 ± 0.03, the Mander’s M1 (Red) was 0.55 ± 0.08, and the Mander’s M2 (Green) was 0.58 ± 0.15.
FIGURE 3.

FeRhoNox-1-labeled Fe2+ stores in endosomes and lysosomes: (a) FeRhoNox-1-positive stores of Fe2+ (FeRhoNox-1, red), EEA1-positive early endosomes (EEA1, green), and DAPI (blue) were observed in U87MG cells. U87MG cells were incubated with EEA1 CellLight overnight at 37°C and then FeRhoNox-1 (10 μM) and DAPI (10 μM) were added, and cells were incubated for an additional 10 min at 37°C. EEA1- and FeRhoNox-1-positive staining showed a very high degree of colocalization; the Pearson correlation coefficient was 0.86 ± 0.07, the Li′s ICQ was 0.38 ± 0.06, the Mander’s M1 (red) was 0.62 ± 0.1, and the Mander’s M2 (green) was 0.55 ± 0.06. (b) FeRhoNox-1-positive stores of Fe2+ (FeRhoNox-1, red), LAMP1-positive late endosomes and lysosomes (LAMP1, green), and DAPI (blue) were observed in U87MG cells. U87MG cells were incubated with LAMP1 CellLight overnight at 37°C and after adding FeRhoNox-1 (10 μM) and DAPI (10 μM) cells were further incubated for 10 min at 37°C. LAMP1- and FeRhoNox1-positive staining showed a moderate degree of colocalization; the Pearson correlation coefficient was 0.62 ± 0.05, the Li′s ICQ was 0.35 ± 0.03, the Mander’s M1 (red) was 0.55 ± 0.08, and the Mander’s M2 (green) was 0.58 ± 0.15. Scale bars were 10 μm. Images were taken from 10 independent cell preparations (n = 10)
To further determine the extent to which FeRhoNox-1-positive staining colocalized with markers for endosomes and lysosomes we reconstructed images using Imaris software confocal images (left panels) of FeRhoNox-1 (red) merged with images of LysoTracker-, EEA1- and LAMP1-positive (green) endolysosomes (right panels) in U87MG cells (Figure 4a–c). FeRhoNox-1-positive staining in endolysosomes colocalized 80% with Lysotracker-, 82% with EEA1- and 70% with LAMP1-stained endolysosomes. We encourage readers to access our Supplemental Figures to see 3-D videos of cells turning on their Y-axis; FeRhoNox-1 colocalization with Lysotracker, EEA1, and LAMP1 staining is clearly observed (Figure S5).
FIGURE 4.
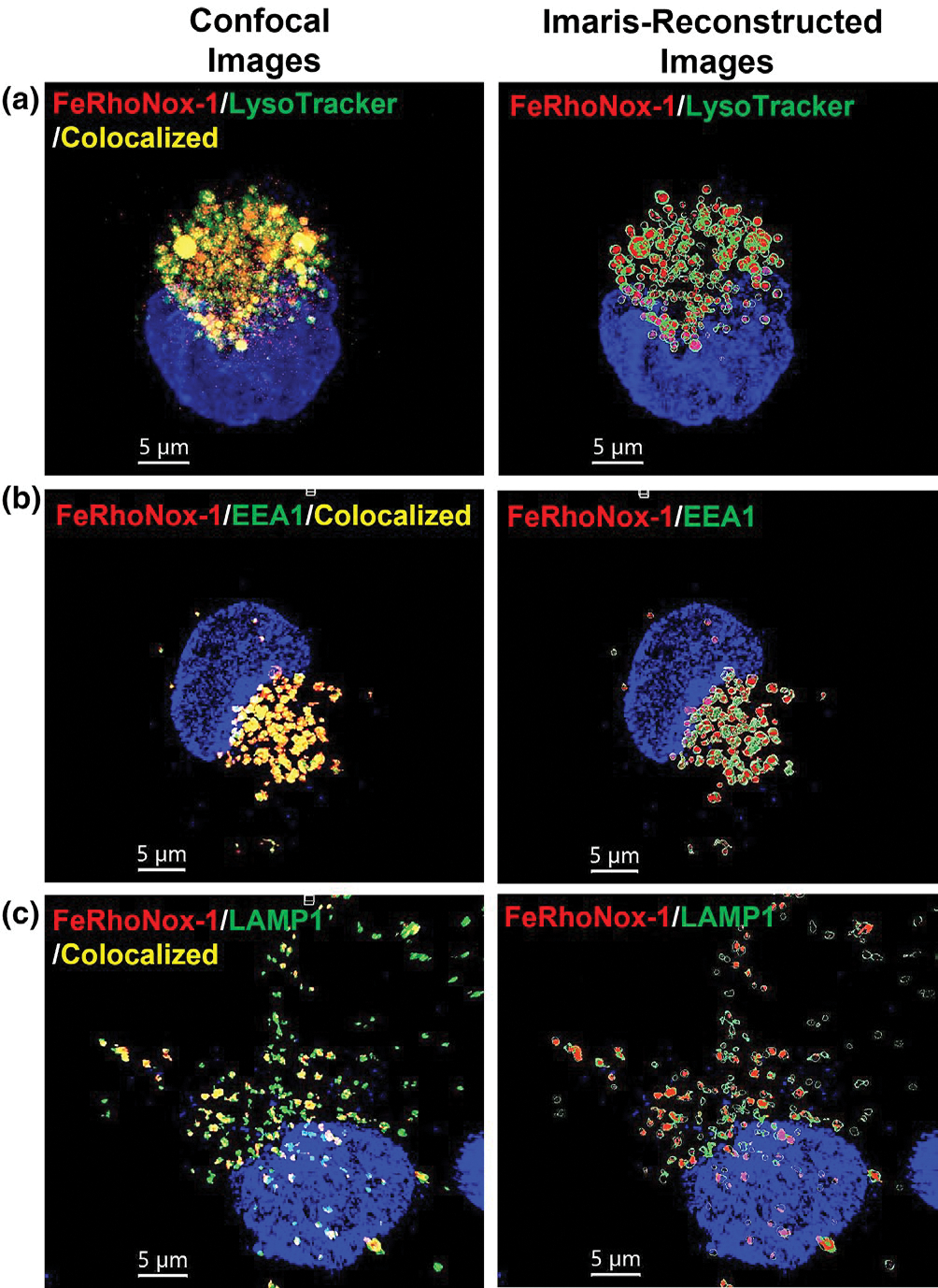
Confocal and Imaris-reconstructed images of FeRhoNox-1 merged with LysoTracker-, EEA1- and LAMP1-positive endolysosomes: (left panel images) Representative confocal images of FeRhoNox-1 (red) and DAPI (blue) relative fluorescence intensity merged with (a) LysoTracker- (green), (b) EEA1- (green) and (c) LAMP1- (green) positive endolysosomes in U87MG cells. (right panel images) Representative Imaris reconstructed images of FeRhoNox-1 (red) and DAPI (blue) relative fluorescence intensity merged with (a) LysoTracker- (green), (b) EEA1- (green) and (c) LAMP1- (green) positive endolysosomes in U87MG cells. FeRhoNox-1 positive endolysosomes colocalized 80% with Lysotracker-, 82% with EEA1- and 70% with LAMP1-positive endolysosomes. Scale bars are 5 μm. Images were taken from 10 independent cell preparations (n = 10) and cells were randomly selected from each culture dish
3.4 |. FeRhoNox-1 positive stores of Fe2+ were not highly expressed in Golgi
Having observed FeRhoNox-1-positive staining in endolysosomes, we next determined the extent to which FeRhoNox-1-positive staining was co-localized in Golgi especially because this stain was marketed as a Golgi-specific marker. Staining patterns in U87MG cells for FeRhoNox-1 and GolgiTracker were distinctly different (Figure 5a). When the images were merged, FeRhoNox-1-positive staining displayed almost no colocalization with GolgiTracker-positive staining (Figure 5a); the Pearson correlation coefficient was 0.09 ± 0.04, the Li′s ICQ was 0.05 ± 0.02, the Mander’s M1 (Red) was 0.05 ± 0.08, and the Mander’s M2 (Green) was 0.03 ± 0.03. Imaris reconstruction of Golgi-GFP CellLight- and FeRhoNox-1-stained images showed that the Golgi staining was distinct from FeRhoNox-1-labeled endolysosomes (Figure 5b, left panel), and that the Golgi-GFP CellLight staining was continuous within a single Golgi apparatus (Figure 5b, right panel). We encourage readers to access our Supplemental Figures to see a 3-D video of a cell turning on its Y-axis and clearly showing the lack of FeRhoNox-1 colocalization with Golgi-GFP CellLight-labeled Golgi (Figure S5).
FIGURE 5.

FeRhoNox-1 positive staining minimally colocalized with Golgi-GFP CellLight staining: Representative confocal images of U87MG cells transduced with Golgi-GFP CellLight (green) and then stained with FeRhoNox-1 (red) and DAPI (blue). (a) When confocal images of the staining were merged, it was obvious that the FeRhoNox-1-positive staining minimally colocalized with the Golgi-positive staining; the Pearson correlation coefficient was 0.09 ± 0.04, the Li′s ICQ was 0.05 ± 0.02, the Mander’s M1 (red) was 0.05 ± 0.08, and the Mander’s M2 (green) was 0.03 ± 0.03. (b) When images were reconstructed using Imaris software (version 9.5, Oxford instruments), it was obvious that Golgi staining was distinct from FeRhoNox-1 staining (left panel), and that the Golgi-positive staining was within Golgi-positive organelles). Scale bars were 1 or 3 μm. Images were taken from 10 independent cell preparations (n = 10) and cells were randomly selected from each culture dish
3.5 |. FeRhoNox-1 fluorescence intensity and endolysosome sizes were increased with ferric ammonium citrate and were decreased with deferoxamine
We next examined the labile nature of endolysosome pools of Fe2+ by determining the extent to which FeRhoNox-1 MFI increased in cells treated with FAC, decreased in cells treated with the endolysosome-specific iron chelator deferoxamine (DFO), and whether the treatments with FAC and DFO altered colocalizations between FeRhoNox-1- and LysoTracker-positive endolysosomes. As expected, under control conditions we observed a high level of colocalization between FeRhoNox-1 and LysoTracker staining of endolysosomes in U87MG cells; the Pearson correlation coefficient was 0.84 ± 0.03, the Li′s ICQ was 0.37 ± 0.03, the Mander’s M1 (Red) was 0.70 ± 0.1, and the Mander’s M2 (Green) was 0.68 ± 0.05 (Figure 6a). Incubation of U87MG cells for 1 h with FAC (50 μM) resulted in statistically significant (p < 0.001) increases in FeRhoNox-1 MFI from 101 ± 5% in control cells to 146 ± 6% in cells pre-treated with FAC (Figure 6b,d), and the degree of colocalization between FeRhoNox-1 and LysoTracker staining remained high; the Pearson correlation coefficient was 0.77 ± 0.04, the Li′s ICQ was 0.36 ± 0.04, the Mander’s M1 (Red) was 0.68 ± 0.2, and the Mander’s M2 (Green) was 0.67 ± 0.07 (Figure 6b). Incubation of U87MG cells for 1 h with DFO (50 μM) decreased significantly (p < 0.0001) FeRhoNox-1 MFI to 51 ± 5% of control values (Figure 6c,d). Because of the decreased red fluorescence, colocalization between FeRhoNox-1 and LysoTracker staining appeared lower for the Pearson correlation coefficient (0.54 ± 0.03), the Li′s ICQ (0.25 ± 0.04), and for the Mander’s M1 (Red, 0.31 ± 0.10). However, for the colocalization between FeRhoNox-1 and LysoTracker, the fluorescence intensity of LysoTracker remained high (0.70 ± 0.05) for Mander’s M2 (Green) (Figure 6c,d) because DFO decreases endolysosome pH and LysoTracker localizes with acidic endolysosomes and is unaffected by DFO.
FIGURE 6.

FeRhoNox-1 fluorescence intensity increased with ferric ammonium citrate (FAC) and decreased with deferoxamine (DFO), and FAC did not change the relative extent of colocalization with LysoTracker-positive labeling of endolysosomes; DFO did change the extent of relative colocalization with LysoTracker-positive labeling of endolysosomes: (a) Tnder control conditions (CTL), there was a high level of colocalization between FeRhoNox-1- and LysoTracker-positive staining of endolysosomes in U87MG cells; the Pearson correlation coefficient was 0.84 ± 0.03, the Li′s ICQ was 0.37 ± 0.03, the Mander’s M1 (red) was 0.70 ± 0.1, and the Mander’s M2 (green) was 0.68 ± 0.05. (b) Pre-incubation of U87MG cells with FAC (50 μM) for 1 h increased FeRhoNox-1 fluorescence staining in endolysosomes but did not change the extent of colocalization between FeRhoNox-1 positive and LysoTracker-positive staining; the Pearson correlation coefficient was 0.77 ± 0.04, the Li′s ICQ was 0.36 ± 0.04, the Mander’s M1 (red) was 0.68 ± 0.2, and the Mander’s M2 (green) was 0.67 ± 0.07. (c) Incubation of U87MG cells for 1 h with DFO (50 μM) decreased FeRhoNox-1 MFI. Because of the decreased red fluorescence, colocalization between FeRhoNox-1 and LysoTracker staining appeared to be lower for the Pearson correlation coefficient (0.54 ± 0.03), the Li′s ICQ (0.25 ± 0.04), and for the Mander’s M1 (red, 0.31 ± 0.10). However, the colocalization between FeRhoNox-1 and LysoTracker staining remained high (0.70 ± 0.05) for Mander’s M2 (green) (Figure 6c). (d) Semi-quantitative FeRhoNox-1 mean fluorescence intensity increased significantly (p < 0.001) with ferric ammonium citrate (FAC) and decreased significantly (p < 0.001) with the iron chelator deferoxamine (DFO). Scale bars were 5 μm. Images were taken from ten independent cell preparations (n = 10) and cells were randomly selected from each culture dish. Each data point represents the mean of the fluorescence from 10 cells (n = 50). (d) F(2, 12) = 614, p < 0.0001
3.6 |. FeRhoNox-1 was used to measure quantitatively levels of Fe2+ in endolysosomes
Using a cell lysate system, we determined MFI of FeRhoNox-1 and LysoTracker for concentrations of Fe2+ ranging from 0 to 200 μM by confocal microscopy. With those data, we generated a standard curve for FeRhoNox-1/Lysotracker fluorescence intensity ratios. For the generated linear relationship between FeRhoNox-1/Lysotracker ratios and [Fe2+], the Pearson correlation coefficient was 0.96 ± 0.02 (Figure 7a). The [Fe2+]el in U87MG cells was 50.4 ± 6.4 μM in control cells, 73.6 ± 5.3 μM in FAC-treated cells, and 12.4 ± 4.1 μM in DFO-treated cells (Figure 7b). As well, we determined the relative numbers (frequency) of endolysosomes containing [Fe2+]; the values for [Fe2+]el were arbitrarily placed in the following bins 0 to 25, 26 to 50, 51 to 75, 76 to 100, 101 to 150, 151 to 200, and 201+ μM. Typically, 300–400 endolysosomes were observed in control cells and 75% of these contained [Fe2+] in the range 0–50 μM (Figure 7c). Pre-treatment with FAC increased the percentage of endolysosomes containing higher [Fe2+]; only 10% contained [Fe2+] in the range 0–50 μM (Figure 7c). Pre-treatment with DFO increased the percentage of endolysosomes containing lower [Fe2+]; 100% contained [Fe2+] in the range of 0–50 μM (Figure 7c). The [Fe2+]el in primary rat cortical neurons was 32.7 μM in control cells (Figure 7d).
FIGURE 7.

Quantitative measures of [Fe2+]el increased with ferric ammonium citrate and decreased with deferoxamine: (a) A standard concentration/response curve for FeRhoNox-1/Lysotracker fluorescence intensity ratios at known concentrations of Fe2+ using a cell lysate solution; the linear relationship generated had a Pearson correlation coefficient of 0.96 ± 0.03. (b) Quantitative measures of [Fe2+]el; levels were 50.4 ± 6.3 μM in control cells, 73.6 ± 5.1 μM in cells pre-incubated with FAC, and 12.4 ± 4.2 μM in cells pre-incubated with DFO. (c) Numbers of endolysosomes containing low (0 to 50 μM), medium (51 to 150 μM), and high (151 to 250 μM) [Fe2+] were altered by treatments with FAC and DFO. Under control conditions (open circles) 78% of endolysosomes contained 0 to 50 μM. When cells were treated with FAC (open triangles), 9% of endolysosomes contained 0 to 50 μM. When cells were treated with DFO (open squares), 100% of endolysosomes contained 0 to 50 μM. (d) Quantitative measures of [Fe2+]el; levels were 50.4 ± 6.3 μM in astrocytoma control cells and 32.7 ± 7.4 μM in primary rat cortical neurons. (a, c) Data represent the means ± SD of three experiments of independent preparations performed in three technical replicates (n = 9). (b, d) Each data point represents the mean fluorescence intensity from 10 cells (n = 50). (b) F(2, 12) = 308.1, p < 0.0001; (d) p = 0.0003, df = 8, t = 6.09
3.7 |. Heterogeneity of Fe2+-containing endolysosomes relative to the nucleus and plasma membranes
Endolysosomes are mobile and heterogeneously distributed with cells. Accordingly, we next determined intracellular distributions of endolysosomes based on [Fe2+]el. Under control conditions, endolysosomes were mainly near the nucleus (perinuclear); as the distance from the nucleus increased there were significantly fewer endolysosomes (Figure 8a,b). Similar results were observed in primary rat neurons (Figure S10a,c). With regards to [Fe2+]el, levels were highest in endolysosomes closest to the nucleus, and significantly less [Fe2+]el was found as distances from the nucleus increased (Figure 8c,d). Similar results were observed in primary rat neurons (Figure S10b,d). With regards to plasma membranes, more endolysosomes were located furthest away from plasma membranes and significantly fewer were found close to plasma membranes (Figure 9a,b). With regards to Fe2+, levels were highest in endolysosomes furthest away from plasma membranes and significantly less [Fe2+] was found in endolysosomes closest to plasma membranes (Figure 9c,d).
FIGURE 8.

Endolysosomes were more numerous and contained higher levels of Fe2+ when located close to the nucleus: (a) Representative confocal image of an U87MG cell labeled with FeRhoNox-1 for [Fe2+]el, LysoTracker for endolysosomes, and DAPI for nuclei. (b) Endolysosomes were more abundant when located closest to the nucleus. (c) The cell depicted in (a) above was reconstructed with Imaris software (version 9.5, Oxford instruments) to demonstrate [Fe2+]el relative to endolysosome locations. (d) Endolysosomes contained higher levels of Fe2+ when located closest to the nucleus. Scale bars are 5 μm. Each data point represents the mean fluorescence intensity of endolysosomes per cell measured from 10 cells (n = 50). (b) F(4, 20) = 433.2, p < 0.0001; (d) F(4, 20) = 191.8, p < 0.0001
FIGURE 9.
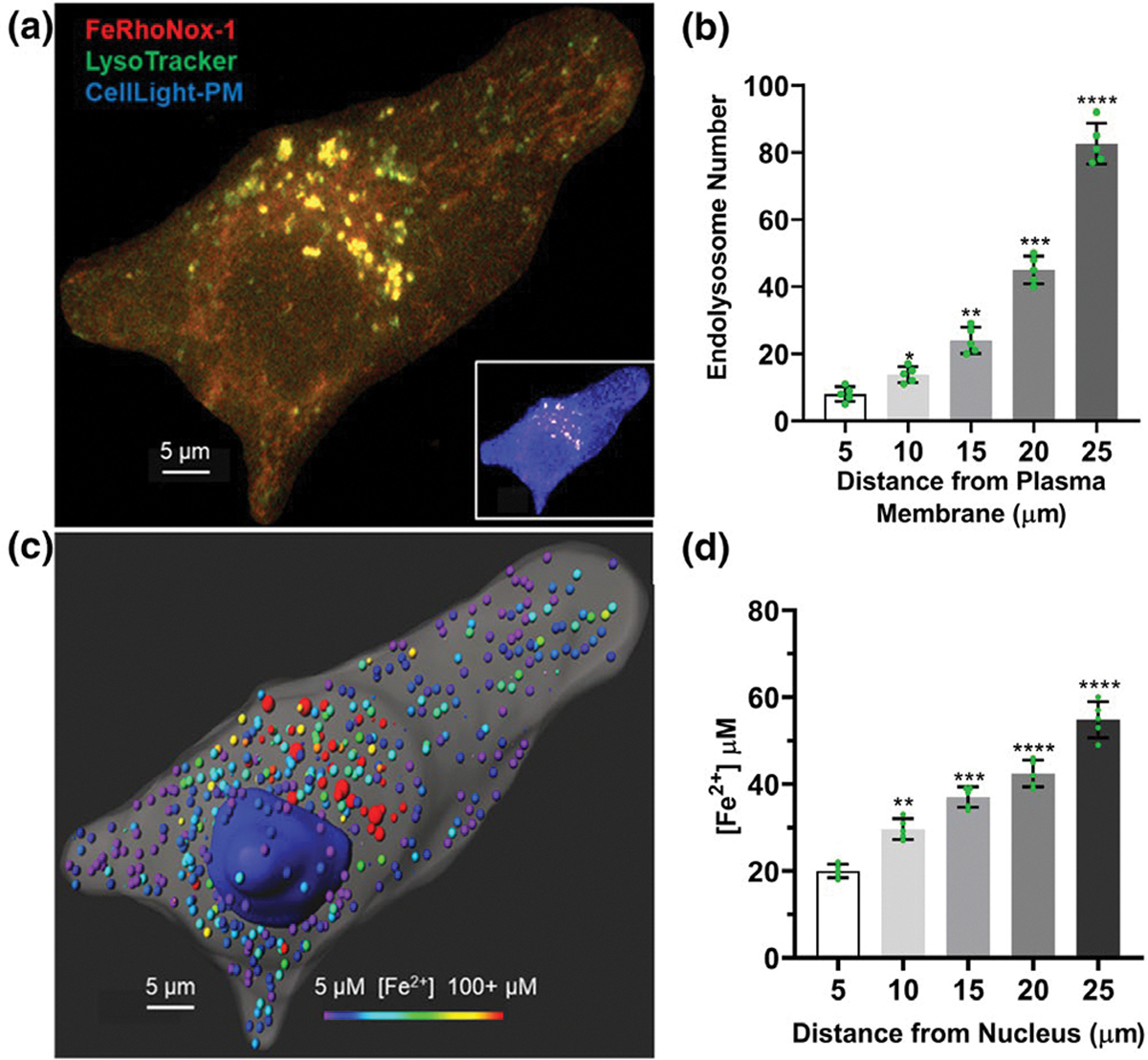
Endolysosomes were more abundant and contained higher levels of Fe2+ when located further away from plasma membranes: (a) Representative magnified confocal image (inset) of an U87MG cell labeled with FeRhoNox-1 for [Fe2+]el, LysoTracker for endolysosomes, and CellLight-PM for plasma membranes. (b) Endolysosomes were more abundant when located furthest away from plasma membranes. (c) The cell depicted in (a) above was reconstructed with Imaris software (version 9.5, Oxford instruments) to demonstrate [Fe2+]el relative to endolysosome locations. (d) Endolysosomes contained higher levels of Fe2+ when located furthest away from plasma membranes. Scale bars are 5 μm. Each data point represents the mean fluorescence intensity of endolysosomes per cell measured from 10 cells (n = 50). (b) F(4, 20) = 286.3, p < 0.0001; (d) F(4, 20) = 107, p < 0.0001
3.8 |. Effects of DFO and FAC on endolysosome numbers, sizes (area), pH, and intracellular distribution relative to the nucleus
We next determined the effects of DFO (50 μM) and FAC (50 μM) on endolysosome numbers, sizes (area), pH, and intracellular distribution relative to the nucleus. Using LysoTracker to label endolysosomes, chelating endolysosome iron with DFO significantly decreased the numbers (p < 0.01) and area (p < 0.05) of endolysosomes as well as significantly (p < 0.01) changed the intracellular distribution of endolysosomes such that they were on average 1.57 μm closer to the nucleus. In comparison, increasing levels of endolysosome Fe2+ with FAC significantly increased the numbers (p < 0.0001) and area (p < 0.01) of endolysosomes as well as significantly (p < 0.001) changed the intracellular distribution of endolysosomes such that they were on average 2.63 μm further away from the nucleus (Figure 10a–d). Virtually identical data were obtained using the red LysoTracker dye DND-99 (Figure S6). Similar results were also observed in primary rat neurons (Figure S11a–c). Because endolysosome pH affects endolysosome size and intracellular distribution patterns, we determined the possible effects of DFO and FAC on endolysosome pH. Using LysoSensor DND-160 as described previously (Hui, Soliman et al. 2019), DFO (50 μM) significantly (p < 0.01) decreased endolysosome pH from 4.84 to 4.57 (54% gain of H+), and FAC (50 μM) increased significantly (p < 0.05) endolysosome pH from 4.84 to 5.02 (37% loss of H+) (Figure 10e). FAC increased endolysosome pH and increased FeRhoNox-1 MFI concurrently, and DFO decreased endolysosome pH and decreased FeRhoNox-1 MFI concurrently (Figure S7).
FIGURE 10.

Effects of DFO and FAC on endolysosome numbers, sizes, pH, and intracellular distribution relative to the nucleus: (a–d) Representative images and bar graphs showing DFO (50 μM) significantly decreased the number of LysoTracker positive vesicles per cell and their sizes (area in μm2) as well as significantly decreased their distance by on average 1.57 μm to the nucleus. FAC (50 μM) significantly increased the numbers and sizes of endolysosomes as well as significantly changed the intracellular distribution of endolysosomes such that they were on average 2.63 μm away from the nucleus. (e) Using LysoSensor DND-160 to measure levels of pH in endolysosomes, 5 min incubations with DFO (50 μM) decreased and FAC increased endolysosome pH (n = 30). Scale bars are 3 μm. Each data point represents the mean fluorescence intensity from 10 cells performed from five technical replicates (n = 50). (b) F(2, 12) = 686.1, p < 0.0001; (c) F(2, 12) = 96.7, p < 0.0001; (d) F(2, 12) = 1063, p < 0.0001; (e) F(2, 12) = 98.5, p < 0.0001
4 |. DISCUSSION
Iron metabolism is physiologically important and pathologically relevant. Iron in endolysosomes is particularly important because they are “master regulators of iron metabolism” central to iron trafficking; iron homeostasis is linked to endolysosome acidity (Rizzollo et al., 2021; Weber et al., 2020). These acidic organelles contain high levels of transition metals including iron (Xu & Ren, 2015), which is essential for a variety of biological events and has implications in the pathogenesis of cancer and neurodegenerative diseases (Halcrow, Lynch, et al., 2021b; Padmanabhan, Brookes, & Iqbal, 2015; Rockfield, Raffel, Mehta, Rehman, & Nanjundan, 2017). Furthermore, levels of intracellular iron are controlled by a complex array of iron transport, iron binding, and iron release mechanisms. Therefore, the current study adds significantly to this field because it is important to measure levels of Fe2+ in endolysosomes, to know the extent to which [Fe2+]el is labile and regulated, and what happens to endolysosome morphology and positioning within cells following increases or decreases in [Fe2+]el.
The fluorescence “turn-on” probe FeRhoNox-1 and other recent Fe2+-specific probes have pronounced specificity for Fe2+ (Hirayama, 2018; Hirayama et al., 2017; Hirayama, Miki, & Nagasawa, 2019; Mukaide et al., 2014). In agreement with previous studies (Hirayama et al., 2013; Mukaide et al., 2014), we found that FeRhoNox-1 was highly selective for Fe2+. No significant increases were observed in FeRhoNox-1 fluorescence with Fe3+, zinc, magnesium, or calcium; these same cations did not interfere with Fe2+-induced increases in FeRhoNox-1 fluorescence. Furthermore, we examined how pH levels would affect FeRhoNox-1 fluorescence. We found that pH values 7.5 or higher significantly reduced FeRoNox-1 fluorescence intensity, which is consistent with a previous study (Hirayama et al., 2013). Because FeRhoNox-1 is Fe2+ dependent, these findings are also consistent with previous studies demonstrating that Fe2+ oxidation was incomplete at pH 6.5–7.5, and at pH 8.5 was complete (Morgan and Lahav, 2007; Lakshmanan, Clifford and Samanta, 2009). Thus, results from others and us confirm that FeRhoNox-1 is a very selective probe with which studies can be conducted to determine [Fe2+] over a wide range of physiologically relevant endolysosome pH conditions.
It is important to understand the mechanisms underlying the FeRhoNox-1 complex, which is a rhodamine B-based derivative. Based on their photophysical properties, rhodamine dyes are used for detecting metal ions and for intracellular signaling. FeRhoNox-1 is similar to rhodamine B containing a xanthene ring, a spirolactone and quinoid structure, and an N-oxide moiety or tertiary amine. The most important characteristic of FeRhoNox-1 is the fluorescence-switching mechanism: the N-oxide moiety is converted to a tertiary amine via Fe2+-mediated deoxygenation. This mechanism converts non-fluorescence FeRhoNox-1 into fluorescence rhodamine B (Hirayama et al., 2013). FeRhoNox-1 selectivity for Fe2+ is likely because of reduction potentials because Fe3+/Fe2+ cycling has the most negative redox potential (Hirayama et al., 2013). However, the mechanism of deoxygenation of N-oxides by Fe2+ remains unknown but is suggested to occur by forming iron-oxo complexes via oxygen abstraction by Fe2+ to supply alkyl amine (Bratsch, 1989; Moens, Roos, Jaque, De Proft, & Geerlings, 2007).
Moreover, our findings are consistent with a previous study demonstrating FeRhoNox-1 showed high fluorescence under mildly acidic to mildly basic pH conditions (Hirayama et al., 2013). Notably, the spirocyclic structure of these complexes is sensitive to pH. Under acidic conditions rhodamine yields strong fluorescence, but emission intensity decreases at highly basic pH values when the spirocyclic form is closed (Shen, Chen, Zhang, Miao, & Zhao, 2015). However, another study determined that FeRhoNox-1 is present as a non-fluorescent closed spirolactone only in highly basic ranges of pH >11.5 (Niwa, Hirayama, Okuda, & Nagasawa, 2014). Thus, our studies in mildly acidic to mildly basic pH conditions suggests that as pH increases, the decrease in FeRhoNox-1 MFI is dependent on Fe2+ to Fe3+ oxidation. Additionally, rhodamine B exists as a fluorescence opened quinoid structure independent of pH (Niwa et al., 2014). Thus, the addition of KOH in real-time decreases FeRhoNox-1 MFI yet does not oxygenate the tertiary amine on rhodamine B to generate an N-oxide on FeRhoNox-1. These data suggest the fluorescence rhodamine B complex is dependent on Fe3+/Fe2+ cycling for an on/off switch. Furthermore, enzymes in cell lysate can convert Fe3+ to Fe2+ (Ohgami et al., 2006), and the observed increase in FeRhoNox-1 MFI, following the KOH-induced decrease in MFI, is likely because of Fe2+-mediated deoxygenation of FeRhoNox-1. Correspondingly, after KOH-induced Fe2+-oxidation in the citrate buffer, the fluorescence signal remains low as the citrate buffer “locks-up” the Fe3+ in a Fe(III)-citrate complex not allowing Fe2+ cations to return to solution (Hamm, Shull, & Grant, 1954). Thus, these data suggest that FeRhoNox-1 in cell lysate can likely cycle between on and off fluorescence under physiologically relevant conditions.
Regarding intracellular localization of FeRhoNox-1, a pattern consistent with FeRhoNox-1-positive Golgi staining was reported in human hepatocellular carcinoma cells (HepG2) and human breast cancer cells (MCF-7) (Hirayama et al., 2013). However, no co-distribution studies were conducted with markers for endolysosomes. Here, we report that FeRhoNox-1-stained endolysosome-positive stores of Fe2+ with high correlation coefficients for colocalization with markers for endolysosomes (LysoTracker), early endosomes (EEA1), and late endosomes/lysosomes (LAMP1). Notably, LysoTracker favors acidic environments, thus, additional experiments with LAMP1 and EEA1 transfections were performed and similar colocalizations were observed independently of organelle pH. The correlations between LysoTracker and FeRhoNox-1 staining were virtually identical to those reported for a different endolysosome-specific Fe2+ probe (Hirayama, 2018). Furthermore, our data are consistent with others that LysoTracker staining colocalized with FeRhoNox-1 staining in human fibroblasts (Toyokuni, Ito, Yamashita, Okazaki, & Akatsuka, 2017), and depending on cell type 40 to 80% of the organelles stained FeRhoNox-1-positive were lysosomes (Ito et al., 2016). Even qualitatively, the FeRhoNox-1 staining observed was consistent with the punctate nature of endolysosomes and different from the wispy nature of Golgi staining.
FeRhoNox-1 MFI increased with FAC and decreased with DFO, but FAC did not change the relative extent of colocalization with LysoTracker-positive endolysosomes; DFO did change the relative extent of colocalization. DFO is endocytosed and was confirmed by us. FAC is endocytosed in a transferrin-dependent and -independent manner (Morgan, 1981; Richardson & Baker, 1992). According to our data, FeRhoNox-1 is not endocytosed/pinocytosed like DFO. However, FeRhoNox-1 has a high pKa value 11.4 and is attracted to the acidic nature of endolysosomes and likely becomes trapped inside endolysosomes as studies have shown (de Duve et al., 1974; Larsen, Escargueil, & Skladanowski, 2000; MacIntyre & Cutler, 1988; Martínez-Zaguilán et al., 1999; Simon, Roy, & Schindler, 1994). Furthermore, a study demonstrated weak basic compounds are sequestered into endolysosomes and not Golgi because of pH partitioning (i.e., cytosolic pH vs. endolysosome pH). In contrast, zwitterionic molecules are sequestered into Golgi and not endolysosomes through a drug transporter-mediated process. These two distinct mechanisms for intracellular compartmentalization occur simultaneously (Gong, Duvvuri, & Krise, 2003), and, at cytosolic levels of pH, FeRhoNox-1 is not a zwitterionic molecule (Hirayama et al., 2013). Interestingly, the developers of FeRhoNox-1 made a Golgi-specific Fe2+ dye, Gol-SiRhoNox, by modifying FeRhoNox-1 with a myristoyl motif (Hirayama et al., 2013; Hirayama et al., 2019; Ishida, Nayak, Mindell, & Grabe, 2013). All of these studies suggest FeRhoNox-1 unlikely stains Golgi and could explain why FeRhoNox-1 is localizing with endolysosomes. Qualitatively, the Golgi staining from the Gol-SiRhoNox and the zwitterionic studies look very similar to our Golgi staining (Gong et al., 2003; Hirayama et al., 2019).
Under normal conditions, [Fe2+]el in astrocytoma cells and primary neurons were determined to be 50.4 μM and 32.7 μM, respectively. We found one report of [Fe2+]el using an indirect measure of chelatable iron; in rat liver endothelial cells a subpopulation of endolysosomes contained 16 μM [Fe2+] (Petrat, Groot, & de, Rauen U., 2001). Semi-quantitative X-ray microanalysis showed iron concentrated in lysosomes and iron concentrations increased nine-fold in hepatocyte lysosomes from iron-loaded rats; the iron-loaded lysosomes were enlarged and misshapen (LeSage, Kost, Barham, & LaRusso, 1986; Myers, Prendergast, Holman, Kuntz, & LaRusso, 1991). Relative to other divalent cations in endolysosomes, our value for [Fe2+]el was eight times less than Ca2+; 400–600 μM (Christensen, Myers, & Swanson, 2002; Hui et al., 2015; Lakpa, Halcrow, Chen, & Geiger, 2020; Morgan, Platt, Lloyd-Evans, & Galione, 2011). Pre-loading cells with Fe2+ by FAC resulted in a 1.5-fold increase in [Fe2+]el, while pre-incubation with the Fe2+-chelating compound DFO decreased [Fe2+]el to 12.4 μM. In addition, cancer cells contain higher levels of [Fe2+] (Torti & Torti, 2013) in cytosol and mitochondria (Ito et al., 2016), and our data demonstrate that [Fe2+]el levels in U87MG astrocytoma cells are significantly higher than [Fe2+]el levels in primary neurons.
Lysosomes make up ~5% of the total intracellular volume, are heterogeneous structurally (ranging in diameter from 100 to 500 nm), are electron dense, and are trafficked dynamically in cells; features likely impacting their homeostatic regulatory actions including proteolysis (Araki, Yokota, Takashima, & Ogawa, 1995; Ballabio & Bonifacino, 2020; Berg, Gjoen, & Bakke, 1995; Cabukusta & Neefjes, 2018; Cuervo, Dice, & Knecht, 1997; Groh & von Mayersbach, 1981; Holtzman, 1989; Kelly, Waheed, Van Etten, & Chang, 1989; Lie & Nixon, 2019; Terman, Kurz, Gustafsson, & Brunk, 2006; Xu & Ren, 2015). Additionally, links between endolysosomes and disease pathogenesis raise the possibility that heterogeneous populations of endolysosomes are selectively linked to specific diseases (Kett & Dauer, 2016).
Endolysosome positioning is important for their ability to sense and respond to nutrients, growth factors, and cellular stress (Lie & Nixon, 2019). Perinuclear endolysosomes are more acidic and comprise ~80% of the total number of endolysosomes in cells, whereas, when located near plasma membranes the pH values are higher (Johnson, Ostrowski, Jaumouille, & Grinstein, 2016). Clearly, pH values differ depending on the subcellular compartmentalization of endolysosomes, and endolysosome trafficking is altered by changes in endolysosome pH. This endolysosome trafficking is regulated by microtubule-mediated bidirectional movement controlled by GTPase binding to kinesin and dynesin motors (Ballabio & Bonifacino, 2020; Gowrishankar & Ferguson, 2016). Bidirectional movement of endolysosomes occurs in a “stop-and-go” fashion; a process that may occur because of organelle-to-organelle interactions or the motor proteins become exhausted (Cabukusta & Neefjes, 2018).
Here, we found that endolysosome subpopulations exist and are differentiated based on their Fe2+ content. While it is unclear why the Fe2+ content is variable, one possibility is the differences in endolysosome pH depending on their intracellular positions and that endolysosome de-acidification causes Fe2+ efflux from endolysosomes (Halcrow, Lakpa, et al., 2021a; Johnson et al., 2016; Uchiyama et al., 2008). Our findings demonstrating [Fe2+]el was highest near the nucleus is consistent with findings that lipid ROS production was highest perinuclearly and this generated ROS participates in ferroptosis (Torii et al., 2016). Additionally, [Fe2+] might be highest in perinuclear lysosomes where autophagy of iron-rich compounds has recently occurred (Kurz et al., 2011). Also, perinuclear regions are where autophagosome–lysosome fusion occurs primarily (Xu & Ren, 2015) and where endolysosomes are mainly localized intracellularly (Li et al., 2016). We found that DFO and FAC change the pH at a similar time point that they change [Fe2+]el. Increasing iron with FAC and decreasing iron with DFO altered the size, numbers, and intracellular distribution of endolysosomes. When DFO chelates iron it releases protons which would explain the change in endolysosome pH becoming more acidic, and this acidic nature could cause the endolysosomes to move closer to the nucleus. Whereas FAC de-acidifies endolysosomes, is reduced to Fe2+, and shifts the endolysosomes on average 2.63 μm away from the nucleus, which is consistent with previous studies demonstrating that a higher luminal pH of endolysosomes is closer to the plasma membranes (Chen et al., 2013; Fleming et al., 1998; Gunshin et al., 1997; Johnson et al., 2016; Ohgami et al., 2005). Also, because of how FAC is taken up by cells, the FeRHoNox-1 intensity increases are likely easier to see near the plasma membrane. Indeed, endolysosome iron homeostasis is necessary for balancing endolysosome acidification, endolysosome movement, biogenesis, and membrane trafficking (Du et al., 2021; Lin, Epstein, & Liton, 2010; Rizzollo et al., 2021). Alternatively, [Fe2+]el might be lower in “resting” non-perinuclearly localized lysosomes that had not recently participated in autophagy of iron-rich compounds (Kurz, Terman, Gustafsson, & Brunk, 2008).
Together, our findings and the findings of others suggest strongly that FeRhoNox-1 is a useful probe to detect relative and absolute levels of endolysosome Fe2+, and with this probe, we and others can investigate further the physiological and pathological roles of this releasable and biologically active store of Fe2+.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences under award numbers P30GM100329 and U54GM115458, the National Institute of Mental Health under award numbers R01MH100972, R01MH105329 and R01MH119000, the National Institute of Neurological Diseases and Stroke under award number 2R01NS065957, and the National Institute of Drug Abuse under award number 2R01DA032444.
All experiments were conducted in compliance with the ARRIVE guidelines.
Abbreviations:
- [Fe2+]el
endolysosome concentration of Fe2+
- DAPI
4′,6-diamidino-2-phenylindole
- DFO
deferoxamine
- DMEM
Dulbecco’s Modified Eagle Medium
- FAC
ferric ammonium citrate
- Fe2+
ferrous iron
- Fe3+
ferric iron
- FeCl2
iron(II) chloride tetrahydrate
- FeCl3
iron(III) chloride
- HepG2
hepatocellular carcinoma cells
- KOH
potassium hydroxide
- MCF-7
Michigan Cancer Foundation-7 human breast carcinoma cells
- MES
2-(N-morpholino) ethane sulfonic acid
- MFI
mean fluorescence intensity
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- STEAP3
six-transmembrane epithelial antigen of prostate 3
Footnotes
CONFLICT OF INTEREST
The authors declare that this manuscript was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
Data available on request from the authors
REFERENCES
- Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, & Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase-and mTOR-dependent regulation of amino acid efflux from lysosomes. Science, 358, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelmans F, Wattiaux R, & De Duve C (1955). Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochemical Journal, 59, 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki N, Yokota S, Takashima Y, & Ogawa K (1995). The distribution of cathepsin D in two types of lysosomal or endosomal profiles of rat hepatocytes as revealed by combined immunocytochemistry and acid phosphatase enzyme cytochemistry. Experimental Cell Research, 217, 469–476. [DOI] [PubMed] [Google Scholar]
- Bainton DF (1981). The discovery of lysosomes. Journal of Cell Biology, 91, 66s–76s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio A, & Bonifacino JS (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nature Reviews Molecular Cell Biology, 21, 101–118. [DOI] [PubMed] [Google Scholar]
- Berg T, Gjoen T, & Bakke O (1995). Physiological functions of endosomal proteolysis. The Biochemical Journal, 307(Pt 2), 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann M, Schütt F, Holz FG, & Kopitz J (2004). Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. The FASEB Journal, 18, 562–564. [DOI] [PubMed] [Google Scholar]
- Bratsch SG (1989). Standard electrode potentials and temperature coefficients in water at 298.15 K. Journal of Physical and Chemical Reference Data, 18, 1–21. [Google Scholar]
- Cabukusta B, & Neefjes J (2018). Mechanisms of lysosomal positioning and movement. Traffic, 19, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Garcia-Santos D, Ishikawa Y, Seguin A, Li L, Fegan KH, Hildick-Smith GJ, Shah DI, Cooney JD, Chen W, King MJ, Yien YY, Schultz IJ, Anderson H, Dalton AJ, Freedman ML, Kingsley PD, Palis J, Hattangadi SM, … Paw BH (2013). Snx3 regulates recycling of the transferrin receptor and iron assimilation. Cell Metabolism, 17, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KA, Myers JT, & Swanson JA (2002). pH-dependent regulation of lysosomal calcium in macrophages. Journal of Cell Science, 115, 599–607. [DOI] [PubMed] [Google Scholar]
- Colacurcio DJ, & Nixon RA (2016). Disorders of lysosomal acidification-the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Research Reviews, 32, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF, & Knecht E (1997). A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. The Journal of Biological Chemistry, 272, 5606–5615. [DOI] [PubMed] [Google Scholar]
- De Duve C, & Wattiaux R (1966). Functions of lysosomes. Annual Review of Physiology, 28, 435–492. [DOI] [PubMed] [Google Scholar]
- Du W, Gu M, Hu M, Pinchi P, Chen W, Ryan M, Nold T, Bannaga A, & Xu H (2021). Lysosomal Zn2+ release triggers rapid, mitochondria-mediated, non-apoptotic cell death in metastatic melanoma. Cell Reports, 37, 109848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, & Van Hoof F (1974). Commentary. Lysosomotropic agents. Biochemical Pharmacology, 23, 2495–2531. [DOI] [PubMed] [Google Scholar]
- Fenton HJH (1894). LXXIII.—Oxidation of tartaric acid in presence of iron. Journal of the Chemical Society, Transactions, 65, 899–910. [Google Scholar]
- Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, & Andrews NC (1998). Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proceedings of the National Academy of Sciences, 95, 1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Duvvuri M, & Krise JP (2003). Separate roles for the Golgi apparatus and lysosomes in the sequestration of drugs in the multidrug-resistant human leukemic cell line HL-60*. Journal of Biological Chemistry, 278, 50234–50239. [DOI] [PubMed] [Google Scholar]
- Gowrishankar S, & Ferguson SM (2016). Lysosomes relax in the cellular suburbs. The Journal of Cell Biology, 212, 617–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh V, & von Mayersbach H (1981). Enzymatic and functional heterogeneity of lysosomes. Cell and Tissue Research, 214, 613–621. [DOI] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, & Hediger MA (1997). Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature, 388, 482–488. [DOI] [PubMed] [Google Scholar]
- Halcrow PW, Lakpa KL, Khan N, Afghah Z, Miller N, Datta G, Chen X, & Geiger JD (2021a). HIV-1 gp120-induced Endolysosome de-acidification leads to efflux of Endolysosome iron, and increases in mitochondrial iron and reactive oxygen species. Journal of Neuroimmune Pharmacology. 10.1007/s11481-021-09995-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halcrow PW, Lynch ML, Geiger JD, & Ohm JE (2021b). Role of endolysosome function in iron metabolism and brain carcinogenesis. Seminars in Cancer Biology, 76, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RE, Shull CM, & Grant DM (1954). Citrate complexes with iron(II) and iron(III)1. Journal of the American Chemical Society, 76, 2111–2114. [Google Scholar]
- Hill JM (1985). Iron concentration reduced in ventral pallidum, globus pallidus, and substantia nigra by GABA-transaminase inhibitor, gamma-vinyl GABA. Brain Research, 342, 18–25. [DOI] [PubMed] [Google Scholar]
- Hirayama T (2018). Development of chemical tools for imaging of Fe(II) ions in living cells: A review. Acta Histochemica et Cytochemica, 51, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama T, Miki A, & Nagasawa H (2019). Organelle-specific analysis of labile Fe(ii) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics, 11, 111–117. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Okuda K, & Nagasawa H (2013). A highly selective turn-on fluorescent probe for iron(II) to visualize labile iron in living cells. Chemical Science, 4, 1250–1256. [Google Scholar]
- Hirayama T, Tsuboi H, Niwa M, Miki A, Kadota S, Ikeshita Y, Okuda K, & Nagasawa H (2017). A universal fluorogenic switch for Fe(ii) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chemical Science, 8, 4858–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman E (1989). Lysosomes. Springer Science & Business Media. [Google Scholar]
- Hughes CE, Coody TK, Jeong M-Y, Berg JA, Winge DR, & Hughes AL (2020). Cysteine toxicity drives age-related mitochondrial decline by altering iron homeostasis. Cell, 180, 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Chen X, & Geiger JD (2012a). Endolysosome involvement in LDL cholesterol-induced Alzheimer’s disease-like pathology in primary cultured neurons. Life Sciences, 91, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Chen X, Haughey NJ, & Geiger JD (2012b). Role of endolysosomes in HIV-1 tat-induced neurotoxicity. ASN Neuro, 4, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Geiger NH, Bloor-Young D, Churchill GC, Geiger JD, & Chen X (2015). Release of calcium from endolysosomes increases calcium influx through N-type calcium channels: Evidence for acidic store-operated calcium entry in neurons. Cell Calcium, 58, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Nayak S, Mindell JA, & Grabe M (2013). A model of lysosomal pH regulation. The Journal of General Physiology, 141, 705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito F, Nishiyama T, Shi L, Mori M, Hirayama T, Nagasawa H, Yasui H, & Toyokuni S (2016). Contrasting intra- and extracellular distribution of catalytic ferrous iron in ovalbumin-induced peritonitis. Biochemical and Biophysical Research Communications, 476, 600–606. [DOI] [PubMed] [Google Scholar]
- Jiang L, Zheng H, Lyu Q, Hayashi S, Sato K, Sekido Y, Nakamura K, Tanaka H, Ishikawa K, Kajiyama H, Mizuno M, Hori M, & Toyokuni S (2021). Lysosomal nitric oxide determines transition from autophagy to ferroptosis after exposure to plasma-activated Ringer’s lactate. Redox Biology, 43, 101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DE, Ostrowski P, Jaumouille V, & Grinstein S (2016). The position of lysosomes within the cell determines their luminal pH. The Journal of Cell Biology, 212, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, & Ward DM (2013). The essential nature of iron usage and regulation. Current Biology, 23, R642–R646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BM, Waheed A, Van Etten R, & Chang PL (1989). Heterogeneity of lysosomes in human fibroblasts. Molecular and Cellular Biochemistry, 87, 171–183. [DOI] [PubMed] [Google Scholar]
- Kett LR, & Dauer WT (2016). Endolysosomal dysfunction in Parkinson’s disease: Recent developments and future challenges. Movement Disorders, 31, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempner MS, & Styrt B (1983). Alkalinizing the intralysosomal pH inhibits degranulation of human neutrophils. The Journal of Clinical Investigation, 72, 1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J-Y, Kim HN, Hwang JJ, Kim Y-H, & Park SE (2019). Lysosomal dysfunction in proteinopathic neurodegenerative disorders: Possible therapeutic roles of cAMP and zinc. Molecular Brain, 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Eaton JW, & Brunk UT (2011). The role of lysosomes in iron metabolism and recycling. The International Journal of Biochemistry & Cell Biology, 43, 1686–1697. [DOI] [PubMed] [Google Scholar]
- Kurz T, Terman A, Gustafsson B, & Brunk UT (2008). Lysosomes in iron metabolism, ageing and apoptosis. Histochemistry and Cell Biology, 129, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakpa KL, Halcrow PW, Chen X, & Geiger JD (2020). Readily releasable Stores of Calcium in neuronal Endolysosomes: Physiological and pathophysiological relevance. Advances in Experimental Medicine and Biology, 1131, 681–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen AK, Escargueil AE, & Skladanowski A (2000). Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacology & Therapeutics, 85, 217–229. [DOI] [PubMed] [Google Scholar]
- LeSage GD, Kost LJ, Barham SS, & LaRusso NF (1986). Biliary excretion of iron from hepatocyte lysosomes in the rat. A major excretory pathway in experimental iron overload. The Journal of Clinical Investigation, 77, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W, Gao Q, Cheng X, & Xu H (2016). A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nature Cell Biology, 18, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie PPY, & Nixon RA (2019). Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiology of Disease, 122, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Epstein DL, & Liton PB (2010). Intralysosomal iron induces lysosomal membrane permeabilization and cathepsin D-mediated cell death in trabecular meshwork cells exposed to oxidative stress. Investigative Ophthalmology & Visual Science, 51, 6483–6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntyre AC, & Cutler DJ (1988). The potential role of lysosomes in tissue distribution of weak bases. Biopharmaceutics & Drug Disposition, 9, 513–526. [DOI] [PubMed] [Google Scholar]
- Martínez-Zaguilán R, Raghunand N, Lynch RM, Bellamy W, Martinez GM, Rojas B, Smith D, Dalton WS, & Gillies RJ (1999). pH and drug resistance. I. Functional expression of plasma-lemmal V-type H+-ATPase in drug-resistant human breast carcinoma cell lines. Biochemical Pharmacology, 57, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Mindell JA (2012). Lysosomal acidification mechanisms. Annual Review of Physiology, 74, 69–86. [DOI] [PubMed] [Google Scholar]
- Moens J, Roos G, Jaque P, De Proft F, & Geerlings P (2007). Can electrophilicity act as a measure of the redox potential of first-row transition metal ions? Chemistry –A European Journal, 13, 9331–9343. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Platt FM, Lloyd-Evans E, & Galione A (2011). Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. The Biochemical Journal, 439, 349–374. [DOI] [PubMed] [Google Scholar]
- Morgan EH (1981). Transferrin, biochemistry, physiology and clinical significance. Molecular Aspects of Medicine, 4, 1–123. [Google Scholar]
- Mukaide T, Hattori Y, Misawa N, Funahashi S, Jiang L, Hirayama T, Nagasawa H, & Toyokuni S (2014). Histological detection of catalytic ferrous iron with the selective turn-on fluorescent probe RhoNox-1 in a Fenton reaction-based rat renal carcinogenesis model. Free Radical Research, 48, 990–995. [DOI] [PubMed] [Google Scholar]
- Myers BM, Prendergast FG, Holman R, Kuntz SM, & LaRusso NF (1991). Alterations in the structure, physicochemical properties, and pH of hepatocyte lysosomes in experimental iron overload. The Journal of Clinical Investigation, 88, 1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash B, Tarn K, Irollo E, Luchetta J, Festa L, Halcrow P, Datta G, Geiger JD, Meucci O (2019) Morphine-induced modulation of Endolysosomal iron mediates upregulation of ferritin heavy chain in cortical neurons. eNeuro 6, 6, ENEU19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Hirayama T, Okuda K, & Nagasawa H (2014). A new class of high-contrast Fe(II) selective fluorescent probes based on spirocyclized scaffolds for visualization of intracellular labile iron delivered by transferrin. Organic & Biomolecular Chemistry, 12, 6590–6597. [DOI] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, & Fleming MD (2005). Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nature Genetics, 37, 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, McDonald A, & Fleming MD (2006). The Steap proteins are metalloreductases. Blood, 108, 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan H, Brookes MJ, & Iqbal T (2015). Iron and colorectal cancer: Evidence from in vitro and animal studies. Nutrition Reviews, 73, 308–317. [DOI] [PubMed] [Google Scholar]
- Perera RM, & Zoncu R (2016). The lysosome as a regulatory hub. Annual Review of Cell and Developmental Biology, 32, 223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrat F, Groot H, & de Rauen U (2001). Subcellular distribution of chelatable iron: A laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. The Biochemical Journal, 356, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D, & Baker E (1992). Two mechanisms of iron uptake from transferrin by melanoma cells. The effect of desferrioxamine and ferric ammonium citrate. Journal of Biological Chemistry, 267, 13972–13979. [PubMed] [Google Scholar]
- Rizzollo F, More S, Vangheluwe P, & Agostinis P (2021). The lysosome as a master regulator of iron metabolism. Trends in Biochemical Sciences, 46, 960–975. [DOI] [PubMed] [Google Scholar]
- Rockfield S, Raffel J, Mehta R, Rehman N, & Nanjundan M (2017). Iron overload and altered iron metabolism in ovarian cancer. Biological Chemistry, 398, 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, & Ballabio A (2013). Signals from the lysosome: A control Centre for cellular clearance and energy metabolism. Nature Reviews. Molecular Cell Biology, 14, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S-L, Chen X-P, Zhang X-F, Miao J-Y, & Zhao B-X (2015). A rhodamine B-based lysosomal pH probe. Journal of Materials Chemistry B, 3, 919–925. [DOI] [PubMed] [Google Scholar]
- Simon S, Roy D, & Schindler M (1994). Intracellular pH and the control of multidrug resistance. PNAS, 91, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, Kurz T, Gustafsson B, & Brunk UT (2006). Lysosomal labilization. IUBMB Life, 58, 531–539. [DOI] [PubMed] [Google Scholar]
- Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, & Yamada K (2016). An essential role for functional lysosomes in ferroptosis of cancer cells. The Biochemical Journal, 473, 769–777. [DOI] [PubMed] [Google Scholar]
- Torti SV, & Torti FM (2013). Iron and cancer: More ore to be mined. Nature Reviews. Cancer, 13, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S, Ito F, Yamashita K, Okazaki Y, & Akatsuka S (2017). Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radical Biology & Medicine, 108, 610–626. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Sumiya T, Tachikawa M, Yamakawa T, Murata S, Yagi Y, Sato K, Stephan A, Ito K, Ohtsuki S, Couraud PO, Suzuki T, & Terasaki T (2019). Involvement of Claudin-11 in disruption of blood-brain, -spinal cord, and -arachnoid barriers in multiple sclerosis. Molecular Neurobiology, 56, 2039–2056. [DOI] [PubMed] [Google Scholar]
- Uchiyama A, Kim JS, Kon K, Jaeschke H, Ikejima K, Watanabe S, & Lemasters JJ (2008). Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology, 48, 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber RA, Yen FS, Nicholson SPV, Alwaseem H, Bayraktar EC, Alam M, Timson RC, La K, Abu-Remaileh M, Molina H, & Birsoy K (2020). Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Molecular Cell, 77, 645–655. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, & Ren D (2015). Lysosomal physiology. Annual Review of Physiology, 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request from the authors