

SUMMARY
Williams-Beuren syndrome (WBS) is a rare disorder caused by hemizygous microdeletion of ~27 contiguous genes. Despite neurodevelopmental and cognitive deficits, individuals with WBS have spared or enhanced musical and auditory abilities, potentially offering insight into the genetic basis of auditory perception. Here we report that mouse models of WBS have innately enhanced frequency-discrimination acuity and improved frequency coding in the auditory cortex (ACx). Chemogenetic rescue showed frequency-discrimination hyperacuity is caused by hyperexcitable interneurons in ACx. Haploinsufficiency of one WBS gene, Gtf2ird1, replicated WBS phenotypes by downregulating the neuropeptide receptor VIPR1. VIPR1 is reduced in ACx of individuals with WBS and in cerebral organoids derived from human induced pluripotent stem cells with the WBS microdeletion. Vipr1 deletion or overexpression in ACx interneurons mimicked or reversed, respectively, the cellular and behavioral phenotypes of WBS mice. Thus, the Gtf2ird1–Vipr1 mechanism in ACx interneurons may underlie the superior auditory acuity in WBS.
Keywords: Williams-Beuren syndrome, frequency-discrimination acuity, auditory cortex, inhibitory interneurons, Gtf2ird1, VIPR1
In Brief:
Williams-Beuren syndrome is a neurodevelopmental disorder that is also associated with spared or superior auditory abilities. This condition is caused by a down- regulation of the neuropeptide receptor VIPR1, driven by Gtf2ird1 haploinsufficiency.
Graphical Abstract
INTRODUCTION
The ability to distinguish acoustic frequencies from each other or from the surrounding auditory scene has been essential for survival throughout evolution, and in humans remains fundamental to everyday hearing, linguistics, and musicality (Feng and Ratnam, 2000; Gervain and Geffen, 2019; Peretz, 2016; Stewart, 2008). Musical training and ability are associated with superior frequency discrimination (Micheyl et al., 2006; Spiegel and Watson, 1984). Conversely, poor frequency discrimination may impair language abilities (Kleindienst and Musiek, 2011; Mengler et al., 2005). Yet the neural and genetic mechanisms underlying frequency discrimination are not well understood. To better understand frequency-discrimination acuity, we turned to Williams-Beuren syndrome (WBS).
WBS is a neurodevelopmental disorder usually caused by a 1.55- to 1.83-Mb hemizygous microdeletion containing 25–27 contiguous genes in chromosomal locus 7q11.23 (Bayés et al., 2003; Kozel et al., 2021; Meyer-Lindenberg et al., 2006; Schubert, 2009). Music and language abilities of persons with WBS are preserved or enhanced, despite developmental delays, intellectual disability (average IQ <70), and other cognitive and learning deficits (Bellugi et al., 2000; Mervis et al., 2000; Morris and Braddock, 2020). WBS is associated with emotional response to certain sounds, particularly music (Levitin et al., 2004; Thakur et al., 2018); love of/interest in music is noted in the earliest descriptions of WBS (von Arnim et al., 1964). Enhanced musicality, language skills, and auditory acuity are seen in WBS (Bellugi et al., 2000; Don et al., 1999; Lenhoff, 1998, 2006; Levitin et al., 2004; Udwin and Yule, 1990), and even prevalent “absolute pitch” in some musically trained patients with WBS (Lenhoff, 2006; Lenhoff et al., 2001) [Note, absolute pitch requires training in early childhood; therefore, it may be no more prevalent in the WBS population than in healthy subjects (Martínez-Castilla et al., 2013; Pober, 2010)].
Humans with WBS have atypical neuroanatomy related to auditory processing. Frequency discrimination (Kumar et al., 2019; Tramo et al., 2005) and perception of music and speech (Brauchli et al., 2019; Stewart, 2008) are partially attributed to the auditory cortex (ACx). Despite lower overall cortical volume (Reiss et al., 2000), ACx is spared or increased in WBS (Holinger et al., 2005; Martens et al., 2010). Those with WBS also have elevated auditory-evoked potentials (Zarchi et al., 2015) and atypical activation of cortical areas by sound (Levitin et al., 2003; Thornton-Wells et al., 2010), suggesting that ACx abnormalities underlie WBS hyperacuity or other auditory enhancements in WBS.
Preclinical studies also support ACx involvement in frequency discrimination (Aizenberg and Geffen, 2013; Dykstra et al., 2012; Talwar and Gerstein, 2001; Tramo et al., 2002; but see Gimenez et al., 2015; Ohl et al., 1999). Optogenetic activation or inhibition of parvalbumin-positive (PV+) GABAergic interneurons in the ACx improved or worsened, respectively, behavioral performance, depending on frequency discrimination (Aizenberg et al., 2015).
Here we sought to examine the mechanistic underpinnings of frequency-discrimination capability in mouse models of WBS (WBS mice) carrying a hemizygous microdeletion spanning Fkbp6–Gtf2i equivalent to the human 1.55-Mb WBS locus (Osborne, 2010; Segura-Puimedon et al., 2014; Valero et al., 2000).
RESULTS
WBS mice have enhanced innate frequency discrimination
We compared innate frequency-discrimination abilities in 6– to 12–week-old WBS models (CD+/− mice) (Segura-Puimedon et al., 2014) (Figure 1A) to wild-type (WT) mice using a pre-pulse inhibition (PPI)–based test of the auditory startle response (ASR) (Figure 1B; Aizenberg and Geffen, 2013; Aizenberg et al., 2015; Clause et al., 2011). PPI of the ASR is proportional to the frequency difference between background and pre-pulse tones; greater PPI indicates more robust frequency discrimination. In both genotypes, larger frequency shifts caused more PPI, but CD+/− mice exhibited greater PPI at pre-pulse frequencies closer to background (Figure 1C). We quantified frequency-discrimination threshold (FDT) using 16.4-kHz background frequency in both genotypes as a measure of frequency-discrimination. FDT in CD+/− mice was ~50% lower than that in WT mice (Figure 1D). Male and female CD+/− mice were equally affected (two-way ANOVA, Pgenotype = 0.02, Psex = 0.8). Frequency-discrimination hyperacuity (auditory hyperacuity) was not unique to 16.4-kHz background; discrimination was also enhanced at 9.8-kHz background (Figure S1A, B). Thus, WBS mice discriminate acoustic frequencies better than WT mice.
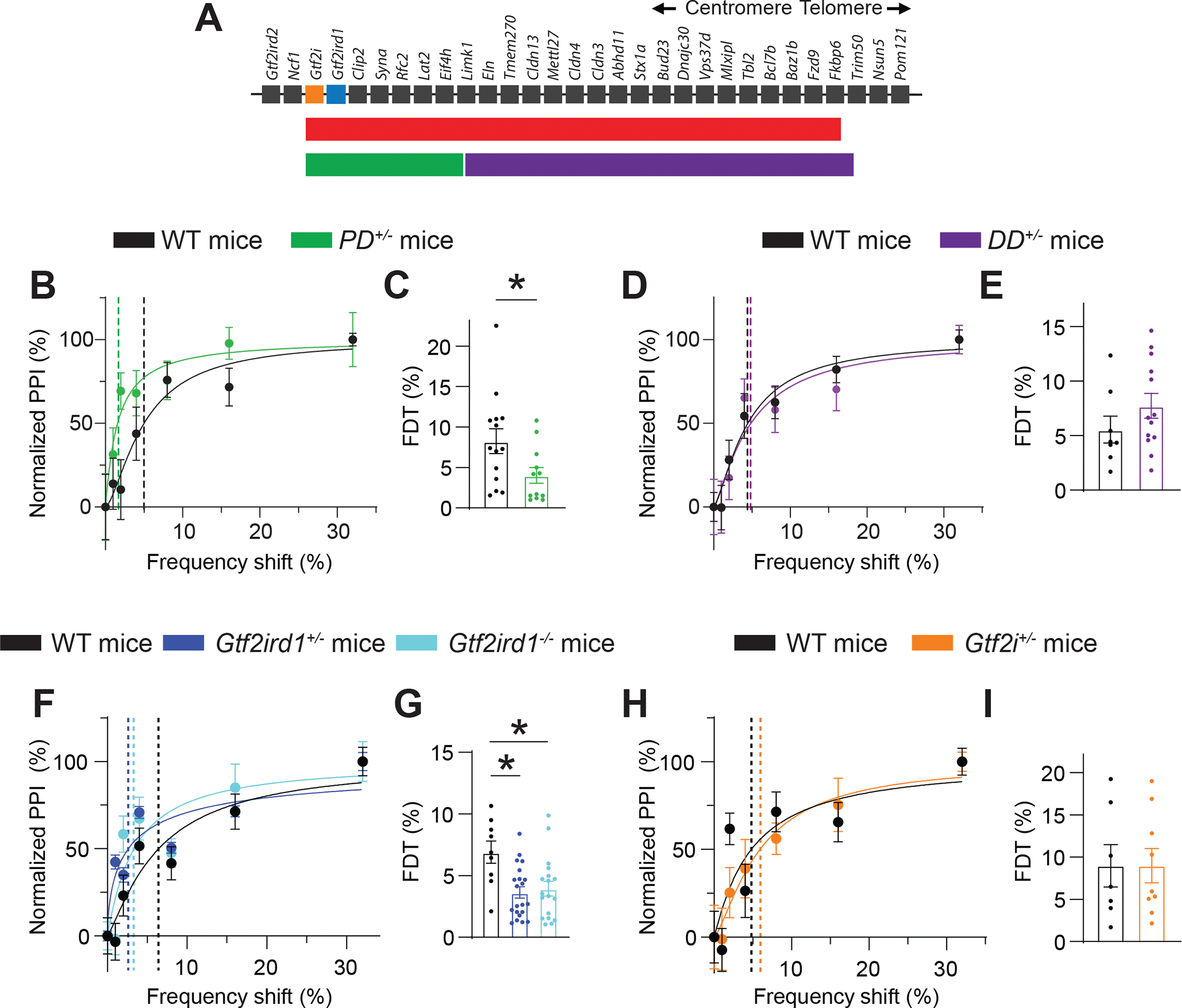
Figure 1: WBS mice have frequency-discrimination (auditory) hyperacuity and excess cortical inhibition from hyperexcitable FS interneurons.
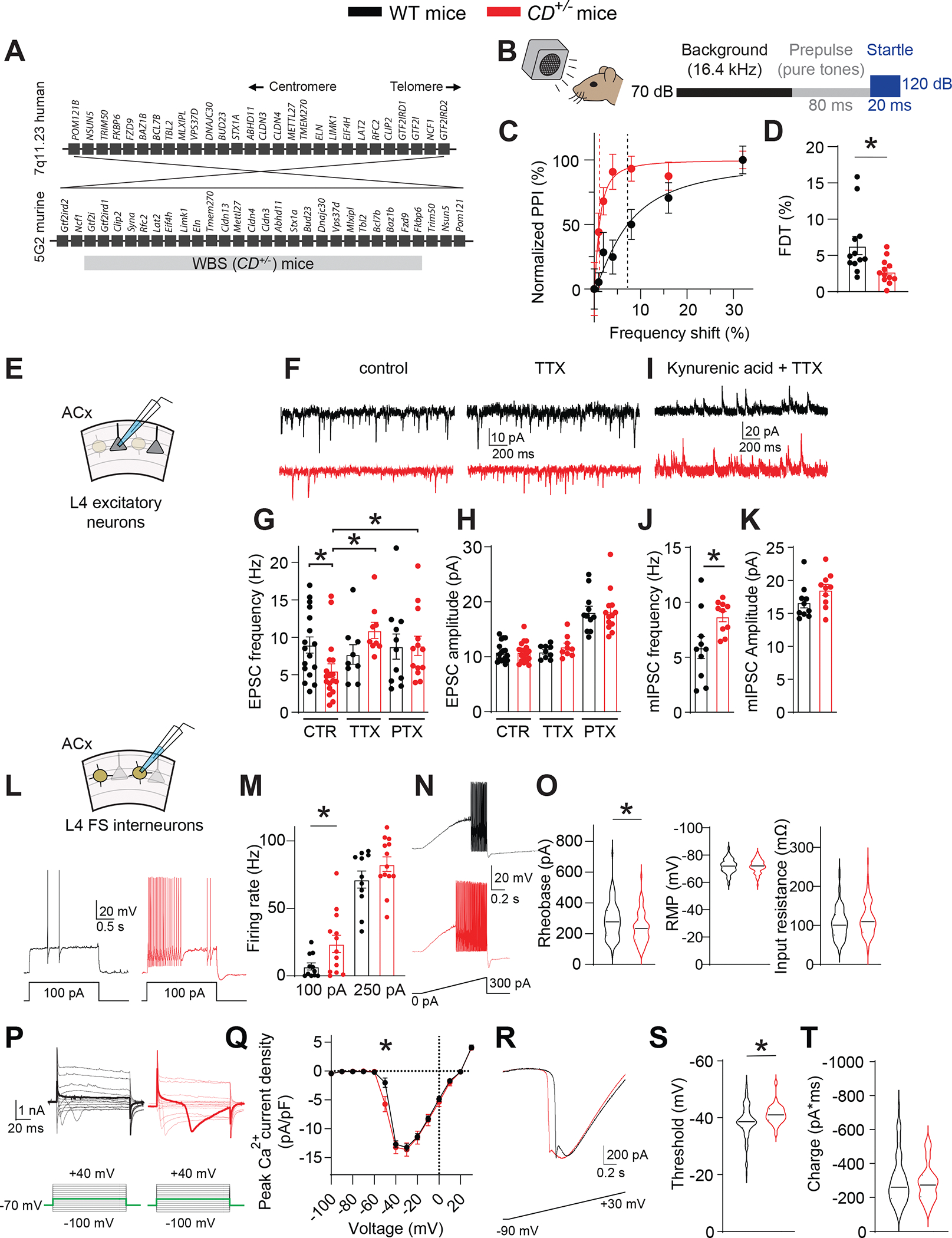
A. The 7q11.23 locus in humans and the syntenic region of mouse chromosome 5. Also shown is the WBS (CD+/−) mouse with hemizygous microdeletion spanning the syntenic Gtf2i–Fkbp6 region.
B. Scheme for testing frequency discrimination. A background tone (16.4 kHz, 70 dB) is present during the experiment. Variable frequency pre-pulse tones (80 ms, 70 dB) are presented before the startle stimulus (white noise, 20 ms, 120 dB).
C. Recordings from single WT and CD+/− mice of normalized PPI magnitude as a function of frequency difference between background and pre-pulse tones. Points and error bars are mean ± SEM of 10 repeated measurements in the same animal. Solid lines are linear-regression fits to these points; dotted lines are frequency-discrimination thresholds (FDTs).
D. The FDT is smaller in CD+/− (n=12) vs WT (n=12) mice. Rank sum test, *P = 0.006.
E. Scheme of whole-cell voltage-clamp recording from L4 excitatory (pyramidal) neurons in the ACx.
F. Traces showing sEPSCs in WT and CD+/− neurons in control ACSF and in the presence of 0.5 μM TTX.
G. EPSC frequency in WT and CD+/− mice. In control ACSF (CTR), EPSC frequency is lower in CD+/− (n=18) vs WT (n=17) cells. Rank sum test, *P = 0.014. TTX (0.5 μM) and PTX (100 μM) reverse the decreased EPSC frequency in CD+/− neurons. ANOVA on ranks: control vs TTX, *P <0.001; control vs PTX, *P = 0.048. In TTX, EPSC frequency is the same in WT (n=14) vs CD+/− (n=12) mice. In PTX, EPSC frequency is the same in WT (n=11) vs CD+/− (n=13) mice.
H. EPSC amplitude does not differ between WT and CD+/− mice (same n as above) in control, TTX, or PTX.
I. Traces showing mIPSCs in WT and CD+/− mice in the presence of 3 mM kynurenic acid and 0.5 μM TTX.
J, K. Comparison of mIPSC frequency (J) and amplitude (K) in WT and CD+/− cells. mIPSC frequency is higher in CD+/− (n=10) vs WT (n=10) cells; t-test, *P = 0.019. mIPSC amplitude did not differ in CD+/− vs WT cells (same n as above).
L. (Top) Scheme of whole-cell current-clamp recording from L4 FS inhibitory interneurons in the ACx. Traces of interneuron voltage in response to a 100-pA depolarizing current step in WT and CD+/− mice.
M. AP firing rate is higher after a 100-pA step in CD+/− (n=13) vs WT (n=11) cells. Rank sum test, *P = 0.039. However, the AP firing rate is the same after a 250-pA step.
N. FS interneuron AP firing after a current ramp (0–300 pA, 1 s) in WT and CD+/− mice.
O. Rheobase (left) is lower in CD+/− (n=92) vs WT (n=72) cells. Rank sum test, *P = 0.007. Input resistance (right)and resting RMP (middle) do not differ between WT and CD+/− cells.
P. (Left) Scheme of whole-cell voltage-clamp recording from L4 FS interneurons in the ACx. (Right) Current responses to depolarizing voltage steps from −90 mV in the presence of 0.5 μM TTX, internal Cs+, and 3 mM Ba2+ in WT and CD+/− interneurons. Inward current is present at lower voltage steps (lower threshold) in CD+/− mice. Bold traces (upper), representative responses at −50 mV; (lower), current steps to −50 mV.
Q. Current–voltage relation of inward current density in WT (n=21) and CD+/− (n=20) cells. Inward voltage-gated current was larger in CD+/− vs WT interneurons after a step to −50 mV. Rank sum test, *P = 0.006.
R. Traces of current after a voltage ramp (−90 mV to +30 mV, 1s) in WT and CD+/− interneurons.
S. Inward voltage-gated current threshold is lower (more hyperpolarized) in CD+/− (n=30) vs WT (n=71) interneurons. Rank sum test, *P = 0.008.
T. Total integrated inward current (charge) does not differ between CD+/− (n=30) and WT (n=71) FS interneurons.
Data are presented as the mean ± SEM with individual measurements overlaid as dots. When n >50, the data are presented as violin plots.
When we attempted to assess frequency discrimination with an alternative auditory-cued Go/No-go task that required mice learn to pair a tone and reward and discern it from nonrewarded tones, only 1 of 5 CD+/− mice learned the task itself after 2 weeks of training, compared to 7 or 12 WT mice (Figure S1C), consistent with known learning deficits in WBS mice (Li et al., 2009; Zhao et al., 2005). Even if some CD+/− mice proceeded to the auditory-discrimination task, the contribution of learning vs auditory acuity would be equivocal.
ASR alone did not differ between WT and CD+/− mice (Figure S1D), suggesting that auditory hyperacuity was not caused by altered startle reflex. The auditory brainstem response (ABR) (Figure S1E), which measures initial sound processing (e.g., cochlear transduction, brainstem nuclei responsiveness), did not differ between genotypes. Thus, CD+/− mice have intact peripheral hearing, suggesting that their auditory hyperacuity arises in the central auditory system.
Increased cortical inhibition in the ACx of WBS mice
Synaptic interactions in the ACx affect frequency-discrimination acuity in mice (Aizenberg et al., 2015). We examined cellular and circuit properties of neurons in the ACx, the ventral division of the medial geniculate (MGv, auditory thalamus), and connections between the two in acute brain slices from WT and CD+/− mice.
Spontaneous excitatory synaptic currents (sEPSCs) in Layer (L) 4 (thalamorecipient) excitatory neurons in the ACx (Richardson et al., 2009; Smith and Populin, 2001) were significantly less frequent in CD+/− mice compared to WT mice (Figure 1E–G). sEPSC frequency was comparable in MGv excitatory (relay) neurons, from each genotype (Figure S1F). The sEPSC amplitude was preserved in CD+/− mice in both regions (Figures 1H, S1F). Reduced sEPSC frequency in the CD+/− ACx was not caused by reduced thalamic input. Postsynaptic currents in L4 cortical excitatory neurons elicited by stimulating ascending thalamocortical axons did not differ in amplitude or paired-pulse ratio between WT and CD+/− mice (Figure S1G–J). It was also not caused by altered intrinsic properties of L4 cortical excitatory neurons (Figure S2A–I), or of excitatory (relay) neurons in the MGv (Figure S2J–Q). These results imply abnormality in the ACx local synaptic circuitry.
Reduced sEPSC frequency in the CD+/− ACx was caused by increased cortical inhibition. The difference in sEPSC frequency recorded from ACx excitatory neurons was abolished by the GABAA receptor antagonist picrotoxin (PTX; Figure 1G) or voltage-gated Na+-channel blocker tetrodotoxin (TTX; Figure 1F, G). The EPSC amplitude was not different between genotypes under any condition (Figure 1H), indicating that the postsynaptic glutamate receptors at excitatory synapses are unchanged. Thus, decreased sEPSC frequency in CD+/− mice was not a property of presynaptic glutamatergic inputs but rather a consequence of increased inhibition in the ACx circuit.
Consistent with this notion, the frequency of miniature inhibitory postsynaptic currents (mIPSCs) in excitatory neurons in the CD+/− ACx was higher (Figure 1I, J), but their amplitude did not differ (Figure 1K). Thus, in WBS mice, cortical excitatory neurons receive similar direct excitatory inputs but stronger inhibitory inputs, resulting in reduced spontaneous excitatory synaptic activity in the ACx.
Hyperexcitability of inhibitory interneurons in the WBS ACx
To identify the source of elevated inhibition in the ACx of WBS mice, we recorded from L4 fast-spiking (FS) interneurons in auditory TC slices; most FS interneurons are PV+ and comprise the major subclass of cortical interneurons (Scala et al., 2019; Tremblay et al., 2016). In response to current injection, FS interneurons fired more action potentials (Aps) in CD+/− than in WT ACx (Figure 1L, M), as evident at smaller (100 pA) but not larger (250 pA) currents, suggesting that the threshold for eliciting APs (rheobase) was reduced in CD+/− mice. To measure rheobase, current ramps were delivered in the presence of kynurenic acid and PTX to block ionotropic glutamate receptors and GABA receptors, respectively. APs were evoked at lower currents in CD+/− than in WT interneurons, indicating reduced rheobase (Figure 1N, O). Thus, inhibitory FS interneurons in the WBS ACx are hyperexcitable.
The persistence of hyperexcitability in the presence of synaptic blockers implied that an intrinsic property of FS interneurons accounts for their hyperexcitability. However, the resting membrane potential and input resistance was comparable between genotypes (Figure 1O), suggesting that hyperexcitability originated from active properties, such as voltage-dependent conductance. Screens for changes in voltage-gated channels showed no difference between WT and CD+/− FS interneurons, in the amplitude of voltage-gated Na+ currents, K+ currents, or hyperpolarization-activated currents (Ih) (Figure S3). However, when using a protocol to isolate voltage-gated Ca2+ currents (Olson et al., 2005), depolarizing voltage steps delivered to FS interneurons resulted in inward voltage-gated current activated at more hyperpolarized voltages in CD+/− mice than in WT mice (Figure 1P, Q). The threshold at which the inward conductance was activated in response to a voltage ramp was ~2.5 mV lower in CD+/− mice than in WT mice (Figure 1R, S), but the total inward current did not differ (Figure 1T). The activation voltage of the inward current in CD+/− interneurons was close to the threshold for AP generation; therefore, it may cause the hyperexcitability of ACx interneurons.
Chemogenetic inhibition of ACx interneuron hyperexcitability reverses frequency-discrimination hyperacuity in CD+/− mice
If interneuron hyperexcitability underlies frequency-discrimination hyperacuity in CD+/− mice, then decreasing interneuron excitability in the ACx should reverse the phenotype. To reduce ACx interneuron excitability, we used the designer receptor exclusively activated by designer drug (DREADD) hM4Di, which hyperpolarizes neurons after activation by Compound 21 (C21) (Thompson et al., 2018). To express hM4Di in ACx interneurons, we injected recombinant AAVs (rAAVs) that express Cre-dependent hM4Di (rAAV-hSyn-DIO-hM4Di-IRES-mCitrine) into the ACx of Gad2Cre;WT mice and Gad2Cre;CD+/− mice (Gad2, glutamic acid decarboxylase 2), which express Cre recombinase in most interneurons (Ledri et al., 2014). Immunochemical validation in Gad2Cre;Ai14 mice that expresses tdTomato in a Cre-dependent manner, showed colocalization of tdTomato and GABA in cortex (Figure 2A); the rAAV injection sites were localized to the ACx (Figure 2B). Recording from hM4Di-expressing ACx cells in acute slices confirmed that C21 decreased the number of APs elicited by current injections in cortical FS interneurons (Figure 2C).
Figure 2: Chemogenetic inhibition of ACx interneurons reverses frequency-discrimination hyperacuity in CD+/− mice.
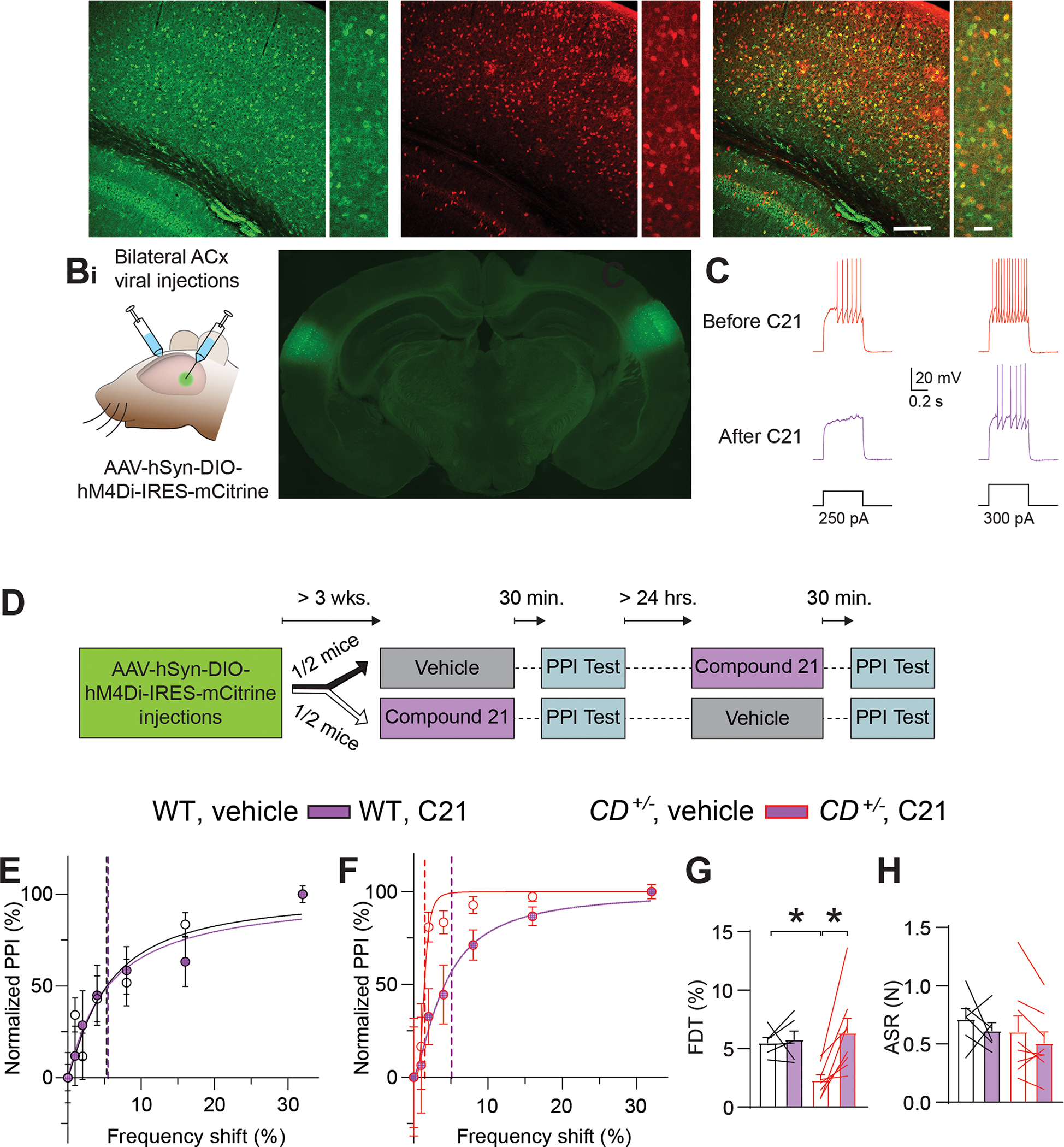
A. Images of GABA+ cells in the ACx (Ai), Gad2Cre–dependent tdTomato fluorescence (Aii), or both (Aiii). Lower (left) and higher (right) magnification images are shown for each.
B. Chemogenetic experiments. Bi. Scheme showing bilateral stereotactic injection of virus into the ACx. Bii. Coronal brain section with targeted bilateral injection of rAAVs encoding GFP into the ACx.
C. Compound 21 (C21) inhibits hM4Di+ interneurons. Responses to two intensities of depolarizing current injection in an hM4Di-expressing FS interneuron before (top) and after (bottom) C21 application in CD+/− mice.
D. Experimental timeline for behavioral testing of the effect of chemogenetic inhibition of interneurons.
E, F. Normalized PPI magnitude in individual WT (E) and CD+/− (F) mice, as a function of frequency difference between background and pre-pulse tones after intraperitoneal injection of vehicle or C21. Symbols and error bars are mean ± SEM of 10 repeated measurements in the same animal. Solid lines are linear-regression fits; dotted lines are frequency-discrimination thresholds (FDTs).
G. The FDT is reversed in CD+/− mice (n=8) after C21 injection, but C21 had no effect in WT mice (n=5). RM ANOVA CD+/−;vehicle vs CD+/−;C21 *P = 0.002. WT;vehicle vs CD+/−;vehicle *P = 0.029. Unpaired t-test WT:vehicle vs. CD+/−;C21 P = 0.574.
H. ASR is unchanged after C21 injection in WT (n=5) or CD+/− mice (n=8).
Averaged data are presented as mean ± SEM, with individual animals overlaid as lines connecting measured values in vehicle and C21.
To determine whether chemogenetic reduction of interneuron excitability reversed frequency-discrimination hyperacuity in WBS mice, we injected vehicle or C21 into Gad2Cre;WT or Gad2Cre;CD+/− mice that expressed hM4Di in ACx interneurons; ~30 min later, we tested frequency discrimination using PPI. Several days later, we injected the same animals with the opposite drug (C21 or vehicle) and tested frequency discrimination again (Figure 2D). In Gad2Cre;CD+/− mice, C21 but not vehicle restored FDT to WT levels; in Gad2Cre;WT mice, FDT was unchanged (Figure 2E–G). ASR was unaffected by C21 in either genotype (Figure 2H). Thus, hyperexcitability of ACx inhibitory interneurons mediates frequency-discrimination hyperacuity in WBS mice.
Improved frequency coding by the ACx in WBS mice
To examine how altered ACx circuitry in WBS mice affects frequency encoding we measured sound-evoked activity in the ACx of awake mice (Figure 3A) by performing simultaneous two-photon imaging in hundreds of individual L4 excitatory neurons expressing the genetically encoded fluorescent Ca2+ indicator GCaMP6f (Figure 3B) (Chen et al., 2013; Romano et al., 2015). Several weeks after installing cranial windows, we delivered tones at multiple frequencies and intensities in a pseudo-random order to awake GCaMP6fExN-L4;WT mice and GCaMP6fExN-L4;CD+/− mice and analyzed tone-evoked changes in GCaMP6f fluorescence. We collected data from 7130 cells in 36 mice (GCaMP6fExN-L4;WT; 5726 cells, 30 mice; GCaMP6fExN-L4;CD+/−; 1404 cells, 6 mice).
Figure 3: Improved frequency coding by the ACx in awake WBS mice.
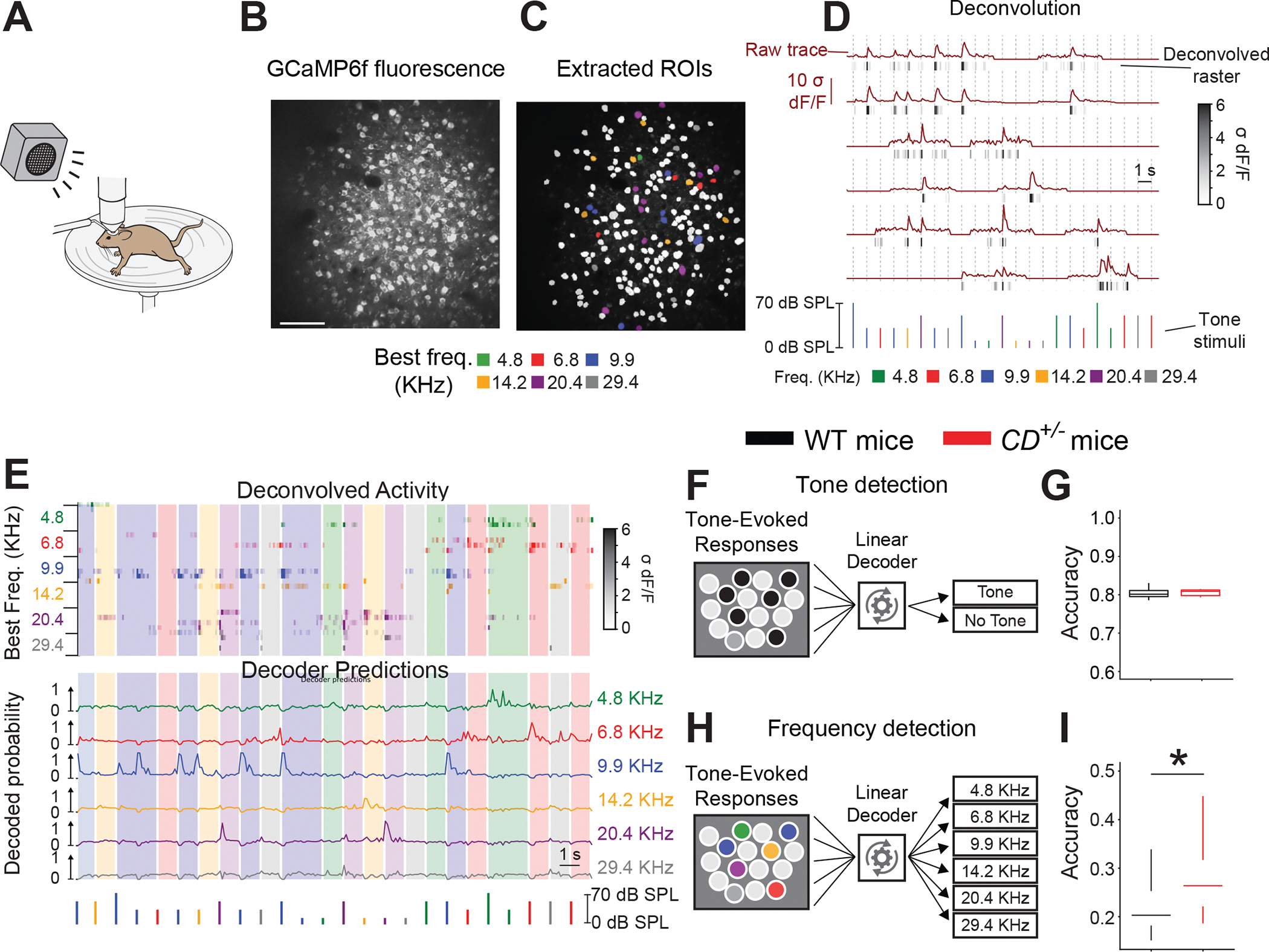
A. Scheme showing two-photon Ca2+ imaging and sound delivery to an awake, head-fixed mouse on a running wheel.
B, C. (B) Images of L4 excitatory cells responding to sound with changes in GCaMP6f fluorescence in the ACx. (C) Segmented ROIs representing individual excitatory neurons. Neurons are color-coded for their best frequency. Noncolored neurons indicate spontaneously active cells that did not respond to sound cues.
D. GCaMP6f fluorescence responses to pure tones (brown, “Raw trace,” top) that are processed to produce temporally discrete raster plots (gray/black, “Deconvolved raster,” bottom). Pure tone stimuli are shown below. Frequency is indicated by color, and intensity is indicated by height. Times of tone delivery are indicted by dotted vertical lines.
E. (Top) Responses of 25 cells after tone stimuli and decoder predictions. Stimuli are shown below. Each deconvolved activity row is a raster representing 1 cell. Cells are sorted by best frequency, as indicated by the raster color and frequency label at the left. Response intensity is indicated by color saturation, as shown in the scale bar (right). Vertical shading indicates the frequency last presented. (Bottom) Each decoder prediction row indicates the predicted probability of one frequency tone (indicated by line color and label at right) having been presented.
F. Scheme of the decoder, which pools responses from all imaged neurons to predict whether any tone was presented.
G. Equal accuracy of the tone-detection decoder was based on the activity of neurons in WT (n=30) and CD+/− (n=6) mice.
H. Scheme of decoder, which pools responses from all imaged neurons to predict which frequency of tone was presented.
I. Frequency detection is more accurate based on the activity of CD+/− (n=6 mice) vs WT (n=30 mice) neurons. Rank sum test *P = 0.034.
Data are presented as interquartile ranges (quantiles 5-25-50-75-95).
We identified sound-responsive L4 excitatory neurons and measured (Blundon et al., 2017; Klibisz et al., 2017) and deconvolved (Friedrich et al., 2017) their Ca2+ responses to categorize each neuron’s receptive field and best frequency (Figure 3C, D). Frequency tuning was heterogeneous in the ACx (Figure 3C). Frequency encoding, as it relates to perception, likely involves groups of neurons (Downer et al., 2021; Micheyl et al., 2013; See et al., 2018). To determine the ACx’s frequency-coding capacity, we trained linear decoders via machine learning to predict tones and their frequency from the deconvolved Ca2+ responses of all imaged neurons from each mouse (Figure 3E). The linear decoder for tone prediction performed equally well (>80%) in both genotypes (Figure 3F, G), but a second decoder for frequency prediction was more accurate in CD+/− neurons (Figure 3H, I) indicating enhanced frequency information present in the activity of CD+/− neurons.
To understand why the frequency decoder was more accurate in CD+/− than WT mice, we restricted the analysis window from 400 ms to 100 ms after tone presentation. Under those conditions, the frequency decoder performed equally well between genotypes (Figure S4A), suggesting that later components of the responses in CD+/− mice provide additional frequency information. In fact, sound responses in CD+/− neurons, were more sustained (Figure S4B), as evidenced by longer temporal autocorrelation range of responses in CD+/− vs WT neurons (Figure S4C). To ensure that differences in the total number of cells or the fraction of tone-responsive cells did not affect frequency coding, we randomly selected neuronal populations from WT and CD+/− mice that matched those variables. The improved accuracy persisted in CD+/− mice (Figure S4D, E). Thus, prolonged temporal components of tone-evoked neural responses in the ACx may enable WBS mice to better encode frequency information, which could underlie frequency-discrimination hyperacuity.
Haploinsufficiency of Gtf2ird1 replicates the frequency-discrimination hyperacuity phenotype of CD+/− mice
To determine which WBS gene(s) causes the auditory-hyperacuity phenotype of WBS mice, we measured PPI in mice with smaller microdeletions within the WBS-critical region. The mice had a proximal deletion (PD) spanning Limk1–Gtf2i or a distal deletion (DD) spanning Trim50–Limk1 (Li et al., 2009) that together encompassed the CD microdeletion (Figure 4A). The phenotype of PD+/− mice was like that of CD+/− mice (Figure 4B, C), but DD+/− mice resembled WT mice (Figure 4D, E) suggesting that the causal gene(s) for frequency-discrimination hyperacuity is within the PD region. Despite innate auditory hyperacuity, PD+/− mice had deficits in learning an auditory-discrimination task, like those in CD+/− mice. None of the PD+/− mice tested learned the Go/No-go task after 2 weeks of training (Figure S5A).
Figure 4: Gtf2ird1 deletion alone replicates the frequency-discrimination hyperacuity phenotype of WBS mice.
A. The syntenic region of mouse chromosome 5 corresponding to the locus deleted in humans with WBS. Also shown are the microdeletion regions for CD+/−, PD+/−, and DD+/− mice.
B. Normalized PPI magnitudes from individual WT and PD+/− mice, as a function of frequency difference between background and pre-pulse tones. Points and error bars are mean ± SEM of 10 repeated measurements in the same animal. Solid lines are linear-regression fits; dotted lines are frequency-discrimination thresholds (FDTs).
C. The FDT is lower in PD+/− (n=12) than in WT (n=14) mice. Unpaired t-test *P = 0.034.
D. Normalized PPI magnitudes in individual WT and PD+/− mice, as a function of frequency difference between background and pre-pulse tones.
E. Average FDT does not differ between WT (n=8) and DD+/− (n=18) mice.
F. Normalized PPI magnitudes from individual WT, Gtf2ird1+/−, and Gtf2ird1−/− mice as a function of frequency difference between background and pre-pulse tones.
G. FDT is lower in Gtf2ird1+/− and Gtf2ird1−/− mice than in WT mice. One-way ANOVA: WT vs Gtf2ird1+/− *P = 0.004; WT vs Gtf2ird1−/− *P = 0.008.
H. Normalized PPI magnitudes from individual WT and Gtf2i+/− mice, as a function of frequency difference between background and pre-pulse tones.
I. FDT does not differ between WT (n=7) vs Gtf2i+/− (n=9) mice.
Averaged data are presented as the mean ± SEM, with individual measurements overlaid as dots.
Within the PD deletion, haploinsufficiency of Gtf2ird1 (Howard et al., 2012; Proulx et al., 2010; Schneider et al., 2012; Young et al., 2008) and Gtf2i (Barak et al., 2019) has been implicated in WBS cognitive symptoms. Individuals with microdeletions including GTF2IRD1 and GTF2I have cognitive deficits like those of persons with WBS (Broadbent et al., 2014; Tassabehji et al., 2005); conversely, those with deletions that exclude these genes have more preserved cognitive function (Antonell et al., 2010; van Hagen et al., 2007; Hirota et al., 2003).
Gtf2ird1+/− and Gtf2ird1−/− mice performed better than WT mice in frequency-discrimination tests (Figure 4F, 4G, S5B). Conversely, FDT in Gtf2i+/− mice was indistinguishable from WT mice (Figure 4H, I). ASR was unaffected in the PD+/−, DD+/−, Gtf2ird1+/−, Gtf2ird1−/−, or Gtf2i+/− mice (Figure S5C–F). Thus, hemizygous deletion of Gtf2ird1 may cause frequency-discrimination hyperacuity in WBS mouse models.
Gtf2ird1 haploinsufficiency downregulates Vipr1 in mice
The putative transcription factor GTF2IRD1 has many gene targets (Kopp et al., 2020) that may influence frequency discrimination. Hyperexcitability of cortical interneurons causes auditory hyperacuity; thus, we isolated those cells from Gtf2ird1−/− mice and screened for differentially regulated genes using RNA-seq. (Figure 5A). RNA-seq analysis revealed several genes differentially expressed in Gtf2ird1−/− vs WT interneurons (Figure 5B). Gene ontology (GO) enrichment analysis identified other transcription factors as the largest group of differentially regulated genes (Figure S6A).
Figure 5: Gtf2ird1 deletion reduces VIPR1 in murine and human WBS neurons.
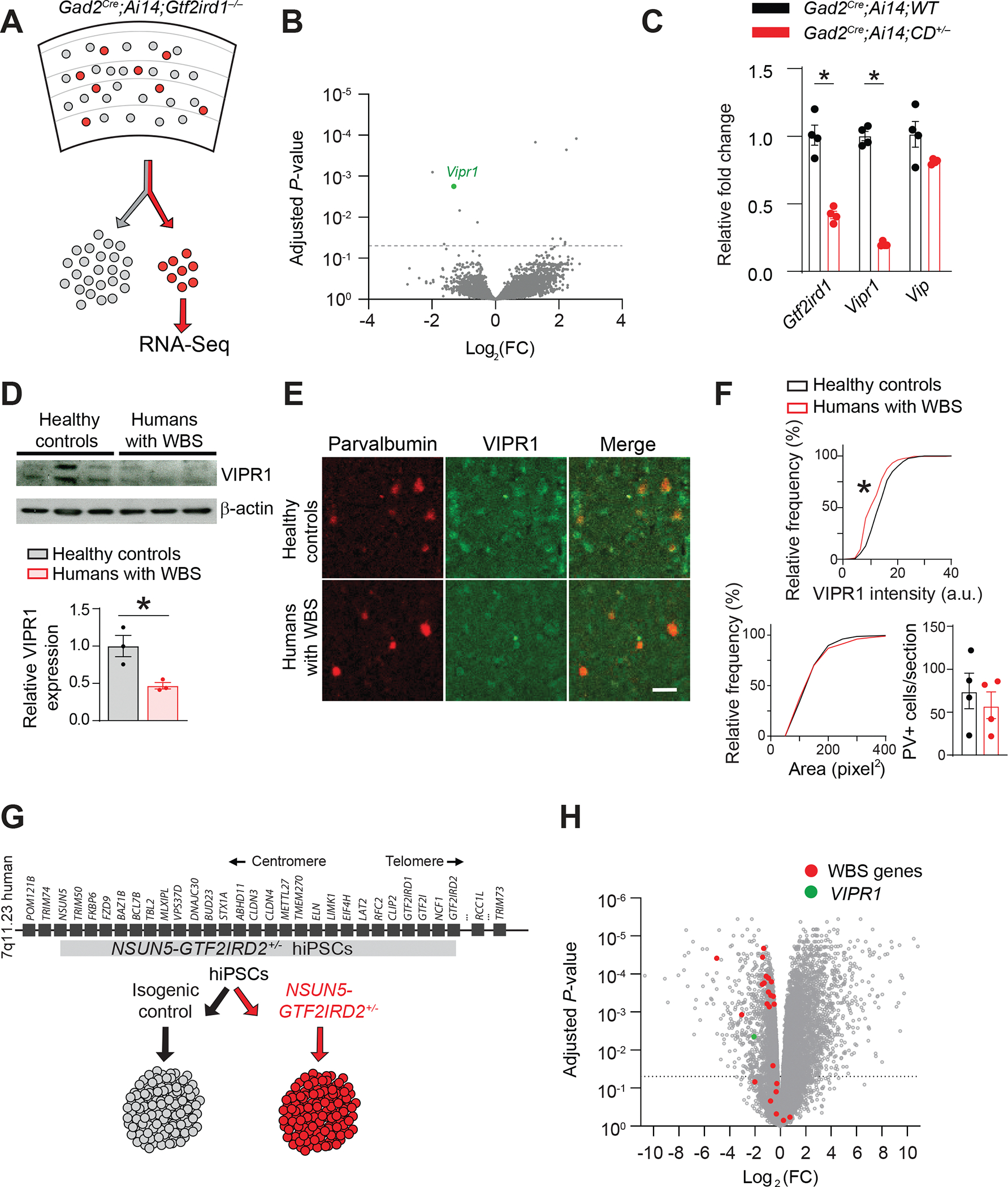
A. Scheme of GAD2+ inhibitory neurons (red) sorted from the mouse cortex for RNA-seq analysis.
B. Volcano plot of RNA-seq data comparing Gtf2ird1−/− and WT GAD2+ inhibitory neurons. The dotted line represents adjusted P = 0.05. Vipr1 expression is significantly reduced in Gtf2ird1−/− inhibitory neurons. Data are from Gtf2ird1+/+ (n=3) and Gtf2ird1−/− (n=3) mice.
C. Gtf2ird1 and Vipr1 transcript levels in GAD2+ cortical interneurons from CD+/− mice (n=4 mice) are lower than in WT controls (n=4 mice), and the VIPR1 ligand, Vip, is unchanged. Gtf2ird1: unpaired t-test, *P = 0.002. Vipr1: unpaired t-test *P <0.001.
D. Western blotting (top) and quantification (bottom) of VIPR1 levels in postmortem human brain samples from the superior temporal gyrus of patients with WBS (n=3) are lower than that in healthy control (n=3) subjects. Unpaired t-test, *P = 0.023.
E. VIPR1 expression is decreased in the ACx interneurons of postmortem brains of humans who had WBS compared to healthy controls. Images of sections of the superior temporal gyrus containing the ACx immunohistochemically stained for PV (left) and VIPR1 (middle). Overlapping staining is shown in yellow (right).
F. (Top) Cumulative probability histogram showing lower VIPR1 fluorescence intensity within PV+ cell bodies of WBS (n=4) compared to healthy control (n = 4) sections. Kolmogorov-Smirnov test, *P <0.0001. (Below, left) Cumulative probability histogram showing the area of PV+-labeled cell bodies does not differ between WBS and healthy sections. (Below, right) The number of PV+ cells also did not differ between WBS and healthy controls.
G. Scheme of the generation of cerebral organoids from an isogenic hiPSC line carrying a hemizygous microdeletion spanning NSUN5–GTF2IRD2 in the WBS critical locus. The resulting NSUN5–GTF2IRD2+/− hiPSC line is shown in gray. Organoids generated from NSUN5–GTF2IRD2+/− and isogenic control hiPSC lines were used for RNA-seq analysis.
H. Volcano plot representing differentially expressed genes between NSUN5–GTF2IRD2+/− and isogenic control organoids. The dotted line represents adjusted P = 0.05. WBS genes are those mapped within the WBS critical locus in humans. Data are from four control and four NSUN5–GTF2IRD2+/− organoids.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
One down-regulated gene, vasoactive intestinal polypeptide receptor gene, Vipr1, was particularly relevant because CD+/− cortical interneurons have altered voltage-gated current and VIPR1 influences multiple voltage-gated channels (Gherghina et al., 2017; Hayashi et al., 2002; Tang et al., 2019; Zhu and Ikeda, 1994). Decreased Vipr1 expression in cortical interneurons isolated from Gad2Cre;Ai14;CD+/− mice was confirmed by qPCR analysis, but Vip, the gene that encodes the VIPR1 ligand, was unchanged (Figure 5C). Unlike Vipr1, other genes identified by RNA-seq in Gtf2ird1−/− mice were either not differentially expressed in CD+/− interneurons or were differentially expressed in the opposite direction (Figure S6B). The consistent Vipr1 downregulation and its connection to voltage-gated currents merited further examination of its role in auditory hyperacuity.
VIPR1 is downregulated in the ACx interneurons of persons with WBS and in cerebral organoids derived from hiPSCs with an isogenic WBS microdeletion
To examine whether VIPR1 downregulation occurs in humans with WBS, we obtained postmortem ACx samples from patients with WBS. VIPR1 levels were lower in WBS brain lysate than control lysate, based on Western blot analysis (Figure 5D). To examine VIPR1 expression in interneurons, we immunolabelled cortical sections with VIPR1 and the FS interneuron marker PV and quantified VIPR1 in PV+ cells; VIPR1 staining was less intense in PV+ WBS interneurons than in controls (Figure 5E, F), but the size and number of PV+ neurons were the same (Figure 5F).
As an alternative model of human WBS, we used bulk RNA-seq to compare gene expression in cerebral organoids generated from NSUN5–GTF2IRD2+/− hiPSCs to isogenic hiPSCs (Figure 5G). The expression of almost all WBS genes within the NSUN5–GTF2IRD2 microdeletion was reduced in mutant organoids, suggesting that NSUN5–GTF2IRD2+/− organoids model WBS at the transcriptional level (Figures 5H and S6E). VIPR1 expression was also reduced in NSUN5–GTF2IRD2+/− organoids (Figures 5H and S6E). Per GO enrichment analysis, synaptic genes and biological pathways relevant to neuronal activity, GABAergic neurons, and neurodevelopment were also downregulated in WBS cerebral organoids (Figure S6F).
Acute inhibition of VIPR1 mimics WBS interneuron phenotypes
Blocking VIPR1 with the VIPR1-specific antagonist PG 97–269 lowered the AP induction threshold in response to a current ramp in FS interneurons in WT cortex, mimicking the CD+/− phenotype (Figure 6A, B). In the CD+/− cortex, interneurons were hyperexcitable, and PG 97–269 had no additional effect (Figure 6A, B). In WT mice, applying PG 97–269 also mimicked the CD+/− voltage-gated channel phenotype, shifting the threshold of inward current activation to more hyperpolarized potentials (Figure 6C, D). The shift was 2.1 mV ± 0.4 mV, like the difference between CD+/− and WT interneurons (Figure 2D). The shift was smaller in CD+/− interneurons than WT interneurons (Figure 6C, D). Thus, in WT brain slices, tonic VIPR1 activity limits interneuron excitability; this activity is absent in CD+/− mice, possibly due to decreased VIPR1 levels. The VIPR1-specific agonist [Ala11,22,28]-VIP did not affect WT or CD+/− interneurons [WT (n = 13): paired t-test P = 0.637; CD+/− (n = 11): paired t-test P = 0.727] suggesting that an endogenous VIPR1 ligand is present at high enough concentration to saturate VIPR1. Thus, interneuron hyperexcitability in the WBS ACx may reflect reduced VIPR1 signaling.
Figure 6: Pharmacologic block of VIPR1 causes FS interneuron hyperexcitability and lowers the threshold for inward voltage-gated current.
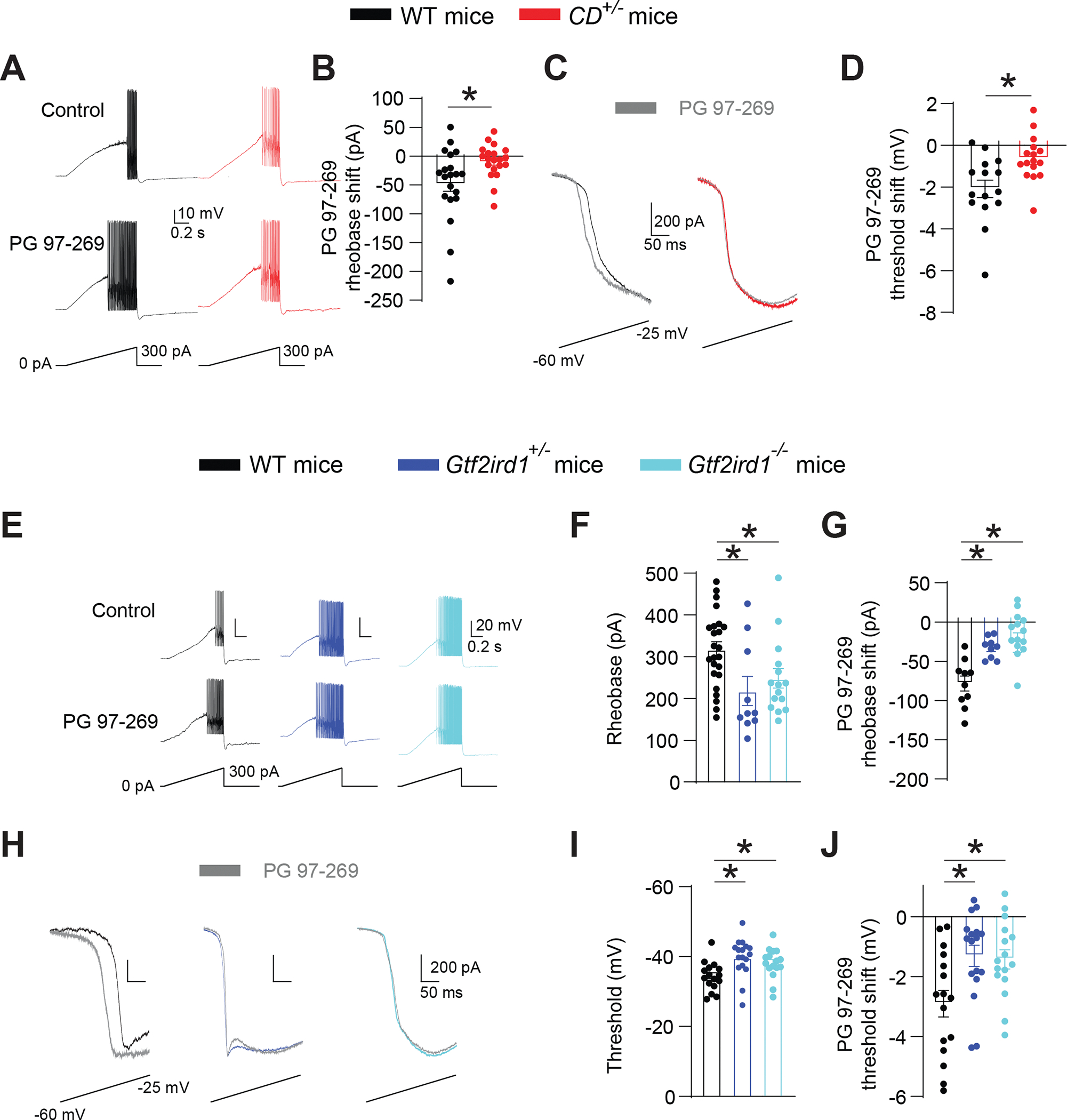
A, B. The VIPR1-specific antagonist PG 97–269 elevates excitability in WT but not in CD+/− FS interneurons in the ACx. (A) Traces of voltage responses to a current ramp before and after local application of PG 97–269 in WT and CD+/− FS interneurons. (B) PG 97–269 lowers the rheobase in WT (n=20; paired t-test, *P = 0.003) but not in CD+/− (n=20) cells. WT vs CD+/−, rank sum test, *P = 0.018.
(C) Traces of current response to a voltage ramp.
(D) PG 97–269 causes a larger threshold shift in inward voltage-gated current in WT (n=15) vs CD+/− (n=16) interneurons. Unpaired t-test, *P = 0.006.
E-G. Gtf2ird1-deficient ACx FS interneurons are hyperexcitable, and PG 97–269 has a diminished effect on their excitability. (E) Traces showing voltage and AP responses to current ramps in WT, Gtf2ird1+/−, and Gtf2ird1−/− FS interneurons, before and after local application of PG 97–269. (F) Ramp rheobase is lower in Gtf2ird1+/− (n=10) and Gtf2ird1−/− (n=15) interneurons than in WT (n=25) cells. One-way ANOVA: WT vs. Gtf2ird1+/− *P = 0.008; WT vs Gtf2ird1−/− *P = 0.022. (G) PG 97–269 has a weaker effect on rheobase in Gtf2ird1+/− (n=9) and Gtf2ird1−/− (n=14) FS interneurons than in WT (n=10) cells. One-way ANOVA: WT vs Gtf2ird1+/− *P = 0.021; WT vs Gtf2ird1−/− *P <0.001.
H-J. Threshold for the inward voltage-gated current is more hyperpolarized, less sensitive to PG 97–269 in FS cortical interneurons from Gtf2ird1-deficient mice. (H) Traces of current responses to voltage ramps in WT, Gtf2ird1+/−, and Gtf2ird1−/− interneurons, before and after local application of PG 97–269. (I) Inward current threshold is more hyperpolarized in Gtf2ird1+/− (n=17) and Gtf2ird1−/− (n=16) interneurons compared to WT (n=16) cells. One-way ANOVA: WT vs Gtf2ird1+/− *P = 0.005; WT vs Gtf2ird1−/− *P = 0.046.
(J) PG 97–269 has a weaker effect on inward voltage-gated current threshold in Gtf2ird1+/− (n=17) and Gtf2ird1−/− (n=16) interneurons than in WT (n=16) cells. One-way ANOVA: WT vs Gtf2ird1+/− *P = 0.012; WT vs Gtf2ird1−/− *P = 0.016.
Averaged data are presented as the mean ± SEM, with individual measurements overlaid as dots.
Gtf2ird1 deletion causes interneuron hyperexcitability and lowers the threshold of inward voltage-gated current in ACx interneurons
Do CD+/− interneurons and Gtf2ird1+/− interneurons have the same phenotype? FS interneurons in the ACx of Gtf2ird1+/− mice and Gtf2ird1−/− mice showed hyperexcitability; their rheobases did not differ from that in CD+/− interneurons (Figure 6E, F). As in CD+/− mice, PG 97–269 had a small or no effect on the excitability of FS interneurons in the ACx of Gtf2ird1+/− mice and Gtf2ird1−/− mice (Figure 6G).
The inward voltage-gated current threshold in Gtf2ird1+/− and Gtf2ird1−/− cortical interneurons was also shifted compared to WT interneurons and was less sensitive to PG 97–269, like results from CD+/− mice (Figure 6H–J). The PG 97–269 threshold shift was also reduced in Gtf2ird1+/− and Gtf2ird1−/− cortical interneurons (Figure 6I). The consistency of the cellular phenotypes and PG-97–269 sensitivity between Gtf2ird1-deficient mice and CD+/− mice supports Gtf2ird1 regulating ACx interneuron excitability and frequency-discrimination acuity in WBS mice via reduction of VIPR1.
Vipr1 reduction in interneurons is necessary and sufficient for the frequency-discrimination hyperacuity and interneuron hyperexcitability in WBS mice
To test if reduced VIPR1 in interneurons underlies frequency-discrimination hyperacuity in WBS mice, we genetically reduced the VIPR1 level in ACx interneurons of WT mice and replenished it in those of CD+/− or Gtf2ird+/− mice.
We generated mice with chromic reduction of Vipr1 only in GAD2+ interneurons (Gad2Cre;Vipr1fl/+ mice) (Figure S7A). The Vipr1 transcript decreased in a dose-dependent manner in the cortex of Gad2Cre;Vipr1fl/+ mice and Gad2Cre;Vipr1fl/fl mice compared to WT (Gad2Cre;Vipr1+/+) mice (Figure S7B).
Rheobase was reduced in FS interneurons of Gad2Cre;Vipr1fl/+ mice and Gad2Cre;Vipr1fl/fl mice compared to that in WT littermates (Figure 7A, B). Gad2Cre;Vipr1fl/+ mice and Gad2Cre;Vipr1fl/fl mice had normal ASR (Figure S7C) but improved frequency discrimination like that of CD+/− mice (Figure 7C, D). Thus, chronically decreased expression of Vipr1 only in interneurons, which is likely representative of WBS, was sufficient to mimic the behavioral and cellular phenotypes of WBS mice.
Figure 7: Reduced Vipr1 in ACx interneurons is necessary and sufficient for the cell hyperexcitability and frequency-discrimination hyperacuity in WBS mice.
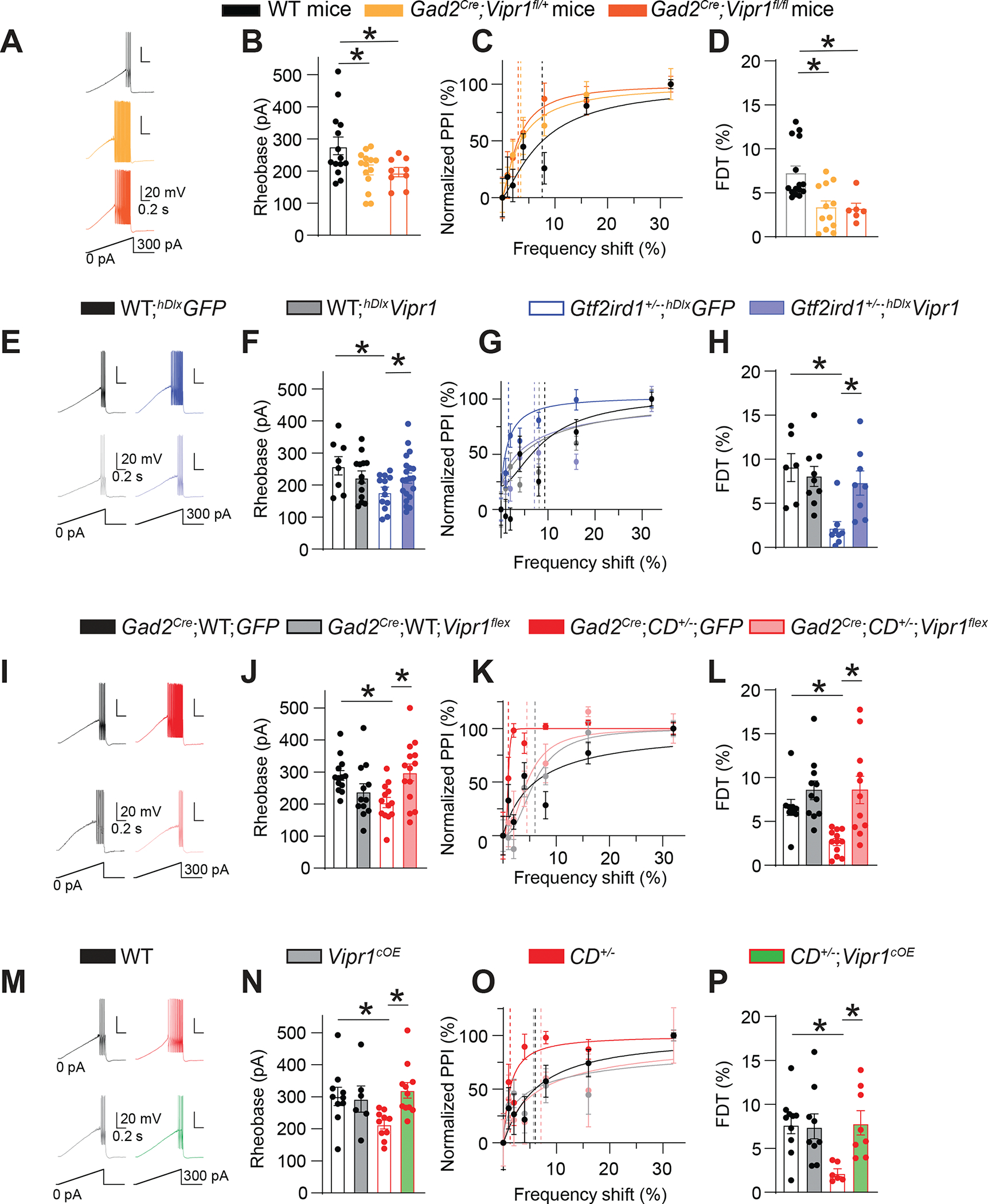
A-D. Conditional knockout of Vipr1 in interneurons mimics interneuron hyperexcitability and frequency-discrimination hyperacuity phenotypes of WBS mice. (A) Traces of voltage responses to a current ramp in WT, Gad2Cre;Vipr1fl/+, and Gad2Cre;Vipr1fl/fl FS interneurons. (B) Ramp rheobase is lower in ACx FS interneurons in Gad2Cre;Vipr1fl/+ (n=14) and Gad2Cre;Vipr1fl/fl (n=9) mice than in WT mice (n=14). One-way ANOVA: WT vs Gad2Cre;Vipr1fl/+ *P = 0.04; WT vs Gad2Cre;Vipr1fl/fl *P = 0.033. (C) Normalized PPI magnitudes in individual WT, Gad2Cre;Vipr1fl/+, and Gad2Cre;Vipr1fl/fl mice, as a function of frequency difference between background and pre-pulse tones. Points and error bars are mean ± SEM of 10 repeated measurements in the same animal. Solid lines are linear-regression fits; dotted lines are frequency-discrimination thresholds (FDTs). (D) FDT is lower in Gad2Cre;Vipr1fl/+ (n=12) and Gad2Cre;Vipr1fl/fl (n=6) mice vs WT (n=15) mice. One-way ANOVA: WT vs Gad2Cre;Vipr1fl/+ *P = 0.002; WT vs Gad2Cre;Vipr1fl/fl *P = 0.012.; Gad2Cre;Vipr1fl/+ vs. Gad2Cre;Vipr1fl/fl P = 0.992.
E-H. Viral expression of Vipr1 in ACx interneurons reverses interneuron hyperexcitability and frequency-discrimination hyperacuity phenotypes of Gtf2ird1-deficient mice. (E) Traces of voltage responses to a current ramp in WT, Gtf2ird1+/−, and Gtf2ird1−/− FS interneurons after viral expression of GFP (above) or Vipr1 using AAV-hDlx-Vipr1-GFP (hDlxVipr1) or AAV-hDlx-GFP (hDlxGFP) (below). (F) Ramp rheobase is restored to WT levels in ACx FS interneurons from Gtf2ird1-deficient mice after viral expression of Vipr1. Two-way ANOVA: WT;hDlxGFP vs WT;hDlxVipr1 P = 0.06; Gtf2ird1+/−,−/−;hDlxGFP vs Gtf2ird1+/−,−/−;hDlxVipr1 *P = 0.024; WT;hDlxGFP vs Gtf2ird1+/−,−/−;hDlxGFP *P = 0.002 (n=18 WT;hDlxGFP, n=12 WT;hDlxVipr1, n=25 Gtf2ird1+/−,−/−;hDlxGFP, and n=19 Gtf2ird1+/−,−/−;hDlxVipr1). (G) Normalized PPI magnitudes from individual WT;hDlxGFP, WT;hDlxVipr1, Gtf2ird1+/−;hDlxGFP and Gtf2ird1+/−;hDlxVipr1 mice, as a function of frequency difference between background and pre-pulse tone. (H) FDT is restored to WT levels in Gtf2ird1-deficient mice after viral overexpression of Vipr1 in ACx interneurons. Two-way ANOVA: Gtf2ird1+/−,−/−;hDlxGFP vs Gtf2ird1+/−,−/−;hDlxVipr1 *P = 0.019; WT;hDlxGFP vs Gtf2ird1+/−,−/−;hDlxGFP *P = 0.005 WT;hDlxGFP vs. Gtf2ird1+/−,−/−;hDlxVipr1 P = 0.723 (n=6 WT;hDlxGFP, n=9 WT;hDlxVipr1, n=8 Gtf2ird1+/−,−/−;hDlxGFP, and n=8 Gtf2ird1+/−,−/−;hDlxVipr1).
I-L. Viral overexpression of Vipr1 in GAD2+ interneurons in the ACx reverses interneuron hyperexcitability and frequency-discrimination hyperacuity phenotypes of CD+/− mice. (I) Traces of voltage responses to a current ramp in WT and CD+/− FS interneurons after Gad2Cre-dependent viral expression of GFP (above) or Vipr1 (below). (J) Ramp rheobase is restored to WT levels in ACx FS interneurons from CD+/− mice after Gad2Cre-dependent expression of Vipr1. Two-way ANOVA: CD+/−;GFP vs CD+/−;Vipr1flex *P = 0.019; WT;GFP vs CD+/−;GFP *P = 0.031 WT;GFP vs. CD+/−;Vipr1flex P = 0.988 (n=13 WT;GFP, n=12 WT;Vipr1flex, n=13 CD+/−;GFP, and n=14 CD+/−;Vipr1flex). (K) Normalized PPI magnitudes from individual WT;GFP, WT;Vipr1flex, CD+/−;GFP, and CD+/−;Vipr1flex mice, as a function of frequency difference between background and pre-pulse tones. (L) FDT is restored to WT levels in CD+/− mice after overexpression of Vipr1 in ACx interneurons. CD+/−;GFP vs CD+/−;Vipr1flex rank sum test *P = 0.001; WT;GFP vs CD+/−;GFP rank sum test *P = 0.001, WT;GFP vs. CD+/−;Vipr1flex rank sum test P = 0.704; (n=9 WT;GFP, n=11 WT;Vipr1flex, n=12 CD+/−;GFP, n=11 CD+/−;Vipr1flex).
M-P. Transgenic overexpression of Vipr1 in GAD2+ interneurons reverses interneuron hyperexcitability and frequency-discrimination hyperacuity phenotypes of CD+/− mice. (M) Traces of voltage responses to a current ramp in WT and CD+/− FS interneurons after Gad2Cre-dependent transgenic overexpression of Vipr1 in WT (left) or CD+/− mice (right). (N) Ramp rheobase is restored to WT levels in ACx FS interneurons from CD+/− mice after Gad2Cre-dependent transgenic overexpression of Vipr1 (Vipr1cOE). All data are from littermates of the following genotypes: WT: WT;WT;WT; Vipr1cOE: Gad2Cre;WT;Vipr1OE; CD+/−: WT;CD+/−;WT; CD+/−;Vipr1cOE: Gad2Cre;CD+/−;Vipr1OE. Two-way ANOVA: CD+/− vs CD+/−;Vipr1cOE *P = 0.004, WT vs CD+/− *P = 0.018, WT vs. CD+/−;Vipr1cOE P = 0.873 (n=10 WT, n=6 Vipr1cOE, n=10 CD+/−, n=11 CD+/−;Vipr1cOE). (O) Normalized PPI magnitudes from individual WT, Vipr1cOE, CD+/−, and CD+/−;Vipr1cOE mice, as a function of frequency difference between background and pre-pulse tones. (P) FDT is restored to WT levels in CD+/− mice after transgenic overexpression of Vipr1. Two-way ANOVA: CD+/− vs CD+/−;Vipr1cOE *P = 0.028; WT vs CD+/− *P = 0.025 WT vs. CD+/−;Vipr1cOE P = 0.999 (n=10 WT, n=9 Vipr1cOE, n=6 CD+/−, and n=8 CD+/−;Vipr1cOE).
Averaged data are presented as the mean ± SEM, with individual measurements overlaid as dots.
To determine if Vipr1 depletion in FS interneurons mediates FS interneuron hyperexcitability and frequency-discrimination hyperacuity in WBS mice, we increased Vipr1 expression in those cells using three strategies: (1) We used rAAVs expressing Vipr1 and GFP under control of human Dlx5/6 enhancer (hDlx) (AAV-hDlx-Vipr1-GFP), which restricts expression to GABAergic interneurons (Dimidschstein et al., 2016). GFP and tdTomato fluorescence were highly co-localized in GAD2+ cells when AAV-hDlx-Vipr1-GFP was injected into the ACx of Gad2Cre;Ai14 mice (Figure S7D). After bilateral injection of AAV-hDlx-Vipr1-GFP (or AAV-hDlx-GFP as control) into the ACx, WT interneurons showed no difference in excitability (Figure 7E, F);, but Gtf2ird1+/− FS interneurons with overexpressed Vipr1 (but not GFP) showed reduced excitability (increased rheobase) (Figure 7E, F). No viruses altered the ASR (Figure S7E). AAV-hDlx-Vipr1-GFP increased FDT (reducing frequency-discrimination hyperacuity) in Gtf2ird1+/− mice but not in WT mice; AAV-hDlx-GFP expression alone did not (Figure 7G, H). Thus, Vipr1 replenishment in ACx interneurons reversed the cellular and frequency-discrimination phenotypes in Gtf2ird1+/− mice to WT levels.
(2) Again using a viral strategy, we injected rAAVs encoding Cre-dependent Vipr1 (AAV-CAG-Flex-Vipr1-GFP) or GFP control (AAV-CAG-Flex-GFP) into the ACx of Gad2Cre mice crossed with CD+/− mice. Overexpressing Vipr1 did not alter GFP+ FS interneurons in WT mice (Figure 7I, J), but in CD+/− mice, it elevated rheobase to WT levels, while FS interneuron hyperexcitability was maintained with control GFP virus (Figure 7I, J). No viral injection altered the ASR (Figure S7F), while Vipr1 overexpression increased FDT in CD+/− mice but not WT mice; GFP expression had not effect (Figure 7K, L).
(3) We generated transgenic mice with conditional overexpression of Vipr1 in interneurons (Vipr1cOE mice) (Figure S7G, H). Transgenic overexpression of Vipr1 in interneurons did not affect the ASR in WT or CD+/− mice (Figure S7I). It also did not alter the rheobase or frequency discrimination in WT mice (Figure 7M–P), but Vipr1cOE;CD+/− mice cellularly and behaviorally resembled WT mice. Vipr1 overexpression in CD+/− interneurons reversed the rheobase phenotype in ACx interneurons (Figure 7M, N) and the frequency-discrimination phenotype (Figure 7O, P). Thus, reduced Vipr1 expression in ACx interneurons was necessary for the behavioral and cellular phenotypes of WBS mice.
DISCUSSION
Frequency-discrimination hyperacuity may underlie spared or enhanced auditory abilities in persons with WBS. Lenhoff et al. found increased prevalence of absolute pitch in five individuals with WBS who were selected for their musical training (Lenhoff et al., 2001). Some studies found pitch discrimination [i.e., the ability to distinguish the notes of a musical scale) (Plack et al., 2005)] in individuals with WBS equal to that of control subjects (Don et al., 1999; Levitin, 2005), suggesting a relative strength in WBS. However, others found worse pitch discrimination in patients with WBS (Hopyan et al., 2001; Martínez-Castilla and Sotillo, 2014). This heterogeneity may reflect the small number of subjects tested, variability in pitch discrimination among individuals (Mosing et al., 2014; Seesjärvi et al., 2016; Smith et al., 2017), or differences in testing conditions. That individuals with WBS may not possess increased pitch discrimination does not preclude that they may possess increased frequency discrimination; previous work suggests that pitch and frequency are encoded in different areas of the human ACx (Bendor and Wang, 2006).
Auditory information processing in WBS appears atypical based on responses in the brains of persons with WBS by using functional magnetic resonance imaging (fMRI) (Levitin et al., 2003; Thornton-Wells et al., 2010). However, these studies did not correlate their findings with auditory symptoms. Thus, how abnormal auditory processing contributes to specific WBS symptoms is unclear. Our results suggest that increased inhibition in the ACx affects innate auditory behavior in WBS. Modulation of inhibitory interneuron activity specifically in the ACx via targeted expression of DREADDs reversed the frequency-discrimination hyperacuity phenotype in WBS mice. Replenishing VIPR1 only in FS interneurons in the ACx also reversed the phenotype. Thus, increased cortical inhibition may cause differences in fMRI studies of patients, though changes in frequency encoding are probably more subtle than would be evident in fMRI. Although this shows the importance of the ACx in frequency discrimination, it does not discount the contribution of other brain areas to the mouse or human auditory phenotypes in WBS.
How interneuron hyperexcitability changes frequency tuning in the ACx is unclear. VIP+ interneurons have been linked to cortical disinhibition i.e., increased activity of VIP+ neurons inhibits other interneurons and synaptically increases or prolongs the activity of principal excitatory neurons (Kullander and Topolnik, 2021). Longer duration responses of excitatory neurons to a tone in CD+/− mice appeared to improve frequency coding in vivo, which might be a consequence of polysynaptic disinhibitory interactions. In WT animals, the excitability of FS interneurons increased after blocking VIPR1, suggesting that VIPR1s are tonically active due to high levels of ambient VIP or another endogenous VIPR1 agonist (e.g., PACAP) in brain slices. VIP+ interneurons are spontaneously active (Mesik et al., 2015; de Vries et al., 2020), which could cause tonic VIP release. Optogenetically increasing VIP+ interneuron activity decreases the efficiency of encoding sound information (Bigelow et al., 2019). This may be the inverse of our results, with improved frequency coding by the ACx correlated with decreased VIPR1 activity. Responses of VIP+ interneurons also differ across sensory cortices (Mesik et al., 2015), and varied effects of diminished VIPR1 signaling may add to the heterogeneous cognitive symptoms of WBS. VIP+ neurons have mostly been studied as GABAergic interneurons, presumed to signal through postsynaptic GABA receptors. Our results suggest that at least some of their neuromodulatory action is mediated via VIP receptors (e.g., VIPR1) rather than GABA receptors, which could reflect an underappreciated diversity of postsynaptic actions of VIP+ interneurons.
The identity of the elevated inward voltage-gated current in WBS interneurons is unclear. VIPR1 signals mainly via Gs and protein kinase A activation (Couvineau and Laburthe, 2012), but also via Gi/o and protein kinase C (Cunha-Reis et al., 2017). VIPR1 regulates multiple types of ion channels, including voltage-gated Ca2+ channels (Hayashi et al., 2002; Zhu and Ikeda, 1994), Ca2+-activated K+ channels (Taylor et al., 2014), TRP channels (Tang et al., 2019), and others (Johnson et al., 2019), any of which could affect neuronal excitability alone or in combination. The voltage-gated inward current activated at more hyperpolarized potentials in CD+/− interneurons may reflect a change in the activation or activity of voltage-gated Ca2+ channels, but any other channel that conducts Ba2+ and is insensitive to TTX and internal Cs+ block could mediate the additional current.
The full scope of the WBS phenotype, even in the auditory system, probably involves GTF2IRD1-regulated genes other than Vipr1. However, the reversal of FS interneuron excitability and frequency discrimination by Vipr1 replenishment only in interneurons of WBS and Gtf2ird1+/− mice argues that the Gtf2ird1–Vipr1 axis is crucial for these phenotypes. Gtf2ird1 and Vipr1 may have other roles in neurodevelopment, but because pharmacologic and genetic interventions reversed the cellular and behavioral phenotypes in adult WBS mice, auditory hyperacuity is not caused by irrevocable neurodevelopmental changes.
In summary, we identified the Gtf2id1–Vipr1 pathway in WBS mice that, if diminished, increases the excitability of GABAergic interneurons and improves frequency coding by the ACx. ACx interneuron hyperexcitability leads to improved innate auditory perception. Because reducing Vipr1 expression in ACx interneurons causes auditory hyperacuity in WBS mice, it is tempting to speculate possible VIPR1-targeting interventions for improving auditory-perceptual acuity in individuals without WBS.
Limitations of the current study
Besides musical fascination and auditory hyperacuity, persons with WBS have hyperacusis. One behavioral proxy for testing sound sensitivity in mice is the ASR test. Reports on ASR in WBS mice are inconsistent (Li et al., 2009; Segura-Puimedon et al., 2014). We found in the presence of a pure-tone background, WBS mice showed normal ASR across all conditions, but this setting may not be ideal for testing hyperacusis, and more precisely tailored studies are needed.
A VIPR1-selective antagonist, PG 97–269, mimicked the cellular phenotypes of WBS mice in brain slices. PG 97–269 and other VIPR1 targeting drugs are peptides with weak bioavailability and short half-lives, limiting their use to target the brain in vivo (Latek et al., 2019). VIPR1 is broadly expressed, raising concerns about negative effects of systemic delivery. Developing stable small-molecule agonists and antagonists to manipulate VIPR1 activity in vivo would aid in the study of its role in auditory and other behaviors. Alternatively, identifying downstream signaling components and ion channels by which VIPR1 affects ACx interneurons may identify more accessible pharmacologic targets (Langer et al., 2022).
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stanislav S. Zakharenko (stanislav.zakharenko@stjude.org)
Materials availability
Materials generated in this study are available upon request from the lead contact.
Data and code availability
RNA-seq data are available in the NCBI GEO database under accession number GSE195491 (tdTomato+ cells from the cortex of Gad2Cre;Ai14;Gtf2ird1+/+ or Gad2Cre;Ai14;Gtf2ird1−/− mice) and GSE195505 (organoids derived from the hiPSCs with the WBS microdeletion and isogenic controls). All other data and code generated are available upon request from the lead contact.
Any additional information required to reanalyze the data in this paper are available from the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice (6–12 weeks old) of both sexes were used. Mice were group housed by sex on a 12–hour light:dark cycle. Generation of CD+/−, PD+/−, and DD+/− murine models of WBS (Li et al., 2009; Segura-Puimedon et al., 2014), Gtf2ird1+/− mice (Young et al., 2008) and Gtf2i+/− mice (Sakurai et al., 2011) have been described previously. Gad2Cre, PVCre, Ai14, Ai93, CaMKIIαttA, and Scnn1aCre mice were purchased from the Jackson Laboratory (JAX) and maintained on mixed CBA-C57BL/6 background mice. CD+/−, PD+/−, and DD+/− mice were backcrossed with C57BL/6J mice. Gtf2ird1+/− mice were obtained from the University of Toronto on 129/CO1 background and backcrossed with CBA mice. Gtf2i+/− mice were obtained from the Icahn School of Medicine at Mount Sinai on C57BL/6J background and backcrossed with CBA mice. Gad2Cre;CD, Gad2Cre;Ai14;Gtf2ird1, and Gad2Cre;Vipr1fl/+ mice were, therefore, on mixed CBA-C57BL/6 background. The care and use of animals were reviewed and approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital.
Postmortem human brain samples
Material from the superior temporal gyrus of eight subjects with WBS (five males, three females; age 17–69 years) and eight age- and sex-matched control subjects was obtained from the NIH NeuroBioBank at the University of Maryland. Three fresh-frozen samples each (one male, two females) from the WBS brains and control brains were used for Western blot analysis. Five fixed samples each from the WBS brains and control brains were received, and four from each condition (three males, one female) were of sufficiently good quality to be used for immunohistochemical analysis.
METHOD DETAILS
Generation of Vipr1–conditional knockout mice
The Vipr1-cKO mouse model was engineered using CRISPR/Cas9 technology and direct embryo injection. Briefly, prior to embryo injection, chemically modified single-guide (sgRNAs; Synthego) were tested for activity in mouse Neuro2a cells stably expressing Cas9 and assayed by targeted next-generation sequencing (NGS) as previously described (Sentmanat et al., 2018). Resulting NGS data were analyzed using CRIS.py (Connelly and Pruett-Miller, 2019). For animal model generation, ten 3– to 4–week-old C57BL/6J female mice from JAX were superovulated with 5 units of pregnant mare’s serum gonadotropin (PMSG; ProSpec) and 48 h later, with 5 units of human chorionic gonadotrophin (hCG; Sigma). After overnight mating with C57BL/6J males, the females were euthanized, and oocytes were harvested from the ampullae. The protective cumulus cells were removed using hyaluronidase, and the oocytes were washed and graded for fertilization by observing the presence of two pronuclei. A mixture of the sgRNAs, Cas9, and ssODNs (single-stranded oligodeoxyribonucleotide) consisting of 60 ng/μL Cas9 protein (St. Jude Protein Production Core), 20 ng/μL of each sgRNA, and 5–10 ng/μL of each ssODN (IDT) were injected into the pronucleus of oocytes. The injected oocytes were then returned to culture media (M16 or Advanced-KSOM, both from Millipore) and later the same day transferred to Day 0.5 pseudo-pregnant fosters. Pups were born after 19 days gestation and were sampled at Days 7–10 for genotyping via targeted NGS. Animals positive for both LoxP-site integration events were weaned at Day 21. At 6 weeks of age, they were backcrossed to C57BL/6J mice and then bred to homozygosity. Editing construct sequences and relevant primers are listed in Table: Materials for generation of Vipr1-cKO mice.
Table:
Materials for generation of Vipr1-cKO mice.
| Name | Sequence (5’ to 3’) |
|---|---|
|
| |
| CAGE289.Vipr1.g3 spacer | AAGUGGGAUAAGAGUUCAUC |
| CAGE289.g3.sense.ssODN | *GGTTTTTGTAGGGGACAATTTAGAAGTGGGAT |
| *AltR modifications | AAGAGTTCATAACTTCGTATAATGTATGCTATAC GAAGTTATGGATCCATCTGGGCCTAGGATGGG TTATAGCCTGGGTTGGGGTTGG |
| CAGE289.DS.F | GGAGCCAAGAGTCCTGAGAAGGCCC |
| CAGE289.DS.R | CACAGGCTTTCGGAGTAGGGGGCCA |
| CAGE290.Vipr1.g10 spacer | AGCCACAGCUAGACCCUUAA |
| CAGE290.DS.F | CCCTCACGTCACGAGCCCAGTCCAA |
| CAGE290.DS.R | TTTGTGCTGATGGGCTGCTGCAGGG |
| CAGE290.g10.anti.ssODN | *CCTCCCTCCTTGGGTAGCCCAGCAGCCACAGC |
| *AltR modifications | TAGACCCTATAACTTCGTATAGCATACATTATAC GAAGTTATGGATCCTAAAGGTAGTTTCCAGATA AGAGCTGGGAACTCCCCAGAT |
Generation of Vipr1-OE transgenic mice
For the generation of the Vipr1-OE vector, full-length mVipr1 cDNA was subcloned into the multiple cloning site of a pCAGGs-LSL-IRES-EGFP backbone by using the following primer sets: mVipr1 F (5′-TAGTGGATCCCCCGGATGCGCCCTCCGAGC-3′) and mVipr1 R (5′-CGAGGTTAACGAATTTCAGACCAGGGAGACCTCCGC-3′) and linearized with restriction enzyme PvuI for pronuclear microinjection. Female C57BL/6J mice (3– to 4–weeks-old) were superovulated with gonadotrophin injections 1 and 2 days prior to the experiment; the first 5 units of gonadotrophin were isolated from pregnant mare serum P, then 48 h later, they were injected with 5 units of hCG. Dams were then mated to C57BL/6J males. Fertilized zygotes were collected the following morning in M2 or Advanced-KSOM media, and cumulus cells were stripped from the zygotes with hyaluronidase. The cytoplasm of each zygote was microinjected with 1–5 ng/μL linearized pCAG-LSL-Vipr1-IRES-eGFP DNA diluted in IDTE (a Tris-EDTA buffer at pH 7.5). After being maintained in culture in M16 or Advanced-KSOM media, the injected zygotes were transferred into the oviducts of pseudo-pregnant females. At 7–10 days of age, pups were sampled for genotyping and fluorescence in situ hybridization (FISH) confirmation of the genomic insertion of the pCAG-LSL-Vipr1-IRES-eGFP transgene. FISH was performed as follows: purified pCAG-LSL-Vipr1-IRES-eGFP DNA was labeled by nick translation using a red dUTP (AF594, Molecular Probes), and control probes were labeled with a green dUTP (AF488, Molecular Probes). Mouse lung fibroblasts from transgenic mice were grown in culture and harvested by conventional cytogenetic methods as a source of metaphase chromosomes. The labeled transgene probe was first hybridized to transgenic metaphases to identify the site of insertion. A second hybridization using the transgene probe and a chromosome-specific control probe was performed to confirm the identity of the chromosome bearing the transgene insertion. Hybridizations were carried out using a hybridization buffer containing 50% formamide, 10% dextran, and 2× saline-sodium citrate buffer (SSC). Fixed slides were denatured in 70% formamide and 2× SSC, at 80 °C. Posthybridization washes were done using 50% formamide and 2× SSC at 37 °C. Slides were mounted in Vectashield mounting medium containing DAPI, and images were acquired using a Nikon Eclipse 80i with a ×100, 1.40-NA Plan Apo objective and CytoVision version 7.7 (Leica Biosystems). CAG-LSL-Vipr1-IRES-GFP mice were crossed with Gad2Cre mice and then with CD+/− mice resulting in Vipr1cOE;CD+/− and Vipr1cOE;WT mice.
Mouse behavioral tests
Innate Frequency Discrimination (Auditory) Acuity test.
Frequency-discrimination acuity was assessed via PPI of the ASR and using a hardware–computer interface (SM1000-II; Kinder Scientific), as previously described (Aizenberg et al., 2015; Blundon et al., 2017). In brief, a background pure tone (16.4 or 9.8 kHz) was played at a sound pressure level (SPL) of 70 dB throughout the session, unless otherwise noted. Each session was split into four blocks. Block 1 consisted of a 5-min acclimation period in which the background tone was played. Block 2 consisted of nine startle trials in which a 120-dB SPL, 20-ms white noise (WN) burst was played. Block 3 consisted of prepulse trials and 10 startle-only trials in a pseudo-random order. Each pre-pulse trial consisted of a 70-dB SPL 80-ms pre-pulse (pure-tone frequency was 0%, 1%, 2%, 4%, 8%, 16%, or 32% lower than that of the background tone), followed by a 120-dB SPL, 20-ms WN startle pulse, and then returned to the background tone after the startle. Every trial in Block 3 was presented 10 times. Block 4 consisted of three startle trials to identify any habituation over the session. The intertrial interval was 10–20 s, and the startle magnitude was the maximum force exerted immediately after the startle pulse. For all trials, .wav files were created using Audacity 2.1.2 (Audacity, open source). PPI percentage was calculated from Block 3 data as follows: [1 − (pre-pulse trial/average startle only trial)] * 100. Values in Block 2 trials were compared with those in Block 4 as an internal control for startle attenuation over the course of the session. Each animal then had a 3-parameter logistic regression curve fitted to the PPI percentages at each pre-pulse frequency to determine the frequency at which 50% of the total acoustic startle inhibition was achieved, subsequently called the FDT; animals with an r2 <0.7 were excluded from further analyses. FDT values were then analyzed using a t-test, a one-way ANOVA, or a paired t-test, as appropriate. Pure tone frequencies and sound intensities were calibrated daily by using the sound level meters NL-52 (Rion Co., LTD) and SMSPL Rev B (Kinder Scientific), respectively. In chemogenetic experiments administered on consecutive days, animals were intraperitoneally (i.p.) injected with DREADD agonist C21 (1 mg/kg in 0.9% saline; Tocris) or vehicle 30 minutes before undergoing the PPI test. Injections were randomized using a within-subject, counterbalanced design to control for treatment order
Auditory Acuity Cued Go/No-go Task.
We attempted to assess frequency discrimination using an auditory-cued Go/No-go task, based on a previously published protocol (Froemke et al., 2012) with a modification that required the mouse to initiate each tone presentation. In brief, we food restricted animals to 90%–85% of their body mass and used 10% sucrose solution as a reward. Mice were weighed 3 times/week and given access to ad lib food for 2 hours after each session. Each session was run using an operant chamber (ENV-307W, MedAssociates Inc., St. Albans, VT) enclosed in a sound-attenuating chamber (ENV-022V). The operant chamber was equipped with a nose poke and a food trough with a dipper that was used to present the reward upon successful performance of the task (see below). Each animal was required to progress through two training stages before starting the Go/No-go task. Both training stages and the Go/No-go task sessions last for 1 hour.
Training Stage 1 (food trough training): Each mouse was given a reward (3-s access) for each head entry into the food trough; access was accompanied by the playing of the target tone (8 kHz, 0.5-s duration, at 80-db SPL). To progress to Training Stage 2, mice were required to have 2 consecutive days of receiving at least 20 rewards.
Training Stage 2 (nose poke to initiate trial): To learn trial initiation, mice were required to put their nose into the nose poke hole, after which the target tone played (8 kHz, 0.5-s duration, at 80-db SPL), and the reward receptacle was raised until the mouse received the reward. The mouse was moved to the second part of Training Stage 2 after obtaining 50 rewards. To progress to the Go/No-go task, the mice were required to initiate a trial and obtain the reward within 3 seconds in at least 50 trials, with a successful reward rate of at least 80% for 2 consecutive days. Animals that did not reach this level of performance after 14 days were eliminated from the experiment.
Go/No-go task: When trials were initiated, as in Stage 2, one of five tones was played (2, 4, 8, 16, or 32 kHz, 0.5-s duration, at 80-db SPL) randomly. To receive a reward, the mouse had to enter the reward trough within 3 seconds of the 8-kHz tone playing; entry after other tones did not receive a reward and were punished with a 7-s time-out period, where all lights were turned off and no trials could be initiated. To assess frequency discrimination, the no-go tones were brought closer to the go tone after the mouse achieved greater than 85% accuracy, such that the second phase was 4, 6, 8, 12, or 16 kHz with the tones of each subsequent phase getting closer to the go tone in the same pattern.
Auditory Brainstem Response test.
ABR experiments were performed as previously described (Chun et al., 2017; Ingham et al., 2011; Mellado Lagarde et al., 2014). Briefly, mice were anesthetized with Avertin (0.6 mg/g bodyweight, i.p.), and ABR was measured using a Tucker Davis Technology (TDT) System III with RZ6 Multiprocessor and BioSigRZ software. Sounds were delivered via the MF-1 speaker in the open-field configuration. ABR waveforms were recorded using subdermal needles placed at the vertex of the skull, below the pinna of the ear, and at the base of the tail. The needles were connected to a low-impedance head stage (RA4LI, TDT) and fed into the RZ6 multiprocessor through a preamplifier (RA4PA, Gain 20×, TDT). ABR waveforms were averaged from 500 presentations of a tone (21 tones/s) in the alternating phase and were band-pass filtered (300 Hz-3 kHz). The ABR threshold was defined as the minimum sound intensity that elicited a wave above the noise level. All ABR experiments were conducted in a sound booth (Industrial Acoustic Company, IAC, Model 120A double wall).
Single-cell electrophysiology
Auditory TC brain slices.
Acute primary TC slices (400-μm thick) containing the left ACx and the left ventral part of the MGv of the thalamus were prepared as previously described mice (Bayazitov et al., 2013; Blundon et al., 2011; Chun et al., 2013; Cruikshank et al., 2002). Briefly, mouse brains were removed and placed in cold (4 °C) dissecting media containing (in mM) 125 choline-Cl, 2.5 KCl, 0.4 CaCl2, 6 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose (300–310 mOsm), equilibrated with 95% O2/5% CO2. TC slices were obtained from the left hemisphere by using a slicing angle of 15° to horizontal. Slices were transferred to ACSF containing (in mM) 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose (300–310 mOsm), equilibrated with 95% O2/5% CO2 at 34 °C for 30 min followed by 1 h at room temperature prior to use. Slices were transferred to a recording chamber mounted on an upright microscope (Olympus) and superfused (~2 mL/min) with warm (30–32 °C) ACSF. Slices were viewed with a CCD camera (Rolera-XR, QImaging) using IR-DIC optics. Thalamorecipient pyramidal neurons in L4 (~ 300 μm from the pia) were identified by soma shape and size and by a large visible apical dendrite projecting toward the pia. If recorded in current-clamp mode, pyramidal neurons were additionally verified as regularly spiking. FS interneurons were identified as having nonpyramidal shape and multipolar dendritic projections from the soma. The FS phenotype was verified by recording in current-clamp mode. Mice with tdTomato genetically expressed in PV+ cells (PVCre;Ai14;CD+/− and PVCre;Ai14;WT) mice or by assessing their soma size, shape, and location. were used in a subset of experiments in which fluorescently labelled soma were targeted using the microscope’s epifluorescence.
Whole-cell recording.
Whole-cell recordings were made with patch pipettes (3–5 MOhm) using a Multiclamp 700B amplifier, digitized (10 kHz) with a Digidata 1440, and recorded using pCLAMP 10 software (all Molecular Devices). In all experiments, membrane potentials were corrected for a liquid junction potential of −10 mV. In voltage-clamp recordings, series resistance, input resistance, and holding current were monitored for stability. During current-clamp recordings, pipette capacitance and series resistance were compensated using the amplifier’s circuits. Input resistance and membrane-resting voltage were monitored during recordings. Cells with series resistance greater than 40 MOhms in voltage-clamp recordings and 30 MOhms in current-clamp recordings or cells that changed resistance values more than 20% over the duration of recordings were rejected. Drugs were added to ACSF or locally applied via continuous pressure ejection from a large-diameter pipette placed in the slice near the recorded cell. Pressure ejection of control ACSF caused no detectable effect on neurons.
For standard voltage-clamp recordings, patch pipettes were filled with an internal solution containing (in mM) 125 CsMeSO3, 2 CsCl, 10 HEPES, 0.1 EGTA, 4 ATP-Mg2, 0.3 GTP-Na, 10 creatine phosphate-Na2, 5 QX-314, and 5 TEA-Cl (pH 7.4, 290–295 mOsm). For current-clamp recordings, internal solution contained (in mM) 115 potassium gluconate, 20 KCl, 10 HEPES, 4 MgCl2, 0.1 EGTA, 4 ATP-Mg2, 0.4 GTP-Na, and 10 creatine phosphate-Na2 (pH7.4, 290–295 mOsm). For voltage-clamp recording of voltage-gated Ca2+ currents, external CaCl2 was replaced with 3 mM BaCl2, 0.5 μM TTX was included in the ACSF, and EGTA and QX-314 were omitted from the internal solution. For voltage clamp I-V curves, cells were hyperpolarized to −90 mV followed by steps of increasing depolarization amplitude (duration as indicated in text/figures). Current intensity was corrected for linear leak current, as determined from a brief −5-mV step from rest. Na+ and Ca2+ current density was quantified as the peak inward current divided by the membrane capacitance. K+ current density was quantified as the steady-state outward current divided by the membrane capacitance. The Ih density was determined by delivering 2-s hyperpolarizing pulses from rest and measuring the inward current “sag” divided by the membrane capacitance. To determine the Ca2+ current threshold, 1-s ramps from −90 to +30 mV were delivered. Responses were leak-subtracted, and the threshold was quantified as the peak of the second derivative of the current signal.
Spontaneous synaptic inputs were recorded with neurons held at −70 mV for excitatory postsynaptic potentials (with or without inhibitory inputs blocked by 100 μM PTX, as indicated) and 0 mV for inhibitory synaptic inputs (with or without excitatory inputs blocked with 3 mM kynurenic acid as indicated). For miniature synaptic events, 0.5 μM TTX was included in the ACSF. Spontaneous activity was recorded for 5–10 min beginning at least 2 min after whole-cell break-in. EPSCs were automatically detected using miniAnalysis (Synaptosoft) as deviations of more than 5× the baseline root mean squared noise level.
Current clamp input–output curves were obtained by delivering 1-s current pulses of increasing amplitude. Rheobase was determined by delivering a current ramp at 300 pA/s and measuring the current intensity that elicited the first spike. Input resistance was calculated either from a small hyperpolarizing test pulse or from the slope of the initial linear response to the ramp. Individual spike properties (threshold, after-hyperpolarization potential [AHP], half-width, etc.) were measured using MiniAnalysis (Synaptosoft). Threshold was determined as the peak of the second differential of the voltage signal. AHP was determined as the negative peak voltage relative to the threshold.
To generate TC input–output curves, TC postsynaptic currents (PSCs) were evoked by current pulses (intensity 0.1–1 mA, duration, 100 μs) delivered to the thalamic radiation via tungsten concentric bipolar electrodes (FHC) using a stimulus isolator (Isoflex; A.M.P.I.). Monosynaptic EPSC amplitude was quantified as the initial slope of the inward current response.
In vivo viral injections
Generation of pAAV-hDLX-Vipr1-T2A-eGFP and pAAV-hDLX-Vipr1-T2A-tdTomato plasmids.
Coding sequences of the mVipr1 (Genbank Accession number: NM_011703.4) were amplified with primers, Vipr1 F (5’-CTTAAGAAAGGTCGACCACCATGCGCCCTCCGAGCCT-3’) and Vipr1 R (5’-TGCCCTCTCCGGATCCGACCAGGGAGACCTCCGC-3’) from cDNA, generated from reverse-transcribed mouse whole-brain RNA using the Superscript First-Strand Synthesis RT-PCR Kit (Invitrogen), inserted into pAAV-hDLX-T2A-eGFP vector plasmid (modified from Addgene plasmid 83895) by infusion cloning (638933, Takara Bio Inc.).
Generation of pAAV-hDlx-Vipr1-T2A-TdTomato plasmid.
The protein-coding sequence of tdTomato was PCR-amplified from pGP-AAV-CAG-FLEX-jGCaMP7s-WPRE (Addgene 104495) by using two PCR primers, tdTomato F (5′-CTTAAGAAAGGTCGACCACCATGGTGAGCAAGGGCGAG-3′) and tdTomato R (5′-CCGCTATCACAGATCACTAGTCTTGTACAGCTCGTCC-3′) and replaced the eGFP-coding sequence of pAAV-hDLX-VIPR1-T2A-EGFP by infusion cloning. The pAAV-hDlx-Flex-GFP-Fishell_6 plasmid was a gift from Dr. Gordon Fishell’s lab (Addgene plasmid # 83895). The pAAV-hSyn-DIO-HA-hM4D(Gi)-IRES-mCitrine was a gift from Bryan Roth (Addgene plasmid # 50455; RRID:Addgene_50455)
Surgery.
Mice were anesthetized with 2% isoflurane (in pure oxygen). Under aseptic conditions, a midline incision was made in the scalp. Virus was injected bilaterally into the primary ACx (250 nL per site at a rate of 30 nL/min; coordinates: 2.2 mm caudal to bregma, 0.3 mm medial to the dorsal insertion of the temporalis muscle onto the skull, and injection depth 0.8 mm).
In vivo two-photon calcium imaging
We used this method in GCaMP6fExN-L4;WT and GCaMP6fExN-L4;CD+/− mice as previously described (Blundon et al., 2017). To selectively express GCaMP6f in L4 excitatory neurons, we crossed Ai93 mice (TIGRE-Ins-TRE-LSL-GCaMP6f) (Madisen et al., 2015) with CamKIIαtTA mice (excitatory neurons specificity) and with Scnn1aCre mice (L4 specificity). We refer to the resultant transgenic mice as GCaMP6fExN-L4 mice. We then crossed those mice with CD+/− mice.
Surgery.
Mice were anesthetized with a mixture of ketamine/xylazine (100/10 mg/kg body weight) and subsequent injections of 50 mg/kg ketamine. Under aseptic conditions, a 2- to 3-g stainless steel headpost was fixed to three miniature screws in the skull and cemented into place with dental cement. Using the headpost to secure the animal’s head, the lateral temporalis muscle was removed to reveal the skull overlying the ACx. A craniotomy was made using a 1.5-mm biopsy punch, and a plastic well was cemented around the craniotomy to hold saline. The overlying dura was carefully removed, and a 3-mm glass coverslip was cemented over the cranial window. To reduce postoperative pain, decrease inflammation, and eliminate infection, each mouse was given subcutaneous injections of meloxicam (2 mg/kg), Baytril (5 mg/kg), dexamethasone (2 mg/kg), and amoxicillin (0.3 mg/mL) in the drinking water. The animals received this postoperative care for the duration of the experiments.
Imaging.
After recovery, mice were acclimated to the head-fixed setup. For at least 3 days prior to imaging, the animal was stabilized on a rotating disc under the two-photon microscope while the head was secured in place with the headpost. Acclimation began with 15-min intervals and progressed to 1-h intervals. During acclimation and imaging, animals were in the dark, surrounded by a sound-attenuating chamber. To determine differences in spontaneous firing patterns between genotypes and sound-evoked firing patterns, animals were imaged during 30 min of silence and 30 min of sound delivery. GCaMP6f fluorescence in L4 neurons located 300–400 μm beneath the pial surface was monitored with the Olympus multiphoton imaging system (FVMPE-RS, FluoView FV1000) and an Insight tunable femtosecond-pulsed laser unit (Spectra-Physics). Neurons expressing GCaMP6f were imaged with a 25× water immersion objective (NA 1.05, Olympus XPlan N) using an excitation wavelength of 930 nm with a resonant scanner at a rate of 10 frames/s with a field of view of 512 μm × 512 μm. Tones were generated with OpenEx software and an RZ6 signal processor (TDT) with 100-MHz processing speed and delivered through a free-field electrostatic speaker placed 10 cm from the contralateral ear of the animal. During sound-delivery experiments, the sound-stimulation software triggered the start of the microscope-scanning software. GCaMP6f fluorescence was measured in response to pure tones, with frequencies ranging from 4.8 to 29.4 kHz, intensities of 10- to 70-dB SPL (60- to 0-dB attenuation, respectively), and duration of 50 ms played at 1 Hz in pseudo-random order. Cells were included in the analysis if they were in focus during both the sound and silent conditions.
Video and calcium data processing.
Videos were corrected for movement artifacts with a custom Matlab routine. Each frame was aligned to a stable reference frame using a nonrigid image-registration algorithm, as previously described (Blundon et al., 2017; Rueckert et al., 1999). Following stabilization, video segments with excessive movement artifacts were pruned from the video sequence by using a custom Matlab code. For automatic cell identification, a custom Fiji (Schindelin et al., 2012) macro was developed that first involved image background subtraction followed by image down-sampling. To identify regions of interest (ROIs) corresponding to active cell soma, we used the Ilastik software package (Berg et al., 2019) to train a classifier to segment all cell bodies, frame by frame, that had calcium intensities above the local background. The background levels were estimated by the classifier by the paired manual annotations from inside and outside cell bodies. We used a background-subtracted image as an input for the cell segmentation. We used a temporal moving median filter with a 10-s window to remove the background intensity for all pixels in the 512 × 512 time-lapse image. We used the ROIs defined by the segmented cell bodies in each frame to calculate the mean intensity of the calcium signal. The mean fluorescence intensity and the frame number, location, and area of the ROIs of the image sequence were stored in comma-separated value (CSV) files.
Fluorescence signals from active cell soma were normalized to the baseline, and ΔF/F of the peak amplitudes was calculated as the change in fluorescence over baseline fluorescence levels × 100%. We calculated the ΔF/F image by using the following equation:
where F was the raw calcium signal image, and Fmean was the corresponding temporal moving mean filter with a 10-s window.
We next assigned cell ID numbers to active cell soma by using a custom R script. The XY coordinates of the active soma detected in the entire recording of a cell were typically clustered within a few pixels. The spread of the coordinates of the centers depended on the degree-of-motion artifact. We used hierarchical clustering of the XY coordinates to identify individual cells. We adjusted the hierarchical tree-cut height parameter to minimize over-segmentation (multiple cell IDs assigned to one biological cell) and under-segmentation (single-cell ID assigned to multiple biological cells).
Deconvolution.
We used the OASIS software package (Friedrich et al., 2017) to deconvolve raw calcium ΔF/F traces. We used 45 ms as the rise time and 142 ms as the decay time parameters (Chen et al., 2013). We used all the local maxima of the deconvolved trace as cell-firing events.
Decoder.
To analyze the differences in sound discrimination based on L4 excitatory activity, we constructed a linear frequency and cue decoder based on the deconvolved calcium traces, inspired by the linear decoder (Kingsbury et al., 2020). Deconvolved traces were z-scored. To reduce the dimensionality of the training data, principal component analysis (PCA) was performed, and the top 20 principal components (PCs) were extracted. A logistic-regression model that includes the projections onto the PCs as input was used. For the frequency decoder, the model was trained to predict the frequency of the cue that was most recently presented. Unless otherwise noted, this was trained on and applied to only the cue frame and the four frames after the cue (extending 400 ms after tone presentation). The sound–no sound decoder was trained to predict whether a frame was a cue frame or not. For this decoder, only cue frames and the five frames preceding a cue frame were used. Five-fold cross-validation was used to estimate the decoder performance as follows: data were divided into five blocks and from these blocks, five train/validation splits were constructed, where each split used one block for validation and the remaining four blocks for training. Mean validation accuracies over all splits were reported in the text. To avoid leakage, splits were contiguous periods of the entire recording and were performed such that no individual cue-presentation period was split between train and validation.
To determine what factors most contributed to the significant difference in frequency discrimination between WT and WBS, we performed the same decoder analysis using modifications or subsets of the original data described above. First, to investigate if the difference in discriminability was the result of different numbers of cells being reliably imaged and analyzed in WBS vs WT, we balanced the data sets such that the input to the decoder for all recordings was a set of 50 randomly selected cells of the entire population. Second, to investigate if the difference in discriminability was a result of different proportions of sound-responsive cells in WBS vs WT, we matched these proportions in the following way: a cell was considered sound responsive if its mean deconvolved signal at cue frames was more than 1.96 standard errors above its mean baseline level (mean activity over the five pre-cue frames). For each recording, cells were randomly selected, such that 20% were sound-responsive. Finally, to investigate if the difference in discriminability was a result of the choice of frames over which the decoding was performed, we repeated the frequency-decoding analysis using only the cue frame and the frame immediately after it (100 ms after tone presentation). This decoder analysis was performed with custom code in python using the scikit-learn package (Pedregosa et al., 2011).
RNA-seq analysis
Isolation of GAD2+ cortical interneurons.
To isolate interneurons, we generated Gtf2ird1−/− mice that express tdTomato under control of the interneuron-selective Gad2 promoter (Gad2Cre;Ai14;Gtf2ird1−/− mice) and sorted tdTomato+ cells from the cortex of Gad2Cre;Ai14;Gtf2ird1+/+ and Gad2Cre;Ai14;Gtf2ird1−/− mice. Mice were euthanized via cervical dislocation and decapitated. The cortex was isolated and washed with cold Earle’s Balanced Salt Solution (LK003188, Worthington Biochemical Company) and then placed in plain neurobasal medium (21103049, Thermo). The tissue was dissociated with activated papain (LK003178, Worthington Biochemical Company) and DNAse I (DN25, Sigma-Aldrich) for 30 min at 37 °C. Then it was triturated by repeated gentle pipetting with a 2-mL glass pipette. Tissue digestion was stopped by adding reconstituted BSA-ovalbumin solution (LK003182, Worthington Biochemical Company). The resulting single-cell suspension was filtered through a 40-μm cell strainer (BD 352350), centrifuged at 300 ×g for 5 min at room temperature, washed once, and resuspended with cold Earle’s Balanced Salt Solution. The single-cell suspension was then FACS-sorted by an Aria Fusion cytometer (BD Biosciences) equipped with blue (488 nm), yellow/green (561 nm), red (640 nm), and violet (405 nm) lasers to isolate tdTomato+ cells. A 100-μm nozzle was used for sorting, and BD FACS Diva Software (BD Biosciences) was used for data acquisition and analysis.
Stranded total RNA-seq.
Total RNA was isolated from brain tissue or organoids by using mirVana RNA isolation kit (ThermoFisher), quantified using the Quant-iT RiboGreen RNA assay (ThermoFisher), and quality checked by the 2100 Bioanalyzer RNA 6000 Nano assay (Agilent) or 4200 TapeStation High Sensitivity RNA ScreenTape assay (Agilent) prior to library generation. Libraries were prepared from total RNA with the TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer’s instructions (Illumina, PN 20020599). Libraries were analyzed for insert-size distribution using the 2100 BioAnalyzer High Sensitivity kit (Agilent), 4200 TapeStation D1000 ScreenTape assay (Agilent), or 5300 Fragment Analyzer NGS fragment kit (Agilent). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (ThermoFisher) or by low-pass sequencing with a MiSeq nano kit (Illumina). Paired-end 100-cycle sequencing was performed on a NovaSeq 6000 (Illumina).
RNA-seq data analysis.
Total stranded RNA-seq data were processed by the internal AutoMapper pipeline. Briefly, the raw reads were first trimmed (Trim-Galore version 0.60), then mapped to the human genome assembly GRCh38 (STAR v2.7; Dobin et al., 2013). The gene-level values were then quantified (RSEM v1.31; Li and Dewey, 2011) based on GENCODE annotation (v31). Low-count genes were removed from the analysis by using a CPM cutoff corresponding to a count of 10 reads and only confidently annotated (levels 1 and 2 gene annotation), and protein-coding genes were used for differential-expression analysis. Normalization factors were generated using the TMM method (Robinson and Oshlack, 2010); counts were normalized using voom (Law et al., 2014); and normalized counts were analyzed using the lmFit and eBayes functions (R limma package version 3.42.2; Smyth, 2005). The significantly up- and downregulated genes were defined by an adjusted p-value <0.05.
Gene Ontology enrichment analysis.
GO enrichment analysis was performed as previously described (Reimand et al., 2019). Briefly, differentially expressed mRNAs (FDR <0.05) were ranked by p-value. Enrichment analysis was performed using g:Profiler (version e105_eg52_p16_e84549f) (Raudvere et al., 2019), with a custom background gene list consisting of all mRNAs detected (> 10 counts) in each sequencing experiment. Enriched GO terms relevant to neurobiology were selected for graphing. Dot plots were prepared in R using ggplot2.
Quantitative RT-PCR
Total RNA was isolated from the tissue or cells with Aurum Total RNA Mini kit (7326820, Bio-Rad). The iScript kit (1708840, Bio-Rad) was used to synthesize cDNA from the isolated total RNA and the quantitative RT-PCR was performed using SYBR Green (4309155, Life Technologies) according to manufacturer’s instructions. Primers (Table: qPCR primers) were designed using Beacon Designer (Premier Biosoft), and the specificity of each primer pair was manually verified using ClustalOmega (EMBL). Absence of primer-dimers and contamination with genomic DNA was verified with melting curves for each run. Expression levels of all genes were normalized to GAPDH or U6 for each biological replicate. Samples from each mouse were run in two or three technical replicates.
Table:
qPCR primers
| Gene | Primer | Sequence |
|---|---|---|
|
| ||
| mm Gapdh | F | GAGAAACCTGCCAAGTATG |
| R | CTCAGTGTAGCCCAAGATG | |
| mm B3glct | F | CCTTGTTACCGCACTTTTCT |
| R | TGTAGTCTCGTCTCTTCTTCAC | |
| mm Cdk5rap1 | F | ATGCGGAGAGGATATTCAAGA |
| R | TAAGGCTCACACCTGGGATA | |
| mm Chek2 | F | ATTGTCTAATCAAGATCACTGA |
| R | CCACATAAGGTTCTCATCAA | |
| mm Dgkb | F | AACTTAATCCGATCCTTCAT |
| R | GATAGTTGTCATTCCTCCTT | |
| mm Dusp6 | F | AATTCCTATCTCGGATCACT |
| R | GGCTTCATCTATGAAAGAAATG | |
| mm Fry | F | CTGGAAAGCATTGAAATCAC |
| R | TTCTTGTTCTCTGGTCTTCT | |
| mm Fzd2 | F | GCCTGTGGAAGCTGTTGGATA |
| R | GGAGCGAGGAGAAAGG GAAAT | |
| mm Gtf2ird | F | AGAGATAGCAATGTTGAGG C |
| R | TGAAGGATCTGAGACCGTAA | |
| mm Idua | F | TGGAACTTTGAGACTTGGAA |
| R | GTAATTCAGGAAGCCTTGTG | |
| mm Lyzl4 | F | CAGGGCATAGGAGAACATTC |
| R | GATCCTGCTCCATGAGAAAC | |
| mm Mtus 2 | F | GTCCAAGAACTGATGTCTACTC |
| R | CACCTGGTCCTGTAATGTCA | |
| mm Plcd3 | F | TACTTTATCTCGTCCTCTCA |
| R | CAAAGGCCCTAATATAAGC C | |
| mm Plcel | F | AAGCATCCATCTCAGAATCC |
| R | AATCTCATCACAAGGTCTTCAA | |
| mm Vegfa | F | CAGATGTGAATGCAGACCAA |
| R | TTTGACCCTTTCCCTTTCCT | |
| mm Vip | F | AAGCAGACTCTGACATCTTG |
| R | CTGGCATTTCTTGACACATC | |
| mm Vipr1 | F | ACCATCATCAACTCCTCACT |
| R | CAGGATGAAGTTCACCAAGAT | |
| mm U6 | F | CGCTTCGGCAGCACATATAC |
| R | TTCACGAATTTGCGTGTCAT | |
| GFP | F | CTACGGCAAGCTGACCCTGAAGTT |
| R | CTCGGCGCGGGTCTTGTAGTT | |
Abbreviations: F, forward; mm, Mus musculus; R reverse
Western blot analysis
Brain tissue was resuspended with RIPA buffer containing protease inhibitors and sonicated twice at 15% amplitude for 10 s in the sonifier (Bronson) on ice. Supernatant was collected from total-protein lysate by centrifugation at 13,000 ×g for 10 min at 4 °C. After quantification of the supernatant fraction by BCA assay (23225, ThermoFisher), 10 mg of the protein sample was fractionated using the SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a PVDF membrane (88518, ThermoFisher). After incubation with 5% (wt/vol) nonfat dry milk in TBST (10 mM Tris. pH 8.0, 150 mM NaCl, and 0.5% (vol/vol) Tween 20) for 30 min, membranes were incubated with anti-VIPR1 (1:250 dilution; PA3–113, ThermoFisher) or anti-ACTB (actin) (1:5,000 dilution; A5316, Sigma-Aldrich) antibodies at room temperature for 1 h. Membranes were washed for 5 min three times and incubated with a 1:3,000 dilution of horseradish peroxidase–conjugated anti-rabbit or anti-mouse antibodies (SC-2054 or SC-2005, Santa Cruz Biotechnology) at room temperature for 1 h. Blots were washed with TBST three times and developed using the ECL system (34075, Pierce Biotechnology Inc.).
Histology and immunohistochemistry
Mice were deeply anesthetized and intracardially perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4), and brains were fixed overnight. Each brain was sliced (50 μm) coronally with a vibratome (Leica). The brain sections were preincubated in sodium citrate buffer (10 mM citrate buffer, pH 6.0) at 80 °C for 20 min, cooled to room temperature, and washed in 1× PBS for 20 min twice. Sections were incubated in PBS-blocking buffer (5% goat serum, 3% BSA, 0.2% Triton X100, in PBS) for 1 h at room temperature and incubated with the following primary antibodies: VIPR1 (1:250, PA3-113, Invitrogen), parvalbumin (PV) (1:5000, PV235, Swant), GFP (1:1000, ab13970, Abcam), GABA (1:1000, A2052, Sigma Aldrich) for 48 h at 4 °C. Appropriate Alexa dye–conjugated secondary antibodies (1:1000, Thermo Fisher Scientific) were used to detect primary antibody binding for 48 h at 4 °C. DAPI (Invitrogen) was used as the nuclear counterstain. Images of immunostained postmortem human brain sections were quantified using Fiji (ImageJ). PV-stained sections were thresholded and used to automatically generate ROIs around PV+ cell bodies. Average pixel intensity of VIPR1 staining of the same slices was quantified within those ROIs.
Generation of hiPSCs with isogenic WBS microdeletion in culture
The NSUN5–GTF2IRD2 heterozygous microdeletion was introduced into TP-190a hiPSC clones by using the CRISPR/Cas9 method. Three TP-190a hiPSC clones were obtained by reprogramming dental pulp stem cells from a healthy male (ALSTEM) by using episomal plasmids. All three clones expressed pluripotency markers, had a normal karyotype (G-Banding and SNP Microarray), and displayed high neural differentiation potential. One clone (#2) was selected for differentiation experiments. TP190a hiPSCs were pretreated with StemFlex (Thermo Fisher Scientific) supplemented with 1× RevitaCell (Thermo Fisher Scientific) for 1 h. Then, approximately 2×106 cells were transiently co-transfected with precomplexed ribonuclear proteins consisting of 150 pmol of each chemically modified sgRNA, 100 pmol SpCas9 protein (St. Jude Protein Production Core), and 500 ng pMaxGFP (Lonza). The transfection was performed via nucleofection (Lonza, 4D-Nucleofector™ X-unit) using solution P3 and program CA-137 in a large (100 μL) cuvette according to the manufacturer’s recommended protocol. At 12 days posttransfection, cells were sorted based on viability and plated onto Vitronectin XF (Stem Cell Technologies)–coated plates into prewarmed (37 °C) StemFlex media supplemented with 1× CloneR (Stem Cell Technologies). Clones were expanded, screened, and verified for the desired deletion via Sanger sequencing. Zygosity was confirmed using 5’, 3’, and internal primers. Editing construct sequences and relevant primers are listed in Table: Materials for generating an isogenic WBS microdeletion in hiPSCs. Of note, the CAGE865.GTF2IRD2.g3 sgRNA was designed to a unique sequence in the TP190a genome that differs from the reference genome.
Table:
Materials for generating an isogenic WBS microdeletion in hiPSCs.
| Name | Sequence (5’ to 3’) |
|---|---|
|
| |
| CAGE636.NSUN5.g11 spacer | UUGAACGGGUCGAGGUGCCA |
| CAGE865.GTF2IRD2.g3 spacer | AAUGGCGGCGUCGGCGGCGU |
| CAGE865.GTF2IRD2.DS.Deletion.F2 | CCCCGAAGCGTGCTCGT |
| CAGE636.NSUN.DS.Deletion.R2 | GCGGCTCTTTGCTGTCTCTT |
| CAGE636.DS.internal.F | ACTGACCAGCACACCAACAA |
| CAGE636.DS.internal.R | GCTCAACGGTGGAAAGAGGA |
| CAGE636.DS.5’.F | GGGGCCGTTTCTCTTGCAGGCTAGC |
| CAGE636.DS.5’.R | TCTTTCTCTTTGGGGCTGGGCTGGG |
| CAGE865.DS.3’.F | AAAAAGGAGGGCGAGTGGCGAGCAG |
| CAGE865.DS.3’.R | CCCCACCCTCACACCTCTGGTCCTG |
The control hiPSC line TP-190a and the isogenic microdeletion line TP-190a-NSUN-GTF2IRD2-DEL clone 2F9 were maintained in culture on hES-qualified Matrigel (4354277, Corning) in complete mTeSR1 (85850, STEMCELL Technologies) at 37 °C. The cultures were passaged with Versene (15040066, ThermoFisher).
Organoids
Human organoids were generated using a method adapted from a previously published protocol for cerebral organoid production (Rai et al., 2021). Briefly, hiPSC cultures were dissociated into single cells with Accutase (AT-104, Innovative Cell Technologies) and plated into low-attachment 96-well V-bottom plates (MS-9096VZ, Sbio) at 9000 cells/well, in EB media (DMEM:F12, 20% Knockout Serum Replacement (10828, Life Technologies), 3% ES-FBS (ES-009-C, SIGMA), 1× Glutamax (Gibco), 1× β-mercaptoethanol (2020-07-30, Gibco), 1× antibiotic-antimycotic (Gibco) supplemented with 5 μM SB-431542 (TGFβ inhibitor, 1614, Tocris), 2 μM dorsomorphin (3093, Tocris), 3 μM IWR1e (Wnt inhibitor, 681669, EMD Millipore), 1% v/v growth factor–reduced Matrigel (354230, Corning), and 2 μM thiazovivin (72254, STEMCELL Technologies). Half the media was replaced on Day 2. On Days 4 and 6, half the media was replaced with GMEM KSR media (GMEM, 20% KSR, 1× NEAA (Gibco), 1× pyruvate (Gibco), 1× β-mercaptoethanol, 1× antibiotic-antimycotic) supplemented with 5 μM SB-431542, 3 μM IWR1e, 2.5 μM cyclopamine (72074, STEMCELL Technologies) and 2 μM thiazovivin. On Day 8, half the media was replaced with GMEM KSR media supplemented with 5 μM SB-431542, 3 μM IWR1e, and 2.5 μM cyclopamine. On Days 10, 12, 14, and 16, half the media was replaced with GMEM KSR media supplemented with 5 μM SB-431542 and 3 μM IWR1e. On Days 18 and 20, half the media was replaced with CBO N2 media (DMEM:F12, 1× chemically defined lipid concentrate (11905-031, Life Technologies), 1× N2 supplement (17502-048, Gibco) and 100× antibiotic-antimycotic) supplemented with 1× B27 supplement without vitamin A (12587-010, Gibco), 10 ng/mL bFGF (78003.1, STEMCELL Technologies), and 10 ng/mL EGF (AF-100-15-100UG, Peprotech). On Day 22, organoids were transferred to a magnetic stir bioreactor (BWS-S03N0S-6, ABLE Corporation) in CBO N2 media supplemented with 1× B27 supplement without vitamin A, 10 ng/mL bFGF, and 10 ng/ml EGF (AF-100-15-100UG, Peprotech), and agitated at 4 rpm. Half the media was replaced on Days 24, 26, and 28. On Day 30, the media was changed to CBO FBS media (DMEM:F12, 1× chemically defined lipid concentrate (11905-031, Life Technologies), 1× N2 supplement, 10% ES-FBS, 5 μg/mL heparin, and 1× antibiotic-antimycotic) supplemented with 1× B27 supplement without vitamin A. Complete media was replaced every 4 days. On Days 42 and 46, the media was changed to CBO FBS media supplemented with 1× B27 supplement without vitamin A, 10 ng/mL BDNF (450-02, Peprotech), and 10 ng/mL GDNF (450-10, Peprotech). Starting Day 50, the media was changed to BrainPhys media (05790, STEMCELL technologies) supplemented with 1× N2 supplement, 1× B27 supplement without vitamin A, 10 ng/mL BDNF, and 10 ng/mL GDNF. Complete media was replaced every 4 days. Starting at Day 35, large cerebral organoids were pinched into two halves by using a pair of sterile forceps; this was repeated once every 5–7 days to avoid large necrotic centers.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistics were calculated using Excel (Microsoft), Sigmaplot (Systat), Prism (Graphpad), R, or Python. Bar graphs, box plots, and violin plots show means ± SEM; overlaid dots show individual measurements. Statistical comparisons with significant results are noted in the text or figure legends. Unless otherwise noted, distributions were tested for normality (Shapiro-Wilk test) and equal variance (Brown-Forsythe test). If the distribution passed, a paired or unpaired t-test was performed. If it failed, a rank sum test or signed rank test was performed. To compare more than two distributions, one-way, two-way, or repeated measures ANOVAs were performed. To compare cumulative distributions, a Kolmogorov-Smirnov test was used. Significance was designated as P < 0.05. Comparisons with P > 0.05 were not reported.
Supplementary Material
A. Recordings from individual WT and CD+/− mice of normalized PPI magnitude, as a function of frequency difference between 9.8-kHz background and pre-pulse tones. Points and error bars are mean ± SEM of 10 repeated measurements in the same animal. Solid lines are linear-regression fits to these points; dotted lines are frequency-discrimination thresholds (FDTs).
B. The FDT is smaller in CD+/− (n = 6) vs WT (n = 5) mice. Rank sum test, *P = 0.009.
C. Time course of trial-initiation learning (stages 1 and 2) of a Go/No-go auditory-discrimination task. All WT (n = 12) and CD+/− (n = 5) mice learned the location of a food trough, but only 1 CD+/− mouse learned trial initiation after 2 weeks of training, compared to 7 WT mice.
D. The acoustic startle response (ASR) is the same in WT (n = 12) and CD+/− mice (n = 12).
E. The threshold sound level needed to elicit the acoustic brainstem response (ABR) is not different between WT (n = 19) and CD+/− mice (n = 17).
F. Spontaneous excitatory activity in thalamic relay neurons (Left, scheme) is normal in CD+/− mice. (Right, top) Traces of sEPSCs from WT and CD+/− thalamic relay neurons. (Right, bottom) The sEPSC frequency and amplitude are not different in WT (n = 7) and CD+/− (n = 8) neurons.
G. Scheme of stimulating (stim.) ascending TC input from the auditory thalamus (MGv) and recording from an L4 pyramidal neuron from the ACx in an auditory TC brain slice.
H. Representative traces showing evoked EPSC in response to a pair of presynaptic stimuli (arrows) in WT and CD+/− mice.
I. The EPSC slope in response to the first stimulus, as a function of stimulus amplitude, is not different between WT (n = 19) and CD+/− (n = 25) mice.
J. The paired-pulse ratio is not different between WT (n = 9) and CD+/− (n = 9) mice.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
A. (Top) Scheme of whole-cell pyramidal neuron recording in the auditory cortex (ACx). (Bottom) Traces showing voltage and action potential (AP) responses to depolarizing current injection in WT and CD+/− pyramidal neurons.
B. All intensities of current injection elicit similar frequency AP responses in WT (n = 15) and CD+/− (n = 12) pyramidal neurons.
C. Traces showing voltage and AP responses to a current ramp (0–300 pA, 1 s) in WT and CD+/− pyramidal neurons.
D. The ramp current threshold that elicits APs (rheobase), resting membrane potential (RMP), and input resistance measured from the initial linear ramp response are the same in WT (n = 16) and CD+/− (n = 21) mice.
E. Traces of individual APs from a train of APs elicited by current injection in WT and CD+/− pyramidal neurons.
F-I. AP peak amplitude (F), half-width (G), threshold (H), and after-hyperpolarization amplitude (AHP) (I) do not differ between WT (n = 15) and CD+/− (n = 12) pyramidal neurons.
J. (Top) Scheme of thalamic excitatory (relay) neuron recording location in the MGv. (Bottom) Traces showing voltage and AP responses to depolarizing current injection in WT and CD+/− neurons.
K. All intensities of current injection elicit similar frequency AP responses in WT (n = 9) and CD+/− (n = 8) neurons.
L. Rheobase measured as the threshold current step to elicit APs (Left: WT, 9 neurons and CD+/−, 8 neurons), RMP (Middle: WT, 12 neurons and CD+/−, 12 neurons), and input resistance (Right: WT, 12 neurons and CD+/−, 12 neurons) measured from small subthreshold current steps did not differ between genotypes.
M. Traces of individual APs from a train of APs elicited by depolarizing current injection in WT and CD+/− neurons.
N-Q. AP peak amplitude (N), half-width (O), threshold (P), and AHP (Q) did not differ between WT (n = 9) and CD+/− (n = 8) neurons.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
(Left) Scheme of whole-cell voltage-clamp recording from L4 fast-spiking (FS) interneurons in the ACx.
A. Representative current responses to increasing amplitude voltage steps from −90 mV. Na+ current was present as a fast inward current (arrow).
B. Current–voltage relations of peak Na+ current density in WT (n = 16) and CD+/− (n = 20) interneurons. Na+ current density did not differ at any voltage.
C. Representative current responses to increasing amplitude voltage steps from −90 mV in the presence of 0.5 μM TTX, 3 mM KYN, and 100 μM PTX. K+ current was present as a sustained outward current.
D. Current–voltage relations of steady-state K+ current density in WT (n = 19) and CD+/− (n= 14) interneurons. K+ current density did not differ at any voltage.
E. Representative current responses to hyperpolarizing voltage steps from rest in the presence of 0.5 μM TTX. Ih current was present as a slowly developing sag inward current.
F. Current–voltage relations of Ih current density in WT (n = 5) and CD+/− (n = 6) interneurons. Ih current density did not differ at any voltage. RM ANOVA P = 0.906.
Averaged data are presented as the mean ± SEM.
A. Time course of trial-initiation learning (stages 1 and 2) of a Go/No-go auditory-discrimination task. All WT (n = 10) and PD+/− (n = 6) mice learned the location of a food trough; 0 PD+/− mice and 6 WT mice learned trial initiation after 2 weeks of training.
B. Comparison of normalized FDT values across all WBS and single-gene–deficient genotypes. Comparable decreases in FDT are observed in CD+/−, PD+/−, Gtf2ird1+/−, and Gtf2ird1−/− mice.
C-F. The ASR did not differ between WT (n = 14) and PD+/− (n = 12) mice (C); between WT (n = 8) and DD+/− (n = 13) mice (D); and across WT (n = 9), Gtf2ird1+/− (n = 21), and Gtf2ird1−/− (n = 13) mice (E); or between WT (n = 7) and Gtf2i+/− (n = 9) mice (F).
Averaged data are presented as mean ± SEM with individual measurements overlaid as dots.
A. Tone-frequency decoder fails to identify a difference in accuracy between WT and CD+/− neurons after the window for analysis was reduced from 400 ms to 100 ms after tone presentation.
B. Deconvoluted GCaMP6f responses in WT and CD+/− excitatory neurons show more sustained activity in mutant cells.
C. CD+/− excitatory neurons have a longer temporal autocorrelation range of individual responses than do WT neurons. Rank sum test *P =0.004.
D, E. Improved accuracy of frequency decoding in the ACx of CD+/− mice persisted in random subsets of cells with matching proportions of tone-responsive to nonresponsive cells (D, rank sum test *P = 0.009) or random subsets with matching numbers of cells (E, rank sum test *P = 0.002).
Data are presented as interquartile ranges (quantiles 5-25-50-75-95).
A. Dot plot showing Gene Ontology (GO) term enrichment among upregulated mRNAs in Gtf2ird1−/− interneurons relative to WT. GO:BP, biological processes; GO:MF, molecular functions.
B. Relative fold change of several genes, as measured by qPCR analysis of GAD2+ cortical interneurons from CD+/− mice (n = 4 mice) compared to WT controls (n = 4 mice). Genes that were downregulated in RNA-seq from Gtf2ird1−/− mice are to the left of the graph, and those that were upregulated are to the right. Unpaired t-test values: Fry *P <0.001; Mtus2 *P = 0.001; Idua P = 0.193; Dusp6 P = 0.087; Cdk5rap1 P = 0.137; Chek2 *P = 0.003; Plce1 *P <0.001; and Dgkb *P <0.001.; Fzd2 P = 0.994; B3glct P = 0.376; Vegfa P = 0.325; Plcd3 P = 0.389.
C. An ~1.57-Mb heterozygous microdeletion in human chromosome 7q11.23 was created in hiPSCs from a healthy control by using two single-guide RNAs (sgRNAs, small arrows).
D. The resulting heterozygous clones have one NSUN5–GTF2IRD2 microdeletion allele and one intact allele.
E. Relative fold–change values for individual genes within the NSUN5–GTF2IRD2 locus (shaded area), flanking genes, and VIPR1 in a healthy control organoid and in NSUN5–GTF2IRD2+/− organoids. All unpaired t-test values, *P <0.05. WBS genes with no expression in organoids are not shown.
F. Dot plots showing GO term enrichment among downregulated mRNAs in brain organoids carrying the WBS deletion, relative to isogenic control organoids. GO:BP, biological processes; GO:CC, cellular components; GO:MF, molecular functions.
Averaged data are presented as the mean ± SEM.
A. Scheme for engineering the Vipr1–conditional knockout (cKO) mouse. The sgRNAs were designed to target the 5’- and 3’-ends of exon 2 of the mVipr1 gene. An ssODN consisting of 40-bp homology arms flanking a LoxP site (arrowhead) and BamHI-restriction site (for downstream genotyping) was designed to insert the LoxP site directly into each corresponding sgRNA cut site (two ssODNs total). Founder animals containing both LoxP sites on the same allele were bred to homozygosity. Floxed Vipr1 animals were crossed with Gad2Cre mice to create Gad2Cre;Vipr1fl/+ mice. Excision of exon 2 after Cre expression results in a premature stop codon (stop sign) in exon 3, which stimulates nonsense-mediated decay and tissue-specific knockout.
B. Vipr1 is reduced in the cortex of Vipr1 cKO mice (Gad2Cre;Vipr1fl/+ and Gad2Cre;Vipr1fl/fl mice). The qPCR quantification of the Vipr1 level in WT (n = 24 replicates; 8 mice), Gad2Cre;Vipr1fl/+ (n = 15 replicates; 5 mice), and Gad2Cre;Vipr1fl/fl mice (n = 12 replicates; 4 mice). One-way ANOVA: WT vs Gad2Cre;Vipr1fl/+ *P = 0.012; WT vs Gad2Cre;Vipr1fl/fl *P <0.001; Gad2Cre;Vipr1fl/+ vs Gad2Cre;Vipr1fl/fl *P = 0.012.
C. Acoustic startle response (ASR) is normal in Vipr1 cKO mice.
D. Low-magnification (top) and high-magnification (bottom) images of the ACx of Gad2Cre;Ai14 mice expressing tdTomato in interneurons injected with AAV-hDlx-Vipr1-GFP.
E. ASR is unchanged in Gtf2ird1-deficient mice after overexpression of Vipr1 in the ACx interneurons by using AAV-hDlx-Vipr1-GFP.
F. ASR is unchanged in Gad2Cre;CD+/− mice after overexpression of Vipr1 in the ACx interneurons by using AAV-CAG-Flex-Vipr1-GFP.
G. Scheme for engineering the Vipr1 conditional overexpression (cOE) mouse. Transgenic mice expressing the CAG-LoxP-Stop-LoxP-Vipr1-IRES-GFP construct were crossed with Gad2Cre mice to produce Vipr1cOE mice.
H. Validation of Vipr1cOE mice by qRT-PCR. Rank sum test *P = 0.024 (WT [Cre−] n=3 replicates; Vipr1cOE n=6 replicates).
I. ASR is normal in CD+/− mice crossed with Vipr1cOE mice.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-VIPR1 | ThermoFisher | Cat#: PA3-113; RRID: AB_2273050 |
| Anti-β-actin | Sigma Aldrich | Cat#: A5316; RRID: AB_476743 |
| Anti-rabbit (western) | Santa Cruz Biotechnology | Cat#:SC-2054; RRID: AB_631748 |
| Anti-mouse | Santa Cruz Biotechnology | Cat#: SC-2005; RRID: AB_631736 |
| Anti-parvalbumin | Swant | Cat#: PV235; |
| Anti-GFP | Abcam | Cat#: ab13970; RRID: AB_300798 |
| Anti-GABA | Sigma Aldrich | Cat#: A2052; RRID: AB_477652 |
| Alexa fluor 568 goat anti-mouse | ThermoFisher | Cat#: A11004; RRID: AB_2534072 |
| Alexa fluor 488 goat anti-chicken | ThermoFisher | Cat#: A11039; RRID: AB 2534096 |
| Alexa fluor 488 goat anti-rabbit | ThermoFisher | Cat#: A11008; RRID: AB_143165 |
| Bacterial and virus strains | ||
| pAAV-hDLX-Vipr1-T2A-eGFP | This paper | n/a |
| pAAV-hDLX-Vipr1-T2A-tdTomato | This paper | n/a |
| rAAV-hSyn-DIO-hM4Di-IRES-mCitrine | Addgene | Plasmid#: 50455; RRID: Addgene_50455 |
| AAV-CAG-Flex-Vipr1-GFP | This paper | n/a |
| AAV-CAG-Flex-GFP | This paper | n/a |
| Biological samples | ||
| Human superior temporal lobe samples | NIH NeuroBioBank | n/a |
| Chemicals, peptides, and recombinant proteins | ||
| PG-97-269 | Bachem | Cat#: 4048647 |
| Critical commercial assays | ||
| mirVana RNA isolation kit | ThermoFisher | Cat#: AM1561 |
| TruSeq Stranded Total RNA Library Prep | Illumina | Cat#: 20020599 |
| Aurum Total RNA Mini kit | Bio-Rad | Cat#: 7326820 |
| Deposited data | ||
| RNA-seq (mouse cortical interneurons) | NCBI GEO | Accession #: GSE195491 |
| RNA-seq (hIPSC organoids) | NCBI GEO | Accession #: GSE195505 |
| Experimental models: Cell lines | ||
| NSUN5–GTF2IRD2+/− hIPSC | This paper | n/a |
| Experimental models: Organisms/strains | ||
| Gtf2ird1 mice | Young, et al., 2008 | n/a |
| CD+/− mice | Segura-Puimedon et al., 2014 | n/a |
| PD+/− mice | Jackson Laboratory | Strain#: 023885; RRID:IMSR_JAX:02 3885 |
| DD+/− mice | Jackson Laboratory | Strain#: 023888; RRID:IMSR_JAX:02 3888 |
| Gtf2i mice | MMRC | Stock#: 034666-UCD; RRID: MMRRC_034666-UCD |
| Vipr1 cKO mice | This paper | n/a |
| Vipr1-OE mice | This paper | n/a |
| Gad2Cre | Jackson Laboratory | Strain#: 019022; RRID: IMSR_JAX: 019022 |
| PVCre | Jackson Laboratory | Strain#: 017320; RRID: IMSR_JAX: 017320 |
| Ai14 | Jackson Laboratory | Strain#: 007914; RRID: IMSR_JAX: 007914 |
| Ai93 | Jackson Laboratory | Strain#: 024107; RRID: IMSR_JAX: 024107 |
| CaMKIIαttA | Jackson Laboratory | Strain#: 24108; RRID: IMSR_JAX: 24108 |
| Scnn1aCre | Jackson Laboratory | Strain#: 009613; RRID: IMSR_JAX:009613 |
| Oligonucleotides | ||
| See tables in methods. | ||
| Recombinant DNA | ||
| n/a | ||
| Software and algorithms | ||
| pClamp | Molecular Devices | n/a |
| miniAnalysis | Synaptosoft | n/a |
| Prism | Graphpad | n/a |
| Sigmaplot | Systat | n/a |
| Fiji | Schindelin et al., 2012 | n/a |
| Matlab | Mathworks | n/a |
| Ilastik | Berg et al., 2019 | n/a |
| OASIS | Friedrich et al., 2017 | n/a |
| Linear Decoder | This paper | n/a |
Highlights.
WBS mice have innate frequency-discrimination hyperacuity
Hyperexcitable interneurons in the ACx account for auditory hyperacuity
Gtf2ird1 haploinsufficiency causes auditory hyperacuity via VIPR1 downregulation
VIPR1 is reduced in human WBS ACx and WBS hiPSC-derived brain organoids
ACKNOWLEDGEMENTS
This work was supported, in part, by the National Institutes of Health (R01 MH097742, R01 DC012833), the Stanford Maternal and Child Health Research Institute Uytengsu-Hamilton 22q11 Neuropsychiatry Research Program, and the American Lebanese Syrian Associated Charities (ALSAC) to S.S.Z. We thank Zakharenko lab members for constructive comments; Dr. Valery Stewart for mouse production; Drs. Shibiao Wan, Yiping Fan, Gang Wu, and Kristen Thomas for RNA-seq data analysis; Andrew Schild for qPCR; Dr. Anjana Nityanandam for organoids; Dr. Victoria Campuzano Uceda and the Universitat Pompeu Fabra (Barcelona, Spain) for CD+/− mice; Damian Kaminski and Molly Lancaster for in vivo viral injections; Kimberly Lowe and Dr. Richard Ashmun for cell sorting; Elizabeth Stevens for graphical abstract design, and Dr. Angela McArthur for manuscript editing. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other granting agencies.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aizenberg M, and Geffen MN (2013). Bidirectional effects of aversive learning on perceptual acuity are mediated by the sensory cortex. Nat. Neurosci. 16, 994–996. doi: 10.1038/nn.3443. [DOI] [PubMed] [Google Scholar]
- Aizenberg M, Mwilambwe-Tshilobo L, Briguglio JJ, Natan RG, and Geffen MN (2015). Bidirectional Regulation of Innate and Learned Behaviors That Rely on Frequency Discrimination by Cortical Inhibitory Neurons. PLOS Biol. 13, e1002308. doi: 10.1371/journal.pbio.1002308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonell A, Del Campo M, Magano LF, Kaufmann L, De La Iglesia JM, Gallastegui F, Flores R, Schweigmann U, Fauth C, Kotzot D, et al. (2010). Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williamse-Beuren syndrome neurocognitive profile. J. Med. Genet. 47, 312–320. doi: 10.1136/jmg.2009.071712. [DOI] [PubMed] [Google Scholar]
- von Arnim G, Engel P, Arnim G, Engel P, von Arnim G, and Engel P (1964). Mental Retardation Related to Hypercalcaemia. Dev. Med. Child Neurol. 6, 366–377. doi: 10.1111/j.1469-8749.1964.tb08138.x. [DOI] [PubMed] [Google Scholar]
- Barak B, Zhang Z, Liu Y, Nir A, Trangle SS, Ennis M, Levandowski KM, Wang D, Quast K, Boulting GL, et al. (2019). Neuronal deletion of Gtf2i, associated with Williams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug. Nat. Neurosci. 22, 700–708. doi: 10.1038/s41593-019-0380-9. [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Westmoreland JJ, and Zakharenko SS (2013). Forward suppression in the auditory cortex is caused by the Ca3.1 calcium channel-mediated switch from bursting to tonic firing at thalamocortical projections. J. Neurosci. 33, 18940–18950. doi: 10.1523/JNEUROSCI.3335-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayés M, Magano LF, Rivera N, Flores R, and Pérez Jurado A, L. (2003). Mutational Mechanisms of Williams-Beuren Syndrome Deletions. Am. J. Hum. Genet. 73, 131–151. doi: 10.1086/376565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellugi U, Lichtenberger L, Jones W, Lai Z, and St. George M. (2000). I. The neurocognitive profile of Williams syndrome: A complex pattern of strengths and weaknesses. J. Cogn. Neurosci. 12, 7–29. doi: 10.1162/089892900561959. [DOI] [PubMed] [Google Scholar]
- Bendor D, and Wang X (2006). Cortical representations of pitch in monkeys and humans. Curr. Opin. Neurobiol. 16, 391–399. doi: 10.1016/J.CONB.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 2019 1612 16, 1226–1232. doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- Bigelow J, Morrill RJ, Dekloe J, and Hasenstaub AR (2019). Movement and VIP Interneuron Activation Differentially Modulate Encoding in Mouse Auditory Cortex. ENeuro 6, 164–183. doi: 10.1523/ENEURO.0164-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Bayazitov IT, and Zakharenko SS (2011). Presynaptic gating of postsynaptically expressed plasticity at mature thalamocortical synapses. J. Neurosci. 31, 16012–16025. doi: 10.1523/JNEUROSCI.3281-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Roy NC, Teubner BJW, Yu J, Eom T-Y, Sample KJJ, Pani A, Smeyne RJ, Han SB, Kerekes RA, et al. (2017). Restoring auditory cortex plasticity in adult mice by restricting thalamic adenosine signaling. Science 356, 1352–1356. doi: 10.1126/science.aaf4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauchli C, Leipold S, and Jäncke L (2019). Univariate and multivariate analyses of functional networks in absolute pitch. Neuroimage 189, 241–247. doi: 10.1016/J.NEUROIMAGE.2019.01.021. [DOI] [PubMed] [Google Scholar]
- Broadbent H, Farran EK, Chin E, Metcalfe K, Tassabehji M, Turnpenny P, Sansbury F, Meaburn E, and Karmiloff-Smith A (2014). Genetic contributions to visuospatial cognition in Williams syndrome: insights from two contrasting partial deletion patients. J. Neurodev. Disord. 6, 18. doi: 10.1186/1866-1955-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 499, 295–300. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Bayazitov IT, Blundon JA, and Zakharenko SS (2013). Thalamocortical Long-Term Potentiation Becomes Gated after the Early Critical Period in the Auditory Cortex. J. Neurosci. 33, 7345–7357. doi: 10.1523/JNEUROSCI.4500-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Du F, Westmoreland JJ, Han SB, Wang Y-D, Eddins D, Bayazitov IT, Devaraju P, Yu J, Mellado Lagarde MM, et al. (2017). Thalamic miR-338–3p mediates auditory thalamocortical disruption and its late onset in models of 22q11.2 microdeletion. Nat. Med. 23, 39–48. doi: 10.1038/nm.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Nguyen T, and Kandler K (2011). An acoustic startle-based method of assessing frequency discrimination in mice. J. Neurosci. Methods 200, 63–67. doi: 10.1016/j.jneumeth.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly JP, and Pruett-Miller SM (2019). CRIS.py: A Versatile and High-throughput Analysis Program for CRISPR-based Genome Editing. Sci. Rep. 9, 4194. doi: 10.1038/s41598-019-40896-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvineau A, and Laburthe M (2012). VPAC receptors: structure, molecular pharmacology and interaction with accessory proteins. Br. J. Pharmacol. 166, 42–50. doi: 10.1111/j.1476-5381.2011.01676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, and Metherate R (2002). Auditory thalamocortical synaptic transmission in vitro. J.Neurophysiol. 87, 361–384. . [DOI] [PubMed] [Google Scholar]
- Cunha-Reis D, Ribeiro JA, de Almeida RFM, and Sebastião AM (2017). VPAC 1 and VPAC 2 receptor activation on GABA release from hippocampal nerve terminals involve several different signalling pathways. Br. J. Pharmacol. 174, 4725–4737. doi: 10.1111/bph.14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi G-A, Guo L, Xu Q, Liu R, Lu C, Chu J, et al. (2016). A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci. 2016 1912 19, 1743–1749. doi: 10.1038/nn.4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/BIOINFORMATICS/BTS635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don AJ, Schellenberg GE, and Rourke BP (1999). Music and Language Skills of Children with Williams Syndrome. Child Neuropsychol. 5, 154–170. doi: 10.1076/chin.5.3.154.7337. [DOI] [Google Scholar]
- Downer JD, Verhein JR, Rapone BC, O’Connor KN, and Sutter ML (2021). An Emergent Population Code in Primary Auditory Cortex Supports Selective Attention to Spectral and Temporal Sound Features. J. Neurosci. 41, 7561–7577. doi: 10.1523/JNEUROSCI.0693-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra AR, Koh CK, Braida LD, and Tramo MJ (2012). Dissociation of Detection and Discrimination of Pure Tones following Bilateral Lesions of Auditory Cortex. PLoS One 7, e44602. doi: 10.1371/journal.pone.0044602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng AS, and Ratnam R (2000). Neural Basis of Hearing in Real-World Situations. Annu. Rev. Psychol. 51, 699–725. doi: 10.1146/annurev.psych.51.1.699. [DOI] [PubMed] [Google Scholar]
- Friedrich J, Zhou P, and Paninski L (2017). Fast online deconvolution of calcium imaging data. PLOS Comput. Biol. 13, e1005423. doi: 10.1371/JOURNAL.PCBI.1005423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Carcea I, Barker AJ, Yuan K, Seybold BA, Martins ARO, Zaika N, Bernstein H, Wachs M, Levis PA, et al. (2012). Long-term modification of cortical synapses improves sensory perception. Nat. Neurosci. 2012 161 16, 79–88. doi: 10.1038/nn.3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervain J, and Geffen MN (2019). Efficient Neural Coding in Auditory and Speech Perception. Trends Neurosci. 42, 56–65. doi: 10.1016/j.tins.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherghina FL, Tica AA, Deliu E, Abood ME, Brailoiu GC, and Brailoiu E (2017). Effects of VPAC1 activation in nucleus ambiguus neurons. Brain Res. 1657, 297–303. doi: 10.1016/j.brainres.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez TL, Lorenc M, and Jaramillo S (2015). Adaptive categorization of sound frequency does not require the auditory cortex in rats. J. Neurophysiol. 114, 1137–1145. doi: 10.1152/jn.00124.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hagen JM, van der Geest JN, van der Giessen RS, Lagers-van Haselen GC, Eussen HJFMM, Gille JJP, Govaerts LCP, Wouters CH, de Coo IFM, Hoogenraad CC, et al. (2007). Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams Syndrome. Neurobiol. Dis. 26, 112–124. doi: 10.1016/j.nbd.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Endoh T, Ahibukawa Y, Yamamoto T, and Suzuki T (2002). VIP and PACAP Inhibit L-, N- and P/Q-Type Ca2+ Channels of Parasympathetic Neurons in a Voltage Independent Manner. Bull. Tokyo Dent. Coll. 43, 31–39. doi: 10.2209/tdcpublication.43.31. [DOI] [PubMed] [Google Scholar]
- Hirota H, Matsuoka R, Chen X-N, Salandanan LS, Lincoln A, Rose FE, Sunahara M, Osawa M, Bellugi U, and Korenberg JR (2003). Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on Chromosome 7q11.23. Genet. Med. 5, 311–321. doi: 10.1097/01.GIM.0000076975.10224.67. [DOI] [PubMed] [Google Scholar]
- Holinger DP, Bellugi U, Mills DL, Korenberg JR, Reiss AL, Sherman GF, and Galaburda AM (2005). Relative sparing of primary auditory cortex in Williams Syndrome. Brain Res. 1037, 35–42. doi: 10.1016/j.brainres.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Hopyan T, Dennis M, Weksberg R, and Cytrynbaum C (2001). Music Skills and the Expressive Interpretation of Music in Children with Williams-Beuren Syndrome: Pitch, Rhythm, Melodic Imagery, Phrasing, and Musical Affect. Child Neuropsychol. 7, 42–53. doi: 10.1076/chin.7.1.42.3147. [DOI] [PubMed] [Google Scholar]
- Howard ML, Palmer SJ, Taylor KM, Arthurson GJ, Spitzer MW, Du X, Pang TYC, Renoir T, Hardeman EC, and Hannan AJ (2012). Mutation of Gtf2ird1 from the Williams–Beuren syndrome critical region results in facial dysplasia, motor dysfunction, and altered vocalisations. Neurobiol. Dis. 45, 913–922. doi: 10.1016/j.nbd.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Ingham NJ, Pearson S, and Steel KP (2011). Using the Auditory Brainstem Response (ABR) to Determine Sensitivity of Hearing in Mutant Mice. Curr. Protoc. Mouse Biol. 1, 279–287. doi: 10.1002/9780470942390.mo110059. [DOI] [PubMed] [Google Scholar]
- Johnson GC, May V, Parsons RL, and Hammack SE (2019). Parallel signaling pathways of pituitary adenylate cyclase activating polypeptide (PACAP) regulate several intrinsic ion channels. Ann. N. Y. Acad. Sci. 1455, 105–112. doi: 10.1111/nyas.14116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanold PO, Nelken I, and Polley DB (2014). Local versus global scales of organization in auditory cortex. Trends.Neurosci. 37, 502–510. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury L, Huang S, Raam T, Ye LS, Wei D, Hu RK, Ye L, and Hong W (2020). Cortical Representations of Conspecific Sex Shape Social Behavior. Neuron 107, 941–953.e7. doi: 10.1016/J.NEURON.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleindienst L, and Musiek F (2011). Do frequency discrimination deficits lead to specific language impairments? Hear. J. 64, 10–11. doi: 10.1097/01.HJ.0000396582.67365.34. [DOI] [Google Scholar]
- Klibisz A, Rose D, Eicholtz M, Blundon J, and Zakharenko S (2017). Fast, simple calcium imaging segmentation with fully convolutional networks. In Deep Learning in Medical Image Analysis and Multimodal Learning for Clinical Decision Support, Cardoso MJ, Arbel T, Carnero G, Syeda-Mahmood T, Tavares JMRS, Moradi M, Bradley A, Greenspan H, Papa JP, Madabhushi A, et al. , eds. (Springer International Publishing; ), pp. 285–293. [Google Scholar]
- Kopp ND, Nygaard KR, Liu Y, McCullough KB, Maloney SE, Gabel HW, and Dougherty JD (2020). Functions of Gtf2i and Gtf2ird1 in the developing brain: Transcription, DNA binding and long-term behavioral consequences. Hum. Mol. Genet. 29, 1498–1519. doi: 10.1093/hmg/ddaa070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozel B, Barak B, Kim C, Mervis C, Osborne L, Porter M, and Pober B (2021). Williams syndrome. Nat. Rev. Dis. Prim. 7, 1–22. doi: 10.1038/S41572-021-00276-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullander K, and Topolnik L (2021). Cortical disinhibitory circuits: cell types, connectivity and function. Trends Neurosci. 44, 643–657. doi: 10.1016/J.TINS.2021.04.009. [DOI] [PubMed] [Google Scholar]
- Kumar M, Xiong S, Tzounopoulos T, and Anderson CT (2019). Fine Control of Sound Frequency Tuning and Frequency Discrimination Acuity by Synaptic Zinc Signaling in Mouse Auditory Cortex. J. Neurosci. 39, 854–865. doi: 10.1523/JNEUROSCI.1339-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer I, Jeandriens J, Couvineau A, Sanmukh S, and Latek D (2022). Signal Transduction by VIP and PACAP Receptors. Biomedicines 10, 406. doi: 10.3390/BIOMEDICINES10020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latek D, Langer I, Krzysko KA, and Charzynski L (2019). A Molecular Dynamics Study of Vasoactive Intestinal Peptide Receptor 1 and the Basis of Its Therapeutic Antagonism. Int. J. Mol. Sci. 20, 4348. doi: 10.3390/IJMS20184348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014 152 15, 1–17. doi: 10.1186/GB-2014-15-2-R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D, and Kokaia M (2014). Global optogenetic activation of inhibitory interneurons during epileptiform activity. J. Neurosci. 34, 3364–3377. doi: 10.1523/JNEUROSCI.2734-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhoff HM (1998). Insights Into the Musical Potential of Cognitively Impaired People Diagnosed with Williams Syndrome. Music Ther. Perspect. 16, 33–36. doi: 10.1093/mtp/16.1.33. [DOI] [Google Scholar]
- Lenhoff HM (2006). Absolute pitch and neuroplasticity in Williams-Beuren syndrome. In Williams-Beuren Syndrome: Research, Evaluation, and Treatment, Morris C, Lenhoff HM, and Wang PP, eds. (Baltimore, MD: John Hopkins University Press; ), pp. 325–342. [Google Scholar]
- Lenhoff HM, Perales O, and Hickok G (2001). Absolute pitch in Williams syndrome. Music Percept. 18, 491–503. doi: 10.1525/mp.2001.18.4.491. [DOI] [Google Scholar]
- Levitin DJ (2005). Musical Behavior in a Neurogenetic Developmental Disorder: Evidence from Williams Syndrome. Ann. N. Y. Acad. Sci. 1060, 325–334. doi: 10.1196/ANNALS.1360.027. [DOI] [PubMed] [Google Scholar]
- Levitin DJ, Menon V, Schmitt JEE, Eliez S, White CD, Glover GH, Kadis J, Korenberg JR, Bellugi U, and Reiss AL (2003). Neural Correlates of Auditory Perception in Williams Syndrome: An fMRI Study. Neuroimage 18, 74–82. doi: 10.1006/nimg.2002.1297. [DOI] [PubMed] [Google Scholar]
- Levitin DJ, Cole K, Chiles M, Lai Z, Lincoln A, and Bellugi U (2004). Characterizing the musical phenotype in individuals with Williams Syndrome. Child Neuropsychol. 10, 223–247. doi: 10.1080/09297040490909288. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011 121 12, 1–16. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HHH, Roy M, Kuscuoglu U, Spencer CMM, Halm B, Harrison KCC, Bayle JHH, Splendore A, Ding F, Meltzer LAA, et al. (2009). Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol.Med. 1, 50–65. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. doi: 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens MA, Reutens DC, and Wilson SJ (2010). Auditory cortical volumes and musical ability in Williams syndrome. Neuropsychologia 48, 2602–2609. doi: 10.1016/j.neuropsychologia.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Martínez-Castilla P, and Sotillo M (2014). Pitch Processing in Children with Williams Syndrome: Relationships between Music and Prosody Skills. Brain Sci. 4, 376. doi: 10.3390/BRAINSCI4020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Castilla P, Sotillo M, and Campos R (2013). Do individuals with Williams syndrome possess absolute pitch? Child Neuropsychol. 19, 78–96. doi: 10.1080/09297049.2011.639755. [DOI] [PubMed] [Google Scholar]
- Mellado Lagarde MM, Wan G, Zhang L, Gigliello AR, McInnis JJ, Zhang Y, Bergles D, Zuo J, and Corfas G (2014). Spontaneous regeneration of cochlear supporting cells after neonatal ablation ensures hearing in the adult mouse. Proc.Natl.Acad.Sci.U.S.A 111, 16919–16924. doi: 10.1073/pnas.1408064111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengler ED, Hogben JH, Michie P, and Bishop DVM (2005). Poor frequency discrimination is related to oral language disorder in children: a psychoacoustic study. Dyslexia 11, 155–173. doi: 10.1002/DYS.302. [DOI] [PubMed] [Google Scholar]
- Mervis CB, Robinson BF, Bertrand J, Morris CA, Klein-Tasman BP, and Armstrong SC (2000). The Williams Syndrome Cognitive Profile. Brain Cogn. 44, 604–628. doi: 10.1006/brcg.2000.1232. [DOI] [PubMed] [Google Scholar]
- Mesik L, Ma W, Li L, Ibrahim LA, Huang ZJ, Zhang LI, and Tao HW (2015). Functional response properties of VIP-expressing inhibitory neurons in mouse visual and auditory cortex. Front. Neural Circuits 09, 22. doi: 10.3389/fncir.2015.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Mervis CB, and Berman KF (2006). Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat. Rev. Neurosci. 7, 380–393. doi: 10.1038/nrn1906. [DOI] [PubMed] [Google Scholar]
- Micheyl C, Delhommeau K, Perrot X, and Oxenham AJ (2006). Influence of musical and psychoacoustical training on pitch discrimination. Hear. Res. 219, 36–47. doi: 10.1016/J.HEARES.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Micheyl C, Schrater PR, and Oxenham AJ (2013). Auditory Frequency and Intensity Discrimination Explained Using a Cortical Population Rate Code. PLOS Comput. Biol. 9, e1003336. doi: 10.1371/JOURNAL.PCBI.1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C, and Braddock S (2020). Health Care Supervision for Children With Williams Syndrome. Pediatrics 145, e20193761. doi: 10.1542/PEDS.2019-3761. [DOI] [PubMed] [Google Scholar]
- Mosing MA, Pedersen NL, Madison G, and Ullén F (2014). Genetic Pleiotropy Explains Associations between Musical Auditory Discrimination and Intelligence. PLoS One 9, e113874. doi: 10.1371/journal.pone.0113874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl FW, Wetzel W, Wagner T, Rech A, and Scheich H (1999). Bilateral ablation of auditory cortex in Mongolian gerbil affects discrimination of frequency modulated tones but not of pure tones. Learn. Mem. 6, 347–362. . [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, and Surmeier DJ (2005). G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci 25, 1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LR (2010). Animal models of Williams syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 154C, 209–219. doi: 10.1002/ajmg.c.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830. doi: 10.48550/arXiv.1201.0490. [DOI] [Google Scholar]
- Peretz I (2016). Neurobiology of Congenital Amusia. Trends Cogn. Sci. 20, 857–867. doi: 10.1016/j.tics.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Plack CJ, Oxenham A, Fay R, and Popper A (2005). Pitch. Neural coding and perception (New York, NY: Springer New York; ). [Google Scholar]
- Pober BR (2010). Williams-Beuren syndrome. N. Engl. J. Med. 362, 239–252. doi: 10.1056/NEJMRA0903074. [DOI] [PubMed] [Google Scholar]
- Proulx É, Young EJ, Osborne LR, and Lambe EK (2010). Enhanced prefrontal serotonin 5-HT1A currents in a mouse model of Williams-Beuren syndrome with low innate anxiety. J. Neurodev. Disord. 2, 99–108. doi: 10.1007/s11689-010-9044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Coleman Z, Curley M, Nityanandam A, Platt A, Robles-Murguia M, Jiao J, Finkelstein D, Wang YD, Xu B, et al. (2021). Proteasome stress in skeletal muscle mounts a long-range protective response that delays retinal and brain aging. Cell Metab. 33, 1137–1154.e9. doi: 10.1016/J.CMET.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198. doi: 10.1093/NAR/GKZ369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, et al. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 14, 482–517. doi: 10.1038/S41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AL, Eliez S, Schmitt JE, Straus E, Lai Z, Jones W, and Bellugi U (2000). IV. Neuroanatomy of Williams Syndrome: A High-Resolution MRI Study. J. Cogn. Neurosci. 12, 65–73. doi: 10.1162/089892900561986. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Blundon JA, Bayazitov IT, and Zakharenko SS (2009). Connectivity patterns revealed by mapping of active inputs on dendrites of thalamorecipient neurons in the auditory cortex. J. Neurosci. 29, 6406–6417. doi: 10.1523/JNEUROSCI.0258-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010 113 11, 1–9. doi: 10.1186/GB-2010-11-3-R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano SA, Pietri T, Perez-Schuster V, Jouary A, Haudrechy M, and Sumbre G (2015). Spontaneous neuronal network dynamics reveal circuit’s functional adaptations for behavior. Neuron 85, 1070–1085. doi: 10.1016/j.neuron.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueckert D, Sonoda LI, Hayes C, Hill DL, Leach MO, and Hawkes DJ (1999). Nonrigid registration using free-form deformations: application to breast MR images. IEEE Trans.Med.Imaging 18, 712–721. doi: 10.1109/42.796284. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Dorr NP, Takahashi N, McInnes LA, Elder GA, and Buxbaum JD (2011). Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 4, 28–39. doi: 10.1002/aur.169. [DOI] [PubMed] [Google Scholar]
- Scala F, Kobak D, Shan S, Bernaerts Y, Laturnus S, Cadwell CR, Hartmanis L, Froudarakis E, Castro JR, Tan ZH, et al. (2019). Layer 4 of mouse neocortex differs in cell types and circuit organization between sensory areas. Nat. Commun. 2019 101 10, 1–12. doi: 10.1038/s41467-019-12058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012 97 9, 676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Skitt Z, Liu Y, Deacon RMJ, Flint J, Karmiloff-Smith A, Rawlins JNP, and Tassabehji M (2012). Anxious, hypoactive phenotype combined with motor deficits in Gtf2ird1 null mouse model relevant to Williams syndrome. Behav. Brain Res. 233, 458–473. doi: 10.1016/j.bbr.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Schubert C (2009). The genomic basis of the Williams-Beuren syndrome. Cell. Mol. Life Sci. 66, 1178–1197. doi: 10.1007/s00018-008-8401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See JZ, Atencio CA, Sohal VS, and Schreiner CE (2018). Coordinated neuronal ensembles in primary auditory cortical columns. Elife 7, e35587. doi: 10.7554/eLife.35587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seesjärvi E, Särkämö T, Vuoksimaa E, Tervaniemi M, Peretz I, and Kaprio J (2016). The Nature and Nurture of Melody: A Twin Study of Musical Pitch and Rhythm Perception. Behav. Genet. 46, 506–515. doi: 10.1007/s10519-015-9774-y. [DOI] [PubMed] [Google Scholar]
- Segura-Puimedon M, Sahún I, Velot E, Dubus P, Borralleras C, Rodrigues AJ, Valero MC, Valverde O, Sousa N, Herault Y, et al. (2014). Heterozygous deletion of the Williams–Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum. Mol. Genet. 23, 6481–6494. doi: 10.1093/hmg/ddu368. [DOI] [PubMed] [Google Scholar]
- Sentmanat MF, Peters ST, Florian CP, Connelly JP, and Pruett-Miller SM (2018). A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 8, 888. doi: 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PH, and Populin LC (2001). Fundamental differences between the thalamocortical recipient layers of the cat auditory and visual cortices. J Comp Neurol. 436, 508–519. doi: 10.1002/cne.1084. [DOI] [PubMed] [Google Scholar]
- Smith LM, Bartholomew AJ, Burnham LE, Tillmann B, and Cirulli ET (2017). Factors affecting pitch discrimination performance in a cohort of extensively phenotyped healthy volunteers. Sci. Rep. 7, 16480. doi: 10.1038/s41598-017-16526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2005). limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Gentleman R, Carey VJ, Huber W, Irizarry RA, and Dudoit S, eds. (New York, NY: Springer New York; ), pp. 397–420. [Google Scholar]
- Spiegel MF, and Watson CS (1984). Performance On Frequency-discrimination Tasks By Musicians And Nonmusicians. J. Acoust. Soc. Am. 76, 1690–1695. doi: 10.1121/1.391605. [DOI] [Google Scholar]
- Stewart L (2008). Fractionating the musical mind: insights from congenital amusia. Curr. Opin. Neurobiol. 18, 127–130. doi: 10.1016/J.CONB.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Talwar SK, and Gerstein GL (2001). Reorganization in awake rat auditory cortex by local microstimulation and its effect on frequency-discrimination behavior. J.Neurophysiol. 86, 1555–1572. doi: 10.1152/jn.2001.86.4.1555. [DOI] [PubMed] [Google Scholar]
- Tang B, Wu J, Zhu MX, Sun X, Liu J, Xie R, Dong TX, Xiao Y, Carethers JM, Yang S, et al. (2019). VPAC1 couples with TRPV4 channel to promote calcium-dependent gastric cancer progression via a novel autocrine mechanism. Oncogene 38, 3946–3961. doi: 10.1038/s41388-019-0709-6. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, et al. (2005). GTF2IRD1 in Craniofacial Development of Humans and Mice. Science 310, 1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- Taylor RDT, Madsen MG, Krause M, Sampedro-Castañeda M, Stocker M, and Pedarzani P (2014). Pituitary adenylate cyclase-activating polypeptide (PACAP) inhibits the slow afterhyperpolarizing current sI AHP in CA1 pyramidal neurons by activating multiple signaling pathways. Hippocampus 24, 32–43. doi: 10.1002/hipo.22201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur D, Martens MA, Smith DS, and Roth E (2018). Williams Syndrome and Music: A Systematic Integrative Review. Front. Psychol. 9, 2203. doi: 10.3389/fpsyg.2018.02203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KJ, Khajehali E, Bradley SJ, Navarrete JS, Huang XP, Slocum S, Jin J, Liu J, Xiong Y, Olsen RHJ, et al. (2018). DREADD Agonist 21 Is an Effective Agonist for Muscarinic-Based DREADDs in Vitro and in Vivo. ACS Pharmacol. Transl. Sci. 1, 61–72. doi: 10.1021/acsptsci.8b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton-Wells TA, Cannistraci CJ, Anderson AW, Kim C-Y, Eapen M, Gore JC, Blake R, and Dykens EM (2010). Auditory Attraction: Activation of Visual Cortex by Music and Sound in Williams Syndrome. Am. J. Intellect. Dev. Disabil. 115, 172–189. doi: 10.1352/1944-7588-115.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramo MJ, Shah GD, and Braida LD (2002). Functional Role of Auditory Cortex in Frequency Processing and Pitch Perception. J. Neurophysiol. 87, 122–139. doi: 10.1152/jn.00104.1999. [DOI] [PubMed] [Google Scholar]
- Tramo MJ, Cariani PA, Koh CK, Makris N, and Braida LD (2005). Neurophysiology and Neuroanatomy of Pitch Perception: Auditory Cortex. Ann. N. Y. Acad. Sci. 1060, 148–174. doi: 10.1196/annals.1360.011. [DOI] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292. doi: 10.1016/j.neuron.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udwin O, and Yule W (1990). Expressive language of children with Williams syndrome. Am. J. Med. Genet. 37, 108–114. doi: 10.1002/AJMG.1320370620. [DOI] [PubMed] [Google Scholar]
- Valero MC, De Luis O, Cruces J, and Pérez Jurado LA (2000). Fine-Scale Comparative Mapping of the Human 7q11.23 Region and the Orthologous Region on Mouse Chromosome 5G: The Low-Copy Repeats That Flank the Williams–Beuren Syndrome Deletion Arose at Breakpoint Sites of an Evolutionary Inversion(s). Genomics 69, 1–13. doi: 10.1006/GENO.2000.6312. [DOI] [PubMed] [Google Scholar]
- de Vries SEJ, Lecoq JA, Buice MA, Groblewski PA, Ocker GK, Oliver M, Feng D, Cain N, Ledochowitsch P, Millman D, et al. (2020). A large-scale standardized physiological survey reveals functional organization of the mouse visual cortex. Nat. Neurosci. 23, 138–151. doi: 10.1038/s41593-019-0550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EJ, Lipina T, Tam E, Mandel A, Clapcote SJ, Bechard AR, Chambers J, Mount HTJ, Fletcher PJ, Roder JC, et al. (2008). Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes Brain Behav. 7, 224–234. doi: 10.1111/j.1601-183X.2007.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarchi O, Avni C, Attias J, Frisch A, Carmel M, Michaelovsky E, Green T, Weizman A, and Gothelf D (2015). Hyperactive auditory processing in Williams syndrome: Evidence from auditory evoked potentials. Psychophysiology 52, 782–789. doi: 10.1111/psyp.12407. [DOI] [PubMed] [Google Scholar]
- Zhao C, Avilés C, Abel RA, Almli CR, McQuillen P, and Pleasure SJ (2005). Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development 132, 2917–2927. doi: 10.1242/dev.01871. [DOI] [PubMed] [Google Scholar]
- Zhu Y, and Ikeda SR (1994). VIP inhibits N-type Ca2+ channels of sympathetic neurons via a pertussis toxin-insensitive but cholera toxin-sensitive pathway. Neuron 13, 657–669. doi: 10.1016/0896-6273(94)90033-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Recordings from individual WT and CD+/− mice of normalized PPI magnitude, as a function of frequency difference between 9.8-kHz background and pre-pulse tones. Points and error bars are mean ± SEM of 10 repeated measurements in the same animal. Solid lines are linear-regression fits to these points; dotted lines are frequency-discrimination thresholds (FDTs).
B. The FDT is smaller in CD+/− (n = 6) vs WT (n = 5) mice. Rank sum test, *P = 0.009.
C. Time course of trial-initiation learning (stages 1 and 2) of a Go/No-go auditory-discrimination task. All WT (n = 12) and CD+/− (n = 5) mice learned the location of a food trough, but only 1 CD+/− mouse learned trial initiation after 2 weeks of training, compared to 7 WT mice.
D. The acoustic startle response (ASR) is the same in WT (n = 12) and CD+/− mice (n = 12).
E. The threshold sound level needed to elicit the acoustic brainstem response (ABR) is not different between WT (n = 19) and CD+/− mice (n = 17).
F. Spontaneous excitatory activity in thalamic relay neurons (Left, scheme) is normal in CD+/− mice. (Right, top) Traces of sEPSCs from WT and CD+/− thalamic relay neurons. (Right, bottom) The sEPSC frequency and amplitude are not different in WT (n = 7) and CD+/− (n = 8) neurons.
G. Scheme of stimulating (stim.) ascending TC input from the auditory thalamus (MGv) and recording from an L4 pyramidal neuron from the ACx in an auditory TC brain slice.
H. Representative traces showing evoked EPSC in response to a pair of presynaptic stimuli (arrows) in WT and CD+/− mice.
I. The EPSC slope in response to the first stimulus, as a function of stimulus amplitude, is not different between WT (n = 19) and CD+/− (n = 25) mice.
J. The paired-pulse ratio is not different between WT (n = 9) and CD+/− (n = 9) mice.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
A. (Top) Scheme of whole-cell pyramidal neuron recording in the auditory cortex (ACx). (Bottom) Traces showing voltage and action potential (AP) responses to depolarizing current injection in WT and CD+/− pyramidal neurons.
B. All intensities of current injection elicit similar frequency AP responses in WT (n = 15) and CD+/− (n = 12) pyramidal neurons.
C. Traces showing voltage and AP responses to a current ramp (0–300 pA, 1 s) in WT and CD+/− pyramidal neurons.
D. The ramp current threshold that elicits APs (rheobase), resting membrane potential (RMP), and input resistance measured from the initial linear ramp response are the same in WT (n = 16) and CD+/− (n = 21) mice.
E. Traces of individual APs from a train of APs elicited by current injection in WT and CD+/− pyramidal neurons.
F-I. AP peak amplitude (F), half-width (G), threshold (H), and after-hyperpolarization amplitude (AHP) (I) do not differ between WT (n = 15) and CD+/− (n = 12) pyramidal neurons.
J. (Top) Scheme of thalamic excitatory (relay) neuron recording location in the MGv. (Bottom) Traces showing voltage and AP responses to depolarizing current injection in WT and CD+/− neurons.
K. All intensities of current injection elicit similar frequency AP responses in WT (n = 9) and CD+/− (n = 8) neurons.
L. Rheobase measured as the threshold current step to elicit APs (Left: WT, 9 neurons and CD+/−, 8 neurons), RMP (Middle: WT, 12 neurons and CD+/−, 12 neurons), and input resistance (Right: WT, 12 neurons and CD+/−, 12 neurons) measured from small subthreshold current steps did not differ between genotypes.
M. Traces of individual APs from a train of APs elicited by depolarizing current injection in WT and CD+/− neurons.
N-Q. AP peak amplitude (N), half-width (O), threshold (P), and AHP (Q) did not differ between WT (n = 9) and CD+/− (n = 8) neurons.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
(Left) Scheme of whole-cell voltage-clamp recording from L4 fast-spiking (FS) interneurons in the ACx.
A. Representative current responses to increasing amplitude voltage steps from −90 mV. Na+ current was present as a fast inward current (arrow).
B. Current–voltage relations of peak Na+ current density in WT (n = 16) and CD+/− (n = 20) interneurons. Na+ current density did not differ at any voltage.
C. Representative current responses to increasing amplitude voltage steps from −90 mV in the presence of 0.5 μM TTX, 3 mM KYN, and 100 μM PTX. K+ current was present as a sustained outward current.
D. Current–voltage relations of steady-state K+ current density in WT (n = 19) and CD+/− (n= 14) interneurons. K+ current density did not differ at any voltage.
E. Representative current responses to hyperpolarizing voltage steps from rest in the presence of 0.5 μM TTX. Ih current was present as a slowly developing sag inward current.
F. Current–voltage relations of Ih current density in WT (n = 5) and CD+/− (n = 6) interneurons. Ih current density did not differ at any voltage. RM ANOVA P = 0.906.
Averaged data are presented as the mean ± SEM.
A. Time course of trial-initiation learning (stages 1 and 2) of a Go/No-go auditory-discrimination task. All WT (n = 10) and PD+/− (n = 6) mice learned the location of a food trough; 0 PD+/− mice and 6 WT mice learned trial initiation after 2 weeks of training.
B. Comparison of normalized FDT values across all WBS and single-gene–deficient genotypes. Comparable decreases in FDT are observed in CD+/−, PD+/−, Gtf2ird1+/−, and Gtf2ird1−/− mice.
C-F. The ASR did not differ between WT (n = 14) and PD+/− (n = 12) mice (C); between WT (n = 8) and DD+/− (n = 13) mice (D); and across WT (n = 9), Gtf2ird1+/− (n = 21), and Gtf2ird1−/− (n = 13) mice (E); or between WT (n = 7) and Gtf2i+/− (n = 9) mice (F).
Averaged data are presented as mean ± SEM with individual measurements overlaid as dots.
A. Tone-frequency decoder fails to identify a difference in accuracy between WT and CD+/− neurons after the window for analysis was reduced from 400 ms to 100 ms after tone presentation.
B. Deconvoluted GCaMP6f responses in WT and CD+/− excitatory neurons show more sustained activity in mutant cells.
C. CD+/− excitatory neurons have a longer temporal autocorrelation range of individual responses than do WT neurons. Rank sum test *P =0.004.
D, E. Improved accuracy of frequency decoding in the ACx of CD+/− mice persisted in random subsets of cells with matching proportions of tone-responsive to nonresponsive cells (D, rank sum test *P = 0.009) or random subsets with matching numbers of cells (E, rank sum test *P = 0.002).
Data are presented as interquartile ranges (quantiles 5-25-50-75-95).
A. Dot plot showing Gene Ontology (GO) term enrichment among upregulated mRNAs in Gtf2ird1−/− interneurons relative to WT. GO:BP, biological processes; GO:MF, molecular functions.
B. Relative fold change of several genes, as measured by qPCR analysis of GAD2+ cortical interneurons from CD+/− mice (n = 4 mice) compared to WT controls (n = 4 mice). Genes that were downregulated in RNA-seq from Gtf2ird1−/− mice are to the left of the graph, and those that were upregulated are to the right. Unpaired t-test values: Fry *P <0.001; Mtus2 *P = 0.001; Idua P = 0.193; Dusp6 P = 0.087; Cdk5rap1 P = 0.137; Chek2 *P = 0.003; Plce1 *P <0.001; and Dgkb *P <0.001.; Fzd2 P = 0.994; B3glct P = 0.376; Vegfa P = 0.325; Plcd3 P = 0.389.
C. An ~1.57-Mb heterozygous microdeletion in human chromosome 7q11.23 was created in hiPSCs from a healthy control by using two single-guide RNAs (sgRNAs, small arrows).
D. The resulting heterozygous clones have one NSUN5–GTF2IRD2 microdeletion allele and one intact allele.
E. Relative fold–change values for individual genes within the NSUN5–GTF2IRD2 locus (shaded area), flanking genes, and VIPR1 in a healthy control organoid and in NSUN5–GTF2IRD2+/− organoids. All unpaired t-test values, *P <0.05. WBS genes with no expression in organoids are not shown.
F. Dot plots showing GO term enrichment among downregulated mRNAs in brain organoids carrying the WBS deletion, relative to isogenic control organoids. GO:BP, biological processes; GO:CC, cellular components; GO:MF, molecular functions.
Averaged data are presented as the mean ± SEM.
A. Scheme for engineering the Vipr1–conditional knockout (cKO) mouse. The sgRNAs were designed to target the 5’- and 3’-ends of exon 2 of the mVipr1 gene. An ssODN consisting of 40-bp homology arms flanking a LoxP site (arrowhead) and BamHI-restriction site (for downstream genotyping) was designed to insert the LoxP site directly into each corresponding sgRNA cut site (two ssODNs total). Founder animals containing both LoxP sites on the same allele were bred to homozygosity. Floxed Vipr1 animals were crossed with Gad2Cre mice to create Gad2Cre;Vipr1fl/+ mice. Excision of exon 2 after Cre expression results in a premature stop codon (stop sign) in exon 3, which stimulates nonsense-mediated decay and tissue-specific knockout.
B. Vipr1 is reduced in the cortex of Vipr1 cKO mice (Gad2Cre;Vipr1fl/+ and Gad2Cre;Vipr1fl/fl mice). The qPCR quantification of the Vipr1 level in WT (n = 24 replicates; 8 mice), Gad2Cre;Vipr1fl/+ (n = 15 replicates; 5 mice), and Gad2Cre;Vipr1fl/fl mice (n = 12 replicates; 4 mice). One-way ANOVA: WT vs Gad2Cre;Vipr1fl/+ *P = 0.012; WT vs Gad2Cre;Vipr1fl/fl *P <0.001; Gad2Cre;Vipr1fl/+ vs Gad2Cre;Vipr1fl/fl *P = 0.012.
C. Acoustic startle response (ASR) is normal in Vipr1 cKO mice.
D. Low-magnification (top) and high-magnification (bottom) images of the ACx of Gad2Cre;Ai14 mice expressing tdTomato in interneurons injected with AAV-hDlx-Vipr1-GFP.
E. ASR is unchanged in Gtf2ird1-deficient mice after overexpression of Vipr1 in the ACx interneurons by using AAV-hDlx-Vipr1-GFP.
F. ASR is unchanged in Gad2Cre;CD+/− mice after overexpression of Vipr1 in the ACx interneurons by using AAV-CAG-Flex-Vipr1-GFP.
G. Scheme for engineering the Vipr1 conditional overexpression (cOE) mouse. Transgenic mice expressing the CAG-LoxP-Stop-LoxP-Vipr1-IRES-GFP construct were crossed with Gad2Cre mice to produce Vipr1cOE mice.
H. Validation of Vipr1cOE mice by qRT-PCR. Rank sum test *P = 0.024 (WT [Cre−] n=3 replicates; Vipr1cOE n=6 replicates).
I. ASR is normal in CD+/− mice crossed with Vipr1cOE mice.
Averaged data are presented as the mean ± SEM with individual measurements overlaid as dots.
Data Availability Statement
RNA-seq data are available in the NCBI GEO database under accession number GSE195491 (tdTomato+ cells from the cortex of Gad2Cre;Ai14;Gtf2ird1+/+ or Gad2Cre;Ai14;Gtf2ird1−/− mice) and GSE195505 (organoids derived from the hiPSCs with the WBS microdeletion and isogenic controls). All other data and code generated are available upon request from the lead contact.
Any additional information required to reanalyze the data in this paper are available from the lead contact.