

Abstract
Background:
The transient outward current (Ito) that mediates early (phase 1) repolarization is conducted by the KCND3-encoded Kv4.3 pore-forming α-subunit. KCND3 gain-of-function mutations have been reported previously as a pathogenic substrate for J wave syndromes (JWS), including the Brugada syndrome (BrS) and Early Repolarization syndrome (ERS), as well as autopsy-negative sudden unexplained death (SUD). Acacetin, a natural flavone, is a potent Ito current blocker. Acacetin may be a novel therapeutic for KCND3-mediated JWS.
Methods:
KCND3-V392I was identified in an 18-year-old male with JWS/ERS, and a history of cardiac arrest including ventricular tachycardia/ventricular fibrillation and atrial fibrillation/atrial flutter. Pathogenic KCND3 mutation was engineered by site directed mutagenesis and co-expressed with wild type KChIP2 in TSA201 cells. Gene-edited/variant-corrected isogenic control and patient-specific pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from the p. Val392Ile-KCND3-positive patient were generated. Ito currents and action potentials (APs) were recorded before and after treatment with Acacetin using the whole cell patch-clamp and multielectrode array (MEA) technique. Western blot and immunocytochemistry were performed to investigate KCND3 expression.
Results:
KCND3-V392I demonstrated a marked gain-of-function phenotype, increasing peak Ito current density by 92.2% (p<0.05 vs. KCND3-WT). KCND3 expression was significantly increased in KCND3-V392I-derived iPSC-CMs (p<0.05 vs. isogenic control). While KCND3-WT revealed an IC50 of 7.2±1.0 μM for Acacetin effect, 30 μM Acacetin dramatically inhibited KCND3-V392I peak Ito current density by 96.2% (p<0.05 vs. before Acacetin). Ito was also increased by 60.9 % in Kv4.3-V392I iPSC-CM (p<0.05 vs. isogenic control iPSC-CM). 10 μM Acacetin, a concentration approaching its IC50 value, inhibited Ito by approximately 50% in patient-derived iPSC-CMs and reduced the accentuated AP notch displayed in KCND3-V392I-derived iPSC-CMs.
Conclusions:
This pre-clinical study provides pharmacological and functional evidence to suggest that Acacetin may be a novel therapeutic for patients with KCND3 gain-of-function-associated JWS by inhibiting Ito and abolishing the accentuated AP notch in patient-derived iPSC-CMs.
Keywords: Acacetin, Arrhythmia, J Wave Syndrome/ Early Repolarization Syndrome, Gene mutation, Ion channel
Introduction
The transient outward current (Ito) is a rapidly activating potassium channel current that is responsible for phase 1 of the cardiac action potential. Ito is produced by KCND3-encoded Kv4.3 channels. KCND3 gain-of-function mutations generate a pathogenic substrate responsible for the J wave syndromes (JWS), including the Brugada syndrome (BrS)1 and Early Repolarization syndrome (ERS)2, as well as autopsy-negative sudden unexplained death (SUD)3.
The J point denotes the junction of the QRS complex and the ST segment on the ECG, marking the end of depolarization and the beginning of repolarization. The term J wave or Osborn wave denotes an elevated J-point or ST-segment elevation immediately following the QRS complex of the surface ECG4, 5. A prominent Ito-mediated action potential notch in ventricular epicardium but not endocardium produces a transmural voltage gradient during early ventricular repolarization that registers as a J wave or J-point elevation on the ECG4. We refer to the “syndrome” when a J wave pattern in the ECG is accompanied by clinical symptoms including arrhythmic manifestations and sudden cardiac death6. JWS were proposed as a unifying definition for 2 clinical entities, namely BrS and ERS5,6. BrS and ERS have similar cellular and genetic pathophysiology underlying the genesis of arrhythmias secondary to and outward shift in the balance of current in the early phases of the ventricular action potential. The resulting defects give rise to closely coupled extrasystoles via a “phase 2 reentry” mechanism, which in turn precipitates polymorphic ventricular tachycardia and/or fibrillation (VT/VF). The outward shift in the balance of current can be due to reduced inward currents, such as sodium or calcium currents or to increased Ito or ATP-dependent potassium current (KATP)5,6. The imbalance between depolarizing and repolarizing forces in the early phase of cardiac action potential (AP) is the current hypothesis for the generation of a pathologic J wave in the right ventricular outflow tract (RVOT) in the case of BrS or in the inferior/lateral wall in the case of ERS5 6.
Implantation of a cardioverter-defibrillator (ICD) is an effective therapeutic strategy for those JWS patients with either resuscitated sudden cardiac arrest (SCA) or arrhythmic syncope5. However, an ICD may be problematic in infants or young children because of the high complication rate5. Quinidine by virtue of its effect to inhibit Ito effectively prevents/reduces ventricular fibrillation (VF) in patients with BrS as well as ERS and has proven to be an alternative to ICD therapy7, 8. Owing to its action to inhibit IKr, quinidine predisposes to the development of QT interval prolongation and the development of life-threatening Torsade de Pointes arrhythmias9. These actions of quinidine have prompted the search for other pharmacologic approaches to the treatment of JWS.
Acacetin, a natural flavone effectively blocks human atrial ultra-rapidly delayed rectifier potassium (IKur), acetylcholine-activated potassium (IK-ACh), and Ito currents and therefore has been viewed as a promising agent for the treatment of atrial fibrillation (AF)10. Since IKur and IK-ACh are expressed at negligible levels in ventricular myocardium, we hypothesize that acacetin, may function as an Ito specific channel blocker in the ventricles and serve as a novel therapeutic for JWS caused by an increased, unopposed Ito current in the ventricular epicardium.
As such, in the present study, we investigated the potential Ito-blocking effects of Acacetin in TSA201cells heterologously expressing a KCND3 gain-of-function variant (p. Val392Ile-KCND3) as well as patient-specific pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from a p. Val392Ile-KCND3-positive patient with recurrent ventricular arrhythmias and a clinical diagnosis of JWS-ERS subtype.
Methods
Written informed consent was obtained for this Mayo Clinic Institutional Review Board approved study (09–006465). The data that support the findings of this study are available from the corresponding author upon reasonable request. A full description of methods, including drug and statistical analysis are available in supplemental material.
Case Report
An 18-year-old adopted male of Asian descent with an unremarkable medical history aside from three “febrile seizures” during early childhood presented to a local emergency department after suffering a witness collapsed with seizure-like activity while camping outdoors with his family. Preceding his collapse, the patient awoke from sleep with extreme dizziness, diaphoresis, and nausea and vomiting and alerted his mother. In the local emergency department, the patient once again lost consciousness and seizure-like activity was noted. On telemetry, polymorphic ventricular tachycardia/ventricular fibrillation (VF) was noted (Figure 1A). After a pulse was subsequently lost, cardiopulmonary resuscitation (CPR) was initiated promptly and spontaneous return of circulation achieved following several rounds of CPR and six external defibrillator shocks.
Figure 1.
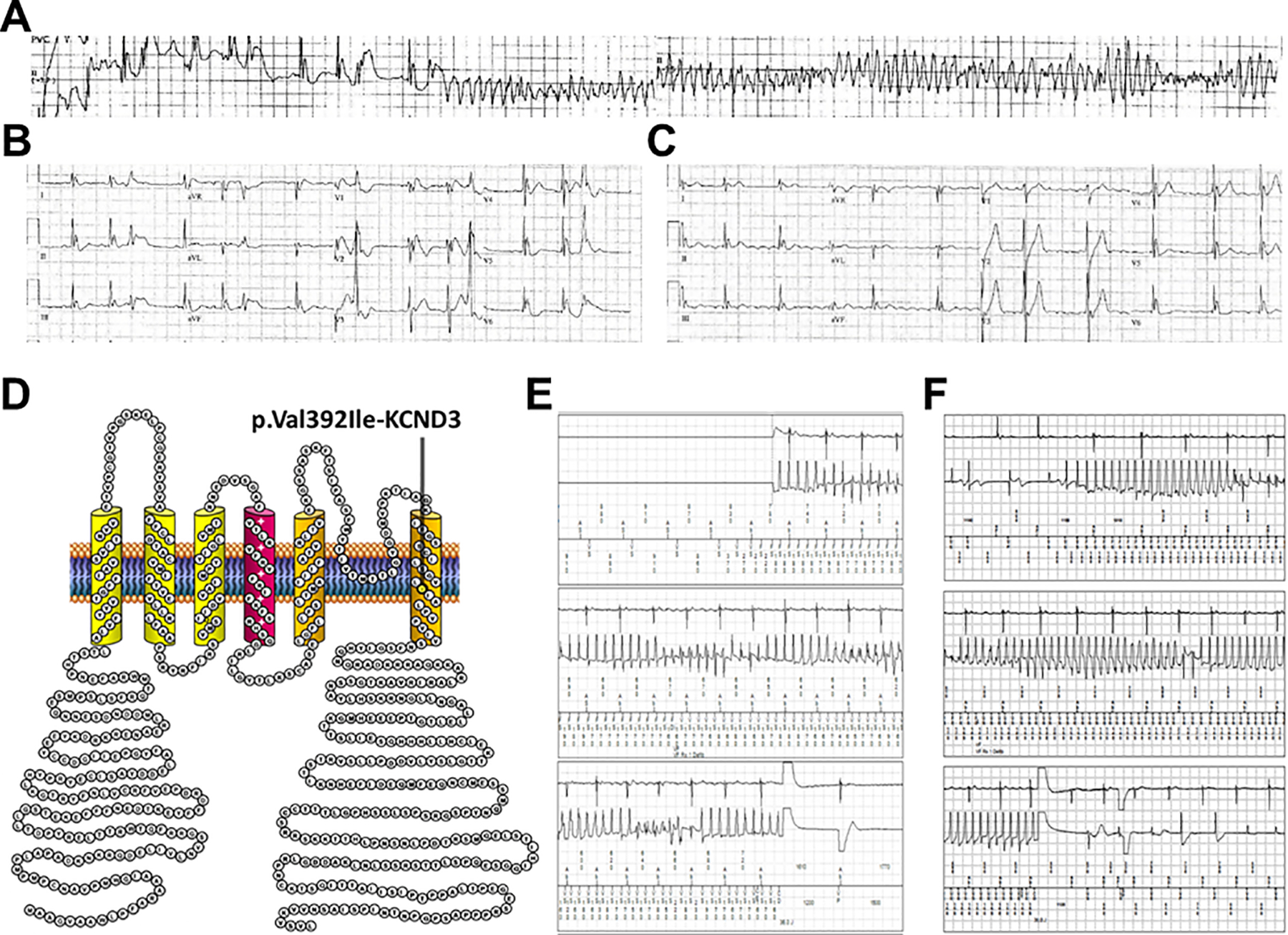
Electrographic findings observed in a p. Val392Ile-KCND3-positive patient with a J-wave spectrum disorder. A. Telemetry rhythm strips obtained during resuscitation efforts after the p. Val392Ile-KCND3-positive patient experienced an in-hospital cardiac arrest. B. Immediate post-cardiac arrest 12-lead ECG displaying a global early repolarization pattern. C. Post-cardiac arrest 12-lead ECG displaying an inferolateral early repolarization pattern. D. Localization of the p. Val392Ile-KCND3 variant on the linear topology of the Kv4.3 voltage-gate potassium channel. E. Device EGM displaying the patient’s first appropriate ventricular fibrillation-terminating (pre-quinidine) ICD therapy. F. Device EGM displaying the patient’s second (after quinidine cessation for gastrointestinal symptoms/transaminitis) appropriate ventricular fibrillation-terminating ICD therapy.
Following his in-hospital cardiac arrest, the patient was admitted and immediate post-SCA work-up was remarkable for serial 12-lead ECGs displaying global and inferolateral global J point elevation and paroxysmal atrial flutter (Figure 1B and 1C). Prior to discharge, the patient received a dual-chamber ICD for secondary prevention.
Due to concerns for possible BrS, the patient was referred to the Mayo Clinic Windland Smith Rice Genetic Heart Rhythm Clinic and subsequently underwent commercial BrS/ERS genetic testing which identified a likely pathogenic variant (p. Val392Ile-KCND3) that localizes to the transmembrane domain of the Kv4.3 voltage-gate potassium channel (Figure 1D). Importantly, p. Val392Ile-KCND3 was identified previously in a young SUD victim, found to confer an in vitro gain-of-function electrophysiological phenotype, and is the only putative BrS/SUD-associated KCND3 genetic variant absent in the Genome Aggregation Database (gnomAD) of public exomes/genomes3.
Of note, the patient’s first post-discharge device interrogation revealed two episodes of atrial fibrillation/atrial flutter shortly after discharge. Over the next three years, there were several episodes of symptomatic atrial flutter with rapid ventricular response and one inappropriate ICD shock due to T-wave over sensing.
Unfortunately, roughly four years after his sentinel in-hospital cardiac arrest, the patient received an appropriate VF-terminating ICD shock while sleeping (Figure 1E). Given concern for p. Val392Ile-KCND3-mediated ERS (i.e., Ito gain-of-function), the patient was admitted and loaded with quinidine. However, approximately one week after quinidine was initiated, the patient developed nausea and diffuse muscle aches. Work-up at this time revealed elevated liver enzymes and due to concern for quinidine-induced lupus, quinidine was held with subsequent resolution of both symptoms and transaminitis. In this setting, the patient experienced a second appropriate ventricular fibrillation-terminating ICD shock while sleeping (Figure 1F). He was again admitted and continued to have episodes of short-coupled torsades de pointes/premature ventricular contraction (PVC)-triggered VF.
With limited viable pharmacologic options, the patient ultimately underwent a PVC-targeted ablation as the inciting PVC was felt to be originating from the left posterior fascicle and was discharged subsequently on mexiletine. However, he experienced two additional appropriate VF-terminating ICD shocks over a 48-hour period and was once again admitted. After considerable discussion, the decision was ultimately made to trial flecainide for PVC suppression. In the event that flecainide was unsuccessful or exacerbated the patient’s underlying genetic substrate, patient-specific iPSC-CMs were generated to produce a “disease in the dish” model for which experimental agents such as Acacetin could be tested for therapeutic efficacy in vitro before consideration of compassionate use in the patient should the need arise.
Results
KCND3-V392I was reported previously as a gain-of-function variant in a sudden death patient3. Acacetin is a potent Ito channel blocker11. In order to examine the optimal concentration for acacetin, we performed a dose-response assessment of the effect of Acacetin to block Ito in HEK293 cells transfected with WT or mutant KCND3 together with KChIP2-WT. Figure 2A shows representative Ito traces recorded before and after exposure to 7.5 μM Acacetin. Acacetin blocked Ito in a concentration –dependent manner with an IC50 of 7.2±1.0 μM (n=3–5) (Figure 2B). Previous studies revealed that 30 μM Acacetin potently blocked Kv4.3-WT and variants in segment 6, V392A, I395A and V399A11. Here, we tested whether 30 μM Acacetin has similar effects on Ito in Kv4.3-WT as well as channels expressing the gain-of-function variant, V392I in TSA201 cells.
Figure 2.
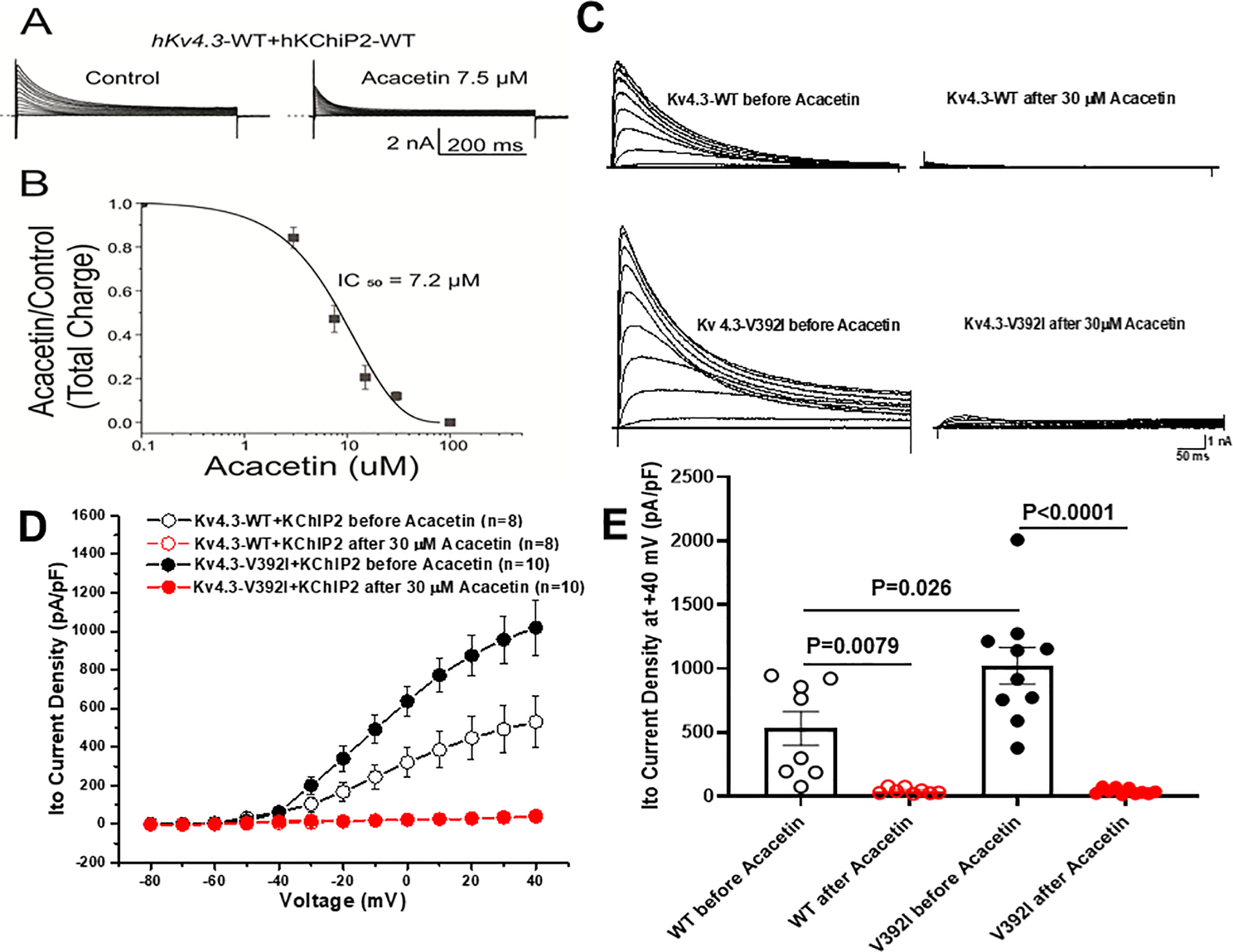
Acacetin dramatically blocked Ito KCND3-WT and –V392I currents expressed in HEK293/TSA201 cells. A. Representative whole-cell Kv4.3-WT plus KChIP2-WT traces before and after 7.5 μM Acacetin from HEK293 cells. B. Concentration-response relationships for Ito total charge normalized to the current recorded at +50 mV under control and Acacetin conditions (room temperature). The IC50 value of Acacetin block of the Ito measured with KCND3-WT was 7.2±1.0 μM (n=3–5). C. Representative Ito KCND3-WT/V392I traces before and after 30 μM Acacetin from TSA201 cells elicited by step depolarization of 500 ms duration to +40 mV from a holding potential of −80 mV in 10 mV increments. D. The current voltage relationship for Ito KCND3-WT (n= 8) and V392I (n=10) before and after 30 μM Acacetin. All values represent mean±SEM. E. Bar graph showing Ito peak current density at +40 mV for KCND3-WT (n= 8) and V392I (n=10) before and after 30 μM Acacetin.
Consistent with our previous report3, Kv4.3-V392I plus KChIP2 increased Ito current, whereas perfusion of 30 μM Acacetin dramatically blocked Kv4.3-WT and V392I Ito channels (Figure 2C). Analysis of the current-voltage relationship indicated that Kv4.3-V392I plus KChIP2-WT significantly increased Ito density from −10 mV to +40 mV (n=10, p<0.05) compared with Kv4.3-WT plus KChIP2-WT (n=8, Figure 2D). At +40 mV, Ito density was increased significantly by 92.2% from 530.8±132.2 pA/pF (WT, n=8) to 1020.3±143.4 pA/pF (V392I, n=10, p=0.026) (Figure 2E). 30 μM Acacetin almost completely blocked Kv4.3-WT and V392I current from −30 mV to +40 mV (p<0.05 vs. before Acacetin) reaching a level of 42.8±7.9 pA/pF (WT at +40 mV, n=8, p=0.0079 vs. before Acacetin) and 39.1±7.1 pA/pF (V392I at +40 mV, n=10, p<0.0001 vs. before Acacetin) respectively (Figure 2D, 2E).
In order to examine whether an Ito gain-of-function is observed in Kv4.3-V392I iPSC-CMs and the effect of Acacetin, we recorded Ito in iPSC-CMs derived from the Kv4.3-V392I patient and gene-edited/variant-corrected isogenic control cell lines. Similar to the findings in TSA201 cells, Ito was greater in Kv4.3-V392I iPSC-CM compared to the isogenic control (Figure 3). At +40 mV, Ito density was 60.9% greater in iPSC-CMs from 20.2±2.6 pA/pF (isogenic control, n=10) to 32.5±4.6 pA/pF (V392I, n=11, p=0.034 vs. isogenic control) (Figure 3C). 10 μM Acacetin, a concentration approaching its IC50 value, significantly inhibited Ito by 53.8% (n=11, p=0.0028 vs. before Acacetin) and 39.1% (n=10, p=0.0004 vs. before Acacetin) in case- and isogenic control-derived iPSC-CMs respectively.
Figure 3.
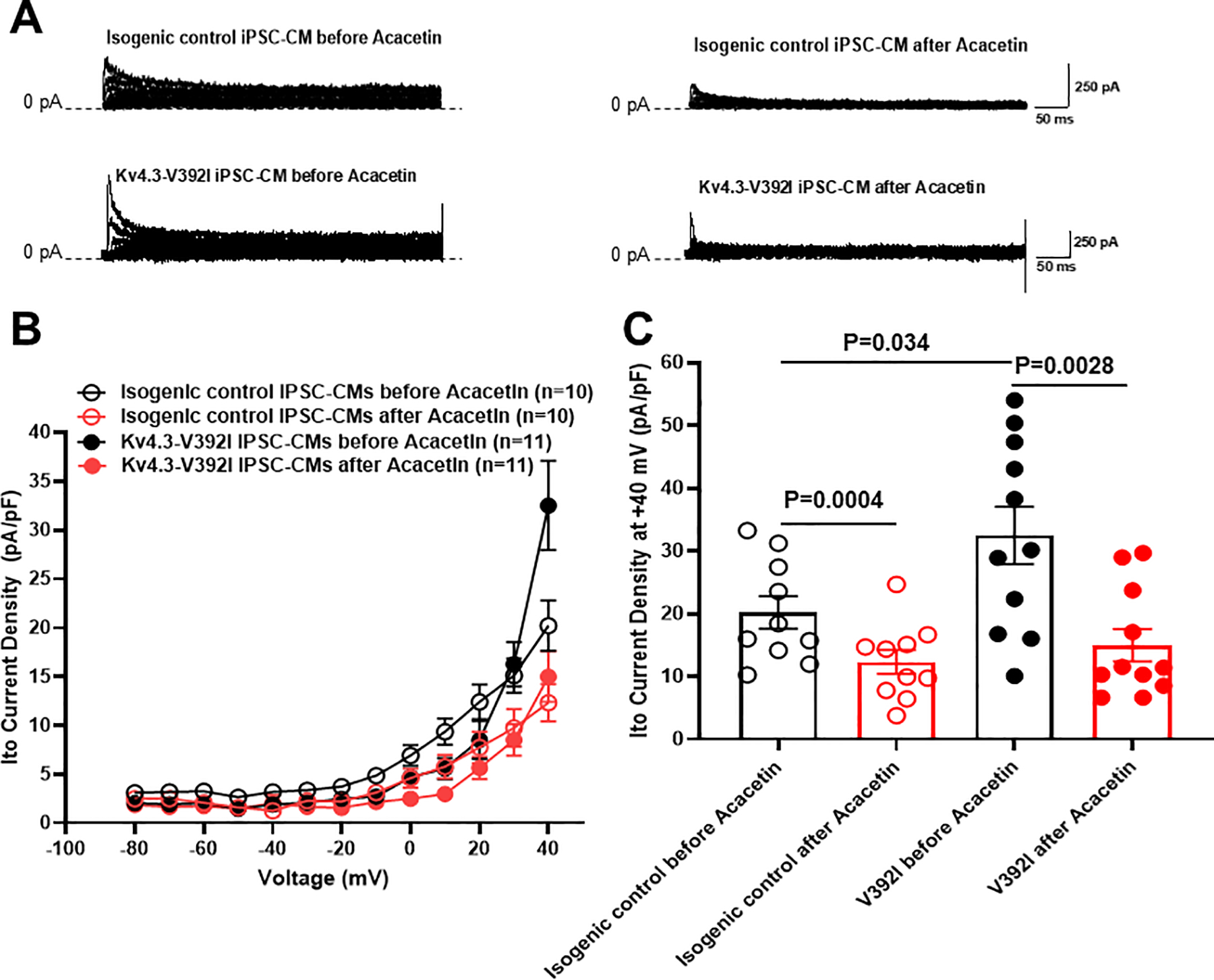
Acacetin significantly inhibited Ito currents from isogenic control and KCND3-V392I iPSC-CMs. A. Representative Ito traces before and after 10 μM Acacetin from isogenic control and KCND3-V392I iPSC-CMs. B. The current voltage relationship for Ito from isogenic control (n= 10) and V392I (n=11) iPSC-CMs before and after 10 μM Acacetin. All values represent mean±SEM. C. Bar graph showing Ito peak current density at +40 mV from isogenic control (n=10) and V392I (n=11) iPSC-CMs before and after 10 μM Acacetin.
To investigate whether KCND3 expression level is altered in KCND3-V392I-derived iPSC-CMs and where KCND3 is located, we carried out western blot and immunofluorescence study on iPSC-CMs. Our data showed KCND3 expression relative to vinculin was significantly increased in KCND3-V392I iPSC-CMs (2.1 ± 0.18, n=6) compared with that in control (1 ± 0.20, n=6, p=0.0087) (Figure 4A, 4B). KCND3 was located in nucleus, plasma membrane (images on the left panel, Figure 4C), endoplasmic reticulin (images on the right panel, Figure 4C), and KCND3 fluorescent intensity was significantly increased in KCND3-V392I-derived iPSC-CMs (44.4±2.1 A.U., n=201) compared with the control cells (27.4±1.1 A.U., p<0.0001) (Figure 4D).
Figure 4.
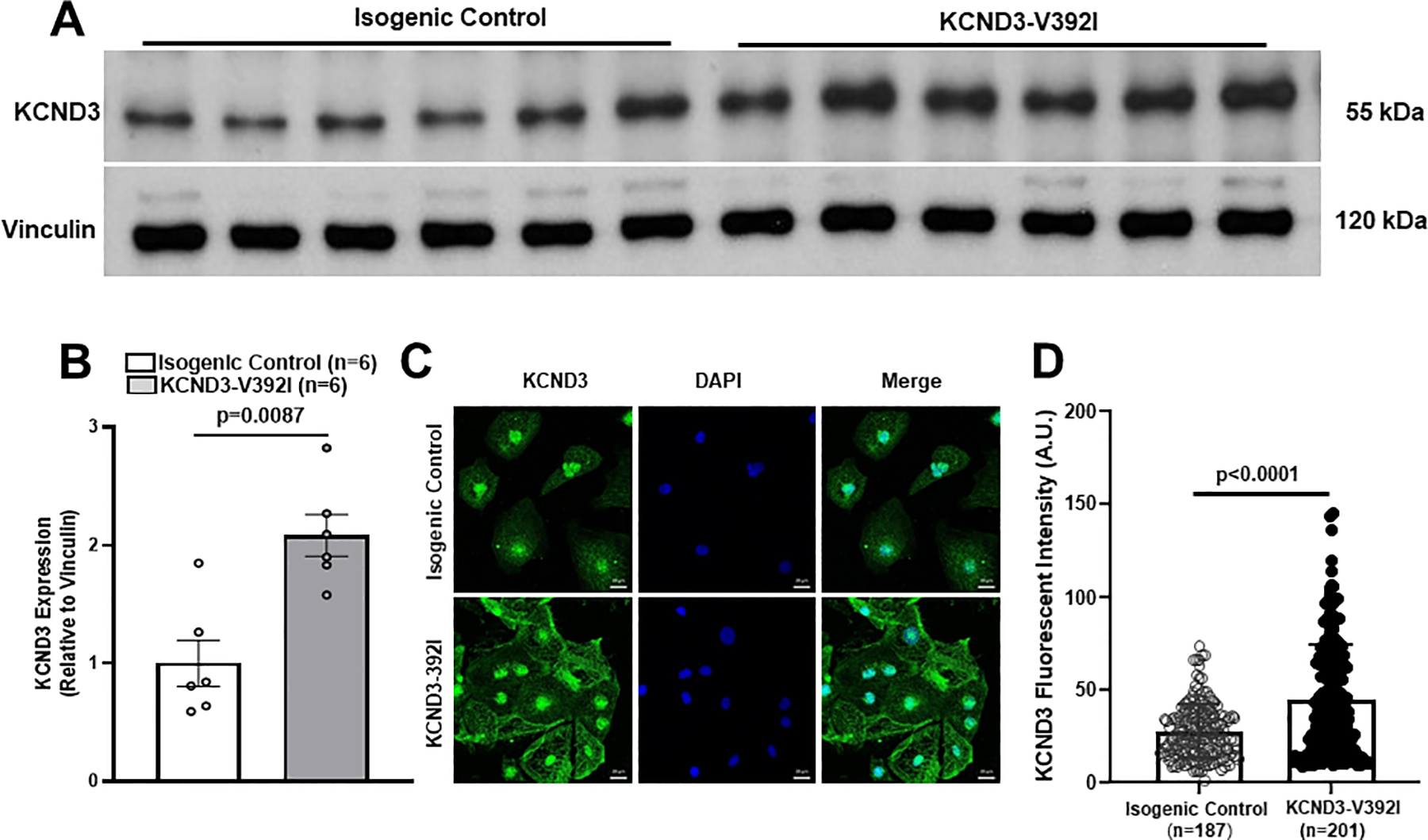
KCND3 expression was significantly increased in KCND3-V392I iPSC-CMs. A. Western blot demonstrating KCND3 expression (relative to Vinculin) in isogenic control and KCND3-V392I iPSC-CMs. B. Bar graph showing KCND3 expression in isogenic control (n=6) and KCND3-V392I iPSC-CMs (n=6) by western blot data analysis. All values represent mean±SEM. Un-paired nonparametric (Mann-Whitney) test was used to determine statistical significance of KCND3 protein expression between two groups. C. Immunofluorescence study demonstrating KCND3 (green) localization in isogenic control and KCND3-V392I iPSC-CMs. Scale bars equal 20μm. D. Bar graph showing KCND3 fluorescent intensity in isogenic control (n=187) and KCND3-V392I iPSC-CMs (n= 201). All values represent mean±SEM.
Next, we tested whether Ito gain-of-function caused by Kv4.3-V392I could demonstrate pro-arrhythmic activities in patient cells using patch clamp current configuration. Action potential (AP) measurements were performed in both isogenic control and Kv4.3-V392I-derived iPSC-CMs. Interestingly, Kv4.3-V392I-iPSC-CMs (not isogenic control-derived iPSC-CMs) revealed an accentuated AP notch. Not surprisingly, 10 μM Acacetin reduced the AP notch, restored AP dome in Kv4.3-V392I-derived iPSC-CMs indicating its potential anti-arrhythmic effect (Figure 5A). However, action potential duration (APD50 and APD90) remained unchanged between isogenic control and Kv4.3-V392I-derived iPSC-CMs before the treatment of Acacetin. 10 μM Acacetin did not affect APD50 and APD90 in both isogenic control and Kv4.3-V392I-derived iPSC-CMs (Figure 5B, 5C) (Supplemental Table III in Supplemental material).
Figure 5.
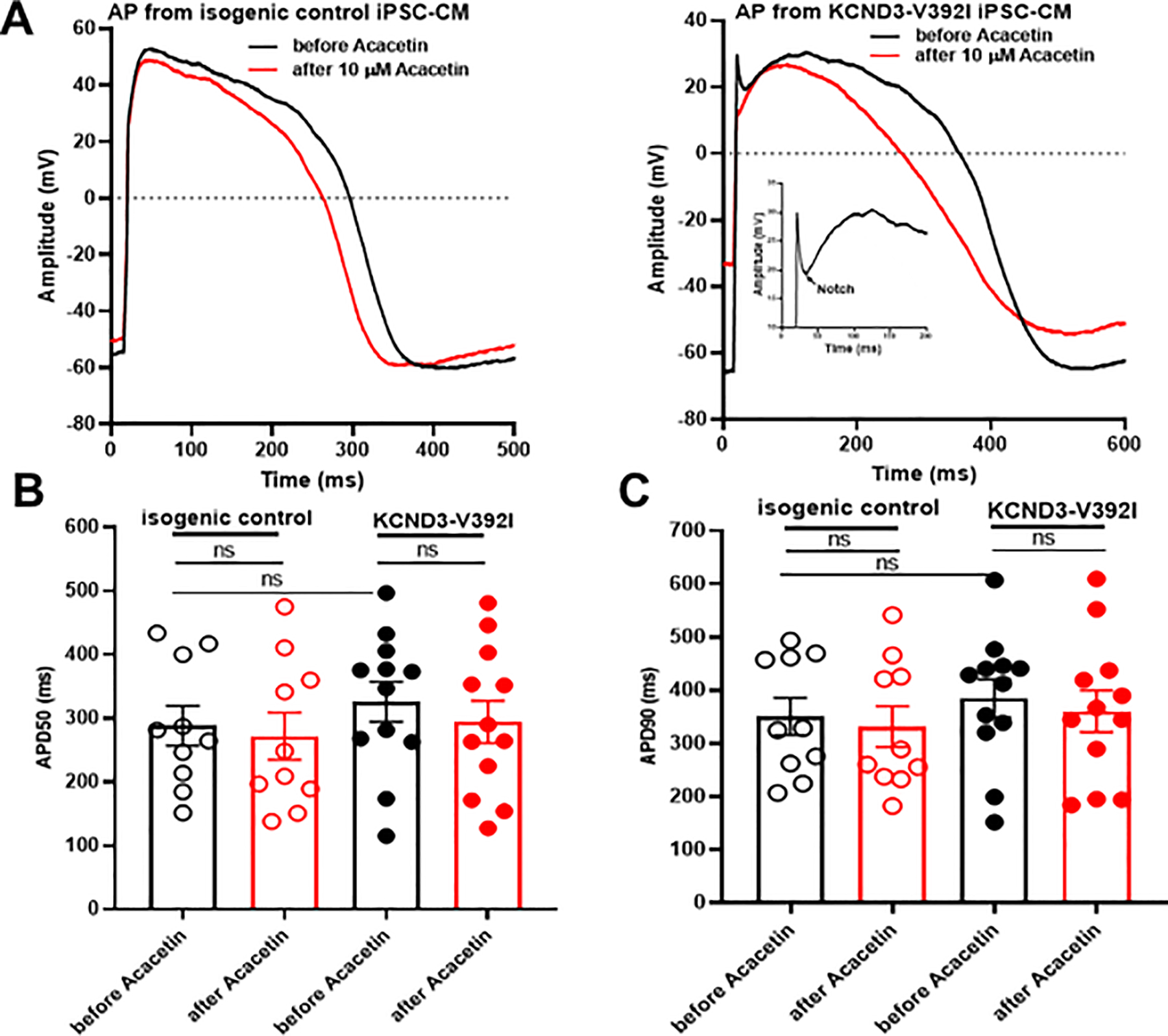
Acacetin reduced accentuated AP notch in Kv4.3-V392I iPSC-CMs. A. Representative patch clamp AP traces before and after 10 μM Acacetin from isogenic control and KCND3-V392I iPSC-CMs. B. Bar graph showing APD50 from isogenic control (n= 10) and V392I (n=12) iPSC-CMs before and after 10 μM Acacetin. All values represent mean±SEM. ns represents no statistical significance. C. Bar graph showing APD90 from isogenic control (n= 10) and V392I (n=12) iPSC-CMs before and after 10 μM Acacetin. All values represent mean±SEM. ns represents no statistical significance.
Since Ito current and AP phase 1 notch were not displayed in every iPSC-CM, it is hard to obtain enough pro-arrhythmic activities using patch clamp assay for statistical analysis. To confirm whether KCND3-V392I could induce increased phase I notch, and whether Acacetin could abolish this increased phase I notch, we used a high-throughput assay, the LEAP technology, to quantify action potential phase I notch. Our data showed that there were 16.7% of phase I notch-like traces in KCND3-V392I iPSC-CMs compared with 3.3% in isogenic control cells (56 out of 335 vs. 14 out of 422 respectively, p=0.00001). After treatment of KCND3-V392I iPSC-CMs with 10 μM of Acacetin for 30 min, phase I notch-like traces were reduced to 11.0% (58 out of 528, p=0.016 vs. before Acacetin) (Figure 6A, 6B). Consistent with patch clamp AP measurements, Acacetin didn’t affect APD30, APD50 and APD90 when using MEA assay (Figure 6A, 6C) (Supplemental Table IV in Supplemental material).
Figure 6.
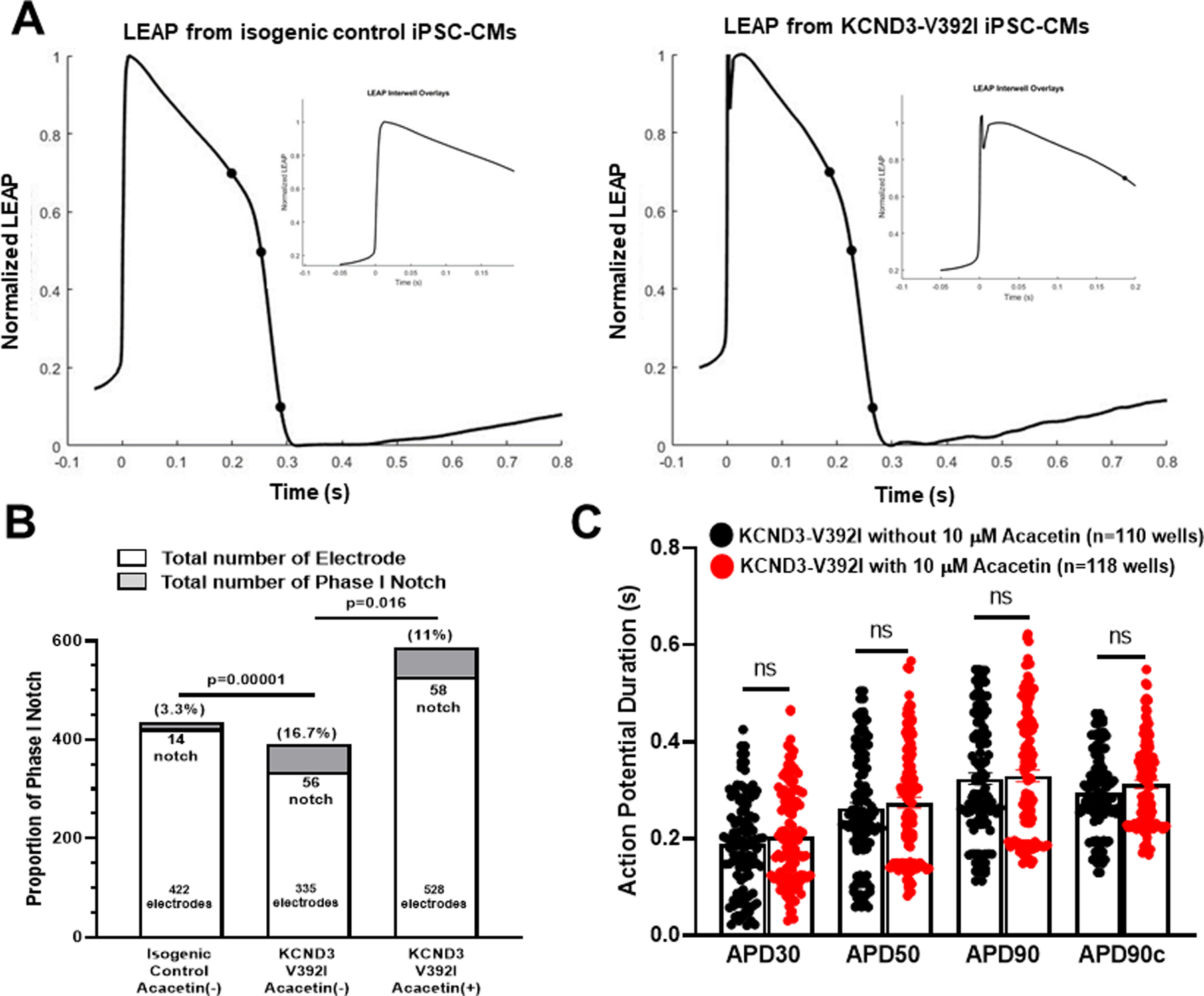
Acacetin reduced total number of phase 1 notch in Kv4.3-V392I iPSC-CMs. A. Representative LEAP traces from isogenic control and KCND3-V392I iPSC-CMs. B. Bar graph showing LEAP proportion of phase I notch in isogenic control and KCND3-V392I iPSC-CMs without and with 10 μM Acacetin. Percentage of Phase I Notch among total electrodes in isogenic control vs. that in KCND3-V392I without Acacetin (p=0.00001), percentage of Phase I Notch among total electrodes in KCND3-V392I without Acacetin vs. that in KCND3-V392I with Acacetin (p=0.016). C. Bar graph showing LEAP APD30, APD50 and APD90 in KCND3-V392I iPSC-CMs without (n=110) and with (n=118) 10 μM Acacetin. All values represent mean±SEM. APD90 (Fridericia s) represents corrected APD90. ns represents no significant difference.
Discussion
The JWSs, consisting of BrS and ERS, have captured the interest of the cardiology community over the past 2 decades and are associated with vulnerability to development of polymorphic VT and VF leading to SCD5. BrS can be diagnosed in the presence of a “type 1 Brugada ECG,” consisting of a J-point elevation ≥2 mm with descending ST segment and negative T wave in the right precordial leads V1 and V2. ERS is generally diagnosed in patients who display ER in the inferior and/or lateral leads presenting with aborted cardiac arrest, documented VF, or polymorphic VT with ER defined as the presence of (1) an end-QRS notch or slur on the downslope of a prominent R wave. The notch/slur should be entirely above the baseline. (2) A peak of the J wave (notched or slurred) ≥0.1 mV in 2 or more contiguous leads of the 12-lead ECG, excluding leads V1 to V35 6.
Kv4.3-V392I was first reported in a 20-year-old male autopsy-negative sudden death victim without any premortem ECGs and a negative family history of sudden death or arrhythmia-related cardiac events3. Here, we reported Kv4.3-V392I mutation in an adopted 18-year-old patient diagnosed clinically as ERS with the J-point elevation in serial 12-lead ECGs displaying global and inferolateral global J point elevation and polymorphic VT, VF, atrial fibrillation and atrial flutter. Our patch clamp studies revealed increased levels of Ito in both TSA201 cells transfected with KCND3-V392I and iPSC-CMs derived from the patient with the KCND3-V392I variant, which is consistent with our previous findings. Further, our western blot study revealed that KCND3 expression was increased in KCND3-V392I-derived iPSC-CMs compared with control iPSC-CMs. Immunofluorescence study exhibited that KCND3 was localized in nucleus, cytosol, endoplasmic reticulum and plasma membrane. KCND3 fluorescent intensity was also increased in KCND3-V392I iPSC-CMs indicating KCND3 expression was accentuated in patient cells. These two findings supported that our Ito gain-of-function observed in patch clamp study was mainly due to a biogenic increase in KCND3 expression, rather than a biophysical gain-of-function in the KCND3-encoded Kv4.3 channels from the patient with the KCND3-V392I variant.
Our group previously reported that gain-of-function pathogenic variants in KCND3 can serve as a pathogenic substrate for BrS1. Since BrS and ERS have similar cellular and genetic pathophysiology for the genesis of arrhythmias5, our findings point to the gain of function of Ito caused by the KCND3-V392I mutation as providing the pathogenic substrate in this patient diagnosed with JWS/ERS.
Acacetin is a flavone compound (5, 7-dihydroxy-4-methoxyflavone) that is broadly distributed in plant pigments and responsible for many of the colors in nature10. Acacetin blocks the Kv1.3 channel, inhibits human T cell activation, and serves as an attractive therapeutic target to treat inflammatory and immunological disorders12. Acacetin suppresses IKur, I-ACh and Ito thereby prolonging the action potential duration in human atrial myocytes without affecting sodium, L-type calcium, or inward-rectifier potassium currents in guinea pig cardiac myocytes. Although Acacetin causes a weak reduction in hERG and hKCNQ1/hKCNE1 channels stably expressed in HEK 293 cells, it did not prolong the corrected QT interval in rabbit hearts. In anesthetized dogs, Acacetin is reported to prolong the atrial effective refractory period without prolonging the corrected QT interval and to effectively prevent atrial fibrillation (AF)10.
Acacetin inhibits hKv4.3 channels by binding to their P-loop filter helix and S6 domain at both the closed and open state of the channels and decreases the recovery from inactivation11. Blockade of hKv4.3 wild type channel by Acacetin is use- and frequency-dependent with IC50 of 7.9 μM. hKv4.3 mutants T366A and T367A affecting the P-loop helix, and V392A, I395A and V399A affecting the S6-segment have been shown to reduce channel blocking efficacy of Acacetin (IC50, 44.5 μM for T366A, 25.8 μM for T367A, 17.6 μM for V392A, 16.2 μM for I395A, and 19.1 μM for V399A)11.
Here, we report blockade of Kv4.3-WT Ito by Acacetin with IC50 of 7.2 μM in HEK293 cells. In order to examine whether KCND3-V392I causes reduced efficacy of Ito block effect by Acacetin, we utilized 30 μM concentration which was close to the IC50 of Acacetin for previously reported KCND3 variants located in S6-segment. Surprisingly, 30 μM Acacetin almost completely inhibited Ito carried by WT Kv4.3 channels as well as mutant Kv4.3 channels (V392I - located in the S6-segment) in which the current exhibited a prominent gain of function. In order to test whether a 10 μM IC50 concentration of Acacetin might be appropriate to treat this patient, we generated isogenic control iPSC-CMs and patient-derived iPSC-CMs harboring the KCND3-V392I pathogenic variant. We found that 10 μM Acacetin inhibited Ito by approximately 50% in iPSC-CMs from the KCND3-V392I-positive patient and approximately 40% from isogenic control iPSC-CMs. In the patient-derived iPSC-CMs, Acacetin brought Ito back to normal levels, thus correcting the gain-of-function caused by the pathogenic variant.
Recently, Di Diego et al reported that Acacetin (5–10 μM) reduced Ito density, AP notch and J wave area and totally suppressed the electrocardiographic and arrhythmic manifestation of both BrS and ERS. In wedge and whole-heart models of JWS, increasing Ito with NS5806, decreasing INa or ICa (with ajmaline or verapamil) or hypothermia all resulted in accentuation of epicardial AP notch and ECG J waves, resulting in characteristic BrS and ERS phenotypes. All repolarization defects giving rise to VT/VF in the BrS and ERS models were reversed by acacetin, resulting in total suppression of VT/VF indicating Acacetin as a promising new pharmacologic treatment for JWS13. Here, we report that KCND3-V392I-derived iPSC-CMs demonstrated accentuated AP notch, whereas 10 μM Acacetin corrected the increased AP notch and reduced the total number of AP notches observed in patient-derived iPSC-CMs harboring the KCND3-V392I pathogenic variant without affecting APD which was similar to the findings observed in the pharmacological mimic-JWS animal model13. These findings suggest that this action of Acacetin to reduce Ito may be of therapeutic benefit by producing an inward shift in the balance of current in the early phases of the epicardial action potential, thus reversing the substrate responsible for the development of prominent J waves that reflect the substrate for development of phase 2 reentry and VT/VF4. Acacetin may thus serve as a novel therapeutic for both BrS and ERS providing an alternative to quinidine for these sudden death syndromes and avoiding the development of QT interval prolongation and the development of life-threatening Torsade de Pointes arrhythmias induced by quinidine9. Besides VT/VF, this patient also presented atrial fibrillation and atrial flutter. Since Acacetin prolonged the atrial effective refractory period without prolonging the corrected QT interval which has been shown to effectively prevent atrial fibrillation10, this compound may benefit this patient with atrial arrhythmias treatment as well. However, because our patient has remained event free, we have not yet pursued compassionate use status of Acacetin.
Conclusions
This pre-clinical study provides pharmacological and functional evidence to suggest that Acacetin may be a novel therapeutic for patients with JWS particular those stemming from KCND3 gain of function by inhibiting Ito and abolishing the accentuated AP notch in patient-derived iPSC-CMs harboring KCND3 gain-of-function pathogenic variant.
Supplementary Material
Acknowledgments:
This work was supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program.
Sources of Funding:
Supported by NIH grants HL47678, HL138103, HL152201 (CA), W.W. Smith Trust (CA) and the Wistar and Martha Morris Fund (CA).
Disclosures:
Dr. Ackerman is a consultant for Abbott, ARMGO Pharma, Boston Scientific, Daiichi Sankyo, Invitae, LQT Therapeutics, Medtronic, and UpToDate. Dr. Ackerman and Mayo Clinic are involved in an equity/royalty relationship with AliveCor, Anuman, and Pfizer. These relationships are all modest, and none of these entities have contributed to this study in any manner. Dr. Antzelevitch is a consultant to Trevena Pharmaceuticals, Novartis Institutes for BioMedical Research and Praxis Pharmaceuticals and has received grant funds from Trevena Pharmaceuticals and Novartis Institutes for BioMedical Research. The other authors report no conflicts.
Nonstandard Abbreviations and Acronyms:
- Ito
The transient outward current
- JWS
J wave syndromes
- BrS
Brugada syndrome
- ERS
Early Repolarization syndrome
- SUD
autopsy-negative sudden unexplained death
- iPSC-CMs
pluripotent stem cell-derived cardiomyocytes
- AP
action potential
- VT/VF
ventricular tachycardia and/or fibrillation
- KATP
ATP-dependent potassium current
- RVOT
right ventricular outflow tract
- ICD
implantation of a cardioverter-defibrillator
- SCA
sudden cardiac arrest
- IKur
ultra-rapidly-delayed rectifier potassium
- IK-ACh
acetylcholine-activated potassium
- AF
atrial fibrillation
- CPR
cardiopulmonary resuscitation
- gnomAD
Genome Aggregation Database
- PVC
premature ventricular contraction
Footnotes
References:
- 1.Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, and Ackerman MJ. Transient outward current (I (to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011; 8:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antzelevitch C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J. 2012; 76:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giudicessi JR, Ye D, Kritzberger CJ, Nesterenko VV, Tester DJ, Antzelevitch C, Ackerman MJ. Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Hum Mutat. 2012; 33:989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antzelevitch C and Yan GX. J wave syndromes. Heart Rhythm. 2010; 7:549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, Gussak I, Hasdemir C, Horie M, Huikuri H et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace. 2017; 19:665–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priori SG and Napolitano C. J-Wave Syndromes: Electrocardiographic and Clinical Aspects. Card Electrophysiol Clin. 2018; 10:355–369. [DOI] [PubMed] [Google Scholar]
- 7.Belhassen B, Glick A and Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004; 110:1731–1737. [DOI] [PubMed] [Google Scholar]
- 8.Yan GX and Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999; 100:1660–1666. [DOI] [PubMed] [Google Scholar]
- 9.Barrows B, Cheung K, Bialobrzeski T, Foster J, Schulze J and Miller A. Extracellular potassium dependency of block of HERG by quinidine and cisapride is primarily determined by the permeant ion and not by inactivation. Channels. 2009; 3:4, 240–249. [PubMed] [Google Scholar]
- 10.Li GR, Wang HB, Qin GW, Jin MW, Tang Q, Sun HY, Du XL, Deng XL, Zhang XH, Chen JB et al. Acacetin, a natural flavone, selectively inhibits human atrial repolarization potassium currents and prevents atrial fibrillation in dogs. Circulation. 2008; 117:2449–2457. [DOI] [PubMed] [Google Scholar]
- 11.Wu HJ, Sun HY, Wu W, Zhang YH, Qin GW and Li GR. Properties and molecular determinants of the natural flavone acacetin for blocking hKv4.3 channels. PLoS One. 2013; 8: e57864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao N, Dong Q, Fu XX, Du LL, Cheng X, Du YM, Liao YH. Acacetin blocks kv1.3 channels and inhibits human T cell activation. Cell Physiol Biochem. 2014; 34:1359–1372. [DOI] [PubMed] [Google Scholar]
- 13.Di Diego JM, Patocskai B, Barajas-Martinez H, Borbáth V, Ackerman MJ, Burashnikov A, Clatot J, Li GR, Robinson VM, Hu D et al. PLos One. 2020; Nov 24;15(11): e0242747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.