

Abstract
Polycystic kidney disease (PKD) is characterized by the formation and progressive enlargement of fluid-filled cysts due to abnormal cell proliferation. Cyclic AMP agonists, including arginine vasopressin, stimulate ERK-dependent proliferation of cystic cells, but not normal kidney cells. Previously, B-Raf proto-oncogene (BRAF), a MAPK kinase kinase that activates MEK-ERK signaling, was shown to be a central intermediate in the cAMP mitogenic response. However, the role of BRAF on cyst formation and enlargement in vivo had not been demonstrated. To determine if active BRAF induces kidney cyst formation, we generated transgenic mice that conditionally express BRAFV600E, a common activating mutation, and bred them with Pkhd1-Cre mice to express active BRAF in the collecting ducts, a predominant site for cyst formation. Collecting duct expression of BRAFV600E (BRafCD) caused kidney cyst formation as early as three weeks of age. There were increased levels of phosphorylated ERK (p-ERK) and proliferating cell nuclear antigen, a marker for cell proliferation. BRafCD mice developed extensive kidney fibrosis and elevated blood urea nitrogen, indicating a decline in kidney function, by ten weeks of age. BRAFV600E transgenic mice were also bred to Pkd1RC/RC and pcy/pcy mice, well-characterized slowly progressive PKD models. Collecting duct expression of active BRAF markedly increased kidney weight/ body weight, cyst number and size, and total cystic area. There were increased p-ERK levels and proliferating cells, immune cell infiltration, interstitial fibrosis, and a decline in kidney function in both these models. Thus, our findings demonstrate that active BRAF is sufficient to induce kidney cyst formation in normal mice and accelerate cystic disease in PKD mice.
Keywords: mitogen activated protein kinase, MAPK, ERK, cell proliferation, polycystic kidney disease, ADPKD
Graphical Abstract

INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disorder characterized by incessant growth of fluid-filled cysts, leading to loss of functional nephrons, inflammation, fibrosis, and progressive decline in renal function. Approximately one-half of ADPKD patients reach end-stage renal disease by the 6th decade of life, accounting for 5–10% of patients on renal replacement therapy.1 The majority of ADPKD cases are caused by mutations in PKD1 or PKD2, which encode polycystin-1 (PC1) and polycystin-2 (PC2), respectively.2 PC1 and PC2 interact to form a Ca2+ permeable, non-selective cation channel; however, it remains unclear how PKD mutations induce renal cyst formation.3
The mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway is an important signaling cascade involved in epithelial cell proliferation. The family of RAF kinases, consisting of RAF-1 (also called CRAF), BRAF, and ARAF, phosphorylate the MAPK kinase MEK, which in turn phosphorylates and activates ERK.4, 5 Phosphorylated ERK (P-ERK) translocates into the nucleus where it activates transcription factors involved in cell proliferation, including Fos and Elk-1. RAF-1 and BRAF share homology in amino acid sequence; however, their regulation by cAMP is different, and the mitogenic response to cAMP stimulation depends on whether the signaling is through RAF-1 or BRAF.6–8 In some cells, protein kinase A (PKA) phosphorylates inhibitory sites on RAF-1, decreasing P-ERK levels and cell proliferation in response to cAMP. By contrast, cAMP activates BRAF and ERK-dependent proliferation of other cell types, including thyroid cells, hepatocytes, and neuronal cells.9, 10
Human ADPKD kidneys and PKD mouse kidneys have elevated P-ERK levels compared to normal kidneys.11–13 In cultured human ADPKD cells, cAMP agonists increase BRAF activity, leading to ERK-dependent cell proliferation.11, 12, 14–18 By contrast, cAMP has no effect on BRAF activity in normal human kidney (NHK) cells, but rather decreases proliferation through RAF-1 inhibition.11, 12 These data indicate that BRAF is a key molecular switch responsible for cAMP-dependent proliferation of cyst epithelial cells; however, the impact of active BRAF specifically on cyst formation and enlargement has not been demonstrated in vivo.
To determine if active BRAF is sufficient to initiate cell proliferation and cystogenesis, we generated a transgenic mouse (BRAFLSL-V600E) to conditionally express BRAFV600E (valine to glutamic acid substitution at position 600), a common activating mutation in BRAF.19 BRAFLSL-V600E mice were crossed with Pkhd1-Cre mice20 to express constitutively active BRAF specifically in collecting ducts (CD), a predominant site for cyst formation.21–24 We also determined if expression of active BRAF caused a further increase in ERK activation and accelerated cystic disease in Pkd1RC/RC mice, an orthologous model of ADPKD, and pcy/pcy mice, a well-characterized slowly progressive model of renal cystic disease. Our results demonstrate, for the first time, that active BRAF is sufficient to induce cystogenesis in normal mice and accelerate the progression of cystic disease in two relevant models of PKD.
METHODS
Full methods are available in Supplemental Material. We generated a transgenic mouse strain BRAFLSL-V600E which has the human BRAFV600E coding sequence downstream of a splice acceptor and the loxP-flanked-mCherry STOP cassette under the transcriptional control of the endogenous Rosa26 transcriptional regulatory elements (Supplemental Figure S1a). BRAFLSL-V600E mice were bred with Pkhd1-Cre mice to selectively overexpress active BRAF in CDs of otherwise normal mice (BRafCD mice). In all experiments, BRAFLSL-V600E mice were heterozygous for the transgene.
The BRAFLSL-V600E allele was crossed into the Pkd1RC/RC mouse, a slowly progressive model of ADPKD and Pkhd1-Cre mice to generate Pkd1RC/RC; BRAFV600E; Pkhd1-Cre (Pkd1RC/RC; BRafCD) mice. We did not observe any difference in BRAFV600E-induced cystogenesis between wildtype mice compared to phenotypically normal Pkd1RC/+ mice. BRAFLSL-V600E mice were also bred into the pcy/pcy mouse, a well-characterized slowly progressive non-orthologous model of PKD that is caused by a mutation in the murine homolog of NPHP3.25–29 Pkhd1-Cre mice were crossed to these mice to generate pcy/pcy; BRAFLSL-V600E; Pkhd1-Cre (pcy/pcy; BRafCD) mice. The Institutional Animal Care and Use Committee of University of Kansas Medical Center approved the protocols for the use of these mice (protocol no. 2021–2605).
We measured total body weight (BW) and collected blood and kidneys for analysis of blood urea nitrogen (BUN), kidney weight/body weight (KW/BW) (%), renal cyst number, cystic index, fibrosis (Masson’s trichrome), BRAF expression, P-ERK, P-LKB1, cell proliferation (PCNA and Ki-67), apoptosis (cleaved-caspase-3), senescence marker p16INK4A, and macrophage marker CD68.
RESULTS
CD-specific expression of BRAFV600E induces renal cyst formation in normal mice.
Overexpression of human BRAFV600E was confirmed by immunoblot analysis by probing with an antibody for the V600E mutant form of human BRAF (Supplemental Figure S1b). BRafCD mice appeared normal; however, BW was lower and KW/BW (%) was elevated by 3 weeks (Supplemental Table S1). Histological analysis revealed the presence of dilated tubules and de novo cyst formation in the cortex and medulla (Figure 1a). BRAFV600E localization was visualized by immunofluorescence using a BRAF antibody and Dolichos biflorus agglutinin (DBA), a CD marker (Supplemental Figure S2). We observed staining for BRAFV600E in DBA-positive tubules of BRafCD kidneys. Collecting ducts and cysts of BRafCD kidneys lacked staining for mCherry protein, consistent with Cre induced removal of the lox-stop-lox (LSL) cassette. DBA staining confirmed that 98.8 ± 0.6% of cysts originated from CD cells (n = 403 cysts in representative kidney sections from 6 mice per group), and no cysts were positive for Lotus tetragonolobus (LTA), a marker for proximal tubules (Figure 1d,e and Supplemental Figure S3)
Figure 1. Collecting duct (CD)-specific expression of constitutively active BRAF induces cell proliferation and cyst formation in wildtype kidneys.
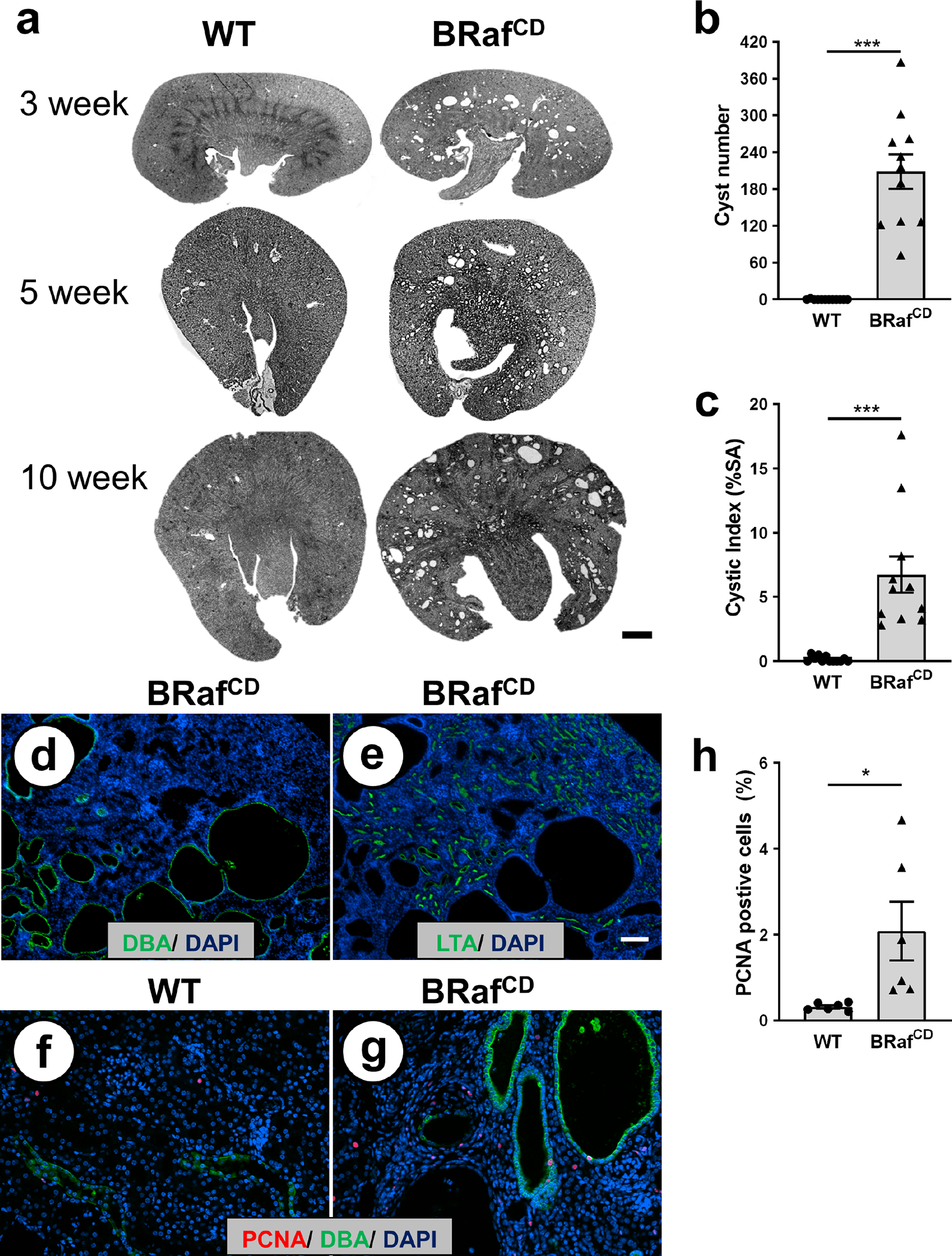
(a) Representative kidney sections from 3-, 5-, and 10-week-old BRAFLSL-V600E; Pkhd1-Cre (BRafCD) and phenotypic normal (BRAFLSL-V600E, No Cre; wildtype [WT]) mice. Scale bar = 1 mm. Bar graphs are mean ± SEM for (b) number of cysts and (c) the percentage of cystic area per total kidney cross-sectional surface area (cystic index) for N = 11 mice per group. ***P < 0.001, compared to WT kidneys. Representative fluorescent images of adjacent kidney sections (5 μm) from 10-week-old BRafCD mice stained with (d) CD marker Dolichos biflorus agglutinin (DBA; green) or (e) Lotus tetragonolobus (LTA; green). DAPI (blue) was used to visualize nuclei. Scale bar = 100 μm. Fluorescent images of (f) WT and (g) BRafCD kidneys stained with an antibody to PCNA (red) and DBA (green). Scale bar = 5 μm. (h) Bar graph represents the mean ± SEM for the percentage of PCNA-positive cells in 10-week-old kidney sections (N = 6 per group) normalized to total nuclei. *P < 0.05, compared to WT kidneys.
Histological analysis of representative kidney sections confirmed the presence of numerous cysts by 10 weeks of age (Figure 1b). Cystic index was 6.7 ± 1.4% for BRafCD mice compared to 0.2 ± 0.1% for phenotypically normal Pkd1RC/+ (WT) kidneys (Figure 1c). Kidney sections were stained with DBA (green), an antibody for PCNA (red) to measure the number of proliferating cells, and DAPI (blue) to determine total nuclei (Figure 1f, g). As expected, there were few proliferating cells in the normal kidneys and expression of active BRAF resulted in a 3.4-fold higher number of PCNA-positive cells (Figure 1h). Elevated PCNA levels in BRafCD kidneys were confirmed by immunoblot analysis (Supplemental Figure S4). There were increased numbers of proliferating cells in DBA-positive tubules and cysts, as well as the interstitium; however, there were few DBA-negative epithelial cells that stained positive for PCNA (Supplemental Figure S5a). The number of cells staining positive for Ki-67, another proliferation marker, was 8.5-fold higher in the BRafCD compared to wildtype kidneys (Supplemental Figure S6), confirming the PCNA results. Kidneys of 10-week-old BRafCD mice had elevated P-ERK (Supplemental Figure S5b), and both cysts and dilated tubules of BRafCD kidneys contained tightly packed cells and multi-layered sheets of cells (Figure 1g, Supplemental Figure S6b).
Effect of BRAFV600E expression on fibrosis and renal function.
Representative 10-week-old kidney sections were stained with Masson’s trichrome to visualize fibrosis (Figure 2a, b). Fibrotic areas with distinct blue staining of collagen deposits and pre-fibrotic areas with interstitial edema that stained light blue were increased 13.6-fold in BRafCD compared to normal kidneys (Figure 2c). There was pitting of the surface of the kidneys, indicative of focal fibrosis and scarring, and contraction of the cortex (Supplemental Figure S7). BRafCD kidneys had an accumulation of immune cells, determined by staining with the macrophage marker CD68 (Supplemental Figure S8). Blood urea nitrogen (BUN) was significantly elevated in BRafCD mice (Figure 2d), indicating a decline in renal function.
Figure 2. CD-specific expression of active BRAF leads to renal fibrosis and a decline in kidney function in wildtype mice.

Representative images of kidney sections from 10-week-old (a) wildtype (WT) and (b) BRafCD mice stained with Masson’s trichrome to visualize pathogenic collagen deposits (blue) and pre-fibrotic interstitial edema (pale blue appearance, arrows). Scale bar = 100 μm. (c) Data are mean ± SEM for fibrotic index (percentage of interstitial fibrosis and edema per total area of the section). Slides containing tissue sections were coded to conceal the group assignment before being visually scored (N = 4 kidneys per group). (d) Data are mean ± SEM for levels of blood urea nitrogen (BUN), a marker of renal function, in WT and BRafCD mice at 10 weeks of age (N = 4). *P < 0.05 and ***P < 0.001, compared to WT.
Effect of CD-specific expression of BRAFV600E on renal cystic disease in Pkd1RC/RC mice.
Kidneys of Pkd1RC/RC; BRafCD mice were clearly cystic and notably larger than kidneys of Pkd1RC/RC littermates (Figure 3a). At 10 weeks, Pkd1RC/RC; BRafCD mice had significantly lower BW compared to Pkd1RC/RC littermates (Figure 3b); while KW/BW (%) was more than 2-fold higher (Figure 3c). Tissue sections of Pkd1RC/RC; BRafCD kidneys showed that activated BRAF accelerated cystic disease such that noticeably larger cysts were present as early as 5 weeks (Figure 3d). By 10 weeks, cyst number increased from 152 ± 25 to 354 ± 45 (P < 0.001) and cystic area increased from 11.6% ± 2.5 to 37.5 ± 2.8% (P < 0.001) in Pkd1RC/RC; BRafCD kidneys compared to Pkd1RC/RC kidneys (Figures 3e, f). There was also a corresponding increase in PCNA (Figure 4a–c, Supplemental Figure S4) and P-ERK (Figure 4d–f, Supplemental Figure S9a) in 10-week-old Pkd1RC/RC; BRafCD kidneys compared to Pkd1RC/RC kidneys; however, we did not detect differences in phosphorylated S6 (P-S6), a downstream target of mTOR (data not shown) in either the male or female kidneys. However, at 5 weeks, we observed a small but significant increase in P-S6/S6 in the male Pkd1RC/RC; BRafCD kidneys compared to Pkd1RC/RC kidneys (Supplemental Figure S10). By contrast, there was no difference in P-S6 levels in the kidneys of female mice. Despite increased cell proliferation and cyst growth, there were no indications of solid neoplastic lesions due to BRAFV600E expression. We found that BRAFV600E expression increased levels of p16INK4a, a tumor suppressor expressed by senescent cells and cleaved caspase-3, a marker for apoptosis (Supplemental Figures S11, S12)
Figure 3. CD expression of active BRAF accelerates renal cystic disease in Pkd1RC/RC mice.

(a) Representative images of kidneys of Pkd1RC/RC (RC/RC) and Pkd1RC/RC; BRAFLSL-V600E;Pkhd1-Cre (RC/RC; BRafCD) mice at 10 weeks of age. Scale bar = 5 mm. Bar graphs are mean ± SEM for (b) body weight (BW) and (c) kidney weight, as a percentage of body weight [KW/BW (%)], for Pkd1RC/RC (RC/RC; N = 11) and Pkd1RC/RC; BRAFLSL-V600E; Pkhd1-Cre (RC/RC; BRafCD, N = 10) mice at 10 weeks of age. (d) Images of tissue sections from RC/RC and RC/RC; BRafCD kidneys at 5 and 10 weeks of age, demonstrating an acceleration of cystic disease. Scale bar = 1 mm. Bar graphs are mean ± SEM for (e) the number of cysts (> 50 μm diameter) per section and (f) percentage of total surface area of the section (cystic index). ***P < 0.001, compared to RC/RC.
Figure 4. CD expression of active BRAF increases ERK phosphorylation and cell proliferation in Pkd1RC/RC kidneys.

Representative 10-week-old (a) RC/RC and (b) RC/RC; BRafCD kidney sections stained with an antibody to PCNA (red) and DBA (green). There were increased PCNA-positive cells in the cyst-lining epithelia as well as the interstitium (Supplemental Figure S5). Scale bar = 500 μm. (c) Percentage of PCNA-positive nuclei, normalized to total nuclei that were visualized with DAPI (blue). A minimum of 4 × 103 cells per kidney section were counted. P < 0.01, compared to RC/RC (N = 6). (d) Representative immunoblots for P-ERK, ERK, and BRAF in 10-week-old kidneys without (−) and with (+) CD expression of the BRAFV600E transgene. GAPDH was used as a loading control. The number above the blots are P-ERK/ERK and BRAF/GAPDH, respectively. Bar graphs are mean ± SEM for levels of (e) P-ERK/ERK and (f) BRAF/GAPDH for RC/RC and RC/RC; BRafCD kidneys. *P < 0.05, compared to RC/RC (N = 3).
Pkd1RC/RC mice had significant renal fibrosis at 10 weeks and the expression of active BRAF increased fibrosis by 2.6-fold (Figure 5a–c). To investigate a potential mechanism for increased fibrosis, we measured levels of phosphorylated acetyl-CoA carboxylase (ACC), a target for the energy sensor AMP-activating protein kinase (AMPK), which contributes importantly to fatty acid metabolism and development of renal fibrosis.30 ERK phosphorylates a key inhibitor site (Ser 428) on LKB1 (P-LKB1), the main upstream kinase involved in AMPK activation. We found that the expression of active BRAF in WT and Pkd1RC/RC kidneys increased P-LKB1 and caused a trend for decreased P-ACC (Supplemental Figure S9, S10).
Figure 5. Effect of CD expression of active BRAF on renal fibrosis and function, and the survival of Pkd1RC/RC mice.

Tissue sections from 10-week-old (a) RC/RC and (b) RC/RC; BRafCD were stained with Masson’s trichrome to visualize fibrosis. Scale bar = 50 μm. (c) The percentage of fibrosis to the surface area (SA) of the entire kidney section (fibrotic index) for RC/RC and RC/RC; BRafCD kidneys. (d) Bar graph represents the mean ± SEM for BUN levels from RC/RC and RC/RC; BRafCD mice. **P < 0.01 and ***P < 0.001, compared to RC/RC. (e) Gehan-Breslow survival curves for BRafCD (N = 14; 8 males and 6 females), RC/RC (N = 10; 7 males and 3 females), and RC/RC; BRafCD (N = 10; 7 males and 3 females) mice for 60 weeks. Wildtype mice lacking transgenic BRAF expression were not included since there is 100% survival during this period. Mice were given standard chow and water ad libitum and monitored regularly until a humane end point was reached. Average survival age for BRafCD mice (26 ± 1 weeks) and RC/RC; BRafCD mice (31 ± 1 weeks) were significantly different from that of the RC/RC mice (61 ± 4 weeks, P < 0.001).
Expression of active BRAF in Pkd1RC/RC mice increased the number of CD68 positive cells consistent with macrophage accumulation (Supplemental Figure S8) and the fibrotic index (Figure 5c). There was a corresponding increase in BUN from 25.7 ± 1.0 to 44.5 ± 2.2 mg/dl, P < 0.01 (Figure 5d), indicating a more rapid decline in kidney function in PKD mice expressing the active BRAF.
Effect of CD-specific expression on BRAFV600E on survival of wildtype and Pkd1RC/RC mice.
In a survival study, mice expressing active BRAF died at 20–30 weeks of age, regardless of the Pkd1 genotype (Figure 5e). The kidneys were massively enlarged at the time of death and examination of a kidney of BRafCD mice revealed that the kidneys had a severe dilated renal pelvis and distortion of the renal papilla, indicating hydronephrosis (Supplemental Figure S13). To determine if ureteral obstruction contributed to early tubule dilation and cyst formation, ureters of BRafCD mice were examined by a renal pathologist (T.F). By 2 weeks of age, BRafCD kidneys had pre-cystic tubule dilations; however, there was no evidence of ureteral obstruction, suggesting that increased cell proliferation and cyst formation were not consequences of intraluminal pressure. Furthermore, progressive loss of the renal papilla, which typically occurs in hydronephrosis, was not observed at 10 weeks (Figures 1a, 3d and 6a), a time nearly 8 weeks after initial cyst formation.
Figure 6. CD-selective expression of active BRAF increased renal cystic disease and fibrosis and accelerated the decline in kidney function in pcy/pcy mice.

(a) Representative kidney sections from 10-week-old pcy/pcy and pcy/pcy; BRAFLSL-V600E; Pkhd1-Cre (pcy/pcy; BRafCD) mice. Scale bar = 1 mm. Bar graphs represent the mean ± SEM for (b) body weight (BW) and (c) kidney weight (KW), as a percentage of BW [KW/BW (%)], (d) cyst area, relative to total surface area [cystic index (% SA)], and (e) number of cysts in kidney sections from pcy/pcy and pcy/pcy; BRafCD mice. **P < 0.01 and ***P < 0.001, compared to pcy/pcy mice (N = 4–6). (f) Fluorescent images of kidney sections from pcy/pcy and pcy/pcy; BRafCD mice that were stained with PCNA (red), DBA (green) and DAPI (blue). (g) Bar graph displays the mean ± SEM for PCNA-positive cells as a % of total nuclei. *P < 0.05, compared to pcy/pcy kidneys (N = 4) (h) Representative immunoblots for P-ERK, ERK, BRAF, and GAPDH in kidney lysates from pcy/pcy mice without (−) or with (+) CD expression of active BRAF. The numbers above the bands are P-ERK/ERK and BRAF/GAPDH, respectively. (i) The percentage of fibrosis to the surface area (SA) of the entire kidney section (fibrotic index) for pcy/pcy and pcy/pcy; BRafCD kidneys. ***P < 0.001, compared to pcy/pcy (N = 6). (j) BUN levels in pcy/pcy and pcy/pcy; BRafCD mice at 10 weeks of age **P < 0.01, compared to pcy/pcy mice (N = 4).
Effect of CD-specific expression on BRAFV600E on renal cystic disease in pcy/pcy mice.
Next, we determined the effect of activated BRAF on disease progression in pcy/pcy mice. These mice develop renal cysts by 4 weeks of age and total kidney volume increases exponentially up to 20 weeks of age.28 We found that pcy/pcy; BRafCD mice had a pronounced increase in KW/BW (%) and a 2.3-fold increase in cystic area at 10 weeks compared to pcy/pcy littermates (Figure 6a–e). pcy/pcy; BRafCD kidneys had elevated levels of P-ERK, a 2.5-fold increase in proliferating cells (Figure 6f–h), a pronounced increase in interstitial fibrosis, and a decline in renal function, as evidenced by a marked increase in BUN (Figure 6i, j).
DISCUSSION
The role of cell proliferation in renal cyst enlargement is well-established; however, it is unclear if aberrant proliferation alone is sufficient for cyst initiation. We generated a conditional allele for tissue-specific control of the expression of constitutively active BRAFV600E, allowing for the investigation of ERK-dependent cell proliferation on cyst formation in normal kidneys. Our data indicate that CD expression of active BRAF increased renal P-ERK levels and induced cell proliferation and de novo cyst formation in BRafCD mice by 3 weeks of age. These mice developed enlarged cystic kidneys with extensive fibrosis and elevated BUN by 10 weeks. We also showed that CD expression of active BRAF increased P-ERK and cell proliferation, accelerated renal cyst growth, fibrosis, and the decline of function in Pkd1RC/RC and pcy/pcy mice, two relevant models of slowly progressive PKD.
Kidneys express BRAF and RAF-1, kinases that are activated by small GTPases within the RAS family. Inactive RAF is held in an auto-inhibited state, stabilized by 14–3-3 protein. Upon stimulation, RAF is recruited to the plasma membrane by activated RAS, which relieves RAF auto-inhibition with concurrent phosphorylation of various activating residues. Of the RAF kinases, BRAF is the easiest to be stimulated by RAS31, 32 and has the highest basal kinase activity.33, 34 Consequently, mutational activation of BRAF is frequently found in human cancers.35 Multiple pathways regulate RAF activity through phosphorylation of serine and threonine residues,36 and the balance between the phosphorylation states of stimulatory and inhibitory sites.
BRAF has two critical phosphorylation sites (Thr598 and Ser601) that determine the extent to which cAMP stimulates BRAF activity and MEK/ERK pathway. Substitution of glutamic acid for valine at codon 600 of BRAFV600E causes insensitivity to negative feedback mechanisms resulting in constitutive, cAMP-independent activation of the kinase.35 BRAFV600E is a common mutation in tumors and account for more than 50% of melanomas. BRAFV600E is also present in benign tumors, including melanocytic nevi (moles), which rarely progress to melanoma37 since it is thought that persistent activate BRAF limits the cell’s proliferative capacity by induction of cellular senescence.
Evidence has shown that cAMP agonists, including arginine vasopressin (AVP), play a central role in ADPKD.14, 38–40 Intracellular cAMP stimulates the proliferation of ADPKD cells but inhibits the proliferation of NHK cells.11, 12 This difference in the mitogenic response to cAMP appears to be at the level of BRAF. 11, 15 Under normal conditions, BRAF is repressed, possibly by a Ca2+-dependent mechanism, and cAMP does not activate BRAF signaling. By contrast, BRAF is de-repressed in ADPKD cells, allowing cAMP-dependent activation of the BRAF-MEK-ERK pathway and cyst epithelial cell proliferation.41 Ca2+ restriction in normal kidney cells using Ca2+ channel blockers caused a phenotypic switch such that cAMP was able to activate BRAF-MEK-ERK and cell proliferation. Conversely, treatment of ADPKD cells with a Ca2+ channel activator prevented cAMP-dependent activation of BRAF and cell proliferation, restoring a normal cellular response to cAMP.42 While these and other studies indicate cAMP-dependent BRAF activation contributes to the proliferation of cystic cells, the capacity for active BRAF and ERK-dependent cell proliferation to initiate cyst formation in otherwise normal cells has not been demonstrated.
Using a novel conditional transgenic mouse model, we showed that CD expression of active BRAF was sufficient to cause PKD in mice without Pkd1 or Pkd1 mutations. While this observation, by itself, does not establish a causal relationship between cAMP-dependent ERK-mediated cell proliferation and the cystic phenotype in ADPKD, it does clearly demonstrate that active BRAF is sufficient to trigger cyst initiation in collecting ducts. It remains unclear if cell proliferation is the only requisite stimulus for renal cyst formation. Transgenic mice overexpressing murine c-myc displayed abnormal renal cell proliferation and formation of cysts throughout the cortex and medulla, leading to death due to renal failure. 43 By contrast, transgenic overexpression of Cux-1, a homeobox gene that increases cell proliferation through p27 inhibition, caused multiorgan hyperplasia, but not renal cyst formation.43 Despite increased cell proliferation and a greater than 40% increase in renal mass by 6 weeks of age, there was no evidence of renal cysts by 6 months in Cux-1 transgenic mice.44 Sharma et al. also showed that increased proximal tubule cell proliferation by Cux1 expression did not accelerate cystic disease in mice after induction of cilia loss.45 These apparently discordant results may be due to differences in the signaling pathways that drive cell proliferation or that proliferation alone is not the only stimulus for cyst formation. It is possible that changes in other cellular phenotypes are involved, such as cell differentiation, planar cell polarity, or fluid secretion. The impact of the proliferative signal may also depend on the cell type. Additional studies are needed to determine if expression of activated BRAF in proximal tubules induces renal cyst formation.
CD expression of BRAFV600E caused an increase in renal P-ERK levels and exacerbated cystic disease in both Pkd1RC/RC and pcy/pcy mice. The ability of activated BRAF to further promote cystogenesis in these mice suggests that endogenous wildtype BRAF is not fully activated, or its abundance is too low for full stimulation of the MEK-ERK pathway in cystic cells. Similarly, Ye et al. showed that activation of protein kinase A (PKA) signaling by kidney-specific knockout of the PKA-regulatory subunit Riα (Prkar1a gene) caused an upregulation of ERK signaling and cell proliferation, and aggravated cystic disease in Pkd1RC/RC mice.46
Active BRAF was sufficient to stimulate cell proliferation and induce cyst formation in wildtype mice, displaying an ADPKD pattern. The focal nature of the cysts suggests additional contributing factors are involved in the initial phase of cystogenesis. One possibility is that tubule obstruction due to cellular hyperplasia is necessary for intracavitary accumulation of fluid. Previous work showed that most cysts in end-stage ADPKD kidneys lack any connections to the nephron and contained hyperplastic outgrowths within the cyst epithelium.47 It is also possible that there is variable expression of factors involved in tissue injury and remodeling within the renal microenvironment (e.g. matrix metalloproteases, matricellular proteins, chemokines, cytokines) that contribute to the focal nature of cystogenesis.
Transepithelial fluid secretion plays a critical role in cyst growth in ADPKD. There is no evidence that active BRAF directly stimulates net solute and fluid secretion. Previously, we showed that CDs have an endogenous fluid secretory mechanism that is normally masked by net fluid absorption. 48–50 Active BRAF may lead to incomplete cellular differentiation and reduced Na+-dependent fluid absorption, unmasking Cl−-dependent fluid secretion. In addition, we found that BRAF expression increased the phosphorylation of an inhibitory residue on LKB1 (Supplemental Figures S9, S10), reducing AMPK signaling (decreased P-ACC). During periods of low cellular energy, AMPK activation has been shown to inhibit CFTR Cl channels. 51 We speculate that BRAF inhibition of LKB1-AMPK signaling contributes to increased CFTR-dependent Cl− secretion in CD cysts of BRafCD mice.
CD-specific expression of BRAFV600E in PKD mice increased PCNA-staining of DBA-positive cystic cells as expected; however, unlike WT kidneys expressing BRAFV600E, there was increased PCNA staining of DBA-negative cysts (Supplemental Figure S14). cAMP activation of wildtype BRAF and expression of active BRAFV600E may lead to poor cellular differentiation of the cystic epithelium. A lack of lectin staining has been demonstrated in cysts of ADPKD kidneys, in situ, where a significant percentage of cysts failed to stain tubule-specific lectins. 21, 52 We also found an increase in interstitial cell proliferation in BRAF-expressing kidneys. While we cannot rule out the possibility that other cell types in the kidneys expressed BRAFV600E, it is likely that the cysts injure the surrounding tissue causing changes in the microenvironment that stimulate myofibroblast recruitment, proliferation and activation, and macrophage infiltration, leading to renal fibrosis and a decline in renal function. Cellular senescence and a senescence-associated secretory phenotype are also known to contribute to renal fibrosis through expression of inflammatory cytokines and immune cell modulators, fibroblast activation, and matrix accumulation. We found that CD expression of active BRAF increased the expression of the senescence marker p16INK4a in wildtype and Pkd1RC/RC mice. Additional studies are needed to provide insight into the role of cellular senescence in inflammation and fibrosis in ADPKD.
BRAFCD mice had lower BW than normal littermates, suggesting a delay in body growth that was unrelated to renal insufficiency. It is possible that other tissues express Pkhd1, leading to Cre-mediated expression of the BRAF transgene; however, we did not observe any obvious changes in the appearance of other organs. BRAFCD mice developed hydronephrosis and died at ~27 to weeks of age. There was no evidence of obstruction in serial sections of the ureters of BRafCD mice at 3 weeks of age, the earliest time in which tubule dilation and cysts were observed. Hydronephrosis is frequently associated with effacement of the renal medulla and dilation of the renal pelvis. We did not observe a dilated renal pelvis in the wildtype or PKD mice that expressed BRAFV600E at 10 weeks (Figures 1a, 3d, and 6a). As such, we speculate that proteinaceous cast material and/or masses of epithelial cells shed from the tubules during periods of rapid cell proliferation may have led to ureteral obstruction resulting in severe hydronephrosis and death in both Pkd1+/+ and Pkd1RC/RC mice. The persistent expression of activated BRAF may cause a phenotype similar to Erdheim-Chester disease, a rare systemic form of histiocytosis associated a high prevalence of BRAFV600E mutations (57–70% of cases), and ureteral obstruction and hydronephrosis caused by fibrosis and infiltrating histiocytes. 53, 54 Despite a more severe renal phenotype in Pkd1RC/RC mice by 10 weeks, the survival rates for BRAFCD and Pkd1RC/RC; BRafCD mice were similar, suggesting that the cause of death was due to hydronephrosis unrelated to the severity of the cystic disease.
An important question is if BRAF is a druggable target to inhibit cyst growth in PKD. Previously, we showed that Sorafenib (BAY 43–9006, Nexavar), a RAF inhibitor, blocked cAMP-dependent ERK activation, cell proliferation and in vitro cyst growth of human ADPKD cells.16 In a subsequent study, treatment with the RAF inhibitor PLX5568 caused a dose-dependent inhibition of cell proliferation and in vitro cyst growth, and attenuated PKD in the Han:SPRD Cy/+ rat; although, it also resulted in elevated renal and hepatic fibrosis.55 These studies suggest that BRAF may be a therapeutic target to slow PKD progression; however, side-effects and resistance to BRAF inhibitors remain significant clinical challenges.56 Evidence has shown that RAF inhibitors have a paradoxical effect on ERK activation due to drug-induced BRAF-RAF-1 dimerization, and this activation is not compromised if the kinase activity of one of the RAF partners is destroyed.57, 58 Another factor to consider in targeting BRAF as an anti-PKD therapy is the effect of epidermal growth factor receptor (EGFR) expression by cystic epithelial cells. EGFR is capable of undergoing feedback activation in response to down-stream inhibition of MAPK signaling.59, 60 This phenomenon renders cells with high levels of EGFR refractory to BRAF-specific inhibition, as subsequent activation of EGFR signaling leads to activation of RAF-1. As such, inhibition of MAPK signaling as an anti-PKD therapy may require dual-blockade of BRAF and RAF-1 or inhibition of BRAF and MEK. Alternatively, therapies designed to reduce renal cAMP production, such as tolvaptan, may be potentiated by combination therapy designed to inhibit BRAF kinase.
Supplementary Material
Supplemental Figure S1. Schematic for collecting duct (CD)-specific overexpression of active BRAFV600E and levels of BRAF expression in BRAFLSL-V600E; Pkhd1-Cre (BRAFCD) kidneys.
Supplemental Figure S2. Expression and localization of human BRAFV600E and mCherry in wildtype (WT) and BRAFCD kidneys.
Supplemental Figure S3. CD selective expression of active BRAF induced cyst formation in the collecting ducts of WT kidneys.
Supplemental Figure S4. CD expression of active BRAF increased renal PCNA levels in WT and RC/RC mice.
Supplemental Figure S5. CD expression of active BRAF increased P-ERK and proliferation of CD cells and interstitial cells in the kidneys.
Supplemental Figure S6. CD expression of active BRAF increased Ki-67 expression and induced hyperproliferation, tubule dilation, and cyst formation in WT kidneys.
Supplemental Figure S7. CD expression of active BRAF leads to pitting of the surface of BRafCD kidneys.
Supplemental Figure S8. CD expression of active BRAF increased the percentage of CD68 positive cells in WT and RC/RC kidneys.
Supplemental Figure S9. CD expression of BRAF increased the phosphorylation of LKB1 and decreased phosphorylation of ACC in 10-week-old WT and RC/RC kidneys.
Supplemental Figure S10. Effect of active BRAF on renal ERK, LKB1/AMPK, and mTOR signaling, and PCNA expression in 5-week-old RC/RC kidneys.
Supplemental Figure S11. CD expression of active BRAF increased cleaved caspase-3, a marker of apoptosis, in WT and RC/RC kidneys.
Supplemental Figure S12. CD expression of active BRAF increased p16INK4a, a marker of cellular senescence, in WT and RC/RC kidneys
Supplemental Figure S13. Hydronephrosis was observed in kidneys of mice expressing active BRAF at the time of death.
Supplemental Figure S14. Immunofluorescence images of PCNA, DBA, and LTA staining of RC/RC and RC/RC; BRafCD kidneys.
Supplemental Table S1. The effect of CD expression of BRAFV600E on kidney and body weight.
References
TRANSLATIONAL STATEMENT.
ADPKD is a genetic disorder characterized by relentless growth of numerous fluid-filled cysts in the kidneys and progressive decline in renal function. In ADPKD, cAMP agonists, including arginine vasopressin (AVP), stimulate BRAF, a kinase upstream of ERK, leading to the proliferation of cyst-lining cells. We show that expression of a constitutively active BRAF induced ERK-mediated cell proliferation and renal cyst formation in normal mice and accelerated disease in PKD mice. These results demonstrate that active BRAF is a sufficient stimulus to induce cystic disease. A better understanding of the mechanisms for BRAF regulation in ADPKD may lead to new therapies.
ACKNOWLEDGMENTS
We thank Dr. James Calvet for helpful suggestions during the preparation of the manuscript and Marsha Danley, Grant Johnson, and Corey White for technical assistance. This work was supported by a grant from the National Institutes of Diabetes and Digestive and Kidney Disease (R01DK081579; D.P. Wallace), a fellowship through the KUMC Biomedical Research Training Program (A. Raman) and grants from the PKD Foundation (D. Wallace, T. Fields, and S. Parnell). The BRAFLSL-V600E transgenic mouse strain was generated with the assistance of the KUMC Transgenic and Gene Targeting Institutional Facility (J. Vivian) supported by the KU Cancer Center, COBRE in Molecular Regulation of Cell Development and Differentiation, and the Kansas IDeA Network of Biomedical Research Excellence (P30 CA168524, P30 GM122731, P20 GM103418). Pkhd1-Cre mice were received from the UTSW O’Brien Center Core (NIH P30DK079328).
Footnotes
DISCLOSURE STATEMENT
The authors declare no competing interests.
Supplementary information is available on Kidney International’s website.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chebib FT, Torres VE. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am J Kidney Dis 2016; 67: 792–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. Journal of internal medicine 2007; 261: 17–31. [DOI] [PubMed] [Google Scholar]
- 3.Hanaoka K, Qian F, Boletta A, et al. Co-assembly of polycystin-1 and −2 produces unique cation-permeable currents. Nature 2000; 408: 990–994. [DOI] [PubMed] [Google Scholar]
- 4.Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–183. [DOI] [PubMed] [Google Scholar]
- 5.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res 1998; 74: 49–139. [DOI] [PubMed] [Google Scholar]
- 6.Dugan LL, Kim JS, Zhang Y, et al. Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J Biol Chem 1999; 274: 25842–25848. [DOI] [PubMed] [Google Scholar]
- 7.Vossler MR, Yao H, York RD, et al. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 1997; 89: 73–82. [DOI] [PubMed] [Google Scholar]
- 8.Erhardt P, Troppmair J, Rapp UR, et al. Differential regulation of Raf-1 and B-Raf and Ras-dependent activation of mitogen-activated protein kinase by cyclic AMP in PC12 cells. Mol Cell Biol 1995; 15: 5524–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacNicol MC, MacNicol AM. Nerve growth factor-stimulated B-Raf catalytic activity is refractory to inhibition by cAMP-dependent protein kinase. J Biol Chem 1999; 274: 13193–13197. [DOI] [PubMed] [Google Scholar]
- 10.Vuchak LA, Tsygankova OM, Prendergast GV, et al. Protein kinase A and B-Raf mediate extracellular signal-regulated kinase activation by thyrotropin. Mol Pharmacol 2009; 76: 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 2003; 63: 1983–1994. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi T, Pelling JC, Ramaswamy NT, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 2000; 57: 1460–1471. [DOI] [PubMed] [Google Scholar]
- 13.Nagao S, Yamaguchi T, Kusaka M, et al. Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63: 427–437. [DOI] [PubMed] [Google Scholar]
- 14.Mangoo-Karim R, Uchic M, Lechene C, et al. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86: 6007–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calvet JP. The Role of Calcium and Cyclic AMP in PKD. In: Li X (ed). Polycystic Kidney Disease: Brisbane (AU), 2015. [PubMed] [Google Scholar]
- 16.Yamaguchi T, Reif GA, Calvet JP, et al. Sorafenib inhibits cAMP-dependent ERK activation, cell proliferation, and in vitro cyst growth of human ADPKD cyst epithelial cells. Am J Physiol Renal Physiol 2010; 299: F944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace DP. Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta 2011; 1812: 1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Yamada S, LaRiviere WB, et al. Generation and phenotypic characterization of Pde1a mutant mice. PLoS One 2017; 12: e0181087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercer K, Giblett S, Green S, et al. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer research 2005; 65: 11493–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams SS, Cobo-Stark P, Hajarnis S, et al. Tissue-specific regulation of the mouse Pkhd1 (ARPKD) gene promoter. Am J Physiol Renal Physiol 2014; 307: F356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verani RR, Silva FG. Histogenesis of the renal cysts in adult (autosomal dominant) polycystic kidney disease: a histochemical study. Mod Pathol 1988; 1: 457–463. [PubMed] [Google Scholar]
- 22.Thomson RB, Mentone S, Kim R, et al. Histopathological analysis of renal cystic epithelia in the Pkd2WS25/- mouse model of ADPKD. Am J Physiol Renal Physiol 2003; 285: F870–880. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi M, Yamaji Y, Monkawa T, et al. Expression and localization of the water channels in human autosomal dominant polycystic kidney disease. Nephron 1997; 75: 321–326. [DOI] [PubMed] [Google Scholar]
- 24.Heggo O A microdissection study of cystic disease of the kidneys in adults. J Pathol Bacteriol 1966; 91: 311–315. [DOI] [PubMed] [Google Scholar]
- 25.Omran H, Haffner K, Burth S, et al. Evidence for further genetic heterogeneity in nephronophthisis. Nephrol Dial Transplant 2001; 16: 755–758. [DOI] [PubMed] [Google Scholar]
- 26.Gattone VH 2nd, Sinders RM, Hornberger TA, et al. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int 2009; 76: 178–182. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi H, Calvet JP, Dittemore-Hoover D, et al. A hereditary model of slowly progressive polycystic kidney disease in the mouse. Journal of the American Society of Nephrology : JASN 1991; 1: 980–989. [DOI] [PubMed] [Google Scholar]
- 28.Wallace DP, Hou YP, Huang ZL, et al. Tracking kidney volume in mice with polycystic kidney disease by magnetic resonance imaging. Kidney Int 2008; 73: 778–781. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi T, Nagao S, Kasahara M, et al. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis 1997; 30: 703–709. [DOI] [PubMed] [Google Scholar]
- 30.Lee M, Katerelos M, Gleich K, et al. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. Journal of the American Society of Nephrology : JASN 2018; 29: 2326–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nature reviews Molecular cell biology 2004; 5: 875–885. [DOI] [PubMed] [Google Scholar]
- 32.Niault TS, Baccarini M. Targets of Raf in tumorigenesis. Carcinogenesis 2010; 31: 1165–1174. [DOI] [PubMed] [Google Scholar]
- 33.Pritchard CA, Samuels ML, Bosch E, et al. Conditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells. Mol Cell Biol 1995; 15: 6430–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emuss V, Garnett M, Mason C, et al. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer research 2005; 65: 9719–9726. [DOI] [PubMed] [Google Scholar]
- 35.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954. [DOI] [PubMed] [Google Scholar]
- 36.Guan KL, Figueroa C, Brtva TR, et al. Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem 2000; 275: 27354–27359. [DOI] [PubMed] [Google Scholar]
- 37.Damsky WE, Bosenberg M. Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene 2017; 36: 5771–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Wu Y, Ward CJ, et al. Vasopressin directly regulates cyst growth in polycystic kidney disease. Journal of the American Society of Nephrology : JASN 2008; 19: 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto CS, Raman A, Reif GA, et al. Phosphodiesterase Isoform Regulation of Cell Proliferation and Fluid Secretion in Autosomal Dominant Polycystic Kidney Disease. Journal of the American Society of Nephrology : JASN 2016; 27: 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rees S, Kittikulsuth W, Roos K, et al. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. Journal of the American Society of Nephrology : JASN 2014; 25: 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamaguchi T, Wallace DP, Magenheimer BS, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279: 40419–40430. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi T, Hempson SJ, Reif GA, et al. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. Journal of the American Society of Nephrology : JASN 2006; 17: 178–187. [DOI] [PubMed] [Google Scholar]
- 43.Trudel M, D’Agati V, Costantini F. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int 1991; 39: 665–671. [DOI] [PubMed] [Google Scholar]
- 44.Ledford AW, Brantley JG, Kemeny G, et al. Deregulated expression of the homeobox gene Cux-1 in transgenic mice results in downregulation of p27(kip1) expression during nephrogenesis, glomerular abnormalities, and multiorgan hyperplasia. Developmental biology 2002; 245: 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma N, Malarkey EB, Berbari NF, et al. Proximal tubule proliferation is insufficient to induce rapid cyst formation after cilia disruption. Journal of the American Society of Nephrology : JASN 2013; 24: 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye H, Wang X, Constans MM, et al. The regulatory 1alpha subunit of protein kinase A modulates renal cystogenesis. Am J Physiol Renal Physiol 2017; 313: F677–F686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grantham JJ, Geiser JL, Evan AP. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int 1987; 31: 1145–1152. [DOI] [PubMed] [Google Scholar]
- 48.Wallace DP, Christensen M, Reif G, et al. Electrolyte and fluid secretion by cultured human inner medullary collecting duct cells. Am J Physiol Renal Physiol 2002; 283: F1337–1350. [DOI] [PubMed] [Google Scholar]
- 49.Wallace DP, Rome LA, Sullivan LP, et al. cAMP-dependent fluid secretion in rat inner medullary collecting ducts. Am J Physiol Renal Physiol 2001; 280: F1019–1029. [DOI] [PubMed] [Google Scholar]
- 50.Grantham JJ, Wallace DP. Return of the secretory kidney. Am J Physiol Renal Physiol 2002; 282: F1–9. [DOI] [PubMed] [Google Scholar]
- 51.Hallows KR, Mount PF, Pastor-Soler NM, et al. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol 2010; 298: F1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faraggiana T, Bernstein J, Strauss L, et al. Use of lectins in the study of histogenesis of renal cysts. Lab Invest 1985; 53: 575–579. [PubMed] [Google Scholar]
- 53.Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease. Blood 2020; 135: 1311–1318. [DOI] [PubMed] [Google Scholar]
- 54.Matzumura M, Arias-Stella J 3rd, Novak JE. Erdheim-Chester Disease: A Rare Presentation of a Rare Disease. J Investig Med High Impact Case Rep 2016; 4: 2324709616663233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchholz B, Klanke B, Schley G, et al. The Raf kinase inhibitor PLX5568 slows cyst proliferation in rat polycystic kidney disease but promotes renal and hepatic fibrosis. Nephrol Dial Transplant 2011; 26: 3458–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kakadia S, Yarlagadda N, Awad R, et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther 2018; 11: 7095–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rushworth LK, Hindley AD, O’Neill E, et al. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol 2006; 26: 2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matallanas D, Birtwistle M, Romano D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer 2011; 2: 232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012; 2: 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483: 100–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Schematic for collecting duct (CD)-specific overexpression of active BRAFV600E and levels of BRAF expression in BRAFLSL-V600E; Pkhd1-Cre (BRAFCD) kidneys.
Supplemental Figure S2. Expression and localization of human BRAFV600E and mCherry in wildtype (WT) and BRAFCD kidneys.
Supplemental Figure S3. CD selective expression of active BRAF induced cyst formation in the collecting ducts of WT kidneys.
Supplemental Figure S4. CD expression of active BRAF increased renal PCNA levels in WT and RC/RC mice.
Supplemental Figure S5. CD expression of active BRAF increased P-ERK and proliferation of CD cells and interstitial cells in the kidneys.
Supplemental Figure S6. CD expression of active BRAF increased Ki-67 expression and induced hyperproliferation, tubule dilation, and cyst formation in WT kidneys.
Supplemental Figure S7. CD expression of active BRAF leads to pitting of the surface of BRafCD kidneys.
Supplemental Figure S8. CD expression of active BRAF increased the percentage of CD68 positive cells in WT and RC/RC kidneys.
Supplemental Figure S9. CD expression of BRAF increased the phosphorylation of LKB1 and decreased phosphorylation of ACC in 10-week-old WT and RC/RC kidneys.
Supplemental Figure S10. Effect of active BRAF on renal ERK, LKB1/AMPK, and mTOR signaling, and PCNA expression in 5-week-old RC/RC kidneys.
Supplemental Figure S11. CD expression of active BRAF increased cleaved caspase-3, a marker of apoptosis, in WT and RC/RC kidneys.
Supplemental Figure S12. CD expression of active BRAF increased p16INK4a, a marker of cellular senescence, in WT and RC/RC kidneys
Supplemental Figure S13. Hydronephrosis was observed in kidneys of mice expressing active BRAF at the time of death.
Supplemental Figure S14. Immunofluorescence images of PCNA, DBA, and LTA staining of RC/RC and RC/RC; BRafCD kidneys.
Supplemental Table S1. The effect of CD expression of BRAFV600E on kidney and body weight.
References