

Abstract
Background—
Obstructive sleep apnea (OSA) and its features, such as chronic intermittent hypoxia (IH), may differentially affect specific molecular pathways and processes in the pathogenesis of coronary artery disease (CAD) and influence the subsequent risk and severity of CAD events. In particular, competing adverse (e.g. inflammatory) and protective (e.g. increased coronary collateral blood flow) mechanisms may operate, but remain poorly understood. We hypothesize that common genetic variation in selected molecular pathways influences the likelihood of CAD events differently in individuals with and without OSA, in a pathway-dependent manner.
Methods—
We selected a cross-sectional sample of 471,877 participants from the UK Biobank, with 4,974 ascertained to have OSA, 25,988 to have CAD, and 711 to have both. We calculated pathway-specific polygenic risk scores (PS-PRS) for CAD, based on 6.6 million common variants evaluated in the CARDIoGRAMplusC4D genome-wide association study (GWAS), annotated to specific genes and pathways using functional genomics databases. Based on prior evidence of involvement with IH and CAD, we tested PS-PRS for the HIF-1, VEGF, NFκB and TNF signaling pathways.
Results—
In a multivariable-adjusted logistic generalized additive model, elevated PS-PRSs for the KEGG VEGF pathway (39 genes) associated with protection for CAD in OSA (interaction odds ratio 0.86, p = 6E-04). By contrast, the genome-wide CAD PRS did not show evidence of statistical interaction with OSA.
Conclusions—
We find evidence that pathway-specific genetic risk of CAD differs between individuals with and without OSA in a qualitatively pathway-dependent manner. These results provide evidence that gene-by-environment interaction influences CAD risk in certain pathways among people with OSA, an effect that is not well-captured by the genome-wide PRS. This invites further study of how OSA interacts with genetic risk at the molecular level, and suggests eventual personalization of OSA treatment to reduce CAD risk according to individual pathway-specific genetic risk profiles.
Introduction
Obstructive sleep apnea (OSA) is a common disorder characterized by recurrent episodes of hypoxemia during sleep, sleep fragmentation, and activation of the sympathetic nervous system, potentially stimulating pro-inflammatory and pro-thrombotic pathways that drive atherosclerosis1. Epidemiological studies implicate OSA as a potentially modifiable risk factor for cardiovascular disease (CVD), stimulating research aimed at quantifying the impact of OSA and its treatment on both coronary artery disease (CAD) and cerebrovascular disease2. While OSA increases the incidence of cerebrovascular disease, weaker and less consistent associations support associations between OSA and incident CAD3. Moreover, large clinical trials (RCTs) of the effect of positive airway pressure (PAP) treatment of OSA on composite CVD outcomes have provided equivocal results, in some cases with point estimates in the direction of increased risk in the treatment group4–7.
Potential explanations for disappointing results of PAP RCTs include treatment non-adherence, non-optimal trial participant selection (e.g. advanced CVD at baseline) and limited statistical power8,9. Another possibility is that putative CVD benefits of OSA treatment may vary across individuals, influenced by factors related to characteristics of OSA-related stress not captured by the traditional metric of OSA severity (apnea hypopnea index, AHI), as well as by underlying host characteristics10,11, including genetic predisposition to CVD and genetic variation within known OSA pathways12,13. Moreover, some features of OSA might even prove protective for CVD in some individuals, and therefore treatment of OSA with PAP could increase CVD risk among those individuals14. This hypothesis derives support from human and animal studies showing that exposure to intermittent hypoxemia - a cardinal feature of OSA - can promote collateral blood flow, reflective of well-known effects of ischemic preconditioning15.
The need to develop a precision medicine-informed approach to CVD risk stratification in OSA agrees with data indicating that OSA-related CVD risk varies across subgroups defined by biomarkers16,17 or clusters of phenotypic traits18. A central question, and the one that we address here, is whether genetic variation in CAD risk interacts with OSA features to modulate CAD risk in a pathway-dependent manner19. We conceptualize repeated intermittent hypoxia (IH) and reoxygenation as a modifying environment for preexisting genetic risk for CAD, locating our analysis within a gene-by-environment (GxE) interaction paradigm. Our pathway-level GxE approach provides a new scale on which to study the genetic architecture of CAD susceptibility and its modification by OSA.
We build on prior work that (1) identified genetic markers for CAD risk involved in atherosclerosis, small vessel disease, inflammatory pathways, and angiogenesis, with evidence from genome-wide association studies (GWAS), expression studies, animal experiments and clinical trials20,21; and (2) showed the influence of IH on a variety of pathways (in positive and negative directions) relevant to CAD, including redox, inflammatory and angiogenic pathways22.
Our approach considers effect modification of genetic risk, aggregated to the pathway level, and investigates pathway-specific polygenic risk scores (PS-PRS) for CAD. PRSs have demonstrated success in identifying high risk populations, but may perform poorly due to overly simplistic dimension reduction when compared with non-linear methods to summarize risk across many alleles23,24. Here we consider a novel PRS approach, where we group markers by pathways to create multiple PS-PRSs aligned logically with genetic architecture, to facilitate investigation of GxE interaction at the pathway level.
We hypothesized that common genetic variation, in specific molecular pathways for CAD that are linked to the pathophysiology of OSA, influences the likelihood of CAD events differently in individuals with and without OSA, in a pathway-dependent manner. Our work was motivated by the potential for differential effects of OSA on multiple processes that impact CAD, such as inflammation and atherosclerosis, thrombosis, collateral vessel formation (via angiogenesis), control of coronary artery blood flow, and plaque disruption. Of central interest is the idea that OSA could be a “double-edged sword” with both positive or negative effects on CAD. In particular, intermittent hypoxia – a physiological stress characteristic of OSA – may exert either positive effects through ischemic preconditioning or negative effects by boosting atherosclerotic processes including inflammation14,15. We pre-selected four regulatory pathways of interest based on our hypotheses of the impacts of chronic IH on angiogenic and inflammatory processes in CAD, the HIF-1, VEGF, NFκB and TNF signaling pathways. We focused on these four representative pathways for their recognized roles in these processes, which have been previously shown to involve differential expression of constituent genes in OSA25–32.
By investigating whether OSA enhances, does not change, or ameliorates genetic risk of developing CAD among individuals with specific risk allele profiles within these pathways, we aim to provide new mechanistic insight into the biological effects of OSA on CAD. In addition, we explore the ability to identify patients with OSA who may have either protection against or increased risk of CAD events from their OSA status according to their specific genetic profile. Ultimately, we propose that this approach provides a novel step on the path toward understanding the complex role of OSA in CAD and formulating a personalized approach to treatment.
Methods
A full description of data and methods, including version and location of software and databases used to conduct the analyses, is available in the supplementary material. Research was conducted using de-identified data made available through collaboration with UK Biobank. Requests for data access can be made via UK Biobank with application materials available at www.ukbiobank.ac.uk. The National Health Service National Research Ethics Service (ref. 11/NW/0382) gave approval for the UK Biobank study, with each participant providing written informed consent. This project has been reviewed and approved by the Mass General Brigham IRB.
Results
Clinical characteristics
Of the 476,851 UK Biobank33 (UKBB) participants in our analytic sample, 4,974 (1.1%) were classified to have OSA, 26,699 (5.6%) to have CAD, and 711 both. Phenotype and covariate definitions are available in Supplemental Table I. The median (IQR) age was 58 (50, 63) years with a similar age distribution by OSA status (Table 1). As expected, OSA cases tended to have a higher prevalence of several CAD risk factors and comorbidities. Additional details are provided in the Supplement.
Table 1.
Sample characteristics
| OSA control | OSA case | |||
|---|---|---|---|---|
| N | 471,877 | 4,974 | ||
| Outcome: | ||||
| CAD, N (%) | 25,988 | (5.50) | 711 | (14.30) |
| Covariates: | ||||
| BMI (median [IQR]) | 26.67 | (24.09, 29.79) | 31.61 | (28.11, 36.39) |
| Age (median [IQR]) | 58 | (50.00, 63.00) | 58 | (51.00, 63.00) |
| Male (%) | 213,700 | (45.30) | 3,622 | (72.80) |
| Smoking (%) | ||||
| current | 36,568 | (7.70) | 519 | (10.40) |
| never | 259,572 | (55.00) | 2,162 | (43.50) |
| occasionally | 12,484 | (2.60) | 183 | (3.70) |
| previous | 163,253 | (34.60) | 2,110 | (42.40) |
| Self-reported white (%) | 397,269 | (84.20) | 4,054 | (81.50) |
| Genetic PC1 (mean (SD)) | −1.53 | (53.33) | 1.95 | (60.96) |
| Genetic PC2 (mean (SD)) | 0.5 | (27.38) | 0.21 | (27.29) |
| Genetic PC3 (mean (SD)) | −0.17 | (14.61) | 0.82 | (15.30) |
| Genetic PC4 (mean (SD)) | 0.13 | (10.32) | −0.2 | (12.07) |
| Genetic PC5 (mean (SD)) | 0.05 | (7.57) | 0.28 | (7.42) |
| BiLEVE genotype platform (%) | 48,528 | (10.30) | 619 | (12.40) |
| COPD (%) | 18,888 | (4.00) | 608 | (12.20) |
| Asthma (%) | 62,630 | (13.30) | 1,126 | (22.60) |
| Standardized PRS and PS-PRS Exposures: | ||||
| KEGG hsa04066 HIF1 (mean (SD)) | −0.0002 | 1.000 | −0.011 | 0.985 |
| KEGG hsa04370 VEGF (mean (SD)) | −0.0014 | 1.000 | 0.029 | 0.982 |
| KEGG hsa04064 NFκB (mean (SD)) | 0.0002 | 1.000 | 0.003 | 1.005 |
| KEGG hsa04668 TNF (mean (SD)) | 0.0002 | 1.000 | 0.015 | 1.017 |
| HIF1 core genes (mean (SD)) | 0.0018 | 1.000 | −0.016 | 0.999 |
| VEGF core genes (mean (SD)) | 0.0003 | 1.000 | −0.010 | 0.991 |
| NFκB core genes (mean (SD)) | 0.0011 | 1.000 | −0.015 | 1.006 |
| TNF core genes (mean (SD)) | −0.0009 | 1.000 | 0.001 | 0.992 |
| CAD PRS (mean (SD)) | 0.0000 | 0.998 | 0.034 | 1.015 |
| OSA genes (mean (SD)) | −0.0016 | 0.999 | −0.005 | 0.997 |
Outcome, covariate and polygenic risk score distributions by OSA status. OSA: Obstructive sleep apnea, CAD: coronary artery disease, BMI: body mass index, PC: principal component, COPD: chronic obstructive pulmonary disease, KEGG: Kyoto encyclopedia of genes and genomes, hsa: Homo sapiens pathway reference number, HIF1: hypoxia inducible factor 1, VEGF: vascular endothelial growth factor, NFκB: nuclear factor kappa- beta, TNF: tumor necrosis factor, PRS: polygenic risk score, PS-PRS: pathway-specific polygenic risk score.
Characteristics of the PS-PRSs
Our gene-annotation procedure (Figure 1) for the 6,630,150 single nucleotide variants in the previously published CAD PRS described by Khera et al34, based on the LDPred algorithm35 and the CARDIoGRAMplusC4D Consortium CAD GWAS36, resulted in the assignment of 70.3% of the 6.6M markers to one or more genes (average: 1.1 genes assigned per annotated variant). Pathway specific polygenic risk scores (PS-PRSs) were calculated for each individual as the effect-weighted sum of the count of risk alleles at each locus across all pathway SNVs. Pathway genes, defined with reference to the Kyoto Encyclopedia of Genes and Genomes37 (KEGG) are shown in Supplemental Table II. After standardization on the cohort as a whole, participants with or without OSA had similar PS-PRS (Table 1).
Figure 1:
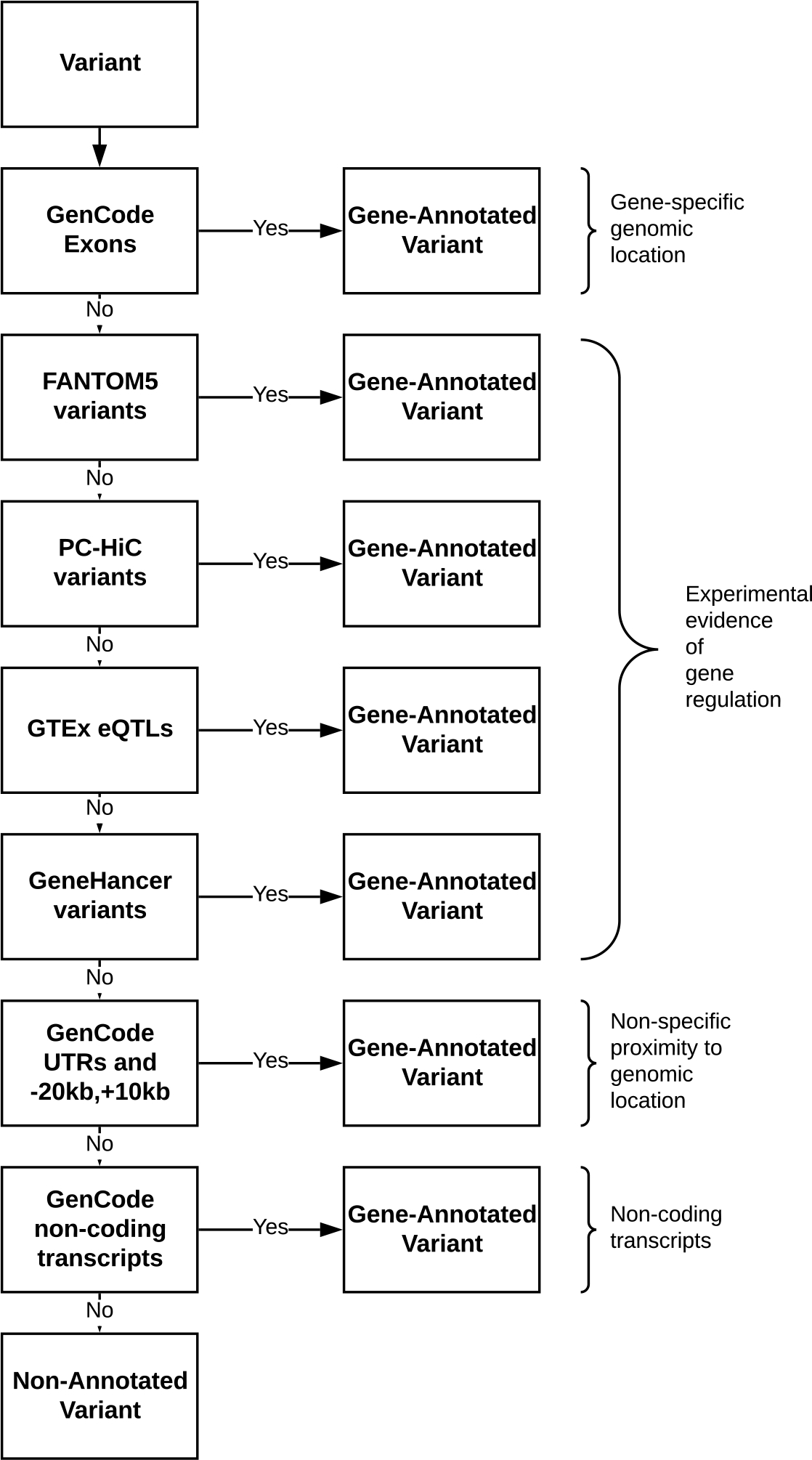
Flow diagram showing the gene annotation process. Gene annotation process, showing data sources and sequential priority. Single nucleotide variants (SNVs) available in a given data set (“yes”) were annotated to one or more genes and set aside without further annotation. Remaining un-annotated SNVs (“no”) advanced to be considered for annotation using potentially less-definitive methods.
Correlations between the PS-PRSs
Due to shared genes between pathway definitions, there was some moderate correlation between the genetic risk scores. The NFκB signaling pathway (KEGG hsa04064: 100 genes) and TNF signaling pathway (KEGG hsa04668: 110 genes) share 29 genes, which induced a correlation of 0.39 in the pathway-specific risk scores (Figure 2). The curated 225-gene OSA/CAD pathway shares 22 genes with the KEGG HIF1 pathway, driving the observed correlation of 0.54. Similarly, because VEGF core-genes VEGFA and FLT1 and HIF1 core-genes HIF1A and ARNT reside within the HIF1 KEGG pathway, they induce correlations of 0.44 and 0.32 respectively. Additional pairwise correlations for pathways, genes, and modules is shown in Figure 2 and Supplemental Figure I, and further discussed in the Supplement.
Figure 2:
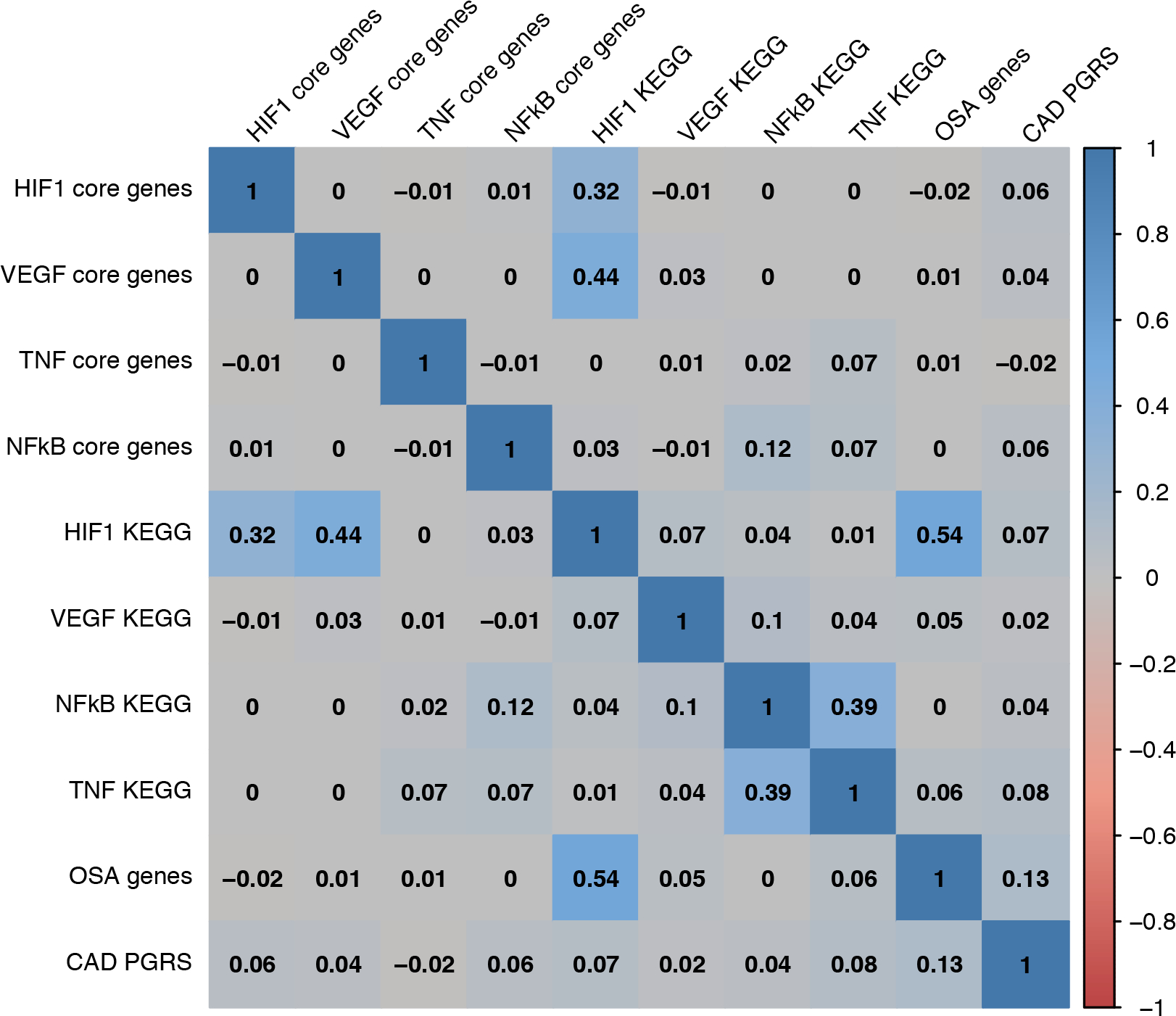
Pairwise correlations among selected pathway-specific polygenic risk scores. Pearson correlations between participants’ gene- and pathway-specific genetic risk scores (GSRS and PS-PRS) in the primary analysis.
Estimated main effects of OSA, PRS and PS-PRSs
As incidental findings, we report estimated OSA and PS-PRS main effects, which were ancillary to our primary GxE analysis. We find that OSA associates with higher CAD risk, with an approximate odds ratio (OR) of 1.5, and 95% confidence interval (CI) of (1.4, 1.6) across all PS-PRS GxE models shown in Table 2, as well as in marginal models fit without PS-PRS or GxE terms (Supplemental Table III). Also, as expected, the full genome-wide CAD PRS associated strongly with increased CAD risk in both a marginal PRS-only and a GxE model. For each standard deviation increase in the CAD PRS, the odds of CAD (in OSA and separately in OSA controls) increased by a factor of approximately 1.70 (Table 2 and Supplemental Table III). Additionally, the PS-PRSs for each KEGG signaling pathway associated strongly with CAD both marginally in the combined sample and as a genetic main effect, estimated in OSA controls, but had notably smaller estimated odds ratios (range 1.040 to 1.062). Core gene modules were also associated both marginally and in OSA controls, but had still smaller odds ratios (range 1.019 to 1.032) (Table 2). Estimated PS-PRS main effects in OSA controls were not appreciably different from the marginal PS-PRS effects from models fit on the combined data without GxE interaction terms (Supplemental Table III).
Table 2:
Primary analysis of selected OSA pathways: Covariate-adjusted odds of CAD per s.d. of standardized PS-PRSs with effect-measure modification by OSA status
| PS-PRS | OSA main effect | PS-PRS effect in OSA Controls | PS-PRS effect in OSA Cases | OSA × PS-PRS interaction effect | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | OR | 95% CI | p-value | OR | 95% CI | p-value | OR | p-value | |
| A) Selected pathways: core-gene modules | ||||||||||
| HIF1 core genes | 1.50 | (1.38, 1.64) | 1.022 | (1.009, 1.035) | 1.06E-03 | 1.114 | (1.025, 1.212) | 1.10E-02 | 1.090 | 4.46E-02* |
| VEGF core genes | 1.51 | (1.38, 1.64) | 1.032 | (1.019, 1.045) | 1.66E-06 | 0.952 | (0.874, 1.037) | 2.58E-01 | 0.922 | 6.70E-02 |
| NFκB core genes | 1.51 | (1.38, 1.64) | 1.031 | (1.018, 1.045) | 4.45E-06 | 0.981 | (0.903, 1.066) | 6.54E-01 | 0.952 | 2.45E-01 |
| TNF core genes | 1.51 | (1.38, 1.64) | 1.019 | (1.006, 1.033) | 4.44E-03 | 1.013 | (0.931, 1.102) | 7.59E-01 | 0.994 | 8.94E-01 |
| B) Selected pathways: KEGG signaling pathways | ||||||||||
| HIF1 KEGG | 1.51 | (1.38, 1.64) | 1.062 | (1.048, 1.076) | 6.81E-20 | 1.089 | (1.001, 1.185) | 4.84E-02 | 1.025 | 5.66E-01 |
| VEGF KEGG | 1.51 | (1.38, 1.64) | 1.040 | (1.027, 1.054) | 4.30E-09 | 0.894 | (0.821, 0.974) | 1.02E-02 | 0.860 | 5.98E-04† |
| NFκB KEGG | 1.51 | (1.38, 1.64) | 1.058 | (1.044, 1.072) | 3.26E-17 | 0.998 | (0.919, 1.085) | 9.71E-01 | 0.944 | 1.74E-01 |
| TNF KEGG | 1.51 | (1.38, 1.64) | 1.052 | (1.038, 1.065) | 1.65E-14 | 0.994 | (0.916, 1.079) | 8.86E-01 | 0.945 | 1.85E-01 |
| C) Selected aggregated pathway comparators | ||||||||||
| CAD PRS | 1.52 | (1.39, 1.67) | 1.695 | (1.672, 1.719) | <1.0E-99 | 1.664 | (1.525, 1.816) | 3.30E-30 | 0.982 | 6.80E-01 |
| OSA genes | 1.50 | (1.37, 1.63) | 1.102 | (1.087, 1.116) | 1.17E-46 | 1.222 | (1.122, 1.330) | 3.81E-06 | 1.109 | 1.83E-02* |
Each row depicts a separate PS-PRS model showing the odds ratio-scale OSA and PS-PRS main effects and the PS-PRS × OSA interaction effect from a model of the form logit(CAD) = OSA β1 + PS-PRS β2 + PS-PRSxOSA β3 + covariates. The OSA main effect (the OSA effect in subjects with a 0 value of the centered PS-PRS) is β1, the PS-PRS main effect (the PS-PRS effect in OSA controls) is β2, and the OSA × PS-PRS interaction effect is β3. The PS-PRS effect in OSA Cases, β4, is therefore defined as β4 = β2 + β3. Odds ratios are obtained by exponentiating the corresponding β’s. The primary analysis tests for GxE, i.e. effect-measure modification of the genetic PS-PRS effect by OSA, in the ten pathways shown, as quantified by the p-value for the OSA × PS-PRS interaction effect. OSA main effects and PS-PRS effects in OSA cases and controls are shown for context. Nominally significant (*) GxE results do not pass Bonferroni-corrected significance (†) level 5E-3. Three groupings representing different types of pathways are included: A) core-gene modules B) KEGG-defined signaling pathways, and C) aggregate pathways. Covariates adjusted in a logistic generalized additive model: mutual interaction of age and BMI by sex (via tensor-product thin plate cubic penalty regression splines) as well as smoking and its interaction with sex, self-reported white race, the first 5 genetic PCs, genotype platform (BiLEVE), asthma and chronic obstructive pulmonary disease. OR: odds ratio; CI: confidence interval; OSA: Obstructive sleep apnea, CAD: coronary artery disease, KEGG: Kyoto encyclopedia of genes and genomes, HIF1: hypoxia inducible factor 1, VEGF: vascular endothelial growth factor, NFκB: nuclear factor kappa- beta, TNF: tumor necrosis factor, PRS: polygenic risk score, PS-PRS: pathway-specific polygenic risk score.
Estimated interaction effects between OSA, and PS-PRSs
In our primary analysis, we find evidence of effect-measure modification of genetic risk by OSA status. Specifically, at Bonferroni-corrected significance-level 5E-3, we see significant qualitative effect modification in the VEGF KEGG pathway (interaction odds ratio 0.86, p = 6E-04), leading to differential genetic effects among participants with and without OSA (Table 2). Concretely, among OSA cases, we estimated the odds of CAD decreased, by a factor of 0.89 per standard deviation increase in the VEGF KEGG PS-PRS, contrasted with an increase of 1.04 among OSA controls. The point estimate for GxE interaction in the VEGF core-genes PS-PRS is qualitatively similar but not statistically significant. We do not find significant evidence of effect-measure modification between OSA and the KEGG HIF1 PS-PRS, TNF PS-PRSs, the NFκB PS-PRSs, nor the full CAD PRS. However, we find a nominally significant trend for risk-amplifying effect-measure modification for both the HIF1 core-genes PS-PRS (interaction OR 1.1, p = 4.46E-02) and the curated 225 gene PS-PRS based on a literature-derived CAD- and OSA-involved genes (interaction OR 1.1, p = 1.8E-02).
In secondary analysis of module- and gene-specific interactions, adjusting for multiple testing, we find statistically significant effect-measure modification in one of the 30 pathway submodules (VEGF endothelial migration: CDC42, MAPK11, MAPK12, MAPK13, MAPK14, MAPKAPK2, MAPKAPK3, PTK2, PXN) and none of the 268 genes tested from the four KEGG pathways of interest (Supplemental Tables IV and V). We also see nominally significant effect-measure modification in several pathway modules and genes, with estimated effects generally consistent with pathways and modules that contain them. In Figure 3, we provide a pathway diagram representing established biological relationships37 of selected genes, modules, and pathways involved in the analysis.
Figure 3:
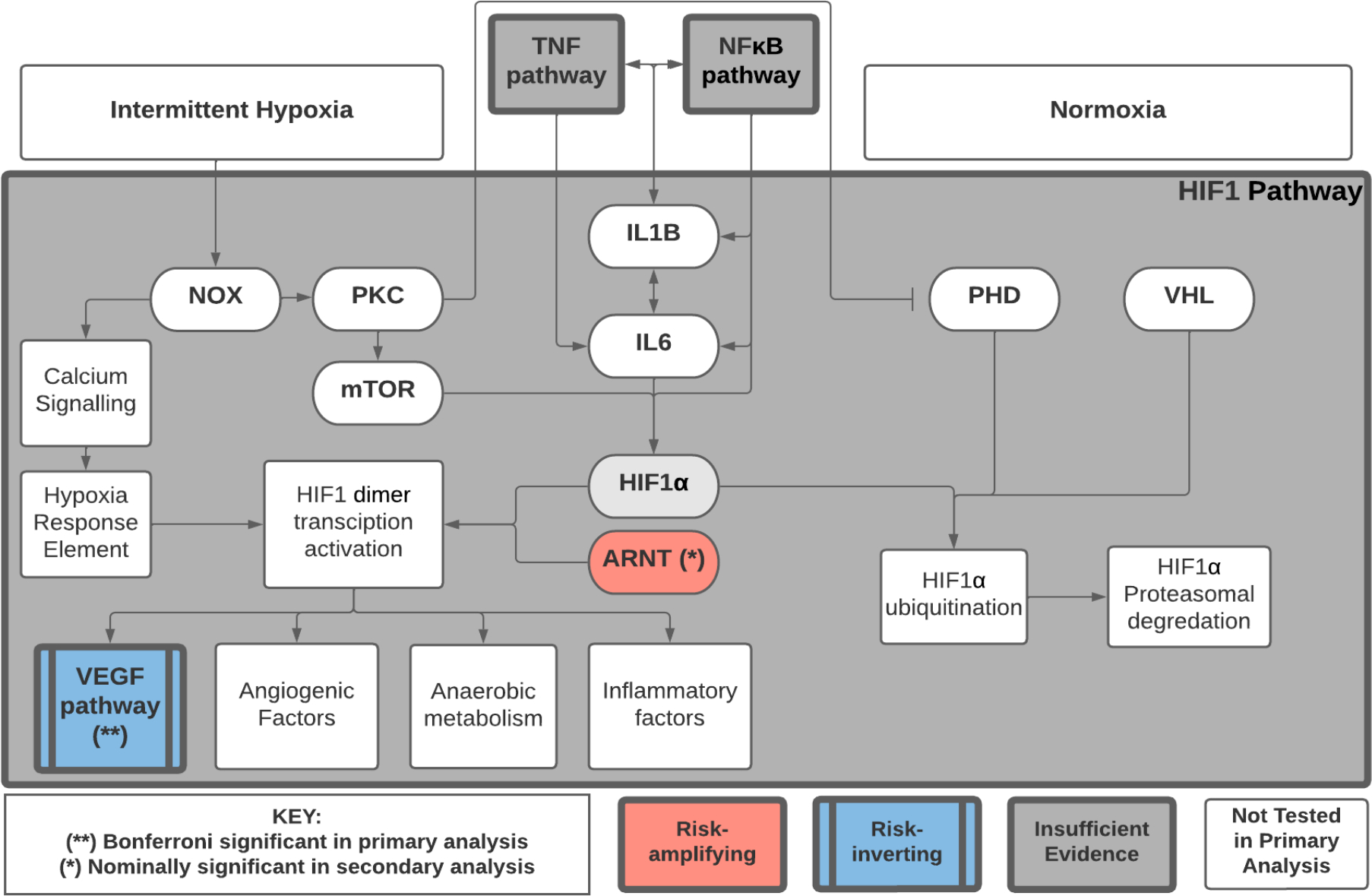
Pathway diagram of selected genes and pathways. Pathway diagram showing the relationship of selected genes and pathways in the analysis of effect-measure modification by OSA on genetic risk of CAD, using pathway-specific polygenic risk scores (PS-PRS) and gene-specific risk scores (GSRS). Risk inverting effect-measure modification by OSA on the VEGF KEGG pathway PS-PRS attained Bonferroni-corrected statistical significance (p<0.005) based on 10 pre-specified tests in the primary analysis. The GSRS for ARNT had nominally significant (p<0.05) risk-amplifying effect-measure modification by OSA. The HIF1 KEGG pathway contains additional genes and modules not shown, and partially overlaps the NFκB and TNF pathways. Estimated PS-PGRS effect modification for HIF-1, NFκB and TNF pathways did not meet nominal significance in primary analysis.
Sensitivity Analyses
We obtained similar results in sensitivity analyses (Supplemental Table VI). For the analyses of males-only, self-reported whites-only, and when adjusting for potentially mediating comorbidities, the KEGG VEGF PS-PGRS had consistently Bonferroni-significant risk-inverting effect-measure modification, while the curated 225 gene CAD- and OSA-related pathway had consistently nominally significant risk-amplifying effect modification; the females-only analysis was underpowered with only 71 joint OSA/CAD cases, nonetheless KEGG VEGF was nominally significant with similar risk-inverting estimated effect.
PS-PRS associations with C-reactive protein
In a supplemental analysis (Supplemental Table VII) we find that CAD risk as quantified by PS-PRSs for the KEGG HIF1 and NFκB pathways is associated with increased CRP, respectively 4.5% (95% CI: 4.3%, 4.8%) and 0.5% (CI: 0.2%, 0.8%) per standard deviation PS-PRS, whereas the VEGF pathway PS-PRS CAD risk is associated with decreased CRP, at −0.8% per standard deviation (CI: −1.0%, −0.5%). Associations between log CRP and core-genes pathway PS-PRSs are weak or negligible. The genome-wide CAD PRS is not associated with CRP, but the PS-PRS for the curated list of 225 OSA-CAD genes is positively associated with CRP levels at 1.1% increase per standard deviation (CI: 0.8%, 1.4%).
Discussion
Overview and context
Using biobank-scale data we tested the hypothesis that genetic variation in specific molecular pathways that modulate risk of CAD may influence the propensity for CAD differently in individuals with and without OSA—an exposure postulated to have complex effects on atherosclerosis, inflammation, and angiogenesis. We demonstrate novel interaction effects between OSA and PS-PRS within one pathway (VEGF) postulated a priori from current biological understanding to play a role in OSA-related hypoxia in CAD. The effect-measure modification of PS-PRS effects suggests that OSA can increase or attenuate genetic risk for CAD in a pathway-dependent manner, and, reciprocally, that subgroups with specific genetic profiles within certain CAD pathways may experience the influence of OSA differently. Our results also suggest heterogeneity of this effect modification across pathways, modules and genes.
Primary and secondary analyses
In our primary analysis, we see Bonferroni-significant evidence of risk-inverting effect-measure modification in the KEGG VEGF pathway. In secondary analysis we also see suggestive evidence of risk-inverting effect-measure modification across multiple independent signals within this pathway. This qualitative effect-measure modification in the VEGF KEGG pathway suggests that among OSA cases, the direction of effect of the VEGF PS-PRS risk alleles, averaged across markers in the pathway, is reversed compared with OSA controls. For example, among OSA cases whose VEGF risk score is one standard deviation above the mean, we estimate that odds of CAD is decreased by approximately 14% compared to those with the mean VEGF risk score.
One explanation consistent with these data would be that OSA perturbs the tissue environment such that SNVs that increase CAD risk in normoxia on average now function to reduce risk under hypoxia. For example, downregulation of a certain gene under normoxia may have been deleterious, but becomes beneficial under hypoxia (or vice versa). The 39 genes in the KEGG VEGF pathway mediate inflammation and angiogenesis, with genes regulating endothelial cell survival, proliferation, and migration as well as vascular permeability. Under intermittent hypoxia, angiogenesis may predominate, potentially conferring protection from clinically observed or fatal CAD events (ascertained in our CAD outcome) due to heart muscle collateralization.
By contrast we see nominally-significant suggestive evidence of risk-amplifying effect-modification in the HIF1 core genes module, apparently driven by the ARNT gene, as well as in the PS-PRS consisting of 225 curated OSA-CAD genes, suggesting a risk-amplifying GxE signal may be more widely distributed. If confirmed these effects are consistent with the idea that OSA-related physiological stressors may amplify the deleterious effects of certain genetic variants on CAD. For example, ARNT, whose gene-product HIF1-β creates a heterodimer with HIF-1α enabling nuclear activation of the HIF1 transcription factor, is a plausible key player in the regulatory network immediately upstream of a host of cellular responses to hypoxia. Although ARNT is not under direct hypoxic regulation, concentration of HIF1-β may serve as a rate-limiter on HIF1’s transcription factor activity specifically under hypoxic conditions when HIF1-α concentration is maximized38. Hence, genetic effects on ARNT plausibly interact mechanistically with IH-induced effects on HIF1-α. Further investigation of OSA’s and IH’s potential for risk-amplifying effect modification appears warranted.
If confirmed, differential positive and negative GxE interaction effects, increasing or decreasing individuals’ prior genetic risk in separate genes and pathways in OSA, would be consistent with the idea of a “double-edged” role of OSA in CAD. In particular, though increased concentrations of HIF1 in OSA is expected to augment VEGF, a number of considerations could explain why the CAD effects of e.g. of ARNT risk alleles may (on average) be either unaffected or amplified by OSA but the effects of VEGF pathway risk alleles may (on average) be inverted, including differential downstream effects. This is supported by the association analysis of log CRP levels, where the qualitatively different effect of the KEGG VEGF PS-PRS CAD risk variants (CRP reducing), as compared to the HIF1, NFκB, and TNFA KEGG pathways of interest (CRP increasing), suggests pathway-specific mediation of genetic risk by biomarkers such as CRP.
These findings support the potential for CAD susceptibility to depend both on physiological stressors, such as those associated with OSA, and underlying genetics, via complex interactions between these two sources of risk. Confirmation of the present findings could enable clinicians to prioritize patients with genetic risk factors that are amplified under response to hypoxia for more aggressive OSA treatment, but manage more conservatively those whose genetic risk profile indicates protection under IH and who have few symptoms.
These results open the way to varied experimental studies to probe the potentially novel molecular mechanisms that may lead to the observed qualitative effect-measure modification and underly the findings. Further study of genes and variants in this pathway and their effect on measured biomarkers and gene expression may provide clarification of the genetic mediators of CAD risk in OSA, and potentially point to molecular targets for ameliorating OSA-related CAD. Additional detail is provided in the Discussion Supplement.
Additional Findings
Testing our primary hypothesis yielded additional interesting findings. We demonstrated that a genome-wide CAD PRS associated with CAD in both the OSA cases and controls was distributed similarly (in mean and standard deviation) and did not reveal significant effect-measure modification by OSA. We also confirmed that the chosen PS-PRSs for CAD associate positively with CAD in OSA controls, and that we see similar distribution of PRS and PS-PRSs when stratifying by OSA status. Additionally, given that nominally significant point estimates of interaction between genetic risk for CAD and OSA differed qualitatively across pathways, this suggests analyses ignoring heterogeneity (such as only assessing the full PRS) may incorrectly lead to the conclusion that there is no evidence of GxE interaction. Lastly, we demonstrated a consistent positive association between OSA and CAD, marginally after adjusting for covariates only, as well as in all PS-PRS models adjusting for covariates and GxE interaction effects, with a consistent effect size (OR 1.5) similar to that previously reported in observational cohort studies39.
Strengths and weaknesses
This study has several notable strengths. Our methodology offers the opportunity to detect distinct positive and negative interaction effects in separate pathways, which might otherwise interfere with each other and prevent detection of a GxE interaction at the genome-wide PRS level. Furthermore, if our hypothesis holds, environments such as OSA-related hypoxemia will perturb gene activity across regulatory pathways or submodules containing thousands of SNVs. So, we expect aggregating SNV risk to genes, modules, or pathways acting together to produce similar downstream biological effects may enhance power and reduce the multiple testing burden, as compared with SNV-level interaction testing. So, PS-PRS may provide useful intermediate levels between individual genetic markers and genome-wide risk scores to study effect-measure modification. And unlike the challenges to studying these regulatory pathways with biological assays in relevant tissues and with appropriate timing, our approach extracts additional insights from existing GWAS and biobank data.
The study also has several limitations. First, we perform a cross-sectional analysis, due to the limited follow-up for incident CAD, as well as the difficulty determining the date of onset of sleep apnea. Cross-sectional analysis is commonly used in genetics, including the GWAS and PRS analyses on which our PS-PRS were based. While in the GxE context ideally the OSA exposure should be established as preceding the CAD outcome, the presence of OSA in the medical record generally is believed to reflect disease that occurred years earlier40. Second, UK Biobank has several sources of selection bias, including healthy volunteer bias, which may limit generalizability. Also, to overcome a limitation of the UK Biobank dataset in which sleep apnea is not subclassified as central or obstructive either in self-reported or medical records data, we attempted to reduce potential CSA cases, which are likely causally downstream of atherosclerosis and CAD, by requiring the presence of self-reported snoring, and we further enhanced specificity in the controls by eliminating subjects with self-reported snoring and daytime sleepiness. The resulting contrast between cases and controls should be interpreted in this context, which we view as indicative of clinically diagnosed OSA vs healthy controls. Additional discussion of ascertainment is provided in the Discussion Supplement. Further, as discussed above, we selected pathways based on literature-supported prior hypotheses, however, we were limited by power and for this reason and to minimize multiple comparisons, we did not comprehensively investigate sources of pathway-level genetic risk for CAD in OSA. We were also limited by power in our ability to look for sex-differences in GxE effects, as further discussed in the Discussion Supplement.
In conclusion, this study provides insight into the influence of OSA on CAD and identifies a novel mechanism that may explain the evident heterogeneity of CAD risk among individuals with OSA. Specifically, we find that OSA status can modify pre-existing genetic risk of CAD within the VEGF pathway, and more generally we provide a plausible approach to assess stratified genetic risk for CAD in OSA by illustrating a novel method for understanding pathway-specific genetic risk and its role in GxE interaction. Future larger PS-PGRS GxE studies may allow probing of dose-dependent effects of OSA-related intermittent hypoxia on a comprehensive set of CAD pathways, while further study of these pathways and genes with additional –omics data or animal studies may further clarify our current findings. Having identified genetic pathways whose effect is modified by OSA status, we may also identify individuals with OSA who are most susceptible (or resilient) to CAD, disentangling their genetic risk, their OSA exposure risk, and the interaction between the two. Together these investigations may lead to personalized management of OSA and the ability to target treatment toward specific impacted pathways in those at heightened risk.
Supplementary Material
Acknowledgments:
This research has been conducted using the UK Biobank Resource (application 6818). We would like to thank the participants and researchers from the UK Biobank who contributed or collected data. Research reported in this publication was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Numbers F32HL152555, R35HL135818 and R21HL145425. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Dr. Rutter would like to acknowledge The University of Manchester (Research Infrastructure Fund).
Disclosures: Dr. Goodman reports grants from National Heart, Lung, and Blood Institute, NIH, during the conduct of the study; Dr. Cade has nothing to disclose; Dr. Shah has nothing to disclose; Dr. Huang has nothing to disclose; Dr. Dashti has nothing to disclose; Dr. Saxena has nothing to disclose; Dr. Rutter reports consulting fees and non-promotional lecture fees from Novo Nordis, outside the submitted work; Dr. Libby reports grants from National Heart, Lung, and Blood Institute, American Heart, RRM Charitable Fund, and Simard Fund; Dr. Libby is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron; Dr. Libby is a member of the scientific advisory board for Amgen, Caristo, Cartesian, CSL Behring, DalCor Pharmaceuticals, Dewpoint, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec, and XBiotech, Inc.; Dr. Libby’s laboratory has received research funding in the last two years from Novartis; Dr. Libby is on the Board of Directors of XBiotech, Inc. Dr. Libby has a financial interest in Xbiotech, a company developing therapeutic human antibodies; Dr. Libby’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies; Dr. Sofer has nothing to disclose. Dr. Redline reports grants from NIH during the conduct of the study; grants and personal fees from Jazz Pharma, personal fees from Eisai Inc, personal fees from Apnimed Inc, outside the submitted work; and Dr. Redline is the first incumbent of an endowed professorship donated to the Harvard Medical School by Dr. Peter Farrell, the founder and Board Chairman of ResMed, through a charitable remainder trust instrument, with annual support equivalent to the endowment payout provided to the Harvard Medical School during Dr. Farrell’s lifetime by the ResMed Company through an irrevocable gift agreement.
Sources of Funding:
National Heart, Lung, and Blood Institute, NIH
Nonstandard Abbreviations and Acronyms
- BMI
body mass index
- CAD
coronary artery disease
- COPD
chronic obstructive pulmonary disease
- CRP
C-reactive protein
- CVD
cardiovascular disease
- GAM
generalized additive model
- GSRS
gene-specific risk score
- GWAS
genome-wide association study
- GxE
gene-by-environment interaction
- HIF-1
Hypoxia-inducible factor 1
- ICD10
International Classification of Diseases, version 10
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- NFκB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- OSA
obstructive sleep apnea
- PAP
positive airway pressure
- PRS
polygenic risk scores
- PS-PRS
pathway-specific polygenic risk scores
- RCT
randomized controlled trial
- SNV
single nucleotide variant
- TNF
Tumor necrosis factor
- UKBB
UK Biobank
- VEGF
Vascular endothelial growth factor
Footnotes
Supplemental Materials:
References:
- 1.Javaheri S, Barbe F, Campos-Rodriguez F, Dempsey JA, Khayat R, Javaheri S, Malhotra A, Martinez-Garcia MA, Mehra R, Pack AI. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. Journal of the American College of Cardiology. 2017;69:841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauters F, Rietzschel ER, Hertegonne KB, Chirinos JA. The link between obstructive sleep apnea and cardiovascular disease. Current atherosclerosis reports. 2016;18:1. [DOI] [PubMed] [Google Scholar]
- 3.Loke YK, Brown JWL, Kwok CS, Niruban A, Myint PK. Association of obstructive sleep apnea with risk of serious cardiovascular events: a systematic review and meta-analysis. Circulation: Cardiovascular Quality and Outcomes. 2012;5:720–728. [DOI] [PubMed] [Google Scholar]
- 4.McEvoy RD, Antic NA, Heeley E, Luo Y, Ou Q, Zhang X, Mediano O, Chen R, Drager LF, Liu Z. CPAP for prevention of cardiovascular events in obstructive sleep apnea. New England Journal of Medicine. 2016;375:919–931. [DOI] [PubMed] [Google Scholar]
- 5.Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. American journal of respiratory and critical care medicine. 2016;194:613–620. [DOI] [PubMed] [Google Scholar]
- 6.Barbé F, Durán-Cantolla J, Sánchez-de-la-Torre M, Martínez-Alonso M, Carmona C, Barceló A, Chiner E, Masa JF, Gonzalez M, Marín JM. Effect of continuous positive airway pressure on the incidence of hypertension and cardiovascular events in nonsleepy patients with obstructive sleep apnea: a randomized controlled trial. Jama. 2012;307:2161–2168. [DOI] [PubMed] [Google Scholar]
- 7.Khan SU, Duran CA, Rahman H, Lekkala M, Saleem MA, Kaluski E. A meta-analysis of continuous positive airway pressure therapy in prevention of cardiovascular events in patients with obstructive sleep apnoea. European heart journal. 2018;39:2291–2297. [DOI] [PubMed] [Google Scholar]
- 8.Peker Y, Wegscheider K, Eulenburg C. Reply: Effect of Continuous Positive Airway Pressure Therapy on Cardiovascular Outcomes: Risk Assessment. American journal of respiratory and critical care medicine. 2017;196:662–663. [DOI] [PubMed] [Google Scholar]
- 9.Drager LF, McEvoy RD, Barbe F, Lorenzi-Filho G, Redline S. Sleep apnea and cardiovascular disease: lessons from recent trials and need for team science. Circulation. 2017;136:1840–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pack AI. Application of personalized, predictive, preventative, and participatory (P4) medicine to obstructive sleep apnea. A roadmap for improving care? Annals of the American Thoracic society. 2016;13:1456–1467. [DOI] [PubMed] [Google Scholar]
- 11.Bonsignore MR, Giron MCS, Marrone O, Castrogiovanni A, Montserrat JM. Personalised medicine in sleep respiratory disorders: focus on obstructive sleep apnoea diagnosis and treatment. European Respiratory Review. 2017;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schultz A, Lavie L, Hochberg I, Beyar R, Stone T, Skorecki K, Lavie P, Roguin A, Levy AP. Interindividual heterogeneity in the hypoxic regulation of VEGF: significance for the development of the coronary artery collateral circulation. Circulation. 1999;100:547–552. [DOI] [PubMed] [Google Scholar]
- 13.Bielicki P, Barnas M, Brzoska K, Jonczak L, Plywaczewski R, Kumor M, Stepkowski T, Chazan R, Kruszewski M, Sliwinski P. Genetic determinants of cardiovascular disease in patients with obstructive sleep apnea (OSA). In: Eur Respiratory Soc; 2015. [Google Scholar]
- 14.Floras JS. Sleep apnea and cardiovascular disease: an enigmatic risk factor. Circulation research. 2018;122:1741–1764. [DOI] [PubMed] [Google Scholar]
- 15.Aronson D, Lavie L, Lavie P. Does OSA upregulate cardioprotective pathways to an ischemic insult? Chest. 2018;153:295–297. [DOI] [PubMed] [Google Scholar]
- 16.Lebkuchen A, Freitas LS, Cardozo KH, Drager LF. Advances and challenges in pursuing biomarkers for obstructive sleep apnea: implications for the cardiovascular risk. Trends in cardiovascular medicine. 2020. [DOI] [PubMed] [Google Scholar]
- 17.Sánchez-de-la-Torre M, Khalyfa A, Sánchez-de-la-Torre A, Martinez-Alonso M, Martinez-García MÁ, Barceló A, Lloberes P, Campos-Rodriguez F, Capote F, Diaz-de-Atauri MJ. Precision medicine in patients with resistant hypertension and obstructive sleep apnea: blood pressure response to continuous positive airway pressure treatment. Journal of the American College of Cardiology. 2015;66:1023–1032. [DOI] [PubMed] [Google Scholar]
- 18.Mazzotti DR, Keenan BT, Lim DC, Gottlieb DJ, Kim J, Pack AI. Symptom subtypes of obstructive sleep apnea predict incidence of cardiovascular outcomes. American journal of respiratory and critical care medicine. 2019;200:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavie L Intermittent hypoxia and obstructive sleep apnea: mechanisms, interindividual responses and clinical insights. In: Atherosclerosis, Arteriosclerosis and Arteriolosclerosis. IntechOpen; 2019. [Google Scholar]
- 20.Mann DL, Zipes DP, Libby P, Bonow RO, Braunwald E. Braunwald’s heart disease : a textbook of cardiovascular medicine. Tenth edition. ed. Philadelphia, PA: Elsevier/Saunders; 2015. [Google Scholar]
- 21.Libby P, Hansson GK. From Focal Lipid Storage to Systemic Inflammation: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;74:1594–1607. doi: 10.1016/j.jacc.2019.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewan NA, Nieto FJ, Somers VK. Intermittent hypoxemia and OSA: implications for comorbidities. Chest. 2015;147:266–274. doi: 10.1378/chest.14-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho DSW, Schierding W, Wake M, Saffery R, O’Sullivan J. Machine learning SNP based prediction for precision medicine. Frontiers in Genetics. 2019;10:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horne B, Anderson J, Carlquist J, Muhlestein J, Renlund D, Bair T, Pearson R, Camp N. Generating genetic risk scores from intermediate phenotypes for use in association studies of clinically significant endpoints. Annals of human genetics. 2005;69:176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavie L, Kraiczi H, Hefetz A, Ghandour H, Perelman A, Hedner J, Lavie P. Plasma vascular endothelial growth factor in sleep apnea syndrome: effects of nasal continuous positive air pressure treatment. American journal of respiratory and critical care medicine. 2002;165:1624–1628. [DOI] [PubMed] [Google Scholar]
- 26.Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, Hirano T, Adachi M. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation. 2003;107:1129–1134. [DOI] [PubMed] [Google Scholar]
- 27.Htoo AK, Greenberg H, Tongia S, Chen G, Henderson T, Wilson D, Liu SF. Activation of nuclear factor κB in obstructive sleep apnea: a pathway leading to systemic inflammation. Sleep and Breathing. 2006;10:43–50. [DOI] [PubMed] [Google Scholar]
- 28.Ryan S, Taylor CT, McNicholas WT. Predictors of elevated nuclear factor-κB–dependent genes in obstructive sleep apnea syndrome. American journal of respiratory and critical care medicine. 2006;174:824–830. [DOI] [PubMed] [Google Scholar]
- 29.Jelic S, Lederer DJ, Adams T, Padeletti M, Colombo PC, Factor PH, Le Jemtel TH. Vascular inflammation in obesity and sleep apnea. Circulation. 2010;121:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xi L, Serebrovskaya TV. Intermittent hypoxia and human diseases. Springer Science & Business Media; 2012. [Google Scholar]
- 31.Zhang X-B, Jiang X-T, Cai F-R, Zeng H-Q, Du Y-P. Vascular endothelial growth factor levels in patients with obstructive sleep apnea: a meta-analysis. European Archives of Oto-Rhino-Laryngology. 2017;274:661–670. [DOI] [PubMed] [Google Scholar]
- 32.Labarca G, Gower J, Lamperti L, Dreyse J, Jorquera J. Chronic intermittent hypoxia in obstructive sleep apnea: a narrative review from pathophysiological pathways to a precision clinical approach. Sleep and Breathing. 2019:1–10. [DOI] [PubMed] [Google Scholar]
- 33.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS medicine. 2015;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature genetics. 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, Genovese G, Loh P-R, Bhatia G, Do R. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. The american journal of human genetics. 2015;97:576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikpay M, Goel A, Won H-H, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nature genetics. 2015;47:1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanehisa M A database for post-genome analysis. Trends in genetics: TIG. 1997;13:375–376. [DOI] [PubMed] [Google Scholar]
- 38.Dengler VL, Galbraith MD, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Critical reviews in biochemistry and molecular biology. 2014;49:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottlieb DJ, Yenokyan G, Newman AB, O’Connor GT, Punjabi NM, Quan SF, Redline S, Resnick HE, Tong EK, Diener-West M, et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the sleep heart health study. Circulation. 2010;122:352–360. doi: 10.1161/CIRCULATIONAHA.109.901801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahaghi F, Basner RC. Delayed diagnosis of obstructive sleep apnea: don’t ask, don’t tell. Sleep and Breathing. 1999;3:119–124. [DOI] [PubMed] [Google Scholar]
- 41.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, Crawford DC. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene–disease associations. Bioinformatics. 2010;26:1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Appelman Y, van Rijn BB, Monique E, Boersma E, Peters SA. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis. 2015;241:211–218. [DOI] [PubMed] [Google Scholar]
- 44.Wood SN. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. Journal of the Royal Statistical Society: Series B (Statistical Methodology). 2011;73:3–36. [Google Scholar]
- 45.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J. GENCODE reference annotation for the human and mouse genomes. Nucleic acids research. 2019;47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Consortium Fantom. A promoter-level mammalian expression atlas. Nature. 2014;507:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–1384. e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferrero E Using regulatory genomics data to interpret the function of disease variants and prioritise genes from expression studies. F1000Research. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.