

Abstract
Introduction
Perioperative alterations in perfusion lead to ischemia and reperfusion injury, and supplemental oxygen is administered during surgery to limit hypoxic injury but can lead to hyperoxia. We hypothesized that hyperoxia impairs endothelium-dependent and -independent vasodilation but not the vasodilatory response to heme-independent soluble guanylyl cyclase activation.
Methods
We measured the effect of oxygen on vascular reactivity in mouse aortas. Mice were ventilated with 21% (normoxia), 60% (moderate hyperoxia), or 100% (severe hyperoxia) oxygen during 30 minutes of renal ischemia and 30 minutes of reperfusion. Following sacrifice, the thoracic aorta was isolated, and segments mounted on a wire myograph. We measured endothelium-dependent and -independent vasodilation with escalating concentrations of acetylcholine (ACh) and sodium nitroprusside (SNP), respectively, and we measured the response to heme-independent soluble guanylyl cyclase activation with cinaciguat. Vasodilator responses to each agonist were quantified as the maximal theoretical response (Emax) and the effective concentration to elicit 50% relaxation (EC50) using a sigmoid model and nonlinear mixed effects regression. Aortic superoxide was measured with dihydroethidium probe and HPLC quantification of the specific superoxide product 2-hydroxyethidium.
Results
Hyperoxia impaired endothelium-dependent (ACh) and -independent (SNP) vasodilation compared to normoxia and had no effect on cinaciguat-induced vasodilation. The median ACh Emax was 76.4% (95% CI: 69.6 to 83.3) in the normoxia group, 53.5% (46.7 to 60.3) in the moderate hyperoxia group, and 53.1% (46.3 to 60.0) in the severe hyperoxia group (p<0.001, effect across groups), while the ACh EC50 was not different among groups. The SNP Emax was 133.1% (122.9–143.3) in normoxia, 128.3% (118.1–138.6) in moderate hyperoxia, and 114.8% (104.6–125.0) in severe hyperoxia (p<0.001, effect across groups), and the SNP EC50 was 0.38 log M greater in moderate hyperoxia than in normoxia (95% CI: 0.18 to 0.58, p<0.001). Cinaciguat Emax and EC50 were not different among oxygen treatment groups (median range Emax 78.0% to 79.4% and EC50 −18.0 to −18.2 log M across oxygen groups). Aorta 2-hydroxyethidium was 1419 pmol/mg protein (25th-75th percentile: 1178–1513) in normoxia, 1993 (1831–2473) in moderate hyperoxia, and 2078 (1936–2922) in severe hyperoxia (p=0.008, effect across groups).
Conclusions
Hyperoxia, compared to normoxia, impaired endothelium-dependent and -independent vasodilation but not the response to heme-independent soluble guanylyl cyclase activation, and hyperoxia increased vascular superoxide production. Results from this study could have important implications for patients receiving high concentrations of oxygen and at risk for ischemia reperfusion-mediated organ injury.
Keywords: Oxygen, normoxia, vascular biology, nitric oxide, vasodilation, vasoconstriction, endothelium, perfusion, vascular reactivity, soluble guanylyl cyclase, myography, dihydroethidium, superoxide
Introduction
Postoperative organ dysfunction is common in the two million patients who undergo cardiac and vascular surgery each year. Following cardiac surgery, 10% of patients may suffer stroke and 25% kidney injury.1,2 These events increase the odds of mortality 5-fold and directly contribute to chronic kidney disease and neurologic impairment. These injuries are impacted by organ perfusion and tissue oxygenation. Changes in blood pressure, cardiac output, circulating blood volume, and tissue manipulation alter perfusion during surgical procedures and lead to tissue hypoxia, anaerobic respiration, reperfusion, oxidative damage, inflammation, and cell death.3 In an effort to avoid tissue hypoxia, supplemental oxygen is frequently administered during surgery.4 Hyperoxia, however, may also precipitate organ injury by increasing production of reactive oxygen species (ROS), intensifying inflammation, and decreasing hyperoxia-inducible factor (HIF) mediated gene transcription.5,6
Hyperoxia also induces vasoconstriction, increases vascular resistance, and decreases blood flow.3,7,8 These alterations in perfusion may further impact tissue ischemia and reperfusion injury. In healthy blood vessels, the nitric oxide (NO) pathway has a major role in mediating vascular tone. Shear stress and other factors induce endothelial nitric oxide synthase (eNOS) to produce NO.9,10 NO diffuses into the adjacent smooth muscle cell and binds soluble guanylyl cyclase at the beta-1 subunit heme-moiety.11 Soluble guanylyl cyclase then catalyzes the conversion of guanosine triphosphate to cyclic guanosine monophosphate (GMP). Cyclic GMP induces vasodilation by stimulating protein kinases to activate myosin phosphatase, calcium-gated potassium channels, and sarcoplasmic calcium ATPases which reduce cytosolic calcium.12,13 Excess ROS can impact this pathway in several ways. ROS can uncouple eNOS, leading to the generation of superoxide rather than NO.14 ROS directly react with and eliminate NO, thus reducing NO bioavailability, and ROS may impair soluble guanylyl cyclase activation by oxidizing the soluble guanylyl cyclase NO-binding heme-moiety.15,16 Because of these potentially deleterious effects of oxygen on NO-signaling, a deeper understanding of the impact of excess oxygen on vascular function and subsequently tissue perfusion and organ injury is needed. We hypothesized that hyperoxia impairs endothelium-dependent and -independent vasodilation but not the response to a heme-independent soluble guanylyl cyclase activator.
Methods
Surgical model and oxygen treatment
We measured the effect of increasing concentrations of oxygen on vascular reactivity in aortas from mice exposed to anesthesia, mechanical ventilation, surgery, and renal ischemia and reperfusion. We chose a model of ischemia reperfusion injury because it simulates surgical stress in patients. Results from this preclinical model may ultimately be tested in patients.
All procedures involving animals were approved by the Vanderbilt Institutional Animal Care and Use Committee and adhere to guidelines set in the National Institutes of Health Guide for the Care and use of Laboratory Animals. Male FVB/NJ mice were obtained from Jackson Laboratories (Bar Harbor, Maine) and acclimated to their environment prior to undergoing surgery at 8 weeks of age. Mice were housed in institutional department of animal care facilities, provided standard chow and water ad libitum and were maintained on a 12-hour light/dark cycle. Mice were anesthetized with an intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine, and the back and flanks of the animals were shaved. We performed fiberoptic endotracheal intubation of each mouse with a 20 ga IV catheter and connected the catheter to a small animal ventilator (SAR-1000 Ventilator, CWE Inc., Ardmore, PA). Mice were randomly assigned to oxygen treatment and mechanically ventilated with 21% (normoxia), 60% (moderate hyperoxia), or 100% (severe hyperoxia) inspired oxygen at a rate of 110 breaths per minute and a tidal volume of 10 mL/kg.
To perform surgery and renal ischemia and reperfusion, animals were secured on a heated pad and prepped with betadine and chlorhexidine. Following a midline dorsal incision, the right kidney was exposed, ligated with 3–0 suture, and excised. The left kidney was then similarly exposed. The renal hilum was dissected, and the renal artery and vein clamped with a 200–240 g pressure Schwartz clamp (RS-5459, Roboz Surgical Instrument Co., Gaithersburg, MD) for 30 minutes. After 30 minutes of renal ischemia, the clamp was removed. Thirty minutes after reperfusion, the chest was opened with a ventral midline incision, the left ventricle was punctured, and arterial blood collected for blood gas measurement and terminal exsanguination. The right atrium was then incised, and the left ventricle was perfused with 10cc of cold physiologic saline solution (PSS) to clear blood from the vasculature. The descending thoracic aorta was dissected and excised from the level of the left subclavian artery to the diaphragmatic hiatus and placed in cold PSS.
Myography
The aorta from each mouse was divided axially into four 2 mm segments to provide four experimental replicates. Each segment was placed on wire myograph pins (620M, Danish Myo Technology A/S, Hinnerup, Denmark) in a separate well with 5 mL cold PSS and warmed to 37°C over 15 minutes while being bubbled with 21%, 60%, or 95% oxygen, continuing the in vivo oxygen treatment ex vivo. Five percent carbon dioxide was used as buffer in all wells. The samples were subjected to two additional washes with warm PSS and normalized to a resting tension of 36 mN. Aortic segments then underwent a wake-up protocol by incubating in 5mL of high-potassium PSS (KPSS) for 15 minutes, washing with PSS three times over 10 minutes, and incubating again in 5 mL of KPSS for 15 minutes. After an additional three washes with PSS over 10 minutes to complete the normalization and wake up protocols, drug treatments were initiated.
Vascular reactivity
Endothelium-dependent vasorelaxation was measured in response to ACh following 20-minute incubation with 5 μL 10−3 M norepinephrine (Sigma-Aldrich, St. Louis, MO, USA).17 ACh (Sigma-Aldrich) was added every one minute in steadily increasing doses from 10−11 M to 3×10−5 M well concentration. The vessels were then washed three times with PSS over 10 minutes, and the resting tension was readjusted to 36 mN. Vessels were again incubated with 10-3 M norepinephrine for 20 minutes, and endothelium-independent vasorelaxation was measured in response to escalating doses of SNP (Sigma-Aldrich) from 10−11 M to 3×10−5 M. The vessels were then washed three times with PSS over 10 minutes, and the resting tension was readjusted again to 36 mN. After 20 minutes of incubation with norepinephrine, we measured the response to the heme-independent soluble guanylyl cyclase activator cinaciguat (Sigma-Aldrich), using a dose range from 10−20 M to 10−9 M.16
Tension measurement
Vascular tension was recorded throughout the experiment using Labchart 8 (ADInstruments, Colorado Springs, CO). Resting tension was defined as the median tension exerted by a vessel immediately prior to the addition of norepinephrine. Active tension was defined as the difference between the force 20 minutes after norepinephrine-induced vasoconstriction and the resting tension. The response to vasodilators was measured as the percent relaxation from the active tension.
Superoxide staining and quantification
Dihydroethidium (DHE) was used to characterize and quantify oxidation products in aortic tissue. DHE reacts with superoxide to generate 2-hydroxyethidium and quantification of 2-hydroxyethidium reflects superoxide production.18,19 Following oxygen treatment and renal ischemia reperfusion surgery as described above, the distal thoracic aorta was isolated and excised as above and placed in cold PBS. Aorta was cleaned of perivascular fat.
For histological staining of oxidation products, one 4 mm segment was placed in OCT embedding compound, frozen in liquid nitrogen, and stored at −80°C until sectioning. 8 μm sections were mounted on a slide, OCT compound was dissolved and removed, and slides were incubated with 5μM DHE in PBS for 30 minutes. Sections from each treatment group were placed on the same slide to ensure homogenous staining across oxygen treatment groups. Positive control slides were treated with 10μM antimycin-A and stained with 5μM DHE. Negative control slides were stained with PBS alone. Slides were light-shielded throughout staining and imaging. Images were collected within 60 minutes using a Zeiss LSM 880 Confocal Laser Scanning Microscope. Laser power, exposure, sensitivity, and resolution were optimized and kept constant across all sections and control slides. DHE fluorescence was imaged using the Alexa 555 channel, and elastin autofluorescence was imaged with the Alexa 488 channel.20
For high-performance liquid chromatography (HPLC) quantification of 2-hydroxyethidium,19 four 2mm aortic rings were excised and placed in cold Krebs-Hepes buffer (KHB). Samples were incubated in 50μM DHE in KHB and incubated at 37°C for 30 minutes. After 30 minutes, samples were centrifuged, KHB removed, tissue placed in methanol, and stored at −80°C. Aortic rings were homogenized with a glass pestle, and the tissue homogenate was passed through a 0.22 μm syringe filter, and analyzed by HPLC according to previously published protocols.21 In brief, a C-18 reverse-phase column (nucleosil 250–4.5 mm) and a mobile phase containing 0.1% trifluoroacetic acid and an acetonitrile gradient (from 37% to 47%) at a flow rate of 0.5 mL/min were used. 2-hydroxyethidium was quantified by fluorescence detection using an emission wavelength of 580 nm and an excitation of 480 nm.22 Superoxide levels are reported as pmol 2-hydroxyethidium per mg protein.
Statistical analysis
Measurements were summarized using median (25th percentile-75th percentile) or estimate (95% confidence interval [CI]). The Jonckheere trend test was used to test for treatment effects among ordinal oxygen exposure groups and unpaired quantitative outcomes including arterial blood gas data, norepinephrine contraction response, and 2-hydroxyethidium. The Wilcoxon rank-sum test was used for non-parametric comparisons between individual oxygen treatment groups.
The relationship between vasodilator concentration and vessel percent relaxation was modeled using a sigmoid Emax model: E(concentration) = Emax ⋅ (1 + e -rate⋅(concentration - EC50))−1, where E(concentration) represents the vessel percent relaxation. Emax represents the maximum relaxation percentage, EC50 represents the concentration at which the relaxation percentage is one half of the maximum relaxation percentage, and rate represents the steepness of the sigmoid curve.
The effect of oxygen exposure on the dose-relaxation relationship was quantified by estimating its effects on each of the three sigmoid Emax model parameters using nonlinear mixed effects regression. Inter-vessel heterogeneity in the Emax and EC50 parameters were modeled using mutually independent random intercepts. Preliminary analyses indicated that allowing for inter-vessel heterogeneity in the rate parameter caused the model to be inestimable or weakly estimable. Thus, the rate parameter was not allowed to vary randomly by vessel. Maximum likelihood methods were used to estimate and summarize uncertainty in the model parameters. A Wald chi-square test was used to test the null hypothesis that oxygen exposure has no effect on any of the three model parameters. For each model parameter and pair of oxygen exposure levels, the difference between the corresponding model parameter was estimated using a 95% CI and a Wald chi-square test was used to test the null hypothesis that the difference is equal to zero.23 All analyses were implemented in R.24 Nonlinear mixed effects regression was implemented using the nlme add-on package.25
Results
Mice and oxygen treatment
Four aortic rings were collected for myography from each mouse exposed to anesthesia, mechanical ventilation, surgery and renal ischemia and reperfusion. Endothelium-dependent and -independent relaxation experiments were performed using five mice (20 rings) for each oxygen treatment, and soluble guanylyl cyclase activator experiments were performed using 3 mice (12 rings) for each oxygen treatment. Additional mice were used for DHE staining and superoxide quantification. The median weight of mice was 25.28 grams (25th percentile-75th percentile: 24.37–26.93). Median PaO2 was 104 mmHg (93.75–112.0, n=8) in mice treated with 21% inspired oxygen, 218 mmHg (211.0–222.0, n=5) in mice treated with 60% inspired oxygen, and 324 mmHg (269.8–400.8, n=4) in mice treated with 100% inspired oxygen (p<0.001; Table 1). Hemoglobin oxygen saturation was also increased in mice treated with hyperoxia, and arterial pH was marginally lower in mice treated with 21% and 60% oxygen compared to mice treated with 100% oxygen. PaCO2 and arterial lactate were not different between oxygen treatment groups.
Table 1.
Arterial blood gas data in mice treated with 21%, 60%, or 100% inspired oxygen. Data are presented as median (25th percentile – 75th percentile). P-values reflect effect of oxygen treatment across groups using the Jonckheere trend test.
| Lab value | 21% | 60% | 100% | P-value |
|---|---|---|---|---|
| n = 8 | n = 5 | n = 4 | ||
|
| ||||
| pH | 7.25 (7.21–7.28) | 7.24 (7.23–7.31) | 7.35 (7.32–7.43) | 0.05 |
| pCO2 (mmHg) | 35.4 (30.3–40.5) | 47.4 (36.4–54) | 38.7 (31.1–48.9) | 0.28 |
| pO2 (mmHg) | 104 (94.5–112) | 218 (214–221) | 318 (275.5–383.5) | <0.001 |
| Hemoglobin O2 Saturation (%) | 96.5 (95.5–97.5) | 100 (100–100) | 100 (100–100) | <0.001 |
| Lactate (mmol/L) | 1.31 (1.03–1.87) | 0.94 (0.66–2.47) | 0.91 (0.44–1.11) | 0.33 |
Impact of oxygen on vasoconstriction
The median vasoconstriction response to norepinephrine (i.e., the active tension) was increased with increased oxygen tension. Active tension was 8.28 mN (25th percentile – 75th percentile: 6.80 – 9.28) in the normoxia group, 8.57 mN (6.78 – 11.89) in the moderate hyperoxia group, and 9.82 mN (8.67 – 11.57) in the severe hyperoxia group (n=20 aortic rings in each oxygen group, p=0.03, effect across groups, Figure 1). When comparing individual treatments, vasoconstriction in severe hyperoxia was greater than in normoxia (p=0.009), but vasoconstriction in moderate hyperoxia was not significantly different than normoxia (p=0.36) or severe hyperoxia (p=0.39).
Figure 1.
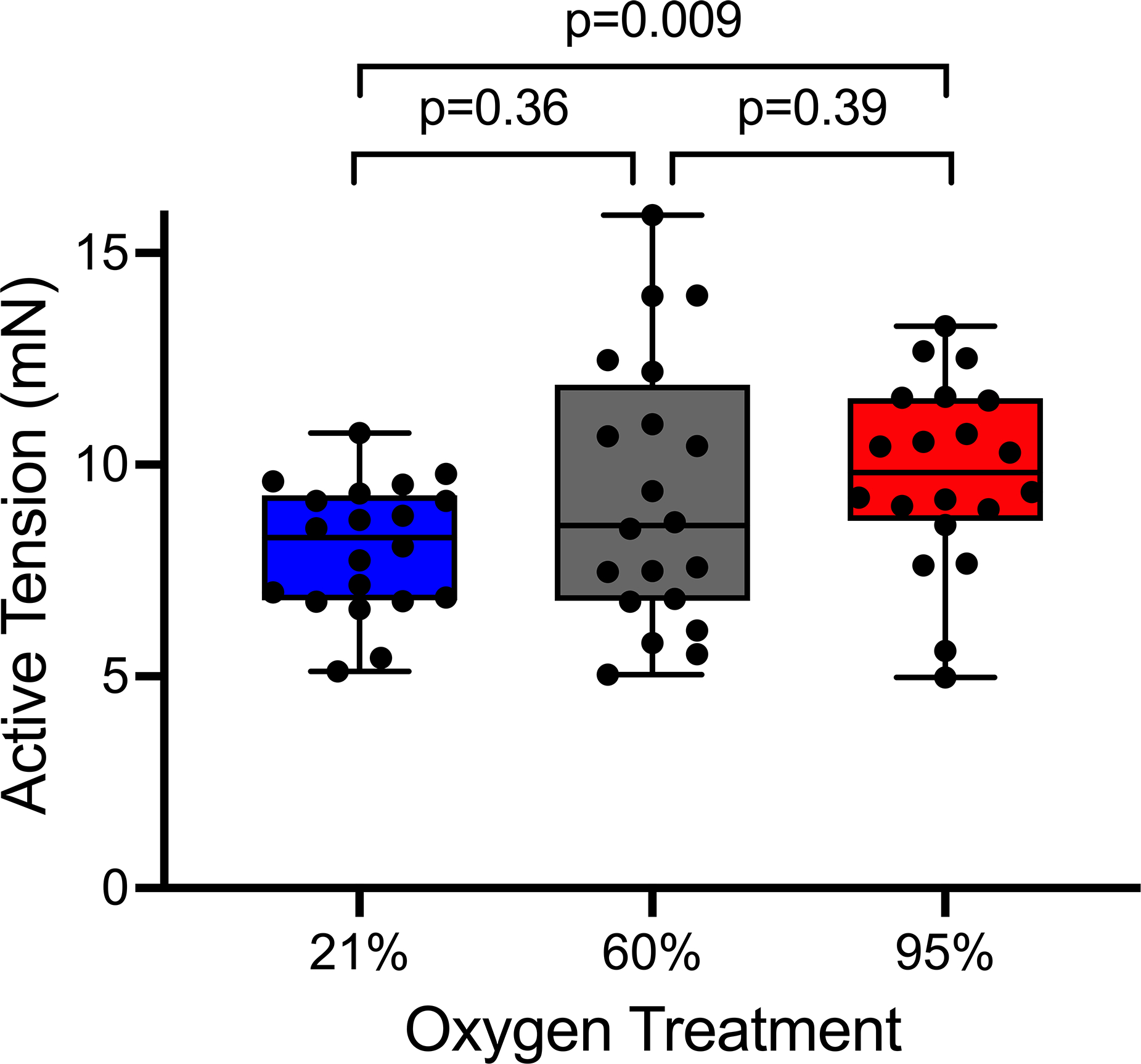
Effect of oxygen treatment on vasoconstriction in response to norepinephrine. Box and whisker plots represent the median, 25th and 75th percentiles in the box, and the minimum and maximum values as whiskers. Each dot represents one aortic ring (n=20 per group). p=0.03, effect of oxygen treatment across groups using the Jonckheere trend test. Comparisons between individual oxygen treatment groups used the Wilcoxon rank-sum test.
Impact of oxygen on vascular relaxation
Hyperoxia impaired endothelium-dependent vasodilation in response to ACh (Table 2, Figure 2). The median ACh Emax in the normoxia group (76.4% [95% CI: 69.6 to 83.3]) was 22.9% greater (95% CI: 13.3 to 32.6, p<0.001) than in the moderate hyperoxia group (53.5% [95% CI: 46.7 to 60.3]) and 23.3% greater (95% CI: 13.7 to 33.0, p<0.001) than in the severe hyperoxia group (53.1% [95% CI: 46.3 to 60.0]). The ACh Emax was not different between moderate hyperoxia and severe hyperoxia groups (p=0.94). The ACh concentration at which half the maximal relaxation was achieved (i.e., the EC50) was not affected by oxygen treatment. Specifically, the ACh EC50 was −7.59 log M (95% CI: −7.72 to −7.47) in the normoxia group, −7.62 log M (95% CI: −7.76 to −7.48) in the moderate hyperoxia group, and -7.63 log M (95% CI: −7.77 to −7.50) in severe hyperoxia (p=ns across treatment groups). The Wald chi-square test that assessed an effect across all parameters in the dose-response curves confirmed an effect of oxygen treatment (p<0.001) on ACh-mediated vasorelaxation.
Table 2.
Vascular reactivity in vessels isolated from mice ventilated with 21%, 60%, or 100% oxygen and then incubated with 21, 60, or 95 % oxygen during myography. Data reported are model estimates (95% CI) for the maximum relaxation percentage (Emax) and the concentration at which the relaxation percentage is one half of the maximum relaxation percentage (EC50). N represents the number of aortic ring replicates in each experiment. P-values test the null hypothesis that oxygen had no effect on any of the three model parameters using a Wald chi-square test.
| Drug | Oxygen (%) | Emax (% relaxation) | EC50 (log molar) | P-value |
|---|---|---|---|---|
|
| ||||
| Acetylcholine (n = 20) | 21 | 76.4 (69.6 to 83.3)*† | −7.59 (−7.72 to −7.47) | <0.001 |
| 60 | 53.5 (46.7 to 60.3) | −7.62 (−7.76 to −7.48) | ||
| 100 | 53.1 (46.3 to 60.0) | −7.63 (−7.77 to −7.50) | ||
|
| ||||
| Sodium nitroprusside (n = 20) | 21 | 133.1 (122.9 to 143.3)† | −7.43 (−7.57 to −7.29)* | <0.001 |
| 60 | 128.3 (118.1 to 138.6) | −7.04 (−7.19 to −6.90) | ||
| 100 | 114.8 (104.6 to 125.0) | −7.31 (−7.45 to −7.16)* | ||
|
| ||||
| Cinaciguat (n = 12) | 21 | 76.4 (68.8 to 84.0) | −18.1 (−18.3 to −17.9) | 0.50 |
| 60 | 79.1 (71.5 to 86.7) | −18.0 (−18.2 to −17.7) | ||
| 100 | 78.0 (70.4 to 85.6) | −18.2 (−18.5 to −18.0) | ||
indicates p<0.05 vs. 60% oxygen, and
indicates p<0.05 vs. 100% oxygen for comparisons of specific parameters between individual treatment groups.
Figure 2.
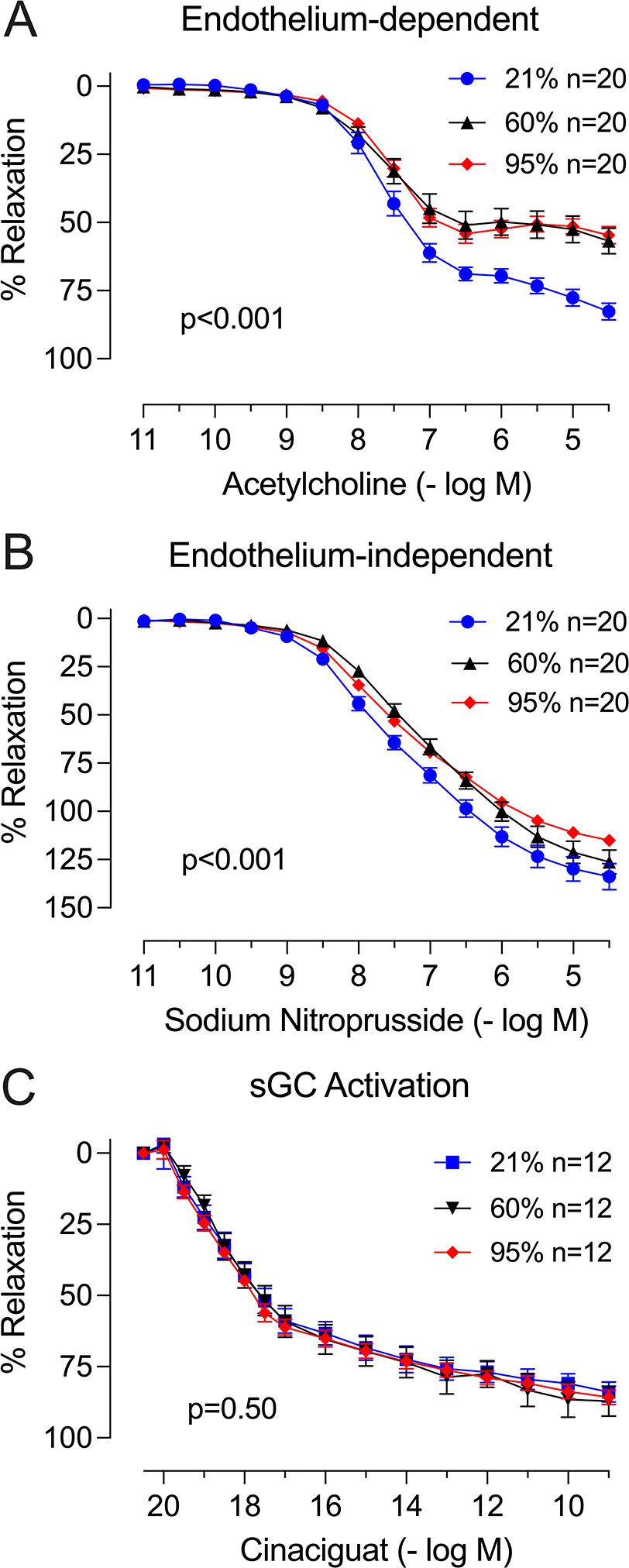
Effect of oxygen treatment on vascular reactivity. Data shown as dose-response curves to (A) acetylcholine (endothelium-dependent response), (B) sodium nitroprusside (endothelium-independent response), and (C) cinaciguat (soluble guanylyl cyclase [sGC] activation). N represents the number of aortic ring replicates subjected to the relaxation protocol. P-values test the null hypothesis that oxygen had no effect on any of the three model parameters using a Wald chi-square test.
Hyperoxia also impaired endothelium-independent vasodilation in response to SNP. The SNP Emax was 133.1% (95% CI: 122.9 to 143.3) in the normoxia group, 128.3% (95% CI: 118.1 to 138.6) in the moderate hyperoxia group, and 114.8% (95% CI: 104.6 to 125.0) in the severe hyperoxia group. The median SNP Emax in the normoxia group was 18.3% greater (95% CI: 3.9 to 32.7, p<0.001) than the median SNP Emax in the severe hyperoxia group. The increase in SNP Emax between normoxia and moderate hyperoxia groups (4.8% [95% CI: −9.7 to 19.2], p=0.52) was not statistically significant, and there was a trend towards higher SNP Emax in the moderate hyperoxia group compared to the severe hyperoxia group (13.6% [95% CI: −0.9 to 28.0], p=0.07)). Vessels from mice treated with moderate hyperoxia required a significantly higher concentration of SNP to achieve half-maximal relaxation compared to vessels from mice treated with normoxia or vessels from mice treated with severe hyperoxia. The SNP EC50 was −7.43 log M (95% CI: −7.57 to −7.29) in the normoxia group, −7.04 log M (−7.19 to −6.90) in the moderate hyperoxia group, and −7.31 log M (95% CI: -7.45 to −7.16) in the severe hyperoxia group. The moderate hyperoxia group had a SNP EC50 0.38 log M greater (95% CI: 0.18 to 0.58, p<0.001) than the SNP EC50 in the normoxia group and 0.26 log M greater (95% CI: 0.06 to 0.47, p=0.01) than the SNP EC50 in the severe hyperoxia group. The SNP EC50 was not different between the severe hyperoxia and normoxia groups. The Wald chi-square test that assessed an effect across all parameters in the dose-response curves confirmed an effect of oxygen treatment (p<0.001) on SNP-mediated vasorelaxation.
Heme-independent soluble guanylyl cyclase activation induced similar vasodilatory responses in all three oxygen treatment groups. The cinaciguat Emax was 79.4% (95% CI: 68.8 to 84.0) in the normoxia group, 79.1% (95% CI: 71.5 to 86.7) in the moderate hyperoxia group, and 78.0% (95% CI: 70.4 to 85.6) in the severe hyperoxia group, and the cinaciguat EC50 was −18.1 log M (95% CI: −18.3 to −17.9) in the normoxia group, −18.0 log M (95% CI: −18.2 to −17.7) in the moderate hyperoxia group, and −18.2 log M (95% CI: −18.5 to −18.0) in the severe hyperoxia group (p=ns for all comparisons). The Wald chi-square test showed no significant effect of oxygen treatment (p=0.50) across all parameters in the cinaciguat dose-response curves.
Aortic tissue oxidation
Representative images showing DHE fluorescence in aortic tissue are shown in Figure 3. Hyperoxia treatment significantly increased superoxide production as measured by 2-hydroxyethidium levels using HPLC. Median 2-hydroxyethidium in the normoxia group was 1419 pmol/mg (25th percentile-75th percentile: 1178–1513, n=4), compared to 1993 pmol/mg (1831–2473, n=4) in the moderate hyperoxia group and 2078 pmol/mg (1936–2922, n=3) in the severe hyperoxia group, (p=0.008, treatment effect across groups, Figure 4). 2-hydroxyethidium was significantly lower in the normoxia group compared to the moderate hyperoxia group (p=0.02) and compared to the severe hyperoxia group (p=0.03). There was no significant difference in 2-hydroxyethidium between the moderate hyperoxia and severe hyperoxia groups (p=0.48).
Figure 3.
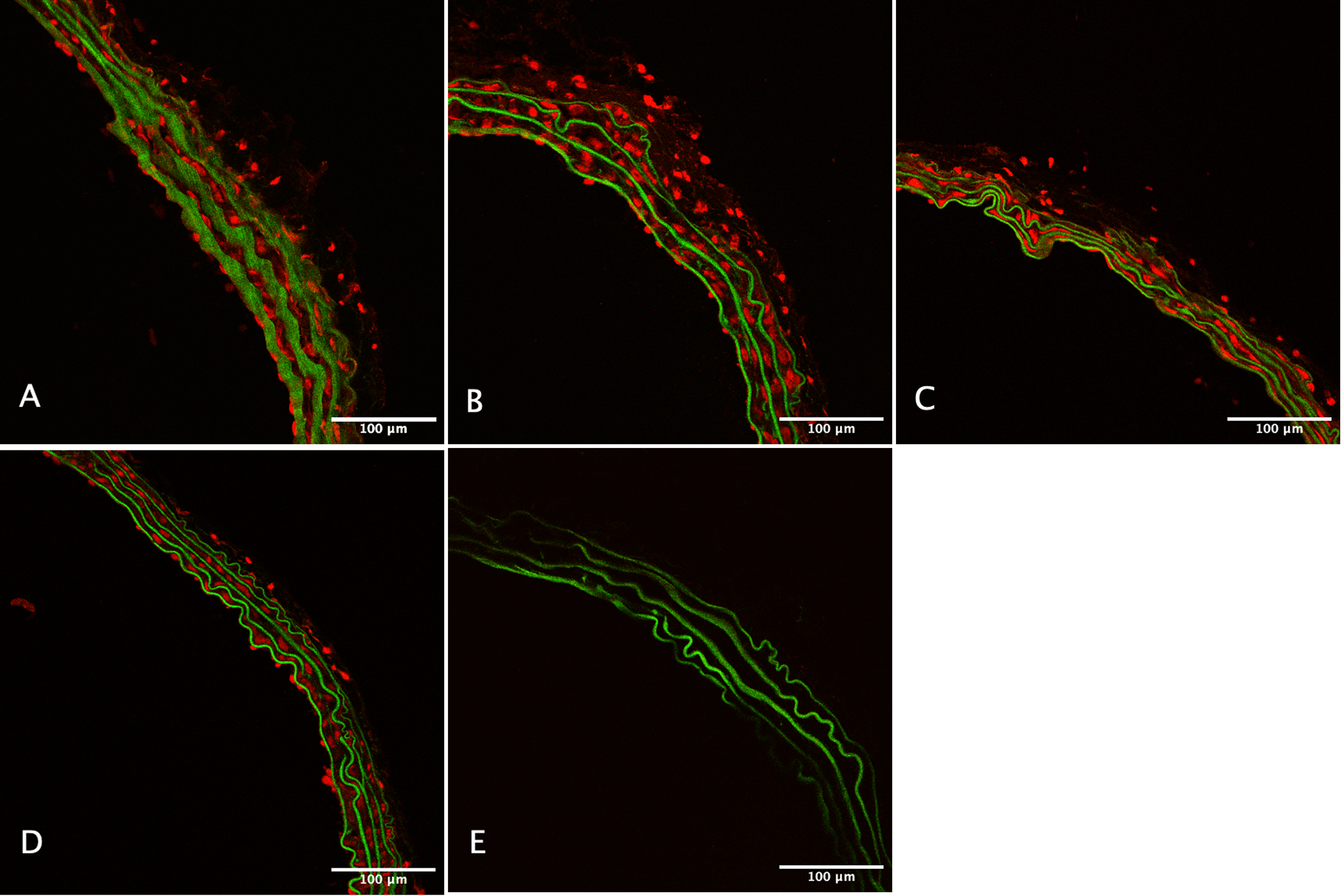
Representative confocal microscopy images of mouse aorta stained with dihydroethidium at 20X magnification after treatment with (A) 21%, (B) 60%, and (C) 100% oxygen. (D) Positive control, and (E) negative control images are also shown.
Figure 4.
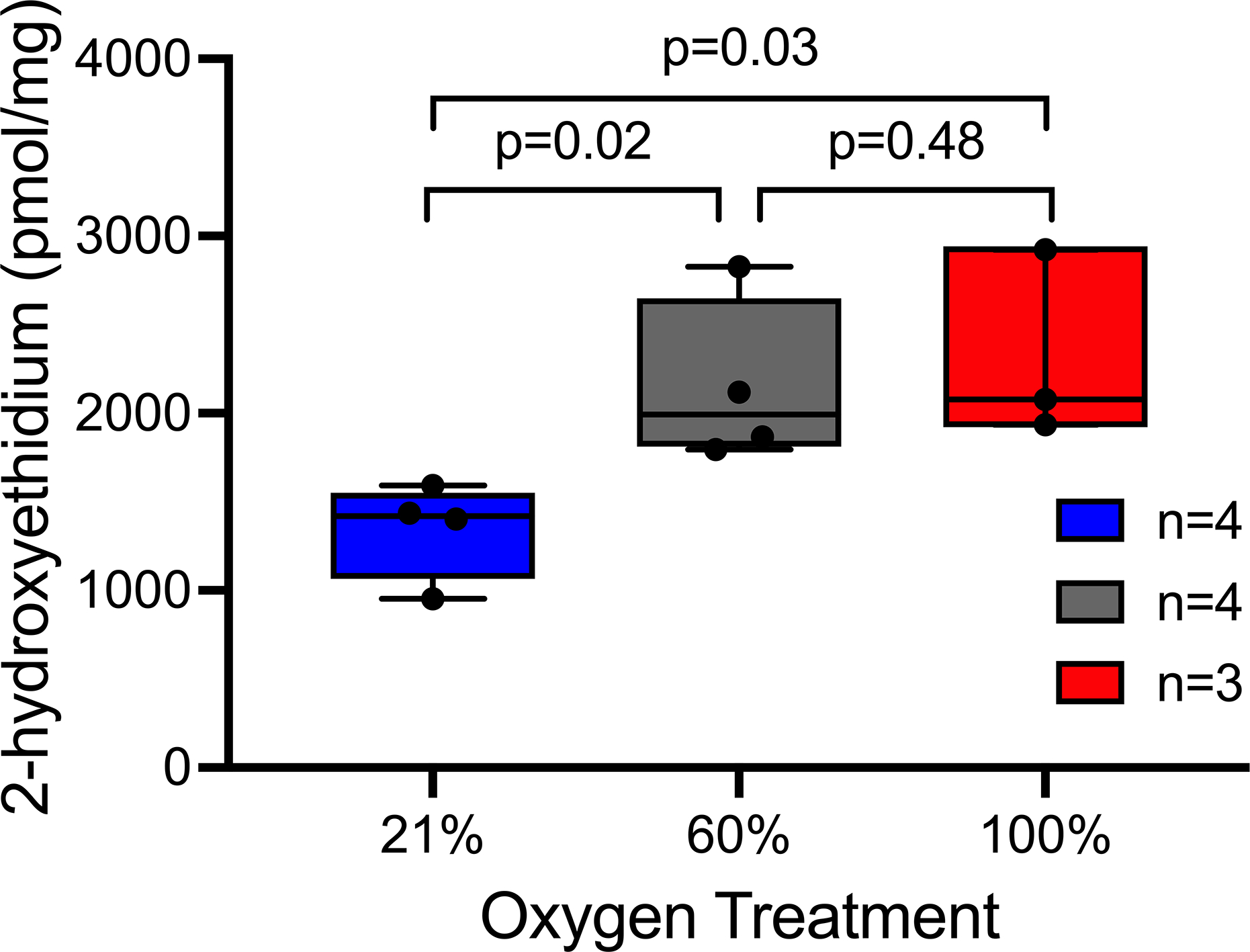
Effect of oxygen treatment on aortic 2-hydroxyethidium production, presented as median ± SD per mg protein. Box and whisker plots represent the median, 25th and 75th percentiles in the box, and the minimum and maximum as whiskers. N represents number of mice in each group. p=0.008 for the effect of oxygen treatment across groups using the Jonckheere trend test. Comparisons between individual oxygen treatment groups used the Wilcoxon rank-sum test.
Discussion
In this pre-clinical study of oxygen exposure and vascular reactivity, hyperoxia impaired endothelium-dependent and -independent vasodilation compared to normoxia, but heme-independent activation of soluble guanylyl cyclase eliminated this impairment. Hyperoxia during surgery also increased superoxide in aortic tissue. The mechanisms by which hyperoxia impairs vascular relaxation are uncertain, although these findings suggest that production of reactive oxygen species and alterations in NO signaling may be involved.
Prior studies provide additional evidence that hyperoxia impairs vascular function and potentially through effects on NO signaling. In coronary vasculature, for example, 100% FiO2 during coronary catheterization in healthy individuals increased coronary artery resistance and decreased coronary blood flow.7 These changes were unaffected by intracoronary ACh, but normal flow and resistance were restored by adenosine, a purinergic receptor agonist that impacts coronary smooth myocytes independent of NO.26 In another study, 100% FiO2 decreased forearm blood flow and this decrease was also unaffected by intraarterial ACh, but the endothelium-independent vasodilator verapamil, which inhibits calcium influx into vascular smooth muscle, restored normal flow.27 In that experiment, the coadministration of vitamin C, an antioxidant, during ACh infusion eliminated the effect of hyperoxia. These studies suggest that hyperoxia-induced vascular impairment is at least partially endothelium-dependent, and that oxidative stress contributes to this dysfunction. Others have noted no impact of hyperoxia on vascular relaxation in ex vivo femoral arteries and gracilis arterioles in healthy mice, although these studies did not administer oxygen treatment in vivo nor induce surgical stress or an ischemia reperfusion event prior to blood vessel isolation and vascular reactivity testing.28 Additional studies found increased systemic vasoconstriction, reduced vasodilator responses, and decreased blood flow in response to hyperoxia, although these responses differed across vascular beds, and molecular mediators of these findings have been unclear.29–31
The current study provides additional evidence that hyperoxia impairs endothelium-mediated vasodilation (reduced ACh Emax in both moderate and severe hyperoxia compared to normoxia) but also that hyperoxia impairs endothelium-independent vasodilation. For example, the SNP EC50 was increased in moderate hyperoxia compared to normoxia. This effect may be a result of elimination of NO (decreased NO bioavailability) or impaired NO binding to soluble guanylyl cyclase, but the similar SNP Emax between these two groups is evidence that increased NO administration may overcome this effect. In severe hyperoxia, however, SNP Emax was reduced compared to normoxia, indicating that increased NO in severe hyperoxic conditions does not result in complete soluble guanylyl cyclase activation. Hyperoxia could impair NO activation of soluble guanylyl cyclase by oxidizing the soluble guanylyl cyclase NO-binding heme moiety,32 as activation of soluble guanylyl cyclase with cinaciguat, which binds independent of the NO-binding site,16 was not affected by oxygen treatment. These results suggest that, once activated, soluble guanylyl cyclase function is not different between hyperoxic and normoxic conditions. Previous studies have observed that ROS such as superoxide uncouple eNOS, directly eliminate NO, and oxidize soluble guanylyl cyclase, but we did not measure these mechanisms directly.14,32 Hyperoxia, compared to normoxia, increased aortic superoxide quantified by measuring 2-hydroxyethidium in the current study, supporting the idea that hyperoxia-induced oxidative damage may contribute to these findings, although additional experiments are required to clarify these mechanisms.
Excess oxygenation also increased the vasoconstrictor response to norepinephrine in a dose-dependent manner. Further study will be required to elucidate the mechanisms involved, but given the observed effects of hyperoxia on vasodilation, it is possible that the effects of hyperoxia on norepinephrine-induced vasoconstriction may be a result of the impairment on the vasodilatory cascade. Other potential mechanisms include enhanced alpha-1 receptor activity. Notably, we examined vascular relaxation responses relative to the achieved active tension (i.e., percent relaxation), so any effect of differences in active tension on vasorelaxation is minimized or eliminated.
Our study has several limitations. Mice were administered oxygen treatments during mechanical ventilation, surgery, and renal ischemia and reperfusion in vivo, but the vascular reactivity studies were performed on isolated aortic segments ex vivo. While this improved our ability to isolate the effect of hyperoxia on vascular reactivity, it limited the extent to which other counterregulatory systems in vivo may respond to changes in vascular oxygen tension and impact vascular reactivity. In addition, we did not measure specific enzymatic activities or substrates along the pathways of interest. Because hyperoxia did not impair vasodilation in response to heme-independent soluble guanylyl cyclase activation, future investigation may examine whether pre-treatment with a soluble guanylyl cyclase activator or stimulator restores normal vascular reactivity in hyperoxic environments. This approach could lead to novel therapies for surgical patients.
In summary, hyperoxia as compared to normoxia impaired endothelium-dependent and -independent vasodilation but not the vasodilator response to heme-independent soluble guanylyl cyclase activation. Increased ROS production in hyperoxic conditions may contribute to these findings. Further understanding of the mechanisms involved will be important for patients receiving high concentrations of supplemental oxygen and prone to ischemia reperfusion-mediated organ injury.
Acknowledgments
We acknowledge Dr. David G. Harrison, Professor of Medicine, Vanderbilt University Medical Center for contributions to study design; the Translational Pathology Shared Resource (supported by NCI/NIH Cancer Center Support Grant P30CA068485) for tissue sectioning; and the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126) for the use of their Zeiss LSM880 microscope (supported by NIH grant S10OD021630 1).
Footnotes
Conflicts of Interest and Sources of Funding:
No author reports a significant conflict of interest. This work was supported by grant support from the National Institutes of Health (T32GM108554 [EHM], R01HL144943 [SID] R01HL157583 [SID, FTB, MGL], R01GM112871 [MSS, FTB], R35GM145375 [MSS, FTB], and K23GM129662 [MGL]), a 2018 Burroughs Wellcome Fund Physician-Scientist Institutional Award to Vanderbilt University (ID: 1018894 [MJK]), and the Vanderbilt Undergraduate Summer Research Program (TJN).
Contributor Information
Eric H. Mace, Department of Surgery at Vanderbilt University Medical Center, Nashville, TN, USA.
Melissa J. Kimlinger, School of Medicine at Vanderbilt University, Nashville, TN, USA.
Tom J. No, College of Arts and Sciences at Vanderbilt University, Nashville, TN, USA.
Sergey I. Dikalov, Department of Medicine at Vanderbilt University Medical Center, Nashville, TN, USA.
Cassandra Hennessy, Department of Biostatistics at Vanderbilt University Medical Center, Nashville, TN, USA..
Matthew S. Shotwell, Department of Biostatistics at Vanderbilt University Medical Center, Nashville, TN, USA.
Frederic T. Billings, IV, Department of Anesthesiology at Vanderbilt University Medical Center, Nashville, TN, USA..
Marcos G. Lopez, Department of Anesthesiology at Vanderbilt University Medical Center, Nashville, TN, USA..
References:
- 1.Billings FT, Lopez MG, Shaw AD. The incidence, risk, presentation, pathophysiology, treatment, and effects of perioperative acute kidney injury. Can J Anaesth J Can Anesth. 68(3):409–422; 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiele RH, Theodore DJ, Gan TJ. Outcome of Organ Dysfunction in the Perioperative Period. Anesth Analg. 133(2):393–405; 2021. [DOI] [PubMed] [Google Scholar]
- 3.Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 17(11):1391–1401; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehrenfeld JM, Funk LM, Van Schalkwyk J, Merry AF, Sandberg WS, Gawande A. The incidence of hypoxemia during surgery: evidence from two institutions. Can J Anaesth J Can Anesth. 57(10):888–897; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helmerhorst HJF, Schultz MJ, van der Voort PHJ, de Jonge E, van Westerloo DJ. Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care Lond Engl. 19:284; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimlinger MJ, Mace EH, Harris RC, Zhang MZ, Barajas MB, Hernandez A, Billings FT. Impact of Inhaled Oxygen on Reactive Oxygen Species Production and Oxidative Damage during Spontaneous Ventilation in a Murine Model of Acute Renal Ischemia and Reperfusion. Med Res Arch. 9(10):2575; 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNulty PH, King N, Scott S, Hartman G, McCann J, Kozak M, Chambers CE, Demers LM, Sinoway LI. Effects of supplemental oxygen administration on coronary blood flow in patients undergoing cardiac catheterization. Am J Physiol-Heart Circ Physiol. 288(3):H1057–H1062; 2005. [DOI] [PubMed] [Google Scholar]
- 8.Nensén O, Hansell P, Palm F. Intrarenal oxygenation determines kidney function during the recovery from an ischemic insult. Am J Physiol-Ren Physiol. 319(6):F1067–F1072; 2020. [DOI] [PubMed] [Google Scholar]
- 9.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 33(7):829–837; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis ME, Grumbach IM, Fukai T, Cutchins A, Harrison DG. Shear Stress Regulates Endothelial Nitric-oxide Synthase Promoter Activity through Nuclear Factor κB Binding. J Biol Chem. 279(1):163–168; 2004. [DOI] [PubMed] [Google Scholar]
- 11.Busker M, Neidhardt I, Behrends S. Nitric Oxide Activation of Guanylate Cyclase Pushes the α1 Signaling Helix and the β1 Heme-binding Domain Closer to the Substrate-binding Site. J Biol Chem. 289(1):476–484; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-Dependent Protein Kinases and the Cardiovascular System: Insights From Genetically Modified Mice. Circ Res. 93(10):907–916; 2003. [DOI] [PubMed] [Google Scholar]
- 13.Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 52(3):375–414; 2000. [PubMed] [Google Scholar]
- 14.Schmidt HHHW, Ghezzi P, Cuadrado A, eds. Reactive Oxygen Species: Network Pharmacology and Therapeutic Applications. Vol 264. Springer International Publishing; 2021. [Google Scholar]
- 15.Schulz E, Gori T, Münzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens Res. 34(6):665–673; 2011. [DOI] [PubMed] [Google Scholar]
- 16.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, HS AK, Meurer S, Deile M, Taye A, Knorr A, Lapp H, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 116(9):2552–2561; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ignarro LJ, Byrns RE, Buga GM, Wood KS, Chaudhuri G. Pharmacological evidence that endothelium-derived relaxing factor is nitric oxide: use of pyrogallol and superoxide dismutase to study endothelium-dependent and nitric oxide-elicited vascular smooth muscle relaxation. J Pharmacol Exp Ther. 244(1):181–189; 1988. [PubMed] [Google Scholar]
- 18.Wang Q, Zou MH. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial ROS in AMPK Knockout Mice Blood Vessels. In: Neumann D, Viollet B, eds. AMPK. Vol 1732. Methods in Molecular Biology. Springer; New York. 507–517; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zielonka J, Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med. 48(8):983–1001; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deshpande P, Gogia N, Chimata AV, Singh A. Unbiased automated quantitation of ROS signals in live retinal neurons of Drosophila using Fiji/ImageJ. BioTechniques. 71(2):416–424; 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertens Dallas Tex 1979. 49(4):717–727; 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 107(1):106–116; 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrell Frank E. Jr. Section 9.3.1. In: Regression Modeling Strategies. Springer-Verlag; 2001. [Google Scholar]
- 24.R Core Team. R: A language and environment for statistical computing. Published online. https://www.r-project.org/ 2001.
- 25.Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. Published online https://cran.r-project.org/package=nlme; 2001.
- 26.Sato A, Terata K, Miura H, Toyama K, Loberiza FR, Hatoum OA, Saito T, Sakuma I, Gutterman DD. Mechanism of vasodilation to adenosine in coronary arterioles from patients with heart disease. Am J Physiol-Heart Circ Physiol. 288(4):H1633–H1640; 2005. [DOI] [PubMed] [Google Scholar]
- 27.Mak S, Egri Z, Tanna G, Colman R, Newton GE. Vitamin C prevents hyperoxia-mediated vasoconstriction and impairment of endothelium-dependent vasodilation. Am J Physiol-Heart Circ Physiol. 282(6):H2414–H2421; 2002 [DOI] [PubMed] [Google Scholar]
- 28.Smit B, Smulders YM, de Waard MC, Oudemans-van Straaten HM, Girbes ARJ, Eringa EC, Spoelstra-de Man AME, Bachschmid MM. Hyperoxia does not directly affect vascular tone in isolated arteries from mice. PLOS ONE. 12(8):e0182637; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daly WJ, Bondurant S. Effects of oxygen breathing on the heart rate, blood pressure, and cardiac index of normal men—resting, with reactive hyperemia, and after atropine. J Clin Invest. 41(1):126–132; 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crawford P, Good PA, Gutierrez E, Feinberg JH, Boehmer JP, Silber DH, Sinoway LI. Effects of supplemental oxygen on forearm vasodilation in humans. J Appl Physiol. 82(5):1601–1606; 1997. [DOI] [PubMed] [Google Scholar]
- 31.Pedersen PK, Kiens B, Saltin B. Hyperoxia does not increase peak muscle oxygen uptake in small muscle group exercise: Hyperoxia and V˙o2peak in small muscle group exercise. Acta Physiol Scand. 166(4):309–318; 1999. [DOI] [PubMed] [Google Scholar]
- 32.Shah RC, Sanker S, Wood KC, Durgin BG, Straub AC. Redox regulation of soluble guanylyl cyclase. Nitric Oxide. 76:97–104; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]