

Abstract
Homeostatic plasticity (HP) encompasses a suite of compensatory physiological processes that counteract neuronal perturbations, enabling brain resilience. Currently, we lack a complete description of the homeostatic processes that operate within the mammalian brain. Here, we demonstrate that acute, partial AMPAR-specific antagonism induces potentiation of presynaptic neurotransmitter release in adult hippocampus, a form of compensatory plasticity that is consistent with the expression of presynaptic homeostatic plasticity (PHP) documented at peripheral synapses. We show that this compensatory plasticity can be induced within minutes, requires postsynaptic NMDARs and is expressed via correlated increases in dendritic spine volume, active zone area, and docked vesicle number. Further, simultaneous postsynaptic genetic reduction of GluA1, GluA2 and GluA3 in triple heterozygous knockouts induces potentiation of presynaptic release. Finally, induction of compensatory plasticity at excitatory synapses induces a parallel, NMDAR-dependent potentiation of inhibitory transmission, a cross-modal effect consistent with the anti-epileptic activity of AMPAR-specific antagonists used in humans.
ETOC Paragraph:
Chipman and colleagues characterize NMDA receptor-dependent compensatory plasticity in the adult hippocampus, consistent with expression presynaptic homeostatic plasticity (PHP). Mechanistically, the compensatory plasticity is driven by NMDAR-dependent spine growth and active zone expansion. Remarkably, compensatory plasticity at excitatory synapses are linked to potentiation of inhibitory transmission, ultimately favoring net inhibition.
Introduction
A variety of compensatory physiological processes have been described within the mammalian central nervous system (Aoto et al., 2008; Burrone et al., 2002; Davis, 2006; Desai et al., 1999; Jakawich et al., 2010; Kim and Ryan, 2010; Li et al., 2020; Mitra et al., 2011; Murthy et al., 2001; O’Brien et al., 1998; Turrigiano et al., 1998). Among these, prolonged activity blockade can induce compensatory changes in postsynaptic neurotransmitter receptor abundance, a process termed quantal scaling that has been documented both in vitro following activity blockade (Aoto et al., 2008; O’Brien et al., 1998; Turrigiano et al., 1998) and in vivo following sensory deprivation (Desai et al., 2002). Another example occurs at the neuromuscular junctions (NMJ) of Drosophila, rodents, and humans. At the NMJ, disruption of postsynaptic neurotransmitter receptors (pharmacologically or genetically) induces a compensatory increase in presynaptic neurotransmitter release that offsets the magnitude of postsynaptic neurotransmitter receptor disruption and restores synaptic gain to baseline values (Cull-Candy et al., 1980; Delvendahl et al., 2019; Müller et al., 2012; Plomp et al., 1992; Wang et al., 2016). This process is referred to as presynaptic homeostatic plasticity (PHP) (Davis, 2006; Dickman and Davis, 2009; Frank et al., 2009; Harris et al., 2018; Hauswirth et al., 2018)
To date, it remains generally unknown whether a compensatory process resembling PHP is expressed at synapses in the mammalian central nervous system (CNS). A recent study demonstrated that partial antagonism of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors, or chronic deletion of the GluA4 AMPAR subunit (encoded by the Gria4 gene) induces a potentiation of excitatory presynaptic neurotransmitter release at the cerebellar mossy fiber synapse (Delvendahl et al., 2019). It remains unknown whether this occurs at synapses that do not utilize the sparsely expressed Gria4 receptor subunit.
Here, we characterize a form of compensatory presynaptic plasticity at excitatory synapses in the CA1 region of adult hippocampus that is induced following pharmacological or genetic disruption of postsynaptic AMPARs. We provide multiple lines of evidence that this compensatory plasticity requires the action of postsynaptic NMDARs and is mediated by a coordinated, trans-synaptic expansion of active zone area, docked vesicle number and postsynaptic spine volume. Finally, we demonstrate that the compensatory modulation of excitatory transmission induces a parallel up-regulation of inhibitory neurotransmitter release. We propose a model that is consistent with expression of peripheral PHP, but which encompass expression mechanisms not observed at the NMJ including NMDAR-dependence as well as the coupling of excitatory and inhibitory synaptic transmission.
Results
We sought to selectively antagonize postsynaptic AMPARs in CA1 region of hippocampus. GYKI 53655 (hereafter referred to as GYKI) is a highly selective AMPAR antagonist (Frerking et al., 2001; Paternain et al., 1995). We determined the concentration-dependence of GYKI-mediated AMPAR antagonism in adult hippocampal brain slice (~P60-120) (Figure 1A–C). Acute application of 5μM GYKI partially antagonizes AMPAR-mediated mEPSC and stimulus-evoked EPSCs in the medial aspect of stratum oriens (SO) by ~60% (Figure 1B–D), without altering waveform kinetics (Supplemental Figure 1A).
Figure 1. Partial inhibition of AMPARs is accompanied by rapid compensatory recovery of excitatory neurotransmission at adult mouse CA1 synapses.
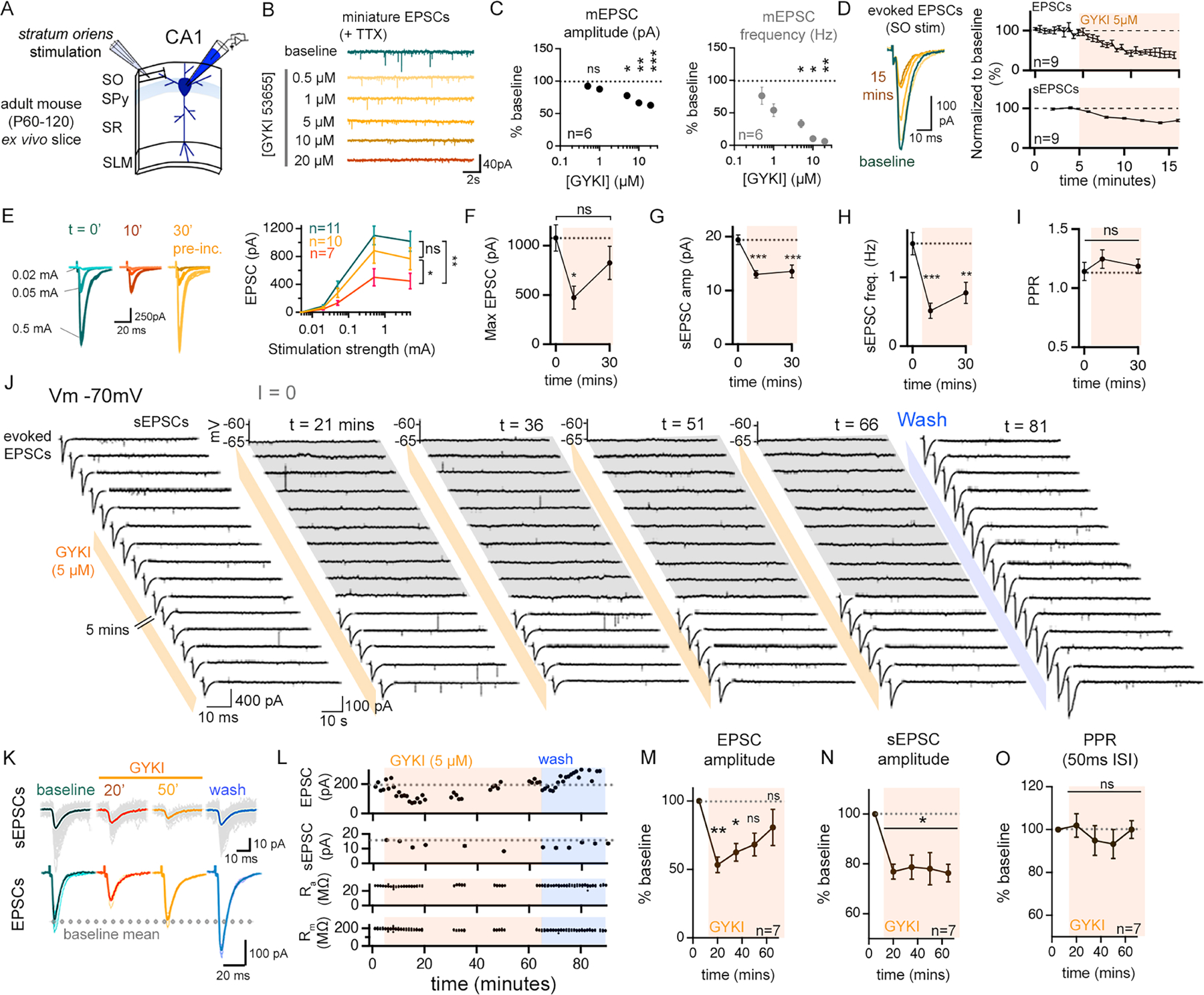
(A) Schematic of the recording configuration. SO, stratum oriens; SPy, stratum pyramidale; SR, stratum radiatum; SLM, stratum lacunosum moleculare. (B) Representative traces (TTX; 500nM), GYK concentration indicated (5-min per concentration). (C) Quantification for data as in (B), normalized to the baseline (zero GYKI). (D) EPSCs and sEPSCs during acute GYKI application. (E) Stimulus input/output in control (blue) and GYKI-treated slices. 10-minute data is paired to control (t=0) data. 30-minute data from separate GYKI pre-incubated slices. (F) Mean maximum EPSCs from (E). (G-H) Quantification for recordings as in (E). (I) Paired pulse ratios (PPR; 50ms ISI) from (E). (J) Representative traces of EPSCs (left) and sEPSCs (right) from a single continuous experiment. GYKI application indicated by orange bars, and wash-off by blue bars. Membrane potential was recorded in current clamp (I=0), indicated in grey. (K-L) Representative (grey traces; colour indicates mean waveform) synaptic events (K) and quantification (L) of EPSC and sEPSCs prior to GYKI wash-on (baseline), following GYKI wash-on (20 min, 50 min), and after wash-off. Membrane resistance (Rm) and access resistance (Ra) are stable (L, bottom). (M-O) Average EPSC amplitudes (M), sEPSC amplitudes (N), and PPRs (O) normalized to baseline.
n = # cells from at least 3 mice. Data are mean (± SEM). ns p>.05; * p<.05; ** p<.01; ***p<.001, one-way repeated measures ANOVA or Friedman test with Dunnett’s or Benjamini, Krieger, and Yekutieli post-hoc test (C, E, M, N, O), one-way Kruskal-Wallis ANOVA with Dunn’s post-hoc test (F-I).
In order to assess synaptic gain, we generated stimulus input/output curves by evoking EPSCs in the stratum oriens with progressively stronger stimulus intensities until EPSCs reached a plateau. Acute wash-on of GYKI for 10 minutes reveals the sub-blocking effect of GYKI (5μM) on plateau EPSC amplitude. Next, we pre-incubated slices for 30 minutes in GYKI (5μM) and recorded in the continued presence of GYKI (5μM). We find that input/output curves are significantly larger than the acute GYKI condition, but not significantly different from controls (Figure 1E–F). The recovery of plateau EPSC amplitudes toward baseline (Figure 1E, F) occurs despite a persistent decrease in sEPSC amplitude and frequency (Figure 1G, H). It is important to note that the adult hippocampal ex vivo slice preparation generally lacks spontaneous action potentials, rendering the sEPSC amplitude and frequency statistically identical to mEPSC amplitude and frequency (recorded in the presence of TTX) (Supplemental Figure 2E–H). Thus, we can use sEPSC amplitude to monitor the continued action of GYKI, ensuring sustained partial AMPAR antagonism.
In order to chart the recovery EPSC amplitudes in the continued presence of GYKI, patch recordings were achieved and sustained for up to 90 minutes allowing continual assessment of GYKI application and washout. An example recording (Figure 1J) demonstrates a rapid decrease of both spontaneous and evoked EPSC amplitudes following the application of GYKI (5μM). Over the next 30–40 minutes, EPSC amplitudes recover to baseline values in the continued presence of GYKI, whereas sEPSC amplitudes remain depressed (Figure 1J–N; Supplemental Figure 2A–D). We note that the recovery of EPSCs occurred without evidence of postsynaptic action potentials (Figure 1J). Finally, GYKI washout was achieved in a subset of recordings that we were able to sustain for the necessary length of time (>80min). Upon washout, sEPSCs recover towards baseline values while EPSC amplitudes potentiate above baseline (Figure 1J–L; wash-off sEPSCs, 95.9 ± 6.76% of baseline amplitude, EPSCs 134.4 ± 12.8% of baseline amplitude n= 4).
Two additional data sets are worth noting. Elevated excitability cannot account for the restoration of EPSC amplitudes (Supplemental Figure 1B–D). In addition, the recovery of EPSC amplitudes to baseline values occurs without a change in paired-pulse ratio (PPR) (Figure 1I, O). Changes in PPR are generally interpreted to reflect alterations in the presynaptic release mechanism (Dobrunz and Stevens, 1997; Zucker and Regehr, 2002). However, it is well established that PHP at the NMJ occurs without a change in PPR, an effect that is attributed to expansion of the readily releasable pool (RRP) of synaptic vesicles (Davis and Müller, 2015).
NMDAR-mediated synaptic currents are potentiated by AMPAR-specific antagonism
Because GYKI is an AMPAR-specific antagonist, it is possible to assess whether NMDAR-mediated synaptic currents are altered by partial GYKI-mediated AMPAR antagonism. We prepared control and GYKI pre-incubated slices and generated stimulus input-output curves (Figure 2A). To isolate NMDAR-mediated EPSCs we acutely blocked all AMPAR-mediated neurotransmission by bath application of NBQX (10μM; see Supplemental Figure 4F) immediately prior to assessing synaptic currents at a holding potential of +40mV in low extracellular Mg2+ (0.5 mM). We find that slices pre-incubated in GYKI (5μM for 30 mins) have significantly potentiated NMDAR-mediated input/output curves without a change in the amplitude or frequency of the underlying NMDAR-mediated mEPSC (Figure 2A,B; Supplemental Figure 3A–D). Notably, the potentiation of evoked NMDAR EPSCs occurs without a change in waveform kinetics or receptor subtype contribution (Supplemental Figure 3E–G). Finally, we show that the magnitude of AMPAR inhibition by GYKI is well correlated with the magnitude of the potentiated NMDAR-mediated EPSC (Figure 2C). Given that NMDAR EPSC amplitudes are potentiated without an underlying change in NMDAR mEPSCs, the data are consistent with GYKI inducing a compensatory increase in presynaptic release. This conclusion is supported by data demonstrating that pre-incubation of slices in either of two additional AMPAR-selective antagonists, perampanel (PMP) (Yelshanskaya et al., 2016) and JNJ55511118 (JNJ) (Maher et al., 2016) also drive the potentiation of NMDAR-mediated neurotransmission (Figure 2D, F). And, notably, the degree of AMPAR antagonism and NMDAR potentiation place the action of perampanel and JNJ55511118 on the same dose-response curve as GYKI (Figure 2F).
Figure 2. AMPAR-specific antagonism results in a graded potentiation of NMDAR-mediated neurotransmission.
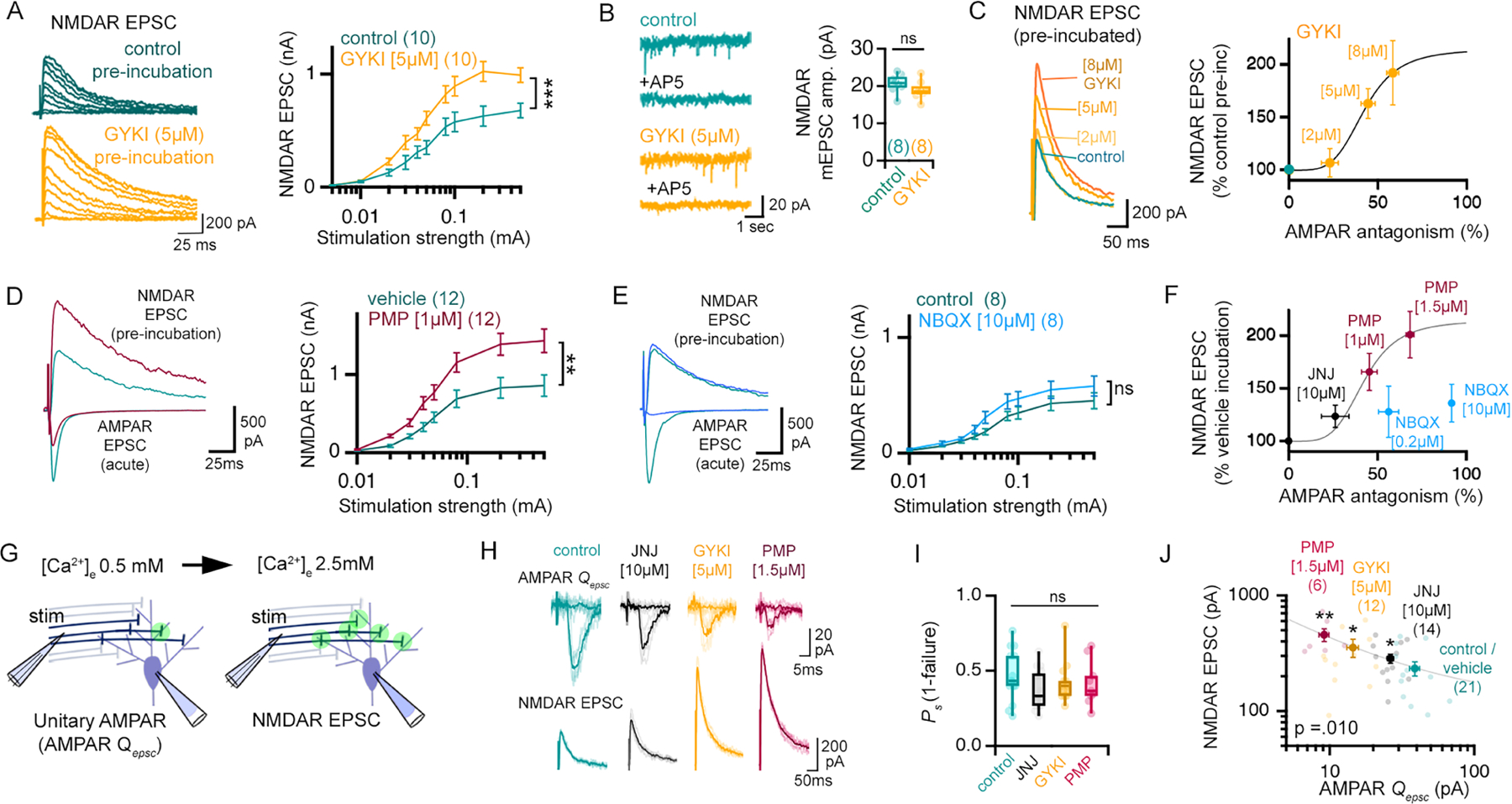
(A) Representative traces and input/output curves in control and 30-minute GYKI (5 μM), as indicated. (B) Representative NMDAR-mediated mEPSCs and quantification, GYKI incubation as in (A). (C) Normalized NMDAR-mediated EPSCs after GYKI pre-incubation (concentrations shown). Graph relates NMDAR EPSC following GYKI pre-incubation to AMPAR antagonism observed following acute GYKI wash-on (Hill coefficient = 4.45). (D) AMPAR EPSCs acquired in the absence (green) or immediately following PMP application (magenta). NMDA EPSCs following pre-incubation (30min) in ACSF (blue; 0.01% DMSO) or PMP (magenta). Stimulation input/output curves (right) acquired following 30-minute pre-incubation (PMP; 1 μM) or vehicle (0.01% DMSO). (E) Same as D but using NBQX (dark blue; 10 μM). (F) Normalized NMDAR-mediated EPSCs after pre-incubation with PMP, JNJ55511118 (JNJ), or NBQX compared to AMPAR-mediated EPSCs following acute wash-on of indicated drug concentrations. Curve is reproduced from (C). (G) Illustration of stimulus standardization paradigm. Green circles represent actively releasing synapses. (H) Unitary AMPA-mediated EPSCs and failures in 0.5mM [Ca2+]e / 2.5mM [Mg2+]e (top) and evoked NMDAR EPSCs in 2.5 mM [Ca2+]e / 0.5mM [Mg2+]e (bottom). (I) Probability of release success in 0.5mM [Ca2+]e / 2.5mM [Mg2+]e. (J) Single experiments (light markers) and means (dark markers) for each pre-incubation condition showing the relationship between Qepsc amplitudes (0.5mM [Ca2+]e), and evoked NMDAR EPSCs (2.5 mM [Ca2+]e). Pearson’s correlation (p=0.010).
n = # cells is shown in brackets in figure panels and were obtained from at least 3 mice. Data are mean (± SEM). ns p>.05; * p<.05; ** p<.01; ***p<.001 one-way repeated measures ANOVA (A, D, E) or Student’s t-test (B, I, J). Different drug treatments are always paired with vehicle (i.e. 0.01% DMSO for PMP) or control aCSF (i.e. water for GYKI) experiments.
We subsequently repeated experiments analyzing the effects of GYKI, perampanel, and JNJ55511118, using a unique method to normalize stimulation across cells and slices. We begin each recording in low external calcium (0.5mM) and adjust the placement of a fine-tipped, theta glass bipolar stimulus electrode to achieve a constant average failure rate of approximately 60% (Figure 2G–H; Supplemental Figure 4A; see methods). The preparation is then switched to high external calcium (2.5mM) and NMDAR-mediated EPSC amplitudes are assessed (as above). There are two advantages to this approach: 1) we normalize the stimulus to minimize cell-to-cell variability and 2) we attain estimates of failure rate and evoked unitary release event amplitude for each condition, designated hereafter as Qepsc (Figure 2H–I). It is important to note any possible expression of compensatory plasticity at low external calcium would cause us to under-estimate the magnitude of compensatory plasticity after switching to high calcium. We demonstrate that JNJ55511118, GYKI, and perampanel each significantly reduce Qepsc amplitudes under low external calcium and each drug potentiates the NMDAR-mediated EPSC recorded at high external calcium (Figure 2J). Notably, a statistically significant (p = 0.01) negative correlation exists between the magnitude of AMPAR antagonism and the potentiation of NMDAR-mediated EPSCs. These data underscore our conclusion that sub-blocking concentrations of AMPAR-specific antagonists induce a compensatory enhancement of presynaptic release.
Next, we addressed the action of a commonly used non-selective (non-NMDAR) AMPAR antagonist. The quinoxaline derivative, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX) antagonizes AMPA and kainate receptors (KARs) (Yu and Miller, 1995). In contrast to GYKI and perampanel, sub-blocking (0.2μM) and full-blocking (10μM) concentrations of NBQX only weakly potentiate NMDAR EPSCs (Figure 2E). Notably, these data points clearly reside off the dose-response curve populated by GYKI, perampanel and JNJ55511118 (Figure 2F). These data are consistent with a large literature using NBQX for electrophysiological assessments of synaptic transmission, which do not show evidence of rapid, compensatory plasticity of NMDAR currents.
NMDAR and KAR antagonists oppose compensatory plasticity following AMPAR antagonism
The differential activity of AMPAR-specific antagonists (GYKI, perampanel, JNJ55511118) versus NBQX prompted us to test whether kainate receptors (KAR) and/or NMDAR function might participate in the rapid induction of compensatory synaptic plasticity. For these experiments, we return to the measurement of AMPAR-mediated EPSCs, again using failure rates to normalize stimulation and assess unitary EPSC amplitudes (Qepsc) (Figure 3A–H). We then measure AMPAR-mediated EPSC amplitudes throughout the transition from low-to-high [Ca2+]e and refer to this as a ‘calcium input-output’ paradigm and the resulting graphs as ‘calcium input-output plots’. As expected, GYKI pre-incubation significantly reduced Qepsc amplitudes, while EPSC amplitudes are identical to controls at high [Ca2+]e (Figure 3B–D; see also Supplemental Figure 4B–D). By contrast, NBQX similarly diminished Qepsc amplitudes but EPSC amplitudes remain significantly smaller than control at high [Ca2+]e (Figure 3E). Once again, these data are consistent with GYKI inducing compensatory plasticity, while NBQX does not.
Figure 3. KAR and NMDAR antagonists impair the compensatory potentiation of AMPAR-mediated neurotransmission.
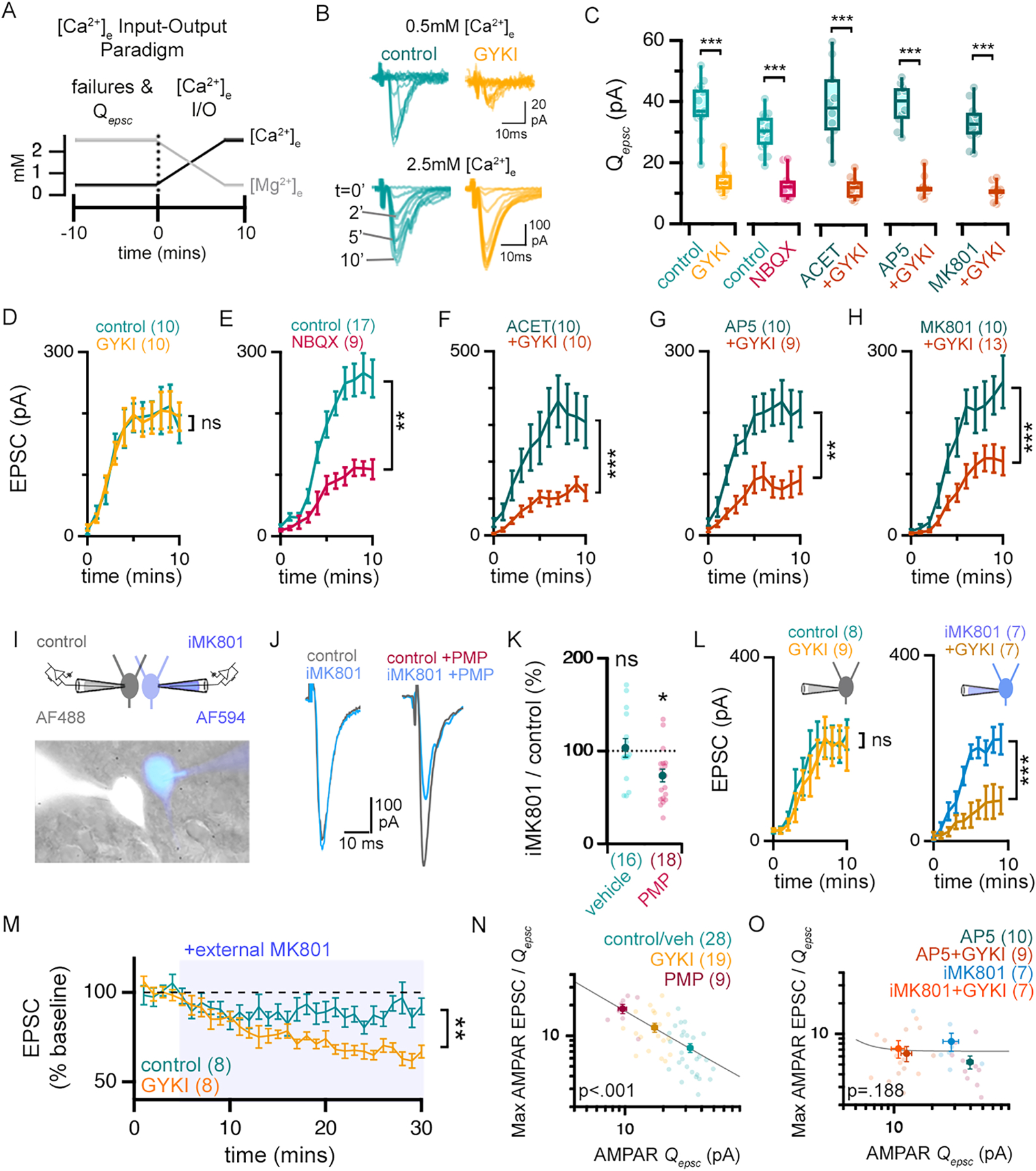
(A) Schematic of calcium input/output (I/O) paradigm. (B) Representative AMPAR-mediated currents in low [Ca2+]e (0.5mM) (top), and during 2.5mM [Ca2+]e wash-on (bottom) wash-on time indicated. (C) Average amplitudes of unitary AMPAR EPSCs (i.e. Qepsc) in the presence or absence of indicated antagonists. (D-H) EPSC amplitudes as a function of time following onset of high [Ca2+]e perfusion. (I) Recording configuration and image of patched cells (± iMK801 (1mM) and AlexaFluo488 (AF488) or AlexaFluo594 (AF594)). (J) Representative AMPAR-mediated EPSCs in control and iMK801-filled neurons after incubation in vehicle (0.01% DMSO; left traces) or PMP (0.5μM; right traces). (K) The relative effect of iMK801 on AMPAR-EPSC amplitudes (EPSCMK801/EPSCcontrol*100) as in (J). (L) Graphs as in (D) for control internal (left) or iMK801 internal solution (right). (M) EPSC amplitudes (normalized to baseline) during MK801 (20 μM) application (indicated, blue) following pre-incubation ± GYKI (5 μM, 30 min). (N-O) Single experiments (light markers) and means (dark markers) of each pre-incubation drug combination. Qepsc determined for each cell during 10-minutes in (0.5mM) calcium. Max AMPAR EPSC is the plateau average in the calcium I/O experiment. Data fit with a Power function.
n = # of cells shown in brackets and obtained from at least 3 different mice. ns p>.05; * p<.05; ** p<.01; ***p<.001 one-way ANOVA and Tukey’s multiple comparisons test (C), two-way repeated measures ANOVA (D-H, L, M), paired Student’s t-test two-tailed (K). P values in N&O are the result of Pearson’s correlation. Data are mean ± SEM except boxplots.
We then tested co-incubation of GYKI with NMDAR antagonists (MK801 or AP5) and with the KAR antagonist ACET (Dargan et al., 2009). Pre-incubation in MK801 (10μM), AP5 (20μM) or ACET (1μM) alone had little effect on either Qepsc amplitude or EPSC amplitude (Figure 3C–H). And, when co-incubated with GYKI, each of the three antagonists (MK801, AP5 or ACET) revealed a similar antagonism of Qepsc amplitude compared to GYKI alone (Figure 3C). However, compared to GYKI alone, each of these antagonists prevented EPSCs from reaching control levels at high [Ca2+]e.(Figure 3F–H). Similarly, co-incubation with ACET prevents the potentiation of NMDAR EPSCs induced by perampanel (Supplemental Figure 4G–H). Together, these data suggest that both NMDAR and KAR function are required for the restoration of EPSC amplitudes following GYKI antagonism.
A few additional points are worth noting regarding the potential contribution of postsynaptic NMDARs and KAR to measured EPSCs at resting voltages. Consistent with prior reports (Frerking and Nicoll, 2000), ACET-sensitive evoked currents in CA1 pyramidal cells are negligible (~4% of total EPSC amplitude) and do not change with GYKI treatment (Supplemental Figures 4F and 5). Similarly, a direct impact of AP5 and MK801 on measured EPSCs is unlikely since residual NMDAR-mediated currents at a Vm of −70mV are negligible (Supplemental Figure 4F).
To determine whether postsynaptic NMDARs are necessary for compensatory plasticity, we took advantage of the ability of MK801 to block NMDARs from the interior of the cell. We performed a dual patch experiment with one pipette containing MK801 (iMK801; 1mM) and the other without (control). EPSCs were measured simultaneously in both cells immediately following application of AP5 (20 μM) to isolate AMPAR-mediated neurotransmission (Supplemental Figure 4I). Evoked EPSCs are unaltered by the presence of postsynaptic iMK801. But, in the presence of GYKI, the iMK801 EPSCs are consistently smaller than the paired control (Figure 3J&K). We repeated this experiment using single patch electrodes that either contained iMK801 or not, and generated calcium input-output plots, in the presence or absence of GYKI pre-incubation. iMK801 alone had little effect, but prevented EPSCs from compensating back to control levels in the presence of GYKI (Figure 3L). Finally, we confirmed that bath application of MK801 reverses the potentiation of EPSCs after GYKI pre-incubation (Figure 3M). Together, these data support the conclusion that postsynaptic NMDARs are required for the maintenance of AMPAR-mediated neurotransmission after partial AMPAR antagonism.
Finally, we take advantage of the fact that we acquire both the average unitary amplitude (AMPA Qepsc) and maximal EPSC amplitude (AMPA EPSC-max) for each recording. We calculate the ratio of EPSC-max to Qepsc (a proxy for presynaptic release) and document a strong negative correlation between this ratio and the extent of Qepsc antagonism when data obtained with perampanel, GYKI, and control conditions are plotted together (Figure 3N; p<0.001). Importantly, this observed negative correlation is completely blocked in the presence of AP5 (bath application) or iMK801 (presented in patch pipette) (Figure 3O; p=0.188). The negative correlation is also blocked in the presence of ACET and when NBQX is substituted for GYKI (Supplemental Figure 4E). These data support the presence of a graded, compensatory response following postsynaptic AMPAR-specific antagonism.
Maintenance of synaptic gain following simultaneous postsynaptic depletion of GluA1, GluA2, GluA3
In order to unambiguously and persistently perturb postsynaptic AMPARs, we genetically depleted the three major contributing AMPAR subunits (GluA1, GluA2 and GluA3) from CA1 pyramidal neurons (Granger et al., 2011). To do so, we injected retrograde transporting adeno-associated viruses (i.e. retroAAVs) (Tervo et al., 2016) harboring GFP-Cre into the subiculum of the dorsal hippocampus of triple heterozygous floxed mice (GRIA1flx/wt,2 flx/wt,3 flx/wt) (Figure 4A–B). This strategy restricts the expression of Cre to postsynaptic CA1 pyramidal neurons (Figure 4C). AAVs expressing only GFP were used as control.
Figure 4. Genetic depletion of GluA1, GluA2, and GluA3 engages persistent synaptic compensation.
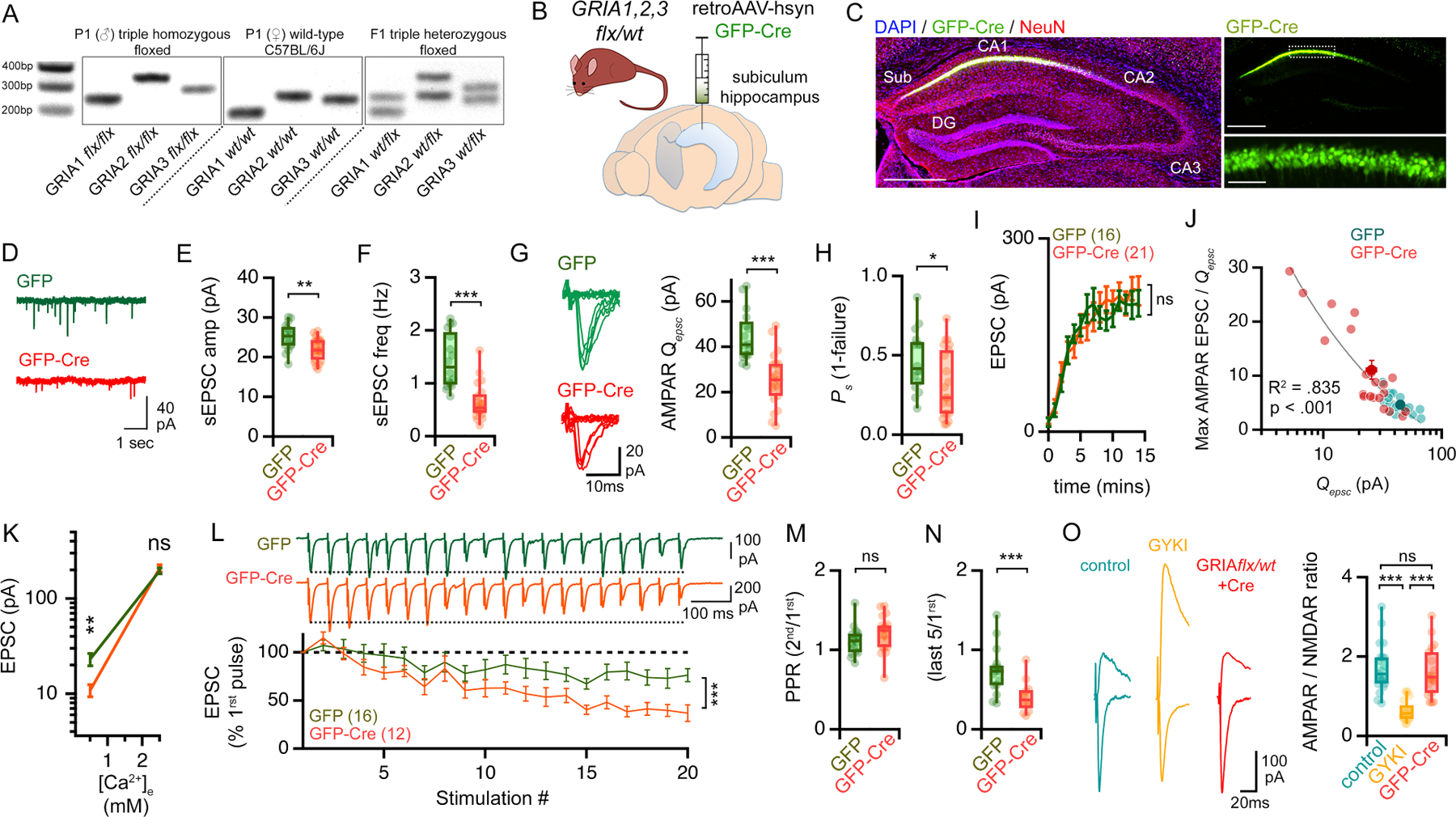
(A) Representative PCR-based genotyping results for triple heterozygous GRIA1, 2, 3 floxed mice. (B) Delivery of retrograde AAVs (retroAAV-hsyn;GFP-Cre) to the dorsal subiculum. (C) Images 4 weeks after virus injection. Slices immunolabeled anti-GFP (green) and neurons (NeuN; red). Nuclei are DAPI (blue). Scale bars are 500μm (left and top right) and 50 μm (bottom right). (D-F) Representative traces (D), average amplitude (E) and frequency (F) of sEPSCs recorded in GFP-Cre+ cells (red) and GFP controls (green). (G) Representative traces and amplitudes of Qepsc recorded in 0.5mM [Ca2+]e. (H) Success rates (Ps) of synaptic events for experiments shown in G, I & J. (I) Calcium input/output plots (transition from 0.5mM to 2.5mM [Ca2+]e). (J) Single experiments (light markers) and means (dark markers). Qepsc were determined for each cell during 10-minute in low (0.5mM) calcium. Max EPSC is the average plateau from calcium I/O. Data fit with a Power function. (K) Average of evoked EPSC amplitudes, including failures, at indicated [Ca2+]e. (L) Representative traces and quantification of average EPSCs (± SEM), normalized to EPSC1 (20Hz). (M-N) Paired pulse ratios (PPRs) (M) and the average of the last 5 EPSCs /1st (N). (O) Representative traces of AMPAR- and NMDAR-mediated EPSCs (left) and AMPAR/NMDAR ratios (right).
n = # of cells is shown in brackets and is obtained from at least 3 different mice. ns p>.05; * p<.05; ** p<.01; ***p<.001 Student’s t-test two-tailed (E, F, G, H, J, K, M, N), one way ANOVA and Dunnett’s multiple comparisons test, two-way repeated measures ANOVA (I, L).
One month after virus injection, Cre+ pyramidal neurons revealed smaller and less frequent spontaneous EPSCs compared to GFP+ controls (Figure 4D–F). In addition, Qepsc amplitudes recorded in low [Ca2+]e were smaller compared to Cre+ cells (Figure 4G–H). Thus, postsynaptic AMPAR function is impaired in the triple heterozygous knock-down condition. Next, we standardized our stimulation to achieve near equivalent failure rates in low [Ca2+]e and generated calcium input-output plots. EPSC amplitudes recorded at high [Ca2+]e were equivalent to controls (Figure 4I, K). We also document a significant negative correlation between the extent disruption of Qepsc amplitude versus estimated presynaptic release (ratio of EPSC-max to Qepsc ) (Figure 4J). And, consistent the induction of compensatory presynaptic potentiation, we observe enhanced synaptic depression in triple heterozygous neurons during prolonged stimulus trains (Figure 4L–N). Taken together, these data are consistent with compensatory potentiation of presynaptic neurotransmitter release following GluA1,2,3 depletion.
Finally, we assessed NMDAR-mediated transmission. Whereas GYKI or perampanel treatment strongly potentiates NMDAR-mediated EPSCs, genetic depletion of GluA1,2,3 does not (Figure 4O). This represents a fundamental difference comparing the acute versus persistent disruption of AMPARs. The absence of a change in NMDAR EPSCs is consistent with previously published data from hippocampal slice cultures examining homozygous GRIA1,2,3 mutant neurons (Lu et al., 2009). It is worth noting that different molecular mechanisms contribute to PHP the NMJ when neurotransmitter receptors are acutely antagonized versus genetically depleted (Harris et al., 2018; see below and discussion).
Enhanced presynaptic release offsets the magnitude of AMPAR perturbation.
Presynaptic vesicle release can be described as a binomial process (Korn et al., 1984; Malinow and Tsien, 1990; Saviane and Silver, 2007). As such, it can be adjusted via changes in either of the two binomial variables; ‘P’ inferred to represent the probability of vesicle fusion, or ‘N ‘ inferred to represent the number of functional release sites. In order to estimate these parameters, we employed a multiple probability fluctuation analysis (MPFA, see methods). We measured EPSC amplitude and variance at three concentrations of extracellular [Ca2+]e and [Mg2+]e, doing so in the presence or absence of GYKI pre-incubation. We fit a multinomial to the data in a mean-variance plot of EPSC amplitudes to extract the mean quantal amplitude (designated here as Qmpfa) and N (Figure 5C–E). GYKI treatment is associated with an expected significant decrease in Qmpfa (to 41.0 ± 2.3% of control values) (Figure 5D) as well as a significant increase in N (226.2 ± 34.1% increased compared to control) (Figure 5E). There is no change in estimates of P (Supplemental Figure 6G). Finally, the effect on N is blocked by MK801 (Figure 5A, B, G–J).
Figure 5. Compensatory plasticity achieved by increased release site number and expansion of the RRP.
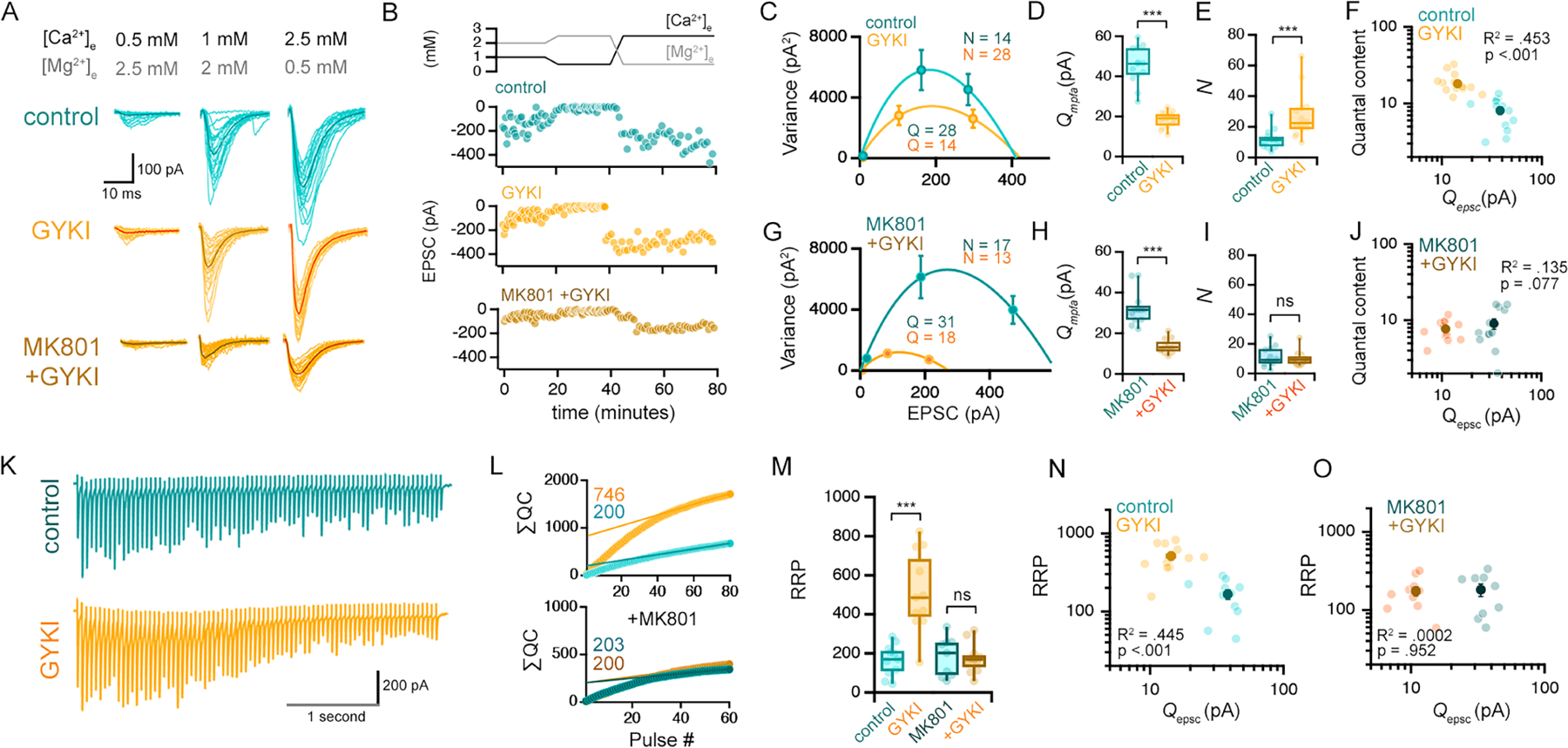
(A-B) Representative EPSCs for indicated drug and divalent concentrations (C) Representative mean EPSC vs. variance plots from a single experiment (see methods). N and Q values derived from the shown fits. (D-E) Quantal amplitudes (Qmpfa) and release site number (N) for all cells. (F) Relationship between estimated quantal content at 2.5mM [Ca2+]e (QC = EPSC2.5/Qmpfa) and Qepsc. Individual experiments (light markers) and mean ± SEM (dark markers) are shown. (G-J) Data as in (C-F), with MK801 (10 μM) in recording aCSF. (K) Representative traces (20Hz), 2.5mM [Ca2+]e recorded after MPFA (above). (L-M) Estimations for RRP size based on linear back-extrapolation of cumulative quantal contents (∑QC). (N-O) Relationship between RRP size and unitary AMPAR (Qepsc) amplitudes.
Control n = 13 cells from 12 mice; GYKI n =14 cells from 12 mice; Control + MK801 n =12 cells from 9 mice; GYKI + MK801 n=12 from 7 mice. *** p<.001. One-way ANOVA and Tukey’s multiple comparisons tests (M), two-tailed Student’s t-test (D, H), or Kruskal-Wallis ANOVA and/or Mann-Whitney non-parametric tests (E, I). R2 and P values in F, J, N, O are the result of Pearson correlations.
Next, we pursued a second method to estimate presynaptic release. We demonstrate a high degree of correlation between Qepsc and Qmpfa (correlation R2 = .835; Supplemental Figure 6H, I), attesting to the accuracy of these measurements. Since estimates of Qepsc and Qmpfa are achieved following axon stimulation at low [Ca2+]e, these values can be used to estimate presynaptic release (quantal content) when EPSCs are subsequently recorded following a shift to high [Ca2+]e. Using this approach we demonstrate that quantal content (QC = EPSC/Qmpfa) is consistently larger in GYKI-incubated slices than controls (Figure 5F). Importantly, the values of QC exhibit a strong negative correlation when plotted against Qepsc (Figure 5F; p < 0.001), again supporting the existence of compensatory plasticity. Finally, as predicted, the GYKI-dependent potentiation of presynaptic release is blocked by the presence of MK801 (Figure 5J; p = .077). Taken together, our statistical analyses support NMDAR-dependent compensatory potentiation of presynaptic release sites following AMPAR antagonism.
Homeostatic expansion of the readily releasable pool of synaptic vesicles
We estimated the size of the readily releasable pool (RRP) of synaptic vesicles using the ‘SMN’ model (Schneggenburger et al., 1999) (Figure 5L–N) (see methods). Immediately following an MPFA assay (above) and under conditions of high [Ca2+]e we provided stimulus trains (3–4 seconds at 20Hz). The amplitude and short-term dynamics of EPSC amplitudes in GYKI-incubated slices are similar to controls (Figure 5K; Supplemental Figure 6A). The data reveal a large, GYKI-dependent increase in the RRP (Figure 5L,M). Once again, we demonstrate that the NMDAR antagonist MK801 blocks potentiation of the RRP (Figure 5L,M). Because our estimates of RRP were obtained directly following an MPFA analysis (above) we are also able to plot RRP versus an estimate of quantal size Qepsc (Figure 3N,O) obtained at the same synapses. The data, once again, reveal a strong negative correlation that is blocked by MK801. Finally, we provide further validation of our RRP estimates using a separate model, the ‘EQ’ model (Elmqvist and Quastel, 1965). This alternative approach provides similar estimates of RRP potentiation following AMPAR antagonism (Supplemental Figure 6F).
Optical analyses confirm GYKI-dependent potentiation of glutamate release
We next turned to dissociated hippocampal cultures where individual presynaptic release events can be resolved in space and time using an optical reporter of presynaptic glutamate release (iGluSnFR-A184S) (Marvin et al., 2013). Optical events were collected across a field of view and synaptic release sites were visualized during action potential stimulation (Figure 6A, B). We restricted our initial analysis to presumed single synaptic boutons (diffraction limited ROI) in which stimulus-locked, action potential-mediated release events were obtained as well as spontaneous fusion events (Figure 6C–E). By normalizing average evoked event amplitudes to average spontaneous amplitudes, we achieved an optical estimate of quantal content (QC; Evoked/Spontaneous) at each synaptic ROI (Figure 6F). GYKI induced a significant potentiation of optical quantal content per ROI, as compared to CNQX (Figure 6F). When this analysis was repeated, determining the average QC per cell inclusive of all synaptic ROI within a field of view, we arrived at the same conclusion (Supplemental Figure 7D). We subsequently deployed the most recent version of GluSnFR3 (Aggarwal et al., 2022), which has greater photo-stability, enabling two additional analyses. We assessed the ratio of success to failures across a field of view, allowing calculation of release probability per ROI (Figure 6G). We also calculated a standard ∆F/F for all ROI (Figure 6H). Application of GYKI caused a significant increase in release probability and ∆F/F amplitudes per ROI compared to application of CNQX. Finally, two additional experiments were performed. First, MK801 (1μM) application to GYKI pre-incubated cultures causes a rapid depression of the integrated evoked GluSnFR signal (Figure 6I, J). Second, we co-incubated cultures with CNQX and GYKI. The presence of CNQX significantly reduces the effect of GYKI on release probability and abolishes the effect of GYKI on ∆F/F amplitude (Figure 6G, H). These data argue that CNQX antagonizes the compensatory modulation of presynaptic release, consistent with non-specific antagonism of KARs (see above). Taken together, these data further support the compensatory potentiation of presynaptic release following partial, AMPAR-specific antagonism.
Figure 6. Optical quantal analysis confirms an NMDAR-dependent potentiation of glutamate release.
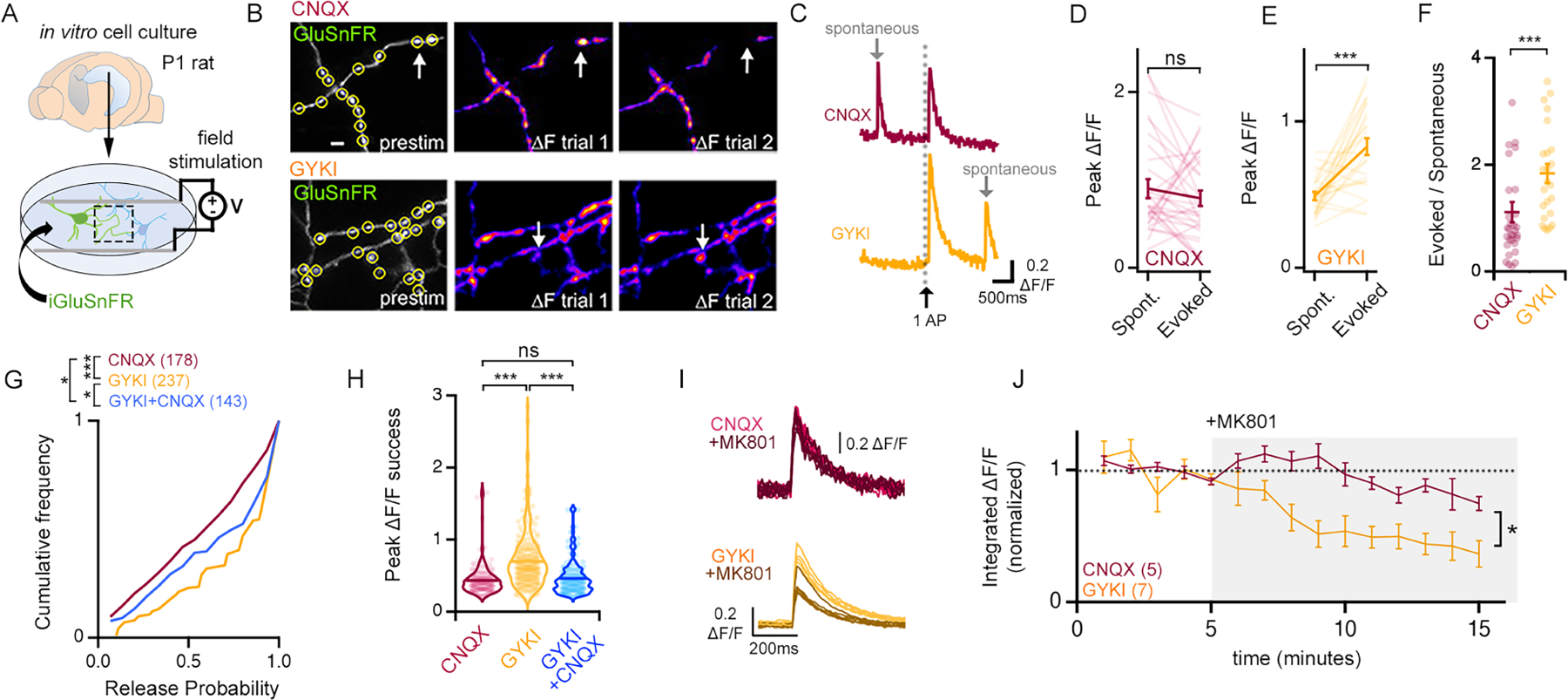
(A) Illustration of rat primary cell culture. (B) Example images of GluSnFR fluorescence in CNQX (10 μM) or GYKI (5 μM). Arrows identify boutons with stochastic glutamate release events. Scale bar is 2 μm. (C) Example spontaneous (grey arrow) and evoked GluSnFR ∆F/F waveforms following single APs (black arrow). (D-E) Individual (light lines) and mean ± SEM (dark lines) ∆F/F amplitudes of co-captured spontaneous and evoked GluSnFR events. CNQX n = 31 boutons, 8 cells; GYKI n= 25 boutons, 9 cells. (F) Optical quantal contents (Evoked/Spontaneous) of single boutons. (G) Cumulative distribution plots of the probability of a successful release event (see methods). (H) Average peak ∆F/F of successful events for each measured bouton. CNQX n = 178 boutons; GYKI n = 237 boutons; GYKI+CNQX n=143 boutons. (I-J) Single action potential evoked GluSnFR ∆F/F events (I) and average integrated ∆F/F ± SEM (J) before and during MK801 (1 μM).
ns p>.05; * p<.05; *** p<.001 Paired Student’s t-test two tailed (D, E) or two-tailed Mann-Whitney test (F), Kruskal-Wallis and Dunn’s multiple comparisons test (G, H), two-way repeated measures ANOVA (J).
AMPAR-specific antagonism drives synapse growth
We performed serial section transmission EM and analyzed 50μm x 50μm x 2μm volumes of stratum oriens. Samples were prepared from adjacent hippocampal slices (± GYKI in one animal and ± perampanel in a second) as per electrophysiology (see methods). Two methods were used to generate systematic and unbiased quantification. First, we reconstructed dendritic segments including spines, active zones and docked vesicles in control and GYKI treated samples (Figure 7D). Dendrites were chosen based upon: 1) orientation with respect to cell bodies and 2) maximal inclusion within the slice volume. Upon reconstruction, dendrite diameter, spine density, spine shapes and paucity of shaft-spines, are all consistent with the identification of secondary or tertiary dendrites of CA1 pyramidal neurons (Katz et al., 2009; Menon et al., 2013). A second method was applied to the perampanel treated samples. We identified every active zone present in a single plane at the mid-point of the 2μm EM volume (Figure 7B, red markers). Then, each synapse was fully reconstructed including spine, bouton, active zone and docked vesicle number. Examples of docked vesicles are shown for individual cross sections and a fully reconstructed bouton (Figure 7E, F). The two methods produced nearly identical estimates of average active zone area (GYKI control = 0.054μm2 vs. perampanel control = 0.051μm2; p=0.39, Student’s t-test, two tailed) and spine volumes (GYKI control = 0.031μm3 vs perampanel control = 0.037μm3; p=0.167, Student’s t-test, two tailed), and these estimates are quantitatively similar to data in the literature (Katz et al., 2009; Menon et al., 2013).
Figure 7. Serial-section reconstruction electron microscopy identifies NMDAR-dependent synaptic growth as a structural correlate of compensatory synaptic plasticity.
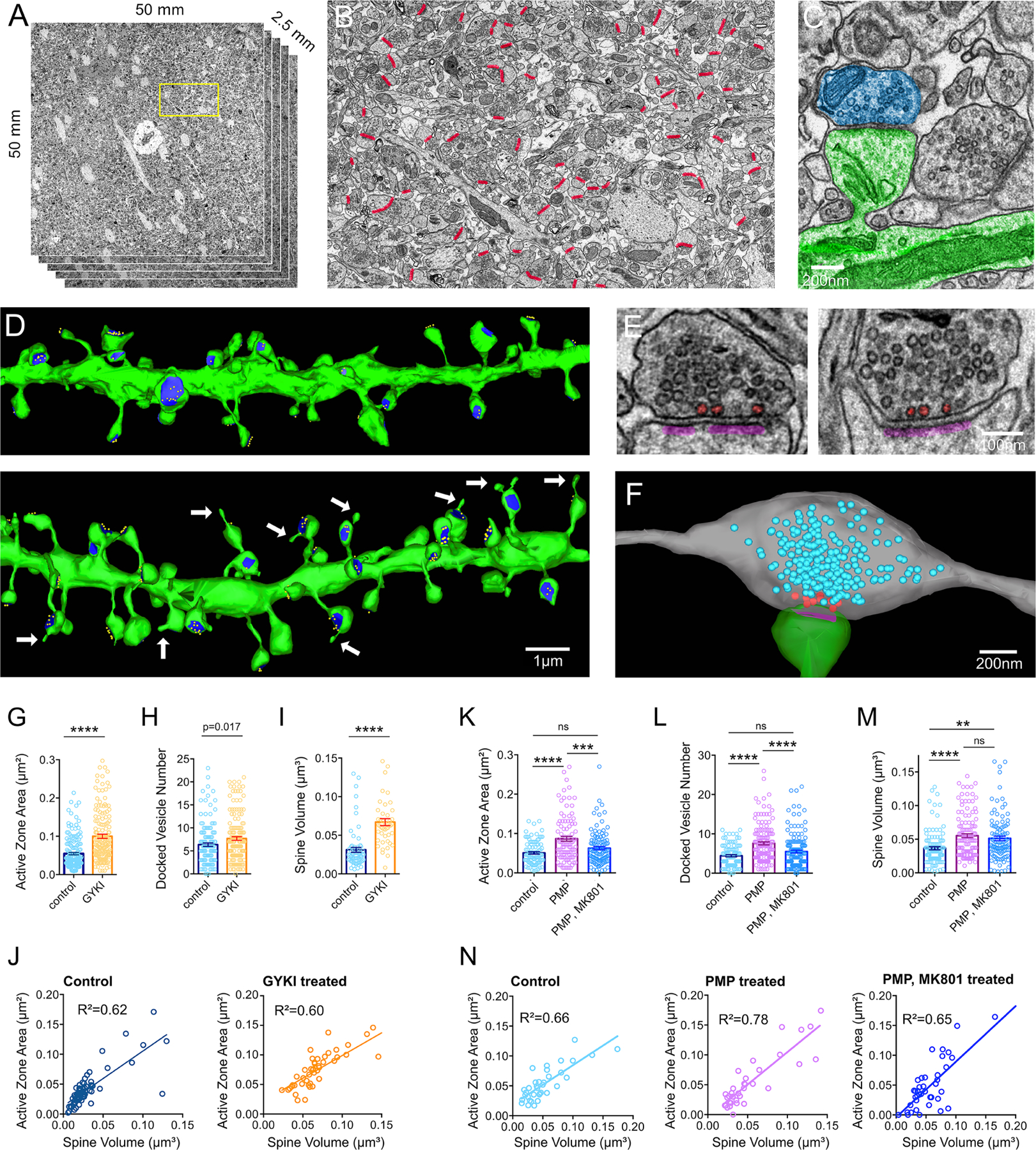
(A) Overview of ROI for hippocampal volumes. Yellow box indicates region in (B). (B) Mid-volume section with every active zone indicated (red). (C) Representative dendritic shaft and spine (green) with associated bouton (magenta). (D) Dendrites from serial sections (green), including active zones (blue) and docked vesicles (yellow) from acute brain slices incubated in control solutions (top) or GYKI (10μM) (bottom) for 30 minutes prior to fixation for EM. Arrows identify spine head protrusions. (E) Representative synaptic profiles showing postsynaptic densities (magenta) and docked vesicles (red). (F) Reconstructed bouton (grey) and spine (green) with postsynaptic density (magenta). Synaptic vesicles are blue, docked vesicles are red. (G-I) Active zone area (G), docked vesicle number (H), and spine volume (I) from slices ± GYKI (10μM), 30 minutes. (J) Spine volume versus active zone area. R2 values are from Pearson correlations. (K-N) Same as G-J, but for slices treated ± perampanel (PMP; 2 μM), and/or MK801 (10μM), or control (0.01% DMSO).
ns p>.05; ** p<.01; *** p<.001, **** p<0.0001 and p-values shown are the result of Student’s two-tailed t-tests (G-I) or one-way ANOVA and Bonferroni’s multiple comparisons test (K-M).
First, we present evidence of synapse growth in the presence of GYKI. We document abundant sprouting events, originating from spine heads (Figure 7D, white arrows). Quantitatively, sprouts occur in the presence of GYKI at a rate of 0.499/μm of dendrite length (n=10 dendrites, average 3.56 spines/μm) and were never observed in control (n=10 dendrites, average 3.80 spines/μm). We also document statistically significant increases in active zone area, docked vesicle number and spine volume in the presence of GYKI (Figure 7G–I). Measurements of linear spine length indicate that there is no significant change in spine neck length (GYKI vs control; p=0.07, Student’s t-test, two tails), demonstrating that the spine volume change is primarily due to expansion of the spine head. In addition, active zone area and spine volume remain highly correlated (Figure 7J). Finally, live two-photon imaging of dendritic segments in acute slices is consistent with a rapid phase of spine growth (Supplemental Figure 7F–I).
Next, we analyzed the effects of perampanel incubation, as well as co-incubation with perampanel plus MK801. The presence of perampanel induces an increase in active zone area, docked vesicle number, and spine volume that directly parallel the effects of GYKI (Figure 7K–M). Importantly, the presence of MK801 blocks the change in active zone area and docked vesicle number, but does not alter the perampanel-dependent increase in spine volume. Again, active zone area and spine volume remain correlated (Figure 7N). Taken together, these data suggest that AMPAR-specific antagonists initiate compensatory NMDAR-dependent synapse growth that is consistent with the observed NMDAR-dependent enhancement of presynaptic release and potentiation of the RRP (see also discussion).
AMPAR-specific antagonism rapidly potentiates inhibitory synaptic transmission.
GYKI is a 2,3-benzodiazapine with anticonvulsive activity (Donevan and Rogawski, 1993; Goulton et al., 2010; Zorumski et al., 1993). Perampanel is an anti-epileptic that was first developed to treat partial and tonic-clonic seizures (Frampton, 2015). Here, we demonstrate that sub-blocking concentrations of both drugs induce a compensatory potentiation of presynaptic release that rapidly restores excitatory transmission to baseline. This should obviate the anti-epileptic activity of these drugs. To investigate this discrepancy, we assayed inhibitory synaptic transmission.
We assessed both excitatory and inhibitory synaptic transmission under identical stimulus conditions for each recorded cell. To do so, we sequentially recorded synaptic activity while the cell was clamped at the reversal potential for synaptic inhibition (−70mV) followed by clamping at the reversal potential for synaptic excitation (0mV), and we did so in the presence or absence of GYKI (5μM) or perampanel (0.5μM) (Figure 8A, B). To ensure that we completely isolate synaptic inhibition, we added AP5 (20μM) and NBQX (10μM) to the recording chamber immediately prior to acquiring IPSC amplitudes. As expected, GYKI and perampanel decreased sEPSC amplitude and frequency without altering spontaneous sIPSCs (Figure 8C–E). As a consequence, the ratio of excitatory to inhibitory spontaneous amplitudes is significantly decreased (Figure 8F).
Figure 8. AMPAR antagonism initiates an NMDAR-dependent cross-modal potentiation of inhibitory neurotransmission.
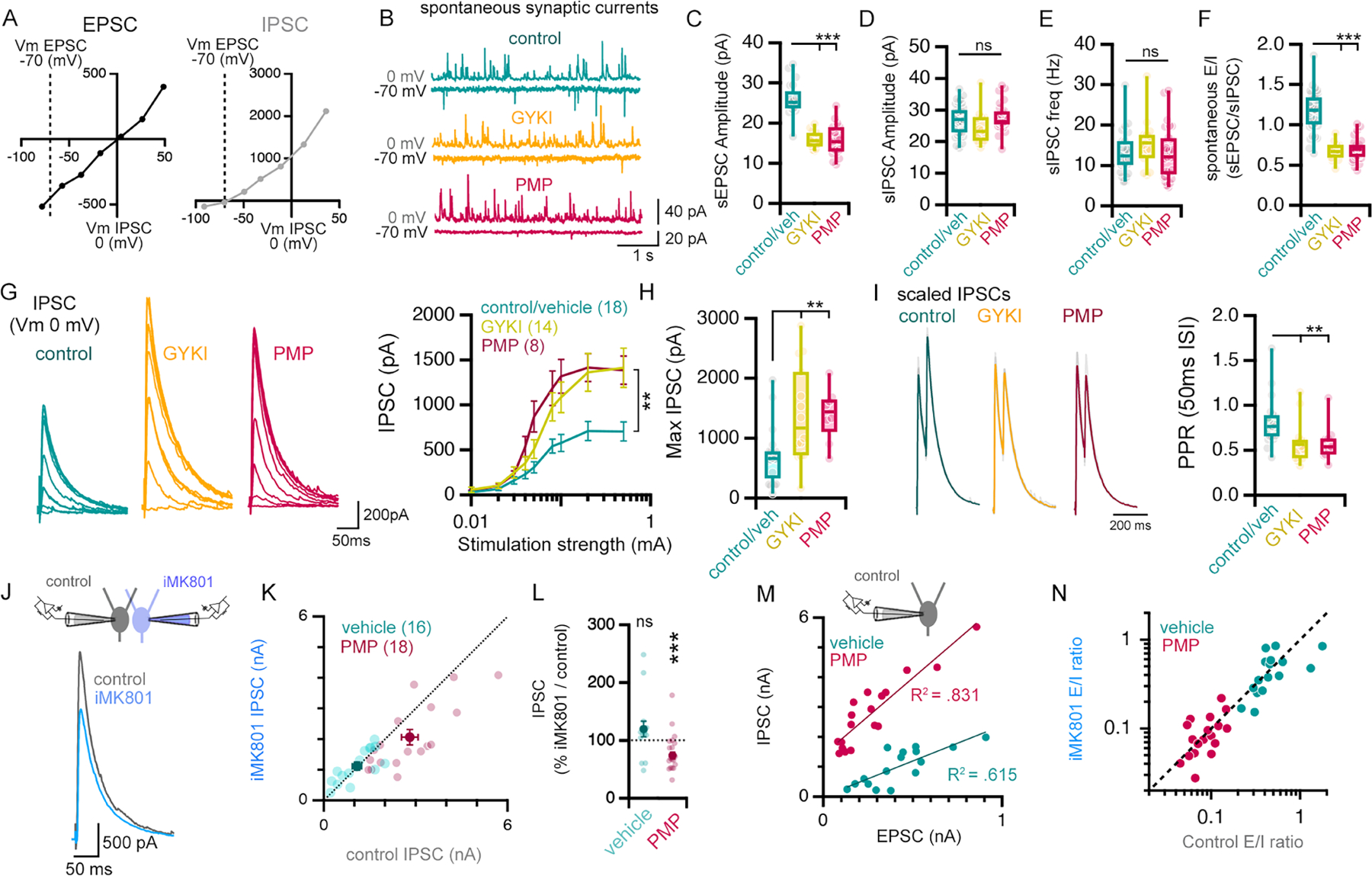
(A) Current/voltage relationships for EPSCs and IPSCs. (B-E) Representative traces (B), amplitude (C, D) and frequency (E) of recordings of spontaneous EPSCs and IPSCs. (F) The relative amplitude of spontaneous EPSCs and IPSCs (sE/I). (G) Representative traces of evoked IPSCs in response to a step increases in stimulation strength. n = # of cells from 10 (control), 7 (GYKI), and 3 (PMP) mice. (H) IPSCs evoked by maximal stimulation (mean of 0.1 and 0.5mA). (I) Paired pulse ratios (PPR) of IPSCs (50 ms inter-stimulus intervals), maximal stimulation (0.5mA). (J) Illustration of experimental design (top) and representative IPSC traces (bottom). (K) IPSCs measured in control- and iMK801-filled neurons after pre-incubation in vehicle (0.01% DMSO) or perampanel (PMP; 0.5μM). (L) Influence of iMK801 on IPSC amplitudes in pairs (IPSCMK801/IPSCcontrol*100), conditions as in (K). Control, n = 16 pairs from 3 mice; PMP, n = 18 pairs from 5 mice. (M) Correlation of excitation (AMPAR EPSC) and inhibition (IPSC) measured in slices pre-incubated in vehicle (0.01% DMSO) or PMP (0.5μM). R2 values are the result of Pearson correlations. (N) Excitation/inhibition ratio measured with control and iMK801 pipettes in PMP (0.5μM) or vehicle (0.01% DMSO).
ns p>.05; **p<.05, *** p<.01 One-way ANOVA or two-way repeated measures ANOVA and Tukey’s multiple comparisons test (C-G), Kruskal Wallis and Dunn’s post-hoc test (H, I), or two-tailed paired Student’s t-test (vehicle vs iMK801; L). Vehicle (0.01% DMSO) and control aCSF experiments are binned in (B-I).
Next, we generated stimulus input/output curves to assess action-potential evoked synaptic transmission. Remarkably, pre-incubation of slices in GYKI (5μM) or perampanel (0.5μM) caused a large, highly statistically significant increase in IPSC amplitudes compared to controls (Figure 8G–H). When we assessed paired pulse ratio for inhibitory synaptic transmission, the presence of GYKI or perampanel significantly reduced the PPR compared to controls (Figure 8I), suggesting that enhanced IPSC amplitude is caused, at least in part, by elevated presynaptic release probability. Thus, potentiation of evoked synaptic inhibition parallels the compensatory potentiation of release at excitatory synapses, the net effect being enhanced evoked synaptic inhibition and a decrease in E/I ratio.
Postsynaptic NMDARs are necessary for potentiation of inhibitory transmission
We asked whether the MK801-dependent block of compensatory plasticity also disrupts potentiation of inhibitory synaptic transmission. We employed a dual patch clamp approach and recorded simultaneously from a cell with normal internal pipette solution (control) and a cell with iMK801 (1mM) (Figure 8J). Excitatory neurotransmission was first sampled and is presented (above) in Figure 3J and K. Here, we present the subsequent analysis of inhibitory neurotransmission, which was isolated by the addition of NBQX (10μM) to the recording bath, allowing IPSCs to be sampled at 0mV. When recording from aCSF pre-incubated slices, equivalent IPSCs are recorded in the control and iMK801 pipettes and data reside on a line of unity (Figure 8K, L). However, perampanel pre-incubated slices demonstrate a large potentiation of IPSC amplitudes in the control pipette, an effect that is suppressed in the iMK801 pipette such that the data fall off the line of unity (Figure 8K, L). In Figure 8M, we demonstrate that EPSC and IPSC amplitudes remain highly correlated when assayed within single pyramidal neurons, but IPSC amplitudes are strongly potentiated in the presence of perampanel.
Finally, we plot E/I ratios comparing control and perampanel pre-incubated slices, assessing how these ratios are affected by the presence of iMK801. Data recorded from perampanel pre-incubated slices (pink) are completely separated from those recorded in ACSF (green), yet remain on a line of unity (Figure 8N). How is this explained? First, perampanel application initiates the restoration of excitatory EPSCs and simultaneous potentiation of IPSCs, with a net effect of diminished E/I ratio. Then, consider that MK801 blocks the permapanel-dependent potentiation of both EPSCs and IPSCs. So, in the presence of MK801, perampanel also leads to diminished E/I ratio. Since E/I ratio is diminished for conditions plotted on both the X and Y-axes, the net effect is to shift data to the left along the line of unity. The complete separation of data (green from pink) is a clear demonstration of effect size, demonstrating the potent capacity of compensatory plasticity to simultaneously adjust both excitatory and inhibitory synaptic transmission.
Discussion
We demonstrate that pharmacological or genetic perturbation of postsynaptic AMPARs initiates an offsetting, compensatory potentiation of presynaptic neurotransmitter release and synapse growth. This form of compensatory plasticity shares fundamental characteristics with presynaptic homeostatic plasticity (PHP) documented at peripheral synapses of flies, mice, and humans (Davis, 2013), including a rapid induction phase (minutes) induced pharmacologically, and a persistent phase (days to months) induced by genetic disruption of postsynaptic AMPARs. Additional parallels include expression via potentiation of the RRP and a significant negative correlation between postsynaptic receptor antagonism and enhanced presynaptic release. However, we also define novel elements compared to the NMJ including a requirement for postsynaptic NMDARs (see below for further discussion) and an expression mechanism that includes trans-synaptic growth regulation, defined ultrastructurally. Thus, we propose the existence of hippocampal PHP with expression mechanisms that are unique to central synaptic circuitry.
A novel role for NMDARs
In all of our assays, MK801 strongly impairs hippocampal PHP. How do NMDARs participate given the absence of substantive postsynaptic depolarization to relieve a Mg2+ block (Figure 1)? Previously, residual Ca2+ influx at the spine head was documented under conditions of AMPAR blockade and normal resting membrane potentials during single synapse activation (Bloodgood et al., 2009). This NMDAR-dependent, AMPAR-independent Ca2+ influx could represent a source of signaling that contributes to NMDAR-dependent presynaptic homeostatic plasticity. If so, the phenomenon identified by Boodgood and colleagues (2009) might represent a signaling capacity of NMDARs that is essential for the induction of PHP following AMPAR antagonism.
Preservation of short-term plasticity
The rapid potentiation of presynaptic release during hippocampal PHP occurs without a change in paired-pulse plasticity, a finding that is in agreement with work in peripheral neuromuscular synapses (Ortega et al., 2018; Wang et al., 2016; Weyhersmüller et al., 2011). We show that hippocampal PHP is achieved through a change in the number of neurotransmitter release sites, represented statistically as an increase in the binomial parameter ‘N’, and represented ultra-structurally as an increase in the number of docked synaptic vesicles and active zone area. This mechanism of expression is not predicted to alter paired-pulse plasticity (Ortega et al., 2018). This feature of compensatory plasticity seems particularly relevant to the stabilization of information transfer within neural circuits (Abbott and Regehr, 2004; Davis and Murphey, 1994; Tsodyks and Markram, 1997).
Genetic induction
We observe differences comparing pharmacological and genetic induction of hippocampal PHP. Most notably, NMDAR-mediated neurotransmission is unchanged by persistent GluA1,2,3-depletion, but is strongly potentiated by pharmacological AMPAR antagonism. Two explanations seem plausible. GluA1,2,3-depleted synapses may not physically enlarge, perhaps due to limited AMPAR availability. As a consequence, PHP may be primarily achieved via enhanced release probability (Supplemental Figure 8). Alternatively, active zone growth during the early stages of PHP may be transient, eventually resolving such that synapses solely express enhanced presynaptic release probability. In support of this possibility, we note that different phases of structural LTP have been documented, including early spine expansion followed by spine contraction (Sun et al., 2021). Further, we note that different mechanisms can be engaged during the induction versus long-term expression of diverse types of neural plasticity including PHP at peripheral synapses (Harris et al., 2017) and LTP in the mammalian CNS (Nicoll, 2017; Bliss and Collingridge, 2013; Chang et al., 2017; Bosch et al., 2014). Accordingly, we postulate that compensatory plasticity in hippocampus, identified here, may transition between mechanisms responsible for the rapid induction versus sustained expression.”
We also note that a previously published study examined GluA1,2,3 triple homozygous knockouts, as well as double knockouts combinations, using a neonatal slice culture preparation. There was no evidence of compensatory presynaptic plasticity (Lu et al., 2009). Several points are worth considering, beyond the fact that neonatal slice cultures differ substantively from the acute adult slice preparation used here. First, it is possible that a complete knockout of individual receptor subunits actually blocks compensatory plasticity. Consistent with such a possibility, other forms of compensatory plasticity including ‘synaptic consolidation’ and quantal scaling require the GluA2 receptor subunit (Gainey et al., 2009; Levy et al., 2015). It is also possible that compensatory presynaptic changes do occur in double and triple GluA knockouts, but remain difficult to assess due to the large decreases in AMPAR-mediated transmission (Lu et al., 2009) For example, at the mossy fiber synapse in cerebellum, knockout of the sole postsynaptic GluA subunit (GluA4) abolishes AMPA-mediated postsynaptic currents and invokes compensatory presynaptic changes, but documenting the presynaptic changes required measurement of presynaptic capacitance and calcium conductance (Delvendahl et al., 2019), assays that are extremely challenging at the smaller en passent synapses in hippocampus.
Cross-modal potentiation of inhibitory neurotransmission
During PHP, the cross-modal potentiation of inhibitory synaptic transmission occurs downstream of postsynaptic NMDARs within individual CA1 neurons, arguing for the spread of a hetero-synaptic signaling mechanism. The potentiation of inhibitory synaptic transmission is most likely expressed as an increase in presynaptic release probability, as evidenced by a change in paired-pulse plasticity. Importantly, recent data have defined a similar connection between the activation of postsynaptic NMDARs and potentiation of inhibitory synaptic transmission (Chiu et al., 2018; Gu et al., 2016). We postulate that similar signaling mechanisms could be engaged to link the cross-modal modulation of excitation and inhibition, regardless of whether this signaling is initiated by the induction of homeostatic versus Hebbian plasticity.
Ultimately, these data raise important questions regarding the stabilization of neural circuitry, higher brain function and animal behavior. We note that hippocampal PHP is defined by the restoration of excitatory synaptic gain and does not, necessarily, ensure stabilization of neural circuit function. For example, it has been postulated that homeostatic modification of excitatory synaptic transmission could induce maladaptive effects associated with altered circuit function and disease (Bourgeron, 2015; Davis, 2006; Davis and Goodman, 1998). On the other hand, the complexity necessary to stabilize animal behavior following a perturbation is likely to require coordination among many homeostatic different homeostatic signaling systems such as those that stabilize synaptic gain, neuronal excitability and neural circuit activity. Finally, as suggested previously, it remains plausible that some elements of neural circuits are simply not under homeostatic control (Davis, 2006).
Star Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Graeme W. Davis (Graeme.Davis@ucsf.edu).
Materials Availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate standardized datatypes for public repositories. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines and knockout genetics
Male and female C57BL6/J (IMSR_JAX:000664) were obtained as adults (8–10 weeks old) from The Jackson Laboratory and used for experiments between the ages of P60-120.
Male and female Thy1-GFP-M (IMSR_JAX:007788) were obtained from the UCSF Mouse Inventory Database and bred in-house on a C57BL6/J background and used for experiments between the ages of P60-120. Genotyping of transgenic mice was performed via Transetyx automated genotyping service (https://www.transnetyx.com) on genomic DNA isolated from ear biopsies using the following primers: 5’-CCACAGAATCCAAGTCGGAACTC-3’ and 5’-CTCGCCCTTGCTCACCAT-3’.
Triple floxed GRIA1flx/flx, GRIA2flx/flx, GRIA3flx/flx (GRIA1,2,3flx/flx) mice were described previously (Lu et al., 2009) and were obtained as a kind gift from Dr. R. Nicoll at the University of California, San Francisco. Male GRIA1,2,3flx/flx were bred with female C57BL6/J to obtain GRIA1,2,3flx/wt progeny that were used for experiments. Genotyping of transgenic mice was performed using PCR on genomic DNA isolated from ear biopsies using the following primers: GRIA1flx, 5’-CAC TCA CAG CAA TGA AGC AGG AC-3’ and 5’-CTG CCT GGG TAA AGT GAC TTG G-3’. GRIA2flx, 5’-GCG TAA GCC TCT GAA ATA CCT-3’ and 5’-GTT GTC TAA CAA GTT GTT GAC C-3’. GRIA3flx, 5’-CCA ATG TTG TTT AGC CTT TGC-3’ and 5’-GGT ATA TCT TCC CAG CCC CAA G-3’.
All experiments were matched to mouse age and sex. All procedures were performed in accordance with UCSF (protocol # AN108729-02B) IACUC guidelines.
Rat primary neuron cultures
Sprague-Dawley rats of either sex (mixed litter) at P1 were used for the preparation of primary hippocampal neuron cultures. All procedures were performed in accordance with UCSF (protocol # AN108729-02B) and Dartmouth College (protocol # 00002115) IACUC guidelines.
METHODS DETAILS
Acute slice electrophysiology
All experiments were matched to mouse sex, brain hemisphere, and rostral-caudal slice position across experimental conditions. Briefly, mice were deeply anesthetized with isoflurane and transcardially perfused with ice-cold cutting aCSF solution containing (in mM): 93 N-methyl D-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 20 glucose, 5 Na ascorbate, 2 thiourea, 3 sodium pyruvate, 12 N-acetyl L-cysteine, 10 MgSO4, 0.5 CaCl2, pH adjusted to 7.4 with HCl and bubbled with 95% O2 / 5% CO2, ~300 mOsm. Brains were extracted, blocked, and fixed to the cutting stage with Vetbond tissue adhesive positioned at a ~30–40° angle from horizontal along the rostral/caudal axis using a 4% agar block. 350 μm transverse hippocampal sections of were obtained in ice-cold cutting aCSF with a ceramic blade (Cadence blades #EFINZ10), and a Leica VT1200 vibrating microtome. Hemispheres were separated and small cuts were made near the CA2/CA1 border in each hippocampus to prevent recurrent activity. Slices were incubated for 12 minutes in cutting aCSF warmed to 34°C, then placed in holding aCSF solution containing (in mM) 81.2 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 20 D-glucose, 5 Na ascorbate, 2 thiourea, 3 sodium pyruvate, 12 N-acetyl L-cysteine, 2 MgSO4, 2 CaCl2, pH 7.4, bubbled with 95% O2 / 5% CO2, ~300 mOsm at room temperature (~20°C) for up to 8 hours until used in experiments.
Whole-cell patch clamp recordings were obtained from CA1 pyramidal neurons using an Olympus BX51W1 microscope equipped with IR-DIC optics and a motorized stage. Pyramidal neurons were visually identified by their large cell bodies and position within the pyramidal cell layer. Post hoc visualization of cell morphology confirmed pyramidal cell identity in a subset of experiments (not shown). Voltage clamp and current clamp experiments were carried out using Multiclamp 700B amplifiers and Clampex10.7 acquisition software (Molecular Devices). Analysis was performed using Clampfit10.7 software. Patch pipettes (borosilicate glass, OD 1.5mm, ID 0.86mm, tip resistance 2–4 MΩ) were pulled using a Sutter P-97 micropipette puller. Slices were constantly perfused with recording aCSF at 1.5–2mL/min containing (in mM) 119 NaCl, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2, 20 D-glucose and 0.5 Na ascorbate pH 7.4, bubbled with 95% O2 / 5% CO2, ~295–305 mOsm and maintained at 32–34°C using an in-line heater (Harvard Instruments). Picrotoxin (100 μM; Tocris #1128) was added to the recording aCSF to block GABAA-receptors and isolate glutamatergic synaptic transmission unless otherwise stated. Internal pipette solution contained (in mM) 130 CsMeSO3, 8 NaCl, 4 Mg-ATP, 0.3 Na-GTP, 0.5 EGTA, 10 HEPES, pH 7.3, 5 QX314-bromide (Tocris #2555), ~290–295 mOsm, liquid junction potential ~−12mV or 142 K-gluconate, 10 HEPES, 1 EGTA, 2.5 Mg2Cl, 4 Mg2-ATP, 0.3Na3-GTP, 10 Na-phosphocreatine, pH 7.3, 290–295 mOsm, liquid junction potential ~ −8mV. In some experiments MK801 (1mM, Tocris #0924) was added to the internal solution. Liquid junction potentials were corrected for experiments measuring excitation and inhibition in parallel (i.e. Figure 8). Patch solutions were allowed >10 minutes to equilibrate through the cell before experiments were performed. Pipette series resistances were ~20 MΩ and were compensated by ~30–60% in some experiments to achieve Rs values of <10 MΩ (see below). Experiments in which uncompensated Rs was >30 MΩ or changed by >20% were discarded. For experiments that required sampling EPSCs in a high external calcium concentration (2.5mM [Ca2+]/0.5mM [Mg2+]) slices were first placed into a recording aCSF that contained low concentrations of calcium and high concentrations of magnesium (0.5mM [Ca2+]/2.5mM [Mg2+]). This approach was used to protect against excitotoxicity and the induction of use-dependent synaptic plasticity. After patch formation the recording aCSF was quickly switched to one containing high concentrations of calcium and low magnesium (2.5mM [Ca2+]/0.5mM [Mg2+]). EPSC were sampled and reached a plateau after ~10–15 minutes (i.e. Figure 3D–H; see descriptions of calcium input/output experiments below).
AMPAR antagonists; preincubation experiments
Slices were immersed in recording aCSF (2mM [Ca2+]/1mM [Mg2+]) bubbled with 95% O2/ 5% CO2 and warmed to 34° C and containing 2, 5, 8, or 10 μM GYKI 53655 (Tocris #2555), 0.1–10 μM NBQX (Tocris #1044), 0.5–1.5 μM perampanel (Adooq Bioscience #A12489), or 10μM JNJ55511118 (Tocris # 6278) for 10–30 minutes. Where indicated, the NMDAR-specific antagonists AP5 (20 μM; Tocris # 0106), or MK801 (10μM; Tocris # 0924), or the KAR-specific antagonist, ACET (10μM; Tocris # 2728) were incubated along with GYKI or perampanel. Control experiments were always conducted with slices that had been incubated in the same warmed aCSF solutions lacking drug for the same duration. For drugs that were dissolved in DMSO (i.e. perampanel; JNJ55511118), the same volume of DMSO (i.e. 0.01%) was used as controls (i.e. vehicle condition). Experimental and control experiments were always interleaved and hemispheres were paired. After pre-incubation, slices were placed in the recording chamber and continually perfused with control or drug-containing solution in the presence of picrotoxin (100 μM), unless otherwise stated, at 32–34°C for an additional ~10–15 minutes before whole cell patch formation.
EPSC input/output experiments
Electrical input/output stimulation experiments were performed in physiological concentrations of extracellular calcium and magnesium (2mM [Ca2+]e/1mM [Mg2+]e) and in the presence of picrotoxin (100 μM). Patch pipettes were filled with K-gluconate-based internal solutions and pipette series resistance was left uncompensated (Rs control mean 20.76 ± 1.35 MΩ, n=11; GYKI mean 23.76 ± 0.09 MΩ, n=10). Only stable experiments (<20% change from baseline Rs) were analyzed. Monopolar tungsten wire stimulation electrodes were inserted into the opposite end of pulled patch electrodes (borosilicate glass, OD 1.5mm, ID 0.86mm, tip resistance 2–4 MΩ), filled with aCSF and placed in the proximal/medial aspect of the stratum oriens ~100 μm from the pyramidal cell layer and ~200μm from the perpendicular axis of the patched cell at a depth of ~20–50 μm. One pole of the stimulus isolation unit (A.M.PI. ISO-Flex; Jerusalem, Israel) was connected to the tungsten monopolar stimulation electrode, and the other was connected to a AgCl ground wire inserted into the recording bath. Stimulus strength was increased in small increments ranging from 0.005–5mA, pulse duration remained constant (0.1ms). Excitatory postsynaptic currents (EPSCs) were evoked once every 30 seconds (i.e. 0.0333 Hz) or once every minute (0.0167Hz). Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded during the intervals between evoked stimuli and measured using the template matching algorithm in Clampfit10.7 software.
GYKI wash-on and EPSC recovery experiments
Experiments that involved the acute application of GYKI and long duration monitoring of EPSCs (i.e. Figure 1J–O) were performed in physiological concentrations of calcium and magnesium (2mM [Ca2+]e/1mM [Mg2+]e) and in the presence of picrotoxin (100 μM). Patch pipettes were filled with K-gluconate-based internal solutions described above. Pipette series resistance was left uncompensated (mean 25.06 ± 0.89 MΩ, n=16). Only stable experiments (<20% change from baseline Ra) were analyzed. The positive and negative poles of a stimulus isolation unit (A.M.PI. ISO-Flex; Jerusalem, Israel) were connected to two tungsten wire stimulation electrodes and inserted into each barrel of theta glass pipettes (borosilicate glass, OD 1.5 mm, ID 1.00 mm, SEP 0.2 mm, tip diameter ~ 1–3 μm) and filled with aCSF. Theta glass stimulation electrodes were placed in the proximal/medial aspect of the stratum oriens ~100 μm from the pyramidal cell layer and ~200μm from the perpendicular axis of the patched cell at a depth of ~20–50 μm. The stimulus strengths were maximal (0.5mA/0.1ms) and were kept constant throughout the experiment. Stable EPSCs were achieved by carefully adjusting the position of stimulation electrodes during a pre-experiment baseline sampling period. Once stable responses were achieved, the position and strength of the stimulation electrode was fixed and not further altered for the duration of the experiment. EPSCs were sampled once every minute (i.e. 0.0167Hz) in voltage clamp mode (Vm −70mV) for 20 mins during GYKI application, then in 5-minute segments, interspersed by 10-minute intervals during the recovery period. Cells were unclamped and allowed to fluctuate around their membrane potentials during these 10 minute ‘rest’ periods. Recorded cells had stable membrane potentials, access resistance, and membrane resistances throughout the duration of the experiment. Only cells in which stable recordings were achieved for at least 45 minutes and where sEPSC events could be clearly resolved above noise were included in analysis (n=16). For a subset of cells accepted for subsequent analysis (n=7), the amplitude of sEPSCs rapidly depressed upon GYKI wash-on and remained stable throughout the remaining duration of the recording (Figure 1M; sEPSCt=20mins = 76.86 ± 2.99% of baseline, sEPSCt=45mins = 78.06 ± 6.48% of baseline, p= .908 two-tailed paired Student’s t-test; n=7 out of 16 cells). Conversely, the amplitude of sEPSCs in the remaining cells continued to decline throughout of the duration of the recording (sEPSCt=20mins = 80.06 ± 3.12% of baseline, sEPSCt=45mins = 69.35 ± 1.46% of baseline, p= .0015 two-tailed paired Student’s t-test; n=9 out of 16 cells), suggesting progressive inhibition of AMPARs by GYKI or possible run-down of postsynaptic responses. Because we could not interpret the effects of a progressively changing baseline, these cells were omitted from further analysis.
Isolation and measurement of NMDAR currents
To isolate NMDAR-mediated EPSCs we acutely blocked all AMPAR-mediated neurotransmission by bath application of the AMPAR/KAR antagonist, NBQX (10μM; Supplemental Figure 4F) immediately before measurements of NMDAR EPSCs at a holding potential of +40mV in low extracellular Mg2+ (0.5 mM). Monopolar tungsten wire stimulation electrodes were inserted into the opposite end of pulled patch electrodes (borosilicate glass, OD 1.5mm, ID 0.86mm, tip resistance 2–4 MΩ), filled with aCSF and placed in the proximal/medial aspect of the stratum oriens ~100 μm from the pyramidal cell layer and ~200μm from the perpendicular axis of the patched cell at a depth of ~20–50 μm. One pole of the stimulus isolation unit (A.M.PI. ISO-Flex; Jerusalem, Israel) was connected to the tungsten monopolar stimulation electrode, and the other was connected to a AgCl ground wire inserted into the recording bath. Stimulation electrode position was finely adjusted until a threshold response was elicited between at 0.01 mA, but not at 0.005mA. Stimulus strength was increased in small increments ranging from 0.005–5mA, pulse duration remained constant (0.1ms). We then generated stimulus-evoked input/output curves and measured NMDAR-mediated synaptic currents (Figure 2A). We recorded NMDAR-mediated mEPSCs in the presence of TTX at a holding potential of −70mV, in zero Mg2+, and immediately following the application of NBQX (10μM) to fully block AMPARs. Subsequent application of the NMDAR antagonist AP5 completely abolished these events, confirming that they are NMDAR-mediated (Figure 2D).
Multiple Probability Fluctuation Analysis (MPFA)
Quantal analysis was performed according to previously published protocols (Rothman and Silver, 2018; Saviane and Silver, 2007). Patch pipettes were filled with CsMeSO3-based internal solutions and pipette Rs was compensated by 30–60% (compensated Rs control mean 5.67 ± 0.28 MΩ, n=13; GYKI pre-incubated mean 5.74 ± 0.31 MΩ, n=14). Experiments in which uncompensated Rs was >30 MΩ or changed by >20% throughout the duration of the experiment were discarded. Bipolar tungsten wire stimulation electrodes were inserted into each barrel of theta glass pipettes (borosilicate glass, OD 1.5 mm, ID 1.00 mm, SEP 0.2 mm, tip diameter ~ 1–3 μm) and filled with aCSF (2mM [Ca2+]/1mM [Mg2+]). Stimulation electrodes were placed in the proximal/medial aspect of the stratum oriens ~100 μm from the pyramidal cell layer and ~200μm from the perpendicular axis of the patched cell at a depth of ~20–50 μm. Stable EPSCs were achieved during a pre-experiment baseline sampling period in 1mM [Ca2+]e/2mM [Mg2+]e. The stimulus strengths were kept constant (0.5mA/0.1ms), and once stable, the position and strength of the stimulation electrode was fixed and not further altered. EPSCs were evoked in three different concentrations of extracellular calcium [Ca2+]e and magnesium [Mg2+]e; the total concentration of divalent ions was kept constant at 3mM. aCSF solutions were applied to slices in the following sequence; 1mM [Ca2+]e/2mM [Mg2+]e, 0.5mM [Ca2+]e/2.5mM [Mg2+]e, and 2.5mM [Ca2+]e/0.5mM [Mg2+]e. At least 30 sweeps of EPSCs were sampled for each [Ca2+]e/[Mg2+]e condition, once they stabilized after solution exchange. EPSCs were sampled at a holding potential of −70mV. The following stimulation frequencies were used to sampled EPSCs at each [Ca2+]e/[Mg2+]e condition to avoid synaptic potentiation or depression; 1mM [Ca2+]e/2mM [Mg2+]e 0.033Hz, 0.5mM [Ca2+]e/2.5Mm [Mg2+]e 0.05Hz, 2.5mM [Ca2+]e/0.5mM [Mg2+]e 0.0167Hz. Mean peak EPSCs and background-subtracted variances were calculated for each [Ca2+]e/[Mg2+]e condition once EPSCs stabilized following each solution exchange, and fit to the following multinomial model using IgorPro8 and NeuroMatic v3.0 (Rothman and Silver, 2018):
where Qmpfa is the mean quantal amplitude, N is the mean number of active release sites, α represents a probability density function that approximates the distribution of release probabilities across release sites (constrained at 2 to approximate the heterogeneous distribution of Prs at hippocampal synapses (Branco and Staras, 2009), CVQI is the coefficient of variation of quantal amplitudes within a single release site, and CVQII is the coefficient of variation of quantal amplitudes across release sites. CVQI was not directly measured and was constrained to 0.36, based on previous measurement at hippocampal synapses (McAllister and Stevens, 2000). The total quantal variance (CVQT) was estimated as the CV of successful EPSC amplitudes recorded in the 0.5 mM [Ca2+]e/2.5mM [Mg2+]e condition (i.e. the variance of Qespc amplitudes). CVQII was subsequently estimated from the following equation (Saviane and Silver, 2007):
We obtained mean values of 0.40 ± 0.03 for control and 0.38 ± 0.04 for GYKI treated synapses, in close agreement with previously reported values in hippocampal slices (i.e. 0.42) (Bekkers et al., 1990). The error in estimating the sample variance (δsample variance) was calculated as follows (Saviane and Silver, 2007):
Where n is the number of sweeps, and δI is the standard deviation of EPSC amplitudes across sweeps. Multinomial fits were weighted according to the estimated δsample variance. The mean probability of vesicle release from any given active release site (N) in each [Ca2+]e/[Mg2+]e condition was determined based on the following equation:
Where Pr is the probability of a vesicle fusion event at any given release site (N). We independently verified the accuracy of our estimation by measuring the amplitude of successful EPSCs obtained in the 0.5 mM [Ca2+]e/2.5mM [Mg2+]e. We refer to this empirical estimation as Qepsc throughout.
Calcium input/output experiments
In order to standardize stimulation conditions across different slices from different mice, we developed an approach in which the failure rate of EPSCs in low calcium aCSF (0.5mM [Ca2+]e/2.5mM [Mg2+]e) was used as an indicator of axon recruitment. EPSC amplitude distributions in control conditions were less variable using this method (CVstim I/O = 0.64; CVcalcium I/O = 0.38), suggesting that this is a more consistent approach for estimating synaptic strength.
Slices were preincubated in aCSF ± GYKI (2mM [Ca2+]e/1mM [Mg2+]e) for 10–30 minutes before being placed in the recording chamber containing low calcium aCSF (0.5mM [Ca2+]e/2.5mM [Mg2+]e). Bipolar theta glass stimulation electrodes were first positioned in the proximal/medial aspect of the stratum oriens as described above for MPFA. Cells were voltage clamped at −70mV. Pipette series resistance was ~15–20 MΩ and was compensated by 20–60% (compensated Rs pipette control mean 6.29 ± 0.42 MΩ, n=7; pipette control GYKI pre-incubated mean 6.44 ± 0.97 MΩ, n=7; iMK801 control mean 5.84 ± 0.28 MΩ, n=7; iMK801 GYKI pre-incubated mean 5.54 ± 0.47 MΩ, n=7). Experiments in which uncompensated Rs was >30 MΩ or changed by >20% were discarded. The patched cell was allowed to equilibrate for at least 10 minutes before the experiment began. For experiments in which MK801 was included in the patch pipette, a pre-experimental stimulation period involved delivering pairs of pulses at a frequency of 0.05 Hz for 10 minutes to fully block NMDARs. A total of ~20 minutes elapsed between break-in and the beginning of an experiment. This approach fully blocks NMDARs in the sampled inputs (see Supplemental Figure 4I). Stimulation strength was kept constant at 0.5mA/0.1ms duration. EPSC events were achieved by carefully adjusting the position of stimulation electrodes to achieve a consistent average failure rate of ~30–70% in 0.5mM [Ca2+]e/2.5mM [Mg2+]e (see Supplemental Figure 4A&B). Once the occurrence of successes and failures was stable, the position of the stimulation electrode was fixed and was not altered for the remaining duration of the experiment. At least 30 sweeps were sampled at 0.05Hz (i.e. for 10 minutes) under these conditions at 0.5mM [Ca2+]e/2.5mM [Mg2+]e, then the bath perfusion was switched to one containing 2.5mM [Ca2+]e/0.5mM [Mg2+]e. During the aCSF transition period, EPSCs were sampled at 0.0167Hz. The average EPSC of 5 plateaued responses was normalized to the amplitude of the successful EPSCs in low calcium conditions (i.e. Qepsc) to obtain an estimate of quantal contents shown in Figure 3N&O. In some experiments NBQX (10 μM) was applied immediately following plateaued EPSC measurements to acutely isolate NMDAR EPSCs as described above. After verifying that AMPARs were fully blocked (this occurred within <5 minutes of NBQX application; Supplemental Figure 4F), cells were voltage clamped at +40mV and NMDAR-mediated EPSCs were sampled. In some cases, EPSCs were subsequently blocked with AP5 (20 μM; data not shown), to confirm their NMDAR-dependence.
Double patch experiments
Slices were first incubated in control or perampanel (0.5μM) containing aCSF (in 2mM [Ca2+]e, 1mM [Mg2+]e) for 15 minutes at 34°C, then placed in the recording and perfused with low [Ca2+]e (0.5mM), high [Mg2+]e (2.5mM) aCSF. The control electrode (labeled with AlexaFluo488, 10 μM) was lowered and positioned close to one of the desired cells. The second electrode was tip filled with normal internal solution lacking dye (<0.2μl) and back-filled with internal solution containing MK801 (1mM) and AlexaFluo594 (10μM). Inclusion of normal solution in the tip, along with the presence of high [Mg2+]e prevents undesired spillage of internal solution and blockade of external NMDARs. Once the whole-cell configuration was achieved with the iMK801 electrode, a second neighboring cell was patched with the control electrode. Pipette series resistance was ~20 MΩ and compensated by 10–40% (uncompensated Rs control incubation control pipette mean 23.14 ± 1.30 MΩ, control incubation iMK801 pipette mean 24.54 ± 1.24 MΩ, n=16; perampanel incubation control pipette 22.95 ± 1.55 MΩ, n=18; iMK801 GYKI pre-incubated mean 24.82 ± 1.27 MΩ, n=18). Internal solution was allowed to perfuse into the cell for 10 minutes while high [Ca2+]e, low [Mg2+]e aCSF was perfused into the recording chamber. A pre-experiment stimulation period (pairs of pulses at 0.05Hz for 40–60 sweeps) was applied to fully block NMDARs in the iMK801 cell. Overall, patched cells were held for ~20 minutes before the beginning of the experiment. Once NMDARs were fully blocked, cells were rested for a further 5 minutes and AP5 (20μM) was added to the recording chamber to fully block NMDARs throughout the entire slice (Supplemental Figure 4I). EPSCs were then sampled (0.0167Hz) at the reversal potential for inhibition (−70mV) in both neurons to stimulation of axons in the stratum oriens. Immediately following sampling of EPSCs, NBQX (10μM) was added to the recording chamber to fully block AMPARs. Any residual inward current at −70mV was subtracted from the sampled EPSCs to obtain pure AMPAR-mediated EPSCs. IPSCs were sampled (0.0167Hz) at the reversal potential for inhibition (0mV) in both neurons using the same stimulation electrode.
Live imaging of dendritic spines
Acute slices of hippocampus were obtained from adult thy1-GFP-M mice (Feng et al., 2000) as described above for ex vivo electrophysiology experiments. Live imaging of spine dynamics was performed in 350μm thick acute slices prepared from adult male and female (8–10 weeks) thy1-GFP-M mice as described above. Tertiary dendritic segments of basal dendrites in the stratum oriens were imaged using a multiphoton microscope (Intelligent Imaging Innovations – 3i system) and SlideBook software (Denver, CO, USA) equipped with an Olympus water immersion 60X, 1.00 NA objective. Slices were perfused with aCSF containing (in mM) 119 NaCl, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2, 20 D-glucose and 0.5 Na ascorbate, 0.5 Trolox, pH 7.4, bubbled with 95% O2 / 5% CO2, ~295–305 mOsm and maintained at 32–34° C using an in-line heater (Harvard Instruments) and heated bath chamber (Luigs and Neumann). Dendrite segments (length range 17.32–34.67μm) were imaged as 3D Z-stacks (20–30 μm, optical section 0.55 μm) in 2-photon scanning mode (910 nm, ~15% laser power, 4 μs dwell time). Z-stacks of dendrites were imaged once every 10 minutes during the application of GYKI (5μM) for 1 hour. Average 2D projection images were computed from 3D Z-stacks using Fiji software and the areas of spine heads (range 24–49 spines/dendrite) were fit with ellipsoids and measured by an experimenter blinded to treatment condition. Values were normalized to the average size of spines in the first two image stacks (i.e. the baseline spine size). Overall spine size changes (shown in Supplemental Figure 7J) are computed as the fractional difference between the average spine size in the last three image sessions (t = 40, 50, 60 mins) to the average of the first two image sessions, prior to GYKI application (t = 0, 10 mins).
Stereotaxic surgeries
Adult GRIA1,2,3flx/wt mice of both sexes (age P60-100) were deeply anesthetized with isoflurane and positioned on a stereotaxic frame. Retrograde serotype AAVs expressing GFP-Cre (pENN.AAVrg.hSyn.HI.eGFP-Cre.WPRE.SV40; Addgene #105540-AAVrg) or only GFP (pAAVrg-hSyn-EGFP; Addgene #50465-AAVrg) were injected into the following coordinates: X (posterior from bregma) 2.1mm, Y (lateral from bregma) ± 0.6mm, Z (ventral from pia) 1.55mm. 200–250 nLs of virus at a titre of ~1X1012 was injected bilaterally. Viruses were expressed for 3–4 weeks prior to slice physiology experiments.
Immunohistochemistry
Perfusion fixed sections of mouse brains were cut on a cryostat (70um thick), dried on slides and washed in PBS containing 0.1M glycine. Slices were blocked and permeabilized in PBS containing 1% Triton X-100 (PBT) and 10% goat serum for 1 hour at room temperature and incubated overnight at 4°C in PBT containing goat serum (10%) and polyclonal primary antibodies against GFP (rabbit anti-GFP, ThermoFisher A-6455, 1:1000) and NeuN (guinea pig anti-NeuN, Synaptic Systems 266 004, 1:500). Slices were washed again in PBS and incubated in secondary antibodies (goat anti-rabbit IgG AlexaFluor488 and goat anti-guinea pig IgG AlexaFluo594, each 1:500) for 1 hour at room temperature and mounted in Vectashield containing DAPI (Vector Laboratories, Burlingame, CA, USA).
Cell culture and glutamate imaging
Hippocampal CA1-CA3 regions were dissected with dentate gyrus removed from P1 Sprague-Dawley rats of either sex (mixed litter), dissociated (bovine pancreas trypsin; 5 min at room temperature), and plated on polyornithine-coated coverslips (Carolina Biological; item 633095; 22×22×0.17 mm borosilicate glass) inside a 6 mm diameter cloning cylinder (Ace Glass) as previously described (Hoppa et al., 2012). Calcium phosphate mediated transfection was performed on 5-day-old cultured neurons with the described plasmids (below). Glutamate release was measured using intensity-based glutamate-sensing fluorescent reporter (iGluSnFR) GluSnFr variant A184S (Addgene #106198) (Marvin et al., 2013) and iGluSnFR3 v857 (a kind gift from Dr. K. Podgorski, Jenalia Research Campus). All experiments were performed at 35° C using a custom-built objective heater. Coverslips were mounted in a rapid-switching, laminar-flow perfusion and stimulation chamber on the stage of a custom-built laser microscope. The total volume of the chamber was ~ 750 μl and was perfused at a rate of 400 μl/min. During imaging, cells were continuously perfused in a standard saline solution containing the following in mM: 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, 30 glucose, solutions were supplemented with either 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (Sigma) or 5 μM GYKI (Tocris). For high glutamate calibration, a 500μl volume of 10 μM glutamate was rapidly perfused into the cell imaging chamber. For measuring exocytosis, specimens of iGluSnFr A184S-transfected neurons were illuminated by a 488 nm laser 2 mW (Coherent OBIS laser) with ZET488/10x and ZT488rdc dichroic (Chroma) through a Zeiss EC Plan-Neofluar 40 × 1.3 NA Objective. GluSnFr florescent emission was collected through an ET525/50m filter (Chroma) and captured with an IXON Ultra 897 EMCCD (Andor). GluSnFr fluorescence was collected with an exposure time of 9.83 ms and images were acquired at 100 Hz. Stimulation for firing action potentials for evoked vesicle fusion were evoked by passing 1 ms current pulses, yielding fields of ~12 V/cm2 (unless otherwise noted) using platinum/iridium electrodes. Spontaneous release was easily identified by eye and were found to have a normal and quantal distribution. Not shown, these same sized signals can be identified in the presence of TTX (3 μM) confirming that they are spontaneous fusion. Images were analyzed in ImageJ (http://rsb.info.nih.gov/ij) by using custom-written plugins (http://rsb.info.nih.gov/ij/plugins/time-series.html).
Electron Microscopy
Acute brain slices were prepared as described above for electrophysiology experiments. After recovery for 1.5 hours, slices were incubated for 30 minutes at 34° C in aCSF solutions with or without GYKI (5–10 μM), cooled to ~4 °C then fixed by immersion up to 2 hours in 2% glutaraldehyde in 0.1M Na-cacodylate buffer, pH 7.4 at room temperature (RT) followed by overnight at 4° C. Fixed slices were then post-fixed with 1% OsO4/1.5% KFe(CN)6/0.1 M Na-cacodylate for 1 hr at RT, followed by 1% OsO4/0.1M Na-cacodylate for 1 hr at RT, en bloc staining in 5% uranyl acetate in water for 1 hr at RT, dehydration, infiltration and polymerization in Eponate 12 resin (Ted Pella, Inc., Redding, CA). Serial sections (35 or 50nm thickness) of the stratum oriens were cut with a Leica UCT ultramicrotome using a Diatome diamond knife, picked up on Pioloform coated slot grids and stained with uranyl acetate and Sato’s lead (Sato, 1968). Sections were imaged with an FEI Tecnai T12 TEM at 120 kV using a Gatan U895 4k x 4k camera. Synaptic profiles were selected based on active zone membranes that were precisely perpendicular to the plane section. 50 μm x 50 μm montages of 40–50 sections per sample were imaged using SerialEM (Mastronarde, 2005) and aligned with TrakEM2/Fiji (Cardona et al., 2012; Schindelin et al., 2012). Modeling and analysis were performed with IMOD (Kremer et al., 1996).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis were performed using Origin Pro 9, Prism 8, or Igor Pro 8. When means are shown, error bars indicate standard error. Box plots represent median and Tukey interquartile range, whiskers represent max and min. Parametric or non-parametric statistical analyses were performed when data were normally distributed and when normality could be rejected, respectively. Statistical tests used are indicated in figure legends. When shown in figures, n = the number of cells. For each experiment, data from at least three separate animals was collected.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-GFP polyclonal (1:1000) | ThermoFisher | Cat # A6455 |
| Guinea pig anti-NeuN polyclonal (1:500) | Synaptic Systems | Cat # 266 004 |
| Alexa Fluo 488 AffiniPure Goat Anti-Rabbit IgG (H+L) (1:500) | Jackson Immuno-Research Laboratories | Cat # 111-545-003 RRID: AB_2338046 |
| Alexa Fluo 594 AffiniPure Goat Anti-Guinea Pig IgG (H+L) (1:500) | ThermoFisher | Cat # A-11076 |
| Chemicals, peptides, and recombinant proteins | ||
| GYKI 53655 | Tocris | Cat # 2555 |
| Perampanel | Adooq Biosciences | Cat # A12489 |
| JNJ 55511118 | Tocris | Cat # 6278 |
| D-AP5 | Tocris | Cat # 0106 |
| (+)-MK801 maleate | Tocris | Cat # 0924 |
| ACET | Tocris | Cat # 2728 |
| NBQX disodium salt | Tocris | Cat # 1044 |
| Picrotoxin | Tocris | Cat # 1128 |
| CNQX disodium salt | Alomone | Cat # G-141 |
| Bacterial and virus strains | ||
| pENN.AAVrg.hSyn.HI.eGFP-Cre.WPRE.SV40 | Addgene plasmid #105540-AAVrg was a gift from James M. Wilson | Addgene Cat # #105540-AAVrg |
| pAAVrg-hSyn-EGFP | Addgene plasmid #50465-AAVrg was a gift from Bryan Roth. | Addgene Cat # 50465-AAVrg |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: Tg(Thy1-EGFP)MJrs/J | The Jackson Laboratory | JAX: 007788 |
| Mouse: GRIA1,2,3 flx/flx | Dr. R. Nicoll lab - Lu et al. 2009 | N/A |
| Rat: Sprague Dawley (SAS SD) | Charles River | CR: 400SASSD |
| Oligonucleotides | ||
| GRIA1 forward primer: 5’-CAC TCA CAG CAA TGA AGC AGG AC-3’ | Elim Biopharmaceuticals | Oligo ID: 348107-1 |
| GRIA1 reverse primer: 5’-CTG CCT GGG TAA AGT GAC TTG G-3’ | Elim Biopharmaceuticals | Oligo ID: 348107-2 |
| GRIA2 froward primer: 5’-GCG TAA GCC TCT GAA ATA CCT-3’ | Elim Biopharmaceuticals | Oligo ID: 348107-3 |
| GRIA2 reverse primer: 5’-GTT GTC TAA CAA GTT GTT GAC C-3’ | Elim Biopharmaceuticals | Oligo ID: 348107-4 |
| GRIA3 froward primer: 5’-CCA ATG TTG TTT AGC CTT TGC-3’ | Elim Biopharmaceuticals | Oligo ID: 348107-5 |
| GRIA3 reverse primer: 5’-GGT ATA TCT TCC CAG CCC CAA G-3’ | Elim Biopharmaceuticals | Oligo ID: 348107-6 |
| Thy1 EGFP forward primer: 5’-CCACAGAATCCAAGTCGGAACTC-3’ | Transnetyx | N/A |
| Thy1 EGFP reverse primer: 5’-CTCGCCCTTGCTCACCAT-3’ | Transnetyx | N/A |
| Recombinant DNA | ||
| iGluSnFR.A184S | Marvin et al. 2013 | Addgene Cat # 106198 |
| iGluSnFR3 v857 | Agarwall et al. 2022 | Addgene Cat # 178333 |
| Software and algorithms | ||
| GraphPad Prism (9.4.0) | GraphPad | https://www.graphpad.com |
| Igor Pro 8 (8.04) | WaveMetrics | https://www.wavemetrics.com/software/igor-pro-8 |
| Fiji | NIH | https://imagej.net/software/fiji/ |
| Axon pClamp10 | Molecular Devices | https://support.moleculardevices.com/s/article/Axon-pCLAMP-10-Electrophysiology-Data-Acquisition-Analysis-Software-Download-Page |
| NeuroMatic v3.0 | Rothman and Silver, 2018 | http://www.neuromatic.thinkrandom.com/NMInstall.html |
Highlights:
AMPAR antagonism induces rapid compensatory presynaptic plasticity in adult hippocampus.
Postsynaptic NMDARs are required for the expression of compensatory plasticity.
Plasticity includes expansion of active zones, spine volumes and the docked vesicle pool.
Compensation at excitatory synapses is linked to potentiation of synaptic inhibition.
ACKNOWLEDGEMENTS
Supported by NINDS grant number R35NS097212 and Simons Foundation P0529284 to GWD, NINDS Grants F31NS110192 to L.C.P. and 1R01NS112365 to M.B.H., and NSF Integrative Organismal Systems Grant 1750199 to SJB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Graeme W. Davis is a member of the advisory board of the journal Neuron. The authors declare no other competing interests.
References
- Abbott LF, and Regehr WG (2004). Synaptic computation. Nature 431, 796–803. 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Aggarwal A, Liu R, Chen Y, Ralowicz AJ, Bergerson SJ, Tomaska F, Hanson TL, Hasseman JP, Reep D, Tsegaye G, et al. (2022). Glutamate indicators with improved activation kinetics and localization for imaging synaptic transmission. Biorxiv 2022.02.13.480251 10.1101/2022.02.13.480251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, and Chen L (2008). Synaptic Signaling by All-Trans Retinoic Acid in Homeostatic Synaptic Plasticity. Neuron 60, 308–320. 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, and Stevens CF (1990). Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proceedings of the National Academy of Sciences 87, 5359–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Giessel AJ, and Sabatini BL (2009). Biphasic Synaptic Ca Influx Arising from Compartmentalized Electrical Signals in Dendritic Spines. PLoS Biology 7, e1000190–10. 10.1371/journal.pbio.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 16, 551–563. 10.1038/nrn3992. [DOI] [PubMed] [Google Scholar]
- Branco T, and Staras K (2009). The probability of neurotransmitter release: variability and feedback control at single synapses. Nature Reviews. Neuroscience 10, 373–383. [DOI] [PubMed] [Google Scholar]
- Burrone J, O’Byrne M, and Murthy VN (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418. 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Cardona A, Saalfeld S, Schindelin J, Arganda-Carreras I, Preibisch S, Longair M, Tomancak P, Hartenstein V, and Douglas RJ (2012). TrakEM2 Software for Neural Circuit Reconstruction. PloS One 7, e38011–8. 10.1371/journal.pone.0038011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CQ, Martenson JS, Yamazaki M, Natsume R, Sakimura K, Tomita S, Tavalin SJ, and Higley MJ (2018). Input-Specific NMDAR-Dependent Potentiation of Dendritic GABAergic Inhibition. Neuron 97, 368–377.e3. 10.1016/j.neuron.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, and Uchitel OD (1980). On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J Physiology 299, 621–638. 10.1113/jphysiol.1980.sp013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargan SL, Clarke VRJ, Alushin GM, Sherwood JL, Nisticò R, Bortolotto ZA, Ogden AM, Bleakman D, Doherty AJ, Lodge D, et al. (2009). ACET is a highly potent and specific kainate receptor antagonist: Characterisation and effects on hippocampal mossy fibre function. Neuropharmacology 56, 121–130. 10.1016/j.neuropharm.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW (2006). HOMEOSTATIC CONTROL OF NEURAL ACTIVITY: From Phenomenology to Molecular Design. Annu Rev Neurosci 29, 307–323. 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW (2013). Homeostatic Signaling and the Stabilization of Neural Function. Neuron 80, 718–728. 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, and Goodman CS (1998). Genetic analysis of synaptic development and plasticity: homeostatic regulation of synaptic efficacy. Curr Opin Neurobiol 8, 149–156. 10.1016/s0959-4388(98)80018-4. [DOI] [PubMed] [Google Scholar]
- Davis GW, and Müller M (2015). Homeostatic Control of Presynaptic Neurotransmitter Release. Annual Review of Physiology 77, 251–270. 10.1146/annurev-physiol-021014-071740. [DOI] [PubMed] [Google Scholar]
- Davis GW, and Murphey RK (1994). Long-term regulation of short-term transmitter release properties: retrograde signaling and synaptic development. Trends Neurosci 17, 9–13. 10.1016/0166-2236(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Delvendahl I, Kita K, and Müller M (2019). Rapid and sustained homeostatic control of presynaptic exocytosis at a central synapse. Proceedings of the National Academy of Sciences 21, 201909675. 10.1073/pnas.1909675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, and Turrigiano GG (1999). Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nature Neuroscience 2, 515–520. 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, and Turrigiano GG (2002). Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 5, 783–789. 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Dickman DK, and Davis GW (2009). The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science (New York, N.Y.) 326, 1127–1130. 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, and Stevens CF (1997). Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18, 995–1008. [DOI] [PubMed] [Google Scholar]
- Donevan SD, and Rogawski MA (1993). GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron 10, 51–59. 10.1016/0896-6273(93)90241-i. [DOI] [PubMed] [Google Scholar]
- Elmqvist D, and Quastel DM (1965). A quantitative study of end-plate potentials in isolated human muscle. J Physiology 178, 505–529. 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, and Sanes JR (2000). Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron 28, 41–51. 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Frampton JE (2015). Perampanel: A Review in Drug-Resistant Epilepsy. Drugs 75, 1657–1668. 10.1007/s40265-015-0465-z. [DOI] [PubMed] [Google Scholar]
- Frank CA, Pielage J, and Davis GW (2009). A Presynaptic Homeostatic Signaling System Composed of the Eph Receptor, Ephexin, Cdc42, and CaV2.1 Calcium Channels. Neuron 61, 556–569. 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, and Nicoll RA (2000). Synaptic kainate receptors. Curr Opin Neurobiol 10, 342–351. 10.1016/s0959-4388(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Frerking M, Schmitz D, Zhou Q, Johansen J, and Nicoll RA (2001). Kainate Receptors Depress Excitatory Synaptic Transmission at CA3→CA1 Synapses in the Hippocampus via a Direct Presynaptic Action. J Neurosci 21, 2958–2966. 10.1523/jneurosci.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, and Turrigiano GG (2009). Synaptic Scaling Requires the GluR2 Subunit of the AMPA Receptor. Journal of Neuroscience 29, 6479–6489. 10.1523/jneurosci.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulton CS, Patten AR, Kerr JR, and Kerr DS (2010). Pharmacological Preconditioning with GYKI 52466: A Prophylactic Approach to Neuroprotection. Front Neurosci-Switz 4, 54. 10.3389/fnins.2010.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Gray JA, Lu W, and Nicoll RA (2011). Genetic analysis of neuronal ionotropic glutamate receptor subunits. The Journal of Physiology 589, 4095–4101. 10.1113/jphysiol.2011.213033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Zhou L, and Lu W (2016). An NMDA Receptor-Dependent Mechanism Underlies Inhibitory Synapse Development. CellReports 14, 471–478. 10.1016/j.celrep.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris N, Fetter RD, Brasier DJ, Tong A, and Davis GW (2018). Molecular Interface of Neuronal Innate Immunity, Synaptic Vesicle Stabilization, and Presynaptic Homeostatic Plasticity. Neuron 1–22. 10.1016/j.neuron.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth AG, Ford KJ, Wang T, Fetter RD, Tong A, and Davis GW (2018). A postsynaptic PI3K-cII dependent signaling controller for presynaptic homeostatic plasticity. ELife 7, e31535. 10.7554/elife.31535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, and Ryan TA (2012). α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125. 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJL, and Sutton MA (2010). Local Presynaptic Activity Gates Homeostatic Changes in Presynaptic Function Driven by Dendritic BDNF Synthesis. Neuron 68, 1143–1158. 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Menon V, Nicholson DA, Geinisman Y, Kath WL, and Spruston N (2009). Synapse Distribution Suggests a Two-Stage Model of Dendritic Integration in CA1 Pyramidal Neurons. Neuron 63, 171–177. 10.1016/j.neuron.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, and Ryan TA (2010). CDK5 Serves as a Major Control Point in Neurotransmitter Release. Neuron 67, 797–809. 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber D, Burnod Y, and Triller A (1984). Regulation of efficacy at central synapses. J Neurosci 4, 125–130. 10.1523/jneurosci.04-01-00125.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology 116, 71–76. 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Levy JM, Chen X, Reese TS, and Nicoll RA (2015). Synaptic Consolidation Normalizes AMPAR Quantal Size following MAGUK Loss. Neuron 87, 534–548. 10.1016/j.neuron.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Suutari BS, Sun SD, Luo Z, Wei C, Chenouard N, Mandelberg NJ, Zhang G, Wamsley B, Tian G, et al. (2020). Neuronal Inactivity Co-opts LTP Machinery to Drive Potassium Channel Splicing and Homeostatic Spike Widening. Cell 181, 1547–1565.e15. 10.1016/j.cell.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, and Nicoll RA (2009). Subunit Composition of Synaptic AMPA Receptors Revealed by a Single-Cell Genetic Approach. Neuron 62, 254–268. 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher MP, Wu N, Ravula S, Ameriks MK, Savall BM, Liu C, Lord B, Wyatt RM, Matta JA, Dugovic C, et al. (2016). Discovery and Characterization of AMPA Receptor Modulators Selective for TARP-γ8. J Pharmacol Exp Ther 357, 394–414. 10.1124/jpet.115.231712. [DOI] [PubMed] [Google Scholar]
- Malinow R, and Tsien RW (1990). Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature 346, 177–180. 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen T-W, Bargmann CI, et al. (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nature Methods 10, 162–170. 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology 152, 36–51. 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- McAllister AK, and Stevens CF (2000). Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proceedings of the National Academy of Sciences 97, 6173–6178. 10.1073/pnas.100126497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon V, Musial TF, Liu A, Katz Y, Kath WL, Spruston N, and Nicholson DA (2013). Balanced Synaptic Impact via Distance-Dependent Synapse Distribution and Complementary Expression of AMPARs and NMDARs in Hippocampal Dendrites. Neuron 80, 1451–1463. 10.1016/j.neuron.2013.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Mitra SS, and Tsien RW (2011). Heterogeneous reallocation of presynaptic efficacy in recurrent excitatory circuits adapting to inactivity. Nature Neuroscience 15, 250–257. 10.1038/nn.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Liu KSY, Sigrist SJ, and Davis GW (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. Journal of Neuroscience 32, 16574–16585. 10.1523/jneurosci.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, and Zhu Y (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32, 673–682. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, and Huganir RL (1998). Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Ortega JM, Genç Ö, and Davis GW (2018). Molecular mechanisms that stabilize short term synaptic plasticity during presynaptic homeostatic plasticity. Elife 7, e40385. 10.7554/elife.40385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternain AV, Morales M, and Lerma J (1995). Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron 14, 185–189. 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, Kempen GT van, and Molenaar, P.C. (1992). Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. The Journal of Physiology 458, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JS, and Silver RA (2018). NeuroMatic: An Integrated Open-Source Software Toolkit for Acquisition, Analysis and Simulation of Electrophysiological Data. Frontiers in Neuroinformatics 12, 1159–21. 10.3389/fninf.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T (1968). A modified method for lead staining of thin sections. Journal of Electron Microscopy 17, 158–159. [PubMed] [Google Scholar]
- Saviane C, and Silver RA (2007). Estimation of quantal parameters with multiple-probability fluctuation analysis. Methods in Molecular Biology (Clifton, N.J.) 403, 303–317. 10.1007/978-1-59745-529-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, and Neher E (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23, 399–409. 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Sun Y, Smirnov M, Kamasawa N, and Yasuda R (2021). Rapid Ultrastructural Changes in the PSD and Surrounding Membrane after Induction of Structural LTP in Single Dendritic Spines. J Neurosci 41, 7003–7014. 10.1523/jneurosci.1964-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DGR, Hwang B-Y, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 92, 372–382. 10.1016/j.neuron.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsodyks MV, and Markram H (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proceedings of the National Academy of Sciences 94, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, and Nelson SB (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wang X, Pinter MJ, and Rich MM (2016). Reversible Recruitment of a Homeostatic Reserve Pool of Synaptic Vesicles Underlies Rapid Homeostatic Plasticity of Quantal Content. Journal of Neuroscience 36, 828–836. 10.1523/jneurosci.3786-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmüller A, Hallermann S, Wagner N, and Eilers J (2011). Rapid Active Zone Remodeling during Synaptic Plasticity. Journal of Neuroscience 31, 6041–6052. 10.1523/jneurosci.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshanskaya MV, Singh AK, Sampson JM, Narangoda C, Kurnikova M, and Sobolevsky AI (2016). Structural Bases of Noncompetitive Inhibition of AMPA-Subtype Ionotropic Glutamate Receptors by Antiepileptic Drugs. Neuron 91, 1305–1315. 10.1016/j.neuron.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, and Miller RF (1995). NBQX, an improved non-NMDA antagonist studied in retinal ganglion cells. Brain Res 692, 190–194. 10.1016/0006-8993(95)00665-d. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Yamada KA, Price MT, and Olney JW (1993). A benzodiazepine recognition site associated with the non-NMDA glutamate receptor. Neuron 10, 61–67. 10.1016/0896-6273(93)90242-j. [DOI] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-Term Synaptic Plasticity. Annual Review of Physiology 64, 355–405. 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate standardized datatypes for public repositories. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.