

Abstract
Defective DNA repair pathways contribute to the development of chronic kidney disease (CKD) in humans. However, the molecular mechanisms underlying DNA damage induced CKD pathogenesis are not well understood. Here, we investigated the role of tubular cell DNA damage in the pathogenesis of CKD using mice in which the DNA repair protein Fan1 was knocked out. The phenotype of these mice is orthologous to the human DNA damage syndrome; karyomegalic interstitial nephritis (KIN). Inactivation of Fan1 in kidney proximal tubule cells sensitized the kidneys to genotoxic and obstructive injury characterized by replication stress and persistent DNA damage response activity. Accumulation of DNA damage in Fan1 tubular cells induced epithelial dedifferentiation and tubular injury. Characteristic to KIN, cells with chronic DNA damage failed to complete mitosis and underwent polyploidization. In vitro and in vivo studies showed that polyploidization was caused by the overexpression of DNA replication factors CDT1 and CDC6 in FAN1 deficient cells. Mechanistically, inhibiting DNA replication with Roscovitine reduced tubular injury, blocked the development of KIN and mitigated kidney function in these Fan1 knockout mice. Thus, our data delineate a mechanistic pathway by which persistent DNA damage in the kidney tubular cells leads to kidney injury and development of CKD. Furthermore, therapeutic modulation of cell cycle activity may provide an opportunity to mitigate the DNA damage response induced CKD progression.
Keywords: FAN1, karyomegalic interstitial nephritis, DNA damage, DNA re-replication, chronic kidney disease
Graphical Abstract
INTRODUCTION
Chronic kidney disease (CKD) is a major public health concern worldwide, with a global prevalence of >10% of population1. CKD is a major indicator for the progression to end-stage kidney disease and is associated with significant complications including cardiovascular disease2. Thus, understanding the mechanisms that drive the progression of CKD is paramount for developing impactful therapies. Recent work has demonstrated that CKD can rise from delayed or incomplete resolution of kidney tubular epithelial cell damage after kidney injury3. Indeed, the surviving, unrepaired cell has a distinct molecular identity that reflects its impaired physiological function and reduced proliferative capacity, underscoring its inability to contribute to the repair of kidney tubular injury4, 5. One of the molecular characteristics of such “failed-repair” cell is genomic instability and persistent DNA damage response (DDR) activity5, 6.
While DDR appears to be involved in any type of kidney injury5, its role in CKD is best understood in models where the kidneys are exposed to genotoxic agents, such as cancer therapeutics, which have significant nephrotoxicity and frequently lead to the development of chronic kidney disease in humans7, 8. Cisplatin is a widely used chemotherapeutic whose long-term use is known to underlie progression to CKD in humans9. Cisplatin covalently binds to DNA and forms stable adducts between the opposite strands of DNA double helix, known as interstrand cross-links (ICL), which impede DNA transcription and replication in proliferating cells10. Collision of DNA replisome with an ICL during DNA replication stalls replication machinery in front of the lesion and activates the DDR signaling11. Subsequently, DDR recruits specialized repair complexes, such as the Fanconi anemia (FA) and homologous recombination (HR) proteins to the sites of DNA damage to resolve the lesions12. However, a mechanism by which a defect in ICL repair leads to the progression of CKD is not known.
FAN1 is a DNA endonuclease whose deficiency underlies karyomegalic interstitial nephritis (KIN) (OMIM: 614817)13 and predisposition to cancer in humans14–16. KIN is a genetic form of CKD, characterized by tubulointerstitial fibrosis and formation of enlarged nuclei in the kidneys and other tissues17, 18. FAN1 has been shown to be essential for DNA interstrand crosslink repair19–22 and stabilization of DNA replication forks in response to different forms of replication stress15, 23, 24. However, how loss of FAN1 results in onset of KIN is not understood. While karyomegaly in FAN1-deficient kidney tubular epithelial cells are thought to arise from DNA polyploidization due to mitotic errors25 and result in aneuploidy26, 27, the underlying molecular mechanisms leading to cell division defects has not been elucidated. In this study, we generated a proximal tubule specific knockout of Fan1 and show that Fan1 is indispensable for an intact DDR response and tubular regeneration after genotoxic and obstructive kidney injury. Loss of Fan1 results in persistent DDR signaling in proximal tubule cells due to the accumulation of unresolved ICL and replication stress-induced DNA damage, which in turn induces aberrant DNA re-replication. Blocking cell cycle activity with roscovitine protects Fan1 mouse kidneys from DNA replication stress, tubular cell damage and mitigates kidney function. Together, these findings provide the first mechanistic explanation for the formation of KIN in FAN1-deficient kidneys and other tissues and demonstrate how persistent DDR leads to CKD progression.
METHODS
Mouse lines used and study approval
Fan1loxP/loxP mice were bred with γGt1-Cre mice (#012841, Tg(Ggt1-cre)M3Egn/J)28 to generate proximal tubule specific knockout of Fan1 (Fan1 KO). γGt1-Cre expression in the proximal tubule was demonstrated by crossing γGt1-Cre mice with NuTRAP mice (Jackson Laboratories #029899, B6;129S6-Gt(ROSA)26Sortm2(CAG-NuTRAP)Evdr/J). Cre-negative littermates with γGT1-Cre−;Fan1loxP/loxP or γGT1-Cre−;Fan1+/loxP genotype were used as controls. The experimental protocol was reviewed and approved by the IACUC of the University of Pittsburgh.
Statistical Methods
Statistical analysis was performed using GraphPad Prism 9 (GraphPad software). Normal distribution was confirmed by Shapiro-Wilk normality test, followed by quantile-quantile plot of residuals. Statistical tests are two-tailed, unpaired Student’s t-tests or two-way
ANOVA followed by Tukey’s post-hoc test for multiple group comparisons. When sample size n>30, Kruskal-Wallis test was used for multiple group comparisons. All results are reported as means±SEM. Significance was determined at p<0.05 and represented by * to denote p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Data sharing statement
RNAseq data supporting the findings of this study are openly available in the Gene Expression Omnibus repository at URL https://www.ncbi.nlm.nih.gov/geo/, reference number GSE163862.
See Supplementary Methods for more details on animal treatment protocols, histological and immunofluorescence staining protocols, cell culture techniques and other materials and methods used in this manuscript.
RESULTS
Proximal tubule-specific deletion of Fan1 sensitizes kidneys to genotoxic and oxidative damage
To investigate the molecular mechanism underlying KIN pathogenesis we used Fan1 knockout mice (Fan1 KO) (Supplementary Figure 1A–D and Figure 1A,B). The mice appeared healthy and showed no histological abnormalities in the kidneys. However, repeated administration of low dose cisplatin (2mg/kg) (Figure 1C) was sufficient to trigger histological changes associated with KIN6, and kidney fibrosis in Fan1 KO mice, but not in control mice (Figure 1D,E,F and Supplementary Figure 2A–D), demonstrating the critical function of Fan1 in resolving genotoxic damage in the proximal tubule epithelial cells (PTECs). Importantly, the development of KIN was associated with extensive DNA damage in PTECs, demonstrated by the upregulation of γH2AX, phosphorylated RPA32 (S4/8), 53BP1 and phosphorylated TRIM28/KAP1 (S824)29–32 (Supplementary Figure 3A–F). The expression of these DNA damage markers as well as interstitial fibrosis remain unresolved in cisplatin treated Fan1 KO mice even after a 4-week “recovery” phase, highlighting the long-term impact of unresolved tubular DNA damage to the progression of tubular atrophy and development of fibrosis in KIN (Supplementary Figure 4A–H).
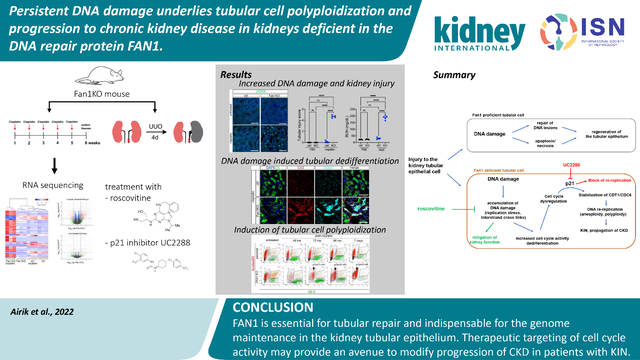
Figure 1. Loss of Fan1 sensitizes kidneys to genotoxic and obstructive tubular injury.
(A) Schematics of the generation of proximal tubule-specific Fan1 knockout (Fan1 KO) mice, by crossing Ggt1Cre;Fan1+/loxP mice with Fan1loxP/loxP mice.
(B) Ggt1-Cre activity is restricted to the proximal segment of the nephron, demonstrated by the co-staining of Lotus tetranogolobus agglutinin (LTL) and anti-GFP antibody in double transgenic mice but not in Ggt1Cre- mice. Scale bar 25 μm.
(C) Schematic diagram of the repeated low dose cisplatin injury protocol. Mice were administered cisplatin weekly at 2mg/kg for 5 weeks, and tissues were collected for analysis 1 week after the last treatment dose.
(D) Histological analysis of kidney sections by Periodic acid–Schiff (PAS) staining demonstrated the formation of KIN in Fan1 KO mice, characterized by tubular atrophy, formation of karyomegalic nuclei (green arrowheads) and segmental basement membrane thickening (black arrowheads) in the proximal tubules. Scale bars 50 μm.
(E) Tubular injury scores in control mice compared with Fan1 KO kidneys after low dose cisplatin administration, cisplatin ctrl 0.1±0.1 vs cisplatin Fan1 KO 4.7±0.1, ****p<0.0001, n=5 each.
(F) Blood urea nitrogen (BUN) measurements in ctrl and Fan1 KO mice show loss of kidney function in Fan1 KO mice after induction of KIN; cisplatin ctrl 31.2±2.9 vs cisplatin Fan1 KO 188.6±7.2, ****p<0.0001, n=5 each.
(G) Schematics of the unilateral ureteral obstruction (UUO) kidney injury model.
(H) PAS staining of sham and UUO kidneys at day 4 reveals more extensive tubular dilations in Fan1 KO kidneys compared with control kidneys. Scale bar 100 μm.
(I) Tubular injury scores in control mice compared to Fan1 KO kidneys after 4 days of UUO, ctrl 1.6±0.2 vs Fan1 KO 4.2±0.1, ****p<0.0001, n=5 each. (E,F,I) Data are presented as the mean ± SEM. A 2-way ANOVA with Tukeys’ post hoc analysis.
To address whether KIN can be caused by other types of tubular stress we employed the unilateral ureteral obstruction (UUO) injury model (Figure 1G). Indeed, Fan1 KO mice displayed extensive tubular atrophy, dilation, and detectable interstitial fibrosis already 4 days after UUO compared to control mice (Figure 1H and 1I, Supplementary Figure 5A). Again, the more severe UUO injury phenotype in Fan1 KO kidneys was associated with increased DNA damage in PTECs (Supplementary Figure 5B). The accumulation of DNA damage in Fan1 KO kidneys was likely caused by their increased sensitivity to oxidative stress, secondary to UUO tubular injury, as revealed by 8-OHdG staining (Supplementary Figure 6A,B). This would be consistent with other reports demonstrating that oxidative DNA damage plays a key role in UUO pathogenesis33, 34. Together, our data show that 1) FAN1 is essential for the repair of diverse DNA damage in PTECs after genotoxic or obstructive kidney injury; and 2) persistent DNA damage in PTECs leads to the development of KIN.
KIN is associated with tubular dedifferentiation and upregulation of tubular injury biomarkers
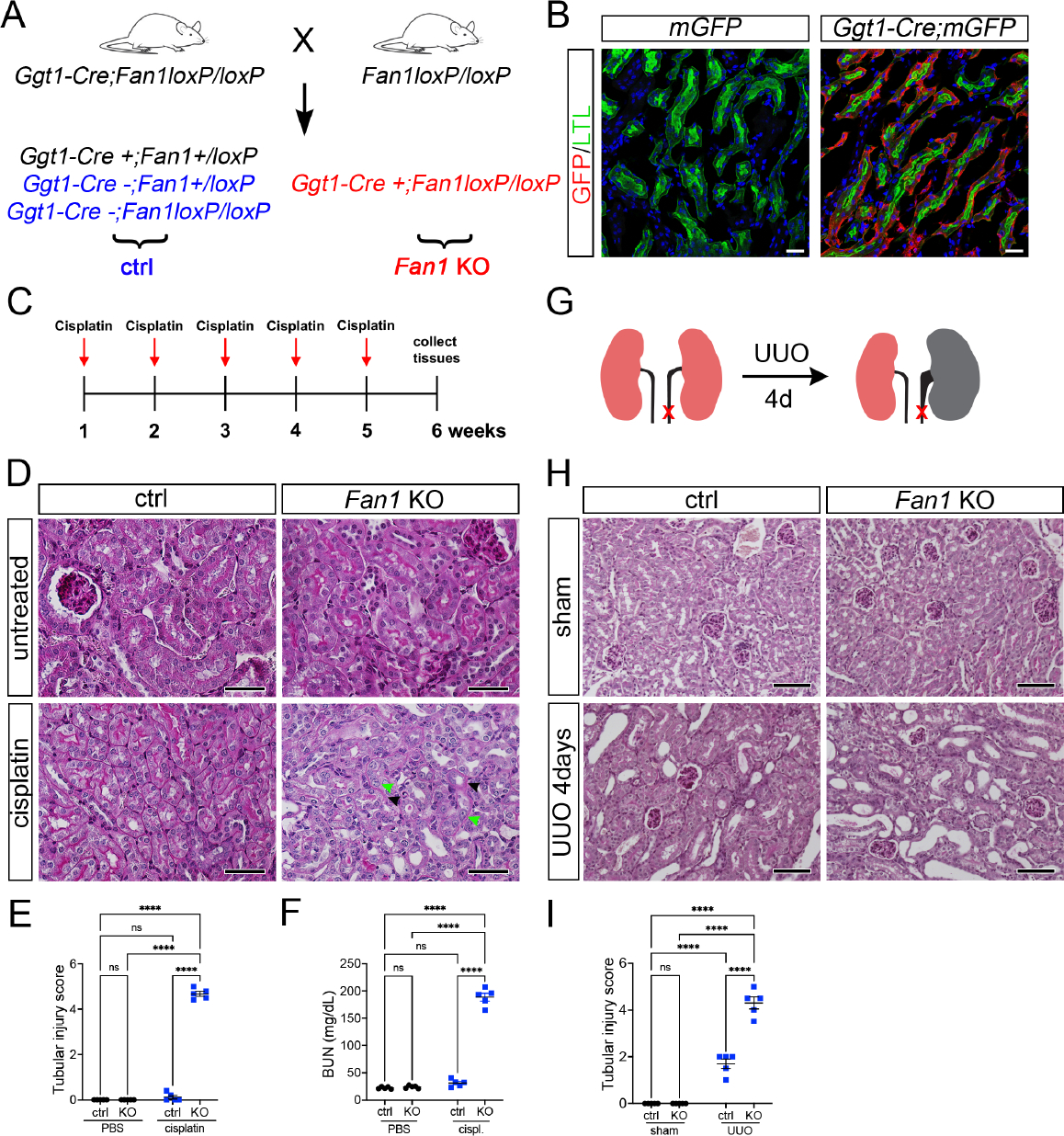
To characterize the effect of persistent DNA damage on PTEC phenotype, we examined the expression of tubular injury (KIM1/Havcr1, NGAL/Lcn2) and dedifferentiation (SOX9, PAX2 and vimentin) biomarkers4, 35–38 in Fan1 KO kidneys after cisplatin or UUO injury. Expression of these markers was increased in Fan1 KO PTECs both after short-term (Figure 2A–C, Supplementary Figure 7A and 7B), and long-term cisplatin injury (Supplementary Figure 7D and 7E), suggesting that persistent DNA damage leads to tubular dedifferentiation and epithelial injury in the kidney. KIM1 and SOX9 were also elevated in Fan1 KO kidneys 4 days after UUO compared to control kidneys (Figure 2D,E and Supplementary Figure 7C). However, the expression of Lcn2, a marker of distal tubule damage39, 40, remained unchanged between control vs. Fan1 KO UUO kidneys (Figure 2F), suggesting that Fan1 is dispensable for distal tubule homeostasis.
Figure 2. Persistent DNA damage induces the expression of tubular injury biomarkers in Fan1 KO kidneys.
(A) KIM1 expression analysis by immunofluorescence staining in control and Fan1 KO kidneys after cisplatin injury. Scale bar 75 μm. Quantification of the KIM1-positive area in LTL-positive proximal tubules shows a significant upregulation of KIM1 in Fan1 KO kidneys (ctrl 2.5±0.4% vs Fan1 KO 17.8±1.6%, ****p<0.0001), n=5 each.
(B) Increased Havcr1/KIM1 expression in cisplatin treated Fan1 KO kidneys was confirmed by qPCR analysis (ctrl 1.6±0.3 vs Fan1KO 6.9±0.6, ****p<0.0001), n=5 each.
(C) qPCR analysis revealed increased Lcn2/NGAL expression in cisplatin treated Fan1 KO kidneys (ctrl 6.1±1.8 vs Fan1 KO 110.7±8.4, ****p<0.0001), n=5 each.
(D) KIM1 expression analysis by immunofluorescence staining in control and Fan1 KO kidneys 4 days after unilateral ureteral obstruction (UUO). Scale bar 100 μm. Quantification of the KIM1-positive area in LTL-positive proximal tubules shows higher KIM1 expression in Fan1 KO UUO kidneys (ctrl 23.5% ± 4.7% vs Fan1 KO 47.4% ± 8.4% %, *p<0.05, ****p<0.0001), n=5 each.
(E) Increased Havcr1/KIM1 expression in Fan1 KO UUO kidneys compared to control UUO kidneys was confirmed by qPCR (ctrl 44.3±2.7 vs Fan1 KO 80.3±7.5, ****p<0.0001), n=5 each.
(F) qPCR analysis did not reveal significant alterations in Lcn2/Ngal expression between Fan1 KO and control kidneys after 4 days of UUO (ctrl 32.9±4.3 vs Fan1 KO 32.6±2.1, ns), n=5 each. (A-F) Data are presented as the mean ± SEM. A 2-way ANOVA with Tukeys’ post hoc analysis.
We next examined the expression of tubular markers associated with more advanced CKD (VCAM1/Vcam1, CCL2/Ccl2 and Dock10) in humans and mice5, 41, and confirmed their upregulation in Fan1 KO kidneys but not in controls (Supplementary Figure 8A,B). Together, these data demonstrate that accumulation of DNA damage underlies epithelial dedifferentiation and tubular injury in KIN.
Transcriptional profiling reveals overrepresentation of DNA repair and cell cycle regulators in KIN
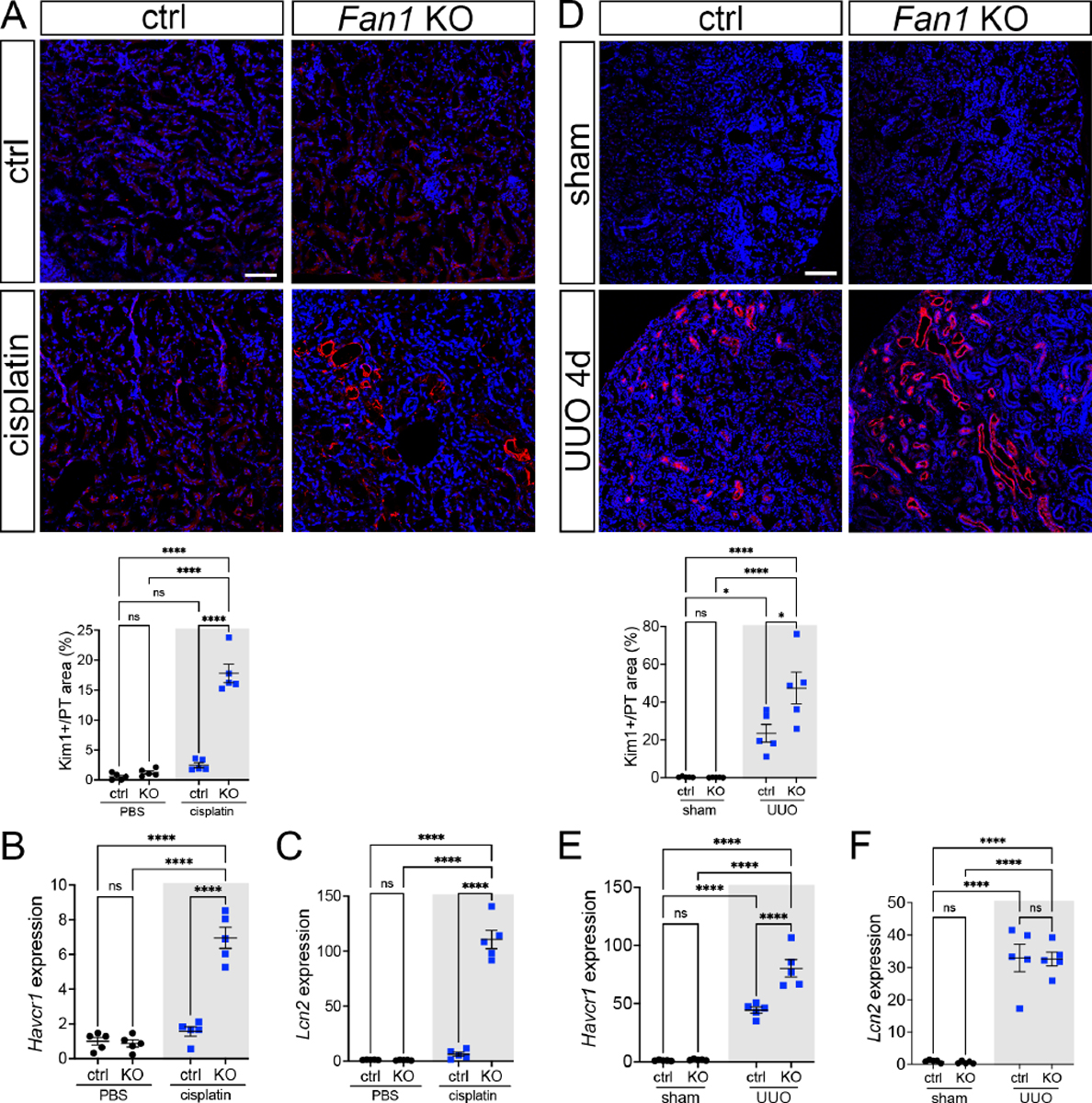
To identify the molecular pathways that are altered in Fan1 KO kidneys in response to persistent DNA damage, we performed a genome-wide transcriptome profiling in whole kidney samples from untreated and cisplatin-treated control and Fan1 KO mice. Hierarchical clustering of the RNA-seq data in the representative heatmap showed a high degree of transcriptional similarity between untreated control, untreated Fan1 KO and cisplatin-treated control mice, demonstrating that Fan1 inactivation in the kidney proximal tubule epithelium in the absence of KIN does not alter basal gene expression (Figure 3A,B and Supplementary Table 1,2). In contrast, we identified 2505 differentially expressed genes (DEGs) after correction for multiple hypothesis testing by Benjamini-Hochberg method, and observed gender-based differences, between cisplatin-treated control and Fan1 KO kidneys, with 1435 transcripts upregulated and 1070 transcripts down-regulated (Figure 3C, Supplementary Figure 9A and Supplementary Table 3).
Figure 3. Transcriptional profiling of Fan1 KO kidneys after induction of KIN.
(A) Heatmap of the DEG list for all conditions. n=3 mice for each experimental group. (B, C) Volcano plots of the DEG list for untreated control and Fan1 KO kidneys (B), and cisplatin treated control and Fan1 KO kidneys (C). The thresholds were set at the values of fold change > 2 and FDR < 0.05.
(D) Top-ranked functional clusters that were differentially expressed in control vs. Fan1 KO kidneys in response to cisplatin treatment.
(E) Gene-set enrichment signature of DNA repair genes in cisplatin-treated Fan1 KO vs control kidneys.
(F) qPCR analysis of the interstrand cross-link repair genes Exo1 (ctrl 2.6±0.4 vs Fan1 KO 15.8±1.0, ****p<0.0001) and Ercc1 (ctrl 1.1±0.1 vs Fan1 KO 1.8±0.1, ****p<0.0001) in cisplatin Fan1 KO kidneys. n=5 each.
(G) qPCR analysis of the homologous recombination genes Brca1 (ctrl 1.0±0.2 vs Fan1 KO 2.9±0.3, ***p<0.001) and Rad51 (ctrl 0.9±0.2 vs Fan1 KO 4.3±0.6, ****p<0.0001) in cisplatin Fan1 KO kidneys. n=5 each.
(H) Gene-set enrichment signature of G2/M checkpoint genes in cisplatin-treated Fan1 KO vs control kidneys.
(I) qPCR analysis of the cell cycle position genes AurkB (ctrl 1.5±0.3 vs Fan1 KO 6.9±0.6, ****p<0.0001), Pkmyt1 (ctrl 1.0±0.2 vs Fan1 KO 2.5±0.2, ***p<0.001) and Birc5 (ctrl 1.7±0.2 vs Fan1 KO 8.6±1.0, ****p<0.0001) in cisplatin Fan1 KO kidneys, n=5 each. (F,G,I) Data are presented as the mean ± SEM. A 2-way ANOVA with Tukeys’ post hoc analysis.
Gene Ontology analysis by DAVID and Gene Set Enrichment Analysis (GSEA)42 showed that most of the upregulated functional clusters in cisplatin-treated Fan1 KO kidneys are related to cell cycle regulation, DNA damage response and DNA replication (Figure 3D,E,H), consistent with the known roles of FAN1 in ICL repair19, 21, 22 and replication fork stabilization15, 23. Using qPCR analysis, we confirmed the dysregulation of selected genes that are representative of specific DNA repair pathways, including interstrand cross-link repair (Exo1, Ercc1) (Figure 3F); homologous DNA repair (Brca1, Rad51) (Figure 3G), as well as cell cycle positioning genes (AurkB, Pkmyt1, Birc5) (Figure 3I and Supplementary Figure 9B). Together, transcriptional profiling demonstrated a broad activation of multiple DNA repair pathways in injured Fan1 KO kidneys, which likely led to overexpression of G2/M regulators43, and promoted PTEC dedifferentiation44.
DNA damage triggers aberrant cell cycle activity in Fan1 KO PTECs
Eight of the top 10 upregulated GO functional terms in cisplatin Fan1 KO kidneys are related to cell cycle activity, mitosis or DNA replication (Figure 3D). The top changed genes included the MCM, GINS genes and the replication factors Cdc6 and Cdt1 (Figure 4A), whose products are involved in regulating DNA replication licensing at the chromatin45. Molecular analysis using western blotting, qPCR and antibody staining on Fan1 KO kidneys confirmed the upregulation of hyperphosphorylated pRB (S807/811), PCNA, CDK6 and several cyclins (Figure 4A–D and Supplementary Figure 10A,B), indicating that DNA damage promotes cell cycle entry and S phase activity in Fan1-deficient PTECs. Importantly, this aberrant cell cycle activity was associated with nuclear enlargement in Fan1 KO kidneys (Figure 4D). In particular, cyclinD1 appeared to exclusively label the enlarged nuclei in Fan1 KO kidneys (Figure 4D), and intriguingly, cyclinD1 expression was maintained in the karyomegalic nuclei in Fan1 KO kidneys even 28 days after cisplatin treatment (Supplementary Figure 10C), suggesting that cyclinD1 is either required for nuclear enlargement and/or for the survival of the enlarged karyomegalic cells. Additionally, expression analysis of replicative helicase proteins MCM2/MCM6 demonstrated their accumulation in karyomegalic nuclei (Figure 4E and Supplementary Figure 10D–F), suggesting that nuclear enlargement in KIN is caused by aberrant replication licensing which leads to DNA endoreplication, a process in which DNA is replicated more than once per cell cycle without intervening mitosis, resulting in giant nuclei46–48. Indeed, karyomegalic nuclei in FAN1-deficient tissues have been shown to contain an aneuploid set of chromosomes25, 26, a hallmark of DNA endoreplication49. Consistent with the findings in human tissues, karyomegalic cells in Fan1 KO kidneys stained strongly positive for γH2AX (Figure 4E), demonstrating underlying genomic instability. Together, the results suggest that persistent DNA damage in Fan1 KO PTECs induces aberrant cell cycle activity and abnormal replication licensing which results in cellular polyploidization.
Figure 4. DNA damage causes aberrant cell cycle activity in Fan1 KO kidneys.
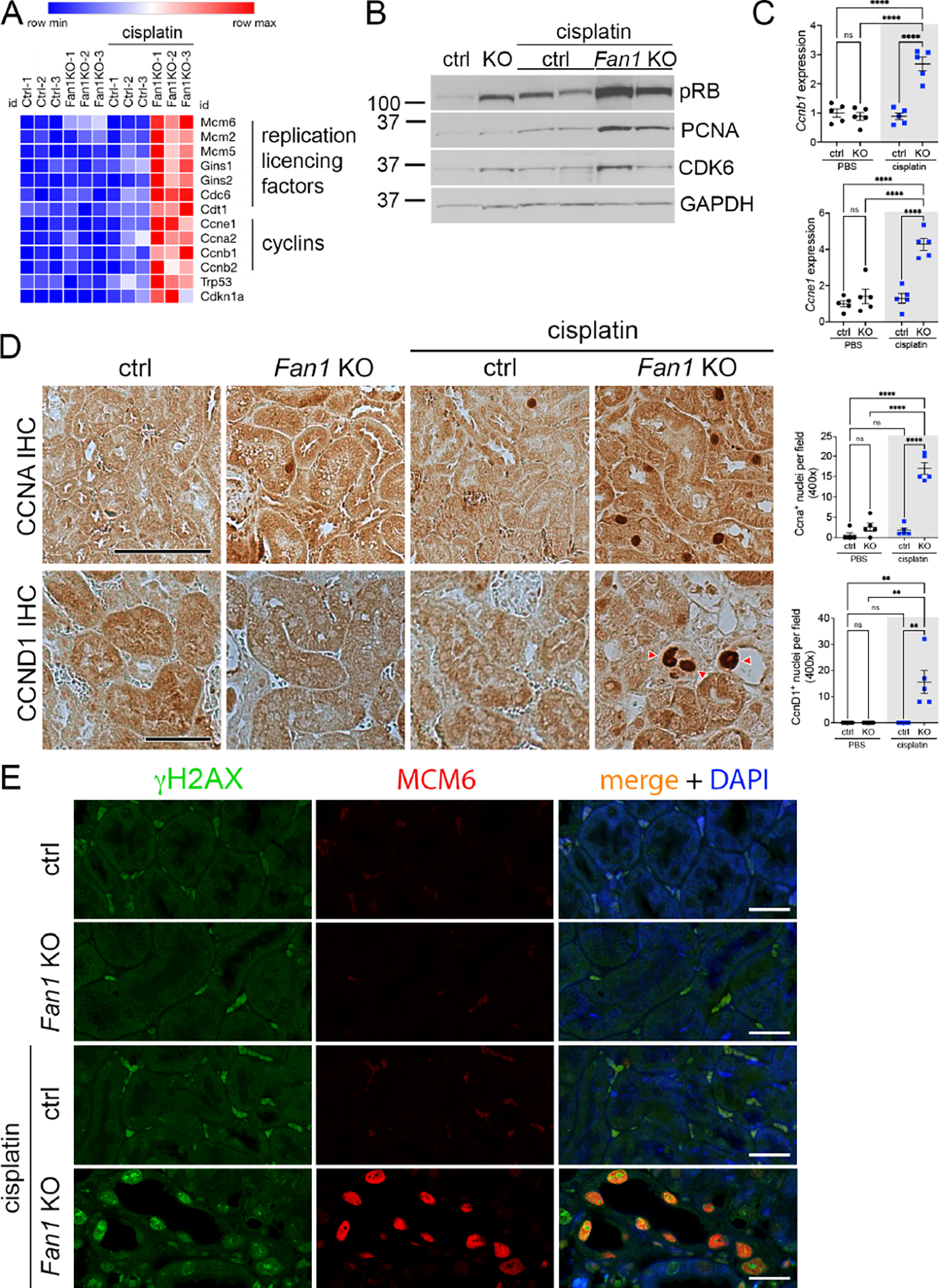
(A) Heatmap of a selection of key cell cycle regulating genes that were upregulated in the RNAseq analysis of Fan1 KO kidneys after cisplatin injury.
(B) Western blot of cell proliferation markers - pRB (S807/811), PCNA and CDK6, in untreated and cisplatin treated kidneys. Gapdh was used as a loading control.
(C) qPCR validation of the increased expression of Ccne1 and Ccnb1 in cisplatin Fan1 KO kidneys. Ccne1 (ctrl 1.3±0.4 vs Fan1 KO 5.2±0.8, ****p<0.0001), Ccnb1 (ctrl 0.7±0.1 vs Fan1 KO 2.6±0.4, ****p<0.0001), n=5 each.
(D) Immunohistochemistry against CCNA and CCND1. The number of CCNA+ cells is increased in cisplatin Fan1 KO kidneys compared with controls. Quantification of CCNA+ nuclei in in 6 random cortical fields (cisplatin ctrl 2.2±0.9 vs cisplatin Fan1 KO 16.0±0.9, ****p<0.0001, n=5 each cohort). Nuclear accumulation of Cyclin D1 (red arrowheads) was observed only in cisplatin treated Fan1 KO kidneys. Quantification of CCND1+ nuclei in 6 random cortical fields (cisplatin treated ctrl 0.0±0.0 vs cisplatin Fan1 KO 14.0±2.6, n=5 each cohort, **p<0.01). Scale bar 50 μm.
(E) Immunofluorescent (IF) analysis of the DNA damage marker γH2AX (green) and minichromosome maintenance protein 6, MCM6 (red) revealed their co-expression in the giant nuclei in cisplatin treated Fan1 KO kidneys, but not in control kidneys. Scale bar 25 μm.
(C,D) Data are presented as the mean ± SEM. A 2-way ANOVA with Tukeys’ post hoc analysis.
Karyomegalic tubular cells fail to complete mitosis
DNA endoreplication can be induced by p21 overexpression in the cell50, 51. Our RNA-seq and confirmatory analysis revealed an increased expression of the tumor suppressor protein Tp53 and its transcriptional target Cdkn1a/p21 in Fan1 KO kidneys (Figure 4A and Figure 5A,B). Importantly, p21 staining was detected only in the karyomegalic nuclei in injured Fan1 KO kidneys (Figure 5A,B), and its expression was maintained in these cells 28 days after the last cisplatin administration (Supplementary Figure 11A). Co-staining of Fan1 KO kidneys with antibodies against Ki67 and p21 (Figure 5C) or MCM2 and p21 (Supplementary Figure 11B), revealed that a significant fraction of PTECs stained positive for Ki67 or MCM2 and p21 in cisplatin treated Fan1 KO kidneys, providing support to the notion that p21 may drive DNA endoreplication in Fan1 KO PTECs. Quantification of the nuclear area in Ki67/p21 double-positive cells confirmed that they overwhelmingly contain enlarged nuclei (Figure 5D). To determine whether p21 upregulation blocks mitosis in Fan1 KO epithelia, as expected for cells undergoing endoreplication, we stained kidneys with antibodies against phosphorylated histone3 (pH3). Indeed, Fan1 KO kidneys displayed increased numbers of PTECs with punctate (G2-specific) vs global (M-specific) pH3 expression pattern (Figure 5E,F), demonstrating that Fan1 KO PTECs fail to complete mitosis after polyploidization.
Figure 5. Karyomegalic cells fail to complete mitosis.
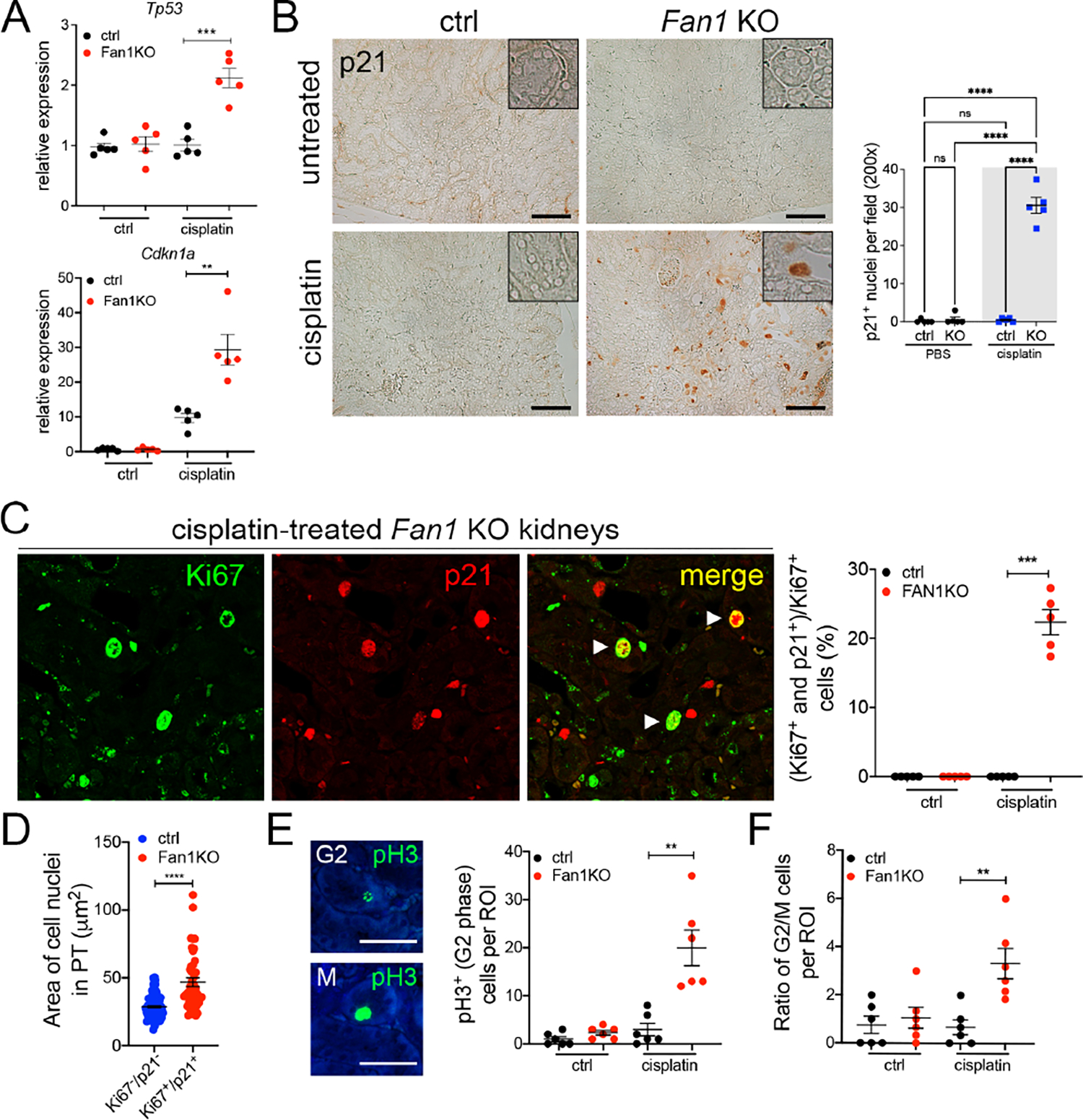
(A) qPCR analysis shows that Tp53 and Cdkn1a/p21 expression is upregulated in Fan1 KO kidneys after cisplatin injury, Tp53 (ctrl 1.0±0.1 vs Fan1 KO 2.1±0.2, ****p<0.001) and Cdkn1a (ctrl 9.7±1.3 vs Fan1 KO 29.4±4.4, ****p<0.01), n=5 each.
(B) Immunohistochemistry against p21 reveals that p21 is expressed in the karyomegalic nuclei Fan1 KO kidneys. The number of p21-positive nuclei were counted in 5 cortical 200x fields per sample (n=5 samples per each cohort, ****p<0.0001). Insets show a magnified view of a proximal tubule. Scale bar 100 μm.
(C) Immunofluorescence staining of Fan1 KO kidneys with Ki67 (green) and p21 (red) shows that a subset of Ki67-positive cells which co-stain with p21. Quantification of the double positive cells shows that ~22% of Ki67+ cells are positive for p21 (0.00 vs 22.36±1.83, ****p<0.0001).
(D) Nuclear area measurement of Ki67/p21 double positive cells in cisplatin treated Fan1 KO kidney proximal tubules (PT) compared to randomly selected nuclei in the PT of control kidneys demonstrates increased nuclear area of the double positive cells (28.7±0.8 n=100 vs 46.8±3.2 n=44, ****p<0.0001).
(E) Detection of G2 and M cell cycle positions with anti-phospho-histone 3 (pH3) antibody in untreated and cisplatin-treated control and Fan1 KO kidneys sections. Quantification of cells in G2 cell cycle position (untreated ctrl 1.0±0.5, untreated Fan1 KO 2.3±0.5, cisplatin ctrl 3.0±1.3, cisplatin Fan1 KO 20.0±3.7, **p<0.01, n=5, each dot = 1ROI). Scale bar 50 μm.
(F) Quantification of the ratio of cells in G2 vs M cell cycle phases demonstrates that ~5 fold more cells are in G2 phase vs M phase in cisplatin treated Fan1 KO kidneys compared to other conditions (untreated ctrl 0.8±0.4, untreated Fan1 KO 1.1±0.4, cisplatin ctrl 0.7±0.3, cisplatin Fan1 KO 3.3±0.6, **p<0.01, n=5, each dot = 1ROI).
(A,B,C,E,F) Data are presented as the mean ± SEM. A 2-way ANOVA with Tukeys’ post hoc analysis.
Blocking cell cycle entry prior to cisplatin treatment prevents karyomegaly and improves kidney function in Fan1 KO mice
To examine whether inhibiting cell cycle activity and DNA replication before cisplatin administration would limit the amount of DNA damage and prevent karyomegaly, we treated Fan1 KO mice with the broad spectrum CDK inhibitor, roscovitine, one hour before cisplatin administration (Figure 6A). Indeed, reduced cell cycle activity in roscovitine pretreated Fan1 KO kidneys (Figure 6B,C,E and Supplementary Figure 12A,B) resulted in significant reduction in γH2AX expression (2.8 fold, Figure 6D), absence of p21-positive cells (Supplementary Figure 12C) and karyomegaly (Figure 6F), demonstrating that roscovitine treatment prevents DNA endoreplication and the accumulation of DNA damage in Fan1 KO kidneys. Importantly, reduced DNA damage in the tubular cells was associated with diminished proximal tubule injury, as measured by KIM1 expression (Figure 6F), as well as reduced BUN levels (Figure 6G). Together, these data demonstrate that blocking cell cycle activity before cisplatin administration limits replication stress, accumulation of DNA damage and preserves kidney function.
Figure 6. Roscovitine administration mitigates cisplatin injury in Fan1 KO mice.
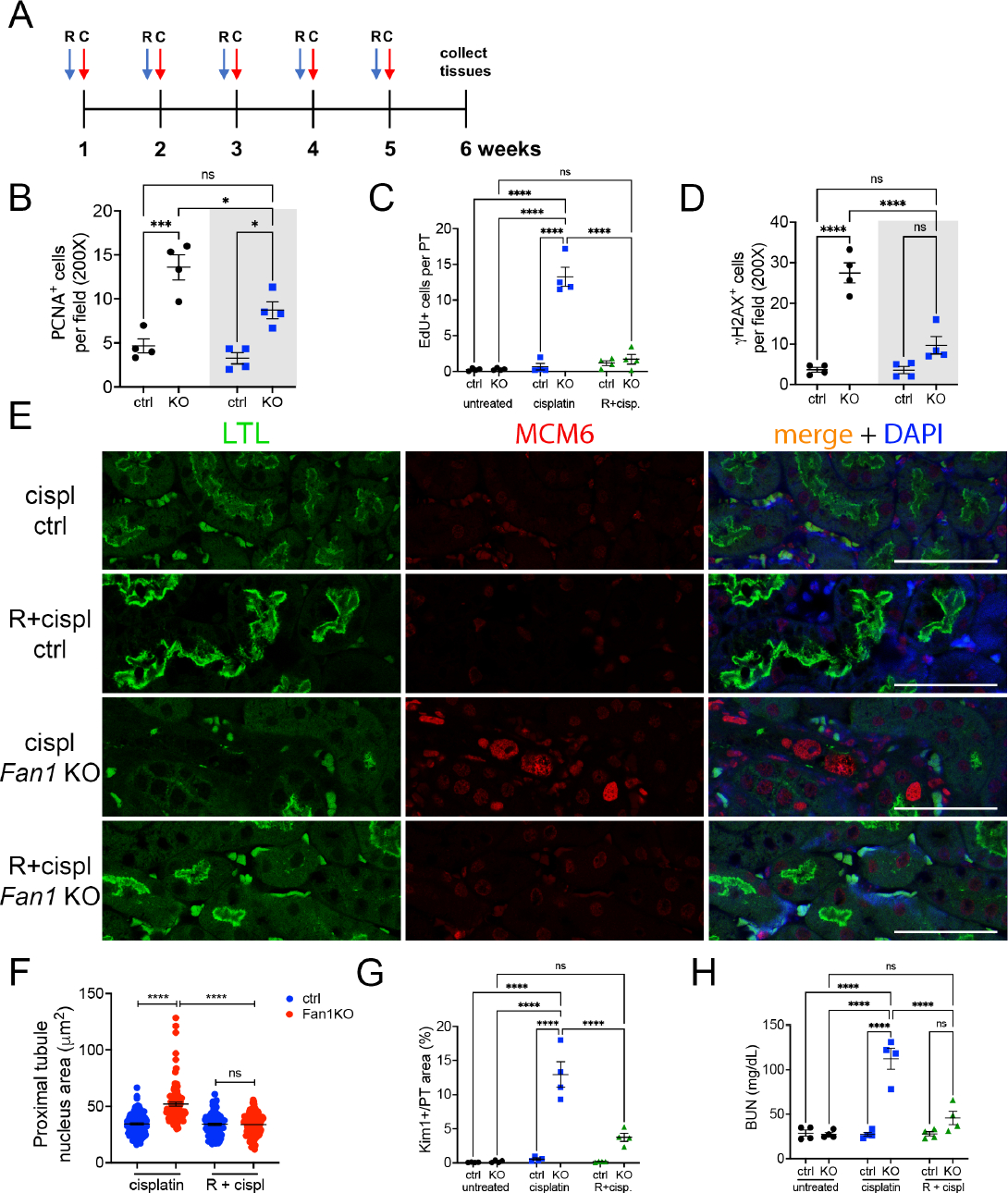
(A) Overview of roscovitine and low dose cisplatin administration protocol. Roscovitine (150 mg/kg) was administered via ip 1 hour before cisplatin (2 mg/kg), and tissues collected on week 6 after the start of the procedure. R – roscovitine, C – cisplatin.
(B) Quantification of PCNA-positive cells in cisplatin and roscovitine/cisplatin treated kidneys reveals that roscovitine treatment blocks S phase cell cycle activity in cisplatin-treated Fan1 KO mice (n=4 mice; *p<0.01, ***p<0.001).
(C) Quantification of EdU-positive cells in cisplatin and roscovitine/cisplatin treated kidneys reveals that roscovitine treatment effectively blocks DNA replication in cisplatin-treated Fan1 KO mice (n=4 mice; ****p<0.0001).
(D) Quantification of γH2AX-positive cells in cisplatin and roscovitine/cisplatin treated kidneys reveals that roscovitine treatment reduces DNA damage in cisplatin-treated Fan1 KO mice (n=4 mice; ****p<0.0001).
(E) IF staining of LTL (green) and MCM6 (red) in cisplatin and roscovitine/cisplatin treated Fan1 KO kidneys. Roscovitine treatment blocks the expression of MCM6 and the formation of karyomegalic nuclei in Fan1 KO proximal tubule cells. Scale bar 50 μm.
(F) Quantification of the nuclear area in proximal tubules of cisplatin and roscovitine/cisplatin treated kidneys demonstrates that roscovitine treatment prevents karyomegaly in Fan1 KO kidneys (cisplatin ctrl 34.4±0.9; cisplatin Fan1 KO 51.9±1.7; R+cispl ctrl 34.1±0.9; R+cispl Fan1 KO 33.7±0.9; ****p<0.0001, n=100 nuclei each).
(G) Quantification of KIM1-positive area in proximal tubules demonstrates that roscovitine administration leads to significant reduction in KIM1 expression in cisplatin treated Fan1 KO kidneys (cisplatin Fan1 KO 12.9±1.9% vs R+cisplatin Fan1 KO 3.7±0.6%, ****p<0.0001), n=4 each.
(H) Blood urea nitrogen measurements in control, cisplatin treated, and roscovitine/cisplatin treated mice. Roscovitine improves kidney function in cisplatin treated Fan1 KO mice, (n=3–4 each; ****p<0.0001).
(B,C,D,G,H) Data are presented as the mean ± SEM. A 2-way ANOVA with Tukeys’ post hoc analysis.
Human FAN1 knockout kidney proximal tubule cells recapitulate KIN in vitro
To investigate the mechanism by which loss of FAN1 results in endoreplication, we established a FAN1 knockout cell line (FAN1 KO hPTEC) (Supplementary Figure 13A). After a pulse of cisplatin the parental and FAN1 KO hPTECs were cultured in a fresh media (Figure 7A), and stained for γH2AX or phospho-ATM (S1871) to follow the kinetics of DNA damage signaling (Figure 7B,C). DDR activity was detected within the first 24 hours after cisplatin treatment and peaked by 48 hours in the parental cell line (Figure 7B,D). Thereafter, the number of γH2AX and pATM nuclear foci declined to baseline and the nuclear morphology returned to normal by 96 hours, indicating successful DNA repair (Figure 7B–E). In contrast, the number of γH2AX and pATM nuclear foci was increased in FAN1 KO hPTECs at 24 hours and 48 hours, respectively, and remained elevated throughout the 96-hour period (Figure 7B–E), demonstrating a failure in DNA damage repair. Importantly, we observed characteristic features of KIN, including increased replication stress, nuclear enlargement, chromosomal fragmentation and aneuploidy in FAN1 KO hPTECs 48 hours after cisplatin (Figure 7B,C,E,F and Supplementary Figure 13B). To follow the cell cycle dynamics of nuclear polyploidization in detail, we combined flow cytometry-based DNA content measurement with Fucci technology52, 53. These data show that injured FAN1 KO hPTECs undergo polyploidization in late-S/G2/M (Figure 7G, Supplementary Figures 14 and 15). In contrast to cellular polyploidization in other kidney injury models54, 55, polyploidization in FAN1 KO hPTECs led to a higher proportion of >4N cells (Figure 7G), suggesting that distinct mechanisms underlie the respective phenotypes. Ultimately, the polyploid FAN1 KO hPTECs exited the cell cycle and maintained a G1-specific Fucci reporter gene expression (Figure 7G, Supplementary Figures 14 and 15).
Figure 7. Human FAN1 knockout proximal tubular epithelial cells undergo DNA re-replication in response to genotoxic damage.
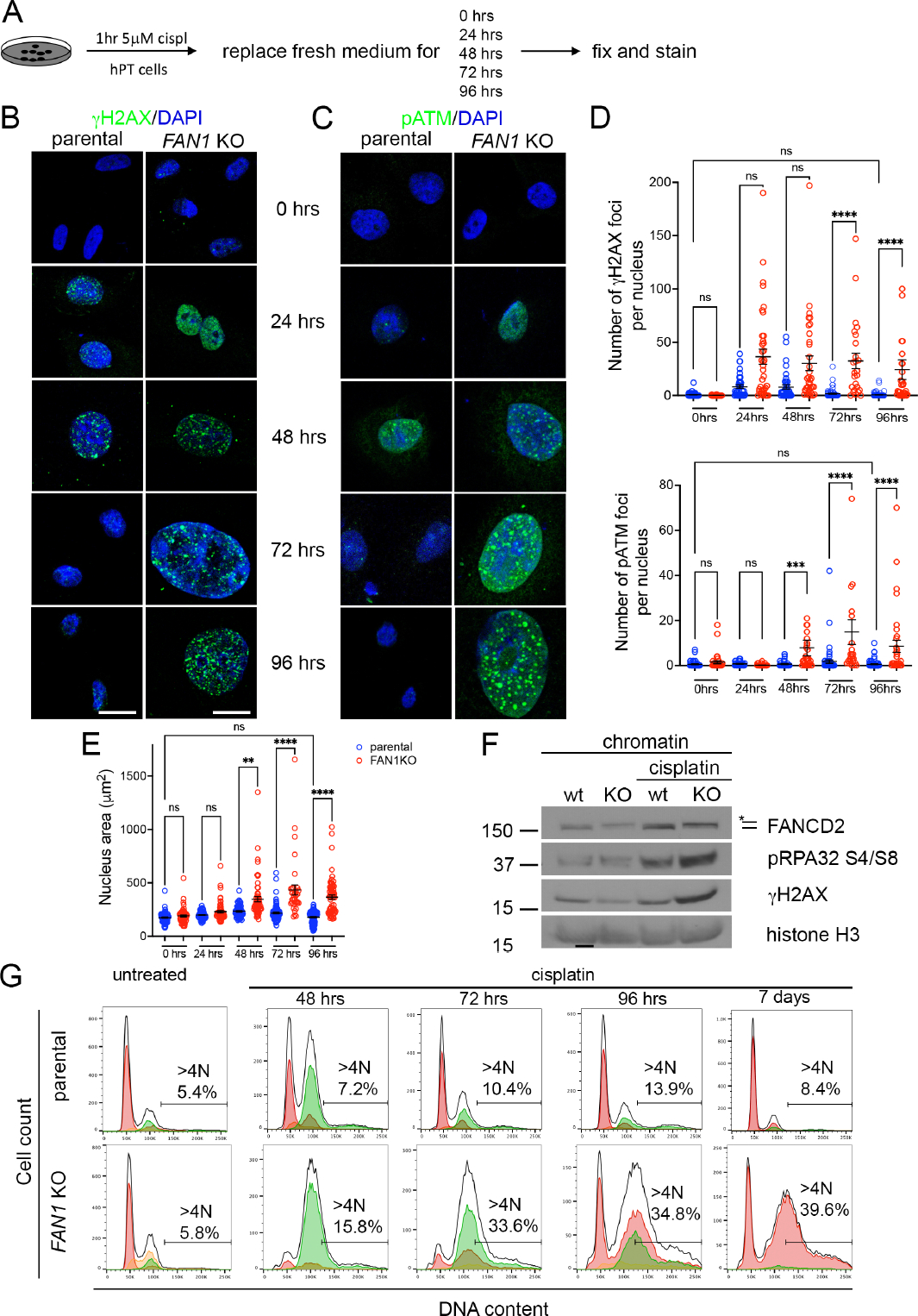
(A) Overview of cisplatin treatment in FAN1 KO hPTEC cells.
(B) γH2AX staining on parental and FAN1 KO hPTEC cells. Scale bar 20 μm.
(C) pATM staining on parental and FAN1 KO hPTEC cells. Scale bar 20 μm.
(D) Quantification of γH2AX and pATM foci numbers per nucleus in the parental and FAN1 KO hPTEC cells, based on the experiments in B and C. n=100 nuclei each condition. ***p<0.001, ****p<0.0001. Data are presented as the mean ± SEM, non-parametric Kruskal-Wallis test.
(E) Increased nuclear area of FAN1 KO hPTEC cells vs the parental cell line after cisplatin treatment. DAPI staining was used to measure the nuclear area, based on the experiments in B and C. **p<0.01, ****p<0.0001. Data are presented as the mean ± SEM, non-parametric Kruskal-Wallis test.
(F) Western blotting of DNA repair pathway markers in hPTECs chromatin preparation reveal the activation of Fanconi anemia repair pathway (ubiquitination of FANCD2, marked by a star), increased levels of replication stress (pRPA32 S4/S8) and DNA double-strand breaks (γH2AX) in cisplatin treated FAN1 KO cells. Histone H3 is used as a chromatin loading control.
(G) Parental and FAN1 KO tFucci(SA)5 hPTECs were treated with cisplatin at 5 μM for 1 hour, as in (A) and cell cycle distribution analyzed by flow cytometry. Cells showing red fluorescence (AzaleaB5(+) and h2–3(−)) are in G1, yellow (AzaleaB5(+) and h2–3(+)) in G1/S, and green (AzaleaB5(−) and h2–3(+)) in late-S/G2/M cell cycle phases. Black contour lines indicate DNA content based on DAPI staining. N values denote DNA content as a multiple of the normal haploid genome. The percentage of cells with a DNA content >4N (polyploid cells) is shown. Untreated parental and FAN1 KO hPTECs have similar cell cycle profiles. Exposure to cisplatin results in FAN1 KO hPTEC accumulation in late-S/G2/M cell cycle phases during which aberrant endoreplication or re-replication occurs, giving rise to polyploidy. Subsequently (by day 7 after cisplatin), polyploid hPTECs exit the cell cycle and express only the G1-specific Fucci reporter gene. These data demonstrate that polyploidization in injured FAN1 KO hPTECs does not result in a simple duplication of the genome that would be detectable by the presence of distinct peaks corresponding to 4N, 8N, 16N etc in the DNA content profile. Instead, the cells show a DNA content profile that is skewed to the right, indicative of unequal chromosome amplification (aneuploidy). It is possible, however, that a subset of the polyploid cells are in a tetraploid G1 state (4N). These data represent three independent experiments.
Next, we examined the expression of key proteins associated with replication stress and DNA licensing to determine whether loss of FAN1 indeed causes aberrant DNA re-replication in hPTECs. Analysis of whole cell lysates and chromatin-bound proteins from parental and FAN1 KO hPTCs (Figure 8A) showed that exposure to cisplatin led to a dramatic increase in the chromatin-bound pATR (S428), ubiquitinated FANCD2, pRPA32 (S4/S8) and γH2AX in FAN1 KO hPTCs, but not in parental cells (Figure 8B), indicating a profound failure of the mutant cells to resolve interstrand crosslinks and the resulting replication stress. FAN1 KO cells also displayed phosphorylation of CHK1 on S317, as well as an increase in phospho-p53 on S15 and its target p21, suggesting a mitotic block (Figure 8B). Consistent with this notion, FAN1 KO hPTECs displayed reduced phospho-histone 3 levels. Importantly, key replication licensing factors CDT1 and CDC6 were dramatically increased at the chromatin of FAN1 KO cells, providing compelling evidence that FAN1 KO cells undergo unscheduled DNA re-replication after genotoxic injury (Figure 8B). Critically, roscovitine treatment mitigated the extent of DNA damage and p21 expression, and blocked CDT1/CDC6 upregulation in KO cells (Figure 8B), providing an explanation for its effect in blocking karyomegaly in FAN1 KO kidneys (Figure 6). Finally, to address whether p21 upregulation is required for DNA endoreplication in FAN1 KO cells after genotoxic injury, we treated cells with the p21 inhibitor UC2288 (Figure 8C). Indeed, inhibiting p21 expression after cisplatin treatment blocked CDT1/CDC6 upregulation (Figure 8D) and nuclear enlargement in FAN1 KO cells (Supplemental Figure 16A), consistent with the notion that p21 upregulation drives polyploidization and karyomegaly in FAN1 KO cells and kidneys.
Figure 8. Inhibiting cell cycle activity or p21 expression blocks DNA re-replication in FAN1 KO hPTECs.
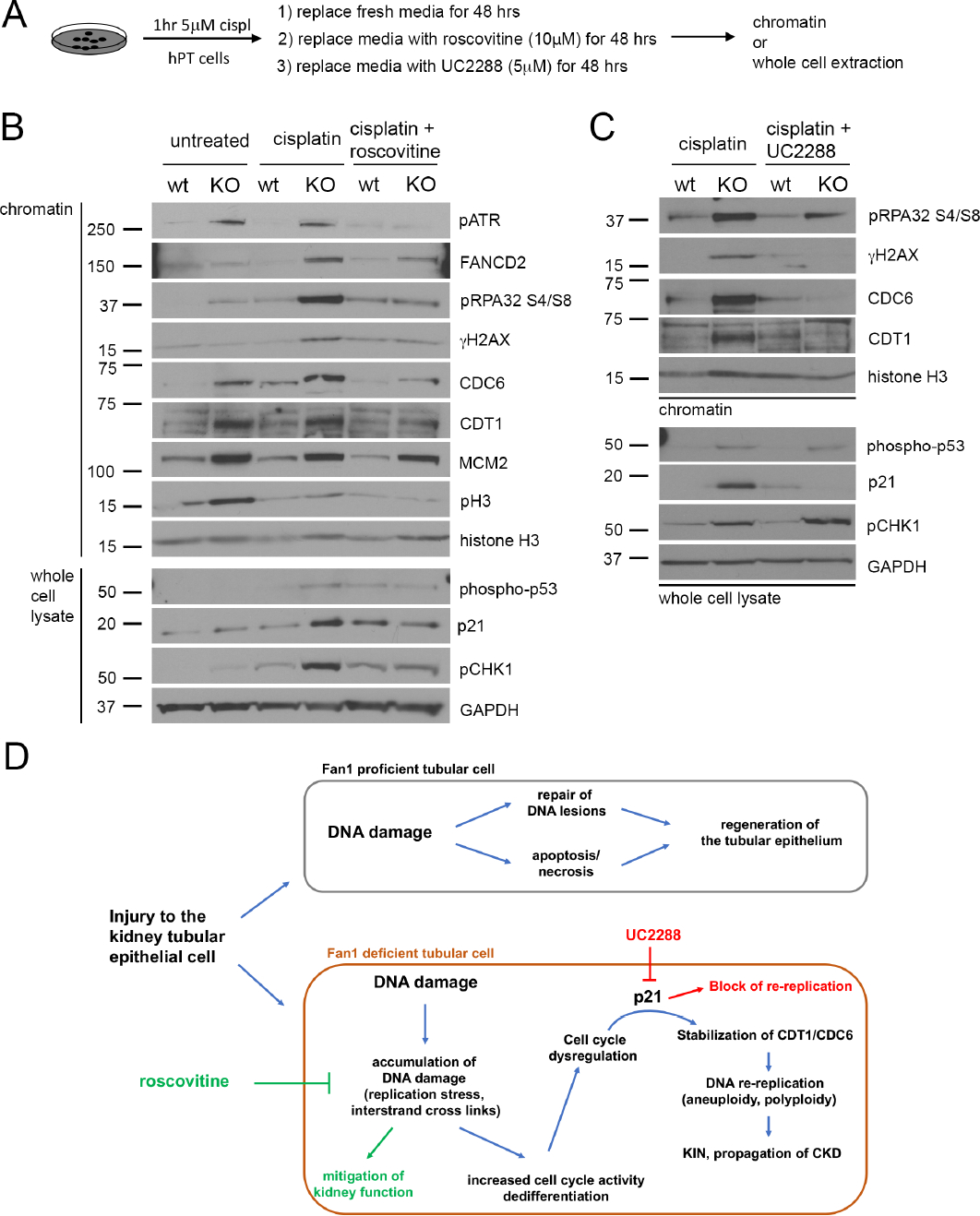
(A) Schematics of roscovitine and UC2288 treatment in FAN1 KO hPTEC cells.
(B) Representative Western blot images of DNA replication licensing proteins and DNA damage markers in control, cisplatin treated and cisplatin + roscovitine treated parental and FAN1 KO hPTECs. Proteins were detected either in chromatin extractions or total cell lysates, as indicated. Histone H3 was used as a loading control for chromatin and GAPDH for whole cell lysate.
(C) Representative Western blot images of DNA replication licensing proteins and DNA damage markers in cisplatin treated and cisplatin + UC2288 treated parental and FAN1 KO hPTECs. Proteins were detected either in chromatin extractions or total cell lysates, as indicated. Histone H3 was used as a loading control for chromatin and GAPDH for whole cell lysate.
(D) Model of DNA damage induced cell cycle abnormalities in a DNA repair deficient proximal tubular cell. Kidney proximal tubule cells with proficient DNA repair will resolve non-lethal DNA damage and regenerate the injured tubule. In contrast, DNA repair deficient cells will accumulate DNA damage through replication stress, which leads to abnormal cell cycle activity and p21-dependent stabilization of CDT1 and CDC6. p21 expressing tubular cells undergo DNA re-replication, which propagates further genomic instability and progression to CKD.
Together, the in vitro findings in hPTECs corroborate in vivo data and provide a mechanistic insight into how impaired DNA repair in kidney tubular cells leads to accumulation of DNA damage, cellular polyploidization and progression to CKD in FAN1-deficient kidneys (Figure 8C).
DISCUSSION
In this study, we have used the DNA repair deficient Fan1 knockout mouse model to investigate the biological function of FAN1 in the kidney and to characterize the long-term effects of persistent DNA damage in the renal tubular epithelium. This work extends our previous studies on the role of Fan156 and provides novel mechanistic insights into the role of impaired DNA repair as an underpinning of CKD, an emerging paradigm in the field of CKD research57–61. Subclinical cisplatin and short-term UUO both induced a strong DDR activity in Fan1 KO but not in control kidneys, demonstrated by the expression of replication stress markers (ubiquitinated-FANCD2, pRPA, γH2AX)15, 23, and in the case of cisplatin also double-strand DNA brake markers (53BP1, pKAP1). Our analysis showed that sensitivity of Fan1 KO kidneys to UUO was likely due to increased oxidative stress in the mutant kidneys, revealing a novel function of Fan1 in the repair of oxidative DNA damage, a common “secondary injury” in non-genotoxic kidney damage34, 62–64. In addition, modeling of the long-term effects of impaired DNA repair in the kidney demonstrated that persistent DNA damage affects tubular repair and drives CKD progression by maintaining tubular VCAM1 and CCL25, 41.
One key finding here is that persistent DNA damage in the PTECs causes cellular injury whose biomarker phenotype is indistinguishable from other forms of kidney injury, including high dose cisplatin61, 65, UUO66, folic acid67, 5/6 nephrectomy68, and IRI69, 70. Importantly, some of these injury types have also been shown to elicit the DNA damage-related repair signaling and checkpoint activation in their pathophysiology71–73, suggesting that the DDR might be the apical determinant for the long-term outcome to many (if not all) renal insults and dictates whether injured kidneys progress to chronic kidney disease or recover from the tubular cell injury.
A characteristic feature of the loss of FAN1 function in kidney tubular epithelial cells is the formation of karyomegalic nuclei with aneuploid chromosome count13, 25. This aberrant karyotype is recapitulated in other mouse models of Fan1-deficiency26, 27, 56 and in human FAN1 knockout PTECs, as demonstrated in this study. However, the cellular mechanism leading to chromosomal abnormalities in FAN1 KO cells had not been investigated previously. Our pharmacological studies with the CDK inhibitor roscovitine and p21 inhibitor UC2288, together with FACS and FUCCI analyses, revealed that chromosomal abnormalities in FAN1-deficient cells arise due to unscheduled DNA re-replication and are attributed to persistent p21 overexpression in response to accumulation of DNA damage during the preceding S-phase. Accordingly, treatment with roscovitine suppressed DNA replication and accumulation of DNA damage after cisplatin injury, prevented p21 upregulation and improved kidney function in Fan1 KO mice. Mechanistically, we showed that increased p21 expression triggered pathological DNA endoreplication in FAN1 KO cells by stabilizing the replication licensing proteins CDT1 and CDC6, which has been shown to induce aberrant MCM2–7 chromatin loading and DNA re-replication in other models50, 51. Mitotic block in FAN1KO cells may have been reinforced by the increased levels of Pkmyt1, a member of the WEE kinase family, which inhibits Cdk1 kinase activity in G2 cells74. Polyploidization in FAN1 KO PTECs is likely irreversible, as demonstrated by the long-term presence of karyomegalic cells with chronic DNA damage in cisplatin Fan1 KO kidneys as well as in patients with FAN1 mutations16, 25.
Our finding that FAN1-deficiency leads to DNA endoreplication suggests that targeted therapy against this pathway has the potential to improve renal outcome after injury. Roscovitine is an experimental drug with described function in blocking precocious replication origin firing in cells with damaged DNA75, 76 and delaying cell cycle entry, thereby allowing for FAN1-independent DDR mechanisms to compensate for the loss of FAN1 activity. While we showed that Roscovitine therapy effectively blocked cell cycle activity and reduced DNA damage (i.e. replication stress) in Fan1 KO kidneys when administered before cisplatin, our in vitro studies with human FAN1KO cells suggest that Roscovitine treatment post-injury may also lead to improved cellular outcomes. Together these observations demonstrate the potential clinical utility of Roscovitine in DDR-associated renal injury and delineate the role of FAN1 in renal tubular regeneration. In addition, in vivo studies with the p21 inhibitor UC2288 should be performed in Fan1 KO mice to assess its impact on tubular regeneration, in the future.
In conclusion, FAN1 is essential for tubular repair and indispensable for genome maintenance in PTECs, consistent with its role as a putative tumor suppressor27. We propose that KIN patients with FAN1 mutations should avoid exposure to agents that may induce further genomic instability. The findings in Fan1 KO mouse and cell culture models identified the molecular mechanism by which impaired DNA repair leads to failed tubular repair and promotes the progression of chronic kidney disease, outcomes that may be modifiable by therapeutic targeting of cell cycle activity.
Supplementary Material
TRANSLATIONAL STATEMENT.
Faulty DNA repair leads to the development of chronic kidney disease (CKD) in humans. However, the mechanism by which DNA damage affects tubular cell function remains unknown. Here, we show that accumulation of DNA damage causes aberrant DNA endoreplication and polyploidization in cells deficient for the DNA repair protein FAN1. Tubular cell polyploidization is associated with epithelial dedifferentiation, tubular injury and loss of function. Blocking DNA replication in FAN1-deficient kidneys mitigates these defects and preserves function, demonstrating that therapeutic targeting of cell cycle activity may provide avenues to prevent kidney injury and/or development of CKD in patients with FAN1 mutations.
ACKNOWLEDGEMENTS
We thank Drs. David Emlet and John Kellum for human primary renal proximal tubule cells. This research was supported by a grant from the National Institutes of Health to R.A. (DK115403). RJT is supported by an ASN Gottschalk Award. YLP is supported by grants from UPMC Children’s Hospital of Pittsburgh and North American Mitochondrial Disease Consortium. The Statistics Consulting Center at the University of Pittsburgh provided assistance with verifying the assumptions of the two-way ANOVA.
Footnotes
DISCLOSURES
None.
Supplementary information is available on Kidney International’s website.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Lv JC, Zhang LX. Prevalence and Disease Burden of Chronic Kidney Disease. Adv Exp Med Biol 2019; 1165: 3–15. [DOI] [PubMed] [Google Scholar]
- 2.Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension 2003; 42: 1050–1065. [DOI] [PubMed] [Google Scholar]
- 3.Canaud G, Bonventre JV. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol Dial Transplant 2015; 30: 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang-Panesso M, Kadyrov FF, Lalli M, Wu H, et al. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J Clin Invest 2019; 129: 5501–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirita Y, Wu H, Uchimura K, Wilson PC, et al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A 2020; 117: 15874–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Airik R, Schueler M, Airik M, Cho J, et al. A FANCD2/FANCI-Associated Nuclease 1-Knockout Model Develops Karyomegalic Interstitial Nephritis. J Am Soc Nephrol 2016; 27: 3552–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer 1998; 34: 1522–1534. [DOI] [PubMed] [Google Scholar]
- 8.Shiraishi F, Curtis LM, Truong L, Poss K, et al. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 2000; 278: F726–736. [DOI] [PubMed] [Google Scholar]
- 9.Latcha S, Jaimes EA, Patil S, Glezerman IG, et al. Long-Term Renal Outcomes after Cisplatin Treatment. Clin J Am Soc Nephrol 2016; 11: 1173–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol 2013; 5: a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, Zhang J, Bellani MA, Pokharel D, et al. Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Rep 2019; 27: 1794–1808 e1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Martinez D, Liang CC, Cohn MA. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci 2016; 73: 3097–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashraf S, Gee HY, Woerner S, Xie LX, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest 2013; 123: 5179–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segui N, Mina LB, Lazaro C, Sanz-Pamplona R, et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015; 149: 563–566. [DOI] [PubMed] [Google Scholar]
- 15.Lachaud C, Moreno A, Marchesi F, Toth R, et al. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016; 351: 846–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray SL, Connaughton DM, Fennelly NK, Kay EW, et al. Karyomegalic Interstitial Nephritis: Cancer Risk Following Transplantation. Nephron 2020; 144: 49–54. [DOI] [PubMed] [Google Scholar]
- 17.Burry AF. Extreme dysplasia in renal epithelium of a young woman dying from hepatocarcinoma. J Pathol 1974; 113: 147–150. [DOI] [PubMed] [Google Scholar]
- 18.Mihatsch MJ, Gudat F, Zollinger HU, Heierli C, et al. Systemic karyomegaly associated with chronic interstitial nephritis. A new disease entity? Clin Nephrol 1979; 12: 54–62. [PubMed] [Google Scholar]
- 19.Liu T, Ghosal G, Yuan J, Chen J, et al. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010; 329: 693–696. [DOI] [PubMed] [Google Scholar]
- 20.Kratz K, Schopf B, Kaden S, Sendoel A, et al. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 2010; 142: 77–88. [DOI] [PubMed] [Google Scholar]
- 21.MacKay C, Declais AC, Lundin C, Agostinho A, et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 2010; 142: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smogorzewska A, Desetty R, Saito TT, Schlabach M, et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 2010; 39: 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaudhury I, Stroik DR, Sobeck A. FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol Cell Biol 2014; 34: 3939–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porro A, Berti M, Pizzolato J, Bologna S, et al. FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat Commun 2017; 8: 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spoendlin M, Moch H, Brunner F, Brunner W, et al. Karyomegalic interstitial nephritis: further support for a distinct entity and evidence for a genetic defect. Am J Kidney Dis 1995; 25: 242–252. [DOI] [PubMed] [Google Scholar]
- 26.Thongthip S, Bellani M, Gregg SQ, Sridhar S, et al. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev 2016; 30: 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lachaud C, Slean M, Marchesi F, Lock C, et al. Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev 2016; 30: 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwano M, Plieth D, Danoff TM, Xue C, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002; 110: 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cleaver JE, Feeney L, Revet I. Phosphorylated H2Ax is not an unambiguous marker for DNA double-strand breaks. Cell Cycle 2011; 10: 3223–3224. [DOI] [PubMed] [Google Scholar]
- 30.Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 2014; 34: 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Czerwinska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. J Biomed Sci 2017; 24: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams RM, Zhang X. Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog Biophys Mol Biol 2020. [DOI] [PubMed] [Google Scholar]
- 33.Dendooven A, Ishola DA Jr., Nguyen TQ, Van der Giezen DM, et al. Oxidative stress in obstructive nephropathy. Int J Exp Pathol 2011; 92: 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aamann MD, Norregaard R, Kristensen ML, Stevnsner T, et al. Unilateral ureteral obstruction induces DNA repair by APE1. Am J Physiol Renal Physiol 2016; 310: F763–F776. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Liu J, Pang P, Krautzberger AM, et al. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep 2015; 12: 1325–1338. [DOI] [PubMed] [Google Scholar]
- 36.Kang HM, Huang S, Reidy K, Han SH, et al. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep 2016; 14: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 1994; 93: 2175–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imgrund M, Grone E, Grone HJ, Kretzler M, et al. Re-expression of the developmental gene Pax-2 during experimental acute tubular necrosis in mice 1. Kidney Int 1999; 56: 1423–1431. [DOI] [PubMed] [Google Scholar]
- 39.Kuwabara T, Mori K, Mukoyama M, Kasahara M, et al. Urinary neutrophil gelatinase-associated lipocalin levels reflect damage to glomeruli, proximal tubules, and distal nephrons. Kidney Int 2009; 75: 285–294. [DOI] [PubMed] [Google Scholar]
- 40.Viau A, El Karoui K, Laouari D, Burtin M, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest 2010; 120: 4065–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerhardt LMS, Liu J, Koppitch K, Cippa PE, et al. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci U S A 2021; 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, et al. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 2002; 30: 285–289. [DOI] [PubMed] [Google Scholar]
- 44.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 2015; 21: 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Remus D, Beuron F, Tolun G, Griffith JD, et al. Concerted loading of Mcm2–7 double hexamers around DNA during DNA replication origin licensing. Cell 2009; 139: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bermejo R, Vilaboa N, Cales C. Regulation of CDC6, geminin, and CDT1 in human cells that undergo polyploidization. Mol Biol Cell 2002; 13: 3989–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev 2007; 21: 497–518. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y, Pozo PN, Oh S, Stone HM, et al. Distinct and sequential re-replication barriers ensure precise genome duplication. PLoS Genet 2020; 16: e1008988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Truong LN, Wu X. Prevention of DNA re-replication in eukaryotic cells. J Mol Cell Biol 2011; 3: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev 2008; 22: 2507–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galanos P, Vougas K, Walter D, Polyzos A, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016; 18: 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakaue-Sawano A, Yo M, Komatsu N, Hiratsuka T, et al. Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle. Mol Cell 2017; 68: 626–640 e625. [DOI] [PubMed] [Google Scholar]
- 53.Ando R, Sakaue-Sawano A, Shoda K, Miyawaki A. Two new coral fluorescent proteins of distinct colors for sharp visualization of cell-cycle progression. bioRxiv 2020: 2020.2003.2030.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manolopoulou M, Matlock BK, Nlandu-Khodo S, Simmons AJ, et al. Novel kidney dissociation protocol and image-based flow cytometry facilitate improved analysis of injured proximal tubules. Am J Physiol Renal Physiol 2019; 316: F847–F855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lazzeri E, Angelotti ML, Peired A, Conte C, et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat Commun 2018; 9: 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braun DA, Rao J, Mollet G, Schapiro D, et al. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat Genet 2017; 49: 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Airik R, Slaats GG, Guo Z, Weiss AC, et al. Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J Am Soc Nephrol 2014; 25: 2573–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaki M, Airik R, Ghosh AK, Giles RH, et al. Exome Capture Reveals ZNF423 and CEP164 Mutations, Linking Renal Ciliopathies to DNA Damage Response Signaling. Cell 2012; 150: 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lans H, Hoeijmakers JH. Genome stability, progressive kidney failure and aging. Nat Genet 2012; 44: 836–838. [DOI] [PubMed] [Google Scholar]
- 60.Canaud G, Brooks CR, Kishi S, Taguchi K, et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci Transl Med 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kishi S, Brooks CR, Taguchi K, Ichimura T, et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J Clin Invest 2019; 129: 4797–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu J, Li X, Zhang M, Chen Z, et al. Regulation of MUTYH, a DNA Repair Enzyme, in Renal Proximal Tubular Epithelial Cells. Oxid Med Cell Longev 2015; 2015: 682861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nezu M, Souma T, Yu L, Suzuki T, et al. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int 2017; 91: 387–401. [DOI] [PubMed] [Google Scholar]
- 64.Saraswati S, Martínez P, Graña-Castro O, Blasco MA. Short and dysfunctional telomeres sensitize the kidneys to develop fibrosis. Nature Aging 2021; 1: 269–283. [DOI] [PubMed] [Google Scholar]
- 65.Landau SI, Guo X, Velazquez H, Torres R, et al. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int 2019; 95: 797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Humphreys BD, Xu F, Sabbisetti V, Grgic I, et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 2013; 123: 4023–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He S, Liu N, Bayliss G, Zhuang S. EGFR activity is required for renal tubular cell dedifferentiation and proliferation in a murine model of folic acid-induced acute kidney injury. Am J Physiol Renal Physiol 2013; 304: F356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang WC, Wright AF, Roman AJ, Cideciyan AV, et al. RPGR-Associated Retinal Degeneration in Human X-Linked RP and a Murine Model. Investigative Ophthalmology & Visual Science 2012; 53: 5594–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang Y, Jiang T, Ouyang J, Zhou Q, et al. Cell atavistic transition: Paired box 2 re-expression occurs in mature tubular epithelial cells during acute kidney injury and is regulated by Angiotensin II. PLoS One 2014; 9: e93563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng C, Wang Q, Wang J, Liu F, et al. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: Case reports and literature review. Medicine (Baltimore) 2017; 96: e8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang L, Besschetnova TY, Brooks CR, Shah JV, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010; 16: 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsuruya K, Furuichi M, Tominaga Y, Shinozaki M, et al. Accumulation of 8-oxoguanine in the cellular DNA and the alteration of the OGG1 expression during ischemia-reperfusion injury in the rat kidney. DNA Repair (Amst) 2003; 2: 211–229. [DOI] [PubMed] [Google Scholar]
- 73.Ma Z, Wei Q, Dong G, Huo Y, et al. DNA damage response in renal ischemia-reperfusion and ATP-depletion injury of renal tubular cells. Biochim Biophys Acta 2014; 1842: 1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther 2004; 3: 305–313. [DOI] [PubMed] [Google Scholar]
- 75.Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005; 122: 915–926. [DOI] [PubMed] [Google Scholar]
- 76.Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A 2010; 107: 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.