

Abstract
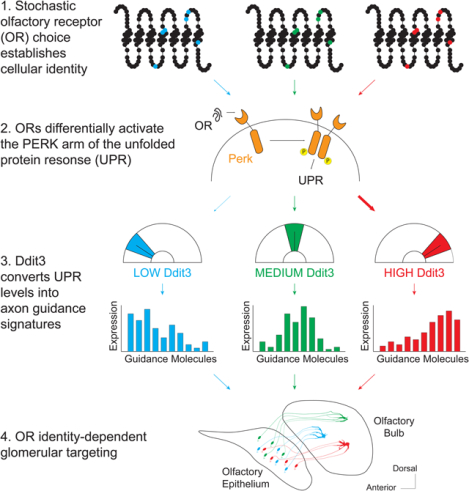
Olfactory sensory neurons (OSNs) convert the stochastic choice of one out of >1000 olfactory receptor (OR) genes into precise and stereotyped axon targeting of OR-specific glomeruli in the olfactory bulb. Here, we show that the PERK arm of the unfolded protein response (UPR) regulates both the glomerular coalescence of like axons, and the specificity of their projections. Subtle differences in OR protein sequences lead to distinct patterns of endoplasmic reticulum (ER) stress during OSN development, converting OR identity into distinct gene expression signatures. We identify the transcription factor Ddit3 as a key effector of PERK signaling that maps OR-dependent ER stress patterns to the transcriptional regulation of axon guidance and cell adhesion genes, instructing targeting precision. Our results extend the known functions of the UPR from a quality control pathway that protects cells from misfolded proteins, to a sensor of cellular identity that interprets physiological states to direct axon wiring.
Graphical Abstract
In Brief
ER stress levels in olfactory sensory neurons is influenced by olfactory receptor protein sequences, which lead to distinct axon guidance gene expression and specific axon targeting in glomeruli.
Introduction
To detect and discriminate between vast numbers of volatile chemicals, terrestrial vertebrates express more than 1000 olfactory receptor (OR) genes in olfactory sensory neurons (OSNs) of their main olfactory epithelia (MOE)(Buck and Axel, 1991). The transformation of odor detection into odor perception requires two key features: singular OR expression in each OSN (Chess et al., 1994) and singular OR representation in the glomeruli of the olfactory bulb (OB) (Mombaerts et al., 1996). The former is achieved by the stochastic, monogenic and monoallelic OR transcription in each mature OSN, while the latter is accomplished by the coalescence of all the OSN axons with the same OR to 2–4 stereotypic glomeruli in the OB. These two processes transform chemical information into distinct and reproducible patterns of glomerular activation that encode for odor identity. As important they are for odor perception, however, the “one receptor per neuron” and “one receptor per glomerulus” principles pose extreme regulatory challenges that require extreme molecular solutions.
Singular OR expression requires the assembly of interchromosomal compartments (Clowney et al., 2012), which place one OR allele into a multi-enhancer hub (Markenscoff-Papadimitriou et al., 2014; Monahan et al., 2019). Translation of the chosen OR in the ER then activates PERK signaling and Atf5 translation, stabilizing this choice for the life of the OSN(Dalton et al., 2013; Lyons et al., 2013). OSN axon guidance, is defined by two factors: the position of the OSN in anatomical regions (“zones”) of the MOE (Ressler et al., 1994; Sullivan et al., 1995) and the sequence of the expressed OR protein(Wang et al., 1998). These factors act hierarchically to shape axon guidance, as zonal boundaries preserve a coarse topographic representation between the MOE and OB, while OR identity instructs targeting of specific, zonally-delimited glomeruli. “Swap” experiments where OSNs choosing a given OR gene express a different OR protein revealed that both identity components control circuit formation (Feinstein and Mombaerts, 2004; Wang et al., 1998). The OR protein regulates axon targeting by controlling the expression of guidance and adhesion molecules (Takeuchi and Sakano, 2014). Specifically, OR basal activity couples to Gs to determine the levels of Nrp1/PlxnA1/Sema3a facilitating targeting across the AP axis of the OB (Imai et al., 2006; Nakashima et al., 2013), while zonal identity broadly organizes the DV axis of the OB through Nrp2 and Sema3F(Takeuchi et al., 2010). Finally, odor-evoked GOlf activity patterns map OR identity to the expression of cell adhesion proteins, instructing glomerular segregation (Nakashima et al., 2019; Serizawa et al., 2006).
Neural activity provides a compelling mechanism for mapping each OR to unique patterns of extracellular guidance molecules. But is activity the sole determinant of targeting? Several observations argue that additional mechanisms must exist, especially for the remarkably precise OR-dependent glomerular segregation step. For one, deleting factors that mediate odor-evoked OR signaling (Belluscio et al., 1998)) and axon potential (Brunet et al., 1996; Lin et al., 2000)) does not disrupt overall glomerular targeting (Zheng et al., 2000). Further, genetically altering odor selectivity via OR mutagenesis does not necessarily alter glomerular specificity (Zhang et al., 2012). Finally, OSNs from the same MOE expressing the same OR have vastly different responses to the same odor (Grosmaitre et al., 2006). Adding to these experimental observations the simple fact that the sensory environment is constantly changing, we reasoned that odor-evoked mechanisms alone cannot account for the stability and precision of OR-instructed glomerular targeting.
Seeking activity-independent axon guidance mechanisms, we asked if the propensity of an OR protein to activate the PERK arm of the unfolded protein response (UPR) (Dalton et al., 2013) could influence targeting specificity. We report that OSNs interrogate the OR protein sequence, tiling certain amino acids to induce different patterns of ER stress that correlate with the expression of axon guidance molecules. Deleting both copies of Perk after OR choice perturbs glomerular coalescence, while altering PERK signaling levels shifts glomerular locations. Using in silico network reconstruction algorithms we identified Ddit3 as an ER stress-responsive regulator that controls OR-directed axon guidance in a concentration-dependent fashion. Our data identify the UPR as a mechanism that couples receptor identity to axon guidance, proposing an alternative regulatory paradigm for the assembly of neural circuits.
Results
Generating a translational fluorescent reporter for ER stress
If OSN identity maps to axon guidance via distinct patterns of ER stress, then PERK signaling must vary with OR identity. To test this, we replaced the coding sequence (CDS) of Atf5 with iRFPp2a-Cre (Figure 1A). Atf5 is a PERK induced transcription factor that is highly transcribed in the OSN lineage (Supplemental Figure S1A) but only translated at the onset of singular OR expression (Dalton et al., 2013). Atf5 translation requires OR-induced, PERK-mediated eIF2a phosphorylation, which bypasses two inhibitory upstream open reading frames (uORFs) contained in the 5’ UTR of its mRNA (Figure 1B) (Dalton et al., 2013). The two Atf5 uORFs are intact in our reporter, placing iRFP fluorescence under the same translational regulation as ATF5 (Figure 1A, B). Heterozygous, Atf5(rep/+) mice, display normal OR expression patterns (Supplemental Figure S1B), allowing use of this reporter for UPR quantification.
Figure 1: OSN types have distinct ER stress patterns.
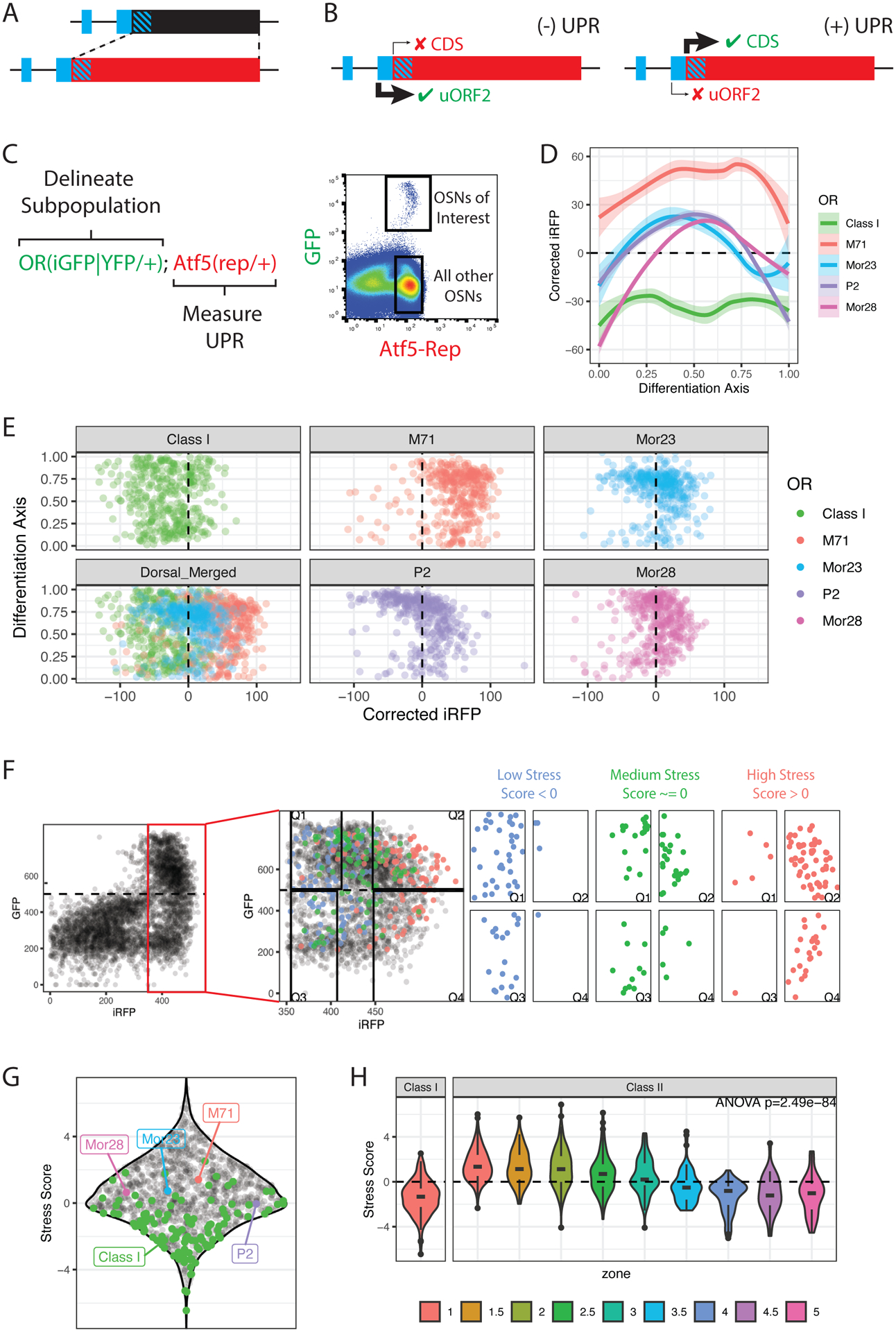
(A) Design of the Atf5(rep) allele. uORFs are shown in blue, diagonal stripes indicate overlap between uORF2 and CDS/reporter.
(B) Regulation of the Atf5(rep) in the presence or absence of UPR (Also see Supplemental Figure S1).
(C) The strategy to measure ER stress in OSN subpopulations. OR loci are tagged with iresGFP or YFP to delimit an OSN subpopulation of interest. The Atf5(rep) encoded iRFP is used to measure UPR levels. Representative flow cytometry data shows the GFP+ OSNs of interest and the iRFP+GFP− population used for normalization between experiments.
(D) Flow cytometry measurements of ER stress using the Atf5(rep) allele in various OSN types. Loess smoothed curves and standard errors of corrected iRFP levels (y-axis) versus differentiation stage (x-axis) are shown for each subpopulation.
(E) Point plots of differentiation stage (y-axis) vs corrected iRFP levels (x-axis) for 400 cells sampled from each OSN type.
(F) The strategy to measure ER stress scores (ERS) in all OSN types. Representative flow cytometry data from Atf5(rep/+); Omp(iresGFP/+) mice shown in black. iOSN (iRFP+GFP−) and mOSN (iRFP+GFP+) populations are delimited by the dashed lines, boxed in red, and shown at higher magnification on the right. iRFP measurements in class I OSNs (blue- low stress), Mor28 OSNs (green- medium stress) and M71 OSNs (red- high stress) are superimposed. We segment iOSN and mOSN populations into high (Q4 and Q2) and low-stress (Q1 and Q3) bins FAC-sorted for RNA-seq. ERS for each OR are then calculated as ERS=1/2(log2(Q2/Q1) + log2(Q4/Q3)).
(G) Sina plot showing ERS for all OR identities (black dots). OSN types that were previously analyzed by flow cytometry are highlighted. Data are averages from 4 biological replicates. Also see Supplemental Figure S2, Supplemental Table S1.
(H) Violin plots of stress scores in class I and class II ORs, colored by zone. P value is for one-way ANOVA of stress scores across zone and class groupings. Data represent averages of 4 biological replicates.
We validated our reporter by immunofluorescence (IF), flow cytometry, and RNA-seq. IF shows that CRE protein is restricted to the OSN lineage of the MOE with expression patterns mimicking Atf5 translation (Supplemental Figure S1C) (Dalton et al., 2013). Flow cytometry verifies iRFP absence before OR expression, peak expression in immature OSNs (iOSNs), and age-dependent decline in mature OSNs (mOSNs) (Supplemental Figure S1D–F). RNA-seq on FAC-sorted cells corroborates that iRFP translation coincides with a strong increase of OR expression and identifies a set of ER stress-associated genes that closely follow the observed iRFP patterns (Supplemental Figure S1G–J). Thus, Atf5(rep) faithfully reports OR induced UPR, allowing a quantitative assessment of the relationship between OSN identity and PERK signaling.
Different OSN identities have distinct levels of ER stress
To test if ER stress patterns differ across OSNs, we crossed the Atf5(rep/+) mice to mice in which either the M71, P2, Mor23, or Mor28 OR genes were tagged with -iresGFP (Figure 1C, Supplemental Tables S1). We also crossed Atf5(rep/+) mice to mice where YFP replaces the S50 OR CDS (Bozza et al., 2009) marking a collection of ~100 OSN subtypes expressing class I ORs. Using flow cytometry, we measured iRFP levels in each GFP+ or YFP+ population using the GFP or YFP levels as a proxy for differentiation (Figure 1C). We observe distinct patterns for the five OSN types: M71+ OSNs have the highest levels of UPR throughout differentiation; Mor23+, and Mor28+ OSNs have moderate levels that attenuate with maturation; P2+ OSNs exhibit the sharpest UPR attenuation (Figure 1D, E). Class I OSNs, whose axons target a distinct region of the dorsal OB (Bozza et al., 2009; Tsuboi et al., 2006), display the lowest levels of iRFP (Figure 1D, E).
To extend our findings, we developed a high-throughput approach to map ER stress trajectories across all OSNs. We used Atf5(rep/+); Omp(iresGFP/+) mice to define iOSN and mOSN populations by flow cytometry, sorting in separate bins OSNs with the top and bottom 25% of iRFP intensities, followed by RNA-seq (Figure 1F, Supplemental Figure S2A–B). Since these OSNs predominately express one OR, OR mRNA abundance in each library is proportional to the number of cells expressing it. We measured the differential expression of every OR between high and low stress bins across development, computing “ER stress scores” (ERS) for each OR identity (Figure 1G) and confirming that ERS recapitulate patterns observed by flow cytometry (Figure 1G, E). Gene set enrichment analysis (GSEA) demonstrates that high-stress OSNs express UPR-associated genes at higher levels than low-stress cells (paj = 0.002) (Supplemental Figure S2C), corroborating that differences in reporter intensity represent meaningful UPR differences.
Since targeting is the product of both zonal and OR identity, we explored the relative contribution of each factor to ERS. Using reported OR-zone mappings(Tan and Xie, 2018), we find that zone 1 OSNs tend to have higher ERS than OSNs from ventral zones (zones 4,5) (Figure 1H). DV origin of the OSN is not a strict correlate of stress, however, since dorsal OSNs expressing class I ORs have the lowest ERS of all (Figure 1H). Furthermore, we observe wide variations in ERS within each zone, suggesting that both OR and zonal identities contribute to the overall stress levels of developing OSNs (Figure 1H). Indeed, we find that ERS also correlate with zonally independent OR features. For example, ORs whose expression is independent of Atf5 (Dalton et al., 2013), and/or of ER chaperons RTP1/2 (Saito et al., 2004; Sharma et al., 2017), have significantly lower ERS than the rest, despite no obvious zonal trends in ATF5 and RTP1/2-dependency (Supplemental Figure S2E–H).
OR identity influences ER stress patterns
To test if OR proteins determine ERS, we used a previously described P2(M71iresLacZ) allele (Feinstein and Mombaerts, 2004) to force moderate-stress P2 OSNs to express the high-stress OR protein M71 (Figure 2A, Supplemental Table S2). We crossed these mice to our Atf5(rep);Omp-iresGFP mice and determined ERS for all ORs by FACS and RNA-seq. ERS for WT ORs are highly correlated with our previous measurements (Supplemental Figure S3A, B), however, stress levels in M71→P2 “swap” OSNs are significantly higher than stress levels in endogenous P2 OSNs (Figure 2B, Supplemental Figure S3C), consistent with the altered targeting specificity of these OSNs. Thus, ERS are predominantly determined by the chosen OR protein, not the choosing OSN identity.
Figure 2: OR protein sequence determines ER stress levels.
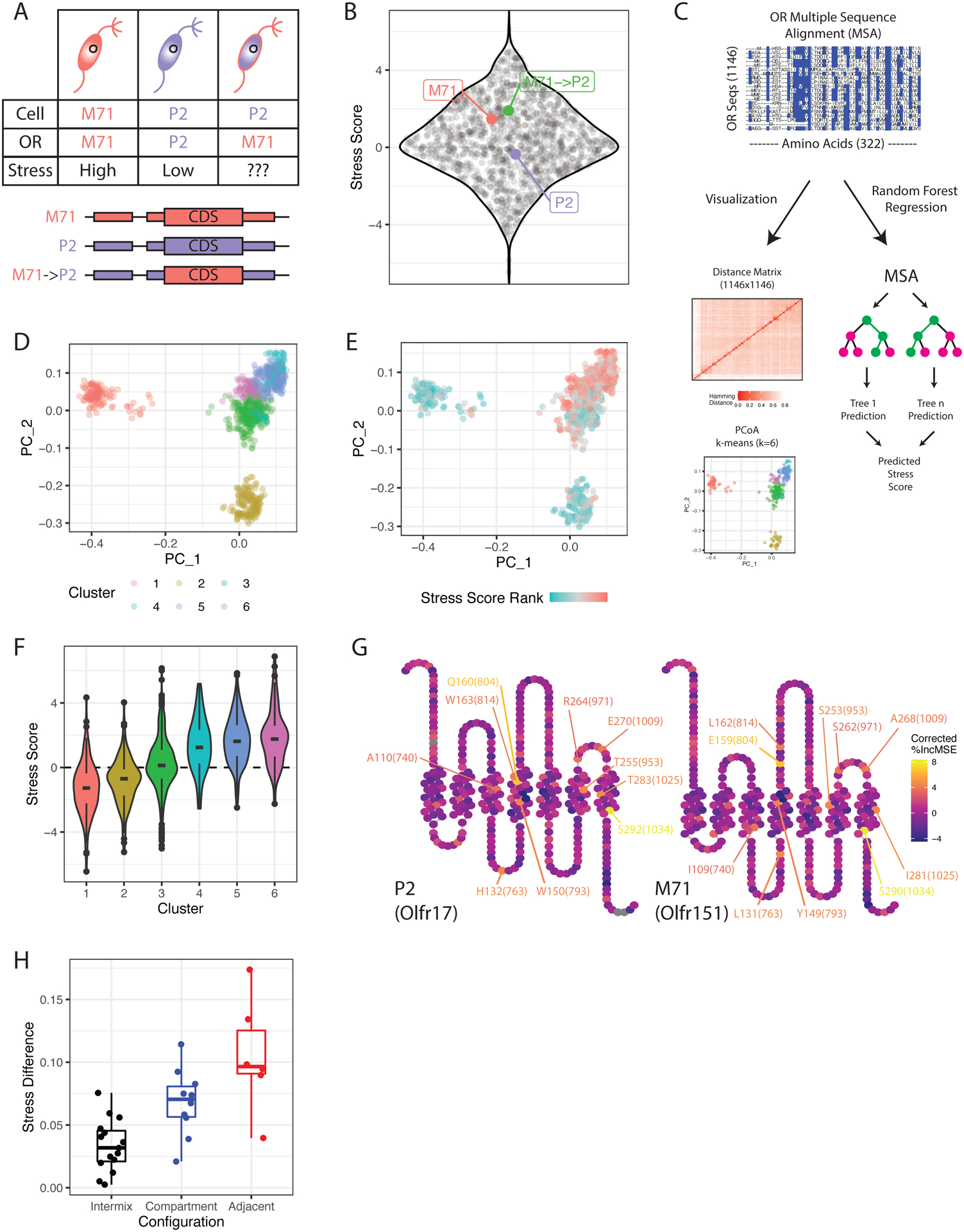
(A) Schematic of the M71→P2 swap experiment. Cellular and OR identities are shown in endogenous M71, P2 and M71→P2 swap OSNs.
(B) Sina plot showing average ERS for all OR identities (black dots) in the M71→P2 swap (n=3 for M71→P2 swap, n=6 for M71 and P2, n=9 for all others, see methods). Padj = 5.5E-4 for M71→P2 vs. P2 comparison (one-way ANOVA with Tukey’s post-test). Also see Supplemental Figure S3, Supplemental Tables S2, S3.
(C) Schematic of the OR amino acid multiple sequence alignment (MSA), visualization and random forest regression (see Supplemental Figure S3).
(D) Principal coordinates analysis (PCoA) of all OR aa sequences. Each point is an OR sequence, distances between points represent similarity to other ORs in the MSA, and colors represent cluster identity.
(E) Same plot as (D), but with colors representing the ranked ERS for each OR.
(F) Violin plot of ERS for each cluster of OR sequences in (D).
(G) Snakeplots of the moderate-stress OR P2 (left) and high-stress OR M71 (right). Colors reflect variable importance in the random forest regression model, regressed by Shannon entropy at each MSA position (see Supplemental Figure S3G). The top 10 most important residues are labeled in each plot as amino acid [position in OR sequence]([position in MSA]). Also see Supplemental Figure S3I.
(H) Predicted ERS difference by random forest regression for pairwise comparisons of M71/M72 mutant ORs versus glomerular configurations. Int=intermixed, Comp = compartmentalized, Adj = adjacent (see methods). Padj Comp-Int = 0.0167, Adj-Int = 4.58E-5, Adj-Comp = 0.0441 (one-way ANOVA with Tukey’s post-test).
To explore how the OR protein sequence influences UPR, we performed a multiple sequence alignment (MSA) of all OR proteins, computed Hamming distances between all sequence pairs, and used principal coordinates analysis (PCoA) to visualize the dataset in two dimensions (Figure 2C). Remarkably, k-means clustering reveals that ORs with similar sequences tend to have similar ERS (Figure 2D–F). One of the low stress clusters contains class I ORs, whereas the five class II-containing clusters form based on stress levels rather than zonal biases (Figure 2E, Supplemental Figure S3D). Therefore, ERS must predominantly be encoded by the primary amino acid (aa) sequence.
We next performed random forest regression using the positions in our OR MSA as predictor variables to model OR ERS (Figure 2C, Supplemental Figure S3E–F). Strikingly, aa sequence alone explains 51% of the variance in ERS (Supplemental Figure S3F). Using permutation, we assessed the importance of each position in the MSA as a predictor variable, regressing the resulting values by the Shannon entropy at each MSA position (Supplemental Figure S3G). This identifies a series of aa scatted throughout the transmembrane and extracellular domains of the OR protein predicted to drive ERS differences (Figure 2G), displaying simple trends in aa composition moving from low to high stress ORs (Supplemental Figure S3I).
If ERS influence axon guidance, then mutating aa with higher predicted ERS input should have a stronger impact on targeting. We took advantage of experiments where residues of OR M72 were introduced into the highly similar OR M71, followed by assessment of axon guidance shifts (Feinstein and Mombaerts, 2004). Reassuringly, our random forest model predicts larger differences in ERS for M71 mutants that induce stronger axon guidance phenotypes (Figure 2H). Further, OR aa polymorphisms between the C57BL/6 and 129 mouse strains also cause targeting differences (Feinstein and Mombaerts, 2004). Of the 4 studied ORs with 129/B6 polymorphisms, M50 displayed the strongest glomerular segregation phenotype (Feinstein and Mombaerts, 2004). Once again, our model predicts that the two aa substitutions in the M50 sequence have bigger impact in ERS than the two substitutions in M72 or the single aa substitutions in P2 and P4 (Supplemental Figure S3H), supporting a causal relationship between OR sequence, ERS, and axon guidance.
ER stress correlates with expression patterns of axon guidance genes and instructs targeting specificity
To explore the link between UPR and OSN targeting we asked if ERS differences correlate with axon guidance gene expression variations. First, we collapsed our OR ERS mappings into two groups: “high stress” (ERS > 0) and “low stress” (ERS < 0) ORs (Figure 3A). We then used scRNA-seq to transcriptionally profile 8,815 Omp-expressing mOSNs, determining the chosen OR in each OSN and using OR-ERS mappings to annotate the cells as “high” or “low” stress (Figure 3B–C). We identified differentially expressed genes between high and low stress OSNs within each zone, Stouffer integrating across zones to isolate genes whose expression depends on OR identity irrespective of zone (Figure 3D). ERS correlate with expression differences for many axon guidance molecules (Figure 3E). Some of these genes, like Nrp2, regulate DV segregation of OSN projections, while others, like Kirrel and Eph control the final sorting of OSN axons to distinct glomeruli. All the “low stress” axon guidance molecules are known to be upregulated by activity, whereas most of the “high stress” axon guidance genes are activity independent (Figure 3E).
Figure 3. ER stress differences correlate with differential expression of axon guidance molecules.
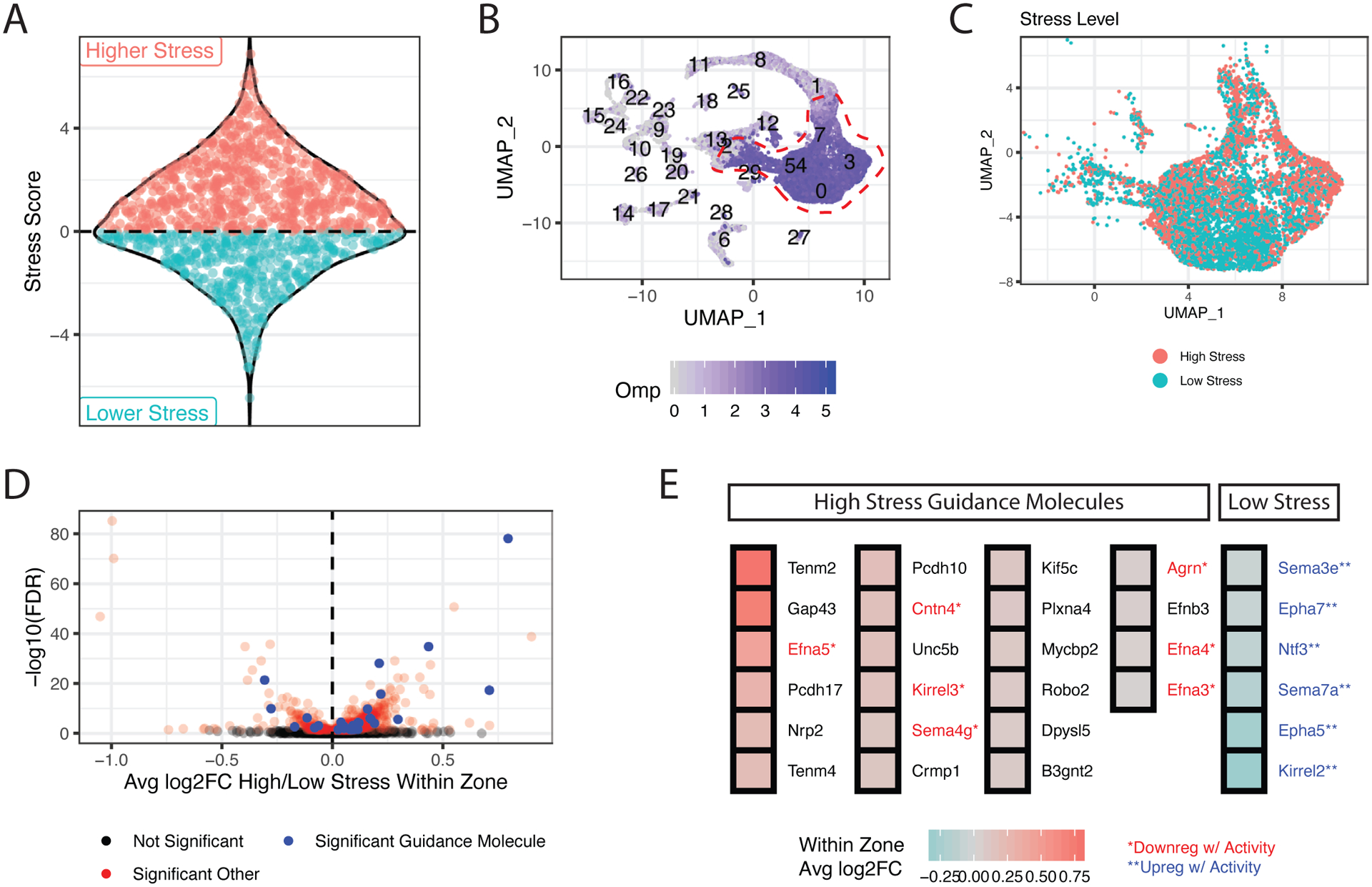
(A) Sina plot showing the decomposition of OR ERS into high (red) and low-stress (blue) groupings. Each point represents an OR.
(B) scRNA-seq MOE analysis, visualized using uniform manifold approximation and projection (UMAP). Normalized Omp counts (blue) identify mOSNs (circled in red).
(C) mOSN clusters from B colored by ERS group of the chosen OR (red=high stress, blue = low stress).
(D) Differential expression of all genes between “high” and “low-stress” OSNs within each zone. Log2 fold changes (log2FC) (x-axis) are the weighted average of high/low-stress expression in each zone. Adjusted p values (y-axis) are computed by Stouffer integration of bright/dim comparisons across zones (see methods). Significant axon guidance molecules are shown in blue, other significant genes in red.
(E) Average log2FC within zone for axon guidance molecules with statistically significant differential expression between high and low-stress OSNs. Genes are colored red or blue and labeled with an asterisk if previously reported to respond to activity.
To test the role of ER stress in targeting specificity, we employed a monoallelic deprivation strategy that provides an internally controlled assessment of axon guidance. We generated OR(iresCre/iresGFP); Rosa26(LSL-tdTomato/+) mice for ORs M71 and Mor28. Since OR choice is monoallelic, OSNs expressing the given OR will chose either the iresGFP allele (green, functionally WT) or the iresCre-tagged allele (red, recombine any floxed allele) (Figure 4A). Crossing one or two Perk floxed alleles (Zhang et al., 2002) into this background enables selective Perk deletion, only in the red, CRE+ OSNs. Because CRE is expressed with the chosen OR, PERK depletion will likely occur after this OR is chosen, disentangling OR choice from axon guidance. Indeed, we observe only a subtle decrease in the ratio of OR+tdtomato+ to OR+GFP+ OSNs in Perk cKO mice relative to WT and Het animals at p5 (Figure 4B, Supplemental Figure S4A–E), in sharp contrast with the effects of the germline Perk deletion, where OR choice is unstable (Dalton et al., 2013).
Figure 4. Continuous PERK signaling is required for OSN axon coalescence.
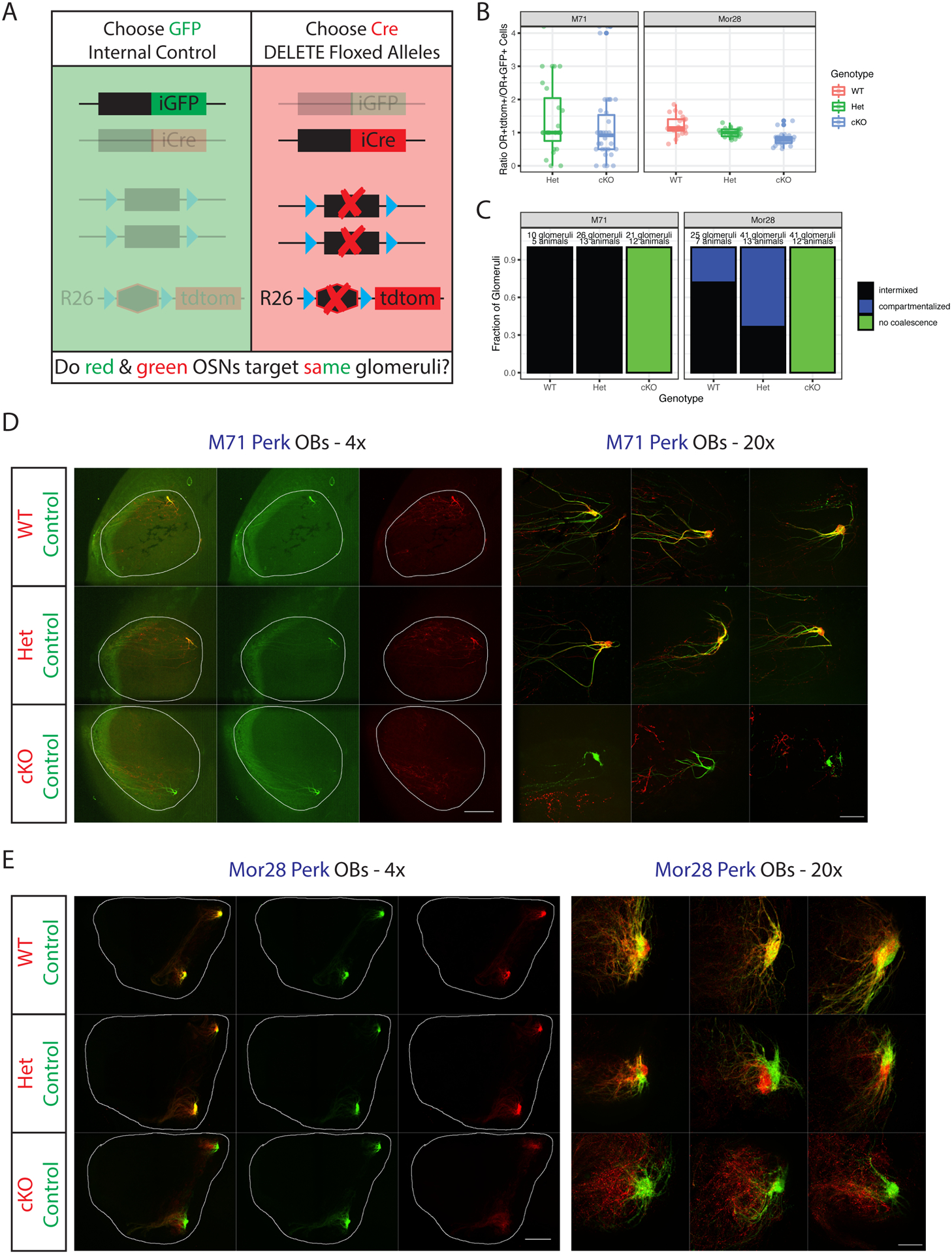
(A) Schematic of the monoallelic deprivation strategy. Mice have the genotype OR(iresCre/iresGFP); Rosa26(LSL-tdtom/+) with or without additional floxed alleles. Targeting of red axons is compared to internal control green axons.
(B) Ratios of OR+tdtomato+ to OR+GFP+ cells in the MOE of p5 mice. Data facetted by OR type and colored by genotype of the tdtomato+ cells. Each point is a section, n=3 mice per genotype except for M71 Het (where n=2). P>0.05 for all comparisons except Mor28 cKO-WT (p=0.0038) by one-way ANOVA with Tukey’s post-test. Also see Supplemental Figure S4A–B.
(C) Blinded quantification of glomerular configurations in the OB of p5 M71 and Mor28 Perk mice, grouped by genotype of the tdtomato+ cells. Also see Supplemental Figure S4D–E, Supplemental Table S4.
(D) Whole mount OB views of M71 Perk mice at p5. Internal control axons are green, experimental axons red with the indicated genotypes. Magnification as indicated, A=anterior, P = posterior. Scale bars 500μm (4x), 100μm (20x).
(E) Same as D but for Mor28 Perk. Also see Supplemental Figure S4C.
In Perk WT mice, red and green neurons target the same glomeruli (Figure 4C–E). Fibers typically intermix to form yellow glomeruli, but occasional segregation of red and green fibers is observed in the Mor28 glomeruli (Figure 4C, E). Deleting both copies of Perk in either M71 or Mor28 OSNs disrupts glomerular coalescence without altering the coarse axon guidance properties of the cKO axons (Figure 4C–E). The phenotype is particularly dramatic in Mor28 Perk cKO mice, where a disorganized meshwork of red fibers is observed just anterior to the control (green) glomeruli (Figure 4E). Failure to coalesce has protracted consequences on this circuit, as cKO neurons are subsequently depleted from the OE and OB by p28 (Supplemental Figure S5A–E). Importantly, the age-related loss of Perk cKO OSNs is likely secondary to the lack of glomerular coalescence(Katidou et al., 2018), since deleting Perk specifically in mOSNs neither disrupts glomeruli nor significantly increases apoptosis (Supplemental Figure S5F–H). Interestingly, loss of just one Perk allele, which slightly reduces UPR levels (Supplemental Figure S4C), increases the fraction of glomeruli that display segregation, and the degree of segregation of red and green axons (Figure 4C, E, Supplemental Figure S4D). Similar levels of MOR28 protein (Barnea et al., 2004) are detected in both the red and green compartments of Mor28 Perk Het glomeruli (Supplemental Figure S4E, Supplemental Table S4), excluding impaired OR trafficking as a cause of this phenotype. This segregation persists in adult mice (Supplemental Figure S5B–C) but is not observed in M71 Perk Het mice.
The Mor28 Perk Het phenotype suggests that slight reduction in PERK signaling could cause targeting shifts without disrupting axon coalescence. To test if the reciprocal also alters targeting, we used the monoallelic deprivation strategy to delete a floxed allele of Hspa5 (Luo et al., 2006), an ER chaperone that attenuates PERK signaling (Bertolotti et al., 2000; Kawaguchi et al., 2020; Li et al., 2008), which we confirmed in OSNs (Figure 5A, Supplemental Figure S6A). Remarkably, deletion of only one Hspa5 allele from M71+ OSNs causes segregation of red and green M71+ axons, which reproducibly innervate different glomeruli (Figure 5B–C). This demonstrates that axon guidance responds to bidirectional UPR modulation and illustrates that sensitivity to subtle ER stress differences extends beyond Mor28+ OSNs. Thus, if we could increase ER stress to saturation levels across all OSNs, OR-instructed signatures of extracellular guidance molecules will become homogeneous between axons, preventing distinction between “like” and “other” axons. To test this, we generated a tetO-regulated, chemically activated Fv2E-Perk fusion transgene(Lu et al., 2004), which we induced with OMPirestTA (Supplemental Figure S6B). Consistent with our prediction, saturating PERK-signaling levels across OSN types completely disrupts glomerular coalescence across the OB (Supplemental Figure S6C). Intriguingly, this dramatic disruption of the glomerular map is reminiscent of the effects of homogeneous clustered protocadherin expression across all OSNs(Mountoufaris et al., 2017), further highlighting that ER stress variations facilitate molecular distinction and segregation of OSN axons. Once more, failure of OSNs to innervate glomeruli also results in increased apoptosis (Supplemental Figure S6D), which may be further accelerated by the molecular consequences of sustained high PERK signaling.
Figure 5. Network reconstruction predicts that Ddit3 controls UPR-responsive axon guidance.
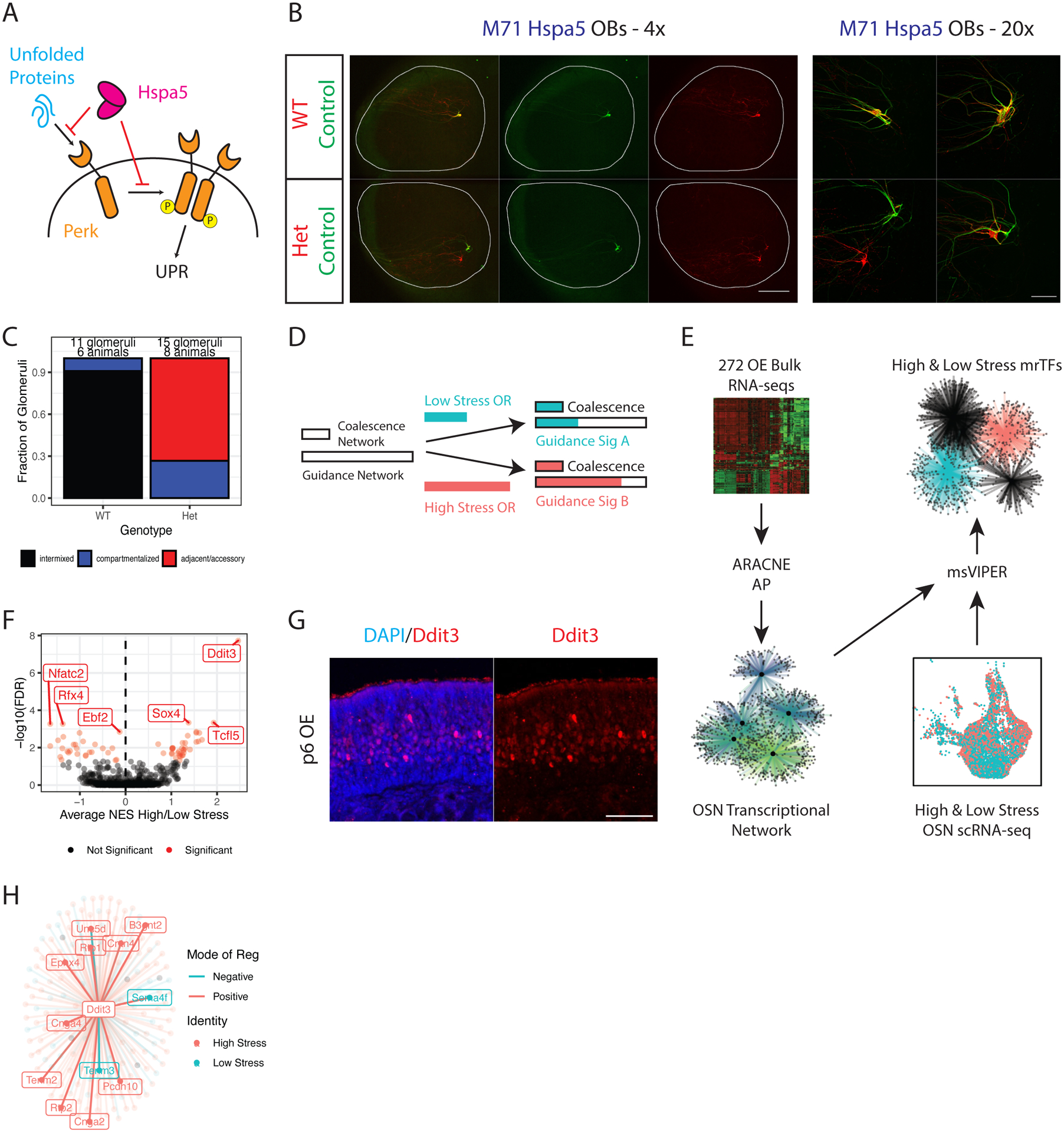
(A) Schematic of PERK activation by unfolded proteins, and the inhibition of the UPR by HSPA5.
(B) Whole mount OB views of M71 Hspa5 mice at p5. Internal control axons in green, experimental axons in red. Magnification as indicated, A = anterior, P = posterior. Scale bars 500μm (4x), 100μm (20x). See also Supplemental Figure S6A.
(C) Blinded quantification of glomerular configurations in the OB of p5 M71 Hspa5 mice, grouped by genotype of the tdtomato+ cells.
(D) Depiction of the role for the UPR in glomerular coalescence and axon guidance. Two UPR responsive networks are proposed with different saturation thresholds (indicated by length of empty bars). A low stress (blue bar) and high stress OR (red bar) both induce enough UPR to saturate the coalescence network, but differentially activate the guidance network.
(E) Schematic of the in silico analysis to identify master regulators of the proposed OSN axon guidance network. 272 RNA-seq libraries from various OSN populations and experimental conditions were input into ARACNE-AP to reconstruct an OSN transcriptional network. The network was input to msVIPER along with scRNA-seq data from high and low stress OSNs to identify UPR-dependent master regulator transcription factors (mrTFs).
(F) msVIPER identifies differentially active mrTFs between high and low stress OSNs. The x axis shows the weighted average of normalized enrichment scores (NES) in high vs low stress OSNs across all zones. The y axis shows the −log10 false discovery rate (FDR) derived from Stouffer integrating msVIPER p values for high/low stress comparisons across all zones and correcting for multiple testing of all TFs. Weights were set as the square root of the number of OSNs in each zone. Significant mrTFs are shown in red, top mrTFs labeled.
(G) Representative IF for DDIT3 in MOE sections of wild type mice at p6. Scale bar 50μm.
(H) Representation of the Ddit3 regulon (genes that the ARACNE-AP algorithm infers are controlled by Ddit3). Points (and labels) for each gene are colored by whether that gene is differentially expressed in high stress OSNs (red) or low stress OSNs (blue) in our scRNA-seq dataset. Connecting line colors denote mode of regulation (blue = negative, red = positive).
Ddit3 mediates UPR-instructed axon targeting specificity
OSNs may have two distinct PERK-dependent regulatory networks with different saturation thresholds (Figure 5D): a “glomerular coalescence” network activated at low UPR levels in every OSN; and an “axon guidance” network activated at higher UPR levels with large dynamic range that controls targeting specificity. In this model, Perk cKO disrupts both networks, while Perk and Hspa5 heterozygote mutations affect only the guidance network by altering the homotypic properties of like axons. To identify UPR effectors of the guidance network, we deployed the ARACNE-AP algorithm (Lachmann et al., 2016) to reconstruct OSN transcriptional networks using mutual information estimators from our RNA-seq datasets (Figure 5E). These networks were input to msVIPER (Alvarez et al., 2016), which scored the activity of master regulator transcription factors (mrTFs) in our scRNA-seq data (Figure 5E). We searched for differential mrTF activity between OSNs choosing a higher-stress (ERS >0) or lower-stress (ERS <0) OR, regressing out zonal identity of these cells as a covariate (Figure 5E). This approach isolates differentially active networks, while removing shared networks. Remarkably, msVIPER identified the UPR-responsive transcription factor Ddit3 ((Hu et al., 2018; Nishitoh, 2012; Ron and Habener, 1992) as the top putative mrTFs in high-stress OSNs (Figure 5F). IF for DDIT3 revealed a “salt and pepper” protein expression pattern consistent with the expectation that Ddit3 levels are determined by OR-instructed ER stress levels (Figure 5G). Ddit3 RNA levels are significantly higher in our bulk-sorted high vs. low stress OSNs (log2FC = 0.9, padj = 4.7E-6) and in high vs. low stress single cells (log2FC = 0.283, padj = 2.6E-8), indicating that this factor is UPR and OR identity-dependent. Indeed, DDIT3 protein levels increase with PERK signaling levels in Fv2E-Perk+ OSNs (Supplemental Figure S6E). Finally, ARACNE-AP predicted that several high stress axon-guidance genes are part of Ddit3’s regulon, i.e. the direct targets of this UPR effector (Figure 5H).
If Ddit3 specifically controls the UPR-responsive “axon targeting” program, Ddit3 deletion should shift glomerular positions without preventing axon coalescence. We crossed a floxed Ddit3 allele (Zhou et al., 2015) into the monoallelic deprivation paradigm. Removing 2 copies of Ddit3 in both M71 and Mor28 Ddit3 cKO OSNs shifts the red cKO glomeruli relative to controls, without compromising glomerular coalescence (Figure 6A–C). The observed shift for M71 cKO OSNs is larger than the shift for Mor28 cKO OSNs (Figure 6A, B), consistent with M71+ OSNs having “higher stress” levels than Mor28+ OSNs. IF confirms that Ddit3 cKO OSNs express normal OR levels, with proper trafficking to the glomeruli (Supplemental Figure S6F–G). As with the Mor28 Perk Het, we find that deleting one copy of Ddit3 increases the fraction of Mor28 glomeruli with segregation of red and green fibers and the degree of segregation (Figure 6B–D). In contrast, we do not observe segregation between M71 Ddit3 Het and control axons, mimicking the effects of heterozygote Perk deletion (Figure 6A, C). Thus, we disentangled the effects of UPR on axon guidance from the effects of UPR on glomerular coalescence, supporting a role of Ddit3 as a specific regulator of UPR-directed glomerular sorting.
Figure 6. Ddit3 deletion alters OSN axon guidance specificity.
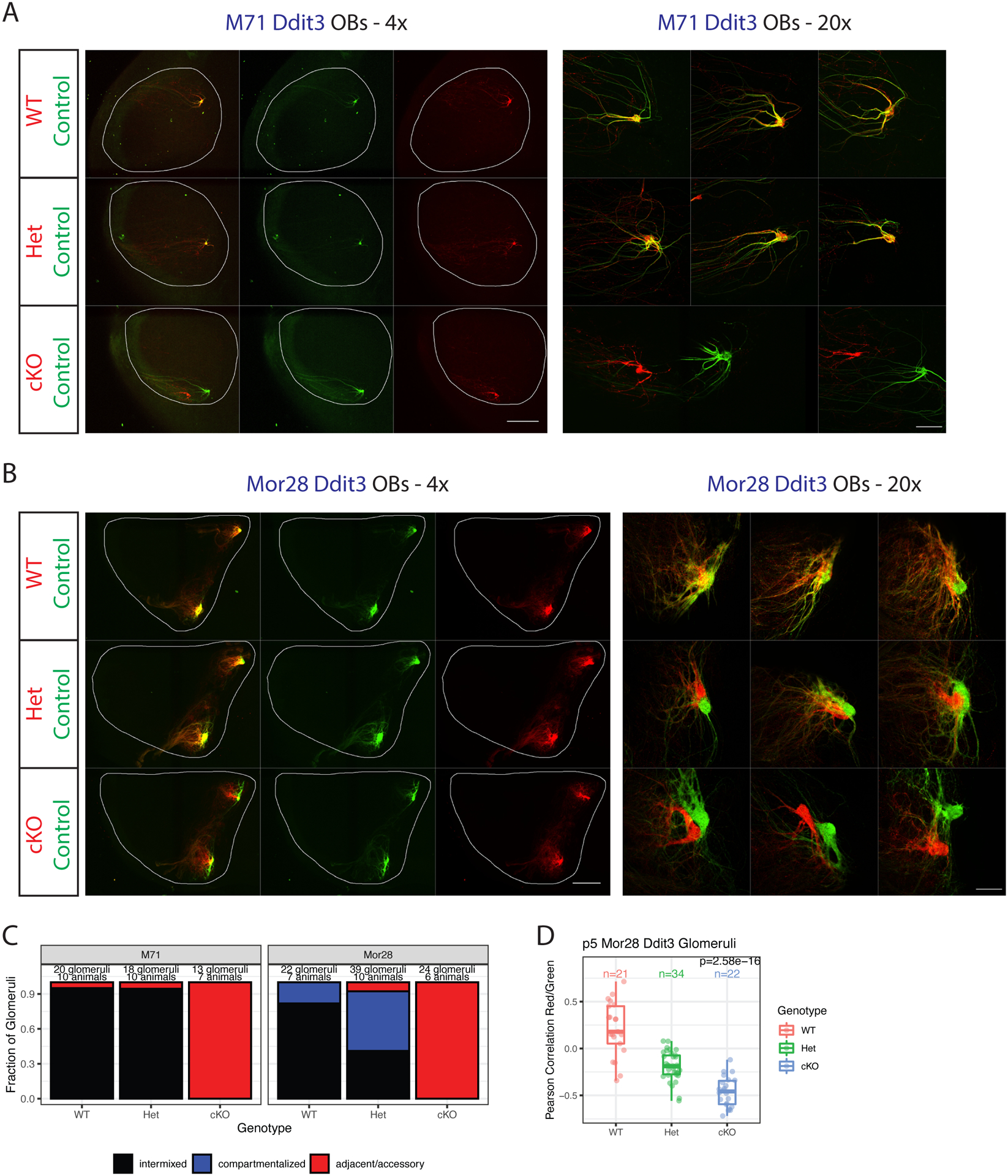
(A-B) Whole mount OB views of M71 (A) and Mor28 (B) Ddit3 mice at p5. Internal control neurons in green, experimental neurons in red. Magnification as indicated, A=anterior, P = posterior. Scale bars 500μm (4x), 100μm (20x). Also see Supplemental Figure S6.
(C) Blinded quantification of glomerular configurations in p5 M71 and Mor28 Ddit3 mice, grouped by genotype of the tdtomato+ cells.
(D) Pearson correlations between red and green signals in Mor28 Ddit3 glomeruli at p5, colored and grouped by genotype of the tdtomato+ cells. Each point is a glomerulus. P value is from one-way ANOVA across genotypes. Number of glomeruli is as indicated.
Deciphering the molecular mechanisms of Ddit3-regulated axon guidance specificity
Since Ddit3 cKO OSNs form glomeruli, we reasoned that these OSNs will survive till p28. Indeed, red axons continue to form intact, but notably shifted glomeruli in M71 and Mor28 Ddit3 cKO p28 mice (Figure 7A–C). Further, the segregation between red and green axons in Mor28 Ddit3 Het mice also persists at p28 (Figure 7B–D). Taking advantage of the abundance of Mor28+ OSNs at p28, we used FACS to isolate Mor28+GFP+ and Mor28+tdtomato+ OSNs for RNA-seq (Supplemental Figure S7A, Supplemental Table S5). Consistent with a subtle targeting phenotype, only a handful of genes were significantly different between WT, Het and cKO OSNs (Figure 7E, Supplemental Figure S7B); strikingly, among them, the high stress guidance molecules Nrp2 and Tenm2 were significantly downregulated upon Ddit3 manipulation (Figure 7E, Supplemental Figure S7B–D). Transcriptional changes are reflected in protein levels, as NRP2 protein is significantly reduced in Mor28+ Ddit3 cKO glomeruli (Supplemental Figure S7E).
Figure 7. Ddit3 regulates the expression of high stress axon guidance molecules.
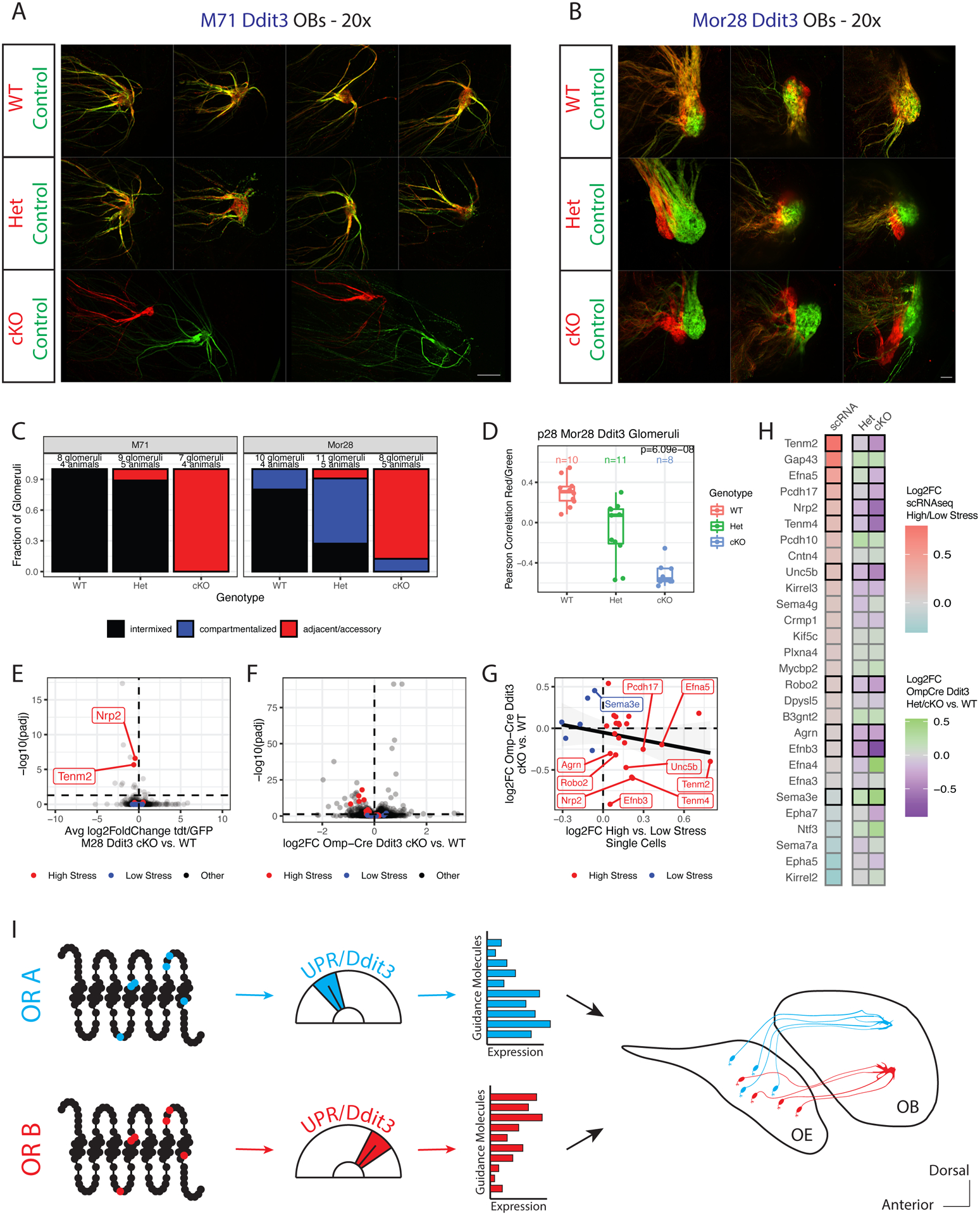
(A, B) Whole mount, zoomed in glomerular views of M71 (A) or Mor28 (B) Ddit3 mice at p28. Internal control neurons in green, experimental neurons in red. Magnification as indicated, A=anterior, P = posterior. Scale bars 100μm (20x).
(C) Blinded quantification of glomerular configurations in M71 and Mor28 Ddit3 mice at p28, grouped by genotype of the tdtomato+ cells.
(D) Pearson correlation between red and green axons in Mor28 Ddit3 WT, Het and cKO mice at p28. Each point is a glomerulus, colored by genotype. P value is from one-way ANOVA across genotypes.
(E) Volcano plot showing the difference in average log2FC tdtomato/GFP in Ddit3 cKO vs. WT cells (x-axis) and −log10 adjusted p values (y-axis) for all genes in Mor28 Ddit3 mice at p28. P values are from a Likelihood Ratio Test (LRT) for whether the tdtomato/GFP ratio significantly changes with genotype. High stress axon guidance molecules are in red, low stress molecules are in blue, others in black. The horizontal dashed line indicates the significance cutoff of padj= 0.05, the vertical line shows a log2FC of zero. Data from n=11 mice. See Supplemental Figure S7A–E, Supplemental Table S5.
(F) Volcano plot for all genes in Omp-iresCre Ddit3 mice. Colors and dashed lines as in E. The x-axis shows log2FC for each gene in Ddit3 cKO vs. WT OSNs, −log10 adjusted p values (y-axis) are from LRT for whether counts significantly change with genotype. Also see Supplemental Figure S7F–J.
(G) Comparison of log2FC in Omp-iresCre Ddit3 cKO vs. WT OSNs (y-axis, from F) versus average log2FC in high vs. low stress OSNs within zone by scRNAseq (x-axis, from Figure 3D–E). Genes with significant (padj < 0.05) changes in the Omp-iresCre Ddit3 cKO dataset are labeled and a linear regression line is shown in black. Red = high stress axon guidance molecules, blue = low stress axon guidance molecules.
(H) Heatmap comparing log2FC between high/low stress single cells within zone (left column, blue → red) to log2FC in Omp-iresCre Ddit3 Het vs. WT OSNs (middle column, purple → green) and cKO vs. WT OSNs (right column, purple → green). Genes are ordered by log2FC from the single cell data and bordered with a black box if their expression significantly changes with genotype by LRT. Also see Supplemental Figure S7J.
(I) Model showing the role of the UPR in axon guidance. Amino acid polymorphisms at critical OR residues lead to differential induction of the UPR and DDIT3. DDIT3 then controls expression of an axon guidance molecule network that orchestrates OSN segregation into OB glomeruli. Additional, zone- and UPR-responsive transcription factors likely contribute to this process.
We expanded our analyses in all mOSNs with Omp-iresCre mediated Ddit3 deletion and RNA-seq, which revealed significant reduction in the expression of 9 high-stress axon guidance molecules, without changes in ORs or most other genes (Figure 7F–H, Supplemental Figure S7F–I). Indeed, guidance genes drive most of the transcriptional changes upon Ddit3 ablation, as GSEA shows that our high-stress axon guidance molecules set is the most significantly depleted gene set in Ddit3 cKO vs. WT mOSNs (NES = −1.86, padj = 2.16E-3 compared to all hallmark mouse set genes). Perhaps most strikingly, heterozygote Ddit3 deletion leads to intermediate levels of axon guidance molecules (between WT and cKO), supporting the notion that levels of ER stress act as a “rheostat” to control OSN axon guidance (Figure 7H, Supplemental Figure S7J).
Discussion
We describe a surprising UPR function in the establishment of neural circuits. Here, the ER-resident kinase PERK interprets the identity of the chosen OR to establish OR-specific extracellular patterns of guidance molecules by modulating DDIT3 levels. Several observations indicate that OR-induced ER signaling facilitates the sensitive distinction between “like” and “other” axons, controlling the glomerular sorting stage of OSN axon guidance (Figure 7I). For one, most molecules in our PERK-dependent guidance network are associated with glomerular segregation. Further, although increasing and decreasing ER stress shifts OSN axons into new glomeruli, these reciprocal manipulations do not result in reciprocal shifts along any OB axes. Rather, subtle transcriptional changes induced by alterations to ER signaling modify the homotypic properties that partition like axons into distinct glomeruli. The ability of an OR to induce ER stress is encoded, to a significant extent, in its primary aa sequence. ORs with similar sequences induce similar levels of ER stress, while aa substitutions induce axon guidance shifts that follow predicted ERS changes. Recent spatial transcriptomic experiments showed that OSNs expressing ORs with similar sequences tend to target proximal glomeruli (Wang et al., 2022). The OR propensity to introduce ER stress may underly this organization, generalizing the phenotypes we observe for Mor28 and M71 to the entire OR repertoire.
ER stress may play a role DV patterning of the OB as well, since the classical DV patterning genes Nrp2 and Robo2 (Takeuchi et al., 2010) are Ddit3-dependent. Furthermore, we observe a trend of decreasing ER stress levels along the dorsoventral axis, suggesting that DV patterning and glomerular segregation are less dichotomous than previously thought. On the other hand, the relationship between AP targeting and ER stress is weaker, as we do not identify Nrp1, Sema3a or Plxna1 (Imai et al., 2006; Nakashima et al., 2013) in our guidance network. Furthermore, Gs, which controls AP targeting through basal activity, is expressed early in the OSN lineage, preceding the burst of ER stress that follows OR choice. A developmentally sequential and functionally independent contribution of these two pathways would explain why M71-driven Gs deletion does not alter glomerular coalescence (Movahedi et al., 2016), whereas M71-driven Ddit3 and Hspa5 deletions have profound consequences on glomerular segregation.
Previous work implicated odor-evoked activity in glomerular sorting and elegantly showed that activity patterns can influence the expression of axon guidance molecules (Nakashima et al., 2019; Serizawa et al., 2006). However, it is not clear how the natural, constantly changing odorant environment provides sufficiently stable sensory input to >1000 ORs, both at the timeframe of one OSN’s projection to the OB, and over an animal’s life. We uncovered an alternative signaling pathway that maps many of the same axon guidance molecules to OR identity without the need for sensory input. Thus, deploying OR-directed UPR levels for axon guidance offers two distinct advantages over odor-evoked activity: First, it assures that the circuit can form without odors, during embryonic development or upon sensory deprivation, for example. Second, by insulating axon guidance from odor stimulation, ER signaling transforms the OR aa sequence, an internal, constant, and genetically hardwired OSN feature, into persistent and invariant targeting. This explains how this regenerating circuit maintains a stable glomerular map in a variable sensory world. Consequently, while OSNs have the transcriptional plasticity to modulate the intensity of an odor (Tsukahara et al., 2021), its perceived identity shall remain stable for life. Consistent with this, our manipulations support a more critical role of ER stress than odor-evoked activity in the assembly of the olfactory circuit, as deleting PERK in select OSNs (or saturating PERK signaling in most OSNs) fully disrupts glomerular coalescence, contrasting the subtle effects of Golf/Cnga2 deletions and of naris occlusion (Belluscio et al., 1998; Brunet et al., 1996; Lin et al., 2000; Zou et al., 2004). That said, both odor-evoked activity and ER stress may cooperate to regulate glomerular segregation, especially in the case of odors with constant high abundance in an animal’s habitat, which can stably influence axon guidance gene expression.
Why deploy such elaborate mechanisms to transform OR identity into guidance specificity? The OR gene family underwent a remarkable degree of evolutionary expansion from ~70 ORs in zebrafish to >1000 ORs in mouse, all while maintaining both the one OR per OSN and the one OR per glomerulus principles (Braubach et al., 2012). This illuminates a fascinating constraint placed on the evolutionary process: the evolution of a novel OR protein with a distinct sensory spectrum must have co-evolved with distinct guidance signatures that allowed meaningful circuit integration. This constraint could be minimized by the ability of PERK and ER stress to act as a molecular conduit between OR identity and axon guidance, bypassing the need to co-evolve new cis regulatory elements that match receptors with axon guidance genes. Our modeling implies that PERK can capture subtle aa changes, thus, ER stress could potential “tile” the OR protein, ensuring that any new sensory “channel” is automatically represented in a new glomerulus.
A homeostatic mechanism adapted to axon guidance
Non canonical, uORF-independent PERK signaling controls retinotectal connections in Xenopus laevis (Cagnetta et al., 2019). Our data extend these findings to the canonical, uORF-dependent PERK signaling pathway. Thus, while prolonged PERK signaling may be toxic and lead to Ddit3-induced apoptosis, OSNs established a window of physiological PERK signaling that regulates axon guidance without killing the OSN. The requirement for low Ddit3 expression may explain why subtle changes to Ddit3 levels still lead to transcriptional and guidance consequences, rendering the OR-PERK-guidance axis exquisitely responsive to OR polymorphisms. However, cooption of UPR in axon guidance may come with a price, as pharmacological PERK stimulation beyond a certain threshold leads to Ddit3 overexpression and may cause apoptosis. Thus, Ddit3 can function as a “molecular timer”, that restricts OSN lifespan to 30–90 days as DDIT3 eventually activates apoptosis. In this vein, the role of Ddit3 in OSN axon guidance could also explain why OSN targeting is disrupted by pathological states involving unfolded proteins (Cao et al.,2012), why APP mutations induce OSN apoptosis before detection of amyloid plaques (Cao et al., 2012; Cheng et al., 2016), and why anosmia constitutes a prodrome symptom of Alzheimer’s disease (Albers et al., 2006; Devanand et al., 2015).
Limitations of the study
While our experiments indicate that the OR protein sequence drives OSN stress levels, future studies will explore the mechanisms by which these residues control stress. Moreover, our ERS calculation was based only on two ER stress FACS bins (top and bottom 25% of iRFP intensity), resulting in low resolution ERS definition; future studies should be able to identify ERS differences for ORs with very similar ER stress patterns. Regarding the mapping of ERS to axon guidance, PERK signaling may also contribute to axon guidance through Ddit3-independent mechanisms, either via other mrTFs, or direct translational regulation of guidance molecules. Further, the causal relationship between OR identity, ER stress, and axon guidance is based on computational analyses and the direct genetic manipulations of M71+, Mor28+, and P2+ OSNs. While our Omp-iresCre Ddit3 RNA-seq results support a general role of UPR in axon guidance, additional studies will be required to formally generalize our findings. Finally, our manipulations explored the effect of PERK signaling only to the establishment of the olfactory circuity and not its maintenance during adult neurogenesis, when the “critical period” of olfactory circuit formation is closed (Ma et al., 2014; Tsai and Barnea, 2014; Wu et al., 2018).
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact Stavros Lomvardas (sl682@cumc.columbia.edu)
Materials availability
This study generated two mouse lines, Atf5(rep) and tetO-Perk2amCherry, which will become available upon request.
Data and code availability
All RNA-seq datasets described in this study have been uploaded to GEO with accession number: GSE198886.
Original code can be found at https://github.com/hshayya/2022_Shayya_UPR_Guidance
Images/other resources may be downloaded from Zenodo (doi: 10.5281/zenodo.6448088).
Any additional information required to re-analyze the data is available upon request.
Experimental Model and Subject Details
Mouse protocols were approved by the Columbia University IACUC under protocol number AABG6553. All mice were housed in standard conditions with a 12-hour light/dark cycle and access to food and water ad libitum. Strains used are indicated in Supplemental Table 2. All animals were on a mixed genetic background and littermate controls were used for all comparisons. Animals were sacrificed by decapitation (if younger than postnatal day 14) or CO2 followed by cervical dislocation, and the main olfactory epithelium (MOE) was isolated by dissection into ice-cold 1xPBS without calcium or magnesium. For experiments requiring single cell suspensions (flow cytometry, FACS, RNA-seq), tissue was prepared using a papain dissociation system (Worthington) following a modified version of the manufacturer’s protocol. Dissociations were for 30–45 minutes at 37°C, centrifugation steps for 5min at 400g and the discontinuous density gradient was not performed. Cells were re-suspended for flow cytometry/FACS in a buffer containing 2%FBS (Gibco), 5mM MgCl2, DAPI (1:10k, Invitrogen) and the remaining DNAase I from the dissociation kit. For all experiments where iRFP intensity was measured to determine stress levels in mice containing the Atf5-reporter allele, cycloheximide (Sigma, 100μg/mL) was added to all solutions during dissociation and cytometry to block new protein synthesis and minimize the effects of dissociation or sorting-related stress on these measurements.
Generation of the Atf5(rep) allele
Atf5(rep) mice were generated by Biocytogen. The targeting design was based on Ensembl transcript ENSMUST00000107893 (GRCm38, accessed December, 2015). Three modifications were made to an Atf5 genomic clone to construct the targeting vector: (1) the entire Atf5 coding sequence (CDS) was replaced with an iRFP713-p2a-Cre cassette, (2) a FRT-Neo-FRT positive selection cassette was introduced into a non-conserved region roughly 300nt downstream of the annotated Atf5 3’UTR and (3) a DTA negative selection cassette was introduced downstream of the 3’ homology arm. The 5’ homology arm was 5.1kb and the 3’ homology arm was 4.9kb. The targeting vector was electroporated into C57BL/6 ES cells and 200 clones were isolated after selection with G418. Correct targeting was verified in 8 clones by long-range PCR and Southern blot. Chimeric mice were generated by injection into BALB/c blastocysts and the line was established from a male with germline transmission.
Generation and analysis of the tetO-Fv2E-Perk-t2amCherry Transgene
The tetO-Fv2E-Perk construct was assembled using NEBuilder HiFi DNA Assembly master mix and PCR amplicons from a plasmid expressing the Fv2E-Perk fusion protein (gift from David Ron) and the pTRE Tight ArchT-GFP plasmid (gift from Yasunori Hayashi). A t2a-mCherry sequence and NheI restriction sites flanking the insert were added in subsequent assembly steps. NheI restriction digest released a fragment containing the tetO-Fv2E-Perk-t2amCherry construct along with the intron and polyA sequences from pTRE Tight ArchT-GFP, which was used for pronuclear injection in B6CBAF1 zygotes. Tail biopsy and PCR was used to identify six founder mice containing the transgene, which were crossed to Omp-irestTA animals to screen for both germline transmission and tTA-dependent transgene expression in mOSNs. Five founders transmitted the transgene and each of those lines expressed to varying extents in mOSNs. The two founder lines with the best expression in Omp(irestTA/+); Tg animals were used for subsequent experiments.
Consistent with previous observations(Lu et al., 2004) leakiness from the Fv2E-Perk fusion protein increased UPR levels and subtly increased apoptosis in the mOSNs of Omp(irestTA/+); Tg mice (Supplemental Figure S6D–E). Higher levels of Fv2E-Perk dimerization and UPR activation were induced by injecting mice with the synthetic molecule AP20187(Serizawa et al., 2006). AP20187 was diluted to 62.5mg/mL in 100% ethanol and frozen at −20°C until the day of injection. Drug was then thawed and diluted in an injection solution containing 4% Ethanol, 10% PEG-400 and 2% Tween 20 (Lu et al., 2004). Mice were given intraperitoneal (IP) injections of 0.1mg/kg AP20187 at p6 and p7 prior to analysis on p8.
Method Details
Flow Cytometry and Fluorescence Activated Cell Sorting (FACS)
Dissociated cells were filtered through a 40μm cell strainer and analyzed on a Beckman Coulter MoFlo Astrios EQ sorter. Laser and detector settings were as described in Supplemental Table 3. Fluorescence compensation was not performed because all fluorochromes of interest utilized different laser sources and had clearly separated emission spectra. Sheath pressure was set at 28 PSI and sample pressure was kept below 28.5 PSI. A 100μm nozzle was used and event rates were between 5,000–15,000 cells per second. For cell sorting experiments, a droplet drive frequency of 49.6 kHz was used, cells were sorted in “purify” mode and the drop envelope was “1–2”. Gating was performed to isolate cells, remove doublets (using the area, height and width side scatter measurements from the 488 laser), and remove DAPI+ dead cells. A gating control sample (age-matched mouse without fluorescence in any of the channels to be measured) was prepared for every experiment and used to set gates for the GFP, tdtomato and/or iRFP channels. For experiments where iRFP levels were measured (Figure 1D–E, Supplemental Figure S1E–F, Supplemental Figure S4C and Supplemental Figure S6A), fcs files were exported from Summit (BD) and analyzed in FlowJo (v10.8.0). After gating in FlowJo, csv files containing channel values for each fluorophore of interest were exported for all cells in the population(s) of interest and analyzed using custom R scripts. For experiments where cells were sorted, sorting was performed directly into Trizol LS (Sigma) or into centrifuge tubes which were subsequently centrifuged for 10 minutes at 800g to recover cell pellets. Cell pellets or Trizol LS samples were snap-frozen in liquid nitrogen and stored at −80°C until further analysis.
Correcting technical variation in iRFP fluorescence measurements
Small variations in flow cytometer setup across days could create technical noise that might confound the direct comparison of fluorescence values measured in different sessions. We used the same instrument for all measurements in this study and kept all configuration parameters constant across days. Though we found that our resulting raw measurements were remarkably stable, we nevertheless implemented a simple normalization approach where we compared the population of interest in each sample to an internal control population that was present in all samples. Briefly, we computed the mean fluorescence intensity for each parameter of interest in the internal control population and subtracted those values from the measurements for each cell in our population of interest. We then compared the corrected measurements for the population of interest across experimental days, ensuring that any changes between samples were the result of biological differences rather than technical variation.
To assess the performance of our normalization approach, we made repeated measurements on cells from the same p11 Atf5(rep/+); Omp(iresGFP/+) mouse, systematically varying the PMT voltages for the GFP and iRFP713 detectors in a 50V range. The iRFP+GFP− population (ii- Supplemental Figure S1E) was used as an internal control to correct the iRFP and GFP fluorescence measurements in all iRFP+ and GFP+ populations across the various PMT settings. We found that corrected measurements were identical across PMT manipulations, demonstrating that PMT fluctuations in the tested range linearly scale and can be removed by our analysis approach (data not shown). Importantly, the range of fluorescence values observed in these control experiments far exceed the fluctuations observed in the internal control population across experimental days (data not shown). This argues that our normalization approach is likely sufficient to correct for the small levels of at least one kind of technical noise that could be present in our experiments. Lastly, we note that the conclusions of this study are identical when uncorrected data is used, further supporting the fact that technical noise occurs on a significantly smaller scale than the biological signal of interest.
iRFP713 measurements in OSN subpopulations
For measurements of Atf5-reporter intensity across OSN differentiation (Supplemental Figure S1F), the iRFP+GFP− population (ii, Supplemental Figure S1E) was the internal control population and was used to correct the iRFP+GFP+ population of interest (i/iii, Supplemental Figure S1E). For measurements in OSN subtypes with different OR identities (Figure 1C–E), the iRFP+GFP− population represents all OSNs other than the subtype of interest and serves as the internal control to the iRFP+GFP+ or iRFP+YFP+ population of interest (Figure 1C). In the OR-iresGFP experiments, the differentiation stage of the GFP+ OSNs was inferred by the corrected GFP fluorescence in each cell, since GFP is expressed via an IRES element inserted 3’ to the OR of interest and OR levels increase with differentiation (Supplemental Figure S1H). In Class I OR experiments, however, YFP is expressed via knock-in to the Olfr545 locus (S50). YFP levels therefore decrease with differentiation, as cells switch from Olfr545 to express another Class I OR(Bozza et al., 2009). Differentiation stage in class I ORs was therefore inferred by inverting the corrected YFP fluorescence values. Importantly, different OSN subpopulations express their chosen ORs at different levels, so GFP and inverted YFP values must be normalized before cross-population comparisons are made. Normalization within subtype was performed by subtracting the minimum GFP or -YFP value, dividing all values by the value at the 99.65th percentile and setting values above 1 to 1 (ie. saturated). Normalized values (differentiation_axis) are presented for all cross-population comparisons. Measurements were made in each OSN subpopulation at least twice on different days, with excellent internal agreement (data not shown). iRFP measurements were made on mice aged between age p11-p14.
iRFP713 measurements in Perk and Hspa5 mutant OSNs
In Atf5(rep) mice, CRE is expressed via a p2a self-cleaving peptide downstream of iRFP713 and is thus subject to the same regulation as the endogenous Atf5 (Supplemental Figure S1). While iRFP713 and CRE expression in the postnatal OE of Atf5(rep) mice is specifically restricted to the Atf5-expressing olfactory lineage (Supplemental Figure S1C), mice containing both the Atf5(rep) and Rosa26(LSL-tdtomato) Cre reporter alleles display widespread tdtomato fluorescence, including in tissues where Atf5 is not found postnatally (data not shown). Atf5 is thus likely to be expressed by early progenitor populations during development. Early expression creates an important caveat: Atf5(rep/+); Perk(fl/+) (Supplemental Figure S4C) and Atf5(rep/+); Hspa5(fl/+) animals (Supplemental Figure S6A) recombine the alleles of interest not only in OSNs, but in many other cell types as well.
iRFP713 measurements in Perk Het OSNs (Supplemental Figure S4C) and Hspa5 Het mOSNs (Supplemental Figure S6A) were made in the same sessions as their respective WT controls, so corrections for technical variation were not made and only iRFP+GFP+ mOSNs were measured. For visualization, the iRFP channel was centered for each replicate by subtracting the mean value in WT OSNs from all measurements and the differentiation axis was computed using the approach described for the OR-iresGFP experiments.
Olfactory Epithelium Immunofluorescence
Dissected MOEs were fixed in 4%(w/v) PFA in 1xPBS for 1hr at 4°C and then washed 3 times for 10 minutes each in 1xPBS. OEs extracted from animals p14 or older were decalcified overnight at 4°C in 0.5M EDTA (pH=8) and washed again in 1xPBS. The decalcification process was not necessary in younger mice. MOEs were cryoprotected overnight at 4°C in 30%(w/v) sucrose in 1xPBS, embedded in OCT, frozen over an ethanol/dry ice slurry and stored at −80°C until sectioning. To ensure full coverage of the MOE, tissue was serially sectioned in the coronal plane, moving from the flat posterior surface to the anterior surface. Two series (10 slides/series, 4 sections/slide) of 15μm sections were collected starting at the moment when turbinate 3 separated from the dorsal-most aspect of the epithelium(Alvites et al., 2018). Slides were frozen at −80°C until the day of staining experiments when they were thawed, washed for 5min in 1xPBS and post-fixed for 10 minutes at room temperature (RT) in 4%(v/v) formaldehyde (Thermo Fisher) in 1xPBS. Tissue was then washed 3 times (5 minutes each, 1xPBS + 0.1% Triton X-100 (Sigma)) and blocked for 1hr at RT in 4%(v/v) Donkey Serurm (Sigma) + 1% Triton X-100 in 1xPBS. Primary antibodies (Supplemental Table 4) were diluted in block solution and incubated overnight at 4°C. The following day, sections were washed, incubated with secondary antibodies (Jackson Immunoresearch, 1:500 in block solution) for 1hr at RT, washed again, and mounted using VECTASHIELD Vibrance (Vector Labs) mounting medium. Imaging was performed in a semi-automated fashion using a Nikon Ti2E microscope outfitted with a W1-Yokogawa spinning disk module and Prime BSI camera (Photometrics). Autofocus was used to set the Z location for each tile using the DAPI channel.
Whole Mount Olfactory Bulb Imaging
Whole OBs were dissected along with the rest of the brain into ice-cold 1xPBS. Brains were placed on 35mm dishes with No 1.5 coverslip bottoms (MatTek), dorsal (M71) or ventral (Mor28) surface facing down depending on the glomeruli being imaged. Excess PBS was blotted away, and OBs were imaged using a Nikon Ti2E microscope outfitted with a W1-Yokogawa spinning disk module and Prime BSI camera (Photometrics). Tiled Z-stacks through entire glomeruli were taken at low (4x) and high (20x) magnification. M71 and Mor28 form two glomeruli in each OB, one lateral and one medial. The medial glomeruli for M71 were rarely visualized at p5 or p28, so whole mount analysis was restricted to the lateral glomeruli in these animals. In contrast, Mor28 glomeruli were readily visible at p5, though the lateral glomeruli were sometimes obscured by the curvature of the OB in p28 animals. Lateral and medial Mor28 glomeruli behaved identically under all manipulations tested, so all data is presented as pools containing both. After imaging, some OBs were fixed in 4% PFA (v/v, in 1xPBS) for 1hr at 4°C, washed 3 times in 1xPBS, embedded overnight in 30% (w/v) sucrose in 1xPBS and frozen in OCT for immunofluorescence experiments.
Olfactory Bulb Immunofluorescence
15μm-thick coronal sections were taken throughout the entire OB, moving anterior to posterior. OB immunostaining was performed using an identical protocol to the MOE. Slides were imaged at 20x in semi-automated fashion using a Nikon Ti2E microscope outfitted with a W1-Yokogawa spinning disk module and Prime BSI camera (Photometrics). Glomeruli were identified by visual inspection of the GFP and tdtomato channels of the resulting image stacks. ROIs were extracted for visualization and further analysis using custom jython scripts in FIJI.
Annotation of glomerular configurations
Configuration of the M71 and Mor28 glomeruli was assessed in a blinded fashion using Z-stacks taken of whole mount OBs at high magnification (20x). Blinding was accomplished using custom jython scripts, run in FIJI (Schindelin et al., 2012). Glomeruli were annotated as “intermixed” if the red and green fibers intertwined with one another, creating yellow pixels or many small patches of red and green. “Compartmentalized” glomeruli showed clear partitioning of red and green fibers into 2 connected regions of the glomerulus. Glomeruli where red and green fibers coalesced separately were annotated as “adjacent/accessory”. Rare instances where a red glomerulus was observed separate from a compartmentalized red/green glomerulus were also annotated as “adjacent/accessory”. Finally, glomeruli where green fibers coalesced, but red fibers were either absent or failed to form a defined structure were annotated as “no_coalescence”. There were exceedingly rare cases in which green fibers failed to form a glomerulus or where the collected images were not focused well enough to determine glomerular configurations. These glomeruli were annotated as “NA” and omitted from subsequent analysis.
Pearson correlations in Mor28 glomeruli
Correlations between red and green pixel intensities in Mor28 glomeruli were computed using whole mount images taken at high magnification (20x). Custom jython scripts were run in FIJI to display the Z stacks in a blinded fashion, facilitate ROI delineation on each Z slice based on the region of the glomerulus that was in focus for that slice and extract red and green pixel values for all pixels in each ROI. Pixel values were read into R, log-10 transformed, and Pearson correlations were taken between red/green values across all Z slices of each glomerulus.
Mor28 and M71 olfactory epithelium cell counts
Tissue sections from M71/Mor28-Perk or Ddit3 mice were immunostained for the appropriate OR and entire sections were imaged at 20x. OR+tdtomato+, OR+tdtomato− and OR+GFP+ cells were counted in a blinded fashion across 2–3 animals per genotype (8–12 sections per animal). Sections were selected such that they spanned the entire anterior-posterior breadth of the OE, in a manner that was identical across animals. Ratios of OR+tdtomato+/OR+GFP+ cells and total tdtomato+/OR+GFP+ cells were computed for each section.
Mor28 immunofluorescence intensity in the olfactory epithelium
OE sections (n=4) from Mor28 Ddit3 WT and cKO mice were immunostained for MOR28 and imaged at 20x. Mor28+tdtomato+ and Mor28+GFP+ cells were counted using the CellCounter plugin in FIJI. Custom jython scripts were used to extract 60 pixel2 ROIs around every identified cell, segment the cell boundaries using GFP or tdtomato signal, and record both the X,Y positions and average signal intensity in the Mor28 channel for each cell. Since immunofluorescence signal can vary across tissue and between tissue sections, only local comparisons were made. This was accomplished by using the hierarchical DBSCAN algorithm implemented in the hdbscan function from the dbscan R package (v1.1–8)(Hahsler et al., 2019) to cluster cells based on their X,Y positions within a section (minPts = 6 cells/cluster). The average log10-transformed Mor28 intensity was computed in tdtomato and GFP+ cells within each cluster, and the resulting local differences in Mor28 intensity between the two cell types were reported.
Bulk RNA Sequencing
Input cell numbers, sequencing depth and alignment parameters for all bulk RNA-seq libraries are provided in Supplemental Table 5. For RNA-seq of OSN developmental populations, cell pellets were lysed in an RNA lysis buffer (20mM Tris pH=7.5, 150mM NaCl, 5mM MgCl2, 1mM DTT, 100μg/mL cycloheximide (Sigma), 1%(v/v) Triton X-100 and 25U/mL TURBO DNAse (Invitrogen)) for 10 minutes on ice(McGlincy and Ingolia, 2017). Lysates were cleared by centrifugation for 10 minutes at 20,000xg at 4°C and RNA was quantified against a standard curve using the Quant-it RiboGreen RNA assay (Thermo Fisher Scientific). Lysate containing 100ng of RNA was mixed with 300μL of Trizol LS and frozen to −80°C until further processing. For all other RNA-seq experiments, 400–20k cells were directly sorted into 500μL Trizol LS, snap frozen in liquid N2, and stored at −80°C until further processing (see Supplemental Table 5).
Trizol LS mixtures were thawed and RNA isolated using the Zymo Direct Zol kit and the manufacturer’s instructions. Eluted RNA was precipitated overnight at −80°C with 300mM sodium acetate, 1.5 volumes of isopropanol and 2uL of Glycoblue (Thermo Fisher). RNA was pelleted by centrifugation at 20,000g for 30minutes at 4°C, washed twice with 70% ethanol and eluted into water. Genomic DNA was removed with TURBO DNAse (Invitrogen) for 30 minutes at 37°C. RNA was then recovered using 1.8x Ampure XP Beads (Beckman Coulter). For RNA-seq of OSN developmental populations, purified RNA yields were assessed by Quant-it RiboGreen RNA assay (Thermo Fisher Scientific) and 6–10ng of RNA was used for library prep (see Supplemental Table 5). For all other RNA-seq experiments, all recovered RNA was used for library prep. Library preparation was performed using the SMARTER Stranded Total RNA-Seq Kit- Pico Input Mamallian v2 (Takara Bio USA). Library quality was assessed using a Bioanalyzer 2100 (Aglient) and libraries were quantified using Qubit HS Assay (Thermo Fisher Scientific). Paired-end Illumina sequencing was performed using the NextSeq 550 or Novaseq 6000 systems (see Supplemental Table 5).
RNA Sequencing General Bioinformatics
Reads were quantified using Salmon (version 0.13.1) against a custom transcriptome based on mm10, with extended olfactory receptor annotations(Ibarra-Soria et al., 2014), EGFP (Addgene #2485), LacZ (Addgene #32642) and tdtomato (Addgene #22799) sequences. Only mm10 transcripts with an annotated CDS were retained in this transcriptome. Quantified reads were summarized to gene level and imported to R using tximport (Soneson et al., 2015)). Analysis used DESeq2(Love et al., 2014) and custom R code. Principal component analysis (PCA) was performed as an initial step to look for sample outliers or batch effects. No outliers were excluded in these experiments, batch effects were modelled into the design formulas when relevant. Normalized counts (using DESeq2’s median of ratios method) were used for all downstream analysis. Where relevant, gene ontology (GO) analysis was performed using the gost function from the gprofiler2 R package (v0.1.8,(Raudvere et al., 2019)), sources = ‘GO’, and a custom background containing all non-OR genes in our genome annotation. Where relevant, gene set enrichment analysis (GSEA) was performed using the fgsea function from the fgsea R package (v1.12.0) (Korotkevich et al., 2021)and the MSigDB mouse-ortholog hallmark gene sets (v7.5.1, downloaded from https://www.gsea-msigdb.org/gsea/msigdb/index.jsp in June 2022). The Wald statistic computed for the contrast of interest in DESeq2 was used as the input to fgsea and nperm was set to 10,000.
OSN developmental RNA-seq
For RNA-seq experiments in INPs, iOSNs and mOSNs (Figure S1G–J), data was analyzed using a model of counts ~ differentiation stage. P values were computed via likelihood ratio test (LRT) against a model of counts ~ 1. A variance stabilizing transformation (VST) was applied to the counts using DESeq2’s vst function and the rows of the resulting matrix were scaled for some visualizations. Clusters of genes with similar patterns of expression across differentiation were identified by applying the base R dist, hclust and cutree (with k=6) functions to the scaled, vst-transformed count data matrix.
Stress Score Definition
iOSNs (iRFP+GFP−) and mOSNs (iRFP+GFP+) were identified via FACS in p6-p11 Atf5(rep/+); Omp(iresGFP/+) mice. High and low iRFP sub-bins were defined as the top and bottom quarter of cells in each developmental population by iRFP fluorescence. 10–20k cells were sorted by FACS from each of these four populations (iOSN high/low iRFP, mOSN high/low iRFP), n=4 biological replicates. RNA-seq libraries were prepared and analyzed in DESeq2 using a model ~population + iRFP intensity. The iRFP high/low contrast was extracted and stress scores were defined for each OR as the log2 fold change associated with this term. Mathematically, this is equal to the average log2 fold change high/low stress across populations. Stress scores were cross-referenced with previously published OR-zone annotations (Tan and Xie, 2018), Rtp1/2 cKO RNA-seq data (Sharma et al., 2017) and Atf5 cKO RNA-seq data (Dalton et al., 2013).
M71→P2 Swap Experiments
The M71→P2 swap experiments used a total of n=6 “WT” animals (Atf5(rep/+); Omp(iresGFP/+), n=4 from stress score definition experiments, n=2 additional) and n=3 “Swap” animals (P2(M71iresLacZ/+); Atf5(rep/+); Omp(iresGFP/+)). FACS and library preparation were as described for the stress score definition experiments. For figures where a single stress score was presented for each gene (Figure 2B, Supplemental Figure S3B), stress score calculations were performed in R using a similar approach to the original stress score calculation. Specifically, normalized counts were first averaged for each population:iRFP intensity grouping, then stress scores were defined as ERS = mean(log2(iOSN High/Low stress), log2(mOSN High/Low stress), na.rm = T). Stress scores for the M71→P2 swap allele were computed using lacZ in “Swap” animals (n=3). Endogenous scores for M71 and P2 were computed only in “WT” animals (n=6) to avoid confounding by the hybrid M71→P2 allele. Stress scores for all other ORs were computed across all libraries (n=9). Importantly, our analysis approach is mathematically equivalent to the DESeq2 approach used in initial stress score definition experiments, which is designed to test whether a given gene is statistically significantly differentially expressed across experimental groups. To ask whether stress scores significantly differed between the M71, P2 and M71→P2 genes, we adjusted our approach by omitting the initial averaging step across libraries in each group. We thus calculated stress scores on a per-library basis, using the formula ERS = mean(log2(iOSN High/Low stress), log2(mOSN High/Low stress, na.rm = T). Only finite stress scores were kept in the final analysis (Supplemental Figure 3C) and one-way ANOVA with Tukey’s post-test was used to determine whether stress score differed across replicate measurements for these genes.
Mor28-Ddit3 RNA-seq
RNA-seq experiments in Mor28 Ddit3 animals were performed in 4 week old mice. GFP+ and tdtomato+ cells were FAC-sorted from WT (n=3), Het (n=5), and cKO (n=4) animals and libraries prepared as above. Three terms were modelled: Genotype (WT/Het/cKO), Population (GFP/tdtomato) and ind (index, 1–5 within each genotype). A likelihood ratio test (LRT) was used to compare the full model of counts ~ Genotype + Genotype:ind + Genotype:Population to a reduced model of counts ~ Genotype + Genotype:ind + Population in DESeq2. All-zero columns of the full and reduced model matrices (which result from the unbalanced design) were removed prior to DESeq2 analysis. P values reflect whether the ratio of counts in tdtomato/GFP cells differs across genotypes. The average difference in the log2 fold change tdtomato/GFP in Ddit3 cKO – WT OSNs was extracted using the DESeq2∷results function, setting contrast = list(c(‘GenotypecKO.Populationtdtomato’), c(‘GenotypeWT.Populationtdtomato’)). The average difference in the log2 fold change tdtomato/GFP in Ddit3 Het – WT OSNs was extracted similarly, but with contrast = list(c(‘GenotypeHet.Populationtdtomato’), c(‘GenotypeWT.Populationtdtomato’)).
Omp(iresCre/+) Ddit3 RNA-seq
RNA-seq experiments in Omp-Cre Ddit3 animals were performed at p9–10. Tdtomato+ OSNs were FAC-sorted from WT (n=4), Het (n=5), and cKO (n=5) animals and libraries prepared as above. A batch effect was observed on PCA (Supplemental Figure S7G), so two terms were modeled: genotype (WT/Het/cKO) and batch (a/b/c). A likelihood ratio test (LRT) was used to compare a full model of counts ~ batch + genotype to a reduced model of counts ~ batch in DESeq2. P values reflect whether counts differ across genotype. Log2 fold changes were computed for cKO vs. WT and Het vs. WT using the relevant terms in DESeq2.
RNA-seq genomic alignments and coverage visualization
Genomic alignments were performed only to visualize RNA-seq coverage, all quantitative comparisons were made using pseudoalignment (see above). STAR (v2.5.3a,(Dobin et al., 2013)) was used to align reads to mm10. During genome generation, our custom set of annotations (see above) were passed to –sjdbGTFfile and – sjdbOverhang was set to 74. Noncanonical unannotated splice junctions were filtered during alignment and -- outFilterMultimapNmax was set to 10. Samtools (v1.7) (Danecek et al., 2021) was used to remove low quality alignments (-q 30). To determine scale factors for coverage plots, reads were counted in R using the featureCounts function from the Rsubread package (v2.0.1,(Liao et al., 2019)), our custom annotations and setting GTF.featureType = ‘exon’, allowMultiOverlap = T, useMetaFeatures = T, readShiftType = ‘downstream’, readShiftSize = 0, read2pos = NULL, strandSpecific = 2 and isPairedEnd = T. The resulting count matrix was read into DESeq2 and the DESeq function was used to compute sizeFactors. Coverage was computed using bamCoverage (v3.1.1,(Ramirez et al., 2016)) with –scaleFactor set to 1/the appropriate sizeFactor from DESeq2 and –binSize 1. Bigwig files for the forward and reverse strands were generated using the relevant -- filterRNAstrand arguments. Bigwig files were read into R using the import function from the rtracklayer package (v1.46.0, (Lawrence et al., 2009)) and coverage plots were generated using custom R code.
OR Protein Multiple Sequence Alignment and PCoA
Extended OR gene models (Ibarra-Soria et al., 2014) were used to extract OR CDSs from the mm10 genome assembly. In cases where multiple transcripts were annotated for a given OR, we used the top-expressed transcript based on Salmon-aligned RNA-seq data from Omp-GFP positive mOSNs. CDS sequences were translated and the resulting protein sequences aligned using the DECIPHER R package (v2.14.0). Alignments were staggered using DECIPHER’s StaggerAlignment function and distance matrices computed using the DistanceMatrix function. Principal coordinates analysis (PCoA) was performed by applying the cmdscale function from the stats package (v3.6.1) to the distance matrix, setting k=30. K-means clustering was performed on the PCoA-reduced data using the kmeans function from the stats package (v3.6.1), setting centers = 6. Clusters were arranged by the average ER stress score for their constituent ORs.
Random Forest Regression
We performed a second OR MSA on a total of 1,161 OR sequences: all the sequences used for PCoA visualization above plus all mutant OR sequences from(Feinstein and Mombaerts, 2004). The resulting MSA contained 2,176 positions, 1,854 (85.2%) of which were empty (“−“) for >90% of the OR sequences. We filtered the MSA to retain the 322 positions in which at least 10% of OR sequences had an aligned amino acid, using these as predictor variables for random forest regression modeling OR stress scores. For model training, we omitted 30 OR sequences without defined stress scores (this includes all of the mutant sequences from(Feinstein and Mombaerts, 2004)). Stress scores were later predicted for these sequences using the trained model.
Model training was performed using the caret (v6.0–86) and randomForest (v4.6–12) packages in R. 10-fold cross validation was repeated 3 times to evaluate model fit across a grid of ntree (100,500,1000) and mtry (3,20,50,100) values. RMSE was used to assess model performance and the importance = T argument was passed to the randomForest function to compute the importance of predictor variables by permutation. We used 500 trees and mtry = 50 for our final model, which provided good balance between model performance and run time. Caret reported an r2 of 0.51 for the final model using the held-out data.
We observed a strong relationship between feature importance in the random forest regression (percent increase in mean squared error, %IncMSE, measured by permutation) and the Shannon entropy at these positions in the MSA (Supplemental Figure S3G). To identify positions with the strongest relationships to stress score without simply biasing to positions with the highest variability in the MSA, we defined corrected %IncMSE as the residuals of a linear model %IncMSE ~ Shannon entropy and used these values for all downstream analysis. Corrected %IncMSE values were visualized by snakeplots, which were computed by creating lookup tables between amino acid positions in P2 and M71 as model ORs vs. positions in the MSA. Uniprot annotations (P2 accession: Q9JKA6, M71 accession: Q60893, accessed June 2022) and a modified version of snakeplotter (https://github.com/Yue-Jiang/snakeplotter) were used to create the snakeplots for each model OR.
Stress score predictions in M71/M72 mutant
The zone 1 ORs M71 and M72 project to adjacent glomeruli in the dorsal OB and differ by just 11 amino acids (Feinstein and Mombaerts, 2004). Feinstein and Mombaerts generated a series of 10 mutant ORs by incorporating subsets of these mutations into the M71 OR locus, making hybrid ORs that were closer to the M72 sequence(Feinstein and Mombaerts, 2004). They then crossed mice containing these hybrid ORs together, making pairwise comparisons as to whether sets of amino acid mutations affected glomerular targeting. We coded the phenotypes from Figure 4 of (Feinstein and Mombaerts, 2004) into three categories: intermixed, compartmentalized (targeting the same glomerulus, but forming distinct sub-structures) and adjacent. Only mutations knocked into the M71 locus were analyzed (ie. endogenous M72 was omitted in favor of the M72→M71 mutant). For lacZ-lacZ comparisons, we considered the configuration compartmentalized if the circles were touching in Figure 4, and adjacent if space could be observed between them. Partial co-mingling in GFP/lacZ experiments was considered compartmentalized. If multiple phenotypes were reported for a given comparison, we used the stronger phenotype. Finally, we used our random forest regression model to predict stress scores for each mutant OR sequence and calculated the absolute value of the difference in stress scores for each pairwise comparison studied(Feinstein and Mombaerts, 2004). We omitted homozygous animals since these all had a phenotype of “intermixed” and a predicted stress score difference of zero.
Stress score predictions in polymorphic C57BL/6 vs 129 ORs
The P2, P4, M72 and M50 OR sequences all display 1–2 polymorphic amino acid residues between the C57BL/6 and 129 mouse strains. As described in Figure 7 of (Feinstein and Mombaerts, 2004), the M50 (129) axons segregate from M50 (B6) axons in vivo, while compartmentalized configurations are observed for the other 3 OR sequences. We used our random forest regression model to predict the stress score associated with each polymorphic OR sequence and calculated the absolute difference in stress scores between B6 and 129 for each of the 4 ORs. To visualize the polymorphic residues on each OR sequence in relation to the transmembrane domains of the OR, we used the following Uniprot annotations: P34986 (M50), Q9JKA6 (P2), Q7TRN0 (P4) and Q60893 (M71, used also for M72 since the M72 annotation Q8VGE3 had only 6 TM domains).
scRNA-seq library construction
scRNAseq data was aggregated from six experiments conducted over several years in the Lomvardas laboratory. Experimental animals were functionally wild type (though some mice contained tTA drivers without a corresponding tetO allele or Cre drivers without corresponding floxed alleles). Animals ranged in age from p14 to adult. MOE were isolated, cells were dissociated using papain (Worthington), dead cells were identified using DAPI, and libraries were prepared using the Chromium Single Cell 3’ Reagent Kit v2 or v3 (10X Genomics). Libraries were sequenced on a Novaseq6000
scRNA-seq alignment and clustering
Fastq files were analyzed using cellranger v2.1.1–3.1.0 (10X Genomics) and a custom transcriptome based on mm10 with extended olfactory receptor annotations (Ibarra-Soria et al., 2014). Filtered count matrices from cellranger were read into R and analyzed using the Seurat package (v3.1.3). A cell was included in the analysis if it contained greater than 200 features and a feature was included in the analysis if it was expressed in greater than 3 cells. The sctransform (SCT) normalization method (Hafemeister and Satija, 2019) was used to correct for sequencing depth in all samples. A standard integration analysis was performed to merge the six datasets using the top 3000 features ranked by SelectIntegrationFeatures. Principal component analysis was performed, and 30 principal components were used for the subsequent UMAP visualization and graph-based clustering steps of the Seurat pipeline (resolution = 0.5). SCT-corrected counts were used for differential expression testing, cluster identification and OR choice annotations.
scRNA-seq OR choice annotations
mOSN-containing clusters were identified by expression of the marker gene Omp. In each cell, SCT-corrected counts were extracted for the top two expressed ORs. The top-expressed OR was considered the chosen OR if the corrected counts for this gene were 2x higher than corrected counts for the second highest expressed OR in that cell. OR choice calls were successfully made for 92% (8112/8815) of cells. The chosen OR was then used to determine the zone of each single cell (Tan and Xie, 2018) and annotate whether the cell was high-stress (chosen OR stress score >0) or low-stress (chosen OR stress score <0).
scRNA-seq differential expression
To identify genes differentially expressed between high and low stress OSNs while simultaneously regressing out zone as a covariate, we employed the pairwiseWilcox function from the scran R package (v1.14.6, (Lun et al., 2016). SCT-corrected, normalized counts were compared between high and low stress OSNs, blocking across zone and adjusting p values for multiple comparisons. Log2 fold changes between high and low-stress OSNs were computed by averaging the expression for each gene in high and low stress OSNs within each zone, taking the log2 fold change high/low stress within each zone, and computing a weighted mean across zones. The weight for each zone was defined as the number of high stress cells * number of low stress cells in that zone. Relationships between the UPR and activity were assessed by cross-referencing our differential expression data with a previous meta-analysis of activity dependent genes in the OE (Wang et al., 2017) and a recent dataset where activity patterns were inferred in a large scRNA-seq dataset (Tsukahara et al., 2021).
Axon Guidance Network Definition
The Amigo2 web application (v2.5.17,(Gene Ontology, 2001, 2021), (Carbon et al., 2009) was used to identify 59 GO terms based on the keyword search “axon+guidance” and the filters idspace: GO and is_obsolete: false. The getBM function from the biomaRt package (v2.42.1, (Durinck et al., 2009)) was used to query the Ensembl database (v97) for all genes associated with these GO terms. 340 genes were identified, and the list was manually curated to include additional olfactory axon guidance genes (Kirrel2/3, Tenm2/4, Pcdh10/17/19 and Nrp) and remove genes whose products are not predicted to be on the cell surface. To identify the network of UPR-dependent axon guidance molecules, the general list of axon guidance molecules was cross-referenced with the list of genes that were statistically significantly differentially expressed between high and low stress OSNs within zone in the scRNA-seq data.
ARACNE-AP Network Reconstruction
272 paired-end bulk RNA-seq samples prepared in the Lomvardas laboratory over the last ten years were quantified using Salmon, as above. These libraries reflect a wide range of sorted cell and whole OE preparations under a variety of genetic and experimental manipulations. A variance stabilizing transformation was applied to the resulting count matrix using the DESeq2∷vst function in R. A list of 919 mouse transcription factors was compiled by using the biomaRt R package to query Ensembl for genes annotated in the “DNA-binding transcription factor activity” GO category (GO:0003700) or annotated as “DNA binding” (GO:0003677) and “transcription, DNA-templated” (GO:0006351) or “transcription factor binding“ (GO:0008134). The vst-transformed count matrix and transcription factor list were fed into ARACNE-AP (v1.0, (Lachmann et al., 2016)), and 100 bootstraps were run using a p value threshold of 1e-8. The bootstraps were consolidated using the – consolidate flag to generate the final network.
msVIPER
The ARACNE-AP-generated OSN transcriptional network was loaded into R using the aracne2regulon function in the viper package (v1.20.0, (Alvarez et al., 2016)). The inferred direction of regulation (tfmode) contained in the regulon object was used for visualization when generating graphical representations of the predicted Ddit3 regulatory network. For identification of differentially active TFs, scRNA-seq data was subset to include only mOSNs where the chosen OR could be determined, and both the stress level and zone of the OR annotated. Count matrices were variance-stabilized by re-running the SCTransform function from the Seurat package, requesting all Pearson residuals instead of the default 3000. OSNs were then split by zone and viper was run on high vs. low stress OSNs within each zone, as per the package vignette. Briefly, gene expression signatures (GES) were computed by performing Student’s t-test on the Pearson residuals (from the @assays$SCT@scale.data slot of the Seurat object) from high vs. low stress OSNs within each zone using viper’s rowTtest function. T statistics were converted to z scores for downstream analysis. Viper’s ttestNull function was used to generate a null model by permuting the samples 1000 times. Viper’s msviper function was then used to identify differentially active TFs using the GES, null model, and regulon object. To aggregate the results across zone, a weighted average of normalized enrichment scores (NES) was used. P values were Stouffer integrated across zones using the sumz function from the metap R package (v1.3) and corrected for multiple testing using the p.adjust function from the stats R package (v3.6.1), method = ‘fdr’. Weights were proportional to the square-root of the number of cells in each zone for NES and p value integration.
QUANTIFICATION AND STATISTICAL ANALYSIS
All bulk RNA-seq data was quantified using salmon(Patro et al., 2017) and the DESeq2 R package(Love et al., 2014). Single-cell RNA-seq data was quantified using cellranger (10x Genomics) as well as the Seurat(Stuart et al., 2019) and scran(Lun et al., 2016) and R packages. msVIPER analysis was performed using the viper R package(Alvarez et al., 2016). Qualitative assessment of axon targeting phenotypes was performed in a blinded fashion, as outlined in the methods details. Quantitative assessment of glomerular configurations was performed using FIJI (Schindelin et al., 2012) as well as custom python and R code. All other quantification was performed using custom python and R code, as outlined in the methods details.
All statistical tests were performed in R. A significance cutoff of p=0.05 was used for all analyses and p values were adjusted for multiple comparisons where relevant (denoted as padj in the text, figures and figure legends). Multiple types of statistical hypothesis tests were employed, and numbers of biological replicates varied across experiments. Specifics are described for each experiment in the accompanying figures, figure legends, and methods details. No methods were used to pre-determine sample sizes.
Supplementary Material
Supplemental Table S1 (relevant to Figure 1): Common ORs referenced in this study
Supplemental Table S2 (relevant to Figure 2): Mouse strains used in this study
Supplemental Table S3 (relevant to Figure 2): Laser and detector settings for FACS experiments
Supplemental Table S4 (relevant to Figure 4): Antibodies used in this study
Supplemental Table S5 (relevant to Figure 7): Summary of all RNA-seq Libraries
Supplemental Figure S1 (relevant to Figure 1). Atf5(rep) is a translational reporter for ER stress that follows the expression properties of the intact Atf5 gene in the OSN lineage.
(A) Single Cell RNA-seq (scRNA-seq) analysis of wild-type OE, visualized using uniform manifold approximation and projection (UMAP). Atf5 is highly transcribed throughout the OSN lineage. Neurog1, Gng8, and Omp expression defines GBCs/INPs, INPs/iOSNs, and mOSNs, respectively.
(B) Log-10 normalized RNA-seq counts for all ORs in Omp-iresGFP+ mOSNs isolated from mice heterozygous or wild-type for the Atf5(rep) allele. Spearman correlation and linear regression line (blue) shown relative to the line y=x (black, dashed).
(C) CRE (green) immunofluorescence in MOE sections from heterozygous Atf5(rep/+) mice at postnatal day 13 (p13). DAPI nuclear marker in blue. Scale bar 50μm.
(D) Schematic of OSN differentiation from immediate neuronal precursors (INPs) to immature OSNs (iOSNs) and mature OSNs (mOSNs). Marker lines for INPs and mOSNs as shown, UPR is maximal in iOSNs.
(E) Representative flow cytometry plots defining (i) INP, (ii) iOSN and (iii) mOSN populations using GFP signal intensity (y- axis) from Ngn1-GFP mice (GFP highest in INPs, at p6) and Omp-iresGFP mice (GFP in mOSNs, at p11 and p56). iRFP intensity (x-axis) is used to quantify levels of ER stress in each population.
(F) iRFP intensity (from the Atf5 reporter allele) in INPs, early mOSNs (p11) and late mOSN (p56), each measured relative to iOSNs (iRFP+GFP− cells in each plot). Each point is one replicate, the red line connects the means in each group.
(G) PCA plot of RNA-seq libraries generated from INP (red), iOSN (green) and mOSN (blue) sorted cells.
(H) Line plot of centered, scaled and variance stabilizing transformation (VST)-transformed OR expression values in INPs, iOSNs and mOSNs by RNA-seq. Each black line is an OR, red line connects medians across all ORs (error bars span first and third quantiles).
(I) Heatmap showing centered, scaled and VST-transformed expression values for all genes with significant differential expression across INPs, iOSNs and mOSNs by RNA-seq (n=2 biological replicates per population). Genes (rows) are ordered by hierarchical clustering and cut into six clusters shown in the color bar at right. Summary medians, first and third quantiles are shown for each cluster in the line plots.
(J) Top significantly-enriched Gene Ontology (GO) terms for clusters 5 and 6 (from I), GO terms are on the y-axis, −log10-false discovery rate (FDR) is on the x. GO terms of interest for this study are highlighted in blue (cluster 5) or pink (cluster 6).
Supplemental Figure S2 (relevant to Figure 1). High-throughput measurement of ER stress scores and correlation with OR trafficking properties
(A) PCA of RNA-seq data used in the high-throughput determination of OR stress levels. The experiment was performed n=4 times, with four libraries prepared for each replicate. Color represents stress bin (high = red, low = blue), shape represents developmental population (iOSN = iRFP = circles, mOSN = iRFPGFP = triangles).
(B) MA plot showing log2 fold changes between high and low stress OSNs (y axis) as a function of the log10-transformed mean of the normalized counts in all libraries (x axis). Significantly differentially expressed (DE) genes are shown in red, non-significant genes are in black, and OR genes are facetted out from all others. Numbers of genes in each category are indicated in the labels, p value is for a chi-squared test of whether OR representation among statistically significant DE genes deviates from the null. Plots depict averages from 4 biological replicates.
(C) Gene set enrichment analysis (GSEA) for 50 hallmark gene sets in high versus low-stress bulk sorted OSNs. Y-axis denotes gene set, x-axis shows normalized enrichment score in high versus low stress cells, colored by significance (red = significant). The hallmark unfolded protein response gene set is highlighted in red on the y axis.
(D) ER stress score values for all ORs in mOSNs only (y-axis) or mOSNs and iOSNs (x-axis) isolated by FACS from Atf5(rep/+); Omp(iresGFP/+) mice. The latter definition is used for all other analyses in the paper. Spearman correlation and linear regression line (blue) shown relative to the line y=x (black, dashed).
(E) Cumulative density plots of ER stress scores in Atf5-dependent (red) versus Atf5-independent (blue) ORs. Data represent averages of 4 biological replicates.
(F) Cumulative density plots of stress scores in Rtp1/2-dependent (red) versus Rtp1/2-independent (blue) ORs. Data represent averages of 4 biological replicates.
(G) Violin plots showing log2 fold changes for all ORs in Omp-iresGFP+ mOSNs isolated from global Atf5 KO vs. WT mice, grouped/colored by zone and facetted by class. Log2 fold changes are similar across these subpopulations. RNA-seq data is from Dalton et al., 2013.
(H) Violin plots showing log2 fold changes for all ORs in the whole olfactory epithelium (OE) of global Rtp1/2 KO vs. WT mice, grouped/colored by zone and facetted by class. Log2 fold changes are similar across these subpopulations. RNA-seq data is from Sharma et al., 2017.
Supplemental Figure S3 (relevant to Figure 2). Random forest regression predicts ER stress scores from OR amino acid sequences.
(A) PCA of RNA-seq libraries used to compute stress scores in the M71→P2 swap experiment. There are four libraries per mouse, colored by the interaction between population (iOSN = iRFP, mOSN = iRFPGFP) and stress level (low vs. high) (see Figure 1F for an overview of stress score calculation and methods for additional details). Circles represent “WT” mice (n=4 animals from Supplemental Figure S2A + n=2 animals added for this experiment), triangles represent M71→P2 swap mice (n=3).
(B) Stress scores computed for all ORs in P2(M71iresLacZ/+); Atf5(rep/+); Omp(iresGFP/+) (y-axis, n=3) versus Atf5(rep/+); Omp(iresGFP/+) (x-axis, n=6) mice (see methods). Spearman correlation and linear regression line (blue) shown.
(C) Stress scores computed on a per-library basis for the M71→P2 swap experiment. Scores for M71 (red) and P2 (purple) were computed using “WT” (Atf5(rep/+); Omp(iresGFP/+), n=6) animals. Scores for the M71→P2 swap allele (green) were computed using “Swap” (P2(M71iresLacZ/+); Atf5(rep/+); Omp(iresGFP/+), n=3) animals. Non-finite scores omitted. Padj P2-M71 = 4.07E-3, Swap-M71 = 0.29, Swap-P2 = 5.55E-4 (one-way ANOVA with Tukey’s post-test).
(D) Principal coordinates analysis (PCoA) for all OR amino acid sequences. Each point represents an OR, distances between points reflect the similarity between OR sequences in the MSA. Colors indicate zonal identity of each OR.
(E) Random forest regression model training using root mean square error (RMSE, y-axis) to evaluate model fit over a grid of ntree (color) and mtry (x-axis) values. 10-fold cross validation was repeated 3 times for each set of parameters. The final model used 500 trees and mtry = 50.
(F) Random forest predicted ER stress scores (y-axis) versus experimentally measured ER stress scores (x-axis, see also Figure 1G) for all OR sequences. Linear regression (blue) is shown relative to the line y=x (dashed, black). R2 is calculated from held-out data in the repeated cross-validation procedure (see methods).
(G) Percent increase in mean squared error (%IncMSE, y-axis) for random forest regression model after permuting each predictor variable (position in the OR MSA) versus the Shannon entropy at each position in the OR MSA. The blue line shows a linear regression of %IncMSE by Shannon entropy, the residuals of which comprise the corrected %IncMSE values plotted in Figure 2G. The top 10 positions by corrected %IncMSE are shown in red and labeled in Figure 2G.
(H) Summary of 4 ORs with polymorphisms between the C57BL/6 (B6) and 129 mouse strains(Feinstein and Mombaerts, 2004). Boxes indicate transmembrane domains; red points indicate the polymorphisms. The heatmaps on the right summarize the RF regression-predicted differences in ER stress scores between the B6 and 129 sequence for each OR (left column) and the configuration observed in vivo by Feinstein and Mombaerts (right).
(I) Amino acid composition at the top 10 residues identified as important for OR stress scores by the random forest regression model. Positions are named as follows: amino acid [position in P2 sequence] ([position in MSA]) (also see Figure 2G). OR sequences are binned into deciles by stress score (1= lowest stress scores, 10 = highest), and fractional amino acid composition at each position across the ten deciles is shown by the colored bars. (−) indicates a gap in alignment at the indicated position. Facets are arranged by descending variable importance.
Supplemental Figure S4 (relevant to Figure 4). Conditional Perk deletion prevents OSN axon coalescence without disturbing the stability of OR gene choice at p5.
(A) IF for M71 (purple) in the MOE of M71 Perk Het (top row) or cKO (bottom row) mice at p5. Endogenous GFP (green) and tdtomato (red) signals are shown. Outlines of M71+GFP+ (green) and M71+tdtomato+ (red) cells are traced on the M71 panel. Scale bar 50μm.
(B) IF for MOR28 (purple) in the MOE of Mor28 Perk WT, Het or cKO mice at p5. Colors as in A. Scale bar 50μm.
(C) Loess smoothed curves and standard errors of corrected iRFP signal (y-axis) vs. differentiation (x-axis) for Omp-iresGFP+ mOSNs in Perk WT (solid black line) vs. global Perk Het (dashed black line) mice. iRFP measurements in OSN subpopulations (from Figure 1D) are shown for comparison.
(D) Pearson correlations between red and green axons in Mor28 Perk WT and Het glomeruli at p5. Each point is a glomerulus, colored and grouped by genotype. P value calculated by two-sample Wilcoxon test.
(E) IF for MOR28 protein (purple) in OB glomeruli of Mor28 Perk WT or Perk Het mice at p5. Endogenous tdtomato (red) and GFP (green) signals are shown. Scale bar 50μm.
Supplemental Figure S5 (relevant to Figure 4). Conditional Perk deletion results in OSN depletion at p28.
(A) Whole mount views of olfactory bulbs of M71 Perk mice at postnatal day 28 (p28). Internal control neurons are shown in green, experimental neurons in red with the indicated genotypes. Magnification as indicated, A=anterior, P = posterior. Scale bars 500μm (4x), 100μm (20x).
(B) Whole mount views of olfactory bulbs of Mor28 Perk mice at postnatal day 28 (p28). Abbreviations and scale bars as in A.
(C) Blinded quantification of glomerular configurations in the OB of p28 M71 (left facet) and Mor28 Perk animals (right facet), grouped by genotype of the tdtomato+ cells. Colors indicate glomerular configurations. Number of animals and glomeruli quantified as labeled.
(D) IF for MOR28 (purple), endogenous GFP (green) and tdtomato (red) in the MOE of Mor28 Perk Het (top row) vs. cKO (bottom row) mice at p28. Outlines of M28+GFP+ (green) and Mor28+tdtomato+ (red) cells are traced on the MOR28 panel. Scale bar 50μm.
(E) Ratios of Mor28+tdtomato+ to Mor28+GFP+ cells in the MOE of Mor28 Perk Het (green) or cKO (blue) mice at p28 (left). Ratios of total tdtomato+ cells to Mor28+GFP+ cells in the same MOEs (right). Each point is a section, data is grouped by biological replicate (n=2 mice per genotype).
(F) IF for activated Caspase 3 (red) in MOE sections from Perk(fl/fl) (top) or Omp(Cre/+); Perk(fl/fl) (bottom) mice at p28.
(G) Quantification of activated Caspase 3+ mOSNs per mm in MOE sections from Perk(fl/fl) (red), Omp(Cre/+); Perk(fl/fl) (green) and Gg8-Cre; Perk(fl/fl) (blue) mice. Gg8-Cre drives Perk deletion at the INP stage, before axonal coalescence, and is included for reference. Each point is a section (n=3 biological replicates/condition, 4–5 sections/replicate). Padj = 0.1122 for Omp-Cre; Perk(fl/fl) vs. Perk(fl/fl), Padj <0.05 for all other comparisons by one-way ANOVA with Tukey’s post-test.
(H) IF for the mOSN marker OMP (green) and iOSN marker GAP43 (red) in coronal OB sections from Perk(fl/fl) (left) or Omp(Cre/+); Perk(fl/fl) (right) mice at p28. Glomeruli are present in both genotypes, albeit less well-defined in Omp-Cre Perk cKO animals.
Supplemental Figure S6 (relevant to Figure 5) ER stress levels determine axon guidance specificity via DDIT3.
(A) Loess smoothed curves and standard errors of corrected iRFP signal (y-axis) vs. differentiation (x-axis) for Omp-iresGFP+ mOSNs in Hspa5 WT (solid black line) vs. global Hspa5 Het (dashed black line) mice. iRFP measurements in OSN subpopulations (from Figure 1D) are shown for comparison.
(B) Diagram of the drug-inducible PERK experiment. tTA is expressed via an IRES element from the Omp locus and drives transcription of the tet-dependent FV2E-Perk-t2amCherry transgene. The Fv2E domain replaces the luminal domain of PERK, placing PERK dimerization and UPR activation under control of the drug AP20187.
(C) Dorsal OBs from p8 mice with indicated genotypes. AP20187 was injected IP at 0.1mg/kg 24 and 48 hours prior to analysis. Scale bar 500um.
(D) IF for caspase 3 (green) in the MOE of p8 mice with the labeled genotypes. AP20187 injection was as in C. DAPI (blue) and mCherry fluorescence from the transgene (red) are shown in the indicated panels.
(E) IF for DDIT3 (purple) in the MOE of p8 mice. Genotypes, drug treatment and counterstains as in D.
(F) IF for MOR28 protein (purple), endogenous GFP (green) and tdtomato (red) in the MOEs of Mor28 Ddit3 WT (top) or cKO (bottom) mice at p5. Outlines of Mor28+GFP+ (green) and Mor28+tdtomato+ (red) cells are traced on the Mor28 panel. Scale bar 50μm.
(G) Ratios of Mor28 IF signal in Mor28+tdtomato+ vs. Mor28+GFP+ cells in the MOE of p5 Mor28 Ddit3 WT (red) or cKO (blue) mice. Each data point is from one cluster of closely-positioned GFP/tdtomato cells from a single section (see methods), colored by genotype of the tdtomato+ cells (n=4 sections/genotype, 5–9 clusters/section). P=0.579 by two-sample Wilcoxon test.
(H) IF for MOR28 in the glomeruli of Mor28 Ddit3 WT (left) or cKO (right) mice at p5. Endogenous GFP (green) and tdtomato (red) shown in top row, MOR28 (purple) in bottom row. ROIs containing the red and green fibers are traced on the Mor28 panels. Data are representative of n=2 biological replicates. Scale bar 50μm.
Supplemental Figure S7 (relevant to Figure 7). Conditional Ddit3 deletion causes stable axon guidance changes by altering the transcription of high stress axon guidance genes.
(A) PCA plot of RNA-seq libraries from GFP+ (green) and tdtomato+ (red) sorted cells from p28 Mor28 Ddit3 WT (circles), Het (triangles) or cKO (squares) mice.
(B) MA plot for the p28 Mor28 Ddit3 RNA-seq experiment. The y-axis shows the difference in average log2 fold changes between tdtomato and GFP OSNs in Ddit3 cKO vs. WT mice. The x-axis shows the log10-transformed average of normalized counts across all samples. ORs are facetted out, significant genes (padj<0.05) are shown in red and genes of interest are labeled.
(C) Log2 fold changes in normalized counts of Nrp2 (top) and Tenm2 (bottom) between tdtomato and GFP cells in p28 Mor28 Ddit3 animals by RNA-seq. Data are colored by genotype (red = WT, green = Het, blue = cKO), and each point represents a biological replicate.
(D) Heatmap showing all genes from the stress-dependent axon guidance network described in Figure 3E. The top row shows the log2 fold change within zone for each gene in high versus low stress OSNs by scRNA-seq (colored blue → red). Genes are arranged in descending order based on these changes. The middle row shows the average log2 fold change tdtomato/GFP in Mor28 Ddit3 Het vs. WT animals (colored purple → green). The bottom row shows the same measurement, but in Mor28 Ddit3 cKO vs. WT animals. Genes where genotype significantly affects the differences in expression between GFP and tdtomato cells in the 4week Mor28 experiment are boxed in black.
(E) IF for NRP2 protein in the OB of p28 Mor28 Ddit3 cKO animals. Endogenous GFP (green, internal controls, functionally WT) and tdtomato (red, cKO) signal is shown in the top row, NRP2 staining (purple) is shown in the bottom row, with GFP (green) and tdtomato (red) glomeruli outlined. Images are representative of n=2 biological replicates. Boxplot shows quantification of the difference in mean log10-transformed NRP2 signal in tdtomato – GFP glomeruli. Each point represents a single section with adjacent GFP and tdtomato glomeruli (p = 2.29E-5 by one-sample Wilcoxon test against the null hypothesis that data is symmetric around 0).
(F) Normalized RNA-seq tracks showing coverage at the Ddit3 locus in the Omp-Cre Ddit3 experiments. Tracks are colored by genotype and represent averages across biological replicates (red = WT [n=4], green = Het [n=5], blue = cKO [n=5]). A gene model of the Ddit3 locus is shown at the bottom, thin boxes represent exons and thick boxes represent the CDS. LoxP sites are shown as red triangles on the gene model and as vertical dashed lines that extend through the RNA-seq tracks.
(G) PCA plot of sorted mOSN RNA-seq libraries from Omp(iresCre/+); Ddit3 WT (circles), Het (triangles) or cKO (squares) animals. Libraries were prepared in three batches (indicated by colors) and the observed batch effect was modeled in downstream analyses.
(H) MA plot depicting the log2 fold change in Omp-iresCre Ddit3 cKO vs. WT OSNs (y-axis) versus the log10 average normalized counts across all conditions (x-axis) in the Omp-iresCre Ddit3 RNA-seq experiment. Points are colored by significance (red = padj < 0.05), genes of interest are labeled, and ORs are shown in a separate facet.
(I) Kernel density estimates of the log2 fold changes for all ORs (black), high stress ORs only (stress score >0, red) or low stress ORs only (stress score <0, blue) in Omp(iresCre/+); Ddit3 cKO vs. WT OSNs. The dashed vertical black line denotes a log2 fold change of zero.
(J) Normalized RNA-seq counts for genes in the identified stress-dependent axon guidance network (Figure 3E) whose expression significantly changes in the Omp-iresCre Ddit3 dataset. Points are colored by genotype (red = WT, green = Het, blue = cKO) and facets are in order of decreasing significance.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cre (rabbit polyclonal) | Novagen | (#D00136944) |
| OMP (goat polyclonal) | Wako | (#544–10001) |
| GAP43 (rabbit polyclonal) | Novus Biologicals | (#143) |
| Cleaved Caspase-3 (rabbit polyclonal) | Cell Signaling Technology | (#9661) |
| Ddit3 (rabbit polyclonal) | Proteintech | (#5204–1-AP) |
| Nrp2 (goat polyclonal) | R&D Systems | (#AF2215) |
| Experimental Models (mice) | ||
| Atf5(rep) | This Study | |
| S50(YFP), Olfr545 tm4Mom | (Bozza et al., 2009) | Jax #006716 |
| M71(iresGFP), olfr151 tm26Mom | (Feinstein and Mombaerts, 2004) | Jax #006676 |
| Mor23(iresGFP), Olfr16 tm2Mom | (Vassalli et al., 2002) | Jax #006643 |
| P2(iresGFP) | (Shykind et al., 2004) | Donated by PI |
| Mor28(iresGFP) | (Shykind et al., 2004) | |
| Ngn-GFP, Tg(Neurog1-EGFP)DF148Gsat | MMRRC | #010618-UCD |
| Omp(iresGFP), | (Shykind et al., 2004) | Donated by PI |
| P2(M71iresLacZ, Olfr17 tm4Mom | (Feinstein and Mombaerts, 2004) | Jax #006651 |
| Perk(fl), Eif2ak3 tm1.2Drc | (Zhang et al., 2002) | Jax #023066 |
| Mor28(iresCre) | (Shykind et al., 2004) | Donated by PI |
| Ddit3(fl), Ddit3 tm1.1Irt | (Zhou et al., 2015) | Jax #030816 |
| Hspa5(fl), Hspa5 tm1.2Alee | (Luo et al., 2006) | Jax #035444 |
| Omp(iresCre), Omp tm1(cre)Jae | (Eggan et al., 2004) | Donated by PI |
| Omp(Cre) | (Li et al., 2004) | Jax #006668 |
| Rosa26(LSL-tdtomato), Gt(ROSA)26Sortm14(CAG-tdTomato)Hze | (Madisen et al., 2010) | Jax #007914 |
| Deposited data | ||
| RNA-seq | GEO | GSE198886 |
| IF Images | Zenodo | (doi: 10.5281/zenodo.6448088) |
| Software and algorithms | ||
| Original code | Github | https://github.com/hshayya/2022_Shayya_UPR_Guidance |
Highlights.
Olfactory receptor (OR) protein sequences determine neuronal ER stress levels
ER stress levels correlate with axon guidance gene expression differences
Manipulation of ER stress levels alter axon targeting specificity
ER stress-responsive Ddit3 levels transform OR identity into targeting specificity
Acknowledgments
Mice were treated in compliance with the rules and regulations of the Columbia University IACUC under protocol number AABG6553. Imaging was performed with the help of the Cellular Imaging Team at the Zuckerman Institute. We thank members of the Lomvardas lab for critical reading of the manuscript. We thank Dr. Tom Bozza for mice and Dr. David Ron for the Fv2E-Perk plasmid. This work was funded by R01DC015451 (SL), R21DC017823 (RD), and F30 DC019263 (HS).
Inclusion and Diversity
We worked to assure sex balance in the inclusion of non-human subjects. One or more authors of this paper self identifies as member of the LGBTQ+ community. While citing references scientifically relevant for this work we worked to promote gender balance in our reference list.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References Cited
- Albers MW, Tabert MH, and Devanand DP (2006). Olfactory dysfunction as a predictor of neurodegenerative disease. Curr Neurol Neurosci Rep 6, 379–386. [DOI] [PubMed] [Google Scholar]
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature genetics 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvites RD, Caseiro AR, Pedrosa SS, Branquinho ME, Varejao ASP, and Mauricio AC (2018). The Nasal Cavity of the Rat and Mouse-Source of Mesenchymal Stem Cells for Treatment of Peripheral Nerve Injury. Anat Rec (Hoboken) 301, 1678–1689. [DOI] [PubMed] [Google Scholar]
- Barnea G, O’Donnell S, Mancia F, Sun X, Nemes A, Mendelsohn M, and Axel R (2004). Odorant receptors on axon termini in the brain. Science 304, 1468. [DOI] [PubMed] [Google Scholar]
- Belluscio L, Gold GH, Nemes A, and Axel R (1998). Mice deficient in G(olf) are anosmic. Neuron 20, 69–81. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, and Ron D (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2, 326–332. [DOI] [PubMed] [Google Scholar]
- Bozza T, Vassalli A, Fuss S, Zhang JJ, Weiland B, Pacifico R, Feinstein P, and Mombaerts P (2009). Mapping of class I and class II odorant receptors to glomerular domains by two distinct types of olfactory sensory neurons in the mouse. Neuron 61, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braubach OR, Fine A, and Croll RP (2012). Distribution and functional organization of glomeruli in the olfactory bulbs of zebrafish (Danio rerio). The Journal of comparative neurology 520, 2317–2339, Spc2311. [DOI] [PubMed] [Google Scholar]
- Brunet LJ, Gold GH, and Ngai J (1996). General anosmia caused by a targeted disruption of the mouse olfactory cyclic nucleotide-gated cation channel. Neuron 17, 681–693. [DOI] [PubMed] [Google Scholar]
- Buck L, and Axel R (1991). A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65, 175–187. [DOI] [PubMed] [Google Scholar]
- Cagnetta R, Wong HH, Frese CK, Mallucci GR, Krijgsveld J, and Holt CE (2019). Noncanonical Modulation of the eIF2 Pathway Controls an Increase in Local Translation during Neural Wiring. Molecular cell 73, 474–489 e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Schrank BR, Rodriguez S, Benz EG, Moulia TW, Rickenbacher GT, Gomez AC, Levites Y, Edwards SR, Golde TE, et al. (2012). Abeta alters the connectivity of olfactory neurons in the absence of amyloid plaques in vivo. Nature communications 3, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, Ami GOH, and Web Presence Working, G. (2009). AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, Jiao S, Gumaste A, Bai L, and Belluscio L (2016). APP Overexpression Causes AbetaIndependent Neuronal Death through Intrinsic Apoptosis Pathway. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess A, Simon I, Cedar H, and Axel R (1994). Allelic inactivation regulates olfactory receptor gene expression. Cell 78, 823–834. [DOI] [PubMed] [Google Scholar]
- Clowney EJ, LeGros MA, Mosley CP, Clowney FG, Markenskoff-Papadimitriou EC, Myllys M, Barnea G, Larabell CA, and Lomvardas S (2012). Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell 151, 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton RP, Lyons DB, and Lomvardas S (2013). Co-opting the unfolded protein response to elicit olfactory receptor feedback. Cell 155, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanand DP, Lee S, Manly J, Andrews H, Schupf N, Doty RL, Stern Y, Zahodne LB, Louis ED, and Mayeux R (2015). Olfactory deficits predict cognitive decline and Alzheimer dementia in an urban community. Neurology 84, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein P, and Mombaerts P (2004). A contextual model for axonal sorting into glomeruli in the mouse olfactory system. Cell 117, 817–831. [DOI] [PubMed] [Google Scholar]
- Gene Ontology, C. (2001). Creating the gene ontology resource: design and implementation. Genome research 11, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology, C. (2021). The Gene Ontology resource: enSupplemental Table S1(relevantriching a GOld mine. Nucleic acids research 49, D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosmaitre X, Vassalli A, Mombaerts P, Shepherd GM, and Ma M (2006). Odorant responses of olfactory sensory neurons expressing the odorant receptor MOR23: a patch clamp analysis in gene-targeted mice. Proceedings of the National Academy of Sciences of the United States of America 103, 1970–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahsler M, Piekenbrock M, and Doran D (2019). dbscan: Fast Density-Based Clustering with R. Journal of Statistical Software 91, 1–30. [Google Scholar]
- Hu H, Tian M, Ding C, and Yu S (2018). The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Frontiers in immunology 9, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Soria X, Levitin MO, Saraiva LR, and Logan DW (2014). The olfactory transcriptomes of mice. PLoS genetics 10, e1004593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Suzuki M, and Sakano H (2006). Odorant receptor-derived cAMP signals direct axonal targeting. Science 314, 657–661. [DOI] [PubMed] [Google Scholar]
- Katidou M, Grosmaitre X, Lin J, and Mombaerts P (2018). G-protein coupled receptors Mc4r and Drd1a can serve as surrogate odorant receptors in mouse olfactory sensory neurons. Molecular and cellular neurosciences 88, 138–147. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Hagiwara D, Miyata T, Hodai Y, Kurimoto J, Takagi H, Suga H, Kobayashi T, Sugiyama M, Onoue T, et al. (2020). Endoplasmic reticulum chaperone BiP/GRP78 knockdown leads to autophagy and cell death of arginine vasopressin neurons in mice. Scientific reports 10, 19730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. bioRxiv, 060012. [Google Scholar]
- Lachmann A, Giorgi FM, Lopez G, and Califano A (2016). ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ni M, Lee B, Barron E, Hinton DR, and Lee AS (2008). The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 15, 1460–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic acids research 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DM, Wang F, Lowe G, Gold GH, Axel R, Ngai J, and Brunet L (2000). Formation of precise connections in the olfactory bulb occurs in the absence of odorant-evoked neuronal activity. Neuron 26, 69–80. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, and Harding HP (2004). Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. The EMBO journal 23, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Mao C, Lee B, and Lee AS (2006). GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Molecular and cellular biology 26, 5688–5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DB, Allen WE, Goh T, Tsai L, Barnea G, and Lomvardas S (2013). An epigenetic trap stabilizes singular olfactory receptor expression. Cell 154, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Wu Y, Qiu Q, Scheerer H, Moran A, and Yu CR (2014). A developmental switch of axon targeting in the continuously regenerating mouse olfactory system. Science 344, 194–197. [DOI] [PubMed] [Google Scholar]
- Markenscoff-Papadimitriou E, Allen WE, Colquitt BM, Goh T, Murphy KK, Monahan K, Mosley CP, Ahituv N, and Lomvardas S (2014). Enhancer interaction networks as a means for singular olfactory receptor expression. Cell 159, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Wang F, Dulac C, Chao SK, Nemes A, Mendelsohn M, Edmondson J, and Axel R (1996). Visualizing an olfactory sensory map. Cell 87, 675–686. [DOI] [PubMed] [Google Scholar]
- Monahan K, Horta A, and Lomvardas S (2019). LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountoufaris G, Chen WV, Hirabayashi Y, O’Keeffe S, Chevee M, Nwakeze CL, Polleux F, and Maniatis T (2017). Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science 356, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi K, Grosmaitre X, and Feinstein P (2016). Odorant receptors can mediate axonal identity and gene choice via cAMP-independent mechanisms. Open Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Ihara N, Shigeta M, Kiyonari H, Ikegaya Y, and Takeuchi H (2019). Structured spike series specify gene expression patterns for olfactory circuit formation. Science 365. [DOI] [PubMed] [Google Scholar]
- Nakashima A, Takeuchi H, Imai T, Saito H, Kiyonari H, Abe T, Chen M, Weinstein LS, Yu CR, Storm DR, et al. (2013). Agonist-independent GPCR activity regulates anterior-posterior targeting of olfactory sensory neurons. Cell 154, 1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H (2012). CHOP is a multifunctional transcription factor in the ER stress response. J Biochem 151, 217–219. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids research 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic acids research 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Sullivan SL, and Buck LB (1994). Information coding in the olfactory system: evidence for a stereotyped and highly organized epitope map in the olfactory bulb. Cell 79, 1245–1255. [DOI] [PubMed] [Google Scholar]
- Ron D, and Habener JF (1992). CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes & development 6, 439–453. [DOI] [PubMed] [Google Scholar]
- Saito H, Kubota M, Roberts RW, Chi Q, and Matsunami H (2004). RTP family members induce functional expression of mammalian odorant receptors. Cell 119, 679–691. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizawa S, Miyamichi K, Takeuchi H, Yamagishi Y, Suzuki M, and Sakano H (2006). A neuronal identity code for the odorant receptor-specific and activity-dependent axon sorting. Cell 127, 1057–1069. [DOI] [PubMed] [Google Scholar]
- Sharma R, Ishimaru Y, Davison I, Ikegami K, Chien MS, You H, Chi Q, Kubota M, Yohda M, Ehlers M, et al. (2017). Olfactory receptor accessory proteins play crucial roles in receptor function and gene choice. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SL, Ressler KJ, and Buck LB (1995). Spatial patterning and information coding in the olfactory system. Current opinion in genetics & development 5, 516–523. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Inokuchi K, Aoki M, Suto F, Tsuboi A, Matsuda I, Suzuki M, Aiba A, Serizawa S, Yoshihara Y, et al. (2010). Sequential arrival and graded secretion of Sema3F by olfactory neuron axons specify map topography at the bulb. Cell 141, 1056–1067. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, and Sakano H (2014). Neural map formation in the mouse olfactory system. Cell Mol Life Sci 71, 3049–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, and Xie XS (2018). A Near-Complete Spatial Map of Olfactory Receptors in the Mouse Main Olfactory Epithelium. Chemical senses 43, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai L, and Barnea G (2014). A critical period defined by axon-targeting mechanisms in the murine olfactory bulb. Science 344, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi A, Miyazaki T, Imai T, and Sakano H (2006). Olfactory sensory neurons expressing class I odorant receptors converge their axons on an antero-dorsal domain of the olfactory bulb in the mouse. The European journal of neuroscience 23, 1436–1444. [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Brann DH, Pashkovski SL, Guitchounts G, Bozza T, and Datta SR (2021). A transcriptional rheostat couples past activity to future sensory responses. Cell 184, 6326–6343 e6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Nemes A, Mendelsohn M, and Axel R (1998). Odorant receptors govern the formation of a precise topographic map. Cell 93, 47–60. [DOI] [PubMed] [Google Scholar]
- Wang IH, Murray E, Andrews G, Jiang HC, Park SJ, Donnard E, Duran-Laforet V, Bear DM, Faust TE, Garber M, et al. (2022). Spatial transcriptomic reconstruction of the mouse olfactory glomerular map suggests principles of odor processing. Nature neuroscience 25, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Titlow WB, McClintock DA, Stromberg AJ, and McClintock TS (2017). Activity-Dependent Gene Expression in the Mammalian Olfactory Epithelium. Chemical senses 42, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Ma L, Duyck K, Long CC, Moran A, Scheerer H, Blanck J, Peak A, Box A, Perera A, et al. (2018). A Population of Navigator Neurons Is Essential for Olfactory Map Formation during the Critical Period. Neuron 100, 1066–1082 e1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Huang G, Dewan A, Feinstein P, and Bozza T (2012). Uncoupling stimulus specificity and glomerular position in the mouse olfactory system. Molecular and cellular neurosciences 51, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, and Cavener DR (2002). The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Molecular and cellular biology 22, 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Feinstein P, Bozza T, Rodriguez I, and Mombaerts P (2000). Peripheral olfactory projections are differentially affected in mice deficient in a cyclic nucleotide-gated channel subunit. Neuron 26, 81–91. [DOI] [PubMed] [Google Scholar]
- Zhou AX, Wang X, Lin CS, Han J, Yong J, Nadolski MJ, Boren J, Kaufman RJ, and Tabas I (2015). C/EBP-Homologous Protein (CHOP) in Vascular Smooth Muscle Cells Regulates Their Proliferation in Aortic Explants and Atherosclerotic Lesions. Circ Res 116, 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou DJ, Feinstein P, Rivers AL, Mathews GA, Kim A, Greer CA, Mombaerts P, and Firestein S (2004). Postnatal refinement of peripheral olfactory projections. Science 304, 1976–1979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 (relevant to Figure 1): Common ORs referenced in this study
Supplemental Table S2 (relevant to Figure 2): Mouse strains used in this study
Supplemental Table S3 (relevant to Figure 2): Laser and detector settings for FACS experiments
Supplemental Table S4 (relevant to Figure 4): Antibodies used in this study
Supplemental Table S5 (relevant to Figure 7): Summary of all RNA-seq Libraries
Supplemental Figure S1 (relevant to Figure 1). Atf5(rep) is a translational reporter for ER stress that follows the expression properties of the intact Atf5 gene in the OSN lineage.
(A) Single Cell RNA-seq (scRNA-seq) analysis of wild-type OE, visualized using uniform manifold approximation and projection (UMAP). Atf5 is highly transcribed throughout the OSN lineage. Neurog1, Gng8, and Omp expression defines GBCs/INPs, INPs/iOSNs, and mOSNs, respectively.
(B) Log-10 normalized RNA-seq counts for all ORs in Omp-iresGFP+ mOSNs isolated from mice heterozygous or wild-type for the Atf5(rep) allele. Spearman correlation and linear regression line (blue) shown relative to the line y=x (black, dashed).
(C) CRE (green) immunofluorescence in MOE sections from heterozygous Atf5(rep/+) mice at postnatal day 13 (p13). DAPI nuclear marker in blue. Scale bar 50μm.
(D) Schematic of OSN differentiation from immediate neuronal precursors (INPs) to immature OSNs (iOSNs) and mature OSNs (mOSNs). Marker lines for INPs and mOSNs as shown, UPR is maximal in iOSNs.
(E) Representative flow cytometry plots defining (i) INP, (ii) iOSN and (iii) mOSN populations using GFP signal intensity (y- axis) from Ngn1-GFP mice (GFP highest in INPs, at p6) and Omp-iresGFP mice (GFP in mOSNs, at p11 and p56). iRFP intensity (x-axis) is used to quantify levels of ER stress in each population.
(F) iRFP intensity (from the Atf5 reporter allele) in INPs, early mOSNs (p11) and late mOSN (p56), each measured relative to iOSNs (iRFP+GFP− cells in each plot). Each point is one replicate, the red line connects the means in each group.
(G) PCA plot of RNA-seq libraries generated from INP (red), iOSN (green) and mOSN (blue) sorted cells.
(H) Line plot of centered, scaled and variance stabilizing transformation (VST)-transformed OR expression values in INPs, iOSNs and mOSNs by RNA-seq. Each black line is an OR, red line connects medians across all ORs (error bars span first and third quantiles).
(I) Heatmap showing centered, scaled and VST-transformed expression values for all genes with significant differential expression across INPs, iOSNs and mOSNs by RNA-seq (n=2 biological replicates per population). Genes (rows) are ordered by hierarchical clustering and cut into six clusters shown in the color bar at right. Summary medians, first and third quantiles are shown for each cluster in the line plots.
(J) Top significantly-enriched Gene Ontology (GO) terms for clusters 5 and 6 (from I), GO terms are on the y-axis, −log10-false discovery rate (FDR) is on the x. GO terms of interest for this study are highlighted in blue (cluster 5) or pink (cluster 6).
Supplemental Figure S2 (relevant to Figure 1). High-throughput measurement of ER stress scores and correlation with OR trafficking properties
(A) PCA of RNA-seq data used in the high-throughput determination of OR stress levels. The experiment was performed n=4 times, with four libraries prepared for each replicate. Color represents stress bin (high = red, low = blue), shape represents developmental population (iOSN = iRFP = circles, mOSN = iRFPGFP = triangles).
(B) MA plot showing log2 fold changes between high and low stress OSNs (y axis) as a function of the log10-transformed mean of the normalized counts in all libraries (x axis). Significantly differentially expressed (DE) genes are shown in red, non-significant genes are in black, and OR genes are facetted out from all others. Numbers of genes in each category are indicated in the labels, p value is for a chi-squared test of whether OR representation among statistically significant DE genes deviates from the null. Plots depict averages from 4 biological replicates.
(C) Gene set enrichment analysis (GSEA) for 50 hallmark gene sets in high versus low-stress bulk sorted OSNs. Y-axis denotes gene set, x-axis shows normalized enrichment score in high versus low stress cells, colored by significance (red = significant). The hallmark unfolded protein response gene set is highlighted in red on the y axis.
(D) ER stress score values for all ORs in mOSNs only (y-axis) or mOSNs and iOSNs (x-axis) isolated by FACS from Atf5(rep/+); Omp(iresGFP/+) mice. The latter definition is used for all other analyses in the paper. Spearman correlation and linear regression line (blue) shown relative to the line y=x (black, dashed).
(E) Cumulative density plots of ER stress scores in Atf5-dependent (red) versus Atf5-independent (blue) ORs. Data represent averages of 4 biological replicates.
(F) Cumulative density plots of stress scores in Rtp1/2-dependent (red) versus Rtp1/2-independent (blue) ORs. Data represent averages of 4 biological replicates.
(G) Violin plots showing log2 fold changes for all ORs in Omp-iresGFP+ mOSNs isolated from global Atf5 KO vs. WT mice, grouped/colored by zone and facetted by class. Log2 fold changes are similar across these subpopulations. RNA-seq data is from Dalton et al., 2013.
(H) Violin plots showing log2 fold changes for all ORs in the whole olfactory epithelium (OE) of global Rtp1/2 KO vs. WT mice, grouped/colored by zone and facetted by class. Log2 fold changes are similar across these subpopulations. RNA-seq data is from Sharma et al., 2017.
Supplemental Figure S3 (relevant to Figure 2). Random forest regression predicts ER stress scores from OR amino acid sequences.
(A) PCA of RNA-seq libraries used to compute stress scores in the M71→P2 swap experiment. There are four libraries per mouse, colored by the interaction between population (iOSN = iRFP, mOSN = iRFPGFP) and stress level (low vs. high) (see Figure 1F for an overview of stress score calculation and methods for additional details). Circles represent “WT” mice (n=4 animals from Supplemental Figure S2A + n=2 animals added for this experiment), triangles represent M71→P2 swap mice (n=3).
(B) Stress scores computed for all ORs in P2(M71iresLacZ/+); Atf5(rep/+); Omp(iresGFP/+) (y-axis, n=3) versus Atf5(rep/+); Omp(iresGFP/+) (x-axis, n=6) mice (see methods). Spearman correlation and linear regression line (blue) shown.
(C) Stress scores computed on a per-library basis for the M71→P2 swap experiment. Scores for M71 (red) and P2 (purple) were computed using “WT” (Atf5(rep/+); Omp(iresGFP/+), n=6) animals. Scores for the M71→P2 swap allele (green) were computed using “Swap” (P2(M71iresLacZ/+); Atf5(rep/+); Omp(iresGFP/+), n=3) animals. Non-finite scores omitted. Padj P2-M71 = 4.07E-3, Swap-M71 = 0.29, Swap-P2 = 5.55E-4 (one-way ANOVA with Tukey’s post-test).
(D) Principal coordinates analysis (PCoA) for all OR amino acid sequences. Each point represents an OR, distances between points reflect the similarity between OR sequences in the MSA. Colors indicate zonal identity of each OR.
(E) Random forest regression model training using root mean square error (RMSE, y-axis) to evaluate model fit over a grid of ntree (color) and mtry (x-axis) values. 10-fold cross validation was repeated 3 times for each set of parameters. The final model used 500 trees and mtry = 50.
(F) Random forest predicted ER stress scores (y-axis) versus experimentally measured ER stress scores (x-axis, see also Figure 1G) for all OR sequences. Linear regression (blue) is shown relative to the line y=x (dashed, black). R2 is calculated from held-out data in the repeated cross-validation procedure (see methods).
(G) Percent increase in mean squared error (%IncMSE, y-axis) for random forest regression model after permuting each predictor variable (position in the OR MSA) versus the Shannon entropy at each position in the OR MSA. The blue line shows a linear regression of %IncMSE by Shannon entropy, the residuals of which comprise the corrected %IncMSE values plotted in Figure 2G. The top 10 positions by corrected %IncMSE are shown in red and labeled in Figure 2G.
(H) Summary of 4 ORs with polymorphisms between the C57BL/6 (B6) and 129 mouse strains(Feinstein and Mombaerts, 2004). Boxes indicate transmembrane domains; red points indicate the polymorphisms. The heatmaps on the right summarize the RF regression-predicted differences in ER stress scores between the B6 and 129 sequence for each OR (left column) and the configuration observed in vivo by Feinstein and Mombaerts (right).
(I) Amino acid composition at the top 10 residues identified as important for OR stress scores by the random forest regression model. Positions are named as follows: amino acid [position in P2 sequence] ([position in MSA]) (also see Figure 2G). OR sequences are binned into deciles by stress score (1= lowest stress scores, 10 = highest), and fractional amino acid composition at each position across the ten deciles is shown by the colored bars. (−) indicates a gap in alignment at the indicated position. Facets are arranged by descending variable importance.
Supplemental Figure S4 (relevant to Figure 4). Conditional Perk deletion prevents OSN axon coalescence without disturbing the stability of OR gene choice at p5.
(A) IF for M71 (purple) in the MOE of M71 Perk Het (top row) or cKO (bottom row) mice at p5. Endogenous GFP (green) and tdtomato (red) signals are shown. Outlines of M71+GFP+ (green) and M71+tdtomato+ (red) cells are traced on the M71 panel. Scale bar 50μm.
(B) IF for MOR28 (purple) in the MOE of Mor28 Perk WT, Het or cKO mice at p5. Colors as in A. Scale bar 50μm.
(C) Loess smoothed curves and standard errors of corrected iRFP signal (y-axis) vs. differentiation (x-axis) for Omp-iresGFP+ mOSNs in Perk WT (solid black line) vs. global Perk Het (dashed black line) mice. iRFP measurements in OSN subpopulations (from Figure 1D) are shown for comparison.
(D) Pearson correlations between red and green axons in Mor28 Perk WT and Het glomeruli at p5. Each point is a glomerulus, colored and grouped by genotype. P value calculated by two-sample Wilcoxon test.
(E) IF for MOR28 protein (purple) in OB glomeruli of Mor28 Perk WT or Perk Het mice at p5. Endogenous tdtomato (red) and GFP (green) signals are shown. Scale bar 50μm.
Supplemental Figure S5 (relevant to Figure 4). Conditional Perk deletion results in OSN depletion at p28.
(A) Whole mount views of olfactory bulbs of M71 Perk mice at postnatal day 28 (p28). Internal control neurons are shown in green, experimental neurons in red with the indicated genotypes. Magnification as indicated, A=anterior, P = posterior. Scale bars 500μm (4x), 100μm (20x).
(B) Whole mount views of olfactory bulbs of Mor28 Perk mice at postnatal day 28 (p28). Abbreviations and scale bars as in A.
(C) Blinded quantification of glomerular configurations in the OB of p28 M71 (left facet) and Mor28 Perk animals (right facet), grouped by genotype of the tdtomato+ cells. Colors indicate glomerular configurations. Number of animals and glomeruli quantified as labeled.
(D) IF for MOR28 (purple), endogenous GFP (green) and tdtomato (red) in the MOE of Mor28 Perk Het (top row) vs. cKO (bottom row) mice at p28. Outlines of M28+GFP+ (green) and Mor28+tdtomato+ (red) cells are traced on the MOR28 panel. Scale bar 50μm.
(E) Ratios of Mor28+tdtomato+ to Mor28+GFP+ cells in the MOE of Mor28 Perk Het (green) or cKO (blue) mice at p28 (left). Ratios of total tdtomato+ cells to Mor28+GFP+ cells in the same MOEs (right). Each point is a section, data is grouped by biological replicate (n=2 mice per genotype).
(F) IF for activated Caspase 3 (red) in MOE sections from Perk(fl/fl) (top) or Omp(Cre/+); Perk(fl/fl) (bottom) mice at p28.
(G) Quantification of activated Caspase 3+ mOSNs per mm in MOE sections from Perk(fl/fl) (red), Omp(Cre/+); Perk(fl/fl) (green) and Gg8-Cre; Perk(fl/fl) (blue) mice. Gg8-Cre drives Perk deletion at the INP stage, before axonal coalescence, and is included for reference. Each point is a section (n=3 biological replicates/condition, 4–5 sections/replicate). Padj = 0.1122 for Omp-Cre; Perk(fl/fl) vs. Perk(fl/fl), Padj <0.05 for all other comparisons by one-way ANOVA with Tukey’s post-test.
(H) IF for the mOSN marker OMP (green) and iOSN marker GAP43 (red) in coronal OB sections from Perk(fl/fl) (left) or Omp(Cre/+); Perk(fl/fl) (right) mice at p28. Glomeruli are present in both genotypes, albeit less well-defined in Omp-Cre Perk cKO animals.
Supplemental Figure S6 (relevant to Figure 5) ER stress levels determine axon guidance specificity via DDIT3.
(A) Loess smoothed curves and standard errors of corrected iRFP signal (y-axis) vs. differentiation (x-axis) for Omp-iresGFP+ mOSNs in Hspa5 WT (solid black line) vs. global Hspa5 Het (dashed black line) mice. iRFP measurements in OSN subpopulations (from Figure 1D) are shown for comparison.
(B) Diagram of the drug-inducible PERK experiment. tTA is expressed via an IRES element from the Omp locus and drives transcription of the tet-dependent FV2E-Perk-t2amCherry transgene. The Fv2E domain replaces the luminal domain of PERK, placing PERK dimerization and UPR activation under control of the drug AP20187.
(C) Dorsal OBs from p8 mice with indicated genotypes. AP20187 was injected IP at 0.1mg/kg 24 and 48 hours prior to analysis. Scale bar 500um.
(D) IF for caspase 3 (green) in the MOE of p8 mice with the labeled genotypes. AP20187 injection was as in C. DAPI (blue) and mCherry fluorescence from the transgene (red) are shown in the indicated panels.
(E) IF for DDIT3 (purple) in the MOE of p8 mice. Genotypes, drug treatment and counterstains as in D.
(F) IF for MOR28 protein (purple), endogenous GFP (green) and tdtomato (red) in the MOEs of Mor28 Ddit3 WT (top) or cKO (bottom) mice at p5. Outlines of Mor28+GFP+ (green) and Mor28+tdtomato+ (red) cells are traced on the Mor28 panel. Scale bar 50μm.
(G) Ratios of Mor28 IF signal in Mor28+tdtomato+ vs. Mor28+GFP+ cells in the MOE of p5 Mor28 Ddit3 WT (red) or cKO (blue) mice. Each data point is from one cluster of closely-positioned GFP/tdtomato cells from a single section (see methods), colored by genotype of the tdtomato+ cells (n=4 sections/genotype, 5–9 clusters/section). P=0.579 by two-sample Wilcoxon test.
(H) IF for MOR28 in the glomeruli of Mor28 Ddit3 WT (left) or cKO (right) mice at p5. Endogenous GFP (green) and tdtomato (red) shown in top row, MOR28 (purple) in bottom row. ROIs containing the red and green fibers are traced on the Mor28 panels. Data are representative of n=2 biological replicates. Scale bar 50μm.
Supplemental Figure S7 (relevant to Figure 7). Conditional Ddit3 deletion causes stable axon guidance changes by altering the transcription of high stress axon guidance genes.
(A) PCA plot of RNA-seq libraries from GFP+ (green) and tdtomato+ (red) sorted cells from p28 Mor28 Ddit3 WT (circles), Het (triangles) or cKO (squares) mice.
(B) MA plot for the p28 Mor28 Ddit3 RNA-seq experiment. The y-axis shows the difference in average log2 fold changes between tdtomato and GFP OSNs in Ddit3 cKO vs. WT mice. The x-axis shows the log10-transformed average of normalized counts across all samples. ORs are facetted out, significant genes (padj<0.05) are shown in red and genes of interest are labeled.
(C) Log2 fold changes in normalized counts of Nrp2 (top) and Tenm2 (bottom) between tdtomato and GFP cells in p28 Mor28 Ddit3 animals by RNA-seq. Data are colored by genotype (red = WT, green = Het, blue = cKO), and each point represents a biological replicate.
(D) Heatmap showing all genes from the stress-dependent axon guidance network described in Figure 3E. The top row shows the log2 fold change within zone for each gene in high versus low stress OSNs by scRNA-seq (colored blue → red). Genes are arranged in descending order based on these changes. The middle row shows the average log2 fold change tdtomato/GFP in Mor28 Ddit3 Het vs. WT animals (colored purple → green). The bottom row shows the same measurement, but in Mor28 Ddit3 cKO vs. WT animals. Genes where genotype significantly affects the differences in expression between GFP and tdtomato cells in the 4week Mor28 experiment are boxed in black.
(E) IF for NRP2 protein in the OB of p28 Mor28 Ddit3 cKO animals. Endogenous GFP (green, internal controls, functionally WT) and tdtomato (red, cKO) signal is shown in the top row, NRP2 staining (purple) is shown in the bottom row, with GFP (green) and tdtomato (red) glomeruli outlined. Images are representative of n=2 biological replicates. Boxplot shows quantification of the difference in mean log10-transformed NRP2 signal in tdtomato – GFP glomeruli. Each point represents a single section with adjacent GFP and tdtomato glomeruli (p = 2.29E-5 by one-sample Wilcoxon test against the null hypothesis that data is symmetric around 0).
(F) Normalized RNA-seq tracks showing coverage at the Ddit3 locus in the Omp-Cre Ddit3 experiments. Tracks are colored by genotype and represent averages across biological replicates (red = WT [n=4], green = Het [n=5], blue = cKO [n=5]). A gene model of the Ddit3 locus is shown at the bottom, thin boxes represent exons and thick boxes represent the CDS. LoxP sites are shown as red triangles on the gene model and as vertical dashed lines that extend through the RNA-seq tracks.
(G) PCA plot of sorted mOSN RNA-seq libraries from Omp(iresCre/+); Ddit3 WT (circles), Het (triangles) or cKO (squares) animals. Libraries were prepared in three batches (indicated by colors) and the observed batch effect was modeled in downstream analyses.
(H) MA plot depicting the log2 fold change in Omp-iresCre Ddit3 cKO vs. WT OSNs (y-axis) versus the log10 average normalized counts across all conditions (x-axis) in the Omp-iresCre Ddit3 RNA-seq experiment. Points are colored by significance (red = padj < 0.05), genes of interest are labeled, and ORs are shown in a separate facet.
(I) Kernel density estimates of the log2 fold changes for all ORs (black), high stress ORs only (stress score >0, red) or low stress ORs only (stress score <0, blue) in Omp(iresCre/+); Ddit3 cKO vs. WT OSNs. The dashed vertical black line denotes a log2 fold change of zero.
(J) Normalized RNA-seq counts for genes in the identified stress-dependent axon guidance network (Figure 3E) whose expression significantly changes in the Omp-iresCre Ddit3 dataset. Points are colored by genotype (red = WT, green = Het, blue = cKO) and facets are in order of decreasing significance.
Data Availability Statement
All RNA-seq datasets described in this study have been uploaded to GEO with accession number: GSE198886.
Original code can be found at https://github.com/hshayya/2022_Shayya_UPR_Guidance
Images/other resources may be downloaded from Zenodo (doi: 10.5281/zenodo.6448088).
Any additional information required to re-analyze the data is available upon request.