

Abstract
Most invasive lobular breast cancers (ILC) are of the luminal A subtype and are strongly hormone receptor positive. Yet, ILC is relatively resistant to tamoxifen and associated with inferior long-term outcomes compared to invasive ductal cancers (IDC). In this study, we sought to gain mechanistic insights into these clinical findings that are not explained by the genetic landscape of ILC and to identify strategies to improve patient outcomes. A comprehensive analysis of the epigenome of ILC in pre-clinical models and clinical samples showed that, compared to IDC, ILC harbored a distinct chromatin state linked to gained recruitment of FOXA1, a lineage-defining pioneer transcription factor. This resulted in an ILC-unique FOXA1-estrogen receptor (ER) axis that promoted the transcription of genes associated with tumor progression and poor outcomes. The ILC-unique FOXA1-ER axis led to retained ER chromatin binding after tamoxifen treatment, which facilitated tamoxifen resistance while remaining strongly dependent on ER signaling. Mechanistically, gained FOXA1 binding was associated with the auto-induction of FOXA1 in ILC through an ILC-unique FOXA1 binding site. Targeted silencing of this regulatory site resulted in the disruption of the feed-forward loop and growth inhibition in ILC.
In summary, ILC is characterized by a unique chromatin state and FOXA1-ER axis that is associated with tumor progression, offering a novel mechanism of tamoxifen resistance. These results underscore the importance of conducting clinical trials dedicated to patients with ILC in order to optimize treatments in this breast cancer subtype.
Keywords: chromatin accessibility, lobular breast cancer, FOXA1, estrogen receptor, endocrine resistance
Introduction
Invasive lobular carcinoma (ILC) is the second most common histological subtype of breast cancer (BC), accounting for 10–15% of all cases. The classical variant of ILC is characterized by relatively uniform and non-cohesive cells that grow as a single file infiltrating the stroma. This growth pattern is attributed to the loss of E-cadherin, the hallmark of ILC (1,2), and can render physical exam and mammographic diagnosis challenging (3,4). Although the majority of ILCs are of low or intermediate grade, express high levels of estrogen receptor (ER), and of luminal A subtype, several studies suggest that ILC long-term outcomes are inferior to stage-matched invasive ductal carcinoma (IDC) (5). Additionally, the retrospective analysis of the BIG-1–98 adjuvant endocrine trial demonstrated that the magnitude of the inferior benefit from tamoxifen compared with an aromatase inhibitor (AI) was greater in ILC compared with IDC (6), indicative of relative resistance to tamoxifen treatment in early-stage treatment naïve ILC.
Several studies have revealed differences in the mutational landscape of ILC compared to IDC. In addition to loss of E-cadherin, ILC is characterized by a higher frequency of FOXA1 mutations (7,8), which are found in 7–9% of primary ILC versus 2% in primary IDC. In primary, treatment naïve, ILC tumors, FOXA1 mutations are associated with increased expression of FOXA1 and unique transcriptional profiles (7). More recently, FOXA1 mutations were shown to be associated with endocrine resistance in metastatic ER-positive (ER+) BC (9). FOXA1 is a pioneer transcription factor (TF) that mediates ER transcriptional activity (10–13). Thus, the increase in FOXA1 mutations in ILC and the disparate patterns of response to the different classes of endocrine treatment suggest that ILC may have a unique ER axis compared to IDC. In support of this notion, previous work has shown that ILC cells are characterized by a unique transcriptional response to estrogen (14). However, the mechanism by which the ER transcriptional axis is altered in ILC models lacking FOXA1 mutations remains elusive. In this study, we performed a comprehensive analysis of the epigenome of ILC in pre-clinical models and clinical samples with the aim to provide mechanistic insights to explain the distinctive responses to estradiol and tamoxifen in ILC versus IDC and to inform us on new potential therapeutic approaches for ILC.
Materials and Methods
Cell lines
MCF7, T47D and MDA-MB-134-VI (MDAMB134) cells were purchased from ATCC; SUM44PE (SUM44) cells were a gift by Dr. Stephen Ethier. All the cells were authenticated by short tandem repeat (STR) profiling at Bio-Synthesis (USA) and regularly tested for mycoplasma contamination by Mycoalert detection Kit (Lonza). The MCF7 cells were maintained in DMEM and T47D cells in RPMI supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). MDAMB134 cells were maintained in L-15 with 20%FBS and 1% P/S. SUM44 were maintained in Ham’s F-12 supplemented with insulin (5ug/mL), hydrocortisone (1ug/mL), fungizone (2.5ug/mL), transferrin (5ug/mL), T3 (6.6ng/mL), ethanolamin (5mM), NaSe (8.7ng/mL) (all from Sigma-Aldrich, St. Louis, MO), gentamicin (25ug/mL), HEPES (10mM), BSA (0.1%) (all from Thermo Fisher Scientific). For hormone deprived (HD) conditions, cells were kept in phenol-red free medium supplemented with 10% heat-inactivated charcoal-stripped (CS)-FBS (with the exception of SUM44) (20% for MDAMB134) and 1% P/S. MCF7, T47D and SUM44 were incubated at 37°C in 5% CO2, MDAMB134 were incubated at 37°C without CO2.
Human tissue studies
The ILC metastatic ascitic fluid collection was conducted in accordance with the Declaration of Helsinki ethical guidelines and approved by the Dana-Farber/Harvard Cancer Center institutional review board (Protocol 93–085). Participant signed a written informed consent form before the collection. Red blood cells and dead cells were removed by Ficoll (Sigma-Aldrich) followed by selection of cancer epithelial cells using EpCam selective beads (Dynabeads). Cancer epithelial cells were subjected to FOXA1, ER and H3K27ac ChIP-seq.
ATAC-seq
For the global chromatin accessibility experiments ductal and lobular cell lines were cultured for three days in HD conditions and then treated with 10nM estradiol for 45 minutes. Nuclei of 100,000 freshly fixed in 1% formaldehyde cells were processed as previously reported (15). Briefly, cells were resuspended in 1mL of cold ATAC-seq resuspension buffer (RSB; 10mM Tris-HCl pH 7.4, 10mM NaCl, and 3mM MgCl2 in water) and centrifuged. Cell pellets were then resuspended in ATAC-seq RSB (0.1% NP40, 0.1% Tween-20, and 0.01% digitonin) and incubated on ice. After lysis, ATAC-seq RSB containing 0.1% Tween-20 (without NP40 or digitonin) was added. Nuclei were centrifuged and then were resuspended in 50μl of transposition mix (25μl 2× TD buffer, 2.5μl transposase (100nM final), 16.5μl PBS, 0.5μl 1% digitonin, 0.5μl 10% Tween-20, and 5μl water). Transposition reactions were incubated at 37°C for 30 min. Reactions were cleaned up with Zymo DNA Clean and Concentrator5 columns.
Chromatin Immunoprecipitation (ChIP)-Sequencing
ChIP experiments were conducted as described previously (16) and were done in duplicates. MCF7, T47D, MDAMB134 and SUM44 cells were cultured for three days in HD conditions then treated with 10nM estradiol for 45 minutes. For the ChIP experiments with tamoxifen we used 10nM 4-hydroxytamoxifen for 45 minutes. Chromatin from 20 million formaldehyde-fixed cells was sonicated to a size range of 200–300 bp. Solubilized chromatin was immunoprecipitated with a mix of the ER antibodies Ab10 (Thermo Fisher Scientific) and SC-543 (Santa Cruz), a mix of the FOXA1 antibodies ab5089 and ab23738 (Abcam), GATA3 antibody D13C9 (Cell signaling) or H3K27Ac antibody (C15410196, Diagenode). The same antibodies were used in each experiment for all the cell lines. The samples were reversed crosslinked, treated with proteinase K, and DNA was extracted. Libraries were sequenced using 75 bp paired-end reads on the Illumina Nextseq500 at the Dana-Farber Cancer Institute.
RNA sequencing
Ductal and lobular cell lines were cultured for three days in HD conditions. After washing the cells with PBS, cells were incubated for 12 hours in HD medium with 10nM estradiol, or DMSO treatment. Total RNA was extracted using the QIAGEN RNeasy kit with DNAse digestion. RNA concentrations were measured by nanodrop and quality of RNA was determined by a Bioanalyzer. For all cell line studies, samples were analyzed in at least duplicates. RNA-seq libraries were made using the TruSeq RNA Sample Preparation Kit (Illumina). Samples were sequenced on an Illumina Nextseq500.
Generation of knock-out cells (CRISPR, ShRNA and siRNA)
-CRISPR/Cas9 cells
Construction of lenti-CRISPR/Cas9 vectors targeting E-cadherin (CDH1) in the two ductal cell lines was performed following the protocol associated with the backbone vector (49535, Addgene). The following sgRNA for CDH1 sequences were used:
gRNA1 F:5′ CACCGAGCTGGCTGACATGTACGG 3′
gRNA1 R: 5′ AAACCCGTACATGTCAGCCAGCTC 3′
-DOX inducible ShFOXA1
FOXA1 shRNA constructs (Novartis) were generated by inserting annealed oligonucleotides into EcoRI/AgeI-digested pLKO-Tet-On plasmid.
The FOXA1 sequence to target was: 5’ GCAGATGTCTTTAAATGAAAT.
Top oligo: CCGGGCAGATGTCTTTAAATGAAATCTCGAGATTTCATTTAAAGACATCTGCTTTTT
Bottom oligo: AATTAAAAGCAGATGTCTTTAAATGAAATCTCGAGATTTCATTTAAAGACATCTGC
Lentivirus was produced in 293T cells to infect cells in media containing polybrene (8–10μg/mL). Cells were selected with puromycin treatment after transduction. 1ug/mL of doxycycline was used for the induction of the shFOXA1.
-siRNA
MCF7 and MDA134 cells were seeded in 6- or 24-well plates and transfected with 20nM siRNA oligos by Lipofectamine RNAiMax reagent (Life Technology). Knockdown efficiency was determined after 72 hours of transfection. Cell counting was performed after 3 and 7 days of transfection. The ON-TARGET siRNA oligos targeting ESR1 (LQ-003401–00), FOXA1 (J-010319–05 and J-010319–06), CASZ1 (LQ-020764–02) or non-targeting control (D-001810–01) were all purchased from Dharmacon.
CRISPRi FOXA1 enhancer
Stable dCas9-KRAB expressing MCF7 and SUM44 cell lines were generated by lentiviral transduction using Lenti-dCas9-KRAB-blast (Addgene #89567) transfer vector and pMD2.G (Addgene #12259) and psPAX (Addgene#12259) lentiviral packaging vectors. Transduced cells were selected and maintained in blasticidin (10μg/mL) selective culture media. Guide RNAs (gRNAs) were designed against peak 1 and non-human targeting control (NTC) gRNA was included and cloned as described previously (detailed protocol available at http://www.broadinstitute.org/rnai/public/resources/protocols). Briefly, for each gRNA complementary single-stranded oligonucleotides were synthetized (Invitrogen), phosphorylated, annealed, and gRNA cassettes were ligated into pXPR_BRD003 gRNA expression vector containing puromycin selection marker. After bacterial transformation (Stbl3 cells, Invitrogen) individual clones were picked and regenerated, and correct gRNA sequence were confirmed by Sanger-sequencing.
Lentiviral particles were generated for each sgRNA experiment by transforming HEK-293T cells with gRNA transfer vectors and lentiviral packaging mix (pMD2.G psPAX). Lentiviral particle containing media were collected and filtered using 0.45μm pore size syringe filters (Corning) after 48h post transfection and used for treatment of previously plated BC cell lines. Media was changed after 24h and replaced by puromycin (2μg/mL) containing selective culture medium. Puromycin resistant cells were collected after 7 days of selection for proliferation studies and RNA for cDNA.
The coordinates of the region of interest (P1): chromosome 14 from 38,067,100 to 38,071,000, Human hg19. Working guides were:
gP1: caccgAGGAGCTACTAGACCAGTAA and aaacTTACTGGTCTAGTAGCTCCTC
NTC: caccGCGACCCAAATGCACCCTTT NTC and aaacAAAGGGTGCATTTGGGTCGC
Quantification and Statistical analysis
Statistical analyses for cell proliferation studies were performed using two-sided Student’s t-tests, and p- values less than 0.05 were considered statistically significant. Error bars represent the ± SEM. Cell line experiments testing cell proliferation were all performed in triplicates.
Survival Analysis
Calculation of Average Modified Z-score (AveMZ)
Breast tumor gene expression profiles with clinicopathologic data were obtained from METABRIC (17,18) via authorized access in Synapse (synapse.sagebase.org). To calculate the FOXA1-ILC_120 gene set expression in the METABRIC data set we used the modified Z-score to transform the gene expression data because it relies on the median and is less influenced by outliers when compared to the standard Z-score based on the mean. The score was calculated by the formula of (X-MED)/(1.486*MAD), where X is the log-transformed gene expression value. MED is the median level of X across samples, and MAD (≠ 0) is the median absolute deviation calculated by MAD = median (|X - median(X)|). The signature score of target gene set was presented as AveMZ by calculating the mean of modified Z-scores of the signature genes.
Kaplan-Meier plots were produced using the R survminer v0.4.8 package to display the survival probabilities per group as a function of time. Groups of ER+ BC cohorts were stratified by the median cutoff of the gene signature score. Hazard ratio (HR) of stratum and P value were calculated using the Wald estimates and likelihood ratio test, respectively, in the Cox proportional hazards model using the R survival v3.2–3 package.
Additional methods are in the supplemental file.
Data availability
The whole-genome sequencing, RNA-seq, ChIP-seq, ATAC-seq, and Hi-ChIP-seq data have been deposited in the Gene Expression Omnibus database under GSE152367.
Results
ILC has a unique chromatin state that is tightly linked to FOXA1 recruitment
To study the epigenetic landscape and identify the genome-wide active regulatory elements accessible to TF binding in ILC versus IDC we first conducted transposase-accessible chromatin sequencing (ATAC-seq) in cell line models of ER+ ILC ((MDA-MB-134-VI (MDAMB134) and SUM44PE (SUM44)) and ER+ IDC (MCF7 and T47D). Unsupervised sample-to-sample correlation analysis segregated the ILC from IDC models, revealing a fundamental unique chromatin state of ILC (Figure 1A). Differential analysis (L2FC>1 and <−1, q-value<0.01) showed 11,777 sites significantly gained in ILC and 5,444 sites significantly gained in IDC (Figures 1B and S1A). The top enriched motifs in the ILC gained sites were FOXA1 motifs, followed by motifs of AP2γ, a key regulator of ER (19), and the estrogen response element (ERE) (Figures 1C and S1A). In contrast, the CTCF motif was the top motif in the IDC gained and the non-differentiated accessible sites (Figures 1D and S1A).
Figure 1. ILC has a unique chromatin cell state.
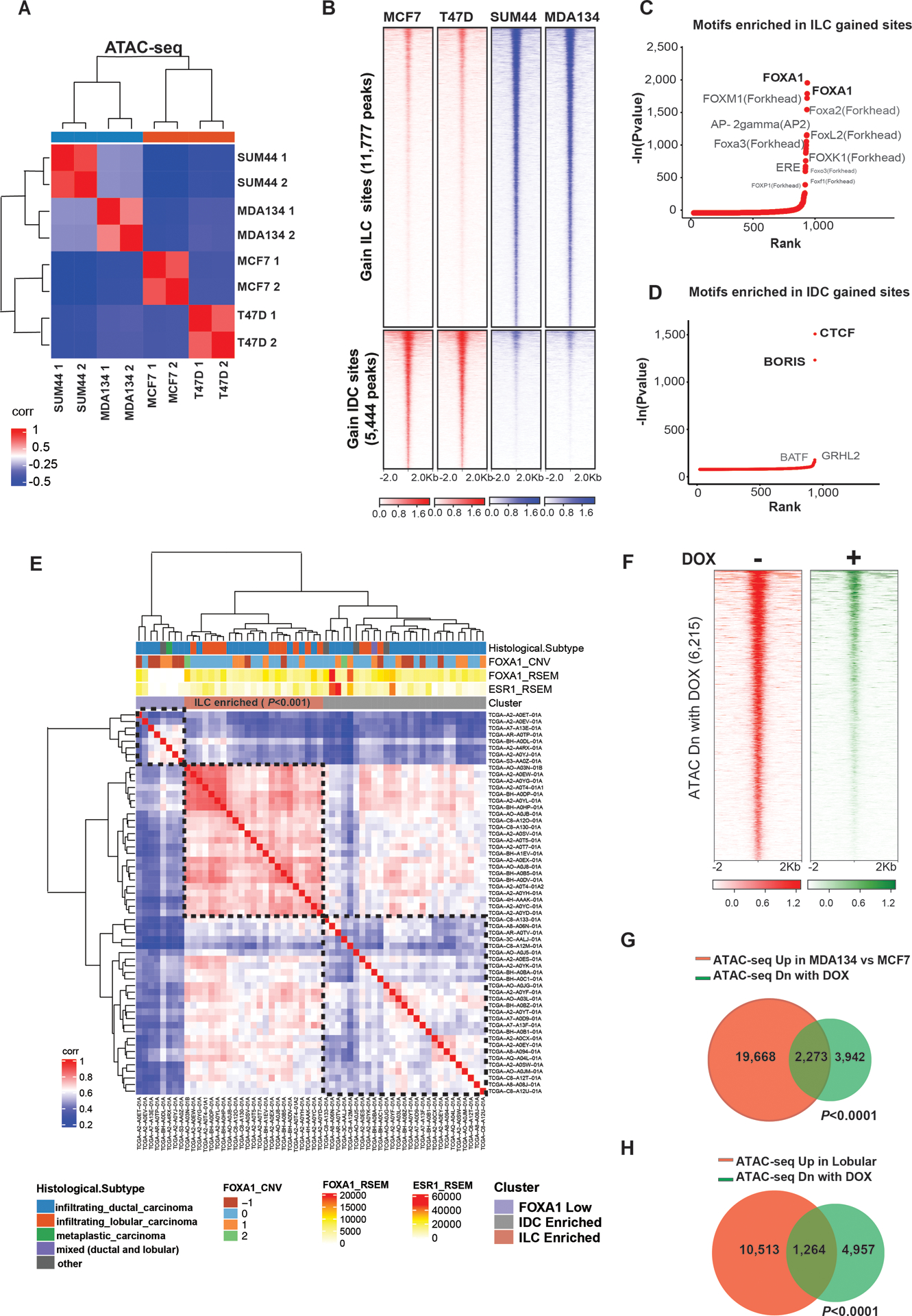
(A) Sample to sample correlation of chromatin accessibility based on transposase-accessible chromatin followed by sequencing (ATAC-seq) by the Euclidean distance between rows/columns and Ward’s method of invasive lobular cancer (ILC) cells (MDAMB134 (MDA134) and SUM44) and invasive ductal cells (IDC) cells (MCF7 and T47D) cells after 10nM β-estradiol (E2) stimulation (cells were grown in hormone deprived (HD) conditions for 3 days followed by 45-minute treatment with 10nM E2). Shown in the plot are results of replicates. (B) Tornado plots of chromatin accessible sites gained in ILC cells (11,777 peaks) in blue and gained in the IDC cells (5,444 peaks) in red (Log 2FC >1 or <−1, Q<0.01). Chromatin accessible sites are shown in a horizontal window of ±2 kb from the peak center. (C-D) Ranking of motifs enriched in the ILC (C) and IDC (D) gained accessible sites based on p-value. (E) Sample to sample correlation heatmap of open chromatin sites in TCGA ER+ BC tumors applying only the chromatin accessible sites gained in the ILC cell line models (11,777 peaks). Samples are clustered by the Euclidean distance between rows/columns and Ward’s method. Samples cluster to three groups including an ILC enriched group (Fisher’s exact test). (F) Tornado plots of chromatin accessible sites lost when FOXA1 is downregulated by a doxycycline (DOX)-inducible shRNA after 3 days of DOX in presence of HD and 45min 10nM E2. (G-H) Venn diagrams of chromatin accessible sites upregulated in MDA134 in comparison to MCF7 (in red) (G) or upregulated in lobular cells in comparison to ductal cells (in red) (H) and the chromatin accessible sites lost by downregulation of FOXA1 by shRNA (in green).
To test the clinical relevance of these findings, we analyzed the ER+ BC samples of The Cancer Genome Atlas Program (TCGA) that were included in the Pan Cancer ATAC-seq study (20). This analysis consisted of 58 ER+ BCs with different histological subtypes (IDC: N=38, ILC: N=14 and other histological subtypes: N=6). None of these samples harbored FOXA1 mutations. Unsupervised sample-to sample correlation of the chromatin accessible sites gained in the ILC model cell lines segregated the samples to three main clusters: a FOXA1 low cluster (biologically an ER-negative cluster), ILC enriched cluster (including 11 of the 14 ILC samples, Fisher exact test p-value <0.001) and IDC enriched cluster (Figure 1E). Thus, ILC can be characterized and identified by the unique chromatin accessible sites.
Because FOXA1 was the top motif enriched in the ILC gained chromatin accessible sites, we next tested the impact of FOXA1 silencing on chromatin accessibility by engineering MDAMB134 cells that stably express a doxycycline (DOX) inducible shFOXA1 (Figure S1B). Silencing of FOXA1 resulted in the loss of 6,215 accessible sites (L2FC <−1, q-value<0.01) without a gain in accessible sites (Figure1F). In control cells not expressing the shFOXA1 construct, DOX treatment had no significant effect on chromatin accessibility (L2FC <−1, q-value<0.01) (Figure S1C). Importantly, the lost sites significantly overlapped with the chromatin accessible sites gained in MDAMB134 compared to MCF7 cells or gained in ILC versus IDC cell models (Figures 1G and 1H).
Since we showed that FOXA1 has a role in facilitating the ILC unique chromatin state, we performed FOXA1 ChIP-seq in all four models and primary ILC cells obtained from a malignant peritoneal effusion from a patient with metastatic ER+ ILC (primary ILC Met). The majority of FOXA1 binding sites were in enhancer regions, including intronic and intergenic regions (Figure S2A). FOXA1 binding clustered the ILC models, including the primary ILC Met cells, and separated them from the IDC models (Figure 2A). This clustering was driven by 12,247 ILC gained peaks (L2FC>1, q-value <0.01) (Figure 2B). In contrast, there was a marginal number of peaks gained in IDC. As expected, FOXA1 motifs were the most significantly enriched motifs in the gained and non-differentiated peaks (Figure 2B). In addition, the ERE motif was among the enriched motifs (Figure 2B). In support of the role of FOXA1 in the ILC unique chromatin state, the sites with gained chromatin accessibility in ILC had increased FOXA1 binding in ILC cells (Figure 2C). In addition, the chromatin accessible sites that were lost in the MDAMB134 cells after FOXA1 silencing had increased FOXA1 binding in MDAMB134 cells compared to MCF7 cells (Figure 2D). Strikingly, all the chromatin accessible sites gained in ILC overlapped with the ILC gained FOXA1 chromatin recruitment (Figure 2E). Moreover, there was a significant correlation between the intensity of the ILC gained FOXA1 binding and the intensity of gained chromatin accessibility in MDAMB134 and SUM44 cells (Spearman correlation coefficient=0.327, P<8.33e-98 for MDAMB134 and Spearman correlation coefficient=0.24, P<1.72e-50 for SUM44 (Figures 2F and S2B)). In aggregate, these results indicate that ILC has a unique chromatin state that is interconnected with the reprogramming of FOXA1 recruitment.
Figure 2. FOXA1 Reprogramming in ILC is linked to the ILC unique chromatin state.
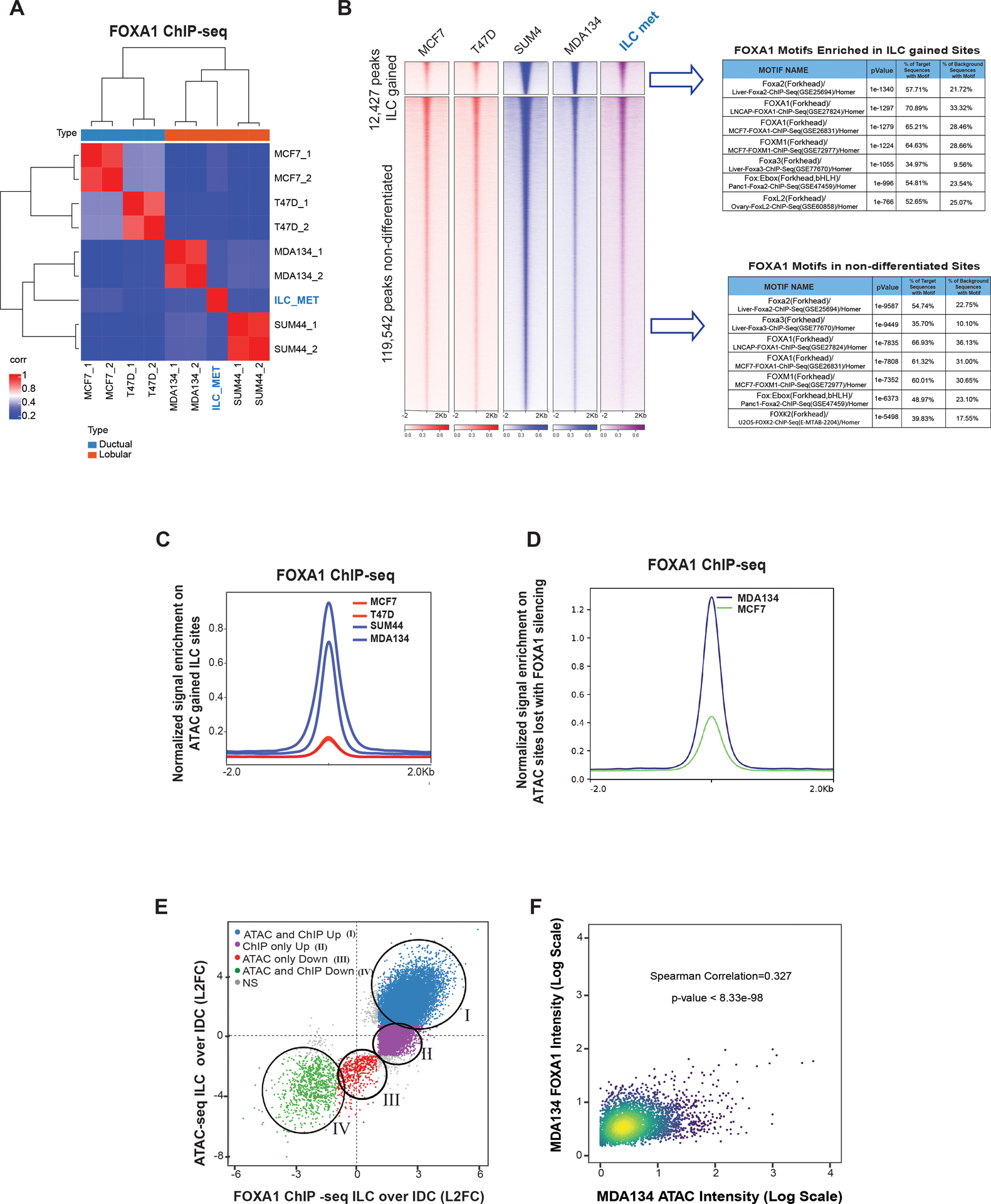
(A) Sample to sample correlation (Euclidean distance between rows/columns and Ward’s method) of FOXA1 binding sites correlation plots between all four cell lines in replicates (MCF7, T47D, MDA134 (MDAMB134) and SUM44) and the primary ILC cells isolated from a malignant peritoneal effusion from a patient with ER positive (ER+) metastatic ILC (ILC met). (B) Tornado plots of FOXA1 binding sites (12,427 sites) gained in ILC (MDA134 and SUM44) compared to IDC cells (MCF7 and T47D) and the union of the non-differentiated sites (Log 2FC >1 or <−1, Q<0.01). Table of the top motifs enriched in the FOXA1 binding sites gained in ILC versus IDC cells (Top) and FOXA1 binding sites non-differentiated between (Bottom) comparing the ILC and IDC sites are reported. (C) Quantitative normalized signal of FOXA1 binding based on FOXA1 ChIP-seq in the ILC gained chromatin gained accessible sites based on the ATAC-seq analysis. (D) Quantitative normalized signal of FOXA1 binding based on FOXA1 ChIP-seq in the chromatin accessible sites lost in MDA134 after FOXA1 silencing based on the ATAC-seq analysis. (E) Comparison of log2FC between ILC (MDA134 and SUM44) and IDC (MCF7 and T47D), FOXA1 binding sites (FOXA1 ChIP-seq, x-axis) versus the log2FC comparing chromatin accessibility (ATAC-seq, y-axis) between ILC (MDA134 and SUM44) and IDC (MCF7 and T47D). (F) Intensity of binding in the intersecting sites of ILC gained FOXA1 binding and ATAC-seq in MDA134 versus MCF7 cells. Spearman correlation and p-value are reported.
The ILC unique FOXA1 cistrome reprograms the ER transcriptional network
Given the known role of FOXA1 in facilitating ER binding (10–13), we next sought to determine how the ER axis is impacted by the reprogramming of the FOXA1 cistrome in ILC. To this end, we performed ER ChIP-seq in the ILC and IDC models. As shown previously (14), ligand dependent ER ChIP-seq of all four cell models showed that majority of ER binding sites were in enhancer regions (Fig. S3A). Unsupervised clustering of the ER binding sites segregated the ILC models, including the primary ILC Met, from the IDC models (Figure 3A). This clustering was driven by 6,885 peaks that were shared between MDAMB134 and SUM44 cells and gained compared to the IDC models (Log 2FC >1 or <−1, q-value <0.01) (Figure 3B). As expected, ILC ER gained binding sites were enriched in the ERE motif, followed by FOXA1 motifs (21) (Figures 3A). Importantly, comparison of ER with FOXA1 ChIP-seq showed that the ER binding sites gained in ILC had increased FOXA1 binding in ILC versus IDC, and there was a significant overlap between the ER sites gained in ILC and the FOXA1 ILC gained sites (44% of the gained ER binding sites overlapped with FOXA1 gained binding sites, p-value<0.0001) (Figures 3C and 3D). There were 9,367 ILC unique FOXA1 peaks that did not overlap with ER binding, which implies that FOXA1 has functions that are ER independent. Thus, the ILC gained FOXA1 binding sites are tightly linked to the ER gained binding but likely have functions that are independent of ER binding.
Figure 3. The ER cistrome in ILC cell lines models.
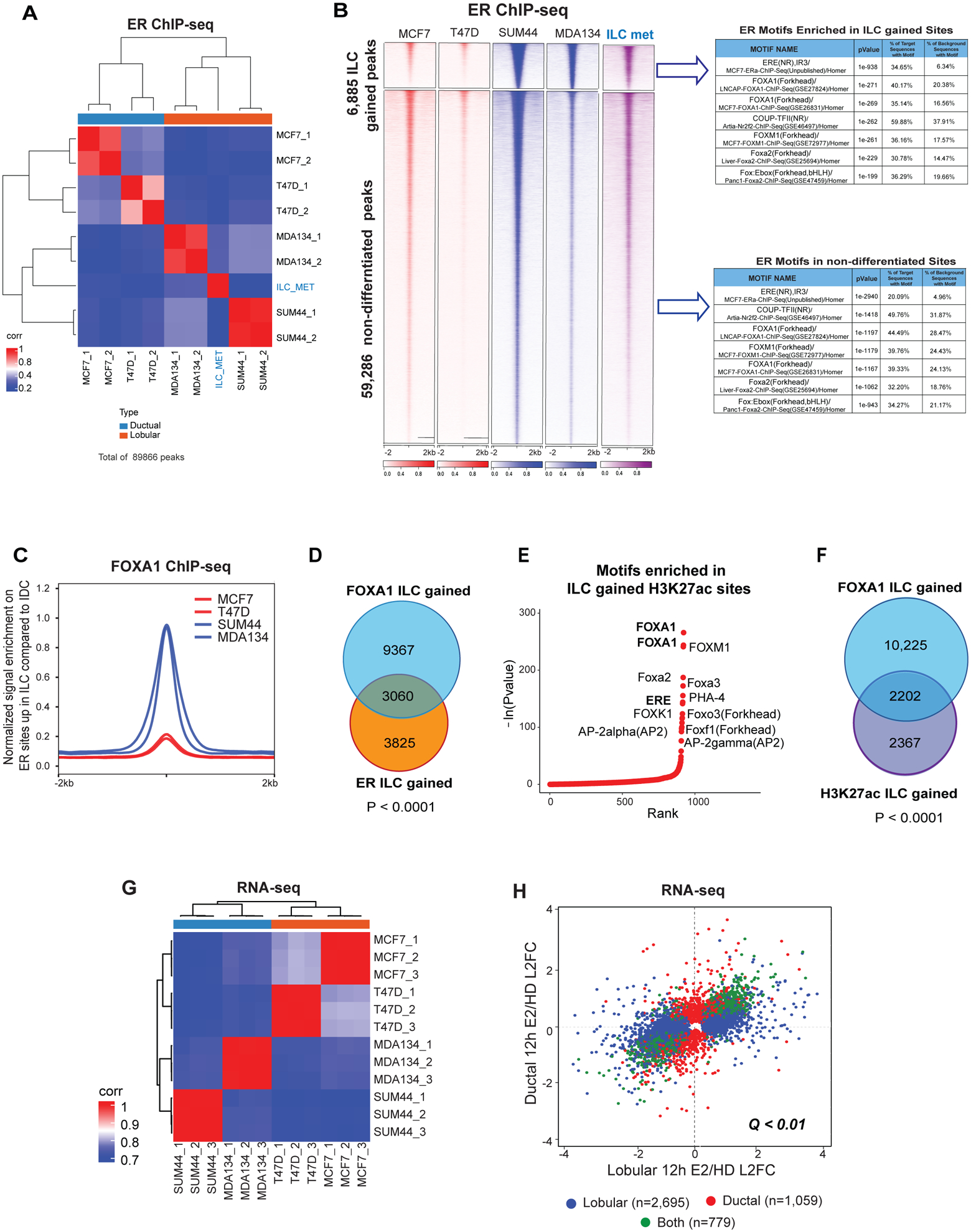
(A) ER ChIP-seq sample to sample correlation plot. IDC (MCF7 and T47D), ILC (MDA134 and SUM44) and primary ILC metastatic cells isolated from malignant peritoneal effusion from a patient with ER+ metastatic ILC (ILC MET) samples are clustered by the Euclidean distance between rows/columns and Ward’s method. (B) Tornado plots of ER binding sites gained in ILC cells compared to IDC cells (6,885 peaks) and the union of the non-differentiated sites (59,286) (Log 2FC >1 or <−1, Q<0.01). ER binding sites is shown in a horizontal window of ±2Kb from the peak center. Table of the top motifs enriched in the ER binding sites gained in ILC versus IDC cells (Top) and ER binding sites non-differentiated between (Bottom) comparing the ILC and IDC sites are reported. (C) Quantitative normalized signal of FOXA1 binding sites (FOXA1 ChIP-seq) on sites of ILC gained ER binding based on ER ChIP-seq in ILC cells (SUM44 and MDA134) and IDC cells (MCF7 and T47D). (D) Venn diagram of FOXA1 ILC gained peaks (12,427) in blue, and the ER ILC gained peaks (6,885) in orange, showing 3,060 peaks overlapping (sharing at least 1 bp) between the two factors. (E) Ranking of the motifs enriched in H3K27ac binding sites gained in ILC cells (SUM44 and MDA134) versus IDC cells (MCF7 and T47D) based on p-values of enrichment analysis. (F) Overlap between FOXA1 ILC gained peaks (12,427) compared to IDC in blue, and the H3K27ac ILC gained peaks (4,569) compared to IDC in purple. (G) RNA-seq sample to sample correlation based on Euclidean distance between rows/columns and Ward’s method of IDC and ILC cells after hormone deprived (HD) and 12 hours of β-estradiol treatment. (H) Comparison of differentially expressed genes between hormone deprived (HD) and 12 hours of β-estradiol (E2) treatment in ILC (SUM44 and MDA134) and IDC (MCF7 and T47D) cells. Genes with differential expression of Q<0.01 are assigned to each category by the color scheme.
To start to elucidate the transcriptional significance of the ILC unique chromatin state and the reprogramming of the FOXA1-ER axis, we first performed H3K27ac ChIP-seq, a histone mark of active enhancers. Sample-to-sample correlation of H3K27ac ChIP-seq segregated ILC from IDC. This segregation was driven by 4,569 ILC gained sites (Figures S3B and S3C). The number of sites gained in IDC was limited. The top motif in the ILC gained H3K27ac sites was FOXA1 followed by ERE (Figures 3E and S3C). Close to 50% of the ILC H3K27ac gained sites overlapped with the FOXA1 ILC gained sites (Figure 3F). Consistent with these findings, RNA-seq analysis of the four cell lines in estradiol stimulated conditions segregated the ILC from the IDC models (Figure 3G) and most of the estradiol regulated genes were different in ILC compared to IDC (Figure 3H). Taken together, these results support a previous study that showed disparate ligand stimulated transcription in ILC versus IDC (14) and provide initial evidence for the role of FOXA1 in the ILC unique transcription.
The ILC-FOXA1 gene set is associated with increased risk of recurrence in ILC tumors of luminal A molecular subtype
To test if the ILC unique FOXA1 chromatin recruitment is driving the ligand stimulated transcriptional differences between ILC and IDC, we integrated the RNA-seq and ChIP-seq data applying Binding and Expression Target Analysis (BETA) (22). BETA basic showed that the FOXA1 binding sites gained in ILC are significantly associated with the upregulation of genes with increased expression in ILC versus IDC in estradiol stimulated conditions (Figure S4A). This association is highlighted at the level of single genes in the volcano plot in figure 4A that shows the overlap between genes predicted to be gained by FOXA1 binding based on BETA minus and the genes upregulated in ILC versus IDC based on the RNA-seq differential expression analysis (Figures 4A and S4B). To further substantiate the link between the FOXA1 gained sites and FOXA1 dependent transcription in ILC, we tested the association between the ILC gained binding sites and the genes regulated by the silencing of FOXA1 using the transcriptomic analysis of MDAMB134 cells with and without DOX induced silencing of FOXA1. BETA basic analysis showed that the ILC FOXA1 gained sites were significantly association with the genes that were down regulated after FOXA1 silencing (Figures S4C and S4D).
Figure 4. FOXA1 drives the ILC unique transcriptome.
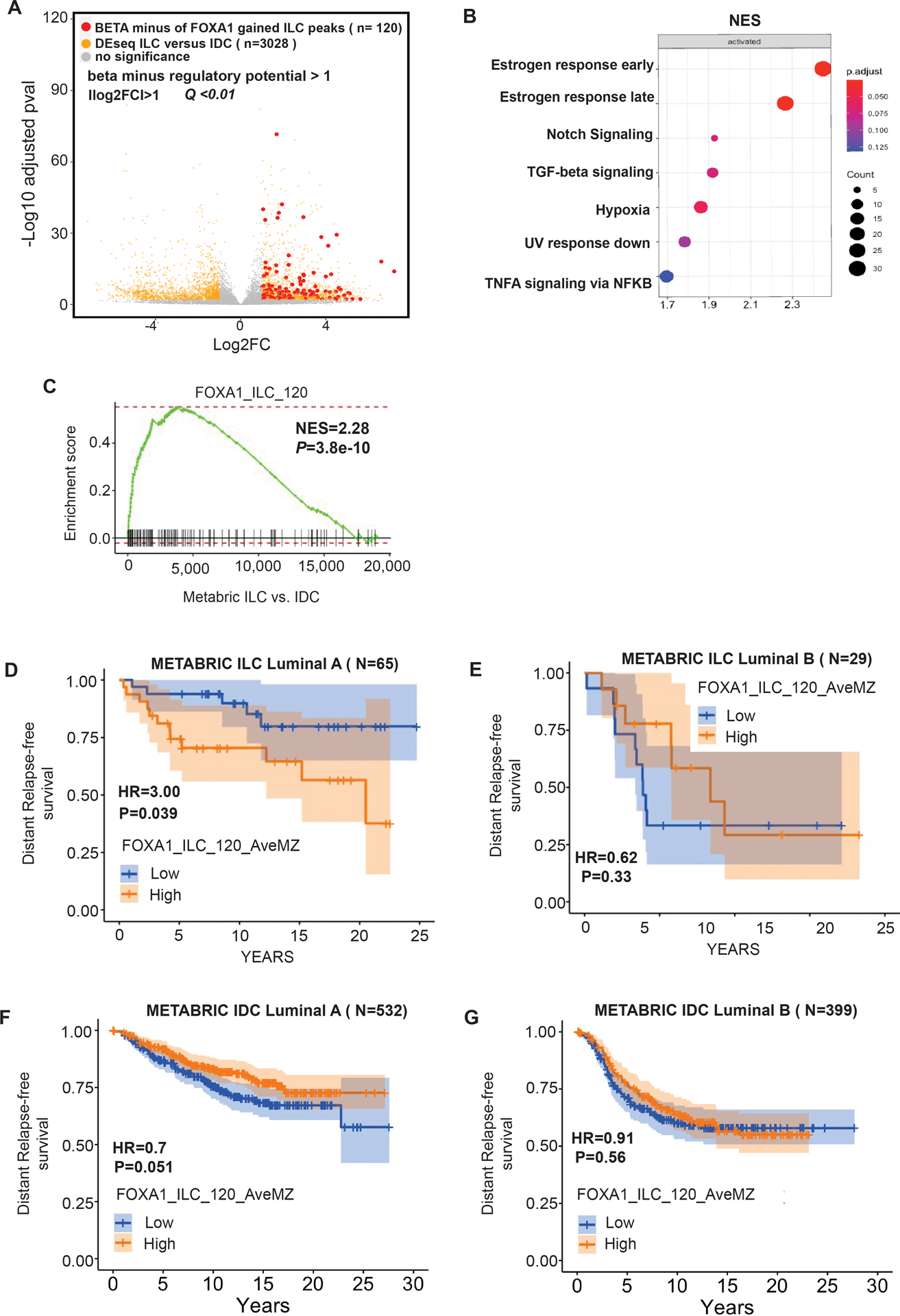
(A) Volcano plot depicting differential (DEseq2) expression comparing RNA-seq of ILC cells (MDA134 and SUM44) versus IDC cells (MCF7 and T47D) after β-estradiol stimulation (E2). Shown in yellow are the genes with significant differential expression ([log2FC] >1, Q<0.01. DESeq2). Red dots represent the genes with significant differential expression ([log2FC] >1, Q<0.01. DESeq2) and are regulated by FOXA1 ILC gained binding sites based on Binding and Expression Target (BETA) minus analysis. (B) Hallmark pathways enriched in the genes regulated by the ILC gained FOXA1 sites based on BETA basic. The normalized enrichment score (NES) is represented in the X-axis, the number of genes in the dataset is the count number represented by the circle size, q-value <0.25. (C) Enrichment plot from Gene Set Enrichment Analysis (GSEA) showing enrichment of the FOXA1_ILC_120 gene set derived from the ILC gained binding sites in ILC versus IDC estrogen receptor positive breast cancers in the METABRIC cohort. (D) Distant free survival in patients with Luminal A molecular subtype ILC from the METABRIC cohort comparing patients with high versus low expression of the FOXA1_ILC_120 gene set. (E) Distant free survival in patients with Luminal B molecular subtype type ILC from the METABRIC cohort comparing patients with high versus low expression of the FOXA1_ILC_120 gene set. (F) Distant free survival in patients with Luminal A molecular subtype IDC from the METABRIC cohort comparing patients with high versus low expression of the FOXA1_ILC_120 gene set. (G) Distant free survival in patients with Luminal B molecular subtype IDC from the METABRIC cohort comparing patients with high versus low expression of the FOXA1_ILC_120 gene set.
We next employed BETA basic to identify the genes that are direct targets of FOXA1 unique to ILC in estradiol stimulated conditions and generated a ranked product gene set termed the FOXA1-ILC gene set (Supplemental Table 1A). Ranked GSEA revealed that the FOXA1-ILC gene set is enriched in genes involved in key cancer pathways (ER response pathways signaling (23,24), TGF-beta signaling (25,26), hypoxia (25), and NFKB signaling (27,28) (Q<0.25)) (Figure 4B). We next generated a more stringent gene set of 120 FOXA1-ILC genes by selecting the genes with a ranked product p-value <0.001 (FOXA1_ILC 120). To study the clinical significance of this gene set we turned to the METABRIC cohort. We first determined that the FOXA1_ILC 120 is enriched in primary ER+ ILC tumors compared to primary ER+ IDC tumors (Figure 4C). Importantly, high versus low expression of the FOXA1_ILC 120 gene set was associated with a significantly lower distant relapse free survival (HR=3, p-value < 0.039) in patients with ILC and not IDC luminal A tumors. This association was also not seen in patients with luminal B BCs. Thus, the FOXA1_ILC 120 is a potential signature of high-risk ILC tumors that are driven by ER but not of tumors that are less dependent on ER signaling, which are enriched in genetic alterations or other pathways associated with treatment resistance, such as P53 mutations (29) (Figures 4D, 4E, 4F and 4G). Hence, this signature could potentially identify the ILC patients that might benefit from improved endocrine treatment strategies.
We next looked at the overlapping FOXA1/ER/H3K27ac sites gained in ILC and integrated these gained sites with the RNA-seq differential expression comparing ILC and IDC in estradiol stimulated conditions by applying BETA basic to identify single genes that are direct targets of FOXA1 and ER and are upregulated in ILC. CASZ1 was the top ranked gene, and RET, and SNAI1 were among the top ranked genes that met these criteria (Supplemental Table 1B and Figure S4E). The protein levels of these three genes were increased in MDAMB134 versus MCF7 cells and downregulated by the silencing of ER or FOXA1 (Figure S4F). Previous studies have shown that RET and SNAIL are ER targets with roles in resistance to endocrine treatment (30–32). In contrast, CASZ1, a zinc finger TF that regulates transcription by binding to the nucleosome remodeling and histone deacetylase (NuRD) complex (33), has not been implicated as an important gene in BC. We first confirmed that CASZ1 expression is significantly higher in ER+ ILC versus ER+ IDC in the METABRIC cohort (Figure S4G). In the MDAMB134 model, silencing of CASZ1 resulted in cell growth inhibition (Figure S4H) and down regulation of genes with key roles in tumor growth, including CDK2 and genes related to receptor tyrosine kinase signaling such as FGFRL1, FGFR4, MAPK9 and PIK3R2 (Figures S4I and S4J). Collectively, these results show that the ILC unique FOXA1 cistrome mediates the unique response to estradiol and the transcription of genes involved in tumor progression and poor outcomes.
Down regulation of E-cadherin in IDC or up regulation of E-cadherin in ILC is not sufficient to induce a unique ER transcriptional program
Although, the loss of E-cadherin in ILC leads to the degradation of beta-catenin, p120 translocates to the cytoplasm or nucleus and in these compartments, it has a role in signaling and transcription regulation (34). We therefore asked if loss of E-cadherin and the release of membrane bound p120 contribute to the ILC unique ER cistrome and transcriptional response to estradiol. To test this, we silenced CDH1 in MCF7 and T47D cells with a CRISPR cas9-gRNA (Figures S5A and S5B). The decrease in E-cadherin expression led to an increased migratory capacity but did not affect cell growth (Figures S5C and S5D). In addition, decreased E-cadherin expression resulted in limited transcriptional changes in MCF7 and T47D cells and the down-regulated genes were involved in pathways of cell adhesion molecules among other pathways (Figures S5E, S5F, S5G and S5H). In addition, silencing of E-cadherin did not lead to significant changes in ER binding or H3K27 acetylation in estrogen stimulated conditions (Figures S5I and S5J). Consistent with these results, there were limited transcriptional changes after estrogen stimulation when comparing the cells with and without E-cadherin down regulation (Figures S5K and S5L). Specifically, FOXA1 and ER expression were not altered after E-cadherin silencing. Additionally, stable overexpression of E-cadherin in MDAMB134 cells with a DOX-inducible construct resulted in a limited number of transcriptional changes at 3 days and 3 weeks (Figures S5M, S5N and S5O). Taken together, silencing of E-cadherin by itself was not sufficient in these models for the rewiring of the FOXA1-ER axis and the altered response to estrogen seen in ILC compared to IDC.
A circuitry of FOXA1 upregulation mediated by a unique super-enhancer
Since we showed that aberrant FOXA1 activity is central to the epigenetic landscape in ILC, we sought to understand the mechanism of the altered FOXA1 recruitment in ILC. Mutations in TFs can contribute to altered binding and tumor progression (9,16), however, whole genome sequencing of the ILC models did not identify FOXA1 mutations (Figure S6A). The overexpression of TFs can also alter the regulatory activity of cancer cells (35) and, in fact, we observed mRNA and protein overexpression of FOXA1 in ILC compared to IDC (Figures 5A and S4F). Since we did not identify FOXA1 copy number variations or mutations in the promoter region to explain the upregulation of FOXA1 (Figure S6A) (24,36), we interrogated the TFs that regulate FOXA1 expression (cistromeDB toolkit (37)). Similar to other key lineage-defining TFs that have been found to be auto-induced (38), the TF with the highest FOXA1 regulatory potential was FOXA1 itself. ER and GATA3, the other two key determinants of ER+ BC, are also potent FOXA1 regulators (Figure 5B). Interestingly, examination of FOXA1 binding upstream to the FOXA1 transcription start site (TSS) revealed a FOXA1 peak in the enhancer region (>10Kb from the TSS) that was unique to the ILC models (P1) (Figure 5C). H3K27 acetylation in this region was also different in the ILC models compared to IDC and extended at least 3KB upstream in ILC overlapping with the P1 peak (Figure 5D). Based on the H3K27ac ChIP-seq analysis this region upstream to the FOXA1 promoter is a super-enhancer region in all four models (Supplemental Table 1C). However, the boundaries of the super-enhancer region in the ILC models extended beyond those of the IDC super-enhancer and in only the ILC models the super-enhancer region overlapped with P1. We also detected the ILC unique FOXA1 P1 peak and super-enhancer region in the primary ILC Met, demonstrating clinical relevance of the P1 peak and unique super-enhancer region (Figures 5C and 5D). Since super-enhancers are characterized by the binding of multiple tissue-specific TFs (39) we looked at the binding of ER and GATA3, the other top ranked FOXA1 regulators, and saw increased ER and GATA3 binding in the ILC unique super-enhancer region (Figure 5E). Furthermore, H3K27ac Hi-ChIP detected ILC unique loops in this region (Figure S6B). Next, we tested the regulatory function of P1 by selective targeting of P1 with dCAS9-KRAB and a gRNA targeting the summit of this peak (details of the coordinates are in the methods). In the SUM44 ILC cells inactivation of P1 decreased the expression of FOXA1 at the mRNA and protein level, led to the regression of cell growth and decreased the expression of canonical ER target genes including PgR, MYC and CCND1 (Figures 5F, 5G, 5H 5I and S6C). Among these genes, MYC and CCDN1 have key roles in cell proliferation. The expression of ER was also downregulated. This is consistent with the downregulation of ER expression when FOXA1 was silenced with siFOXA1 in this study (Figure S4F) and a previous study that demonstrated the downregulation of ER in response to FOXA1 silencing (40). In contrast, inactivation of P1 in MCF7 cells did not impact FOXA1 expression or cell growth despite the sensitivity of MCF7 to a siFOXA1 (Figures 5J, 5K and S6D). The activity of dCAS9-KRAB in MCF7 cells was confirmed by infecting the MCF7 cells engineered to stably express dCAS9-KRAB with a BFP-GFP reporter construct that includes a gRNA that targets the GFP cassette. In these cells, expression of dCAS9-KRAB along with the gGFP resulted in the loss of expression of BFP and GFP (Figure S6E). Collectively, our results support FOXA1 overexpression in the ILC models at least partly through auto-induction by a unique FOXA1 binding site within a super-enhancer.
Figure 5. FOXA1 auto-induction in ILC through a unique super-enhancer and FOXA1 binding site.
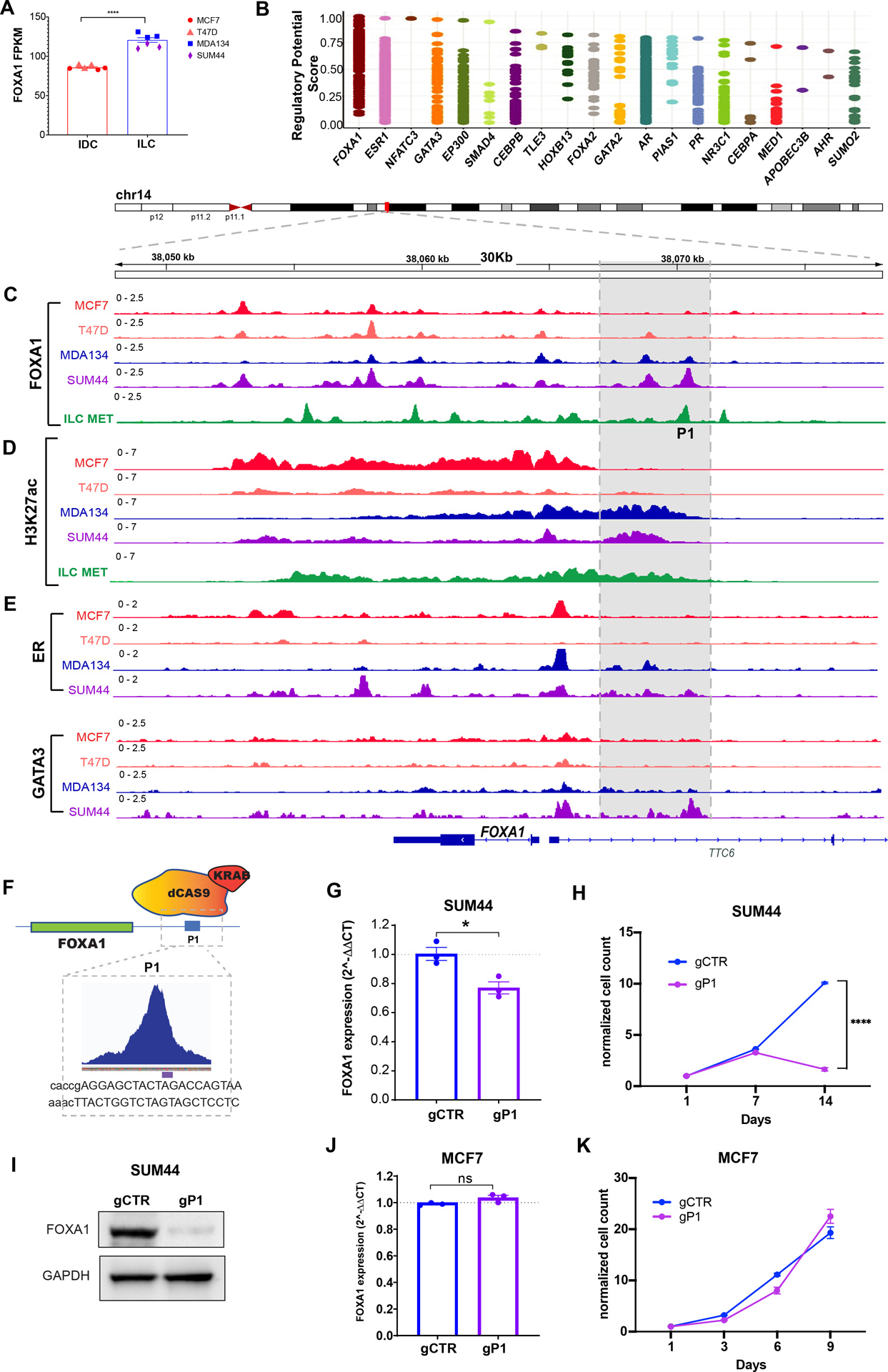
(A) RNA expression (FPKM values) of FOXA1 in IDC (MCF7 and T47D) and ILC (MDA134 and SUM44) cells. ****, t-test p-value <0.00001 (B) Regulatory potential of the transcription factors that regulate FOXA1 based on the CistromeDB toolkit (dbtoolkit.cistrome.org). (C-D) ChIP-seq tracks showing FOXA1 (C) and H3K27acetylation (D) in the IDC (MCF7 and T47D (red and orange tracks) and ILC (MDA134 and SUM44) (blue and purple tracks) cell lines and primary cells from ILC metastasic peritoneal effusion (ILC met, green tracks). (E) ER and GATA3 ChIP-seq tracks for the IDC (MCF7 and T47D) (red and orange tracks) and ILC (MDA134 and SUM44) (blue and purple tracks) cell lines. (F) Cartoon of CRISPRi action at the P1 site. Enlargement showing the gRNA used to target the FOXA1 binding region. (G) mRNA levels of FOXA1 in SUM44 lobular cells by reverse transcription quantitative PCR (RT qPCR) in presence of the gCTR and gP1 at day 7 after transduction. The mRNA expression was normalized to the GAPDH housekeeping gene, and expression levels are presented as 2^–ΔΔCT compared with control. (H) Cell proliferation assay after 14 days in SUM44 cells of control cells and after suppression of the FOXA1 P1 (gP1) using CRISPRi. Error bars represent ±SEM, n=3. (I) Western blot of whole cell lysates for FOXA1 in SUM44 cells in presence of the control guide (gCTR) and the guide RNA targeting P1 (gP1) at day 9 after transduction. (J) FOXA1 mRNA levels of MCF7 in presence of the gCTR and the gP1. (K) Cell proliferation assay in MCF7 cells in presence of the gCTR and the gP1. *p < 0.05; ****p < 0.0001; ns: not significant.
The reprogrammed FOXA1 cistrome drives Tamoxifen resistance in ILC
Similar to the clinical observation of relative resistance to tamoxifen in ILC (6), MDAMB134 and SUM44 cells were resistant to tamoxifen (IC50 of close to 1uM for both ILC models compared to an IC50 of 1nM and 2.2nM for T47D and MCF7, respectively) (Figure 6A). Since the ILC models have a longer doubling time compared to the IDC models, we confirmed the resistance to tamoxifen by growth rate metrics, which are independent of the division rate of the assayed cells (41). The GR50 for tamoxifen was 8–30-fold higher in the ILC models compared to IDC (Figure 6B). Tamoxifen resistance was not due to ligand or ER independent growth, as evidenced by the high sensitivity of MDAMB134 cells to estrogen deprivation and ER silencing, as in MCF7 cells (Figures 6C, 6D, S7A and S7B).
Figure 6. The mechanism of Tamoxifen Resistance in ILC.
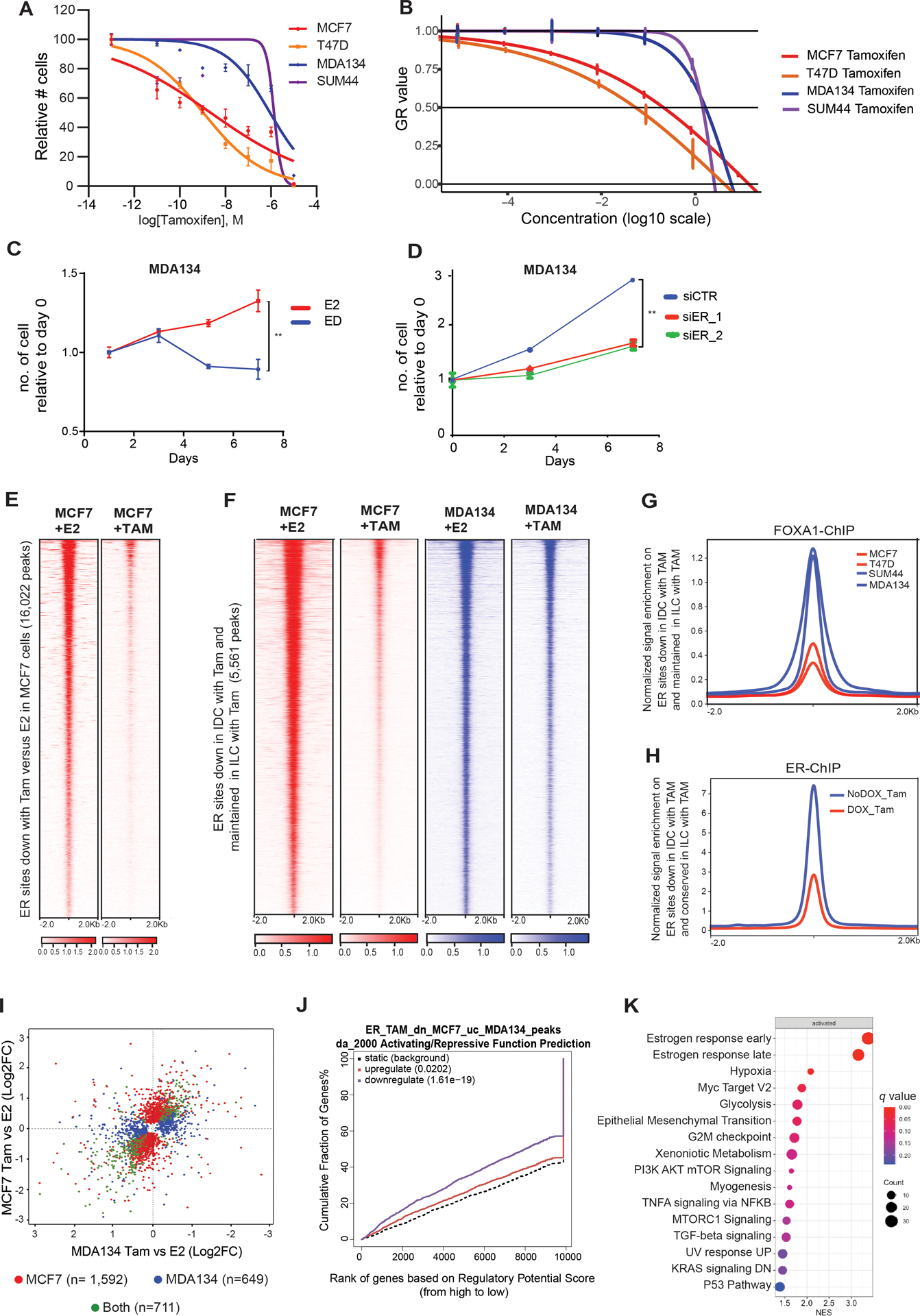
(A-B) Dose response curves of 4-hydroxytamoxifen (tamoxifen) treatment in ductal and lobular cell lines. (A) Curves normalized to vehicle control. IC50 values is in the range of 1uM for both ILC models (MDA134 and SUM44), compared to an IC50 of 1nM and 2.2nM for T47D and MCF7, respectively. (B) GR50s values are 240nM for MCF7 60nM for T47D, 1920nM for MDA134 and 1680μM for SUM44. (C) Cell proliferation curves of MDA134 cells followed for seven days without (ED) or with estradiol (E2, 10nM). Error bars represent ±SEM, n= 3. **, p-value <0.01. (D) Cell proliferation studies of MDA134 cells in full medium conditions including cells transfected with an siControl (siCTR) and cells with silencing of ER (siER_1 and siER_2). Error bars represent ±SEM, n = 3. ****, p-value <0.0001. (E) Tornado plots of ER binding sites lost in MCF7 treated with 4-hydroxytamoxifen (TAM) compared to β-estradiol (E2) treatment. (F) Tornado plots of ER binding sites lost in MCF7 treated with TAM compared to E2 treatment and unchanged in the MDA134 cells in E2 and TAM treated conditions (Log 2FC >1 or <−1, q-value<0.01). (G) Quantitative normalized signal of FOXA1 ChIP-seq binding at ER binding sites lost in MCF7 cells with TAM treatment but unchanged in MDA134 cells. (H) Quantitative normalized signal of ER ChIP-seq binding on ER sites lost in MCF7 cells but retained in MDA134 cells, in MDA134 cells treated with TAM (with and without DOX induction of FOXA1 silencing (shFOXA1). (I) Comparison of differentially expressed genes between β-estradiol (E2) and 4-hydroxytamoxifen (Tam) in MCF7 and MDA134. Genes with FDR <0.05 are assigned to each category by the color scheme. There were 711 shared differentially expressed genes, 1,592 genes differentially expressed in MCF7 cells only, and 649 genes differentially expressed exclusively in MDA134. (J) Binding and Expression Target Analysis (BETA) basic plot of the activating and repressive function of the ER binding sites lost in MCF7 but conserved in MDA134 after tamoxifen treatment. The red line represents the genes upregulated and the purple line the genes downregulated with 4hydroxytamoxifen versus β-estradiol treatment in MCF7 cells. The black dashed line indicates the non-differentially expressed genes as background. The p-value is based on the Kolmogorov-Smirnov test. (K) Hallmark pathways enriched in the genes determined to be downregulated by 4-hydroxytamoxifen compared to E2 treatment and regulated by the ER binding sites lost in MCF7 cells after tamoxifen treatment but unchanged in MDA134. The normalized enrichment score (NES) is represented in the X-axis, the number of genes in the dataset is the count number represented by the circle size, q-value <0.25.
To start to understand the mechanism of relative resistance to tamoxifen in ILC, we performed ER ChIP-seq after tamoxifen or estradiol treatment in MCF7 and MDAMB134 cells. In MCF7 cells, tamoxifen compared to estradiol led to the loss of 16,022 ER binding sites, whereas in MDAMB134 cells there were 37 lost and 236 gained ER binding sites (Log 2FC >1 or <−1 and q-value<0.01) (Figures 6E and S7C). When comparing the two cell lines and two treatment conditions, we identified 5,561 peaks that were lost in MCF7 cells but retained in MDAMB134 cells after tamoxifen treatment (Log 2FC >1 or <−1 and q-value<0.01) (Figure 6F). These ER peaks had increased FOXA1 binding and chromatin accessibility in ILC cells compared to IDC cells (Figures 6G and S7D). Furthermore, down regulation of FOXA1 expression with DOX-induced shFOXA1 in MDAMB134 cells (Figure S1F) resulted in growth inhibition and a loss of ER binding in the sites maintained after tamoxifen treatment (Figures S7E and 6H). Of note, we showed that FOXA1 silencing leads to decreased ER expression (Figure S4F), hence, these functional consequences of FOXA1 silencing can be attributed to its role in facilitating ER binding and regulating the expression of ER.
To study the transcriptional consequences of the disparate effects of tamoxifen treatment on ER binding in ILC versus IDC, we performed RNA-seq with and without tamoxifen treatment in MCF7 and MDAMB134 cells. In line with the differences in the changes in ER binding, there were differences in the transcriptional changes in response to tamoxifen treatment. Comparison of the transcriptional changes induced by tamoxifen in MCF7 and MDAMB134 revealed 1,592 genes differentially expressed in MCF7 cells only, 649 genes differentially expressed in MDAMB134 only, and 711 genes differentially expressed in both cell models (Figure 6I). BETA basic analysis showed that the ER binding sites lost in MCF7 cells but retained in MDAMB134 after tamoxifen treatment had a significant association with the genes downregulated with tamoxifen treatment in MCF7 cells (Figure 6J). GSEA of the ranked genes regulated by the MDAMB134 tamoxifen-maintained ER binding sites in MCF7 cells showed that these genes are involved in pathways that are key therapeutic targets of tamoxifen, such as response to estrogen, MYC targets, and epithelial mesenchymal transition (42) (Figure 6K, Supplemental Table 1D). In aggregate, in MCF7 cells tamoxifen treatment results in the loss of a set of ER binding sites and this is associated with the downregulation of genes important for the therapeutic response to tamoxifen. In contrast, in MDAMB134 cells, ER binding at these sites is retained in a FOXA1 dependent manner.
Discussion
In this study, we found that ILC has a unique chromatin state associated with the altered FOXA1 recruitment. The ILC unique epigenetic state provides one mechanism to explain the disparate transcriptional responses to estrogen and resistance to tamoxifen detected in the ILC cell models despite the high ER expression and ER dependency. These results provide new fundamental insights to the unique biology of ILC and may have important therapeutic implications.
Several mechanisms of aberrant FOXA1 signaling have been described previously, including FOXA1 mutations and amplifications (7–9,24). Though the increased frequency of FOXA1 mutations in ILC suggests that altered FOXA1 signaling is important in the biology of ILC, the majority of ILCs do not harbor a FOXA1 mutation. This implies that other mechanisms of aberrant FOXA1 signaling may be enriched in ILC. Indeed, we identified another mechanism of altered FOXA1 signaling in cell lines and metastatic ILC cells isolated from a patient with a metastatic peritoneal effusion. Herein, we provided evidence for a feed-forward circuit of FOXA1 auto-induction involving a unique super-enhancer and FOXA1 binding site with the co-recruitment of other TFs including ER and GATA3. The co-recruitment of these TFs may suggest that these TFs regulate the transcriptional activity of each other. This is supported by previous studies that showed an interplay between the FOXA1, ER and GR TFs in which both ER and GR can alter the distribution of FOXA1 binding (43,44). Our study demonstrates how the overexpression of a single key TF can lead to sustained effects on an entire transcriptional program and can potentially facilitates tumor progression and treatment resistance.
Previous studies identified several mechanisms of tamoxifen resistance unique to ILC models (45,46). In this study we showed another mechanism of tamoxifen resistance in ILC. Tamoxifen is a selective estrogen receptor modulator that inhibits the transcriptional activity of ER by shifting the balance between co-activator and co-repressor binding (47). Resistance specific to tamoxifen has been attributed mainly to a shift towards increased co-activator versus co-repressor binding, which can lead to ER agonistic activity and explain tumor regression after tamoxifen withdrawal (48,49). Here we show that in MCF7 cells tamoxifen treatment leads to loss of ER binding in selective sites and reduces ER mediated transcription. Our results suggest that gained FOXA1 recruitment in ILC can counteract this activity of tamoxifen and preserve ER binding. In addition, FOXA1 silencing was sufficient to inhibit this ER binding either through downregulation of ER expression or by the perturbation of ER binding. Together, these results highlight the importance of developing approaches for targeting traditionally undruggable targets such as FOXA1.
We provide proof of principle for tumor growth inhibition by selective targeting of a unique FOXA1 binding site associated with a super-enhancer that regulates the expression of FOXA1 itself. The advantage of this type of therapeutic approach over current epigenetic strategies is twofold: 1. Substantial efficacy because of the disruption of an auto-regulatory loop that is self-sustained and regulates multiple genes involved in tumor progression. 2. High precision given the disruption of a selective super-enhancer in selective cells as opposed to current therapeutic approaches that target epigenetic regulators and modulate the transcription of multiple genes in a non-selective manner (50). Importantly, selective inhibition of a regulatory region with a small molecule has been shown to be feasible (51). However, of more immediate clinical relevance, we show that ILC is highly dependent on a unique ER transcriptional axis and cell growth remains ligand dependent. These results support pre-clinical studies of ILC models and clinical trials dedicated to patients with ILC to investigate the oral selective estrogen receptor degraders and other novel endocrine treatments currently in clinical development.
Supplementary Material
Significance.
A unique FOXA1-estrogen receptor axis in invasive lobular breast cancer promotes disease progression and tamoxifen resistance, highlighting a potential therapeutic avenue for clinical investigations dedicated to this disease.
Acknowledgements
The work was conducted with support from the Maor Foundation (to E. Winer, O. Metzger Filho and R. Jeselsohn) and NIH support (5RO1CA237414-02 and 1K08CA191058-01A1 to R. Jeselsohn, 5RO1CA193910-03 to M.L. Freedman).
We thank Ms. Cheri Fox for her support, important insights and helpful discussion.
Conflict of interest disclosure statement:
R.S. received previous research funding from AstraZeneca, GlaxoSmithKline, Gilead Sciences, Eli Lilly, and PUMA Biotechnology and consulting/advisory role with compensation for Macrogenics and Eli Lilly. E.P.W. is a consultant for Carrick Therapeutics, G1 Therapeutics, Genentech/Roche, Genomic Health, GSK, Jounce, Leap, Lilly, Novartis, Seattle Genetics, Syros. O.M.F. receives research funding (institutional) from Abbvie, Cascadian Therapeutics, Eisai, Pfizer, Roche/Genentech, and Susan G. Komen for the Cure and is a consultant for Abbvie, G1 Therapeutics, and Groupo Oncoclinicas (Brazil) and receives honoraria from Roche (Brazil) and travel/accommodations/expenses from Grupo Oncoclinicas. R.J. receives research funding from Pfizer and Lilly and is a consultant for Carrick Therapeutics and Luminex.
References:
- 1.Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, et al. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 1995;14(24):6107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006;10(5):437–49 doi 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 3.McCart Reed AE, Kutasovic JR, Lakhani SR, Simpson PT. Invasive lobular carcinoma of the breast: morphology, biomarkers and ‘omics. Breast Cancer Res 2015;17:12 doi 10.1186/s13058-015-0519-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arpino G, Bardou VJ, Clark GM, Elledge RM. Infiltrating lobular carcinoma of the breast: tumor characteristics and clinical outcome. Breast Cancer Res 2004;6(3):R149–56 doi 10.1186/bcr767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pestalozzi BC, Zahrieh D, Mallon E, Gusterson BA, Price KN, Gelber RD, et al. Distinct clinical and prognostic features of infiltrating lobular carcinoma of the breast: combined results of 15 International Breast Cancer Study Group clinical trials. J Clin Oncol 2008;26(18):3006–14 doi 10.1200/JCO.2007.14.9336. [DOI] [PubMed] [Google Scholar]
- 6.Metzger Filho O, Giobbie-Hurder A, Mallon E, Gusterson B, Viale G, Winer EP, et al. Relative Effectiveness of Letrozole Compared With Tamoxifen for Patients With Lobular Carcinoma in the BIG 1–98 Trial. J Clin Oncol 2015;33(25):2772–9 doi 10.1200/JCO.2015.60.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163(2):506–19 doi 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J Clin Oncol 2016;34(16):1872–81 doi 10.1200/JCO.2015.64.0334. [DOI] [PubMed] [Google Scholar]
- 9.Arruabarrena-Aristorena A, Maag JLV, Kittane S, Cai Y, Karthaus WR, Ladewig E, et al. FOXA1 Mutations Reveal Distinct Chromatin Profiles and Influence Therapeutic Response in Breast Cancer. Cancer Cell 2020;38(4):534–50 e9 doi 10.1016/j.ccell.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res 2007;67(13):6477–83 doi 10.1158/0008-5472.CAN-07-0746. [DOI] [PubMed] [Google Scholar]
- 11.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005;122(1):33–43 doi 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 12.Eeckhoute J, Lupien M, Meyer CA, Verzi MP, Shivdasani RA, Liu XS, et al. Cell-type selective chromatin remodeling defines the active subset of FOXA1-bound enhancers. Genome Res 2009;19(3):372–80 doi 10.1101/gr.084582.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res 2013;23(1):12–22 doi 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sikora MJ, Cooper KL, Bahreini A, Luthra S, Wang G, Chandran UR, et al. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res 2014;74(5):1463–74 doi 10.1158/0008-5472.CAN-13-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watt AC, Cejas P, DeCristo MJ, Metzger-Filho O, Lam EYN, Qiu X, et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat Cancer 2021;2(1):34–48 doi 10.1038/s43018-020-00135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018;33(2):173–86 e5 doi 10.1016/j.ccell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486(7403):346–52 doi 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rueda OM, Sammut SJ, Seoane JA, Chin SF, Caswell-Jin JL, Callari M, et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 2019;567(7748):399–404 doi 10.1038/s41586-019-1007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, et al. AP-2gamma regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J 2011;30(13):2569–81 doi 10.1038/emboj.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science 2018;362(6413) doi 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung E, Acevedo ML, Cole PA, Kraus WL. Altered pharmacology and distinct coactivator usage for estrogen receptor-dependent transcription through activating protein-1. Proc Natl Acad Sci U S A 2005;102(3):559–64 doi 10.1073/pnas.0407113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 2013;8(12):2502–15 doi 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu M, Bao W, Wang J, Yang T, He X, Liao Y, et al. FOXA1 promotes tumor cell proliferation through AR involving the Notch pathway in endometrial cancer. BMC Cancer 2014;14:78 doi 10.1186/1471-2407-14-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci U S A 2016;113(43):E6600–E9 doi 10.1073/pnas.1612835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu X, Pereira R, De Angelis C, Veeraraghavan J, Nanda S, Qin L, et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc Natl Acad Sci U S A 2019. doi 10.1073/pnas.1911584116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song B, Park SH, Zhao JC, Fong KW, Li S, Lee Y, et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J Clin Invest 2019;129(2):569–82 doi 10.1172/JCI122367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franco HL, Nagari A, Kraus WL. TNFalpha signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell 2015;58(1):21–34 doi 10.1016/j.molcel.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sikora MJ, Jacobsen BM, Levine K, Chen J, Davidson NE, Lee AV, et al. WNT4 mediates estrogen receptor signaling and endocrine resistance in invasive lobular carcinoma cell lines. Breast Cancer Res 2016;18(1):92 doi 10.1186/s13058-016-0748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70 doi 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horibata S, Rice EJ, Mukai C, Marks BA, Sams K, Zheng H, et al. ER-positive breast cancer cells are poised for RET-mediated endocrine resistance. PLoS One 2018;13(4):e0194023 doi 10.1371/journal.pone.0194023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherbakov AM, Andreeva OE, Shatskaya VA, Krasil’nikov MA. The relationships between snail1 and estrogen receptor signaling in breast cancer cells. J Cell Biochem 2012;113(6):2147–55 doi 10.1002/jcb.24087. [DOI] [PubMed] [Google Scholar]
- 32.Gattelli A, Garcia Sola ME, Roloff TC, Cardiff RD, Kordon EC, Chodosh LA, et al. Chronic expression of wild-type Ret receptor in the mammary gland induces luminal tumors that are sensitive to Ret inhibition. Oncogene 2018;37(29):4046–54 doi 10.1038/s41388-018-0235-y. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z, Lam N, Thiele CJ. Zinc finger transcription factor CASZ1 interacts with histones, DNA repair proteins and recruits NuRD complex to regulate gene transcription. Oncotarget 2015;6(29):27628–40 doi 10.18632/oncotarget.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Ven RA, Tenhagen M, Meuleman W, van Riel JJ, Schackmann RC, Derksen PW. Nuclear p120-catenin regulates the anoikis resistance of mouse lobular breast cancer cells through Kaiso-dependent Wnt11 expression. Dis Model Mech 2015;8(4):373–84 doi 10.1242/dmm.018648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 2012;22(2):209–21 doi 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017;547(7661):55–60 doi 10.1038/nature22992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng XD Rongbin, Wan Changxin, Shi Xiaoying, Zhang Xiaoyan, Meyer Clifford A.. Cistrome Data Browser and Toolkit: analyzing human and mouse genomic data using compendia of ChIP-seq and chromatin accessibility data. Quant Biol 2020;8(3):267–76 doi 10.1007/s40484-020-0204-7. [DOI] [Google Scholar]
- 38.Young RA. Control of the embryonic stem cell state. Cell 2011;144(6):940–54 doi 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153(2):307–19 doi 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fei T, Li W, Peng J, Xiao T, Chen CH, Wu A, et al. Deciphering essential cistromes using genome-wide CRISPR screens. Proc Natl Acad Sci U S A 2019;116(50):25186–95 doi 10.1073/pnas.1908155116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods 2016;13(6):521–7 doi 10.1038/nmeth.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Cheng Y, Wang Y, Fan Y, Li C, Zhang Y, et al. Tamoxifen reverses epithelial-mesenchymal transition by demethylating miR-200c in triple-negative breast cancer cells. BMC Cancer 2017;17(1):492 doi 10.1186/s12885-017-3457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paakinaho V, Swinstead EE, Presman DM, Grontved L, Hager GL. Meta-analysis of Chromatin Programming by Steroid Receptors. Cell Rep 2019;28(13):3523–34 e2 doi 10.1016/j.celrep.2019.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, et al. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell 2016;165(3):593–605 doi 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riggins RB, Lan JP, Zhu Y, Klimach U, Zwart A, Cavalli LR, et al. ERRgamma mediates tamoxifen resistance in novel models of invasive lobular breast cancer. Cancer Res 2008;68(21):8908–17 doi 10.1158/0008-5472.CAN-08-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sottnik JL, Bordeaux EK, Mehrotra S, Ferrara SE, Goodspeed AE, Costello JC, et al. Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Coregulator in Invasive Lobular Carcinoma of the Breast. Mol Cancer Res 2021;19(8):1270–82 doi 10.1158/1541-7786.MCR-21-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000;103(6):843–52 doi 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 48.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science 2002;295(5564):2465–8 doi 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 49.Howell A, Dodwell DJ, Anderson H, Redford J. Response after withdrawal of tamoxifen and progestogens in advanced breast cancer. Ann Oncol 1992;3(8):611–7 doi 10.1093/oxfordjournals.annonc.a058286. [DOI] [PubMed] [Google Scholar]
- 50.Ediriweera MK, Tennekoon KH, Samarakoon SR. Emerging role of histone deacetylase inhibitors as anti-breast-cancer agents. Drug Discov Today 2019;24(3):685–702 doi 10.1016/j.drudis.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Kurmis AA, Dervan PB. Sequence specific suppression of androgen receptor-DNA binding in vivo by a Py-Im polyamide. Nucleic Acids Res 2019;47(8):3828–35 doi 10.1093/nar/gkz153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole-genome sequencing, RNA-seq, ChIP-seq, ATAC-seq, and Hi-ChIP-seq data have been deposited in the Gene Expression Omnibus database under GSE152367.