

Abstract
Triosephosphate isomerase (TPI) performs the 5th step in glycolysis, operates near the limit of diffusion, and is involved in “moonlighting” functions. Its dimer was found singly phosphorylated at Ser20 (pSer20) in human cells, with this post-translational modification (PTM) showing context-dependent stoichiometry and loss under oxidative stress. We generated synthetic pSer20 proteoforms using cell-free protein synthesis that showed enhanced TPI activity by 4-fold relative to unmodified TPI. Molecular dynamics simulations show that the phosphorylation enables a channel to form that shuttles substrate into the active site. Refolding, kinetic, and crystallographic analyses of point mutants including S20E/G/Q indicate that hetero-dimerization and subunit asymmetry are key features of TPI. Moreover, characterization of an endogenous human TPI tetramer also implicates tetramerization in enzymatic regulation. S20 is highly conserved across eukaryotic TPI, yet most prokaryotes contain E/D at this site, suggesting that phosphorylation of human TPI evolved a new switch to optionally boost an already fast enzyme. Overall, complete characterization of TPI shows how endogenous proteoform discovery can prioritize functional versus bystander PTMs.
Graphical Abstract
INTRODUCTION
The post-translationally modified forms of a single protein, or its proteoforms, determine the composition and function of protein complexes.1,2 However, finding post-translational modifications (PTMs) on endogenous proteins that regulate enzymatic function is challenging, even for abundant glycolytic enzymes. To detect and assign a function to PTMs, their stoichiometry and context-dependent proteoform dynamics can help prioritize them for study. This approach was taken to the 5th enzyme in glycolysis, human triosephosphate isomerase (TPI), well known to run at or near the diffusion limit for its isomerization reaction and has 20 PTMs of 6 different types on 17 residues reported in UniProt.3–6
TPI, present in almost every organism, functions as a dimer of TIM barrels (a fold named after TPI) and is an ancient enzyme that isomerizes dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P) in glycolysis and gluconeogenesis.4 The TPI reaction also lies at the intersection of glycolysis and the pentose phosphate pathway, thereby mediating lipid metabolism and the cellular response to oxidative stress.7 With >200 crystal structures deposited in the PDB,8 TPI is the best studied glycolytic enzyme with roles in metabolism, aging and disease.8,9 TPI expression levels have been correlated with the progression of different types of cancer.10,11 Additionally, TPI aggregation and nitrotyrosination have been implicated in Alzheimer’s disease.12,13 Point mutations in TPI have been shown to drive TPI-deficiency disorder,14 linked to the accumulation of DHAP and formation of toxic methylglyoxal.15 TPI deficiency manifests clinically as congenital hemolytic anemia and symptoms of neuromuscular degeneration. Non-enzymatic and “moonlighting” functions for TPI have also been described in virulence, immunity, spermatozoa differentiation, cell cycle signaling and neuronal function.16
Advances in mass spectrometry (MS) of intact proteoforms (i.e., “Top-Down”, or TDMS) now enable systematic discovery of the modification landscape of whole multi-proteoform complexes.17 Unlike many structural biology techniques, TDMS run in the “native” mode detects PTMs on endogenous protein complexes with low bias.18–20 Native TDMS first measures the intact masses of modified states of an endogenous complex; then, subsequent stages of tandem MS liberate the subunits and create fragment ions that localize PTMs on subunit sequences.21 The untargeted application of “native proteomics” determined 125 protein assemblies and metalloenzymes in a 2018 report, including a curious phosphorylation on Serine 20 (Ser20) of the TPI dimer.20
TPI is the original example of a “perfect enzyme”, as its catalytic efficiency (kcat/KM), ranging from 107 to 1010 M−1s−1 across organisms, is limited by the diffusion rate of the substrate into the enzyme active site.3,5 Hence, an abundant phosphorylation on a single subunit of the TPI dimer raised structural, functional, and evolutionary questions about one of nature’s most efficient—and best studied—biocatalysts. Phosphorylation at Ser20 (pSer20) is an annotated PTM of unknown function; however, CDK2 is the only kinase shown to act on TPI thus far.22 One study examined TPI inactivation by CDK2 phosphorylation but did not find pSer20 as the phospho-site responsible for their observations.23 In a separate study, phosphorylation at Ser79 by CDK2 was correlated with TPI translocation to the nucleus and implicated with nuclear nutrient and cell-cycle signals through the regulation of nuclear acetate and histone acetylation.24
Given that less than 3% of phosphorylation events have annotated functions,25 we investigated the structural and enzymatic effects of this phosphorylation using pSer20 proteoforms from cell-free protein synthesis,26,27 recombinant point mutants, and X-ray crystallography. We find that asymmetric phosphorylation at Ser20, that is, on only one subunit, can induce structural changes to promote substrate diffusion into the TPI active site, resulting in significant activity enhancement. We further combine biochemical and computational methods to inform a structural model and functional assignment for this reversible phosphorylation.
RESULTS
Localizing TPI Phosphorylation at Ser20.
We performed targeted analyses in multiple cell lines, including colorectal cancer, HEK, Ramos, and HeLa cells and revealed the predominance of three major forms of TPI dimers (Figure 1A). One dimer, present in all cell lines, was characterized as the canonical homodimer of unmodified TPI (with loss of initiator methionine, i.e., Met-Off). Intriguingly, the two other dimers were found to be entirely asymmetric. Namely, one dimer was phosphorylated on one subunit but unmodified on the other (Met-Off). No homodimers of the modified subunits were detected (Figures S1 and S2).
Figure 1.
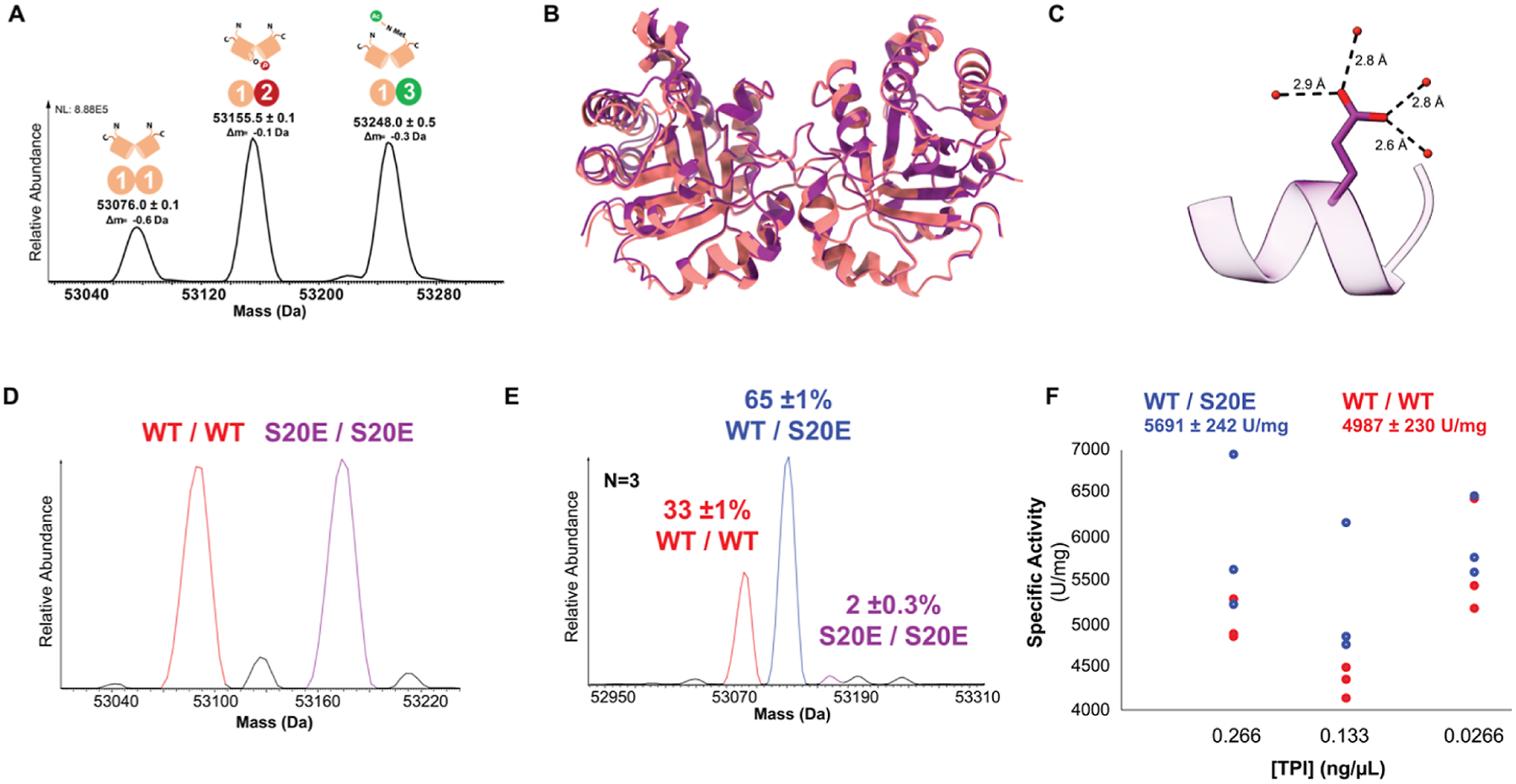
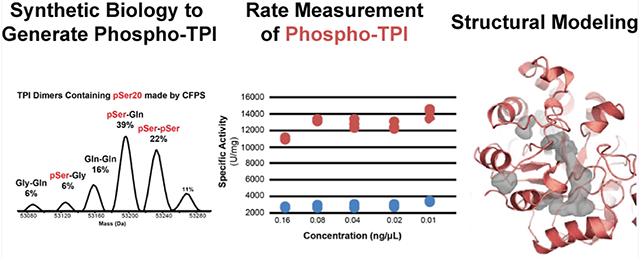
MS, structural, and biochemical characterization of endogenous pSer20 TPI and of recombinant WT and S20E TPI constructs. (A) Native-mode MS revealed three endogenous multi-proteoform complexes (MPC) of TPI (MS1). MPC [1–1] is an unmodified homodimer; MPC [1–2] is the asymmetric pSer20; MPC [1–3] contains asymmetric Met-ON N-terminal acetylation. Their complete characterization is offered in Figure S1. (B) Solved crystal structures for TPI-S20E and WT homodimers, colored in purple and red, respectively. (C) S20E residue in crystal structure of S20E homodimer. The S20E residue is depicted in blue, and water molecules are colored red. (D) Native mass spectrum of WT and S20E TPI homodimers mixed in 1:1 ratio to confirm their relative abundances prior to denaturation and refolding. (E) Representative spectrum of refolded TPI dimers reveals 65 ± 1% heterodimerization preference of WT with S20E (n = 3), whereas 33 ± 1 and 2 ± 0.3% of TPI refold as the WT/WT and S20E/S20E homodimer, respectively. (F) Activity assay of WT/WT (red) and refolded dimers containing the S20E/WT heterodimer at 50% abundance (blue).
Tandem MS fragmentation of phospho-TPI from these cells localized the asymmetric phosphorylation to position Ser20 (pSer20; Figures S1C and S2). Quantitative results from various approaches indicate phosphorylation stoichiometry from asynchronous cells in culture to be ~10% in the various tested cell lines (Figure S3). As a preliminary probe of the function of pSer20, we measured the TPI activity of chromatographically separated, MS-validated endogenous TPI dimers. Controlled for TPI concentration, we found that a fraction enriched for pSer20-TPI was 1.8 times more active than a fraction containing only unmodified TPI (Fig. S3D).
Heteromeric S20E Mutant Is a Pseudo-Mimetic of Phospho-TPI.
To interrogate the kinetic effects of pSer20 with a synthetic mixture, we mimicked the phosphorylation event at position 20 with a Ser-to-Glu mutation.28 The S20E homodimer—wherein both subunits are mutated—did not show a significant activity difference relative to the wild-type (Figure S4A). Moreover, we crystallized and solved the 1.54 Å crystal structure of the S20E homodimer and did not find meaningful differences in their structures (Figure 1B and Table S1). The S20E crystal structure aligns tightly with the WT structure (PDB: 4POC) with an RMSD of 0.250 Å and exhibits high tertiary structural conservation. The bulkier functional group of the glutamate residue does not seem to perturb nearby structural regions, most likely due to its flexibility conferred by the glutamate Cβ and Cγ backbone. Instead, we observe the glutamate residue pointing toward a solvent exposed area and coordinated by four water molecules (Figure 1C and Table S1).
Considering the asymmetry imposed by endogenous hemi-phosphorylation, the S20E mutation would have to be present asymmetrically (a S20E/WT heterodimer) to recapitulate the activity enhancement observed for the endogenous dimers. To detect this hypothesized effect, we denatured the wild-type (WT) and S20E homodimers and refolded them together in a 1:1 ratio (Figure 1D). As a dimer, TPI has a low dissociation constant and off rates ranging from 40 days to 1000 years, which suggest that once folded, TPI does not dissociate during its lifetime in the cell.29 TDMS analysis of the mixture refolded in a narrow protein concentration range showed that there was reproducible and preferential formation of the S20E/WT heterodimer (65 ± 1%, n = 3). The WT and S20E homodimers made up 33 ± 1 and 2 ± 0.3% of the mixture, respectively (Figures 1E and S4B). Activity measurement of a mixture containing 50% heterodimer of S20E/WT showed a modest 14 ± 6% increase relative to WT/WT homodimer (Figure 1F). This heterodimerization behavior has also been reported for the hybridization of TPI from Trypanosoma cruzi (Tc) and Trypanosoma brucei (Tb).30 Interestingly, asymmetric behavior of the two WT subunits has been examined before in the context of proteolysis and biochemical refolding, suggesting that asymmetry may be an important aspect of TPI allosteric regulation.31 Moreover, the E104D point mutant, associated with TPI deficiency disorder, did not fully redimerize after denaturation and its MS charge state distribution became denatured-like, resembling that of an intrinsically disordered protein32 (Figure S5).
pSer20 Proteoform Is >4-fold More Active Than Unmodified TPI.
To further examine the kinetic effects of the phosphorylation, we generated the pSer20 TPI proteoform using co-translational incorporation of phosphoserine via a cell-free protein synthesis (CFPS)33 (Figures 2A and S6). The MS analysis of the CFPS-generated pSer20 showed that it can in fact homodimerize in vitro (Figure 2A), even though the doubly phosphorylated dimer was never observed endogenously. Moreover, CFPS resulted in the misincorporation of glutamine and glycine at position 20, yielding a population of TPI dimers (Figures 2A and S6A,B). Consistent with our refolding studies, the predominant dimer species in the CFPS mixture (39%) was a hetero-dimer of the pSer20 and S20Q TPI proteoforms. Overall, >60% of the dimers present in the mixture contained pSer20 (Figure S6B,C). We then compared the TPI activity of the CFPS-generated dimers to that of unmodified TPI and found that the CFPS sample containing pSer20 was 4.4 ± 0.2 times more active than unmodified TPI (n = 6, Figures 2B and S6D).
Figure 2.
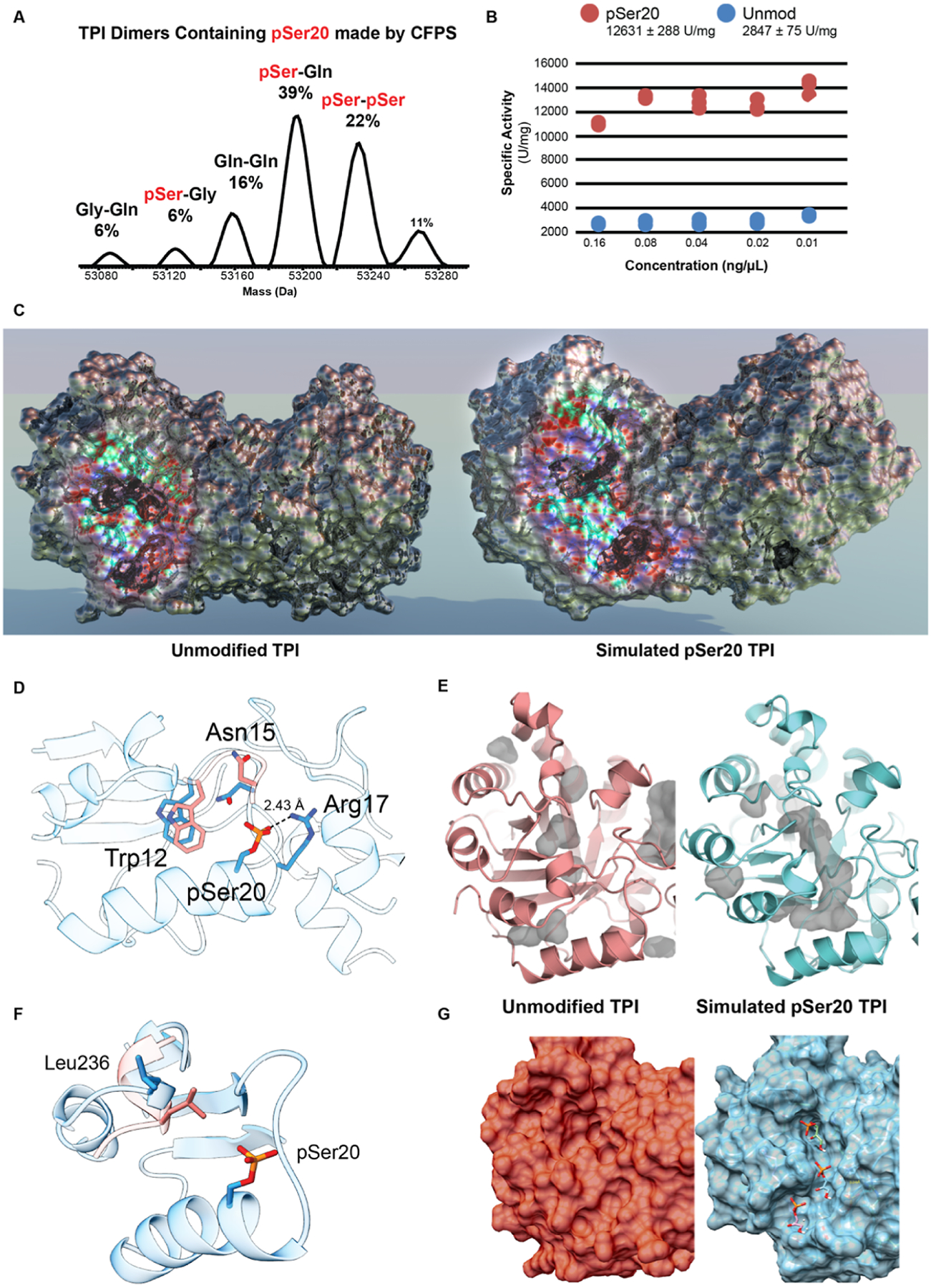
Proteoform synthesis to test activity of pSer20-containing TPI dimers. (A) Intact MS analysis of TPI dimers containing pSer20 and mutant dimers generated by mis-incorporation of Gln and Gly during cell-free proteoform synthesis (CFPS). (B) TPI activity assay comparing the specific activity (U/mg) of pSer20-containing samples vs unmodified, WT TPI. (C) Comparing the crystal structure of unmodified, WT TPI (PDB: 4POC) to the molecular dynamics-simulated structure of TPI-pSer20 (sTPI-pSer20) exposed the formation of an opening into the active site. Area of interest that undergoes structural change has been colorized to differentiate it from the rest of the protein. (D) Structural changes of loop region near the S20 phosphorylation site. The residues for sTPI-pS20 are colored in pink, and WT TPI residues are depicted in blue. (E) Visualization of cavities present in the sTPI-pSer20 model (right) and the WT structure (left). (F) Residue Leu236 and its effect on substrate cavity. sTPI-pS20 is colored in pink and TPI-WT L236 is colored in green. Gray areas represent cavities in the structure. (G) Substrate docking simulation on sTPI-pSer20 model (right) shows substrate diffusing through channel into the active site. WT structure (left) for comparison. sTPI-pSer20 model available in augmented reality in Figure S8.
Facilitated Substrate Diffusion into Active Site Is Consistent with pSer20 Rate Enhancement.
To inform a structural hypothesis for the higher activity of asymmetric TPI dimers with pSer20, we carried out small molecule ligand docking of the substrate DHAP in the active site of unmodified human TPI crystal structure (4POC.pdb) to obtain the binding energy (BE) and Kd. We then phosphorylated the TPI structure at Serine 20 on one or both subunits and performed molecular dynamics simulations at 5–50 ns to visualize the conformational changes in the protein structure. With the energy-minimized structure of the phosphorylated protein, we again docked the substrate into the active site of the protein to compute the BE and Kd.
The simulated structure of the asymmetrically phosphorylated dimer (sTPI-pSer20) exhibits structural differences to the WT dimer near the phosphorylation site, with an RMSD value of 1.74 Å (Figures 2C and S7). Because a bulkier phosphorylated S20 would clash with Gly16 in the WT structure, there is a structural shift in the sTPI-pSer20 model for the helix that contains pSer20 and for the loop region Asn11-Lys18 (loop 1), which is part of the dimer interface. Due to steric hindrance, the loop shifts 1.9 Å away from pSer20, and this conformation is stabilized via a salt bridge between Arg17 and pSer20 (Figure 2D; sTPI-pSer20 model available in augmented reality in Figure S8). Within the loop region, Trp12 moves 1.5 Å away from the substrate channel and Asn15 shifts 110° toward Trp12 (Figures 2D and S8). These key changes that accommodate the bulkier pSer20 result in further changes to the hydrogen bonding interactions at the dimer interface of loops 1–3 and result in the formation of a channel into the active site (Figures 2E,F and S9 and Tables S1 and S2).
Snapshots of three timepoints in the substrate docking simulation reveal that the substrate is shuttled into the active site by residues lining the emergent channel (Figure 2G, right). After being stabilized by water molecules near the opening of the channel, the substrate interacts with Asn15 and Leu236, then with Gly232 and Lys13 near the active site. Consistent with a model of facilitated diffusion, the energetics of the substrate docking simulation with DHAP showed a ΔG of −4.28 and −13.5 kcal/mol for unmodified and hemi-phosphorylated TPI, respectively. Moreover, the strain energy for unmodified was 11.25 kcal/mol, compared to 1.56 kcal/mol for sTPI-pSer20. The simulation of the doubly phosphorylated TPI dimer did not converge on a structure within a 50 ns timeframe.
Evolutionary Analysis of TPI Reveals Co-evolution of Interfacial Residues with Position 20.
TPI sequence identity across all organisms ranges from 18 to 98%, with strict conservation of the active site residues (Asn11, Lys13, His95, Glu165) and of the three-dimensional TIM-barrel structure.8 54,722 TPI sequences were identified in the UniRef90 database, in which highly similar sequences are represented by a single member to avoid overrepresentation of frequently sequenced strains, and that revealed several striking trends in connection to Ser20. A total of 42.4% of sequences contain an amino acid with a carboxylate side chain (Asp, Glu) in that position, while 13.7% feature a serine, 10.8% have an asparagine or glutamine, and 33.1% have another residue (Figures 3A and S10A and S11). The presence of a serine or threonine residue at this position is highly correlated with the presence of an amine-containing lysine or arginine residue two or three positions prior (Figures 3B,C and S10), prompting the identification of a key salt bridge in hemi-phosphorylated TPI. In the MD-simulated TPI structure, the basic residue at position 17 is predicted to form a salt bridge with pSer20. Supporting this view, 53.4% of sequences with a serine at position 20 have a lysine or arginine at position 18, compared to just 7.4% with acidic glutamates or aspartates at that position.
Figure 3.
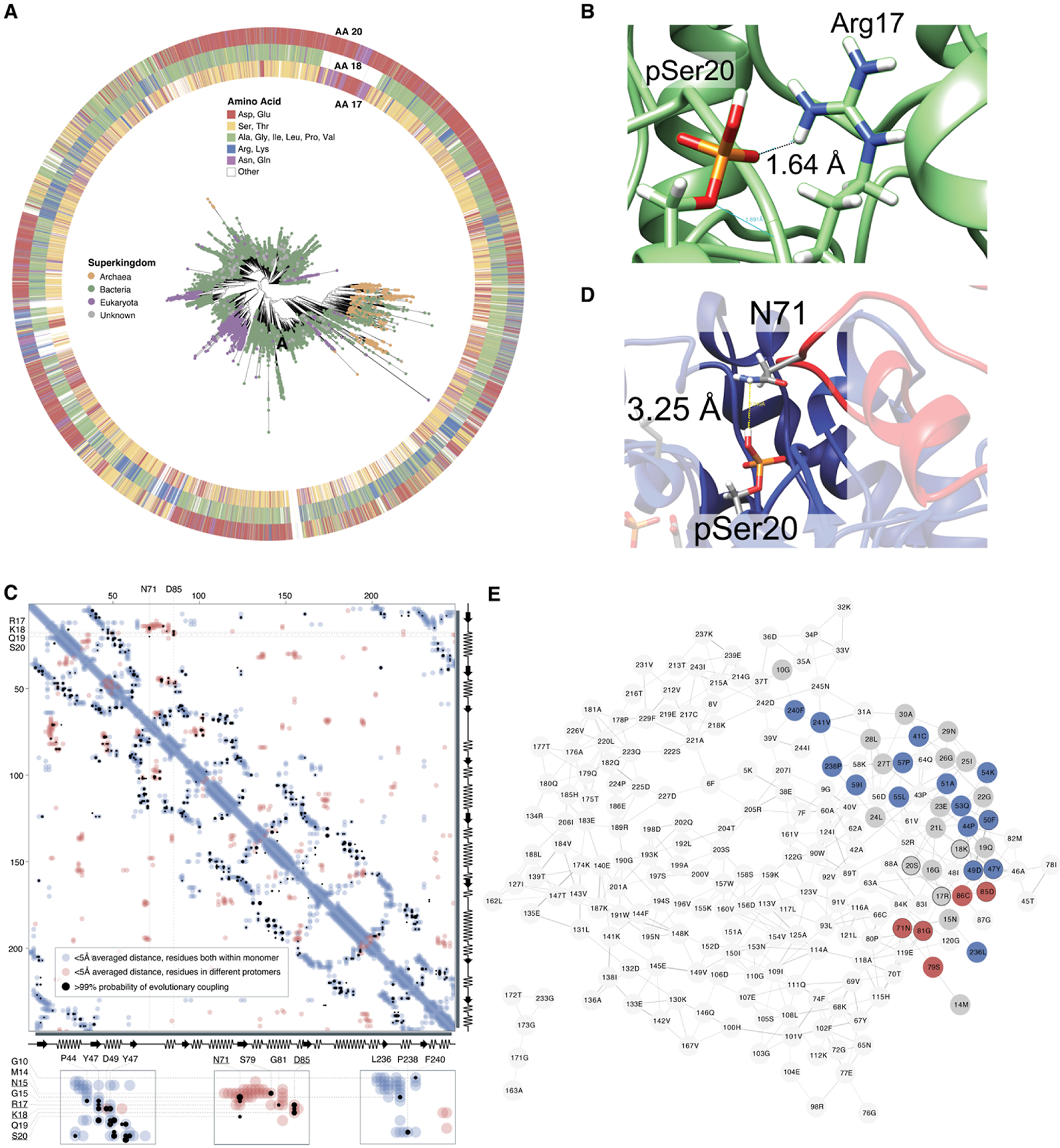
Sequence-based analysis of 54722 TPI sequences across the tree of life. (A) All TPI sequences in the UniRef90 database were used to construct sequence similarity networks (SSNs), and for visualization, representative protein sequences were chosen at 70% identity (Figure S10A–D). These representative sequences were additionally aligned against the TPI Pfam hidden Markov model (PF00121), and this alignment was used to construct an unrooted phylogenetic tree. Nodes are colored according to superkingdom membership, and residue identity at positions 17/18 and 20 is illustrated surrounding the tree. (B) Interaction between Arg17 and pSer20 in our structural model. (C) Evolutionary coupling of residues in TPI. Analyses were carried out using EVCouplings V2, at a bitscore of 0.7, but the same couplings were observed at lower bitscores. Residues within a 5 Å distance within the structure of the monomeric protein are indicated in light blue, while residues on separate protomers within a homomultimeric unit that are within an average 5 Å are highlighted in red. Residues with a >99% likelihood of evolutionary coupling are indicated in black. (D) Notably, despite a predicted distance that may preclude direct hydrogen bonding, Ser20 and Asn71 are proposed as an evolutionarily coupled pair, likely as part of interactions at or near the homodimer interface. Coupling between Arg17, Lys18, and Asp85 is additionally likely to be directly related to the formation of the homodimer interface. (E) Residue co-evolution network for long-distance co-evolutionary partners, organized by connectivity with unweighted edges (edges represent >99% likelihood for evolutionary coupling). Residues with intramolecular proximity to Asn15-Ile25 are shown in gray; more distant residues with evolutionary coupling on the same subunit as Asn15-Ile25 are highlighted in blue; distant residues on the subunit opposite to Asn15-Ile25 are highlighted in red.
Further analysis of TPI sequences provides other connections between pSer20 and oligomer formation. Evolutionary coupling is predicted between Ser20 and Asn71 (Figure 3C), and this is potentially explained by an interaction at the homodimeric interface. In our MD-simulated structure, pSer20 is separated by 3.5 Å from Asn71, part of the opposite subunit’s “interdigitating loop”, which mediates the dimeric interface (Figure 3D).8 Arg17 and Lys18 are also proposed to exhibit evolutionary coupling with residues involved in homodimer formation, particularly Asp85 (Figure 3E).8 Interestingly, the residues Asn15 and Leu236 that are predicted to guide the substrate into the active site through the pSer20-enabled channel also exhibit >99% chance of evolutionary coupling (Figure 3E), suggesting the presence of an evolutionary “cassette”.
Oxidative Stress Decreases pSer20 Stoichiometry and Alters TPI Oligomerization.
Previous reports demonstrate that TPI plays a role in mediating the cellular response to oxidative stress by helping to re-route carbohydrate flux from glycolysis to the pentose phosphate pathway.34 Specifically, TPI-deficient cells containing a loss-of-function I170V mutation exhibit more resistance to diamide, an oxidizing reagent that depletes cellular glutathione.35 We thus interrogated the phosphorylation stoichiometry on endogenous TPI dimers in HEK cells treated with hydrogen peroxide (H2O2, 1 mM and 100 mM) and diamide (5 mM) (Figure 4A–D; Figure S12). Interestingly, ion-exchange fractionation of treated cell lysates exhibited altered but reproducible TPI elution patterns compared to control cells (Figure 4B,C). MS quantitation of TPI using a methionine-tagged TPI standard, spiked in at several concentrations, shows that phosphorylated dimer abundance decreases in both diamide- and H2O2-treated cells compared to untreated cells. Of note, we observed 6-fold and 13-fold decreases in dimer phosphorylation relative to unmodified TPI in cells treated with 1 mM and 100 mM H2O2, respectively (Figures 4D and S13).
Figure 4.
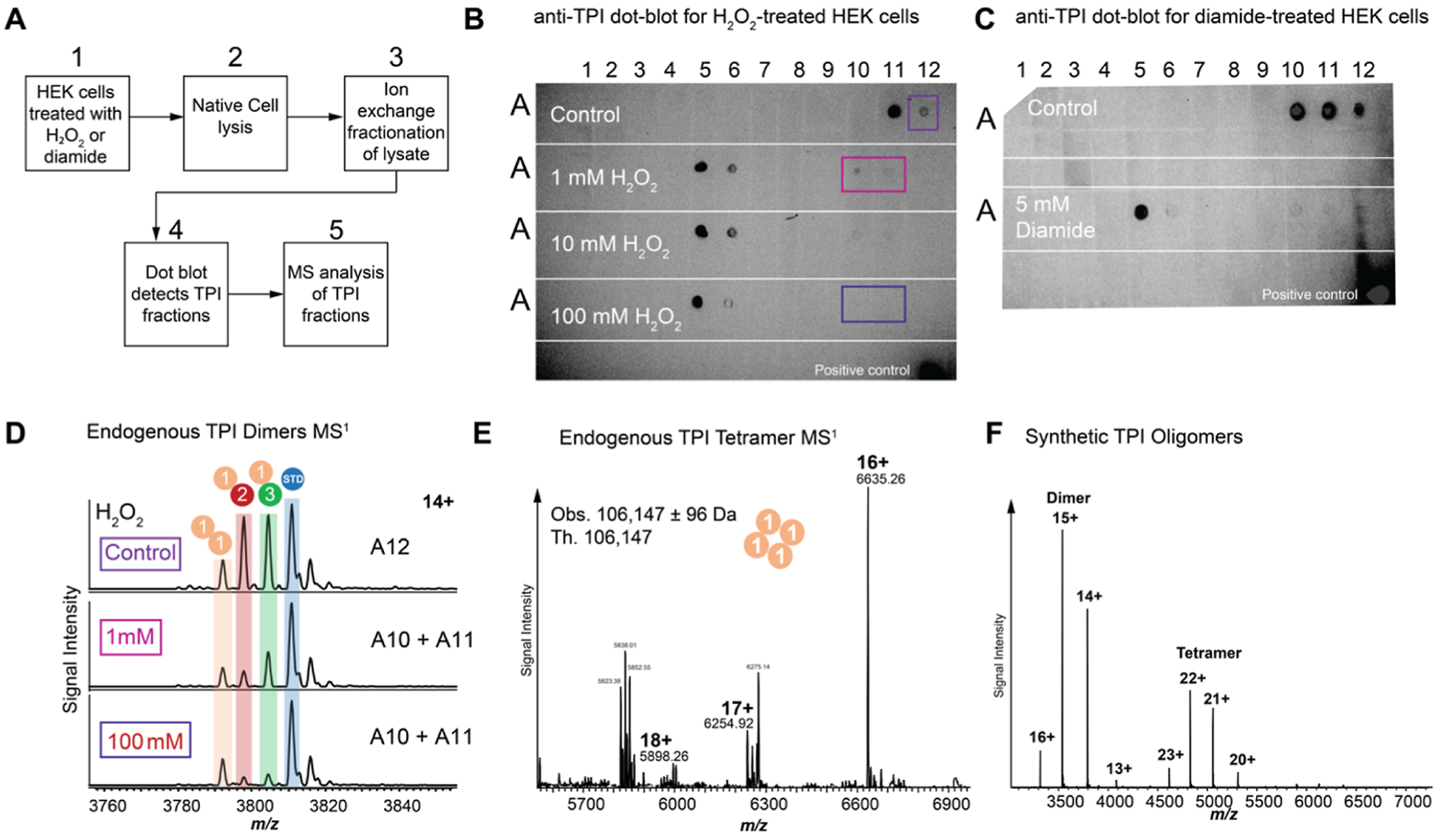
TPI phosphorylation and tetramerization status during oxidative stress. (A) Workflow for purification and characterization of endogenous TPI dimers. (B,C) Dot blots showing TPI-containing fractions upon chromatographic separation of HEK cell lysates after treatment with increasing concentration of hydrogen peroxide or diamide. (D) MS analysis of TPI-containing fractions showing dimer composition at three hydrogen peroxide concentrations. (E) Analysis of endogenous TPI tetramer observed in cells treated with hydrogen peroxide and diamide. (F) Concentration-dependent oligomerization of recombinant TPI in vitro at 10 μM.
Discovery-mode MS analysis of TPI-containing fractions from diamide- and H2O2-treated cells revealed the presence of a TPI tetramer (Figures 4E and S14). TDMS characterization of the endogenous tetramer showed the non-covalent oligomerization of four unmodified, Met-Off TPI subunits (Figures 4E and S14). We examined the oligomerization behavior of recombinant, unmodified TPI using MS and detected the formation of higher-order complexes at 10 μM TPI concentration, suggesting concentration-dependent oligomerization under conditions of oxidative stress (Figure 4F).
DISCUSSION
Advances in MS now enable the characterization of endogenous complexes with complete molecular specificity, providing definitive information on the oligomerization state of proteoforms and the stoichiometry of their PTMs. Despite hundreds of references pointing at >20 modification sites throughout the human TPI sequence, native TDMS acted as a “filter”, prioritizing the Ser20 phosphorylation for further study.
It is challenging to produce a >90% pure hemi-phosphorylated complex in sufficient quantities for structural and kinetic work, like determination of Michaelis–Menten kinetics. Functional investigation of modifications to Ser20 PTM required mimicry of the hemi-phosphorylated form for crystallography, activity, and refolding studies. While noncanonical amino acid incorporation and CFPS36 was used to create a mixture of TPI dimers containing the pSer20 proteoform with >4-fold higher activity, these results are indicative of rate enhancement due to Ser20 phosphorylation. This highlights the current difficulty of functional assignment of proteoforms, requiring a new generation of tools for chemo- or bio-synthesis to provide “knock-in” and “knock-out” to enhance proteoform-level knowledge.
Molecular dynamics simulation of the asymmetric pSer20-TPI dimer captures the structural significance of the modification. The formation of a discernible channel on the phosphorylated subunit facilitates substrate diffusion into the active site, explaining how a “catalytically perfect” enzyme can overcome the limit of diffusion.37,38 Consistent with Chou’s model for diffusion-controlled enzymatic reactions, the substrate docking simulation shows how the modified protein scaffold can actively guide the substrate into the TPI active site.39 The residues involved in substrate shuttling, in particular Asn15 and Leu235, also have some degree of co-evolution.
The analysis of >54,000 TPI sequences revealed the overwhelming prevalence of Asp and Glu at position 20 in bacteria compared to Ser20 in eukaryotes and the high co-occurrence of Ser20 with basic residues (Arg and Lys) at position 18. From an evolutionary perspective, these results suggest that ancestral TPI had an acidic side chain at position 20 and escaped the central dogma by evolving a post-translational regulatory switch, pSer20. Interestingly, about 5% of known phospho-serines are thought to have derived from sites with carboxylate side chains that create salt bridges with nearby conserved basic residues;40 the phosphorylation of Ser20 thus has the potential to restore the salt bridge in a context-dependent manner. In our MD simulations, pSer20 and Arg17 are predicted to form a salt bridge, and co-evolutionary coupling between these residues, Lys18, Asn71, and Asp85, which are part of the homodimeric interface, provides additional support for the structural perturbation associated with pSer20. The evolutionary coupling of Asn15 and Leu235, which help shuttle the substrate through the pSer20-enabled channel, is further indicative of structural fine-tuning near the phosphorylation site.
In the context of glycolysis, the function of TPI is to maintain its isomerization reaction near thermodynamic equilibrium and has not been shown to be a key regulatory enzyme in metabolic flux.41 Our discovery of a phosphorylation-activated mechanism for TPI rate enhancement now suggests that the enzyme’s influence on glycolytic metabolic flux may not be mediated by changes in its transcription or translation, as shown in overexpression systems.41 Instead, modified TPI proteoforms and oligomeric state could potentiate primary metabolism without the need for upregulating protein expression. Consistent with such a model, loss of function due to TPI tetramerization42–44 may explain why previous studies that measure metabolic flux upon TPI overexpression do not identify it as a key regulator.41 Moreover, the dramatic decrease of the activating pSer20 modification under oxidative stress is consistent with previous reports that show that TPI-deficient cells redirect carbohydrate flux away from glycolysis into the pentose phosphate pathway, altering the redox equilibrium of cytoplasmic NADP(H) and thus alleviating oxidative stress.34,35,45
CONCLUSIONS
TPI serves as a model system for understanding the effect of a single phosphorylation on the structure and function of an enzyme. Moreover, reversible regulation of the TPI structure via symmetry breaking highlights a regulatory motif with implications for other dimeric enzymes. This work provides an example of how exact biochemical composition from endogenous multi-proteoform complexes complements atomic-resolution structural studies via crystallography, cryo-electron microscopy, or in silico simulation. While native top-down MS provides far less coverage of the proteome than protease-based proteomics workflows, it offers a new filter to help discern functionally important versus bystander PTMs on even the best studied enzymes.
MATERIALS AND METHODS
Strains and Plasmids.
C321.ΔA.759.T7.D is a derivative of C321.ΔA.75946 to which a modified version of the 1 gene (expressing the viral T7 RNA polymerase) has been added to enable transcription through the T7 promoter in CFPS without the need for exogenous supplementation.47 Lysates derived from this strain were used for the cell-free synthesis of all TPI proteoforms lacking pSer. C321.ΔA.759.ΔserB is a derivative of C321.ΔA.759 that features the additional inactivation of SerB to stabilize pSer proteoforms. This strain was transformed with plasmid B40 OTS (encoding orthogonal translation components for pSer incorporation), and lysates derived from this strain were used for the cell-free synthesis of all TPI proteoforms featuring pSer. Carbenicillin (50 μg/mL) was used for culturing C321.ΔA.759 and its derivatives; kanamycin (50 μg/mL) was used for maintaining pJL1 plasmids and the pSer B40 orthogonal translation system (OTS) plasmid.
Plasmid DNA Purification.
For applications requiring large amounts of plasmid DNA (templating cell-free protein synthesis reactions), plasmid DNA was purified from cells using Hi-Speed Plasmid Maxi Kit (Qiagen, Venlo, the Netherlands).
TPI Expression Vector Assembly Using Gibson Assembly.
To assemble plasmids encoding TPI variants from linear DNA pieces, throughout this work we use the method of Gibson et al.48 DNA inserts encoding the variants were designed such that they had ~20 bp of flanking homology with the linearized back of expression vector pJL1(REF). Plasmids were assembled individually by incubating 50 ng of each insert (WT, S20X, S20E, and E104D) with an assembly mix [6.7 mM PEG-8000, 107 mM Tris–HCL pH 7.5, 10.7 mM magnesium chloride, 213 μM dATP, 213 μM dGTP, 213 μM dCTP, 213 μM dTTP, 10.7 mM dithiothreitol, 1 mM nicotinamide adenine dinucleotide (NAD), 0.0043 U/μL T5 exonuclease (New England Biolabs, Ipswich, MA), 4.3 U/μL Taq ligase (New England Biolabs, Ipswich, MA), and 0.023 U/μL Phusion polymerase (New England Biolabs, Ipswich, MA)] for 1 h at 50 °C. Following this incubation, 2 μL of reaction volume was transformed into electrocompetent DH5α cells using a Micropulse electroporator (Bio-Rad, Hercules, CA). Transformed cells were recovered for 1 h in LB media at 37 °C and 250 rpm. 100 μL of recovered culture was spread onto selective plates and put at 37 °C overnight. Resulting colonies contained the assembled plasmids of interest.
Functional Inactivation of serB in Strain C321.ΔA.759.
To facilitate co-translational incorporation of pSer, the Escherichia coli pSer-specific phosphatase SerB was functionally inactivated in strain C321.ΔA.759 following a previously described approach.46 In brief, multiplex advanced genome engineering (MAGE)49 was employed to insert frameshift and nonsense mutations into the open reading frame of the serB gene in C321.ΔA.759. After several cycles of MAGE, individual colonies were plated and screened via colony PCR to identify clones that had taken up the desired mutations. Sanger sequencing confirmed the generation of strain C321.ΔA.759.ΔserB.
Cell Extract Preparation.
Chassis strain cells were grown in 1 L of 2xYTPG media (pH 7.2) in a 2.5 L Tunair shake flask and incubated at 34 °C at 220 rpm. Cultures were induced with 1 mM IPTG at an OD600 of 0.6, and cells carrying B40 OTS plasmid were supplemented with 2 mM pSer and 50 μg/mL of kanamycin at this point. After induction, cultures were permitted to continue to grow to an OD600 of 3.0. Cells were pelleted by centrifuging for 15 min at 5000×g at 4 °C, washed three times with cold S30 buffer (10 mM tris-acetate pH 8.2, 14 mM magnesium acetate, 60 mM potassium acetate, 2 mM dithiothreitol),50 and stored at −80 °C. To make the cell extract, cell pellets were thawed and suspended in 0.8 mL of S30 buffer per gram of wet cell mass and 1.4 mL of cell slurry was transferred into 1.5 mL microtubes. The cells were lysed using a Q125 Sonicator (Qsonica, Newtown, CT) with 3.175 mm diameter probe at a 20 kHz frequency and 50% amplitude for three cycles of 45 s ON/59 s OFF. To minimize heat damage during sonication, samples were placed in an ice-water bath. The extract was then centrifuged at 12,000×g at 4 °C for 10 min. This was followed by a run-off reaction (37 °C at 250 rpm for 1 h) and second centrifugation (10,000×g at 4 °C for 10 min).51 The supernatant was flash-frozen using liquid nitrogen and stored at −80 °C until use.
CFPS Reaction.
A modified PANOx-SP system was utilized for CFPS reaction testing incorporation of Sep.52,53 Briefly, a 15 μL CFPS reaction in a 2.0 mL microtube was prepared by mixing the following components: 1.2 mM ATP; 0.85 mM each of GTP, UTP, and CTP; 34 μg/mL folinic acid; 170 μg/mL of E. coli tRNA mixture; 13.3 μg/mL plasmid; 16 μg/mL T7 RNA polymerase; 2 mM for each of the 20 standard amino acids; 0.33 mM NAD; 0.27 mM coenzyme-A; 1.5 mM spermidine; 1 mM putrescine; 4 mM sodium oxalate; 130 mM potassium glutamate; 10 mM ammonium glutamate; 12 mM magnesium glutamate; 57 mM HEPES, pH 7.2; 33 mM phosphoenolpyruvate (PEP); and 27% v/v of cell extract. For co-translational pSer incorporation, 2 mM pSer was supplemented to cell-free reactions. Each CFPS reaction was incubated for 20 h at 30 °C unless noted otherwise. E. coli total tRNA mixture (from strain MRE600) and phosphoenolpyruvate were purchased from Roche Applied Science (Indianapolis, IN). ATP, GTP, CTP, UTP, 20 amino acids, and other materials were purchased from Sigma (St. Louis, MO) without further purification. For scaled-up synthesis of TPI proteoforms, 250 μL reactions were assembled in a 24-well plate (Falcon 351147; Corning, NY). To reduce loss of reaction volume by evaporation, unused wells were filled with 2 mL water, and the chamber was sealed with Parafilm M (Bemis, Neenah, WI). Plates were shaken at 300 RPM for 20 h at 30 °C.
Purification and Protease Treatment of Synthesized TPI Proteoforms.
TPI synthesized in CFPS was purified using Qiagen Ni-NTA agarose (Qiagen, Valencia, CA) according to the manufacturer’s manual. Following washes, bound TPI was eluted in 500 mM imidazole. 5 μL of Ulp1 protease produced in-house was added to the sample, and the entire volume was dialyzed against 50 mM Tris–HCl, 150 mM NaCl, and 1 mM DTT in a Slide-A-Lyzer Dialysis Cassette with a MWCO of 3.5 kDa. Dialysis proceeded for up to 48 h at 4 °C. Following dialysis, the cleaved sample was removed from the cassette for further use.
Native MS.
Protein samples were measured at a concentration of 2 μM and desalted into 150 mM ammonium acetate using 10–30 kDa MWCO 0.5 mL spin filters (Millipore-Sigma). Samples were analyzed using a Q Exactive HF mass spectrometer with Extended Mass Range (QE-EMR) by Thermo Fisher Scientific. Data were collected using XCalibur Qual Browser 4.0.27.10 (Thermo Fisher Scientific). The native electrospray platform was coupled to a three-tiered tandem MS process. First, the analysis of the intact complex (MS1) provides the total complex mass (reported as a deconvoluted neutral average mass value).21 In stage two, the complex is activated by collisions with nitrogen gas to eject subunits (MS2). In stage three, further vibrational activation of the ejected subunits via collisions with nitrogen gas yields backbone fragmentation products from each monomer (MS3) that are recorded at isotopic resolution (120,000 resolving power at m/z 400). These fragments can be mapped onto the primary sequence of the subunits in order to localize PTMs.
QE-EMR Parameters.
The native top-down MS workflow utilizes native electrospray ionization (nESI) source held at +2 kV, C-trap entrance lens voltage setting between 1.8 and 4 V, HCD gas pressure setting between 2 and 4 V, and CID voltage set at 15–25 V for desalting and 75–100 V for subunit ejection. HCD energy was set to 100–120 V for subunit fragmentation with a pressure of 2. Microscans were set to 20 and max injection time to 2000 ms for collection of fragmentation data.
MS Data Analysis.
Intact mass values for protein complexes and ejected subunits, the MS1 and MS2 measurements, were determined by deconvolution to convert data from the m/z to the mass domain using MagTran 1.0354 (mass range: 15,000–300,000 Da; max no. of species: 10–15; S/N threshold: 1; mass accuracy: 0.05 Da; charge determined by charge envelop only). Intact mass measurements were reported as neutral average masses; errors represent 1σ deviation from the mean of the masses calculated for all sampled charge states.
High-resolution fragmentation data were processed using Xtract (signal-to-noise threshold ranging from 1 to 30, Thermo Fisher Scientific), mMass 5.5.0 (www.mmass.org), ProSight Lite 1.455 (precursor mass type: average; fragmentation method: HCD; fragmentation tolerance: 10–15 ppm), and TDValidator 1.056 (max ppm tolerance: 25 ppm; cluster tolerance: 0.35; charge range: 1–10; minimum score: 0.5; S/N cutoff: 3; Mercury7 Limit: 0.0001; minimum size: 2) to assign recorded fragment ions to the primary sequence of the subunits. Specifically, ProSight Lite and TDValidator were used to analyze fragmentation spectra in medium throughput to assign and validate b and y fragment ions to the sequences and for generating a p-score. mMass was used to interrogate individual fragment ions within a spectrum not identified by TDValidator or ProSight Lite. Proteoforms were identified by mapping backbone fragment ions to their amino acid sequence using ProSight Lite.55 Unexplained mass shifts (Δm) observed at the MS1, MS2, and MS3 levels for the intact complex and subunits, respectively, were manually interrogated using the UNIMOD database (http://www.unimod.org/modifications_list.php) as a reference for candidate modifications.
Cell-Free Protein Synthesis.
CFPS reactions were prepared as reported previously.57 TPI synthesized by CFPS purified using Qiagen Ni-NTA agarose (Qiagen, Valencia, CA). After washing, TPI was eluted with 500 mM imidazole. 5 μL of Ulp1 protease was added to the sample, and the entire volume was dialyzed against 50 mM Tris–HCl, 150 mM NaCl, and 1 mM DTT in a Slide-ALyzer Dialysis Cassette with a MWCO of 3.5 kDa. Dialysis proceeded for up to 48 h at 4 °C. Following dialysis, the cleaved sample was removed from the cassette for further use.
TPI Activity and Refolding Assays.
Kinetic rates of DLD1 fractionated cell lysates and purified TPI (by either bacterial expression or cell-free) were compared using a commercial activity assay (Abcam product, ID#: ab197001) based on the enzyme-coupled assay published by Plaut and Knowles.58 Fractions of interest contained equal concentrations of triose phosphate isomerase (TPI) but differing ratios of phosphorylated and unmodified TPI. The assay couples TPI and its downstream partner glyceraldehyde-3 phosphate dehydrogenase (GAPDH), which reduces NAD+ to NADH. Given sufficient concentration of TPI substrate and GAPDH (roughly ×5000 times more concentrated than TPI), the formation of NADH will be solely dependent on the concentration and activity of TPI. NADH formation was monitored via UV–Vis absorbance at 450 nm using a Neo Synergy2 plate reader (Biotek Instruments, Inc.) in kinetic mode for 40 min. All assays were run at 37 °C.
Refolding assays were performed on 1:1 mixtures of TPI species as validated by MS. Specifically, MS was used to determine relative concentrations to ensure refolding at exact ratios. 3 M guanidinium HCl was used to denature TPI samples, and for refolding, the dilution buffer consisted of 10 mM Tris HCl, 25 mM NaCl, 10% glycerol, and 1 mM DTT.
Molecular Dynamics Simulations.
Using the physics-based docking simulation programs implemented in Schrodinger platform, the small molecule ligand docking of the substrate dihydroxy acetone phosphate was carried out in the active site of human TPI (4POC.pdb) crystal structure to obtain the BE or Kd of the substrate. Then, the TPI structure was phosphorylated at Serine 20 residue, and molecular dynamics simulations at 5–50 ns were performed to visualize the conformational change to the protein structure. Considering the energy-minimized structure of the phosphorylated protein, again the substrate was docked into the active site of the protein and the BE/Kd was computed.
Sequence-Based Analysis of the TPI Family.
Protein sequences and metadata for all members of the Pfam protein family59 for TPI (PF00121) were downloaded from the curated UniRef90 database, a subset of the UniProt database60 in which representative sequences are chosen for clusters of proteins above a specific level of similarity, in order to decrease the number of sequences for bioinformatics analyses while retaining sequence diversity. Homologs under 200 aa in length were removed from analysis as potentially truncated sequences. Where possible, analyses were performed with full data sets, but due to the computational resources required for visualization, Uniref90 data were used for construction of phylogenetic trees and a further trimmed data set was used for construction of SSNs. The Uniprot metadata for these sequences were used for visualization of traits such as superkingdom membership in both phylogenetic trees and SSNs.
The resulting protein sequences were aligned against the TPI PFAM using hmmalign,61 and residues at positions not present in the model were removed. This alignment was used to identify conservation of specific residues of interest and to calculate their co-occurrence. An unrooted approximate-maximum-likelihood phylogenetic tree was constructed using FastTree.62 For tree construction, the WAG model for amino acid evolution was used, as were subtree-prune-regraft moves; calculation of a gamma-based likelihood was also enabled. The resulting tree was visualized in R using the ggtree package,63 highlighting traits associated with specific sequences such as phylum membership or sequence conservation.
In parallel, SSNs were constructed using the online EFI-EST toolset.64 The trimmed UniRef90 sequences for members of the TPI Pfam family were used as an input data set. Representative nodes were chosen at 50% ID, and a final expectation value cutoff of 1E-60 was used for edges. The resulting networks were visualized with Cytoscape,65 with metadata regarding sequence conservation mapped onto nodes.
Evolutionary Coupling Analysis.
Analysis of coupled residue evolution was carried out using EVCouplings 2.66 The Met-off form of human TPI isoform 1 (UniProt: P60174–1) was used as a reference to preserve residue numbering. While the data in the figures used a bitscore of 0.7 (recommended by the software), the same couplings involving residues of interest were observed at lower bitscores. Inter-residue distances were calculated using averaging over PDB entries 1hti:A (1–248), 6upf:B (2–248), 4unk:B (4–245), 4unl:A (3–248), 4unl:B (3–248), 4zvj:A (4–248), 4zvj:B (2–248), 6c2g:A (4–247), 6c2g:B (4–247), 6c2g:C (4–247), 6c2g:D (4–247), 6d43:A (4–248), 6d43:B (3–248), 6nlh:A (5–248), 6nlh:B (4–248), 6nlh:C (4–248), 6nlh:D (4–248), 6nlh:E (5–248), 6nlh:F (4–248), 6nlh:G (4–248), 6nlh:H (5–248), 6up1:A (4–248), 6up1:B (3–248), 6up5:A (3–248), and 6up5:B (2–248)).
TPI S20E and WT Expression and Crystallography.
Transformed E. coli BL21(DE3) cells were inoculated in 2xYTPG media. When the OD reached around 0.7, the culture was induced with 1 mM IPTG, followed by 4 h of protein expression at 37 °C. Cells were harvested via centrifugation at 8000×g for 30 min, and the pellet was frozen with liquid nitrogen. His-SUMO-TPI purification was performed by Histag purification using Qiagen Ni-NTA agarose (Qiagen, Valencia, CA). After washing, TPI was eluted with 500 mM imidazole. 5 μL of Ulp1 protease was added to the sample, and the entire volume dialyzed against 50 mM Tris–HCl, 150 mM NaCl, and 1 mM DTT in a Slide-ALyzer Dialysis Cassette with a MWCO of 3.5 kDa. Dialysis proceeded for up to 48 h at 4 °C. Following dialysis, the cleaved sample was removed from the cassette and concentrated using a 3 kDa MWCO spin filter (Millipore-Sigma).
TPI S20E crystals were obtained by the sitting drop vapor diffusion method by mixing 1 μL of 35 mg/mL TPI S20E with 1 μL of reservoir solution containing 0.2 M magnesium chloride hexahydrate, 0.1 M TRIS hydrochloride pH 8.5, and 35% PEG 4000 at 18 °C, as previously described.67 Data collection was performed at the Advanced Photon Source at Argonne National Laboratory. Xia2 was used to process and integrate all data sets.68 Phaser69 was used to obtain molecular replacement solutions using the structure of WT TPI (PDB: 4POC) as the search model (99% amino acid sequence identity). Starting from the initial model, COOT70 was used to manually build the structure, followed by refinement using Phenix.71 The model quality was assessed using MolProbity.72 The final model for the TPI S20E structure includes 2 dimer molecules in the asymmetric unit with residues 2–248 and 426 water molecules. The server MoleOnline was used to identify cavities and tunnels in TPI WT and TPI S20.73
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences P41 GM108569 for the National Resource for Translational and Developmental Proteomics at Northwestern University and NIH grants S10OD025194 and RF1AG063903 (Kelleher lab). L.F.S. is a Gilliam Fellow of the Howard Hughes Medical Institute. Research in this publication is also supported by Thermo Fisher Scientific and a fellowship associated with the Chemistry of Life Processes Predoctoral Training Grant T32GM105538 at Northwestern University. We acknowledge R. Mishra and CMIDD for molecular dynamics simulation work. M.C.J. gratefully acknowledges the Army Research Office Grants W911NF-16-1-0372 and W911NF-18-1-0200, the David and Lucile Packard Foundation and the Camille Dreyfus Teacher-Scholar Program. KD-SepRS-EFSep-5x tRNASep (B40 OTS) was a gift from J. Rinehart (Addgene plasmid # 52054; http://n2t.net/addgene:52054; RRID:Addgene_52054).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00324.
Findings reported in this paper, 3D model of sTPI-pSer20 in augmented reality, and hydrogen bonding distances calculated from the crystallographic and computational results (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.2c00324
The authors declare the following competing financial interest(s): M.C.J. has a financial interest in SwiftScale Biologics, Design Pharmaceuticals, Induro Therapeutics, and Pearl Bio. N.L.K. serves as a consultant to Thermo Fisher Scientific. M.C.J. and N.L.K.s interests are reviewed and managed by Northwestern University in accordance with their conflict-of-interest policies. All other authors declare no competing interests.
Contributor Information
Luis F. Schachner, Department of Chemistry, the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States;.
Benjamin Des Soye, Department of Chemistry, the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States; Department of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois 60208, United States;.
Soo Ro, Department Molecular and Biological Sciences, Northwestern University, Evanston, Illinois 60208, United States.
Grace E. Kenney, Department Molecular and Biological Sciences, Northwestern University, Evanston, Illinois 60208, United States; Department of Chemistry, Harvard University, Cambridge, Massachusetts 02140, United States;.
Ashley N. Ives, Department of Chemistry, the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States;.
Taojunfeng Su, Department of Chemistry, the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Young Ah Goo, Department of Chemistry, the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Michael C. Jewett, Department of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois 60208, United States;.
Amy C. Rosenzweig, Department Molecular and Biological Sciences, Northwestern University, Evanston, Illinois 60208, United States;.
Neil L. Kelleher, Department of Chemistry, the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States; Department Molecular and Biological Sciences, Northwestern University, Evanston, Illinois 60208, United States;.
REFERENCES
- (1).Smith LM; Kelleher NL Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Skinner OS; Havugimana PC; Haverland NA; Fornelli L; Early BP; Greer JB; Fellers RT; Durbin KR; Do Vale LHF; Melani RD; et al. An informatic framework for decoding protein complexes by top-down mass spectrometry. Nat. Methods 2016, 13, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Blacklow SC; Raines RT; Lim WA; Zamore PD; Knowles JR Triosephosphate isomerase catalysis is diffusion controlled. Biochemistry 1988, 27, 1158–1165. [DOI] [PubMed] [Google Scholar]
- (4).Knowles JR; Albery WJ Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc. Chem. Res 1977, 10, 105–111. [Google Scholar]
- (5).Stroppolo ME; Falconi M; Caccuri AM; Desideri A Superefficient enzymes. Cell. Mol. Life Sci 2001, 58, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Davidi D; Longo LM; Jabłońska J; Milo R; Tawfik DS A Bird’s-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations. Chem. Rev 2018, 118, 8786. [DOI] [PubMed] [Google Scholar]
- (7).Wierenga RK; Kapetaniou EG; Venkatesan R Triosephosphate isomerase: a highly evolved biocatalyst. Cell. Mol. Life Sci 2010, 67, 3961–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Olivares-Illana V; Riveros-Rosas H; Cabrera N; Tuena de Gómez-Puyou M; Pérez-Montfort R; Costas M; Gómez-Puyou A A guide to the effects of a large portion of the residues of triosephosphate isomerase on catalysis, stability, druggability, and human disease. Proteins: Struct., Funct., Bioinf 2017, 85, 1190–1211. [DOI] [PubMed] [Google Scholar]
- (9).Verlinde CLMJ; Hannaert V; Blonski C; Willson M; Périé JJ; Fothergill-Gilmore LA; Opperdoes FR; Gelb MH; Hol WGJ; Michels PAM Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resistance Updates 2001, 4, 50–65. [DOI] [PubMed] [Google Scholar]
- (10).Dang Y; Wang Z; Guo Y; Yang J; Xing Z; Mu L; Zhang X; Ding Z Overexpression of triosephosphate isomerase inhibits proliferation of chicken embryonal fibroblast cells. Asian Pac. J. Cancer Prev 2011, 12, 3479–3482. [PubMed] [Google Scholar]
- (11).Jiang H; Ma N; Shang Y; Zhou W; Chen T; Guan D; Li J; Wang J; Zhang E; Feng Y; et al. Triosephosphate isomerase 1 suppresses growth, migration and invasion of hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun 2017, 482, 1048–1053. [DOI] [PubMed] [Google Scholar]
- (12).Roland BP; Zeccola AM; Larsen SB; Amrich CG; Talsma AD; Stuchul KA; Heroux A; Levitan ES; VanDemark AP; Palladino MJ Structural and Genetic Studies Demonstrate Neurologic Dysfunction in Triosephosphate Isomerase Deficiency Is Associated with Impaired Synaptic Vesicle Dynamics. PLoS Genet. 2016, 12, No. e1005941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Guix FX; Ill-Raga G; Bravo R; Nakaya T; de Fabritiis G; Coma M; Miscione GP; Villà-Freixa J; Suzuki T; Fernàndez-Busquets X; et al. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 2009, 132, 1335–1345. [DOI] [PubMed] [Google Scholar]
- (14).Schneider AS Triosephosphate isomerase deficiency: historical perspectives and molecular aspects. Best Pract. Res. Clin. Haematol 2000, 13, 119–140. [DOI] [PubMed] [Google Scholar]
- (15).Ahmed N; Battah S; Karachalias N; Babaei-Jadidi R; Horányi M; Baróti K; Hollan S; Thornalley PJ Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim. Biophys. Acta 2003, 1639, 121–132. [DOI] [PubMed] [Google Scholar]
- (16).Mónica R-B; Ruy P-M Medical and Veterinary Importance of the Moonlighting Functions of Triosephosphate Isomerase. Curr. Protein Pept. Sci 2019, 20, 304–315. [DOI] [PubMed] [Google Scholar]
- (17).Schachner LF; Tran DP; Lee AS; McGee JP; Jooss K; Durbin KR; Seckler HS; Adams L; Cline EN; Melani RD; et al. Reassembling protein complexes after controlled disassembly by top-down mass spectrometry in native mode. Int. J. Mass Spectrom 2021, 465, 116591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kenney GE; Dassama LMK; Pandelia M-E; Gizzi AS; Martinie RJ; Gao P; DeHart CJ; Schachner LF; Skinner OS; Ro SY; et al. The biosynthesis of methanobactin. Science 2018, 359, 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Park YJ; Kenney GE; Schachner LF; Kelleher NL; Rosenzweig AC Repurposed HisC Aminotransferases Complete the Biosynthesis of Some Methanobactins. Biochemistry 2018, 57, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Skinner OS; Haverland NA; Fornelli L; Melani RD; Do Vale LHF; Seckler HS; Doubleday PF; Schachner LF; Srzentic K; Kelleher NL; Compton PD Top-down characterization of endogenous protein complexes with native proteomics. Nat. Chem. Biol 2018, 14, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Belov ME; Damoc E; Denisov E; Compton PD; Horning S; Makarov AA; Kelleher NL From protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal. Chem 2013, 85, 11163–11173. [DOI] [PubMed] [Google Scholar]
- (22).Huang JX; Lee G; Cavanaugh KE; Chang JW; Gardel ML; Moellering RE High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat. Methods 2019, 16, 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lee W-H; Choi J-S; Byun M-R; Koo K.-t.; Shin S; Lee S-K; Surh Y-J Functional inactivation of triosephosphate isomerase through phosphorylation during etoposide-induced apoptosis in HeLa cells: Potential role of Cdk2. Toxicology 2010, 278, 224–228. [DOI] [PubMed] [Google Scholar]
- (24).Zhang J-J; Fan T-T; Mao Y-Z; Hou J-L; Wang M; Zhang M; Lin Y; Zhang L; Yan G-Q; An Y-P; et al. Nuclear dihydroxyacetone phosphate signals nutrient sufficiency and cell cycle phase to global histone acetylation. Nat. Metab 2021, 3, 859–875. [DOI] [PubMed] [Google Scholar]
- (25).Needham EJ; Parker BL; Burykin T; James DE; Humphrey SJ Illuminating the dark phosphoproteome. Sci. Signaling 2019, 12, No. eaau8645. [DOI] [PubMed] [Google Scholar]
- (26).Silverman AD; Karim AS; Jewett MC Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet 2020, 21, 151–170. [DOI] [PubMed] [Google Scholar]
- (27).Oza JP; Aerni HR; Pirman NL; Barber KW; ter Haar CM; Rogulina S; Amrofell MB; Isaacs FJ; Rinehart J; Jewett MC Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat. Commun 2015, 6, 8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Thorsness PE; Koshland DE Jr. Inactivation of isocitrate dehydrogenase by phosphorylation is mediated by the negative charge of the phosphate. J. Biol. Chem 1987, 262, 10422–10425. [PubMed] [Google Scholar]
- (29).Rietveld AWM; Ferreira ST Deterministic Pressure Dissociation and Unfolding of Triose Phosphate Isomerase: Persistent Heterogeneity of a Protein Dimer. Biochemistry 1996, 35, 7743–7751. [DOI] [PubMed] [Google Scholar]
- (30).Zomosa-Signoret V; Hernández-Alcántara G; Reyes-Vivas H; Martínez-Martínez E; Garza-Ramos G; Pérez-Montfort R; Tuena de Gómez-Puyou M; Gómez-Puyou A Control of the Reactivation Kinetics of Homodimeric Triosephosphate Isomerase from Unfolded Monomers. Biochemistry 2003, 42, 3311–3318. [DOI] [PubMed] [Google Scholar]
- (31).De La Mora-De La Mora I; Torres-Larios A; Mendoza-Hernández G; Enriquez-Flores S; Castillo-Villanueva A; Mendez ST; Garcia-Torres I; Torres-Arroyo A; Gómez-Manzo S; Marcial-Quino J; et al. The E104D mutation increases the susceptibility of human triosephosphate isomerase to proteolysis. Asymmetric cleavage of the two monomers of the homodimeric enzyme. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 2702–2711. [DOI] [PubMed] [Google Scholar]
- (32).Santambrogio C; Natalello A; Brocca S; Ponzini E; Grandori R Conformational Characterization and Classification of Intrinsically Disordered Proteins by Native Mass Spectrometry and Charge-State Distribution Analysis. Proteomics 2019, 19, 1800060. [DOI] [PubMed] [Google Scholar]
- (33).Oza JP; Aerni HR; Pirman NL; Barber KW; ter Haar CM; Rogulina S; Amrofell MB; Isaacs FJ; Rinehart J; Jewett MC Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat. Commun 2015, 6, 8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ralser M; Wamelink MM; Kowald A; Gerisch B; Heeren G; Struys EA; Klipp E; Jakobs C; Breitenbach M; Lehrach H; Krobitsch S Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol 2007, 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ralser M; Heeren G; Breitenbach M; Lehrach H; Krobitsch S Triose Phosphate Isomerase Deficiency Is Caused by Altered Dimerization–Not Catalytic Inactivity–of the Mutant Enzymes. PLoS One 2006, 1, No. e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kofman C; Lee J; Jewett MC Engineering molecular translation systems. Cell Syst. 2021, 12, 593–607. [DOI] [PubMed] [Google Scholar]
- (37).Getzoff ED; Cabelli DE; Fisher CL; Parge HE; Viezzoli MS; Banci L; Hallewell RA Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature 1992, 358, 347. [DOI] [PubMed] [Google Scholar]
- (38).Schreiber G; Fersht AR Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol 1996, 3, 427–431. [DOI] [PubMed] [Google Scholar]
- (39).Zhou G-Q; Zhong W-Z Diffusion-controlled reactions of enzymes. A comparison between Chou’s model and Alberty-Hammes-Eigen’s model. Eur. J. Biochem 1982, 128, 383–387. [PubMed] [Google Scholar]
- (40).Pearlman SM; Serber Z; Ferrell JE A Mechanism for the Evolution of Phosphorylation Sites. Cell 2011, 147, 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tanner LB; Goglia AG; Wei MH; Sehgal T; Parsons LR; Park JO; White E; Toettcher JE; Rabinowitz JD Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Deb I; Poddar R; Paul S Oxidative stress-induced oligomerization inhibits the activity of the non-receptor tyrosine phosphatase STEP61. J. Neurochem 2011, 116, 1097–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kohlhoff M; Dahm A; Hensel R Tetrameric triosephosphate isomerase from hyperthermophilic Archaea. FEBS Lett. 1996, 383, 245–250. [DOI] [PubMed] [Google Scholar]
- (44).Walden H; Bell GS; Russell RJ; Siebers B; Hensel R; Taylor GL Tiny TIM: a small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol 2001, 306, 745–757. [DOI] [PubMed] [Google Scholar]
- (45).Grüning N-M; Du D; Keller MA; Luisi BF; Ralser M Inhibition of triosephosphate isomerase by phosphoenolpyruvate in the feedback-regulation of glycolysis. Open Biol. 2014, 4, 130232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Martin RW; Des Soye BJ; Kwon Y-C; Kay J; Davis RG; Thomas PM; Majewska NI; Chen CX; Marcum RD; Weiss MG; et al. Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nat. Commun 2018, 9, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Des Soye BJ; Gerbasi VR; Thomas PM; Kelleher NL; Jewett MC A Highly Productive, One-Pot Cell-Free Protein Synthesis Platform Based on Genomically Recoded Escherichia coli. Cell Chem. Biol 2019, 26, 1743–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gibson DG; Young L; Chuang RY; Venter JC; Hutchison CA 3rd; Smith HO Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (49).Wang HH; Isaacs FJ; Carr PA; Sun ZZ; Xu G; Forest CR; Church GM Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Swartz JR; Jewett MC; Woodrow KA Cell-free protein synthesis with prokaryotic combined transcription-translation. Methods Mol. Biol 2004, 267, 169–182. [DOI] [PubMed] [Google Scholar]
- (51).Kwon YC; Jewett MC High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep 2015, 5, 8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jewett MC; Swartz JR Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng 2004, 86, 19–26. [DOI] [PubMed] [Google Scholar]
- (53).Jewett MC; Calhoun KA; Voloshin A; Wuu JJ; Swartz JR An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol 2008, 4, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhang Z; Marshall AG A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom 1998, 9, 225–233. [DOI] [PubMed] [Google Scholar]
- (55).Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Fornelli L; Srzentić K; Huguet R; Mullen C; Sharma S; Zabrouskov V; Fellers RT; Durbin KR; Compton PD; Kelleher NL Accurate sequence analysis of a monoclonal antibody by top-down and middle-down orbitrap mass spectrometry applying multiple Ion activation techniques. Anal. Chem 2018, 90, 8421–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Des Soye B Cell-free Platforms for Synthesis of Non-standard Polypeptides In Vitro; Northwestern University: Evanston, IL, 2018. [Google Scholar]
- (58).Plaut B; Knowles JR pH-dependence of the triose phosphate isomerase reaction. Biochem. J 1972, 129, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Mistry J; Chuguransky S; Williams L; Qureshi M; Salazar GA; Sonnhammer ELL; Tosatto SCE; Paladin L; Raj S; Richardson LJ; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Consortium TU UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Eddy SR Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [DOI] [PubMed] [Google Scholar]
- (62).Price MN; Dehal PS; Arkin AP FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One 2010, 5, No. e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Yu G; Smith DK; Zhu H; Guan Y; Lam TT-Y ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol 2017, 8, 28–36. [Google Scholar]
- (64).Zallot R; Oberg N; Gerlt JA The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Hopf TA; Green AG; Schubert B; Mersmann S; Schärfe CPI; Ingraham JB; Toth-Petroczy A; Brock K; Riesselman AJ; Palmedo P; Kang C; Sheridan R; Draizen EJ; Dallago C; Sander C; Marks DS The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 2018, 35, 1582–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Cabrera N; et al. Differential effects on enzyme stability and kinetic parameters of mutants related to human triosephosphate isomerase deficiency. Biochim. Biophys. Acta 2018, 1862, 1401–1409. [DOI] [PubMed] [Google Scholar]
- (68).Evans P Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Struct. Biol 2006, 62, 77–82. [DOI] [PubMed] [Google Scholar]
- (69).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2009, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Berka K; Hanak O; Sehnal D; Banas P; Navratilova V; Jaiswal D; Ionescu CM; Svobodova Varekova R; Koca J; Otyepka M MOLEonline 2.0: interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res. 2012, 40, W222–W227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.