

Abstract
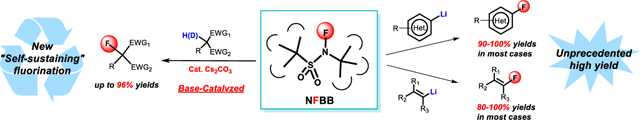
Fluorination of carbanions is pivotal for the synthesis of fluorinated compounds, but the current N-F fluorinating agents have significant drawbacks due to many reactive locations that surround the reactive N-F site. By developing a sterically hindered N-fluorosulfonamide reagent, namely N-fluoro-N-(tert-butyl)-tert-butanesulfonamide (NFBB), we discovered a conceptually novel base-catalyzed, self-sustaining fluorination of active methylene compounds and achieved the high-yielding fluorination of the hitherto difficult highly basic (hetero)aryl and alkenyl lithium species. In the former, the mild and high yield fluorination of active methylene compounds exhibited wide functional group tolerance and its novel catalytic fluorination-deprotonation cycle mechanism was demonstrated by deuterium-tracing experiments. In the latter, NFBB reacted with a variety of highly basic (hetero)aryl and alkenyl lithium species to provide the desired fluoro (hetero)arenes and alkenes in unprecedented high or quantitative yields.
By developing a sterically hindered fluorinating agent, N-fluoro-N-(tert-butyl)-tert-butanesulfonamide (NFBB), we discovered a conceptually novel base-catalyzed, self-sustaining fluorination of active methylene compounds and achieved an unprecedented high-yield fluorination of highly basic (hetero)aryl and alkenyl lithium species.
Keywords: fluorination, catalysis, N-F reagents, active methylene compounds, organolithium species
Graphical Abstract
Introduction
Fluorinated active methylene compounds have recently gained increased attention as hit-to-lead compounds in life-science programs[1] and as versatile building blocks for the synthesis of valuable pharmaceutical products.[2] The fluorination of active methylene compounds is typically made by treating their metal salts with N-F fluorinating agents or using Lewis acidic transition metal catalysts.[3–7] However, there is no report on base catalyzed fluorination approaches to carry out the selective fluorination of active methylene compounds despite the drawbacks of using equimolar amounts of strong bases—which reduce the functional group tolerance of the substrates—or using environmentally unfriendly metals. (Hetero)aryl and alkenyl fluorides are considered key synthetic precursors in medicine,[8,9] agrochemicals,[8a,10] and novel functional materials.[8a,11] The obvious choice for their synthesis are (hetero)aryl- and alkenyl lithium species or Grignard reagents because they are readily available,[12] but methods for the fluorination of highly basic organolithium species have only produced incremental advances in the last 50 years since the first fluorination of phenyl lithium was conducted using dangerous FClO3, which produced fluorobenzene in just 42% yield.[13]
The most commonly used NF reagents, N-fluorobenzenesulfonimide (NFSI)[3] and N-chloromethyl-N-fluoro-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate (Selectfluor™),[4] can fluorinate nucleophiles such as enolate anions, but their performance with strongly basic (hetero)aryl and alkenyl lithium species is subpar, giving unsatisfactory yields in the case of NFSI or failing altogether in the case of Selectfluor.[3–5,14–16] [Figure 1(1)]. We speculated that the shortcomings observed with existing fluorinating reagents were caused by their many reactive sites susceptible to deprotonation or nucleophilic attack by strong bases. Hence, we designed a molecule consisting of the smallest possible fluorine core, -SO2NF-, sandwiched between two bulky inert tert-butyl groups, namely N-fluoro-N-(tert-butyl)-tert-butanesulfonamide (NFBB) [Figure 1(2)]. We expected that the pronounced steric hindrance of the tert-butyl groups would prevent side reactions on the sulfone site (a blockade strategy) and allowed only the fluorine atom to participate in the reaction. Herein we report the high yield synthesis of a readily made, easily handled NFBB reagent and its application in base-catalyzed fluorination of active methylene compounds with a novel self-sustaining fluorination-deprotonation cycle mechanism, and in the high-yielding fluorination of (hetero)aryl and alkenyl lithium species [Figure 1(2)].
Figure 1.

Fluorination of active methylene compounds and (hetero)aryl and alkenyl lithium species
Results and Discussion
To prepare NFBB (1) we needed to overcome two synthetic challenges. The first one was the thermal instability of the C(tBu)-S bond, likely caused by the C-S bond cleavage promoted by the electron-withdrawing NF substituent. For example, it is known that tBuSO2Cl decomposes at room temperature with a half-life ca. 34 h at 35 °C.[17] The second challenge was the anticipated low chemical yield expected in the fluorination of the NFBB precursor 2. Although many N-fluorosulfonamides have been synthesized by fluorination of the corresponding sulfonamides with molecular fluorine (F2) diluted with N2[3,14,18,19], the sulfonamide of an amine possessing a tertiary butyl group has given a very low yield (14%) of the N-F product because the S-N bond of RSO2NHtBu was cleaved during the fluorination.[14] To our delight, we found that the readily available precursor, N-(tert-butyl)-tert-butanesulfonamide[20] (2), was fluorinated with 5% F2/N2 in acetonitrile in the presence of NaF to produce NFBB 1 in a good conversion yield (Scheme 1). This result can be justified by claiming that the pronounced steric hindrance of the two bulky tert-butyl groups permits F2 to react with NH, but blocks attack on the S-N bond.
Scheme 1.

Synthesis of NFBB with molecular fluorine or NFSI
Although F2 is readily generated through electrolysis, and current safety measures make its use appropriate in industrial environments,[8a,21] the use of F2 is rare in standard research laboratories because of its explosiveness and toxicity. Thus, we investigated the synthesis of NFBB using safe alternative fluorine sources. In 2000, it was reported that N-fluoro sulfonamides could be prepared from sulfonamides using the innocuous and readily available NFSI.[22] However, in addition to the low yields reported, the method required large excess of NFSI (3 eq) and KH (6 eq) and dilute conditions (0.1 M). Despite a plethora of optimization efforts, this reaction was regarded as impractical.[23]
After many attempts (see SI), we found that when 2 reacted with a significantly lesser amount of inexpensive NaH in 1,2-dimethoxyethane (DME) in the presence of tetrabutylammonium chloride, followed by fluorination with an almost equimolar amount of NFSI, the reaction produced NFBB in a high isolated yield (85%) after distillation (96% NMR yield before isolation) (Scheme 1). The surprisingly facile distillation of NFBB, high chemical yield and trouble-free preparation make this method suitable for scalable production. NFBB is air and thermally stable, stable to acid and alkali, and has very low oxidation power: NFBB did not decompose with Et3N in a week and did so very slowly with KI; its half-life was ca. 8 h (see SI, p S6–9). These features are in sharp contrast to the current N-F reagents such as NFSI and Selectfluor, which decompose immediately with Et3N (see SI, p S9–10) and KI. NFBB is exceedingly soluble in non-polar and polar organic solvents. The aforementioned properties of NFBB are the results of its special structure: a SO2NF moiety flanked by two very bulky, lipophilic, and σ-electron-donating tert-butyl groups.
The conventional methods for the fluorination of active methylene compounds rely on the fluorination of their sodium or potassium salts, or through direct fluorination, either in the presence or absence of Lewis acid catalysts, with a strong N-F fluorinating agent such as NFSI[3], Selectfluor[4], N-fluoropyridinium salts (NFPY)[5], or N-fluoromethanesulfonimide (Me-NFSI)[6]. For asymmetric fluorination, optically active Lewis acid catalysts containing transition metals such as Zn, Ti, Cu, Ni, Pd, Sc, and Ru were used.[7] The direct, or the Lewis acid-catalyzed fluorination generate a considerably strong acidic environment, which reduce the functional group tolerance.
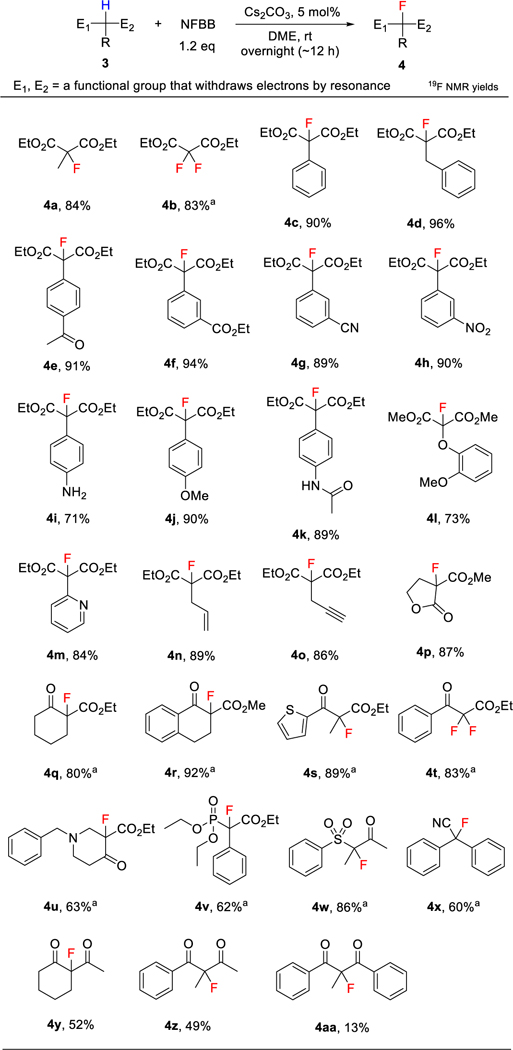
We examined the fluorination of active methylene compounds with NFBB and found that the yields were independent of the amount of the base used and rather increased with lesser amount of the base. We also noticed an effective deprotonating ability of the bulky sulfonamido anion, [tBuSO2NtBu]−, the by-product of NFBB after the fluorination. This behavior is altogether different from another N-F reagent like NFSI, which gives a very stable anion, [PhSO2NSO2Ph]− after fluorination. This observation led to a conceptually new approach, namely, the base-catalyzed “self-sustaining fluorination” of active methylene compounds, by which NFBB acted as both fluorinating and deprotonating agent. We found Cs2CO3 as the best catalyst because of its better solubility in organic solvents and the expected enhanced reactivity of carbanions.[24] The optimal conditions are shown in Table 1. DME proved to be better for this reaction than other solvents such as acetonitrile, dichloromethane, diethyl ether, and toluene.
Table 1.
Cs2CO3-catalyzed fluorination of active methylene compounds with NFBB.
|
10 mol% Cs2CO3 was used. See SI for detailed procedures.
We examined a broad range of malonate derivatives 3a-p and other active methylene compounds 3q-z,aa (Table 1). Diethyl 2-phenylmalonate (3c) was fluorinated in excellent 90% yield. A variety of functional groups on the benzene ring of 3c were tolerated. Acetyl (3e), ethoxycarbonyl (3f), cyano (3g), nitro (3h), methoxy (3j), and acetamido (3k) functional groups on the benzene ring did not lower the yields. It is worth noting that the amino group (3i), which is prone to oxidation and is a strong activator of the aromatic ring, also worked well, furnishing 4i in 71% yield. NFSI and Selectfluor decompose with easily oxidizable aniline (see p S9–10 in SI). Other 2-substituents of malonates such as benzyl (3d), phenoxy (3l), pyridyl (3m), allyl (3n), and propynyl (3o) were tolerated and efficiently converted to the fluoro products in high to excellent yields. Keto esters (3q-u), diketones (3y,z) and other types of active methylene compounds (3v-x) were also suitable for this fluorination reaction. The low fluorination yield (13%) of diketone 3aa could be ascribed to the low reactivity of its salt, a more stabilized anionic species caused by two considerably electron-withdrawing benzoyl groups. Cyclic malonate (3p), cyclic keto esters (3q,r) and thienyl keto ester (3s) produced the desired products in very good yields.
The nipecotic acid derivative (3u), which is a potent inhibitor of γ-aminobutyric acid (GABA) uptake[25], was selectively fluorinated by our method in 63% yield, though it has an easily oxidizable amine moiety. Difluorination of 2,2-unsubstituted malonate (3b) and keto ester (3t) were also obtained in high yields when 2.2 eq of NFBB was used, demonstrating the high applicability of this method. This new method needs only a catalytic amount of a mild and safe base and hence, is highly economical and environmentally friendly.
In our proposed reaction mechanism (Scheme 2), Cs2CO3 deprotonates the active methylene compound I, producing the anionic species II and CsHCO3. Intermediate II reacts with NFBB to produce the fluoro product III and sulfonamido anion species IV, which deprotonates I to regenerate the anion II with sulfonamide V (deprotonation A process) and then resumes the fluorination-deprotonation cycle. In an alternative scenario, the anion IV could deprotonate the newly formed CsHCO3 to regenerate Cs2CO3 (deprotonation B) because CsHCO3 (pKa = ~10 in H2O) is more acidic than the active methylene compound (diethyl 2-ethylmalonate pKa = 15; diethyl malonate pKa = 13.3 in H2O).[26] Cs2CO3 then resumes the fluorination and deprotonation cycle to produce the fluoro product III. However, since the solubility of Cs2CO3 in DME solvent is very low, the actual concentration of resulting CsHCO3 may be very low, whereas that of the active methylene compound I is high because it is soluble, so we expected that IV is the major contributor to the deprotonation of I. The basicity of Cs2CO3 is weaker than that of many active methylene compounds and it is present only in catalytic amounts, so we believe that the relatively strong basic sulfonamido anion (IV) plays the leading role in this efficient Cs2CO3-catalyzed fluorination reaction.
Scheme 2.

Proposed reaction mechanism for Cs2CO3-catalyzed fluorination of active methylene compounds with NFBB
To confirm our proposed fluorination-deprotonation mechanism, we carried out the reaction of 2-deuterated malonate 5 with 87% D purity. As seen in Scheme 3, we found that sulfonamide 6 had 92% D purity by 1H NMR, which clearly supported the fluorination-deprotonation cycle mechanism. Since cesium carbonate (Cs2CO3) is a dibasic substance, the second base, CsHCO3, can participate in the catalytic cycle as discussed above. We investigated whether the presence of CsHCO3 was crucial for the base-catalyzed fluorination process by conducting the reaction using NaH as a mono-basic substance and found that the malonate was fluorinated in good yield (75%) using a catalytic amount of NaH under the same conditions (Scheme 3 bottom).
Scheme 3.

Probing the mechanism of fluorination of active methylene compounds
NaH does not form a second base, only the anionic species II by the reaction of active methylene compound I. This result revealed that deprotonation B process is not the predominant path. The efficient Cs2CO3-catalyzed reaction is the result of a superb combination of the mild fluorination ability of NFBB and the effective deprotonation ability of the resulting bulky sulfonamide base IV. This self-sustaining fluorination could not be applied to less acidic ketone compounds (see detailed discussion and results in SI, p S19).
As discussed in the introduction, the fluorination of (hetero)aryl and alkenyl lithium species has produced unsatisfactory results. With phenyl lithium, NFSI produced phenyl fluoride in only 37% yield,[15b] while Selectfluor failed.[15c] Only two examples in which NFSI delivered substituted phenyl fluorides in higher than 80% yields have been reported, and these occurred when special flow microreactor technology was used.[15d] Conventional fluorinating agents such as NFSI and Selectfluor have many reactive sites (⇒) other than the N-F site (⇒) as seen in Figure 2. In contrast, sterically hindered NFBB has only one available site: the N-F center.
Figure 2.

Reactive sites of NFSI, Selectfluor and sterically hindered NFBB against strong bases such as organolithium species: “⇒” means deprotonation or nucleophilic attack; “⇒” means fluorination reaction.
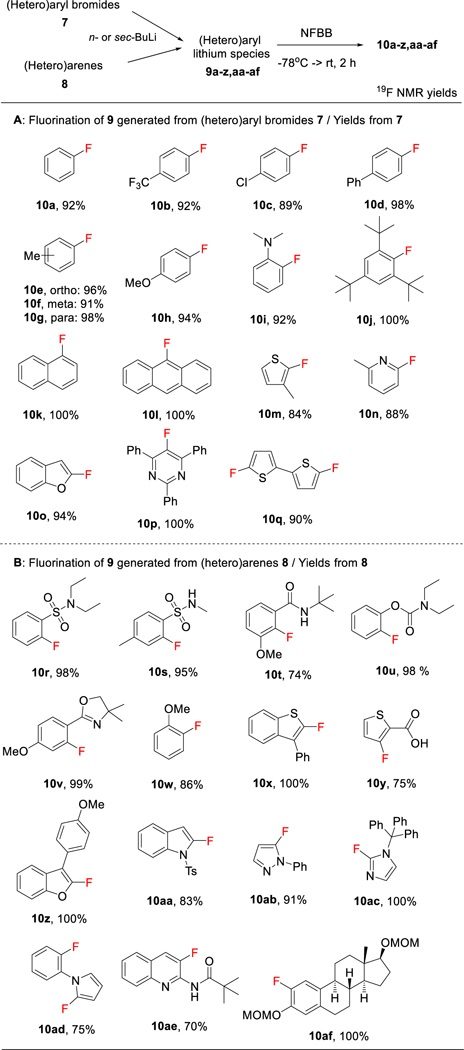
Table 2A illustrates the NFBB fluorination of many (hetero)aryl lithium species 9—prepared in situ from (hetero)aryl bromides 7 with n-BuLi—producing (hetero)aryl fluorides 10a-q in extremely high overall yields. Quantitative yield (100%) or close to it (>90%) were obtained in most cases. Electron-withdrawing (10b,c) and -donating (10e-j) substituents were well tolerated. It should be noted that NFBB was able to fluorinate a site that was hindered by bulky tert- butyl groups in quantitative fashion (10j). Difluorination of dilithio bithiophene proceeded in excellent (90%) yield (10q). The lower yields observed with 10c, m, and n were likely due to the difficulty in generating (hetero)aryl lithium species 9 from bromoarenes 7. Table 2B shows the fluorination of the lithium species 9 generated by the regioselective deprotonation of (hetero)arenes 8 using n- or sec-BuLi. Many fluoro(hetero)arenes were obtained in quantitative yield or close to it. We attributed the lesser yields obtained for 10t, y, aa, ad, and ae to the difficulty in generating the lithium species 9 from the substrates 8. It is worth noting that 10af—a precursor readily derived to bioactive 2-fluoro estradiol—was obtained in quantitative yield by NFBB-fluorination using the MOM-protected estradiol. This method is simple and much better than the two recently reported multi-step methods for the preparation of 2-fluoro estradiol using Selectfluor/Et3N·3HF[27] and CsF under photoactivation.[28]
Table 2.
Fluorination of (hetero)aryl lithium species 9 with NFBB.
|
See SI for detailed procedures.
We conducted a gram-scale preparation of Boc-protected 2-amino-5-fluorothiazole 10ag (Scheme 4 top), an important building block for glucokinase activators that are potential medicaments for Type 2 Diabetes.[29] 2-(N-Boc-amino)thiazole 8ag was treated with 2.2 eq of n-BuLi to yield dilithium 9ag, followed by treatment with NFBB to give the desired 5-fluorinated 2-aminothiazole 10ag in a 90% isolated yield. In contrast, the fluorination of the dilithium 9ag with NFSI gave a mixture of 10ag (70%), 5-phenylsulfonyl-2-(N-Boc-amino)thiazole (15%), and the starting 8ag (10%), from which fluoro product 10ag was isolated in only 36% yield after repeated crystallization.[30]
Scheme 4.

Reactivity of NFBB to lithium species
In 1986, Schwartz first reported the fluorination of alkenyl lithium species—formed in situ from alkenyl iodides with tert-BuLi—with N-fluoro-N-(tert-butyl)benzenesulfonamide at −120 °C.[31] However, a considerable amount of the protonated products formed in this reaction, and protonation became the major reaction when the temperature rose above −120 °C. After NFSI was introduced in 1991, this method, using NFSI, has been applied for the preparation of many biologically attractive alkenyl fluorides.[32] However, these preparations suffered from protonated byproducts or low yields. Although alkenyl bromides 11 can undergo Br/Li exchange with tert-BuLi at −120 °C to generate alkenyl lithium species 12,[33] the conversion of easily available alkenyl bromides to alkenyl fluorides via lithiation has only been reported in few cases, and only in low yields.[34] We have successfully carried out the fluorination of alkenyl lithium species 12 from bromides 11 with NFBB at −90 °C to rt to give the corresponding fluoroalkenes (13a-j) in high yields and in stereo- and regiospecific manner (Scheme 4 middle) (Table 3A).
Table 3.
Fluorination of alkenyl lithium species 12 with NFBB.

|
The data are 19F NMR yields with NFSI reported in ref. [15c]. See SI for detailed procedures.
Arylvinyl fluorides 13a, c, d, and f-h were obtained in excellent overall yields (81–100%) from bromides 11. The main cause for the lower yields of 13b and e was ascribed to the difficulty in generating alkenyl lithium species 12 from 11. Alkylvinyl fluoride 13i was also obtained in excellent yield. Moreover, the fluorinated steroid derivative 13j was obtained in an excellent 90% yield, indicating the high potential of this method in the medicinal field.
In 2013, Altman and coworkers reported the NSFI fluorination of alkenyl lithium species 12—generated in situ by treatment of 2,4,6-triisopropylbenzenesulfonyl hydrazone 15 derived from ketone 14 with n-BuLi (Shapiro reaction).[15c] We treated the alkenyl lithium species 12—prepared in the same way as Altman’s—with NFBB (Scheme 4 bottom) and delivered aryl fluoroalkenes (13k-o) and alkyl fluoroalkenes (13p,q) in remarkably higher yields compared to NFSI (Table 3B),[15c] demonstrating the viability of NFBB for preparing fluoroalkenes. These outstanding results for the fluorination of the organolithium species with NFBB could be attributed to the blockade effect of the two inert bulky tert-butyl groups, as seen in Figure 2. As shown in Scheme 5, we found that NFBB did not react with alkynyl lithium 16 and remained intact (top). This unexpected chemoselectivity was clearly proved by the reaction of dilithium 17, which gave only product 18 fluorinated at the aryl lithium moiety (bottom). This unexpected chemoselectivity cannot be explained by the steric hindrance of NFBB because alkynyl lithium species are sterically smaller than aryl lithium species and NFBB can fluorinate sterically hindered sites as demonstrated in the fluorination of 9j. The chemoselectivity could be attributed to the mild reactivity of NFBB and to the unusual N-F fluorination mechanism, which is still unsolved.[35]
Scheme 5.

Reactivity of NFBB to alkynyl lithium species.
In addition to organolithium compounds, Grignard reagents are also important precursors to (hetero)aryl fluorides. Since there are no systematic reports on the fluorination of traditional standard Grignard reagents without LiCl-mediation, we developed a convenient pathway to serve this purpose. Standard aryl magnesium chlorides were generated via I/Mg exchange in DME with iPrMgCl from the corresponding aryl iodides at rt, or low temperature for electron rich, or electron deficient aromatic derivatives, respectively. Then NFBB, dissolved in twice the volume of tert-butyl methyl ether (TBME), was added and the reaction mixture was stirred at rt, giving the fluorinated products in moderate to high yields (see detailed discussion and results, and Table S5 in SI section). Thus, NFBB demonstrated the usefulness of the standard Grignard reagents.
Conclusion
We synthesized NFBB—a reagent with high fluorine content, stable to air and heat—in high yield via standard distillation. NFBB is a mild and highly selective reagent for fluorination of active methylene compounds through a conceptually new base-catalyzed fluorination-deprotonation which we call “self-sustaining” fluorination. NFBB also fluorinated highly basic (hetero)aryl and alkenyl lithium species in unprecedented high or quantitative yields. NFBB showed extraordinary chemoselectivity against alkynyl lithium species. Standard Grignard reagents were fluorinated by NFBB in good yields and with high functional group tolerance.
Supplementary Material
Acknowledgements
The authors are grateful to the National Institutes of Health (NIGMS R01GM121660) for financially supporting this work.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- [1].Harsanyi A; Sandford G. Org. Process Res. Dev 2014, 18, 981–992. [Google Scholar]
- [2].Massa A. (ed) A special issue for “Recent Advances in the Chemistry of Active Methylene Compounds”, Curr. Org. Chem 2012, Vol. 16, No. 19. [Google Scholar]
- [3].Differding E; Ofner,H.Synlett1991,187–189. [Google Scholar]
- [4].Banks RE; Mohialdin-Khaffaf SN; Lal GS; Sharif I; Syvret RG J. Chem. Soc., Chem. Commun 1992, 595–596. [Google Scholar]
- [5].Umemoto T; Fukami S; Tomizawa G; Harasawa K; Kawada K; Tomita KJ Am. Chem. Soc 1990, 112, 8563–8575. [Google Scholar]
- [6].Fukushi K; Suzuki S; Kamo T; Tokunaga E; Sumii Y; Kagawa T; Kawada K; Shibata N. Green Chem. 2016,18,1864–1868. [Google Scholar]
- [7].Liang T; Newmann CN; Ritter T. Angew. Chem. Int. Ed, 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]
- [8].a) Selected books: Kirsch P. Modern Fluoroorganic Chemistry - Synthesis, Reactivity, Application/2nd edition; Wily-VCH: Weinheim, 2013. [Google Scholar]; b) Ojima I. (ed) Fluorine in Medicinal Chemistry and Chemical Biology; Wily-Blackwell: Chichester, U.K., 2009. [Google Scholar]
- [9].a) Inoue M; Sumii Y; Shibata N. ACS Omega. 2020, 5, 10633–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mei H; Remete AM; Zou Y; Moriwaki H; Fustero S; Kiss L; Soloshonok VA; Han J. Chin. Chem. Lett 2020, 31, 2401–2413. [Google Scholar]; c) Meanwell NA J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- [10].a) Ogawa Y.; Tokunaga E; Kobayashi O; Hirai K; Shibata N. iScience. 2020, 23, 101467. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fujiwara T; O’Hagan D. J. Fluorine Chem 2014, 167, 16–29. [Google Scholar]; c) Jeschke P. ChemBioChem. 2004, 5, 570–589. [DOI] [PubMed] [Google Scholar]
- [11].Berger R; Resnati G; Metrangolo P; Weber E; Hullinger J. Chem. Soc. Rev 2011, 40, 3496–3508. [DOI] [PubMed] [Google Scholar]
- [12].a) Wakerfield BJ Organolithium Methods; Academic Press: London, 1988. [Google Scholar]; b) Snieckus V. Chem. Rev 1990. 90, 879–933. [Google Scholar]; c) Whisler MC; MacNeil S; Snieckus V; Beak P. Angew. Chem. Int. Ed 2004, 43, 2206–2225. [DOI] [PubMed] [Google Scholar]; d) Schlosser M, Angew. Chem. Int. Ed 2005, 44, 376–393. [DOI] [PubMed] [Google Scholar]; e) Brandsma L; Zwikker JW, Sci. Synth, 2006, 8a, 253–270. [Google Scholar]; f) Lai Y-H Synthesis 1981, 8, 585–604. [Google Scholar]; g) Knochel P; Dohle W; Gommermann N; Kneisel FF; Kopp F; Korn T; Sapountzis I; Vu VA Angew. Chem. Int. Ed 2003, 42, 4302–4320. [DOI] [PubMed] [Google Scholar]
- [13].Schlosser M; Heinz G. Chem. Ber 1969, 102, 1944–1953. [Google Scholar]
- [14].Barnette WE J. Am. Chem. Soc 1984, 106, 452–454. [Google Scholar]
- [15].a) Snieckus V; Beaulieu F; Mohri K; Ham W; Murphy CK; Davis FA Tetrahedron Lett. 1994, 35, 3465–3468. [Google Scholar]; b) Davis FA; Han W; Murphy CK J. Org. Chem 1995, 60, 4730–4737. [Google Scholar]; c) Yang M-H; Matikonda SS; Altman RA Org. Lett 2013, 15, 3894–3897. [DOI] [PubMed] [Google Scholar]; d) Nagaki A; Uesugi Y; Kim H; Yoshida J-I Chem. Asian J 2013, 8, 705–708. [DOI] [PubMed] [Google Scholar]
- [16].a) Hu Jingyu; Hu J. (2019) “NFSI and Its Analogs Fluorination for Preparing Aryl Fluorides” In: Hu J; Umemoto T. (eds) Fluorination. Synthetic Organofluorine Chemistry; Springer: Singapore. [Google Scholar]; b) de Azambujia F; Altman RA (2018) “NFSI and Its Analogs Fluorination for Preparing Alkenyl Fluorides” In: Hu J; Umemoto T. (eds) Fluorination. Synthetic Organofluorine Chemistry; Springer: Singapore. [Google Scholar]
- [17].van Aller RT; Scott RB Jr; Brockelbank EL J. Org. Chem 1966, 31, 2357–2365. [Google Scholar]
- [18].Differing E; Lang RW Helv. Chim. Act 1989, 72, 1248–1252. [Google Scholar]
- [19].Umemoto T; Yang Y; Hammond GB Beilstein J. Org. Chem 2021, 17, 1752–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gontcharov AV; Liu H; Sharpless KB Org. Lett 1999, 1, 783–786. [DOI] [PubMed] [Google Scholar]
- [21].Banks RE; Sharp DWA; Tatlow JC (eds) Fluorine: The First Hundred Years (1886–1986); Elsevier: 1986. [Google Scholar]
- [22].Taylor DM; Meier GP Tetrahedron Lett. 2000, 41, 3291–3294. [Google Scholar]
- [23].a) Wang D; Wu L; Wang F; Wan X; Chen P; Lin Z; Liu G. J. Am. Chem. Soc 2017, 139, 6811–6814. [DOI] [PubMed] [Google Scholar]; b) Meyer D; Jangra H; Walther F; Zipse H; Renaud P. Nat. Commun 2018, 9, 4888. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Modak A; Pinter EN; Cook SP J. Am. Chem. Soc 2019, 141, 18405–18410. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bafaluy D; Muñoz-Molina JM; Funes-Ardoiz I; Herold S; de Aguirre AJ; Zhang H; Maseras F; Belderrain TR; Pérez PJ; Muñiz K. Angew. Chem. Int. Ed 2019, 58, 8912–8916. [DOI] [PubMed] [Google Scholar]; e) Zhang Z; Stateman LM; Nagib DA Chem. Sci 2019, 10, 1207–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dijkstra G; Kruizinga WH; Kellogg RM J. Org. Chem 1987, 52, 4230–4234. [Google Scholar]
- [25].a) Shaikh NS; Deshpande VH; Bedekar AV Tetrahedron 2001, 57, 9045–9048. [Google Scholar]; b) Ali FE; Bondinell WE; Dandridge PA; Frazee JS; Garvey E; Girard GR; Kaiser C; Ku TW; Lafferty JJ; Moonsammy GI; Oh H-J; Rush JA; Setler PE; Stringer OD; Venslavsky JW; Volpe BW; Yunger LM; Zirkle CL J. Med. Chem 1985, 28, 653–660. [DOI] [PubMed] [Google Scholar]
- [26].Bell RP, The Proton in Chemistry, Ithica, Cornell University Press, 1959. [Google Scholar]
- [27].Yasui N; Mayne C, Katzenellenbogen G, J. A. Org. Lett 2015, 17, 5540–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tay NES; Chen W; Levens A; Pistritto VA; Huang Z; Wu Z; Li Z; Nicewicz DA Nat. Catal, 2020, 3, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cheruvallath ZS; Gwaltney SL II; Sabat M; Tang M; Wang H; Jennings A; Hosfield D; Lee B; Wu Y; Halkowycz P; Grimshaw CE Bio. & Med. Chem. Lett 2017, 27, 2678–2682. [DOI] [PubMed] [Google Scholar]
- [30].Briner PH; Fyfe MCT; Martin P; Murray PJ; Naud F; Procter MJ, Org. Process Res. Dev 2006, 10, 346–348. [Google Scholar]
- [31].Lee SH; Schwartz JJ Am. Chem. Soc 1986, 108, 2445–2447. [DOI] [PubMed] [Google Scholar]
- [32].a) Selected recent papers: Hamon N; Kaci M; Uttaro J-P; Périgaud C; Mathé C. Eur. J. Med. Chem 2018, 150, 642–654. [DOI] [PubMed] [Google Scholar]; b) Yoon J-S; Jarhad DB; Kim G; Nayak A; Zhao LX; Yu J; Kim H-R; Lee JY; Mulamoottil VA; Chandra G; Byun WS; Lee SK; Kim Y-C; Jeong LS Eur. J. Med, Chem 2018, 155, 406–417. [DOI] [PubMed] [Google Scholar]
- [33].Neumann H; Seebach D. Tetrahedron Lett. 1976, 17, 4839–4842. [Google Scholar]
- [34].a) Li X; Singh SM; Luu-The V; Côté J; Laplante S; Labrie F. Bioorg. Med. Chem 1996, 4, 55–60. [DOI] [PubMed] [Google Scholar]; b) Cappelli A; Galeazzi S; Giuliani G; Anzini M; Donati A; Zetta L; Mendichi R; Aggravi M; Giorgi G; Paccagnini E; Vomero S. Macromolecules. 2007, 40, 3005–3014. [Google Scholar]; c) Medvecky M; Istrate A; Leumann CJ J. Org. Chem 2015, 80, 3556–3565. [DOI] [PubMed] [Google Scholar]
- [35].For the fluorination with N-F fluorinating agents, two reaction mechanisms, SET (single electron transfer) and SN2, were proposed but the long dispute whether either happens continues: see ref.19.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.