

Abstract
Lymphocytic choriomeningitis virus (LCMV) is the prototypic arenavirus and a natural mouse pathogen. LCMV Armstrong, an acutely resolved strain, and LCMV Clone 13, a mutant that establishes chronic infection, have provided contrasting infection models that continue to inform the fundamental biology of T cell differentiation, regulation of exhaustion, and response to checkpoint blockade. Here, we report the isolation and characterization of LCMV Minnesota (LCMV-MN), which was naturally transmitted to laboratory mice upon cohousing with pet shop mice and shares 80–95% amino acid homology with previously characterized LCMV strains. Infection of laboratory mice with purified LCMV-MN resulted in viral persistence that was intermediate between LCMV Armstrong and Clone 13, with widely disseminated viral replication and viremia that was controlled within 15–30 days, unless CD4 T cells were depleted prior to infection. LCMV-MN responding CD8+ T cells biased differentiation towards the recently described PD1+ CXCR5+ Tim-3lo stem-like CD8+ T cell population (also referred to as T exhausted progenitors, Tpex) that effectuates responses to PD-1 blockade checkpoint inhibition, a therapy that rejuvenates responses against chronic infections and cancer. This subset resembled previously characterized PD1+ TCF1+ stem-like CD8+ T cells by transcriptional, phenotypic, and functional assays, yet was atypically abundant. LCMV-MN may provide a tool to better understand the breadth of immune responses in different settings of chronic antigen stimulation as well as the ontogeny of T exhausted progenitors and the regulation of responsiveness to PD-1 blockade.
Keywords: LCMV, Pet shop mice, cohousing, T exhausted progenitors, Chronic antigen, stem-like CD8 T cells
Introduction
Lymphocytic choriomeningitis virus (LCMV), the prototypic Arenavirus, has long been used as a model virus to study virus-host interaction and the regulation of adaptive immunity. The common house mouse (Mus musculus) is a natural reservoir of LCMV. Laboratory, wild and pet rodents including rats, hamsters and guinea pigs are also susceptible to infections. Since its first identification in 1933 from a case of fatal encephalitis, now more than 30 different strains of LCMV have been isolated (1–3). The neurotropic strains usually cause an acute infection that is rapidly controlled by cytotoxic CD8 T cells (4), a typical example being LCMV-Armstrong. Splenic-origin strains cause viscerally disseminated infection throughout the host and can lead to lifelong viral persistence (5). The clone 13 variant isolated from the spleen of wild type LCMV-Armstrong infected carrier mice belongs to this latter group of strains.
Key concepts in T cell mediated immunity including MHC restriction, T cell memory, and functional exhaustion of T cells were enabled by pioneering work in the LCMV model system (6–12), and these concepts have been applied broadly to the regulation of immunity in diverse infections and tumors. The striking difference in biological outcome despite minimal genetic variation (5 nucleotide and 2 amino acid changes) between LCMV Armstrong and Cl13 has been oft-exploited to define the role of antigen-persistence in the regulation of T cell exhaustion and response to inhibitory checkpoint blockade, a common immunotherapy for the treatment of cancer (13). Programmed Death-1 (PD-1) is a co-inhibitory receptor expressed on T cells experiencing chronic antigenic stimulation that regulates excessive T cell responses to minimize self-damage. But PD-1/PD-L1 signaling pathways are often co-opted by cancer cells to induce repressive epigenetic states and muted antigen sensitivity in T cells leading to weakened antitumor functions (14). Antibodies that block PD-1’s engagement with PD-L1 can reinvigorate T cell function and potentiate their proliferation (15). While this restorative proliferation event was initially characterized at the population level, more refined analyses revealed that a subset of exhausted T cells (Tex) are more responsive to PD-1 checkpoint blockade (16). These cells are marked by the expression of transcription factor Tcf1 and also express B cell follicle homing chemokine receptor CXCR5 and have been referred to as progenitor exhausted T cells (Tpex) or stem-like CD8 T cells (17–20). Moreover, these cells maintain high expression of PD-1 but have reduced expression of other inhibitory receptors including Tim-3, 2B4 and LAG-3 in comparison to CXCR5neg terminally exhausted cells (17–21). Defining the ontogeny of Tpex is of interest for understanding the regulation of immunity to chronic infections as well as checkpoint blockade therapy of cancer (22).
We developed a model of cohousing pet shop mice with laboratory mice in order to introduce more natural microbial experience to mice raised under specific pathogen-free (SPF) conditions (23). During the course of these studies, we detected a low frequency of LCMV seropositivity among pet shop mice. Isolation, purification, and sequencing revealed this to be an unreported LCMV strain, hereby referred to as LCMV-MN, reflecting its identification at the University of Minnesota. Here, we report a basic virological and immunological characterization, including the finding that infection of C57Bl/6 mice with LCMV-MN results in particularly abundant Tpex. LCMV-MN may provide a useful tool to advance our understanding of immune responses to persistent antigens.
Materials and Methods
Mice and infections
C57BL/6 (B6) and B6.SJL-PtprcaPepcb/BoyCrCrl (CD45.1) female mice were purchased from The National Cancer Institute and were maintained in specific pathogen free (SPF) conditions at the University of Minnesota. Pet store mice were purchased from various pet stores in the greater Minneapolis-St. Paul metropolitan area. Co-housing of SPF mice with sex-matched pet store partners was performed as described (23) within the University of Minnesota ABSL-3 facility. Mice were infected with the indicated strains of LCMV to characterize viral replication and immune responses within the ABSL-3 facility. The dose and route of infection are as follows: LCMV Armstrong-2X105 plaque-forming units (PFU) via intraperitoneal (i.p.) injection, LCMV Clone13–2X106 PFU via intravenous (i.v.) injection, and LCMV MN-2X106 PFU via i.v. injection. All mice were used in accordance with the Institutional Animal Care and Use Committees guidelines at the University of Minnesota.
LCMV-MN isolation and virus genomic sequencing
Kidneys were isolated from a lab mouse that became seropositive for LCMV virus after cohousing, as determined by Charles River EZ spot Multiplexed Fluorometric ImmunoAssay. Homogenized kidney samples were incubated on Vero cell monolayers for three days and the supernatant was passaged through fresh Vero cells. Evidence of LCMV viral growth was confirmed by indirect immunofluorescence using anti-Nucleoprotein monoclonal antibody (VL-4, (24)). The LCMV containing supernatants was triple plaque purified in BHK-21 cells and the virus isolate was sequenced.
Paired end Illumina sequencing was performed and sequencing reads were imported into Geneious Prime® 2021.1.1. The reads were mapped to the two segments corresponding to NCBI reference genome assembly GCA_004789475 using the Geneious mapper, with the medium sensitivity preset customized to use three iterations of fine tuning and a minimum overlap length of 50bp. Gene and open reading frame (ORF) annotations were lifted from the reference sequences to the consensus sequences from each segment. ORF boundaries were then manually adjusted to reflect the longest potential ORF corresponding to each protein.
Phylogenetic analyses
Protein sequences of strain LCMV-MN were compared to published sequences available for other LCMV strains (Supplementary Table-1: Cl13, Armstrong, Docile, WE, and Traub). The Geneious Prime (2021.2.1) sequence analysis package (Biomatters Ltd) was used to compare the sequence homology of the 4 coding regions (the glycoprotein [S], viral RNA-dependent RNA polymerase [LP], nucleocapsid protein [NP], and Z-protein).
Phylogenetic and molecular evolutionary analyses of the S, LP, and NP coding regions were conducted in Geneious Prime. The Geneious aligner was used for global pairwise alignment with free end gaps and BLOSUM matrices were used to score the alignments. Genetic distances were estimated by using the Jukes-Cantor model and phylogenetic trees were constructed by using the neighbor-joining method. Branch bootstrap values were determined by using 1000 iterations.
Lymphocyte isolation and phenotyping
Lymphocyte isolation from secondary lymphoid organs (SLOs) and non-lymphoid tissues (NLTs) was performed as described (25). Briefly, an intravascular staining method was used to discriminate between cells present in the vasculature from cells in the NLT parenchyma as described previously. Animals were injected i.v. with biotin/fluorochrome-conjugated anti-CD8α through the tail vein. Three minutes’ post-injection, animals were sacrificed, and tissues were harvested as described (26). Lymphoid tissues were mashed using the plunger of a 3-mL syringe and filtered through 70 μm mesh before staining. Indicated non-lymphoid tissues were chopped into small pieces and incubated with RPMI+5% FBS containing collagenase type-IV (0.5mg/ml) and Dnase I (2μg/ml) at 37°C with constant shaking for 30 min-1 hour. After the incubation, tissues were further dissociated using a gentleMACS dissociator (Miltenyi Biotec) and filtered twice through a 70 μm mesh before staining.
Isolated lymphocytes were surface-stained with antibodies specific for CD3 (145–2C11), CD45 (30F-11), CD45.1 (A20), MHC II (Ia-Ie) (M5/114.15.2), CD8α (53–6.7), CD8β (YTS156.7.7), CD45.2 (104), CD62L (MEL-14), CD44 (IM7), CD69 (H1.2F3), CD103 (M290), CX3CR1 (SA011F11), CCR7 (4B12), CD160 (7H1), CXCR3 (CXCR3–173), KLRG1 (2F1), CD28 (37.51), CD127 (A7R34), CD27 (LG.3A10), Ly6C (HK1.4), CD11a (M17/4), CD122 (TM-β1), CD49a (Ha31/8), CD5 (57–7.3), PD1 (RMP1–30), Tbet (4B10), Eomes (Dan11mag), TCF1 (S33–966), and Ki67 (B56). The above antibodies were purchased from BD Biosciences, Biolegend or Affymetrix eBiosciences. Cell viability was determined using Ghost Dye 780 (Tonbo Biosciences). Antibody against granzyme-B (GB11) was purchased from Thermofisher. For intracellular transcription factor, granzyme-B and Ki67 staining, the eBioscience FoxP3/Transcription factor staining buffer set was utilized. Stained samples were acquired using LSRII or LSR Fortessa flow cytometers (BD) and analyzed with FlowJo software (Treestar).
Tissue freezing, tissue clearing and immunofluorescence staining
For immunohistochemistry, spleen and lymph nodes were fixed overnight in 2% paraformaldehyde at 4°C, dehydrated in 30% sucrose, and embedded in optimal cutting temperature compound (OCT). 7µm sections were cut, dried, and stained with unconjugated anti-LCMV NP antibody (VL-4, BioXCell), followed by secondary staining with goat anti-rat IgG AF488 (Jackson Immuno Research), and then additional stains were performed with the following conjugated antibodies: F4/80 efluor 570 (eBioscience), CD3 BV421 (Biolegend), and B220 BV480 (BD Horizon). Each staining step was performed for 1h at room temperature. Images were captured by confocal microscopy using the Leica Stellaris 8 at 40x final magnification, using a 20x objective (NA = 0.75).
The tissue clearing procedure was modified based on the protocol described elsewhere (26). Briefly, lymph nodes were fixed in 1% BD cytofix for 24h at 4°C. Tissues were then incubated in blocking buffer (1% BSA, 0.3% Triton X-100, and 1:100 mouse Fc block) for 24h, stained in blocking buffer containing anti-NP CF633, F4/80 ef570 and B220 BV480 for 3d, and washed in PBS containing 0.3% Triton X-100 and 0.5% 1-thioglycerol. Conjugation of VL-4 with CF633 was performed according to manufacturer recommendations (Biotium). The block, stain, and wash steps were performed at 37°C, and at 100 rpm. Tissues were cleared in clearing media containing 0.1% Triton X-100, 0.5% 1-thioglycerol in 86% (w/v) histodenz (Sigma) prepared in 40% (v/v) N-methylacetamide to achieve a final refractive index of 1.49–1.5. Clearing was performed for 8–24h at room temperature prior to image acquisition.
Confocal image acquisition and analysis
Stained and cleared tissues were embedded in clearing media in a 2.5mm silicone gel spacer (Electron Microscopy Sciences) on a microscope slide and sealed with a cover slip. Confocal imaging was performed on an inverted SP8 Stellaris microscope (Leica Microsystems) using a 20× 0.6NA, 1.5mm working distance objective. 512×512 voxel density at 2x optical zoom was used at 4um z-step to perform image acquisition. Collected images were analyzed using Imaris 9 (Bitplane Scientific Software).
Cell sorting, adoptive transfer and RNA preparation
Live CD8a+ H2/Db-gp33 tetramer+ cell populations were isolated from spleen and lymph nodes of LCMV-MN infected mice and separated into Slamf6+/Tim3− and Slamf6-/Tim3+ populations by sorting on a BD FACSAria II. For adoptive transfer, equivalent numbers of sorted cells were intravenously injected into congenically distinct recipient mice as indicated in the results section. The recipient mice were either infected with LCMV-MN soon after or treated with PD-L1 neutralizing antibodies as described in the results section. For PD-L1 blocking, animals were injected with 200 ug of anti-PD-L1 blocking antibody (clone 10F.9G2, Bioxcell) every 3 days. For RNA isolation, sorted cells were homogenized using QIAshredder columns (QIAGEN) and RNA was extracted using a RNeasy micro kit (QIAGEN) per the manufacturer’s instructions. RNA integrity was assessed using capillary electrophoresis with the Agilent 2100 BioAnalyzer system (Agilent Technologies, Santa Clara, CA, USA) which generated an RNA Integrity Number (RIN). Samples with an RNA Integrity Number (RIN) >7 were included in this study. Three biological replicates were included for each cell population.
Library preparation, RNA sequencing and data processing
Sequencing libraries were prepared using the Clontech SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian kit. RNA sequencing (50-bp single-end) with the HiSeq 2500 Illumina was done at the University of Minnesota Genomics Center (UMGC). The sequences were processed with the Collection of Hierarchical UMII-RIS Pipelines (CHURP, v 0.2.2) developed by the Research Informatics Solutions (RIS) group at the Minnesota Supercomputing Institute. The reference implementation of the CHURP package is at: https://github.com/msi-ris/CHURP.git. (27). The Mouse mm10 reference was used for mapping the sequences.
Differential gene expression (DGE) and GSEA enrichment analyses
Within the CHURP pipeline, sequence reads were counted with featureCounts v.1.6.2 and the Empirical analysis of DGE (EDGE) tool was used to identify differentially expressed genes between the Slamf6+/Tim3− and Slamf6-/Tim3+ cell populations (28). Genes were considered differentially expressed if they had an FDR p value ≤ 0.05 and an absolute log fold change ≥ 1.5 (Supplemental Table-2).
A Tpex related ranked gene list was obtained from the output of a GSEA enrichment test (GSEA v3.0 [build:0160]) comparing previously published progenitor exhausted to terminally exhausted CD8 T cells that were generated with LCMV-Cl13 (GSE84105) (17). The limma barcodeplot function was used to plot enrichment of the up-regulated and down-regulated LCMV-MN Slamf6+/Tim3− specific genes in the ranked Tpex gene list.
scRNA-seq analyses comparing Slamf6+ cells 30 days after infection with LCMV-MN to Tpex at early time point after infection with LCMV-Cl13
Single cell transcriptomes for P14 transgenic CD8 T cells (specific for the immunodominant LCMV epitope gp33) collected from mice days 4.5 and 7 after infection by acute (LCMV-Armstrong) or chronic (LCMV-Cl13) were downloaded from GEO. (GSE119940; (29)). An integrated data set of these transcriptomes was created with Seurat v4.0 (30). Cells with a percentage of mitochondrial genes below 0.05% were included. Outlier cells expressing less than 1,000 or greater than 5,000 genes were removed and cells with an RNA library size between 1,000 and 20,000 UMI counts were retained.
The Seurat Convert() function was used to create an AnnData object from the integrated Seurat object for analyses with scvi-tools (single-cell variational inference tools). Prior to modeling with scvi, an initial gene filter removed genes expressed in fewer than four cells and the top 4,000 highly variable genes (HVGs) were selected by the Seurat v3 method (29) as implemented by scvi (30). Scvi normalized gene expression values were used for generating t-distributed stochastic neighborhood embedding (tSNE) plots. Gene scores were computed based on the Scanpy implementation of the Seurat method for scoring genes (31) and then overlayed onto tSNE plots. Upregulated LCMV-MN Slamf6+/Tim3− genes were used for scoring the single cells (Supplemental Table-2). Genes upregulated in d4.5 memory precursor (Mpc) and genes upregulated in d7 LCMV Cl13 Tpex were previously reported (29).
Statistics
If the samples followed normal distribution, then parametric tests (unpaired two-tailed Student’s t-test for two groups and one-way ANOVA with Tukey’s multiple comparison test for more than two groups) were used. Two-way ANOVA with Sidak’s multiple comparison test was used if the effect of two independent variables were being considered among more than two sample groups. If the samples deviated from a Gaussian distribution, non-parametric tests (Mann–Whitney U test for two groups, Kruskal–Wallis with Dunn’s multiple comparison test for more than two groups) were used unless otherwise stated in the figure legend. For paired analyses not conforming to Gaussian distribution, Wilcoxon matched-pair signed rank test was used. D’Agostino and Pearson omnibus normality test was used to determine whether samples adhered to Gaussian distribution or not. Variances between groups were compared using an F test and found to be equal. All statistical analysis was done in GraphPad Prism (GraphPad Software Inc.). p < 0.05 was considered significant.
Data availability
The RNA-sequencing data described in this study have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) and can be downloaded with accession number GSE207983. Public data sets were downloaded from GEO (GSE84105 and Series GSE119940) (https://www.ncbi.nlm.nih.gov/geo/).
Results
Isolation and genomic sequence characterization of LCMV-MN
We purchased mice from local pet shops in the Minneapolis/St. Paul, Minnesota metropolitan area, and co-housed them with adult SPF C57Bl/6 mice within an ABSL-3 facility. A routine serological screen detected evidence of LCMV infection in a single pet shop mouse (subsequently observed in 6% of pet shop mice) (32) and laboratory mice also became seropositive upon co-housing (data not shown). Seventy days after co-housing, we homogenized kidneys from a C57Bl/6 cage mate, incubated clarified homogenate with susceptible Vero cells, and confirmed LCMV presence via indirect immunofluorescence microscopy with a monoclonal (VL4) antibody specific for LCMV nucleoprotein (24) (Fig. 1A and data not shown). Viral supernatant was triple-plaque purified in BHK-21 cells, and a master stock was established for further characterization. To assess the in vitro growth characteristics of this newly isolated virus, named LCMV-MN in reference to it being isolated at the University of Minnesota, we infected BHK-21 cells at 0.01 multiplicity of infection (MOI). LCMV Cl13 and Armstrong strain infections were used for comparison purposes. Virus growth was measured by titration of virus present in tissue culture supernatants at the indicated time post-infection. All viruses tested showed similar growth properties in BHK-21 cells (Fig. 1B). LCMV-MN produced well-defined plaques on Vero cell monolayers with size and appearance more similar to LCMV-Armstrong than LCMV Cl13 (Fig. 1C).
Figure 1. Isolation, in vitro growth and genomic characterization of LCMV-MN.

(A) Schematic of the process depicting isolation of a novel LCMV like virus from kidney homogenates of C57Bl/6 mice that were cohoused with petstore mice. The initial virus stock was confirmed via positive reactivity to LCMV-N monoclonal antibody (VL-4) and was triple plaque purified to establish final stock. (B) Comparative growth kinetics among LCMV-Armstrong, -Cl13 and -MN showing viral output from BHK-21 cells infected at an MOI of 0.1. Supernatant was collected at times indicated and titered on Vero cells using plaque assay. (C) Plaque morphology of the indicated viruses on Vero cells. (D) Amino acid percentage homology among various established LCMV strains and LCMV-MN across the four viral proteins (indicated at the top of the respective checkered box). The scale for each of the protein comparisons is on the right of the panel and represents percent identity. (E) Comparison of amino acid sequences across select stretched of glycoprotein (top) and polymerase protein (bottom) with a particular emphasis on conservation of GP260 and LP1079 residues among LCMV-MN and indicated strains. (F) Phylogenetic tree analysis output of the 4 proteins of LCMV-MN and other LCMV strains based on neighbor-joining method. Data in B are representative of two separate experiments with 3 replicate each per time point. Plaque morphology in ‘D’ is representative of 3 independent repeats. Bars indicate mean ± SEM. Figure in A created with Biorender (Biorender.com).
The viral stock was sequenced using paired-end Illumina sequencing and reads were mapped to NCBI reference genome assembly GCA_004789475. Gene and ORF annotations were based on reference sequences. The sequence was compared to all LCMV strains for which a complete sequence was available in the NCBI database, LCMV Armstrong, Clone 13, WE, Traub, and Docile (Supplementary Table-1). Amino acid sequences of the four proteins shared 80–95% homology, indicating that this isolate represents a previously uncharacterized strain (Fig. 1D). LCMV-MN was most closely related to LCMV-Docile, although still exhibited nonsynonymous variation in all proteins. Previous work has identified two of the amino acid residues that influence LCMV persistence. A leucine at position 260 of the viral glycoprotein GP mediates high-affinity virus binding to the host alpha-dystroglycan receptor resulting in establishment of persistent infection by clone 13 (33, 34). The lysine to glutamine mutation in residue 1079 of polymerase protein has also been noted in several strains that induce persistent infections. Other variations that have not been definitively characterized, including in the Z protein, may also influence viral persistence. LCMV-MN shared the same GP260 residue as Cl13, but the same L1079 residue as Armstrong, a feature in common with other LCMV strains (Fig. 1E). Phylogenetic analysis of 4 proteins, placed LCMV-MN between WE and Traub, but with a greater divergence from these strains than the divergence between Arm and Cl13 (Fig. 1F).
Weight loss and in vivo growth kinetics of LCMV-MN
We next challenged SPF C57Bl/6 mice with 2×106pfu LCMV-MN i.v. and evaluated weight loss and viral load in blood and tissues. Comparisons were made to LCMV Arm (2×105 pfu i.p.) and LCMV Cl13 (2X106 pfu i.v.). Cl13, but not LCMV Arm, induced protracted weight loss. In contrast, LCMV-MN induced significant, yet transient, weight loss that recovered by approximately two weeks after infection (Fig. 2A).
Figure 2. In vivo growth characteristics of LCMV-MN.
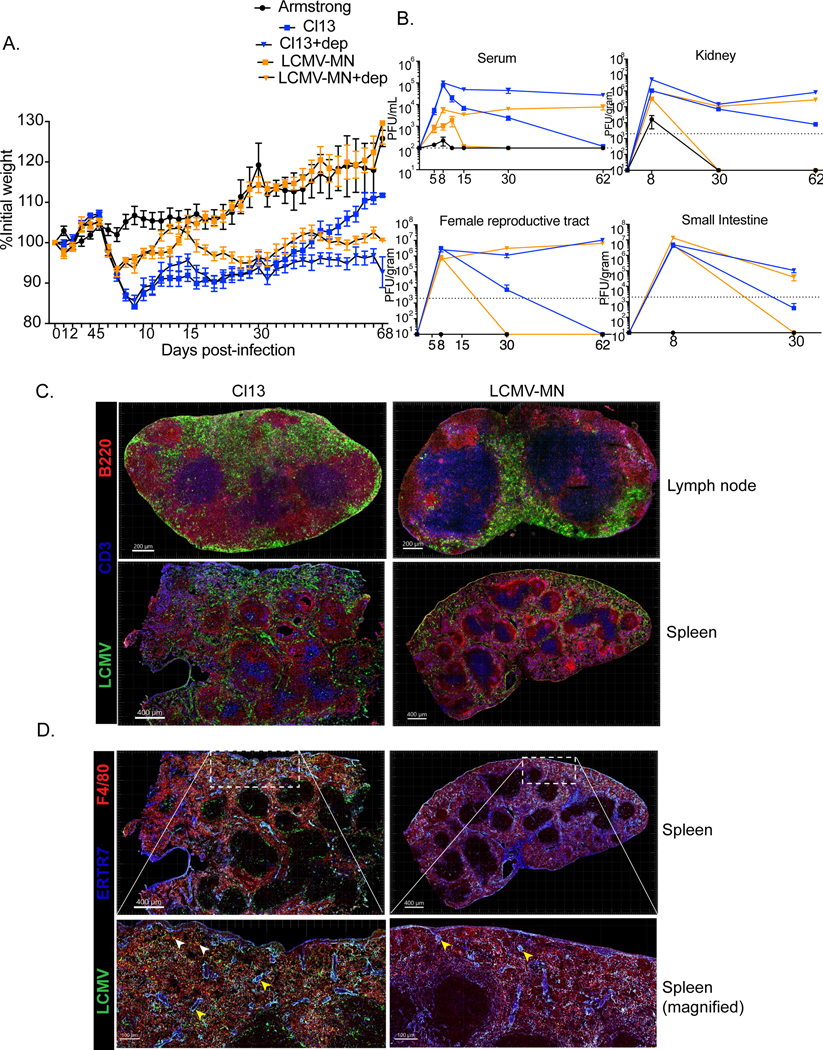
C57Bl/6 mice were either treated with anti-CD4 antibody (0.5mg/mouse 1 day before and 1 day after infection) or remained untreated and were infected with 2 X106 PFU of LCMV-MN or 2 X106 PFU of LCMV-Cl13 i.v. Another group of mice were infected with 2X105 PFU of LCMV-Armstrong i.p. to establish acute infection. (A) Kinetics of weight loss among the 5 groups of mice during the first 68 days post-infection. Weight measurement was done every day for the first 15 days and then every 2–3 days. (B) Kinetic analysis of virus titer in serum (in PFU/ml), kidney, female reproductive tract and small intestine (all in PFU per gram of tissue) for the 5 separate infections. The limit of detection is indicated by the dashed line. (C&D) Mice were infected with LCMV-Cl13 or LCMV-MN and then examined 35 days later by immunofluorescence microscopy. (C) Representative images of lymph node (top) and spleen (bottom) (LCMV-nucleoprotein, green; CD3, blue; B220, red). (D) Comparison of LCMV nucleoprotein distribution among splenic fibroblastic reticular cells and macrophages (LCMV-nucleoprotein, green; ERTR7, blue; F480, red). Yellow arrowhead-colocalization of LCMV-nucleoprotein and ERTR7 signal. White arrowhead- colocalization of LCMV-nucleoprotein and F480 signal. Data are representative of two separate experiments with either n=5 mice/group per experiment (A and B) or n=3 mice/group per experiment (C and D). Bars indicate mean ± SEM.
CD4 T cells are sometimes experimentally depleted before LCMV Cl13 infection to establish durable viremia, so we tested the impact of CD4 depletion prior to LCMV-MN infection (35). In this context, LCMV-MN infection resulted in durable weight loss almost as severe as observed in CD4 T cell-depleted mice infected with LCMV Cl13 (Fig. 2A). CD4 T cell depletion also resulted in durable viremia after LCMV-MN infection. However, LCMV-MN was largely cleared from serum of undepleted mice by 15 days post infection, and undetectable by 30 days post infection. Unlike LCMV Cl13, LCMV-MN was also cleared from visceral organs in undepleted mice, although virus persisted much longer than observed after LCMV Arm infection (Fig. 2B). Collectively, these data indicate that LCMV-MN induced intermediate weight loss, viral load, and persistence compared to LCMV Arm and Cl13, however LCMV-MN chronically infected visceral organs when CD4 T cells were depleted prior to infection.
We then assessed thin sections of spleen and lymph node for distribution of viral protein by staining with anti-LCMV-NP antibody, anti-CD3 (T cells), and anti-B220 (B cells) 35 days after infection with LCMV Cl13 or LCMV-MN. We also performed similar staining after clearing-enhanced 3D microscopy of unsectioned lymph node tissue. Viral protein persisted after both LCMV-Cl13 and MN infections. The LCMV-NP signal was distributed across the subcapsular and medullary sinus as well as interfollicular zones in Cl13 infected lymph nodes (Fig. 2D). In contrast, the subcapsular sinus was largely devoid of nucleoprotein signal in LCMV-MN infected mice. 3D imaging further supported this distinct viral distribution (Video-1 and 2). LCMV Cl13 infected spleen displayed a distinctive reticular pattern of NP antigen distribution as described previously with prominent signals throughout the marginal zone, white pulp and red pulp (36). The viral antigen signal was much more muted in LCMV-MN infected spleen and limited to marginal zone and red pulp (Fig. 2D). To investigate if LCMV-MN targets distinct cell types compared to Cl13 we stained splenic sections with the fibroblastic reticular cell (FRC) marker ERTR7 and the macrophage specific marker F4/80, as both of these cell types are known to be infected by LCMV (34, 36). Whereas LCMV nucleoprotein signal was found in both FRCs and F4/80+ cells in Cl13 infected spleens, it was only detected in the FRCs of LCMV-MN infected spleens (Fig. 2D). Taken together, these data indicate that LCMV Cl13 and LCMV-MN infections result in differences in the magnitude, duration, and distribution of viral antigens and/or replication.
Dynamics and function of CD8 and CD4 T cell response to LCMV-MN infection
C57Bl/6 mice were infected with three strains of LCMV and the frequency of CD8 T cells to two dominant epitopes, H-2Db/gp33 and H-2Db/gp276, were measured in peripheral blood with MHC I tetramers (Fig. 3A, B and E). Each infection induced distinct kinetics, with the response to LCMV-MN not peaking until approximately two weeks after infection, and the response remained robust throughout the 62 days of analysis. However, the CD4 T cell response to the I-Ab-gp66 epitope was similar regardless of LCMV strain (Fig. 3C, D and F).
Figure 3. Dynamics of CD8 and CD4 T cell responses to LCMV infections.

C57Bl/6 mice were infected with the 3 indicated strains of LCMV. (A) The frequency of H-2Db/gp33-specific CD8 T cells were measured in peripheral blood after indicated days post-infection. Plots gated on live CD8α+ lymphocytes. (B) The kinetics of H-2Db/gp33 and H-2Db/gp276-specific CD8 T cell response in peripheral blood. Representative flow plots showing GP66–77: I-Ab MHC II tetramer+ CD4 T cells in blood (C) and their kinetics (D) is shown. Plots gated on live B220-CD8-F480-CD11c- CD4+ T cells. (E, F) Enumeration of H-2Db/gp33-specific CD8 T cells and GP66–77: I-Ab MHC II tetramer+ CD4 T cells in spleen at day 30 post indicated virus infection. Data are representative of three separate experiments with n=3 mice/group per experiment (A, B and E) or 2 independent experiment with n=3 mice/group per experiment (C, D and F). Bars indicate mean ± SEM. *p <0.05, **p <0.001, ns-not significant. Kruskal Wallis one-way ANOVA with Dunn’s multiple comparison test.
Chronic LCMV infection is associated with a hierarchical loss of CD8 T cell-derived IL-2, TNFα, and IFNγ cytokine production. IL-2 production is impaired most significantly, whereas IFNγ production is maintained unless T cells become severely dysfunctional in the presence of sustained high levels of antigen stimulation (8). When we assessed the ability to express cytokines in response to 4 hours of peptide restimulation, LCMV-MN specific CD8 T cells exhibited intermediate functionality compared to LCMV Arm and Cl13, with a partial defect in IL-2 production and reduced mean fluorescent intensity (MFI) of IFNγ staining (Fig. 4A-D). The ability to degranulate, as measured by intracellular CD107a staining in the presence of brefeldin A and monensin, was unimpaired. Next, eight or 30 days after infection, we compared the percent of CD8 T cells that stained with H-2Db/gp33 tetramer to the percent that expressed cytokines after 4 hours of in vitro peptide stimulation. As shown in Fig.4E both LCMV-Cl13 and -MN gp33-specific CD8 T cells exhibited a reduced ability to express IFN-γ and TNFα by 30 days post infection, unlike LCMV-Arm infected animals. Cytokine production by CD4 T cells appeared similar after infection with each LCMV strain 31 days post infection (Fig. 4E). Collectively these data indicate that although the LCMV-MN infection generated a higher magnitude H-2Db/gp33-specific CD8 T cell response compared to Arm and Cl13 infection, functionality was somewhat compromised. However, the CD4 T cell responses were comparable among the infections.
Figure 4. Function of CD8 and CD4 T cell response to LCMV infections.

(A) Splenocytes from C57Bl/6 mice infected 30 days prior with the 3 strains of LCMV were ex vivo stimulated with gp33–41 peptide. Representative flow plots depicting the frequency of indicated cytokines and degranulation marker (CD107a) positive cells are shown. Plots are gated on live CD8α+ cells. (B) Kinetics of IFN-γ+ CD8 T cells in spleen after gp33–41 peptide stimulation. (C) Geometric mean fluorescence intensity (gMFI) of IFN-γ expression on responding cells from spleen at day 30 post indicated viral infection. (D) Frequency of IL-2 secreting CD8 T cells among splenocytes at day 30 post indicated viral infection. (E) Ratio of splenic IFN-γ and TNF-α producing cells to gp33 tetramer+ CD8 T cells at day 8 and day 30 post indicated viral infection. (F) Splenocytes from C57Bl/6 mice infected 8- or 31-days prior with the 3 strains of LCMV were ex vivo stimulated with LCMV gp66–77 peptide. Percentage of CD4 T cells producing IFN-γ, IL-2 and TNF-α is shown. Data are representative of two separate experiments with n=3 mice/group per experiment per group. Bars indicate mean ± SEM. *p <0.05, **p <0.001, ***p <0.0001, ns-not significant. Kruskal Wallis one-way ANOVA with Dunn’s multiple comparison test.
Antigen-specific CD8 T cell differentiation in visceral tissues after LCMV infections
We compared H-2Db/gp33-specific CD8 T cell nonlymphoid tissue distribution and differentiation 30 days after infection with LCMV Arm, Cl13, or MN. As previously reported (37), compared to LCMV Arm, LCMV Cl13 infection resulted in significantly more virus specific T cells that could be isolated from the female reproductive tract and kidney, yet no increase in the small intestine intraepithelial lymphocytes (SI-IEL, Fig. 5A-B). Although LCMV-MN maintained the largest number of H-2Db/gp33-specific CD8 T cells in spleen, that did not extend to nonlymphoid organs. Most nonlymphoid tissues, particularly small intestine epithelium, are dominated by resident memory CD8 T cells (TRM) after acute infections (38). CD8+ SI-IEL TRM have a well-defined and relatively homogenous phenotype after LCMV Arm infection, most notably expressing CD69 and CD103 (39–41). As shown in Fig. 5C, LCMV-MN induced SI-IEL that were impaired in CD103 expression, and a discrete subset that also lacked CD69. This CD69neg CD103neg population (i.e., lacking conventional residence markers) expressed high levels of CD127 and Tim-3, a moderate level of PD-1, and low levels of Lag-3. Thus, LCMV-MN infection exhibited intermediate persistence relative to LCMV Arm and Cl13, it resulted in a phenotypically distinct T cell population within the intestinal epithelium that was rare after both LCMV Arm or Cl13 infections. Whether the noncanonical population represented recent migrants or a transitional state of differentiation would be difficult to assess at this time. However, these observations directed our attention to testing for the presence of the recently described Tpex subset that putatively provides a lymphoid reservoir of cells with the potential to produce differentiated migrating progeny.
Figure 5. Antigen-specific CD8 T cell differentiation in visceral tissues after LCMV infections.

(A) Frequency of H-2Db/gp33-specific CD8 T cells that were present in the extravascular space (protected from intravenous CD8α antibodies) were measured in 2 barrier mucosal tissues and kidneys at 30 days post-infection. Plots are gated on live CD8β+ T cells (CD8a IV-). SI-IEL: small intestine intraepithelial lymphocytes and FRT: female reproductive tract. (B) Enumeration of H-2Db/gp33-specific CD8 T cells in indicated organs. (C) Representative flow plots depicting expression of number of inhibitory receptors, memory markers as well as resident memory T cell marker on H-2Db/gp33-specific CD8 T cells in small intestine IEL. Plots are gated on tetramer+ live CD8β+ T cells. Data are representative of two separate experiments with n=3 mice/group per experiment. Bars indicate mean ± SEM. ***p <0.0001, ns-not significant. Kruskal Wallis one-way ANOVA with Dunn’s multiple comparison test.
LCMV-MN infection enhances development of Slamf6+ progenitor exhausted CD8 T cells
Tpex, characterized by a CXCR5+/Tim-3- phenotype, are localized to spleen and lymph nodes (17–19). This population is largely responsible for maintaining an ongoing response during persistent antigen stimulation and responding most potently to checkpoint inhibition. After PD-1/PD-L1 checkpoint blockade, Tpex proliferate and give rise to a transitional Tim-3- population that migrates to infected sites or tumors, prior to terminal differentiation and more pronounced exhaustion (17, 19, 21, 42). Given the overabundance of a potentially transitional T cell population in the small intestine and virus-specific T cells in spleen after LCMV-MN (Fig. 5 and 3), we examined the presence of Tpex-phenotype cells. LCMV-MN infection induced at least 10-fold more Tpex-phenotype CD8 T splenocytes as compared to either LCMV Cl13 or Armstrong and this population was sustained for at least 60 days (Fig. 6A-B). These CXCR5+/Tim-3lo cells were also highly enriched for Slamf6 expression, as observed in other model systems (43). Further interrogation of H-2Db/gp33-specific cells revealed that the Slamf6+ subset matched previously described Tpex expression profiling of transcription factors, and markers of memory T cell differentiation, effector function, and migration (Fig. 6C). A curious exception is that, unlike previously noted Tpex, this subset contained CD62L+ CD8 T cells.
Figure 6. LCMV-MN infection enhances development of Slamf6+ progenitor exhausted CD8 T cells.

(A) Representative flow plots depicting H-2Db/gp33-specific CD8 T cell frequency (top), expression of Tpex markers on antigen-specific CD8 T cells (middle and bottom) in spleen of animals infected with indicated LCMV strains 30 days before. Top panel is gated on live CD8 T cells whereas the bottom two panels are gated on gp33 tetramer+ CD8 T cells in spleen. (B) Kinetics of Tpex CD8 T cells in spleen (top) and their numbers at day 30 and day 60 post-infection (bottom). (C) Representative flow plots showing expression of various phenotypic markers associated with the progenitor population in LCMV-MN infected animal (30 days post-infection). Plots are gated on gp33 tetramer+ CD8 T cells in spleen. (D) Congenically marked PD1+ Slamf6+ Tim3− and PD1+ Slamf6- Tim3+ CD8 T cells from LCMV-MN infected mice (Day 30 PI) were isolated by cell sorting and transferred i.v. to 2 groups of congenically distinct naive mice followed by LCMV-MN infection. Representative flow plots depicting expansion of transferred cells in blood on indicated days and their frequency and phenotype in spleen at 14 days post-infection. Data are representative of two separate experiments with n=4–5 mice/group per experiment per group. Bars indicate mean ± SEM. **p <0.001, ns-not significant. Kruskal Wallis one-way ANOVA with Dunn’s multiple comparison test.
By phenotypic criteria, these data indicated that LCMV-MN induces a preponderance of Tpex within 30–60 days after infection, compared to LCMV Cl13. Functionally, Tpex are defined in part by the enhanced ability to proliferate and differentiate in response to further antigen stimulation. To test this property, we sorted Slamf6+/Tim3− (Tpex phenotype) vs. Slamf6-/Tim3+ (terminal/exhausted phenotype) CD45.1+ H-2Db/gp33-specific CD8 T cells after LCMV-MN and transferred them i.v. into naïve CD45.2+ C57Bl/6 mice. Recipients were bled the following day immediately prior to infection (day 0) with LCMV-MN. The ensuing response was monitored in blood and spleen, and donor cells were identified by CD45.1 staining (which were almost entirely H-2Db/gp33-tetramer staining positive, not shown). As shown in figure 6D, the Slamf6+/Tim3− subset exhibited the Tpex property of enhanced proliferation potential. Moreover, the Slamf6+/Tim3− subset uniquely had the capacity to self-renew Tpex-phenotype CD8 T cells as well as give rise to more terminally exhausted-phenotype progeny. These data suggest that LCMV-MN induces abundant T cells that conform to phenotypic and functional properties previously defined for Tpex.
LCMV-MN induced Slamf6+/Tim3− CD8 T cells transcriptionally resemble Tpex and expand in response to PD-L1 blockade
To compare the transcriptional program of LCMV-MN induced Slamf6+/Tim3− CD8 T cells with previously described Tpex, we performed RNAseq on sorted H-2Db/gp33-tetramer subsets isolated from spleen and LNs 30 days after infection. We identified 146 up-regulated and 152 down-regulated genes in the Slamf6+/Tim3− population relative to the Slamf6-/Tim3+ population (Supplemental Table-2). These differentially expressed genes were enriched for previously described Tpex genes (Fig.7A) (17). As shown in Figure 7B, the LCMV-MN Slamf6+/Tim3 population had similar up- or down-regulation of many previously reported Tpex genes (17). For example, LCMV-MN induced Slamf6+/Tim3− CD8 T cells showed upregulation of Tcf7, Id3, and P2rx7 and downregulation of Id2, Gzma, Prdm1, and Entpd1 (which encodes CD39). Additionally, transcription factors associated with a Tpex program such as Tox, Bach2, and Hif1a were up-regulated in the LCMV-MN Slamf6+/Tim3 population (Fig 7C).
Figure 7. LCMV-MN induced Slamf6+/Tim3− CD8 T cells transcriptionally resemble Tpex and expand in response to PD-L1 blockade.

RNA-seq sequencing and differential analyses were performed to compare LCMV-MN induced Slamf6+/Tim3− (Slamf6+) and Slamf6-Tim3+ (Tim3+) CD8 T cells 30 days post-infection. (A) GSEA analysis showing enrichment of Tpex progenitor specific transcripts in LCMV-MN induced Slamf6+/Tim3− cells based on a ranked list of genes from a previous study of Tpex (progenitor exhausted T cells) and terminal exhausted T cells generated >45 days after infection with LCMV-Cl13. (GSE84105,(17)). (B) Relative gene expression heatmaps depicting 50 upregulated and 50 downregulated genes that best distinguish Tpex from terminal exhausted T cells (an asterisk (*) indicates that the gene was significantly differentially expressed in LCMV-MN Slamf6+ cells). (C) Relative gene expression heatmaps depicting expression of transcription factors associated with a Tpex transcriptional program. (D) Clustering analysis of published scRNAseq data (29) of d4.5 and d7 LCMV-Arm and -Cl13 specific CD8 T cells revealed 14 transcriptional distinct cell clusters. (E) Gene expression scores overlayed on tSNE plots for left: d30 pi LCMV-MN Slamf6+/Tim3− upregulated genes (Supplemental Table-2), middle: d7 pi Cl13 Tpex up regulated, and right: d4.5 pi memory precursor (Mpc) up regulated (29). A red (Max) gene score indicates high expression and enrichment of the gene set whereas a blue (Min) gene score indicates low to no expression of a gene set in the cell. (F) Infection and time designations overlayed on tSNE plots showing the Mpc and Tpex clusters that correspond to the gene scores given in 7E. (G) Slamf6+/Tim3− and Slamf6-Tim3+ CD8 T cells from LCMV-MN infected mice (Day 15 pi) were isolated by cell sorting and transferred i.v. into two groups of congenically distinct infection-matched mice. Recipients in each group received either anti-PD-L1 neutralizing antibody (0.2 mg/mouse) or PBS i.p. twice a week. Representative dot plots depicting presence of transferred cells and their phenotype in spleen at day 14. Flow data were concatenated from 2–5 animals due to scarcity of cells in certain groups. (H) Number of H-2Db/gp33-specific transferred CD8 T cells in each group. RNA-seq data is derived from 3 repeats/group. Data in G and H are representative of two separate experiments with n=3–5 mice/group per experiment. Bars indicate mean ± SEM. **p <0.001. Two-way ANOVA with Tukey’s multiple comparison test.
LCMV-Arm and -Cl13 specific CD8 T cells appear transcriptionally similar through the first few days of activation, but then diverge (29). This point was confirmed when we interrogated previously published single cell transcriptomes for P14 transgenic CD8 T cells (specific for the immunodominant LCMV epitope gp33) collected from mice 4.5 and 7 days after infection with LCMV-Armstrong or LCMV-Cl13 (Fig. 7D). We next asked how the relatively abundant population of Slamf6+/Tim3− CD8 T cells induced by LCMV-MN infection (30 days after infection) compared transcriptionally to these early CD8 T cells subsets. We first defined the signature of LCMV-MN Slamf6+/Tim3− genes (Supplemental Table-2) and addressed enrichment within cells derived from LCMV-Arm and -Cl13 infections. We found that LCMV-MN Slamf6+/Tim3− genes were particularly enriched within specific clusters of cells (Fig 7E, left tSNE plot). To better understand the identity of these clusters, we asked which Cl13 and Arm specific cells expressed Tpex and memory precursor cell (Mpc) genes (Fig. 7 E, middle and right tSNE plots). As anticipated from our gene set enrichment analysis in Fig 7A, we found that one cluster that with high LCMV-MN Slamf6+ scores was d7 Cl13 Tpex cells (Fig. 7E-F). Interestingly, a cluster of d4.5 Mpc also scored high for LCMV-MN Slamf6+/Tim3− genes (Fig. 7E-F). It should be noted that d4.5 Mpc show similar up- or down-regulation of many d7 Tpex defining genes (Supplemental Fig.1). In conclusion, LCMV-MN Slamf6+/Tim3− cells express transcriptional similarities to both early Tpex and early Mpc after LCMV-C13 and Arm infections.
Enhanced responsiveness to checkpoint blockade is a cardinal functional feature of Tpex. Fourteen days after LCMV-MN infection, Slamf6+/Tim3− and Slamf6-/Tim3+ H-2Db/gp33-tetramer+ CD8 T cells were sorted from spleen and LNs of CD45.1+ mice and 5X104 cells were transferred to separate infection matched CD45.2+ recipients. Immediately following transfer, recipients received 200μg of anti-PD-L1 antibody every 3 days. Control recipients received cells but were treated with PBS. Fourteen days later, donor cells were assessed in spleen. As shown in Fig. 7G and 7H, Slamf6+/Tim3− H-2Db/gp33-specific CD8 T cells responded best to PD-L1 blockade and gave rise to Tim-3+ progeny. Taken together, these observations indicated that LCMV-MN infection induced a particularly abundant subset of cells that exhibited the phenotype, transcriptional and functional properties of Tpex.
Discussion
Here we identified a novel strain of LCMV and performed virological and immunological characterizations. Persistence of LCMV-MN in C57Bl/6 mice was intermediate between LCMV-Armstrong and Cl13. Viremia waned after 15–20 days post initial infection unless CD4 T cells were previously depleted. This infection induced a population of CD69neg CD103neg CD8 T cells in the intestinal mucosa, raising questions about T cell migration and resident memory T cell differentiation. Notably, LCMV-MN established an abundant population of CD8 T cells that correlated with the phenotypic, transcriptional, and functional definition of previously described Tpex.
Understanding the ontogeny and regulation of Tpex is of fundamental interest as it may have bearing on the durability of T cell immunity to chronic antigen stimulation and the maintenance of T cells that respond most vigorously to check point blockade therapy. Previous reports indicate that LCMV Cl13 infection maintains substantially more Tpex cells than LCMV-Armstrong infection, suggesting a role for antigen persistence (44). Notably, Cl13-specific CD8 T cells become dependent on antigen, unlike what has been observed after acute LCMV infections (45, 46). CD4 depletion prior to clone13 infection sustains greater viremia and a higher fraction of Tpex. These data support a simple model whereby Tpex quantity linearly correlate with antigen load.
Our observations support a more complex model. LCMV-MN infection biased towards Tpex differentiation, and numerically maintained greater (>10 fold) Tpex in spleen despite intermediate viral load. In situ examination of viral distribution revealed differences between LCMV-MN and Cl13. In contrast to Cl13 infected animals, viral antigen was largely absent from the LN subcapsular sinus of LCMV-MN infected mice. Similar distinctions in viral antigen distribution were also noticed in the splenic white pulp. These observations could be consistent with the hypothesis that the regulation of Tpex has a spatial component (17). Important questions for the future include determining a) the localization of Tpex in relation to persistent antigen presentation, b) whether LCMV-MN induces a unique environmental milieu, or c) whether LCMV-MN promotes a particular T cell-intrinsic differentiation program that predisposes towards Tpex development. Another interesting observation from our studies is the lack of difference in the gp66-specific CD4 T cell response between LCMV-MN and -Cl13 infections despite the higher magnitude of the CD8 T cell response in LCMV-MN infected mice. The mechanism of why CD8 and CD4 T cell response may be differentially regulated is difficult to discern from the available data. Viral tropism differences might influence MHC-I vs MHC-II antigenic presentation. Alternatively, LCMV-MN might establish a different CD4 T cell epitope immunodominance hierarchy than LCMV-Cl13, leading to perturbations in unknown CD4 T cell epitopes that were not measured.
A strength of the LCMV-MN and -Cl13 models is that CD4 depletion provides additional scenarios of persistent viremia. However, it should be noted that using CD4 depletion to achieve this outcome may not recapitulate physiologic settings where chronic viremia is established in the presence of CD4 T cells. Comparing LCMV-Armstrong and Cl13 has provided an important model to interrogate Tpex biology. However, this subset’s relative scarcity is an impediment to detailed characterization. Including LCMV-MN in these analyses offers numerical advantages. It would also provide an additional context or data point to refine the mechanistic understanding of Tpex ontogeny. Further characterization of Tpex may have ramifications for understanding T cell biology to persistent viral infections and perhaps other settings of chronic antigen stimulations which may include cancer and possibly autoimmune or other chronic T cell-mediated diseases.
Supplementary Material
Key points:
Isolation and characterization of a novel LCMV strain from pet shop mice in Minnesota
LCMV-MN exhibits intermediate viral persistence compared to LCMV-Arm and LCMV-Cl13
LCMV-MN infection generates abundant stem-like exhausted progenitor CD8 T cells
Acknowledgement
The authors acknowledge the assistance of the National Institutes of Health (NIH) Tetramer Core facility for providing LCMV-specific CD8 tetramer reagents. The authors also acknowledge the help of University of Minnesota Flow Cytometry core facility and the Biosafety level 3 program.
Financial support
The study was supported by funding from NIH grants 1R01AI146032, 3R01AI084913 and 1R01AI150600 to D.M. L.K.B is a Searle Scholar and is also supported by NIH grant 2P20GM109035, P20GM121298. MCS is supported by the NIH grant 1F31AI152353–01A1.
References:
- 1.Takagi T, Ohsawa M, Morita C, Sato H, and Ohsawa K. 2012. Genomic analysis and pathogenic characteristics of lymphocytic choriomeningitis virus strains isolated in Japan. Comp. Med. 62: 185–192. [PMC free article] [PubMed] [Google Scholar]
- 2.Muckenfuss RS, Armstrong C, and Webster LT. 1934. Etiology of the 1933 epidemic of encephalitis. JAMA J. Am. Med. Assoc. 103: 731–733. [Google Scholar]
- 3.Barthold SW, and Smith SA. 2007. Lymphocytic Choriomeningitis Virus. In The Mouse in Biomedical Research Elsevier. 179–213. [Google Scholar]
- 4.Ahmed R, Salmi A, Butler LD, Chiller JM, and Oldstone MB. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160: 521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed R, and Oldstone MBA. 1988. Organ-specific selection of viral variants during chronic infection. J. Exp. Med. 167: 1719–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zinkernagel RM, and Doherty PC. 1974. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic chorio- meningitis within a syngeneic or semiallogeneic system. Nature 248: 1973–1974. [DOI] [PubMed] [Google Scholar]
- 7.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439: 682–687. [DOI] [PubMed] [Google Scholar]
- 8.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77: 4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Ramachandran S, Mann M, Popkin DL, and Western C. 2012. Role of Lymphocytic Choriomeningitis Virus (LCMV) in Understanding Viral Immunology: Past, Present and Future. Viruses 4: 2650–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moskophidis D, Lechner F, Pircher H, and Zinkernagel RM. 1993. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362: 758–761. [DOI] [PubMed] [Google Scholar]
- 11.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, and Ahmed R. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188: 2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldstone MBA, and Dixon FJ. 1970. PATHOGENESIS OF CHRONIC DISEASE ASSOCIATED WITH PERSISTENT LYMPHOCYTIC CHORIOMENINGITIS VIRAL INFECTION. J. Exp. Med. 131: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zehn D, and Wherry EJ. 2015. Immune Memory and Exhaustion: Clinically Relevant Lessons from the LCMV Model. In Adv Exp Med Biol 137–152. [DOI] [PubMed] [Google Scholar]
- 14.McLane LM, Abdel-Hakeem MS, and Wherry EJ. 2019. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 37: 457–495. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, and Han X. 2015. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Invest. 125: 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, and Wherry EJ. 2012. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (80-. ). 338: 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, and Ahmed R. 2016. Defining CD8 + T cells that provide the proliferative burst after PD-1 therapy. Nature 537: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, Xu L, Chen X, Hao Y, Wang P, Zhu C, Ou J, Liang H, Ni T, Zhang X, Zhou X, Deng K, Chen Y, Luo Y, Xu J, Qi H, Wu Y, and Ye L. 2016. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 540: 470–470. [DOI] [PubMed] [Google Scholar]
- 19.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, and Held W. 2016. T Cell Factor 1-Expressing Memory-like CD8+T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45: 415–427. [DOI] [PubMed] [Google Scholar]
- 20.Wu T, Ji Y, Ashley Moseman E, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, and Schwartzberg PL. 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, Yan P, Nzingha K, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Huang AC, and Wherry EJ. 2020. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52: 825–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, Schietinger A, Schumacher TN, Schwartzberg PL, Sharpe AH, Speiser DE, Wherry EJ, Youngblood BA, and Zehn D. 2019. Defining ‘T cell exhaustion.’ Nat. Rev. Immunol. 19: 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, and Masopust D. 2016. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532: 512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battegay M, Cooper S, Althage A, Bänziger J, Hengartner H, and Zinkernagel RM. 1991. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J. Virol. Methods 33: 191–198. [DOI] [PubMed] [Google Scholar]
- 25.Thompson E.a, Beura LK, Nelson CE, Anderson KG, and Vezys V. 2016. Shortened Intervals during Heterologous Boosting Preserve Memory CD8 T Cell Function but Compromise Longevity. J. Immunol. 196: 3054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Germain RN, and Gerner MY. 2017. Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc. Natl. Acad. Sci. U. S. A. 114: E7321–E7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baller J, Kono T, Herman A, and Zhang Y. 2019. CHURP: A Lightweight CLI Framework to Enable Novice Users to Analyze Sequencing Datasets in Parallel. In Proceedings of the Practice and Experience in Advanced Research Computing on Rise of the Machines (learning) ACM, New York, NY, USA. 1–5. [Google Scholar]
- 28.Robinson MD, McCarthy DJ, and Smyth GK. 2009. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao C, Sun H-WW, Lacey NE, Ji Y, Moseman EA, Shih H-YY, Heuston EF, Kirby M, Anderson S, Cheng J, Khan O, Handon R, Reilley J, Fioravanti J, Hu J, Gossa S, Wherry EJ, Gattinoni L, McGavern DB, O’Shea JJ, Schwartzberg PL, and Wu T. 2019. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 20: 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, and Satija R. 2021. Integrated analysis of multimodal single-cell data. Cell 184: 3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. 2015. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fay EJ, Balla KM, Roach SN, Shepherd FK, Putri DS, Wiggen TD, Goldstein SA, Pierson MJ, Ferris MT, Thefaine CE, Tucker A, Salnikov M, Cortez V, Compton SR, V Kotenko S, Hunter RC, Masopust D, Elde NC, and Langlois RA. 2022. Natural rodent model of viral transmission reveals biological features of virus population dynamics. J. Exp. Med. 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan BM, Emonet SF, Welch MJ, Lee AM, Campbell KP, de la Torre JC, and Oldstone MB. 2011. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc. Natl. Acad. Sci. 108: 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, De La Torre JC, and Oldstone MBA. 2000. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 192: 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R. 2007. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 27: 670–684. [DOI] [PubMed] [Google Scholar]
- 36.Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, Larsen CP, and Ahmed R. 2007. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. U. S. A. 104: 15430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beura LK, Anderson KG, Schenkel JM, Locquiao JJ, Fraser KA, Vezys V, Pepper M, and Masopust D. 2015. Lymphocytic choriomeningitis virus persistence promotes effector-like memory differentiation and enhances mucosal T cell distribution. J. Leukoc. Biol. 97: 217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, and Masopust D. 2015. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, and Masopust D. 2012. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 188: 4866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang N, and Bevan M. 2013. Transforming Growth Factor-β Signaling Controls the Formation and Maintenance of Gut-Resident Memory T Cells by Regulating Migration and Retention. Immunity 39: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masopust D, Vezys V, Wherry EJ, Barber DL, and Ahmed R. 2006. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J. Immunol. 176: 2079–83. [DOI] [PubMed] [Google Scholar]
- 42.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, Yu S, Kurachi M, Herati RS, Vella LA, Baxter AE, Wu JE, Khan O, Beltra J-C, Giles JR, Stelekati E, McLane LM, Lau CW, Yang X, Berger SL, Vahedi G, Ji H, and Wherry EJ. 2019. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51: 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, and Haining WN. 2019. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, and Kallies A. 2020. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 21: 1256–1266. [DOI] [PubMed] [Google Scholar]
- 45.Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, and Ahmed R. 1999. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science (80-. ). 286: 1377–1381. [DOI] [PubMed] [Google Scholar]
- 46.Wherry EJ, Barber DL, Kaech SM, Blattman JN, and Ahmed R. 2004. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 101: 16004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data described in this study have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) and can be downloaded with accession number GSE207983. Public data sets were downloaded from GEO (GSE84105 and Series GSE119940) (https://www.ncbi.nlm.nih.gov/geo/).