

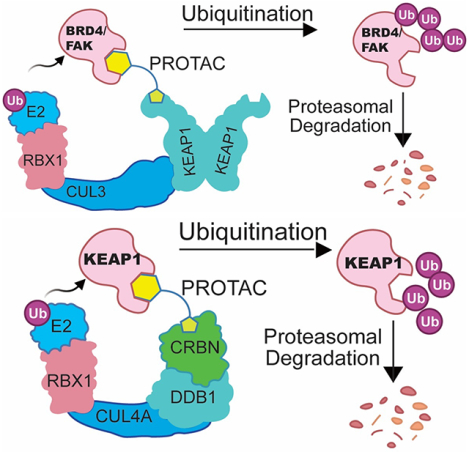
SUMMARY:
Targeted protein degradation (TPD) uses small molecules to recruit E3 ubiquitin ligases into proximity of proteins of interest, inducing ubiquitination-dependent degradation. A major bottleneck in the TPD field is the lack of accessible E3 ligase ligands for developing degraders. To expand the E3 ligase toolbox, we sought to convert the KEAP1 inhibitor KI696 into a recruitment handle for several targets. While we were able to generate KEAP1-recruiting degraders of BET family and murine FAK we discovered that the target scope of KEAP1 was narrow, as targets easily degraded using a cereblon (CRBN)-recruiting degrader were refractory to KEAP1-mediated degradation. Linking the KEAP1-binding ligand to a CRBN-binding ligand resulted in a molecule that induced degradation of KEAP1 but not CRBN. In sum, we characterize tool compounds to explore KEAP1-mediated ubiquitination and delineate the challenges of exploiting new E3 ligases for generating bivalent degraders.
Graphical Abstract
eTOC:
Du et al. explores a variety of bivalent KEAP1-recruiting degraders to assess the generality of KEAP as an E3 ligase for targeted protein degradation They found the targets easily degraded using a cereblon (CRBN) or VHL-recruiting degrader were refractory to KEAP1-mediated degradation.
INTRODUCTION
Targeted protein degradation (TPD) uses small molecules (degraders) to eliminate proteins of interest (POI) from the proteome(Bondeson and Crews, 2017; Sakamoto et al., 2001). Degraders recruit ubiquitin E3 ligases into proximity of the POI, inducing POI ubiquitination and subsequent proteasomal degradation. Advantages of TPD approaches over traditional inhibitors include its differentiated mode of action (MOA) to trigger protein loss, the potential to access “undruggable” targets, the improved selectivity profiles that can be achieved, and the ability to overcome drug resistance(Deshaies, 2020; Gao et al., 2020; Nalawansha and Crews, 2020; Samarasinghe and Crews, 2021). Additionally, given their unique MOA, degrader molecules represent chemical protein knock-down agents that are highly complementary to genetic knock-out and -down technologies (Kostic and Jones, 2020). These advantages heighten interest in TPD strategies for both basic and translational research.
One of the most frequently employed strategies in TPD is the use of Proteolysis Targeting Chimeras (PROTACs)(Salami and Crews, 2017). PROTACs are rationally designed degraders that consist of a ligand specific for a POI, an E3 ubiquitin ligase recruiting ligand, and a linker that connects the two. This strategy has been successfully applied to degrade a wide spectrum of targets, including kinases, transcription factors, and viral proteins (de Wispelaere et al., 2019; Jones, 2018; Lai et al., 2016; Winter et al., 2015). Moreover, several PROTACs are currently progressing through clinical trials, suggesting that PROTACs represent a promising new therapeutic strategy(Flanagan et al., 2019; Khan et al., 2019; Neklesa et al., 2019; Petrylak et al., 2020). Despite the great potential and tremendous interest, only a handful of the more than 600 ubiquitin E3 ligases have been successfully incorporated into the design of functional PROTACs. This limits the degradable POI target space given the evidence that not every POI-E3 ligase combination results in ubiquitination competent complex formation(Donovan et al., 2020). As such, substantial efforts have been invested in generating new E3 ligase ligands, and recent reports have identified novel covalent ligands for DCAF16, RNF114, RNF4, DCAF11, FEM1B and KEAP1(Henning et al., 2022; Lu et al., 2018; Luo et al., 2021; Spradlin et al., 2019; Tong et al., 2020; Zhang et al., 2019; Zhang et al., 2021). Nevertheless, identifying additional ligands for E3 ligases that are suitable for use in PROTACs remains a critical need in the TPD field.
Kelch-like ECH-associated protein 1 (KEAP1) is a key regulator of the cellular response to oxidative and electrophilic stress(Baird and Dinkova-Kostova, 2011; Itoh et al., 1997; Kensler et al., 2007). It is an adaptor protein for the CUL3 E3 ubiquitin ligase, and can ubiquitinate NRF2, a key transcription factor that regulates the expression of antioxidant proteins to protect cells from oxidative damage. Under basal conditions, KEAP1 homodimers bind to both the DLG and ETGE motifs within NRF2 and facilitate NRF2 ubiquitination and subsequent proteasomal degradation, thereby preventing NRF2-mediated transcriptional activity(Hayes and Dinkova-Kostova, 2014; Lo et al., 2006). However, in response to the accumulation of oxidative or electrophilic species, key cysteine residues of KEAP1 are modified, which impairs interaction with NRF2 and blocks its degradation. Thus, upon oxidative or electrophilic stress, NRF2 escapes from KEAP1-mediated degradation and translocates to the nucleus, where it induces expression of antioxidant response element (ARE)-directed genes(Takaya et al., 2012). Activation of NRF2 has been shown to protect against many diseases, including neurodegenerative diseases, aging, diabetes, photo-oxidative stress, cardiovascular disease, inflammation, pulmonary fibrosis disease, acute pulmonary injury, and cancers(O’Connell and Hayes, 2015; Wu and Papagiannakopoulos, 2020).
Given this disease relevance, targeting the KEAP1-NRF2 pathway has been of high interest over the last two decades resulting in numerous disclosed inhibitors(Abed et al., 2015; Hur and Gray, 2011; Jiang et al., 2016; Magesh et al., 2012). Exploiting these existing KEAP1 inhibitors for the development of PROTACs could expand the toolbox of E3 ligase ligands. Jiang and co-workers designed and synthesized peptide ligands of KEAP1 and were able to convert these into peptidic PROTACs that enabled the degradation of tau(Lu et al., 2018). While these PROTACs are valuable as a proof-of-concept, the peptidic nature of the KEAP1 ligand limits their utility in cellular or in vivo contexts. Separately, the Nomura group reported a PROTAC based on bardoxolone, a terpenoid inhibitor that covalently targets Cys151 of the BTB domain of KEAP1, which when conjugated to JQ1 induces BRD4 degradation(Tong et al., 2020). However, they noted that these PROTACs also induced self-degradation of KEAP1 by an unknown mechanism. Recently, Jin group reported BRD4 degraders via hijacking KEAP1 using KI696 as recruiting ligand(Wei et al., 2021). While these early studies suggest that KEAP1 may be a tractable E3 ligase for use in PROTAC design, only tau and BRD4 have been explored, and thus additional studies are necessary to fully characterize the potential of KEAP1-based PROTACs. Here, we describe our efforts to convert the non-covalent KEAP1 inhibitor KI-696(Davies et al., 2016) into bivalent degraders for a diverse range of targets, including transcriptional and epigenetic regulators (BRD4 and BRD9) and kinases (FAK, BTK, EGFR and CDK4/6). While we were able to generate KEAP1 PROTACs that could degrade BRD4 and FAK, they failed to degrade BTK, BRD9, CDK4/6, and EGFR, which have all been successfully degraded using CRBN or VHL-based PROTACs. Additionally, we conjugated KI-696 to a ligand of the CRBN E3 ligase substrate adaptor, thereby obtaining an efficient KEAP1 degrader that may have the potential to potently activate anti-oxidative pathways.
RESULTS
Design and Optimization of BRD4 degraders
As a proof of concept, we assessed whether functional BRD4 degraders could be derived from previously reported reversible KEAP1 binders (KI-696 and DGY-04–091). KI-696 was developed using a fragment and structure-based design strategy resulting in a binder with nanomolar affinity to the KELCH domain of KEAP1(Davies et al., 2016). Based on the reported co-crystal structures of KI-696 and compound 6 bound to KEAP1 (PDB: 5FNU and 5FNT), we observed that the sulfonamide moiety is solvent-exposed and therefore a suitable site to introduce linkers without affecting KEAP1 binding. We coupled either KI-696 or DGY-04–091 to the BRD4 inhibitor (+)-JQ1 with alkyl chain linkers to generate DGY-05–089 and DGY-06–177 (Figure 1A). We used an in vitro TR-FRET displacement assay of KEAP1-NRF2(Nowak et al., 2018) to demonstrate that both DGY-05–089 and DGY-06–177 had similar binding affinities for KEAP1, with IC50 values of 147 nM and 145 nM, respectively (Table S1 and Figure S1). To assess the ability of these compounds to recruit BRD4 to KEAP1, we established a TR-FRET-based KEAP1-BRD4BD2 dimerization assay. Interestingly, DGY-05–089 did not induce ternary complex formation, while DGY-06–177 promoted substantial KEAP1- BRD4BD2 dimerization at concentrations of approximately 100 nM (Figure 1B). To test the ability of these compounds to induce BRD4 degradation in cells, we treated MM.1S cells, a multiple myeloma cell line that is sensitive to (+)-JQ1 treatment, for 24 h, with the CRBN-recruiting BRD4 degrader ZXH-02–043(Nowak et al., 2018) serving as a positive control (Figure 1C). Notably, DGY-06–177 exhibited maximal BRD4 degradation at a concentration of 1 μM and exhibited a “hook effect”(Douglass Jr et al., 2013) at a concentration of 5 μM. DGY-05–089 failed to degrade BRD4, consistent with its inability to induce KEAP1-BRD4BD2 dimerization (Figure 1C).
Figure 1. DGY-06–177 induced degradation of BRD4.
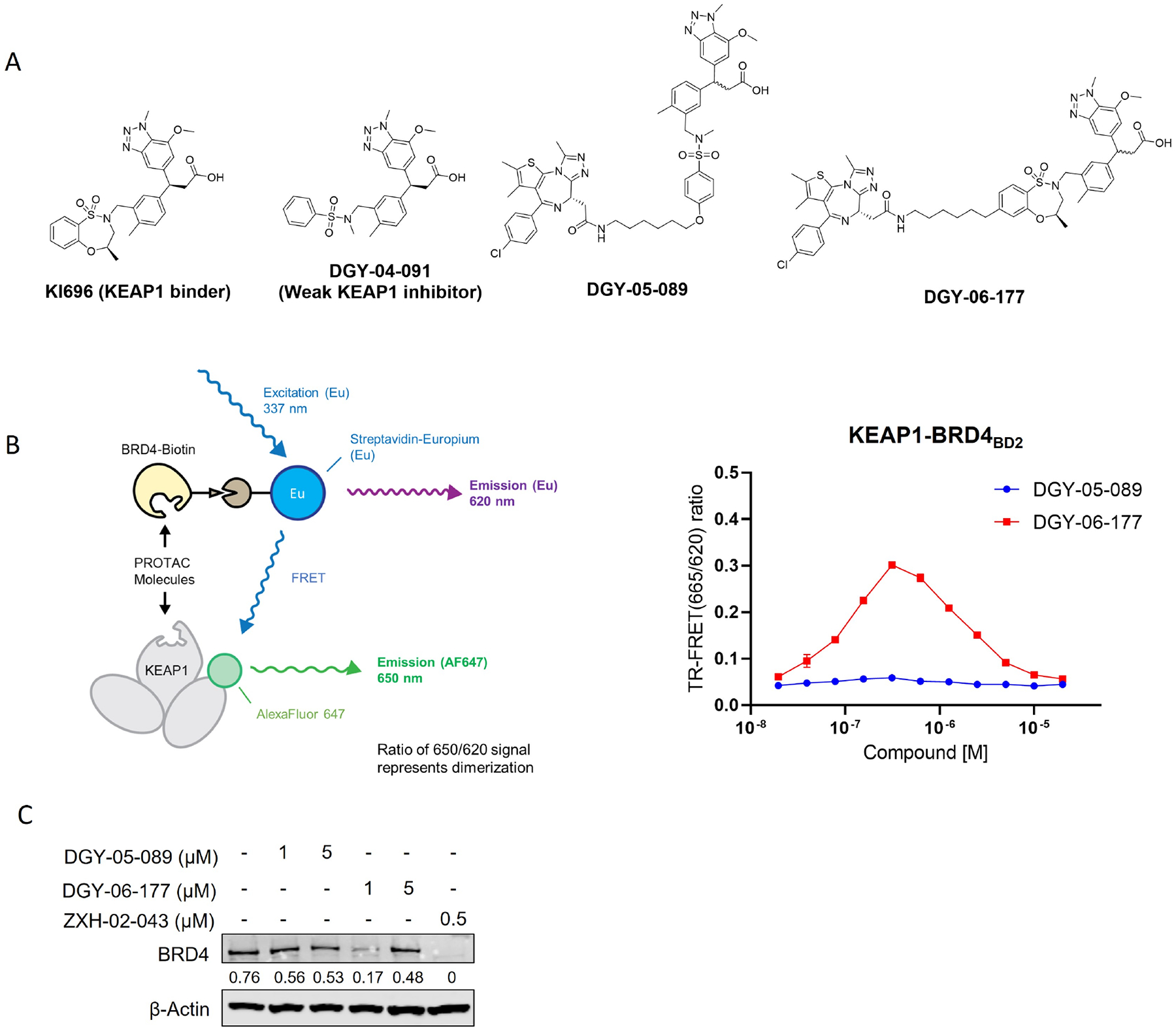
A. Chemical structures of KI-696, DGY-04–091, DGY-05–089 and DGY-06–177. B. KEAP1-BRD4BD2 dimerization TR-FRET assay. Titration of DGY-06–177 or DGY-05–089 to Alexa Fluor™ 647-KEAP1, Eu-streptavidin and biotinylated BRD4BD2. Data are presented as mean ± SD with at least three technical replicates. Representative figure from two independent experiments is shown. C. Immunoblot assessment of MM1.S cells treated with either DMSO or indicated compounds for 24 h. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to β-actin) is shown under each band. See also Figure S1 and Table S1.
To optimize these initial compounds, we first sought to obtain each enantiopure isomer of DGY-06–177. We used chiral HPLC to separate both enantiomers of a key intermediate (compound 5; see supplemental Synthetic Scheme) which were elaborated further to prepare enantiopure compounds DGY-06–177-pk1 and DGY-06–177-pk2 (Figure 2A). We tested these two compounds using a TR-FRET KEAP1-BRD4BD2 dimerization assay and observed that the enantiomers exhibited very different abilities to form the KEAP1-BRD4BD2 complex. DGY-06–177-pk2 exhibited dimerization activity at concentrations of approximately 100 nM, whereas DGY-06–177-pk1 induced dimerization only at high concentrations of ~10 μM (Figure 2B). The difference observable compared to the racemic mixture is marginal, as expected given that it still contains a significant quantity of DGY-06–177-pk2. Our findings are further supported by binding data for KEAP1 for DGY-06–177-pk1 vs DGY-06–177-pk2 (Table S1). We then compared BRD4 expression levels by immunoblot analysis in response to the two enantiomers in MM1.S cells. As expected, the more potent dimerizer, DGY-06–177-pk2, induced stronger degradation of BRD4 compared to its isomer DGY-06–177-pk1 after 24-hour treatment at 1 μM concentration (Figure 2C). We further observed that DGY-06–177-pk2 induces degradation of BRD4 at a concentration of 1 μM in the human ovarian cancer cell line OVCAR8 (Figure S2). Thus, we concluded that DGY-06–177-pk2 possesses the correct stereoisomer to induce productive dimerization and efficient degradation of BRD4.
Figure 2. The enantiomer DGY-06–177-pk2 induced better degradation of BRD4.
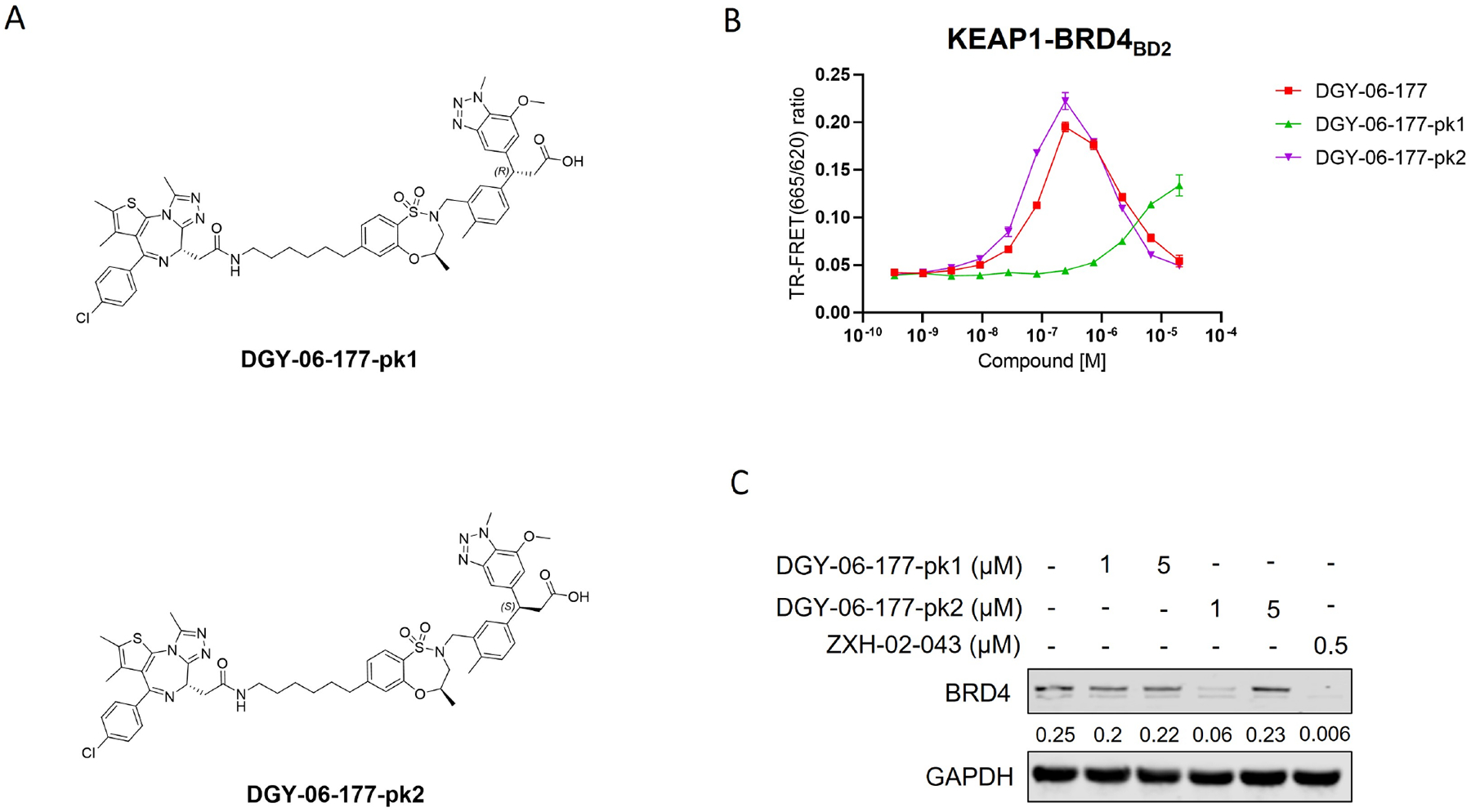
A. Chemical structures of DGY-06–177 pk1/2. B. KEAP1-BRD4BD2 dimerization TR-FRET assay. Titration of DGY-06–177-pk1 or DGY-06–177-pk2 to Alexa Fluor™ 647-KEAP1, Eu-streptavidin and biotinylated BRD4BD2. Data are presented as mean ± SD with at least three technical replicates. Representative figure from two independent experiments is shown. C. Immunoblot assessment of MM1.S cells treated with either DMSO or indicated compounds for 24 h. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to GAPDH) is shown under each band. See also Figure S2.
The linker length is often a critical factor that affects ternary complex formation, transfer of ubiquitin, and subsequent target degradation(Troup et al., 2020). To determine the optimal linker length and composition, we designed and synthesized several derivatives featuring saturated carbon chain or PEG linkers ranging in length from 3 to 12 atoms (Figure 3A). All the compounds engaged KEAP1 in the TR-FRET Nrf2 peptide displacement assay (Figure S1) and induce dimerization of KEAP1 to BRD4BD2 (Figure 3B), but only NJH-05–138 and NJH-05–141 showed cellular degradation of BRD4 to a similar level to DGY-06–177-pk2 (Figure 3C). Taken together, we demonstrated that KEAP1 binder KI-696 can be used to develop PROTAC-based degraders for BRD4.
Figure 3. Screening and evaluation of BRD4 degrader library.
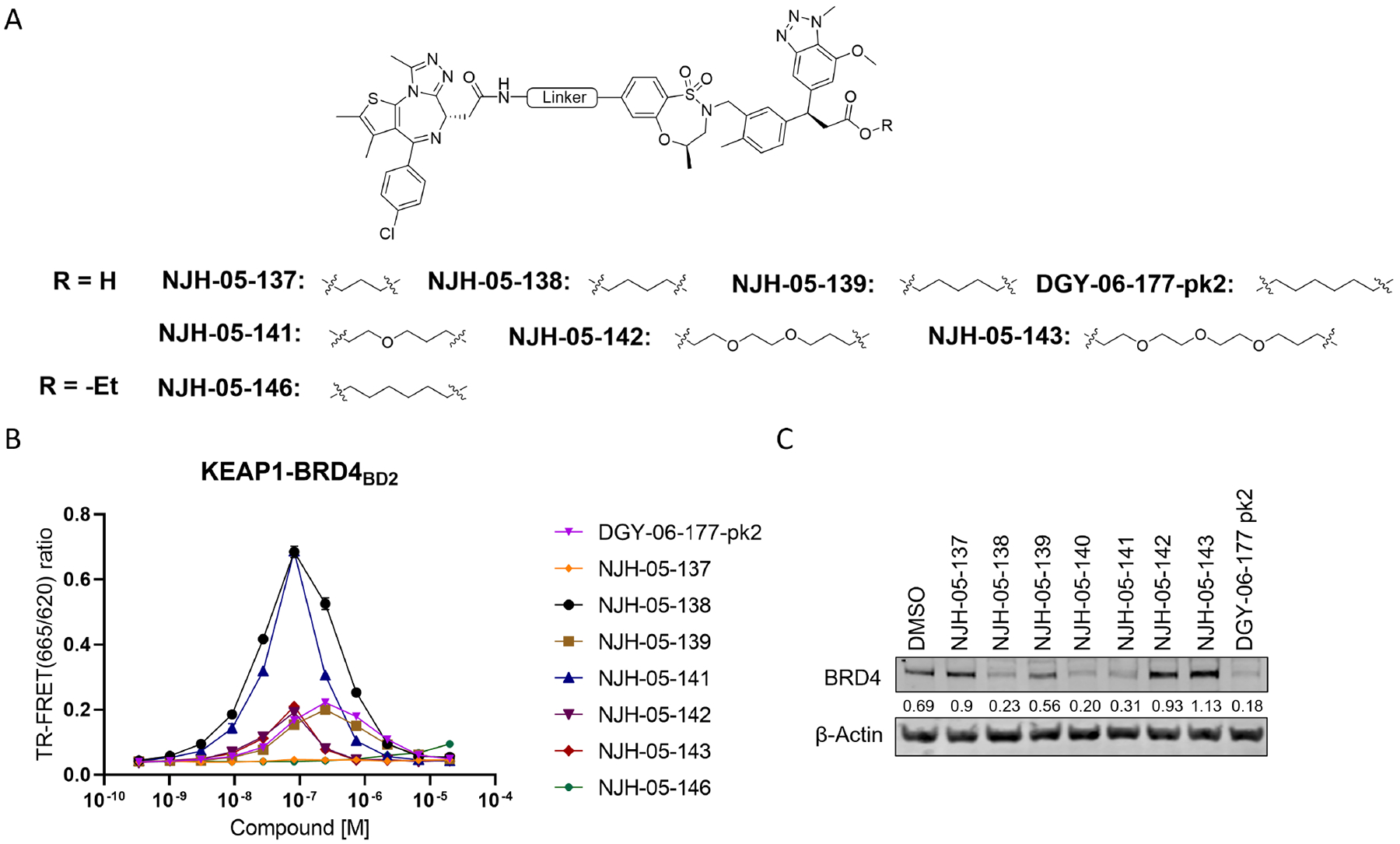
A. Structure of KEAP1-BRD4 compounds B. KEAP1-BRD4BD2 dimerization TR-FRET assay. Titration of indicated compounds to Alexa Fluor™ 647-KEAP1, Eu-streptavidin and biotinylated BRD4BD2. Data are presented as mean ± SD with at least three technical replicates. Representative figure from two independent experiments is shown. C. Immunoblot assessment of MM.1S cells treated with either DMSO or the indicated compounds for 16 h. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to β-actin) is shown under each band (NJH-05–140 was another batch of DGY-06–177-pk2).
DGY-06–177-pk2 is a Selective KEAP1-dependent BET Family Degrader
To confirm that DGY-06–177-pk2-mediated BRD4 degradation was KEAP1-dependent, we compared its degradation profile in parental versus KEAP1 knockdown (KEAP1KD) OVCAR8 cells. In parental OVCAR8 cells, DGY-06–177-pk2 induced potent BRD4 degradation after 16 hours, with maximal degradation observed at 1 μM. By contrast, in KEAP1KD OVCAR8 cells, DGY-06–177-pk2 had no effect on BRD4 abundance, demonstrating that BRD4 degradation was KEAP1-dependent (Figure 4B). To confirm involvement of functional CUL3-KEAP1 E3 ubiquitin complex, we pre-treated cells with the NEDD8-activating enzyme inhibitor MLN4924, which prevents activation of CUL3, and found that BRD4 degradation was blocked (Figure 4C). Finally, pre-treatment of cells with either an excess of (+)-JQ1 or KI-696 abolished DGY-06–177-pk2-induced BRD4 degradation, demonstrating a requirement for both BRD4 and KEAP1 engagement (Figure 4C). To further confirm KEAP1-dependency, we synthesized a corresponding negative control (NJH-05–146) by introducing an ethyl group to mask the carboxylic acid and prevent KEAP1 binding (Figure 4D, Table S1). We confirmed that 16-hour treatment at 0.1, 1, and 10 μM of NJH-05–146 in MM.1S cells did not result in degradation of BRD4. We also performed antiproliferation experiment in MM.1S cells, and found that the negative control compound did not exhibit an anti-proliferative effect (> 10 μM), enabling comparisons with DGY-06–177-pk2 (Figure S2).
Figure 4. Validation of the lead degrader molecule, DGY-06–177-pk2.
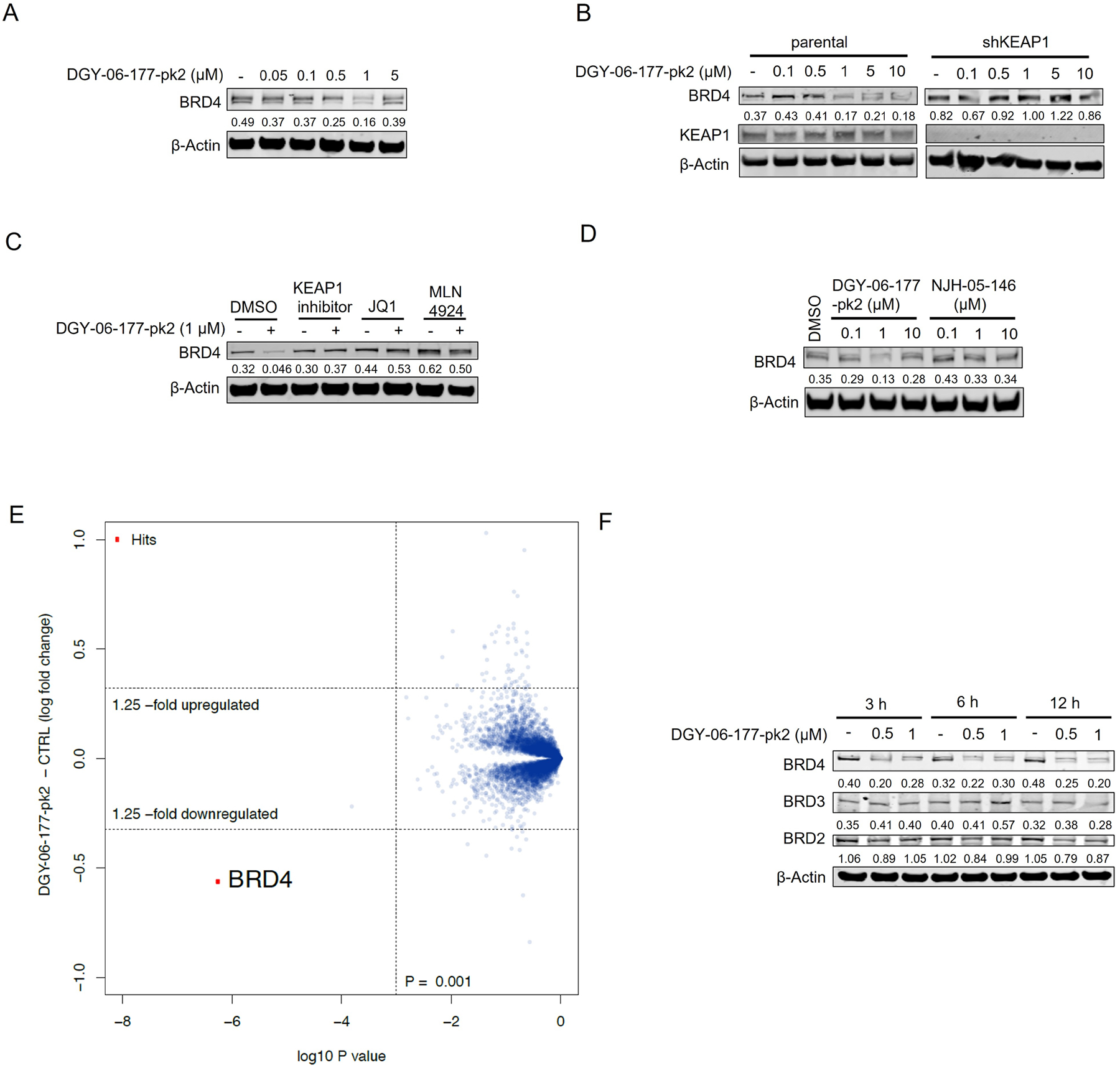
A. Immunoblot assessment of MM1.S cells treated with indicated doses of DGY-06–177-pk2 for 16 h. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to β-actin) is shown under each band. B. Immunoblot assessment of parental and KEAP1 knockdown OVCAR8 cells treated with either DMSO or DGY-06–177-pk2 for 16 h. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to β-actin) is shown under each band. C. Immunoblot assessment of MM.1S cells pretreated with 10 μM of KEAP1 inhibitor, 10 μM of JQ1, 1 μM of MLN4924 for 2 h prior to the addition of DGY-06–177-pk2 for 16 hrs. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to β-actin) is shown under each band. D. Immunoblot assessment of MM.1S cells treated with either DMSO, DGY-06–177-pk2 or its negative control compound at indicated doses for 16 h. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4 normalized to β-actin) is shown under each band. E. Quantitative proteomics profiles of wild-type MM.1S cells treated for 6 h with 0.2 μM of DGY-06–177-pk2. F. Immunoblot assessment of MM.1S cells treated with either DMSO or DGY-06–177-pk2 for the indicated time points. Representative blots from two independent experiments are shown. The relative intensity of each band (BRD4/3/2 normalized to β-actin) is shown under each band. See also Figure S2.
We next assessed the selectivity of degradation by performing multiplexed mass spectrometry-based proteomics in MM.1S cells treated with 200 nM of DGY-06–177-pk2 for 6 hours. Despite the pan-BET family binding of JQ1, quantification of the MM.1S proteome after treatment with DGY-06–177-pk2 identified BRD4 as the only significantly downregulated protein (Figure 4E). Immunoblotting confirmed that DGY-06–177-pk2 preferentially induces BRD4 degradation while largely sparing BRD2&3 (Figure 4F). Our work thus established that DGY-06–177-pk2 represents a BET family selective degrader molecule that functions by hijacking KEAP1.
Multi-kinase degrader NJH-04–098 induces FAK degradation in murine-derived KPC cells
Inspired by the BRD4 degrader, we next sought to investigate activity of KEAP1-based PROTACs against the protein kinome, as kinases represent the largest target family for inhibitor and degrader drug development and discovery. Previously, our group has generated multi-kinase degraders by conjugating a highly promiscuous kinase inhibitor TAE684 to a CRBN-binding ligand(Huang et al., 2018). We employed the same promiscuous kinase inhibitor as the target-recruiting moiety for KEAP1-based PROTACs and synthesized DGY-03–188 by conjugating KI-696 to the TAE684 scaffold (Figure S3A). Surprisingly, DGY-03–188 did not induce any degradation across the whole proteome after a 10 hour treatment in human HEK293T cells (Figure S3B). Species selectivity has been demonstrated to drive the selectivity of degradation with small molecule degraders(Donovan et al., 2018). To explore species-specific degradation with our KEAP1-recruiting molecules, we synthesized additional TAE684-based compounds and tested them in cells derived from the mouse pancreatic cancer LSL-KrasG12D; Trp53lox/+; p48Cre+ (KPC) model.(Parker et al., 2020) Interestingly, proteomics profiling of KPC cells treated with 1 μM of NJH-04–098 for 8 hours revealed that only FAK (PTK2) was significantly downregulated across the proteome (Figure 5A). We then confirmed this result using immunoblotting and observed FAK degradation upon treatment of KPC cells with at doses ranging from 0.5 μM to 10 μM (Figure 5B). To evaluate the kinetics of FAK degradation, we performed time-course experiments upon treatment of using 500 nM and 1 μM NJH-04–098 in KPC cells and observed maximal FAK degradation after 24-hours at 500 nM and 8-hours at 1 μM (Figure 5C). We also assessed KEAP1 dependency by performing a rescue experiment. Degradation of FAK was rescued upon pre-treatment with MLN4924, the parent kinase inhibitor compound (TAE684), and the parent KEAP1 binder (KI696), indicating the requirement for recruiting CUL3-KEAP1 into proximity of FAK for productive degradation (Figure 5D).
Figure 5. Degradation of FAK in murine pancreatic cells.
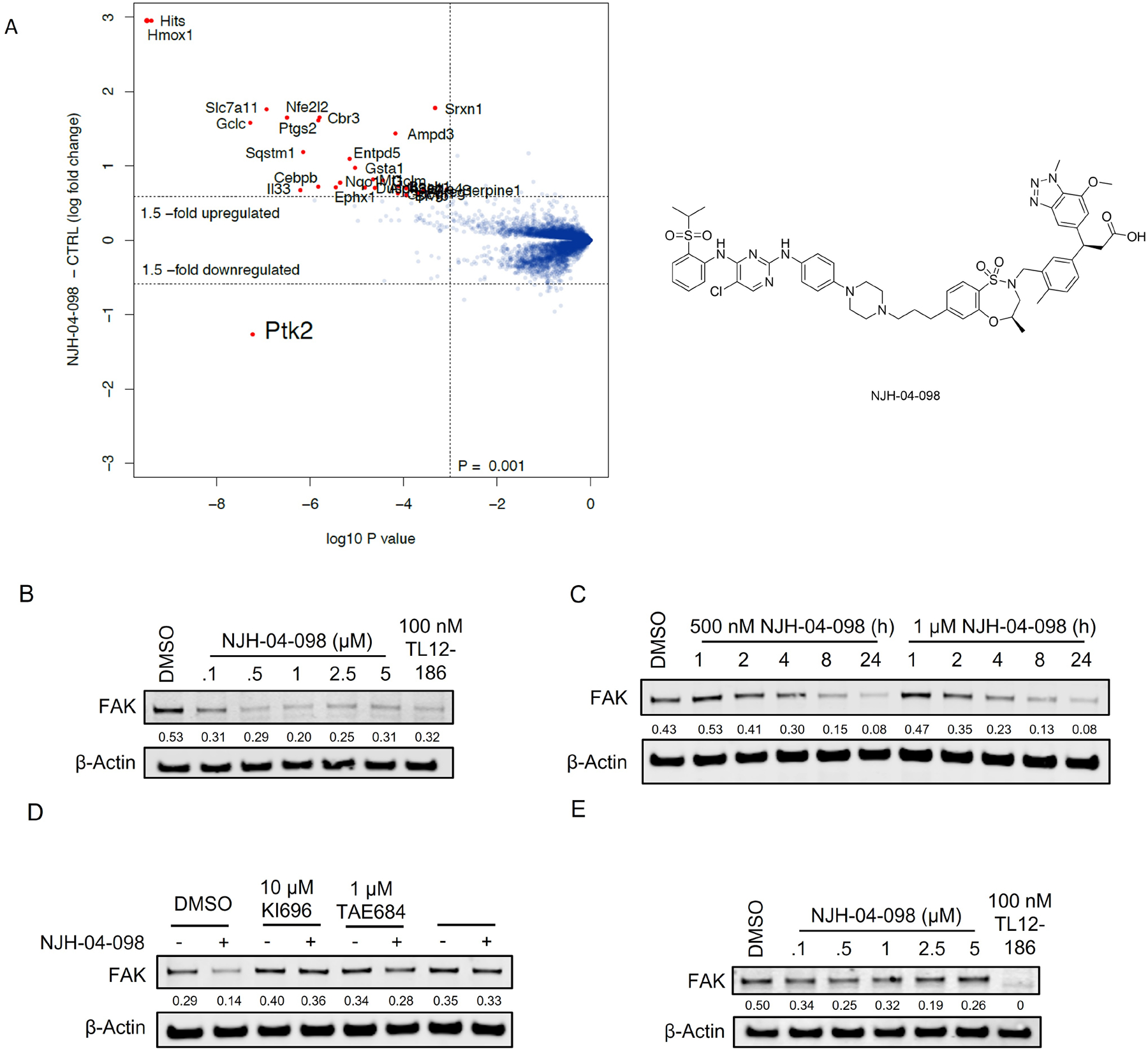
A. Quantitative proteomics profiles of KPC cells treated for 8 h with 1 μM of NJH-04–098. B. Immunoblot assessment of KPC cells treated with the indicated doses of NJH-04–098 for 24 h. Representative blots from three independent experiments are shown. The relative intensity of each band (FAK normalized to β-actin) is shown under each band. C. Immunoblot assessment of KPC cells treated with either DMSO, 500 nM or 1 μM of NJH-04–098 at indicated time points. Representative blots from two independent experiments are shown. The relative intensity of each band (FAK normalized to β-actin) is shown under each band. D. Immunoblot assessment of KPC cells pretreated with 10 μM of KI696, 1 μM of TAE684, 1 μM of MLN4924 for 2 h prior to the addition of 1 μM of NJH-04–098 for 8 h. Representative blots from two independent experiments are shown. The relative intensity of each band (FAK normalized to β-actin) is shown under each band. E. Immunoblot assessment of PATU-8988T cells treated with indicated doses of NJH-04–098 for 24 h. Representative blots from two independent experiments are shown. The relative intensity of each band (FAK normalized to β-actin) is shown under each band. See also Figure S3.
Multi-kinase degrader NJH-04–098 fails to induce FAK degradation in human pancreatic cells
As we observed degradation of FAK in murine-derived KPC cells, we next tested the compound in human-derived PATU-8988T pancreatic cells, which have more profound NRF2 response, at a concentration range of 0.1–5 μM. Consistent with our observations in HEK293T cells, we did not observe degradation of FAK by NJH-04–098 in PATU-8988T cells (Figure 5E). Mouse to human amino acid differences in the E3-POI protein-protein interface have previously been shown to be responsible for species specific degradation(Donovan et al., 2018; Matyskiela et al., 2018). Overall, our data suggests that developing KEAP1-hijacking PROTACs for specific kinases, as illustrated here by FAK, is feasible, but KEAP1 may not be as versatile as other ligases for broad targeting of kinases. Importantly, our data also shows that care needs to be taken when validating PROTACs in several species, as different E3 ligase-POI protein-protein interfaces among species may affect the degradation profile of PROTACs.
KEAP1-based PROTACs are not universally applicable for degradation of kinases
Human kinases have been successfully targeted for degradation using PROTACs hijacking CRBN and/or VHL, and 33% of the kinome has been annotated as degradable(Donovan et al., 2020). However, our attempt to use kinome-wide profiling of KEAP1 degraders suggests that KEAP1-based PROTACs may have a more restricted target space. To expand beyond the FAK degradation that we observed, we used a more targeted approach and generated a series of KEAP1-based PROTACs for Bruton’s Tyrosine kinase (BTK), a kinase shown to be highly degradable with multiple previously reported PROTACs(Arthur et al., 2020; Buhimschi et al., 2018; Dobrovolsky et al., 2019; Gabizon et al., 2020; Jaime-Figueroa et al., 2020; Liu et al., 2020; Schiemer et al., 2021; Shorer Arbel et al., 2021; Sun et al., 2019; Sun et al., 2018; Tinworth et al., 2019; Xue et al., 2020; Xue et al., 2019; Zorba et al., 2018). To design BTK degraders, we first examined a high-resolution crystal structure of BTK bound to ATP-competitive inhibitors CGI-1746(Di Paolo et al., 2011) (PDB: 3OCS), which we had previously successfully employed to generate CRBN-recruiting degraders(Dobrovolsky et al., 2019). We synthesized several KEAP1-recruiting compounds with different KEAP1 binders and various linkers attached via the morpholine moiety (Figure S4). Testing these compounds in the KEAP1-NRF2 TR-FRET displacement assay indicated that all the compounds retained potent binding affinity for KEAP1 and were cell permeable (Figure S1 and Figure S4). Next, we established a TR-FRET-based BTK-KEAP1 dimerization assay to assess the ability of these compounds to induce ternary complex formation and found that all the synthesized compounds were able to do so. To evaluate cellular degradation of BTK, we constructed an inducible cell line expressing the kinase domain of BTK protein fused to eGFP and linked to mCherry through the FMVD 2A ribosomal skipping sequence. In this system, measurement of the ratio of eGFP/mCherry fluorescence can be used to monitor abundance of the BTK-GFP fusion protein. We did not observe degradation of BTK with KEAP1-based PROTACs, whereas our positive control compound DD-03–007(Dobrovolsky et al., 2019), a previously-reported CRBN-based BTK degrader, yielded a clear degradation signal (Figure 6B). To understand whether the GFP fusion to BTK interfered with its degradation, we treated Mino and Jeko cells, two BTK-expressing mantle cell lymphoma cell lines, with the KEAP1-BTK compounds for 24 or 8 hours. Immunoblotting failed to reveal any change in BTK levels (Figure 6C).
Figure 6. Screening and evaluation of BTK degrader library.
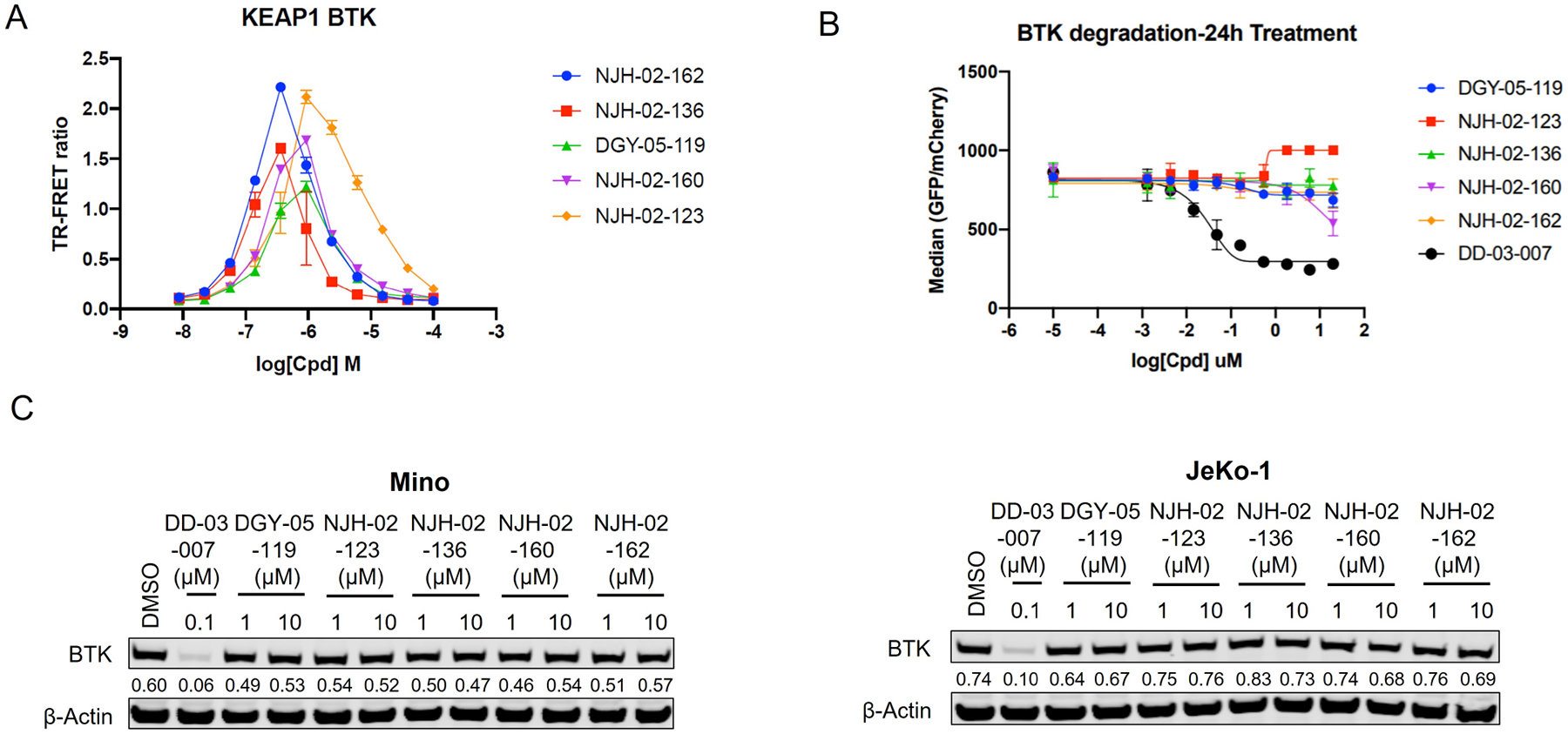
A. KEAP1-BTK dimerization TR-FRET assay. Titration of indicated compounds to Alexa Fluor™ 647-KEAP1, Eu-Streptavidin and biotinylated BTK kinase domain. Data are presented as mean ± SD with at least three technical replicates. Representative figure from two independent experiments is shown B. Expression of BTK-eGFP-2A-mCherry was induced, and cells were treated for 24 h with increasing concentrations of the indicated small molecules. eGFP and mCherry fluorescence were quantified by flow cytometry. Data are presented as mean ± SD of two independent experiments (n = 2). C. Immunoblot analysis of protein abundant of BTK in Mino and JeKo-1 cells. Mino or JeKo-1 cells were treated for 8 h with 0.1 μM, 1 μM or 10 μM of indicated compounds. Representative blots from two independent experiments are shown. The relative intensity of each band (BTK normalized to β-actin) is shown under each band. See also Figure S4 and Figure S5.
Given this negative result, we profiled additional KEAP1 ligands conjugated to CGI-1746, as well as additional targets that have been identified as degradable in previous studies(Burslem et al., 2018; Cheng et al., 2020; Jang et al., 2020; Jiang et al., 2019; Remillard et al., 2017; Su et al., 2019; Yao et al., 2020; Yuan et al., 2021; Zoppi et al., 2018). None of the potential KEAP1-based BTK PROTACs induced degradation in GFP-BTK reporter cell lines or of endogenous BTK in multiple cells lines (Figure S4), despite successful KEAP1-BTK dimerization at around 1 μM. We also explored several other degradable targets, BRD9 (Figure S5A), CDK4/6 (Figure S5B), and EGFR (Figure S5C), but were unable to validate induced degradation of these targets. Taken together, our results demonstrate that KEAP1-based PROTACs do not represent a broadly applicable strategy for kinase degradation, suggesting that E3 ligases may display privileged POI target scope, with CRBN and VHL-based PROTACs being far more effective in degrading kinases.
Development of CRBN-based KEAP1 degraders
The question of whether an E3 ligase could be hijacked to induce degradation of another E3 ligase or itself led to the development of so-called homo- and hetero-PROTACs combining ligands for VHL and CRBN(Girardini et al., 2019; Kim et al., 2019; Maniaci et al., 2017; Powell et al., 2021; Steinebach et al., 2019; Steinebach et al., 2018). To assess the consequences of recruiting KEAP1 to other E3 ligases, we synthesized NJH-04–086 and NJH-04–087 by conjugating KI-696 to two different CRBN binding ligands through 9-atom linkers (Figure 7A). Both compounds exhibited rapid degradation of KEAP1 but spared CRBN (Figure 7B). To validate the mechanism of degradation, we tested these compounds in both wildtype and CRBN −/− MM.1S cells and observed KEAP1 degradation after 2-hour treatment with NJH-04–086 and NJH-04–087 at 5 μM in wildtype but not CRBN−/− MM.1S cells, thereby demonstrating that KEAP1 degradation is CRBN-dependent (Figure 6B). We also synthesized the negative control compound NJH-04–202, which incorporates a methylated glutarimide unable to bind to CRBN; as expected, this compound did not induce KEAP1 degradation (Figure 7C). In addition, pre-treatment of either KI-696 or lenalidomide rescued KEAP1 degradation, and similar rescue was observed with bortezomib and MLN4924 (Figure 7D). We also tested these compounds in several other cell lines including NHBE and IMR32, and observed induced degradation of KEAP1 in both cell lines (Figure S6). Finally, to assess selectivity, we performed proteomics profiling of OVCAR8 cells with treatment of NJH-04–086 and observed clear degradation of KEAP1 and a few zinc finger proteins whose abundance is commonly downregulated with treatment of thalidomide derivatives (Figure 7E). In contrast, treatment with NJH-04–087, the CRBN binder with an expanded 1H-benzo[de]isoquinoline-1,3(2H)-dione moiety, is selective for inducing degradation of KEAP1 across the proteome and does not degrade any zinc finger proteins (Figure 7E), consistent with our previous results(de Wispelaere et al., 2019; Powell et al., 2021). Notably, proteomics profiling of both compounds identified upregulation of HOMX1, a known target gene of NRF2(Ishii et al., 2000). Interestingly, the KEAP1-VHL degrader resulted in degradation of both VHL and KEAP1 at high concentrations and did not result in appreciable degradation of any E3 ligase substrate adaptor (Figure S6). Collectively, we showed that in the context of KEAP1-CRBN PROTACs, KEAP1 is the degradable target, suggesting that compounds like NJH-04–087 can be used as protein-knockdown reagents for KEAP1.
Figure 7. DGY-04–086 and DGY-04–087 induced degradation of KEAP1 in a CRBN dependent manner.
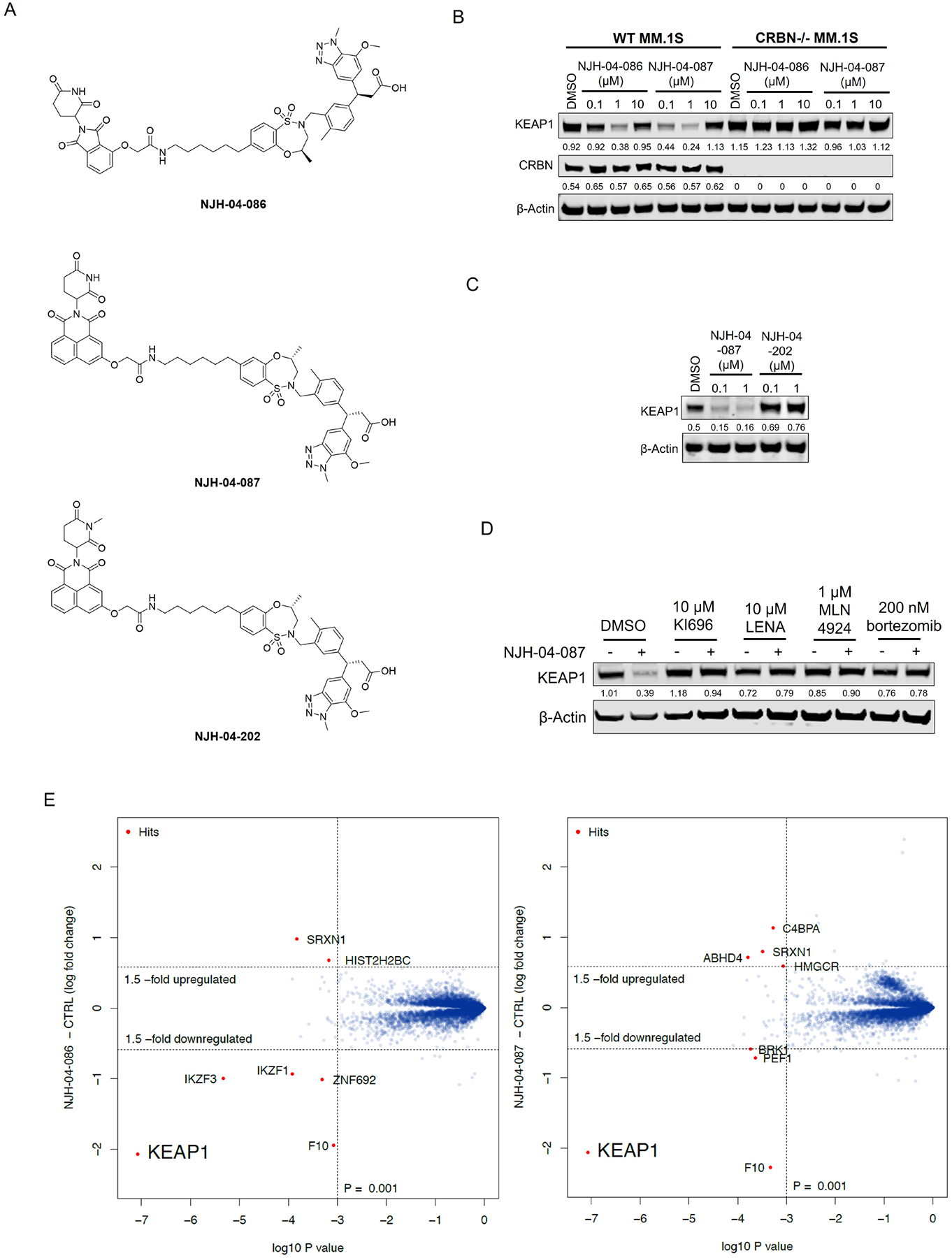
A. Chemical structure of NJH-04–086, NJH-04–087 and NJH-04–202. B. Immunoblot assessment of WT MM.1S and CRBN−/− MM.1S cells treated with NJH-04–086 or NJH-04–087 for 4 h. Representative blots from two independent experiments are shown. The relative intensity of each band (KEAP1 and CRBN normalized to β-actin) is shown under each band. C. Immunoblot assessment of WT MM.1S cells treated with NJH-04–087 or NJH-04–202 for 4 h. Representative blots from two independent experiments are shown. The relative intensity of each band (KEAP1 normalized to β-actin) is shown under each band. D. Immunoblot assessment of MM.1S cells pretreated with 10 μM of KEAP1 inhibitor, 10 μM of lenalidomide, 1 μM of MLN4924 or 200 nM of bortezomib for 2 h prior to the addition of 1 μM of NJH-04–087 for 4 h. Representative blots from two independent experiments are shown. The relative intensity of each band (KEAP1 normalized to β-actin) is shown under each band. E. Quantitative proteomics profiles of MM.1S cells treated for 5 h with 5 μM of NJH-04–086 or NJH-04–087. See also Figure S6.
DISCUSSION
Identifying additional E3 ligases that are capable of being hijacked for TPD has been recognized as one of the major challenges for the field. Here, we address this challenge by exploring KEAP1-based PROTACs that incorporate KI-696 as a KEAP1 recruiting ligand. As an initial proof-of-concept we developed a bromo-domain targeted degrader, DGY-06–177-pk2, that exhibited high potency and BRD4 selective degradation. This suggested that exploring KEAP1-based PROTACs may open additional TPD opportunities. However, given that BRD4 is a highly degradable target, we tested whether KEAP1-based PROTACs are effective as kinase degraders. We initially employed a multi-kinase degrader NJH-04–098 for profiling purposes, and discovered that FAK kinase was the only degraded target. Although we observed that FAK was degraded in mouse pancreatic KPC cells, we did not see the same effects in human cells. A possible explanation is subtle sequence/structure differences between mouse and human KEAP1, resulting in less favourable interactions between human KEAP1 and FAK. Alternatively, KEAP1 activity may be spatially restricted or otherwise differentially regulated between human and mouse cells leading to the observed differences. Therefore, as seen in other examples, each E3 ligase-POI pair represents a unique system with specific characteristics that may or may not allow for PROTAC-mediated productive complex formation. Moreover, our results show that care must be taken when using non-human cell lines as models for degrader validation given that species selectivity, as seen with molecular glue-type degraders, can be a confounding factor and yield results that don’t translate to human systems.
We also assessed the potential for KEAP1-based degraders to induce the removal of additional kinase targets previously shown to be degradable via CRBN-based PROTACs (EGFR, BTK, and CDK4/6), and did not observe any degradation in these systems. The inability to induce degradation could be due to numerous factors. For example, these compounds may not achieve effective poly-ubiquitination even though they are able to form stable ternary complexes(Zeng et al., 2020). Additionally, in comparison with VHL and CRBN, KEAP1 is a dimeric ligase complex, which may contribute to substrate specificity in a poorly understood manner leading to inactive PROTACs. Thus, exploiting KEAP1 for targeted protein degradation approaches is more complex than other ligases such as VHL and CRBN, and additional studies are necessary to elucidate the mechanisms that enable generalization of KEAP1-based PROTACs beyond the targets reported here. Moreover, our results indicate that one may need to be cautious when using BRD4 for PROTAC proof-of-concept work as this protein appears to be especially amenable to PROTAC-induced degradation. Therefore, we recommend that studies aimed at evaluating new E3 recruiting ligands that select BRD4 as a representative target should also include additional confirmatory examples beyond BRD4.
It is important to point out that the PROTAC MS83 that was recently described37, has similarities to our negative control compound NJH-05–146 which was designed with the ester to mask the free carboxylic acid. In our hands, NJH-05–146 was inactive even at prolonged timepoints and did not bind KEAP1 in biochemical assays, while MS83 was active at such prolonged timepoints. We would like to point out, that MS83 was deliberately designed as a pro-drug and differs in aspects of linker design and composition and that the active species is likely closer to MS83A and to our active PROTACs. We cannot rule out that our negative control NJH-05–146 may become active at later timepoints through the activity of esterases, but we were unable to observe such activity and did not attempt to reproduce and directly compare to MS83A. We did not pursue a pro-drug approach because we were able to achieve sufficient cell permeability using typical design principles such as reducing tPSA (190 (DGY-06–177) vs. 208 (MS83) vs. 219 (MS3A) or available H-bond donors and acceptors (Figure S7).
In addition to PROTACs in which the KEAP1 was exploited to ubiquitinate other proteins, we also explored PROTACs that feature both KEAP1 and CRBN ligands. These PROTACs resulted in potent and selective KEAP1 degradation in CRBN-dependent manner. This selective KEAP1 degrader could be a useful probe to further explore pharmacological modulation of the KEAP1/NRF2 pathway.
In summary, we present the discovery of BRD4, FAK and KEAP1 degraders derived from the potent reversible KEAP1 binder KI-696. We envision that these compounds could serve as starting points for developing more powerful tools in TPD field and for further elucidating the mechanism of KEAP1-mediated ubiquitination.
Limitation of the study
The discovery of new E3 ligases which can be hijacked by degraders and assessment of their substrate scope are very important topics which remain amongst the top challenges in the TPD field. Our study addresses these areas for the ligase KEAP1, although many key limitations remain: Cell line dependent degradation is a known issue for all TPD projects in that the molecule may only work in a handful of cell lines but not every cell line, and while we screen multiple lines, no single study can be comprehensive. Inconsistent degradability of targets in different species is another limitation, and as with cell lines no single study including ours can be comprehensive. We cannot exclude that KEAP1-based TPD probes would be highly effective in other species not tested here. It is also important to note that while we utilized highly promiscuous kinase degraders as a surrogate to sample a large target space, this is still just touching a small fraction of the human proteome and it is conceivable that KEAP1 is effective in other areas. While we attempted to investigate 6 distinct protein targets through the synthesis of approximately 50 compounds, our conclusions may be influenced by exploring an insufficient number of compounds. Achieving adequate cell penetration and catalytically competent ternary complex formation often requires extensive structure-activity relationship exploration for each scaffold. Based on the fields’ experience with degraders that recruit CRL4CRBN or CRL4VHL, certain targets require extensive optimization to achieve a degrader of reasonable potency.
SIGNIFICANCE
Targeted protein degradation (TPD) is a popular small molecule drug discovery approach due its ability to be differentiated from conventional occupancy-based inhibitors. Although several degrader molecules are currently in clinical trials, only a very limited number of E3 ligases have been exploited for the development of PROTACs with the vast majority using CRBN or VHL. While several studies have established potential new ligases for TPD, characterization is commonly limited to one target, often the highly degradable BRD4. This study is significant since it lays out a generalizable approach to assess target space for a ligase using multi-targeted degraders and experiments across two species. While the scope of KEAP1, at least for kinase degradation in humans, might be limited, this is important information for the field and the methodologies and approaches provided here will be translatable to new ligases in TPD. There are four significant finds from our study. First, we developed a successful bivalent BRD4 degrader that linked a JQ1-based BRD4 binder to a KEAP binding ligand KI696. Second, we developed a murine-specific Focal Adhesion Kinase (FAK) degrader by linking a promiscuous kinase binder TL13–87 to the KEAP1 recruiter KI696. Third, we demonstrated that it was difficult to prepare KEAP1 recruiting PROTACs that could induce the degradation of BTK, BRD9, CDK4/6, and EGFR despite these proteins being readily degraded by CRBN or VHL-based PROTACs. The data suggests that the proteins that can be degraded by KEAP1 recruiting PROTACs will be distinct from CRBN or VHL-recruiting PROTACS and that further investigation will be required to understand the target-scope of KEAP-based degraders. Fourth, we demonstrated that we could generate efficient CRBN-recruiting degraders of KEAP1 which can serve as useful tools to investigate KEAP1-dependent biology and as controls for characterizing KEAP1-based PROTACs.
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nathanael S. Gray (nsgray01@stanford.edu).
Materials availability
All of the materials support the conclusions relevant to this manuscript are available upon reasonable request from the Lead Contact without restriction. Requested compounds will be provided following completion of an MTA.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. The proteomics data wp-esf_064, wp-esf_214 and wp-esf_102 generated during this study are available at PRIDE archive PXD034424, PXD034423 and PXD034378. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Cell lines
OVCAR8 (donor sex: female), PATU-8988T (DSMZ) (donor sex: female), JeKo-1(donor sex: female) and Mino (donor sex: male) were cultured in DMEM media (Life technologies, Cat #11965118) containing 10% fetal bovine serum (Life technologies, Cat#10437028) and 1% Penicillin/Streptomycin (Life technologies, Cat#10378016). MM.1S (donor sex: female) and KPC (HY15549) (kind gift from Alec Kimmelman) were cultured in RPMI media (Life technologies, Cat# 11875119) containing 10% fetal bovine serum and 1% Penicillin/Streptomycin. All the cell lines were cultured at 37°C in 5% CO2 humidified air and tested for mycoplasma negative. Spodoptera frugiperda (Sf9) cells were cultured in ESF 921 medium (Expression Systems) at 27°C, and High Five™ cells were cultured in Sf-900 II SFM medium (Gibco) at 27°C. Both Sf9 and High Five cells were tested for mycoplasma negative.
Quantification and statistical analysis
For all experiments, the number of replicates and error bars are described in the respective figure legends.
Antiproliferation Assay
Dose-response curves were generated using non-linear regression curve fit in GraphPad Prism 9 (GraphPad Software). Data are presented as mean ± SEM with three technical replicates. Representative figure from two independent experiments is shown.
Quantification of Band Intensity
Quantification of band intensity was determined using Image J. Representative blot from two independent experiments is shown. The relative intensity of each band (target proteins normalized to loading control) is shown under each band.
TR-FRET assay
Data are presented as mean ± SD of two independent experiments (n = 2), each calculated as an average of at least three technical replicates, were plotted, and the half-maximal inhibitory concentration (IC50) values were calculated using the non-linear fit variable slope equation in GraphPad Prism 9.
BTK-GFP reporter cell line assay
Data are presented as mean ± SD of two independent experiments (n = 2) and plotted in GraphPad Prism 9.
Methods Details
Immunoblotting and antibodies
Cells were lysed in RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) (Sigma, Cat# R0278) with protease inhibitor and phosphatase inhibitor (Roche). The protein concentrations were measured by BCA analysis (Thermo Fisher Scientific, Cat # PI23225). Equal amounts of protein were resolved by 4–12% Tris-Base gels (Life Technologies, cat # NW04125BOX), and then transferred to the Immuno-Blot PVDF membrane (BioRad, cat # 1620177). Proteins were probed with appropriate primary antibodies at 4 °C overnight and then with IRDye®800-labeled goat anti-rabbit IgG (LICOR Biosciences, cat # 926–32211), IRDye®800-labeled goat anti-mouse IgG (LICOR Biosciences, cat # 926–32210) or IRDye 680RD goat anti-Mouse IgG (LICOR Biosciences , Cat # 926–68070) secondary antibodies at room temperature for 1 hour. The membranes were detected on Odyssey CLx system.
Antibodies used in this study include anti-following proteins: BRD4 (Bethyl Laboratories, A301–985A-M, 1:1000), BRD3 (Abcam, #ab228936, 1:1000), BRD2 (Bethyl Laboratories, A302–582A, 1:0000), FAK (Cell Signaling Technology, #3285, 1;1000), BTK (Cell signaling Technology, #8547S), KEAP1 (Cell Signaling Technology, #4678, 1;1000), CRBN (Cell Signaling Technology, #71810, 1:1000) and β-Actin (Cell Signaling Technology, #3700, 1:1000). Protein expression levels were quantified and normalized based on the band intensity by using ImageJ 1.53a (http://imagej.nih.gov/ij, RRID:SCR_003070).
Antiproliferation assay
MM.1S cells were seeded at the density of 10000 cells/well in 96-well plates and cultured for 12 hours. The compounds were added to the cells and incubated for 72 hr. Cell viability was determined by using CellTiter-Glo (Promega #G7571) according to the manufacturer’s instructions, measuring luminescence using an Envision plate-reader (PerkinElmer Inc.). Dose-response curves were generated using non-linear regression curve fit in GraphPad Prism 9.2.0(GraphPad Software).
qRT-PCR
1 μg of total RNA from cells was reverse transcribed with oligo(dT) and SuperScript IV Reverse Transcriptase (Life Technologies, #18090050). RT-qPCR was performed using a SYBR Green (Life Technologies, #4367659) and the 7500 Fast Real-Time PCR system (Appiled Biosystems). All runs were accompanied by the internal control GAPDH gene. The samples were run in triplicate and normalized to GAPDH using a ΔΔ cycle threshold-based algorithm to provide arbitrary units representing relative expression levels. The primers for human GCLM (Forward 5’-CAATGATCCAAAAGAACTGC-3’, Reverse 5’-CTCTACTTTTCACAATGACCG-3’), HO1 (Forward 5’- CAACAAAGTGCAAGATTCTG, Reverse 5’-TGCATTCACATGGCATAAAG-3’) and NQO1(Forward 5’- AGTATCCACAATAGCTGACG, Reverse 5’-TTTGTGGGTCTGTAGAAATG-3’) are predesigned primers ordered from Sigma Aldrich. The sequences for human GAPDH gene are: Forward 5’-GTCATGGGTGTGAACCATGAGA-3’, Reverse 5’-GGTCATGAGTCCTTCCACGATAC-3’.
Generation of KEAP1 knockdown cell lines
KEAP1 shRNA plasmid (Sigma, cat# TRCN0000158081) was purchased from Sigma. Lentiviruses were prepared by transfecting 70–80% confluent HEK293T cells together with the lentiviral vector and packing plasmids using Lipofectamine 2000 (Thermo Fisher Scientific, cat# 11668–019) mediated transfection. 48 hours after transfection, the viral supernatant was harvested and filtered with 0.45 μm membrane. OVACR8 cells were then infected by the viral supernatants with polybrene (10 μg/mL) and selected with puromycin (1 μg/mL). The knockdown efficiency was verified by Western blotting.
Protein expression and purification
The human wild-type of Keap1 (residues 321–609) was cloned in N-terminal His6 C-terminal Spy.(Abdulrahman et al., 2009) Baculovirus for protein expression (Invitrogen) was generated by transfection into Spodoptera frugiperda (Sf9) cells at a density of 0.9 × 10*6 cells/ml grown in ESF 921 medium (Expression Systems), and this was followed by three rounds of infection in Sf9 cells to increase viral titer. Recombinant Keap1 protein was expressed, and purified in Trichoplusiani High Five insect cells using the baculovirus expression system (Invitrogen) and was subsequently directly covalently labeled with Alexa Fluor™ 647-SpyCatcherS50C as described previously.(Nowak et al., 2018) The biotinylated human BRD4BD2 and human BTK were expressed and purified as described previously.
KEAP1-NRF2 displacement TR-FRET assay
Competitive titration of compounds was carried out by mixing 100 nM Alexa Fluor™ 647-KEAP1, 100 nM biotinylated Nrf peptide (sequence: GSGAFFAQLQLDEETGEFL) and 2 nM Eu-Streptavidin (Invitrogen, cat# PV5899) in the assay buffer containing 50 mM Tris pH 8.0, 200 mM NaCl, 0.1% Pluronic F-68 solution (Sigma), 0.5% bovine serum albumin (BSA) (w/v) and 1 mM TCEP. After dispensing the assay mixture (15 μl volume), increasing concentrations of compounds were dispensed in a 384-well microplate (Corning, 4514) using a D300e Digital Dispenser (HP) normalized to 1% DMSO. After excitation of europium fluorescence at 337 nm, emission at 620 nm (europium) and 665 nm (Alexa Fluor™ 647) were recorded with a 70-μs delay over 600 μs to reduce background fluorescence, and the reaction was followed over 60 cycles of each data point using a PHERAstar FS microplate reader (BMG Labtech). The TR-FRET signal of each data point was extracted by calculating the 665/620 nm ratio. Data from two independent measurements (n = 2), each calculated as an average of at least three technical replicates per well per experiment, were plotted, and the half-maximal inhibitory concentration (IC50) values were calculated using the non-linear fit variable slope equation in GraphPad Prism 9.
KEAP1-BRD4 and KEAP1-BTK Dimerization assay
The assay mix contained 200 nM biotinylated BRD4BD2 or BTK(residue 396–659), 200 nM Alexa Fluor™ 647-KEAP1, and 2 nM Eu-Streptavidin in the same assay buffer described above. The reactions were incubated for 15 min at room temperature before TR-FRET measurements were conducted. After excitation of europium fluorescence at 337 nm, emission values at 620 nm (europium) and 665 nm (Alexa Fluor™ 647) were recorded with a 70-μs delay over 600 μs to reduce background fluorescence, and the reaction was followed over thirty 200-s cycles of each data point using a PHERAstar FS microplate reader (BMG Labtech). The 665/620 nm ratios were measured as described above. Data from two independent measurements (n = 2), each calculated as an average of at least three technical replicates per well per experiment, were plotted in GraphPad Prism 9.
BTK-GFP reporter cell line assay
BTK was subcloned into mammalian pcDNA5/FRT Vector (Ampicillin and Hygromycin B resistant) modified to contain MCS-eGFP-P2A-mCherry. Stable cell lines expressing eGFP-protein fusion and mCherry reporter were generated using Flip-In 293 system as previously described.(Nowak et al., 2018) Cells were seeded at 30–50% confluency in 96-well plates (3596, Costar) a day before compound treatment. Titrated compounds (were incubated with cells for 24h following trypsinization and resuspension in DMEM media, transferred into 96-well plates (353910, Falcon) and analyzed by flow cytometer (guava easyCyte HT, Millipore). Signal from minimal 3000 events per well was acquired and the eGFP and mCherry florescence monitored. Data was analyzed using FlowJo (FlowJo, LCC). Live cells were gated on forward and side scatter population followed by gating by eGFP and mCherry to remove outliers. The eGFP protein abundance relative to mCherry was quantified for each cell using the formula: 10 × eGFP/mCherry. The median of the eGFP/mCherry ratio was then calculated for final gated population in each well.
Proteomics
TMT-based quantitative LCMS proteomics
HEK239T, MM.1S or KPC cells were treated with DMSO (biological triplicate) or indicated compounds at different concentrations for certain time points and cells were harvested by centrifugation at 4 °C. Cell lysis was performed by the addition of Urea buffer (8 M Urea, 50 mM NaCl, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (EPPS) pH 8.5, Protease and Phosphatase inhibitors) followed by manual homogenization by 20 passes through a 21-gauge (1.25 in. long) needle. Lysate was clarified by centrifugation and protein quantified using bradford (Bio-Rad) assay. 200 μg of protein for each sample was reduced, alkylated and precipitated using methanol/chloroform as previously described. The resulting precipitated protein was resuspended in 4 M Urea, 50 mM HEPES pH 7.4, buffer for solubilization, followed by dilution to 1 M urea with the addition of 200 mM EPPS, pH 8. Proteins were digested for 12 hours at room temperature with LysC (1:50 ratio), followed by dilution to 0.5 M urea and a second digestion step was performed by addition of trypsin (1:50 ratio) for 6 hours at 37 °C. Anhydrous ACN was added to each peptide sample to a final concentration of 30%, followed by addition of Tandem mass tag (TMT) reagents at a labelling ratio of 1:4 peptide:TMT label. TMT labelling occurred over a 1.5 hour incubation at room temperature followed by quenching with the addition of hydroxylamine to a final concentration of 0.3%. Each of the samples were combined using adjusted volumes and dried down in a speed vacuum followed by desalting with C18 SPE (Sep-Pak, Waters). The sample was offline fractionated into 96 fractions by high pH reverse-phase HPLC (Agilent LC1260) through an aeris peptide xb-c18 column (phenomenex) with mobile phase A containing 5% acetonitrile and 10 mM NH4HCO3 in LC-MS grade H2O, and mobile phase B containing 90% acetonitrile and 5 mM NH4HCO3 in LC-MS grade H2O (both pH 8.0). The resulting 96 fractions were recombined in a non-contiguous manner into 24 fractions and desalted using C18 solid phase extraction plates (SOLA, Thermo Fisher Scientific) followed by subsequent mass spectrometry analysis.
Data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with an Proxeon EASY-nLC 1200 LC lump (Thermo FIsher Scientific, San Jose, CA, USA). Peptides were separated on a 50 cm 75 μm inner diameter EasySpray ES803/903 microcapillary column (Thermo Fisher Scientific). Peptides were separated over a 190 min gradient of 6 – 27% acetonitrile in 1.0% formic acid with a flow rate of 300 nL/min.
Quantification was performed using a MS3-based TMT method as described previously (McAlister et al., 2014). The data were acquired using a mass range of m/z 340 – 1350, resolution 120,000, AGC target 5 × 105, maximum injection time 100 ms, dynamic exclusion of 120 seconds for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104 and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with HCD collision energy set to 55%, AGC target set to 2 × 105, maximum injection time of 150 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
LC-MS data analysis
Proteome Discoverer 2.1, 2.2 or 2.4 (Thermo Fisher Scientific) was used for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. The MS/MS spectra were searched against a Swissprot human database (September 2016 or December 2019) containing both the forward and reverse sequences. Searches were performed using a 20 ppm precursor mass tolerance, 0.6 Da fragment ion mass tolerance, tryptic peptides containing a maximum of two missed cleavages, static alkylation of cysteine (57.02146 Da), static TMT labelling of lysine residues and N-termini of peptides (229.1629 Da for 10/11-plex or 304.2071 For 16-plex), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. The peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (summed signal-to-noise across channels < 100 and precursor isolation specificity < 0.5), and the resulting data was filtered to only include proteins with a minimum of 2 unique peptides quantified. Reporter ion intensities were normalized and scaled using in-house scripts in the R framework. Statistical analysis was carried out using the limma package within the R framework.
Chemical synthesis and Structure Validation
Starting materials, reagents and solvents were purchased from commercial suppliers and were used without further purification unless otherwise noted. All reactions were monitored using a Waters Acquity UPLC/MS system (Waters PDA eλ Detector, QDa Detector, Sample manager - FL, Binary Solvent Manager) using Acquity UPLC® BEH C18 column (2.1 × 50 mm, 1.7μm particle size): solvent gradient = 85% A at 0 min, 1% A at 1.7 min; solvent A = 0.1% formic acid in Water; solvent B = 0.1% formic acid in Acetonitrile; flow rate: 0.6mL/min. Reaction products were purified by flash column chromatography using CombiFlash®Rf with Teledyne Isco RediSep® normal-phase silica flash columns (4 g, 12 g, 24 g, 40 g or 80 g) and Waters HPLC system using SunFireTM Prep C18 column (19 × 100 mm, 5 μm particle size): solvent gradient = 80% A at 0 min, 10% A at 25 min; solvent A = 0.035% TFA in Water; solvent B = 0.035% TFA in MeOH; flow rate: 25 mL/min. 1H NMR spectra were recorded on 500 MHz Bruker Avance III spectrometers and 13C NMR spectra were recorded on 125 MHz Bruker Avance III spectrometer. Chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as br (broad), s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). The 13C NMR data of the compounds, NJH-02–160 and NJH-02–162, are not available because of the difficulty in synthesis and poor yield of these compounds.
Synthesis of (S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-(((R)-4methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoic acid (KI696)
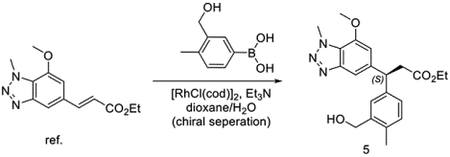
To the solution of ethyl (E)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)acrylate (375 mg, 1.44 mmol)(Davies et al., 2016), (3-(hydroxymethyl)-4-methylphenyl)boronic acid (3.58 mg, 2.16 mmol) and Et3N (218 mg, 2.16 mmol) in dioxane/H2O (10/5 mL). Then [RhCl(cod)]2 (35.4 mg, 0.072 mmol) was added to the mixture. The reaction was heated to 95 °C for 1 hour. The reaction mixture was extracted with ethyl acetate and purified with chromatography to give racemic mixtures ethyl 3-(3-(hydroxymethyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate. Then the mixture was purified by chiral column to get the right isomer ethyl (S)-3-(3-(hydroxymethyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate.
LC/MS m/z calculated for [M+H]+ 384.2, found 384.4.
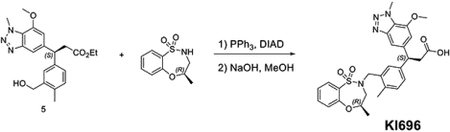
To the solution of (S)-3-(3-(hydroxymethyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (30 mg, 0.078 mmol) and (R)-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide (25 mg, 0.117 mmol) in THF (1 mL), PPh3 (41 mg, 0.156 mmol) and DIAD (31, 0.156 mmol) were added at room temperature. The mixture was stirred for 30 mins. The solvent was removed under vacuum and dissolved in MeOH (2 mL). NaOH (2M aq. Solution, 0.5 mL) was added to the resulting mixture and then heated up to 80 °C for 1 hour. The residue was purified by reverse phase HPLC to give (S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-(((R)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoic acid (26.2 mg, 0.047 mmol, 61%).
LC/MS m/z calculated for [M+H]+ 551.2, found 550.8.
1H NMR (500 MHz, DMSO-d6) δ 7.78 (dd, J = 7.8, 1.7 Hz, 1H), 7.67 (td, J = 7.8, 1.7 Hz, 1H), 7.45 (s, 1H), 7.40 − 7.34 (m, 2H), 7.33 − 7.23 (m, 2H), 7.12 (d, J = 7.8 Hz, 1H), 6.93 (s, 1H), 4.51 (t, J = 8.0 Hz, 1H), 4.43 (d, J = 14.1 Hz, 1H), 4.40 − 4.35 (m, 1H), 4.34 (s, 3H), 3.94 (s, 3H), 3.81 (d, J = 14.0 Hz, 2H), 3.10 (t, J = 7.8 Hz, 2H), 2.79 − 2.72 (m, 1H), 2.24 (s, 3H), 1.10 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.19, 155.39, 147.90, 146.14, 142.52, 142.12, 135.48, 134.08, 133.66, 131.10, 129.42, 129.10, 127.81, 125.05, 124.56, 124.23, 108.62, 107.64, 73.67, 56.66, 52.63, 48.96, 46.86, 37.02, 19.12, 18.51.
The absolute configuration of compound KI696 was determined by comparing its 1H NMR spectrum with the published data.
Synthesis of (S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-((N-methylphenylsulfonamido)methyl)phenyl)propanoic acid (DGY-04–091)
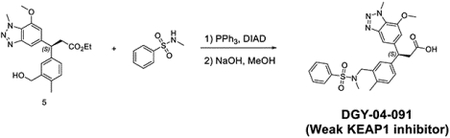
To the solution of (S)-3-(3-(hydroxymethyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (60 mg, 0.16 mmol) and N-methylbenzenesulfonamide (40 mg, 0.23 mmol) in THF (1 mL), PPh3 (84 mg, 0.32 mmol) and DIAD (64.7, 0.32 mmol) were added at room temperature. The mixture was stirred for 30 mins. The solvent was removed under vacuum and dissolved in MeOH (2 mL). NaOH (2M aq. Solution, 0.5 mL) was added to the resulting mixture and then heated up to 80 °C for 1 hour. The residue was purified by reverse phase HPLC to give (S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-((N-methylphenylsulfonamido)methyl)phenyl)propanoic acid (15 mg, 0.029 mmol, 18%).
LC/MS m/z calculated for [M+H]+ 509.2, found 509.2.
1H NMR (500 MHz, DMSO-d6) δ 7.94 − 7.83 (m, 2H), 7.78 − 7.73 (m, 1H), 7.69 (dd, J = 8.4, 6.9 Hz, 2H), 7.43 (s, 1H), 7.29 (s, 1H), 7.24 (dd, J = 7.8, 2.0 Hz, 1H), 7.11 (d, J = 7.8 Hz, 1H), 6.89 (s, 1H), 4.50 (t, J = 8.0 Hz, 1H), 4.33 (s, 3H), 4.09 − 4.04 (m, 2H), 3.90 (s, 3H), 3.07 (d, J = 8.0 Hz, 2H), 2.43 (s, 3H), 2.28 (s, 3H).
Synthesis of 3-(3-(((4-((6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)oxy)-N-methylphenyl)sulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-05–089).
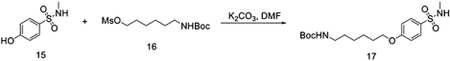
To a solution 4-hydroxy-N-methylbenzenesulfonamide (300 mg, 1.60 mmol) and 6-((tert-butoxycarbonyl)amino)hexyl methanesulfonate (520 mg, 1.76 mmol) in dimethylformamide (DMF) (8 mL), K2CO3 (441 mg, 3.20 mmol) was added at room temperature. Then the mixture was stirred at this temperature overnight. The mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by flash chromatography to yield tert-butyl 6-(4-(N-methylsulfamoyl)phenoxy)hexylcarbamate (600 mg, 1.55 mmol, 97%).
LC/MS m/z calculated for [M+H]+ 387.2, found 387.1.
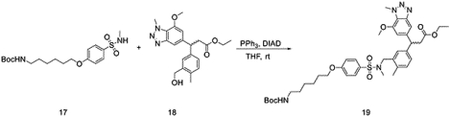
To a solution of tert-butyl 6-(4-(N-methylsulfamoyl)phenoxy)hexylcarbamate (131 mg, 0.34 mmol) and ethyl 3-(3-(hydroxymethyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate(Davies et al., 2016) (100 mg, 0.26 mmol) in THF (2 mL), diisopropyl azodicarboxylate (DIAD) (105 mg, 0.52 mmol) and PPh3 (136 mg, 0.52 mmol) were added at room temperature. The reaction mixture was stirred for 30 mins. The mixture was evaporated under vacuum, and the residue was purified by flash chromatography to yield ethyl 3-(3-((4-(6-(tert-butoxycarbonylamino)hexyloxy)-N-methylphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (27 mg, 0.036 mmol, 14%).
LC/MS m/z calculated for [M+H]+ 752.4, found 752.6.
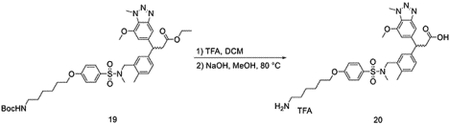
To a solution of ethyl 3-(3-((4-(6-(tert-butoxycarbonylamino)hexyloxy)-N-methylphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (27 mg, 0.036 mmol) in DCM (0.5 mL), TFA (0.5 mL) was added dropwise at room temperature. After 1 hour, the mixture was evaporated under vacuum, and the residue was dissolved in MeOH (1 mL). NaOH (0.2 mL, 2N aq.) was added to the mixture at room temperature. Then the mixture was heated up to 80 °C for 30 mins. After it was allowed to cool down, the mixture was purified by HPLC to yield 3-(3-((4-(6-aminohexyloxy)-N-methylphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (12 mg, 0.017 mmol, 46%) as a TFA salt.
LC/MS m/z calculated for [M+H]+ 624.3, found 624.1.
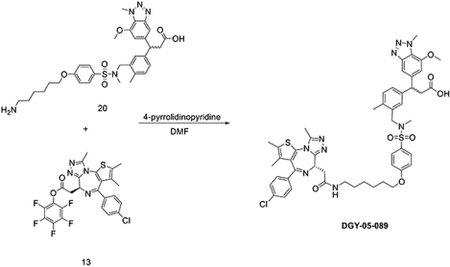
To a solution of 3-(3-((4-(6-aminohexyloxy)-N-methylphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (12 mg, 0.017 mmol) and (S)-perfluorophenyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate (9.6 mg, 0.017 mmol) in DMF (1 mL), 4-pyrrolidinopyridine (5 mg, 0.034 mmol) was added at room temperature. After 3 hours, the mixture was purified by HPLC to yield bifunctional compound DGY-05–089 (11 mg, 0.011 mmol, 63%) as a TFA salt.
LC/MS m/z calculated for [M+H]+ 1006.3, found 1006.2.
1H NMR (500 MHz, DMSO-d6) δ 8.12 (t, J = 5.6 Hz, 1H), 7.76 − 7.60 (m, 2H), 7.44 − 7.31 (m, 5H), 7.20 (d, J = 1.9 Hz, 1H), 7.16 (dd, J = 7.8, 1.9 Hz, 1H), 7.13 − 7.05 (m, 2H), 7.03 (d, J = 7.9 Hz, 1H), 6.81 (s, 1H), 4.50 − 4.39 (m, 2H), 4.25 (s, 3H), 4.01 (t, J = 6.5 Hz, 2H), 3.93 (s, 2H), 3.82 (s, 3H), 3.29 − 2.94 (m, 6H), 2.53 (s, 3H), 2.35 − 2.28 (m, 6H), 2.20 (s, 3H), 1.70 − 1.63 (m, 2H), 1.55 (d, J = 0.9 Hz, 3H), 1.46 − 1.28 (m, 5H). 13C NMR (126 MHz, DMSO) δ 173.18, 169.78, 163.57, 162.62, 155.57, 150.39, 147.90, 146.13, 142.46, 141.81, 137.14, 135.77, 135.60, 133.69, 132.69, 131.30, 131.03, 130.61, 130.31, 130.08, 129.46, 128.93, 127.71, 127.67, 124.24, 115.41, 108.83, 107.51, 68.55, 56.61, 54.36, 52.57, 46.87, 38.86, 38.09, 37.01, 34.53, 29.65, 28.96, 26.57, 25.65, 18.58, 14.51, 13.14, 11.76.
(mixtures of two diastereomers)
Synthesis of 3-(3-(((R)-7-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-06–177).
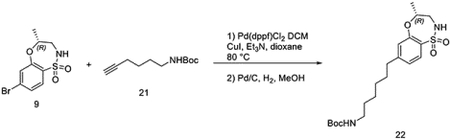
To a solution of (R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide (50 mg, 0.17 mmol), tert-butyl hex-5-yn-1-ylcarbamate (67 mg, 0.34 mmol) and Et3N (171 mg, 1.70 mmol) in dioxane (2 mL), CuI (13 mg, 0.068 mmol) and Pd(dppf)Cl2·DCM (28 mg, 0.034 mmol) were added at room temperature under N2 atmosphere. Then the reaction mixture was heated up to 80 °Cfor 2 hours. The mixture was filtered, and the filtrate was evaporated under vacuum. To a solution of the obtained residue in MeOH (3 mL), Pd/C (8.4 mg) was added slowly. The suspension was stirred overnight under hydrogen atmosphere. The mixture was filtered with celite to get the crude product tert-butyl (R)-(6-(4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)hexyl)carbamate without any further purification.
LC/MS m/z calculated for [M+H]+ 413.2, found 313.2 (de-Boc).
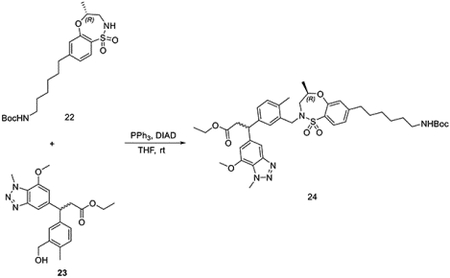
To a solution of tert-butyl (R)-(6-(4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)hexyl)carbamate 22 (88 mg, 0.21 mmol) and ethyl 3-(3-(hydroxymethyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (82 mg, 0.21 mmol) in THF (2 mL), DIAD (85 mg, 0.42 mmol) and PPh3 (110 mg, 0.42 mmol) were added at room temperature. The reaction mixture was stirred for 30 mins before evaporation under vacuum. The residue was purified by flash chromatography to yield ethyl 3-(3-(((R)-7-(6-((tert-butoxycarbonyl)amino)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate 24 (100 mg, 0.13 mmol, 61%).
LC/MS m/z calculated for [M+H]+ 778.4, found 778.4.
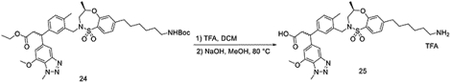
To a solution of ethyl 3-(3-(((R)-7-(6-((tert-butoxycarbonyl)amino)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate 24 (100 mg, 0.13 mmol) in DCM (2 mL), TFA (1 mL) was added dropwise at room temperature. After 1 hour, the mixture was evaporated under vacuum. To a solution of the obtained residue in MeOH (2 mL), NaOH (0.2 mL, 2N aq.) was added at room temperature. Then the mixture was heated up to 80 °C for 30 mins. After it was allowed to cool down, the mixture was purified by HPLC to yield 3-(3-(((R)-7-(6-aminohexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid 25 (67 mg, 0.10 mmol, 79%) as a TFA salt.
LC/MS m/z calculated for [M+H]+ 650.3, found 650.4.
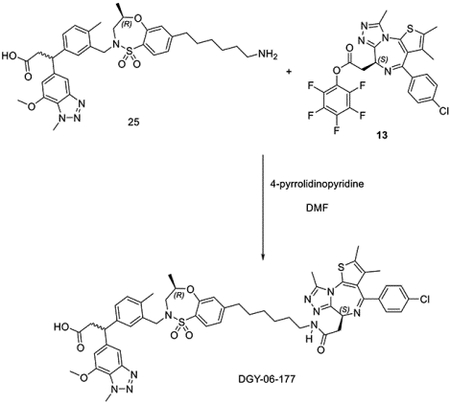
To a solution of 3-(3-(((R)-7-(6-aminohexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid 25 (24 mg, 0.031 mmol) and (S)-perfluorophenyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate 13 (17 mg, 0.031 mmol) in DMF (2 mL), 4-pyrrolidinopyridine (9 mg, 0.062 mmol) was added at room temperature. After 3 hours, the mixture was purified by HPLC to yield bifunctional compound 3-(3-(((R)-7-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid DGY-06–177 (22 mg, 0.021 mmol, 68%) as TFA salt.
LC/MS m/z calculated for [M+H]+ 1032.4, found 1032.5.
1H NMR (500 MHz, DMSO-d6) δ 8.10 (t, J = 5.7 Hz, 1H), 7.58 (dd, J = 7.9, 2.1 Hz, 1H), 7.44 − 7.33 (m, 5H), 7.28 (t, J = 2.5 Hz, 1H), 7.20 (td, J = 7.7, 1.9 Hz, 1H), 7.16 − 7.00 (m, 3H), 6.98 − 6.76 (m, 1H), 4.49 − 4.40 (m, 2H), 4.34 (d, J = 14.2 Hz, 1H), 4.25 (d, J = 4.1 Hz, 3H), 3.85 (d, J = 5.7 Hz, 3H), 3.71 (d, J = 14.0 Hz, 1H), 3.52 (ddd, J = 14.9, 10.1, 4.3 Hz, 1H), 3.27 − 2.95 (m, 6H), 2.69 (dd, J = 39.4, 15.0 Hz, 1H), 2.56 (t, J = 7.8 Hz, 2H), 2.52 (s, 3H), 2.16 (d, J = 5.1 Hz, 3H), 1.53 (m, 4H), 1.38 (d, J = 6.7 Hz, 2H), 1.27 (t, J = 4.2 Hz, 4H), 1.06 (dd, J = 47.9, 6.3 Hz, 3H). (mixtures of diastereomers)
Synthesis of (R)-3-(3-(((R)-7-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-06–177-pk1)
DGY-06–177-pk1 was synthesized starting from R-isomer of compound 5 (Scheme 1) using the same procedure as used for DGY-06–177.
Scheme 1.
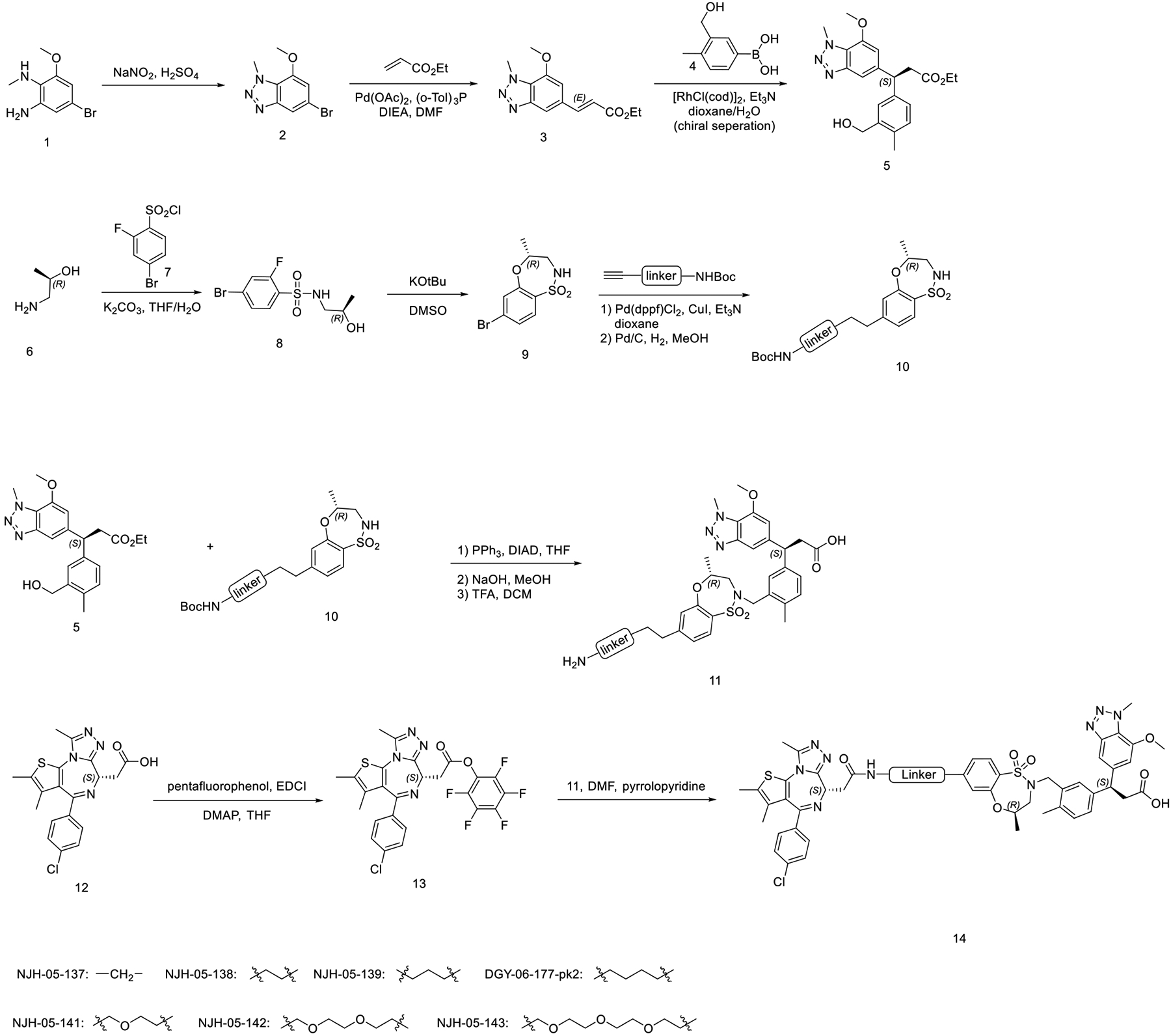
Design and synthesis of KEAP1-BRD4 degraders.
LC/MS m/z calculated for [M+H]+ 1032.4, found 1032.4.
1H NMR (500 MHz, DMSO-d6) δ 12.13 (s, 1H), 8.17 (t, J = 5.6 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.54 − 7.40 (m, 5H), 7.36 (d, J = 1.9 Hz, 1H), 7.33 − 7.25 (m, 1H), 7.18 − 7.08 (m, 3H), 6.91 (s, 1H), 4.51 (dd, J = 8.2, 6.5 Hz, 2H), 4.45 − 4.39 (m, 2H), 4.33 (s, 3H), 3.92 (s, 3H), 3.79 (d, J = 14.0 Hz, 1H), 3.67 − 3.49 (m, 1H), 3.31 − 3.04 (m, 5H), 2.81 (d, J = 15.1 Hz, 1H), 2.64 (t, J = 7.8 Hz, 2H), 2.59 (s, 3H), 2.39 (s, 3H), 2.25 (s, 3H), 1.60 (s, 4H), 1.51 − 1.41 (m, 3H), 1.34 (dt, J = 6.1, 3.5 Hz, 4H), 1.24 (s, 2H), 1.19 (dd, J = 12.8, 6.6 Hz, 4H), 1.16 − 1.10 (m, 5H), 1.06 (d, J = 6.1 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.23, 169.81, 163.46, 155.36, 150.93, 147.93, 146.11, 142.57, 141.92, 137.17, 135.72, 135.59, 133.70, 131.35, 131.17, 131.08, 130.60, 130.25, 130.01, 129.56, 129.03, 128.93, 127.89, 124.75, 124.20, 124.06, 108.76, 107.55, 73.44, 70.07, 56.64, 54.42, 51.04, 48.88, 46.85, 38.86, 38.16, 37.03, 35.24, 30.92, 29.64, 29.57, 28.97, 26.61, 19.30, 18.59, 14.52, 13.12, 11.78.
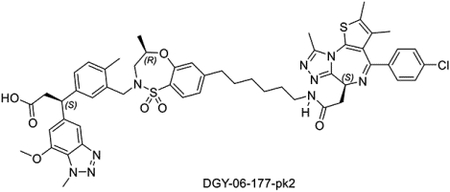
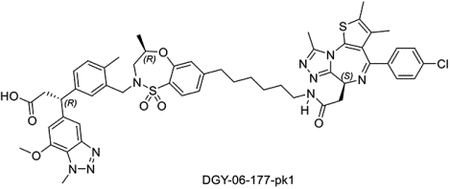
Synthesis of (S)-3-(3-(((R)-7-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-06–177-pk2)
DGY-06–177-pk2 was synthesized starting from S-isomer of compound 5 (Scheme 1) using the same procedure as used for DGY-06–177.
LC/MS m/z calculated for [M+H]+ 1032.4, found 1032.5.
1H NMR (500 MHz, DMSO-d6) δ 11.88 (s, 1H), 7.94 (t, J = 5.6 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.26 (d, J = 8.6 Hz, 2H), 7.23 − 7.17 (m, 3H), 7.12 (d, J = 1.9 Hz, 1H), 7.05 (dd, J = 7.7, 1.9 Hz, 1H), 6.93 (dd, J = 8.0, 1.6 Hz, 1H), 6.90 − 6.85 (m, 2H), 6.70 (s, 1H), 4.28 (ddd, J = 10.0, 6.9, 3.2 Hz, 2H), 4.22 − 4.13 (m, 2H), 4.11 (s, 3H), 3.70 (s, 3H), 3.56 (d, J = 14.0 Hz, 1H), 3.36 (dd, J = 15.4, 10.1 Hz, 1H), 3.08 − 2.87 (m, 6H), 2.50 (d, J = 14.9 Hz, 1H), 2.41 (t, J = 7.8 Hz, 2H), 2.36 (s, 3H), 2.17 (s, 3H), 2.01 (s, 3H), 1.38 (s, 4H), 1.35 (s, 2H), 1.23 (d, J = 9.7 Hz, 4H), 1.18 − 1.06 (m, 4H), 0.86 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.22, 169.82, 163.46, 155.58, 155.36, 150.95, 150.29, 147.95, 146.10, 142.55, 142.18, 137.18, 135.73, 135.52, 133.69, 132.74, 131.35, 131.18, 131.06, 130.61, 130.25, 130.01, 129.61, 129.04, 128.94, 127.83, 124.77, 124.20, 124.08, 108.63, 107.67, 73.41, 56.66, 54.41, 52.48, 48.87, 46.88, 38.86, 38.17, 37.04, 35.24, 30.92, 29.64, 29.50, 28.97, 26.61, 19.19, 18.56, 14.53, 13.13, 11.78.
Synthesis of 3-(7-(2-((2-(2-(2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethoxy)ethoxy)ethyl)amino)-2-oxoethoxy)-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-((4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoic acid (DGY-03–188).
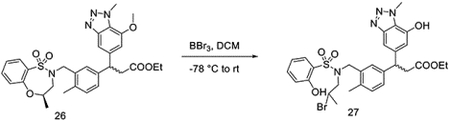
To a solution of 26 (followed the same procedure of KI696 with racemic compound 5) (180 mg, 0.31 mmol) in DCM (2 mL), BBr3 (1.56 mL, 1M in DCM, 1.56 mmol) was added dropwise at −78 °C. The reaction was stirred at this temperature for 1 hour, then allowed to warm to room temperature. The mixture was stirred at room temperature for an additional 6 hours. The reaction was diluted with ethyl acetate, quenched with NaHCO3 (sat. aq.), and extracted with EtOAc. The pooled organic layer was dried over MgSO4, and concentrated under vacuum to yield ethyl 3-(3-((N-(2-bromopropyl)-2-hydroxyphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-hydroxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (27) without any other purification.
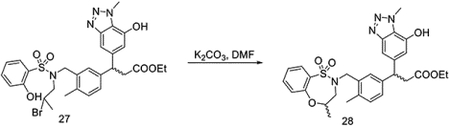
To a solution of 27 (162 mg, 0.25 mmol) in DMF (3 mL), K2CO3 (104 mg, 0.75 mmol) was added in one portion at room temperature. The reaction was stirred at room temperature for 1 hour. The reaction was quenched with water, extracted with EtOAc, and dried with MgSO4. The organic layer was concentrated under vacuum to give the crude 28 without any further purification.
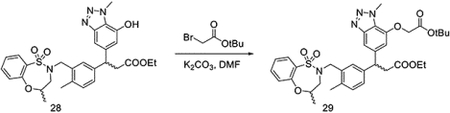
To a solution of 28 (96 mg, 0.17 mmol) in DMF (1 mL), tert-butyl 2-bromoacetate (49.7 mg, 0.26 mmol) and K2CO3 (71.8 mg, 0.52 mmol) were added slowly at room temperature. The reaction was stirred overnight before filtration. The filtrate was concentrated under vacuum and purified by HPLC to yield intermediate 29 (90 mg, 0.13 mmol, 78%).
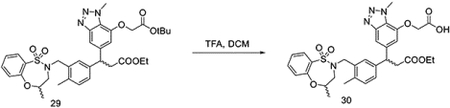
To a solution of 29 (90 mg, 0.13 mmol) in DCM (1 mL), TFA (0.5 mL) was added dropwise at room temperature. The reaction was stirred for 1 hour. The solvent was removed under vacuum to yield the crude 30 without any further purification.
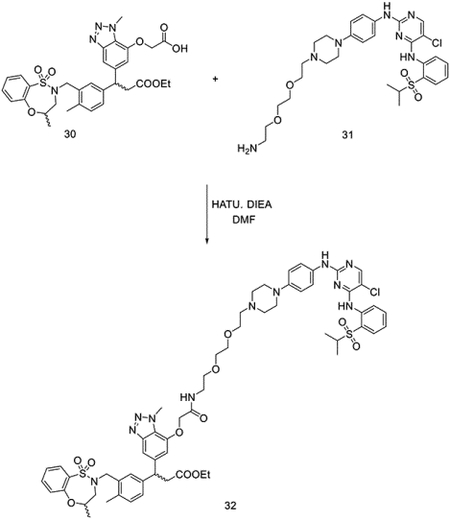
To a solution of 30 (81 mg, 0.13 mmol) and N2-(4-(4-(2-(2-(2-aminoethoxy)ethoxy)ethyl)piperazin-1-yl)phenyl)-5-chloro-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (97 mg, 0.13 mmol) in DMF (2 mL), N, N-diisopropylethylamine (DIEA) (84 mg, 0.65 mmol) and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (99 mg, 0.26 mmol) were added at room temperature. The reaction was stirred for 10 mins before purification by HPLC without any reaction workup to yield bifunctional compound 32 (122 mg, 0.10 mmol, 79%).
LC/MS m/z calculated for [M+H]+ 1222.5, found 1222.3.
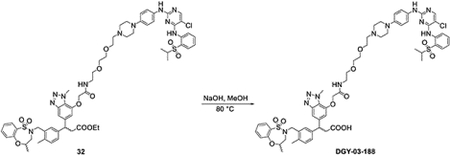
To a solution of bifunctional compound 32 (20 mg, 0.016 mmol) in MeOH (0.5 mL), NaOH (0.04 mL, 2M) was added at room temperature. The mixture was heated to 80 °C for 1 hour. The reaction was purified by HPLC without any reaction workup to yield bifunctional compound DGY-03–188 (11 mg, 0.0092 mmol, 57%).
LC/MS m/z calculated for [M+H]+ 1194.4, found 1194.4.
1H NMR (500 MHz, DMSO-d6) δ 9.69 (s, 1H), 9.45 (s, 1H), 9.33 (s, 1H), 8.57 (s, 1H), 8.18 (s, 1H), 8.05 (q, J = 5.6 Hz, 1H), 7.77 (dd, J = 8.1, 1.5 Hz, 1H), 7.74 − 7.64 (m, 2H), 7.58 (t, J = 7.8 Hz, 1H), 7.41 (d, J = 8.8 Hz, 3H), 7.34 − 7.21 (m, 4H), 7.16 (t, J = 8.7 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.86 (d, J = 8.7 Hz, 2H), 6.79 (d, J = 12.9 Hz, 1H), 4.62 (d, J = 6.3 Hz, 2H), 4.47 − 4.21 (m, 5H), 3.76 − 3.60 (m, 4H), 3.58 − 3.45 (m, 6H), 3.41 − 3.28 (m, 4H), 3.27 − 3.07 (m, 3H), 3.07 − 2.84 (m, 4H), 2.70 (dd, J = 42.7, 15.1 Hz, 1H), 2.17 (d, J = 4.2 Hz, 3H), 1.15 − 0.94 (m, 9H). 13C NMR (126 MHz, DMSO) δ 173.20, 167.84, 159.00, 158.73, 157.96, 155.42, 155.32, 155.11, 148.00, 145.50, 144.48, 138.37, 135.55, 135.42, 133.94, 133.64, 133.31, 131.41, 131.14, 129.06, 125.08, 124.54, 124.42, 121.84, 116.91, 73.74, 69.95, 69.63, 69.11, 68.09, 64.53, 55.50, 55.39, 51.80, 46.75, 46.58, 38.70, 37.09, 19.18, 19.07, 18.51, 18.47, 15.23. (mixtures of diastereomers)
Synthesis of (S)-3-(3-(((R)-7-(3-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–137).
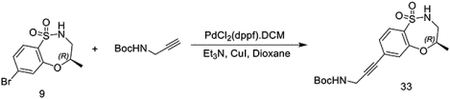
(R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide (300 mg, 1.03 mmol) was dissolved in dioxane (5 mL). N-Boc-propargylamine (191 mg, 1.23 mmol), Et3N (430 μL, 3.09 mmol), CuI (39 mg, 190 mmol), and PdCl2(dppf)•DCM (84 mg, 0.103 mmol) were then added. The mixture was degassed and sparged with N2 and stirred at 80 °C for 5h. The reaction mixture was then filtered, diluted with water and extracted with ethyl acetate. Combined extracts were washed with brine, dried over Na2SO4, and concentrated before purification by silica gel chromatography using a gradient of 0 – 60% EtOAc/Hexanes. The resulting residue was then purified by HPLC to provide tert-butyl (R)-(3-(4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)prop-2-yn-1-yl)carbamate as a colorless oil (279 mg, 0.76 mmol, 74%).
LC/MS m/z calculated for [M+H-COOC(CH3)3]+ 266.1, found 266.9.
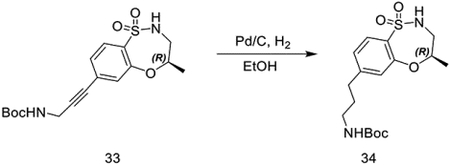
tert-butyl (R)-(3-(4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)prop-2-yn-1-yl)carbamate (279 mg, 0.76 mmol) was dissolved in ethanol (20 mL), and palladium on carbon (50 mg, 10%) was added. The vessel was flushed with H2 and stirred vigorously under H2 (1 atm) overnight. Pd/C was removed by filtration through Celite and concentrated in vacuo to obtain tert-butyl (R)-(3-(4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)propyl)carbamate as a colorless oil (245 mg, 0.65 mmol, 87%).
LC/MS m/z calculated for [M+H-OC(CH3)3]+ 314.2, found 314.8.
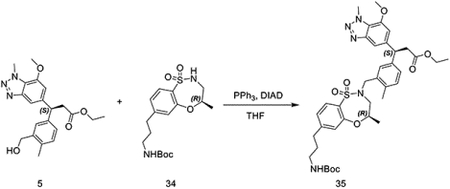
tert-butyl (R)-(3-(4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)propyl)carbamate (56 mg, 0.15 mmol) was dissolved in THF (2 mL) and ethyl (S)-3-(3-(hydroxymethyl)phenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (58 mg, 0.15 mmol) was added. Triphenylphosphine (79 mg, 0.30 mmol) was added, followed by DIAD (61 mg, 0.030 mmol). After 20 minutes, DMSO was added and the solution was purified by HPLC to provide ethyl (S)-3-(3-(((R)-7-(3-((tert-butoxycarbonyl)amino)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate as a brown oil (54 mg, 0.073 mmol, 49%).
LC/MS m/z calculated for [M+H]+ 736.3, found 735.8.
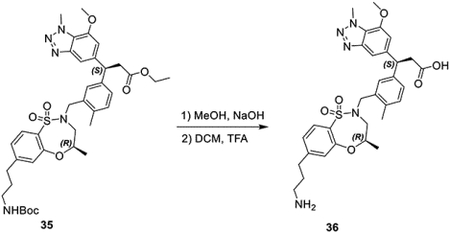
ethyl (S)-3-(3-(((R)-7-(3-((tert-butoxycarbonyl)amino)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (54 mg, 0.073 mmol) was dissolved in MeOH and aqueous NaOH (100 μL, 5M) was added, and the suspension stirred at 80 °C for 3 hours. After cooling, aqueous HCl (100 μL, 6M) was used to acidify the mixture. The solvent was then evaporated, and the solids were suspended in ethanol, then filtered to remove salt. After evaporation, the residue was dissolved in DCM (1 mL), TFA (1 mL) was added and the solution stirred at rt for 2 hours. The solvent was then removed to provide (S)-3-(3-(((R)-7-(3-aminopropyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)-3-(7-methoxy-1-methyl-1Hbenzo[d][1,2,3]triazol-5-yl)propanoic acid as the TFA salt as an amber oil (64 mg, 0.078 mmol, 106%).
LC/MS m/z calculated for [M+H]+ 608.2, found 608.0.
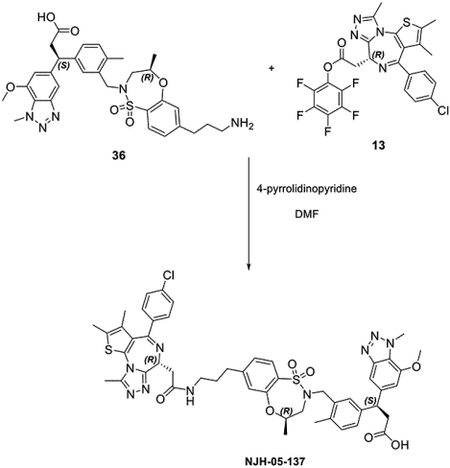
(S)-3-(3-(((R)-7-(3-aminopropyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (18 mg, 0.025mmol) in DMF (1 mL) was combined with 4-pyrrolidinopyridine (15 mg, 0.10 mmol) and perfluorophenyl (S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate (14 mg, 0.025 mmol) in THF (300 μL), was added. The reaction was stirred for 15 minutes, then purified by HPLC to provide NJH-05–137 as a white solid (8.0 mg, 0.0087 mmol, 39%).
LC/MS m/z calculated for [M+H]+ 990.3, found 989.6.
1H NMR (500 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.22 (t, J = 5.6 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.42 − 7.32 (m, 5H), 7.27 (d, J = 2.1 Hz, 1H), 7.21 (dd, J = 7.8, 1.9 Hz, 1H), 7.14 (dd, J = 8.1, 1.6 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.85 (d, J = 1.2 Hz, 1H), 4.49 − 4.38 (m, 2H), 4.35 (d, J = 14.0 Hz, 1H), 4.32 − 4.26 (m, 1H), 4.26 (s, 3H), 3.86 (s, 3H), 3.72 (d, J = 14.0 Hz, 1H), 3.52 (dd, J = 15.4, 10.1 Hz, 1H), 3.20 (dd, J = 7.1, 3.8 Hz, 2H), 3.09 (q, J = 6.5 Hz, 2H), 3.05 − 2.97 (m, 2H), 2.70 − 2.58 (m, 3H), 2.53 (s, 3H), 2.34 (s, 3H), 2.16 (s, 3H), 1.80 − 1.63 (m, 2H), 1.55 (s, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.19, 170.11, 163.67, 155.57, 155.32, 150.43, 147.91, 146.14, 142.51, 142.10, 137.13, 135.76, 135.51, 133.68, 132.66, 131.47, 131.34, 131.10, 130.59, 130.33, 130.08, 129.50, 129.04, 128.92, 127.79, 124.90, 124.23, 124.20, 108.63, 107.64, 73.57, 56.65, 54.36, 52.56, 48.94, 46.87, 38.49, 38.14, 37.02, 32.56, 30.84, 19.15, 18.52, 14.49, 13.13, 11.73.
Synthesis of (S)-3-(3-(((R)-7-(4-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)butyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–138).
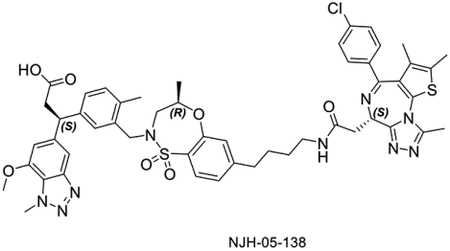
NJH-05–138 was synthesized starting from tert-butyl but-3-yn-1-ylcarbamate and (R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide using the same procedure as used for NJH-05–137.
LC/MS m/z calculated for [M+H]+ 1004.3, found 1003.8. 1H NMR (500 MHz, DMSO-d6) δ 12.00 (s, 1H), 8.15 (t, J = 5.7 Hz, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.41 (d, J = 8.8 Hz, 2H), 7.39 − 7.31 (m, 3H), 7.28 (d, J = 2.0 Hz, 1H), 7.21 (dd, J = 7.8, 1.9 Hz, 1H), 7.10 (d, J = 8.1 Hz, 1H), 7.07 (s, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.85 (s, 1H), 4.48 − 4.38 (m, 2H), 4.38 − 4.27 (m, 2H), 4.26 (s, 3H), 3.86 (s, 3H), 3.71 (d, J = 14.0 Hz, 1H), 3.61 − 3.41 (m, 1H), 3.24 − 2.93 (m, 6H), 2.69 − 2.55 (m, 3H), 2.52 (s, 3H), 2.32 (s, 3H), 2.16 (s, 3H), 1.62 − 1.54 (m, 2H), 1.54 − 1.48 (m, 3H), 1.43 (p, J = 7.1 Hz, 2H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.17, 169.88, 163.57, 155.57, 155.33, 150.79, 150.36, 147.94, 146.12, 142.52, 142.14, 137.15, 135.76, 135.49, 133.70, 132.68, 131.41, 131.25, 131.06, 130.59, 130.32, 130.06, 129.54, 128.95, 127.80, 124.81, 124.22, 124.15, 108.66, 107.67, 73.50, 56.66, 54.36, 52.53, 48.91, 46.88, 38.60, 38.15, 37.02, 34.90, 29.44, 28.15, 19.17, 18.53, 14.49, 13.13, 11.75.
(S)-3-(3-(((R)-7-(5-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)pentyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–139).
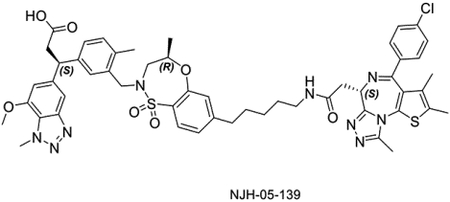
NJH-05–139 was synthesized starting from tert-butyl pent-4-yn-1-ylcarbamate and (R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide using the same procedure as used for NJH-05–137.
LC/MS m/z calculated for [M+H]+ 1018.3, found 1017.6.
1H NMR (500 MHz, DMSO-d6) δ 12.01 (s, 1H), 8.13 (t, J = 5.7 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.46 − 7.39 (m, 2H), 7.39 − 7.33 (m, 3H), 7.27 (s, 1H), 7.21 (dd, J = 7.8, 1.9 Hz, 1H), 7.09 (d, J = 8.0, 1.6 Hz, 1H), 7.07 − 6.99 (m, 2H), 6.85 (d, J = 1.2 Hz, 1H), 4.48 − 4.38 (m, 2H), 4.36 − 4.27 (m, 2H), 4.26 (s, 3H), 3.86 (s, 3H), 3.71 (d, J = 14.0 Hz, 1H), 3.51 (dd, J = 15.4, 10.1 Hz, 1H), 3.22 − 2.96 (m, 6H), 2.64 (d, J = 14.8 Hz, 1H), 2.60 − 2.54 (m, 2H), 2.52 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H), 1.59 − 1.50 (m, 5H), 1.43 (m , 2H), 1.29 (m, 2H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.28, 170.20, 163.78, 155.53, 155.23, 150.97, 150.61, 147.80, 146.18, 142.52, 141.95, 136.99, 135.87, 135.51, 133.71, 132.48, 131.59, 131.27, 131.16, 130.59, 130.34, 130.11, 129.27, 128.92, 127.77, 124.81, 124.24, 124.01, 108.48, 107.60, 73.67, 56.61, 54.24, 52.68, 48.94, 46.82, 38.81, 38.00, 37.02, 35.13, 30.41, 29.25, 26.35, 19.07, 18.46, 14.43, 13.07, 11.59.
Synthesis of (S)-3-(3-(((R)-7-(3-(2-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)ethoxy)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–141).
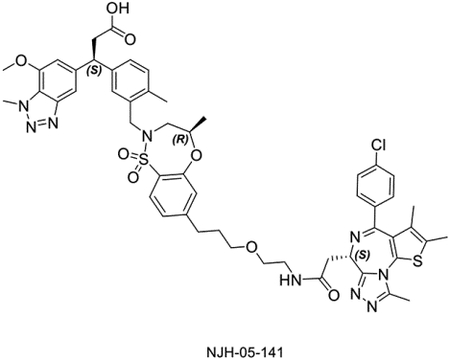
NJH-05–141 was synthesized starting from tert-butyl (2-(prop-2-yn-1-yloxy)ethyl)carbamate and (R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide using the same procedure as used for NJH-05-137.
LC/MS m/z calculated for [M+H]+ 1034.3, found 1034.3. 1H NMR (500 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.22 (t, J = 5.7 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.8 Hz, 2H), 7.38 − 7.32 (m, 3H), 7.27 (s, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.11 (d, J = 7.2 Hz, 1H), 7.07 (s, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.85 (s, 1H), 4.49 − 4.39 (m, 2H), 4.37 − 4.27 (m, 2H), 4.26 (s, 3H), 3.85 (s, 3H), 3.71 (d, J = 14.0 Hz, 1H), 3.51 (dd, J = 15.4, 10.1 Hz, 1H), 3.38 (dh, J = 6.1, 3.2, 2.6 Hz, 4H), 3.31 − 3.12 (m, 4H), 3.04 − 2.98 (m, 2H), 2.69 − 2.59 (m, 3H), 2.51 (s, 3H), 2.33 (s, 3H), 2.16 (s, 3H), 1.83 − 1.73 (m, 2H), 1.54 (s, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.17, 170.14, 163.54, 155.56, 155.36, 150.41, 150.35, 147.94, 146.12, 142.50, 142.14, 137.19, 135.73, 135.48, 133.71, 132.71, 131.49, 131.25, 131.06, 130.64, 130.31, 130.04, 129.53, 129.03, 128.92, 127.79, 124.86, 124.22, 124.17, 108.67, 107.67, 73.53, 69.67, 69.32, 56.66, 54.28, 52.54, 48.91, 46.88, 39.13, 38.00, 37.01, 31.87, 30.74, 19.17, 18.54, 14.50, 13.13, 11.74.
(S)-3-(3-(((R)-7-(3-(2-(2-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)ethoxy)ethoxy)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–142).
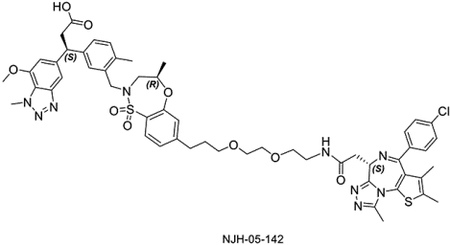
NJH-05–142 was synthesized starting from tert-butyl (2-(2-(prop-2-yn-1-yloxy)ethoxy)ethyl)carbamate and (R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide using the same procedure as used for NJH-05–137.
LC/MS m/z calculated for [M+H]+ 1078.4, found 1078.4. 1H NMR (500 MHz, DMSO-d6) δ 12.03 (s, 1H), 8.22 (t, J = 5.7 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.9 Hz, 2H), 7.38 − 7.32 (m, 3H), 7.27 (s, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.09 (d, J = 8.3 Hz, 1H), 7.07 (s, 1H), 7.04 (d, J = 7.9 Hz, 1H), 6.85 (d, J = 1.1 Hz, 1H), 4.47 − 4.39 (m, 2H), 4.31 (dd, J = 20.9, 12.2 Hz, 2H), 4.26 (s, 3H), 3.85 (s, 3H), 3.71 (d, J = 14.0 Hz, 1H), 3.52 − 3.47 (m, 3H), 3.46 (dd, J = 5.6, 3.3 Hz, 2H), 3.42 (t, J = 5.9 Hz, 2H), 3.35 (t, J = 6.3 Hz, 2H), 3.30 − 3.10 (m, 4H), 3.07 − 2.97 (m, 2H), 2.68 − 2.59 (m, 3H), 2.51 (s, 3H), 2.33 (s, 3H), 2.15 (s, 3H), 1.81 − 1.71 (m, 2H), 1.56 − 1.51 (m, 3H), 1.01 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.17, 170.19, 163.56, 155.54, 155.34, 150.37, 147.93, 146.13, 142.50, 142.12, 137.17, 135.73, 135.48, 133.71, 132.68, 131.48, 131.27, 131.07, 130.64, 130.31, 130.04, 129.49, 129.00, 128.92, 127.78, 124.87, 124.22, 124.16, 108.65, 107.66, 73.54, 70.08, 69.94, 69.84, 69.68, 56.65, 54.27, 52.56, 48.91, 46.87, 39.14, 37.96, 37.01, 31.87, 30.73, 19.16, 18.53, 14.50, 13.13, 11.73.
(S)-3-(3-(((R)-7-(1-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)-2-oxo-6,9,12-trioxa-3-azapentadecan-15-yl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–143).
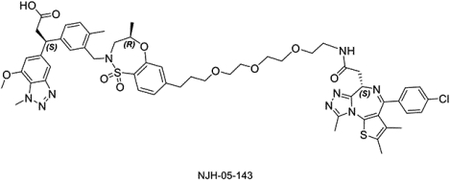
NJH-05–143 was synthesized starting from tert-butyl (2-(2-(2-(prop-2-yn-1-yloxy)ethoxy)ethoxy)ethyl)carbamate and (R)-7-bromo-4-methyl-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide using the same procedure as used for bifunctional compound NJH-05–137.
LC/MS m/z calculated for [M+H]+ 1122.4, found 1122.3. 1H NMR (500 MHz, DMSO-d6) δ 12.03 (s, 1H), 8.20 (t, J = 5.7 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.7 Hz, 2H), 7.38 − 7.31 (m, 3H), 7.27 (s, 1H), 7.21 (d, J = 7.9 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 7.07 (s, 1H), 7.03 (d, J = 7.9 Hz, 1H), 6.85 (s, 1H), 4.48 − 4.36 (m, 2H), 4.36 − 4.27 (m, 2H), 4.26 (s, 3H), 3.85 (s, 3H), 3.71 (d, J = 14.0 Hz, 1H), 3.48 (d, J = 4.7 Hz, 5H), 3.46 − 3.30 (m, 10H), 3.29 − 3.09 (m, 2H), 3.06 − 2.94 (m, 2H), 2.69 − 2.55 (m, 3H), 2.51 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H), 1.81 − 1.71 (m, 2H), 1.54 (s, 3H), 1.01 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.18, 170.22, 163.53, 155.56, 155.33, 150.38, 150.33, 147.92, 146.13, 142.50, 142.11, 137.21, 135.70, 135.48, 133.71, 132.69, 131.47, 131.22, 131.07, 130.62, 130.30, 130.02, 129.48, 129.00, 128.92, 127.78, 124.87, 124.67, 124.22, 124.17, 108.64, 107.65, 73.55, 70.27, 70.11, 69.95, 69.80, 69.66, 56.65, 54.27, 52.57, 48.91, 46.87, 39.13, 37.96, 37.01, 31.85, 30.70, 19.16, 18.53, 14.49, 13.12, 11.72.
Ethyl (S)-3-(3-(((R)-7-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (NJH-05–146).
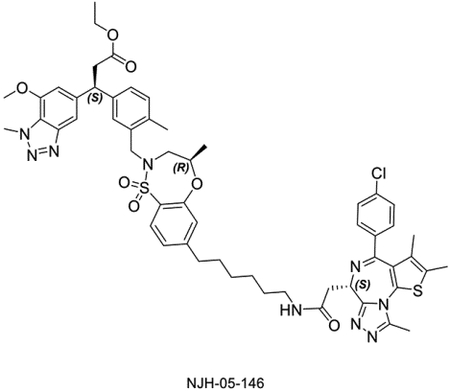
Ethyl (S)-3-(3-(((R)-7-(6-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (14 mg, 0.017 mmol) and (S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetic acid (7 mg, 0.017 mmol) were dissolved in DMF (1 mL). DIEA (15 μL, 0.085 mmol) was added, followed by HATU (13 mg, 0.034 mmol). The mixture was stirred for 20 minutes, then purified by HPLC to provide NJH-05–146 as a white solid (9.4 mg, 0.0089 mmol, 52%).
LC/MS m/z calculated for [M+H]+ 1060.4, found 1059.7. 1H NMR (500 MHz, DMSO-d6) δ 8.18 (t, J = 5.6 Hz, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.53 − 7.39 (m, 5H), 7.36 (d, J = 2.0 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.16 (d, J = 8.1 Hz, 1H), 7.13 − 7.08 (m, 2H), 6.94 (s, 1H), 4.56 − 4.49 (m, 2H), 4.45 − 4.35 (m, 2H), 4.34 (s, 3H), 3.98 − 3.90 (m, 5H), 3.79 (d, J = 14.1 Hz, 1H), 3.60 (dd, J = 15.3, 10.1 Hz, 1H), 3.30 − 3.02 (m, 6H), 2.73 (d, J = 15.0 Hz, 1H), 2.68 − 2.61 (m, 2H), 2.60 (s, 3H), 2.42 − 2.36 (m, 3H), 2.23 (s, 3H), 1.63 − 1.54 (m, 5H), 1.52 − 1.41 (m, 2H), 1.35 (p, J = 3.6 Hz, 4H), 1.10 (d, J = 6.3 Hz, 3H), 1.04 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 171.63, 169.87, 163.59, 158.86, 158.57, 155.55, 155.32, 150.95, 150.40, 147.91, 146.13, 142.22, 141.80, 137.09, 135.79, 135.54, 133.78, 132.64, 131.38, 131.34, 131.08, 130.61, 130.30, 130.07, 129.32, 128.99, 128.92, 127.77, 124.75, 124.24, 124.04, 108.70, 107.61, 73.55, 60.29, 56.67, 54.35, 52.63, 48.92, 46.83, 38.88, 38.11, 37.02, 35.23, 30.86, 29.58, 28.91, 26.57, 21.51, 19.14, 18.98, 18.54, 14.45, 13.11, 11.71.
Synthesis of 3-(3-((4-(6-(2-(4-(4-(6-(3-(4-tert-butylbenzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-ylamino)benzoyl)piperazin-1-yl)acetamido)hexyloxy)-N-methylphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-05–119).
To a solution of tert-butyl 4-(4-(6-(3-(4-tert-butylbenzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-ylamino)benzoyl)piperazine-1-carboxylate(Dobrovolsky et al., 2019) (30 mg, 0.044 mmol) in DCM (0.5 mL), TFA (0.5 mL) was added at room temperature. After 1 hour, the solution was concentrated under vacuum to get the crude product. To the solution of this residue in acetone (1 mL), tert-butyl 2-bromoacetate (6.5 μL, 0.044 mmol) and K2CO3 (12.1 mg, 0.088 mmol) were added at room temperature. After 14 hours, the reaction was quenched with water and extracted with EtOAc (3 × 5 mL). The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum and the residue was purified with flash chromatography to yield tert-butyl 2-(4-(4-(6-(3-(4-tert-butylbenzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-ylamino)benzoyl)piperazin-1-yl)acetate (27.7 mg, 0.040 mmol, 91%).
LC/MS m/z calculated for [M+H]+ 693.4, found 693.1.
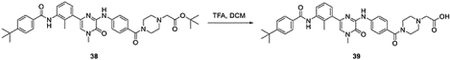
To a solution of tert-butyl 2-(4-(4-(6-(3-(4-tert-butylbenzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-ylamino)benzoyl)piperazin-1-yl)acetate (27.7 mg, 0.040 mmol) in DCM (0.5 mL), TFA (0.5 mL) was added at room temperature. After 1 hour, the solution was concentrated under vacuum to get the crude product without any further purification.
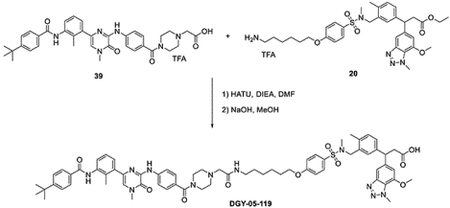
To a solution of 2-(4-(4-(6-(3-(4-tert-butylbenzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-ylamino)benzoyl)piperazin-1-yl)acetic acid TFA salt (30 mg, 0.04 mmol) and ethyl 3-(3-((4-(6-aminohexyloxy)-N-methylphenylsulfonamido)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (30.6 mg, 0.04 mmol) in DMF (1 mL), HATU (30.4 mg, 0.08 mmol) and DIEA (258 mg, 0.20 mmol) were added at room temperature. After 10 mins, the reaction was quenched with water and extracted with EtOAc (3 × 5 mL). The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any further purification. To the solution of resulted residue in MeOH (1 mL), NaOH (200 μL, 2N aq.) was added at room temperature. The reaction was heated to 80 °C for 1 hour. The solution was concentrated under vacuum and purified with HPLC to yield bifunctional compound DGY-05–119 (11 mg, 0.0081 mmol, 20%).
LC/MS m/z calculated for [M+H]+ 1242.6, found 1242.8.
1H NMR (500 MHz, DMSO-d6) δ 12.11 (s, 1H), 9.91 (s, 1H), 9.51 (s, 1H), 8.55 (s, 1H), 8.20 − 8.09 (m, 2H), 8.00 − 7.91 (m, 2H), 7.81 − 7.68 (m, 2H), 7.59 − 7.54 (m, 2H), 7.43 − 7.40 (m, 2H), 7.40 − 7.36 (m, 2H), 7.32 − 7.19 (m, 5H), 7.19 − 7.14 (m, 2H), 7.11 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 1.1 Hz, 1H), 4.49 (t, J = 8.0 Hz, 1H), 4.32 (s, 3H), 4.08 (t, J = 6.4 Hz, 2H), 4.01 (s, 2H), 3.89 (s, 4H), 3.58 (s, 3H), 3.16 (s, 2H), 3.07 (d, J = 8.1 Hz, 2H), 2.38 (s, 3H), 2.28 (d, J = 11.5 Hz, 6H), 1.75 (p, J = 6.5 Hz, 2H), 1.46 (dt, J = 17.2, 8.1 Hz, 5H), 1.39 − 1.34 (m, 1H), 1.33 (s, 9H). (mixtures of isomers) 13C NMR (126 MHz, DMSO) δ 173.18, 169.59, 165.78, 162.59, 154.91, 150.89, 147.90, 146.73, 146.13, 142.46, 142.30, 141.82, 138.71, 137.62, 135.58, 133.67, 133.03, 132.26, 131.50, 131.04, 130.17, 129.43, 128.71, 127.99, 127.90, 127.75, 127.71, 127.66, 127.07, 125.94, 125.67, 124.23, 121.15, 118.94, 115.41, 108.82, 107.54, 68.49, 56.62, 52.56, 52.05, 46.86, 39.12, 37.10, 37.02, 35.14, 34.54, 31.41, 29.23, 28.91, 26.52, 25.60, 18.58, 15.98.
Synthesis of 3-(3-(((R)-7-(6-(4-(4-((6-(3-(4-(tert-butyl)benzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-yl)amino)benzoyl)piperazin-1-yl)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-02–162)
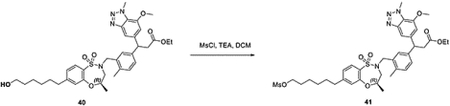
To the solution of ethyl 3-(3-(((R)-7-(6-hydroxyhexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (155 mg, 0.23 mmol) and TEA (64 μL, 0.46 mmol) in DCM (2 mL), was added MsCl (22 μL, 0.28 mmol) at 0 °C. After 20 mins, the reaction was quenched with water and extracted with DCM (3 × 5 mL). The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product which was purified by flash chromatography.
LC/MS m/z calculated for [M+H]+ 757.3, found 757.2.
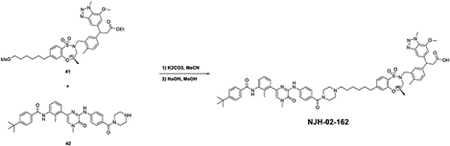
To the solution of ethyl 3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-(((R)-4-methyl-7-(6-((methylsulfonyl)oxy)hexyl)-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoate (23 mg, 0.03 mmol) and 4-(tert-butyl)-N-(2-methyl-3-(4-methyl-5-oxo-6-((4-(piperazine-1-carbonyl)phenyl)amino)-4,5-dihydropyrazin-2-yl)phenyl)benzamide (17 mg, 0.03 mmol) in MeCN (1 mL), was added K2CO3 (17 mg, 0.12 mmol). The reaction mixture was heated to 80 °C for overnight. The reaction was quenched with water and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any purification. The resulted product was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product.
LC/MS m/z calculated for [M+H]+ 1211.6, found 1211.8.
1H NMR (500 MHz, DMSO-d6) δ 12.11 (s, 1H), 9.89 (s, 1H), 9.45 (s, 1H), 8.08 (d, J = 8.3 Hz, 2H), 8.00 − 7.91 (m, 2H), 7.66 (dd, J = 7.9, 2.3 Hz, 1H), 7.61 − 7.50 (m, 2H), 7.44 (d, J = 4.1 Hz, 1H), 7.40 − 7.25 (m, 8H), 7.17 (dd, J = 8.1, 2.6 Hz, 1H), 7.14 − 7.09 (m, 2H), 6.91 (d, J = 11.1 Hz, 1H), 4.59 (d, J = 28.1 Hz, 1H), 4.54 − 4.48 (m, 1H), 4.41 (d, J = 14.4 Hz, 1H), 4.33 (d, J = 4.0 Hz, 3H), 3.92 (d, J = 5.6 Hz, 3H), 3.79 (d, J = 13.9 Hz, 1H), 3.57 (s, 3H), 3.42 (t, J = 5.3 Hz, 1H), 3.09 (t, J = 6.8 Hz, 2H), 2.76 (dd, J = 39.5, 14.9 Hz, 1H), 2.64 (t, J = 7.7 Hz, 3H), 2.29 (s, 3H), 2.24 (d, J = 5.0 Hz, 3H), 1.60 (s, 2H), 1.55 − 1.42 (m, 1H), 1.33 (s, 9H), 1.14 (dd, J = 49.2, 6.3 Hz, 3H).
Synthesis of 3-(3-(((R)-7-(3-(4-(4-((6-(3-(4-(tert-butyl)benzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-yl)amino)benzoyl)piperazin-1-yl)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-02–160)
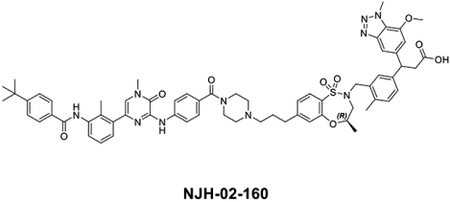
NJH-02–160 was synthesized using the same procedure as used for NJH-02–162.
LC/MS m/z calculated for [M+H]+ 1169.5, found 1169.6.
1H NMR (500 MHz, DMSO-d6) δ 12.12 (s, 1H), 9.89 (s, 1H), 9.45 (s, 1H), 8.08 (d, J = 8.6 Hz, 2H), 7.95 (d, J = 8.3 Hz, 2H), 7.65 (dd, J = 7.9, 2.3 Hz, 1H), 7.61 − 7.47 (m, 2H), 7.44 (d, J = 4.3 Hz, 1H), 7.42 − 7.22 (m, 8H), 7.22 − 7.14 (m, 2H), 7.11 (d, J = 8.3 Hz, 1H), 6.91 (d, J = 11.1 Hz, 1H), 4.86 (d, J = 5.2 Hz, 1H), 4.68 − 4.54 (m, 1H), 4.51 (t, J = 6.7 Hz, 1H), 4.41 (d, J = 13.5 Hz, 2H), 4.33 (d, J = 3.2 Hz, 3H), 4.04 (dd, J = 11.1, 4.2 Hz, 1H), 3.92 (d, J = 5.1 Hz, 4H), 3.79 (d, J = 13.9 Hz, 1H), 3.64 (d, J = 4.6 Hz, 0H), 3.57 (s, 3H), 3.42 (d, J = 5.3 Hz, 1H), 3.09 (t, J = 6.5 Hz, 2H), 2.80 (d, J = 15.0 Hz, 1H), 2.66 (q, J = 7.2, 6.7 Hz, 2H), 2.39 − 2.31 (m, 5H), 2.29 (d, J = 2.7 Hz, 4H), 2.23 (d, J = 4.9 Hz, 4H), 1.82 − 1.73 (m, 2H), 1.56 − 1.42 (m, 2H), 1.32 (s, 11H), 1.14 (dd, J = 49.2, 6.3 Hz, 3H). (mixture of isomers)
Synthesis of 3-(3-(((R)-7-(6-(4-(4-((6-(3-(4-(tert-butyl)benzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-yl)amino)benzoyl)piperazin-1-yl)-6-oxohexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1methyl-1-H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-02–136)
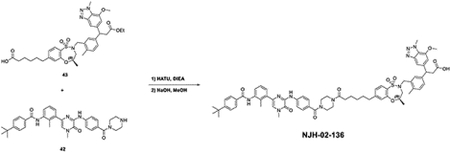
To the solution of 6-((4R)-2-(5-(3-ethoxy-1-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-oxopropyl)-2-methylbenzyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)hexanoic acid (17 mg, 0.025 mmol) and 4-(tert-butyl)-N-(2-methyl-3-(4-methyl-5-oxo-6-((4-(piperazine-1-carbonyl)phenyl)amino)-4,5-dihydropyrazin-2-yl)phenyl)benzamide (14 mg, 0.025 mmol) in DMF (1 mL), was added HATU (19 mg, 0.05 mmol) and DIEA (12 μL, 0.125 mmol). The reaction was quenched with water after 5 mins and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any purification. The resulted product was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product.
LC/MS m/z calculated for [M+H]+ 1225.5, found 1225.7.
1H NMR (500 MHz, DMSO-d6) δ 12.11 (s, 1H), 9.89 (s, 1H), 9.47 (s, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.2 Hz, 2H), 7.66 (dd, J = 7.9, 2.2 Hz, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 4.4 Hz, 1H), 7.37 (dd, J = 10.0, 7.8 Hz, 4H), 7.33 − 7.23 (m, 4H), 7.21 − 7.09 (m, 3H), 6.91 (d, J = 11.2 Hz, 1H), 4.51 (td, J = 7.9, 4.1 Hz, 1H), 4.41 (d, J = 14.3 Hz, 1H), 4.33 (d, J = 3.7 Hz, 3H), 3.92 (d, J = 5.6 Hz, 3H), 3.79 (d, J = 14.0 Hz, 1H), 3.58 (s, 3H), 3.09 (t, J = 7.0 Hz, 2H), 2.76 (dd, J = 39.4, 15.1 Hz, 1H), 2.64 (t, J = 7.8 Hz, 2H), 2.29 (s, 3H), 2.24 (d, J = 5.1 Hz, 3H), 1.58 (dt, J = 37.5, 7.6 Hz, 4H), 1.32 (s, 11H), 1.14 (dd, J = 49.2, 6.3 Hz, 3H). (mixture of isomers) 13C NMR (126 MHz, DMSO) δ 173.25, 171.67, 169.85, 166.06, 155.27, 155.05, 150.96, 150.88, 147.83, 146.65, 146.16, 142.01, 141.72, 138.64, 137.51, 135.57, 135.47, 133.72, 133.08, 132.05, 131.74, 131.30, 131.12, 129.35, 129.02, 128.95, 128.50, 127.96, 127.71, 127.07, 126.05, 125.72, 124.87, 124.23, 124.10, 120.95, 118.97, 108.65, 108.52, 107.63, 107.53, 73.65, 56.62, 52.67, 48.95, 46.83, 37.12, 37.03, 35.09, 32.62, 31.36, 30.53, 28.74, 24.93, 19.22, 19.11, 18.53, 18.49, 16.00.
Synthesis of 3-(3-(((R)-7-(3-(6-(4-(4-((6-(3-(4-(tert-butyl)benzamido)-2-methylphenyl)-4-methyl-3-oxo-3,4-dihydropyrazin-2-yl)amino)benzoyl)piperazin-1-yl)hexanamido)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-02–123)
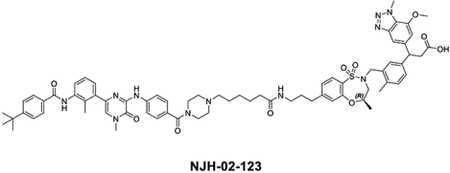
NJH-02–123 was synthesized using the same procedure as used for NJH-02–136.
LC/MS m/z calculated for [M+H]+ 1282.6, exceeded the limit of LCMS, found half peak 642.2.
1H NMR (500 MHz, DMSO-d6) δ 12.11 (s, 1H), 9.89 (s, 1H), 9.47 (s, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.2 Hz, 2H), 7.66 (dd, J = 7.9, 2.2 Hz, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 4.4 Hz, 1H), 7.37 (dd, J = 10.0, 7.8 Hz, 4H), 7.33 − 7.23 (m, 4H), 7.21 − 7.09 (m, 3H), 6.91 (d, J = 11.2 Hz, 1H), 4.51 (td, J = 7.9, 4.1 Hz, 1H), 4.41 (d, J = 14.3 Hz, 1H), 4.33 (d, J = 3.7 Hz, 3H), 3.92 (d, J = 5.6 Hz, 3H), 3.79 (d, J = 14.0 Hz, 1H), 3.58 (s, 3H), 3.09 (t, J = 7.0 Hz, 2H), 2.76 (dd, J = 39.4, 15.1 Hz, 1H), 2.64 (t, J = 7.8 Hz, 2H), 2.29 (s, 3H), 2.24 (d, J = 5.1 Hz, 3H), 1.58 (dt, J = 37.5, 7.6 Hz, 5H), 1.32 (s, 11H), 1.14 (dd, J = 49.2, 6.3 Hz, 3H). (mixture of isomers) 13C NMR (126 MHz, DMSO) δ 173.29, 172.90, 169.83, 166.16, 155.27, 155.13, 150.88, 150.37, 147.81, 146.62, 146.17, 142.61, 142.55, 142.14, 141.99, 141.78, 138.61, 137.46, 135.57, 135.48, 133.70, 133.12, 132.01, 131.71, 131.41, 131.15, 129.31, 129.25, 129.02, 128.71, 127.96, 127.82, 127.79, 127.12, 126.10, 125.75, 124.88, 124.23, 124.14, 121.09, 118.94, 108.61, 108.48, 107.63, 107.51, 73.72, 56.61, 56.11, 52.67, 51.19, 48.96, 46.83, 38.51, 37.15, 37.02, 35.48, 35.09, 32.60, 31.35, 30.62, 25.93, 25.13, 23.40, 19.20, 19.09, 18.50, 18.47, 15.95.
Synthesis of (S)-3-(3-(((R)-7-(6-(2-(4-(3-(N-(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)carbamimidoyl)-5-methyl-4-oxo-4,5-dihydrothieno[3,2-c]pyridin-7-yl)-2-methoxyphenoxy)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-10–077)
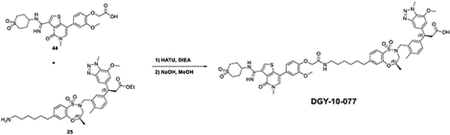
To the solution of 2-(4-(3-(N-(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)carbamimidoyl)-5-methyl-4-oxo-4,5-dihydrothieno[3,2-c]pyridin-7-yl)-2-methoxyphenoxy)acetic acid (7.8 mg, 0.015 mmol)(Remillard et al., 2017) and ethyl (S)-3-(3-(((R)-7-(6-aminohexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (10 mg, 0.015 mmol) in DMF (1 mL), was added HATU (11.4 mg, 0.03 mmol) and DIEA (9.7 mg, 0.075 mmol). The reaction was quenched with water after 5 mins and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any purification. The resulted product was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product.
LC/MS m/z calculated for [M+H]+ 1151.4, found 1151.6.
1H NMR (500 MHz, DMSO-d6) δ 9.47 (s, 1H), 9.41 (d, J = 0.8 Hz, 1H), 8.67 (d, J = 5.7 Hz, 1H), 8.38 (t, J = 5.5 Hz, 1H), 7.85 (s, 1H), 7.65 − 7.50 (m, 2H), 7.37 (d, J = 1.0 Hz, 1H), 7.27 (d, J = 2.0 Hz, 1H), 7.21 (dd, J = 7.8, 1.9 Hz, 1H), 7.08 (dd, J = 8.0, 1.6 Hz, 1H), 7.06 − 7.01 (m, 2H), 6.85 (d, J = 1.1 Hz, 1H), 6.78 (s, 2H), 4.43 (t, J = 8.0 Hz, 1H), 4.31 (q, J = 12.4, 10.9 Hz, 4H), 4.25 (s, 3H), 3.86 (s, 3H), 3.82 − 3.75 (m, 8H), 3.72 (d, J = 14.0 Hz, 1H), 3.54 (s, 4H), 3.15 − 2.91 (m, 4H), 2.73 − 2.61 (m, 4H), 2.56 (t, J = 7.6 Hz, 2H), 2.15 (s, 3H), 1.52 (t, J = 7.5 Hz, 2H), 1.37 (d, J = 7.2 Hz, 2H), 1.25 (d, J = 6.3 Hz, 4H), 1.00 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.23, 164.41, 160.84, 159.83, 158.94, 158.67, 155.25, 150.94, 149.86, 149.34, 147.84, 146.15, 142.54, 142.24, 142.03, 139.77, 138.91, 135.47, 133.70, 131.32, 131.12, 129.30, 128.97, 127.80, 124.80, 124.22, 124.04, 120.34, 118.38, 117.97, 115.94, 115.62, 108.51, 107.64, 105.85, 104.70, 73.65, 56.67, 56.16, 52.65, 48.95, 48.32, 46.83, 41.79, 37.13, 37.02, 35.11, 34.83, 30.64, 29.09, 28.64, 26.51, 19.09, 18.47.
Synthesis of (S)-3-(3-(((R)-7-(6-(2-((2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)(methyl)amino)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-10–075)
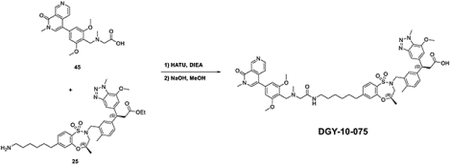
To the solution of N-(2,6-dimethoxy-4-(2-methyl-1-oxo-1,2-dihydro-2,7-naphthyridin-4-yl)benzyl)-N-methylglycine (6 mg, 0.015 mmol)(Remillard et al., 2017) and ethyl (S)-3-(3-(((R)-7-(6-aminohexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoate (10 mg, 0.015 mmol) in DMF (1 mL), was added HATU (11.4 mg, 0.03 mmol) and DIEA (9.7 mg, 0.075 mmol). The reaction was quenched with water after 5 mins and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any purification. The resulted product was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product.
LC/MS m/z calculated for [M+H]+ 1029.4, found 1029.6.
1H NMR (500 MHz, DMSO-d6) δ 9.54 (s, 1H), 9.48 (d, J = 0.8 Hz, 1H), 8.75 (d, J = 5.7 Hz, 1H), 8.45 (t, J = 5.5 Hz, 1H), 7.92 (s, 1H), 7.70 − 7.57 (m, 2H), 7.44 (d, J = 1.0 Hz, 1H), 7.34 (d, J = 2.0 Hz, 1H), 7.29 (dd, J = 7.8, 1.9 Hz, 1H), 7.22 − 7.09 (m, 3H), 6.93 (d, J = 1.2 Hz, 1H), 6.86 (s, 2H), 4.50 (t, J = 8.0 Hz, 1H), 4.44 − 4.36 (m, 6H), 4.33 (s, 3H), 3.93 (s, 3H), 3.89 − 3.83 (m, 8H), 3.79 (d, J = 14.0 Hz, 1H), 3.62 (s, 4H), 3.27 − 3.02 (m, 4H), 2.78 − 2.68 (m, 4H), 2.64 (t, J = 7.7 Hz, 2H), 2.23 (s, 3H), 1.65 − 1.55 (m, 2H), 1.45 (d, J = 7.2 Hz, 2H), 1.32 (s, 4H), 1.08 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.25, 164.40, 160.87, 159.82, 158.98, 158.71, 155.23, 150.96, 149.85, 149.33, 147.82, 146.16, 142.54, 142.24, 142.00, 139.74, 138.89, 135.47, 133.70, 131.30, 131.14, 129.26, 128.96, 127.79, 124.81, 124.22, 124.03, 120.33, 118.38, 118.04, 115.98, 115.69, 108.48, 107.63, 105.83, 104.69, 73.69, 56.66, 56.18, 52.68, 48.96, 48.36, 46.82, 41.84, 37.14, 37.02, 35.09, 30.62, 29.07, 28.61, 26.49, 19.08, 18.46.
Synthesis of (S)-3-(3-(((R)-7-(3-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-04–149)
To the solution of (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (8.3 mg, 0.019 mmol) and ethyl (S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-(((R)-4-methyl-1,1-dioxido-7-(3-oxopropyl)-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoate (17.9 mg, 0.028 mmol) in MeOH (1 mL), was added AcOH (100 μL) and NaBH3CN (5.3 mg, 0.084 mmol) at room temperature. The reaction was quenched with water after the starting material was consumed and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any purification. The resulted product was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product.
LC/MS m/z calculated for [M+H]+ 1035.4, found 1035.0.
1H NMR (500 MHz, DMSO-d6) δ 8.92 (s, 1H), 8.58 (s, 1H), 8.49 (d, J = 7.8 Hz, 1H), 8.19 (s, 1H), 7.64 (t, J = 7.6 Hz, 1H), 7.42 − 7.27 (m, 5H), 7.22 (dd, J = 7.7, 2.0 Hz, 1H), 7.17 − 7.13 (m, 1H), 7.11 (d, J = 1.6 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H), 6.85 (d, J = 1.1 Hz, 1H), 4.88 (t, J = 7.2 Hz, 1H), 4.51 (t, J = 8.5 Hz, 1H), 4.44 (t, J = 8.0 Hz, 1H), 4.34 (d, J = 13.9 Hz, 1H), 4.31 − 4.28 (m, 2H), 4.26 (s, 3H), 4.07 (d, J = 9.2 Hz, 1H), 3.95 − 3.83 (m, 3H), 3.78 (d, J = 11.4 Hz, 1H), 3.76 − 3.70 (m, 1H), 3.40 (dd, J = 11.3, 3.4 Hz, 1H), 3.02 (t, J = 8.3 Hz, 2H), 2.78 (q, J = 7.3, 6.6 Hz, 2H), 2.71 − 2.63 (m, 2H), 2.64 − 2.57 (m, 2H), 2.39 (s, 3H), 2.16 (s, 3H), 2.00 − 1.82 (m, 2H), 1.78 − 1.70 (m, 1H), 1.32 (d, J = 7.1 Hz, 3H), 1.02 (d, J = 8.1 Hz, 12H).
Synthesis of (S)-3-(3-(((R)-7-(6-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (DGY-09–175)
DGY-09–175 was synthesized using the same procedure as used for NJH-04–149.
LC/MS m/z calculated for [M+H]+ 1077.5, found 1077.5.
1H NMR (500 MHz, DMSO) δ 9.01 (s, 2H), 8.64 (d, J = 7.7 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.51 − 7.46 (m, 3H), 7.43 (d, J = 8.3 Hz, 2H), 7.36 − 7.30 (m, 3H), 7.23 (d, J = 7.7 Hz, 1H), 7.17 (t, J = 4.0 Hz, 2H), 6.94 (s, 1H), 4.98 − 4.95 (m, 1H), 4.64 − 4.60 (m, 2H), 4.56 − 4.53 (m, 2H), 4.44 (d, J = 14.2 Hz, 2H), 4.38 (s, 4H), 3.97 (s, 5H), 3.16 − 3.09 (m, 3H), 2.81 (s, 1H), 2.78 (s, 1H), 2.69 (t, J = 7.6 Hz, 2H), 2.50 (s, 3H), 2.27 (s, 3H), 1.44 (d, J = 7.0 Hz, 3H), 1.35 − 1.32 (m, 3H), 1.13 (s, 2H), 1.12 (s, 9H).
13C NMR (126 MHz, DMSO) δ 173.24, 170.60, 166.14, 158.91, 158.65, 155.26, 152.09, 150.93, 148.17, 147.84, 146.16, 144.85, 142.55, 142.00, 135.46, 133.72, 131.60, 131.35, 131.14, 130.18, 129.34, 126.83, 124.86, 124.65, 124.23, 124.07, 108.49, 107.65, 73.71, 69.25, 65.68, 59.38, 56.63, 48.95, 48.28, 47.80, 46.83, 38.03, 37.03, 35.22, 35.04, 30.48, 28.41, 26.54, 26.31, 24.98, 22.78, 19.11, 18.48, 16.32.
Synthesis of (S)-3-(3-(((R)-7-(8-((R)-2-(5-((S)-2-carboxy-1-(7-methoxy-1-methyl-1Hbenzo[d][1,2,3]triazol-5-yl)ethyl)-2-methylbenzyl)-4-methyl-1,1-dioxido-3,4-dihydro-2Hbenzo[b][1,4,5]oxathiazepin-7-yl)octa-1,7-diyn-1-yl)-4-methyl-1,1-dioxido-3,4-dihydro-2Hbenzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1Hbenzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-04–134)
To the solution of intermediate 9 (77 mg, 0.26 mmol), octa-1,7-diyne (17 μL, 0.13 mmol) and Et3N (109 μL, 0.78 mmol) in dioxane (3 mL), was added Pd(dppf)Cl2·DCM adduct (21 mg, 0.026 mmol) and CuI (10 mg, 0.052 mmol). The reaction mixture was heated up to 80 °C for 2 hours. The reaction was quenched with water after the starting material was consumed and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product and the resulted residue was purified with flash chromatography to yield (R)-4-methyl-7-(8-((R)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)octa-1,7-diyn-1-yl)-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide (58 mg, 0.11 mmol, 85%). LC/MS m/z calculated for [M+H]+ 529.1, found 529.2.
To the solution of (R)-4-methyl-7-(8-((R)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-7-yl)octa-1,7-diyn-1-yl)-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepine 1,1-dioxide (58 mg, 0.11 mmol) and intermediate 5 (29 mg, 0.076 mmol) in THF (1 mL), was added PPh3 (39 mg, 0.15 mmol) and DIAD (30 uL, 0.15 mmol). The reaction was quenched after 30 mins and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without purification. The residue was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product (14.4 mg, 0.012 mmol, 11%).
LC/MS m/z calculated for [M+H]+ 1203.4, found 1203.7.
1H NMR (500 MHz, DMSO-d6) δ 12.04 (s, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 1.0 Hz, 2H), 7.32 − 7.18 (m, 8H), 7.04 (d, J = 7.8 Hz, 2H), 6.85 (d, J = 1.1 Hz, 2H), 4.43 (t, J = 8.0 Hz, 2H), 4.36 − 4.30 (m, 4H), 4.25 (s, 6H), 3.85 (s, 6H), 3.75 (d, J = 14.1 Hz, 2H), 3.53 (dd, J = 15.4, 10.2 Hz, 2H), 3.02 (t, J = 7.9 Hz, 4H), 2.67 (d, J = 15.0 Hz, 2H), 2.49 (d, J = 6.2 Hz, 4H), 2.15 (s, 6H), 1.67 (p, J = 3.5 Hz, 4H), 1.02 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, DMSO) δ 171.03, 153.28, 145.82, 144.00, 140.35, 140.04, 133.34, 131.48, 131.46, 128.96, 127.90, 127.37, 127.32, 125.70, 125.61, 124.93, 122.55, 122.10, 106.55, 105.52, 92.89, 77.65, 71.85, 54.53, 50.41, 46.82, 44.75, 34.88, 25.48, 16.91, 16.60, 16.39.
Synthesis of (S)-3-(3-(((R)-7-(3-(4-(4-((6-acetyl-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–032)
NJH-05–032(Jiang et al., 2019) was synthesized using the same procedure as used for NJH-04–149.
LC/MS m/z calculated for [M+H]+ 1037.5, found 1037.5. 1H NMR (500 MHz, DMSO) δ 8.95 (s, 1H), 8.01 (s, 1H), 7.81 − 7.77 (m, 1H), 7.69 − 7.63 (m, 2H), 7.41 (d, J = 3.9 Hz, 1H), 7.32 − 7.29 (m, 1H), 7.27 − 7.24 (m, 1H), 7.22 − 7.18 (m, 1H), 7.17 (s, 1H), 7.13 − 7.11 (m, 1H), 7.10 (s, 1H), 6.89 (d, J = 4.5 Hz, 1H), 4.49 (t, J = 8.1 Hz, 2H), 4.38 (d, J = 14.1 Hz, 1H), 4.32 (d, J = 5.7 Hz, 4H), 4.06 (t, J = 6.2 Hz, 2H), 3.91 (d, J = 5.6 Hz, 4H), 3.19 − 3.10 (m, 4H), 3.10 − 3.00 (m, 4H), 2.77 − 2.73 (m, 2H), 2.73 − 2.65 (m, 3H), 2.55 (s, 1H), 2.42 (s, 1H), 2.30 (s, 1H), 2.21 (d, J = 2.3 Hz, 4H), 1.96 (p, J = 6.8 Hz, 2H), 1.91 − 1.86 (m, 1H), 1.80 − 1.71 (m, 3H), 1.61 − 1.54 (m, 2H), 1.08 (d, J = 6.3 Hz, 3H).
13C NMR (126 MHz, DMSO) δ 173.18, 170.51, 166.11, 155.36, 152.03, 149.00, 148.25, 147.93, 146.13, 144.89, 142.54, 142.13, 135.45, 133.69, 131.85, 131.10, 130.25, 129.35, 126.84, 124.83, 124.23, 108.59, 107.72, 73.71, 69.28, 65.54, 59.37, 57.19, 56.66, 52.60, 48.95, 48.23, 47.22, 46.86,38.20, 37.03, 35.32, 32.18, 26.58, 26.13, 22.86, 19.18, 18.53, 16.43.
Synthesis of (S)-3-(3-(((R)-7-(6-(4-(4-((6-acetyl-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-05–033)
NJH-05–033 was synthesized using the same procedure as used for NJH-05–032.
LC/MS m/z calculated for [M+H]+ 1079.5, found 1079.5.
1H NMR (500 MHz, DMSO-d6) δ 12.13 (s, 1H), 10.41 (s, 1H), 8.97 (s, 1H), 8.06 (d, J = 3.0 Hz, 1H), 7.86 (d, J = 9.1 Hz, 1H), 7.71 − 7.58 (m, 3H), 7.45 (s, 1H), 7.35 (d, J = 2.1 Hz, 1H), 7.28 (dd, J = 7.8, 1.9 Hz, 1H), 7.22 − 7.03 (m, 5H), 6.92 (d, J = 1.1 Hz, 1H), 5.90 − 5.78 (m, 1H), 4.50 (t, J = 8.0 Hz, 1H), 4.41 (d, J = 14.1 Hz, 1H), 4.33 (d, J = 3.7 Hz, 4H), 4.04 (t, J = 6.5 Hz, 2H), 3.93 (d, J = 3.7 Hz, 5H), 3.79 (d, J = 14.0 Hz, 2H), 3.64 − 3.42 (m, 5H), 3.24 − 3.03 (m, 6H), 2.77 − 2.63 (m, 3H), 2.43 (s, 3H), 2.32 (s, 3H), 2.24 (d, J = 3.4 Hz, 5H), 1.90 (s, 2H), 1.79 (d, J = 9.7 Hz, 2H), 1.60 (dt, J = 12.1, 6.3 Hz, 5H), 1.46 − 1.31 (m, 5H), 1.09 (dd, J = 6.3, 2.2 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 203.05, 173.21, 161.12, 158.32, 158.06, 155.31, 155.28, 155.20, 150.94, 147.87, 146.13, 144.19, 143.57, 142.52, 142.30, 142.06, 135.49, 133.69, 131.34, 131.10, 129.42, 128.98, 127.79, 124.84, 124.21, 124.08, 116.23, 108.57, 107.63, 73.59, 65.45, 61.16, 56.63, 53.49, 52.62, 48.94, 48.57, 46.85, 37.01, 35.12, 32.77, 31.72, 30.83, 30.64, 28.97, 28.78, 28.66, 28.07, 25.65, 25.61, 19.13, 18.49, 14.13.
Synthesis of (3S)-3-(3-(((4R)-7-(6-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-5-yl)oxy)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-04–087)
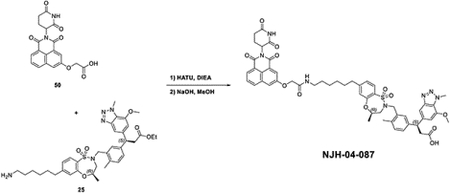
NJH-04–087 was synthesized using the same procedure with intermediates 50(de Wispelaere et al., 2019) and 25 as used for DGY-06–177-pk2.
LC/MS m/z calculated for [M+H]+ 1014.4, found 1014.5.
1H NMR (500 MHz, DMSO-d6) δ 12.11 (s, 1H), 11.02 (d, J = 2.3 Hz, 1H), 8.43 − 8.14 (m, 3H), 7.95 (dd, J = 9.2, 2.6 Hz, 1H), 7.90 − 7.76 (m, 1H), 7.66 (dd, J = 8.8, 7.1 Hz, 1H), 7.45 (s, 1H), 7.35 (d, J = 1.9 Hz, 1H), 7.28 (dd, J = 7.8, 1.8 Hz, 1H), 7.17 − 7.05 (m, 4H), 6.92 (d, J = 3.0 Hz, 1H), 5.84 (ddd, J = 11.8, 7.6, 3.9 Hz, 1H), 4.75 (s, 2H), 4.50 (t, J = 8.0 Hz, 1H), 4.41 (d, J = 14.0 Hz, 1H), 4.36 − 4.28 (m, 4H), 3.93 (d, J = 3.4 Hz, 4H), 3.79 (d, J = 14.0 Hz, 1H), 3.59 (dd, J = 15.4, 10.2 Hz, 1H), 3.16 (dq, J = 13.4, 7.6, 7.2 Hz, 2H), 3.09 (t, J = 7.9 Hz, 2H), 2.94 (ddd, J = 17.6, 14.7, 5.6 Hz, 1H), 2.72 (d, J = 15.1 Hz, 1H), 2.66 − 2.54 (m, 4H), 2.23 (s, 3H), 2.04 (dq, J = 9.9, 4.9, 4.0 Hz, 1H), 1.49 (ddt, J = 33.3, 12.8, 6.4 Hz, 2H), 1.34 − 1.23 (m, 4H), 1.09 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.37, 173.20, 170.66, 167.39, 163.91, 163.57, 163.13, 162.73, 156.64, 155.28, 150.91, 147.90, 146.13, 142.51, 142.09, 135.49, 133.72, 133.39, 131.33, 131.08, 129.45, 129.09, 128.97, 128.41, 127.79, 124.76, 124.21, 124.03, 123.70, 123.55, 115.24, 108.61, 107.64, 73.56, 67.71, 56.64, 52.59, 51.06, 48.93, 46.86, 40.74, 38.73, 37.01, 35.12, 31.27, 30.66, 29.36, 28.78, 26.49, 22.00, 19.15, 18.52.
Synthesis of (3S)-3-(3-(((4R)-7-(6-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)hexyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-04–086)
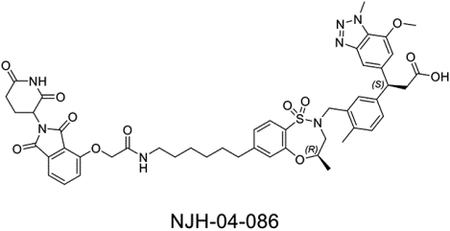
NJH-04–086 was synthesized using the same procedure as used for NJH-04–087.
LC/MS m/z calculated for [M+H]+ 964.4, found 964.6.
1H NMR (500 MHz, DMSO-d6) δ 12.11 (s, 1H), 11.12 (s, 1H), 7.94 (t, J = 5.7 Hz, 1H), 7.81 (dd, J = 8.5, 7.3 Hz, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.45 (s, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.35 (d, J = 1.9 Hz, 1H), 7.28 (dd, J = 7.8, 1.8 Hz, 1H), 7.17 (dd, J = 8.1, 1.6 Hz, 1H), 7.14 − 7.10 (m, 2H), 6.93 (s, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.78 (s, 2H), 4.50 (t, J = 8.0 Hz, 1H), 4.41 (d, J = 14.1 Hz, 1H), 4.39 − 4.35 (m, 1H), 4.33 (s, 3H), 3.93 (s, 3H), 3.80 (d, J = 14.0 Hz, 1H), 3.59 (dd, J = 15.4, 10.2 Hz, 1H), 3.16 (q, J = 6.6 Hz, 2H), 3.09 (t, J = 8.0 Hz, 2H), 2.89 (ddd, J = 17.2, 13.9, 5.4 Hz, 1H), 2.72 (d, J = 15.1 Hz, 1H), 2.64 (dd, J = 8.7, 6.7 Hz, 2H), 2.61 − 2.53 (m, 1H), 2.23 (s, 3H), 2.04 (tt, J = 6.9, 3.9 Hz, 1H), 1.59 (d, J = 8.0 Hz, 2H), 1.46 (t, J = 7.0 Hz, 2H), 1.32 (t, J = 4.2 Hz, 4H), 1.09 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 173.32, 173.23, 170.39, 167.22, 167.16, 166.01, 155.49, 155.32, 150.96, 147.91, 146.11, 142.55, 142.13, 137.44, 135.51, 133.70, 133.47, 131.33, 131.07, 129.52, 129.03, 127.81, 124.82, 124.19, 124.11, 120.80, 117.23, 116.54, 108.58, 107.65, 73.46, 68.04, 56.65, 52.52, 49.24, 48.87, 46.86, 39.32, 38.75, 37.04, 35.15, 31.38, 30.75, 29.32, 28.78, 26.48, 22.44, 19.17, 18.54.
Synthesis of (3S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-(((4R)-4-methyl-7-(6-(2-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-5-yl)oxy)acetamido)hexyl)-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoic acid (NJH-04–202)
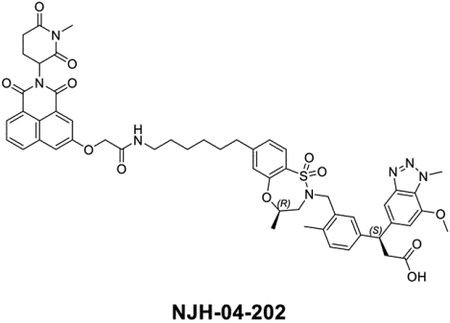
NJH-04–202 was synthesized using the same procedure as used for NJH-04–087.
LC/MS m/z calculated for [M+H]+ 1028.4, found 1028.4.
1H NMR (500 MHz, DMSO-d6) δ 12.10 (s, 1H), 8.45 − 8.16 (m, 4H), 7.96 (dd, J = 9.4, 2.6 Hz, 1H), 7.90 − 7.80 (m, 1H), 7.65 (dd, J = 7.9, 1.9 Hz, 1H), 7.45 (s, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.28 (dd, J = 7.8, 1.9 Hz, 1H), 7.11 (q, J = 8.0, 6.7 Hz, 3H), 6.92 (d, J = 1.1 Hz, 1H), 5.93 (dd, J = 12.2, 5.8 Hz, 1H), 4.75 (s, 2H), 4.50 (t, J = 8.0 Hz, 1H), 4.41 (d, J = 14.0 Hz, 1H), 4.33 (s, 4H), 3.93 (s, 3H), 3.79 (d, J = 14.0 Hz, 1H), 3.59 (dd, J = 15.4, 10.1 Hz, 1H), 3.15 (dt, J = 11.9, 6.9 Hz, 2H), 3.12 − 3.05 (m, 5H), 3.03 − 2.95 (m, 1H), 2.83 − 2.66 (m, 2H), 2.64 − 2.54 (m, 3H), 2.23 (s, 3H), 2.08 (s, 2H), 1.54 − 1.39 (m, 4H), 1.26 (d, J = 14.3 Hz, 4H), 1.09 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 171.09, 170.23, 168.32, 165.21, 165.16, 161.67, 161.35, 161.04, 160.67, 154.47, 153.17, 148.76, 145.78, 143.96, 140.40, 140.02, 133.37, 132.03, 131.55, 131.26, 129.18, 128.92, 127.42, 126.89, 126.28, 125.68, 122.62, 122.05, 121.94, 121.41, 120.89, 106.46, 105.51, 71.29, 65.53, 54.51, 50.36, 49.44, 46.73, 44.72, 36.58, 34.90, 33.01, 29.26, 28.58, 27.30, 27.26, 26.71, 24.84, 24.37, 18.96, 17.04, 16.41.
Synthesis of (S)-3-(3-(((R)-7-(3-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)propyl)-4-methyl-1,1-dioxido-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)-4-methylphenyl)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)propanoic acid (NJH-04–098)
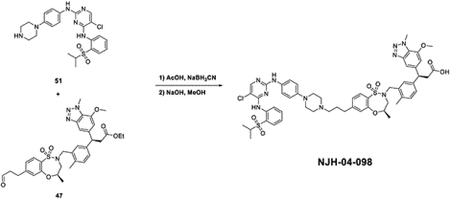
To the solution of 5-chloro-N4-(2-(isopropylsulfonyl)phenyl)-N2-(4-(piperazin-1-yl)phenyl)pyrimidine-2,4-diamine (18 mg, 0.037 mmol) and ethyl (S)-3-(7-methoxy-1-methyl-1H-benzo[d][1,2,3]triazol-5-yl)-3-(4-methyl-3-(((R)-4-methyl-1,1-dioxido-7-(3-oxopropyl)-3,4-dihydro-2H-benzo[b][1,4,5]oxathiazepin-2-yl)methyl)phenyl)propanoate (35 mg, 0.054 mmol) in MeOH (1 mL), was added AcOH (2 drops) and NaBH3CN (7 mg, 0.111 mmol) at room temperature. The reaction was quenched with water after the starting material was consumed and extracted with EtOAc. The organic layer was washed with brine and dried with Na2SO4. The mixture was concentrated under vacuum to get the crude product without any purification. The resulted product was dissolved in MeOH (2 mL) and then NaOH (150 μL, 2M) was added. The reaction was heated to 80°C for 2 hours and the mixture was purified by reverse-HPLC to get titled product (29.7 mg, 0.028 mmol, 75%).
LC/MS m/z calculated for [M+H]+ 1077.4, found 1077.6.
1H NMR (500 MHz, DMSO) δ 9.68 (s, 1H), 9.52 (s, 1H), 9.42 (s, 1H), 8.64 (s, 1H), 8.26 (s, 1H), 7.86 (dd, J = 8.0, 1.6 Hz, 1H), 7.78 − 7.71 (m, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.45 (s, 1H), 7.40 − 7.36 (m, 1H), 7.36 − 7.33 (m, 1H), 7.32 − 7.29 (m, 1H), 7.27 − 7.24 (m, 2H), 7.13 (d, J = 7.8 Hz, 1H), 6.97 − 6.93 (m, 2H), 4.51 (t, J = 8.0 Hz, 1H), 4.42 (d, J = 14.1 Hz, 1H), 4.34 (s, 3H), 3.94 (s, 3H), 3.83 − 3.75 (m, 3H), 3.67 − 3.58 (m, 3H), 3.46 (p, J = 6.8 Hz, 1H), 3.23 − 3.17 (m, 3H), 3.10 (t, J = 8.0 Hz, 1H), 2.95 (t, J = 12.2 Hz, 2H), 2.78 − 2.73 (m, 3H), 2.55 (s, 3H), 2.24 (s, 3H), 2.10 − 2.04 (m, 2H), 1.18 (d, J = 6.7 Hz, 6H), 1.10 (d, J = 6.3 Hz, 3H).13C NMR (126 MHz, DMSO) δ 173.23, 158.82, 158.55, 158.09, 155.44, 155.35, 148.95, 147.92, 146.12, 145.41, 142.57, 142.15, 138.48, 135.48, 133.67, 133.50, 131.90, 131.46, 131.11, 129.41, 129.27, 127.86, 124.90, 124.25, 124.20, 121.65, 116.94, 108.55, 107.70, 73.63, 56.65, 55.44, 55.34, 52.56, 51.41, 48.94, 46.84, 40.76, 40.44, 40.35, 40.27, 40.18, 40.10, 40.02, 39.85, 39.68, 39.52, 39.35, 37.05, 32.00, 24.79, 19.17, 18.54, 15.31.
Supplementary Material
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-KEAP1 | Cell Signaling Technology | Cat # 4678S; RRID: AB_10548196 |
| Anti-CRBN | Cell Signaling Technology | Cat #71810S; RRID: AB_2799810 |
| Anti-BRD4 | Bethyl Laboratories | Cat # A301–985A-M; RRID: AB_2631450 |
| Anti-BRD2 | Bethyl Laboratories | Cat # A302–582A; RRID: AB_2034828 |
| Anti-BRD3 | Abcam | Cat # ab228936; RRID:N/A |
| Anti-β-Actin | Cell Signaling Technology | Cat # 3700; RRID:AB_2242334 |
| Anti-FAK | Cell Signaling Technology | Cat # 3285; RRID: AB_2269034 |
| Anti-BTK | Cell Signaling Technology | Cat # 8547S; RRID: AB_10950506 |
| Goat IRDye 680RD anti-Mouse IgG | LI-COR Bioscience | Cat # 926–68070; RRID: AB_10956588 |
| Goat IRDye 800CW anti-Rabbit IgG | LI-COR Bioscience | Cat # 926–32211; RRID: AB_621843 |
| IRDye®800-labeled goat anti-mouse IgG | LI-COR Bioscience | Cat #926–32210; RRID:AB_621842 |
| Bacterial and Virus Strains | ||
| KEAP1 MISSION shRNA Bacterial Glycerol Stock | Sigma Aldrich | Cat # TRCN0000158081 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DGY-03–188 | This paper | N/A |
| DGY-03–172 | This paper | N/A |
| DGY-04–083 | This paper | N/A |
| DGY-04–087 | This paper | N/A |
| DGY-04–091 | This paper | N/A |
| DGY-04–116 | This paper | N/A |
| DGY-05–089 | This paper | N/A |
| DGY-05–119 | This paper | N/A |
| DGY-06–117 | This paper | N/A |
| DGY-06–117-pk1 | This paper | N/A |
| DGY-06–117-pk2 | This paper | N/A |
| DGY-09–081 | This paper | N/A |
| DGY-09–084 | This paper | N/A |
| NJH-02–123 | This paper | N/A |
| NJH-02–136 | This paper | N/A |
| NJH-02–160 | This paper | N/A |
| NJH-02–162 | This paper | N/A |
| NJH-04–086 | This paper | N/A |
| NJH-04–087 | This paper | N/A |
| NJH-04–098 | This paper | N/A |
| NJH-04–195 | This paper | N/A |
| NJH-04–202 | This paper | N/A |
| NJH-05–137 | This paper | N/A |
| NJH-05–138 | This paper | N/A |
| NJH-05–139 | This paper | N/A |
| NJH-05–141 | This paper | N/A |
| NJH-05–142 | This paper | N/A |
| NJH-05–143 | This paper | N/A |
| NJH-05–146 | This paper | N/A |
| Lenalidomide | Sigma Aldrich | Cat # 901558 |
| MLN4924 | Thermo Fisher Scientific | Cat # 50-547-70001 |
| Bortezomib | MedChemExpress | Cat. No.: HY-10227 |
| KI696 (DGY-09–084) | This paper | N/A |
| TAE684 | MedChemExpress | Cat. No.: HY-10192 |
| TL12–186 | MedChemExpress | Cat. No.: HY-130665 |
| ZXH-02–043 | Nowak, et al., (2018). | N/A |
| JQ1 | Selleckchem | Cat # S7110 |
| Human wild-type of Keap1 (residues 321–609) protein | This study | N/A |
| LanthaScreen™ Eu-Streptavidin | Invitrogen | PV5899 |
| Alexa Fluor™ 647 C2 Maleimide | Invitrogen | A20347 |
| BodipyFL-labeled Spycatcher | Nowak et al., 2018 | N/A |
| Nrf2 peptide | Genscript | N/A |
| Critical Commercial Assays | ||
| BCA protein assay kit | Thermo Fisher Scientific | Cat # 23227 |
| Experimental Models: Cell Lines | ||
| Human: MM.1S | ATCC | Cat # CRL-2974 |
| Human: OVCAR8 | Panagiotis A. Konstantinopoulos’s laboratory | N/A |
| Human: PATU-8988T | DSMZ | Cat # ACC162 |
| Human: JeKo-1 | ATCC | Cat # CRL-3006 |
| Human: Mino | ATCC | Cat # CRL-3000 |
| Insect: Sf9 cells | ATCC | CRL-1711 |
| Insect: High Five™ cells | ThermoFisher Scientific | B85502 |
| Software and Algorithms | ||
| Prism | Graphpad | RRID:SCR_002798 |
| ImageJ | ImageJ | RRID:SCR_003070 |
| Other | ||
| cOmplete, Mini Protease Inhibitor Cocktail | Roche | Cat # 11836153001 |
| PhosSTOP Phosphotase Inhibitor Tablets | Roche | Cat # 04906837001 |
| Odyssey Blocking Buffer | LI-COR Biosciences | Cat # 927–50100 |
| Lipofectamine 2000 Reagent | Thermo Fisher Scientific | Cat # 11668–019 |
| Mycoalert Lonza | Lonza | Cat # LT07–318 |
| CellTiter Glo | Promega | Cat # G7571 |
| FuGene HD transfection reagent | Promega | 4561096 |
| ESF921 insect cell culture medium | Expression Systems | 10437–028 |
| Sf-900 II SFM serum-free medium, complete | Gibco | E2311 |
| Vivaspin Turbo 4, 30,000 MWCO concentrator | Sartorius | 96-001-01 |
| Strep-TactinXT Superflow high capacity | IBA | 10902–088 |
| Superdex 200 Increase 10/300 GL | GE Healthcare | VS04T21 |
| PHERAstar FS microplate reader | BMG Labtech | N/A |
| Deposited data | ||
| wp-esf_064 | This paper | PXD034424 |
| wp-esf_214 | This paper | PXD034423 |
| wp-esf_102 | This paper | PXD034378 |
Highlights:
DGY-06–177-pk2 induced KEAP1-dependent BRD4 degradation
NJH-04–098 results in FAK degradation in mice but not human cells
KEAP1 recruiting PROTACs are less versatile than VHL or CRBN
CRBN-based KEAP1 PROTAC effectively degrades KEAP1
ACKNOWLEDGEMENTS
We thank Milka Kostic and Eric Wang for editing the manuscript. We thank Zhen-Yu Jim Sun for his assistance with NMR data collection.
Grants:
5 R01 CA218278, NIH
Gray/Fischer (MPI) and Hale Center for Pancreatic Cancer.
NCI K22 CA258805 (B.N.) NCI F99 CA253754 (Hojong. Y.)
DECLARATION OF INTERESTS
G.D., J.J., N.J.H., N.S., K.A.D., T.Z., E.S.F. and N.S.G. are inventors on patent applications related to the KEAP1 degraders described in this manuscript. Nathanael Gray is a founder, science advisory board member (SAB) and equity holder in Syros, C4, Allorion, Lighthorse, B2S, Inception, Matchpoint, Larkspur (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Voronoi, Deerfield, Springworks, Interline and Sanofi. E.S.F. is a founder, science advisory board member (SAB), and equity holder in Civetta, Lighthorse, Proximity and Neomorph (board member), an equity holder and SAB in Photys and Avilar, and a consultant to Sanofi, Novartis, Deerfield, and EcoR1. The Fischer lab receives or has received research funding from Novartis, Astellas, Voronoi, Interline and Ajax. J.C. is a scientific founder and equity holder of Matchpoint and M3 bioinformatics & technology Inc, and a consultant and equity holder to Soltego, Allorion, Matchpoint, and receives research funding from Springworks and Interline. T.Z. is a consultant and equity holder for Matchpoint. K.A.D is a consultant to Kronos Bio and Neomorph. The other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdulrahman W, Uhring M, Kolb-Cheynel I, Garnier J-M, Moras D, Rochel N, Busso D, and Poterszman A (2009). A set of baculovirus transfer vectors for screening of affinity tags and parallel expression strategies. Anal. Biochem 385, 383–385. [DOI] [PubMed] [Google Scholar]
- Abed DA, Goldstein M, Albanyan H, Jin H, and Hu L (2015). Discovery of direct inhibitors of Keap1–Nrf2 protein–protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 5, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur R, Valle-Argos B, Steele AJ, and Packham G (2020). Development of PROTACs to address clinical limitations associated with BTK-targeted kinase inhibitors. Exploration of targeted anti-tumor therapy 1, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L, and Dinkova-Kostova AT (2011). The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol 85, 241–272. [DOI] [PubMed] [Google Scholar]
- Bondeson DP, and Crews CM (2017). Targeted protein degradation by small molecules. Annu. Rev. Pharmacol. Toxicol 57, 107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM, Woyach JA, Johnson AJ, Byrd JC, and Crews CM (2018). Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57, 3564–3575. [DOI] [PubMed] [Google Scholar]
- Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, and Wang J (2018). The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol 25, 67–77. e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Yu X, Lu K, Xie L, Wang L, Meng F, Han X, Chen X, Liu J, and Xiong Y (2020). Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem 63, 1216–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, and Saxty G (2016). Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem 59, 3991–4006. [DOI] [PubMed] [Google Scholar]
- de Wispelaere M, Du G, Donovan KA, Zhang T, Eleuteri NA, Yuan JC, Kalabathula J, Nowak RP, Fischer ES, and Gray NS (2019). Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ (2020). Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 580, 329–338. [DOI] [PubMed] [Google Scholar]
- Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, Carano RA, Darrow J, Davies DR, and DeForge LE (2011). Specific Btk inhibition suppresses B cell–and myeloid cell–mediated arthritis. Nat. Chem. Bio 7, 41–50. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, Donovan KA, Yang G, Li Z, and Fischer ES (2019). Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, and Fischer ES (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 7, e38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, and Liu X (2020). Mapping the degradable kinome provides a resource for expedited degrader development. Cell 183, 1714–1731. e1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass EF Jr, Miller CJ, Sparer G, Shapiro H, and Spiegel DA (2013). A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc 135, 6092–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan J, Qian Y, Gough S, Andreoli M, Bookbinder M, Cadelina G, Bradley J, Rousseau E, Willard R, and Pizzano J (2019). Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer (AACR).
- Gabizon R, Shraga A, Gehrtz P, Livnah E, Shorer Y, Gurwicz N, Avram L, Unger T, Aharoni H, and Albeck S (2020). Efficient targeted degradation via reversible and irreversible covalent PROTACs. J. Am. Chem. Soc 142, 11734–11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Sun X, and Rao Y (2020). PROTAC technology: opportunities and challenges. ACS Med. Chem. Lett 11, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardini M, Maniaci C, Hughes SJ, Testa A, and Ciulli A (2019). Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem 27, 2466–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, and Dinkova-Kostova AT (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci 39, 199–218. [DOI] [PubMed] [Google Scholar]
- Henning NJ, Manford AG, Spradlin JN, Brittain SM, Zhang E, McKenna JM, Tallarico JA, Schirle M, Rape M, and Nomura DK (2022). Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J. Am. Chem. Soc 144, 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-T, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho J-H, Ko E, and Jang J (2018). A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol 25, 88–99. e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur W, and Gray NS (2011). Small molecule modulators of antioxidant response pathway. Curr. Opin. Chem. Biol 15, 162–173. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, and Yamamoto M (2000). Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem 275, 16023–16029. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, and Hatayama I (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun 236, 313–322. [DOI] [PubMed] [Google Scholar]
- Jaime-Figueroa S, Buhimschi AD, Toure M, Hines J, and Crews CM (2020). Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett 30, 126877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J, To C, De Clercq DJ, Park E, Ponthier CM, Shin BH, Mushajiang M, Nowak RP, Fischer ES, and Eck MJ (2020). Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew. Chem., Int. Ed. Engl 59, 14481–14489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, and Gray NS (2019). Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem., Int. Ed. Engl 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z-Y, Lu M-C, and You Q-D (2016). Discovery and development of Kelch-like ECH-associated protein 1. nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein–protein interaction inhibitors: achievements, challenges, and future directions. J. Med. Chem 59, 10837–10858. [DOI] [PubMed] [Google Scholar]
- Jones LH (2018). Small-molecule kinase downregulators. Cell Chem. Biol 25, 30–35. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, and Biswal S (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol 47, 89–116. [DOI] [PubMed] [Google Scholar]
- Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, Liu X, Thummuri D, Yuan Y, and Wiegand JS (2019). A selective BCL-X L PROTAC degrader achieves safe and potent antitumor activity. Nat. Med 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Lee DH, Park S, Jo S-H, Ku B, Park SG, Park BC, Jeon YU, Ahn S, and Kang CH (2019). Disordered region of cereblon is required for efficient degradation by proteolysis-targeting chimera. Sci. Rep 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic M, and Jones LH (2020). Critical assessment of targeted protein degradation as a research tool and pharmacological modality. Trends Pharmacol. Sci 41, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, and Crews CM (2016). Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem., Int. Ed. Engl 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Da Y, Wang F, Yan R, Shu Y, Lin P, and Lin J (2020). Targeted selective degradation of Bruton’s tyrosine kinase by PROTACs. Med.Chem. Res 29, 802–808. [Google Scholar]
- Lo SC, Li X, Henzl MT, Beamer LJ, and Hannink M (2006). Structure of the Keap1: Nrf2 interface provides mechanistic insight into Nrf2 signaling. The EMBO journal 25, 3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y, You Q, and Jiang Z (2018). Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem 146, 251–259. [DOI] [PubMed] [Google Scholar]
- Luo M, Spradlin JN, Boike L, Tong B, Brittain SM, McKenna JM, Tallarico JA, Schirle M, Maimone TJ, and Nomura DK (2021). Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol 28, 559–566. e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magesh S, Chen Y, and Hu L (2012). Small molecule modulators of K eap1-N rf2-ARE pathway as potential preventive and therapeutic agents. Med.Res. Rev 32, 687–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C, Hughes SJ, Testa A, Chen W, Lamont DJ, Rocha S, Alessi DR, Romeo R, and Ciulli A (2017). Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Couto S, Zheng X, Lu G, Hui J, Stamp K, Drew C, Ren Y, Wang M, and Carpenter A (2018). SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Bio 14, 981–987. [DOI] [PubMed] [Google Scholar]
- Nalawansha DA, and Crews CM (2020). PROTACs: An emerging therapeutic modality in precision medicine. Cell Chem. Biol 27, 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T, Snyder LB, Willard RR, Vitale N, Pizzano J, Gordon DA, Bookbinder M, Macaluso J, Dong H, and Ferraro C (2019). ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol 37, 1200.30920878 [Google Scholar]
- Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, and Ishoey M (2018). Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Bio 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MA, and Hayes JD (2015). The Keap1/Nrf2 pathway in health and disease: from the bench to the clinic. Biochem. Soc. Trans 43, 687–689. [DOI] [PubMed] [Google Scholar]
- Parker SJ, Amendola CR, Hollinshead KE, Yu Q, Yamamoto K, Encarnación-Rosado J, Rose RE, LaRue MM, Sohn AS, and Biancur DE (2020). Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 10, 1018–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrylak DP, Gao X, Vogelzang NJ, Garfield MH, Taylor I, Dougan Moore M, Peck RA, and Burris III HA (2020). First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI) (American Society of Clinical Oncology; ). [Google Scholar]
- Powell CE, Du G, Bushman JW, He Z, Zhang T, Fischer ES, and Gray NS (2021). Selective degradation-inducing probes for studying cereblon (CRBN) biology. RSC Med. Chem 12, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, Dastjerdi S, Wühr M, Dhe-Paganon S, and Armstrong SA (2017). Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew. Chem., Int. Ed. Engl 56, 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ (2001). Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U S A 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salami J, and Crews CM (2017). Waste disposal—an attractive strategy for cancer therapy. Science 355, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Samarasinghe KT, and Crews CM (2021). Targeted protein degradation: a promise for undruggable proteins. Cell Chem. Biol 28, 934–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemer J, Horst R, Meng Y, Montgomery JI, Xu Y, Feng X, Borzilleri K, Uccello DP, Leverett C, and Brown S (2021). Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat. Chem. Bio 17, 152–160. [DOI] [PubMed] [Google Scholar]
- Shorer Arbel Y, Katz B-Z, Gabizon R, Shraga A, Bronstein Y, Kamdjou T, Globerson Levin A, Perry C, Avivi I, and London N (2021). Proteolysis Targeting Chimeras for BTK Efficiently Inhibit BCell Receptor Signaling and Can Overcome Ibrutinib Resistance in CLL Cells. Front. Oncol 11, 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, To M, Proudfoot A, Ornelas E, and Woldegiorgis M (2019). Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Bio 15, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C, Kehm H, Lindner S, Vu LP, Köpff S, Mármol ÁL, Weiler C, Wagner KG, Reichenzeller M, and Krönke J (2019). PROTAC-mediated crosstalk between E3 ligases. Chem. Commun 55, 1821–1824. [DOI] [PubMed] [Google Scholar]
- Steinebach C, Lindner S, Udeshi ND, Mani DC, Kehm H, Köpff S, Carr SA, Gütschow M, and Krönke J (2018). Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem. Bio 13, 27712782. [DOI] [PubMed] [Google Scholar]
- Su S, Yang Z, Gao H, Yang H, Zhu S, An Z, Wang J, Li Q, Chandarlapaty S, and Deng H (2019). Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J. Med. Chem 62, 7575–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J, and Rao Y (2019). Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 33, 2105–2110. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, Zhao M, Hwang J, Song Y, and Liu W (2018). PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 28, 779–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, and Yamamoto M (2012). Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med 53, 817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinworth CP, Lithgow H, Dittus L, Bassi ZI, Hughes SE, Muelbaier M, Dai H, Smith IE, Kerr WJ, and Burley GA (2019). PROTAC-mediated degradation of Bruton’s tyrosine kinase is inhibited by covalent binding. ACS Chem. Bio 14, 342–347. [DOI] [PubMed] [Google Scholar]
- Tong B, Luo M, Xie Y, Spradlin JN, Tallarico JA, McKenna JM, Schirle M, Maimone TJ, and Nomura DK (2020). Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troup RI, Fallan C, and Baud MG (2020). Current strategies for the design of PROTAC linkers: a critical review. Exploration of Targeted Anti-Tumor Therapy 1, 273–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Meng F, Park K-S, Yim H, Velez J, Kumar P, Wang L, Xie L, Chen H, and Shen Y (2021). Harnessing the E3 Ligase KEAP1 for Targeted Protein Degradation. J. Am. Chem. Soc 143, 37, 15073–15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WL, and Papagiannakopoulos T (2020). The pleiotropic role of the KEAP1/NRF2 pathway in cancer. Annu. Rev. Cancer Biol 4, 413–435. [Google Scholar]
- Xue G, Chen J, Liu L, Zhou D, Zuo Y, Fu T, and Pan Z (2020). Protein degradation through covalent inhibitor-based PROTACs. Chem. Commun 56, 1521–1524. [DOI] [PubMed] [Google Scholar]
- Xue G, Wang K, Zhou D, Zhong H, and Pan Z (2019). Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc 141, 18370–18374. [DOI] [PubMed] [Google Scholar]
- Yao N, Wang C-R, Liu M-Q, Li Y-J, Chen W-M, Li Z-Q, Qi Q, Lu J-J, Fan C-L, and Chen M-F (2020). Discovery of a novel EGFR ligand DPBA that degrades EGFR and suppresses EGFR-positive NSCLC growth. Signal Transduct. Target. Ther 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan K, Wang X, Dong H, Min W, Hao H, and Yang P (2021). Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm. Sin. B 11, 30–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, Nabet B, Gero TW, Feru F, and Li L (2020). Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol 27, 19–31. e16. [DOI] [PubMed] [Google Scholar]
- Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, and Cravatt BF (2019). Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Bio 15, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Luukkonen LM, Eissler CL, Crowley VM, Yamashita Y, Schafroth MA, Kikuchi S, Weinstein DS, Symons KT, and Nordin BE (2021). DCAF11 supports targeted protein degradation by electrophilic proteolysis-targeting chimeras. J. Am. Chem. Soc 143, 5141–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, Koegl M, Riching KM, Daniels DL, and Spallarossa A (2018). Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel–Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem 62, 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J, Zhu H, Farley KA, Ding W, and Schiemer J (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U S A 115, E7285–E7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. The proteomics data wp-esf_064, wp-esf_214 and wp-esf_102 generated during this study are available at PRIDE archive PXD034424, PXD034423 and PXD034378. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.