

Abstract
In this Dispatch from Biotech, we briefly review the urgent need for extensive expansion of newborn screening (NBS) by genomic sequencing, and the reasons why early attempts had limited success. During the next decade transformative developments will continue in society and in the pharmaceutical, biotechnology, informatics, and medical sectors that enable prompt addition of genetic disorders to NBS by rapid whole genome sequencing (rWGS) upon introduction of new therapies that qualify them according to the Wilson and Jungner criteria (Wilson, J. M. G., & Jungner, G., World Health Organization. (1968). Principles and Practice of Screening for Disease. World Health Organization. Retrieved from https://apps.who.int/iris/handle/10665/37650). Herein we describe plans, progress, and clinical trial designs for BeginNGS (Newborn Genome Sequencing to end the diagnostic and therapeutic odyssey), a new international, pre‐competitive, public–private consortium that proposes to implement a self‐learning healthcare delivery system for screening all newborns for over 400 hundred genetic diseases, diagnostic confirmation, implementation of effective treatment, and acceleration of orphan drug development. We invite investigators and stakeholders worldwide to join the consortium in a prospective, multi‐center, international trial of the clinical utility and cost effectiveness of BeginNGS.
Keywords: genetic disease, management guidance, molecular diagnosis, newborn screening, orphan drug, rapid whole genome sequencing
1. INTRODUCTION
Newborn screening (NBS) started in the late 1960s before the birth of molecular biology, human genomics, or even molecular genetics (Peterson et al., 1968; Wilson & Jungner, 1968). At that time phenotypes, proteins, enzymes, biochemicals, and karyotypes were the only biomarkers of childhood genetic disease available. Thus, NBS was based upon these, and was spectacularly successful until the completion of the human genome project, advent of next generation sequencing (NGS), growth in the biotechnology industry, and rare disease pharma. Unfortunately, NBS has not kept pace with the explosion in biotechnology, genomics, molecular genetics, and therapeutic innovations since 2005 (Centers for Disease Control and Prevention [CDC], 2008; Newborn Screening Task Force, 2000; Sontag et al., 2020; Yu et al., in press). Between 2006 and 2022, the number of core disorders recommended for NBS by mass spectrometry (MS, MS‐based NBS) of dried blood spots (DBS)—the United States Recommended Uniform Screening Panel (RUSP)—increased from 29 to only 35, and the number of affected infants identified for these disorders remained static at ~6,500 of ~4 million tested per year (CDC, 2008; Sontag et al., 2020). During that period the number of known genetic diseases swelled to ~7,200, rare disease pharma arose, and hundreds of new targeted treatments were approved or are in clinical trials for orphan childhood genetic diseases (Amberger, Bocchini, Scott, & Hamosh, 2019; Bick et al., 2022; Biesecker, Green, Manolio, Solomon, & Curtis, 2021; Pichini et al., 2022; Woerner, Gallagher, Vockley, & Adhikari, 2021; Yu et al., in press). Thus, today an immense gap has developed between the disorders on the RUSP and those that meet the original criteria for NBS (Wilson & Jungner, 1968). While traditional NBS is not limited to MS, the existence of this gap is egregious since NBS saves lives, avoids suffering, is cost effective, and equitably performed with regard to race, ethnicity, and socioeconomic status (Brosco, Grosse, & Ross, 2015).
Since the advent of NGS, multiple groups have explored replacement of MS‐based NBS with genomic NBS (which is not the intent of BeginNGS) with limited success. The Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) pilot program (2013–2019) found that NBS by whole exome sequencing (WES, NBS‐WES) was less sensitive for RUSP conditions than MS‐based NBS: WES had 88% sensitivity for RUSP disorders in 691 true positive samples by MS‐based NBS (Adhikari et al., 2020; Roman et al., 2020; Wojcik et al., 2021). Separately from NSIGHT, three groups reported similar findings: Bhattacharjee et al. (2015) reported 75% sensitivity of a gene panel in 36 true positive MS‐based NBS children. Bodian et al. (2016) reported 89% concordance of WGS and MS‐based NBS in 1,696 newborns. Cho et al. (2017) reported 93% sensitivity of WES in 81 true positive MS‐based NBS children. An implication of these studies was that genomic NBS was not yet sufficiently sensitive to replace MS‐based NBS. Genomic NBS, however, is ideally suited for filling the disorder gap mentioned above. Specifically, genomic NBS represents a single, comprehensive assay for screening and diagnosis of genetic disorders that meet the original criteria for NBS, but which do not have pathognomonic biochemical markers measurable by MS.
Two NSIGHT projects partly evaluated the utility of genomic NBS to screen for disorders missed by MS. They found that WES identified 3 NBS‐related disorders in 159 infants and 4 “actionable” findings in 106 infants that were missed by MS‐based NBS (Roman et al., 2020; Wojcik et al., 2021). Wojcik et al. (2021) detected mild biotinidase deficiency [Mendelian Inheritance in Man, MIM: 253260], late onset congenital adrenal hyperplasia [MIM: 201910], and glucose 6‐phosphate dehydrogenase deficiency [MIM: 300908]. It is unclear if the benefits of early treatment of any of these disorders outweigh risks. Roman et al. (2020) detected two newborns with disorders that would benefit from early treatment. One had a heterozygous missense variant associated with autosomal dominant familial hypercholesterolemia type 1 [MIM: 606945] with a family history of this disorder, and the other was a female neonate with a heterozygous missense variant associated with mild X‐linked ornithine transcarbamylase deficiency [MIM: 311250] who had normal ammonia levels. Recently, Jian et al. (2022) reported that NBS by WGS for 251 genes (with 16–24‐week turnaround time) had superior analytic performance than MS‐based NBS for 51 disorders in 321 newborns. Confirmatory testing showed a false positive for 3‐methylcrotonyl‐CoA carboxylase 1 deficiency [MIM:210200] identified by traditional MS was correctly identified as an MCCC1 [MIM: 609010] carrier by NBS‐WGS, and six newborns with GJB2 [MIM: 121011]‐associated autosomal recessive deafness 1A [MIM:220290] or MT‐RNR1 [MIM:561000]‐associated aminoglycoside‐induced deafness [MIM:580000] were identified by NBS‐WGS and not by traditional NBS. At present there are at least four commercial NGS panel tests designed for use in newborns. However, there has not yet been broad utilization of genomic NBS either commercially or in the public health service (DeCristo et al., 2021). There are many reasons why this has not yet occurred. First, early efforts faced stiff resistance from traditional NBS practitioners who considered NBS a risk assessment, rather than a diagnostic program. Secondly, there was a dearth of governmental or philanthropic funding for NBS innovation. Thirdly, the many ethical, legal, and social issues (ELSI) of genomic NBS had not been addressed (Clayton, 2010). Fourthly, clinical grade genomic sequencing was too costly relative to the total current cost of NBS in the United States (~$300 per newborn), of which ~$80 was the cost of screening in 2016 (Costich & Durst, 2016; Yu et al., in press). Fifthly, to the best of our knowledge of variant pathogenicity for most genetic diseases was immature. Finally, in addition to external factors, early investigators made two miscalculations. In contrast to the original criteria for NBS, disorder selection was gene‐centric rather than treatment‐centric. Disorders were selected based on “actionability” rather than the Wilson and Jungner criteria (which include broad availability of a treatment with evidence of efficacy in a severe, early childhood onset disease where there was evidence that early treatment improved outcomes) (Andermann, Blancquaert, Beauchamp, & Déry, 2008; Ceyhan‐Birsoy et al., 2017; DeCristo et al., 2021; Milko et al., 2019; Petros, 2012; Wilson & Jungner, 1968). Lastly, groups worked independently and competitively, leading to duplication of effort, inconsistent messaging, inadequate study lengths, and massively underpowered cohorts.
Ten years later, each of the former problems has been mitigated to some extent. First, traditional NBS innovators and practitioners have witnessed the benefit of molecular genetic NBS (or genomic confirmatory testing) for primary immunodeficiencies, cystic fibrosis [MIM: 219700], spinal muscular atrophy type 1 [253300], and Duchenne muscular dystrophy (DMD) [MIM: 310200]. For primary immunodeficiencies and DMD, genomic testing is required in NBS‐positive subjects to define the specific genetic disorder, and, thereby, appropriate specific treatments. Secondly, there is now greater governmental attention to the unmet burden of childhood orphan genetic diseases, leading to large government funded NBS by genomic sequencing efforts in the United Kingdom, the European Union, and Qatar (Bick et al., 2022; Hopkins, Kinsella, & Evans, 2021; Mbarek et al., 2022; Pichini et al., 2022). Thirdly, there is now a large literature regarding the ELSI of NBS by genomic sequencing, as evidenced by an issue of the Hastings Center Report (Johnston et al., 2018). ELSI data from NSIGHT pilot studies has assuaged many of the original concerns regarding genomic sequencing of newborns (Berg et al., 2017; Cakici et al., 2020; Pereira et al., 2019, 2021; Ross & Clayton, 2019; Schwartz et al., 2021). There is also a much more mature regulatory framework regarding individual genomic data security, privacy, and anti‐discrimination protections. Several biotechnology companies are developing human, 30‐fold, third generation whole genome sequencing (WGS) with a consumable cost of $100. Most importantly, rapid clinical WGS (rWGS) and rapid WES (rWES) have proven effective as first tier tests for comprehensive diagnosis of genetic diseases in acutely ill newborns, leading to coverage policies by Medicaid in six states and private payors (reviewed in Kingsmore & Cole, 2022). rWGS has been adapted for NBS and can deliver results in as little as 13 hr, enabling timely treatment initiation even in inborn errors of metabolism with rapidly irreversible brain damage (Kingsmore et al., 2022; Stranneheim et al., 2014). Consensus frameworks now exist for measuring the clinical utility and cost effectiveness of genetic disease diagnosis by NBS (Dimmock et al., 2021; Hinton et al., 2016). Public and private genetic disease variant databases have matured significantly, and allele frequencies are available from very large control cohorts (All of Us, 2022; Gudmundsson et al., 2022; Szustakowski et al., 2021). Lastly, the Orphan Drug Act (1983) created financial benefits to pharmaceutical companies for developing medications for orphan diseases, and the Food and Drug Administration modified regulations to facilitate approval of new drugs for orphan diseases. These have led to massive reallocation of pharma research and development resources to rare genetic diseases.
In summary, we are beginning a new era of genomic NBS. In this Dispatch from Biotech, we will describe plans for and progress with BeginNGS™ (Newborn Genomic Sequencing to end the diagnostic and therapeutic odyssey, Figure 1), a new international, precompetitive, public–private consortium that proposes to implement a self‐learning healthcare delivery system for screening all newborns for hundreds of genetic diseases by rWGS, diagnostic confirmation, implementation of effective treatment, and acceleration of drug development world‐wide (Kingsmore et al., 2022; Owen et al., 2022).
FIGURE 1.

Graphical abstract of the scope and aims of BeginNGS™. From https://radygenomics.org/2022/rcigm‐launches‐program‐to‐advance‐newborn‐screening‐for‐treatable‐genetic‐diseases
2. METHODS
The premise of BeginNGS™ was not to replace MS‐based NBS, but rather to supplement it as a primary screen, diagnostic tool, and management guidance program for disorders that meet the Wilson and Jungner (1968) criteria for NBS but are not currently screened (Figure 1). It will also serve as a supplement and confirmatory molecular diagnostic test for disorders that are currently screened.
2.1. Retrospective BeginNGS clinical study 2
The first retrospective BeginNGS clinical study was recently published (Figure 1, Kingsmore et al., 2022). Retrospective study 2 will occur in the last quarter of 2022 and will have an identical design (Figure 2). The study has two arms. First, qualification of a larger set of variants than in the first retrospective analysis of ~455,000 deidentified United Kingdom Biobank (UKBB) participants and exomes, together with BeginNGS specificity assessment following addition of disorders and new variants. Secondly, BeginNGS sensitivity and clinical utility assessment in ~7,500 critically ill newborns and children and their parents who had received rWGS for molecular diagnosis of a suspected genetic disorder at Rady Children's Institute for Genomic Medicine (RCIGM, compared with 4,374 subjects in study 1).
FIGURE 2.
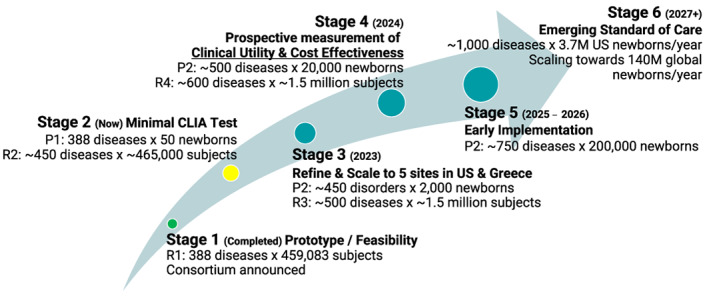
Stages of development of BeginNGS™ and key milestones. Green circle, completed. Yellow circle, in progress. Teal circles, not started. R1, retrospective study 1. R2, retrospective study 2. P1, prospective study 1. P2, prospective study 2. CLIA, clinical laboratory improvements act
We will evaluate ~100 new disorders and ~2,000 new interventions for inclusion in BeginNGS using structured, audited Delphi method developed for the first version (Figure 3, Kingsmore et al., 2022, Owen et al., 2022). We will also evaluate published variants for evidence of pathogenicity using those same procedures. Thus, retrospective study 2 will evaluate ~450 disorders, ~50,000 variants, and ~1,500 treatments (Figure 2).
FIGURE 3.

Methods used to qualify disorders for inclusion in BeginNGS™. GTRx, Genome‐to‐Treatment virtual acute management guidance system (Figure 4). Group A, disorders for which the Delphi panel consensus was that the Phase ii criteria were met. Group B, disorders for which the natural history was not yet well understood or for which early treatment was not yet known to improve outcome but that were retained for prospective clinical study. ICU, intensive care unit; NBS, newborn screening; rWGS, rapid whole genome sequencing
UKBB data was queried through the UKBB Research Analysis Platform under application number 82213. BeginNGS gene regions were extracted from UKBB pVCFs of 454,707 UKBB subjects (Szustakowski et al., 2021). We split multiallelic rows, normalized indels, and filtered out low‐quality variants as described (Kingsmore et al., 2022). We retrieved ClinVar and Mastermind (Genomenon) variants with clinical significance of “Likely_pathogenic” or “Pathogenic” that mapped to the BeginNGS gene regions and had a GNOMAD allele frequency < 0.5% (Chunn et al., 2020; Gudmundsson et al., 2022). We removed ClinVar and Mastermind variants that were block‐listed as a result of the first retrospective study and added missing variants that had been white‐listed as a result of that study (Kingsmore et al., 2022). We intersected the remaining query variant set and UKBB variant set and identified positive individuals based on pattern of inheritance and individual zygosity (heterozygous for dominant disorders, and compound heterozygous, hemizygous, or homozygous for recessive disorders). Where Mendelian Inheritance in Man indicated the pattern of inheritance to be mixed dominant and recessive, we retained only individuals exhibiting recessive patterns of inheritance. We used the aggregated International Statistical Classification of Diseases and Related Health Problems (ICD)‐9/10 codes, Read v2 medication codes, Death Register codes, and self‐reported medical condition data provided for UKBB subjects to identify those affected by specific conditions.
Root cause analysis was performed manually on all BeginNGS positive subjects in the UKBB set to assess the likelihood that they were true or false positives. We first checked gene names, disorder names, and patterns of inheritance to ensure that each variant matched a BeginNGS disorder. We ranked genes by frequency of positive subjects and compared observed frequencies with known incidences of those disorders. Genes with more positive subjects than the population incidence were flagged as outliers. We also ranked variants by proportion and number of positive subjects and those contributing more than 10% were flagged as outliers. Outlier variants identified by these searches underwent: (a) Literature review to assess the quality and quantity of evidence of pathogenicity, including variant effect predictions, the number of citations reporting affected individuals with the BeginNGS disorder in ClinVar or PubMed, and the quality of evidence for pathogenicity in PubMed, including quantitative functional evidence, number of affected subjects, and phenotypes in affected subjects. Well‐established locus‐specific variant databases supplemented reviews of the primary literature. (b) Review of putative compound heterogyzotes to remove those that were either known to occur in cis as recurrent haplotypes or novel haplotypes that were identified by inspection of aligned and phased sequencing reads. (c) Review for evidence that they mapped to regions of the genome that are difficult to genotype with short read sequences. Variants for which root cause analysis identified an artefactual reason for high positivity were block listed. Recurrent variants with strong support for pathogenicity were whitelisted. In retrospective study 2, we anticipate review of ~20,000 variants that did not undergo this analysis in study 1.
Retrospective analysis of genomes and phenotypes of critically ill newborns and children and their parents who had received diagnostic rWGS at RCIGM was approved by the Institutional Review Board of Rady Children's Hospital/University of California—San Diego. The sensitivity and potential clinical utility of BeginNGS were evaluated retrospectively in ~7,500 critically ill children and their parents who had received diagnostic rWGS. For each VCF, we split multiallelic rows, normalized indels, and filtered out low‐quality variants. We intersected the query variant set and RCIGM variant set and identified positive individuals based on pattern of inheritance and individual zygosity. Novel BeginNGS positives underwent clinical interpretation to assess whether they were true or false positives. Sensitivity was assessed by comparison of BeginNGS positives and molecular diagnoses that had been made by clinical rWGS.
In each proband child who had received a molecular diagnosis by rWGS that had been recapitulated by BeginNGS, the observed clinical features were compared with those listed in Mendelian Inheritance in Man, Genetic and Rare Diseases Information Center, and MEDLINE to determine which were attributable to that molecular diagnosis. Based on the assessed efficacy of each indicated intervention for that disorder in GTRx, we compared the impact on the observed, reversible, attributable clinical features of starting those interventions at the actual age of diagnosis by rWGS with that of treatment initiation at the counterfactual age of diagnosis by BeginNGS (day of life 5), as previously described (Kingsmore et al., 2022). The extent to which BeginNGS could have prevented or avoided the occurrence of each of the attributable clinical features was adjudged on a five‐point Likert scale (completely, mostly, partially, none, and uncertain).
2.2. Prospective BeginNGS clinical study 1
The first prospective clinical trial of BeginNGS will be small (n = 50) and designed to refine and validate procedures, rather than test hypotheses related to clinical utility (stage 2, Figure 2). Enrollment will be limited to the Rady Children's Hospital‐San Diego neonatal intensive care unit (NICU) which has participated in several prior clinical studies of diagnostic rWGS. It will enroll from the population of 10–15% of newborns who have an illness, low birthweight, or prematurity that requires transfer to a neonatal intensive care unit, providing time to obtain post‐partum consent (Braun et al., 2020). Fifty families will be enrolled by research staff, who include pediatric research nurses. Genetic counselors will be available to answer questions. The primary endpoint of this pilot study will be to demonstrate the accuracy of results of BeginNGS by comparison with clinical diagnostic rWGS, which will be performed in parallel (Figure 4). This will complete validation according to the Clinical Laboratory Improvements Act (CLIA). The main secondary endpoint will be an assessment of parental perceptions of the benefits and potential harms of BeginNGS. These will be assessed by administration of brief questionnaires at time of consent and return of results. These questionnaires were validated for use in this NICU in the NSIGHT2 study (Cakici et al., 2020). Their reuse will allow quantitative comparison of parental perceptions to diagnostic rWGS and rWGS‐based NBS (Cakici et al., 2020). Another secondary endpoint will be measurement of the rate of identification of variant diplotypes that require confirmatory interpretation. The burden of interpretation of BeginNGS is not yet clear.
FIGURE 4.
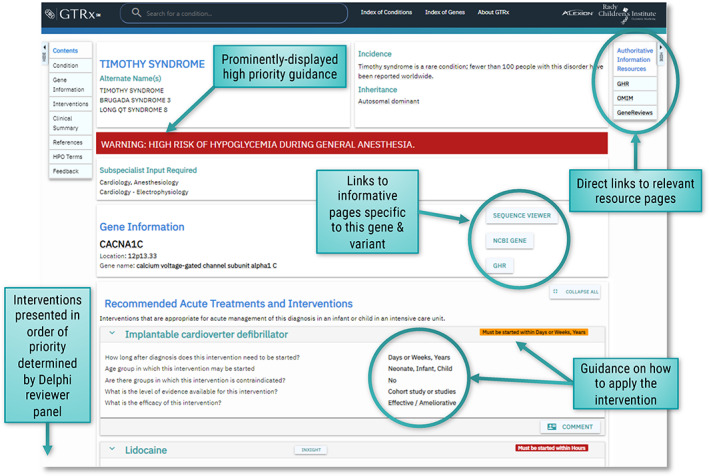
Example of GTRx acute management guidance for Timothy syndrome (MIM: 601005)
2.3. Prospective BeginNGS clinical study 2
Results of the first trial will inform the second, much larger prospective study, which is currently being designed and will start in 2023 (Stages 3–5, Figure 2). It will be international, with initial enrollment sites in the United States and Greece. It will be powered to test the hypotheses that the clinical utility benefits of BeginNGS outweigh any harms, and that BeginNGS has sufficient cost effectiveness for broad implementation in countries currently performing traditional NBS. Enrollment in the United States will include healthy newborns and those with an illness of unknown etiology. In Greece, healthy newborns will be enrolled. The pre‐test probability of genetic disease differs about 60‐fold between these two populations (Kingsmore et al., 2022). Initial site selection for the second study will be designed to facilitate equitable participation of racial, ethnic, and ancestral groups that are minorities in the United States. We propose to use electronic consent and video information about the trial to facilitate consent. We will initially support English, Spanish, and Greek languages. We will add sites and languages iteratively over a 3‐year period.
The primary endpoints of the second clinical study will be the clinical utility and cost effectiveness of BeginNGS (factual) versus standard‐of‐care testing (counterfactual) determined at 1 year of age. The main measure of clinical utility will be the age at implementation of a GTRx intervention upon diagnosis of a genetic disease by BeginNGS versus that by standard‐of‐care testing. Standard‐of‐care includes traditional NBS in that region, and diagnostic testing upon development of symptoms. Other measures of clinical utility will be rates of GTRx intervention, clinically material organ dysfunction, organ failure, death, and expected quality adjusted life years (QALYs). This design and these measures are similar to those of Project Baby Bear (Dimmock et al., 2021) and the framework for NBS outcomes measurement developed by the Advisory Committee on Heritable Disorders in Newborns and Children (Hinton et al., 2016). The second prospective BeginNGS clinical trial will have three types of secondary end points, namely process, perception and phenotype‐driven endpoints. The process measures will be the number of disorders and variants tested, variant block list, variant whitelist, positive screen rate, true positive diagnostic rate (defined by disorders that are fully penetrant in early childhood), false positive rate (including non‐penetrant disorders in early childhood), disease and allele incidence, disease onset age (up to 1 year), and age at disease outcomes measures. These will allow determination of sensitivity, specificity, positive predictive value and negative predictive value. Parental perceptions will be assessed as in the pilot trial. Focus groups are planned in Greece to provide cultural adjustment of the questionnaires for southeastern European populations. Phenotype driven outcomes will be examined by measurement of a common data element set of phenotypes at time of enrollment. Endpoints will be evaluated upon enrollment of 2,000 newborns, 20,000 newborns, and 200,000 newborns.
2.4. Informed parental consent
BeginNGS starts with informed consent from one or both parents either during pregnancy in an obstetrics office or immediately after delivery in a hospital room (Kingsmore et al., 2022). While antepartum enrollment offers unhurried and unstressed decision making, it is logistically more difficult to scale since antenatal care is provided at least at an order of magnitude more community obstetric offices than delivery suites. In addition, rWGS‐based NBS may lack materiality until the baby has been delivered. The average length of hospital stay is 2.2 days in the United States for the two thirds of newborns who undergo vaginal deliveries and 3.5 days for the one third who undergo Cesarean delivery (Rubens et al., 2022). It is at least 1 day longer in Greece.
2.5. Rapid whole genome sequencing from dried blood spots
Dried blood spots are the preferred sample type for BeginNGS since healthy newborns do not have venous access and DBS are well validated for NBS workflows (Ding et al., 2022; Kingsmore et al., 2022; Owen et al., 2022). Nucleic card (ThermoFisher) and Protein Saver 903 (GE Healthcare) cards are acceptable for DBS. Genomic DNA is isolated from DBS with the DNA Flex Lysis Reagent kit (Illumina) from at least five 3 mm2 punches. WGS libraries are prepared using the DNA PCR‐free kit (lllumina). A 2 × 100 nucleotide (nt) rWGS is performed on S2 (6 samples) or S4 (24 samples) flowcells on NovaSeq 6,000 instruments. Sample preparation takes half a day, and rWGS takes 25 hr for S2 flowcells (6 samples) and 36 hr for S4 flowcells (24 samples). The target genome coverage is 30–40× (90–120 GB). rWGS‐based NBS requires batch‐based sample preparation upon arrival of DBS each morning with rWGS that evening.
2.6. BeginNGS bioinformatics
Following rWGS, sequences are aligned to GRCh38 and variants (VCF) identified with DRAGEN v3.10 or later on ICA (Illumina, on Amazon Web Services [AWS] S3, ~40 min per sample). Quality control (QC) metrics are evaluated. The most exacting of these is >10‐fold coverage of every coding nucleotide in >90% of Mendelian Inheritance in Man genes. For rWGS‐based NBS, QC passing VCFs are ingested into a sparse, three‐dimensional array (TileDB v2.8 or later, on AWS S3, ~20 min per sample) and the genotype of each variant in a proband sample automatically refreshes the frequency of each allele in the entire dataset. Results are stored in an additional grouped, variant‐centric TileDB array. Genomic NBS involves screening the ingested VCF against ~30,000 rare (GNOMAD allele frequency < 0.5%), germline, Pathogenic (P) or Likely Pathogenic (LP) ClinVar variants that map to 388 rWGS‐based NBS disease‐gene dyads. The qualification of initial variants, genes and disorders are discussed below, as is their proposed expansion with time (Kingsmore et al., 2022). Approximately 99% of newborns will screen negative, generating a negative screening report akin to those of MS‐based NBS. The one or two positive variants in the remaining 1% of newborns are ingested into Enterprise software (Fabric Genomics), annotated, and manually interpreted, resulting in a diagnostic or negative report. Diagnostic results are returned together with virtual acute management guidance (Genome‐to‐Treatment, GTRx, https://gtrx.rbsapp.net/ or https://gtrx.radygenomiclab.com, Figure 4), either to a medical geneticist (for discharged newborns) or a neonatologist (for hospitalized newborns) (Kingsmore et al., 2022; Owen et al., 2022).
BeginNGS is designed to be self‐learning. Each individual's VCF informs updated allele frequencies for each variant (Kingsmore et al., 2022). Each true and false positive result informs the net pathogenicity assessment for those variants. Each GTRx intervention prescribed and outcome assessed informs the retention of that gene‐disorder dyad in rWGS‐based NBS.
3. DISCUSSION
BeginNGS was informed by a decade of experience with diagnostic rWGS in critically ill neonates with diseases of unknown etiology (reviewed in Kingsmore & Cole, 2022). In 31 clinical studies encompassing 2,433 ill children, 37% were diagnosed with a genetic disease by rWGS or rWES, 29% had an acute change in management, and 18% had a change in outcome (reviewed in Kingsmore & Cole, 2022). The end‐product of this experience was the development of a precision neonatology delivery system for 13‐hr diagnosis of genetic diseases together with acute management guidance for ~450 severe genetic diseases with effective treatments (called Genome‐to‐Treatment, GTRx, Figure 4) (Owen et al., 2022). Even as Medicaid and private payors started to issue coverage policies for rWGS and rWES in critically ill newborns, infants, and children, however, it became clear that prevention of critical illness was the only solution for equitable delivery of neonatal precision medicine regardless of geographic location or ability to pay. The genesis of BeginNGS was the epiphany that diagnostic rWGS and GTRx required relatively modest modifications for rWGS‐based NBS. Diagnostic rWGS in critically ill newborns informed several key decisions regarding BeginNGS:
-
Balancing test cost with time‐to‐treatment initiation. Experience with diagnostic rWGS in thousands of critically ill newborns informed our understanding of rate of disease progression, organ damage, mortality, and real‐world impact of therapy for many genetic diseases (Kingsmore & Cole, 2022). The goal of BeginNGS was to start treatment before symptom onset, or where that was impossible, before irreversible organ damage had occurred. Short read rWGS with GTRx can provide a provisional molecular diagnosis and management guidance in 13 hr, enabling timely treatment initiation even in inborn errors of metabolism with rapidly irreversible neurologic damage for which effective treatments exist (Owen et al., 2022; Stranneheim et al., 2014). Cost, however, is one of the biggest hurdles to national and international adoption of BeginNGS in public health. Ultra‐rapid, short read, diagnostic WGS costs ~$8,000 per infant tested. With automated interpretation and industrial scaling, the cost of 13‐hr time‐to‐treatment can be reduced to ~$2,500 per infant. In the future, with $100 third generation sequencing, automated interpretation, and industrial scaling a cost of $200 per infant tested is feasible. For example, rapid NBS for β‐hemoglobinopathies with Oxford Nanopore long read sequencing was estimated to cost $12 per infant tested (Christopher et al., 2021). Of note, this platform is capable of 8 hr, ~$500 diagnostic rWGS (Goenka et al., 2022).
Until $100 rWGS is available, a 2‐week turnaround would be sufficient for most of the 388 genetic diseases selected for BeginNGS clinical trials. In practice this led to an initial time‐to‐treatment goal of day of life (DOL) five. This, in turn, led to a workflow design that included heel‐prick on DOL one, screening by rWGS in 2 or 3 days, performance of the entire process in conformance with CLIA, and 1 day diagnostic interpretation and return of results accompanied by acute management guidance (Kingsmore et al., 2022). The pilot prospective clinical study will inform an assessment of the relative cost: benefit ratio of treatment on DOL 5 versus earlier and later times.
A reasonable alternative scheme is that healthy babies would receive standard screening with 2‐week turnaround, with 2‐day turnaround (including shipping) reserved for the 10–15% newborns with some indication of illness that is broader than the ICU criteria used for diagnostic rWGS and that includes physician judgment. To this end, we are in the process of building multivariable predictive algorithms for diagnosis of newborn genetic diseases based on univariate phenotypic differences between NICU infants receiving diagnostic rWGS who did or did not receive a diagnosis (Juarez et al., personal communication). In countries other than the United States, cost considerations may require use of targeted rWGS or NGS panels that are customized to also identify known, non‐exonic pathogenic variants (Christopher et al., 2021).
Focus on treatments and outcomes not diseases and diagnoses. Diagnostic rWGS taught us that high rates of parental consent required testing to be focused on maximization of the individual, potential benefit‐to‐harm ratio. Initial efforts at genomic NBS instead focused on panels for which it was clear that gene‐disorder associations were convincing and where diagnostic findings were actionable (Ceyhan‐Birsoy et al., 2017; DeCristo et al., 2021; Milko et al., 2019). This was much broader than the original Wilson and Jungner (1968) criteria for universal NBS. We opted for conservative disorder selection for BeginNGS. We started with 1,527 acute interventions that had evidence of efficacy that mapped to 421 childhood‐onset genetic diseases with sufficient severity to lead to ICU admission (Owen et al., 2022). We evaluated whether these 421 GTRx disorders met the remaining Wilson and Jungner criteria for NBS inclusion, retaining 388 (Figure 3). The advantage of this approach is that it is predicated on having publicly available virtual acute management guidance for each disorder, akin to ACTion sheets, at the outset (ACMG ACT Sheets, 2001–2022, https://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/ACT_Sheets_and_Algorithms.aspx). We propose to expand and update GTRx and the rWGS‐based NBS disorder set annually. The focus on treatments and outcomes informed the primary end point of the main BeginNGS clinical study. In turn, this implies a need for 1 year of follow up of outcomes in BeginNGS enrollees.
Genome sequencing, panel enquiry. Diagnostic rWGS involves ascertainment of all genomic variation, followed by individualized panel enquiry. The panel is the set of disorders with clinical features overlapping each patient's presentation. rWGS‐based NBS allows the panel of disorders, genes, and variants for which results are returned to change with time, region, race, and ethnicity. This is important for at least four reasons. First, the natural history of 25% of the 388 genetic diseases with effective therapies initially selected for rWGS‐based NBS trials was insufficiently clear to unequivocally recommend them for population NBS (Group B in Figure 3, Kingsmore et al., 2022). Inclusion in rWGS‐based NBS trials will allow their natural history to be evaluated in prospective clinical studies that will inform whether they should be retained. Secondly, many new treatments for childhood genetic diseases are being developed, requiring disorders to be added promptly as they are approved (Kingsmore et al., 2022; Owen et al., 2022; Yu et al., in press). Inclusion in rWGS‐based NBS trials will allow the efficacy of newly approved treatments to be evaluated in prospective clinical studies that will inform whether they improve outcomes. Thirdly, different states, regions, and countries have traditionally offered different NBS conditions (Therrell et al., 2015). rWGS enables collection of genome‐wide allele frequency and variant data independent of the conditions for which results are returned, informing population specific incidence that allows rWGS‐based NBS to be regionally tailored. Finally, rWGS enables scoring of ancestry informative markers that could potentially enable allele frequency data to be generated for each racial, ethnic, and ancestral group, informing data‐driven panel selection.
International to ensure full representation of most racial, ethnic, and ancestral groups. Building a strong evidence base for diagnostic rWGS requires multiple clinical studies to be undertaken in many countries (with data standardization and harmonization). Current knowledge of disease‐causing variation and allele frequencies is largely limited to individuals of northern European ancestry. To the best of our knowledge of population‐specific disease incidence is limited to a few highly studied groups such as Finns, Quebecois, Amish, and Ashkenazi Jewish. Site selection for BeginNGS clinical studies will be tailored to ensure representation of every race, ethnicity, and ancestral group (Hobbs et al., personal communication). Initial enrollment will be through the Rady Children's Health System in San Diego, where a majority of newborns are Hispanic, and the University of Tennessee Health System in Memphis (Finkel et al., personal communication), where a majority of newborns are African American. Greece will be the first European country to evaluate BeginNGS at three sites (University of Athens, University of Thessaloniki, University of Thessaly) (Tsipouras et al., personal communication). The ideal solution is enrollment internationally in the ~80 countries that currently perform NBS, with sharing of aggregated, anonymized true positive variants and frequencies (Therrell et al., 2015).
Performance by a pre‐competitive, public–private, biotechnology‐friendly consortium (Figure 3). Pre‐competitive collaboration refers to a group of competing organizations coming together to develop a common solution for a major problem they all share, all can benefit from, and from which none of them would gain a competitive advantage (Institute of Medicine, 2011). These have become relatively common in the pharmaceutical industry and have been successful in genomics. Examples include the SNP consortium and the UKBB. Broad implementation of diagnostic rWGS, which we have been very involved in, required altruistic, time consuming, sharing of knowledge and protocols. The scale and complexity of rWGS‐based NBS mandate multi‐disciplinary, multi‐country, magnanimous collaboration (Figure 5). Academic medicine, however, tends to value intellectual autonomy, rewards competitive behavior, and eschews biotechnology company collaborations. BeginNGS will only be successful if academic groups collaborate and encourage broad participation and shared leadership with biotechnology, information technology, and pharmaceutical companies. Key stakeholders whose opinions must inform BeginNGS are parents of babies affected by genetic diseases (Figure 5). Developing and maintaining an international, public–private, pre‐competitive consortium will probably be one of the most difficult aspects of BeginNGS. Membership in the BeginNGS consortium is open (Benson et al., personal communication). Current participants who have given permission for their names to be used are listed above.
-
Emphasis on informatics not sequencing. Genomic NBS is primarily a knowledge problem, rather than a sequencing problem. To be effective for population screening, rWGS‐based NBS should have an average precision (positive predictive value) of ~50% (Hall et al., 2014; Kingsmore et al., 2022). Since the cumulative incidence of the 388 BeginNGS disorders is estimated to be ~0.9% and recall (sensitivity) will initially be 50–75%, the initial target false positive rate is less than 0.45–0.68% (Kingsmore et al., 2022). Achievement of this specification requires a strong emphasis on informatics to generate a highly qualified set of disease‐causing variants. In turn, this implied a need for large training sets. This was accomplished by limiting variants to pathogenic and likely pathogenic categories, and evaluation of the population frequency of each variant in, for example, the 454,707 UKBB exomes (Kingsmore et al., 2022; Szustakowski et al., 2021). Variants with high allele frequencies that were not known to account for a large proportion of cases in that population were removed. This approach was used with about 10 variant attributes. Following informatic training, the true negative rate (specificity) of ~30,000 pathogenic or likely pathogenic variants associated with 388 disorders was 99.7% by retrospective analysis of UKBB exomes (Kingsmore et al., 2022). The UKBB population, however, are almost all middle‐aged northern Europeans. The true positive rate (sensitivity) was 88.8% in retrospective analysis of 2,208 critically ill children with suspected genetic disorders who had received clinical, diagnostic rWGS (Kingsmore et al., 2022). Feedback‐loop based self‐learning will improve specificity and sensitivity as a function of the number of newborns tested, and thereby decrease the extent of manual variant interpretation. The impending availability of ~1.5 million UKBB and All of Us genome sequences and phenotypes will greatly facilitate this. However, prospective studies are needed to obtain true estimates of positive predictive value and sensitivity in healthy newborns.
Another substantial informatic effort will be the reclassification of variants of uncertain significance (VUS) over time. Mastermind (Genomenon) will play a critical, ongoing role in VUS reclassification by comprehensive curation of the literature and in silico prediction of loss of function variants for disorders where that is the known mechanism of action. Inclusion of data from locus‐specific databases will assist in variant categorization (Chunn et al., 2020; Kingsmore et al., 2022). The increasingly broad use of WGS and WES for diagnosis in affected children will continue to reclassify VUS as P, LP, or likely benign. Lastly, the artificial‐intelligence interpretation tool GEM provides a second automated method for variant prioritization (De La Vega et al., 2021; Kingsmore et al., 2022). Notwithstanding these efforts, however, BeginNGS was predicated on initial optimization of precision (positive predictive value) rather than sensitivity.
Staged development. During the current decade there have been, and will continue to be, transformative developments in society and in the pharmaceutical, biotechnology, informatics, and medical sectors that enable prompt implementation of NBS by rWGS for all disorders that meet the Wilson and Jungner criteria (, 1968). The cost of computation will continue to decline with Moore's law, and cost of WGS will continue to fall at a faster rate. As a result, BeginNGS has a staged, adaptive design (Figure 2). We anticipate annual cycles of development and testing with enrollment in clinical studies increasing by approximately an order of magnitude each year. Stage 1 involved development of a prototype and retrospective testing in ~460,000 subjects (Kingsmore et al., 2022). Stage 2, targeted to be completed in 2022, involves validation of methods to comply as a laboratory developed test under CLIA and safety assessment of enrollment at a single regional NICU site. Stage 2 also includes the addition of ~50 new disorders, ~250 new treatments, ~20,000 new variants, and retrospective testing of each in ~460,000 subjects. Stage 3 (2023) involves enrollment of 2,000 newborns in the United States and Greece and development of a version 2 test with additional disorders, treatments and variants. Stage 4 (2024) involves continued enrollment to reach 20,000 newborns and development of a version 3 test. Stage 5 (2025–2026) involves continued enrollment to reach 200,000 newborns. At each stage, the number of treatments and diseases, range of variant types, sequencing and informatics technologies, and types of results returned will evolve in a data‐driven and stakeholder‐responsive manner. Adaptive design allows BeginNGS to start with a minimal viable scope, to explore alternative approaches to a limited extent, and to defer implementation for some of the more complex decisions until they have been adequately considered by stakeholder workgroups.
Future Integration with NBSTRN. There are substantial synergies between BeginNGS and the work of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD)‐funded NBS Translational Research Network (NBSTRN, nbstrn.org). NBSTRN nominates new conditions for NBS trials. Eight have been studied since 2017 in $7.8 M NICHD‐funded pilots. Data from the BeginNGS clinical study will be deposited in the NBSTRN Longitudinal Pediatric Disease Resource (LPDR) and their Virtual Repository of States, Subjects and Samples (Brower, Chan, Hartnett, & Taylor, 2021). The NBSTRN Conditions Resource, which includes 63 conditions that are high priority candidates for NBS will be integrated with GTRx. Finally, BeginNGS will utilize the NBSTRN “ELSI Advantage” to assist with ethical, legal, and social issues. There are many unanswered ELSI questions related to genomic NBS. Two that are prominent relate to NBS for disorders that meet many, but not all, of the Wilson and Jungner criteria, and secondary uses of genomic NBS data. An ELSI workgroup, led by Dr. Ellen Clayton, will provide guidance for BeginNGS activities, and will suggest add‐on studies for ELSI issues.
FIGURE 5.
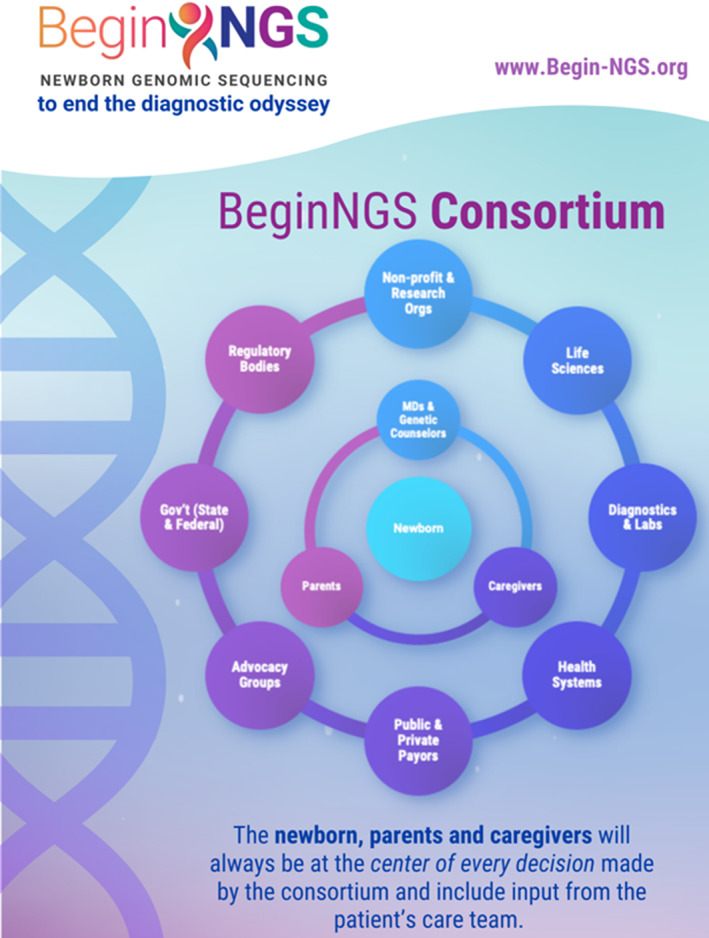
Schematic showing the roles of various stakeholders in BeginNGS™
In conclusion, we invite investigators and stakeholders worldwide to join BeginNGS (Newborn Genomic Sequencing to end the diagnostic and therapeutic odyssey), a new international, pre‐competitive, public–private consortium that proposes to implement a self‐learning healthcare delivery system for screening all newborns for hundreds of genetic diseases, diagnostic confirmation, implementation of effective treatment, and acceleration of drug development.
CONFLICT OF INTEREST
None.
ACKNOWLEDGMENTS
This manuscript was supported by NIH grant grants UL1TR002550 from NCATS to E.J. Topol (with sub‐award to Stephen F. Kingsmore), R01HD101540, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, grants from Alexion Pharmaceuticals, Inozyme Pharma, Inc., and Travere Therapeutics, Inc., Ultragenyx Pharmaceutical, Inc., and in‐kind support from Alexion Pharmaceuticals, Illumina Inc., TileDB Inc., Genomenon Inc., and Fabric Genomics, Inc. A Deo lumen, ab amicis auxilium.
Appendix A.
Members of the BeginNGS Consortium: Saad Abdullah, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Matthew Aujla, Plumcare LLC, Woodbridge, CT 06525, USA. Andrea M. Atherton, Horizon Therapeutics PLC, Deerfield, IL 60015, USA. Dan Averbuj, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Matthew Bainbridge, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Mei W. Baker, University of Wisconsin School of Medicine and Public Health, Madison, WI 53706, USA. Sergey Batalov, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Wendy Benson, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Aaron Besterman, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA; Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Terry Jo V. Bichell, COMBINEDBrain (Consortium for Outcome Measures and Biomarkers for Neurodevelopmental Disorders), Brentwood, TN 37027, USA. Eric Blincow, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Cinnamon Bloss, Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Amy Brower, Newborn Screening Translational Research Network (NBSTRN), American College of Medical Genetics and Genomics, Bethesda, MD 20814, USA. Chester W. Brown, Center for Biomedical Informatics and Department of Pediatrics, The University of Tennessee Health Science Center, and Le Bonheur Children's Hospital, Memphis, TN 38103, USA. Julie Cakici, Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Bryant Cao, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Sara Caylor, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Christina Chambers, Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Kee Chan, Newborn Screening Translational Research Network (NBSTRN), American College of Medical Genetics and Genomics, Bethesda, MD 20814, USA. Ellen W. Clayton, University of Vanderbilt, Nashville, TN 37235, USA. Alexandros Daponte, Department of Obstetrics and Gynecology, University of Thessaly, University Hospital of Larissa, Larissa, Greece. Robert L. Davis, Center for Biomedical Informatics and Department of Pediatrics, The University of Tennessee Health Science Center, and Le Bonheur Children's Hospital, Memphis, TN 38103, USA. Thomas Defay, Alexion, Astra Zeneca Rare Disease, Boston, MA 02210, USA. Guillermo Del Angel, Alexion, Astra Zeneca Rare Disease, Boston, MA 02210, USA. Yan Ding, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Katarzyna Ellsworth, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Annette Feigenbaum, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA; Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Terri H. Finkel, Center for Biomedical Informatics and Department of Pediatrics, The University of Tennessee Health Science Center, and Le Bonheur Children's Hospital, Memphis, TN 38103, USA; Pediatric Medicine, St. Jude Children's Research Hospital, Memphis, TN 38105, USA. Patrick Frias, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Erwin Frise, Fabric Genomics, Inc., Oakland, CA 94612, USA. Robert C. Green, Mass General Brigham, Broad Institute, Ariadne Labs, and Harvard Medical School, Boston, MA 02115, USA. Grigorios Grimbizis, First Department of Obstetrics and Gynecology, Aristotle University of Thessaloniki, Papageorgiou Hospital, Thessaloniki, Greece. Lucia Guidugli, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Kevin P. Hall, Illumina, Inc., San Diego, CA 92122, USA. Rand Haley, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Christian Hansen, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Charlotte A. Hobbs, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Scott D. Kahn, Luna PBC, Inc., San Diego, CA 92121, USA. Mark Kiel, Genomenon, Inc., Ann Arbor, MI 48108, USA. Elizabeth Kiernan, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Stephen F. Kingsmore, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Gail Knight, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Erica S Kobayashi, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Lucita Van Der Kraan, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Chad Krilow, TileDB Inc., Cambridge, MA 02142, USA. Chris M. Kunard, Illumina, Inc., San Diego, CA 92122, USA. Yonghyun Kwon, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Lakshminarasimha Madhavrao, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Kirk Lamoreaux, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Jennie Le, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Jerica Lenberg, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Caloh Leng, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Sebastien Lefebvre, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Rebecca Mardach, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA; Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Chris McReynolds, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Jessica Merritt, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Nicole Miller, Ultragenyx Pharmaceutical, Inc., Novato, CA 94949, USA. William R. Mowrey, Alexion, Astra Zeneca Rare Disease, Boston, MA 02210, USA. Maximillian Muenke, Newborn Screening Translational Research Network (NBSTRN), American College of Medical Genetics and Genomics, Bethesda, MD 20814, USA. Catherine Nester, Inozyme Pharma, Inc., Boston, MA 02210, USA. Russell Nofsinger, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Danny Oh, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Lauren Olsen, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Martin G. Reese, Fabric Genomics, Inc., Oakland, CA 94612, USA. Alexandros Rodolakis, First Department of Obstetrics and Gynecology, National and Kapodistrian University of Athens, Alexandra Hospital, Athens, Greece. Gunter Scharer, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Amy Shapiro, Indiana Hemophilia & Thrombosis Center, Indianapolis, IN 46260, USA. Seth Shelnutt, TileDB Inc., Cambridge, MA 02142, USA. Mari Tokita, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Shyamal S. Mehtalia, Illumina, Inc., San Diego, CA 92122, USA. Albert Oriol, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Stavros Papadopoulos, TileDB Inc., Cambridge, MA 02142, USA. Lynn Perez, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. James Perry, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA; Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Edwin Rosales, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Erica Sanford, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Steve Schwartz, Genomenon, Inc., Ann Arbor, MI 48108, USA. Stacey Seeloff, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Laurie D. Smith, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Jon Soderstrom, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Konstantinos Stamatopoulos, Institute of Applied Biosciences, Centre for Research and Technology Hellas, Thessaloniki, Greece. Jill Strickland, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Nathaly Sweeney, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA; Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Ajay J. Talati, Center for Biomedical Informatics and Department of Pediatrics, The University of Tennessee Health Science Center, and Le Bonheur Children's Hospital, Memphis, TN 38103, USA. Duke Tran, Illumina, Inc., San Diego, CA 92122, USA. Petros Tsipouras, Plumcare LLC, Woodbridge, CT 06525, USA. Meredith Wright, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Leonard A Valentino, National Hemophilia Foundation, New York, New York, USA. Kristen Wigby, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA; Departments of Pediatrics and Neuroscience, University of California San Diego, San Diego, CA 92093, USA. Mary J. Willis, Rady Children's Institute for Genomic Medicine, Rady Children's Hospital, San Diego, CA 92123, USA. Aaron R. Wolen, TileDB Inc., Cambridge, MA 02142, USA.
Kingsmore, S. F. , & The BeginNGS Consortium (2022). Dispatches from Biotech beginning BeginNGS: Rapid newborn genome sequencing to end the diagnostic and therapeutic odyssey. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:243–256. 10.1002/ajmg.c.32005
Funding information Eunice Kennedy Shriver National Institute of Child Health and Human Development; National Center for Advancing Translational Sciences; Alexion Pharmaceuticals; Ultragenyx Pharmaceutical
DATA AVAILABILITY STATEMENT
The data that support the findings will be available in Genome‐to‐Treatment (GTRx) at https://gtrx.rbsapp.net/ following an embargo from the date of publication to allow for commercialization of research findings.
REFERENCES
- ACMG . (2001. –2022). ACT sheets and algorithms. Bethesda, MD: American College of Medical Genetics and Genomics. https://www.ncbi.nlm.nih.gov/books/NBK55832/ [PubMed] [Google Scholar]
- Adhikari, A. N. , Gallagher, R. C. , Wang, Y. , Currier, R. J. , Amatuni, G. , Bassaganyas, L. , … Brenner, S. E. (2020). The role of exome sequencing in newborn screening for inborn errors of metabolism. Nature Medicine, 26, 1392–1397. 10.1038/s41591-020-0966-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- All of US . (2022). Data browser. https://databrowser.researchallofus.org/
- Amberger, J. S. , Bocchini, C. A. , Scott, A. F. , & Hamosh, A. (2019). OMIM.org: Leveraging knowledge across phenotype‐gene relationships. Nucleic Acids Research, 47, D1038–D1043. 10.1093/nar/gky1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann, A. , Blancquaert, I. , Beauchamp, S. , & Déry, V. (2008). Revisiting Wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bulletin of the World Health Organization, 86, 317–319. 10.2471/blt.07.050112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J. S. , Agrawal, P. B. , Bailey, D. B., Jr. , Beggs, A. H. , Brenner, S. E. , Brower, A. M. , … Wise, A. L. (2017). Newborn sequencing in genomic medicine and public health. Pediatrics, 139, e20162252. 10.1542/peds.2016-2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee, A. , Sokolsky, T. , Wyman, S. K. , Reese, M. G. , Puffenberger, E. , Strauss, K. , … Naylor, E. W. (2015). Development of DNA confirmatory and high‐risk diagnostic testing for newborns using targeted next‐generation DNA sequencing. Genetics in Medicine, 17, 337–347. 10.1038/gim.2014.117 [DOI] [PubMed] [Google Scholar]
- Bick, D. , Ahmed, A. , Deen, D. , Ferlini, A. , Garnier, N. , Kasperaviciute, D. , … Scott, R. H. (2022). Newborn screening by genomic sequencing: Opportunities and challenges. International Journal of Neonatal Screening, 8, 4. 10.3390/ijns8030040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker, L. G. , Green, E. D. , Manolio, T. , Solomon, B. D. , & Curtis, D. (2021). Should all babies have their genome sequenced at birth? BMJ, 375, 2679. 10.1136/bmj.n2679 [DOI] [PubMed] [Google Scholar]
- Bodian, D. L. , Klein, E. , Iyer, R. K. , Wong, W. S. , Kothiyal, P. , Stauffer, D. , … Solomon, B. D. (2016). Utility of whole‐genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genetics in Medicine, 18, 221–230. 10.1038/gim.2015.111 [DOI] [PubMed] [Google Scholar]
- Braun, D. , Braun, E. , Chiu, V. , Burgos, A. E. , Gupta, M. , Volodarskiy, M. , & Getahun, D. (2020). Trends in neonatal intensive care unit utilization in a large integrated health care system. JAMA Network Open, 3, e205239. 10.1001/jamanetworkopen.2020.5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosco, J. P. , Grosse, S. D. , & Ross, L. F. (2015). Universal state newborn screening programs can reduce health disparities. JAMA Pediatrics, 169, 7–8. 10.1001/jamapediatrics.2014.2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower, A. , Chan, K. , Hartnett, M. , & Taylor, J. (2021). The longitudinal pediatric data resource: Facilitating longitudinal collection of health information to inform clinical care and guide newborn screening efforts. Interntional Journal of Neonatal Screening, 7, 37. 10.3390/ijns7030037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakici, J. A. , Dimmock, D. P. , Caylor, S. A. , Gaughran, M. , Clarke, C. , Triplett, C. , … Bloss, C. S. (2020). A prospective study of parental perceptions of rapid whole‐genome and ‐exome sequencing among seriously ill infants. American Journal of Human Genetics, 107, 953–962. 10.1016/j.ajhg.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) . (2008). Impact of expanded newborn screening—United States, 2006. Morbidity and Mortality Weekly Report, 57, 1012–1015. [PubMed] [Google Scholar]
- Ceyhan‐Birsoy, O. , Machini, K. , Lebo, M. S. , Yu, T. W. , Agrawal, P. B. , Parad, R. B. , … Rehm, H. L. (2017). A curated gene list for reporting results of newborn genomic sequencing. Genetics in Medicine, 19, 809–818. 10.1038/gim.2016.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, Y. , Lee, C. H. , Jeong, E. G. , Kim, M. H. , Hong, J. H. , Ko, Y. , … Lee, J. S. (2017). Prevalence of rare genetic variations and their implications in NGS‐data interpretation. Scientific Reports, 7, 9810. 10.1038/s41598-017-09247-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher, H. , Burns, A. , Josephat, E. , Makani, J. , Schuh, A. , & Nkya, S. (2021). Using DNA testing for the precise, definite, and low‐cost diagnosis of sickle cell disease and other haemoglobinopathies: Findings from Tanzania. BMC Genomics, 22, 902. 10.1186/s12864-021-08220-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunn, L. M. , Nefcy, D. C. , Scouten, R. W. , Tarpey, R. P. , Chauhan, G. , Lim, M. S. , … Kiel, M. J. (2020). Mastermind: A comprehensive genomic association search engine for empirical evidence curation and genetic variant interpretation. Frontiers in Genetics, 11, 577152. 10.3389/fgene.2020.577152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton, E. W. (2010). Currents in contemporary ethics. State run newborn screening in the genomic era, or how to avoid drowning when drinking from a fire hose. The Journal of Law, Medicine & Ethics, 38, 697–700. 10.1111/j.1748-720X.2010.00522.x [DOI] [PubMed] [Google Scholar]
- Costich, J. F. , & Durst, A. L. (2016). The impact of the affordable care act on funding for newborn screening services. Public Health Reports, 131, 160–166. 10.1177/003335491613100123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Vega, F. M. , Chowdhury, S. , Moore, B. , Frise, E. , McCarthy, J. , Hernandez, E. J. , … Boone, B. (2021). Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Medicine, 13, 153. 10.1186/s13073-021-00965-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCristo, D. M. , Milko, L. V. , O'Daniel, J. M. , Foreman, A. K. M. , Mollison, L. F. , Powell, B. C. , … Berg, J. S. (2021). Actionability of commercial laboratory sequencing panels for newborn screening and the importance of transparency for parental decision‐making. Genome Medicine, 13, 50. 10.1186/s13073-021-00867-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock, D. , Caylor, S. , Waldman, B. , Benson, W. , Ashburner, C. , Carmichael, J. L. , … Farnaes, L. (2021). Project baby bear: Rapid precision care incorporating rWGS in 5 California children's hospitals demonstrates improved clinical outcomes and reduced costs of care. American Journal of Human Genetics, 108, 1231–1238. 10.1016/j.ajhg.2021.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. , Owen, M. , Le, J. , Batalov, S. , Chau, K. , Kwon, Y. H. , … Kingsmore, S. F. (2022). Scalable, high quality, whole genome sequencing from archived, newborn, dried blood spots. medRxiv. 10.1101/2022.07.27.22278102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenka, S. D. , Gorzynski, J. E. , Shafin, K. , Fisk, D. G. , Pesout, T. , Jensen, T. D. , … Ashley, E. A. (2022). Accelerated identification of disease‐causing variants with ultra‐rapid nanopore genome sequencing. Nature Biotechnology, 40, 1035–1041. 10.1038/s41587-022-01221-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson, S. , Singer‐Berk, M. , Watts, N. A. , Phu, W. , Goodrich, J. K. , Solomonson, M. , … O'Donnell‐Luria, A. (2022). Variant interpretation using population databases: Lessons from gnomAD. Human Mutation, 43(8), 1012–1030. 10.1002/humu.24309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, P. L. , Marquardt, G. , McHugh, D. M. , Currier, R. J. , Tang, H. , Stoway, S. D. , & Rinaldo, P. (2014). Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genetics in Medicine, 16, 889–895. 10.1038/gim.2014.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton, C. F. , Homer, C. J. , Thompson, A. A. , Williams, A. , Hassell, K. L. , Feuchtbaum, L. , … Follow‐up and Treatment Sub‐committee of the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) . (2016). A framework for assessing outcomes from newborn screening: On the road to measuring its promise. Molecular Genetics and Metabolism, 118, 221–229. 10.1016/j.ymgme.2016.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, H. , Kinsella, S. , & Evans, G. (2021). Implications of whole genome sequencing for newborn screening. A public dialog. https://s3.eu-west-2.amazonaws.com/ge-production-s3/documents/public-dialogue-wgs-for-nbs-final-report.pdf
- Institute of Medicine (US) Roundtable on Translating Genomic‐Based Research for Health . (2011). Establishing Precompetitive Collaborations to Stimulate Genomics‐Driven Product Development: Workshop Summary. Washington, DC: National Academies Press. [PubMed] [Google Scholar]
- Jian, M. , Wang, X. , Sui, Y. , Fang, M. , Feng, C. , Huang, Y. , … Gao, Y. (2022). A pilot study of assessing whole genome sequencing in newborn screening in unselected children in China. Clinical and Translational Medicine, 12, e843. 10.1002/ctm2.843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J., Lantos, J. D., Goldenberg, A., Chen, F., Parens, E., & Koenig, B. A. (2018). Sequencing Newborns:A Call for Nuanced Use of Genomic Technologies. Hastings Center Report, 48, S2–S6. Portico. 10.1002/hast.874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsmore, S. F. , & Cole, F. S. (2022). The role of genome sequencing in neonatal intensive care units. Annual Review of Genomics and Human Genetics, 23(23. June 8), 427–448. 10.1146/annurev-genom-120921-103442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsmore, S. F. , Smith, L. D. , Kunard, C. M. , Bainbridge, M. , Batalov, S. , Benson, W. , … Defay, T. (2022). A genome sequencing system for universal newborn screening, diagnosis, and precision medicine for severe genetic diseases. American Journal of Human Genetics, 109, 1605–1619. 10.1016/j.ajhg.2022.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbarek, H. , Devadoss Gandhi, G. , Selvaraj, S. , Al‐Muftah, W. , Badji, R. , Al‐Sarraj, Y. , … Qatar Genome Program Research Consortium . (2022). Qatar genome: Insights on genomics from the Middle East. Human Mutation, 43, 499–510. 10.1002/humu.24336 [DOI] [PubMed] [Google Scholar]
- Milko, L. V. , O'Daniel, J. M. , DeCristo, D. M. , Crowley, S. B. , Foreman, A. K. M. , Wallace, K. E. , … Berg, J. S. (2019). An age‐based framework for evaluating genome‐scale sequencing results in newborn screening. The Journal of Pediatrics, 209, 68–76. 10.1016/j.jpeds.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newborn Screening Task Force . (2000). Newborn screening: A blueprint for the future. Pediatrics, 106(Suppl. 2), 383–427. [PubMed] [Google Scholar]
- Owen, M. J. , Lefebvre, S. , Hansen, C. , Kunard, C. M. , Dimmock, D. P. , Smith, L. D. , … Kingsmore, S. F. (2022). An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases. Nature Communications, 13, 4057. 10.1038/s41467-022-31446-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, S. , Robinson, J. O. , Gutierrez, A. M. , Petersen, D. K. , Hsu, R. L. , Lee, C. H. , … The BabySeq Project Group . (2019). Perceived benefits, risks, and utility of newborn genomic sequencing in the BabySeq project. Pediatrics, 143, S6–S13. 10.1542/peds.2018-1099C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, S. , Smith, H. S. , Frankel, L. A. , Christensen, K. D. , Islam, R. , Robinson, J. O. , … Zettler, B. (2021). Psychosocial effect of newborn genomic sequencing on families in the BabySeq project: A randomized clinical trial. JAMA Pediatrics, 175, 1132–1141. 10.1001/jamapediatrics.2021.2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, R. M. , Koch, R. , Schaeffler, G. E. , Wohlers, A. , Acosta, P. B. , & Boyle, D. (1968). Phenylketonuria. Experience at one center in the first year of screening in California. California Medicine, 108, 350–354. [PMC free article] [PubMed] [Google Scholar]
- Petros, M. (2012). Revisiting the Wilson‐Jungner criteria: How can supplemental criteria guide public health in the era of genetic screening? Genetics in Medicine, 14, 129–134. 10.1038/gim.0b013e31823331d0 [DOI] [PubMed] [Google Scholar]
- Pichini, A. , Ahmed, A. , Patch, C. , Bick, D. , Leblond, M. , Kasperaviciute, D. , … Scott, R. H. (2022). Developing a National Newborn Genomes Program: An approach driven by ethics, engagement and co‐design. Frontiers in Genetics, 13, 866168. 10.3389/fgene.2022.866168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman, T. S. , Crowley, S. B. , Roche, M. I. , Foreman, A. K. M. , O'Daniel, J. M. , Seifert, B. A. , … Berg, J. S. (2020). Genomic sequencing for newborn screening: Results of the NC NEXUS project. American Journal of Human Genetics, 107, 596–611. 10.1016/j.ajhg.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, L. F. , & Clayton, E. W. (2019). Ethical issues in newborn sequencing research: The case study of BabySeq. Pediatrics, 144, e20191031. 10.1542/peds.2019-1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubens, M. , Ramamoorthy, V. , Saxena, A. , McGranaghan, P. , Veledar, E. , & Hernandez, A. (2022). Obstetric outcomes during delivery hospitalizations among obese pregnant women in the United States. Scientific Reports, 12, 6862. 10.1038/s41598-022-10786-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, T. S. , Christensen, K. D. , Uveges, M. K. , Waisbren, S. E. , McGuire, A. L. , Pereira, S. , … Holm, I. A. (2021). Effects of participation in a U.S. trial of newborn genomic sequencing on parents at risk for depression. Journal of Genetic Counseling, 31, 218–229. 10.1002/jgc4.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag, M. K. , Yusuf, C. , Grosse, S. D. , Edelman, S. , Miller, J. I. , McKasson, S. , … Shapira, S. K. (2020). Infants with congenital disorders identified through newborn screening—United States, 2015–2017. MMWR. Morbidity and Mortality Weekly Report, 69, 1265–1268. 10.15585/mmwr.mm6936a6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranneheim, H. , Engvall, M. , Naess, K. , Lesko, N. , Larsson, P. , Dahlberg, M. , … Wedell, A. (2014). Rapid pulsed whole genome sequencing for comprehensive acute diagnostics of inborn errors of metabolism. BMC Genomics, 15, 1090. 10.1186/1471-2164-15-1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szustakowski, J. D. , Balasubramanian, S. , Kvikstad, E. , Khalid, S. , Bronson, P. G. , Sasson, A. , … Ye, Z. (2021). Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nature Genetics, 53, 942–948. 10.1038/s41588-021-00885-0 [DOI] [PubMed] [Google Scholar]
- Therrell, B. L. , Padilla, C. D. , Loeber, J. G. , Kneisser, I. , Saadallah, A. , Borrajo, G. J. , & Adams, J. (2015). Current status of newborn screening worldwide. Seminars in Perinatology, 39, 171–187. 10.1053/j.semperi.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Wilson, J. M. G. , & Jungner, G. , World Health Organization . (1968). Principles and Practice of Screening for Disease. World Health Organization. Retrieved from https://apps.who.int/iris/handle/10665/37650 [Google Scholar]
- Woerner, A. C. , Gallagher, R. C. , Vockley, J. , & Adhikari, A. N. (2021). The use of whole genome and exome sequencing for newborn screening: Challenges and opportunities for population health. Frontiers in Pediatrics, 9, 663752. 10.3389/fped.2021.663752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik, M. H. , Zhang, T. , Ceyhan‐Birsoy, O. , Genetti, C. A. , Lebo, M. S. , Yu, T. W. , … Yu, T. W. (2021). Discordant results between conventional newborn screening and genomic sequencing in the BabySeq project. Genetics in Medicine, 23, 1372–1375. 10.1038/s41436-021-01146-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, T. , Kingsmore, S. F. , Green, R. C. , MacKenzie, T. , Wasserstein, M. , Caggana, M. , … Urv, T. K. (in press). Are we prepared to deliver gene‐targeted therapies for rare diseases? American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings will be available in Genome‐to‐Treatment (GTRx) at https://gtrx.rbsapp.net/ following an embargo from the date of publication to allow for commercialization of research findings.