

Abstract
The intermediate oxidation state of sulfoxides is central to the plethora of their applications in chemistry and medicine, yet it presents challenges for an efficient synthetic access, limiting the structural diversity of currently available sulfoxides. Here, we report a data-guided development of direct decarboxylative sulfinylation that enables the previously inaccessible functional group interconversion of carboxylic acids to sulfoxides in a reaction with sulfinates. Given the broad availability of carboxylic acids and the growing synthetic potential of sulfinates, the direct decarboxylative sulfinylation is poised to improve the structural diversity of synthetically accessible sulfoxides. The reaction is facilitated by a kinetically favored sulfoxide formation from the intermediate sulfinyl sulfones, despite the strong thermodynamic preference for the sulfone formation, unveiling the previously unknown and chemoselective radicalophilic sulfinyl sulfone reactivity.
Keywords: carboxylic acids, radical reactions, sulfonates, sulfoxides, visible light
Graphical Abstract

The new metal-free cross-coupling of carboxylic acids and sulfinates provides a direct and modular access to sulfoxides by a photocatalytic process that has unveiled the unexplored chemoselective radicalophilic reactivity of intermediate sulfinyl sulfones and enabled the conjunctive coupling of two carboxylic acids, revealing broad new chemical space accessible by the merger of the two coupling partner classes of emergent synthetic importance.
Introduction
The development of new synthetic transformations facilitating the formation of carbon–heteroatom bonds has played pivotal roles in chemistry and life sciences.[1] Given the importance of the oxidation state of sulfur for the chemical and molecular properties of sulfur-containing organic molecules, the development of methods that enable a selective construction of carbon–sulfur bonds with a specific sulfur oxidation state has remained an important synthetic challenge.[2]
Sulfoxides occupy an intermediate position between sulfides and sulfones. A facile metabolic interconversion with sulfides and sulfones, combined with polarity and increased solubility in water, as well as bioisosterism with the carbonyl group[3] have rendered the sulfoxide functionality indispensable for the discovery of essential medicines and agrochemicals (Figure 1.A).[4] Furthermore, the divergent reactivities of sulfoxides have been used extensively in organic synthesis,[5] while the Lewis basicity of the sulfur atom has recently been exploited for the development of new classes of organocatalysts and ligands for transition metal catalysis.[5a,6] Additionally, the sulfoxide group has increasingly been used in materials science to improve hydrophilicity of functional materials.[7]
Figure 1.
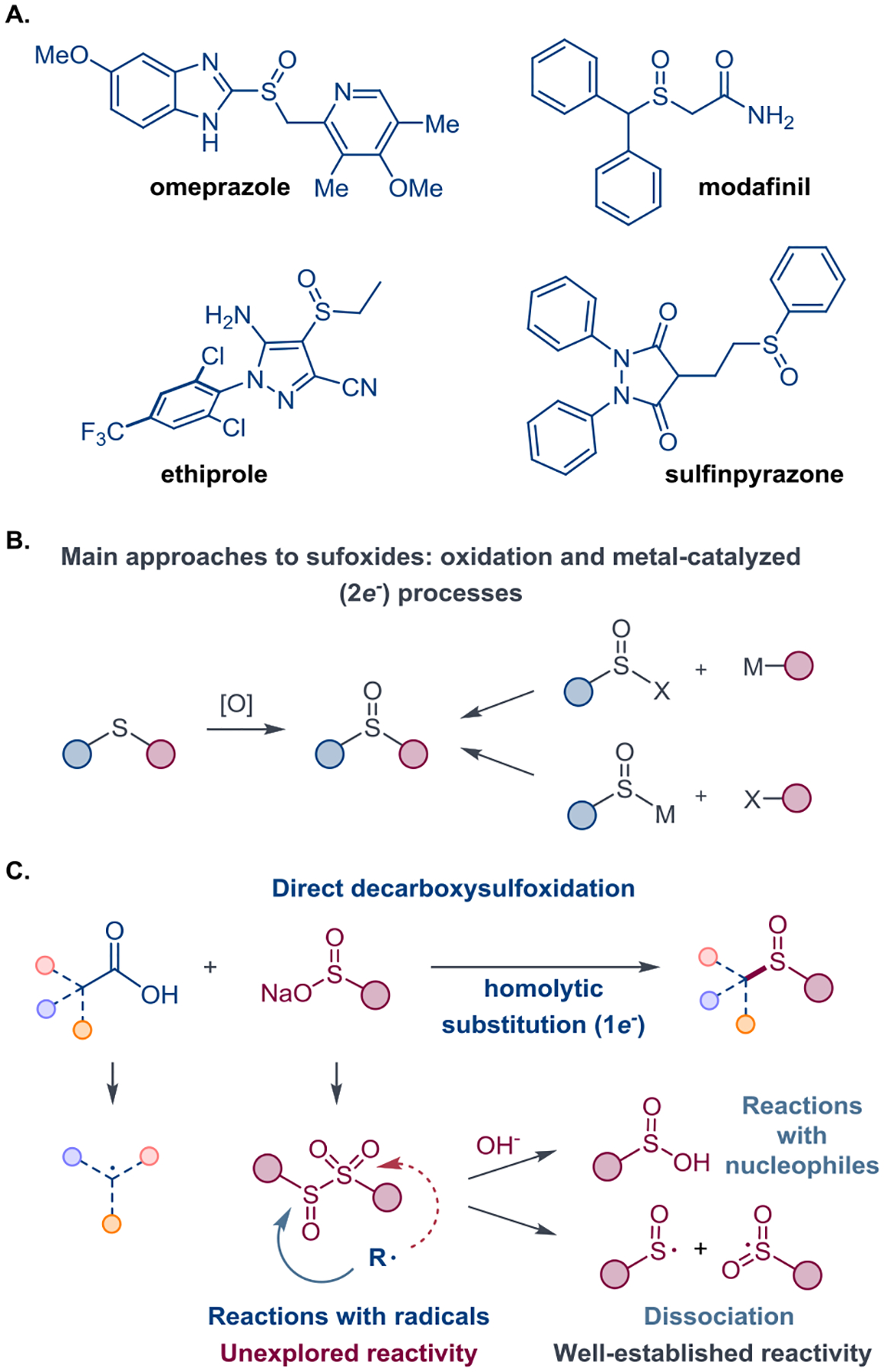
A. Applications of sulfoxides. B. Synthetic approaches to sulfoxides. C. Decarboxylative sulfinylation and the radicalophilc reactivity of sulfinyl sulfones.
Despite the importance of sulfoxides for the chemical and biomedical fields, synthetic approaches to sulfoxides remain limited and largely rely on two-electron processes that include oxidation of sulfides,[8] as well as nucleophilic and electrophilic substitution[9] and transition metal-catalyzed cross-coupling reactions (Figure 1.B).[10] We hypothesized that a one-electron process based on a radical substitution of sulfinyl sulfones can provide a simple entry to sulfoxides (Figure 1.C), facilitating access to the sulfoxide chemical space that has not been accessed with the established methods. Although reactions of sulfinyl sulfones with nucleophiles were previously described,[11] radical displacement reactions remained unexplored (Figure 1.C). Thus, it was unknown if the radical process would demonstrate a high selectivity for the sulfoxide formation relative to the competing reaction at the sulfonyl group, producing a sulfone. Additionally, sulfinyl sulfones readily undergo an unproductive decomposition and a facile homolytic dissociation of the S–S bond[12] that was recently successfully exploited by the Bi group for the construction of sulfinyl vinyl sulfones.[13] These deleterious processes may lead to an unselective formation of the sulfoxide and sulfone products, further underscoring the challenges that had to be circumvented en route to an efficient sulfoxide construction method by radical substitution in sulfinyl sulfones.
We envisioned that the challenges can be addressed through the development of a reaction between carboxylic acids, serving as radical precursors, and sulfinate salts, producing sulfinyl sulfones in situ in the presence of an acylating reagent that can selectively react with sulfinates without engaging carboxylic acids. The direct decarboxylative sulfinylation reaction would for the first time enable a previously unknown one-step functional group interconversion of carboxylic acids to structurally analogous and bioisosteric sulfoxides, provided that a suitable catalytic system is identified for the challenging direct decarboxylation of difficult to oxidize carboxylic acids to by-pass the typically required preactivation to more reactive carboxylic acid derivatives.[14, 15] Furthermore, given the broad availability and structural diversity of carboxylic acids, and the growing synthetic importance of sulfinates,[16] the new reaction was anticipated to provide access to previously unexplored sulfoxide chemical space.
We report herein the development of a direct decarboxylative sulfinylation guided by an inquiry of the reaction accessible chemical space that was facilitated by a new reaction scope analytics tool (PARSE). The reaction enables a one-step, metal-free construction of sulfoxides from carboxylic acids and sulfinates without preactivation of carboxylic acids and for the first time allows for the construction of sulfoxides by a radical substitution of intermediate sulfinyl sulfones. Despite the thermodynamically favored attack at the sulfonyl group, the reaction shows strong kinetic preference for the formation of sulfoxides, underscoring high levels of chemoselectivity that can be obtained with radical substitution reactions.
Results and Discussion
Reaction chemical space mapping can guide the discovery of new reactions and provide important insights into molecular properties of reaction products.[17] To facilitate the analysis of the chemical space that can become accessible by the decarboxylative sulfinylation, we developed a Python-based analytical tool termed Prospective analysis of the reaction scope (PARSE), allowing a facile construction of a dataset of the sulfoxide structures that can become accessible from aliphatic carboxylic acids and sulfinates with subsequent comparison of the newly accessible chemical space with that of the currently available sulfoxides. The structures of the decarboxylative sulfinylation-accessible sulfoxides were generated by combining the SMILES-encoded sulfinyl and alkyl residues derived from the parent structures of the sulfinates and carboxylic acids retrieved from Pubchem.[18] To enable the visualization of the chemical space, the analysis was performed on representative samples (0.25% of the Pubchem-derived datasets for each class of compounds, i.e., carboxylic acids, sulfinates, and sulfoxides), resulting in 805 known sulfoxides and 196749 decarboxylative sulfinylation-accessible sulfoxides (from the representative datasets of 7287 carboxylic acids and 27 sulfinates). The chemical space of the known and decarboxylative sulfinylation-accessible sulfoxides was then compared using molecular weight (MW), molecular complexity (Cm),[19] and fraction of sp3 carbon atoms (Fsp3)[20] as descriptors[21] that were selected based on the importance of the molecule size, as well as structural and functional group content, and the three-dimensional molecular shape for applications in drug discovery, materials science, and organic synthesis. A comparison of the representative set of currently available sulfoxides with the structures of sulfoxides that can become accessible by the direct decarboxylative sulfinylation revealed that the decarboxylative sulfinylation-accessible sulfoxides have a significantly broader distribution with respect to molecular weight (MW), molecular complexity (Cm), and a fraction of sp3 carbon atoms (Fsp3) (Figure 2.A). Similarly, the population density plot obtained by a kernel density estimation (KDE) of the probability density function (PDF)[22] of the distribution of the known sulfoxides in the Fsp3/Cm chemical space point to the greater density of structures in a single region of chemical space (Figure 2.B), as evidenced by a higher peak in the low molecular complexity range. By contrast, decarboxylative sulfinylation-accessible sulfoxides have a lower PDF value and broader distribution, indicating that the set spans a wider range of chemical space (Figure 2.C). Furthermore, a broader distribution across the chemical shape space is also observed for the decarboxylative sulfinylation-accessible sulfoxides (Figure 2.D), especially in the sphere-like region that is underpopulated in the current synthetically accessible medicinal chemical space.[23] Similarly, a broader distribution across the Cm/Fsp3/MW chemical space and greater geometric diversity were also observed for decarboxylative sulfinylation-accessible sulfoxides, when they were compared with the sulfoxides that can become accessible by oxidation of sulfides—one of the most commonly used methods for the preparation of sulfoxides[8] (Figures S20–S25). Taken together, these data indicate that the direct decarboxylative sulfinylation can enable access to structurally diverse sulfoxides with a wide range of molecular properties and facilitate the generation of drug-like molecules and leads, as well as the development of screening libraries. Furthermore, given the simplicity and generality of the SMILES-based strategy[17] outlined in the PARSE tool, it may be applied to the analysis of the chemical space that is accessible with other synthetic methodologies, revealing general patterns of currently available and potentially accessible chemical space that can help guide the development of new methodologies.
Figure 2.
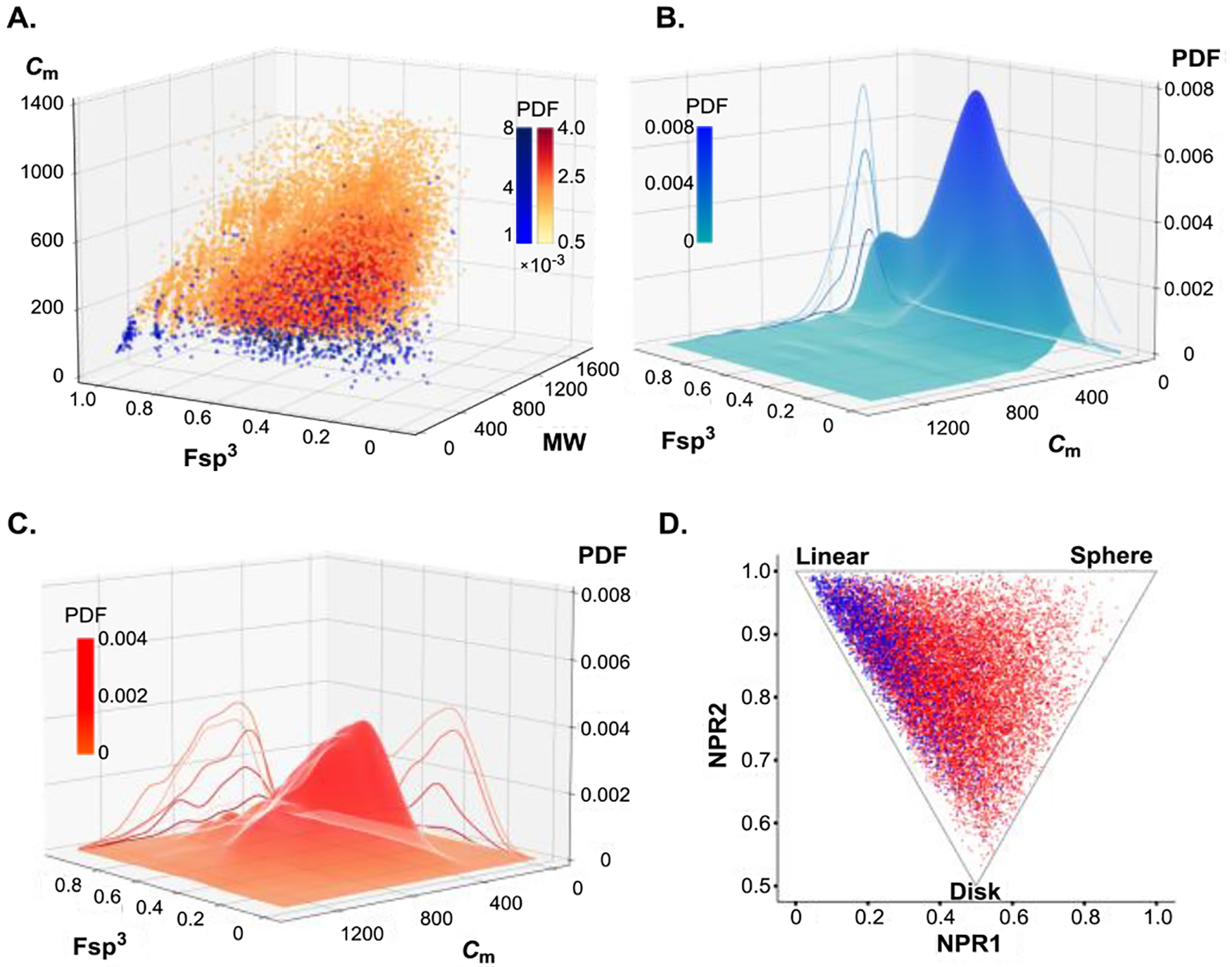
Comparison of the chemical space of the currently available sulfoxides (blue) and the sulfoxides that may become accessible by the direct decarboxylative sulfinylation from known carboxylic acids and sulfinates (red/orange). A. The Cm/Fsp3/MW plot with the darker color representing higher population density (PDF). B. The population density plot for the known sulfoxides. C. The population density plot for the decarboxylative sulfinylation-accessible sulfoxides. D. Geometric diversity population analysis for the currently available and decarboxylative sulfinylation-accessible sulfoxides.
With the results of the PARSE investigation in hand, experiments were carried out to identify the reaction conditions that enable the decarboxylative sulfinylation. Optimization studies revealed that carboxylic acid 1 and sulfinate 2 can be directly converted to sulfoxide 3a in 82% (78% isolated yield) with acridine A1 as a photocatalyst and in the presence of p-bromobenzoyl chloride (PBC) (Figure 2.A,C). Acridines A1-A3[15] were the only photocatalysts that produced sulfoxide 3a, while a wide range of other photocatalysts, including Ir- and Ru-based complexes, as well as 4CzIPN, did not catalyze the decarboxylative sulfinylation (Table S1). Acridine photocatalysts A2 and A3 bearing less sterically encumbered 9-aryl groups were less efficient. The reaction required light to proceed and performed best in dichloromethane as a solvent and at room temperature (Figure 2.B). PBC had a key role in mediating the sulfinylation, outperforming a range of other aromatic and aliphatic acid chlorides and anhydrides. Notably, PBC was superior to structurally related substituted benzoyl chlorides, bearing a variety of electron-donating and electron-withdrawing groups (Figure 2.C and Table S2).
With the optimized reaction conditions in hand, we evaluated the scope of carboxylic acids with sulfinate 2 as a model coupling partner (Scheme 1). Primary carboxylic acids bearing halogen, ester, aryl, ketone, amide, and heterocyclic functionalities produced the corresponding sulfoxides in good yields (3b-3k). Acyclic, as well as carbocyclic and heterocyclic secondary alkylcarboxylic acids were also equally reactive (3a, 3l-3q), and the reaction was amenable to scale-up (3p). Similarly, a range of sulfoxides bearing a variety of tertiary alkyl groups, including cyclobutyl and oxetane ring systems, was readily accessed from the corresponding carboxylic acids (3r-3aa).
Scheme 1.
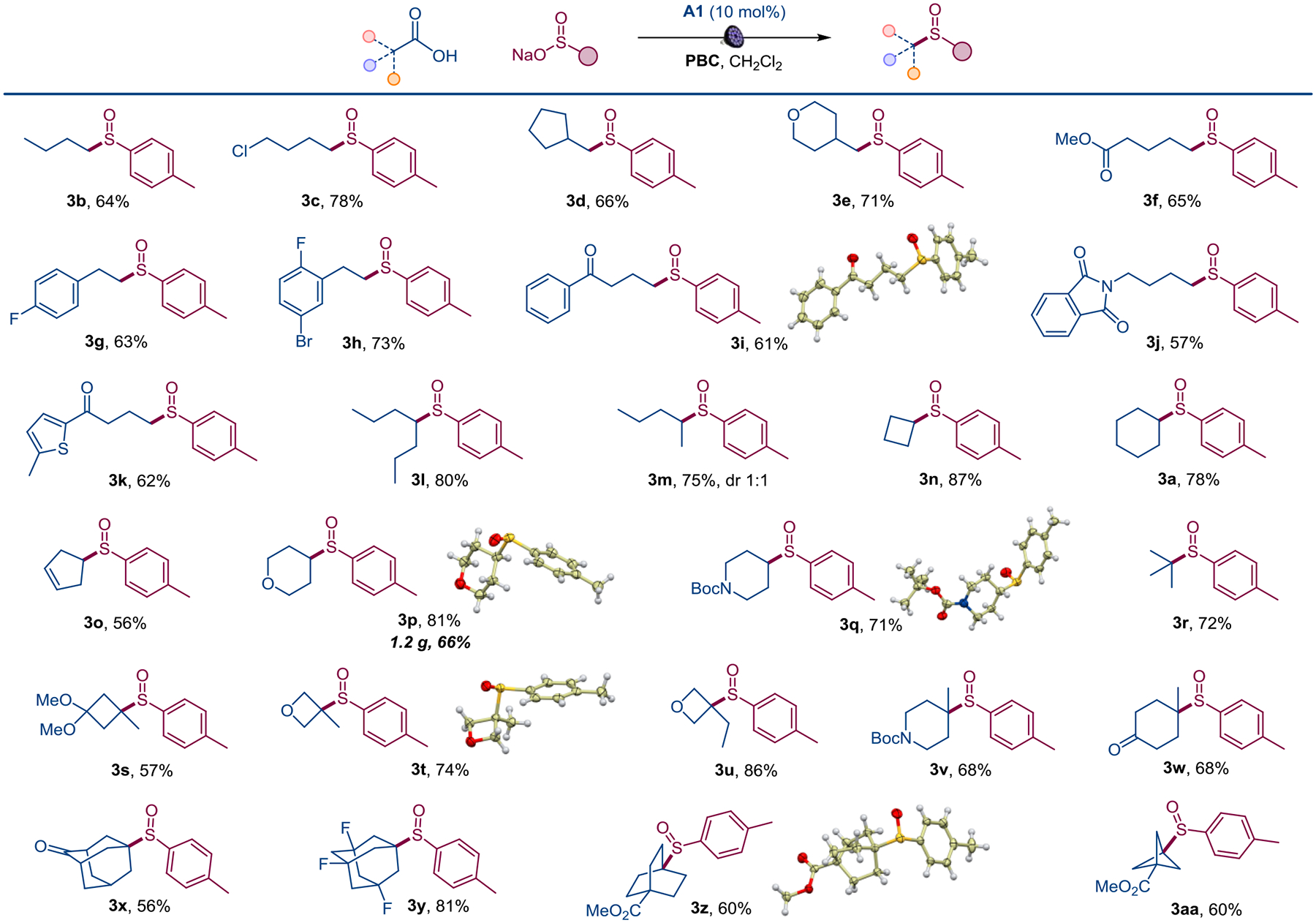
Scope of carboxylic acids in the direct decarboxylative sulfinylation. Reaction conditions: see Figure 3, isolated yields.
The scope of the reaction was further explored with sulfinate salts (Scheme 2). The direct decarboxylative sulfinylation performed well with aliphatic sulfinates, affording a range of unsymmetric dialkyl sulfoxides 4a-4d. We were also pleased to observe that aromatic sulfinate salts were suitable coupling partners. An array of aromatic sulfinates bearing electron-donating and electron-withdrawing substituents, including phenyl, halogen, methoxy, trifluoromethyl, trifluoromethoxy, and amido groups were converted to the corresponding sulfoxides (4e-4n). The reaction can also be extended to heterocyclic sulfinates, producing thiophene and quinoline-derived sulfoxides 4o-4r.
Scheme 2.
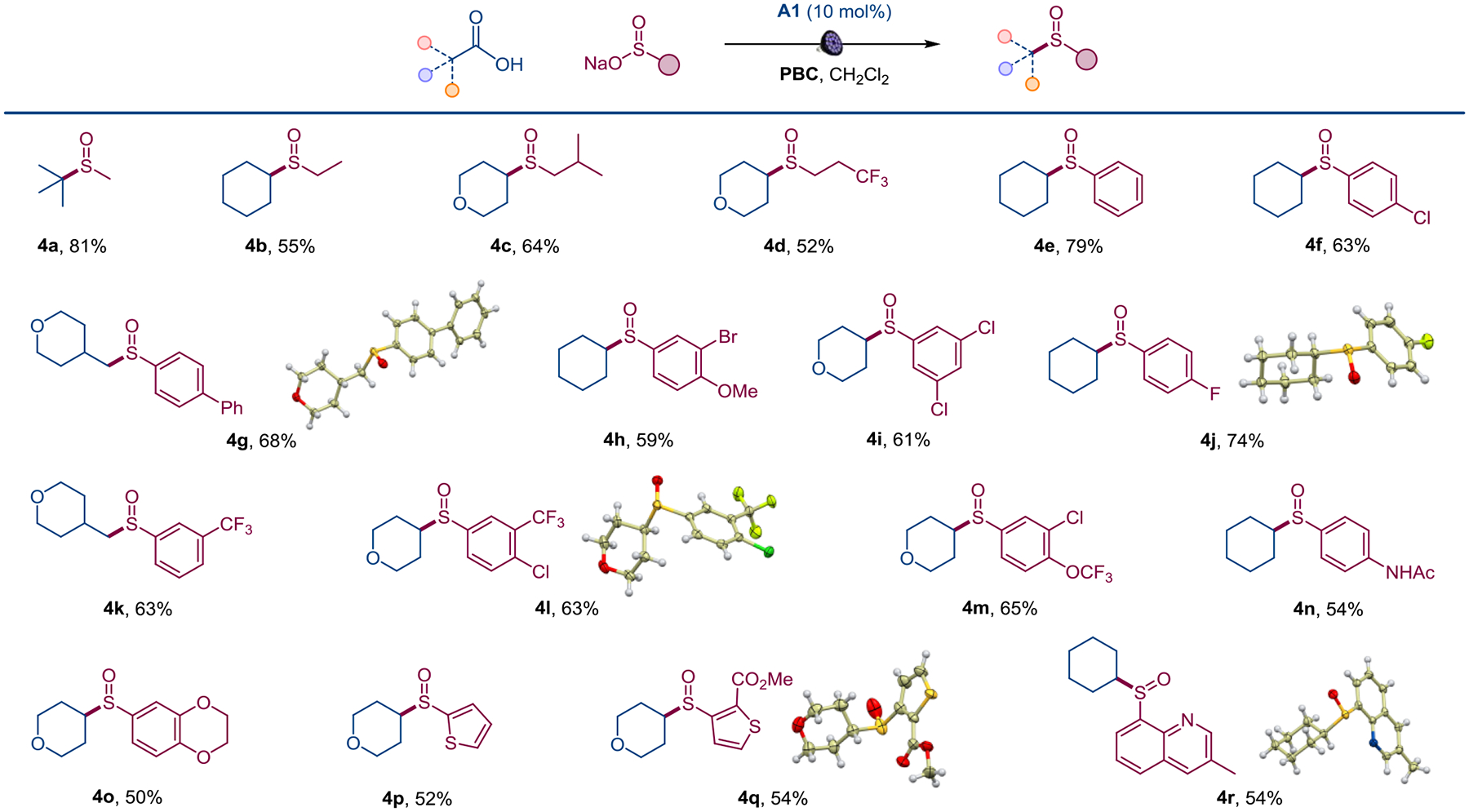
Scope of sulfinates in the direct decarboxylative sulfinylation. Reaction conditions: see Figure 3, isolated yields.
The scope and functional group tolerance of the reaction were further evaluated in the structural settings of natural products and active pharmaceutical ingredients (Scheme 3). Azelaic acid and the cis-epoxyoleic acids were readily converted to the corresponding sulfoxides (5a, 5b), without any detriment to the reactive epoxide ring. Similarly, sulfoxide analogue of antihyperlipidemic gemfibrozil 5c, as well as a para-tolyl analogue of uricosuric drug sulfinpyrazone 5d were readily produced, pointing to potential applications in drug discovery. The decarboxylative sulfinylation of a glutamic acid derivative established a direct route to the aromatic analogue of the medicinally important methionine sulfoxide[24] (5e). Sulfoxides 5f-5h were also successfully derived from santonic acid, D-fructose, and ursodeoxycholic acid. Interestingly, the reaction of gibberellic acid afforded sulfoxide 5i as a single diastereomer, indicating that significant levels of substrate-controlled stereoselectivity can be attained for the sulfur- and carbon-stereocenters.[25]
Scheme 3.
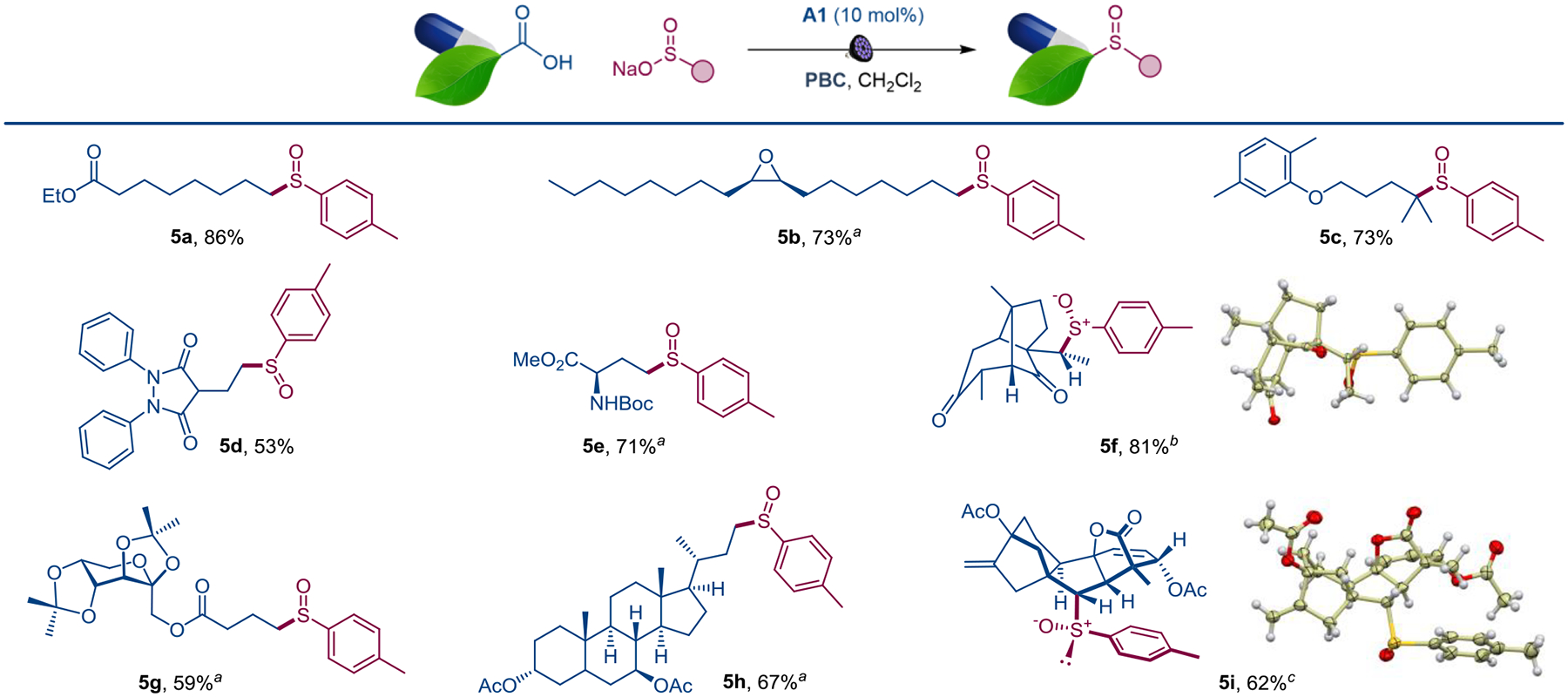
Functionalization of natural products and drugs by the direct decarboxylative sulfinylation. Reaction conditions: see Figure 3, isolated yields. a 1 : 1 dr. b 4.4 : 1.8 : 1 dr. c Single diastereomer.
Given the substantially greater availability and structural diversity of carboxylic acids over sulfinates, we next questioned if a conjunctive cross-decarboxylative sulfinylation could be established, wherein two carboxylic acids are coupled via an intermediacy of a sulfinate derived from one of the coupling partners and a sulfur dioxide source, using the same acridine photocatalyst. The cross-coupling protocol was successfully developed using DABCO–bis(sulfur dioxide) adduct (DABSO) for the sulfinate phase15d (Scheme 4). Application of the conjunctive cross-decarboxylative sulfinylation to a series of carboxylic acid pairs afforded unsymmetric sulfoxides 6a-6e, indicating that it can be leveraged to rapidly build up molecular complexity, drawing from the structural diversity of carboxylic acids. Kinetic studies showed that a reaction of sulfinate 1 with PBC rapidly produced sulfinyl sulfone 7 with a half-conversion time of 20 min, while the half-order in PBC and first order in sulfinate revealed by variable time normalization analysis[26] pointed to the involvement of a PBC-derived acylium intermediate[27] followed by conversion to mixed anhydride 8 (Figure 4.A). Formation and subsequent consumption of intermediate 7 was also observed in the reaction of acid 1 and sulfinate 2 under the optimal conditions (Figure 4.B). Furthermore, the acridine-catalyzed reaction of intermediate 7 with carboxylic acid 1 afforded sulfoxide 3a in 89% yield, while no formation of the corresponding sulfone by-product (cyclohexyl p-tolyl sulfone) was observed (Figure 4.C). These results point to the involvement of intermediate 7 in the decarboxylative sulfinylation process, for example by an alkyl radical-mediated homolytic substitution at the sulfinyl terminus of 7.[28] The results further indicate that the radical substitution occurs in high preference to the homolytic dissociation of 7 to the sulfonyl and sulfinyl radicals (ΔG≠ = 24.3 kcal/mol[12]) that is followed by a reaction of these radicals with the alkyl radical, as this unselective pathway would produce a 1.5 : 1 ratio of the sulfone and sulfoxide products.[29] Additionally, the radical substitution at the sulfinyl terminus also outcompetes the attack at the sulfonyl group, accounting for the absence of sulfones among the reaction products. Computational studies were therefore carried out next to gain insight into the chemoselectivity of the reaction (Figure 4.D). Notably, the alkyl radical attack at the sulfonyl group was significantly more thermodynamically favorable (ΔG = −36.7 kcal/mol for the sulfonyl and −27.3 kcal/mol for the sulfinyl group), driven by the formation of a stronger Me–SO2Ph bond (BDE 71.7 kcal/mol, cf. 51.4 kcal/mol for the Me–S(O)Ph bond). However, the barrier to the reaction at the sulfonyl group was prohibitively high (ΔG≠ = 29.9 kcal/mol). By contrast, the radical substitution at the sulfinyl group was kinetically facile (ΔG≠ = 9.4 kcal/mol), pointing to a strong kinetic preference for the sulfoxide product, in line with the experimental results. While the greater contribution of the sulfinyl sulfur to both the HOMO and the LUMO of intermediate 9 (20.2% and 22.7%, cf. 8.6% and 17.8% for the sulfonyl sulfur) suggested a preferential attack at the sulfinyl group, distortion/interaction activation strain analysis[30] was performed to further clarify the effects that are responsible for the observed divergence in the reactivity of the sulfinyl and sulfonyl groups (Figure 4.E). The analysis revealed that the higher barrier for the reaction at the sulfonyl group is due to a substantially greater distortion of the intermediate 7 fragment in the sulfone pathway, required to accommodate the alkyl radical as it approaches the more substituted sulfur center. This is evidenced by a greater decrease in the pyramidalization angle of the PhSO2 group in TS2 (θp = 2.1° in TS2, θp = 15.2° in 7) compared to the minor change in the pyramidalization angle of the less encumbered PhSO group in TS1 (θp = 12.7° in TS1, θp = 15.5° in 7). Additionally, a less stabilizing interaction curve with a steep inflection past TS1 in the sulfone pathway points to a significant structural adjustment required for the formation of the C–S bond that mostly occurs after traversing TS1, accounting for the disparity in the kinetic and thermodynamic favorability of the sulfone pathway.
Scheme 4.
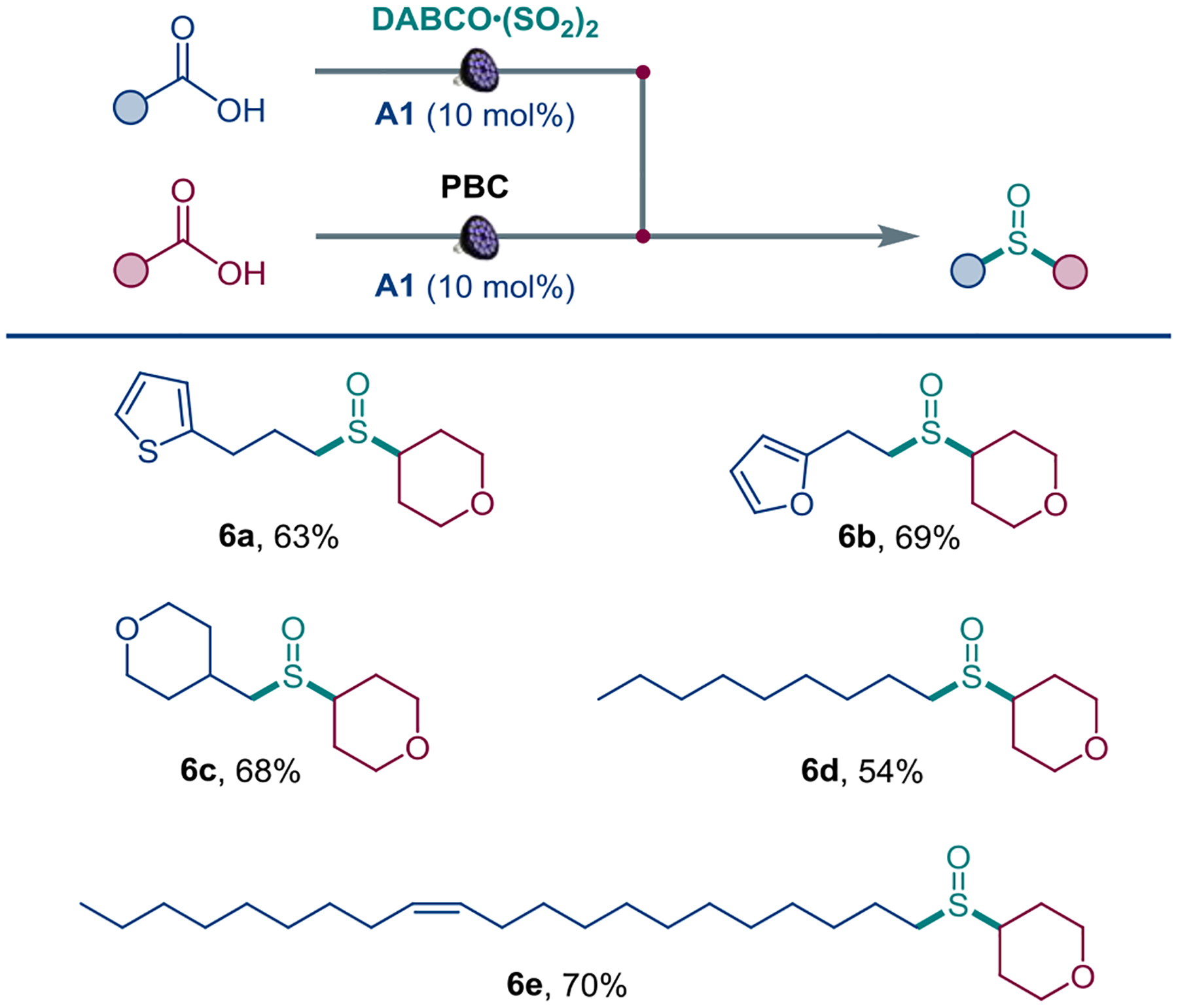
Conjunctive cross-decarboxylative sulfinylation. DABCO = 1,4-diaza-bicyclo[2.2.2]octane.
Figure 4.
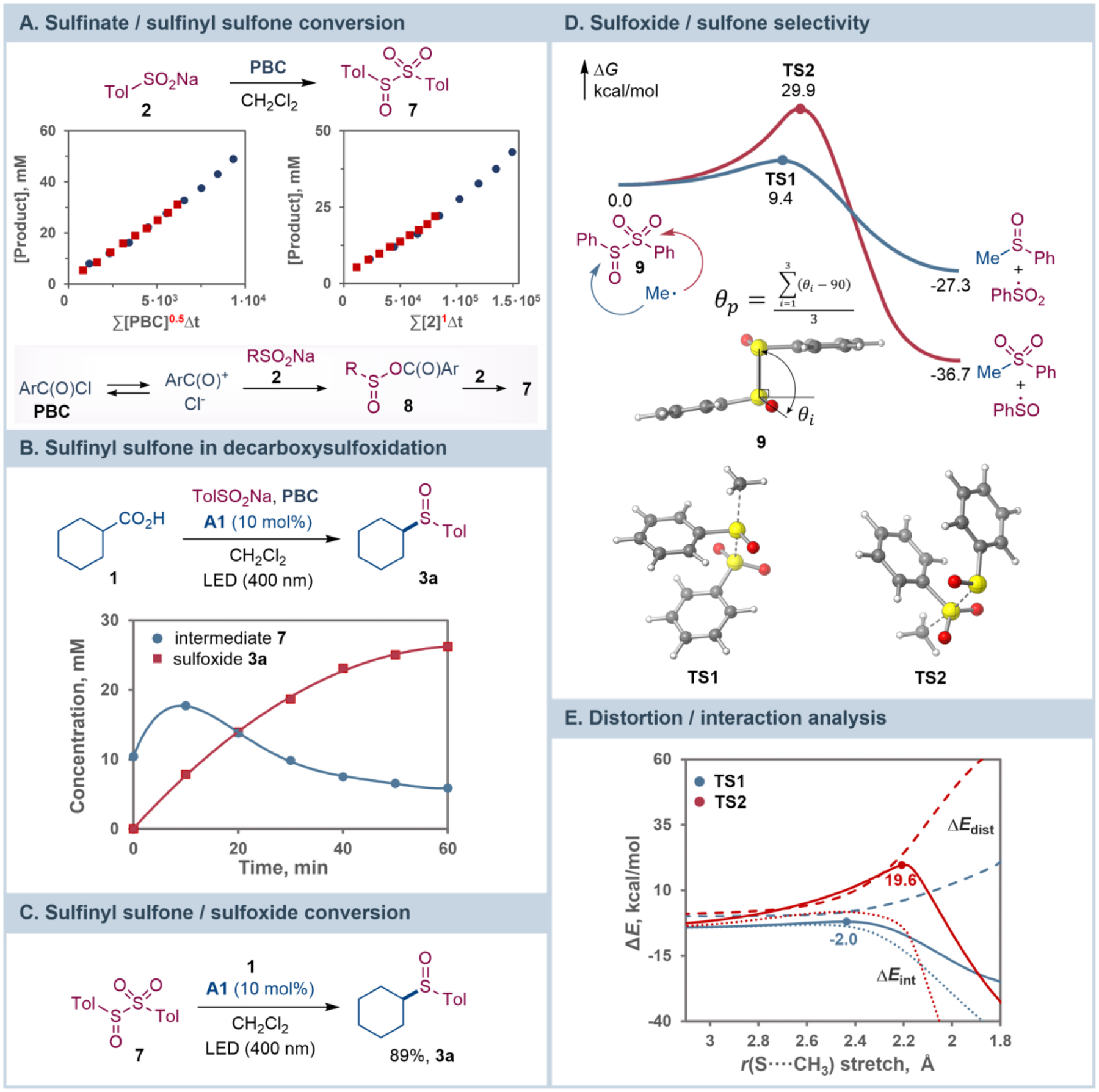
Kinetic and computational studies of the sulfinyl sulfone role in decarboxylative sulfinylation and the sulfoxide/sulfone chemoselectivity.
Taken together, the experimental and computational studies indicate that the direct decarboxylative sulfinylation is enabled by a merger of a photocatalytic decarboxylative cycle, producing an alkyl radical and a kinetically coupled process that entails the generation of a sulfinyl sulfone by a PBC-mediated activation of the sulfinate through mixed anhydride 8, the radical substitution, and the regeneration of the photocatalyst via a hydrogen atom transfer (HAT) between the sulfonyl and acridinyl radicals (Figure 5).
Figure 5.
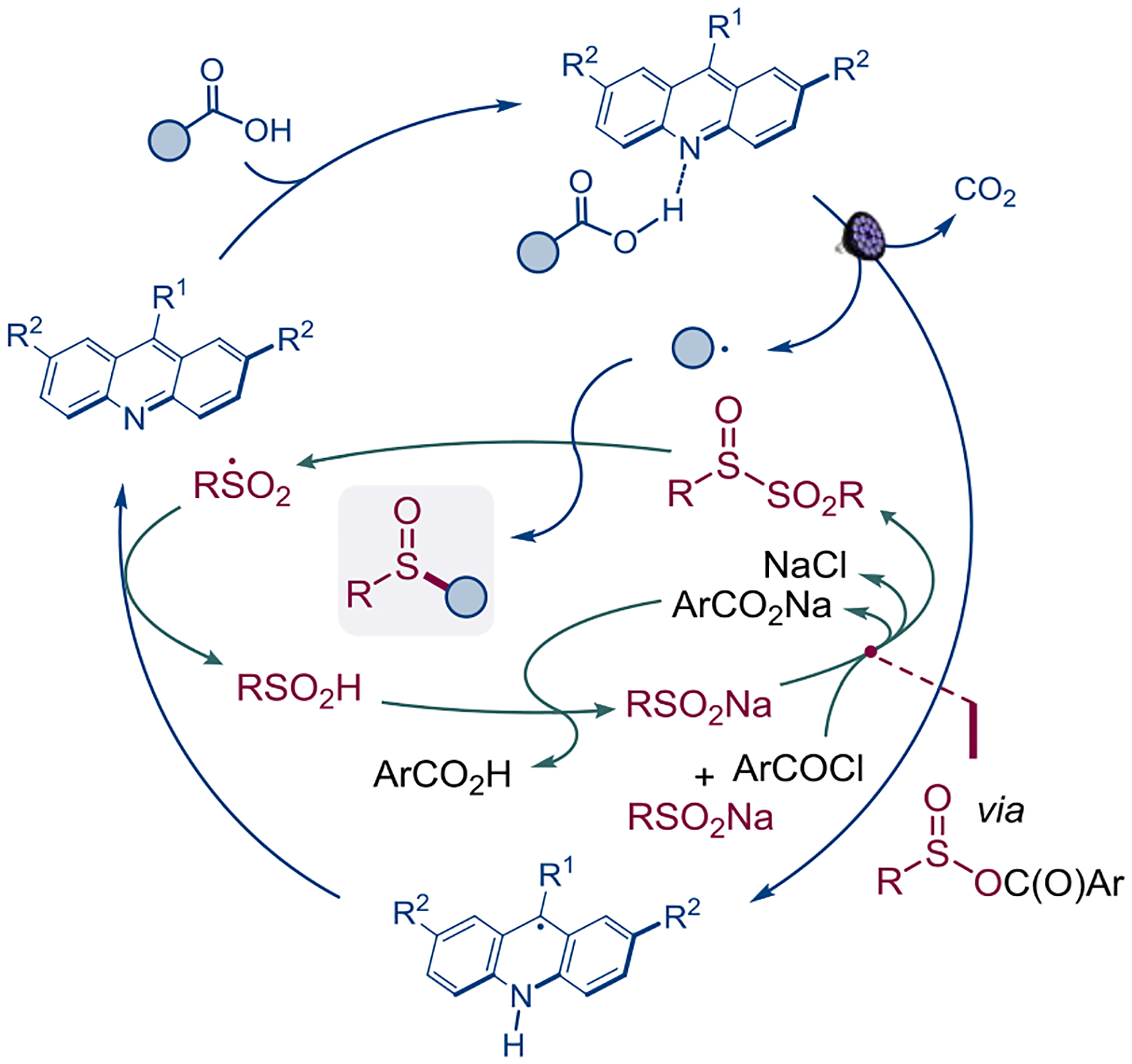
The catalytic system for the direct decarboxylative sulfinylation.
Conclusion
In conclusion, guided by the molecular data analysis pointing to the limited availability of structurally diverse and complex sulfoxides and the improved access to sulfoxides with greater diversity and molecular complexity through a merger of carboxylic acids and sulfinates, we have developed a direct decarboxylative sulfinylation reaction that for the first time allows for a conversion of these abundant feedstocks directly to sulfoxides. The visible light-induced, metal-free reaction is enabled by acridine photocatalysis, and a selective in situ conversion of sulfinates to sulfinyl sulfones that obviates both the preactivation of carboxylic acids and handling unstable sulfinyl sulfones. The study has unveiled the radicalophilic reactivity of sulfinyl sulfones and revealed that they have a kinetic preference for the formation of sulfoxides in a radical substitution, despite the strongly thermodynamically favored formation of sulfones.
Supplementary Material
Figure 3.
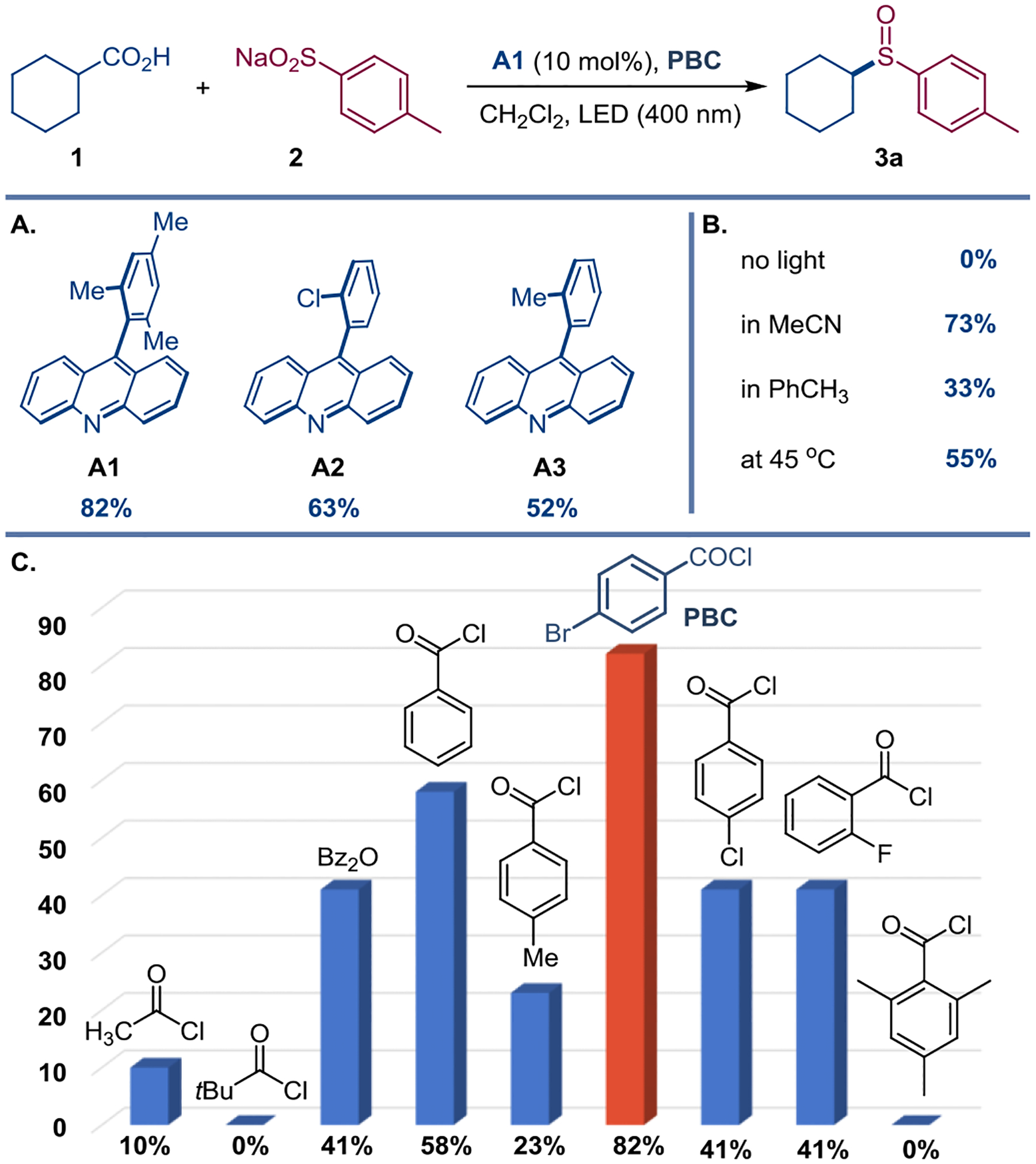
Direct decarboxylative sulfinylation. Reaction conditions: carboxylic acid 1 (0.3 mmol), sulfinate 2 (0.6 mmol), A1 (10 mol%), PBC (0.36 mmol), CH2Cl2 (3.3 mL), LED (400 nm), 12 h. Yields were determined by 1H NMR with 1,4-dimethoxybenzene as an internal standard.
Acknowledgements
Financial support by NIGMS (GM134371) and NSF (CHE-2102646) is gratefully acknowledged. The UTSA NMR and X-ray crystallography facilities were supported by NSF (CHE-1625963 and CHE-1920057). The authors acknowledge the Texas Advanced Computing Center (TACC) and the Extreme Science and Engineering Discovery Environment (XSEDE) for providing computational resources.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Yudin AK, Ed., Catalyzed Carbon-Heteroatom Bond Formation, Wiley-VCH, Weinheim, 2012; [Google Scholar]; b) Liu W, Li J, Huang C-Y, Li C-J, Angew. Chem. Int. Ed 2020, 59, 1786–1796; [DOI] [PubMed] [Google Scholar]; c) Holman KR, Stanko AM, Reisman SE, Chem. Soc. Rev 2021, 50, 7891–7908. [DOI] [PubMed] [Google Scholar]
- [2].a) Oae S, Organic Sulfur Chemistry: Structure and Mechanism, CRC Press, Boca Raton, 1991; [Google Scholar]; b) Patai S, Rapport Z, Eds., The Chemistry of Sulfonic Acids, Esters and their Derivatives, John Wiley & Sons, New York, 1991; [Google Scholar]; c) Lee C-F, Basha RS, Badsara SS, Top. Curr. Chem 2018, 376, 25; [DOI] [PubMed] [Google Scholar]; d) Kaiser D, Klose I, Oost R, Neuhaus J, Maulide N, Chem. Rev 2019, 119, 8701–8780; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhou M, Tsien J, Qin T, Angew. Chem. Int. Ed 2020, 59, 7372–7376; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 7442–7446; [Google Scholar]; f) Hervieu C, Kirillova MS, Suárez T, Müller M, Merino E, Nevado C, Nat. Chem 2021, 13, 327–334; [DOI] [PubMed] [Google Scholar]; g) Zhang Z-X, Willis MC, Chem 2022, 8, 1137–1146. [Google Scholar]
- [3].a) Patani GA, LaVoie EJ, Chem. Rev 1996, 96, 3147–3176; [DOI] [PubMed] [Google Scholar]; b) Brown N, Ed., Bioisosteres in Medicinal Chemistry, Wiley-VCH, Weinheim, 2012. [Google Scholar]
- [4].a) Ilardi EA, Vitaku E, Njardarson JT, J. Med. Chem 2014, 57, 2832–2842; [DOI] [PubMed] [Google Scholar]; b) Scott KA, Njardarson JT, Top. Curr. Chem 2018, 376, 5. [DOI] [PubMed] [Google Scholar]
- [5].a) Patai S, Ed., The Chemistry of Sulfinic Acids, Esters and their Derivatives, John Wiley & Sons, Ltd, New Jersey, 1990; [Google Scholar]; b) Fernandez I, Khiar N, Chem. Rev 2003, 103, 3651–3706; [DOI] [PubMed] [Google Scholar]; c) Feldman KS, Fodor MD, J. Am. Chem. Soc 2008, 130, 14964–14965; [DOI] [PubMed] [Google Scholar]; d) Kaldre D, Klose I, Maulide N, Science 2018, 361, 664–667; [DOI] [PubMed] [Google Scholar]; e) Yan J, Pulis AP, Perry GJP, Procter DJ, Angew. Chem., Int. Ed 2019, 58, 15675–15679; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 15822–15826; [Google Scholar]; f) Leypold M, D’Angelo KA, Movassaghi M, Org. Lett 2020, 22, 8802–8807; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Li G, Nieves-Quinones Y, Zhang H, Liang Q, Su S, Liu Q, Kozlowski MC, Jia T. Nat. Commun 2020, 11, 2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Chen MS, White MC, J. Am. Chem. Soc 2004, 126, 1346–1347; [DOI] [PubMed] [Google Scholar]; b) Mellah M, Voituriez A, Schulz E, Chem. Rev 2007, 107, 5133; [DOI] [PubMed] [Google Scholar]; c) Mariz R, Luan X, Gatti M, Linden A, Dorta R, J. Am. Chem. Soc 2008, 130, 2172–2173; [DOI] [PubMed] [Google Scholar]; d) Trost BM, Rao M, Angew. Chem., Int. Ed 2015, 54, 5026–5043; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 5112–5130; [Google Scholar]; e) Pattillo CC, Strambeanu II, Calleja P, Vermeulen NA, Mizuno T, White MC. J. Am. Chem. Soc 2016, 138, 1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Qiao R, Fu C, Li Y, Qi X, Ni D, Nandakumar A, Siddiqui G, Wang H, Zhang Z, Wu T, Zhong J, Tang S-Y, Pan S, Zhang C, Whittaker MR, Engle JW, Creek DJ, Caruso F, Ke PC, Cai W, Whittaker AK, Davis TP. Adv. Sci 2020, 7, 2000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Imada Y, Iida H, Ono S, Murahashi S-I, J. Am. Chem. Soc 2003, 125, 2868–2869; [DOI] [PubMed] [Google Scholar]; b) Liao S, Čorić I, Wang Q, List B, J. Am. Chem. Soc 2012, 134, 10765–10768; [DOI] [PubMed] [Google Scholar]; c) Newhouse TR, Li X, Blewett MM, Whitehead CMC, Corey EJ, J. Am. Chem. Soc 2012, 134, 17354–17357; [DOI] [PubMed] [Google Scholar]; d) Hussain H, Green IR, Ahmed I, Chem. Rev 2013, 113, 3329–3317; [DOI] [PubMed] [Google Scholar]; e) Sheet D, Paine TK, Chem. Sci 2016, 7, 5322–5331; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Matavos-Aramyan S, Soukhakian S, Jazebizadeh MH, Phosphorus Sulfur Silicon Relat. Elem 2020, 195, 181–193. [Google Scholar]
- [9].a) Olah GA, Nishimura J, J. Org. Chem 1974, 39, 1203–1205; [Google Scholar]; b) Khiar N, Alcudia F, Espartero J-L, Rodríguez L, Fernández I, J. Am. Chem. Soc 2000, 122, 7598–7599; [Google Scholar]; c) Han Z, Krishnamurthy D, Grover P, Fang QK, Su X, Wilkinson HS, Lu Z-H, Magiera D, Senanayake CH, Tetrahedron 2005, 61, 6386–6408; [Google Scholar]; d) Yuste F, Hernández Linares A, Mastranzo VM, Ortiz B, Sánchez-Obregón R, Fraile A, García Ruano JL, J. Org. Chem 2011, 76, 4635–4644; [DOI] [PubMed] [Google Scholar]; e) Yu H, Li Z, Bolm C, Org. Lett 2018, 20, 7104–7106. [DOI] [PubMed] [Google Scholar]
- [10].a) Maitro G, Vogel S, Prestat G, Madec D, Poli G, Org. Lett 2006, 8, 5951–5954; [DOI] [PubMed] [Google Scholar]; b) Jia T, Bellomo A, Montel S, Zhang M, EL Baina K, Zheng B, Walsh PJ, Angew. Chem., Int. Ed 2014, 53, 260–264; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 264–268; [Google Scholar]; c) Jia T, Zhang M, Jiang H, Wang CY, Walsh PJ, J. Am. Chem. Soc 2015, 137, 13887–13893; [DOI] [PubMed] [Google Scholar]; d) Lenstra DC, Vedovato V, Ferrer Flegeau E, Maydom J, Willis MC, Org. Lett 2016, 18, 2086–2089; [DOI] [PubMed] [Google Scholar]; e) Jia T, Zhang M, McCollom SP, Bellomo A, Montel S, Mao J, Dreher SD, Welch CJ, Regalado EL, Williamson RT, Manor BC, Tomson NC, Walsh PJ, J. Am. Chem. Soc 2017, 139, 8337–8345; [DOI] [PubMed] [Google Scholar]; f) Li Y, Wang M, Jiang X, ACS Catal. 2017, 7, 7587–7592; [Google Scholar]; g) Yu H, Li Z, Bolm C, Org. Lett 2018, 20, 2076–2079. [DOI] [PubMed] [Google Scholar]; For other recent approaches to sulfoxides, see:; h) Meyer AU, Wimmer A, König B, Angew. Chem. Int. Ed 2017, 56, 409–412; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 420–423; [Google Scholar]; i) Wang X, Tang Y, Ye S, Zhang J, Kuang Y, Wu J, Org. Lett 2022, 24, 2059–2063. [DOI] [PubMed] [Google Scholar]
- [11].a) Kice JL, Guaraldi G, J. Am. Chem. Soc 1968, 90, 4076–4081; [Google Scholar]; b) Kice JL, Mullan LF, J. Am. Chem. Soc 1976, 98, 4259–4268; [Google Scholar]; c) Kice JL, Wu S-M, J. Org. Chem 1981, 46, 3913–3914; [Google Scholar]; d) Caputo R, Ferreri C, Longobardo L, Palumbo G, Pedatella S, Synth. Commun 1993, 23, 1515–1522. [Google Scholar]
- [12].Kice JL, Pawlowski NE, J. Am. Chem. Soc 1964, 86, 4898–4904. [Google Scholar]
- [13].a) Wang Z, Zhang Z, Zhao W, Sivaguru P, Zanoni G, Wang Y, Anderson EA, Bi X, Nat. Commun 2021, 12, 5244–5253; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Z, Song Q, Feng C, Wang Z, Zhao W, Ning Y, Wu Y, Chem Asian J 2022, 17, e202200299. [DOI] [PubMed] [Google Scholar]
- [14].For recent direct decarboxylative functionalizations, see:; a) Kautzky JA, Wang T, Evans RW, MacMillan DWC, J. Am. Chem. Soc 2018, 140, 6522–6526; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Till NA, Smith RT, MacMillan DWC, J. Am. Chem. Soc 2018, 140, 5701–5705; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sun X, Chen J, Ritter T, Nat. Chem 2018, 10, 1229–1233; [DOI] [PubMed] [Google Scholar]; d) Cartwright KC, Lang SB, Tunge JA, J. Org. Chem 2019, 84, 2933–2940; [DOI] [PubMed] [Google Scholar]; e) Faraggi TM, Li W, MacMillan DWC, Isr. J. Chem 2020, 60, 410–415; [Google Scholar]; f) Li J, Huang CY, Han JT, Li C-J, ACS Catal 2021, 11, 14148–14158; [Google Scholar]; g) Li QY, Gockel SN, Lutovsky GA, DeGlopper KS, Baldwin NJ, Bundesmann MW, Tucker JW, Bagley SW, Yoon TP, Nat. Chem 2022, 14, 94–99; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Kitcatt DM, Nicolle S, Lee A-L, Chem. Soc. Rev 2022, 51, 1415–1453. For other examples, see: [DOI] [PubMed] [Google Scholar]; i) Shang R, Liu L, Sci. China Chem 2011, 54, 1670–1687; [Google Scholar]; j) Wang J, Qin T, Chen T-E, Wimmer L, Edwards JT, Cornella J, Vokits B, Shaw SA, Baran PS, Angew. Chem., Int. Ed 2016, 55, 9676–9679; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 9828–9831; [Google Scholar]; k) Candish L, Teders M, Glorius F, J. Am. Chem. Soc 2017, 139, 7440–7443; [DOI] [PubMed] [Google Scholar]; l) Liu C, Shen N, Shang R. Org. Chem. Front 2021, 8, 4166–4170; [Google Scholar]; m) Sharique M, Majhi J, Dhungana RK, Kammer LM, Krumb M, Lipp A, Romero E, Molander GA, Chem. Sci 2022,13, 5701–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Nguyen VD, Nguyen VT, Haug GC, Dang HT, Jin S, Li Z, Flores-Hansen C, Benavides B, Arman HD, Larionov OV, ACS Catal. 2019, 9, 9485–9498; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nguyen VT, Nguyen VD, Haug GC, Vuong NTH, Dang HT, Arman HD, Larionov OV, Angew. Chem., Int. Ed 2020, 59, 7921–7927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 7995–8001; [Google Scholar]; c) Dang HT, Haug GC, Nguyen VT, Vuong NTH, Arman HD, Larionov OV, ACS Catal. 2020, 10, 11448–11457; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nguyen VT, Haug GC, Nguyen VD, Vuong NTH, Arman HD, Larionov OV, Chem. Sci 2021, 12, 6429–6436; See also: [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nguyen VD, Trevino R, Greco SG, Arman HD, Larionov OV ACS Catal. 2022, 12, 8729–8739; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zubkov MO, Kosobokov MD, Levin VV, Kokorekin VA, Korlyukov AA, Hu J, Dilman AD, Chem. Sci 2020, 11, 737–741; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Dmitriev IA, Levin VV, Dilman AD, Org. Lett 2021, 23, 8973–8977; [DOI] [PubMed] [Google Scholar]; h) Zubkov MO, Kosobokov MD, Levin VV, Dilman AD, Org. Lett 2022, 24, 2354–2358. [DOI] [PubMed] [Google Scholar]
- [16].a) Deeming AS, Russell CJ, Willis MC, Angew. Chem., Int. Ed 2016, 55, 747–750; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 757–760; [Google Scholar]; b) Lo PKT, Chen Y, Willis MC, ACS Catal. 2019, 9, 10668–10673; [Google Scholar]; c) Smith JM, Dixon JA, deGruyter JN, Baran PS, J. Med. Chem 2019, 62, 2256–2264; [DOI] [PubMed] [Google Scholar]; d) Liang S, Hofman K, Friedrich M, Manolikakes G, Eur. J. Org. Chem 2020, 4664–4676; [Google Scholar]; e) Sarver PJ, Bissonnette NB, MacMillan DWC, J. Am. Chem. Soc 2021, 143, 9737–9743; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Jin S, Haug GC, Trevino R, Nguyen VD, Arman HD, Larionov OV, Chem. Sci 2021, 12, 13914–13921; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Andrews JA, Pantaine LRE, Palmer CF, Poole DL, Willis MC, Org. Lett 2021, 23, 8488–8493. [DOI] [PubMed] [Google Scholar]
- [17].a) Mahjour B, Shen Y, Liu W, Cernak T, Nature 2020, 580, 71–75; [DOI] [PubMed] [Google Scholar]; b) Zhang Z, Cernak T, Angew. Chem., Int. Ed 2021, 60, 27293–27298; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2021, 133, 27499–27504; [Google Scholar]; c) Zhang R, Mahjour B, Cernak T, ChemRxiv 2022, DOI: 10.26434/chemrxiv-2022-917k5. [DOI] [Google Scholar]
- [18].Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE, Nucleic Acids Res. 2021, 49, D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Böttcher TJ, Chem. Inf. Model 2016, 56, 462–470. [DOI] [PubMed] [Google Scholar]
- [20].Lovering F, Bikker J, Humblet C, J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- [21].Landwehr EM, Baker MA, Oguma T, Burdge HE, Kawajiri T, Shenvi RA, Science 2022, 375, 1270–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Silverman BW, Density Estimation for Statistics and Data Analysis, Chapman and Hall/CRC, Boca Raton, FL, 1998. [Google Scholar]
- [23].Brown DG, Boström J, J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- [24].Franklin JM, Carrasco GA, Moskovitz J, Neurosci. Lett 2013, 533, 86–89. [DOI] [PubMed] [Google Scholar]
- [25]. CCDC depository numbers for the new structures reported in this work: 2144743 (3i), 2144738 (3p), 2144737 (3q), 2144742 (3t), 2144747 (3z), 2144745 (4g), 2144741 (4j), 2144746 (4l), 2144744 (4q), 2144740 (4r), 2152027 (5fa), 2152041 (5fb), 2149762 (5i).
- [26].Burés J, Angew. Chem., Int. Ed 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 16318–16321. [Google Scholar]
- [27].Bentley TW, J. Org. Chem 2008, 73, 6251–6257. [DOI] [PubMed] [Google Scholar]
- [28]. The alkyl radical involvement was confirmed by experiments with TEMPO, while no TEMPO adducts of sulfinyl and sulfonyl radicals were detected, see Figure S1 in the SI.
- [29]. See Figure S3 and the corresponding discussion in the SI.
- [30].Bickelhaupt FM, Houk KN, Angew. Chem., Int. Ed 2017, 56, 10070–10086; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 10204–10221. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.