

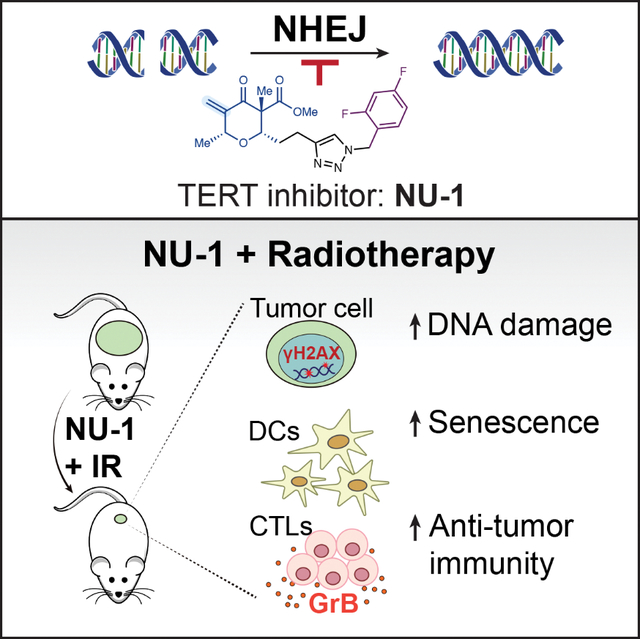
Summary
Beyond synthesizing telomere repeats, the telomerase reverse transcriptase TERT also serves multiple other roles supporting cancer growth. Blocking telomerase to drive telomere erosion appears impractical, but TERT’s non-canonical activities have yet to be fully explored as cancer targets. Here, we used an irreversible TERT inhibitor, NU-1, to examine impacts on resistance to conventional cancer therapies. In vitro, inhibiting TERT sensitized cells to chemotherapy and radiation. NU-1 delayed repair of double strand breaks, resulting in persistent DNA damage signaling and cellular senescence. While NU-1 alone did not impact growth of syngeneic CT26 tumors in BALB/c mice, it dramatically enhanced the effects of radiation, leading to immune-dependent tumor elimination. Tumors displayed persistent DNA damage, suppressed proliferation, and increased activated immune infiltrate. Our studies confirm TERT’s role in limiting genotoxic effects of conventional therapy but also implicate TERT as a determinant of immune evasion and therapy resistance.
In Brief
Liu et al. use telomerase inhibitor NU-1 to further implicate TERT in promoting DNA repair and cell survival after radiation in vitro. In vivo, NU-1 sensitizes tumors to radiation not only by blocking DNA repair, but also by potentiating an adaptive immune response that leads to tumor elimination.
Graphical Abstract
Introduction
Human telomeres consist of 5–15 kb of double stranded 5’-(TTAGGG)n-3’ repeats terminating in a 3’ G-strand overhang at the end of each chromosome that binds the shelterin complex to protect against end-to-end fusions or detection as damaged DNA (de Lange, 2018; Shay and Wright, 2019). TTAGGG repeats lost from the ends during replication are restored by telomerase, an RNA-directed DNA polymerase complex where the telomerase RNA component TERC serves as a template for the telomerase reverse transcriptase TERT (Greider and Blackburn, 1985). TERT expression can be detected in adult stem and progenitor cells but is silenced during somatic cell differentiation. Thus, telomeres can shorten as tissue precursors proliferate, reaching a critical length at the Hayflick limit. Eventually, telomere erosion displaces the shelterin complex, inducing a DNA damage response (DDR). Most cells die or enter replicative senescence, driving inflammation and tissue aging, but some may become genomically unstable and reactivate TERT (Maciejowski and de Lange, 2017). Thereby, the premalignant cells gain immortality, maintaining telomere integrity in the face of unlimited cell division.
The rationale that blocking telomerase might limit the proliferation of cancer cells has led to extensive efforts to target TERT for therapeutic intervention, yielding a wide range of synthetic agents (Guterres and Villanueva, 2020). Prominent examples that have advanced to preclinical and/or clinical studies include oligonucleotide drugs such as Imetelstat (GRN163L) (Herbert et al., 2005), G-quadruplex stabilizers such as TMPyP4 (Wheelhouse et al., 1998), and allosteric inhibitors such as BIBR 1532 (Pascolo et al., 2002). Diverse natural products have also been reported to inhibit telomerase (Ganesan and Xu, 2017). Though few appear promising for clinical development, chemical simplification of the catechin EGCG led to the drug-like competitive inhibitor MST-312 (Seimiya et al., 2002).
Maintaining telomerase suppression long enough to achieve critical erosion has been a challenge in vivo, reflected in the hematopoietic toxicity and resistance that have impacted clinical trials with Imetelstat (Shay, 2016). The complementary strategy of selectively damaging telomeres in TERT-expressing cells via providing altered TERC templates (Kim et al., 2001) or toxic substrates such as 6-thio-dG (Mender et al., 2015) or other dNTPs (Sanford et al., 2020) is promising, but may similarly be limited by toxicity. Beyond repeat synthesis, TERT has long been linked to non-canonical, extra-telomeric activities that enhance stress responses, support stemness, and promote cell growth and survival (Lamy et al., 2013; Ségal-Bendirdjian and Geli, 2019). Multiple studies published over the past two decades confirm early reports (Akiyama et al., 2002; Kondo et al., 1998; Massard et al., 2006) that TERT expression and/or activity significantly impact cancer cell sensitivity to radiation and/or chemotherapy, both in vitro and in vivo (Arndt and MacKenzie, 2016; Fleisig et al., 2016). Early on, this effect was ascribed to a direct role for TERT in repairing double strand breaks (DSBs) (Masutomi et al., 2005), though mechanisms have remained poorly defined. A confounding factor is that telomere dysfunction itself affects sensitivity to therapy, perhaps reflecting a feedback loop where telomeres may serve as oxidative stress sensors (Oikawa and Kawanishi, 1999; von Zglinicki et al., 2000) and that short or damaged telomeres radiosensitize both normal and cancer cells (Goytisolo et al., 2000; Wong et al., 2000). Nonetheless, likely reflecting TERT’s roles in stress response that are telomere-independent, the sensitizing effects of TERT inhibitors do not require prolonged treatment or telomere attrition.
The Streptomyces macrolide antibiotic chrolactomycin was previously identified as a micromolar TERT inhibitor in vitro and proposed to act by covalent binding via a reactive exocyclic methylene group to a nucleophile in the TERT catalytic site (Nakai et al., 2006). Total synthesis has prevented conventional structure-activity analysis to date, inspiring the alternative approach of developing simplified chrolactomycin analogs. This effort yielded NU-1 (Betori et al., 2020), a small-molecule inhibitor bearing an exocyclic methylene that conjugates to a conserved active site cysteine to inhibit TERT catalytic activity in telomerase-positive cancer cell lines. Confirming the mechanism of action, NU-1 has no effects on telomerase-negative cells while the non-reactive des-exomethylene analog NU-2 fails to inhibit TERT or otherwise impact telomerase-positive cells.
Here, we examined the potential of NU-1 to enhance conventional cancer therapy. In vitro, NU-1 and other TERT-binding telomerase inhibitors sensitized cancer cells to radiation and genotoxic chemotherapies. Treating cells with NU-1 prior to radiation delayed DSB repair, prolonged γH2AX foci persistence, extended G1 cell cycle arrest, and promoted cell senescence, suggesting a direct mechanism of radiosensitization. These effects of NU-1 were not observed in a cancer cell line lacking TERT expression. In vivo, when tumors formed by CT26 mouse colon carcinoma cells were treated with an otherwise ineffective radiation dose, most were eliminated when the BALB/c mice were concomitantly treated with NU-1, perhaps reflecting greater persistent DNA damage and/or increased CD8+ T cell infiltrate. Supporting an immune mechanism, CT26 cells treated with NU-1 and radiation form senescent cells that stimulate dendritic cell (DC) maturation/activation, leading to CD8+ T cell proliferation. Considering the potential advantages of targeted covalent inhibitors as probes and drugs (Boike et al., 2022), irreversible inhibition of TERT, as with NU-1, may offer a direct path to tumor-specific sensitizers to improve the efficacy of radiation and other cancer therapies.
Results
NU-1 treatment impacts DNA damage pathway gene expression
Our previous studies demonstrated that at concentrations >1 μM, NU-1 decreases cell viability in telomerase-positive human cancer cell lines, including breast cancer MCF7 (LD50=21 μM) (Betori et al., 2020). The toxic effects of NU-1 appeared within 24 to 48 h, far too rapidly to depend on telomere erosion, given the expected loss of only a few telomeric repeats per population doubling. Towards examining mechanisms of the apparent TERT addiction exposed by NU-1 (Figure 1A), the mRNA expression profiles of MCF7 cells treated with 0.5 μM NU-1 or DMSO vehicle for 48 h were compared, revealing 2117 upregulated and 2069 downregulated genes. Consistent with previous studies (Li et al., 2005), Reactome Gene Ontology (GO) analysis (Fabregat et al., 2018) of the differentially expressed genes identified the pathways Cell Cycle and DNA Repair as the two most downregulated in NU-1 treated cells (−logP=35.9 and 11.7 respectively, Figure 1B). NU-1 also induced down-regulation of the pathways Telomere Maintenance (−logP=3.09). Upregulated GO terms included metabolic, secretion, and membrane-related pathways (Figure 1C). GO analysis with other databases (Huang da et al., 2009; Krämer et al., 2014) revealed complementary patterns, including downregulation of DNA repair (Figure S1A–E).
Figure 1. TERT inhibitor NU-1 modulates cancer cell gene expression.
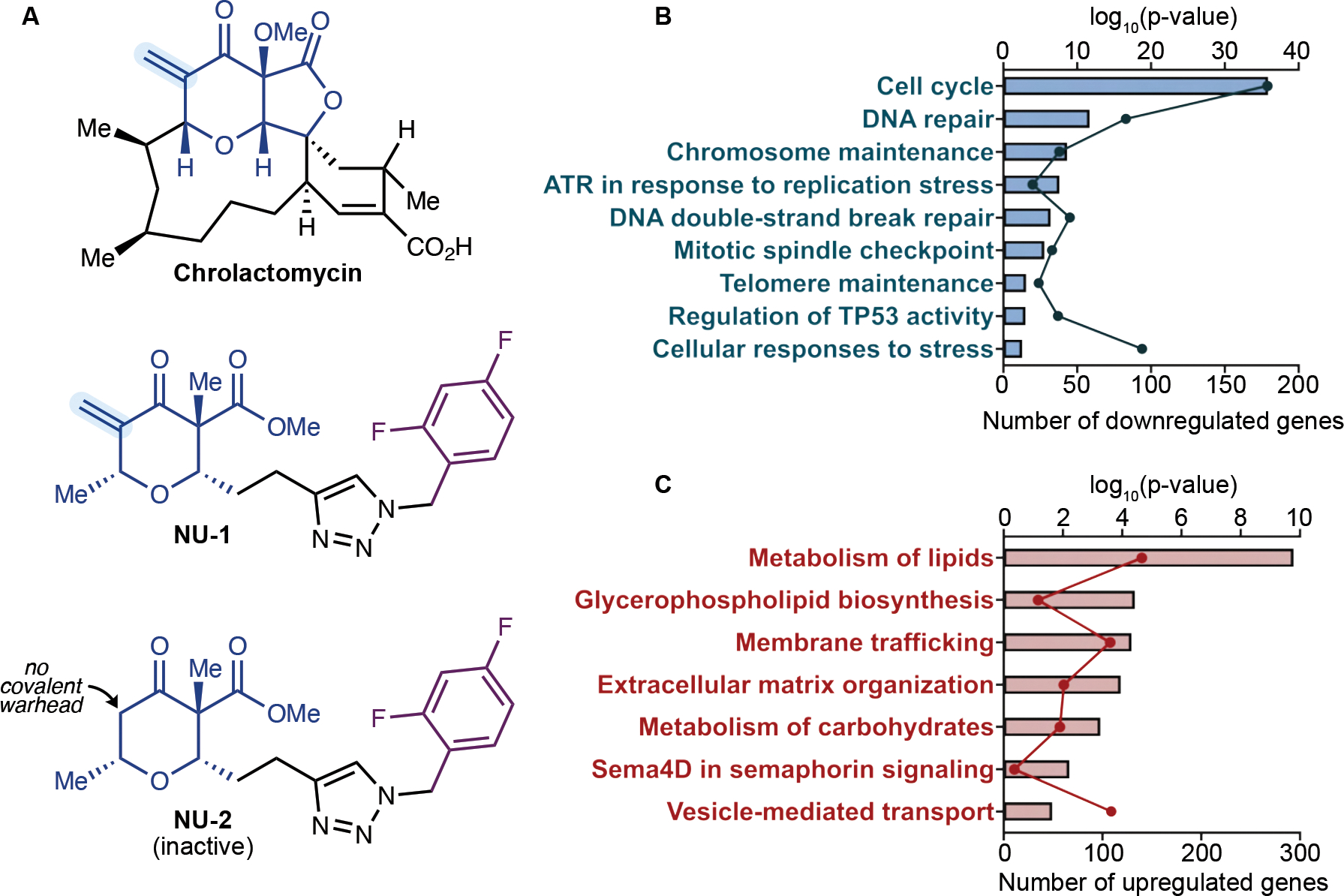
(A) Chemical structures of TERT inhibitors chrolactomycin and NU-1 and the inactive des-exomethylene analog NU-2.
(B-C) Enrichment of downregulated (B) and upregulated (C) pathways in NU-1 treated MCF7 cells through Reactome Gene Ontology (GO) analysis. Dots, number of DEGs. Bars, −Log10(p-value) for each enriched pathway.
See also Figure S1.
NU-1 enhances the chemo-sensitivity of telomerase-positive cancer cells
Toward confirming prior studies and the gene expression analysis, we examined whether NU-1 confers sensitivity to genotoxic stress. Thus, MCF7 cells were incubated for 4 h with NU-1 (0.5 μM), the inactive des-exomethylene analog NU-2 (0.5 μM), the non-competitive inhibitor BIBR 1532 (BIBR, 10 μM), the reversible competitive inhibitor MST-312 (MST, 1 μM) or the vehicle DMSO, and then for 24 h with the topoisomerase I poison irinotecan (1 nM to 100 μM) and analyzed for cell viability. Each of the TERT inhibitors significantly sensitized MCF7 cells to irinotecan. While pretreatment with BIBR or MST decreased the 50% lethal dose (LD50) of irinotecan in MCF7 cells from 0.37 μM to 0.16 and 0.17 μM respectively, NU-1 reduced it to 0.08 μM, displaying the strongest effects. NU-1 also sensitized MCF7 cells to topoisomerase II poisons etoposide and doxorubicin and the spindle poison paclitaxel (Figure 2A, Table S1 and S2). By contrast, no appreciable sensitizing effects of NU-1 or other TERT inhibitors were observed in the ALT cell line Saos-2 (Figure 2B, Table S1 and S2). Another telomerase-positive cell line, A549, displayed a similar pattern to MCF7 (Figure 2C, Table S1 and S2). Calculating a combination index (CI) (Chou and Martin, 2005) confirmed synergy between NU-1 and chemotherapy agents (Figure 2D). Interestingly, the TERT inhibitors lost their sensitizing effects on MCF7 cells when added at the same time as, or 4 h after, irinotecan (Figure 2E and F), suggesting that TERT’s critical protective activity mediates its effects early in the response. These results support NU-1 as a potential sensitizer for genotoxic cancer therapies.
Figure 2. TERT inhibition sensitizes telomerase-positive cells to chemotherapy.
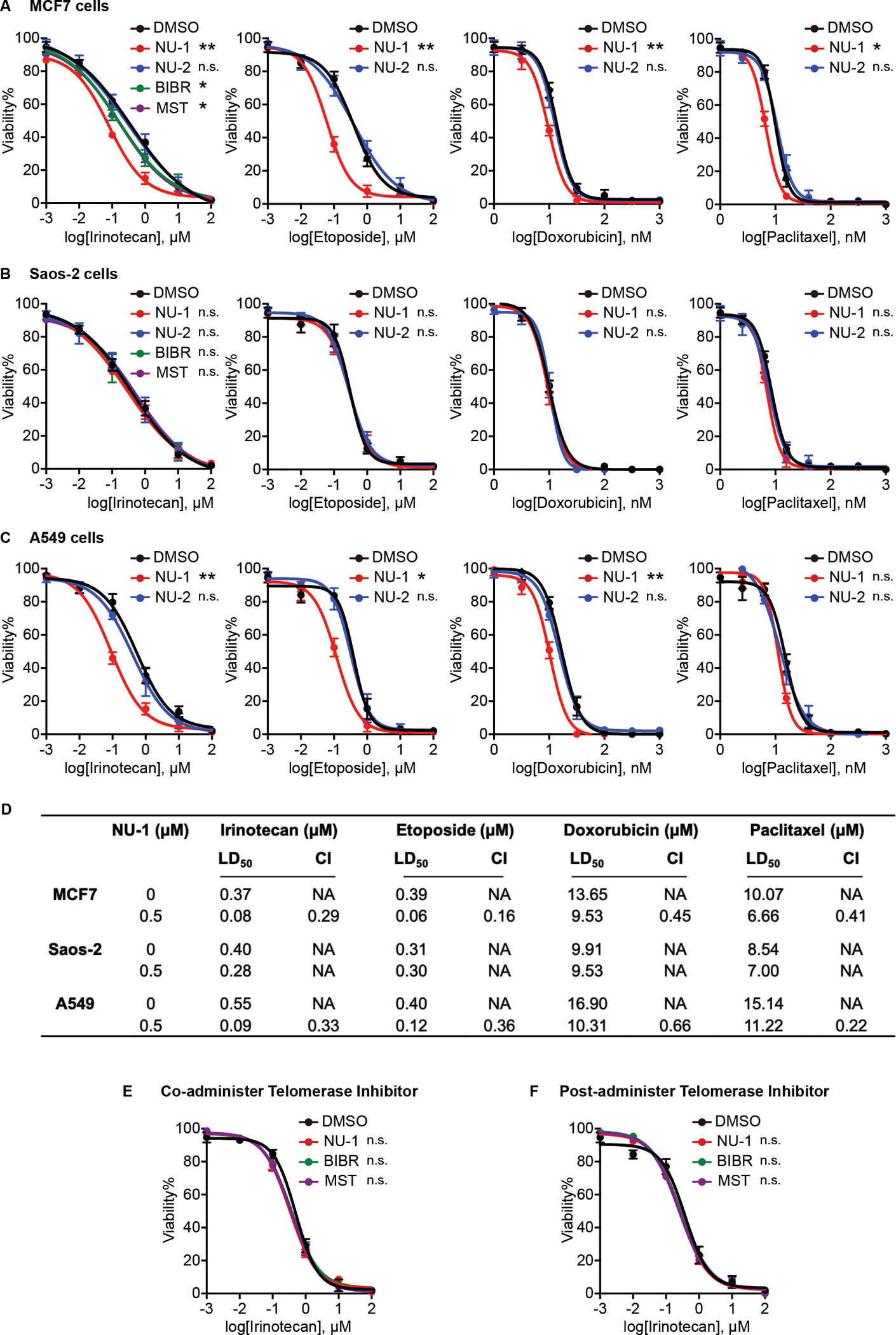
(A-C) Dose-response curves indicating the viability of MCF7 cells (A), ALT Saos-2 cells (B), and A549 cells (C) pretreated for 4 h with DMSO, NU-1 (0.5 μM), or NU-2 (0.5 μM), BIBR 1532 (BIBR, 10 μM), MST-312 (MST, 1 μM), then for 24 h with indicated concentration of irinotecan, etoposide, paclitaxel, or doxorubicin, and cell viability determined.
(D) Lethal Dose 50 (LD50) in cells and Combination Index (CI) were calculated from data from A-C. CI<1, synergistic; CI=1, addictive; CI>1, antagonistic.
(E) Viability of MCF7 cells where DMSO or telomerase inhibitors were co-administered with irinotecan for 24 h.
(F) Viability of MCF7 cells treated with irinotecan for 4 h before adding DMSO or telomerase inhibitors for 24 h.
Data obtained from three replicates, mean ± SEM. LD50 were used for statistical analysis, ** 0.001< P < 0.01, * 0.01< P < 0.05, n.s. P > 0.05 compared to DMSO (unpaired t-test).
NU-1 enhances radiosensitivity in vitro
The non-covalent TERT inhibitors BIBR and MST have been reported as radiosensitizers in human cancer cell lines (Ding et al., 2019; Wang et al., 2015). To examine whether NU-1 has comparable effects, MCF7 cells were treated with DMSO control, NU-1 (0.5 μM), BIBR (10 μM), or MST (1 μM), at concentrations below those that impacted clonogenicity on their own (Figure 3A), followed by increasing doses of ionizing radiation (IR, 0–5 Gy). A clonogenic survival assay demonstrated that each of the TERT inhibitors conferred radiosensitization, with NU-1 displaying the strongest effects (Figure 3B and C, Table S3). Toward further examining the effects of TERT inhibition on radiation response, we treated MCF7 cells with DMSO, NU-2, NU-1, chrolactomycin (CHRO) (Iorio et al., 2012), BIBR, or MST, followed by 0 or 6 Gy and then incubated for 7 days in the continued presence of drugs for time-lapse live-cell imaging. The TERT inhibitors did not limit cell proliferation of unirradiated cells but significantly slowed recovery and/or proliferation following irradiation (Figure 3D). This could not be ascribed to increased cell death as staining cells with the cell membrane integrity probe YO-PRO-1 indicated no increased effects of the TERT inhibitors over radiation alone (Figure 3E). However, examining the time-lapse imaging results revealed that many of the surviving cells treated with both TERT inhibitors and radiation displayed an enlarged and flattened morphology characteristic of senescence. Thus, we examined senescence-associated β-galactosidase (SA-β-Gal) at 7 days after IR. On their own, TERT inhibitors did not increase senescence above background. After irradiation, the DMSO and NU-2 controls displayed a similar increase in SA-β-Gal+ cells (~34%). Addition of TERT inhibitors further increased senescent cells after IR (~55%, Figure 3F and G). As shown in the images, even among the cells not scored as SA-β-Gal+, few displayed the characteristic size and shape of proliferating MCF7 cells. Notably, even 50 nM NU-1 or CHRO was sufficient to enhance senescence after IR, though with less potency (Figure 3H).
Figure 3. TERT inhibition induces radiosensitivity and cellular senescence.
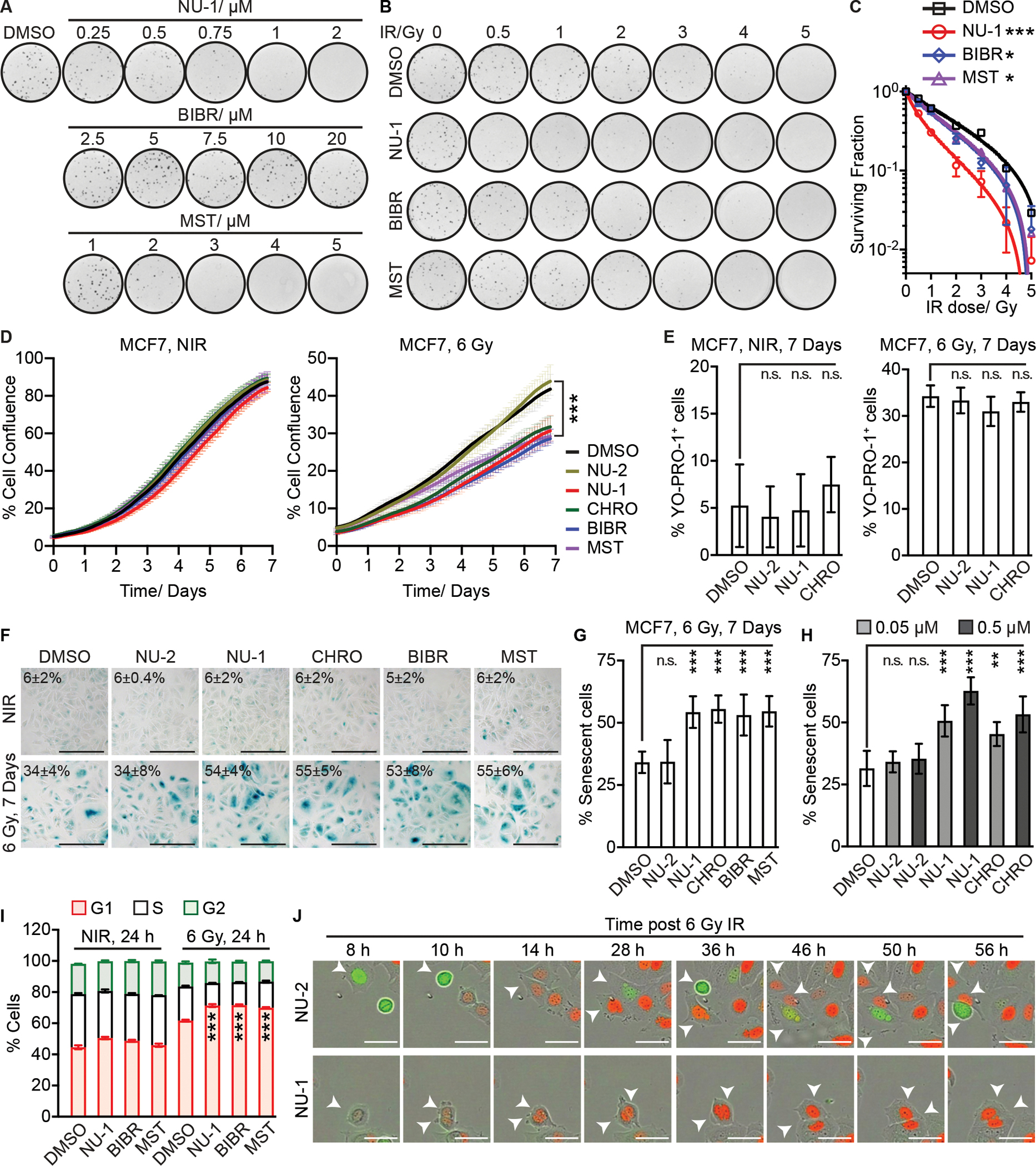
(A) Clonogenic assay of MCF7 cells treated with DMSO control, NU-1, BIBR (BIBR 1532), or MST (MST-312) at indicated concentrations.
(B) Clonogenic survival of MCF7 cells irradiated at indicated doses ± DMSO, NU-1 (0.5 μM), BIBR (10 μM), or MST (1 μM). Representative images from triplicates.
(C) Normalized surviving fractions of cells in B, mean ± SD.
(D) Proliferation analysis over 6 days comparing MCF7 cells treated with TERT inhibitors or controls for 1 h before 0 (left, NIR) or 6 Gy (right) at time 0, mean ± SEM.
(E) Quantification of YO-PRO-1+ cells. Cells were treated with indicated compounds ± IR, followed by staining after 7 days, mean ± SD.
(F) SA-β-Gal staining of MCF7 cells treated as in D, after 7 days. Representative 20X images. Scale bars=200 μm.
(G) Quantification of SA-β-Gal+ cells in F, mean ± SD.
(H) % SA-β-Gal+ MCF7 cells. Cells were treated as indicated for 1 h, followed by IR and staining after 7 days, mean ± SD.
(I) MCF7 cells were treated with indicated compounds ± 6 Gy IR, followed by cell cycle analysis after 24 h. Data from three replicates, mean ± SD.
(J) MCF7-FUCCI cells were treated with NU-1 or NU-2 for 1 h before 6 Gy at time 0 h. Successive representative images are shown. Red, G1 phase. Green, S/G2. Arrows indicate the tracked mother and daughter cells. Scale bars=50 μm. *** P <0.001; ** 0.001< P < 0.01, n.s. P > 0.05 compared to DMSO (unpaired t-test).
Cellular senescence is defined as a state of stable cell cycle arrest. To explore how TERT inhibition might promote senescence, we examined the distribution of cell cycle stages of MCF7 cells based on their DNA content after IR in the presence or absence of TERT inhibitors. Proliferating MCF7 cells were distributed as ~45% G1, 35% S, and 20% G2/M. TERT inhibitors had no appreciable effects on their own after 24 h treatment. Cells treated with IR and then allowed to recover for 24 h displayed an increase in G1 to 62% and decrease in S phase to 22%, consistent with unrepaired DNA damage. Addition of TERT inhibitors further expanded the G1 population (~71%) and reduced S (~15%) (Figure 3I and S2A). To visualize senescent cell cycle arrest and/or mitotic catastrophe, we performed time-lapse live-cell imaging of MCF7-FUCCI cells, which allow individual cells to be tracked through G1 (mCherry-hCdt1, red), S/G2 (mVenus-hGeminin, green), and M phase (Sakaue-Sawano et al., 2008). After IR, most of the surviving NU-2 treated cells eventually resumed proliferation but ~40% remained arrested, displaying persistent expression of the G1 phase marker and developing a senescent morphology (Movie S1). This pattern was enhanced by treating cells with NU-1 (Figure 3J and Movie S2) or CHRO (Movie S3), where ~80% of the surviving cells displayed G1 marker expression and senescent morphology at 6 days. These results confirm that even short-term TERT inhibition can potentiate radiation in promoting accelerated senescence, without waiting for telomere erosion.
Similar experiments were conducted in telomerase negative Saos-2 cells. Cells were assayed for clonogenic survival in the presence of NU-1 (1 μM), BIBR (20 μM), or DMSO control. Neither NU-1 nor BIBR displayed radiosensitization in Saos-2 cells (Figure S3A–C, Table S3). Treating Saos-2 cells with TERT inhibitors for 1 h before 0 or 6 Gy did not change recovery and/or proliferation compared to DMSO or NU-2 control (Figure S3D). Further, at 7 days after IR, ~33% of Saos-2 cells accumulated as SA-β-Gal+ cells with senescent morphology, irrespective of treatment with TERT inhibitors or control compounds (Figure S3E and F).
TERT inhibition delays chromosomal double strand break repair after irradiation
Prior studies have demonstrated persistent DNA damage in cells treated with telomerase inhibitors along with genotoxic stress, suggesting a direct mechanism of radiosensitization. MCF7 cells were treated with NU-2, NU-1, CHRO, BIBR, or MST for 1 h and then irradiated with 0 or 6 Gy, incubated 24 h (roughly one cell cycle for unperturbed MCF7 cells (Sutherland et al., 1983)), and examined by immunofluorescence for 53BP1 and γH2AX foci as markers for persistent DSBs. Unirradiated cells, whether treated with TERT inhibitors or not, yielded a similar average of ~5 foci per nucleus (Figure 4A and C). However, at 24 h after 6 Gy, persistent 53BP1 and γH2AX foci were increased after treatment with TERT inhibitors compared to controls (Figure 4B and D). Cells treated with as low as 100 nM or as high as 1 μM CHRO or NU-1 displayed a similar pattern (Figure S4A and B). To distinguish whether the presumptive chromosomal DSBs might instead be telomere dysfunction induced foci (TIFs), telomeres were costained along with γH2AX. A similar apparent colocalization of ~2% was observed whether cells were treated with 0 or 6 Gy and TERT inhibitors or controls (Figure 4E–G).
Figure 4. TERT inhibition induces persistent DNA damage foci and delays double-strand break repair after irradiation in telomerase positive cells.
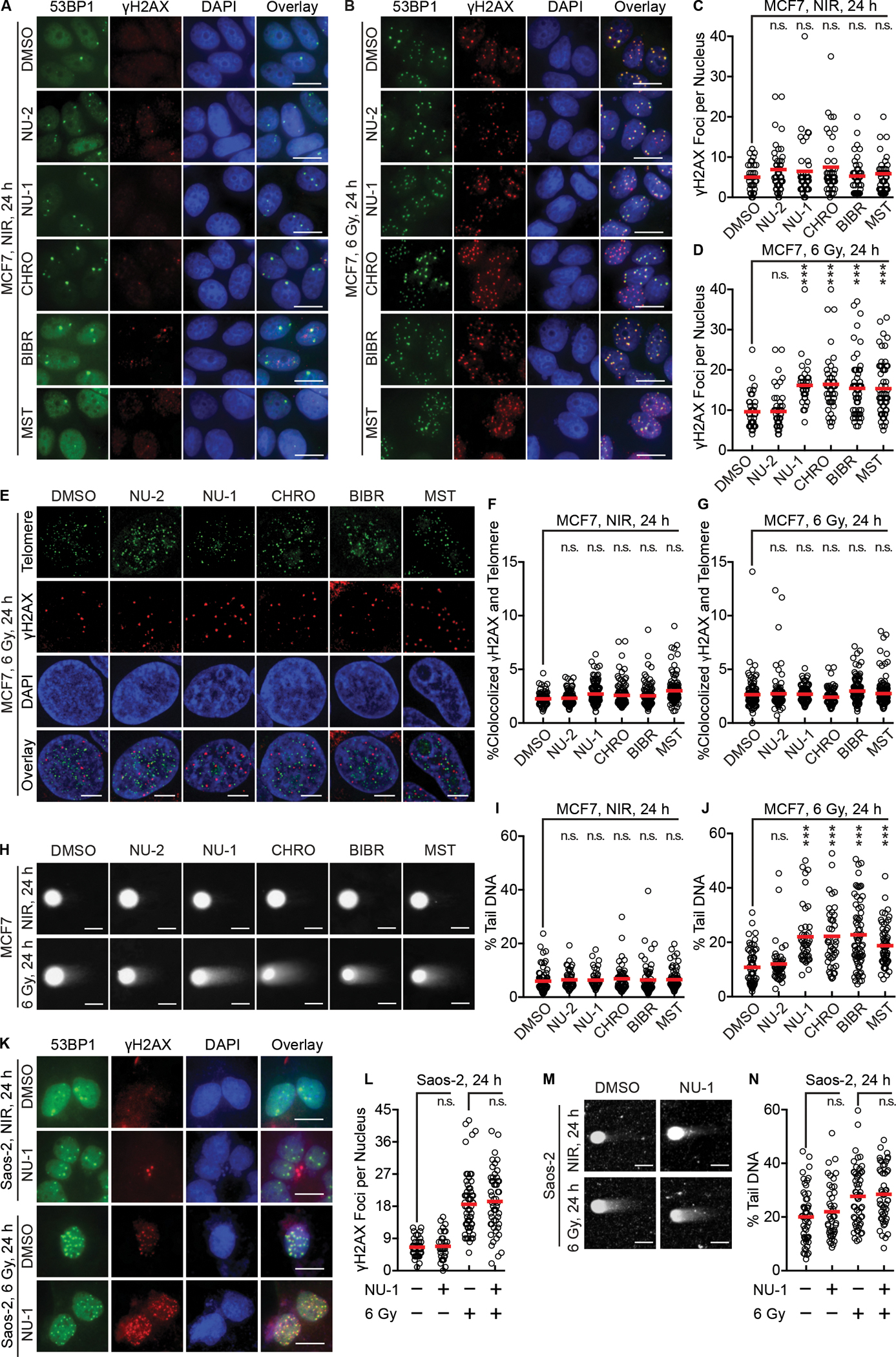
(A and B) Representative pseudo-colored images of staining for DNA damage foci markers 53BP1 (green) or γH2AX (red), DAPI (blue), and a three-color overlay. MCF7 cells were treated with DMSO, NU-2 (0.5 μM), NU-1 (0.5 μM), CHRO (0.5 μM), BIBR (10 μM), or MST (1 μM) for 1 h, followed by 0 (NIR, A) or 6 Gy (B), then fixed and stained after 24 h. Scale bars=20 μm.
(C and D) Quantification of γH2AX foci of cells in A (C) and B (D).
(E) Representative pseudo-colored staining images of cells treated as in B with 6 Gy showing telomere probe (green), γH2AX (red), DAPI (blue), and a three-color overlay. Scale bars=5 μm.
(F and G) Quantification analysis of telomere and γH2AX colocalization after 0 (F) or 6 Gy (G).
(H) Neutral comet assay of MCF7 cells treated as in A and B. Representative images demonstrate “comet tails”. Scale bars=20 μm.
(I and J) Quantification of comet assay results after 0 (I) or 6 Gy (J). % tail DNA indicates proportional of unrepaired chromosomal DSBs.
(K) ALT Saos-2 cells were treated with DMSO or NU-1 (1 μM) for 1 h, then 0 or 6 Gy, fixed and stained after 24 h. Shown are representative images. Scale bars=20 μm.
(L) Quantification of γH2AX foci of cells in K.
(M) Saos-2 cells treated as in H were examined by neutral comet assay. Representative images are shown. Scale bar=20 μm.
(N) Quantification of comet assay results in M. For quantification analysis, >50 cells were analyzed. Shown are individual cells (open circles) and mean (red bar).
*** P < 0.001; ** 0.001 < P < 0.01; n.s. P > 0.05 compared to DMSO (unpaired t-test).
See also Figure S4.
Although the 53BP1 and γH2AX foci serve as useful proxies for DSBs, multiple conditions can uncouple foci persistence from DSB repair (Liu et al., 2019). Thus, we used single-cell electrophoresis (neutral comet assay) to directly evaluate DNA damage in MCF7 cells, where chromosomal fragments form a “comet tail” whose length and intensity correspond to the number of DSBs (Olive and Banáth, 2006). Consistent with foci staining, TERT inhibitors did not increase % tail DNA above background in unirradiated cells (Figure 4H and I). However, in cells examined 24 h after 6 Gy, TERT inhibition significantly enhanced % tail DNA, indicating a defect in DSB repair (Figure 4H and J). In turn, increased % tail DNA in a neutral comet assay cannot be explained by damage to telomeres alone. To examine the cell cycle distribution of damage, the data were plotted against nuclear DNA content (Schindelin et al., 2012), using DAPI for γH2AX foci and total DNA for comet assay. The irradiated cells displayed a high apparent G1 DNA content, as predicted by flow cytometry, but no clear bias in the distribution of persistent foci or unrepaired DSBs (Figure S4C–E).
To evaluate TERT dependence, we similarly examined effects of NU-1 on DSB persistence in telomerase-negative Saos-2 cells. After treating Saos-2 cells with DMSO or 1 μM NU-1 and 0 or 6 Gy, NU-1 appeared not to impact 53BP1 or γH2AX foci on its own or when combined with IR (Figure 4K and L). In turn, neutral comet assays revealed similar levels of unrepaired DSBs with or without NU-1 (Figure 4M and N). Together, these results suggest that the effects of NU-1 that delay DSB repair after irradiation are directly mediated by TERT inhibition.
TERT inhibition targets non-homologous end-joining repair
Radiation-induced DSBs are heterogeneous, occur throughout the cell cycle, and can be repaired by multiple mechanisms, with the majority rejoined by conventional non-homologous end joining (NHEJ) or homologous recombination (HR) (Scully et al., 2019). NHEJ predominates throughout the cell cycle (Mao et al., 2008) and must be partly suppressed to allow HR repair during S and G2 phases, when sister chromatids are available as repair templates (Arnoult et al., 2017). While the pattern of G1 cell cycle arrest and DSB persistence induced by NU-1 point to impacts on NHEJ, we directly examined the impacts of TERT inhibition on DSB repair pathway choice using the Traffic Light Repair Reporter, where repair of an I-Sce-induced DSB by NHEJ results in expression of mCherry while template-directed HR repair leads to expression of eGFP (Figure 5A and S2B) (Certo et al., 2011). After treating with TERT inhibitors or DMSO control, the apparent NHEJ/HR ratio in MCF7 cells was significantly decreased, reflecting reduced NHEJ repair without a significant change to HR (Figure 5B). In telomerase-positive (Akıncılar et al., 2015) 293T human embryonic kidney cells, an even greater decrease in NHEJ/HR ratio was observed upon TERT inhibition (Figure 5C), reflecting not only reduced NHEJ but increased HR, confirming the specificity of the effect. Taken together, our results suggest that targeting TERT leads to slower chromosomal DSB rejoining after radiation due to an NHEJ repair defect.
Figure 5. TERT inhibition targets the non-homologous end-joining DSB repair pathway.
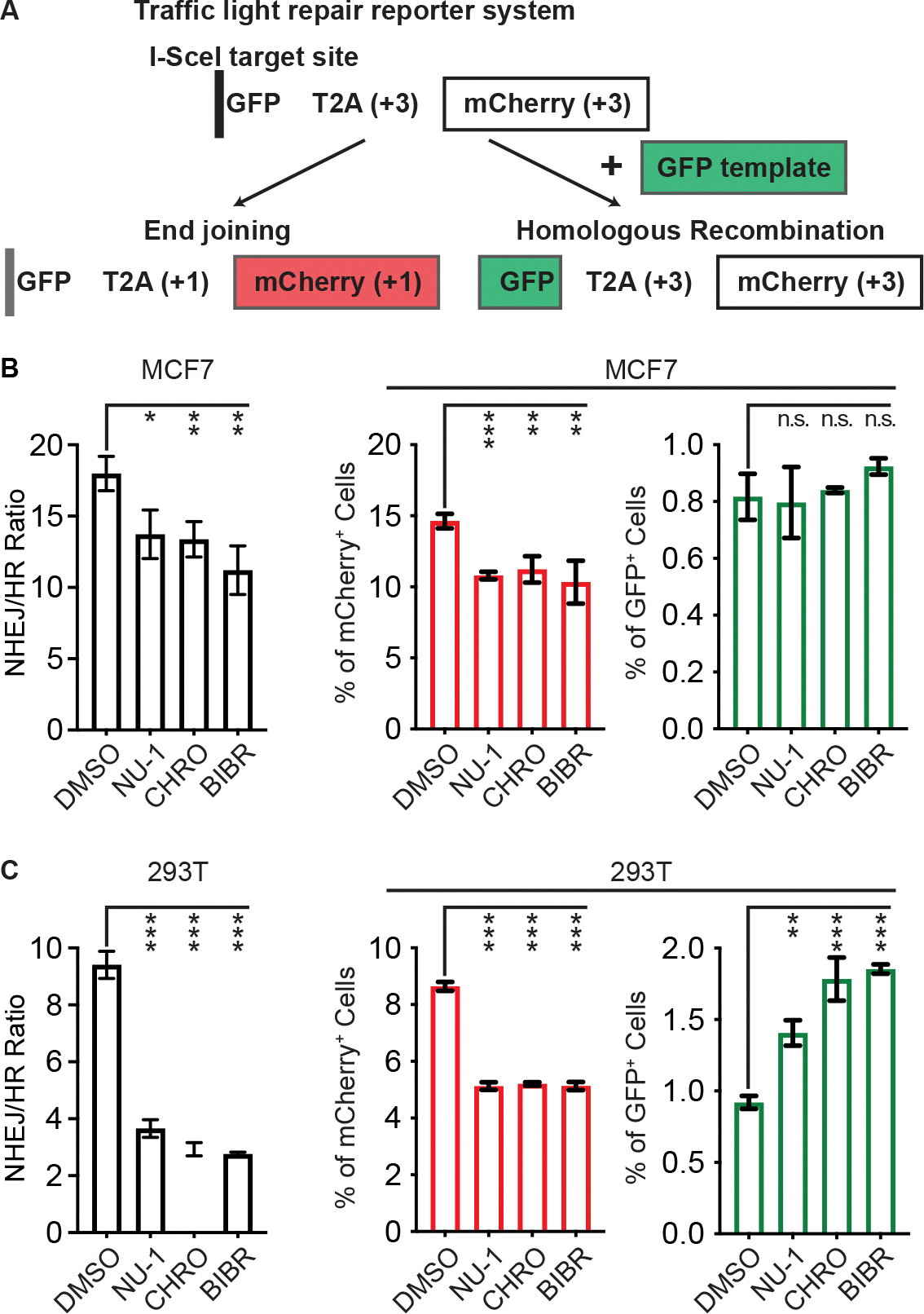
(A) Diagram of the Traffic Light repair reporter system. Repair of an I-Sce-induced DSB in individual cells by end-joining (NHEJ) versus homologous recombination (HR) results in expression of mCherry or GFP, detected by flow cytometry.
(B and C) The ratio of NHEJ to HR (left) and the quantification of mCherry+ or GFP+ cells among single cell population (right) in MCF7 (B) and 293T cells (C). Data from three replicates, mean ± SD.
*** P < 0.001; ** 0.001 < P < 0.01; * 0.01< P < 0.05 compared to DMSO treatment (unpaired t-test).
NU-1 sensitizes CT26 tumors to radiation and enhances anti-tumor immunity
Genetic or pharmaceutical inhibition of TERT can radiosensitize human tumor xenografts in athymic nude mice (Berardinelli et al., 2017). Toward enabling in vivo studies in an immunocompetent host, we assessed NU-1 effects on the BALB/c-derived colon carcinoma cell line CT26. 1 μM NU-1 and 2 μM MST were tolerated in a clonogenic assay while BIBR was non-toxic even at 20 μM, consistent with its known inactivity on murine TERT (Pascolo et al., 2002) (Figure S5A). Non-toxic doses of NU-1 and MST sensitized CT26 cells to radiation and enhanced senescence induction by IR while BIBR had no similar effects (Figure S5B–D, Table S3). After confirming that NU-1 was well-tolerated by BALB/c mice, we then evaluated the growth of subcutaneous CT26 tumors treated with daily intraperitoneal (IP) injections of NU-1 on Days 9 to 13 after tumor inoculation, with or without a single 10 Gy radiation dose on Day 11 (Figure 6A). NU-1 had no appreciable effect on its own while IR produced a moderate growth delay (Figure 6B, C, S6A and B). Tumors treated with IR + NU-1 displayed a marked growth delay, with 7 of 8 tumors being eliminated within two weeks after treatment. Once tumors were cleared, they did not recur (Figure S6C). Histology analysis of tumors removed on Day 18 revealed marked tissue destruction and loss of cellularity after IR + NU-1 treatment while immunofluorescence revealed lower expression of proliferation marker Ki-67 and upregulation of persistent γH2AX compared to IR alone (Figure 6D, E, and S6D).
Figure 6. NU-1 confers immunogenic radiation sensitization that leads to tumor elimination.
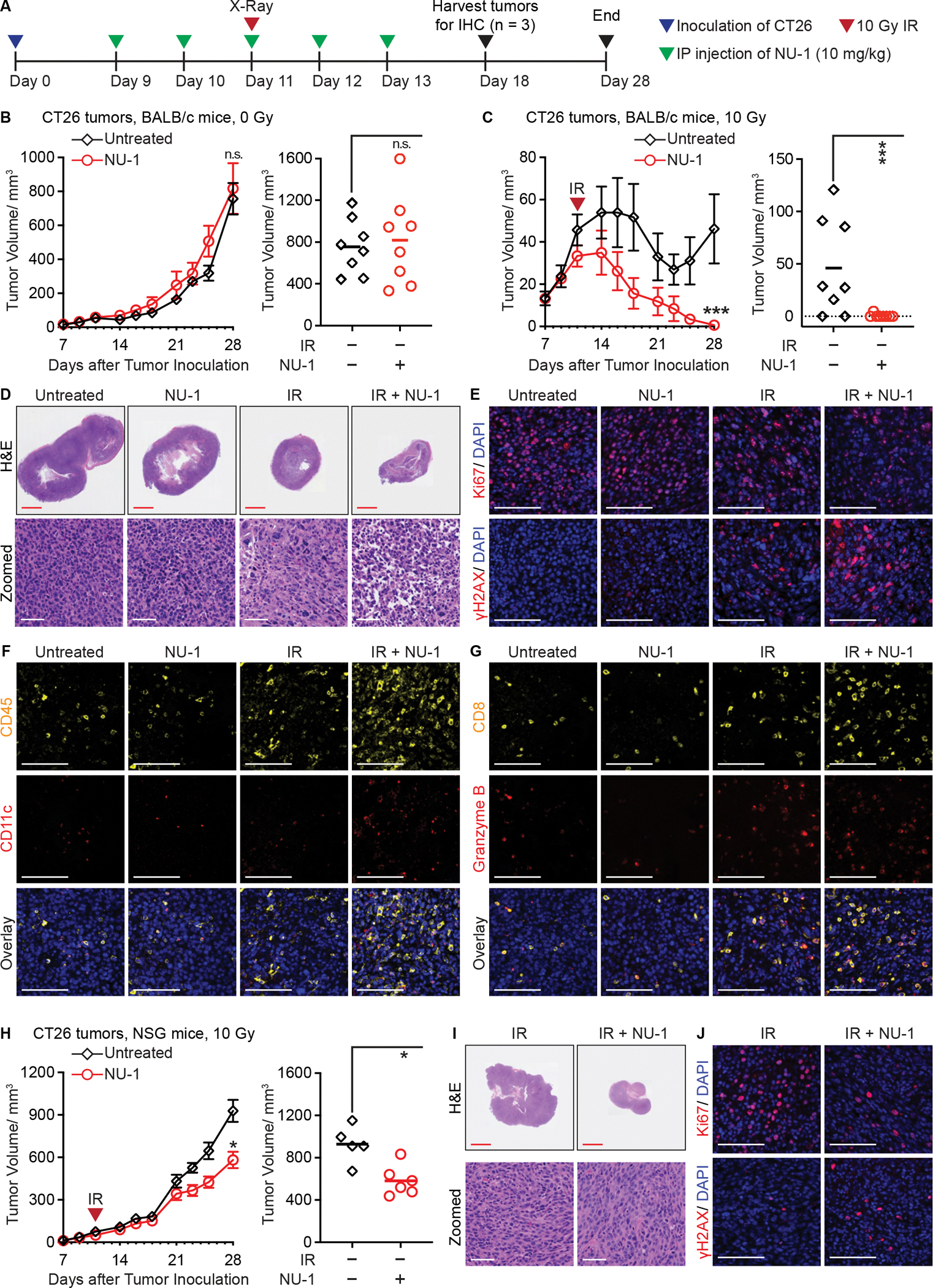
(A) Experimental schema for treating mice bearing CT26 subcutaneous tumors.
(B and C) Tumor growth in BALB/c mice treated with NU-1 alone (B) or in combination with 10 Gy irradiation (IR) (C). Shown are tumor growth kinetics (left, mean ± SEM) and tumor volumes on Day 28 (right, individual volume and mean).
(D) Hematoxylin and eosin (H&E) staining of tumor sections collected on Day 18. Shown are representative whole section scanning (scale bar=2.5 mm) and selected enlarged regions (scale bar=60 μm).
(E-G) Representative pseudo-colored images of staining for Ki67 (red) or γH2AX (red) (E), or CD45 (yellow) and CD11c (red) (F), or CD8 (yellow) and Granzyme B (red) (G), overlaid with DAPI (blue). Serial sections with D were used. Scale bars=20 μm.
(H) Tumor growth in NSG mice treated with IR ± NU-1. Shown are tumor growth kinetics (left, mean ± SEM) and tumor volumes on Day 28 (right, individual volume and mean).
(I) Representative H&E staining examples of tumor sections collected from NSG mice on Day 18. Scale bar=2.5 mm (upper) or 60 μm (lower).
(J) Representative images of staining for Ki67 (red) or γH2AX (red), overlaid with DAPI (blue). Scale bars=20 μm.
*** P < 0.001, * P < 0.05, n.s. P > 0.05 (unpaired t-test).
See also Figure S6.
The elimination of tumors by combined NU-1 and radiation treatment suggested the potentiation of anti-tumor immune response. Therefore, we probed the serial tumor sections to detect CD45+ immune infiltrate, CD11c+ dendritic cells (DCs), CD8+ cytotoxic T lymphocytes (CTLs) and cytotoxic protease granzyme B. IR + NU-1 treated tumors displayed markedly higher tumor immune infiltration, including increased DCs (Figure 6F) along with higher levels of CTLs, many of which appeared activated (CD8+/granzyme B+, Figure 6G). This pattern is consistent with immunogenic radiosensitization by NU-1, enabling an effective anti-tumor immune response. To examine the role of adaptive immunity further, CT26 tumors formed in immunodeficent NSG mice lacking mature B, T or NK cells were treated with IR alone or combined with NU-1, again following the schema in Figure 6A. Unlike CT26 tumors in wildtype BALB/c mice, IR alone failed to induce tumor regression or prolonged growth delay in NSG mice. While concomitant treatment with NU-1 enhanced growth delay in NSG mice (Figure 6H and S6E) and decreased tumor cell proliferation and increased DNA damage response (Figure 6I, J, and S6F), the compound effect was no longer sufficient to eliminate tumors.
Senescent CT26 cells formed by TERT inhibition and radiation activate DCs to prime T cells
A prerequisite for priming CTL-mediated immune response is for DCs to present tumor antigens in the context of the major histocompatibility complex (MHC) (Sánchez-Paulete et al., 2017). To model the process in vitro, we treated CT26 cells with DMSO, NU-1, or MST for 5 days and with 0 or 10 Gy and cocultured them overnight with bone marrow-derived CD11c+/CD103+ dendritic cells (BMDCs) (Mayer et al., 2014) (Figure 7A). Overall, co-culture with senescent irradiated CT26 cells promoted DC surface expression of CD86, CD80, and H-2Kd Class I MHC (Figure 7B and S2C), corresponding to DC maturation/activation. The TERT inhibitors further increased the level of H-2Kd, which may be limiting for CD8+ T cell stimulation.
Figure 7. TERT inhibition and irradiation induces immunogenic senescent CT26 cells capable of stimulating DC function via STING signaling.
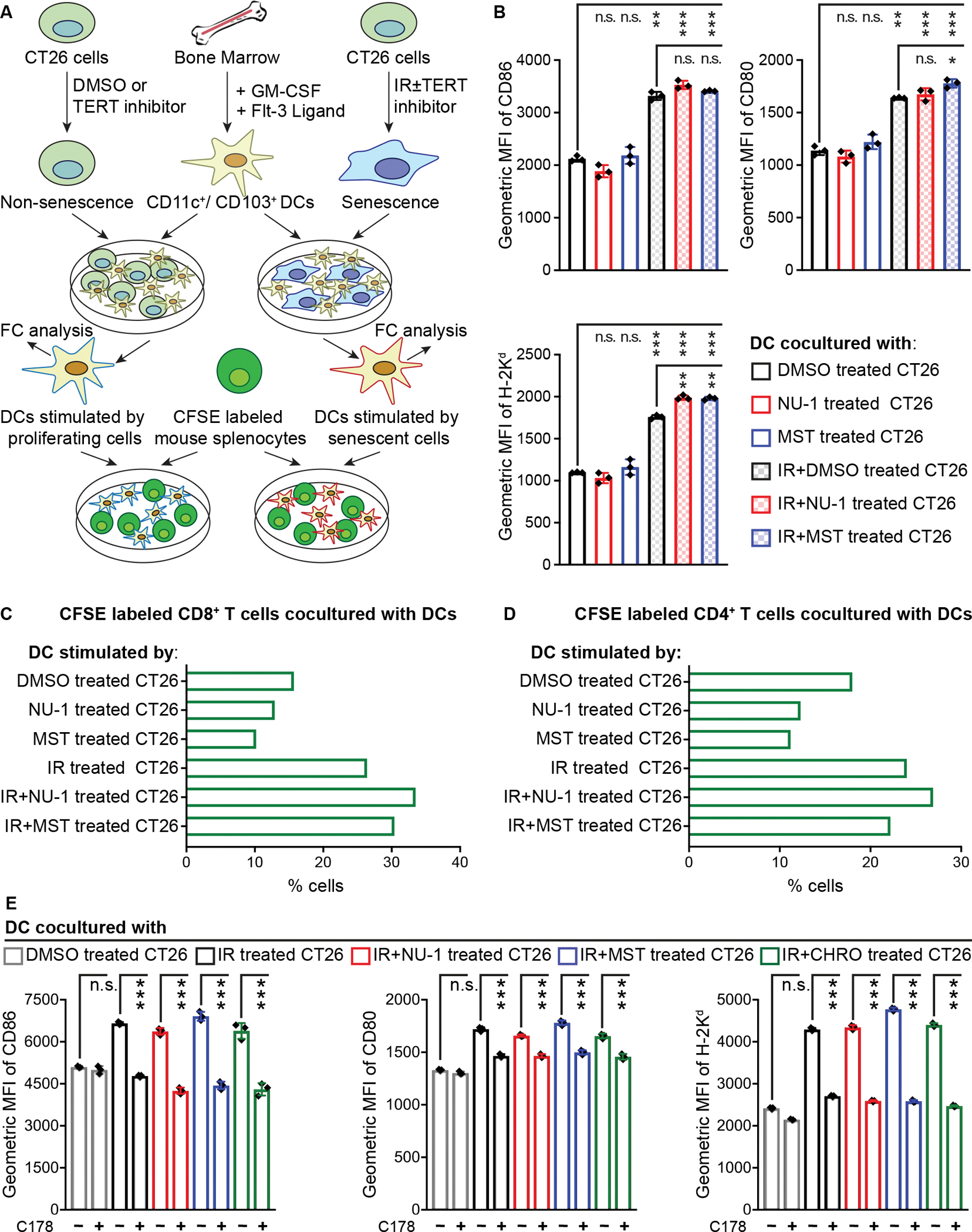
(A) Experimental schema for forming and stimulating BMDCs with CT26 cells and assaying APC function.
(B) Quantitative analysis of DC activation/maturation. CT26 cells treated with DMSO, NU-1, or MST-312 ± IR, incubated 5 days in culture, and combined with BMDCs overnight. Data obtained from live CD11c+/CD103+ DC population in triplicates, mean ± SD.
(C and D) Proliferative rate of live CD8+/CD4− (C) and CD8−/CD4+ (D) T cell population. CFSE labeled murine splenocytes were cocultured for 5 days with DCs pre-stimulated by CT26 cells treated with DMSO, NU-1, or MST ± IR.
(E) CT26 cells treated with indicated compounds ± 10 Gy IR and cocultured with BMDCs as in B. Data from three experiments, mean ± SD. MFI, mean fluorescence intensity.
*** P < 0.001, ** 0.001 < P < 0.01, n.s. P > 0.05 (unpaired t-test).
See also Figures S7.
To examine DC APC function, we combined BMDCs prestimulated by CT26 cells with splenocytes that were obtained from CT26 immunized mice and labeled with carboxyfluorescein succinimidyl ester (CFSE) (Quah et al., 2007). CFSE dilution was examined after 5 days by flow cytometry (Figure S2D). DCs stimulated by unirradiated CT26 cells, treated with TERT inhibitors or not, drove proliferation of 10–16% of CD8+ T cells. DCs co-cultured with senescent CT26 cells increased proliferating CD8+ cells to 26.4% while NU-1 or MST treated and irradiated CT26 cells yielded 33.5% and 30.4% proliferating CD8+ T cells respectively (Figure 7C and S7A). BMDCs cocultured with senescent CT26 cells also induced more CD4+ T cell proliferation, though with little appreciable impact from TERT inhibitors (Figure 7D and S7B). Overall, the pattern confirms the potential for combining NU-1 and radiation to promote a cytotoxic T cell-mediated anti-tumor immune response.
Persistent chromosomal damage and other triggers of senescence can lead to cytoplasmic DNA accumulation and activation of cGAS-STING signaling (Hopfner and Hornung, 2020), an innate immune pathway that drives DC activation and function (Yum et al., 2019). To explore interactions between TERT and STING, we treated proliferating CT26 cells with the covalent STING inhibitor C178 (Haag et al., 2018) prior to IR with or without TERT inhibitors. C178 did not prevent irradiated cells from entering senescence (Figure S7C) but reduced stimulation of DCs to that of unirradiated controls (Figure 7E). These results implicate cytoplasmic DNA sensing and STING activation as mediators of DC activation by senescent cells, but leave open how TERT inhibition may further stimulate anti-tumor immune response.
Discussion
The expression of TERT in adult humans is generally limited to stem and progenitor cells in continuously proliferative tissues (Roake and Artandi, 2020), including lymphoid tissue and bone marrow along with epithelial tissue (Uhlén et al., 2015). A majority of human cancers constitutively express TERT, often resulting from promoter mutations and/or epigenetic changes, and many at comparatively high levels, with a negative impact on overall survival (Wang et al., 2018). A long-standing assumption has been that TERT serves a primary role in maintaining telomere length to support unlimited cell division during tumor growth. Hopes to exploit this potential Achilles heel have long driven development and translation of telomerase inhibitors, with most efforts focused on TERT (Shay and Wright, 2019). Limiting proliferative potential is clearly attractive as a strategy to deplete cancer stem cells and limit recurrence or metastasis but the need for continuous telomerase suppression may incur dose-limiting toxicities such as cytopenias. Of the few agents that remain under clinical development, Imetelstat displays activity in myelofibrosis and myelodysplastic syndrome in Phase II studies (Mascarenhas et al., 2021; Steensma et al., 2021), though the mechanism of action may not depend on telomere erosion (Armanios and Greider, 2015).
As an alternative, multiple TERT inhibitors have been shown to potentiate the effects of radiation or chemotherapy agents (Arndt and MacKenzie, 2016; Berardinelli et al., 2017), apparently independently of telomere length. Indeed, TERT performs multiple non-canonical functions beyond telomere maintenance that contribute to cancer cell survival, proliferation, and immortality (Low and Tergaonkar, 2013; Thompson and Wong, 2020). While cancer cell lines can display TERT addiction and often lose viability upon TERT inhibition in vitro, short-term telomerase inhibition has not demonstrated a similar therapeutic effect on its own in vivo. However, TERT may serve multiple roles in mediating resistance to genotoxic cancer therapy. TERT’s catalytic activity appears to contribute to protection from genotoxic stress (Fleisig et al., 2016; Poynter et al., 2009) via multiple mechanisms, including its mitochondrial functions (Ahmed et al., 2008; Singhapol et al., 2013). Of particular relevance here, TERT has long been implicated in chromosomal DNA repair (Sharma et al., 2003; Shin et al., 2004), though whether this reflects catalytic activity at sites of DNA damage or at telomeres and/or enhanced DNA repair gene expression has remained poorly defined.
In recent work (Betori et al., 2020), we reported NU-1 as an irreversible TERT inhibitor developed via chemical simplification of the natural product chrolactomycin. Docking NU-1 to the tcTERT crystal structure (Mitchell et al., 2010) or the hTERT cryo-EM model (Ghanim et al., 2021) reveals multiple interactions that favor covalent interaction between the NU-1 exomethylene and a conserved cysteine in the primer grip. While an irreversible, covalent inhibitor such as NU-1 might enable long-term, uninterrupted suppression of TERT as may be required to force telomere erosion, this would affect not only tumors but also proliferating normal tissue stem and progenitor cells. Instead, we have explored using NU-1 for short intervals which may expose TERT addiction and/or sensitize tumors to ionizing radiation (IR), potentially allowing tumors to be targeted while sparing other tissues.
Even when applied to cells only one hour prior to IR, NU-1 significantly enhances the effects of radiation in vitro. Cells display persistent DNA damage signaling and delayed DSB repair, leading to prolonged cell cycle arrest and accelerated senescence. The slower DSB repair appears to reflect a specific defect in NHEJ without a similar impact on HR. A direct role in NHEJ is consistent with early studies that implicated TERT in end-joining and the rapid phase of chromosomal DSB repair (Masutomi et al., 2005; Sharma et al., 2003; Shin et al., 2004). Beyond implications for cancer therapy, these results reinforce earlier studies (e.g. (Pirzio et al., 2004)) that raised concerns about using hTERT-immortalized cell lines as “normal cells” for studies of DNA damage response.
Overall, NU-1 may provide a valuable probe to explore DNA repair and/or other non-canonical functions of TERT toward overcoming resistance to conventional therapies in telomerase-positive cancers. A caveat is that catalytic inhibition with NU-1 may have little or no impact on enzyme-independent roles of TERT, such as in protein-protein interactions (Perera et al., 2019) and gene transcription (Koh et al., 2015), which may also contribute to therapy resistance. Nonetheless, NU-1 appeared to impact expression of cell cycle and DNA damage response pathway genes even in undamaged cells, suggesting that TERT catalytic activity might also influence transcription, even if indirectly.
Our most striking results came from examining radiosensitization in vivo. Using CT26 colon carcinoma flank tumors in syngeneic BALB/c mice as an immunogenic tumor model (Lechner et al., 2013), short-term treatment with NU-1 was neither toxic nor effective on its own but markedly enhanced the effects of radiation. Radiation alone induced regression and growth delay, but the combination led to tumor elimination. Notably, there was a greater accumulation of DCs and activated CTLs in combination-treated tumors. A key role for adaptive immunity was consistent with the decreased impact of NU-1 on radiation response of CT26 tumors formed in NSG immunodeficient mice, despite similarly impacting tumor cell proliferation and persistent DNA damage. Modeling the radiosensitization in vitro, coculturing immature DCs with tumor cells driven into senescence by TERT inhibition and irradiation resulted in DC maturation and activation. These DCs displayed increased cytotoxic T cell priming, indicating competence as antigen-presenting cells. A favorable interpretation is that targeting TERT with NU-1 potentiates an effective anti-tumor immune response by promoting immunogenic senescence (Meng et al., 2012) in irradiated tumors and thereby boosting radiation-induced in situ vaccination (Wennerberg et al., 2017).
A prolonged DNA damage response (DDR) was observed both in vitro and in vivo when cells were treated with combined radiation and NU-1, providing a potential link to anti-tumor immunity via cytoplasmic DNA sensing, immunogenic cell death and/or other mechanisms (Chabanon et al., 2021). Considering ongoing efforts to leverage the DDR to enhance immunotherapy, TERT is a promising target, not only given its extra-telomeric roles in DNA repair as studied here but also via activity at telomeres that may impact cGAS-STING-mediated inflammatory signaling (Chen et al., 2017). This latter pathway was recently leveraged to target a cytotoxic T cell response to telomerase-positive tumors in mice using 6-thio-dG to damage telomeres (Mender et al., 2020). Whether localized to chromosome arms or telomeres, unrepairable DNA damage will promote tumor cells to display DAMPs and release inflammatory mediators. In particular, DCs exposed to cancer cells with DNA damage increase surface expression of MHC and costimulatory molecules, enhancing T cell priming capacity (Rad et al., 2003). Indeed, we observed a pattern of persistent DNA damage and infiltration of DCs and activated cytotoxic T cells in sections of tumors treated with NU-1 and radiation. However, TERT’s roles in protecting tumors from radiation and anti-tumor immune response may go beyond DNA repair to involve other non-canonical functions that modulate cell stress response and promote survival. Looking forward, NU-1 and other chrolactomycin analogs can provide useful tools to dissect mechanisms linking telomerase reactivation to tumor immune evasion and to validate TERT as a tumor-specific target to potentiate conventional, targeted or immune therapies.
Limitations of the study
Insofar as the small molecule TERT inhibitors used in this study target TERT catalytic activity, this work does not address enzyme-independent functions that may also affect the response to DNA damage and other cell stresses. This work also leaves open the molecular mechanism of how TERT may promote double strand break repair and does not establish whether the repair defect after TERT inhibition determines the increased immunogenicity of senescent cells. Without additional studies with NU-1 as a chemical probe for TERT in vitro and in vivo, its radiosensitizing effects cannot be ascribed solely to TERT inhibition. While this study implicates dendritic cells and cytotoxic T cells in the enhanced immune response after treatment with NU-1 and radiation, other immune cell types and signals are likely to have an important role. Finally, independent of NU-1, the translational significance of implicating TERT in tumor immune evasion merits further genetic and chemical analysis.
Significance
The telomerase reverse transcriptase (TERT) is required to maintain telomere length for high proliferative demands, but also supports extra-telomeric functions, serving as a significant contributor to intrinsic resistance in the majority of tumors. Despite the lack of efficacy of targeting telomere maintenance and concern about adverse effects of long-term telomerase inhibition, blocking TERT in the short-term to impact stress response and survival may offer a route to tumor-specific sensitization to therapy. This study took advantage of our prior work that developed the covalent TERT inhibitor NU-1. We applied NU-1 alongside other inhibitors to reexamine the roles of TERT in protecting cancer cells and tumors from chemotherapy and radiation. TERT inhibitors synergize with chemotherapy to reduce the viability of cancer cells in vitro. Inhibiting TERT catalytic activity disrupts DNA repair and promotes G1 cell cycle arrest and senescence after radiation. Blocking TERT slows repair by non-homologous end-joining without affecting homologous recombination, setting it apart from other candidate radiosensitizers. Previous studies have documented radiosensitization by TERT inhibitors in vivo, but limited to human xenograft tumors in immunodeficient mice. Here, we demonstrated potent radiosensitization by NU-1 in syngeneic tumors in immune competent hosts. We attributed this not only to increased DNA damage and impacts on cell survival and proliferation, but also to unleashing an adaptive immune response that led to tumor elimination. Suggesting a direct role for TERT in immune evasion, treating tumor cells with TERT inhibitors and radiation forms senescent cells that promote DC activation and T-cell priming. Taken together, our results indicated that TERT is a highly druggable target for tumor-specific immunogenic radiosensitization.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephen J. Kron (skron@uchicago.edu).
Materials availability
This study did not generate new unique reagents.
Data and Code Availability
The RNA sequencing data are available on Sequence Read Archive (SRA): PRJNA663346.
This study did not generate unique code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MCF7 (a breast cancer cell line from a 69-year-old white female in 1970), Saos-2 (an osteosarcoma cell line from an 11-year-old white female in 1973), and CT26 (a colorectal carcinoma cell line from a female BALB/c mouse) cells were obtained from ATCC. 293T (an immortalized cell line exhibiting epithelial morphology isolated from female fetal kidney in 1973) and MCF7 stably expressing the tetracycline-regulated transactivator Tet-On Advanced were obtained from Takara. MCF7-FUCCI cell line with FUCCI cell cycle reporter constructs was reconstructed here by transduction with lentivirus expressing mVenus-hGeminin (1/110)/pCSII-EF-MCS and mCherry-hCdt1 (30/120)/Pcsll-EF-MCS (a kind gift of Atsushi Miyawaki, RIKEN Center for Brain Science, Japan) (Sakaue-Sawano et al., 2008). Cells with positive expression were selected by fluorescence-activated cell sorting (FACS). The cells were maintained at 37°C and 5% CO2 in DMEM containing 4.5 g/l glucose (Thermo Fisher Scientific) supplemented with 10% Tet-approved FBS (Atlanta Biologicals) and 1% penicillin/streptomycin (Thermo Fisher Scientific). The cells were tested for mycoplasma contamination and authenticated by a short tandem repeat profile (IDEXX BioResearch) prior to performing experiments. All experiments were performed within 3 to 10 passages after thawing cells.
Mice
BALB/c wildtype mice were purchased from Envigo. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, NSG mice were originally purchased from The Jackson Laboratory and bred inhouse. Mice of both sexes were used, with no sex difference observed. Naive healthy mice aged 7–9 weeks with an average weight of 20 g were used and maintained according to the guidelines of the Institutional Animal Care and Use Committee. In experiments involving tumor growth, the tumor bearing mice were randomly divided into groups of at least 5 for each treatment condition. No animals died before the final day shown for each experiment except for those sacrificed due to their tumors reaching a humane endpoint. All animal studies were performed as approved by the IACUC of the University of Chicago.
Bone marrow dendritic cells (BMDCs)
Bone marrow dendritic cells (BMDCs) were differentiated as previously described (Mayer et al., 2014). Briefly, bone marrow was isolated from 7–8 week female BALB/c mice, maintained at 37°C and 5% CO2 in basic murine immune cell culture medium supplemented with 1 ng/mL mouse recombinant GM-CSF (PeproTech) and 200 ng/mL mouse recombinant Flt-3 ligand (PeproTech) for 14 days to form immature BMDCs. The basic murine immune cell culture medium was RPMI medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated FBS (Thermo Fisher Scientific), 1% penicillin/streptomycin (Thermo Fisher Scientific), and 50 μM β-mercaptoethanol (Thermo Fisher Scientific). All animal studies were performed as approved by the IACUC of the University of Chicago.
Splenocytes from CT26 cell immunized BALB/c mice
CT26 immunized mice were prepared by subcutaneously injecting 7–9 week female BALB/c mice with 0.5 × 106 irradiated (20 Gy) CT26 cells twice at 10 day intervals. 5 days after the second injection, spleens were isolated from mice, transferred under cell culture hood, quickly immersed in 70% ethanol for 20–30 s for sterilization, then immediately transferred onto the 70 μm strainer in 10 mL cold PBS in 10-cm petri dish. Spleens were then meshed, and the cell strainer was rinsed with 10 mL PBS. The cell suspension was transferred into a 50 mL tube and pelleted by centrifuging for 5 min at 300 g. Red blood cells were lysed using RBC Lysis Buffer (BioLegend, Cat# 420301) according to the manufacturer’s protocol. After washing, the splenocytes were ready for the downstream application. All animal studies were performed as approved by the IACUC of the University of Chicago.
METHOD DETAILS
Chemical probes
Chrolactomycin was isolated from Actinospica (a kind gift of M. Iorio at NAICONS Scrl, Milan, Italy) (Iorio et al., 2012) and NU-1 and the des-exomethylene analog NU-2 were synthesized and purified as described (Betori et al., 2020). BIBR 1532 and MST-312 were obtained from Cayman Chemical. Irinotecan, doxorubicin, etoposide, paclitaxel, and C178 were obtained from Selleck Chemicals.
RNA sequencing analysis
3 × 104/flask MCF-7 cells were seeded in a T25 flask, cultured overnight, and treated with 0.5 μM NU-1 or DMSO for 48 h. Media was removed, cells were washed 2 times with PBS, followed by chemical disassociation with TrypLe (Thermo Fisher Scientific). Media was added to quench the TrypLe, the cell suspension was centrifuged at 300g for 5 min, then the supernatant were removed and the cell pellets were immediately froze in −80 °C. The frozen cell pellets were shipped in cold shipping box with dry ice to Applied Biological Materials for RNA isolation, sequencing, reads alignment and gene abundance estimation. Briefly, RNA was isolated using Trizol (Thermo Fisher Scientific). The quality of the RNA extraction was assessed by gel electrophoresis. All the samples passed internal QC for library preparation. Sequencing libraries were prepared using Illumina TruSeq Stranded mRNA Library Preparation kit following the manufacturer’s recommendations. The quality of the libraries was assessed using Qubit DNA assay, Agilent Bioanalyzer, and qPCR. All libraries passed internal QC. Sequencing was performed with an Illumina NextSeq system. After sequencing, the paired-end reads were aligned with the hg38 human reference genome by Hisat2. The abundance of genes was quantified by Htseq-count. Analysis of differentially expressed genes (DEGs) between NU-1 treated and control cells was performed with EdgeR in R studio in house, with dispersion value estimated as 0.01. Genes were considered significantly upregulated if they displayed fold change > 1.5 and P value < 0.1 and significantly downregulated if fold change < −1.5 and P value < 0.1, comparing NU-1 treated and control cells. The DEG lists were subjected to GO analysis by Reactome (Fabregat et al., 2018), DAVID (Huang da et al., 2009), and Ingenuity Pathway Analysis (IPA) (Krämer et al., 2014) to detect enriched pathways.
Chemosensitization studies
1 × 104/well MCF-7, A549, or Saos-2 cells were seeded in 96-well plates (black wall, flat clear bottom, Corning 3916). Cells were allowed to adhere overnight, then the old medium was replaced by fresh medium containing 0.5 μM NU-1, 0.5 μM NU-2, 10 μM BIBR 1532, 1 μM MST-312 or DMSO vehicle. After incubation for 4 h, DMSO stock solutions were diluted to a final concentration of 0.001, 0.01, 0.1, 1, 10, 100 μM for irinotecan, 0.001, 0.01, 0.1, 1, 10, 100 μM for etoposide, 1, 3, 10, 32, 100, 316, 1000 μM for doxorubicin, or 1, 2.5, 6, 16, 40, 100, 316, 1000 μM for paclitaxel. The final volume of cell culture medium in each well was 100 μL. Then the cells were continuously incubated for 24 h at 37°C and 5% CO2. After that, cell viability was measured by CellTiter-Glo assay (Promega) according to the manufacturer’s protocol. Briefly, after equilibrating plates to room temperature for 30 min, 100 μL of freshly prepared CellTiter-Glo reagent was added to each well, mixed on an orbital shaker for 10 min and luminescence was recorded on a PerkinElmer Enspire multimode plate reader. Data from three independent biological replicates were acquired.
For co-administration chemosensitization studies, 1 × 104 MCF-7 cells were seeded in 96-well plates and allowed to adhere overnight. Then the old medium was replaced by 100 μL fresh medium containing 0.5 μM NU-1 or DMSO + 0.001, 0.01, 0.1, 1, 10, 100 μM irinotecan. Cells were continuously cultured for 24 h at 37°C and 5% CO2. After that, the plates were collected for CellTiter-Glo assay. For post-administration chemosensitization studies, MCF7 cells were seeded and adhered in 96-well plates overnight as mentioned above. Then the old medium was replaced with fresh medium containing 0.001, 0.01, 0.1, 1, 10, 100 μM irinotecan. Cells were incubated for 4 h, followed by addition of either 0.5 μM NU-1 or DMSO and the final volume of cell culture medium in each well brought to 100 μL. Then the plates were incubated for another 24 h before CellTiter-Glo assay. Data from three independent biological replicates were acquired.
Nonlinear regression with variable slope was used to draw the dose response curve and calculate the Lethal Dose 50 for cells (LD50) using GraphPad Prism software. Towards quantitatively measuring the extent of drug interaction, a combination index (CI) was calculated using CompuSyn software (Chou and Martin, 2005). As reported previously, the LD50 of NU-1 in MCF7 and A549 cells is 21 μM and 35 μM respectively, while undetectable in Saos-2 cells (Betori et al., 2020).
Clonogenicity assays
MCF7, Saos-2, or CT26 cells were seeded in 6-well plates at 100 cells per well in triplicate and allowed to attach overnight. MCF7 or CT26 cells were then treated with DMSO vehicle or TERT inhibitors in final concentration of 0.25, 0.5, 0.75, 1, 2 μM for NU-1, 2.5, 5, 7.5, 10, 20 μM for BIBR 1532, or 1, 2, 3, 4, 5 μM for MST-312. Saos-2 cells were treated with DMSO vehicle or TERT inhibitors in final concentration of 0.25, 0.5, 0.75, 1, 2 μM for NU-1 or 2.5, 5, 10, 20, 40 μM for BIBR 1532. The final volume of cell culture medium was 3 mL per well. MCF7 and Saos-2 cells were continuously cultured for 3 weeks and CT26 for 2 weeks in the presence of TERT inhibitors, fixed in methanol for 5 min, then stained with 0.5% crystal violet for 10 min at room temperature. After washing with water, the plates were dried at room temperature and imaged with an iBright Imaging System (Thermo Fisher Scientific).
To assess radiosensitivity, MCF7, Saos-2, or CT26 cells were seeded in 6-well plates at 100 cells per well in triplicate, allowed to attach overnight and treated with DMSO or TERT inhibitors at subtoxic concentrations for 1 h prior to IR. MCF7 cells were treated with 0.5 μM NU-1, 10 μM BIBR 1532, or 1 μM MST-312, Saos-2 cells with 1 μM NU-1 or 20 μM BIBR 1532 and CT26 cells with 1 μM NU-1, 20 μM BIBR 1532 or 2 μM MST-312. Doses of 0, 1, 2, 3, 4, or 5 Gy were applied using a GammaCell 60Co γ-ray source (Nordion) at a dose rate of 7.09 cGy/sec. After that MCF7 and Saos-2 cells were cultured for 3 weeks and CT26 for 2 weeks, then fixed and stained with crystal violet. Colonies of at least 50 cells were counted. Data from three independent biological replicates were acquired. The surviving fraction (SF) was calculated using the following formula:
Time-lapse live-cell analysis of cell proliferation
MCF7 cells were seeded in 6-well plates at 3 × 104 cells per well, recovered overnight, then treated with DMSO, NU-2 (0.5 μM), NU-1 (0.5 μM), chrolactomycin (0.5 μM), BIBR 1532 (10 μM), or MST-312 (1 μM) for 1 h and irradiated with 0 or 6 Gy using the 60Co γ-ray source at 7.09 cGy/sec. Saos-2 cells were similarly seeded and cultured and then treated with DMSO vehicle, NU-1 (1 μM), or BIBR 1532 (20 μM) for 1 h, followed by 0 or 6 Gy IR. Immediately after IR, the plates were analyzed by time-lapse imaging in an IncuCyteS3 (Sartorius) live-cell imaging system. Phase contrast channel images were acquired at 20X magnification with scanning every 2 h for 6–7 days. 25 non-overlapping fields were captured for each well. Quantitative analysis of cell confluency was performed using IncuCyteS3 2019 software.
Apoptosis analysis
MCF7 cells were seeded in 6-well plates with 3 × 104 cells per well, allowed to attach, then treated with DMSO, NU-2 (0.5 μM), NU-1 (0.5 μM) or chrolactomycin (0.5 μM) and irradiated with 0 or 6 Gy. 7 days after IR, cells were incubated with 1 μM YO-PRO-1 iodide (Thermo Fisher Scientific) for 30 min at 37° C and imaged in the IncuCyteS3. Phase contrast and green channel images were acquired at 20X magnification. 25 non-overlapping fields were captured for each well. Quantitative analysis of cell confluency was performed using IncuCyteS3 2019 software.
Senescence-associated beta-galactosidase (SA-β-Gal) assay
MCF7, Saos-2, or CT26 cells were seeded at 3 × 104 per well in 6-well plates and allowed to attach to plate overnight. For TERT inhibition, MCF7 cells were treated with DMSO vehicle, NU-2 (0.5 μM), NU-1 (0.5 μM), chrolactomycin (0.5 μM), BIBR 1532 (10 μM), or MST-312 (1 μM) for 1 h prior to IR, Saos-2 cells were treated with DMSO vehicle, NU-2 (1 μM), NU-1 (1 μM), chrolactomycin (1 μM), BIBR 1532 (20 μM), or MST-312 (1 μM) for 1 h prior to IR, and CT26 cells were treated with MSO vehicle, NU-2 (1 μM), NU-1 (1 μM), chrolactomycin (1 μM), BIBR 1532 (20 μM), or MST-312 (2 μM) for 1 h prior to IR. Then MCF7 and Saos-2 cells were irradiated with 0 or 6 Gy and CT26 cells with 0 or 10 Gy. After IR, MCF7 and Saos-2 cells were continuously cultured for 7 days and CT26 for 5 days, then fixed in 2% PFA and stained with staining buffer (1 mg/ml X-Gal (Golden Bio), 40 mM citric acid/sodium phosphate, 150 mM NaCl, 2 mM MgCl2, 3.3 mM K3[Fe(CN)6], 3.3 mM K₄[Fe(CN)₆], pH=6) for 16 h at 37° C, then washed with warm PBS three times and imaged in PBS within a week. Images were captured on a Zeiss Axiovert 200M microscope with a 20X Plan-NeoFluar objective and Axiocam digital camera. SA-β-Gal-positive and -negative cells were counted in more than 5 fields, yielding an average percentage indicated on each SA-β-Gal image as mean ± SD. Three replicates were performed.
Flow cytometric analysis of cell cycle
MCF7 cells were seeded at 3 × 104 per well in 6-well plates, allowed to attach overnight, then treated with DMSO, NU-1 (0.5 μM), BIBR 1532 (10 μM), or MST-312 (1 μM) for 1 h before irradiation with 0 or 6 Gy. 24 h after IR, cells were trypsinized, fixed with 70% cold ethanol and stained with 1 μg/mL DAPI. Cells were analyzed using a BD Fortessa 4–15 HTS Flow cytometer. DAPI signal was measured using a 405-nm laser for excitation and a 450/50 filter for detection. Data were analyzed by FlowJo univariate cell cycle analysis. Three replicates were performed.
Time-lapse live-cell analysis of cell cycle
MCF7-FUCCI cells were seeded at 3 × 104 per well in 6-well plates, allowed to attach overnight, then treated with 0.5 μM NU-2 control, NU-1 or chrolactomycin for 1 h, followed by 6 Gy. Then, live-cell imaging was conducted using the IncuCyteS3. Phase contrast along with green and red channels were acquired at 20X magnification with scanning every 2 h for 6 days. 16 non-overlapping fields were captured for each well. Representative consecutive images were used to create the movies using Fiji (Schindelin et al., 2012).
DNA damage foci staining
MCF7 or Saos-2 cells were seeded on sterile cover glass at 2.5 × 104 per well in 24-well plates. MCF7 cells were treated with DMSO, NU-2 (0.5 μM), NU-1 (0.5 μM), chrolactomycin (0.5 μM), BIBR 1532 (10 μM), or MST-312 (1 μM) for 1 h, and Saos-2 cells were treated with DMSO or NU-1 (1 μM) for 1 h, followed by 0 or 6 Gy irradiation. 24 h after irradiation, cells were fixed with 4% PFA for 10 min and permeabilized with 0.2% Triton-X 100 diluted in PBS for 10 min at room temperature. After blocking with 5% BSA-PBS for 1 h, primary antibodies for γH2AX (Millipore, 05–636, 1:1000) or 53BP1 (Novus, NB100–304, 1:1000) diluted in 5% BSA-PBS were then incubated on cell slides overnight at 4°C. Following PBS washes, DAPI (1 μg/mL) and fluorescent secondary antibodies (Jackson ImmunoResearch, 1:2000,) diluted in 5% BSA-PBS were applied for 1 h at room temperature. Cell slides were mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific) after PBS washes. Foci images were captured on a Zeiss Axiovert 40CFL with a 40X Plan-NeoFluar objective and pseudo-colored using Fiji. Three replicates were performed.
Telomere PNA and γH2AX double staining
MCF7 cells were seeded onto sterile coverslips at 3 × 104 per well in 24-well plates and cultured overnight, then treated with DMSO, NU-2 (0.5 μM), NU-1 (0.5 μM), chrolactomycin (0.5 μM), BIBR 1532 (10 μM), or MST-312 (1 μM) for 1 h, followed by 0 or 6 Gy IR. 24 h after IR, cells were fixed with 2% PFA for 15 min at room temperature, permeabilized with 0.2% Triton-X100, blocked with 5% BSA-PBS, incubated with anti-γH2AX (Millipore, JBW301, 1:1000) antibodies overnight at 4°C and then 1 h with Alexa Fluor 647-Goat Anti-Mouse antibody (Jackson ImmunoResearch, Cat# 115–605-003) for DNA damage foci staining. After that, cells were washed with PBS three times and fixed again in 2% PFA 15 min at room temperature. Then peptide nucleic acid fluorescence in situ hybridization (PNA FISH) with Alexa488-TelG probe (PNA Bio, F1008) was performed according to the manufacturer’s protocol. Briefly, cells were incubated with 100 μg/ml RNase A solution and dehydrated in 70%, 85% and 100% cold ethanol for 2 min each. 0.5 μM PNA probe in 20 μL hybridization buffer (20 mM Tris, pH 7.4, 60% formamide, 0.5% of blocking reagent (Roche 11096176001)) was heated at 85°C for 5 min. The preheated staining solution was applied to cells at 85°C for 10 min for denaturation followed by overnight hybridization at room temperature in the dark. Cells were washed twice in washing solution (2XSSC/0.1% Tween-20) for 10 min at 55°C, incubated with 1 μg/mL DAPI for 10 min at room temperature, and then mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Images were taken using a 3i Marianas Spinning Disk Confocal with 100X oil objective and pseudo colored with Fiji. Detailed primary antibody information is provided in Key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | Cat#05-636, RRID:AB_309864 |
| 53BP1 Antibody | Novus | Cat#NB100-304, RRID:AB_10003037 |
| Brilliant Violet 421™ anti-mouse CD11c Antibody | BioLegend | Cat#117329, RRID:AB_10897814 |
| Brilliant Violet 711™ anti-mouse CD103 Antibody | BioLegend | Cat#121435, RRID:AB_2686970 |
| APC anti-mouse CD80 Antibody | BioLegend | Cat#104713, RRID:AB_313134 |
| PE/Cyanine7 anti-mouse CD86 Antibody | BioLegend | Cat#105013, RRID:AB_439782 |
| Alexa Fluor® 488 anti-mouse H-2Kd Antibody | BioLegend | Cat#116609, RRID:AB_493066 |
| Purified Rat Anti-Mouse CD45 | BD Biosciences | Cat#550539, RRID:AB_2174426 |
| CD11c (D1V9Y) Rabbit mAb | Cell Signaling Technology | Cat#97585, RRID:AB_2800282 |
| Granzyme B (E5V2L) Rabbit mAb (Mouse Specific) | Cell Signaling Technology | Cat#44153, RRID:AB_2857976 |
| CD8a Monoclonal Antibody (4SM15) | Thermo Fisher Scientific | Cat#14-0808-80, RRID:AB_2572860 |
| Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb | Cell Signaling Technology | Cat#9718, RRID:AB_2118009 |
| Anti-Ki67 antibody (ab15580) | Abcam | Cat#ab15580, RRID:AB_443209 |
| Bacterial and virus strains | ||
| Lentivirus | This paper | NA |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| NU-1 | (Betori et al., 2020) | NA |
| Chrolactomycin | (Iorio et al., 2012) | A kind gift of M. Iorio at NAICONS |
| BIBR 1532 | Cayman Chemical | Cat#16608 |
| MST-312 | Cayman Chemical | Cat#24301 |
| C-178 | Selleck Chemicals | Cat#25860 |
| Irinotecan | Selleck Chemicals | Cat#S1198 |
| Doxorubicin | Selleck Chemicals | Cat#E2516 |
| Etoposide | Selleck Chemicals | Cat#S1225 |
| Paclitaxel | Selleck Chemicals | Cat#S1150 |
| Alexa488-TelG probe | PNA Bio | Cat#F1008 |
| Recombinant Murine GM-CSF | PeproTech | Cat#315-03 |
| Recombinant Murine Flt3-Ligand | PeproTech | Cat#250-31L |
| Critical commercial assays | ||
| CometAssay Single Cell Gel Electrophoresis Assay | R&D systems | Cat#4250-050-K |
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | Cat#G9242 |
| CellTrace™ CFSE Cell Proliferation Kit, for flow cytometry | Thermo Fisher Scientific | Cat#C34554 |
| Deposited data | ||
| RNA sequencing data | This paper | SRA: PRJNA663346 |
| Experimental models: Cell lines | ||
| Human: MCF7 | ATCC | Cat#HTB-22 |
| Human: MCF7 Tet on | Takara | Cat#632108 |
| Human: Saos-2 | ATCC | Cat#HTB-85 |
| Human: A549 | ATCC | Cat#CCL-185 |
| Human: 293T | Takara | Cat# 632180 |
| Mouse: CT26.WT | ATCC | Cat#CRL-2638 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/cAnNHsd | Envigo | Cat#047 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NOD scid gamma (NSG)) | The Jackson Laboratory (Breeding in house) | JAX:005557 |
| Oligonucleotides | ||
| NA | ||
| Recombinant DNA | ||
| pCVL SFFV-EF1s HA.NLS.Sce | (Certo et al., 2011) | Addgene Plasmid #31479 |
| pCVL Traffic Light Reporter 1.1 (Sce target) Ef1a Puro | (Certo et al., 2011) | Addgene Plasmid #31482 |
| pCVL SFFV d14GFP Donor | (Certo et al., 2011) | Addgene Plasmid #31475 |
| mVenus-hGeminin (1/110)/pCSII-EF-MCS | (Sakaue-Sawano et al., 2008) | A kind gift of Atsushi Miyawaki at RIKEN Center for Brain Science, Japan |
| mCherry-hCdt1 (30/120)/pCSII-EF-MCS | (Sakaue-Sawano et al., 2008) | A kind gift of Atsushi Miyawaki at RIKEN Center for Brain Science, Japan |
| Software and algorithms | ||
| Fiji | (Schindelin et al., 2012) | https://imagej.github.io/software/fiji/#publication |
| ImageJ comet assay macro | https://www.med.unc.edu/microscopy/resources/imagej-plugins-and-macros/comet-assay/ | |
| GraphPad Prism | https://www.graphpad.com/ | |
| FlowJo | https://www.flowjo.com/ | |
| Olympus OlyVIA software | https://www.olympus-lifescience.com/en/discovery/image-sharing-made-easy-meet-olyvia/ | https://www.olympus-lifescience.com/en/support/downloads/ |
| IncuCyteS3 2019B | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-software/incucyte-software-v2019b | |
| CompuSyn | (Chou and Martin, 2005) | https://www.combosyn.com/ |
| Reactome | (Fabregat et al., 2018) | https://reactome.org/ |
| DAVID | (Huang da et al., 2009) | https://david.ncifcrf.gov/ |
| Ingenuity Pathway Analysis | (Krämer et al., 2014) | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Other | ||
| NA | ||
Neutral comet assay
For neutral comet assays, MCF7 or Saos-2 cells were seeded at 5 × 104 per well in 6-well plates. MCF7 cells were treated with DMSO, NU-2 (0.5 μM), NU-1 (0.5 μM), chrolactomycin (0.5 μM), BIBR 1532 (10 μM), or MST-312 (1 μM) for 1 h, and Saos-2 cells were treated with DMSO or NU-1 (1 μM) for 1 h, followed by 0 or 6 Gy. 24 h after IR, DNA damage was evaluated by CometAssay Single Cell Gel Electrophoresis (R&D Systems) according to the manufacturer’s protocol. Briefly, cells were trypsinized and resuspended in 1% FBS-PBS at a final concentration of 2 × 105 cells/mL. 20 uL single cell suspension was mixed with 200 uL Comet LM agarose prewarmed at 37°C and transferred onto CometSlides. The CometSlides were incubated at 4°C for 20 min in the dark, immersed in prechilled Lysis Solution for 1 h at 4°C, equilibrated with 1X Neutral Electrophoresis Buffer for 30 min at 4°C, and single cell electrophoresis was performed using fresh 1X Neutral Electrophoresis Buffer at 21 volts for 45 min at 4°C. After electrophoresis, slides were immersed in DNA Precipitation Solution and 70% ethanol for 30 min each at room temperature, then dried overnight at room temperature. Slides were stained with SYBR green (Thermo Fisher Scientific) and imaged on a Zeiss Axiovert 40CFL with a 10X Plan-NeoFluar objective. Two or more replicates were performed. Images were analyzed using an ImageJ comet assay macro (https://www.med.unc.edu/microscopy/resources/imagej-plugins-and-macros/comet-assay/).
Traffic Light Repair Reporter assay
Three constructs for traffic light repair reporter (TLR) system (Certo et al., 2011): pCVL SFFV-EF1s HA.NLS.Sce, pCVL Traffic Light Reporter 1.1 (Sce target) Ef1a Puro and pCVL SFFV d14GFP Donor were obtained from Addgene. Lentivirus-containing supernatant was produced by transfection of the 293T Lenti-X cell line with corresponding plasmids and packaged plasmid mix. Then lentivirus-containing supernatant was applied to MCF7 Tet-On (Takara) cell or 293T cell line. In the meantime, DMSO control, NU-1 (0.5 μM), chrolactomycin (0.5 μM) or BIBR 1532 (10 μM) were added to transfected MCF7 Tet-On cells or 293T cells. Cell were continuously cultured with the presence or absence of TERT inhibitors for 3 days, then collected and analyzed using a BD Fortessa 4–15 HTS Flow cytometer. eGFP fluorescence was measured using a 488-nm laser for excitation and a 530/30 filter for detection, while mCherry fluorescence was measured by using a 561-nm laser for excitation and a 610/20 filter for detection. Data were analyzed using FlowJo software. At least three replicates were performed.
In vivo studies
For tumor inoculation, 7–9 week BALB/c mice or NSG mice were subcutaneously injected in the flank with 0.5 × 106 CT26 cells in 100 μL PBS. On Day 9 (tumor average volume ~ 30 mm3), the tumor bearing BALB/c mice were randomly divided into 4 groups of 8, while the NSG mice were randomly divided into 2 groups of 5–6. For NU-1 treatment, 10 mg/kg of NU-1 in 100 μL PBS was injected intraperitoneally once on each of the 2 days before, the day of and 2 days after irradiation. Mouse weight was measured before treatment. Tumor irradiation was conducted on Day 11 using a RadSource RS-2000 X-Ray generator operating at 160 kV and 25 mA at 10 Gy, calibrated by NIST traceable dosimetry. From Day 7 after tumor inoculation, tumors were measured by calipers every 2–3 days and volume calculated using the formula: Tumor volume (mm3) = length (mm) × width (mm) × width (mm)/2. On Day 18 after tumor inoculation, tumors from 3 mice under each treatment condition were collected for histology analysis. The rest of the mice were continuously maintained and recorded until at least Day 28 or reaching the humane endpoint for tumor volume. No animals that formed tumors after injection with CT26 cells were excluded from any studies reported here.
Histology and immunofluorescence analysis on tumor sections
CT26 tumors were collected 5 days after treatment (Day 18 after tumor inoculation) and fixed in 10% neutral formalin for 24 h. Formaldehyde-fixed paraffin-embedded (FFPE) tumors were sectioned and stained with hematoxylin and eosin (H&E) by the Human Tissue Resource Center at The University of Chicago. To perform immunofluorescence analysis, tumor sections were deparaffinized in xylene for 10 min, then rehydrated with 95%, 70%, 50%, and 0% ethanol for 5 min each. After that, the tumor section was immersed in 10 mM sodium citrate buffer (pH 6.0) for 30 min at 90° C for antigen retrieval. Then the tumor sections were blocked in 5% BSA-TBS, stained with anti-Ki67 (1:1000), anti-γH2AX (1:500), anti-CD45 (1:500), anti-CD11c (1:250), anti-CD8 (1:250), and anti-granzyme B (1:250) primary antibodies diluted in 5% BSA-TBS at 4°C overnight. Followed by washing with TBST, tumor sections were stained with fluorophore-conjugated secondary antibodies (1:2000, Vector Labs) and DAPI (1 μg/mL) diluted in 5% BSA-TBS for 90 min at room temperature. The H&E stained tissue sections were scanned using an Olympus VS200 SlideView Whole Slide Scanner. The fluorescent tumor sections were imaged using a 3i Marianas Spinning Disk Confocal with a 20X objective and pseudo-colored using Fiji. Detailed primary antibody information is provided in Key resources table.
In vitro DC activation assays
To evaluate effects of TERT on tumor immunogenicity, CT26 cells were seeded at 2 × 104 per well in 12-well plates, allowed to attach overnight, and then treated with DMSO, NU-1 (1 μM), or MST-312 (2 μM) for 1 h, followed by 0 or 10 Gy. 5 days after IR, the media were removed, cells washed with PBS, and fresh immune cell culture medium added. CT26 cells were then cultured overnight, followed by coculturing with immature BMDCs for 12–16 h at a 1:2 ratio. After coculture, the suspension BMDCs were collected into round bottom FACS tubes, washed with PBS, and stained with Zombie yellow (1:1000, BioLegend) for 10 min at room temperature. After washing with PBS, the cells were blocked for 10 min on ice with TruStain FcX anti-mouse CD16/32 (1:100, BioLegend) diluted in Cell Staining Buffer, followed by staining with CD11c (1:200), CD103 (1:200), CD86 (1:200), CD80 (1:200), and H-2Kd (1:200) antibodies for 45 min at 4°C. After PBS washing, cells were resuspended in 300 μL Cell Staining Buffer and analyzed using a BD Fortessa 4–15 HTS Flow cytometer within 2 hours. Data were then analyzed by FlowJo software. Detailed antibody information is provided in Key resources table.
To examine roles for STING, CT26 cells were seeded at 2 × 104 per well in 12-well plates and allowed to attach overnight. STING inhibitor C178 (0 or 4 μM) was added, followed by the addition of DMSO vehicle, NU-1 (1 μM), MST-312 (2 μM) or chrolactomycin (1 μM) for 1 h prior to 0 or 10 Gy. CT26 cells were cultured for 5 days, surviving cells were washed twice in PBS and cultured in fresh immune cell culture medium overnight. The CT26 cells were then cocultured with BMDCs for 12–16 h at a 1:2 ratio, and BMDCs were collected and analyzed after coculturing, as described above.
In vitro T cell priming assays
Splenocytes isolated from CT26 immunized mice were stained with 0.5 μM carboxyfluorescein succinimidyl ester (CFSE) (Thermo Fisher Scientific, Cat# C34554) for 10 min at room temperature (Quah et al., 2007). After washing with medium twice, CFSE labeled splenocytes were co-cultured for 5 days in a 20:1 ratio with BMDCs pre-stimulated by CT26 cells as DC activation assays. After coculturing, cells were collected and incubated with Zombie yellow for 10 min at room temperature, followed by PBS washing and blocking with TruStain FcX anti-mouse CD16/32 (1:100, BioLegend). After that, cells were stained with CD4 and CD8 antibodies for 30 min at 4°C. Cells were analyzed using a BD Fortessa 4–15 HTS flow cytometer and FlowJo software. Detailed antibody information is provided in Key resources table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using the non-paired Student’s t-test. Calculations were performed using GraphPad Prism software or Excel. P ≤ 0.05 was considered statistically significant. Power analysis for animal experiments was performed using G*Power software, power (1-β) > 0.85 for all animal experiments. Details on the sample size and p value of independent experiments can be found in the Figure Legends.
Supplementary Material
Movie S1 MCF7-FUCCI cell growth after NU-2 + IR, Related to Figure 3
Movie S2 MCF7-FUCCI cell growth after NU-1 + IR, Related to Figure 3
Movie S3 MCF7-FUCCI cell growth after CHRO + IR, Related to Figure 3
Telomerase inhibition promotes cellular senescence after irradiation. MCF7 cells expressing FUCCI cell cycle indicators (mCherry-hCdt1, red fluorescent G1 reporter; mVenus-hGeminin, green fluorescent S/G2 reporter) were treated with chrolactomycin, NU-1, or NU-2 then with 6 Gy and incubated in an IncuCyte for time-lapse imaging at 2 h intervals for 6 days. In the sample treated with the inactive analog NU-2 (0.5 μM) and 6 Gy (Movie S1), the majority of surviving cells resumed proliferation while a fraction was arrested with the characteristic senescent morphology of enlarged, flattened cells. Presumptive senescent cells were red fluorescent, suggesting a G1 cell cycle arrest. For samples treated with NU-1 (0.5 μM) (Movie S2) or CHRO (chrolactomycin, 0.5 μM) (Movie S3) and then 6 Gy, most cells arrested with a senescent morphology, expressing red fluorescence. Scale bar=100 μm.
Highlights.
Telomerase reverse transcriptase protects cells from chemotherapy and radiation
Inhibiting TERT delays the repair of DSBs mainly through targeting NHEJ
Inhibiting TERT promotes G1 cell cycle arrest and senescence after radiation
TERT inhibitor NU-1 promotes antitumor immunity after radiation
Acknowledgements
These studies were supported by NIH R01 CA217182 to K.A.S. and S.J.K. Additional support to S.J.K. came from NIH R01 CA199663 and the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. Additional funds were provided to S.B.C. by Ernest & Piroska Major Foundation and T.M.B. by Hill Foundation (Australia). This work utilized service cores supported by the University of Chicago Comprehensive Cancer Center support grant P30 CA014599 and the Northwestern University Comprehensive Cancer Center support grant P30 CA060553. We thank Julian Lutze for help with colocalization analysis and our colleagues for helpful support.
Footnotes
Declaration of interests
Y.L., R.C.B., G.B.F., K.A.S., and S.J.K. are co-inventors of IP owned by Northwestern University and the University of Chicago and co-founders and/or co-owners of Riptide Therapeutics, a spin-out from Northwestern University and the University of Chicago, related to applications of chrolog TERT inhibitors in cancer therapeutics. R.C.B. is an employee of Corteva Agriscience. S.J.K. is a co-founder of OncoSenescence.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, and Saretzki G (2008). Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. Journal of cell science 121, 1046–1053. [DOI] [PubMed] [Google Scholar]
- Akıncılar SC, Low KC, Liu CY, Yan TD, Oji A, Ikawa M, Li S, and Tergaonkar V (2015). Quantitative assessment of telomerase components in cancer cell lines. FEBS letters 589, 974–984. [DOI] [PubMed] [Google Scholar]
- Akiyama M, Yamada O, Kanda N, Akita S, Kawano T, Ohno T, Mizoguchi H, Eto Y, Anderson KC, and Yamada H (2002). Telomerase overexpression in K562 leukemia cells protects against apoptosis by serum deprivation and double-stranded DNA break inducing agents, but not against DNA synthesis inhibitors. Cancer Lett 178, 187–197. 10.1016/s0304-3835(01)00838-2. [DOI] [PubMed] [Google Scholar]
- Armanios M, and Greider CW (2015). Treating Myeloproliferation--On Target or Off? N Engl J Med 373, 965–966. 10.1056/NEJMe1508740. [DOI] [PubMed] [Google Scholar]
- Arndt GM, and MacKenzie KL (2016). New prospects for targeting telomerase beyond the telomere. Nat Rev Cancer 16, 508–524. 10.1038/nrc.2016.55. [DOI] [PubMed] [Google Scholar]
- Arnoult N, Correia A, Ma J, Merlo A, Garcia-Gomez S, Maric M, Tognetti M, Benner CW, Boulton SJ, Saghatelian A, and Karlseder J (2017). Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 549, 548–552. 10.1038/nature24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardinelli F, Coluzzi E, Sgura A, and Antoccia A (2017). Targeting telomerase and telomeres to enhance ionizing radiation effects in in vitro and in vivo cancer models. Mutat Res Rev Mutat Res 773, 204–219. 10.1016/j.mrrev.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Betori RC, Liu Y, Mishra RK, Cohen SB, Kron SJ, and Scheidt KA (2020). Targeted Covalent Inhibition of Telomerase. ACS Chem Biol 15, 706–717. 10.1021/acschembio.9b00945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boike L, Henning NJ, and Nomura DK (2022). Advances in covalent drug discovery. Nat Rev Drug Discov. 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, and Scharenberg AM (2011). Tracking genome engineering outcome at individual DNA breakpoints. Nature methods 8, 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, and Postel-Vinay S (2021). Targeting the DNA damage response in immuno-oncology: developments and opportunities. Nat Rev Cancer 21, 701–717. 10.1038/s41568-021-00386-6. [DOI] [PubMed] [Google Scholar]
- Chen YA, Shen YL, Hsia HY, Tiang YP, Sung TL, and Chen LY (2017). Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat Struct Mol Biol 24, 1124–1131. 10.1038/nsmb.3498. [DOI] [PubMed] [Google Scholar]
- Chou T, and Martin N (2005). CompuSyn for drug combinations: PC software and user’s guide: a computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values. ComboSyn, Paramus, NJ. [Google Scholar]
- de Lange T (2018). Shelterin-Mediated Telomere Protection. Annu Rev Genet 52, 223–247. 10.1146/annurev-genet-032918-021921. [DOI] [PubMed] [Google Scholar]
- Ding X, Cheng J, Pang Q, Wei X, Zhang X, Wang P, Yuan Z, and Qian D (2019). BIBR1532, a Selective Telomerase Inhibitor, Enhances Radiosensitivity of Non-Small Cell Lung Cancer Through Increasing Telomere Dysfunction and ATM/CHK1 Inhibition. Int J Radiat Oncol Biol Phys 105, 861–874. 10.1016/j.ijrobp.2019.08.009. [DOI] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res 46, D649–D655. 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisig HB, Hukezalie KR, Thompson CA, Au-Yeung TT, Ludlow AT, Zhao CR, and Wong JM (2016). Telomerase reverse transcriptase expression protects transformed human cells against DNA-damaging agents, and increases tolerance to chromosomal instability. Oncogene 35, 218–227. 10.1038/onc.2015.75. [DOI] [PubMed] [Google Scholar]
- Ganesan K, and Xu B (2017). Telomerase Inhibitors from Natural Products and Their Anticancer Potential. Int J Mol Sci 19. 10.3390/ijms19010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanim GE, Fountain AJ, van Roon AM, Rangan R, Das R, Collins K, and Nguyen THD (2021). Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 593, 449–453. 10.1038/s41586-021-03415-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytisolo FA, Samper E, Martín-Caballero J, Finnon P, Herrera E, Flores JM, Bouffler SD, and Blasco MA (2000). Short telomeres result in organismal hypersensitivity to ionizing radiation in mammals. J Exp Med 192, 1625–1636. 10.1084/jem.192.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW, and Blackburn EH (1985). Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43, 405–413. 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Guterres AN, and Villanueva J (2020). Targeting telomerase for cancer therapy. Oncogene 39, 5811–5824. 10.1038/s41388-020-01405-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, and Ablasser A (2018). Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273. 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, and Gryaznov SM (2005). Lipid modification of GRN163, an N3’-->P5’ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 24, 5262–5268. 10.1038/sj.onc.1208760. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, and Hornung V (2020). Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21, 501–521. 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Iorio M, Maffioli SI, Gaspari E, Rossi R, Mauri P, Sosio M, and Donadio S (2012). Chrolactomycins from the actinomycete actinospica. J Nat Prod 75, 1991–1993. 10.1021/np300470f. [DOI] [PubMed] [Google Scholar]
- Kim MM, Rivera MA, Botchkina IL, Shalaby R, Thor AD, and Blackburn EH (2001). A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci U S A 98, 7982–7987. 10.1073/pnas.131211098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, Li Y, Franzoso G, Li S, Guccione E, and Tergaonkar V (2015). Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest 125, 2109–2122. 10.1172/jci79134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Kondo S, Tanaka Y, Haqqi T, Barna BP, and Cowell JK (1998). Inhibition of telomerase increases the susceptibility of human malignant glioblastoma cells to cisplatin-induced apoptosis. Oncogene 16, 2243–2248. 10.1038/sj.onc.1201754. [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr, and Tugendreich S (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy E, Goetz V, Erlacher M, Herz C, and Mersch-Sundermann V (2013). hTERT: another brick in the wall of cancer cells. Mutat Res 752, 119–128. 10.1016/j.mrrev.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, and Epstein AL (2013). Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother 36, 477–489. 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Crothers J, Haqq CM, and Blackburn EH (2005). Cellular and gene expression responses involved in the rapid growth inhibition of human cancer cells by RNA interference-mediated depletion of telomerase RNA. J Biol Chem 280, 23709–23717. 10.1074/jbc.M502782200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Efimova EV, Ramamurthy A, and Kron SJ (2019). Repair-independent functions of DNA-PKcs protect irradiated cells from mitotic slippage and accelerated senescence. J Cell Sci 132. 10.1242/jcs.229385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low KC, and Tergaonkar V (2013). Telomerase: central regulator of all of the hallmarks of cancer. Trends in biochemical sciences 38, 426–434. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, and de Lange T (2017). Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18, 175–186. 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7, 2902–2906. 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas J, Komrokji RS, Palandri F, Martino B, Niederwieser D, Reiter A, Scott BL, Baer MR, Hoffman R, Odenike O, et al. (2021). Randomized, Single-Blind, Multicenter Phase II Study of Two Doses of Imetelstat in Relapsed or Refractory Myelofibrosis. J Clin Oncol 39, 2881–2892. 10.1200/jco.20.02864. [DOI] [PubMed] [Google Scholar]
- Massard C, Zermati Y, Pauleau AL, Larochette N, Metivier D, Sabatier L, Kroemer G, and Soria JC (2006). hTERT: a novel endogenous inhibitor of the mitochondrial cell death pathway. Oncogene 25, 4505–4514. 10.1038/sj.onc.1209487. [DOI] [PubMed] [Google Scholar]
- Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, and Hahn WC (2005). The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. U S A 102, 8222–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer CT, Ghorbani P, Nandan A, Dudek M, Arnold-Schrauf C, Hesse C, Berod L, Stüve P, Puttur F, and Merad M (2014). Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood, The Journal of the American Society of Hematology 124, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, Gryaznov S, Dikmen ZG, Wright WE, and Shay JW (2015). Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2’-deoxyguanosine. Cancer Discov 5, 82–95. 10.1158/2159-8290.CD-14-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, Zhang A, Ren Z, Han C, Deng Y, Siteni S, Li H, Zhu J, Vemula A, Shay JW, and Fu YX (2020). Telomere Stress Potentiates STING-Dependent Anti-tumor Immunity. Cancer Cell 38, 400–411.e406. 10.1016/j.ccell.2020.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Efimova EV, Hamzeh KW, Darga TE, Mauceri HJ, Fu YX, Kron SJ, and Weichselbaum RR (2012). Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol Ther 20, 1046–1055. 10.1038/mt.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell M, Gillis A, Futahashi M, Fujiwara H, and Skordalakes E (2010). Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol 17, 513–518. 10.1038/nsmb.1777. [DOI] [PubMed] [Google Scholar]
- Nakai R, Ishida H, Asai A, Ogawa H, Yamamoto Y, Kawasaki H, Akinaga S, Mizukami T, and Yamashita Y (2006). Telomerase inhibitors identified by a forward chemical genetics approach using a yeast strain with shortened telomere length. Chem Biol 13, 183–190. 10.1016/j.chembiol.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Oikawa S, and Kawanishi S (1999). Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett 453, 365–368. 10.1016/s0014-5793(99)00748-6. [DOI] [PubMed] [Google Scholar]
- Olive PL, and Banáth JP (2006). The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1, 23–29. 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I, Garin-Chesa P, Rettig WJ, Damm K, and Schnapp A (2002). Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J Biol Chem 277, 15566–15572. Doi 10.1074/Jbc.M201266200. [DOI] [PubMed] [Google Scholar]
- Perera ON, Sobinoff AP, Teber ET, Harman A, Maritz MF, Yang SF, Pickett HA, Cesare AJ, Arthur JW, MacKenzie KL, and Bryan TM (2019). Telomerase promotes formation of a telomere protective complex in cancer cells. Sci Adv 5, eaav4409. 10.1126/sciadv.aav4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirzio LM, Freulet-Marrière MA, Bai Y, Fouladi B, Murnane JP, Sabatier L, and Desmaze C (2004). Human fibroblasts expressing hTERT show remarkable karyotype stability even after exposure to ionizing radiation. Cytogenet Genome Res 104, 87–94. 10.1159/000077470. [DOI] [PubMed] [Google Scholar]
- Poynter KR, Sachs PC, Bright AT, Breed MS, Nguyen BN, Elmore LW, and Holt SE (2009). Genetic inhibition of telomerase results in sensitization and recovery of breast tumor cells. Mol Cancer Ther 8, 1319–1327. 10.1158/1535-7163.MCT-08-0849. [DOI] [PubMed] [Google Scholar]
- Quah BJ, Warren HS, and Parish CR (2007). Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nature protocols 2, 2049–2056. [DOI] [PubMed] [Google Scholar]
- Rad AN, Pollara G, Sohaib SA, Chiang C, Chain BM, and Katz DR (2003). The differential influence of allogeneic tumor cell death via DNA damage on dendritic cell maturation and antigen presentation. Cancer research 63, 5143–5150. [PubMed] [Google Scholar]
- Roake CM, and Artandi SE (2020). Regulation of human telomerase in homeostasis and disease. Nat Rev Mol Cell Biol 21, 384–397. 10.1038/s41580-020-0234-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Sánchez-Paulete A, Teijeira A, Cueto FJ, Garasa S, Pérez-Gracia JL, Sánchez-Arráez A, Sancho D, and Melero I (2017). Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Annals of Oncology 28, xii44–xii55. [DOI] [PubMed] [Google Scholar]
- Sanford SL, Welfer GA, Freudenthal BD, and Opresko PL (2020). Mechanisms of telomerase inhibition by oxidized and therapeutic dNTPs. Nat Commun 11, 5288. 10.1038/s41467-020-19115-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature reviews Molecular cell biology 20, 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségal-Bendirdjian E, and Geli V (2019). Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front Cell Dev Biol 7, 332. 10.3389/fcell.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimiya H, Oh-hara T, Suzuki T, Naasani I, Shimazaki T, Tsuchiya K, and Tsuruo T (2002). Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Mol Cancer Ther 1, 657–665. [PubMed] [Google Scholar]
- Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V, Iliakis G, Shay JW, Young CS, and Pandita TK (2003). hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 22, 131–146. 10.1038/sj.onc.1206063. [DOI] [PubMed] [Google Scholar]
- Shay JW (2016). Role of telomeres and telomerase in aging and cancer. Cancer discovery 6, 584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, and Wright WE (2019). Telomeres and telomerase: three decades of progress. Nat Rev Genet 20, 299–309. 10.1038/s41576-019-0099-1. [DOI] [PubMed] [Google Scholar]
- Shin KH, Kang MK, Dicterow E, Kameta A, Baluda MA, and Park NH (2004). Introduction of human telomerase reverse transcriptase to normal human fibroblasts enhances DNA repair capacity. Clin Cancer Res 10, 2551–2560. 10.1158/1078-0432.ccr-0669-3. [DOI] [PubMed] [Google Scholar]
- Singhapol C, Pal D, Czapiewski R, Porika M, Nelson G, and Saretzki GC (2013). Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS One 8, e52989. 10.1371/journal.pone.0052989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steensma DP, Fenaux P, Van Eygen K, Raza A, Santini V, Germing U, Font P, Diez-Campelo M, Thepot S, Vellenga E, et al. (2021). Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion-Burden Patients With Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J Clin Oncol 39, 48–56. 10.1200/jco.20.01895. [DOI] [PubMed] [Google Scholar]
- Sutherland RL, Hall RE, and Taylor IW (1983). Cell proliferation kinetics of MCF-7 human mammary carcinoma cells in culture and effects of tamoxifen on exponentially growing and plateau-phase cells. Cancer Res 43, 3998–4006. [PubMed] [Google Scholar]
- Thompson CAH, and Wong JMY (2020). Non-canonical Functions of Telomerase Reverse Transcriptase: Emerging Roles and Biological Relevance. Curr Top Med Chem 20, 498–507. 10.2174/1568026620666200131125110. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Pilger R, and Sitte N (2000). Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med 28, 64–74. 10.1016/s0891-5849(99)00207-5. [DOI] [PubMed] [Google Scholar]
- Wang K, Wang RL, Liu JJ, Zhou J, Li X, Hu WW, Jiang WJ, and Hao NB (2018). The prognostic significance of hTERT overexpression in cancers: A systematic review and meta-analysis. Medicine (Baltimore) 97, e11794. 10.1097/md.0000000000011794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sun C, Mao A, Zhang X, Zhou X, Wang Z, and Zhang H (2015). Radiosensitization to X-ray radiation by telomerase inhibitor MST-312 in human hepatoma HepG2 cells. Life Sci 123, 43–50. 10.1016/j.lfs.2014.12.027. [DOI] [PubMed] [Google Scholar]
- Wennerberg E, Lhuillier C, Vanpouille-Box C, Pilones KA, García-Martínez E, Rudqvist NP, Formenti SC, and Demaria S (2017). Barriers to Radiation-Induced In Situ Tumor Vaccination. Front Immunol 8, 229. 10.3389/fimmu.2017.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelhouse RT, Sun D, Han H, Han FX, and Hurley LH (1998). Cationic porphyrins as telomerase inhibitors: the interaction of tetra-(N-methyl-4-pyridyl) porphine with quadruplex DNA. Journal of the American Chemical Society 120, 3261–3262. [Google Scholar]
- Wong KK, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C, Artandi SE, Rudolph KL, Gottlieb GJ, Chin L, et al. (2000). Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet 26, 85–88. 10.1038/79232. [DOI] [PubMed] [Google Scholar]
- Yum S, Li MH, Frankel AE, and Chen ZJJ (2019). Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu Rev Canc Biol 3, 323–344. 10.1146/annurev-cancerbio-030518-055636. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1 MCF7-FUCCI cell growth after NU-2 + IR, Related to Figure 3
Movie S2 MCF7-FUCCI cell growth after NU-1 + IR, Related to Figure 3
Movie S3 MCF7-FUCCI cell growth after CHRO + IR, Related to Figure 3
Telomerase inhibition promotes cellular senescence after irradiation. MCF7 cells expressing FUCCI cell cycle indicators (mCherry-hCdt1, red fluorescent G1 reporter; mVenus-hGeminin, green fluorescent S/G2 reporter) were treated with chrolactomycin, NU-1, or NU-2 then with 6 Gy and incubated in an IncuCyte for time-lapse imaging at 2 h intervals for 6 days. In the sample treated with the inactive analog NU-2 (0.5 μM) and 6 Gy (Movie S1), the majority of surviving cells resumed proliferation while a fraction was arrested with the characteristic senescent morphology of enlarged, flattened cells. Presumptive senescent cells were red fluorescent, suggesting a G1 cell cycle arrest. For samples treated with NU-1 (0.5 μM) (Movie S2) or CHRO (chrolactomycin, 0.5 μM) (Movie S3) and then 6 Gy, most cells arrested with a senescent morphology, expressing red fluorescence. Scale bar=100 μm.
Data Availability Statement
The RNA sequencing data are available on Sequence Read Archive (SRA): PRJNA663346.
This study did not generate unique code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.