

Summary
Postmenopausal women are severely affected by recurrent urinary tract infection (rUTI). The urogenital microbiome is a key component of the urinary environment. However, changes in the urogenital microbiome underlying rUTI susceptibility are unknown. Here, we perform shotgun metagenomics and advanced culture on urine from a controlled cohort of postmenopausal women to identify urogenital microbiome compositional and function changes linked to rUTI susceptibility. We identify candidate taxonomic biomarkers of rUTI susceptibility in postmenopausal women and an enrichment of lactobacilli in postmenopausal women taking estrogen hormone therapy. We find robust correlations between Bifidobacterium and Lactobacillus and urinary estrogens in women without urinary tract infection (UTI) history. Functional analyses reveal distinct metabolic and antimicrobial resistance gene (ARG) signatures associated with rUTI. Importantly, we find that ARGs are enriched in the urogenital microbiomes of women with rUTI history independent of current UTI status. Our data suggest that rUTI and estrogen shape the urogenital microbiome in postmenopausal women.
Keywords: urogenital microbiome, metagenomics, estrogen, urinary tract infection, antibiotic resistance, postmenopausal women, Lactobacillus crispatus, Escherichia coli, dysbiosis, bladder
Graphical abstract
Highlights
-
•
Recurrent UTI changes the composition of the postmenopausal urogenital microbiome
-
•
Network analysis reveals new potential interactions between urogenital microbial taxa
-
•
Urinary estrogen correlates with urogenital Lactobacillus and Bifidobacterium abundance
-
•
Antimicrobial resistance accumulates in urogenital microbiomes with rUTI history
Neugent et al. use metagenomic sequencing, advanced urine culture, and metabolite profiling to survey the urogenital microbiome of postmenopausal women, finding clinically important associations relevant to recurrent UTI susceptibility. These findings provide a foundation for the future development of novel therapeutic and diagnostic strategies for recurrent UTI in postmenopausal women.
Introduction
Urinary tract infection (UTI) is among the most common adult bacterial infections and imparts a particularly significant medical burden on women, with more than 50% of women suffering UTI in their lifetimes.1,2 Historically, UTI has largely been underprioritized in medical research due to low mortalities and the effectiveness of antibiotics. UTI is a disease of disproportionate burden as age is one of the strongest associated risk factors for UTI and the development of recurrent UTI (rUTI).3 Indeed, approximately 50% of UTIs in postmenopausal (PM) women develop into rUTI, which is clinically defined as ≥2 symptomatic UTIs in 6 months or three symptomatic UTI episodes in 12 months.1,4,5 rUTI can last for years, dramatically decreasing quality of life both physically and mentally and, if treatment is unsuccessful, can develop into life-threatening urosepsis.6 Indeed, a 2019 prospective study concluded that rUTI is significantly associated with frailty in American adults 65 years of age and older.7 While preventive strategies, including D-mannose, vaginal estrogen, and methenamine, are often employed, treatment of active UTI primarily relies on the use of antibiotics to achieve urinary tract sterility.3,8, 9, 10 However, increasing rates of antibiotic refractory rUTI make this strategy unsustainable.11 Alternate therapeutic strategies are needed to increase quality of life and reduce adverse outcomes for women with rUTI.
A promising source of therapies for rUTI lies in modulating or restoring the urogenital microbiome.12,13 Decades of medical dogma have assumed sterility of urine and the urinary tract; however, a robust body of work has established the existence of a human urogenital microbiome.14, 15, 16, 17, 18, 19, 20, 21 Taxonomic analyses have associated urogenital microbiome dysbiosis with urinary incontinence, overactive bladder, and bladder cancer.22, 23, 24 A study of 1,600 twins found that increased urinary microbiome diversity is associated with advanced age and that previous UTI, menopause, and host genetics are among the most significant host factors associated with differences in urinary microbiome composition.25 Recent work has used culture-based techniques to shed light on the microbial ecology of the urinary microbiome in PM women with rUTI.26,27 While PM women have been included in prior studies, focused representation in the literature is only now beginning to be established.28 Fundamental knowledge of urogenital microbiome composition and function in the context of rUTI susceptibility is lacking. As a result, the relationship between the urogenital microbiome and rUTI susceptibility is poorly understood in PM women. A 2021 report showed that premenopausal and PM women displayed different core urinary microbiota at the genus level, providing strong rationale to characterize the PM urogenital microbiome in urogenital disease.29
The female urogenital microbiome is reported to be interconnected with the vaginal microbiome.30 For example, D(-)lactate-producing lactobacilli, known to acidify the vagina and protect it from colonization by bacterial and fungal pathogens, have been consistently observed in the female urogenital microbiome in multiple independent studies.23,31 Lactobacilli can directly inactivate urogenital pathogens in vitro and recent work has shown that phenyl-lactic acid produced by Lactobacillus crispatus is bactericidal against uropathogenic bacteria, including uropathogenic Escherichia coli (UPEC).32, 33, 34 These observations beg the question of whether these protective vaginal lactobacilli serve a similar role in the urogenital microbiome. A 2011 clinical trial found a moderate reduction of rUTI incidence among women receiving an intravaginal L. crispatus probiotic.35 While this study was performed in premenopausal women and has yet to be confirmed by a larger study, it does suggest that lactobacilli support urinary tract health. Studies have also begun to establish a relationship between estrogen hormone therapy (EHT) and urogenital populations of lactobacilli.36 In 1993, Raz et al. reported that intravaginal estriol (E3) therapy reduced rUTI incidence and increased vaginal Lactobacillus populations, and a recent randomized clinical trial reported a significant reduction in rUTI incidence among PM women using vaginal EHT (vEHT) compared with placebo.37,38
Given the genomic diversity observed within and among taxonomic clades, metagenomic information beyond 16S rRNA sequence enrichment is needed to assess the functional potential of microbial communities.39 Whole-metagenome analysis of the urogenital microbiome is required to identify the genes and metabolic pathways associated with urinary tract health. Here, we present a whole-genome metagenomic sequencing (WGMS) survey of the urogenital microbiome of a cross-sectional cohort of PM women separated into three groups defined by rUTI history and current UTI status. Our taxonomic biomarker analysis detected a microbial signature that suggests an imprint of past UTI remains in the urogenital microbiome. In concordance with previous reports, in this study we also observed an association between the use of EHT and the presence of lactobacilli in the urogenital microbiome.36 Through measurement of urinary estrogen metabolites, we identified urogenital species whose abundance directly positively or negatively correlated with urinary estrogen concentration and found that distinct taxa correlate with estrogen in PM women with rUTI history compared with those without. Finally, we found that the resistome (i.e., the encoded antimicrobial resistance genes [ARGs]) of the urogenital microbiome is altered in women with rUTI history even in the absence of active infection. Our results suggest that both urogenital microbiome taxonomy and functional potential are shaped by rUTI history and EHT in PM women.
Results
Cohort curation, metagenomic DNA preparation, and whole-genome metagenomic dataset generation
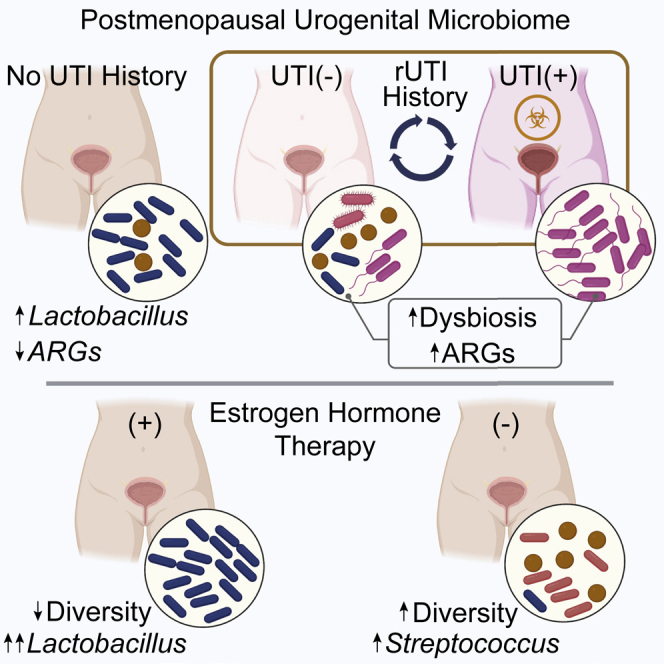
rUTI follows a cyclic pattern of infection (Figure 1A). To model this pattern, PM women were striated into three groups based on rUTI history. Group 1 served as a healthy comparator and consisted of PM women with no lifetime history of symptomatic UTI (No UTI History), group 2 consisted of PM women with a recent history of rUTI but no active UTI at the time of urine donation (rUTI History, UTI(−)), and group 3 consisted of PM women with a history of rUTI and an active, symptomatic UTI at the time of urine donation (rUTI History, UTI(+)) (Figure 1B). All women in the rUTI History, UTI(−), and rUTI History, UTI(+) groups passed strict inclusion criteria for uncomplicated rUTI, meaning they exhibited no compromise of the urinary tract, immune system, or used indwelling or intermittent catheters.3 We determined that 25 women per group were sufficient to balance a priori sample size estimation (Figures S1A and S1B) with clinical feasibility and enrollment rates. The final cohort was balanced for sample size, race, body mass index (BMI), smoking history, EHT use, urine pH, and urinary creatinine concentration. It should be noted as a limitation of this cohort that the women in the rUTI History, UTI(+) group tended to be older, with a median age of 76 years compared with 67 years in the No UTI History and 68 in the rUTI History, UTI(−) groups (p = 0.04) (Table S1). Finally, 37 women in the cohort used EHT, three used D-mannose, and no women used methenamine (Table S2).
Figure 1.

Study design and summary of genera detected by WGMS and advanced urine culture
(A) Illustration of rUTI cycle depicting periods of active, symptomatic UTI with positive urine culture followed by periods of remission with negative urine culture.
(B) Diagram of cohort structure and datasets generated for the study created with BioRender.com.
(C) Taxonomic cladogram of top 20 genera detected in all metagenomes (n = 75) by Metaphlan2. Node size indicates relative abundance and branch length is arbitrary.
(D) Venn diagram depicting the coverage of advanced urine culture calculated at the genus level considering all bacterial genera with >5% WGMS relative abundance in at least one patient.
Urine was collected via the “clean-catch” midstream method, which can also sample the urogenital tract and is therefore representative of the urogenital microbiome, which is inclusive of the bladder, urethral, and, in some cases, vaginal microbiomes.40,41 Metagenomic DNA yields reflected the anticipated biomass of the urogenital microbiome in each (Figure S1C). We observed an average of 67.6% host (human) contamination within the WGMS data (Figure S1D). Previous reports of human contamination in WGMS sequencing of the urogenital microbiome range from 1% to 99% of reads.39,42 To measure potential background and environmental signals, a water sample was randomly inserted into the metagenomic DNA isolation and sequencing workflow.43 Most microbial reads observed in the water mapped to common kit and environmental contaminants (Figure S2A).44 Except for known members of the human microbiome, these background taxa were censored from the data.
Validation of viable urogenital microbiome species through advanced urine culture and WGMS hybrid taxonomic profiling
To validate the presence of living microbiota within the urogenital microbiomes, we coupled WGMS with advanced urine culture, a modification of the previously reported enhanced quantitative urine culture protocol.45 Taxonomic profiling by WGMS detected a total of 276 bacterial, archaeal, and fungal species across 106 genera. The sampled urogenital microbiomes were dominated by the kingdom Bacteria, which represented 99.4% of the detected non-viral, microbial taxa (Figure 1C). Consistent with the observed taxonomic composition of urogenital microbiomes studied to date.15,16, the detected bacterial taxa belonged to four major phyla: Firmicutes (44.7%), Actinobacteria (22.3%), Proteobacteria (20.6%), and Bacteroidetes (12%) (Figure 1C). Advanced urine culture captured 93.9% of bacterial genera detected in WGMS, with observed relative abundance ≥5% in any sample (Figure 1D). Patient-level culture coverage is reported in Figure S2B. The most frequent cultivable genera across all samples were Lactobacillus, Escherichia, Streptococcus, Bifidobacterium, Gardnerella, Klebsiella, Staphylococcus, Finegoldia, Enterococcus, and Facklamia. Pure isolates from every cultivable species were assembled into a biobank of 896 bacterial and fungal isolates (Table S3).
rUTI history is not associated with large-scale alterations of urogenital microbiome ecological structure in the absence of active infection
We analyzed the genus- and species-level taxonomic profiles within the No UTI History, rUTI History, UTI(−) and rUTI History, and UTI(+) groups (Figures 2A, 2B, and S3A; Table S4). The rUTI History, UTI(+) group was mainly dominated by single bacterial uropathogens with little detected Fungi and Archaea (Figures 2B and S3A). The most prevalent bacterium was UPEC (15 out of 25, 60%), the major uropathogen among most types of UTI.3 We also detected known uropathogens, Klebsiella pneumoniae (2 out of 25, 8%), Enterococcus faecalis (1 out of 25, 4%), and Streptococcus agalactiae (1 out of 25, 4%). We observed fungal species including Candida glabrata and Malassenzia globosa. Similarly low relative abundances of archaeal taxa were detected, such as Methanobrevibacter spp. (Figure S3A). The most observed viral taxa were JC polyomavirus (4/25 16%), but human herpes virus 4 (1 out of 25 4%) and Enterobacteria phage lke (1 out of 25 4%) were also detected (Figure S3B).
Figure 2.

The bacterial taxonomic profile of rUTI in PM women
(A and B) (A) Genus- and (B) species-level taxonomic profiles of the top 15 bacterial genera among groups (No UTI History [n = 25], rUTI History, UTI(−) [n = 25], rUTI History, UTI(+) [n = 25]). Remaining genera or species are combined into “Other.”
(C and D) Alpha-diversity of (C) observed species counts and (D) Shannon index between groups (1 = No UTI History, 2 = rUTI History, UTI(−), 3 = rUTI History, UTI(+)). Solid lines represent medians, while dotted lines represent the interquartile range. p value generated by Kruskal-Wallis test with Dunn’s multiple correction post hoc.
(E) Beta diversity by DPCoA. Samples color coded by group. Vectors (gray) represent top loadings (i.e., species).
(F) Volcano plot depicting co-occurrence of genera by Pearson correlation. p value generated by permutation. Red dots represent associations with an FDR-corrected p value < 0.05. Blue dots represent associations with a nominal p value < 0.05, but an FDR-corrected p value > 0.05.
(G) Network analysis of genus-level co-occurrences with nominal p value < 0.05. Nodes represent genera. Edges are defined by Pearson correlation and node size is proportional to the degree of the node.
The most frequently observed bacterial species in women without active UTI (No UTI History and rUTI History, UTI(−)) belonged to the genera Lactobacillus, Bifidobacterium, Gardnerella, Streptococcus, Staphylococcus, and Actinobaculum (Figures 2A and 2B). Fifty-four percent of samples were dominated by one taxon, while others were diverse and exhibited no single predominant taxon. We observed 13 patients (24%) with a >50% relative abundance of various Lactobacillus spp., including L. crispatus, Lactobacillus iners, and Lactobacillus gasseri (Figure 2B). A subset of urogenital microbiomes in the No UTI History and rUTI History, UTI(−) groups was dominated by Bifidobacterium spp., such as Bifidobacterium breve, Bifidobacterium dentium, and Bifidobacterium longum, as well as by Gardnerella vaginalis (Figure 2B). Fungal and archaeal species were observed in low abundance (0%–8.8%) in the No UTI History and rUTI History, UTI(−) urogenital microbiomes and included Candida albicans, C. glabrata, and Candida dubliniensis, as well as M. globosa, Naumovozyma spp., and Eremothecium spp. (Figure S3A). Archaeal species within the No UTI History and rUTI History, UTI(−) urogenital microbiomes included Methanosphaera stadtmanae and Methanobrevibacter spp. Viral taxa were more frequently observed in the No UTI History and rUTI History, UTI(−) groups than in the rUTI History, UTI(+) group and included JC, BK, and Merkel cell polyomaviruses (Figure S3B).
We then calculated alpha-diversity indices including the observed taxa count, Shannon, Simpson, Chao 1, and the abundance-based coverage estimator (ACE) indices. Women in the No UTI History and rUTI History, UTI(−) groups had similarly diverse urogenital microbiomes across different indices (Figures 2C, 2D, and S4A–S4C). These data suggest that, if there are differences in the urogenital microbiomes of PM women who are susceptible to rUTI (rUTI History, UTI(−)) versus those who are not, they are not reflected in alpha-diversity metrics. The rUTI History, UTI(+) group exhibited significantly lower alpha-diversity compared with the rUTI History, UTI(−) cohort (Figures 2C, 2D, and S4A–S4C).
To assess beta diversity, we used double principal coordinate analysis (DPCoA).46 Visualization of the first two principal coordinate analyses (PCoAs) revealed that the urogenital microbiomes of the rUTI History, UTI(+) group clustered along a vector defined by E. coli and were ecologically distinct from the urogenital microbiomes of the No UTI History and rUTI History, UTI(−) groups. The No UTI History and rUTI History, UTI(−) groups exhibited relatively similar clustering patterns (Figure 2E), and clustered along opposing vectors defined by the enrichment of either L. crispatus or G. vaginalis, which are associated with vaginal health or bacterial vaginosis, respectively.47,48 This similar clustering of the No UTI History and rUTI History, UTI(−) cohorts suggests that a history of rUTI does not significantly alter the large-scale taxonomic structure of the urogenital microbiome in PM women.
Taxonomic profile of the PM urogenital microbiome displays co-occurrence structure
Microbial communities can harbor intricate interactions between member taxa.49,50 Exceedingly little is known about interactions and co-occurrence of bacterial species within the urogenital microbiome. We performed taxonomic association analysis to determine the co-occurrence structure of the PM urogenital microbiome and identified 87 statistically significant genus-level associations (p < 0.05) (Figure 2F; Table S5). After multiple hypothesis testing correction, a total of 17 associations exhibited robust statistical significance (Q < 0.05) (Figure 2F). Network visualization of significant positive associations (p < 0.05) revealed three non-interacting microbial clusters (Figure 2G). Cluster 1 taxa included genera associated with vaginal infections, such as Bacteroides and Blautia.51 Cluster 2 exhibited the largest member set and diversity, and captured associations between urogenital microbiome genera (i.e., Peptoniphilus and Finegoldia) but whose association has not yet been reported.52,53 Cluster 2 grouped strongly around the genus Peptoniphilus. Cluster 3 was identified as a pairwise interaction between Gardnerella and Atopobium, taxa associated in vaginal dysbiosis and bacterial vaginosis.54, 55, 56 We also observed anti-correlated taxa (Figures 2F and S4D). The two main hubs of the negative correlation network were Lactobacillus and Escherichia. Of note, the most significant negative association was observed between Lactobacillus and Peptoniphilus (Figures 2F and S4D). These data define patterns of co-occurrence within the urogenital microbiome and suggest candidate taxa that may act as hubs of community structure.
Taxonomic biomarker analysis reveals that rUTI history may alter the species-level taxonomic signature of the urogenital microbiome
Although we did not detect large-scale taxonomic differences between the rUTI History, UTI(−) and No UTI History groups, we hypothesized that small-scale differences may contribute to differential rUTI susceptibility. We therefore performed genus- and species-level differential taxonomic enrichment analysis between the No UTI History and rUTI History, UTI(−) urogenital microbiomes using linear discriminant analysis of effect size (LEfSe), Analysis of Compositions of Microbiomes with Bias Correction (ANCOM-BC), and a Bayesian microbial differential abundance (BMDA) model.57, 58, 59 LEfSe performs a non-parametric assessment of differential abundance, ANCOM-BC adopts a linear regression framework and corrects for latent sampling bias, and BMDA can account for sparsity, over-dispersion, and uneven sampling depth, characteristics widely attributed to the urogenital microbiome. We first sought to validate the ability of BMDA to control type 1 error in our dataset by permuting the rUTI history labels for the No UTI History versus rUTI history comparison (Figure 3A) and the current infection status labels for the UTI(−) versus UTI(+) comparison (Figure 3B). The resulting permuted datasets should exhibit few discriminating taxa if the statistical model employed can control type 1 error. Our results demonstrate that BMDA does control type 1 error on real permuted data like other robust methods (Figures 3A and 3B). Using a permuted synthetic dataset, we further show that the BMDA model may possess more statistical power compared with other commonly used statistical models for differential enrichment analysis in sparse microbiomes such as the skin and urogenital microbiomes (Figure 3C).60 While LEfSe and ANCOM-BC detected no differentially abundant taxa between the No UTI History and rUTI History, UTI(−) groups, BMDA detected multiple differentially abundant taxa, an observation that may be attributable to its higher statistical power. BMDA detected two genera, Aerococcus and Lactobacillus, as well as two species of lactobacilli, L. vaginalis and L. crispatus, as enriched in the No UTI History group (Figures 3D and 3E). At the genus level, Klebsiella, Gemella, Bacteroides, Clostridiales Family XIII Incertae Sedis unclassified, Eggerthella, and Escherichia were among the most enriched in the rUTI History, UTI(−) group. Fifteen species were identified as enriched in the rUTI History, UTI(−) group, namely Ureaplasma parvum, Bacteroides uniformis, Brevibacterium massiliense, Anaerococcus hydrogenalis, Actinomyces turicensis, Prevotella timonensis, E. faecalis, Staphylococcus hominis, Peptoniphilus lacrimalis, Corynebacterium pseudogenitalium, Actinomyces europeaus, Facklamia hominis, Finegoldia magna, Anaerococcus prevotii, and Staphylococcus epidermidis (Figure 3E). Many of these taxa, such as the genera Bacteroides, Streptococcus, Escherichia, Ureaplasma, Finegoldia, and Gemella, have been found in the vaginal microbiome during infection.51,61 Furthermore, A. turicensis and Actinomyces europaeus are known to be associated with UTI.62 Taken together, these results suggest that rUTI history may leave an imprint on urogenital microbiome composition that may be missed by common ecological indices (alpha or beta diversity) and differential abundance pipelines that do not consider the sparsity, over-dispersion, and uneven sampling that is common in low-biomass microbiomes such as the urogenital microbiome.40 However, it will be important to independently validate these findings in future studies involving larger cohorts.
Figure 3.

Bayesian modeling detects the taxonomic imprint of rUTI history on the urogenital microbiome of PM women
(A and B) Analysis of discoveries after permutation (type 1 error) in the taxonomic dataset permuted for (A) rUTI History for the No UTI History versus rUTI History comparison and (B) current infection status for the UTI(+) versus UTI(−) comparison (n = 50 permutations each) for BMDA compared with other commonly used differential enrichment analysis tools.
(C) Comparison of average statistical power as a function of false discovery rate for differential enrichment analysis tools on a synthetic dataset with 1,000 taxa and a sample size of 108 (n = 54 per group).
(D and E) BMDA model comparing genus- (D) and species-level (E) taxonomic enrichment between the No UTI History (n = 25) and rUTI History, UTI(−) (n = 25) groups. Dots, indicating the log10(posterior effect size), are color-coded by group. Lines indicate the 95% credible interval. PPI, posterior probability index.
Urogenital microbiome taxonomic structure differs in women using EHT
Given that many of the urogenital microbiomes of women without active rUTI were dominated by lactobacilli (26%, 13 out of 50) (Figures 2A and 2B), we further characterized this enrichment in the women of the No UTI History and rUTI History, UTI(−) groups, who did not have UTI at the time of urine donation. We screened the clinical metadata for variables associated with Lactobacillus abundance. We observed that EHT was strongly associated with the presence of Lactobacillus in the urogenital microbiome (Figures 4A–4C). Multiple modalities of EHT were represented among the participants of this study, including, systemic EHT (n = 12) and local, vaginal EHT (n = 17). Ecological modeling revealed that the urogenital microbiomes of EHT(+) (aggregated systemic and vaginal) women (n = 29) were significantly less diverse than those of EHT(−) women (n = 21) and tended to be dominated by a single species of lactobacilli (Figures 4D–4F). To identify taxa associated with EHT use, we performed differential taxonomic enrichment analysis using LEfSe and BMDA (Table S6). LEfSe found an enrichment of the genus Lactobacillus in EHT(+) women and the genus Streptococcus in EHT(−) women (Figure 4G). BMDA captured a similar result but further resolved species-level differential enrichment (Figure 4H). L. crispatus and L. vaginalis were significantly enriched in the urogenital microbiomes of EHT(+) women and Streptococcus mitis/oralis/pneumoniae (S. m/o/p) group, Streptococcus infantis, and Atopobium vaginae were enriched in the EHT(−) group (Figure 4H). Separating women by EHT modality, we observed that women using oral EHT (oEHT, n = 6) and patch EHT (pEHT, n = 6) had significant urogenital microbiome enrichment of Lactobacillus. However, Lactobacillus enrichment varied widely in women using vEHT (n = 17) from 0% to >99% relative abundance (Figure S5A).
Figure 4.

Estrogen hormone therapy shapes the urogenital microbiome of PM women
(A and B) (A) Genus- and (B) species-level taxonomic profiles of the relative abundance of the top 22 bacterial genera among EHT(−) (n = 21) and EHT(+) (n = 29) women in the No UTI History and rUTI History, UTI(−) groups. Taxa not within the top 22 are combined into “Other.”
(C) Comparison of Lactobacillus relative abundance between EHT(−) (gray) and EHT(+) (pink) women in the No UTI History and rUTI History, UTI(−) groups. Solid lines represent medians, while dotted lines represent the interquartile range. p values generated by Wilcoxon rank-sum.
(D–F) (D) Observed species count, (E) Shannon index, and (F) Simpson index for EHT(−) (gray) and EHT(+) (pink) women in the No UTI History and rUTI History, UTI(−) groups. Solid lines represent medians, while dotted lines represent the interquartile range. p values generated by Wilcoxon rank-sum.
(G) Two significantly differentially enriched genera (LDA > 4.5) detected by LEfSe between EHT(−) (gray) and EHT(+) (pink). LDA: log10(linear discriminant analysis score). p value was generated by LEfSe.
(H) Differentially enriched taxa between EHT(−) (gray) and EHT(+) (pink) women in the No UTI History and rUTI History, UTI(−) cohorts detected by BMDA. Dots indicate log10(posterior effect size). PPI, posterior probability index. S. m/o/p, Streptococcus mitis/oralis/pneumoniae. EHT(+) is the aggregate of both systemic and vaginal EHT modalities. Lines indicate the 95% credible interval.
Urinary estrogen concentration is positively correlated with urogenital microbiome Lactobacillus abundance in PM women with no UTI history
In order to identify specific taxa-estrogen associations, we optimized a targeted liquid chromatography-mass spectrometry (LC-MS) method to quantify excreted urinary estrogen conjugates of women in the No UTI History and rUTI History, UTI(−) groups (Table S7).63 We limited our analysis to the known major sulfate and glucuronide conjugates of estrone (E1) and 17β-estradiol (E2). We observed significantly higher urinary concentrations of E1 and E2 sulfates and glucuronides in women using oEHT. Women using pEHT had higher urinary concentrations of E1-sulfate (Figure S5G). We then performed exploratory correlation analysis of creatinine-normalized estrogen metabolite concentrations and the species-level taxonomic profile (Table S8). Given that we found evidence that the underlying urogenital microbiome is altered by rUTI history (Figures 3C and 3D), we dichotomized the samples into cohort groups (No UTI History and rUTI History, UTI(−)). We found a striking difference in estrogen-associated taxonomic profiles between the No UTI History and rUTI History, UTI(−) groups (Figures 5A and 5B). We observed correlations between urinary E1 and E2 conjugates and L. crispatus, L. iners, and L. gasseri in the No UTI History group, correlations that were not detected in the rUTI History, UTI(−) group (Figures 5A–5C, S5I, and S5J). B. breve, an Actinobacterium associated with colon health, exhibited the strongest positive correlation across estrogen conjugates in the No UTI History group (Figures 5A and 5C). This correlation was also absent in the rUTI History, UTI(−) group. A. prevotii was consistently and significantly negatively associated with urinary estrogens in the No UTI History group (Figures 5A, 5C, S5I, and S5J). A smaller, distinct set of species correlated with estrogen conjugates in the rUTI History, UTI(−) group (Figures 5B, S5I, and S5J). To assess the robustness of these results, we used a Bayesian correlation approach to account for over-dispersion.64 Consistent with the non-Bayesian analysis, L. gasseri, B. dentium, L. crispatus, and Prevotella disiens abundance were positively correlated with urinary estrogen conjugate sum in the No UTI History group, while A. prevotii was negatively correlated. Bayesian analysis also detected a set of new urinary estrogen-taxa correlations. Of note, E. coli was significantly anti-correlated with summed urinary estrogen concentration in the No UTI History group. Ruminococcus torques, Pantoea spp., Pseudomonas spp., Dorea spp., and Collinsella aerofaciens correlated with estrogen in the rUTI History, UTI(−) group, while Dialister microaerophilus and Corynebacterium aurimucosum were anti-correlated with urinary estrogen in the rUTI History, UTI(−) group (Figures 5D and 5E). These data indicate that distinct urinary taxa correlate with urinary estrogen metabolites in women with no UTI history compared with women with rUTI history.
Figure 5.

Distinct taxa-urinary estrogen metabolite associations between PM women with and without rUTI history
(A and B) Spearman correlation of bacterial species with summed Cr-normalized urinary estrogens in (A) No UTI History and (B) rUTI History, UTI(−) groups. p value generated by permutation. Red and blue dots represent significant (p < 0.05) positive and negative associations, respectively.
(C–E) Taxa-estrogen correlation scatter plots among No UTI History (n = 25) (blue) and rUTI History, UTI(−) women (n = 23) (purple). Linear regression trend line (solid line) is shown with 95% confidence intervals. Bayesian correlation point estimates and 95% credible interval of posterior correlation (Spearman) for the top 10 taxa and Cr-normalized summed urinary estrogen conjugates in the (D) No UTI History and (E) rUTI History, UTI(−) groups. Blue indicates negative, while red indicates positive correlation. Significant correlations also found in the non-Bayesian analysis are bolded. Dots represent the median of the Spearman correlation posterior sampling, and lines indicate the 95% credible interval.
Functional profiling reveals significant differences in the metabolic potential of cohort urogenital microbiomes
We next sought to determine if rUTI leaves a detectable imprint on the functional metabolic potential of the urogenital microbiome. We used the HUMAnN2 pipeline to profile encoded metabolic potential.65 Principal-component analysis (PCA) performed on the relative abundance of metabolic pathways in the three groups identified discriminating clusters that separated the rUTI History, UTI(+) from the rUTI History, UTI(−) and No UTI History urogenital microbiomes (Figure 6A). These results were consistent with the taxonomic beta-diversity analysis (Figure 2E). The rUTI History, UTI(+) group ordinated along vectors defined by the enrichment of lipopolysaccharide (LPS) biosynthesis (n = 4 pathways), demethylmenaquinol-8 biosynthesis, fucose and mannose degradation, D-galacturonate degradation, sucrose degradation, and the tricarboxylic acid (TCA) cycle (Figure 6B). The rUTI History, UTI(−) and No UTI History groups, which were not discriminated, ordinated along vectors defined by the enrichment of nucleotide biosynthesis (n = 8 pathways), L-lysine biosynthesis II, S-adenosyl methionine biosynthesis, and UDP-N-acetyl-glucosamine biosynthesis (Figure 6B). These data suggest that the large-scale genetic potential of the urogenital microbiome is relatively similar between rUTI History, UTI(−) and No UTI History groups but is altered during active rUTI. While we attribute these findings to the predominance of uropathogens in the rUTI History, UTI(+) urogenital microbiome, we also note that the median age of the rUTI History, UTI(+) group was higher than the No UTI History and rUTI History, UTI(−) groups.
Figure 6.

rUTI history and active infection shape the metabolic potential of the urogenital microbiome
(A and B) PCA of metagenome-encoded metabolic pathways. Depiction of ordination and clustering in the first two principal-components (PCs) in (A) and vectors (gray) defining top loadings in (B).
(C and D) Top 40 differentially enriched pathways between No UTI History (blue) and rUTI History, UTI(−) (purple) groups (C) and the No UTI History and rUTI History, UTI(+) (red) groups (D) detected by LEfSe. Pathways met an FDR-corrected p value cutoff of <0.05. LDA, log10(linear discriminant analysis).
Because dimensional reduction techniques often miss fine-scale discriminating features, we next used LEfSe to identify metabolic pathway enrichments between the study groups (Table S9).58 We tested the hypothesis that rUTI history imparts functional changes on the urogenital microbiomes of PM women by comparing the No UTI History and rUTI History, UTI(−) groups. Forty-five discriminatory metabolic pathways were significantly enriched in the rUTI History, UTI(−) and four metabolic pathways significantly enriched in the No UTI History urogenital microbiomes with a false discovery rate (FDR)-corrected p < 0.05 and LDA > 2 (Figure 6C). The top 40 discriminating pathways were carbohydrate metabolism (n = 14), electron carrier biosynthesis (n = 8), amino acid metabolism (n = 5), cell envelope biosynthesis (n = 4), vitamin and cofactor biosynthesis (n = 4), and polysaccharide degradation (n = 3) (Figure 6C). While most carbohydrate metabolic pathways were enriched in the rUTI History, UTI(−) urogenital microbiomes (13 out of 14, 92.9%), we observed an enrichment of D-galactose degradation in the No UTI History group (LDA = 3.06, p = 0.028). Conversely, electron carrier biosynthesis, namely biosynthetic pathways for ubiquinol 7–10 as well as menaquinol 6, 9, and 10 and demethylmenaquinol 9, were enriched in the rUTI History, UTI(−) urogenital microbiomes (Figure 6C). L-lysine biosynthesis, L-threonine biosynthesis, and L-tryptophan degradation were enriched in No UTI History, while L-ornithine biosynthesis and L-arginine degradation were enriched in rUTI History, UTI(−) samples. The remaining discriminating metabolic pathways were enriched in the rUTI History, UTI(−) urogenital microbiomes and included cell envelope biosynthesis, vitamin metabolism, polysaccharide degradation, cinnamate and hydroxy cinnamate degradation, and ppGpp biosynthesis (Figure 6C). These data suggest that the metabolic landscape of the urogenital microbiome may be altered by rUTI history.
Differential enrichment analysis between the No UTI History and rUTI History, UTI(+) groups identified 183 metabolic pathways (Figure 6D). In line with the taxonomic enrichment of gram-negative species, we observed an enrichment of biosynthetic pathways for lipopolysaccharide (LPS) within rUTI History, UTI(+) urogenital microbiomes. Top discriminating pathways included carbohydrate (n = 13), nucleotide (n = 9), and amino acid metabolism (n = 6), as well as cell envelope biosynthesis (n = 5) (Figure 6D). Diverse carbohydrate degradation and central carbon metabolism pathways, including rhamnose, fucose, glyoxylate, and fructuronate degradation, were enriched in rUTI History, UTI(+) samples (Figure 6D). This was coupled with a significant enrichment of TCA cycle metabolism, particularly 2-oxoglutarate decarboxylase and ferroreductase (Figure 6D). Only four metabolic pathways involved in carbohydrate metabolism, including glycolysis from glucose and glucose 6-phosphate, pyruvate fermentation, and N-acetyl glucosamine biosynthesis, were significantly differentially enriched in the No UTI History group (Figure 6D). Nucleic acid biosynthesis pathways were enriched in the No UTI History group, while the rUTI History, UTI(+) group was enriched for nucleic acid degradation pathways (Figure 6D). Differentially enriched amino acid metabolism pathways included L-lysine, L-threonine, and L-isoleucine biosynthesis in the No UTI History group and L-phenylalanine biosynthesis in the rUTI History, UTI(+) group (Figure 6D). These results suggest that the urogenital microbiomes of the rUTI History, UTI(+) group have the potential to utilize a more diverse nutrient set.
Antibiotic resistance genes are enriched in the urogenital microbiomes of women with rUTI history
Resistance to front-line antibiotics, such as trimethoprim-sulfamethoxazole (TMP-SMX), fluoroquinolones, and nitrofurantoin, is becoming a significant barrier to the successful treatment of rUTI.11 Using the Graphing Resistance out of Metagenomes (GROOT) pipeline, we generated a detailed profile of the encoded antimicrobial resistance genes (ARGs) and detected 55 high-confidence ARGs distributed among all three groups (Table S10).66 We observed significantly more ARGs in the urogenital microbiomes of the rUTI History, UTI(−) (p = 0.0455) and rUTI History, UTI(+) (p = 0.0302) groups compared with the No UTI History group (Figure 7A). Interestingly, there was no significant difference in ARG count between rUTI History, UTI(+) and rUTI History, UTI(−) groups. These data suggest that a history of rUTI leaves an imprint on the resistome of the urinary microbiota in PM women even in the absence of active infection.
Figure 7.

rUTI history and active infection shape the resistome of the PM urogenital microbiome
(A) Comparison of ARGs detected within the urogenital microbiomes of the No UTI History, rUTI History, UTI(−), and rUTI History, UTI(+) groups. Solid lines represent median, while dotted lines represent interquartile range. p value generated by Kruskal-Wallis test with uncorrected Dunn’s multiple correction post hoc.
(B) Bayesian differential enrichment analysis of ARGs within cohort urogenital microbiomes. Group comparisons were determined by pairwise differences in ARG(+) proportions. 95% credible intervals, Bayes factor, posterior probability, Fisher exact p values are presented.
(C) Agreement between WGMS ARG detection and antibiotic resistance phenotypes of isolates of the most abundant species present in each rUTI History, UTI(+) patient (E. coli (n = 15), Klebsiella (n = 3), Streptococcus (n = 3), E. faecalis (n = 1), and S. epidermidis (n = 1)). Upper diagonal colors represent WGMS profiling results (blue = ARG (+), white = ARG(−)). Lower diagonal color represents phenotype (red = resistant, yellow = intermediate, white = sensitive, gray = not tested). (1) No UTI History: no history of UTI, no active UTI. (2) rUTI History, UTI(−): history of rUTI, no active UTI. (3) rUTI History, UTI(+): history of rUTI, active UTI.
To assess specific ARG enrichments associated with rUTI history, we used a Bayesian model of proportional enrichment.67 The TEM β-lactamase family, sulfonamide resistance genes sul1 and sul2, and the straA aminoglycoside 3′-phosphotransferase were significantly enriched in the rUTI History, UTI(+) group, while the aminoglycoside 3′-phosphotransferase genes, aph(3′)-III and aph(3′)-Ia; the macrolide resistance gene, ermB; the β-lactam resistance gene, mecA; and the aminoglycoside O-nucleotidyltransferase gene, ant(6)-Ia were enriched in the urogenital microbiomes of the rUTI History, UTI(−) group (Figure 7B). Conversely, no ARGs were significantly enriched in the No UTI History group.
Identification of ARGs in metagenomes is only a prediction of microbiota phenotype.39 To begin to understand how well metagenomic ARG analysis predicts phenotype, we measured antibiotic resistance phenotypes of 22 unique bacterial uropathogens each isolated from an individual rUTI History, UTI(+) patient. Species tested included E. coli, K. pneumoniae, Klebsiella oxytoca, Streptococcus anginosus, S. agalactiae, E. faecalis, and S. epidermidis. Three of the 15 strains with complete or intermediate ampicillin resistance did not have a detected ampicillin resistance gene (Figure 7C). Resistome profiling detected cefixime resistance genes in 50% of the metagenomes (2 out of 4) associated with isolates that were completely or intermediately resistant to cefixime (Figure 7C). The cefixime-resistant strains without detected ARGs were both streptococci. We observed that 50% of isolates with TMP/SMX resistance were isolated from urogenital microbiomes for which resistome profiling detected ARGs sul I/II and drfA1 (Figure 7C). For aminoglycoside resistance, 50% (1 out of 2), 60% (3 out of 5), 100% (1 out of 1), and 88.9% (8 out of 9) of the isolates with complete or intermediate resistance to gentamicin, kanamycin, amikacin, and streptomycin, respectively, had corresponding ARGs in their associated metagenomes (Figure 7C). ARG analysis was relatively poorly predictive of fluoroquinolone resistance, with resistance genes detected in only 27.3% (3 out of 11) and 33.3% (2 out of 6) of the associated metagenomes of isolates with complete or intermediate resistance to ciprofloxacin and levofloxacin, respectively (Figure 7C). This is likely because GROOT does not detect single-nucleotide polymorphisms (SNPs) and fluoroquinolone resistance is often conferred by SNPs in gyrase and topoisomerase I genes.68 While all gram-positive bacterial strains were resistant to erythromycin, macrolide ARGs were only detected in the associated metagenomes of three strains.69 Tetracycline and phenicol ARGs were respectively detected in 42.9% (3 out of 7) and 0% (0 out of 1) of the metagenomes associated with strains with intermediate or resistant phenotypes (Figure 7C).
Discussion
A decade of research has identified and characterized the urogenital microbiome.14, 15, 16, 17, 18, 19 It has become evident that the urogenital microbiome is involved in or affected by urologic disease. Given the connection between host health and microbiome composition, the urogenital microbiome has drawn significant attention in further understanding rUTI susceptibility. Here, we use WGMS to specifically probe urogenital microbiome ecology and function associated with rUTI in PM women. The main uropathogen detected in the rUTI History, UTI(+) group was UPEC, while urogenital microbiomes of the No UTI History and rUTI History, UTI(−) groups were either dominated by a single bacterial species or were diverse. Both the No UTI History and rUTI History, UTI(−) groups exhibited subsets of women with urogenital microbiomes dominated by L. crispatus, L. gasseri, L. iners, B. breve, B. dentium, B. longum, and G. vaginalis. These data support the observations by Thomas-White et al. of an interconnected urogenital microbiome.30 We detected low abundances of fungal and archaeal taxa in all groups; however, little is known about the role of Fungi and Archaea in the female urogenital microbiome.
We identified differentially enriched urogenital microbiome taxa between healthy PM women and those with a history of rUTI that may serve as microbial biomarkers of urogenital tract health. Many of the genera enriched in the rUTI History, UTI(−) group were members of the largest co-occurrence network that clustered strongly around Peptoniphilus, a known member of the vaginal microbiome associated with dysbiosis.70, 71, 72 These data suggest that mutualistic relationships between Peptoniphilus and co-occurring taxa may define community structure in urogenital microbiomes of women with increased rUTI susceptibility.73 Conversely, while lactobacilli were enriched in No UTI History urogenital microbiomes, co-occurrence analysis revealed a negative association between Lactobacillus and Peptoniphilus, suggesting an antagonistic relationship. Taken together, these observations give insight into potentially biologically relevant interactions in the urogenital microbiome that may underlie rUTI susceptibility. These observations are supported by a 2021 report by Vaughan et al. that used 16S rRNA amplicon sequencing to identify taxonomic differences between PM women with rUTI and healthy controls and identified differences in the orders Clostridiales and Prevotellaeceae, which contain the genera Peptoniphilus and Prevotella, respectively.28 Critical future research will validate signatures of urinary dysbiosis associated with rUTI susceptibility in longitudinal studies.
EHT is a common intervention to reduce discomfort associated with menopause.74 EHT, especially vEHT, is also gaining prevalence for rUTI prophylaxis in PM women because estrogen is thought to favor Lactobacillus colonization of the vaginal and urinary microbiomes.37,38,75 Here, we identify L. crispatus and L. vaginalis as associated with EHT use in PM women. Streptococci as well as A. vaginae, a gram-positive species associated with G. vaginalis in bacterial vaginosis, were enriched in women not using EHT.56 Multiple independent studies have evaluated associations between EHT and vaginal and urinary lactobacilli with varying results. For example, Anglim et al. found that vEHT did not significantly alter urinary lactobacilli among PM women with and without rUTI, while Thomas-White et al. reported that vEHT led to a significant enrichment of urinary lactobacilli in PM women with overactive bladder symptoms.36,53 A recent randomized-controlled trial by Lillemon et al. did not find significant changes in urogenital Lactobacillus enrichment following local EHT.76 We observed that Lactobacillus-dominated urogenital microbiomes were associated with women using oEHT (n = 6) and pEHT (n = 6). While reports of systemic EHT-associated urogenital Lactobacillus enrichment are few, a 2016 interventional study using oral conjugated estrogens demonstrated a rapid and significant increase in vaginal Lactobacillus spp.77 Additionally, an independent 2001 report observed an increase in vaginal Lactobacillus spp. in PM women taking a short-term course of oral estriol.78 While the EHT composition and dosage may differ from the cohort studied here, these independent reports are supportive of our findings associating urogenital Lactobacillus enrichment with systemic EHT (oral or transdermal patch).
Although we did observe some Lactobacillus enrichment in women using vEHT (n = 17), this group exhibited much larger variance in Lactobacillus relative abundance than the oral and patch modalities, and 41.2% (7 out of 17) of women using vEHT exhibited a >40% relative abundance of urogenital Lactobacillus. We do not interpret these data as not supporting vEHT-mediated Lactobacillus enrichment. Rather, we hypothesize that, perhaps due to differences in compliance or dosage, the association between vEHT and urogenital Lactobacillus enrichment may be more variable between individuals. Importantly, we identified disease-state-specific taxa-estrogen metabolite correlations. B. breve, L. iners, L. crispatus, and L. gasseri positively correlated with urinary estrogen conjugate concentration in the No UTI History group but not the rUTI History, UTI(−) group. Future mechanistic research in relevant model systems and longitudinal human cohorts is needed to define the effects of rUTI history on EHT urogenital microbiome modulation.
Frequent and repeated treatment of rUTI with antibiotics is thought to spur the evolution of antibiotic resistance among uropathogenic bacteria and perhaps within the urogenital microbiome.11,79,80 Despite the urgent need to understand the impact of antibiotic therapy on the urogenital microbiome, differences in urogenital microbiome ARG prevalence associated with rUTI and rUTI history had not been previously investigated. We found that the urogenital microbiomes of both the rUTI History, UTI(+) and rUTI History, UTI(−) groups contained significantly more ARGs than the No UTI History group, suggesting that rUTI history may enrich for ARG-containing urogenital microbiomes.
This controlled WGMS analysis of urogenital microbiome structure and function in PM women with different histories of rUTI provides a robust foundation for further mechanistic studies of the role of the urogenital microbiome in rUTI susceptibility and disease progression that are necessary for the development of urogenital microbiome-aware alternative therapies for a rUTI.
Limitations of the study
While our observations are in line with previous work and may serve as independent support, it should be noted that methodologies differ between urinary microbiome studies.36,37 Here, we use clean-catch midstream urine, which samples the urinary tract and, in some cases, the vulvovaginal niche. Because different sampling techniques are used to study individual sites within the urinary system and given the observations made by prior comparisons of sampling techniques, studies of the urogenital microbiome may not be completely translatable to studies of specific spatial niches such as those using suprapubic aspiration to study the bladder microbiome.40,41
While we were able to culture most high-abundance genera detected by WGMS, our culturing method used urine cryopreserved in glycerol. As of the writing of this report, culture methods using cryopreserved urine have not been benchmarked against those using fresh samples. Future studies quantitatively assessing culture recovery of diverse urinary microbiota species from fresh versus frozen urine will be of great utility to the field. It should also be noted that while 16S rRNA gene sequencing is able to confidently distinguish species with divergent sequences, some members of the urogenital microbiota, namely Gardnerella spp., possess highly similar 16S rRNA sequences and their speciation requires further genetic information.81
Because taxonomic biomarker analysis in sparse microbiomes, such as that of the urogenital tract, is challenging, we explicitly stress that these identified microbial biomarkers are putative and need to be validated in future independent studies. Limitations of our urogenital microbiome ARG analysis include that the analytical pipeline does not distinguish between TEM alleles and does not detect common SNPs known to confer fluoroquinolone resistance, for example.
Although all participants who used EHT reported being fully compliant with the prescribed treatment regimen, it is a limitation that a detailed history of day-to-day EHT use was not recorded. EHT modalities may also differ in dosage, composition, patient compliance, or primary metabolism. Indeed, we found that both oral and patch EHT were associated with elevated urinary estrogens, while vEHT was not. While it has been shown that vEHT is systemically absorbed at low levels, it is possible that dosage, administration frequency, and sub-physiologic absorption affected the urinary accumulation of excreted estrogens.82, 83, 84, 85 While the variable association between vEHT and urogenital Lactobacillus abundance is in line with previous observations, the relatively small sample size of this study is a limitation.36,53,76 Given the inherent variability in human-derived data, future longitudinal studies with large, controlled cohorts as well as appropriate animal models will be critical to gaining a mechanistic understanding of the relationship between EHT and urogenital microbiome composition.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| PF5 E. coli | This paper | 957 |
| PF19 E. coli | This paper | 1098 |
| C152 E. coli | This paper | 1318 |
| PF20 E. coli | This paper | 1515 |
| PF49 E. coli | This paper | 1679 |
| PF36 E. coli | This paper | 1865 |
| PF42 E. coli | This paper | 1930 |
| PF21 E. coli | This paper | 278 |
| C158 E. coli | This paper | 1361 |
| PF33 E. coli | This paper | 1560 |
| PF34 E. coli | This paper | 1674 |
| PF26 E. coli | This paper | 1669 |
| PF16 E. coli | This paper | 1064 |
| PF22 E. coli | This paper | ECPF22 |
| PF38 E. coli | This paper | ECPF38 |
| PF18 K. pneumoniae | This paper | 200 |
| PF35 K. pneumoniae | This paper | 1080 |
| PF10 K. oxytoca | This paper | 1856 |
| PF8 S. anginosus | This paper | 1593 |
| PF1 S. anginosus | This paper | 1936 |
| PF54 S. agalactiae | This paper | 1689 |
| PF13 E. faecalis | This paper | 262 |
| PF27 S. epidermidis | This paper | 730 |
| Biological samples | ||
| Human-derived urine samples | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sodium 17β-Estradiol-16,16,17-d3 3-Glucuronide | C/D/N isotopes | D-6867 |
| Estrone-2,4,16,16-D4 3-Sulfate Sodium Salt | Millipore Sigma | 524956 |
| Critical commercial assays | ||
| ZymoBIOMICS DNA/RNA Miniprep Kit | Zymo Research | R2002 |
| ZymoBIOMICS Spike-in Control I (High Microbial Load) | Zymo Research | D6320 |
| ZymoBIOMICS Microbial Community Standard II (Log Distribution) | Zymo Research | D6310 |
| Invitrogen Qubit 1X dsDNA High Sensitivity (HS) and Broad Range (BR) Assay Kits | Invitrogen | Q33231 |
| Nextera DNA Flex Library Kit | Illumina | https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/nextera-dna-flex.html |
| Creatinine Urinary Detection Kit | ThermoFisher Scientific | EIACUN |
| Deposited data | ||
| Raw Metagenomic Sequencing Data (human reads removed) | This paper | [NCBI]: PRJNA801448 |
| Oligonucleotides | ||
| Universal 16S rRNA Forward Primer “8F” – AGAGTTTGATCCTGGCTCAG |
Vaishnava et al. 2019 | N/A |
| Universal 16S rRNA Reverse Primer “1492R – GGTTACCTTGTTACGACTT |
Vaishnava et al. 2019 | N/A |
| Universal ITS1 Primer – TCCGTAGGTGAACCTGCGG |
Genewiz | https://www.genewiz.com/en/Public/Services/Molecular-Genetics/Bacterial-and-Fungal-Identification |
| Universal ITS2 Primer – GCTGCGTTCTTCATCGATGC |
Genewiz | https://www.genewiz.com/en/Public/Services/Molecular-Genetics/Bacterial-and-Fungal-Identification |
| Software and algorithms | ||
| fastqc (v0.11.2) | Andrews et al. 2015 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| fastq_screen (v0.4.4) | Wingett and Andrews 2018 | https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ |
| Trim galore | Krueger | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| KneadData | Huttenhower lab | https://huttenhower.sph.harvard.edu/kneaddata/ |
| MetaPhlAn2 | Segata et al. 2012 | https://huttenhower.sph.harvard.edu/metaphlan2/ |
| phyloseq (v1.16.2) | McMurdie and Holmes, 2013 | https://joey711.github.io/phyloseq/ |
| CCREPE (v.1.7.0) | Huttenhower lab | https://huttenhower.sph.harvard.edu/ccrepe/ |
| CytoScape (v 3.8.2) | Cytoscape | https://cytoscape.org |
| regclass R package | Adam Petrie (Author), 2020 | https://cran.r-project.org/web/packages/regclass/index.html |
| rgr R package | Robert G. Garrett (Author), 2018 | https://cran.r-project.org/web/packages/rgr/index.html |
| HUMAnN 2.0 | Franzosa et al., 2018 | https://huttenhower.sph.harvard.edu/humann2/ |
| MetaCyc | Caspi et al., 2018 | https://metacyc.org |
| factoextra R package | Kassamdara & Mundt 2020 | https://cran.r-project.org/web/packages/factoextra/index.html |
| LEfSe | Segata et al., 2011 | https://huttenhower.sph.harvard.edu/lefse/ |
| GROOT | Rowe and Winn, 2018 | https://groot-documentation.readthedocs.io/en/latest/ |
| BMDA | Li et al., 2019 | https://github.com/shuangj00/MicrobiomeBayesDiff |
| DESeq2 | Love et al. 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ANCOM-BC | Lin et al. 2020 | https://www.bioconductor.org/packages/release/bioc/html/ANCOMBC.html |
| MassLynx | Waters | https://www.waters.com/waters/en_US/MassLynx-Mass-Spectrometry-Software-/nav.htm?cid=513164&locale=en_US |
| TargetLynx | Waters | https://www.waters.com/waters/en_US/TargetLynx-/nav.htm?cid=513791&locale=en_US |
| pwr R package | Champely 2020 | https://github.com/heliosdrm/pwr |
| R statistical programing language v4.2.0 | R Core Team 2020 | https://www.r-project.org |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Custom code and data for differential abundance simulation study | This paper | https://github.com/klutz920/BMDA-Simulation |
| Custom code for Bayesian correlation analysis with urinary estrogen metabolites | This paper | https://github.com/klutz920/Bayes-Correlation-Test |
| Custom code for Bayesian proportional enrichment analysis for ARGs | This paper | https://github.com/klutz920/Bayes-Proportion-Test |
| Other | ||
| NextSeq 500 | Illumina | SY-415-1001 |
| Xevo TQ mass spectrometer | Waters | N/A |
| ACQUITY UPLC | Waters | N/A |
| C8 Reverse Phase column (100x2.1mm 1.8μm) | UCT | SLC-8100ID21-18UM |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nicole J. De Nisco (nicole.denisco@utdallas.edu).
Materials availability
All bacterial and fungal strains generated for this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Human subjects
A-priori sample size and power estimation
To estimate the number of patients needed to enroll into each group and predict statistical power, we performed a series of power analyses using the ‘pwr’ package (https://github.com/heliosdrm/pwr) in the R-statistical language based on ranging effect sizes from small to large for both multivariate analysis of 3 groups with equal sample size and pairwise-based comparisons of two groups. Balancing cost and clinical feasibility with predicted statistical power, we chose a sample size of 25 for each group. A priori power analysis found that a sample size of 25 per group was sufficient to generate a power of 0.8 to detect and effect size (f) of 0.366 for an ANOVA and an effect size (d) of 0.808 for a pairwise comparison (T-test) with an alpha of 0.05. Therefore, the cohort is sufficiently powered to observe medium to large effect sizes (Figures S1A and S1B). It should be noted that the associations with EHT were observed on a subset of data following the completion of data collection and were accordingly not part of the a priori sample size calculations used during cohort construction. The No UTI History and rUTI History, UTI(−) groups (n = 50) were grouped by EHT use (EHT(−) n = 21, EHT(+) n = 29) or by EHT modality (EHT(−) n = 21, oEHT n = 6, pEHT n = 6, vEHT n = 17). All reported differences between these groups achieve sufficient statistical significance to reject the null hypothesis of no difference between groups.
Patient recruitment and cohort curation
The current study is approved under IRBs STU032016-006 (University of Texas Southwestern Medical Center) and 19MR0011 (University of Texas at Dallas). Patients were recruited from the Urology Clinic at the University of Texas Southwestern Medical Center between April 2018 and October 2019. Written informed consent was obtained from each patient prior to recruitment into the study cohorts. All patients were PM females. The following set of exclusion criteria were used to initially screen patient’s candidacy for enrollment into the cohort: pre- or perimenopausal status; complicated rUTI; antibiotic exposure within the 4 weeks prior to urine sample donation unless an active infection was detected by culture; pelvic malignancy or history of pelvic radiation within 3 years before urine sample donation; currently receiving chemotherapy; exhibiting renal insufficiency (creatinine >1.5 mg/dL); most recent post void residual (PVR) greater than 100 mL; greater than stage 2 prolapse; pelvic procedure for incontinence within 6 months prior to urine sample donation; use of intermittent catheterization; neurogenic bladder; any upper urinary tract abnormality which may explain rUTI; and Diabetes Mellitus (DM) type 1 or 2. One participant, PF21, from the rUTI History, UTI(+) group had a culture-confirmed, active UTI and antibiotic exposure within the preceding 4 weeks. Among all participants, EHT was either prescribed for rUTI or vaginal atrophy. All urine samples were obtained by “clean-catch” midstream urine collection and therefore were representative of the urogenital microbiome, rather than specifically just the bladder microbiome.41 Patients were educated about the cleaning and urine collection needs for this sampling technique prior to urine collection. Urine samples were stored at 4°C for no more than 3 h before sample processing, aliquoting, and biobanking at −80°C. In total, 258 patients were recruited and screened for enrollment candidacy through interview, clinical assessment, and electronic medical records.
Cohort group definition and curation
Candidates for the No UTI History group both self-reported as having no lifetime history of symptomatic UTI and had no clinically documented history of UTI diagnosis through analysis of electronic clinical records. Urine was screened by both standard clinical urine culture and by plating 100 μL on BD CHROMagar Orientation within 3 h of collection to screen for the presence of uropathogens and asymptomatic bacteriuria.45 Participants were enrolled in the No UTI history groups if they had no self-reported or clinical history of UTI, no UTI symptoms at the time of urine collection, and <104 CFU/mL of known uropathogen by urine culture screening.
Candidates for the rUTI History, UTI(−) group were identified from a pool of patients who had previously sought treatment for clinically diagnosed rUTI in the Urology clinic at the University of Texas Southwestern Medical Center. All candidates for the rUTI History, UTI(−) group had experienced at least 2 UTIs in the preceding 6 months or 3 UTIs in the preceding 12 months to the day of urine sample collection but did not currently present with symptomatic UTI. Culture-based assessment of UTI status was performed as described for the No UTI history cohort. rUTI History, UTI(−) group participants passed the general exclusion criteria, had a clinical history of rUTI within the preceding 12 months, were not experiencing symptomatic UTI at the time of urine collection, and had <104 CFU/mL of known uropathogens by urine culture screening.
Candidates for the rUTI History, UTI(+) group were identified from a pool of patients seeking treatment for clinically diagnosed rUTI in the Urology clinic at the University of Texas Southwestern Medical Center. All candidates for the rUTI History, UTI(+) group had experienced at least 2 UTIs in the preceding 6 months or 3 UTIs in the 12 months prior to urine collection and presented with a symptomatic, culture confirmed UTI on the day of urine collection. Culture-based confirmation of UTI status was performed as described in the No UTI History group section using a cutoff of >104 CFU/mL of known uropathogen.86,87 rUTI History, UTI(+) group participants passed the general exclusion criteria, had a clinical history of rUTI in the last 12 months, and were experiencing symptomatic, culture-proven UTI at the time of sampling.
Bacterial strains
Advanced urine culture and isolate biobanking
Glycerol-stocked urine samples (stored at −80°C) were thawed at room temperature, and then diluted 1:3 and 1:10 in sterile 1X Phosphate Buffered Saline to adjust plating density for high and low biomass samples. 100 μL of urine from each dilution as well as 100 μL of undiluted urine was plated onto blood agar plates (BAP), CHROMagar Orientation, De Man, Rogosa, and Sharpe (MRS) agar, Rabbit BAP (R-BAP), BD BBL CDC anaerobe blood agar (CDC AN-BAP), and Columbia Colistin Naladixic Acid Agar (CNA). Following plating, BAP was incubated in ambient and 5% CO2 atmospheres, CHROMagar Orientation in 5% CO2, MRS and R-BAP in microaerophilic conditions, BD BBL CDC anaerobe blood agar (CDC AN-BAP) in microaerophilic and anaerobic conditions and CNA in all four atmospheric conditions. Plates were incubated at 35°C for 4 days in the respective atmosphere. It should be noted that we were unable to culture Gardnerella spp. using these methods. However, WGMS profiling frequently detected G. vaginalis in the sampled urogenital microbiomes. For targeted isolation of Gardnerella spp., 100 μL urine was plated onto Human polysorbate-80 (HBT) bilayer medium in microaerophilic atmosphere for 3 days. To isolate fungal species, 100 μL urine was plated onto Brain Heart Infusion Agar supplemented with 20 g/L glucose and 50 mg/μL of chloramphenicol (BHIg-Cam) and incubated at 5% CO2 for 3 days.
Bacterial identification was performed by PCR amplification and Sanger sequencing of the 16S rRNA gene from well-isolated colonies as described previously.88 Briefly, the16S rRNA gene was amplified using primers 8F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) by colony PCR89 using DreamTaq Master Mix (ThermoFisher Scientific) and 0.2μM primers. Amplicon size was confirmed on 1% agarose gel, followed by gel purification (Bio basic) and Sanger Sequencing (Genewiz) using the 8F primer. Sequences were analyzed using BLASTn against the NCBI 16S ribosomal RNA (Bacteria and Archaea) database.
For fungal identification, ITS1 and ITS2 regions were amplified using the primer sequences ITS1: 5′-TCCGTAGGTGAACCTGCGG-3′ and ITS2: 5′-GCTGCGTTCTTCATCGATGC-3′ from well-isolated colonies and Sanger sequenced (Genewiz, South Plainfield, NJ, USA). Sequences were analyzed using BLASTn against the NCBI ITS from Fungi type and reference material database.
All the isolated and taxonomically identified isolates (n = 896) were assigned a distinct ID and biobanked at −80°C in glycerol. The isolates were grown in Brain Heart Infusion broth, Tryptic Soy Broth, MRS broth or NYCIII according to their growth preferences and stocked in 16% sterile glycerol for long-term storage at −80C.
Method details
DNA isolation and sequencing
Prior to WGMS, we assessed the quality and reproducibility of 3 metagenomic DNA extraction techniques: a modified genomic DNA (gDNA) isolation based on the Qiagen blood and tissue DNAeasy Kit, the Zymo Research DNA/RNA microbiome miniprep kit, and a modified phenol/chloroform/isoamyl alcohol extraction as demonstrated by Moustafa et al.42 After assessing the quality and yield of metagenomic DNA isolated using the three methods, we chose the Zymo Research kit. Urine samples were allowed to thaw on ice at 4°C. 10–20 mL of urine was centrifuged for 15 min at 4000 × g at 4°C. Urine pellets were resuspended in 750 μL of DNA/RNA Shield (Zymo Research), transferred to a bead beating tube, and subjected to ten 30 s cycles of mechanical bead beating, with 5 min cooling between each cycle. After mechanical lysis, the maximum volume of sample was collected and transferred to a new microcentrifuge tube with DNA/RNA lysis buffer (Zymo). Nucleic acids were purified via the Zymo Research DNA/RNA microbiome miniprep kit per the manufacturer’s instruction. Elution of DNA from the column was performed in nuclease-free water and each column was eluted twice to maximize DNA recovery. As a control to internally assess gDNA extraction efficiency and WGMS limit of detection (LOD), gDNA was concurrently extracted from commercially available community standards (Zymo Research) using the same methods. gDNA was also extracted from nuclease-free water to account for kit and environmental contamination. All DNA samples were subjected to 16S rRNA gene amplification by PCR and visualized by agarose gel electrophoresis to ensure microbial DNA was present before proceeding with WGMS. DNA yield and purity for all samples were assessed by agarose gel electrophoresis, and by fluorescence-based Qubit quantitation of DNA, RNA, and protein. Prior to library preparation the DNA concentration of each sample was normalized and 20pg of spike-in gDNA was added (Zymo Research High Bacterial Load Spike-in), which contains gDNA from the bacterial species Imtechella halotolerans and Allobacillus halotolerans, which are known to not be associated with humans.
WGMS was performed at the University of Texas at Dallas Genome Center using 2 × 150 bp paired-end reads on a Illumina NextSeq 500. Library preparation was performed using Nextera DNA Flex kit. Library preparation of the entire cohort and community standard and water controls was distributed over 2 batches with overlapping samples. All samples were sequenced using 2 × 150 base pair paired-end sequencing in high output mode with a target of ≥50 million paired end reads per sample.
Bioinformatic analyses
All taxonomic, functional, and resistome bioinformatic analyses were performed on an in-house Dell PowerEdge T630 server tower with 256GB RAM, 12 core Intel Xenon processor with 16TB storage capacity or at the Texas Advanced Computing Center (TACC).
Data preprocessing
The fastq files were checked for read quality, adapter content, GC contents, species contamination using fastqc (v0.11.2) and fastq_screen (v0.4.4).90,91 Low-quality reads (a quality score of less than Q20) and adapter were removed using Trim galore (v 0.4.4).92 (Figure S1D). Human DNA sequences were removed using KneadData.93 After host removal, the dataset contained an average of 2.6 × 107 non-human reads per sample.
Taxonomic profiling analyses
The taxonomic assignment and estimation of composition of microbial species present in each sample was performed using MetaPhlAn2.43 MetaPhlAn2 estimates the relative abundance of species by mapping the metagenomic reads against a clade specific marker gene database. The database consists of bacterial, archaeal, viral and eukaryotic genomes. We further used merge_metaphlan_tables module of MetaPhlAn2 to combine the relative abundance estimates of samples in a cohort into one table.
To identify kit, environmental, and background contaminating taxonomic signals, we sequenced a water sample which was randomly inserted into the metagenomic DNA preparation protocol. Sequencing and taxonomic analysis of this sample revealed known kit and environmental contaminants, such as Delftia, Stenotrophomonas, Ralstonia, Bradyrizobium, and others.44 Unless a known member of the human microbiome, these taxa were censored. We observed a small relative abundance of taxa salient to the human urogenital microbiome in the water control, such as Pseudomonas, Escherichia, Klebsiella, Enterococcus, Staphylococcus, and Corynebacterium. These signals ranged from 0.051%–11.5% of approximately 3 million mappable reads observed in the water control (Figure S2A). We further assessed the WGMS limit of reliable detection using a commercially available log community standard (Zymo Research), which is composed of multiple Gram-positive and Gram-negative bacterial and fungi. We observed a strong linear correlation between the theoretical and observed relative abundance above 0.001%. We therefore set a relative abundance threshold of 0.001% for a taxon to be considered as detected within a sample (Figure S1E). Species-level MetaPhlAn 2 taxonomic assignments were not included in analysis if they were “unclassified”.
Alpha-diversity analysis was performed at the species-level using phyloseq (version 1.16.2).94 Beta-diversity analysis was performed using DPCoA on the species-level taxonomic relative abundance dataset using phyloseq (version 1.16.2).94 Taxonomic co-occurrence was performed with CCREPE pipeline using the Pearson correlation and compositionally corrected P-values (https://github.com/biobakery/biobakery/wiki/ccrepe#22-ccrepe-function). Network analysis of taxonomic co-occurrences was performed using CytoScape (Version 3.8.2) with edges defined by the correlation coefficients between taxa nodes in the default prefuse force directed layout.
Taxonomic correlation with urinary estrogens
The Spearman correlation was calculated between a given taxa and urinary estrogen conjugate or conjugate sum using the associate function from the regclass R package. To account for the compositionality of the taxa when computing the correlations, the species-level taxonomic composition dataset was transformed using the centered log ratio (CLR) transformation using the clr function from the rgr R package. Nominal P-values were calculated by permutation. No multiple hypothesis correction was performed on nominal P-values as we considered this an exploratory analysis.95
The Bayesian correlation analysis employed a posterior distribution with the Dirichlet-Multinomial (DM) mode for the full data likelihood and a non-informative uniform prior proportional to one.64 The DM models the non-transformed count data directly and estimates their normalized abundances while accounting for overdispersion in the species-level count data. We then computed the Spearman correlation between each of the estimated normalized abundances and urinary estrogen conjugate sum. For posterior inference, we computed the 95% credible intervals and posterior means for the correlation between each of the normalized abundances and urinary estrogen conjugate sum. The correlation of a taxa-estrogen pair was significant if zero was not contained in the credible interval. Furthermore, for the significant pairs, we calculated the ratio of the proportion of posterior samples with correlations greater than 0.3 to those less than or equal to 0.3. This ratio (Posterior Ratio) indicates correlation strength where a ratio higher than one indicates a more moderate or strong correlation and less than one indicates a weak correlation.
Functional metabolic profiling
Functional metabolic profiling was performed using HUMAnN 2.0.65 HUMAnN2 uses a tiered approach to identify the functional profile of microbial communities. Firstly, it maps the sample reads to clade specific markers and creates a database of pangenomes for each sample. In the second tier, it performs the nucleotide level mapping of sample reads against pangenome database. Lastly, a translated search against Uniref90 is performed for unaligned reads in each sample.96 The output result is the mapping of reads to gene sequences with known taxonomy. The reads are normalized to gene sequence length to give an estimate of per-organism and community total gene family abundance. Next, gene families were analyzed to reconstruct and quantify metabolic pathways using MetaCyc.97 Different modules of HUMAnN such as humann2_join_table and humann2_renorm_table were used to merge the pathway abundance of all the samples in a cohort and normalize the abundance to counts per million (cpm) respectively. We filtered the results to only include pathways whose taxonomic range included bacteria. We further censored pathways which were specifically associated with a particular taxon due to database bias toward commonly isolated and studied species. PCA of functional pathways was performed on the pathway level relative abundance dataset using factoextra (https://cran.r-project.org/web/packages/factoextra/readme/README.html). Pathway differential abundance analysis was performed using LEfSe58 on the pathway-level relative abundance dataset. LEfSe uses Kruskal Wallis and Wilcoxon tests to find the differential pathways between microbial communities. Finally, it uses LDA model to rank the pathways.
Resistome profiling and arg enrichment
We used GROOT (Graphing Resistance Out Of meTagenomes) to generate a profile of antimicrobial resistance genes within the urogenital microbiomes of the present study.66 The default database ARG-ANNOT was used for alignment of the metagenomics reads. Subsequently GROOT report command was used to generate a profile of antibiotic resistance genes at a read coverage of 90%. Filtering of the GROOT results was performed to insure high confidence in ARG presence within the urogenital microbiomes. We used a cutoff of a sufficient amount of reads to generate 10× coverage of an ARG to qualify its detection within a urogenital microbiome. We further collapsed alleles of the β-lactamase genes TEM, CTX, OXA, OXY2, SHV, and cfxA as well as the aminoglycoside ARG Aac3-IIa and Aac3-IIe alleles into single gene-level features to account for multiple-mapping reads.
Bayesian modeling of the resistome data was performed as follows. Resistome data for the three cohorts (No UTI History = 1, rUTI History, UTI(−) = 2, rUTI History, UTI(+) = 3) consisted of 186 antimicrobial resistance genes (ARG) which were collapsed into family-level genes (). Each cell in the data set contained a binary indicator of no detection (0) or detection (1) of the resistance family-level gene within each patient sample such that indicates no detection or detection of resistance family-level gene respectively for sample in cohort .
A Bayesian Beta-Bernoulli model with Jeffreys prior was used to model the posterior distributions of group proportions and pairwise differences for the family-level genes. Two posterior inferences were performed. First, we removed any family-level genes that had no significant pairwise contrast of cohort proportions using 95% credible intervals as criteria. We determined that a significant family-level gene does not have zero contained in a 95% credible interval for at least one pairwise contrast.98 Second, we computed the posterior probability and Bayes Factor (BF) to make inferences on each pairwise contrast of cohort proportions of only the significant family-level genes. The posterior probability of a particular contrast was computed as the proportion of posterior samples satisfying that contrast. The BF computed for each contrast represented the odds of “at least one cohort’s proportion (ω) for gene is different” in favor of .
Taxonomic biomarker analysis
We applied multiple methods of taxonomic differential abundance analysis employing the robust and widely used LEfSe pipeline as well as ANCOM-BS, and BMDA, a recently described Bayesian model of differential abundance.57 LEfSe and ANCOM-BC analyses were performed as previously described.58,59 For the BMDA model we first applied the quality control step (detailed in the supplement of Li et al., 2019) to the raw count data. We then fitted the BMDA model, which is a Bayesian hierarchical framework that uses a zero-inflated negative binomial model to model the raw count data and a Gaussian mixture model with feature selection to identify differentially abundant taxa. The BMDA can fully account for zero-inflation, over-dispersion, and varying sequencing depth. We chose weakly informative priors on all parameters of the model to avoid biased results. For model fitting and posterior inference, BMDA implements the Metropolis-Hastings algorithm within a Gibbs sampler. For prior specification in the top level of the BMDA framework, we used the following default settings. We set the hyperparameters that control the selection of discriminatory features, , resulting in the proportion of taxa expected a priori to discriminate among the groups to be . As for the inverse-gamma priors on the variance components and , we set the shape parameters and the scale parameters to achieve a fairly flat distribution with an infinite variance. We further set the default values of and to 100. For the bottom level of BMDA, we used the following weakly informative settings. The hyperparameters that controlled the percentage of extra zeros a priori were set to . As for the gamma prior on the dispersion parameters, i.e. , we set both and to small values such as 0.001, which led to a vague prior with expectation and variance equal to 1 and 1,000. For the Dirichlet priors on the size factors , we followed Li et al. (2017) by specifying , and .99 For each dataset, we ran a MCMC chain with 10,000 iterations (first half as burn-in). The chain was initialized from a model with 5% randomly chosen set to 1. The marginal posterior probability of inclusion (PPI) was used to identify the set of discriminating taxa between the control and disease groups. Marginal PPI is the proportion of MCMC samples in which a taxon is selected to be discriminatory if it is greater than a pre-specified value. We chose a threshold such that the expected Bayesian false discovery rate (FDR) was less than 0.05.
To assess the ability of BMDA to control for type 1 error, we compared it with commonly used methods for microbial differential abundance analysis, all of which can be implemented in R: DESeq2,100 Wilcoxon, and ANCOM-BC.59 To do so, we generated the negative-control datasets by randomly permuting the two-condition labels (rUTI status or history) our rUTI microbiome data. Specifically we permuted the rUTI status label for the UTI(-)versus UTI(+) comparison and the rUTI history label for the No UTI history versus rUTI history comparison. Any differentially abundant taxa reported from the permuted datasets for each comparison are considered as false discoveries. A method that can control adequately for type 1 error should report few or zero differentially abundant taxa. We conducted this permutation 50 times for each comparison and implemented the ANCOM-BC, BMDA, DESeq2, and Wilcoxon rank-sum tests on the data. DESeq2 employs a Wald test by adopting a generalized linear model based on a negative binomial kernel. ANCOM-BC adopts a linear regression framework and corrects for latent sampling bias. For all competing methods we used default settings. To control for a false discovery rate of 5%, we further adjusted the nominal p-values computed by ANCOM-BC, DESeq2, and Wilcoxon using the Benjamini-Hochberg (BH) method as was performed in Nearing et al. and choose the threshold of posterior probability of inclusion estimated by BMDA following methods outlined in Newton et al.101,102
To further evaluate the performance of BMDA and alternative methods, we generated synthetic data, following the scheme originated by Weiss et al.103 Note that the multinomial distribution-based generative model is substantially different from BMDA, which assumes the counts follow the zero-inflated negative binomial distribution. A brief description of the data generative scheme is described below, while detailed information can be found in the supplement of Weiss et al. Let a -by- count matrix be the simulated taxonomic abundance table, where the number of taxa is set to be and the number of truly discriminatory taxa among two equally sized groups . To incorporate real taxa abundance information, let be a count vector, where , and each was the sum of operational taxonomic unit (OTU) counts for one randomly selected taxon (without replacement) from all the skin samples in a real microbiome study.104 We defined two -by-1 vectors, and , as
where represented the log effect size. Note that . We further drew the observed counts from a multinomial model , where and . Here, denotes the indicator function and is the total sample size. This would yield the first taxa to be truly discriminating between the two equally sized groups. Finally, we permuted the columns of the taxonomic abundance table, , to disperse the taxa. We repeated the steps above to generate 50 replicates. We then assessed the power of each model as a function of FDR.
Antibiotic susceptibility testing (AST)
Assessment of antibiotic (abx) susceptibility was performed via the Kirby-Bauer disk diffusion susceptibility test.105 Antibiotic disks were prepared by aliquoting 10uL of antibiotic stock (GEN 1mg/mL, AMP 1mg/mL, CIP 0.5mg/mL, LVX 0.5mg/mL, ERM 1.5mg/mL, CHL 3mg/mL, TMP/SMX 1.25/23.75mg/mL, NIT 30mg/mL, DOX 3mg/mL) onto the disk in a sterile petri dish and drying at room temperature in the dark. Vehicle control disks were prepared similarly using the diluents of each antibiotic. Strains were streaked from frozen glycerol stocks onto CHROMagar or Blood Agar (species dependent) and incubated overnight at 37°C in ambient conditions or 35°C in 5% CO2. Single, well-isolated colonies were inoculated into 3 mL Brain-Heart-Infusion broth and incubated at the respective atmospheric conditions for 16–18 h. After incubation, cultures were normalized to 0.5 McFarland standard, washed, and resuspended in sterile 1X Phosphate-Buffered Saline (PBS). 150 μL of standardized culture were pipetted onto 150 mm Mueller-Hinton Agar plates and spread using sterile glass beads. Plates were dried in sterile conditions before abx-infused disks were placed on the surface of the agar. E. coli strain ATCC25922 was used for quality and vehicle controls. Sterile 1X PBS was plated as sterility control. Plates were incubated inverted per the recommendation of Clinical and Laboratory Standards Institute (CLSI) M100-ED30: 2020 Performance Standards for AST, 30th Edition (https://clsi.org/standards/products/microbiology/documents/m100/). After incubation, antimicrobial susceptibility was evaluated by measurement of the zone of inhibition and using CLSI established zone diameter breakpoints.
Liquid chromatography mass spectrometry
Direct measurement of urinary estrogen metabolites was performed via a modification of previously reported methods.63 Briefly, urine (500 μL) was diluted and spiked with 100 ng stable isotope-labeled internal standards of d3-Estradiol 3-Glucuronide and d4-Estrone 3-Sulfate. Diluted and spiked samples were loaded onto an equilibrated Phenomenex C18 cartridge for solid phase extraction to separate conjugated estrogens. Following aqueous methanolic extraction of estrogen conjugates and non-polar extraction of free estrogens with methanolic acetone, fractions were dried by vacuum centrifugation and prepared for LC-MS/MS analysis. Estrogen conjugates (sulfates and glucuronides) were directly assayed using a curated and optimized MRM library by LC-MS/MS.
High sensitivity quantitative LC-MS/MS was performed on a Waters Xevo TQ tandem quadrupole MS lined to an ACQUITY UPLC with a Selectra C8 RP column (100 × 2.1 mm 1.8μm, UCT). MRM libraries of estrogen conjugates have been curated to include both analytical and confirmatory transitions for each analyte at optimal retention times to maximize separation. Briefly, data analysis was performed by integrating the peak area of the analytical transition for each analyte. Peak areas were normalized to molecular class-matched internal spike-in standards and mapped to a standard curve to accurately estimate analyte concentration. Urine estrogen metabolite concentrations were then normalized to urinary creatinine, which was measured by colorimetric assay (Sigma).
Quantification and statistical analysis
Statistical analysis was performed using R statistical programing, GraphPad Prism 9, and Microsoft Excel. For hypothesis testing, non-parametric Mann-Whitney U-test was used for pairwise comparisons and the Kruskal-Wallis non-parametric ANOVA with multiple comparison post-hoc was used for non-paired and unmatched comparisons of 3 or more groups. Multiple comparison adjustment was performed using false discover rate (FDR) when appropriate. An alpha of 0.05 was used to control type I error.
Acknowledgments
We wish to acknowledge and sincerely thank the patients who participated in this study. This work was supported by grants from the Welch Foundation (AT-2030-20200401), the Foundation for Women’s Wellness (FWW), and the National Institutes of Health (1R01DK131267-01) to N.J.D., as well as the University of Texas at Dallas New Faculty Research Symposium grant to Q.L. and N.J.D. This work was also supported by the Cecil H. and Ida Green Chair in Systems Biology Science to K.L.P. and The Felicia and John Cain Distinguished Chair in Women’s Health to P.E.Z. We would like to thank all the members of the De Nisco and Palmer labs for their creative input throughout this study. We would further like to acknowledge and thank the Genome Center at the University of Texas at Dallas for their invaluable work and assistance in generating the metagenomic dataset used for this study.
Author contributions
Conceptualization, M.L.N., Q.L., P.E.Z., K.L.P., and N.J.D.; data curation, M.L.N., J.F., A. Kuprasertkul, and A.P.A.; formal analysis, M.L.N, A. Kumar., K.C.L., C.Z., Q.L., C.X., V.S., and N.J.D.; funding acquisition, V.S., Q.L., K.L.P, P.E.Z., and N.J.D.; investigation, M.L.N., N.V.H., V.H.N., A.N., N.N., and T.E.; methodology, M.L.N., A. Kumar., N.V.H., K.C.L., V.H.N., A.N., B.M.S., Q.L., C.X., V.S., P.E.Z., K.L.P., and N.J.D.; project administration, M.L.N., P.E.Z., and N.J.D.; resources, V.S., P.E.Z., K.L.P., and N.J.D.; software, M.L.N., A. Kumar., K.C.L., C.Z., Q.L., and C.X.; supervision, Q.L., C.X., V.S., P.E.Z., K.L.P., and N.J.D.; validation, M.L.N., N.V.H., K.C.L., and C.Z.; visualization, M.L.N., V.H.N., and N.J.D.; writing – original draft, M.L.N., A. Kumar., N.V.H., K.C.L., Q.L., C.X., and N.J.D.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: September 30, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100753.
Supplemental information
Data and code availability
Whole genome metagenomic sequencing read data (FASTQ files) have been deposited onto the NIH Sequence Read Archive (SRA) under the BioProject number [NCBI]: PRJNA801448. Prior to depositing, all human-mapping reads were removed from the data. All original code and simulation study data has been deposited at https://github.com/klutz920/BMDA-Simulation, https://github.com/klutz920/Bayes-Correlation-Test, and https://github.com/klutz920/Bayes-Proportion-Test. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Gaitonde S., Malik R.D., Zimmern P.E. Financial burden of recurrent urinary tract infections in women: a time-driven activity-based cost analysis. Urology. 2019;128:47–54. doi: 10.1016/j.urology.2019.01.031. [DOI] [PubMed] [Google Scholar]
- 2.Jhang J.F., Kuo H.C. Recent advances in recurrent urinary tract infection from pathogenesis and biomarkers to prevention. Ci Ji Yi Xue Za Zhi. 2017;29:131–137. doi: 10.4103/tcmj.tcmj_53_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flores-Mireles A.L., Walker J.N., Caparon M., Hultgren S.J. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015;13:269–284. doi: 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malik R.D., Wu Y.R., Zimmern P.E. Definition of recurrent urinary tract infections in women: which one to adopt? Female Pelvic Med. Reconstr. Surg. 2018;24:424–429. doi: 10.1097/SPV.0000000000000509. [DOI] [PubMed] [Google Scholar]
- 5.Anger J., Lee U., Ackerman A.L., Chou R., Chughtai B., Clemens J.Q., Hickling D., Kapoor A., Kenton K.S., Kaufman M.R., et al. Recurrent uncomplicated urinary tract infections in women: AUA/CUA/SUFU guideline. J. Urol. 2019;202:282–289. doi: 10.1097/JU.0000000000000296. [DOI] [PubMed] [Google Scholar]
- 6.Eriksson I., Gustafson Y., Fagerström L., Olofsson B. Do urinary tract infections affect morale among very old women? Health Qual. Life Outcomes. 2010;8:73. doi: 10.1186/1477-7525-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang M., Quanstrom K., Jin C., Suskind A.M. Recurrent urinary tract infections are associated with frailty in older adults. Urology. 2019;123:24–27. doi: 10.1016/j.urology.2018.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neugent M.L., Hulyalkar N.V., Nguyen V.H., Zimmern P.E., De Nisco N.J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 2020;11:e00218–e00220. doi: 10.1128/mBio.00218-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kranjčec B., Papeš D., Altarac S. D-mannose powder for prophylaxis of recurrent urinary tract infections in women: a randomized clinical trial. World J. Urol. 2014;32:79–84. doi: 10.1007/s00345-013-1091-6. [DOI] [PubMed] [Google Scholar]
- 10.Harding C., Mossop H., Homer T., Chadwick T., King W., Carnell S., Lecouturier J., Abouhajar A., Vale L., Watson G., et al. Alternative to prophylactic antibiotics for the treatment of recurrent urinary tract infections in women: multicentre, open label, randomised, non-inferiority trial. BMJ. 2022;376:e068229. doi: 10.1136/bmj-2021-0068229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malik R.D., Wu Y.R., Christie A.L., Alhalabi F., Zimmern P.E. Impact of allergy and resistance on antibiotic selection for recurrent urinary tract infections in older women. Urology. 2018;113:26–33. doi: 10.1016/j.urology.2017.08.070. [DOI] [PubMed] [Google Scholar]
- 12.Stamm W.E., Norrby S.R. Urinary tract infections: disease panorama and challenges. J. Infect. Dis. 2001;183:S1–S4. doi: 10.1086/318850. [DOI] [PubMed] [Google Scholar]
- 13.Wolfe A.J., Brubaker L. Urobiome updates: advances in urinary microbiome research. Nat. Rev. Urol. 2019;16:73–74. doi: 10.1038/s41585-018-0127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilt E.E., McKinley K., Pearce M.M., Rosenfeld A.B., Zilliox M.J., Mueller E.R., Brubaker L., Gai X., Wolfe A.J., Schreckenberger P.C. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J. Clin. Microbiol. 2014;52:871–876. doi: 10.1128/JCM.02876-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siddiqui H., Nederbragt A.J., Lagesen K., Jeansson S.L., Jakobsen K.S. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 2011;11:244. doi: 10.1186/1471-2180-11-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brubaker L., Wolfe A.J. The female urinary microbiota, urinary health and common urinary disorders. Ann. Transl. Med. 2017;5:34. doi: 10.21037/atm.2016.11.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfe A.J., Toh E., Shibata N., Rong R., Kenton K., Fitzgerald M., Mueller E.R., Schreckenberger P., Dong Q., Nelson D.E., Brubaker L. Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol. 2012;50:1376–1383. doi: 10.1128/JCM.05852-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price T.K., Hilt E.E., Thomas-White K., Mueller E.R., Wolfe A.J., Brubaker L. The urobiome of continent adult women: a cross-sectional study. BJOG. 2020;127:193–201. doi: 10.1111/1471-0528.15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis D.A., Brown R., Williams J., White P., Jacobson S.K., Marchesi J.R., Drake M.J. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front. Cell. Infect. Microbiol. 2013;3:41. doi: 10.3389/fcimb.2013.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khasriya R., Sathiananthamoorthy S., Ismail S., Kelsey M., Wilson M., Rohn J.L., Malone-Lee J. Spectrum of bacterial colonization associated with urothelial cells from patients with chronic lower urinary tract symptoms. J. Clin. Microbiol. 2013;51:2054–2062. doi: 10.1128/JCM.03314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fouts D.E., Pieper R., Szpakowski S., Pohl H., Knoblach S., Suh M.J., Huang S.T., Ljungberg I., Sprague B.M., Lucas S.K., et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012;10:174. doi: 10.1186/1479-5876-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bučević Popović V., Šitum M., Chow C.E.T., Chan L.S., Roje B., Terzić J. The urinary microbiome associated with bladder cancer. Sci. Rep. 2018;8:12157. doi: 10.1038/s41598-018-29054-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearce M.M., Hilt E.E., Rosenfeld A.B., Zilliox M.J., Thomas-White K., Fok C., Kliethermes S., Schreckenberger P.C., Brubaker L., Gai X., Wolfe A.J. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio. 2014;5:e01283-14. doi: 10.1128/mBio.01283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karstens L., Asquith M., Davin S., Stauffer P., Fair D., Gregory W.T., Rosenbaum J.T., McWeeney S.K., Nardos R. Does the urinary microbiome play a role in urgency urinary incontinence and its severity? Front. Cell. Infect. Microbiol. 2016;6:78. doi: 10.3389/fcimb.2016.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adebayo A.S., Ackermann G., Bowyer R.C.E., Wells P.M., Humphreys G., Knight R., Spector T.D., Steves C.J. The urinary tract microbiome in older women exhibits host genetic and environmental influences. Cell Host Microbe. 2020;28:298–305.e3. doi: 10.1016/j.chom.2020.06.022. [DOI] [PubMed] [Google Scholar]
- 26.Burnett L.A., Hochstedler B.R., Weldon K., Wolfe A.J., Brubaker L. Recurrent urinary tract infection: association of clinical profiles with urobiome composition in women. Neurourol. Urodyn. 2021;40:1479–1489. doi: 10.1002/nau.24707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hochstedler B.R., Burnett L., Price T.K., Jung C., Wolfe A.J., Brubaker L. Urinary microbiota of women with recurrent urinary tract infection: collection and culture methods. Int. Urogynecol. J. 2022;33:563–570. doi: 10.1007/s00192-021-04780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaughan M.H., Mao J., Karstens L.A., Ma L., Amundsen C.L., Schmader K.E., Siddiqui N.Y. The urinary microbiome in postmenopausal women with recurrent urinary tract infections. J. Urol. 2021;206:1222–1231. doi: 10.1097/JU.0000000000001940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ammitzbøll N., Bau B.P.J., Bundgaard-Nielsen C., Villadsen A.B., Jensen A.M., Leutscher P.D.C., Glavind K., Hagstrøm S., Arenholt L.T.S., Sørensen S. Pre- and postmenopausal women have different core urinary microbiota. Sci. Rep. 2021;11:2212. doi: 10.1038/s41598-021-81790-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas-White K., Forster S.C., Kumar N., Van Kuiken M., Putonti C., Stares M.D., Hilt E.E., Price T.K., Wolfe A.J., Lawley T.D. Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun. 2018;9:1557. doi: 10.1038/s41467-018-03968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards V.L., Smith S.B., McComb E.J., Tamarelle J., Ma B., Humphrys M.S., Gajer P., Gwilliam K., Schaefer A.M., Lai S.K., et al. The cervicovaginal microbiota-host interaction modulates Chlamydia trachomatis infection. mBio. 2019;10:015488–e1619. doi: 10.1128/mBio.01548-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdul-Rahim O., Wu Q., Price T.K., Pistone G., Diebel K., Bugni T.S., Wolfe A.J. Phenyl-lactic acid is an active ingredient in bactericidal supernatants of Lactobacillus crispatus. J. Bacteriol. 2021;203:e0036021. doi: 10.1128/JB.00360-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juárez Tomás M.S., Ocaña V.S., Wiese B., Nader-Macías M.E. Growth and lactic acid production by vaginal Lactobacillus acidophilus CRL 1259, and inhibition of uropathogenic Escherichia coli. J. Med. Microbiol. 2003;52:1117–1124. doi: 10.1099/jmm.0.05155-0. [DOI] [PubMed] [Google Scholar]
- 34.Osset J., Bartolomé R.M., García E., Andreu A. Assessment of the capacity of Lactobacillus to inhibit the growth of uropathogens and block their adhesion to vaginal epithelial cells. J. Infect. Dis. 2001;183:485–491. doi: 10.1086/318070. [DOI] [PubMed] [Google Scholar]
- 35.Stapleton A.E., Au-Yeung M., Hooton T.M., Fredricks D.N., Roberts P.L., Czaja C.A., Yarova-Yarovaya Y., Fiedler T., Cox M., Stamm W.E. Randomized, placebo-controlled phase 2 trial of a Lactobacillus crispatus probiotic given intravaginally for prevention of recurrent urinary tract infection. Clin. Infect. Dis. 2011;52:1212–1217. doi: 10.1093/cid/cir183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas-White K., Taege S., Limeira R., Brincat C., Joyce C., Hilt E.E., Mac-Daniel L., Radek K.A., Brubaker L., Mueller E.R., Wolfe A.J. Vaginal estrogen therapy is associated with increased Lactobacillus in the urine of postmenopausal women with overactive bladder symptoms. Am. J. Obstet. Gynecol. 2020;223:727.e1–727.e11. doi: 10.1016/j.ajog.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raz R., Stamm W.E. A controlled trial of intravaginal estriol in postmenopausal women with recurrent urinary tract infections. N. Engl. J. Med. 1993;329:753–756. doi: 10.1056/NEJM199309093291102. [DOI] [PubMed] [Google Scholar]
- 38.Ferrante K.L., Wasenda E.J., Jung C.E., Adams-Piper E.R., Lukacz E.S. Vaginal estrogen for the prevention of recurrent urinary tract infection in postmenopausal women: a randomized clinical trial. Female Pelvic Med. Reconstr. Surg. 2021;27:112–117. doi: 10.1097/SPV.0000000000000749. [DOI] [PubMed] [Google Scholar]
- 39.Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017;35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 40.Karstens L., Asquith M., Caruso V., Rosenbaum J.T., Fair D.A., Braun J., Gregory W.T., Nardos R., McWeeney S.K. Community profiling of the urinary microbiota: considerations for low-biomass samples. Nat. Rev. Urol. 2018;15:735–749. doi: 10.1038/s41585-018-0104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brubaker L., Gourdine J.P.F., Siddiqui N.Y., Holland A., Halverson T., Limeria R., Pride D., Ackerman L., Forster C.S., Jacobs K.M., et al. Forming consensus to advance urobiome research. mSystems. 2021;6:e0137120. doi: 10.1128/mSystems.01371-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moustafa A., Li W., Singh H., Moncera K.J., Torralba M.G., Yu Y., Manuel O., Biggs W., Venter J.C., Nelson K.E., et al. Microbial metagenome of urinary tract infection. Sci. Rep. 2018;8:4333. doi: 10.1038/s41598-018-22660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segata N., Waldron L., Ballarini A., Narasimhan V., Jousson O., Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salter S.J., Cox M.J., Turek E.M., Calus S.T., Cookson W.O., Moffatt M.F., Turner P., Parkhill J., Loman N.J., Walker A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price T.K., Dune T., Hilt E.E., Thomas-White K.J., Kliethermes S., Brincat C., Brubaker L., Wolfe A.J., Mueller E.R., Schreckenberger P.C. The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J. Clin. Microbiol. 2016;54:1216–1222. doi: 10.1128/JCM.00044-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pavoine S., Dufour A.B.A.B., Chessel D. From dissimilarities among species to dissimilarities among communities: a double principal coordinate analysis. J. Theor. Biol. 2004;228:523–537. doi: 10.1016/j.jtbi.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 47.Ravel J., Brotman R.M. Translating the vaginal microbiome: gaps and challenges. Genome Med. 2016;8:35. doi: 10.1186/s13073-016-0291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ravel J., Gajer P., Abdo Z., Schneider G.M., Koenig S.S.K., McCulle S.L., Karlebach S., Gorle R., Russell J., Tacket C.O., et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA. 2011;108:4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alteri C.J., Himpsl S.D., Mobley H.L.T. Preferential use of central metabolism in vivo reveals a nutritional basis for polymicrobial infection. PLoS Pathog. 2015;11:e1004601. doi: 10.1371/journal.ppat.1004601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keogh D., Tay W.H., Ho Y.Y., Dale J.L., Chen S., Umashankar S., Williams R.B.H., Chen S.L., Dunny G.M., Kline K.A. Enterococcal metabolite cues facilitate interspecies niche modulation and polymicrobial infection. Cell Host Microbe. 2016;20:493–503. doi: 10.1016/j.chom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ceccarani C., Foschi C., Parolin C., D'Antuono A., Gaspari V., Consolandi C., Laghi L., Camboni T., Vitali B., Severgnini M., Marangoni A. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019;9:14095. doi: 10.1038/s41598-019-50410-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas-White K.J., Kliethermes S., Rickey L., Lukacz E.S., Richter H.E., Moalli P., Zimmern P., Norton P., Kusek J.W., Wolfe A.J., et al. Evaluation of the urinary microbiota of women with uncomplicated stress urinary incontinence. Am. J. Obstet. Gynecol. 2017;216:55.e1–55.e16. doi: 10.1016/j.ajog.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anglim B., Phillips C., Shynlova O., Alarab M. The effect of local estrogen therapy on the urinary microbiome composition of postmenopausal women with and without recurrent urinary tract infections. Int. Urogynecol. J. 2022;33:2107–2117. doi: 10.1007/s00192-021-04832-9. [DOI] [PubMed] [Google Scholar]
- 54.Hardy L., Jespers V., Dahchour N., Mwambarangwe L., Musengamana V., Vaneechoutte M., Crucitti T. Unravelling the bacterial vaginosis-associated biofilm: a multiplex Gardnerella vaginalis and Atopobium vaginae fluorescence in situ hybridization assay using peptide nucleic acid probes. PLoS One. 2015;10:e0136658. doi: 10.1371/journal.pone.0136658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardy L., Jespers V., Abdellati S., De Baetselier I., Mwambarangwe L., Musengamana V., van de Wijgert J., Vaneechoutte M., Crucitti T. A fruitful alliance: the synergy between Atopobium vaginae and Gardnerella vaginalis in bacterial vaginosis-associated biofilm. Sex. Transm. Infect. 2016;92:487–491. doi: 10.1136/sextrans-2015-052475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bradshaw C.S., Tabrizi S.N., Fairley C.K., Morton A.N., Rudland E., Garland S.M. The association of Atopobium vaginae and Gardnerella vaginalis with bacterial vaginosis and recurrence after oral metronidazole therapy. J. Infect. Dis. 2006;194:828–836. doi: 10.1086/506621. [DOI] [PubMed] [Google Scholar]
- 57.Li Q., Jiang S., Koh A.Y., Xiao G., Zhan X. Bayesian modeling of microbiome data for differential abundance analysis. arXiv. 2019 doi: 10.48550/arXiv.1902.08741. Preprint at. [DOI] [Google Scholar]
- 58.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin H., Peddada S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020;11:3514. doi: 10.1038/s41467-020-17041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shipitsyna E., Roos A., Datcu R., Hallén A., Fredlund H., Jensen J.S., Engstrand L., Unemo M. Composition of the vaginal microbiota in women of reproductive age--sensitive and specific molecular diagnosis of bacterial vaginosis is possible? PLoS One. 2013;8:e60670. doi: 10.1371/journal.pone.0060670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Könönen E., Wade W.G. Actinomyces and related organisms in human infections. Clin. Microbiol. Rev. 2015;28:419–442. doi: 10.1128/CMR.00100-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Berg C.L., Venter G., van der Westhuizen F.H., Erasmus E. Data on the optimisation of a solid phase extraction method for fractionating estrogen metabolites from small urine volumes. Data Brief. 2020;29:105222. doi: 10.1016/j.dib.2020.105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tu S. 2014. The Dirichlet-Multinomial and Dirichlet-Categorical Models for Bayesian Inference. [Google Scholar]
- 65.Franzosa E.A., McIver L.J., Rahnavard G., Thompson L.R., Schirmer M., Weingart G., Lipson K.S., Knight R., Caporaso J.G., Segata N., Huttenhower C. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods. 2018;15:962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rowe W.P.M., Winn M.D. Indexed variation graphs for efficient and accurate resistome profiling. Bioinformatics. 2018;34:3601–3608. doi: 10.1093/bioinformatics/bty387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeffreys H. An invariant form for the prior probability in estimation problems. Proc. R. Soc. Lond. A Math. Phys. Sci. 1946;186:453–461. doi: 10.1098/rspa.1946.0056. [DOI] [PubMed] [Google Scholar]
- 68.Correia S., Poeta P., Hébraud M., Capelo J.L., Igrejas G. Mechanisms of quinolone action and resistance: where do we stand? J. Med. Microbiol. 2017;66:551–559. doi: 10.1099/jmm.0.000475. [DOI] [PubMed] [Google Scholar]
- 69.Arsic B., Barber J., Čikoš A., Mladenovic M., Stankovic N., Novak P. 16-membered macrolide antibiotics: a review. Int. J. Antimicrob. Agents. 2018;51:283–298. doi: 10.1016/j.ijantimicag.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 70.Marrazzo J.M., Thomas K.K., Fiedler T.L., Ringwood K., Fredricks D.N. Relationship of specific vaginal bacteria and bacterial vaginosis treatment failure in women who have sex with women. Ann. Intern. Med. 2008;149:20–28. doi: 10.7326/0003-4819-149-1-200807010-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Onderdonk A.B., Delaney M.L., Fichorova R.N. The human microbiome during bacterial vaginosis. Clin. Microbiol. Rev. 2016;29:223–238. doi: 10.1128/CMR.00075-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diop K., Diop A., Michelle C., Richez M., Rathored J., Bretelle F., Fournier P.E., Fenollar F. Description of three new Peptoniphilus species cultured in the vaginal fluid of a woman diagnosed with bacterial vaginosis: Peptoniphilus pacaensis sp. nov., Peptoniphilus raoultii sp. nov., and Peptoniphilus vaginalis sp. nov. Microbiologyopen. 2019;8:e00661. doi: 10.1002/mbo3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faust K., Sathirapongsasuti J.F., Izard J., Segata N., Gevers D., Raes J., Huttenhower C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012;8:e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fait T. Menopause hormone therapy: latest developments and clinical practice. Drugs Context. 2019;8:212551. doi: 10.7573/dic.212551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stamm W.E. Estrogens and urinary-tract infection. J. Infect. Dis. 2007;195:623–624. doi: 10.1086/511526. [DOI] [PubMed] [Google Scholar]
- 76.Lillemon J.N., Karstens L., Nardos R., Garg B., Boniface E.R., Gregory W.T. The impact of local estrogen on the urogenital microbiome in genitourinary syndrome of menopause: a randomized-controlled trial. Female Pelvic Med. Reconstr. Surg. 2022;28:e157–e162. doi: 10.1097/SPV.0000000000001170. [DOI] [PubMed] [Google Scholar]
- 77.Shen J., Song N., Williams C.J., Brown C.J., Yan Z., Xu C., Forney L.J. Effects of low dose estrogen therapy on the vaginal microbiomes of women with atrophic vaginitis. Sci. Rep. 2016;6:24380. doi: 10.1038/srep24380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshimura T., Okamura H. Short term oral estriol treatment restores normal premenopausal vaginal flora to elderly women. Maturitas. 2001;39:253–257. doi: 10.1016/s0378-5122(01)00212-2. [DOI] [PubMed] [Google Scholar]
- 79.Waller T.A., Pantin S.A.L., Yenior A.L., Pujalte G.G.A. Urinary tract infection antibiotic resistance in the United States. Prim. Care. 2018;45:455–466. doi: 10.1016/j.pop.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 80.Abbo L.M., Hooton T.M. Antimicrobial stewardship and urinary tract infections. Antibiotics (Basel) 2014;3:174–192. doi: 10.3390/antibiotics3020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Putonti C., Thomas-White K., Crum E., Hilt E.E., Price T.K., Wolfe A.J. Genome investigation of urinary Gardnerella strains and their relationship to isolates of the vaginal microbiota. mSphere. 2021;6:001544–e221. doi: 10.1128/mSphere.00154-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Labrie F., Cusan L., Gomez J.L., Côté I., Bérubé R., Bélanger P., Martel C., Labrie C. Effect of one-week treatment with vaginal estrogen preparations on serum estrogen levels in postmenopausal women. Menopause. 2009;16:30–36. doi: 10.1097/gme.0b013e31817b6132. [DOI] [PubMed] [Google Scholar]
- 83.Dorr M.B., Nelson A.L., Mayer P.R., Ranganath R.P., Norris P.M., Helzner E.C., Preston R.A. Plasma estrogen concentrations after oral and vaginal estrogen administration in women with atrophic vaginitis. Fertil. Steril. 2010;94:2365–2368. doi: 10.1016/j.fertnstert.2010.03.076. [DOI] [PubMed] [Google Scholar]
- 84.Santen R.J. Vaginal administration of estradiol: effects of dose, preparation and timing on plasma estradiol levels. Climacteric. 2015;18:121–134. doi: 10.3109/13697137.2014.947254. [DOI] [PubMed] [Google Scholar]
- 85.O'Connell M.B. Pharmacokinetic and pharmacologic variation between different estrogen products. J. Clin. Pharmacol. 1995;35:18S–24S. doi: 10.1002/j.1552-4604.1995.tb04143.x. [DOI] [PubMed] [Google Scholar]
- 86.Smith M.A., Puckrin R., Lam P.W., Lamb M.J., Simor A.E., Leis J.A. Association of increased colony-count threshold for urinary pathogens in hospitalized patients with antimicrobial treatment. JAMA Intern. Med. 2019;179:990–992. doi: 10.1001/jamainternmed.2019.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilson M.L., Gaido L. Laboratory diagnosis of urinary tract infections in adult patients. Clin. Infect. Dis. 2004;38:1150–1158. doi: 10.1086/383029. [DOI] [PubMed] [Google Scholar]
- 88.De Nisco N.J., Neugent M., Mull J., Chen L., Kuprasertkul A., de Souza Santos M., Palmer K.L., Zimmern P., Orth K. Direct detection of tissue-resident bacteria and chronic inflammation in the bladder wall of postmenopausal women with recurrent urinary tract infection. J. Mol. Biol. 2019;431:4368–4379. doi: 10.1016/j.jmb.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vaishnava S., Yamamoto M., Severson K.M., Ruhn K.A., Yu X., Koren O., Ley R., Wakeland E.K., Hooper L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andrews S. 2015. FastQC: A Quality Control Tool for High Throughput Sequence Data. [Google Scholar]
- 91.Wingett S.W., Andrews S. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res. 2018;7:1338. doi: 10.12688/f1000research.15931.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krueger Felix, James Frankie, Ewels Phil, Afyounian Ebrahim, Schuster-Boeckler Benjamin. TrimGalore. [DOI]
- 93.McIver Lauren, Abu-Ali Galeb, Franzosa Eric, Schwager Randall, Morgan Xochitl, Waldron Levi, et al. bioBakery: KneadData. https://github.com/biobakery/biobakery [DOI] [PMC free article] [PubMed]
- 94.McMurdie P.J., Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Althouse A.D. Adjust for multiple comparisons? It's not that simple. Ann. Thorac. Surg. 2016;101:1644–1645. doi: 10.1016/j.athoracsur.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 96.Suzek B.E., Wang Y., Huang H., McGarvey P.B., Wu C.H., UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–932. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Caspi R., Billington R., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Midford P.E., Ong Q., Ong W.K., et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46:D633–D639. doi: 10.1093/nar/gkx935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hoff P.D. Spinger; 2009. A First Course in Bayesian Statistical Methods. [DOI] [Google Scholar]
- 99.Li Q., Guindani M., Reich B.J., Bondell H.D., Vannucci M. A Bayesian mixture model for clustering and selection of feature occurrence rates under mean constraints. Stat. Anal. Data Min.: ASA Data Sci. J. 2017;10:393–409. [Google Scholar]
- 100.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Newton M.A., Noueiry A., Sarkar D., Ahlquist P. Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics. 2004;5:155–176. doi: 10.1093/biostatistics/5.2.155. [DOI] [PubMed] [Google Scholar]
- 102.Nearing J.T., Douglas G.M., Hayes M.G., MacDonald J., Desai D.K., Allward N., Jones C.M.A., Wright R.J., Dhanani A.S., Comeau A.M., Langille M.G.I. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 2022;13:342. doi: 10.1038/s41467-022-28034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weiss S., Xu Z.Z., Peddada S., Amir A., Bittinger K., Gonzalez A., Lozupone C., Zaneveld J.R., Vázquez-Baeza Y., Birmingham A., et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Lozupone C.A., Turnbaugh P.J., Fierer N., Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hudzicki J. 2009. Kirby-Bauer Disk Diffusion Susceptibility Test Protocol. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome metagenomic sequencing read data (FASTQ files) have been deposited onto the NIH Sequence Read Archive (SRA) under the BioProject number [NCBI]: PRJNA801448. Prior to depositing, all human-mapping reads were removed from the data. All original code and simulation study data has been deposited at https://github.com/klutz920/BMDA-Simulation, https://github.com/klutz920/Bayes-Correlation-Test, and https://github.com/klutz920/Bayes-Proportion-Test. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.