

Abstract
Vaccines are the primary intervention against influenza. Currently licensed inactivated vaccines focus immunity on viral hemagglutinin (HA). Self-amplifying mRNA (sa-mRNA) vaccines offer an opportunity to generate immunity to multiple viral proteins, including additional neuraminidase (NA). This evaluation of a bicistronic approach for sa-mRNA vaccine development compared subgenomic promoter and internal ribosome entry site strategies and found consistent and balanced expression of both HA and NA proteins in transfected cells. In mice, sa-mRNA bicistronic A/H5N1 vaccines raised potent anti-HA and anti-NA neutralizing antibody responses and HA- or NA-specific CD4+ and CD8+ T cell responses. The addition of NA also boosted the cross-neutralizing response to heterologous A/H1N1. Similar immunogenicity results were obtained for bicistronic seasonal A/H3N2 and B/Yamagata vaccines. In ferrets, sa-mRNA bicistronic A/H1N1 vaccine fully protected lung from infection by homologous virus and showed significant reduction of viral load in upper respiratory tract, warranting further evaluation of sa-mRNA bicistronic vaccine in humans.
Keywords: sa-mRNA, influenza, bicistronic, emagglutinin, neuraminidase, cross-reactive immune response
Graphical abstract
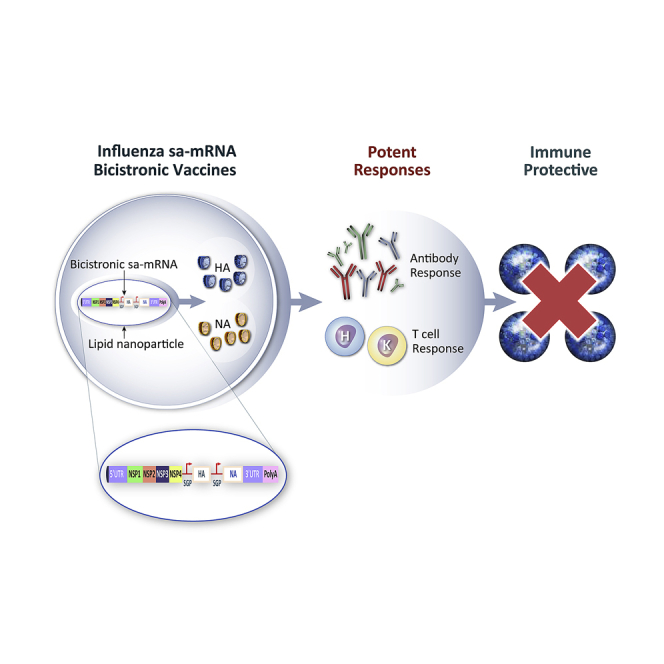
Self-amplifying mRNA bicistronic influenza vaccines offer immunity to both neuraminidase and hemagglutinin. These vaccines raised potent antigen-specific neutralizing antibody and T cell responses in mice. Neuraminidase boosted cross-neutralizing response to heterologous stains. Self-amplifying mRNA bicistronic vaccine fully protected ferrets from lung infection and reduced viral load in nose.
Introduction
Vaccination is considered the most effective means to reduce the substantial morbidity and mortality of influenza infection.1,2 Influenza hemagglutinin (HA) is the dominant viral surface glycoprotein; it mediates viral entry through binding to host cell surface receptors and facilitating viral and cellular membrane fusion.3,4 HA is a major target for virus-neutralizing antibodies and has been identified as the primary antigen in subunit or split influenza vaccines (inactivated influenza vaccine [IIV]).5
Neuraminidase (NA) is the second-most abundant glycoprotein on the surface of influenza viruses.6 NA proteins form a homotetramer, with the sialidase catalytic sites and epitopes for NA-inhibiting (NI) antibodies located on the four upper vertices.7 NA facilitates viral entry by removing decoy receptors from mucins that trap inhaled virus particles, allowing viral respiratory tract penetration.8 More importantly, NA mediates viral release by catalyzing the cleavage of glycosidic linkage to remove sialic acids from the surface of the infected cell, allowing for the release and spread of newly formed virus particles.9
Based on field research10 and human challenge studies,10,11 increased anti-NA antibodies correlate with reduced influenza virus infection and illness in healthy young adults. A weak correlation of vaccine effectiveness with NI antibody titers has also been observed in clinical studies of attenuated and inactivated influenza vaccines.12 In addition, NA from seasonal A/H1N1 vaccines has been shown to afford partial cross-protection against pandemic A/H5N1 infection.13,14 These data suggest that including NA in the flu vaccine formulations would likely increase their effectiveness.
Recently, mRNA vaccines have been evaluated in large human cohorts for efficacy in preventing symptomatic disease and reducing COVID-19 severity.15,16 Compared with conventional protein vaccines, mRNA vaccines present unique advantages, including production of immunogenic antigens in native conformation in human cells and substantial reductions in vaccine developmental and manufactural timelines.
However, the rate of adverse effects (AEs) with these mRNA vaccines is also unprecedentedly high.17 These AEs are partially due to the high dose requirements for mRNA vaccines, which have a short life inside of cells due to constant degradation.18 Instead, self-amplifying RNA (sa-mRNA) has emerged as a promising next-generation mRNA vaccine with the potential for lower dose requirements,19,20 due to a longer duration and higher yield of antigen expression,21 leading to lower AEs and more vaccine production.
In prior research, we showed that sa-mRNA COVID-19 vaccines raised a robust neutralizing antibody immune response and antigen-specific CD4+ and CD8+ T cell response in mice, and protected hamsters against SARS-CoV-2 infection.22 Clinical studies have also shown promising results for sa-mRNA vaccines against COVID-19.23 For the current analysis, we evaluated bicistronic strategies that allowed multiple genes-of-interest (GOI) to be co-expressed by a single sa-mRNA within the same cells, with the objective of developing influenza sa-mRNA vaccines comprising both HA and NA antigens.
Results
Subgenomic promoter (SGP) and internal ribosome entry site (IRES) strategies were selected from five sa-mRNA bicistronic strategies for co-expression of influenza A/H5N1 hemagglutinin and neuraminidase antigens
Five strategies were explored for bicistronic expression of HA and NA antigens from A/turkey/Turkey/1/2005 (H5N1): (1) a minimal 26S SGP for transcription of the second GOI; (2) an EMCV-IRES to initiate the translation of the second GOI; (3) a furin-based proteolytic cleavage site for post-translational cleavage of fused HA and NA proteins; (4) a viral 2A peptide linker for ribosome skipping during translation; and (5) a combined strategy of furin-based cleavage and a 2A peptide linker to reduce the additional amino acid sequences from the 2A-based strategy (Figure S1A).
The five strategies of sa-mRNA bicistronic A/H5N1 HA-NAs were synthesized and 5′-capped in vitro by enzymatic reactions, and electroporated into baby hamster kidney (BHK) cells. SGP and IRES were selected because of higher antigen expression in BHK cells (data not shown) and no introduction of additional amino acid sequences.24,25 Following SGP and IRES, the sa-mRNAs were encapsulated into lipid nanoparticles (LNPs) composed of synthetic lipids and characterized for particle biophysical attributes (Figure S2). The LNP-formulated sa-mRNA bicistronic vaccines were transfected into BHK cells, and flow cytometry using anti-HA and anti-NA antibodies showed the co-expression of both proteins within the same cell as the dominant population (Figure S1B).
SGPv2 was selected as the optimal subgenomic promoter approach for second GOI expression
While the first version of SGP, SGPv1, produced a high percentage of double-positive populations, the protein expression level for the second antigen was lower than for the first antigen, as shown by geometric mean fluorescence intensity (gMFI) analysis measuring the antigen-specific signal in positive cells (Figure S1C). A series of SGPs was designed with increasing lengths (SGPv2-SGPv4) to select the sequence effectively recruiting nonstructural protein (NSP) machinery for optimal subgenomic transcription of second GOI (Figure S1A). SGPv2 and SGPv3 were shown by gMFI analysis to drive improved NA expression as the second GOI without affecting HA expression as the first GOI (Figure S1C). SGPv2 was selected for its shorter length of sequence.
Balanced antigen expression of first and second GOIs by SGPv2 and IRES strategies were observed with sa-mRNA bicistronic A/H5N1 vaccines
To evaluate the impact on both antigen expression by the order of the GOIs on sa-mRNA, sa-mRNA A/H5N1 with HA and NA in both orders, and by SGPv2 or IRES bicistronic strategies, vaccines were produced, formulated in LNP, and transfected into BHK cells (Figure 1A). Flow cytometry showed the co-expression of both antigens by all four sa-mRNA bicistronic vaccines (Figure 1B). gMFI analysis showed that both SGPv2 and IRES strategies co-produced a largely balanced expression for both antigens (Figure 1C), with a minimal advantage for the second antigen. Total protein expression in bulk transfected cells was measured by isotope-dilution mass spectrometry (IDMS), with results presented as the normalized antigen expression against the housekeeping gene (Figure 1D). The IDMS showed that the SGPv2 strategy produced a generally balanced expression of both antigens in both orders and IRES strategy produced a slightly favorable expression for the second antigen.
Figure 1.

sa-mRNA bicistronic influenza A/H5N1 vaccine design and characterization in vitro
(A) Design schematic for subgenomic promoter (SGP) and internal ribosomal entry site (IRES) strategies. GOI, gene of interest; NSP, nonstructural protein; UTR, untranslated region. (B) Baby hamster kidney (BHK) cells were transfected with sa-mRNA bicistronic A/H5N1 vaccines and analyzed with flow cytometry for expression of hemagglutinin (HA) and neuraminidase (NA). Representative flow plots for cells expressing HA (X-axis) and NA (Y-axis) from different sa-mRNA bicistronic vaccines are labeled above the graph and the fraction of cells positive for both HA and NA are circled. Data were representative of at least three independent experiments. (C) Geometric mean fluorescence intensities (gMFI) for HA+ (left panels) and NA+ (right panels) cells for all tested sa-mRNA concentrations. Data were representative of at least three independent experiments. (D) BHK cells were transfected with sa-mRNA bicistronic A/H5N1 vaccines. Expression of NA and HA in cells was quantified by IDMS and results were normalized with GAPDH IDMS; the bar graph indicates the amount of relative antigen expression (fold difference) of the second GOI compared with the first GOI. Expression levels were averaged using four peptides for HA and three peptides for NA.
sa-mRNA bicistronic A/H5N1 vaccines induced potent H5 and N1 neutralizing antibody titers in mice
To evaluate neutralizing antibody response by sa-mRNA bicistronic A/H5N1 vaccines, BALB/c mice were immunized at day 1 and day 22 with sa-mRNA bicistronic A/H5N1, monocistronic H5, or monocistronic N1 vaccines at doses of 1.0 μg RNA or 0.01 μg RNA. Mice were killed at day 43, and serum from day 43 was tested for anti-HA neutralizing antibody response by hemagglutination inhibition (HAI) assay of turkey red blood cells (TRBC) by avian influenza A/turkey/Turkey/01/2005 (H5N1) virus, and by microneutralization (MN) assay against the primary infection of Madin-Darby Canine Kidney (MDCK) cells by A/H5N1 virus. HAI assays showed that all bicistronic A/H5N1 vaccines induced potent anti-H5 neutralizing antibodies regardless of the order of antigens or bicistronic strategy: HAI titer was ∼10ˆ3.0 for the 1 μg dose and ∼10ˆ2.5 for the 0.1 μg dose. This was comparable to results obtained with monocistronic H5 (HAI titer ∼10ˆ3.1 for the 1 μg dose and ∼10ˆ2.5 for the 0.1 μg dose) (Figure 2A). MN assays showed similar results for all bicistronic A/H5N1 vaccines (Figure 2B), with robust MN of ∼10ˆ4.1 for the 1 μg dose and ∼10ˆ3.3 for the 0.1 μg dose. As expected, HAI and MN titers for monocistronic N1 were below the threshold of detection.
Figure 2.

sa-mRNA bicistronic A/H5N1 vaccines induce potent anti-HA and anti-neuraminidase neutralizing titers and cross-reactive response to heterologous A/H1N1 virus in BALB/c mice
Female BALB/c mice, 8–10 weeks old, were immunized (10 mice/group) on days 1 and 22 with bilateral 50 μL intramuscular injections in the rear quadriceps. Serum samples were obtained from bleed-outs of euthanized animals on day 43. Monocistronic and bicistronic sa-mRNA vaccines with different antigen orders were evaluated for their ability to induce anti-H5 neutralizing antibody responses by (A) hemagglutination inhibition (HAI) assay, (B) microneutralization (MN) assay, and (C) anti-N1 neutralizing antibody responses by neuraminidase inhibition (NAI) enzyme-linked lectin assay (ELLA). Cross-reactive anti-NA antibodies against heterologous strain A/H1N1 (A/Delaware/55/2019, striped bars) were examined by NAI ELLA (D) and microneutralization long form (MN-LF, E). Bars represent the geometric mean titer, with each dot denoting an individual titer. n = 10, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Anti-NA antibody responses were evaluated with neuraminidase inhibition (NAI) enzyme-linked lectin assay (ELLA) for the inhibition of enzymatic activity of recombinant A/turkey/Turkey/01/2005 NA protein. All bicistronic A/H5N1 vaccines induced potent anti-N1 neutralizing antibodies, with an NAI ELLA titer ∼10ˆ5.7 for the 1 μg dose and ∼10ˆ5.0 for the 0.1 μg dose. This was comparable to results obtained with monocistronic N1 (NAI ELLA titer ∼10ˆ6.0 for the 1 μg dose and ∼10ˆ5.6 for the 0.1 μg dose), regardless of the order of antigens or the bicistronic strategy used (Figure 2C). For all assays, the effect of sa-mRNA dose (1.0 vs 0.01 μg) was highly significant (p < 0.0001, 2-way ANOVA). The effect of bicistronic strategies, SGPv2 versus IRES, was not significant. The effect of gene order on HAI, MN, and NAI also was not statistically significant.
sa-mRNA bicistronic A/H5N1 vaccines induced cross-reactive neutralizing antibody response against A/H1N1 in mice
To evaluate cross-reactive neutralizing antibody response, the serum raised by the sa-mRNA bicistronic A/H5N1 (H5-SGPv2-N1), monocistronic H5 or N1 vaccines at a dose of 1 μg RNA were tested against seasonal influenza A/Delaware/55/2019 (H1N1) by NAI ELLA and MN-long form (MN-LF), which measures the neutralization of both primary and secondary infection by anti-NA and anti-HA antibodies. NAI ELLA showed that anti-NA titer induced by bicistronic A/H5N1 or monocistronic N1 vaccines against heterologous N1 from A/H1N1 was significantly reduced compared with homologous N1 from A/H5N1 (from 10ˆ6.1 to 10ˆ3.2 for bicistronic A/H5N1 vaccine, and from 10ˆ7.3 to 10ˆ3.7 for monocistronic N1 vaccine). However, these vaccines still raised anti-NA neutralizing titers, while NAI ELLA titers induced by monocistronic H5 vaccine were below the threshold of detection (Figure 2D). MN-LF assay confirmed that the bicistronic A/H5N1 and monocistronic N1 raised significantly higher neutralization titers (10ˆ2.6 by bicistronic A/H5N1, 10ˆ2.7 by monocistronic N1) against heterologous A/H1N1 compared with the monocistronic H5 vaccine (10ˆ2.0) (p < 0.0001 for both; Figure 2E). These results suggested that the additional cross-reactivity was due to the inclusion of NA in the sa-mRNA bicistronic vaccine.
sa-mRNA bicistronic A/H5N1 vaccines elicited robust cellular immune responses in mice
To evaluate T cell immune response, spleens from BALB/c mice immunized by sa-mRNA bicistronic A/H5N1, monocistronic H5, or monocistronic N1 vaccines at doses of 1 μg were collected during euthanization on day 43 and tested for H5- and N1-specific CD4+ and CD8+ T cells by interleukin (IL)-2, tumor necrosis factor (TNF)α, interferon (IFN)γ, IL-5, and/or IL-13 production upon stimulation with H5 or N1 peptides. CD4+ (Figure 3A) and CD8+ (Figure 3B) T cells were observed in response to both H5 (left panels) and N1 antigens (right panels) for all sa-mRNA bicistronic A/H5N1 vaccines. As expected, based on prior research by Pepini et al (2017),26 vaccination with all sa-mRNA vaccines in BALB/c mice led to elevated IL-2, TNFα, and IFN-γ levels compared with adjuvanted protein vaccines, consistent with a Th1 response. The order of gene within the bicistronic construct did not substantially change the magnitude or quality of CD4+ or CD8+ T cell responses; likewise, SGPv2 and IRES led to similar immunogenicity. The CD8+ T response to monocistronic H5 or N1 vaccines was greater than to the same antigen in the bicistronic A/H5N1 vaccines and the monocistronic H5 and N1 combinations, suggesting competition for antigen presentation. Together, these data showed a robust induction of antigen-specific T cells, irrespective of the bicistronic strategies or antigen orders.
Figure 3.

Influenza sa-mRNA bicistronic A/H5N1 vaccines elicit robust cellular immune responses in BALB/c mice
Female BALB/c mice, 8–10 weeks old, were immunized (10 mice/group) on days 1 and 22 with bilateral 50 μL intramuscular injections in the rear quadriceps. Spleens were collected from euthanized animals on day 43. The frequency of intracellular cytokine expression among CD4+ (A) and CD8+ (B) in response to stimulation is displayed. Spleens from five mice/group were pooled and processed, and splenocytes were stimulated with anti-mouse CD28 in the absence or presence of synthetic peptides representing immunodominant epitopes in the H5 or N1 proteins encoded by the vaccine. Cells were stained with fluorescently tagged antibodies to cell surface markers CD3, CD4, CD8, and intracellular cytokines interleukin-2 (IL-2), tumor necrosis factor α (TNFα), interferon γ (IFNγ), IL-5, and IL-13 were analyzed by flow cytometry. Each column represents a mean from duplicate cultures of the five pooled spleens.
sa-mRNA bicistronic A/H3N2 and B/Yamagata vaccines induced potent neutralizing antibody responses and cellular immune responses in mice
To extend our evaluation of bicistronic strategy with SGPv2 (selected due to a shorter sequence than IRES) on seasonal influenza A and B strains, sa-mRNA bicistronic vaccines against A/Delaware/39/2019 (H3N2) and B/Singapore/INFTT-16-0610/2016 (Yamagata) were produced and immunized into BALB/c mice. MN assays showed that the sa-mRNA bicistronic A/H3N2 and B/Yamagata vaccines raised potent neutralizing titers against HA (∼10ˆ3.6 for the 1 μg dose and ∼10ˆ2.8 for the 0.1 μg dose of bicistronic A/H3N2 vaccine, and ∼10ˆ4.1 for the 1 μg dose and ∼10ˆ3.1 for the 0.1 μg dose of bicistronic B/Yamagata vaccine), comparable to levels with monocistronic HAs (10ˆ3.6 for the 1 μg dose of A/H3N2 vaccine and 10ˆ3.8 for the 1μg dose of B/Yamagata vaccine, Figure 4A). This response was dose-dependent (p < 0.0001), with no significant effect of gene order on anti-HA neutralizing response observed for either sa-mRNA vaccine. The anti-NA response measured by NAI ELLA (Figure 4B) was also strongly induced by each sa-mRNA bicistronic vaccine (∼10ˆ3.0 for the 1 μg dose and ∼10ˆ2.5 for the 0.1 μg dose of A/H3N2 vaccines, and ∼10ˆ3.1 for 1 μg dose and ∼10ˆ2.7 for 0.1 μg dose of B/Yamagata vaccines). MN-LF assays for anti-HA and NA response (Figure 4C) showed similar results, with robust MN-LF titers of ∼10ˆ2.9 for the 1 μg dose and ∼10ˆ2.5 for the 0.1 μg dose of A/H3N2 vaccines, and ∼10ˆ2.9 for the 1 μg dose and ∼10ˆ2.4 for the 0.1 μg dose of B/Yamagata vaccines. While no effect on gene order was observed for the anti-N2 response by A/H3N2 vaccine, the anti-B/Yamagata NA response was significantly higher in sa-mRNA NA-HA compared with sa-mRNA HA-NA (NAI: p = 0.0001, MN_LF: p = 0.004).
Figure 4.

sa-mRNA bicistronic A/H3N2 and B/Yamagata vaccines induce potent neutralizing titers to hemagglutinin and neuraminidase from A/H3N2 and B/Yamagata viruses
Female BALB/c mice, 8–10 weeks old, were immunized (10 mice/group) on days 1 and 22 with bilateral 50 μL intramuscular injections in the rear quadriceps. Serum samples were obtained from bleed-outs of euthanized animals on day 43. Bicistronic and monocistronic vaccines were evaluated for their capacity to induce (A) hemagglutinin (HA)-neutralizing antibody titers by microneutralization (MN) assay, (B) neuraminidase (NA)-neutralizing antibody titers by neuraminidase inhibition (NAI) enzyme-linked lectin assay (ELLA), and (C) microneutralization long form (MN-LF). Bars represent the geometric mean titer; each dot denotes an individual titer. n = 10, ∗p < 0.05, ∗∗p < 0.01.
The T cell responses in mice with sa-mRNA bicistronic A/H3N2 and B/Yamagata vaccines were evaluated using similar approaches as for the bicistronic A/H5N1 vaccine. The tests showed robust antigen-specific CD4+ T cell responses to the sa-mRNA bicistronic A/H3N2 and B/Yamagata vaccines (Figure S3A). Due to availability of stimulation for A/H3N2 in BALB/c mice, only the CD8+ T cells by sa-mRNA B/Yamagata vaccine were evaluated with the potent antigen-specific CD8+ T cells (Figure S3B).
sa-mRNA bicistronic A/H1N1 vaccines protect ferrets from homologous A/H1N1 infection
To evaluate the protective effect from sa-mRNA bicistronic vaccines, ferrets were immunized with sa-mRNA bicistronic vaccines co-expressing HA and NA against another seasonal influenza strain, A/Netherlands/602/2009 (H1N1) virus, at doses of 5 μg RNA or 0.5 μg RNA, sa-mRNA monocistronic HA or NA vaccines at 5.0 μg RNA, or PBS as control on day 1 and day 22. Serum collected on day 49 was tested for anti-HA antibody response by HAI and MN assays and anti-NA antibody response by ELLA, respectively. The results confirmed that sa-mRNA A/H1N1 vaccines containing HA raised anti-HA neutralizing HAI and MN titers (Figures S4A and S4B) and vaccines containing NA raised anti-NA ELLA titers (Figure S4C). All ferrets were challenged post bleeding with homologous A/Netherlands/602/2009 (H1N1) virus. Virus was collected by throat and nose swabs on days 50, 51, 52, 53, and 54 (days 0, 1, 2, 3, and 4 post the challenge). High levels of virus were recovered from control ferrets, but a robust reduction of virus recovery was observed in all vaccinated animals (Figures S4D and 5A). Body weight loss during the challenge was measured showing the highest weight loss in control ferrets and much reduced weight loss in all vaccinated animals (Figure S4E).
Figure 5.

sa-mRNA bicistronic A/H1N1 vaccines protect viral infection in lung and nose in ferrets
Female domestic ferrets (n = 6) were immunized twice, 3 weeks apart, with 5.0 μg (blue) or 0.5 μg (red) of sa-mRNA H1-N1, 5.0 μg of sa-mRNA H1 (green) or sa-mRNA N1 (purple), or PBS (black) as control. Ferrets were challenged 4 weeks after the second dose with A/Netherland/602/2009 (H1N1) virus at 106 50% tissue culture infectious dose (TCID50) per animal and killed 4 days later. Lungs and nasal turbinates were collected for recovery of infectious virus. (A) Virus recovery in nose swabs during challenge. (B and C) Total virus recovery from lung (B) and nasal turbinates (C) expressed as TCID50/g of tissue. Each dot represents an individual sample, and the line the geometric mean for the group. The dotted line in each panel represents the lower limit of quantitation for the assay. Statistical analysis by 2-way ANOVA with Tukey’s multiple comparison test was performed using GraphPad Prism 9.1.0. ∗p < 0.05, ∗∗∗∗p < 0.0001.
All ferrets were killed on day 54 (4 days post challenge). Infectious virus was measured in the lungs and nasal turbinates of the euthanized ferrets. In the lung, the viral titer recovered from control ferrets was 10ˆ5.2 50% tissue culture infectious dose per gram (TCID50/g) and from the vaccine-immunized ferrets was <10ˆ1.5 TCID50/g, under the limit of quantitation of the assay and demonstrating full lower respiratory tract protection with all vaccines, sa-mRNA bicistronic A/H1N1 at 0.5 μg and 5 μg doses, as well as monocistronic H1 or N1 at 5 μg dose (Figure 5B). In the upper respiratory tract (Figure 5C), virus recovery from nasal turbinates was 10ˆ6.9 TCID50/g in control ferrets. Viral titers were reduced to 10ˆ4.8 TCID50/g by sa-mRNA monocistronic N1 at 5 μg dose, 10ˆ3.7 TCID50/g by monocistronic H1 at 5 μg dose and 10ˆ3.0 and 10ˆ2.6 TCID50/g by bicistronic A/H1N1 at the 0.5 μg and 5 μg doses, respectively.
Discussion
In this study, we advanced an sa-mRNA platform by incorporating a bicistronic capability to express additional immunologically important antigen combinations. We found pandemic and seasonal influenza sa-mRNA bicistronic HA-NA vaccines produced HA and NA antigens in cells, raised robust serological and cell-mediated immune responses in mice, and protected ferrets from influenza infection.
This analysis compared several bicistronic sa-mRNA strategies (SGP, IRES, furin, 2A, and furin+2A), and selected the SGP and IRES strategies due to their suitability for platform application. Cell-based assays and mouse immunogenicity studies demonstrated that the IRES strategy co-expressed both HA and NA and raised comparable immune response with both antigens regardless of their order on sa-mRNA. With the first version of the SGP strategy, the second GOI was less immunogenic than the first GOI (data not shown) due to lower antigen expression. This prompted the screening of SGPv1-v4, with SGPv2 identified as providing a balanced expression of both GOIs in either order, and its suitability for HA and NA vaccine development.
HA has been the primary or only antigen in licensed inactivated and recombinant influenza vaccine formulations. Our mouse immunogenicity studies demonstrated a robust anti-HA neutralizing response with sa-mRNA vaccines containing HA antigen at doses as low as 0.01 μg, comparable to the response achieved with MF59 adjuvanted protein vaccine (data not shown). sa-mRNA vaccines containing HA also induced HA-specific CD4+ T cells. While MF59 adjuvanted protein vaccines also raised CD4+ T cell response, non-adjuvanted protein vaccines, the majority of licensed inactivated and recombinant influenza vaccines, did not stimulate this type of immune response.27 The sa-mRNA vaccines also stimulated potent CD8+ T cell response, an effect that is lacking in both non-adjuvanted and adjuvanted protein vaccines.28
NA is the second-most abundant influenza viral surface protein. The correlation of anti-NA immunity with the prevention of influenza virus infection and illness has been demonstrated in natural infection and clinical studies.10,11 Our bicistronic strategy provides a controlled approach to express and raise a balanced immune response for both NA and HA antigens in the same cells, while our mouse studies of sa-mRNA bicistronic influenza vaccines confirmed a potent anti-NA neutralizing antibody and an NA-specific CD4+ and CD8+ T cell response. With the additional NA antigen, bicistronic A/H5N1 vaccines not only raised a potent neutralizing response to homologous pandemic A/H5N1, but also to heterologous seasonal A/H1N1. Because NA sequences are more conservative, with less antigenic drift than HA,29 it is likely that the addition of NA antigen improves cross-reactive immune response to mismatched strains. Our ferret challenge study showed that NA alone from A/H1N1 fully protected lung and partially protected nose from homologous A/H1N1 infection. The bicistronic vaccines co-expressing both HA and NA improved the protection in the upper respiratory tract compared with those of monocistronic HA, demonstrating the potential for NA as a robust and additional antigen critical against influenza infection.
In conclusion, next-generational sa-mRNA bicistronic influenza vaccines developed to co-express HA and NA raised robust humoral and cellular response in mice and elicited high levels of cross-neutralization against a heterologous influenza virus in relatively low doses. sa-mRNA bicistronic vaccines also protected ferrets from homologous influenza infection. These findings suggest that these vaccines warrant further evaluation in humans.
Materials and methods
Construct design and cloning
All bicistronic constructs were generated based on the TC-83 Venezuelan equine encephalitis (VEEV) strain (Genbank No. L01443.1) with a chimeric 3′UTR obtained from the Sindbis virus (Genbank No. KT121726.1). The SGPv1 contained 19 nt upstream of TSS of TC-83; SGPv2 contained the identical full SGP sequence as described by Blakney et al (2018)30; SGPv3 contained 50 nt upstream of TSS of TC-83; and SGPv4 contained 71 nt upstream of TSS of TC-83. The IRES sequence was wt encephalomyocarditis virus (EMCV) IRES.31 T2A sequences were adapted from a codon-optimized peptide 2A sequence described by Ibrahimi et al (2009).24 The furin cleavage site was GIRRKRSVSH.25 The furin + T2A combo design used a minimal furin peptide RRKRS, with the N-terminal GS linker and GSG linker followed by the T2A peptide.
H5 and N1 sequences were from A/turkey/Turkey/1/2005 (H5N1), H3 and N2 sequences were from A/Delaware/39/2019 (H3N2), HA and NA sequences were from B/Singapore/INFTT-16-0160/2016 (Yamagata), and H1 and N1 sequences were from A/Netherland/602/2009 (H1N1). All HA and NA sequences were inserted by Gibson assembly into an sa-mRNA DNA construct with a T7-promoter and a poly A (A = 37), followed by a reverse BspQI site for linearization.
In vitro transcription and capping
As described by Palladino et al (2022),22 linearized DNA templates were enzymatically transcribed into RNA with T7 RNA polymerase (New England Biolabs, Ipswich, MA, USA), followed by digestion with Turbo DNase (Life Technologies, Carlsbad, CA, USA) to remove template DNA, and subsequently capped using a Vaccinia capping system (New England Biolabs). RNA from transcription/capping reactions was purified by tangential flow filtration (TFF) and frozen at −80°C.
sa-mRNA/LNP formulation
Again, using methods from Palladino et al (2022),22 RNA in citrate buffer was formulated into LNPs using a proprietary ionizable lipid, 1,2-diastearoylsn-glycero-3-phosphocholine (DSPC; Avanti Polar Lipid), 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG-DMG 2000; NOF America Corporation, White Plains, NY, USA), and cholesterol (Sigma-Aldrich, St. Louis, Mo, USA), dissolved in ethanol through a NanoAssemblr mixing instrument (Precision Nanosystems, Vancouver, Canada). The nanoparticles were buffer-exchanged into a Tris buffer with NaCl and sucrose by TFF, sterile-filtered, and stored at −80°C.
RNA and LNP transfection
Naked sa-mRNA was electroporated with the Gene Pulser Xcell Electroporation System (Bio-Rad, Hercules, CA, USA). Indicated concentrations of 5 μL RNA were mixed with 1∗106 BHK-V cells in 250 μL optiMEM. For LNP-formulated sa-mRNA, 100 μL of sa-mRNA in room-temperature optiMEM (Thermo Fisher Scientific, Waltham, MA, USA) with indicated RNA concentrations was added to 1∗106 BHK-V cells in 250 μL room-temperature optimum. Cells were left overnight and harvested for fluorescence-activated cell sorting (FACS).
Cell culture and FACS staining
Antibodies were labeled with Zenon kits (Z-25451, Z-25402, Z25408; Thermo Fisher Scientific) according to the colors used in the FACS. After transfection, cells were collected, fixed, and permeabilized with Cytofix/Cytoperm (BD Biosciences, Franklin Lakes, NJ, USA), and stained with AF-647 conjugated human anti-HA and/or AF-488 conjugated human anti-NA antibodies. The frequencies of HA- and NA-positive cells were enumerated by flow cytometry using a Fortessa flow cytometer (BD Biosciences) and analyzed by FlowJo software (BD Biosciences). Controls of single-staining for HA or NA only and no stains were performed showing low background. The mean fluorescence intensity was determined using FlowJo to draw a subpopulation of antigen-positive cells; the gMFI of that population was calculated as an indicator of average protein expression levels.
Isotope-dilution mass spectrometry
An amount of 106 transfected cells were resuspended in 200 μL total (50:50% v/v) of 3M Guanidinium chloride (Sigma-Aldrich) and RIPA buffer (Thermo Fisher Scientific) and lysed by probe sonication. The suspension was precipitated with acetone (Sigma-Aldrich) at −20°C overnight and centrifuged to collect the pellet. The pellet was washed twice with cold ethanol and dried by a SpeedVac concentrator (Thermo Fisher Scientific). The pellet was then resuspended in 50 μL 0.1M NH4HCO3 (LC-MS grade H2O, Thermo Fisher Scientific), followed by the addition of 5 μL trypsin (Sigma-Aldrich) at 1 μg/μL. The reaction was carried out overnight at 37°C. Formic acid 5 μL (Thermo Fisher Scientific) was added to inactivate the trypsin. The samples were centrifuged, and the supernatant was further cleared using 0.2 μm polytetrafluoroethylene filters (4 mm, VWR, Radnor, PA, USA).
For 18 μL of filtered sample, a 2 μL aliquot of 1 pmol/mL reconstituted peptide solution was added; this contained VIPELNGKˆ and VGVNGFGRˆ (GAPDH peptides); FLTEKˆ, YNGIITDTIKˆ, and GDVFVIRˆ (N1 peptides); and IQIIPKˆ, LVLATGLRˆ, EFNNLERˆ, and EEISGVKˆ (H5 peptides) as an internal standard. The samples were injected into a BEH C-18 (Waters, Milford, MA, USA) RP-HPLC column. The aqueous mobile phase was 0.1% formic acid in MS-grade water, and the organic phase (B) was MS-grade acetonitrile (Thermo Fisher Scientific) with 0.1% formic acid. The gradients involved initial column conditions of 100% A at 0.2 mL/min for the first 5 min, ramping up to 100% B within 15 min. The eluents were introduced to the TSQ tandem mass spectrometer (Thermo Scientific) via an electrospray interface, with a positive voltage of 3,800, an ion transfer tube with temperature 325°C, an evaporator temperature of 275°C, and sheath gas 35 applied for the MS-method.
For each sample, the absolute values of the two GAPDH peptides (VIPELNGK, VGVNGFGR) were averaged. Absolute quantities of the other peptides were normalized with respect to the calculated GAPDH average value. Next, for each individual peptide, expression ratios were calculated for the GOIs located at the second loci versus the first loci. Estimated ratios from the FLTEK, YNGIITDTIK, GDVFVIR (N1) and IQIIPK, LVLATGLR, EFNNLER, and EEISGVK (H5) peptides were averaged for each GOI to report a final expression ratio between the second and first GOI loci. The error bar for each GOI’s final ratio was estimated as the standard deviation of the expression ratios, calculated from selected peptides of the same GOI.
Viruses
The influenza viruses used in this study, A/Delaware/55/2019 (H1N1); A/Delaware/39/2019 (H3N2); B/Singapore/INFTT-16-0610/2016 (Yamagata), and A/turkey/Turkey/01/2005 (H5N1), were propagated in MDCK cells (Seqirus proprietary, 33016-P) at 34°C for 72 h. Working virus stocks were then fully sequenced and titrated, either in a fluorescent focus-based assay (expressed as fluorescent focus-forming unit [FFU/mL]) or by obtaining a 50% Tissue Culture Infective Dose (TCID50) using MDCK cells.
Mouse immunogenicity studies
The mouse studies were conducted at Biomodels LLC (Waltham, MA, USA). Female BALB/c mice, 8 to 10 weeks old, were immunized (10 mice/group) on days 1 and 22 with bilateral 50 μL intramuscular injections in the rear quadriceps. To evaluate antibody response, serum samples were obtained from bleed-outs of euthanized animals on day 43. To evaluate cell-mediated immunity, spleens were removed from each animal immediately after euthanasia.
Prior to bleeding, mice were anesthetized with 2.5% isoflurane. Mice were euthanized by exsanguination under anesthesia: euthanasia was confirmed by cervical dislocation. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978).
HAI assay
Sera were evaluated for HAI, as described by Heeringa et al (2020).32 Briefly, sera were treated with receptor-destroying enzyme (RDE) (Denka Seiken Co., Ltd., Tokyo, Japan) and diluted with PBS to an initial dilution of 1:10. Serially diluted (2-fold), heat-inactivated, and RDE-treated sera from immunized mice were incubated with an equal volume of viruses (4 hemagglutinin units [HAU] per well) at room temperature for 30 min. After incubation, an equivalent volume of TRBC (Lampire Biological Labs, Pipersville, PA, USA) diluted to 0.5% in PBS was added, and the plates were incubated at room temperature for 45 min. Hemagglutination inhibition was determined by visual inspection, and HAI titer was expressed as the reciprocal of the highest dilution of the samples with hemagglutination inhibition.
Microneutralization assay
The neutralization capacity of the sera was examined against the homologous vaccine strains using a virus fluorescent focus-based MN assay, as previously described by Heeringa et al (2020).32 Briefly, sera were pre-incubated with the virus (∼1,000–2,000 FFU/well) and allowed to react for 2 h at 37°C. Subsequently, monolayers of MDCK cells were inoculated with the virus-sera mixture and incubated overnight at 37°C. The monolayers were fixed, and infected cells were stained for the nucleoprotein of influenza A (clones A1, A3 blend, Millipore) or influenza B (clones B2, B4 blend, Millipore) and labeled with a goat anti-mouse IgG (H + L) secondary antibody conjugated to Alexa Fluor 488 (Invitrogen, Waltham, MA, USA). Fluorescent foci were imaged by an Immunospot analyzer (Cellular Technology Limited, Shaker Heights, OH, USA) and quantified with Immunospot 7.0.12.1 software (Cellular Technology Limited). MN titer was determined by nonlinear regression using GraphPad Prism (GraphPad Software, San Diego, CA, USA) to calculate the reciprocal of the dilution that caused a 50% reduction in viral foci versus no-serum controls.
MN assay long form
To assess MN by anti-NA and anti-HA antibodies, an HA quantification-based MN assay with a longer incubation time was developed. Two-fold serial dilution of RDE and heat-treated test samples were mixed with an equal volume of influenza virus (A/H5N1, A/H1N1, or B Yamagata) solution containing 100 TCID50/well in neutralization medium (33016 MDCK protein free medium (PFM) [GIBCO, Thermo Fisher Scientific]), incubated for 1 h at 37°C with 5% CO2. The MDCK 33016-PFM cells had been seeded on the preceding day as 2.5E4/well (2.5E6/plate) in cell growth medium (DM134; Irvine Scientific, Santa Ana, CA, USA). Next, 100 μL of serum-virus mixture was transferred from each well onto the confluent cell monolayer and incubated for 1 h at 37°C with 5% CO2. The antibody/virus mixture was removed, and cells were washed twice and incubated for 5 days (37°C, 5% CO2) with neutralizing media (100 μL/well) containing 2-fold serially diluted serum samples supplemented with TPCK-trypsin working stock (Sigma-Aldrich).
The plates were examined by HA assay for MN titers. Subsequently, 50 μL of supernatant was transferred to each well and an equal volume of 0.5% TRBC was added and incubated at room temperature for 30 min. The presence of TRBC agglutination (no neutralization) or absence (neutralization) was observed under biosafety level (BSL) 3 practice for A/H5N1 and BSL2 practice for A/H1N1 and B/Yamagata. The highest serum dilution that protected cells from infection was taken used as the neutralization titer.
Enzyme-linked lectin assay
Sera were examined for NAI activity by ELLA.33 Briefly, NA from the homologous or heterologous vaccine strains was mixed with serial dilutions of heat-inactivated sera in buffer containing 33.3 mM MES (pH 6.5; Alfa Aesar, Haverhill, MA, USA), 4 mM calcium chloride (K-D Medical, Columbia, MD, USA), 0.5% Tween-20 (Thermo Fisher Scientific), and 1% BSA fraction V (Calbiochem, Sigma-Aldrich) in plates coated with fetuin (25 μg/mL in PBS). Following overnight incubation at 37°C, the cleavage of sialic acid was detected by peanut agglutinin-HRP conjugate (1 μg/mL in PBS, Thermo Fisher Scientific), developed with TMB (Thermo Fisher Scientific), and stopped with 2 N sulfuric acid (Sigma-Aldrich). Absorbance was measured on a Synergy H1 plate reader (BioTek Instruments, now Agilent Technologies, Winooski, VT, USA). NAI titer was determined by generating a nonlinear regression in GraphPad Prism (GraphPad Software) and calculating the reciprocal of the dilution that resulted in a 50% reduction in NA activity versus the no-serum controls.
Statistics for serology assays
Log-transformed titers were compared between groups by 2-way ANOVA with Tukey’s multiple comparisons test using GraphPad Prism software version 9.1.2 (GraphPad Software).
T cell antigen stimulation and intracellular cytokine staining
Spleens from five mice per group were removed from each animal immediately after euthanasia and pooled, and single-cell suspensions were prepared in RPMI medium (Thermo Fisher Scientific) containing 1× penicillin/streptomycin and 50 μM 2-mercaptoethanol. For each simulation condition, duplicate cultures of 2 × 106 splenocytes were prepared for each splenocyte pool. H5-specific T cells were stimulated with a pool of H5 peptides spanning amino acids 125–139, 156–170, 168–176, 192–206, 212–221, 243–257, and 530–538. N1-specific T cells were stimulated with a pool of N1 peptides spanning amino acids 71–85, 370–384, and 187–196. A/H3N2 and B/Yamagata HA-NA-specific CD4 T cells were stimulated with homologous MDCK-cell-derived monovalent bulk. A CD8-stimulating peptide spanning HA amino acids 551–559 was used as stimulator for B/Yamagata-specific CD8 T cells. The final peptide concentration for each stimulation was 1 μg/mL.
All cultures contained anti-CD28 (BD Biosciences) at a final concentration of 1 μg/mL, with BD GolgiPlug Protein Transport Inhibitor (containing Brefeldin A) (BD Biosciences) added after 2 h. After 6 h in a humidified incubator at 37°C with 5% CO2, cells were stained with LIVE/DEAD fixable aqua dead cell stain kit (Invitrogen), washed, and stained with APC-H7-labeled anti-CD4 (BD Biosciences), and Alexa Fluor 700-labeled anti-CD8 (BD Biosciences). Cells were washed, fixed with Perm/Wash buffer (BD Biosciences), and stained with a mixture of PerCP/Cy5.5-labeled anti-IFNγ (eBioscience, Thermo Fisher Scientific), Brilliant Violet 605-labeled anti-IL-2 (BD Biosciences), allophycocyanin-labeled anti-IL-5 (BioLegend, PerkinElmer, Waltham, MA, USA), and phycoerythrin-labeled anti-IL-13 (eBioscience, Thermo Fisher Scientific). Samples were processed on a Fortessa (BD Biosciences) and analyzed by FlowJo software v10.8.1. The net (%) antigen-specific CD4 or CD8 T cells were calculated as the difference between the percent cytokine-positive cells in the antigen-stimulated and unstimulated cultures, with 95% confidence limits calculated using Microsoft Excel.
Ferret challenge study
The ferret challenge study was approved by the Central Authority for Scientific Procedures on Animals (Centrale Commissie Dierproeven) and conducted in accordance with the European guidelines (EU directive on animal testing 86/609/EEC) and local Dutch legislation on animal experiments. The in-life phase took place at Viroclinics Biosciences BV, Viroclinics Xplore, Schaijk, the Netherlands. Female domestic ferrets, 6–12 months old, were immunized twice, under isoflurane anesthesia, 3 weeks apart, at days 1 and 22, and challenged at day 50, 4 weeks after the second immunization, by intranasal infection with 106 TCID50 per dose of A/Netherland/602/2009 (H1N1) virus in a total dose volume of 3 mL. Animals were euthanized by abdominal exsanguination under anesthesia on day 54.
Tissue samples (lung and nasal turbinates) were collected and homogenized in influenza infection medium (EMEM containing BSA, penicillin, streptomycin, amphotericin-B, L-glutamine, sodium bicarbonate, HEPES, and trypsin) and centrifuged briefly. Quadruplicate 10-fold serial dilutions were used to determine replication competent virus titers in confluent layers of MDCK cells. Serial dilutions of the viral samples were incubated on MDCK monolayers for 6 days at 37°C. After culture supernatants were removed, turkey erythrocytes were added to the plates for incubation for 1 h at 4°C. HA patterns were read, and virus titers were calculated with the Spearman-Karber method.
Throat and nasal swabs analyzed as described.34 Briefly, swabs were collected from all animals before challenge on day 50 and on days 51, 52, 53, and 54 (days 1, 2, 3, and 4 post challenge). Individual swabs were homogenized and resuspended in 3 mL medium and stored at −80°C until analysis. Viral titers were determined by means of virus titration culture on MDCK cells. Data were expressed as log TCID50 per milliliter of swabs.
Acknowledgments
We thank Viroclinics Xplore for performing the ferret challenge study and protection analysis, and Seqirus Research staff, Xiuwen Ma for protein reagents, Formulation and Delivery group for sa-mRNA vaccine formulation, and Annette Ferrari for serology assays. Editorial support was provided by medical consultant C. Gordon Beck and was funded by Seqirus, Inc.
Author contributions
Conceptualization: Y.W., G.P., C.C., G.R.O., E.C.S.; Methodology: C.C., G.P., N.M., G.R.O., Y.W.; Investigation: C.C., S.D., M.S., M.C., N.M., E.R., C.L., H.P.; Writing – original draft: C.C., M.S., M.C., E.R., N.M., G.P., Y.W.; Writing – review & editing: C.C., G.P., Y.W.
Declaration of interests
All authors were employed by Seqirus at the time they completed the work described in this publication. Seqirus funded this research; some authors own CSL stocks.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.09.013.
Supplemental information
Data availability statement
The authors declare that all relevant data supporting the findings of this study are available within the article.
References
- 1.Nichol K.L., Treanor J.J. Vaccines for seasonal and pandemic influenza. J. Infect. Dis. 2006;194:S111–S118. doi: 10.1086/507544. [DOI] [PubMed] [Google Scholar]
- 2.Poland G.A., Rottinghaus S.T., Jacobson R.M. Influenza vaccines: a review and rationale for use in developed and underdeveloped countries. Vaccine. 2001;19:2216–2220. doi: 10.1016/s0264-410x(00)00448-5. [DOI] [PubMed] [Google Scholar]
- 3.Wilson I.A., Skehel J.J., Wiley D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 4.Gamblin S.J., Vachieri S.G., Xiong X., Zhang J., Martin S.R., Skehel J.J. Hemagglutinin structure and activities. Cold Spring Harb. Perspect. Med. 2021;11:a038638. doi: 10.1101/cshperspect.a038638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong S.S., Webby R.J. Traditional and new influenza vaccines. Clin. Microbiol. Rev. 2013;26:476–492. doi: 10.1128/cmr.00097-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krammer F., Fouchier R.A.M., Eichelberger M.C., Webby R.J., Shaw-Saliba K., Wan H., Wilson P.C., Compans R.W., Skountzou I., Monto A.S. NAction! How can neuraminidase-based immunity contribute to better influenza virus vaccines? mBio. 2018;9 doi: 10.1128/mBio.02332-17. e02332-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcelin G., Sandbulte M.R., Webby R.J. Contribution of antibody production against neuraminidase to the protection afforded by influenza vaccines. Rev. Med. Virol. 2012;22:267–279. doi: 10.1002/rmv.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X., Steukers L., Forier K., Xiong R., Braeckmans K., Van Reeth K., Nauwynck H. A beneficiary role for neuraminidase in influenza virus penetration through the respiratory mucus. PLoS One. 2014;9:e110026. doi: 10.1371/journal.pone.0110026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palese P., Tobita K., Ueda M., Compans R.W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61:397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- 10.Couch R.B., Atmar R.L., Franco L.M., Quarles J.M., Wells J., Arden N., Niño D., Belmont J.W. Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J. Infect. Dis. 2013;207:974–981. doi: 10.1093/infdis/jis935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Memoli M.J., Shaw P.A., Han A., Czajkowski L., Reed S., Athota R., Bristol T., Fargis S., Risos K., Powers J.H., et al. Evaluation of antihemagglutinin and antineuraminidase antibodies as correlates of protection in an influenza A/H1N1 virus healthy human challenge model. mBio. 2016;7:e00417. doi: 10.1128/mBio.00417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monto A.S., Petrie J.G., Cross R.T., Johnson E., Liu M., Zhong W., Levine M., Katz J.M., Ohmit S.E. Antibody to influenza virus neuraminidase: an independent correlate of protection. J. Infect. Dis. 2015;212:1191–1199. doi: 10.1093/infdis/jiv195. [DOI] [PubMed] [Google Scholar]
- 13.Rockman S., Brown L.E., Barr I.G., Gilbertson B., Lowther S., Kachurin A., Kachurina O., Klippel J., Bodle J., Pearse M., Middleton D. Neuraminidase-inhibiting antibody is a correlate of cross-protection against lethal H5N1 influenza virus in ferrets immunized with seasonal influenza vaccine. J. Virol. 2013;87:3053–3061. doi: 10.1128/jvi.02434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandbulte M.R., Jimenez G.S., Boon A.C.M., Smith L.R., Treanor J.J., Webby R.J. Cross-reactive neuraminidase antibodies afford partial protection against H5N1 in mice and are present in unexposed humans. PLoS Med. 2007;4:e59. doi: 10.1371/journal.pmed.0040059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Absalon J., Koury K., Gruber W.C. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2021;384:1578. doi: 10.1056/NEJMc2036242. [DOI] [PubMed] [Google Scholar]
- 16.El Sahly H.M., Baden L.R., Essink B., Doblecki-Lewis S., Martin J.M., Anderson E.J., Campbell T.B., Clark J., Jackson L.A., Fichtenbaum C.J., et al. Efficacy of the mRNA-1273 SARS-CoV-2 vaccine at completion of blinded phase. N. Engl. J. Med. Overseas. Ed. 2021;385:1774–1785. doi: 10.1056/NEJMoa2113017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim M.S., Jung S.Y., Ahn J.G., Park S.J., Shoenfeld Y., Kronbichler A., Koyanagi A., Dragioti E., Tizaoui K., Hong S.H., et al. Comparative safety of mRNA COVID-19 vaccines to influenza vaccines: a pharmacovigilance analysis using WHO international database. J. Med. Virol. 2022;94:1085–1095. doi: 10.1002/jmv.27424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Łabno A., Tomecki R., Dziembowski A. Cytoplasmic RNA decay pathways—enzymes and mechanisms. Biochim. Biophys. Acta. 2016;1863:3125–3147. doi: 10.1016/j.bbamcr.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Brito L.A., Chan M., Shaw C.A., Hekele A., Carsillo T., Schaefer M., Archer J., Seubert A., Otten G.R., Beard C.W., et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014;22:2118–2129. doi: 10.1038/mt.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogel A.B., Lambert L., Kinnear E., Busse D., Erbar S., Reuter K.C., Wicke L., Perkovic M., Beissert T., Haas H., et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol. Ther. 2018;26:446–455. doi: 10.1016/j.ymthe.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blakney A.K., Ip S., Geall A.J. An update on self-amplifying mRNA vaccine development. Vaccines (Basel) 2021;9:97. doi: 10.3390/vaccines9020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palladino G., Chang C., Lee C., Music N., De Souza I., Nolasco J., Amoah S., Suphaphiphat P., Otten G.R., Settembre E.C., Wen Y. Self-amplifying mRNA SARS-CoV-2 vaccines raise cross-reactive immune response to variants and prevent infection in animal models. Mol. Ther. Methods Clin. Dev. 2022;25:225–235. doi: 10.1016/j.omtm.2022.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollock K.M., Cheeseman H.M., Szubert A.J., Libri V., Boffito M., Owen D., Bern H., O'Hara J., McFarlane L.R., Lemm N.M., et al. COVAC1 study Group Safety and immunogenicity of a self-amplifying RNA vaccine against COVID-19: COVAC1, a phase I, dose-ranging trial. EClinicalMedicine. 2022;44:101262. doi: 10.1016/j.eclinm.2021.101262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibrahimi A., Vande Velde G., Reumers V., Toelen J., Thiry I., Vandeputte C., Vets S., Deroose C., Bormans G., Baekelandt V., et al. Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Hum. Gene Ther. 2009;20:845–860. doi: 10.1089/hum.2008.188. [DOI] [PubMed] [Google Scholar]
- 25.Izidoro M.A., Gouvea I.E., Santos J.A.N., Assis D.M., Oliveira V., Judice W.A.S., Juliano M.A., Lindberg I., Juliano L. A study of human furin specificity using synthetic peptides derived from natural substrates, and effects of potassium ions. Arch. Biochem. Biophys. 2009;487:105–114. doi: 10.1016/j.abb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pepini T., Pulichino A.M., Carsillo T., Carlson A.L., Sari-Sarraf F., Ramsauer K., Debasitis J.C., Maruggi G., Otten G.R., Geall A.J., et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: implications for vaccine design. J. Immunol. 2017;198:4012–4024. doi: 10.4049/jimmunol.1601877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Hagan D.T., Rappuoli R., De Gregorio E., Tsai T., Del Giudice G. MF59 adjuvant: the best insurance against influenza strain diversity. Expert Rev. Vaccines. 2011;10:447–462. doi: 10.1586/erv.11.23. [DOI] [PubMed] [Google Scholar]
- 28.Gilbert S.C. T-cell-inducing vaccines—what's the future. Immunology. 2012;135:19–26. doi: 10.1111/j.1365-2567.2011.03517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Air G.M. Influenza neuraminidase. Influenza Other Respir. Viruses. 2012;6:245–256. doi: 10.1111/j.1750-2659.2011.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blakney A.K., McKay P.F., Shattock R.J. Structural components for amplification of positive and negative strand VEEV splitzicons. Front. Mol. Biosci. 2018;5:71. doi: 10.3389/fmolb.2018.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bochkov Y.A., Palmenberg A.C. Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques. 2006;41:283–284. doi: 10.2144/000112243. 286, 288 passim. [DOI] [PubMed] [Google Scholar]
- 32.Heeringa M., Leav B., Smolenov I., Palladino G., Isakov L., Matassa V. Comparability of titers of antibodies against seasonal influenza virus strains as determined by hemagglutination inhibition and microneutralization assays. J. Clin. Microbiol. 2020;58 doi: 10.1128/JCM.00750-20. e00750-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Couzens L., Gao J., Westgeest K., Sandbulte M., Lugovtsev V., Fouchier R., Eichelberger M. An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J. Virol. Methods. 2014;210:7–14. doi: 10.1016/j.jviromet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Baras B., Stittelaar K.J., Simon J.H., Thoolen R.J.M.M., Mossman S.P., Pistoor F.H.M., van Amerongen G., Wettendorff M.A., Hanon E., Osterhaus A.D.M.E. Cross-protection against lethal H5N1 challenge in ferrets with an adjuvanted pandemic influenza vaccine. PLoS One. 2008;3:e1401. doi: 10.1371/journal.pone.0001401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data supporting the findings of this study are available within the article.