

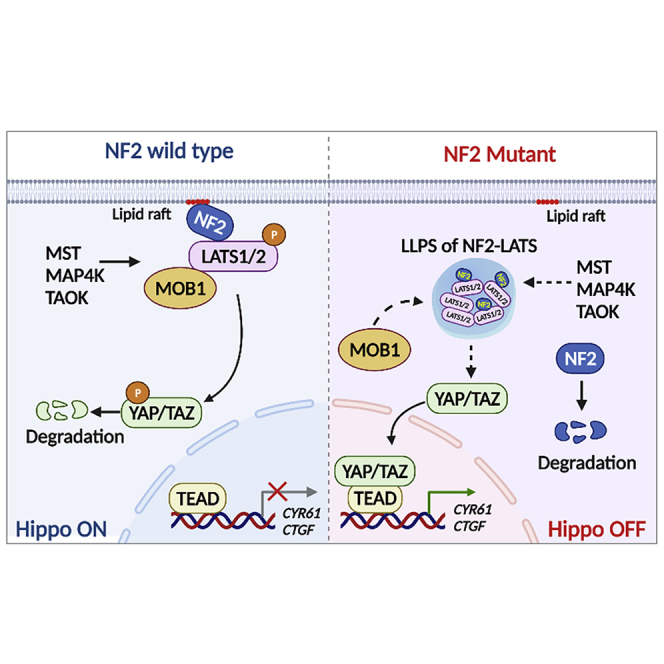
Summary
Neurofibromatosis type 2 is an autosomal dominant multiple neoplasia syndrome and is usually caused by mutations in the neurofibromin 2 (NF2) gene, which encodes a tumor suppressor and initiates the Hippo pathway. However, the mechanism by which NF2 functions in the Hippo pathway isn’t fully understood. Here we identified a NF2 c.770-784del mutation from a neurofibromatosis type 2 family. MD simulations showed that this mutation significantly changed the structure of the F3 module of the NF2-FERM domain. Functional assays indicated that the NF2 c.770-784del variant formed LLPS in the cytoplasm with LATS to restrain LATS plasma membrane localization and inactivated the Hippo pathway. Besides, this deletion partly caused a skipping of exon 8 and reduced the protein level of NF2, collectively promoting proliferation and tumorigenesis of meningeal cells. We identified an unrecognized mechanism of LLPS and splicing skipping for the NF2-induced Hippo pathway, which provided new insight into the pathogenesis of neurofibromatosis type 2.
Subject areas: Clinical genetics, Pathophysiology, Functional aspects of cell biology
Graphical abstract
Highlights
-
•
NF2 c.770-784 deletion is a novel mutation related to Neurofibromatosis type 2
-
•
NF2 variant forms LLPS in the cytoplasm with LATS and inhibits the Hippo pathway
-
•
NF2 variant causes an aberrant skipping of exon 8 and reduces NF2 protein level
-
•
NF2 variant promotes proliferation and tumorigenesis of meningeal cells
Clinical genetics; Pathophysiology; Functional aspects of cell biology
Introduction
Neurofibromatosis type 2 is hereditary autosomal dominant disorder. It is characterized by bilateral vestibular schwannomas that occur together with an increased risk of meningiomas, cranial nerve tumors, optic gliomas, and/or spinal tumors (Coy et al., 2020). Neurofibromatosis type 2 generally presents in young adults and has almost 100% penetrance by the age of 60 with an incidence of about one in 25,000 (Asthagiri et al., 2009; Evans et al., 2010).
The Neurofibromin 2 (NF2) gene was first identified as a potential neurofibromatosis type 2 tumor suppressor in 1993 (Rouleau et al., 1993; Trofatter et al., 1993). The NF2 gene maps on chromosome 22q12.2 and is composed of 16 exons, terminating with exon 17 in its canonical isoform (Cooper and Giancotti, 2014). The mutational frequency of NF2 was estimated to be 6.5 × 10−6. Approximately half of the patients with neurofibromatosis type 2 present with no prior family history of the condition and with de novo mutations of NF2 (Asthagiri et al., 2009; Baser, 2006). At least 450 variants have been noted occurring in the NF2 gene (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=NF2). The NF2 gene encodes the tumor suppressor NF2 protein (also termed “Merlin” for “moesin, ezrin, radixin-like protein”), which consists of 595 amino acids with 70kDa (Bretscher et al., 2002). NF2 contains a conserved N-terminal FERM (4·1-ezrin-radixin-moesin) domain, a central coiled-coil (CC) domain, and a C-terminal domain (CTD) (Li et al., 2014). The FERM domain is composed of three structural modules (F1, F2, F3) to form a compact clover-shaped structure (Hennigan et al., 2010).
NF2 has been shown to act as an upstream regulator of the Hippo pathway, which plays a pivotal role in tumor suppression by restricting cellular proliferation (Havranek and Islam, 2021; Ma et al., 2019; Pan, 2010; Yu et al., 2010). NF2 can initiate the Hippo signal by recruiting large tumor suppressor (LATS) to the plasma membranes for phosphorylation and activation by mammalian Ste20-like kinases ½ (MST1/2) (Yin et al., 2013). LATS mediated the phosphorylation of the key downstream effector YAP (yes association protein), regulating its cytoplasmic localization and transcriptional inactivity (Yin et al., 2013; Rausch and Hansen, 2020). NF2’s deletion or loss-of-function suppresses the Hippo pathway, eventually leading to tumorigenesis (Hong et al., 2020). Phosphorylation of both LATS and YAP has been shown to be dramatically reduced in the NF2 KO cells (Plouffe et al., 2016). Recently, the Lin group reported that LATS1 undergoes liquid-liquid phase separation (LLPS). The interaction of LATS with phosphatidic acids (PA) promoted LATS phase separation and inhibited LATS1-mediated YAP phosphorylation (Li et al., 2021). NF2 form cellular condensates with IRF3 and TBK1 in innate nucleic acid sensing (Meng et al., 2021). However, the mechanism of how NF2 recruits LATS to the plasma membrane and functions in Hippo signaling is not yet fully understood.
In an effort to understand the role of the NF2 gene in the pathogenesis of neurofibromatosis type 2 in the Chinese population, we initiated a systematic NF2 mutational screening. In the present study, we identified a novel c.770-784 deletion of NF2, which changed NF2 localization from the plasma membrane to the cytoplasm. Correspondingly, we also examined the role and mechanism of this NF2 mutant variant in tumorigenesis through a series of functional assays, gene-edited cells, xenograft models, and other techniques. Our results suggest that the observed novel deletion of the NF2 gene is clearly associated with neurofibromatosis type 2.
Results
Clinical presentation
The Han Chinese family included five patients with neurofibromatosis type 2. The whole family consisted of 10 members from four generations (Figure 1A). We collected five family members’ and four related individuals’ samples (peripheral blood and clinical evaluations) with their consent. The sagittal T1-weighted contrast-enhanced MRI of the proband (Ⅳ-3) revealed several isohyperintense lesions at the accessory nerve of the foramen magnum (Figure 1B/a) and at L4-5 and L2-3 levels of the lumbar spine (Figure 1B/b). The abdominal T1-weighted contrast-enhanced MRI also showed a subcutaneous mass to the left of the lumbar region (Figure 1B/c). The hard bumps were about 3 × 4 cm at that time and they presented with clear boundaries. The T1-weighted contrast-enhanced cranial MRI of the proband’s father (Ⅲ-4) revealed multifocal hyperintense lesions involving the cerebral falx, and at the convexity of the dura mater, bilateral cerebellopontine angle regions and lateral medulla oblongata (Figure 1B/d, 1B/e). The enhanced brain MRI also indicated hyperintensity as shown in the left frontal mass displaying a characteristic “dural tail sign” (Figure 1B/f). In addition to the two patients above, the proband’s aunt (Ⅲ-1), grandmother (Ⅱ-2), and grandmother’s sister (Ⅱ-3) had all passed away owing to neurofibromatosis type 2.
Figure 1.

A novel c.770-784del variant of the NF2 gene was identified
(A) The Chinese neurofibromatosis type 2 pedigree. Affected individuals are indicated by filled symbols. The arrow denotes the proband.
(B) MRI features of IV-3 (1B/a, 1B/b, 1B/c) and III-4 (1B/d, 1B/e, 1B/f).
(C) Histological features of IV-3 (1C/a, 1C/b, 1C/c) and III-4 (1C/d, 1C/e, 1C/f).
(D) Partial sequencing chromatograms of PCR products of the NF2 gene. See also Figure S1.
(E) Partial sequencing chromatograms showing the c.770-784del of NF2.
(F) PCR-PAGE analysis for the c.770-784del of NF2.
(G-I) MD simulations on FERM domain. (G) RMSD curves for the Cα atoms of the FERM domain. (H) RMSF for the Cα atoms of the FERM domain. (I) Overlay of the structures of the FERM domain in wild-FERM and mutant-FERM at the end of the simulation. See also Table S1.
(J–M) MD simulations on NF2 protein. RMSD curves versus simulation times for the wild-type NF2 (J) and mutant NF2 (K). Structures of the wild-type NF2 (L) and mutant NF2 (M) at the end of the simulation. See also Table S1.
The lesions of the proband were nodular and firm. The tumor was composed of short fascicles of spindle cells which were seen under microscopy to be haphazardly arranged in a variably collagenized or loosely ground structure. Hematoxylin and eosin (HE) stains revealed compact cellular areas with spindle-shaped cells and with the palisading of nuclei representing Verocay bodies containing areas of cellular myxoid connective tissue (Figure 1C/a, 1C/b). An immunohistochemistry stain tested positive for S100 (Figure 1C/c). The specimen of the proband’s father was also gray and nodular. His HE stain showed proliferating spindle cells with small oval nuclei and fibrous stroma (Figure 1C/d, 1C/e). The hypercellular and hypocellular sclerotic areas could be recognized. Mitotic activity was inconspicuous and necrotic. Hemorrhagic areas were also not observed. The immunohistochemical stain tested positive for vimentin/EMA (Figure 1C/f).
These clinical presentations suggested diagnoses for the proband of a neurofibromatosis type 2 associated neurofibroma, and for the proband’s father of a neurofibromatosis type 2 associated meningioma.
Identification of a novel c.770-784del mutation of the neurofibromin 2 gene
According to the candidate gene strategy, we performed the mutational screening of the NF2 gene with peripheral blood samples. All 17 exons of NF2 were PCR-amplified and sequence analyzed from the patients, unaffected relatives, and unrelated control individuals. The sequencing chromatograms (Figure 1D) showed that the heterozygous peaks began from position c.770 in the forward fragment and from position c.784 in the reverse fragment of exon 8 in two patients (Ⅲ-3, Ⅳ-3). No other sequence changes in this gene were detected among these individuals. To identify the mutation, its PCR products were cloned into a pMD19-T vector and analyzed by sequencing. Results indicated that a 15bp-deletion existed between c.770-784 in one strand (Figure 1E). PCR-PAGE analysis showed that both the affected patients (Ⅲ-3, Ⅳ-3) carried the heterozygous 15bp-deletion, while other unaffected members lacked this deletion (Figure 1F). We did not detect the presence of this 15bp-deletion in any of the 106 control individuals living in the same area. We searched out the sequences of NF2 protein in 15 organisms and made alignments. The five amino acids (Pro-Trp-Asn-Glu-Ile) at position 257-261 of NF2 protein were highly evolutionarily conserved. Trp258 and Ile261 were virtually identical from humans to primates, mammals, amphibians, fish, insects, and nematodes (Figure S1). Based on evaluations of the Mutation Taster website and the statement of the American College of Medical Genetics and Genomics (ACMG), these results suggest that the NF2 c.770-784del (VCV001299301.1) may be involved in neurofibromatosis type 2.
Next, we performed a molecular dynamic (MD) simulation to understand the stability, structure, and dynamics of this mutant variant (Havranek and Islam, 2021). The root-mean-squared deviation (RMSD) curves represent the time evolution of the structural differences between the simulated structures and the initial structures. These curves suggest that the wild-type FERM domain remained close to the initial structure throughout the 200ns simulation (Figure 1G, blue curve). The mutant FERM underwent large structural changes where the most prominent structural change occurred at around 125ns (Figure 1G, green curve). The root-mean-squared fluctuation (RMSF) curves indicate that the wild-type FERM domain remained slightly more flexible than its mutant counterpart (Figure 1H). The superimposition of the wild-type and mutant FERM structures shows that the deletion only causes structural changes in the F3 module, without having much effect on F1 and F2 modules (Figure 1I). These structural changes caused by the deletion altered the surface profile of the FERM domain, further affecting interactions between the FERM domain and other parts of the NF2 protein. We then ran MD simulations on NF2 protein with and without the deletion. The RMSD curves show that from 50ns both the FERM domain and the C-terminal domain became stabilized right through to the end of the simulation (Figures 1J and 1K). In the mutant-NF2 system, the FERM domain became stabilized after around 50ns (Figure 1K, dark-gray line). Intriguingly, the C-terminal domain in the mutant-NF2 system kept changing throughout the simulation (Figure 1K, green line), in contrast to the observation that the C-terminal domain became very stable after 50ns in the wild-NF2 system (Figure 1J, blue line). Figures 1L and 1M show the binding modes of the C-terminal domain with the FERM domain in the wild-NF2 (Figure 1L) and mutant-NF2 (Figure 1M) systems at the end of the simulations. These results suggest that NF2 p.257-261del significantly changes the structure of the F3 module.
Neurofibromin 2 mutant restricted its binding to the plasma membrane
To examine whether the c.770-784 deletion of NF2 affects the subcellular localization of NF2, the pEGFP-NF2 (WT) or pEGFP-NF2 (mut) were transfected into the HEK293T cells for the transient expression. Consistent with the plasma membrane-specific dye CellMask™, the same cells showed a pattern of green fluorescence that coincides with GFP-NF2 (WT) expression. The superimposition of the two panels showed almost a complete overlap of the two patterns, demonstrating that the NF2-WT localizes to the plasma membrane. In contrast, the NF2-mut expressing cells showed typical green fluorescence indicating that most had occurred in the cytoplasm, and only a little in the plasma membrane (Figure 2A). To confirm this situation, we performed a lipid dot-blot assay. Phosphatidic acid (PA) carries a negative charge and is emerging as an important signaling lipid in the plasma membrane. Although the NF2-mut was detected as having interaction with PA and was also PA-dose dependent, the interaction strength was significantly lower than that of the NF2-WT protein (Figure 2B). To confirm the critical amino acid residue of NF2 binding to PA, we constructed some plasmids carrying a single amino acid residue deletion at p.257-261 (Pro-Trp-Asn-Glu-Ile, respectively). Compared with other deletions, the Trp258 deletion significantly changed the subcellular localization of NF2 (Figure S2A) and decreased its interaction with PA (Figure S2B). Both the immunostaining and lipid dot-blot assay suggested that the p.257-261 deletion of NF2 had markedly decreased its binding with PA, leading to the NF2 variant being transferred from the plasma membrane to the cytoplasm, particularly as Trp258 is one of the critical residues of NF2 localizing to the plasma membrane.
Figure 2.

NF2 mutant showed restricted binding to the plasma membrane
(A) pEGFP-NF2-WT, pEGFP-NF2-mut and pEGFP were expressed in HEK293T cells, respectively. The immunofluorescence pattern of transfected HEK293T cells was double-labeled with CellMask™ and GFP. Scale bar, 50μm. See also Figure S2A.
(B) The Lipid dot-blot assay confirmed the interaction between PA with NF2. See also Figure S2B.
(C) Time-series fluorescence microscopy analysis of GFP-NF2 puncta. Bottom row shows zoom-in view of two fusing puncta. Scale bar, 10μm (top) and 1μm (bottom). See also Video S1.
(D) Representative micrographs of GFP-NF2 puncta before and after photobleaching. Scale bar, 0.2μm. See also Figure S3.
(D′) Quantification of fluorescence intensity recovery in the bleached region of GFP-NF2 puncta. Error bars, SEM of three independent experiments.
(E) NF2-mut contained an IDR.
(F) GFP-NF2-mutΔIDR were expressed in HEK293T cells. The immunofluorescence pattern of NF2-mutΔIDR puncta. Scale bar, 20μm.
(F′) Quantification of puncta per cell. Error bars, SEM of three micrographs. nsP>0.05, Student’s t test.
(G) Time-series fluorescence microscopy analysis of NF2-mutΔIDR puncta. Bottom row shows zoom-in view of two fusing puncta. Scale bar, 10μm (top) and 1μm (bottom). See also Video S2.
(H) Representative micrographs of NF2-mutΔIDR puncta before and after photobleaching. Scale bar, 0.2μm.
(H′) Quantification of fluorescence intensity recovery in the bleached region of NF2-mutΔIDR puncta. Error bars, SEM of three independent experiments.
(I) No in vitro liquid droplets of either NF2-WT, NF2-mut or NF2-mutΔIDR proteins.
Strikingly, the NF2-mut was not uniformly distributed in the cytoplasm but formed multiple discrete puncta (Figure 2A). Moreover, in vivo studies revealed that the NF2-mut puncta were dynamic in the cytoplasm and could spontaneously undergo fusion in HEK293T cells (Figure 2C and Video S1). The fluorescence recovery after the photobleaching (FRAP) assay showed that the green fluorescence of NF2-mut puncta recovered within a few minutes after photobleaching (Figures 2D, 2D′). NF2-mut puncta remained in the cytoplasm even after saponin treatment where it disrupted membrane organelles (Figure S3). These results indicate that the NF2-mut puncta present in the dynamic non-membrane-bound compartments, in a manner similar to liquid-liquid phase separation (LLPS). Bioinformatics analysis revealed that the NF2 variant contained an intrinsically disordered region (IDR) in p.308-590 amino acid residues of the α-helical and CTD (Figure 2E). This IDR is suggested to be one of the main driving forces of LLPS. We constructed the pEGFP-NF2-mutΔIDR plasmid lacking an IDR for overexpression in HEK293T cells. The NF2-mutΔIDR still formed droplets. The numbers of NF2-mutΔIDR droplets in each cell seemed to show no significant differences, from those of the NF2-mut (Figure 2F). There was also no significant difference in the number of puncta between NF2-mutΔIDR and NF2-mut (Figure 2F’). These in vivo droplets possessed the LLPS characteristics of spontaneous fusion (Figure 2G and Video S2) and fluorescence recovery after photobleaching (Figures 2H and 2H′). Furthermore, in vitro phase separation assays in a physiological buffer showed that none of the NF2-WT, NF2-mut, or NF2-mutΔIDR proteins could form liquid droplets (Figure 2I). These results suggest that the in vivo LLPS of the NF2-mut might be not driven by its own IDR, but induced by other factors.
Neurofibromin 2 mutant restricted the recruitment of large tumor suppressor to the plasma membrane and formed liquid-liquid phase separation of the neurofibromin 2-large tumor suppressor in the cytoplasm
The formation of LLPS can occur between homologous proteins or heterogeneous proteins. It has been reported that NF2 can interact with LATS and that LATS can form homologous LLPS. To verify whether heterogeneous LLPS forms between NF2 with LATS, we compared the localization of LATS only with the colocalization of both LATS and NF2 in HEK293T cells. LATS was mainly located in the cytoplasm and formed several puncta when only pRFP-LATS plasmid was transfected into HEK293T cells. Strikingly, LATS and NF2-WT were co-located in the cell membrane, while LATS and NF2-mut formed puncta and co-located in the cytoplasm (Figure 3A). Compared with that of LATS puncta, the number of NF2-LATS heterogeneous puncta was significantly increased (p < 0.001) (Figure 3A’), but their volume was significantly decreased (p < 0.0001) (Figure 3A’’). Co-IP assay showed that there was no significant difference in LATS interaction with NF2-WT or NF2-mut (Figure S4). If pEGFP-NF2-mutΔBD without binding domain (BD) to LATS and pRFP-LATS were co-transfected into HEK293T cells, LATS still located in the cytoplasm and formed several puncta. NF2-mutΔBD was evenly distributed in the cytoplasm, which is different not only from the plasma membrane localization of NF2-WT, but also from the cytoplasmic puncta of NF2-mut (Figure 3A). These results suggest that the subcellular localization of LATS is mediated by NF2, which interacts with LATS through its BD region. NF2 and LATS may together form heterogeneous LLPS.
Figure 3.

NF2 mutant restricted the recruitment of LATS to the plasma membrane and formed LLPS of NF2-LATS in the cytoplasm
(A) The pRFP-LATS only, pRFP-LATS and pEGFP-NF2-WT, pRFP-LATS and pEGFP-NF2-mut, pRFP-LATS and pEGFP-NF2-mut BDdel were expressed in HEK293T cells, respectively. The immunofluorescence pattern of in transfected HEK293T cells. The number (A′) and size (A″) of puncta were quantified. Scale bar, 20μm. See also Figure S4.
(B) Phase-separation assays of LATS, LATS and NF2-WT, LATS and NF2-mut proteins in vitro. Scale bar, 20μm.
(C) Time-series fluorescence microscopy analysis of LATS, LATS and NF2-WT, LATS and NF2-mut. Scale bar, 20μm.
(D, D′) in vitro FRAP assay and quantification of LATS and NF2-WT puncta.
(E, E′) in vitro FRAP assay and quantification of LATS and NF2-mut puncta.
(F, F′) in vitro FRAP assay and quantification of LATS puncta. Time 0s indicates the photobleaching pulse. Scale bar, 20μm. Error bars, SEM of three independent experiments. Data are represented as mean ± SEM ∗∗∗p < 0.001, Student’s t test.
We further performed studies to verify the heterogeneous LLPS of NF2 and LATS. The expressed and purified EGFP-NF2 (WT) or EGFP-NF2 (mut) were mixed with RFP-LATS protein in LLPS buffer with PEG-8000. Regardless of whether it was NF2-WT or NF2-mut, both were observed to be co-localized with LATS and formed some droplets in vitro as noted under the fluorescence microscope (Figure 3B). Moreover, these droplets were dynamic and could spontaneously undergo fusion (Figure 3C). An in vitro FRAP assay indicated that the fluorescence of LATS/NF2-WT (Figures 3D and 3D′) or LATS/NF2-mut (Figures 3E and 3E′) co-puncta had recovered within a few minutes after photobleaching, similar to that of LATS alone (Figures 3F and 3F′). These results suggest that LATS mediates the formation of heterogeneous LLPS between NF2 and LATS.
Construction and identification of meningioma cell line carrying neurofibromin 2 c.770-784del mutation
IOMM-Lee cells, a malignant meningioma (MM) cell line, may prove useful as a research model (Lee, 1990). To elucidate whether this NF2 mutant leads to increased tumor formation from meningeal cells, we constructed the IOMM meningioma cell lines with NF2 c.770-784del using the CRISPR/Cas9 system (Figure 4A) and investigated the essential signaling pathways of NF2. Several heterozygous (NF2+/−) clones were identified by sequence analysis of the NF2 gene and PCR-amplification of cDNA from gene-edited cells (Figures 4B and 4C). Using the primers NF2-WT homologous to the 5′ AND-3′ ends of the cDNA, three NF2 products of RT-PCR were detected in the agarose gel (Figure 4C). PCR products were cloned into a PCR 2.1-TOPT vector and analyzed by sequencing. Results showed that these NF2 transcripts were the wild-type full-length with 1785bp, c.770-784del variant with 1770bp, and exon 8 skipping variant with 1650bp, respectively (Figure 4D). Further, we performed exon trapping using the pSPL3 plasmids (Figure 4E). Minigene assays showed that both the pSPL3 and pSPL3-NF2-mut gave rise to a 187bp PCR fragment missing exon 8 of NF2 gene, while pSPL3-NF2-WT only yielded a main RT-PCR product of 322bp containing exon 8, and pSPL3-NF2-mut also has a PCR product of 307bp lacking NF2 c.770-784 (Figure 4F). To sum up, we proved that NF2 c.770-784del is a splicing mutation partly causing the skipping of exon 8 (Figure 4G). These results suggested that we had successfully generated the IOMM meningioma cell lines with NF2 c.770-784del heterozygous (NF2+/−) mutant similar to patients.
Figure 4.

Construction and identification of meningioma cell line with NF2 c.770-784 heterozygous deletion
(A) Schematic diagram of guide RNA and pMD19-NF2-mut construction using the CRISPR/Cas9 system.
(B) Partial sequencing chromatograms of NF2+/− from gene-edited cells.
(C) NF2 mRNA identification of gene-edited cells.
(D) The sequence chromatograms of partial cDNA sequences of WT-IOMM and NF2+/−IOMM.
(E) The schematic diagram of pSPL3-NF2-mut plasmids.
(F) Partial sequence chromatograms and gel electrophoresis of RT-PCR products in pSPL3-NF2 (WT or mut) transfected cells.
(G) The schematic diagram of splicing both in WT-IOMM and NF2+/−IOMM.
Neurofibromin 2 mutant reduced the phosphorylation of large tumor suppressor and yes association protein, therefore inactivating the Hippo pathway
Western blotting showed that NF2+/−IOMM cells express lower levels of NF2 relative to the control IOMM meningioma cells (Figure 5A). The steady-state level of total LATS and YAP showed no significant changes in NF2+/−IOMM and WT-IOMM (Figure 5A). Compared to WT-IOMM cells, the relative phosphorylation levels of LATS1/2 and YAP were reduced to 30 ± 6% (p < 0.001) and 53 ± 8% (p = 0.0015) in NF2+/−IOMM (Figures 5A, 5A′, and 5A″), respectively. Immunofluorescence staining showed that the proportion of YAP that had localized in the nucleus rather than the cytoplasm was far higher in NF2-mut cells than in NF2-WT cells, indicating that this NF2-mut caused YAP translocation into the nucleus (Figure 5B). Both subcellular fractionation and Western blotting results were consistent with the above observation (Figure 5C). Furthermore, the transcriptional enhanced associate domain (TEAD) family of transcription factors was activated (Figures 5A and 5A‴), resulting in marked elevation of expression levels of YAP/TEAD target genes, such as CTGF (Figure 5D) and CYR61 (Figure 5E) in NF2+/−IOMM cells. These results suggest that NF2 deletion caused changes in the subcellular localization of LATS from the cell plasma membrane to the cytoplasm and promoted the formation of heterogeneous LLPS of NF2-LATS, reducing the phosphorylation of LATS and YAP, and thus inhibiting the Hippo pathway in meningioma cells.
Figure 5.

NF2 mutant promoted cell proliferation through inactivating Hippo pathway
(A, A′, A″, A‴) Western blotting and quantification of NF2, LATS1/2, YAP and TEAD. GAPDH is shown as a loading control.
(B) The immunofluorescence pattern of RFP-YAP1 and GFP-NF2 in HEK293T cells, which were transfected with exogenous GFP-NF2 (WT or mut) and RFP-YAP1. Scale bar, 50μm.
(C) Western blotting of YAP subcellular fractionation. Vinculin and Lamin B are shown as loading control of cytoplasm and nucleus, respectively.
(D and E) Relative expression level of CTGF and CYR61 by qPCR.
(F) Observation of WT-IOMM and NF2+/−IOMM meningioma cells under light microscope.
(G) Growth curve of WT-IOMM and NF2+/−IOMM meningioma cells. Error bars, SEM of three micrographs.
(H) Doubling time of WT-IOMM and NF2+/−IOMM meningioma cells.
(I, I′, I″) Observation, quantification on rate of clone formation and clone size of WT-IOMM and NF2+/−IOMM meningioma cells.
(J, J′) Observation and quantification on in vitro wound-healing assay of WT-IOMM and NF2+/−IOMM meningioma cells. Error bars, SEM of three independent experiments. Data are represented as mean ± SEM ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Student’s t test.
Neurofibromin 2 mutant increased cell proliferation and tumorigenicity
To investigate the pathogenicity of the NF2 c.770-784del, we performed cell proliferation and tumorigenicity assays. The IOMM meningioma cells grew by adherence to polygonal shape and containing abundant cytoplasm. The NF2+/−IOMM cells grew faster and had higher density (Figure 5F). After subculture for 4 days, the cell number of WT-IOMM and NF2+/−IOMM cell lines was approximately 0.97×106 and 1.13×106 (p=0.023) (Figure 5G). By calculation, the doubling time was 23.5h of WT-IOMM cells and 20.5h (p=0.015) of NF2+/−IOMM cells (Figure 5H). In addition, an in vitro colony formation assay (Figure 5I) showed that the rate of clone formation for the WT-IOMM and NF2+/−IOMM cell lines was approximately 30.9 and 36.3% (p=0.0097) (Figure 5I’), and the clone size of those cells was 49.7pels and 113.7pels (p < 0.001) (Figure 5I’’). The in vitro wound-healing assay showed that the cell migration rate of NF2+/−IOMM cells was faster than that of wild-type (Figure 5J). 36 h after scratching, the wounding healing percentage of WT-IOMM and NF2+/−IOMM cells was approximately 37.9 and 58.4% (p = 0.021) (Figure 5J’).
Next, we implanted WT-IOMM or NF2+/−IOMM cells into immunocompromised mice which were euthanized 22 days late (Figure 6A). Results indicated that xenograft tumors from NF2+/−IOMM cells grew faster than those from WT-IOMM cells. The volume of xenograft tumors at day 22 were 0.55 ± 0.03 cm3 (p = 0.0116) from NF2+/−IOMM cells and 0.41 ± 0.08 cm3 from WT-IOMM cells (Figure 6B). After harvesting (Figure 6C), the tumors were weighted and shown 0.30 ± 0.05g (p = 0.0024) from NF2+/−IOMM cells and 0.17 ± 0.05g from WT-IOMM cells (Figure 6C’). HE staining showed no significant changes in histomorphology in the NF2+/−IOMM and WT-IOMM groups (Figure 6D). Consistent with that in the IOMM cells (Figures 6D and 6D′), the total YAP level showed no significant changes in the xenograft tumors from WT-IOMM (67.89 ± 2.29%) and NF2+/−IOMM (63.19 ± 1.18%). Assessing the cell proliferation using Ki67 immunohistochemistry, a significant increase of proliferation was evident in NF2+/−IOMM (24.40 ± 3.67%, p=0.012), compared to the WT-IOMM of 15.10 ± 0.47% (Figures 6D and 6D″). There were no significant variations in CD31, which usually represents tumor angiogenesis (Figures 6D and 6D‴). These results from xenograft tumorigenesis models confirmed that the meningioma cells with NF2 c.770-784del presented with increased cell proliferation and more serious tumorigenicity. The NF2 c.770-784del is probably associated with neurofibromatosis type 2.
Figure 6.

NF2 mutant increased tumorigenicity
(A) Immunocompromised mice were implanted with WT-IOMM or NF2+/−IOMM meningioma cells.
(B) Growth curve of xenograft tumors from mice implanted with WT-IOMM and NF2+/−IOMM meningioma cells.
(C, C′) Harvests and quantification of xenograft tumors from mice implanted with WT-IOMM and NF2+/−IOMM meningioma cells.
(D, D′, D″, D‴) Immunohistochemistry and quantification of HE, YAP, Ki67 and CD31 of tumor from mice implanted with WT-IOMM and NF2+/−IOMM meningioma cells. Error bars, SEM of three independent experiments. Data are represented as mean ± SEM nsP>0.05, ∗p < 0.05, ∗∗p < 0.01, Student’s t test.
Discussion
In the present study, we performed clinical, genetic, cellular, and molecular characterization of a pedigree of neurofibromatosis type 2. We identified a novel c.770-784 deletion of NF2 in this pedigree, which encoded five highly conserved amino acids (Pro-Trp-Asn-Glu-Ile) at position 257-261 of NF2 protein. The FERM domain, in the N-terminal of NF2, is composed of three structural modules (F1, F2, F3) and the threee modules together form a compact clover-shaped structure (Hennigan et al., 2010). The deleted peptides included the linkers between the two anti-parallel beta sheets in the F3 module of the FERM domain. There were some known pathogenic mutations in the F3 subdomain (Cooper and Giancotti, 2014), suggesting F3 subdomain is essential in tumor-suppression function. In addition, NF2 can suppress the proliferation of cells, in which FERM play an important role by binding a lot of factors, such as membrane proteins (Sato and Sekido, 2018; Zhou and Hanemann, 2012), transmembrane receptors (Kim et al., 2016) and lipids (Chinthalapudi et al., 2018; Hong et al., 2020). Our MD simulations showed that the mutation caused dramatic structural changes in both the orientation and shape of the beta-barrel of the F3 module where the C′-end alpha helix of CTD binds with the FERM domain, which may change the conformation of NF2 (Hennigan et al., 2010) and affect the NF2’s ability to bind effectors (Chinthalapudi et al., 2018). Our research showed that NF2 c.770-784del changed the interaction between FERM and CTD domain, this may be the reason for causing neurofibromatosis type 2.
In vitro and in vivo studies indicated NF2 as mainly localized to the plasma membrane (Cooper and Giancotti, 2014; Yin et al., 2013). Most notably, NF2 was shown to directly interact with PA, which is an anionic lipid consisting of a negatively charged phosphomonoester head group attached to a hydrophobic diacylglycerol backbone (Han et al., 2018; Shin and Loewen, 2011). The lipid-binding deficient mutants in FERM, such as T59V, W60E, R309Q, and R310Q, led to the disability of NF2 to bind phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] in the plasma membrane (Chinthalapudi et al., 2018). In this report, the NF2 membrane localization was significantly restricted owing to the novel variant of p.257-261del at the F3 module, for which the hydrophobic Trp258 is a critical residue. The lipid dot-blot assay confirmed that the deleted peptides in the FERM domain could interact with PA. In addition to PA, there are other plasma membrane components, such as PI(4,5)P2, phosphatidylserine (PS), capable of interacting with NF2 (Li et al., 2021). Our study is also in agreement with the observation that the FERM domain contains multiple conserved lipid-binding sites (Li et al., 2021). Several previous reports identified NF2 mutations between p.257-261 from patients with neurofibromatosis type 2, such as nonsense mutations of p.Trp258X (c.773G>A) (Wallace et al., 2004) and p.Trp258X (c.774G>A) (Ruttledge et al., 1996), duplication mutation of c.774-778dupGAATG (Laniewski-Wollk et al., 2008). Our data suggest that the p.257-261 motif is probably involved in this newly identified lipid-binding site of NF2 protein, in which the highly conserved hydrophobic residue Trp258 is crucial for its attachment to the plasma membrane.
Previous studies have shown that NF2 directly binds to and recruits the LATS to the plasma membrane where LATS1/2 are phosphorylated by their upstream kinases, thereby initiating the Hippo pathway (Hong et al., 2020; Ma et al., 2019; Yin et al., 2013). The FERM domain mediates NF2’s bindings to its various targets, of which the Q178del mutant could directly interfere with the interaction between NF2 with LATS (Li et al., 2015). In addition, LATS has also been reported to directly combine with PA (Li et al., 2021). Here the p.257-261del variant did not directly affect the binding of NF2 to LATS, but restricted the localization of the NF2-LATS to the plasma membrane. These observations suggest that the NF2-FERM domain includes multiple sites for NF2 binding with LATS or PA and that the subcellular localization of LATS is mediated by NF2. The phosphorylation of LATS being reduced owing to the disability of NF2-LATS localization to the plasma membrane in NF2+/−IOMM, suggested that the Hippo pathway had become inactive. The elevated unphosphorylated YAP is then translocated into the nucleus, interacting with the TEAD to induce the expression of the downstream target genes CTGF and CYR61 (Verma et al., 2019; White et al., 2019).
Strikingly, the NF2 variant caused the formation of NF2-LATS heterogeneous LLPS in the cytoplasm. The LLPS of biological macromolecules is crucial for signal transduction and gene regulation (Brangwynne et al., 2009; Li et al., 2012; Sun et al., 2018). Our previous reports showed that LATS alone undergoes LLPS (Li et al., 2021). The NF2-FERM domain has been shown to bind with LATS and PI(4,5)P2 (Stickney et al., 2004). In this study, and differing from LATS, NF2 does not alone undergo LLPS, but it interacts with LATS through the FERM binding domain (p.69-100) (Li et al., 2015) and forms LLPS of NF2-LATS. If NF2 is wild type, it mediates the LATS to the lipid raft of the plasma membrane, where MST1/2 phosphorylates and activates LATS, in turn, to phosphorylate YAP, and thereby activating the Hippo pathway. However, the NF2 p.257-261del variant impairs its binding site with PA and restricts the LATS to the plasma membrane, which leads to the forming of the LLPS of NF2-LATS in the cytoplasm and thus reducing the phosphorylation of LATS. Our observation confirmed that both NF2 binding and the corresponding mediation of the localization of the LATS at the plasma membrane are necessary for LATS activation. Any mutation that affects NF2 binding to LATS or NF2 membrane localization may therefore lead to LATS inactivation. These results suggest that LATS undergoes LLPS by interacting with a variety of molecules or forming complexes, which then affects its activity and regulates downstream signaling pathways. Therefore, NF2 plays a crucial role in regulating LATS by the formation of the NF2-LATS phase separation.
Minigene assay and Sanger sequence results of NF2 cDNA proved that NF2 c.770-784 deletion is also a splicing mutation resulting in the skipping of exon 8 (Figures 4E–4I). Splicing mutation is a common mutation in inherited diseases, may occur in both introns and exons and disturb existing splice sites or splicing regulatory sequences (intronic and exonic splicing silencers and enhancers), create new ones, or activate the cryptic ones (Anna and Monika, 2018). Using Human Splicing Finder (HSF) Website, we predicted that CCGTGGA (NF2 c.770-776) may be an exonic splicing enhancers (ESE) site. In normal situation, an exon with a majority of ESE will favor the exon recognition by the spliceosome. In our study, NF2 c.770-784 deletion removes ESE in Exon 8 and significantly alters the ESE/ESS ration, finally makes spliceosome can’t recognize exon and results the skipping of Exon 8. Western blotting shows that the skipping of exon 8 reduced the protein level of NF2 significantly, which can further inhibit the Hippo signaling pathway. Furthermore, consistent with recent report that the IOMM-Lee cells lacking NF2 formed bigger colonies compared to control IOMM-Lee cells (Waldt et al., 2021), here we found that the gene-edited IOMM cells carrying the NF2 c.770-784del heterozygous mutation similar to patients showed increased cell proliferation and enhanced tumorigenicity.
In conclusion, we have identified a novel NF2 c.770-784del mutation associated with neurofibromatosis type 2. This mutation impairs NF2 localization to the plasma membrane and forms LLPS of NF2-LATS in the cytoplasm, while cause the skipping of exon8 and reduction of NF2 expression level. This then inactivates LATS and further inhibits the Hippo pathway, eventually promotes the proliferation and tumorigenesis of meningeal cells. Our findings thus provide insight into the pathogenesis of neurofibromatosis type 2 that occurs via NF2 variant.
Limitations of the study
Our findings demonstrate that NF2 c.770-784 deletion is a novel mutation associated with Neurofibromatosis type 2, which can partly cause the skipping of Exon 8 and reduce the protein level of NF2. However, we did not analyze mutation and NF2 variants in the patient tumor materials, only in peripheral blood and gene-edited cell lines. In addition, NF2 as a tumor suppressor gene requires a LOH for the consequences of Merlin loss to come into play. We did not further explore the mechanism of how NF2 c.770-784del mutant functions in a dominant-negative fashion. Therefore, we will confirm the NF2 mutant and variants in tumor materials once we get the tumor samples. And further studies on how NF2 c.770-784del functions in a dominant-negative fashion may help us to better understand the occurrence of Neurofibromatosis type 2.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Anti-GAPDH | Abcam | Cat# ab181602; RRID: AB_2630358 |
| Rabbit polyclonal Anti-flag | Protein tech | Cat# 20543-1-AP; RRID:AB_11232216 |
| Mouse monoclonal Anti-HA | Sino Biological | Cat# 100028-MM10; RRID:AB_2860039 |
| Mouse monoclonal Anti-GST | Protein tech | Cat# HRP-66001; RRID:AB_2883833 |
| Rabbit monoclonal Anti-YAP | Cell Signaling Technology | Cat# 14074; RRID:AB_2650491 |
| Rabbit polyclonal Anti-phospho-YAP(S127) | Cell Signaling Technology | Cat# 4911S; RRID:AB_2218913 |
| Rabbit monoclonal Anti-LATS1 | Cell Signaling Technology | Cat# 3477S; RRID:AB_2133513 |
| Rabbit monoclonal Anti-LATS1(T1027) | Cell Signaling Technology | Cat# 8654S; RRID:AB_10971635 |
| Rabbit monoclonal Anti-NF2 | Abcam | Cat# ab109244; RRID:AB_10866511 |
| Rabbit polyclonal Anti-TEAD1 | Affinity | Cat# DF3141; RRID:AB_2835518 |
| Rabbit monoclonal Anti-Ki67 | Cell Signaling Technology | Cat# 9027; RRID:AB_2636984 |
| Mouse monoclonal Anti-CD31 | Cell Signaling Technology | Cat# 3528; RRID:AB_2160882 |
| Rabbit monoclonal Anti-Vinculin | Cell Signaling Technology | Cat# 13901T; RRID:AB_2728768 |
| Rabbit monoclonal Anti-Lamin B | Cell Signaling Technology | Cat# 13435S; RRID:AB_2737428 |
| House polyclonal Anti-mouse IgG-HRP | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Goat polyclonal Anti-rabbit IgG-HRP | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Chemicals | ||
| Trizol | Invitrogen | Cat # 15596026 |
| NucleoBond® CB 100 | TaKaRa | Cat# 740508 |
| PrimeSTAR® Max DNA Polymerase | TaKaRa | Cat# R045A |
| SuperScript III First-Strand Synthesis SuperMix | Invitrogen | Cat# 18080400 |
| RNase H | Thermo Scientific | Cat# EN0201 |
| Lipofectamine® 3000 Transfection Kit | Invitrogen | Cat# L3000075 |
| CellMask™ Deep Red Plasma membrane Stain | Invitrogen | Cat# C10046 |
| DAPI | Thermo Scientific | Cat# 62248 |
| 1,2-Dipalmitoyl-sn-glycero-3-phosphate | MedChemExpress | Cat# HY-113437 |
| Subcellular Protein Fractionation Kit | Thermo Scientific | Cat# 78840 |
| ClonExpress ® MultiS One Step Cloning Kit | Vazyme | Cat# C113 |
| SuperReal PreMix Plus (SYBR Green) | TIANGEN | Cat# FP205 |
| Dulbecco’s Modified Eagle Medium | Gibco | Cat# 11965118 |
| Fetal bovine serum | Gibco | Cat# 16000-044 |
| Bacterial and virus strains | ||
| TOP10 Chemically Competent Cell | YEASEN | Cat# 11801ES80 |
| Rosetta Competent Cell | TIANGEN | Cat# CB108 |
| Experimental models: cell lines | ||
| HEK293T cell lines | American Type Culture Collection | Cat# CRL-3216;RRID:CVCL_0063 |
| IOMM-Lee cell lines | American Type Culture Collection | Cat# CRL-3370; RRID:CVCL_5779 |
| Experimental models: Organisms/strains | ||
| BALB/c nude mice | Shanghai Laboratory Animal Center | N/A |
| Oligonucleotides | ||
| NF2-Exon1 Forward: CTGCCACTGTAGCCACTCAA | This paper | N/A |
| NF2-Exon1 Reverse: AGCCTGACACTGAAATCCTGAA | This paper | N/A |
| NF2-Exon2 Forward: TGTCCAAGGGATGTATTGTGTATG | This paper | N/A |
| NF2-Exon2 Reverse: CTGCATGACTTAATTCAGGAATGTG | This paper | N/A |
| NF2-Exon3 Forward: CTTAATCCTAGCTCTGCTCACTTC | This paper | N/A |
| NF2-Exon3 Reverse: ATTCTTCACCATCACACCTTCAC | This paper | N/A |
| NF2-Exon4 Forward: ACTGAACTCTCCACCTGTCTG | This paper | N/A |
| NF2-Exon4 Reverse: GTCCACAAGTCCCATAACATCTC | This paper | N/A |
| NF2-Exon5 Forward: GACTTGTCATCGGAATTGAATTGC | This paper | N/A |
| NF2-Exon5 Reverse: CCCACACTGTTTACTGGAGATTAC | This paper | N/A |
| NF2-Exon6 Forward: TGAGTTGCTCCTCCAATAGAATG | This paper | N/A |
| NF2-Exon6 Reverse: CTTGACAGGCAGTACACATACC | This paper | N/A |
| NF2-Exon7 Forward: AAGGGCAAGGTGGGAAACTAA | This paper | N/A |
| NF2-Exon7 Reverse: AGCAGAACCAGGTCCAAGTC | This paper | N/A |
| NF2-Exon8 Forward: CAGAAGTCACAAGAATGGAACAGT | This paper | N/A |
| NF2-Exon8 Reverse: ATCAACAACCACACCCTCAAAG | This paper | N/A |
| NF2-Exon9 Forward: CCAGGACAAGGCATAACTTCAT | This paper | N/A |
| NF2-Exon9 Reverse: CGCCAAGTGAGATACCATTCTATA | This paper | N/A |
| NF2-Exon10 Forward: TCCAGAGCTGACGGCACTA | This paper | N/A |
| NF2-Exon10 Reverse: GTAGGCATCGGCAAATGAAGAT | This paper | N/A |
| NF2-Exon11 Forward: TGTTCATTCACCACTAGACTGT | This paper | N/A |
| NF2-Exon11 Reverse: AATCTGCTTCTGCCAACACA | This paper | N/A |
| NF2-Exon12 Forward: GGCTTGTCATTTCTTGTCAGAGG | This paper | N/A |
| NF2-Exon12 Reverse: AAGAGACACAAGTATGACCAGAGA | This paper | N/A |
| NF2-Exon13 Forward: TGTTGTCCTTGCTTGGAAGTTC | This paper | N/A |
| NF2-Exon13 Reverse: GGCAGACCAGGATGTTCTACC | This paper | N/A |
| NF2-Exon14 Forward: GGGAGTGAAGTGGAGGGATG | This paper | N/A |
| NF2-Exon14 Reverse: TGAATGCTGATCTGTTGTCCTT | This paper | N/A |
| NF2-Exon15 Forward: CCTAGATCGCACACCAAGCA | This paper | N/A |
| NF2-Exon15 Reverse: CTCAACTTCCATCCAGCCAATG | This paper | N/A |
| NF2-Exon16 Forward: CCAGGAACAGTCTCACCATT | This paper | N/A |
| NF2-Exon16 Reverse: GGATAACGACCACAAGTAGGAT | This paper | N/A |
| NF2-Exon17a Forward: GACAGCCAACTTCTTGAGCAT | This paper | N/A |
| NF2-Exon17a Reverse: GGAGGGAAGGTGAAGCAGAT | This paper | N/A |
| NF2-Exon17b Forward: CATAGACGCTCCTTCCTGTGT | This paper | N/A |
| NF2-Exon17b Reverse: CAACATCCATACTTACGCTCTAGG | This paper | N/A |
| NF2-Exon17c Forward: ATCCTGCCTTCTTCCTACTATCA | This paper | N/A |
| NF2-Exon17c Reverse: GAGGTGCGTGCCTGTAATC | This paper | N/A |
| NF2-Exon17d Forward: TGGAGTGCAATGGCGTGAT | This paper | N/A |
| NF2-Exon17d Reverse: GCAGTGAGAGACAGCAGAGG | This paper | N/A |
| NF2-mut-test Forward: GACCCTGAGAACAGACTGAC | This paper | N/A |
| NF2-mut-test Reverse: ACCTCCTTGTCACTGTACGA | This paper | N/A |
| CRISPR-NF2-del-test Forward: TCAGCTGGCGCTTACAGTAG | This paper | N/A |
| CRISPR-NF2-del-test Reverse: GGGAAAGATCTGCTGGACCC | This paper | N/A |
| pSPL3-test Forward: TCACCTGGACAACCTCAAAG | This paper | N/A |
| pSPL3-test Reverse: TGAGGAGTGAATTGGTCGAA | This paper | N/A |
| NF2-WT Forward: ATGGCCGGGGCCATCGCTTC | This paper | N/A |
| NF2-WT Reverse: GAGCTCTTCAAAGAAGGCCAC | This paper | N/A |
| NF2-mut Forward: CGAAACATCTCGTACAGTGAC | This paper | N/A |
| NF2-mut Reverse: CGAGATGTTTCGGAA GGAGATCTTGGGGGTCAG |
This paper | N/A |
| NF2-p257del Forward: TGGAATGAAATCCGAAACATC | This paper | N/A |
| NF2-p257del Reverse: GATTTCATTCCAGAA GGAGATCTTGGGGGTCAG |
This paper | N/A |
| NF2-p258del Forward: AATGAAATCCGAAACATCTCG | This paper | N/A |
| NF2-p258del Reverse: TCGGATTTCATTCGGGA AGGAGATCTTGGGGGT |
This paper | N/A |
| NF2-p259del Forward: GAAATCCGAAACATCTCGTAC | This paper | N/A |
| NF2-p259del Reverse: GTTTCGGATTTCCCA CGGGAAGGAGATCTTGGG |
This paper | N/A |
| NF2-p260del Forward: ATCCGAAACATCTCGTACAGT | This paper | N/A |
| NF2-p260del Reverse: GATGTTTCGGATATTCCA CGGGAAGGAGATCTT |
This paper | N/A |
| NF2-p261del Forward: CGAAACATCTCGTACAGTGAC | This paper | N/A |
| NF2-p261del Reverse: CGAGATGTTTCGTTCATTC CACGGGAAGGAGAT |
This paper | N/A |
| NF2-skip Forward: GAACTACTTTGCAATCCGGT TTACTATTAAACCACTGG |
This paper | N/A |
| NF2-skip Reverse: CCAGTGGTTTAATAGTAAAC CGGATTGCAAAGTAGTTC |
This paper | N/A |
| YAP Forward: GAGGCAGAAGCCATGGATC | This paper | N/A |
| YAP Reverse: AATTCAGTCTGCCTGAGGGC | This paper | N/A |
| LATS1 Forward: ATGAAGAGGAGTGAAAAGCC | This paper | N/A |
| LATS1 Reverse: GTTAAACATATACTAGATCGC | This paper | N/A |
| pGEX-4T2-NF2 Forward: ATCTGGTTCCGCGTGGATC CATGGCCGGGGCCATCGCTTC |
This paper | N/A |
| pGEX-4T2-NF2 Reverse: GTCAGTCACGATGCGGCCG CGAGCTCTTCAAAGAAGGCCA |
This paper | N/A |
| pX459-NF2-del Forward: CACCGTCGGATTTCATTCCACGGGA | This paper | N/A |
| pX459-NF2-del Reverse: AAACTCCCGTGGAATGAAATCCGAC | This paper | N/A |
| pSPL3-NF2 Forward: ACGGGATCACCAGAATTC AGCCAGTGACAGAAGCCCTCAG |
This paper | N/A |
| pSPL3-NF2 Reverse: ACCAGATATCTGGGATCC CACACCCTCAAAGCCTGGGAAT |
This paper | N/A |
| EGFP-NF2 Forward: CGAGCTCAAGCTTCGAATT ATGGCCGGGGCCATCGCTTC |
This paper | N/A |
| EGFP-NF2 Reverse: GACCGGCCGGTGGATCGC GAGCTCTTCAAAGAAGGCCAC |
This paper | N/A |
| RFP-LATS1 Forward: GATCTCGAGCTCAAGCTT CGATGAAGAGGAGTGAAAAGC |
This paper | N/A |
| RFP-LATS1 Reverse: CTCGGAGGAGGCCATGG ATCCAACATATACTAGATCGCG |
This paper | N/A |
| 3×HA-NF2 Forward: ATTGAATTCCCCGGGGAT CCATGGCCGGGGCCATCGCTTC |
This paper | N/A |
| 3×HA-NF2 Reverse: TATGGGTAGTCGACTC TAGAGAGCTCTTCAAAGAAGGCCAC |
This paper | N/A |
| 3×Flag-LATS1 Forward: AAGCTTGCGGCCGCG AATTCATGAAGAGGAGTGAAAAGCC |
This paper | N/A |
| 3×Flag-LATS1 Reverse: TTGTAGTCAGCCCGGGAT CCAACATATACTAGATCGCGAT |
This paper | N/A |
| RFP-YAP Forward: CGAGCTCAAGCTTCGAATTCA TGGATCCCGGGCAGCAGCC |
This paper | N/A |
| RFP-YAP Reverse: CGGAGGAGGCCATGGAT CCTAACCATGTAAGAAAGCTTTC |
This paper | N/A |
| 3×Flag-YAP Forward: GCTTGCGGCCGCGAATTC ATGGATCCCGGGCAGCAGCC |
This paper | N/A |
| 3×Flag-YAP Reverse: GTAGTCAGCCCGGGATCC TAACCATGTAAGAAAGCTTTC |
This paper | N/A |
| GAPDH Forward: CAGGGCTGCTTTTAACTCTGGTA | This paper | N/A |
| GAPDH Reverse: CATGGGTGGAATCATATTGGAAC | This paper | N/A |
| CTGF-qPCR Forward: CCAATGACAACGCCTCCTG | This paper | N/A |
| CTGF-qPCR Reverse: TGGTGCAGCCAGAAAGCTC | This paper | N/A |
| CYR61-qPCR Forward: AGCCTCGCATCCTATACAACC | This paper | N/A |
| CYR61-qPCR Reverse: TTCTTTCACAAGGCGGCACTC | This paper | N/A |
| Recombinant DNA | ||
| pEGFP-NF2-WT | This paper | N/A |
| pEGFP-NF2-mut | This paper | N/A |
| pEGFP-NF2-p.257del | This paper | N/A |
| pEGFP-NF2-p.258del | This paper | N/A |
| pEGFP-NF2-p.259del | This paper | N/A |
| pEGFP-NF2-p.260del | This paper | N/A |
| pEGFP-NF2-p.261del | This paper | N/A |
| pGEX-NF2-WT | This paper | N/A |
| pGEX-4T2-NF2-mut | This paper | N/A |
| pGEX-4T2-NF2-p.257del | This paper | N/A |
| pGEX-4T2-NF2-p.258del | This paper | N/A |
| pGEX-4T2-NF2-p.259del | This paper | N/A |
| pGEX-4T2-NF2-p.260del | This paper | N/A |
| pGEX-4T2-NF2-p.261del | This paper | N/A |
| pRFP-LATS1 | This paper | N/A |
| pRFP-YAP1 | This paper | N/A |
| p3×HA-NF2-WT | This paper | N/A |
| p3×HA-NF2-mut | This paper | N/A |
| p3×Flag-LATS1 | This paper | N/A |
| p3×Flag-YAP1 | This paper | N/A |
| pX459-NF2-mut | This paper | N/A |
| pMD19-NF2-mut | This paper | N/A |
| Software and algorithms | ||
| Snapgene | Snepgene | N/A |
| ImageJ | ImageJ | N/A |
| GraphPad Prism 8 | GraphPad | N/A |
| Adobe Photoshop | Adobe Photoshop | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Qingfeng Yan (qyan@zju.edu.cn)
Materials availability
Antibodies were obtained from the commercial sources described in the STAR Methods key resources table. Material generated in this study will be made available upon reasonable request.
Experimental model and subject details
Cell culture
HEK293T cell line and IOMM-Lee cell line were purchased from the American Type Culture Collection (ATCC, Manassas, VA). They were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin and maintained in a humidified atmosphere containing 5% CO2 at 37°C. All cell culture reagents were purchased from GIBCO (Invitrogen, Grand Island, NY).
Animals
BALB/c nude mice were purchased from Shanghai Laboratory Animal Center. All experimental male animals were conducted 6-8 weeks of age, and its protocol was approved by the Animal Welfare and Ethics Committee of Zhejiang University.
Method details
Subjects
The study was approved by the Ethics Committee of the Sir Run Run Shaw Hospital, School of Medicine Zhejiang University, China, and conducted in agreement with the Principles of the Declaration of Helsinki. Informed consent, blood samples, and clinical evaluations were obtained from all of the participating family members. Members of this family were interviewed and evaluated to identify both personal or medical histories and any other clinical abnormalities.
Clinical evaluations
Evaluations of the proband IV-3 and relatives were assessed at a number of levels including medical history, physical examination, laboratory tests, magnetic resonance imaging (MRI), histology and immunohischemistry (Gehlhausen et al., 2015). Phenotype-positive neurofibromatosis type 2 was defined by the following criteria for relatives: (1) bilateral eighth nerve masses revealed with appropriate imaging techniques; or (2) a first-degree relative with neurofibromatosis type 2 and either unilateral eighth nerve mass, or the presence of two of the following: neurofibroma, meningioma, glioma, schwannoma or juvenile posterior subscapular lenticular opacity (Evans, 2009).
Mutation analysis of the NF2 gene
Genomic DNA was isolated from the peripheral blood of participants using a TaKaRa NucleoBond® CB 100 kit (TaKaRa Biotechnology). 20 pairs of primers for PCR-amplifying exons and their flanking sequences were used for this analysis. The sequences were as follows. NF2-Exon1, forward: 5’-CTGCCACTGTAGCCACTCAA-3’, reverse: 5’-AGCCTGACACTGAAATCCTGAA-3’; NF2-Exon2, forward: 5’-TGTCCAAGGGATGTATTGTGTATG-3’, reverse: 5’-CTGCATGACTTAATTCAGGAATGTG-3’; NF2-Exon3, forward: 5’-CTTAATCCTAGCTCTGCTCACTTC-3’, reverse: 5’-ATTCTTCACCATCACACCTTCAC-3’; NF2-Exon4, forward: 5’-ACTGAACTCTCCACCTGTCTG-3’, reverse: 5’-GTCCACAAGTCCCATAACATCTC-3’; NF2-Exon5, forward: 5’-GACTTGTCATCGGAATTGAATTGC-3’, reverse: 5’-CCCACACTGTTTACTGGAGATTAC-3’; NF2-Exon6, forward: 5’-TGAGTTGCTCCTCCAATAGAATG-3’, reverse: 5’- CTTGACAGGCAGTACACATACC-3’; NF2-Exon7, forward: 5’- AAGGGCAAGGTGGGAAACTAA-3’, reverse: 5’-AGCAGAACCAGGTCCAAGTC-3’; NF2-Exon8, forward: 5’-CAGAAGTCACAAGAATGGAACAGT-3’, reverse: 5’-ATCAACAACCACACCCTCAAAG-3’; NF2-Exon9, forward: 5’-CCAGGACAAGGCATAACTTCAT-3’, reverse: 5’-CGCCAAGTGAGATACCATTCTATA-3’; NF2-Exon10, forward: 5’-TCCAGAGCTGACGGCACTA-3’, reverse: 5’-GTAGGCATCGGCAAATGAAGAT-3’; NF2-Exon11, forward: 5’-TGTTCATTCACCACTAGACTGT-3’, reverse: 5’-AATCTGCTTCTGCCAACACA-3’; NF2-Exon12, forward: 5’-GGCTTGTCATTTCTTGTCAGAGG-3’, reverse: 5’-AAGAGACACAAGTATGACCAGAGA-3’; NF2-Exon13, forward: 5’-TGTTGTCCTTGCTTGGAAGTTC-3’, reverse: 5’-GGCAGACCAGGATGTTCTACC-3’; NF2-Exon14, forward: 5’-GGGAGTGAAGTGGAGGGATG-3’, reverse: 5’-TGAATGCTGATCTGTTGTCCTT-3’; NF2-Exon15, forward: 5’-CCTAGATCGCACACCAAGCA-3’, reverse: 5’-CTCAACTTCCATCCAGCCAATG-3’; NF2-Exon16, forward: 5’-CCAGGAACAGTCTCACCATT-3’, reverse: 5’-GGATAACGACCACAAGTAGGAT-3’; NF2-Exon17a, forward: 5’-GACAGCCAACTTCTTGAGCAT-3’, reverse: 5’-GGAGGGAAGGTGAAGCAGAT-3’; NF2-Exon17b, forward: 5’-CATAGACGCTCCTTCCTGTGT-3’, reverse: 5’-CAACATCCATACTTACGCTCTAGG-3’; NF2-Exon17c, forward: 5’-ATCCTGCCTTCTTCCTACTATCA-3’, reverse: 5’-GAGGTGCGTGCCTGTAATC-3’; NF2-Exon17d, forward: 5’-TGGAGTGCAATGGCGTGAT-3’, reverse: 5’-GCAGTGAGAGACAGCAGAGG-3’. Fragments spanning 17 exons and flanking sequences from the proband and his father, two unaffected relatives (IV-1, III-5), and two unrelated control individuals, were PCR amplified, purified, and sequencing analyzed (Liu et al., 2014). These sequence results were compared with the NF2 genomic sequence (NG_009057.1).
MD simulation system
Four systems (wild-FERM, mutant-FERM, wild-Merlin, and mutant-Merlin) were constructed for the molecular dynamics (MD) simulations. The starting coordinates of wild-FERM were taken from the crystal structure of the human NF2-FERM domain (PDB entry: 1H4R). The intact structure of the NF2 protein was built using homology modeling in I-TASSER, with the homologous meosin structure (PDB entry: 2I1K) serving as a template. Detailed information of the four simulation systems is listed in Table S1. MD simulations were carried out using the GROMACS 4.5.5 package with a CMAP-modified CHARMM36 force field (Cang et al., 2013). Electrostatic interactions were calculated using a particle-mesh Ewald (PME) algorithm.
Cell lines, culture conditions, and RNA extraction
HEK293T cells and the IOMM-Lee human meningioma cells (ATCC) were grown in regular Dulbecco’s Modified Eagle’s Medium (Gibco), supplemented with 10% fetal bovine serum (Gibco) (Liu et al., 2014). Total RNA was isolated using TRIzol reagent (Invitrogen).
Subcellular localization
The coding regions of cDNA lacking their natural stop codons were obtained by PCR using high fidelity PrimeSTAR Max Premix (TaKaRa). The products were cloned into the pEGFP-N2 or pRFP-N2 (Clontech). Resultant constructs were transfected into HEK293T cells using the Lipofectamine® 3000 Transfection Kit (Invitrogen). The chamber slides were stained with CellMask™ Plasma Membrane Stains (Life technologies), then observed under confocal fluorescence microscope.
Recombinant protein expression and purification
To create bacterial vectors expressing recombinant protein, the corresponding regions were amplified by PCR using cDNA as a template and cloned into pGEX-4T2 vector (Addgene). All constructs were identified by Sanger sequence and expressed in Rosetta (DE3) cells and purified with glutathione beads (BBI) for GST-fused proteins.
Protein-lipid binding assay
As for the PA membrane, PA (MCE) was dissolved in chloroform at different concentrations and dotted on the nitrocellulose membrane (ThermoFisher). Lipid membrane was blocked in TBST buffer containing 3% fatty acid-free BSA (BBI) at 4°C overnight. 0.5 ug/mL purified protein was incubated with the lipid membrane at room temperature for 2 hours with gentle agitation. After that, the lipid membrane was washed three times in TBST buffer and subjected to primary antibody incubation. Western blotting was performed to detect the lipid-associated protein with indicated antibodies.
Cell lysis, fractionation, immunoprecipitation and immunoblotting
Cells were homogenized in RIPA buffer supplemented with Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail, Panobinostat, and Methylstat. Lysates were cleared by centrifugation at 13,000 rpm for 15 min at 4 °C. Supernatants were analyzed for immunoblotting or for immunoprecipitation with the indicated antibodies. The blotting signals were detected using Clarity Western ECL Substrate (Bio-Rad). Cell fractionation was conducted using Subcellular Protein Fractionation Kit (ThermoFisher). Fractionation fidelity was verified by detection of the cytosolic protein Vinculin in the soluble fraction, and Lamin B in the nuclear fraction.
Phase separation assay in cells
HEK293T cells expressing pRFP-LATS1 and pEGFP-NF2 (WT, mut, mut BDdel) were grown on coverslips. After adhering to coverslips, cells were washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 10 min. After two more washes in PBS, cells were analyzed by confocal microscopy (OLYMPUS, FV3000), and the visible puncta with diameter more than 0.5 μm were defined as LATS1 puncta.
In vitro phase separation assay
In vitro phase separation assays were performed in a physiological LLPS buffer (20mmol/L Tris-HCl, pH 7.5, 15mmol/L NaCl, 130mmol/L KCl, 5mmol/L KH2PO4, 1.5mmol/L MgCl2, and 1mg/mL BSA). The droplet assembly for pEGFP-NF2 or pRFP-LATS was mixed to a 10%(w/v) final concentration of PEG8000 (Sigma). A phase separation assay was carried out on glass-bottomed 35mm dishes (NEST), sealed with optically clear adhesive film to prevent evaporation. These were then observed under confocal fluorescence microscope (Li et al., 2021).
FRAP
FRAP experiments were performed on an OLYMPUS FV3000 confocal microscope with a 60× oil immersion objective. For the in vivo experiments, FRAP assays were carried out on an OLYMPUS FV3000 confocal microscope at 37 °C in a live-cell-imaging chamber. Droplets were bleached with a 488-nm laser pulse (50% intensity, 0.5 s). The recovery from photobleaching was recorded for the indicated time. Analysis of the recovery curves was carried out with ImageJ software.
Saponin permeabilization assay
HEK293T cells growing on coverslips were transfected with pEGFP-NF2 and/or pRFP-LATS1 by lipofectamine 3000 for 8 h. After washing twice with PBS, cells were stained with plasma membrane dye. Then cells were washed twice with PBS and incubated with 0.05% saponin in PBS and fixed with 4% paraformaldehyde in PBS for 10 min. After two more washes in PBS, cells were imaged using confocal microscope (OLYMPUS, FV3000).
Construction of NF2+/--IOMM-Lee cell lines using CRISPR/Cas9
Gene-specific sgRNAs were designed according to primer design principles and named pX459-NF2-del. These were then cloned into pSpCas9(BB)-2A-Puro (Addgene). Either pX459-NF2-del or pMD19-NF2-mut were transfected to IOMM-Lee cells using a Lipofectamine® 3000 Transfection Kit (Invitrogen), selected with puromycin (Amresco) for 2 days. Single clones were sorted and expanded, then confirmed by Sanger sequencing.
Minigene assay
To confirm the probable splice mutation, in vitro analysis was performed using a minigene splicing assay based on the pSPL3 exon trapping vector. Fragments with the NF2-WT (135bp) or NF2-mut (120bp) containing exon 8, flanked by upstream intron 7 (248bp) and downstream intron 8 (132bp), were cloned into splicing vector pSPL3 using homologous recombination method with ClonExpress ® MultiS One Step Cloning Kit (Vazyme). After Sanger sequencing, the pSPL3, pSPL3-NF2-WT and pSPL3-NF2-mut were transferred respectively into HEK293T cell line using the Lipofectamine® 3000 Transfection Kit (L3000-008, Invitrogen, USA) according to the manufacturer’s protocol. After 48h transfection, cells were harvested, and total RNA was extracted with TRIzol reagent (Invitrogen). The vector-specific pSPL3-test primers were used for RT-PCR to confirm the splicing patterns.
Cell growth and colony formation assay
WT-IOMM、NF2+/--IOMM cells were seeded in 12-well plates at a density of 4 × 104 cells/well, and were digested and counted using 0.4% trypan blue staining under inverse light microscope for 4 consecutive days. Doubling time of cells was calculated according to the formula DT=t×lg2/(lgNt-lgN0). Cells were seeded into 6-well plates at a low density (200 cells/well) and cultured for 10 days. Colonies were fixed in formalin, and strained with 0.1% crystal violet (Sigma-Aldrich). The colonies were counted, relative colony numbers and sizes were shown.
Wound-healing assay
WT-IOMM、NF2+/--IOMM cells were plated into the 12-well plates at a density of 2 × 105 cells/well and cultured overnight in complete medium, and when a confluent monolayer was formed, transferred to 1% serum-free medium for 2 days. The confluent monolayers were wounded using a disposable plastic pipette tip (approximately 1mm in size) and washed with serum-free medium, incubated until they were nearly confluent. Then a wound track was scored in each well; every 12hr after scratching, the cells were observed under an inverted microscope.
Quantitative real-time PCR
Cells were collected and total RNA preparations were obtained using Trizol reagent (Invitrogen). The expression levels of RNA were quantified by real-time PCR using an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The primers used for PCR amplification are as follows. CTGF-qPCR, forward: 5’-CCAATGACAACGCCTCCTG-3’, reverse: 5’-TGGTGCAGCCAGAAAGCTC-3’; CYR61-qPCR, forward: 5’-AGCCTCGCATCCTATACAACC-3’, reverse: 5’-TTCTTTCACAAGGCGGCACTC-3’.
Xenograft tumorigenesis model
Twenty 6-8-week-old BALB/c nude mice (Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China) were maintained under specific pathogen-free condition and fed verified mouse chow, and were injected subcutaneously with 5×106 IOMM-Lee cells. Tumor volumes were calculated from the length (a, mm) and the width (b, mm) by using the following formula: V(mm3)=ab2/2. Tumor volume was detected every 3 days. According to the Institutional Animal Care and Use Committee (IACUC) protocol for these experiments, once any tumor exceeded a volume of 1,000 mm3, 1.5 cm in diameter or 10% of body weight, the mice would immediately be euthanized. At the 21 days after cell injection, mice were sacrificed and tumors were harvested (measured and weighted) and fixed in 4% paraformaldehyde. Tumor weight was calculated as mean weight ± SEM in each group (n=6).
Immunohistochemistry
Formalin-fixed, paraffin-embedded specimens were collected. Immunohistochemical staining was conducted on 5μm thick paraffin-embedded sections using Ki67, YAP and CD31 antibodies on the standard HRP detection system (R&D systems). Tissues were counterstained with haematoxylin, dehydrated and mounted.
Quantification and statistical analysis
Where indicated, experiments were repeated at least three times and statistical analysis was performed using unpaired t tests. Data is represented as mean±SEM (n=3). ns, not statistically significant; ∗P< 0.05; ∗∗ P< 0.01; ∗∗∗ P< 0.001.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32070584, 31771398), and Zhejiang Provincial Natural Science Foundation of China (LZ19C060001). We thank Chris Wood of the College of Life Sciences, Zhejiang University for checking the English of this article.
Author contributions
Q.Y conceived and designed the research with the assistance from S.Y and A.L. Z.J and M.L performed the biochemical, molecular and xenograft experiments with the assistance from Z.L, X.D, M.F, D.W, D.X, H.Z, Y.Q, Q.Z, D.L, and W.Y. S.Y and X.Q ascertained and processed clinical specimens. X.C conducted the MD simulation system. Q.Y, Z.J, S.Y, and M.L contributed to discussion and data interpretation with assistance from J.W.Z, W.W and A.L. Q.Y, Z.J and M.L wrote the article.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research. We avoided “helicopter science” practices by including the participating local contributors from the region where we conducted the research as authors on the paper.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105275.
Contributor Information
Shuxu Yang, Email: yang.sx@163.com.
Aifu Lin, Email: linaifu@zju.edu.cn.
Qingfeng Yan, Email: qfyan@zju.edu.cn.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Anna A., Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet. 2018;59:253–268. doi: 10.1007/s13353-018-0444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthagiri A.R., Parry D.M., Butman J.A., Kim H.J., Tsilou E.T., Zhuang Z., Lonser R.R. Neurofibromatosis type 2. Lancet (London, England) 2009;373:1974–1986. doi: 10.1016/S0140-6736(09)60259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baser M.E., Contributors to the International NF2 Mutation Database The distribution of constitutional and somatic mutations in the neurofibromatosis 2 gene. Hum. Mutat. 2006;27:297–306. doi: 10.1002/humu.20317. [DOI] [PubMed] [Google Scholar]
- Brangwynne C.P., Eckmann C.R., Courson D.S., Rybarska A., Hoege C., Gharakhani J., Jülicher F., Hyman A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Bretscher A., Edwards K., Fehon R.G. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Cang X., Du Y., Mao Y., Wang Y., Yang H., Jiang H. Mapping the functional binding sites of cholesterol in β2-adrenergic receptor by long-time molecular dynamics simulations. J. Phys. Chem. B. 2013;117:1085–1094. doi: 10.1021/jp3118192. [DOI] [PubMed] [Google Scholar]
- Chinthalapudi K., Mandati V., Zheng J., Sharff A.J., Bricogne G., Griffin P.R., Kissil J., Izard T. Lipid binding promotes the open conformation and tumor-suppressive activity of neurofibromin 2. Nat. Commun. 2018;9:1338. doi: 10.1038/s41467-018-03648-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J., Giancotti F.G. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014;588:2743–2752. doi: 10.1016/j.febslet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coy S., Rashid R., Stemmer-Rachamimov A., Santagata S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. 2020;139:643–665. doi: 10.53347/rid-31082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D.G.R. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J. Rare Dis. 2009;4:16. doi: 10.1186/1750-1172-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D.G., Howard E., Giblin C., Clancy T., Spencer H., Huson S.M., Lalloo F. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am. J. Med. Genet. 2010;152A:327–332. doi: 10.1002/ajmg.a.33139. [DOI] [PubMed] [Google Scholar]
- Gehlhausen J.R., Park S.J., Hickox A.E., Shew M., Staser K., Rhodes S.D., Menon K., Lajiness J.D., Mwanthi M., Yang X., et al. A murine model of neurofibromatosis type 2 that accurately phenocopies human schwannoma formation. Hum. Mol. Genet. 2015;24:1–8. doi: 10.1093/hmg/ddu414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H., Qi R., Zhou J.J., Ta A.P., Yang B., Nakaoka H.J., Seo G., Guan K.L., Luo R., Wang W. Regulation of the hippo pathway by phosphatidic acid-mediated lipid-protein interaction. Mol. Cell. 2018;72:328–340.e8. doi: 10.1016/j.molcel.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havranek B., Islam S.M. Prediction and evaluation of deleterious and disease causing non-synonymous SNPs (nsSNPs) in human NF2 gene responsible for neurofibromatosis type 2 (NF2) J. Biomol. Struct. Dyn. 2021;39:7044–7055. doi: 10.1080/07391102.2020.1805018. [DOI] [PubMed] [Google Scholar]
- Hennigan R.F., Foster L.A., Chaiken M.F., Mani T., Gomes M.M., Herr A.B., Ip W. Fluorescence resonance energy transfer analysis of merlin conformational changes. Mol. Cell Biol. 2010;30:54–67. doi: 10.1385/1-59259-052-7:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong A.W., Meng Z., Plouffe S.W., Lin Z., Zhang M., Guan K.L. Critical roles of phosphoinositides and NF2 in Hippo pathway regulation. Genes Dev. 2020;34:511–525. doi: 10.3410/f.737466547.793581629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Kim S., Lee S.H., Kim W., Sohn M.J., Kim H.S., Kim J., Jho E.H. Merlin inhibits Wnt/β-catenin signaling by blocking LRP6 phosphorylation. Cell Death Differ. 2016;23:1638–1647. doi: 10.1038/cdd.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łaniewski-Wołłk M., Gos M., Koziarski A., Szpecht-Potocka A. Identification of mutations in the NF2 gene in Polish patients with neurofibromatosis type 2. J. Appl. Genet. 2008;49:297–300. doi: 10.1007/bf00218846. [DOI] [PubMed] [Google Scholar]
- Lee W.H. Characterization of a newly established malignant meningioma cell line of the human brain: IOMM-Lee. Neurosurgery. 1990;27:389–395. doi: 10.1097/00006123-199009000-00008. discussion 396. [DOI] [PubMed] [Google Scholar]
- Li P., Banjade S., Cheng H.C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J.V., King D.S., Banani S.F., et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.3410/f.717960640.793463709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.H., Tian T., Ge Q.W., He X.Y., Shi C.Y., Li J.H., Zhang Z., Liu F.Z., Sang L.J., Yang Z.Z., et al. A phosphatidic acid-binding lncRNA SNHG9 facilitates LATS1 liquid-liquid phase separation to promote oncogenic YAP signaling. Cell Res. 2021;31:1088–1105. doi: 10.1038/s41422-021-00530-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Wei Z., Zhang J., Yang Z., Zhang M. Structural basis of the binding of Merlin FERM domain to the E3 ubiquitin ligase substrate adaptor DCAF1. J. Biol. Chem. 2014;289:14674–14681. doi: 10.1074/jbc.m114.551184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhou H., Li F., Chan S.W., Lin Z., Wei Z., Yang Z., Guo F., Lim C.J., Xing W., et al. Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res. 2015;25:801–817. doi: 10.1038/cr.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Song Y., Li D., He X., Li S., Wu B., Wang W., Gu S., Zhu X., Wang X., et al. 2014. The Novel Mitochondrial 16S rRNA 2336T>C Mutation Is Associated with Hypertrophic Cardiomyopathy; pp. 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Meng Z., Chen R., Guan K.L. The hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 2019;88:577–604. doi: 10.1036/1097-8542.318880. [DOI] [PubMed] [Google Scholar]
- Meng F., Yu Z., Zhang D., Chen S., Guan H., Zhou R., Wu Q., Zhang Q., Liu S., Venkat Ramani M.K., et al. Induced phase separation of mutant NF2 imprisons the cGAS-STING machinery to abrogate antitumor immunity. Mol. Cell. 2021;81:4147–4164.e7. doi: 10.1016/j.molcel.2021.07.040. [DOI] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010;19:491–505. doi: 10.1007/978-1-4614-6220-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe S.W., Meng Z., Lin K.C., Lin B., Hong A.W., Chun J.V., Guan K.L. Characterization of hippo pathway components by gene inactivation. Mol. Cell. 2016;64:993–1008. doi: 10.1007/978-1-4939-8910-2_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch V., Hansen C.G. The hippo pathway, YAP/TAZ, and the plasma membrane. Trends Cell Biol. 2020;30:32–48. doi: 10.1016/j.tcb.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Rouleau G.A., Merel P., Lutchman M., Sanson M., Zucman J., Marineau C., Hoang-Xuan K., Demczuk S., Desmaze C., Plougastel B., et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- Ruttledge M.H., Andermann A.A., Phelan C.M., Claudio J.O., Han F.Y., Chretien N., Rangaratnam S., MacCollin M., Short P., Parry D., et al. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am. J. Hum. Genet. 1996;59:331–342. doi: 10.1136/jmg.33.12.1050-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Sekido Y. NF2/Merlin inactivation and potential therapeutic targets in mesothelioma. Int. J. Mol. Sci. 2018;19:E988. doi: 10.3390/ijms19040988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.J.H., Loewen C.J.R. Putting the pH into phosphatidic acid signaling. BMC Biol. 2011;9:85. doi: 10.1186/1741-7007-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickney J.T., Bacon W.C., Rojas M., Ratner N., Ip W. Activation of the tumor suppressor merlin modulates its interaction with lipid rafts. Cancer Res. 2004;64:2717–2724. doi: 10.1158/0008-5472.can-03-3798. [DOI] [PubMed] [Google Scholar]
- Sun D., Wu R., Zheng J., Li P., Yu L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018;28:405–415. doi: 10.1038/s41422-018-0017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofatter J.A., MacCollin M.M., Rutter J.L., Murrell J.R., Duyao M.P., Parry D.M., Eldridge R., Kley N., Menon A.G., Pulaski K., et al. A novel moesin-ezrin-radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;75:826. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- Verma S., Yeddula N., Soda Y., Zhu Q., Pao G., Moresco J., Diedrich J.K., Hong A., Plouffe S., Moroishi T., et al. BRCA1/BARD1-dependent ubiquitination of NF2 regulates Hippo-YAP1 signaling. Proc. Natl. Acad. Sci. USA. 2019;116:7363–7370. doi: 10.1073/pnas.1822155116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldt N., Kesseler C., Fala P., John P., Kirches E., Angenstein F., Mawrin C. Crispr/Cas-based modeling of NF2 loss in meningioma cells. J. Neurosci. Methods. 2021;356 doi: 10.1016/j.jneumeth.2021.109141. [DOI] [PubMed] [Google Scholar]
- Wallace A.J., Watson C.J., Oward E., Evans D.G.R., Elles R.G. Mutation scanning of the NF2 gene: an improved service based on meta-PCR/sequencing, dosage analysis, and loss of heterozygosity analysis. Genet. Test. 2004;8:368–380. doi: 10.1089/gte.2004.8.368. [DOI] [PubMed] [Google Scholar]
- White S.M., Avantaggiati M.L., Nemazanyy I., Di Poto C., Yang Y., Pende M., Gibney G.T., Ressom H.W., Field J., Atkins M.B., Yi C. YAP/TAZ inhibition induces metabolic and signaling rewiring resulting in targetable vulnerabilities in NF2-deficient tumor cells. Dev. Cell. 2019;49:425–443.e9. doi: 10.1016/j.devcel.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F., Yu J., Zheng Y., Chen Q., Zhang N., Pan D. Spatial organization of hippo signaling at the plasma membrane mediated by the tumor suppressor merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.3410/f.718102176.793484267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Zheng Y., Dong J., Klusza S., Deng W.M., Pan D. Kibra functions as a tumor suppressor protein that regulates hippo signaling in conjunction with merlin and expanded. Dev. Cell. 2010;18:288–299. doi: 10.3410/f.2456975.2093077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Hanemann C.O. Merlin, a multi-suppressor from cell membrane to the nucleus. FEBS Lett. 2012;586:1403–1408. doi: 10.1016/j.febslet.2012.03.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.