

Abstract
Activation of the receptor for advanced glycation end products (RAGE) has been shown to play an active role in the development of multiple neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and Amyotrophic Lateral Sclerosis. Although originally identified as a receptor for advanced glycation end products, RAGE is a pattern recognition receptor able to bind multiple ligands. The final outcome of RAGE signaling is defined in a context and cell type specific manner and can exert both neurotoxic and neuroprotective functions. Contributing to the complexity of the RAGE signaling network, different RAGE isoforms with distinctive signaling capabilities have been described. Moreover, multiple RAGE ligands bind other receptors and RAGE antagonism can significantly affect their signaling. Here, we discuss the outcome of cell-type specific RAGE signaling in neurodegenerative pathologies. In addition, we will review the different approaches that have been developed to target RAGE signaling and their therapeutic potential. A clear understanding of the outcome of RAGE signaling in a cell type- and disease-specific manner would contribute to advance the development of new therapies targeting RAGE. The ability to counteract RAGE neurotoxic signaling while preserving its neuroprotective effects would be critical for the success of novel therapies targeting RAGE signaling.
Keywords: Advanced Glycation End Products Receptor, Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, Astrocytes, Microglia, Neurons, Parkinson’s Disease
Graphical Abstract
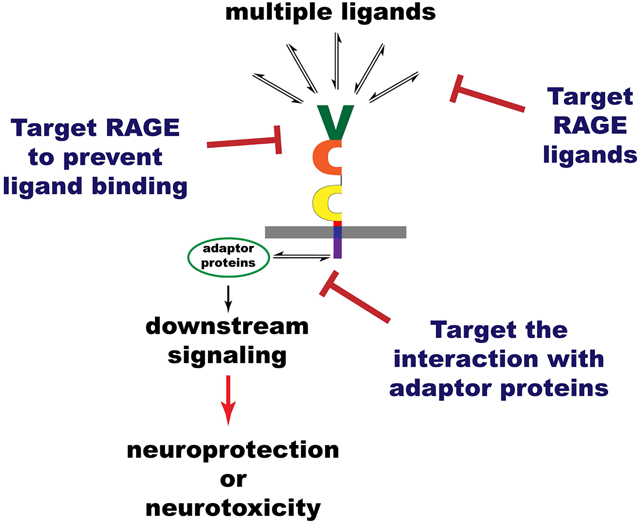
This review examines the beneficial and detrimental effects of RAGE signaling in neurodegenerative conditions. Several methods have been developed to target RAGE signaling with therapeutic purposes. Strategies to specifically counteract RAGE signaling-detrimental effects while preserving its potential beneficial effects would be needed to harness their therapeutic potential.
Introduction.
RAGE, the receptor for advanced glycation end products, is a single-pass type I membrane protein and a member of the immunoglobulin superfamily of cell surface molecules. The protein contains an extracellular region with one variable (V-type) and two constant (C2-type) immunoglobulin-like domains (C1 and C2), a transmembrane domain, and a highly charged cytoplasmic domain [1] (Figure 1). It was originally isolated from bovine lungs based on its ability to bind glycated-albumin [1, 2]. Further research has revealed that RAGE is a pattern recognition receptor that interacts with multiple ligands in addition to advanced glycation end products (AGEs) [3]. Moreover, RAGE has been shown to act as a cargo transporter through the blood brain barrier [4–6], to participate as a cell adhesion molecule enhancing cell-matrix and cell-cell adhesion [7], and/or to activate multiple cell signaling pathways, depending on the cell type/tissue considered and/or the ligand and its concentration [8]. In physiological conditions, RAGE is ubiquitously expressed at low levels, except for the lung, where it is expressed at high levels and has been shown to play important homeostatic roles [9–11]. The expression of RAGE increases in pathological conditions and RAGE signaling has been shown to play an active role in chronic pathological conditions, including late diabetic complications, cardiovascular disease, cancer, and neurodegenerative diseases [12–14]. However, it is important to note that RAGE activation does not always have detrimental consequences. Indeed, it has been shown that in cases of acute injury, such as nerve crush, administration of soluble RAGE (a scavenger of RAGE ligands) or RAGE blocking antibodies significantly impair nerve regeneration [15]. Remarkably, this was not observed in RAGE knockout mice, which display no significant beneficial or adverse effects on nerve fiber regeneration or restoration of conduction velocities after acute nerve crush [16]. As discussed below, because most RAGE ligands can interact with other receptors, the approach used to target RAGE signaling could have profound consequences on the signaling of its ligands through RAGE or other receptors. In this review, we will focus on the role of cell-type specific RAGE signaling in the central nervous system and its potential contribution to neurodegeneration in pathological conditions. In addition, we will discuss the different approaches that have been developed to target RAGE signaling and their therapeutic potential.
Figure 1. RAGE variants generated by alternative splicing or ectodomain shedding.
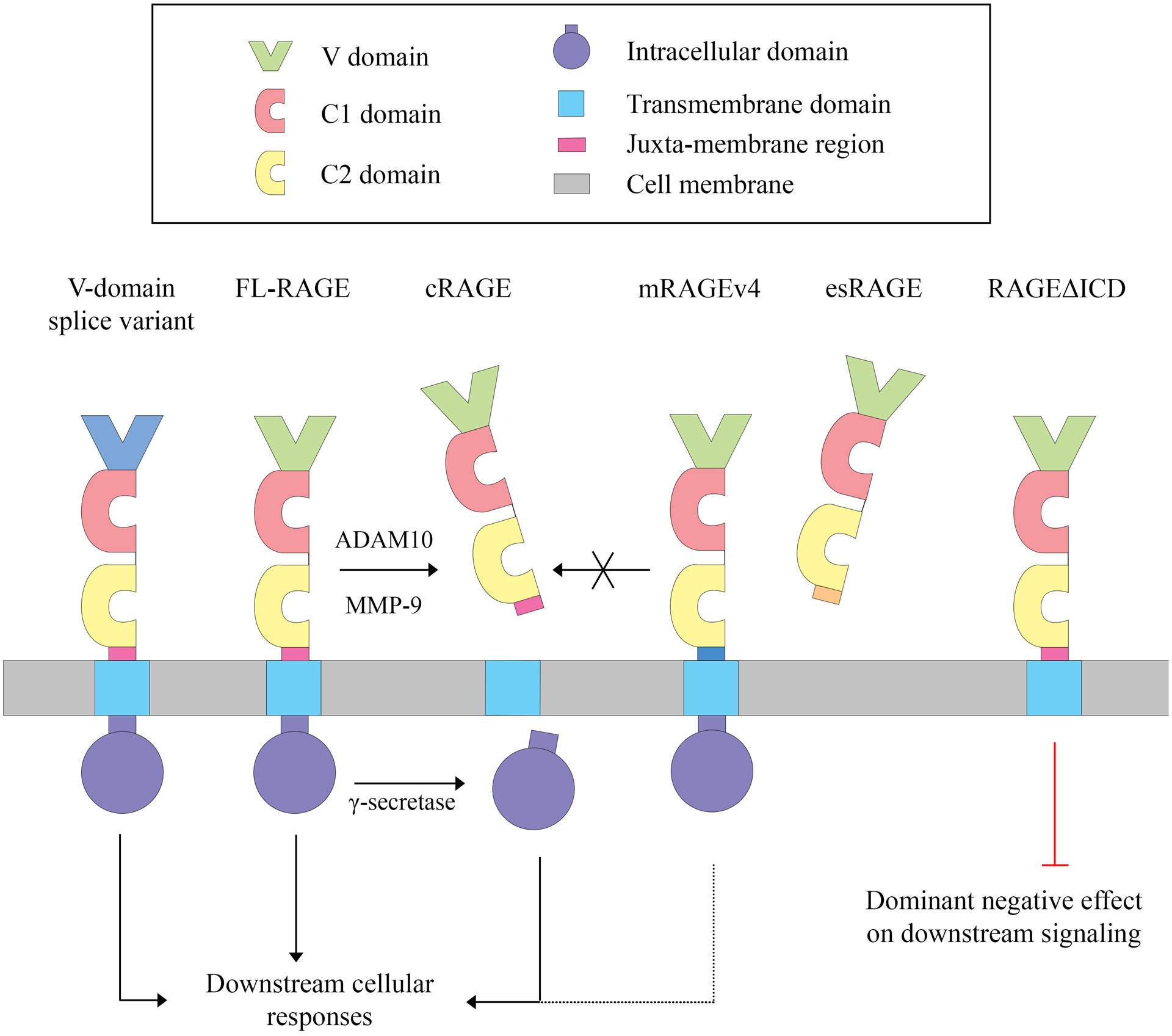
RAGE is a member of the immunoglobulin superfamily of cell surface molecules. It is a single-pass type I membrane protein that contains one variable (V) and two constant (C1 and C2) immunoglobulin-like domains in the extracellular domain, followed by a transmembrane domain and a highly charged intracellular domain (ICD). The most abundant RAGE isoform, in both humans and mice, is the full-length protein (FL-RAGE). Alternative splicing that affects the V-domain produces splice variants that are predicted, but not confirmed, to affect ligand binding. Another splice variant present in murine and human tissues generates a RAGE protein that lacks most of the intracellular domain (RAGEΔICD) and has a dominant negative function on RAGE signaling. Alternative splicing also generates a soluble RAGE isoform known as endogenous secretory (es) RAGE or RAGEv1, which is the second most abundant RAGE transcript variant in humans. Soluble esRAGE has an intact extracellular domain that acts as a decoy receptor blocking FL-RAGE signaling. Ectodomain shedding by ADAM10 or MMP-9 can generate another form of soluble RAGE, cleaved (c) RAGE, that has decoy functions similar to esRAGE. The two soluble RAGE isoforms can be distinguished by the presence of a unique C-terminal sequence in esRAGE. Additionally, following ectodomain shedding, the remaining membrane-bound C-terminal fragment of RAGE is cleaved by γ-secretase to release RAGE ICD, which may then be involved in further RAGE signal transduction pathways. Rodents express a distinct transcript variant (mRAGEv4), not observed in humans or other primates, missing nine amino acid residues located in the extracellular region of the protein, proximal to the transmembrane domain. This isoform is resistant to shedding, but it is not yet clear how mRAGEv4 impacts RAGE signaling and downstream cellular responses.
1. Determinants of RAGE signaling outcome.
RAGE activation leads to a wide array of cellular responses, including cell survival or cell death, activation of a proinflammatory response, oxidative stress, neurite outgrowth, cell migration, and cell proliferation. Among the factors determining the final outcome of RAGE signaling are: i) expression of different RAGE isoforms generated by alternative splicing or ectodomain shedding; ii) glycosylation; iii) interaction with adaptor proteins and co-receptors; and iv) the ligand and its concentration.
i). Alternative splicing of RAGE transcripts and ectodomain shedding.
Multiple RAGE variants generated by alternative splicing have been identified at the transcript level in mouse and human tissues [17–27]. Although several of those variants have not been confirmed to be translated in vivo, different RAGE isoforms have been observed to be differentially expressed at the protein level in different cell types/tissues in physiological and pathological conditions [19, 22, 27]. The exon/intron splice pattern of the different RAGE variants and the relative expression of the different isoforms differ between humans and mice, which could have important consequences in defining species-specific regulation of RAGE signaling and function [25].
A summary of the best characterized RAGE variants is presented in Figure 1. Some splice variants alter the V-domain and are predicted to affect ligand binding, although functional characterization of these variants is still awaited [23, 25]. Another splice variant, which has been shown to be expressed at mRNA level in human and murine tissues, generates a RAGE isoform with a truncated intracellular domain (RAGEΔICD) [26]. Although confirmation that RAGEΔICD is expressed in vivo at protein level is still missing, RAGEΔICD has been shown to exert a dominant negative function on RAGE signaling after experimental overexpression in glioma cells, suggesting that it could have a key role in the regulation of RAGE signaling [26]. In humans, the second most abundant RAGE transcript variant after full-length RAGE (FL-RAGE) is endogenous secretory (es) RAGE (also known as RAGEv1), which is produced by alternative splicing of intron 9 and the removal of exon 10. This leads to a truncated protein with a unique C-terminal sequence of 16 amino acids lacking the transmembrane and cytoplasmic domain [19, 23]. Soluble esRAGE/RAGEv1 has an intact extracellular domain with the ability to bind RAGE ligands and acts as a decoy receptor blocking RAGE signaling [19, 28].
In addition to being generated by alternative splicing of mRNA transcripts, soluble RAGE (sRAGE) can also be produced by ectodomain shedding of the cell-surface receptor by ADAM10 (A Disintegrin And Metalloproteinase Domain 10) or MMP-9 (Matrix MetalloProteinase-9) [29–32]. The shedding of FL-RAGE is induced by ligand binding [31, 33], activation of protein kinase PKCα/PKCβI by treatment with phorbol myristate acetate [31], increased cytosolic calcium levels and activation of phosphoinositide 3- kinase [32, 33], activation of multiple G protein-coupled receptors signaling through different cascades [34], inhibitors of serine/threonine or tyrosine phosphatases [33], and activators of latent metalloproteinases such as 4-aminophenylmercuric acetate (APMA) [33]. The soluble RAGE produced by ectodomain shedding [cleaved RAGE (cRAGE)] can be distinguished from esRAGE by the presence of the unique C-terminal sequence generated by alternative splicing in esRAGE. However, both isoforms are expected to bind the same ligands and exert the same function. In mice, shedding seems to be the major mechanism leading to the production of soluble RAGE [27, 31]. Low levels of transcript variants coding for esRAGE are expressed under normal physiological conditions in mouse tissues [25], while murine FL-RAGE displays a higher rate of constitutive shedding after overexpression in cell lines, compared to human FL-RAGE [27]. Interestingly, rodents express a distinct transcript variant (mRAGEv4), not observed in humans and other primates, which lacks the complete exon 9 encoding nine amino acid residues located in the extracellular region of the protein, proximal to the transmembrane domain [25, 35]. This transcript variant is exclusively localized to the plasma membrane and is resistant to shedding when overexpressed in cells [27, 33, 35]. Thus, humans and mice appear to use different mechanisms to regulate and generate sRAGE isoforms. This is an important observation to consider when performing preclinical studies in mouse models, because the relative abundance of different isoforms (some species-specific) may have a significant impact on RAGE signaling capabilities.
The mechanism regulating FL-RAGE ectodomain shedding is conserved in humans and mice [33]. In addition to generating cRAGE with decoy functions, ectodomain shedding also leads to the release of RAGE intracellular domain (ICD), by processing of the remaining membrane-bound C-terminal fragment of RAGE by γ-secretase [30, 32]. RAGE-ICD has been shown to translocate to the nucleus and to play a critical role in RAGE signal transduction, in particular in the regulation of cell migration [30, 32].
ii). Glycosylation.
RAGE has two N-glycosylation sites on the V-domain (Asn25 and Asn81 in human RAGE and Asn25 and Asn80 in mouse RAGE), and although glycosylation of Asn81 occurs less efficiently than glycosylation of Asn25, both sites have been shown to be modified by complex glycans and hybrid glycans [36, 37]. Glycosylation affects RAGE-ligand interaction differentially depending on the ligand considered. The AGE Nε-(carboxymethyl)lysine binds to glycosylated RAGE but no binding was detected with non-glycosylated RAGE [38]. In addition, glycosylation and further modification of glycans by carboxylation (glutamic acid residues linked to core N-glycans) have been shown to positively affect the ability of RAGE to interact with certain ligands, including HMGB1, S100A8/A9 and S100A12 [36, 39, 40]. On the other hand, non-glycosylated RAGE has a higher affinity for glycolaldehyde-derived AGEs, although this effect was not observed for glyceraldehyde-derived AGEs [41]. However, the binding of other ligands, such as S100B, S100A6, or S100A11, is not significantly affected by the status of RAGE glycosylation [36, 42]. The RAGE polymorphic variant G82S, containing a glycine to serine substitution at position 82 within the second N-glycosylation site, is fully glycosylated, while wild-type RAGE is only partially glycosylated at Asn81 [37]. Interestingly, despite being glycosylated, the RAGE variant G82S displays an increased affinity for glycolaldehyde-derived AGEs, similar to non-glycosylated wild-type RAGE [41]. Molecular dynamics simulations showed that the binding pocket in the glycosylated RAGE G82S variant is more exposed and accessible to external ligands when compared to wild-type RAGE [43]. This may explain the increased affinity of the polymorphic variant for amyloid-beta (Aβ42) when compared to wild-type RAGE [43].
iii). Interaction with adaptor proteins and co-receptors.
RAGE is a type I membrane protein with a short cytoplasmic domain that lacks catalytic activity, but it is essential for signal transduction. Indeed, a truncated isoform of RAGE lacking its cytoplasmic domain behaves as a dominant-negative mutant, able to bind ligands but lacking the ability to activate signaling pathways [26]. The intracellular domain of RAGE has been shown to interact with different adaptor proteins.
Using an affinity chromatography technique, Ishihara et al. [44] identified the interaction of RAGE-ICD with extracellular signal-regulated protein kinases-1 and −2 (ERK-1/2). Direct binding was confirmed for ERK-2 in an in vitro assay and was independent of RAGE phosphorylation status, indicating that RAGE is not an ERK substrate but stabilizes ERK to regulate its activation and substrate interaction under the proximal region of the plasma membrane [44].
In addition, a yeast-two-hybrid screening identified Diaphanous-1 (DIAPH1) as a RAGE-ICD interacting protein [45]. DIAPH1 interacts through its formin homology 1 (FH1) domain with two residues (Arg366 and Gln367) in RAGE-ICD and controls the activation of multiple signaling cascades [46]. Interestingly, RAGE is able to constitutively bind DIAPH1. It has been proposed that it is the clustering of RAGE receptors, induced by ligand binding, that leads to DIAPH1 disinhibition and activation of downstream effectors [46]. Depending on the cell type and cellular context, multiple downstream effectors have been shown to be activated by RAGE-DIAPH1 interaction, including members of the Rho family of GTPases such as RAC1, CDC42 and RHOA; protein kinases such as AKT, GSK3β (glycogen synthase kinase 3b) and ROCK (Rho-associated, coiled-coil-containing protein kinase); and transcription factors such as Egr-1 (early growth response protein-1) and NF-κB (nuclear factor-kappa B) [11].
Another RAGE-ICD interacting protein identified using a yeast two-hybrid approach is the Ser/Thr protein kinase PRAK (p38-regulated/activated protein kinase) [47]. Aβ treatment induces the phosphorylation of PRAK and promotes its direct interaction with RAGE-ICD, leading to the activation of a Rheb-mTORC1/p70s6k pathway that regulates the induction of autophagy in SH-SY5Y neuroblastoma cells [47]. RAGE-ICD has also been shown to interact with TIRAP (Toll-Interleukin 1 Receptor domain-containing Adaptor Protein) and MyD88 (Myeloid Differentiation protein 88), known adaptor proteins for Toll-like receptor-2 and −4 (TLR2/4), after ligand-induced phosphorylation of residue Ser391 in RAGE-ICD by PKCζ [48]. This indicates a potential functional interaction between RAGE and TLRs to regulate the activation of inflammatory pathways [48]. Moreover, a recent study using a T7 phage display system identified SLP76 (Src homology 2 domain-containing leukocyte protein of 76 kDa) as a protein interacting with RAGE-ICD [49]. SLP76 interacts with RAGE-ICD through its sterile α motif (SAM) to activate a downstream signal cascade involving activation of p38 MAPK, Erk1/2, and NF-κB signaling in macrophages [49].
Thus, by interacting with different adaptor proteins, RAGE is able to activate multiple signaling pathways, including activation of NADPH oxidase, MAP kinases (ERK1/2, p38, or JNK), rho-GTPases, phosphoinositol-3-kinase, JAK/STAT, and transcription factors [including NF-κB, activator protein-1 (AP-1), cAMP response element-binding (CREB) protein, signal transducer and activator of transcription family 3 (STAT3), nuclear factor of activated T-cells 1 (NFAT1), and myogenin] [9, 11]. The outcome of RAGE signaling is in part defined by the pool of adaptor proteins expressed by the cell and the physiological context, including the level of antioxidant defenses, energy metabolism, and expression of downstream effectors [9, 11].
RAGE also interacts with other cell surface receptors to activate specific signaling pathways. RAGE signals together with the p75 neurotrophin receptor (p75NTR) to induce motor neuron death in response to oxidatively modified nerve growth factor (NGF) [50]. Post-translational oxidative modifications, such as nitration or glycation, promote NGF oligomerization and confer upon the neurotrophin the ability to signal through RAGE-p75NTR at physiological concentrations (picomolar range) [50, 51]. Blocking either p75NTR or RAGE completely prevented the motor neuron death induced by oxidatively modified NGF, indicating that the simultaneous binding to both receptors is essential to induce death signaling [50, 51]. Crosslinking and co-immunoprecipitation studies indicated that p75NTR and RAGE interact in the cell surface of primary cortical neurons and NSC-34 motor neuron-like cells expressing endogenous levels of both receptors, while an in situ proximity ligation assay confirmed RAGE-p75NTR interaction in the soma and neurites of mouse primary motor neurons [50]. RAGE has also been shown to interact with the β2-integrin Mac-1. In addition to function as a counterreceptor for Mac-1, mediating intercellular contacts to promote leukocyte recruitment [52], RAGE also interacts with Mac-1 in a cis fashion on leukocytes [53]. High mobility group protein box-1 (HMGB1) stimulates the interaction between RAGE and Mac-1 on leukocytes, and both receptors are required for HMGB1 to activate NF-κB signaling [53].
Other reports have shown the interaction between RAGE and leukocyte-associated Ig-like receptor-1 (LAIR-1) in monocytes/macrophages. RAGE and LAIR-1 form a multimeric protein complex together with HMGB1 and the complement component C1q. The tetramolecular complex is held together by C1q, which binds RAGE and HMGB1 through its globular domain and LAIR-1 through the collagen-like tail [54, 55]. The interaction between both receptors was confirmed in a proximity ligation assay. In this assay, RAGE-LAIR-1 interaction was only detected when C1q was present, and it occurred in the presence or absence of HMGB1 [54]. While HMGB1 by itself signals through RAGE to induce a proinflammatory phenotype, HMGB1 together with C1q signals through RAGE-LAIR-1 to induce an anti-inflammatory response in monocytes, involving increased production of proresolving lipid mediators [54, 55]. Other groups have also shown that RAGE interacts with G-protein-coupled receptors, including the leukotriene B4 (LTB4) receptor 1 (BLT1), the formyl peptide receptors FPR1 and FPRL1, and the type 1 angiotensin II receptor (AT1) [56–58]. This interaction has been shown to increase the spectrum of ligands interacting with FPR1 and FPRL1 in glial cells [56], and to modulate the output of LTB4-BLT1 signaling in both positive and negative ways. By enhancing ERK phosphorylation, RAGE-BLT1 promotes neutrophil migration and suppresses NF-κB-driven production of proinflammatory cytokines in response to LTB4 [57]. Pickering et al. [58] showed evidence that RAGE and the AT1 receptor form a heteromeric complex at the plasma membrane. Moreover, they showed that the ability of angiotensin II to activate NF-κB signaling and subsequently induce inflammation through the AT1 receptor does not involve classical Gq-induced signaling, but rather a RAGE ligand-independent transactivation of RAGE signaling following AT1 receptor activation. Accordingly, the proinflammatory effects of the AT1 receptor were enhanced in the presence of RAGE and blocked after RAGE deletion, while AT1 receptor-dependent signaling via classical Gq pathways was not affected by RAGE expression [58]. Furthermore, Yokoyama et al. [59] showed that the interaction between RAGE and the AT1 receptor is bidirectional, and that RAGE ligands are able to activate AT1 receptor signaling inducing Gi, but not Gq activation.
iv). Ligand-receptor interaction.
RAGE is considered a pattern recognition receptor and binds a wide array of ligands, including AGEs, amyloid and fibrillar protein aggregates, members of the S100/calgranulin protein family, heme, C1q, and the DNA binding protein HMGB1 (also known as amphoterin) [3, 9, 60–62]. Structural studies of RAGE extracellular domain have shown that it is arranged in two independent structural units, one formed by the V and C1 domains (VC1 unit) and the other formed by the C2 domain, which is attached to VC1 by a flexible linker [63]. Most ligands have been shown to interact with the V domain or the VC1 structural unit, which displays a positively charged surface [3]. Only two members of the S100 protein family, S100A6 and S100A13, have been shown to interact with the C2 domain of RAGE and both appear to induce apoptosis [64, 65].
The oligomerization of RAGE ligands has been proposed to be key for activation of RAGE signaling [3]. RAGE constitutively oligomerizes on the plasma membrane and the binding of its oligomeric ligands stabilizes the formation of higher order RAGE oligomeric complexes with signaling capabilities [66–69]. In this model of RAGE signaling activation, the higher expression level of RAGE occurring in pathological conditions will promote receptor preassembly, shifting the equilibrium towards the formation of higher order oligomerization states that facilitate RAGE signaling hyperactivation [67]. On the other hand, expression of RAGE isoforms lacking the intracellular domain, including RAGEΔICD and soluble RAGE, would lead to the formation of signaling incompetent hetero-complexes lacking the ability to recruit intracellular adaptor molecules, thus preventing RAGE signaling [67].
Interestingly, the outcome of S100B signaling through RAGE seems to depend not only on the cell type considered but also on the concentration of the ligand, which could also promote its oligomerization in the non-reducing conditions found in the extracellular space [70]. In N18 neuroblastoma cells overexpressing RAGE, nanomolar concentrations of S100B trigger neurite outgrowth, while micromolar concentrations of S100B induce apoptosis [71]. Activation of RAGE signaling by low (nanomolar) concentrations of S100B has also been shown to protect hippocampal neurons from N-methyl-d-aspartate (NMDA)-induced excitotoxicity [72], and LAN-5 neuroblastoma cells from Abeta25–35 toxicity [73]. Conversely, as observed in N18 cells, micromolar concentrations of S100B also induce cell death in LAN-5 neuroblastoma cells and primary dorsal root ganglia neurons [73, 74]. A similar effect was observed in cortical neurons exposed to excitotoxic concentrations of glutamate. Villareal et al. observed that nanomolar concentrations of S100B rescued cortical neurons from glutamate-induced neuronal death, while micromolar S100B levels had a detrimental effect on glutamate-exposed neurons [75]. These authors also showed that the basal NF-κB activity in neurons and the level of NF-κB signaling induction mediated by S100B are key determinants for the prevalence of pro-survival or pro-death S100B effects; with overactivation of NF-κB signaling contributing to neuronal degeneration [75]. It is important to mention that in other neuroblastoma cell lines, such as SH-SY5Y and Neuro2a cells, micromolar concentrations of S100B promote survival and proliferation, respectively [64, 76]. It is also worth mentioning that S100B has been shown to signal in a RAGE-independent manner in myoblasts and microglia, adding yet another level of complexity to the regulation of these intricate signaling pathways [42]. Taken together, these results stress the fact that the outcome of RAGE signaling not only depends on the ligand, its concentration and duration of the stimulus, but also on the cellular context and the inherent characteristics of the cell type considered (e.g., ability to counteract oxidative stress, metabolic flexibility, etc).
2. RAGE signaling in neurodegenerative diseases.
Current evidence indicates an active participation of RAGE signaling in chronic neurodegenerative diseases such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD) and Parkinson’s disease (PD). RAGE is expressed by different cell types in the nervous system, including neurons, astrocytes, microglia, oligodendrocytes, vascular endothelial cells and pericytes [77, 78]. The outcome of RAGE signaling in each cellular compartment is different and the interplay between the different cellular responses defines the final effect on disease progression. A summary of the effect of modulating RAGE expression/signaling, ubiquitously or in a cell-type specific manner, in different models of neurodegeneration is provided in Table 1.
Table 1.
Summary of the effects of modulating RAGE signaling in models of neurodegeneration.
| Pathology | Cell type-specific changes in RAGE expression [selected references] | Effects of altering RAGE signaling in mouse models [selected references] |
|---|---|---|
| Alzheimer’s disease | Increased expression in: |
|
| Amyotrophic lateral sclerosis | Increased expression in: |
|
| Parkinson’s disease | Increased expression in: | |
| Huntington’s disease | Increased expression in: | No data available. |
| Multiple sclerosis | Increased expression in: |
|
Alzheimer’s disease.
An increase in RAGE expression in neurons, glial cells, and brain vasculature has been observed in the brain of AD patients and mouse models [79–83]. RAGE was identified as one of the receptors for amyloid-β peptide (Aβ), inducing cellular stress and microglia activation in response to soluble or fibrillar Aβ [79, 80]. In neuronal cells, Aβ signaling through RAGE induces p38 MAPK activation and impairs synaptic plasticity, blocking the induction of long-term potentiation (LTP) without affecting long-term depression (LTD), paired-pulse facilitation, or basal synaptic transmission [84, 85]. At higher concentrations (micromolar range), in primary neuronal cultures and SHSY-5Y neuroblastoma cells, Aβ oligomers and aggregates induce apoptosis in a RAGE-dependent manner, while the toxicity induced by Aβ fibrils does not appear to involve RAGE signaling [86]. Interestingly, Aβ oligomers and aggregates seem to interact with distinct nonoverlapping regions of RAGE extracellular domain, since neutralizing antibodies targeting the V-domain can only prevent the toxicity induced by Aβ oligomers while neutralization of the C1-domain is required to prevent the toxicity induced by Aβ aggregates [86].
Further support for the involvement of neuronal RAGE signaling in AD pathology was obtained by the group of Dr. Shirley ShiDu Yan using a transgenic mouse model with neuronal-specific RAGE overexpression. These investigators showed that the overexpression of RAGE in neurons aggravates the phenotype of J20 transgenic mice, a widely used AD mouse model overexpressing human amyloid precursor protein (APP) with the Swedish and Indiana mutations [87]. While neuronal overexpression of RAGE hastened synaptic deficits and spatial learning/memory impairment, the overexpression of a dominant negative form of RAGE lacking the intracellular domain exerted a neuroprotective effect, preserving spatial learning/memory and reducing pathological changes in J20 mice [87]. Moreover, it was shown that RAGE signaling promotes APP processing by regulating β- and/or γ- secretase activity [88, 89]. Accordingly, genetic RAGE deletion in J20 mice reduces amyloid pathology and ameliorates the development of learning/memory deficits [89]. However, it is important to mention that the neuroprotective effect of genetic RAGE ablation observed in J20 AD mice was not reproduced in ArcAβ mice, which overexpress human APP with the Swedish and Arctic mutations [90]. Moreover, RAGE signaling has been shown to promote adult hippocampal neurogenesis. Meneghini et al. observed that activation of RAGE signaling by HMGB1 and Aβ oligomers induces NF-κB signaling and promotes the differentiation of adult hippocampal neural precursor cells toward the neuronal lineage [91]. However, they observed that in an AD mouse model, this neurogenic response mediated by RAGE signaling appears to be insufficient to attenuate the development of cognitive impairment, likely by decreased survival and/or lack of complete maturation/integration of the adult newborn neurons [91]. These results illustrate the functional complexity of RAGE signaling and highlight the fact that RAGE signaling inhibition could have a detrimental effect on adult hippocampal neurogenesis.
The group of Dr. Shirley ShiDu Yan also identified an important contribution of microglial RAGE signaling in the development of AD pathology in mouse models [92]. They observed that in J20 mice, the overexpression of RAGE in microglial cells accelerated and increased the expression of proinflammatory cytokines and the development of gliosis and amyloid pathology, hastening the development of spatial learning/memory deficits [92]. Importantly, microglia-specific overexpression of a dominant negative form of RAGE lacking the intracellular domain exerted a neuroprotective effect, reducing neuroinflammation, Aβ accumulation, and attenuating the development of cognitive impairment [92]. These results suggest that inhibiting RAGE signaling specifically in microglial cells could be a valid therapeutic approach to preventing or delaying neurodegeneration in AD.
The increased expression of RAGE at the level of the brain vasculature also appears to play an important role in the development of AD pathology. In the brain vasculature, RAGE functions as a cell-signaling receptor and as a cargo transporter through the blood-brain barrier (BBB). RAGE mediates transcytosis and accumulation of amyloid-β peptide (Aβ) into the brain and increases the expression of proinflammatory mediators and endothelin (ET-1), which promotes vasoconstriction [4, 93]. Accordingly, blocking Aβ-RAGE interaction at the level of the BBB reduced Aβ accumulation in the brain, reduced neuroinflammation, and improved cerebral blood flow in AD mouse models [4]. Moreover, Aβ signaling through RAGE in endothelial cells facilitates the transendothelial migration of monocytes, which may contribute to the increased presence of this inflammatory cell type in the brain of AD patients [94].
Further support for an involvement of RAGE signaling in AD pathology comes from the observation of a significant association between the G82S allele polymorphism and the development of the pathology in Chinese and European populations [95, 96]. This is consistent with the fact that this RAGE variant displays increased affinity for Aβ42 when compared to wild-type RAGE [43]. Moreover, when compared to control subjects, a significant reduction in the level of sRAGE in plasma was observed in patients with vascular dementia and AD; while the AD group showed significantly lower levels when compared to patients with vascular dementia [97]. These results were confirmed by Liang et al. [98], who also observed a decrease in plasma sRAGE levels in patients with non-AD neurodegenerative dementias. A decrease in plasma sRAGE levels in AD patients was also observed by Xu et al. [99] in a smaller sample population, which may explain why the authors did not find a significant difference in the level of plasma sRAGE between control patients and patients with vascular dementia. In a more recent study, Chen et al. [100] observed that high sRAGE levels were associated with a lower prevalence of dementia, although in a longitudinal analysis sRAGE levels were not associated with the incidence of dementia after 18.7 years of follow-up. Further stressing the relevance of these studies, the presence of the RAGE polymorphic variant G82S was associated with a greater reduction in the levels of sRAGE in plasma and a faster rate of cognitive decline [95]. This is particularly relevant considering the ability of sRAGE to act as a decoy receptor to prevent RAGE signaling activation and the neuroprotective effect of increasing sRAGE levels in AD mouse models [4, 101].
Amyotrophic Lateral Sclerosis.
Increased levels of RAGE and several of its ligands, including glycated proteins, oxidatively modified NGF, HMGB1, and S100B, have also been observed in the spinal cord of ALS patients and mouse models [50, 102–108]. An important role of RAGE signaling in ALS pathology is supported by the fact that RAGE expression level in the cervical spinal cord of ALS patients negatively correlates with the age at onset and age at death or tracheostomy [108]. Histological analysis revealed that RAGE is expressed by astrocytes, microglia, and motor neurons in the degenerating spinal cord of ALS patients [103, 104, 108]. A potential pathophysiological role of RAGE signaling in each one of these cell types has been proposed. Astrocytes isolated from rodents overexpressing different ALS-linked mutant human superoxide dismutase 1 (hSOD1) variants induce motor neuron death in co-cultures [109, 110]. We showed that the motor neuron death induced by ALS-astrocytes in co-culture involves the activation of RAGE signaling in motor neurons [50]. Accordingly, the neuronal death induced by astrocytes overexpressing hSOD1G93A was prevented by RAGE blocking antibodies, while motor neurons isolated from RAGE-knockout mice were not sensitive to the neurotoxic factor/s derived from ALS-astrocytes [50]. Moreover, spinal cord extracts from symptomatic hSOD1G93A-mice, which display increased expression of multiple RAGE ligands, induce the death of cultured hSOD1G93A-motor neurons by a mechanism involving RAGE signaling [50]. These results obtained in cell culture models suggest that RAGE signaling could play a role in the death of motor neurons occurring in ALS pathology.
Interestingly, Serrano et al. [111] observed that in an ALS rat model overexpressing hSOD1G93A, the progression of the disease is accompanied by a shift in the expression of RAGE from motor neurons to astrocytes [111]. The decrease in neuronal RAGE staining observed with the progression of the disease could be explained by the loss of RAGE-expressing motor neurons; while the increased expression of RAGE observed in astrocytes could play an important pathophysiological role, especially in relation to neuroinflammation. Silencing of the RAGE ligand S100B decreases the expression of inflammatory markers in primary astrocytes isolated from hSOD1G93A mice [111]. Additionally, in primary astrocyte cultures, micromolar concentrations of S100B induce, in a RAGE-dependent manner, a proinflammatory phenotype that is neurotoxic for neurons exposed to oxygen–glucose deprivation [112]. However, Brambilla et al. [113] showed that RAGE signaling activation in astrocytes can also increase the production of trophic factors that promote motor neuron survival. In primary spinal cord astrocyte cultures, activation of RAGE and TLR4 signaling by HMGB1 increases the expression of BDNF (brain-derived neurotrophic factor) and GDNF (glial cell-derived neurotrophic factor), two important trophic factors for motor neurons [113]. Remarkably, astrocytes overexpressing hSOD1G93A display impaired ability to increase BDNF and GDNF production in response to HMGB1 stimulation, suggesting that ALS astrocytes may have impaired neurotrophic functions [113]. Hence, while inhibition of RAGE signaling in motor neurons could be neuroprotective, the effect of inhibiting RAGE signaling in astrocytes carries a greater risk of negatively impacting their ability to provide enhanced trophic support in response to HMGB1 released by damaged cells.
A potential neuroprotective role for RAGE signaling in ALS is also supported by the observation that hSOD1G93A mice with partial or complete RAGE ablation display a shorter lifespan [114]. By crossing hSOD1G93A mice with RAGE knockout mice, we observed that hSOD1G93A mice with RAGE haploinsufficiency [hSOD1G93A;RAGE(+/−)] display a shorter lifespan compared to hSOD1G93A mice in a wild-type background [hSOD1G93A;RAGE(+/+)]. Despite the detrimental effect in lifespan, hSOD1G93A;RAGE(+/−) mice preserved hind-limb grip strength throughout most of the disease progression. On the other hand, animals with complete and ubiquitous RAGE deletion showed no significant improvement in grip strength and only the detrimental effect on overall survival was observed [114]. Although these results could reflect important neuroprotective functions of RAGE signaling at central level, they could also be explained, at least in part, by the critical homeostatic role of RAGE signaling in lung function [115, 116]. The potential detrimental effect of RAGE deletion in the lungs could be aggravated by the overexpression of mutant hSOD1G93A in this ALS mouse model. This ALS-linked hSOD1 variant retains catalytic activity [117], and it was previously shown that the overexpression of wild-type SOD1 accelerates the development of asbestos-induced pulmonary fibrosis [118]. Together, these results further support the idea that inhibition of RAGE signaling in a cell type-specific manner could improve the overall neuroprotective effect observed.
It is important to highlight that a different study analyzing the effect of global RAGE deletion in the progression of the disease in hSOD1G93A mice reported different results. In this study, the authors observed an overall increase in the survival of hSOD1G93A mice after RAGE deletion [107]. These disparate results could be explained by the fact that in this study, Lee et al. [107] used a RAGE knockout mouse line that displays a genomic duplication that leads to an approximately two-fold upregulation of glyoxalase-1 (Glo1) expression [119], a key enzyme in the detoxification of methylglyoxal, the most reactive glycating agent in vivo [120]. This increase in Glo1 expression is expected to lessen the production of glycated proteins, one of the main RAGE ligands, and it could significantly contribute to the neuroprotective effects observed in this RAGE knockout mouse model.
The relevance of RAGE signaling in microglia cells in the context of ALS was recently established using transgenic mouse models. Using RAGEflox/flox mice crossed with mice expressing Cre recombinase under the control of the Cx3cr1 promoter in a tamoxifen-inducible manner, MacLean et al. [108] showed that RAGE deletion in microglial cells (and monocytes/macrophages) starting at 90 days of age significantly reduced gliosis and motor neuron loss, and preserved motor function in male hSOD1G93A mice, but not female mice. Moreover, a transcriptomic analysis revealed that changes in multiple pathways linked to RAGE signaling were significantly ameliorated in hSOD1G93A male mice with microglia RAGE deletion [108]. Although the outcome of the study by MacLean et al. [108] could be confounded by the fact that the mouse model used has hemizygous Cx3cr1 deletion and the deletion of RAGE is not restricted to microglia but also occurs in monocytes/macrophages, the data compiled exposed a specific effect on microglia cell reactivity. The authors observed that in the spinal cord of male hSOD1G93A mice with microglia RAGE deletion, the acquisition of the damage/disease-associated microglia phenotype observed in this ALS mouse model is reduced [108].
A sex-specific effect of RAGE signaling inhibition in hSOD1G93A mice was also observed after daily treatment with the RAGE antagonist FPS-ZM1, a BBB permeant RAGE antagonist [121], starting at the age of 60 days [114]. Although no statistically significant effect in the onset of the disease or lifespan was observed, FPS-ZM1 treatment significantly improved hind-limb grip strength in both male and female hSOD1G93A mice during the progression of the disease. In female mice, FPS-ZM1 treatment slowed down early disease progression, as evidenced by a delay in the age at which they displayed 5% and 10% peak body weight loss. However, the disease progressed at a faster rate at late stages, leading to a lack of a significant effect in overall survival [114]. On the other hand, in male mice, FPS-ZM1 treatment significantly extended the duration of the disease at late stages, although the two-week extension in the median survival observed in male mice did not reach statistical significance [114]. These results suggest that the timing of RAGE signaling inhibition could also significantly impact the final therapeutic effect and, for example, limiting the treatment window to an early disease stage might prove to have a better functional outcome in female hSOD1G93A mice.
As observed in other neurodegenerative conditions, ALS patients display decreased levels of soluble RAGE in serum, although no correlation with clinical parameters of the disease was evident [122]. Since soluble RAGE has the ability to antagonize RAGE signaling, a reduction in its levels could impact the neurodegenerative process. Accordingly, daily treatment with sRAGE by intraperitoneal injection, starting at 8 weeks of age, improved motor function and modestly extended the survival in hSOD1G93A male mice [123]. sRAGE does not gain access into the central nervous system (CNS) after intraperitoneal administration [4], suggesting that the neuroprotection conferred by the treatment depends on its effects in the periphery, likely at the neuromuscular junction level.
Parkinson’s disease.
RAGE signaling has also been implicated in the pathophysiology of PD. Increased expression of RAGE and its ligands was observed in the substantia nigra and frontal cortex of patients with incidental Lewy Body disease (considered a presymptomatic stage of PD), and in PD patients and mouse models [124–127]. In the 1-methyl-4-phenyl-1,2,3,6-tetrahy-dropyridine (MPTP) model of PD, increased RAGE expression was observed in dopaminergic neurons and glial cells and was linked to activation of NF-κB signaling [127]. Moreover, in RAGE knockout mice, MPTP-induced neurodegeneration is attenuated and animals displayed reduced microgliosis and astrogliosis, as well as an increased number of surviving dopaminergic neurons in the substantia nigra pars compacta [127]. In addition, pharmacological inhibition of RAGE signaling using FPS-ZM1 reduced astrocyte and microglia activation, prevented the loss of dopaminergic neurons, and attenuated locomotor and exploratory deficits induced by intracranial administration of 6-hydroxydopamine in rats [128]. Importantly, in both studies there was a limit to the neuroprotective effects of inhibiting RAGE signaling. This indicates that RAGE-independent mechanisms are involved in the neurodegeneration induced by MPTP or 6-hydroxydopamine treatment. The canonical RAGE ligands S100B and HMGB1 have also been implicated in the pathophysiology of PD, particularly through a RAGE/TNFα neuroinflammatory pathway [124, 129]. Interestingly, Viana et al. [130] observed that at early stages of MPTP-induced toxicity there is a selective upregulation of inhibitory RAGE isoforms (lacking the intracellular domain) in striatal astrocytes. The authors proposed that this upregulation of C-terminal-truncated RAGE variants could be part of a neuroprotective response attempting to limit the detrimental effects of RAGE ligand signaling that could lead to the establishment of chronic inflammation [130].
Other pathologies.
Increased RAGE expression was also observed in the caudate nucleus of HD patients. It occurs predominantly in astrocytes, but also in medium spiny neurons, and it has been associated with the extent of neurodegeneration [131, 132]. Kim et al. [132] observed that RAGE expression colocalized with two of its ligands, S100B and carboxymethyllysine. Moreover, they observed a direct correlation between the level of expression and colocalization of RAGE and its ligands with the pathological grade of the disease [132]. However, the functional consequences of RAGE signaling in astrocytes in the context of HD have not been determined, and either detrimental or beneficial effects on the survival of neighboring neurons have been envisioned [132].
An upregulation in the expression of RAGE and its ligands was also observed in active lesions in multiple sclerosis (MS) patients and in the animal model of experimental autoimmune encephalomyelitis (EAE) [133, 134]. Moreover, blocking RAGE signaling by administration of sRAGE has a protective effect in the EAE mouse model, decreasing immune cell infiltration into the CNS [133]. Further support for the involvement of RAGE signaling in the development of this disease comes from the observation of decreased plasma sRAGE levels in MS patients when compared to healthy controls. In MS patients, sRAGE levels correlated with the severity of the disease, i.e., lower levels of sRAGE in plasma were linked to more severe forms of the disease and higher rate of annual relapse [135]. In addition, the presence of the RAGE G82S allele polymorphic variant was associated with an increased risk of developing MS in a Chinese population [136]. As observed in AD patients (see above), the carriers of the variant allele presented a faster progression of disability and reduced serum sRAGE levels [136].
3. Targeting RAGE with therapeutic purposes.
As described above, multiple lines of evidence support an involvement of RAGE signaling in various pathological conditions. However, the specific targeting of RAGE signaling for therapeutic purposes has proven to be extremely complex. This is in part due to the fact that RAGE can exert both beneficial and detrimental effects, depending on the physiological context, the presence of multiple ligands and its concentration level, and on the cell type considered. Figure 2 summarizes the different approaches that have been used to target RAGE signaling for therapeutic purposes.
Figure 2. Different strategies to target RAGE signaling with therapeutic purposes.
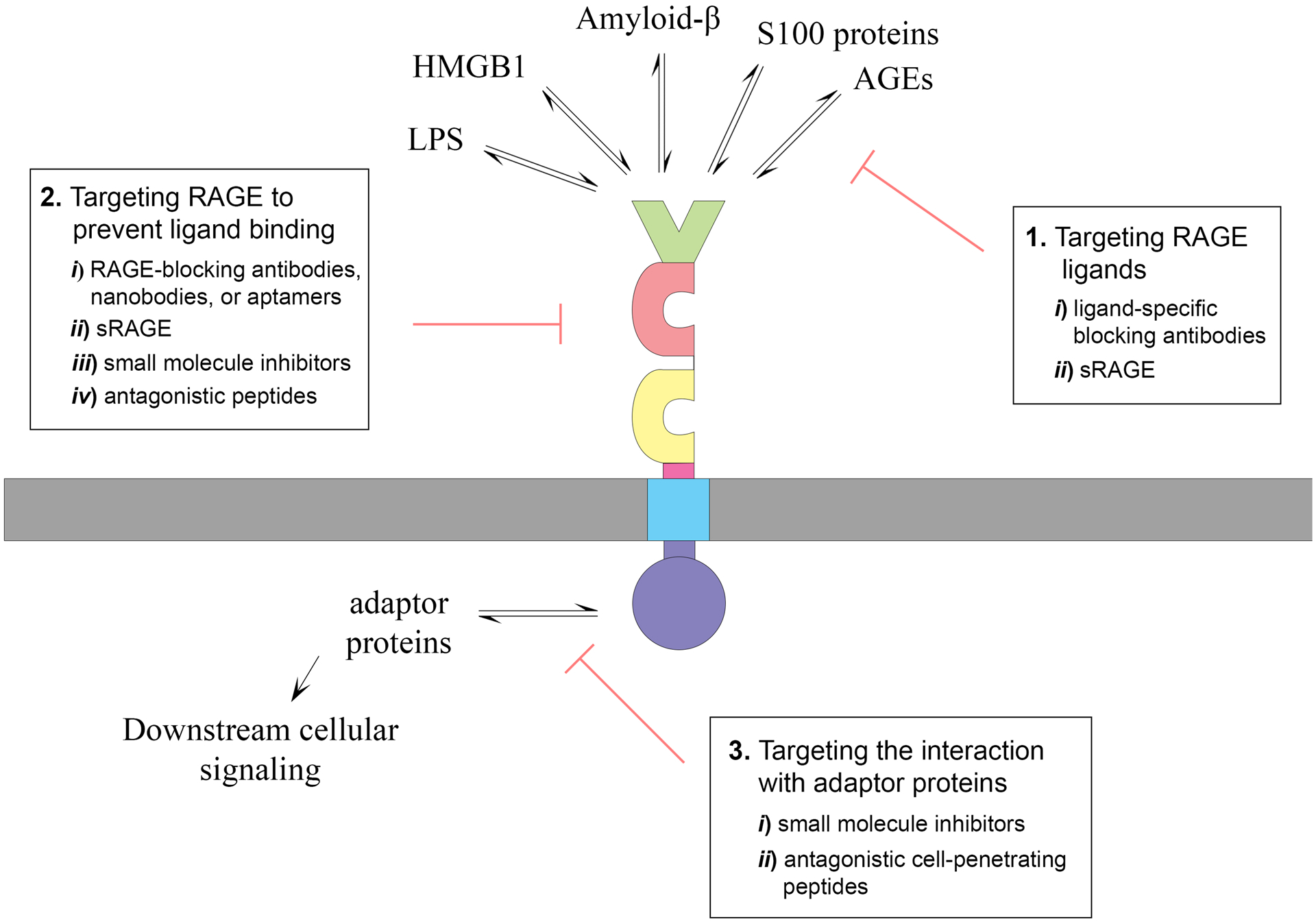
(1) One approach to block RAGE signaling is to target its ligands. This can be done using ligand-specific blocking antibodies (i), or by increasing sRAGE levels (ii) either, by administration of the recombinant protein or by increasing its expression levels. However, RAGE is a pattern recognition receptor that, in addition to bind AGEs, it interacts with multiple DAMPs and PAMPs, including S100 proteins, HMGB1, amyloid-β, and lipopolysaccharide (LPS). Multiple RAGE ligands coexist in pathological conditions, decreasing the effectivity of this approach. (2) Another strategy to block RAGE signaling is to target the receptor in order to prevent ligand binding. This can be done using i) RAGE-specific blocking antibodies, nanobodies or aptamers; ii) sRAGE; iii) small molecule inhibitors; or iv) RAGE antagonistic peptides designed based on the structure of known RAGE ligands. The use of antibodies targeting specific extracellular domains in RAGE can confer a certain degree of ligand specificity. Nanobodies and aptamers offer several advantages over conventional antibodies in terms of therapeutic potential, including their small size, stability, limited immunogenicity, and strong tissue penetration. In addition to sequestering ligands, sRAGE prevents RAGE signaling by preventing the formation of signaling-competent RAGE complexes. The use of small molecule inhibitors to prevent ligand binding is the approach that has been advanced further along the therapeutic development pipeline. (3) The identification of RAGE adaptor proteins essential for signal transduction has allowed the development of a different strategy that could confer more specificity, due to the differential expression of the adaptor proteins in different cell types and their involvement in specific cellular responses. Two approaches have been used to block the interaction of adaptor proteins with RAGE intracellular domain: i) small molecule inhibitors that act intracellularly, and ii) cell-penetrating peptides designed based on structural information regarding the interaction interface of the adaptor protein with RAGE-ICD.
One approach to block receptor signaling is to target its ligands. However, RAGE is a pattern recognition receptor that interacts with multiple damage-associated and pathogen-associated molecular patterns (DAMPs and PAMPs, respectively), including S100 proteins, HMGB1, serum amyloid A and amyloid-β and lipopolysaccharide (LPS) [3, 137, 138]. As indicated above, neurodegenerative conditions are characterized by concurrent upregulation of multiple RAGE ligands, hence limiting the possibility and effectiveness of ligand targeting as an approach to inhibit receptor signaling activation. Moreover, RAGE shares multiple ligands with other pattern recognition receptors, including Toll-like receptors (TLRs), and the signaling pathways activated by RAGE and TLRs converge at multiple levels to coordinate the final response to an insult [139]. Thus, pharmacological RAGE antagonists that prevent the binding of multiple ligands to the receptor could alter this intricate regulatory network and promote signaling through other receptors. In addition, soluble RAGE isoforms, by competing for ligand binding, prevent signaling not only through RAGE but could also affect signaling through other receptors that share the same ligands. Indeed, this seems to be the most plausible explanation for the disparate results obtained when analyzing the effect of RAGE signaling in nerve regeneration after crush injury in nondiabetic conditions. While RAGE knockout mice display no significant beneficial or adverse effect on nerve fiber regeneration after acute nerve crush [16], administration of soluble RAGE (a scavenger of RAGE ligands) or expression of signal transduction-deficient mutant RAGE in mononuclear phagocytes or peripheral neurons, significantly impair nerve regeneration [15, 140]. These results suggest that certain RAGE ligands could signal through other receptors to convey regenerative responses. These beneficial responses would be prevented when scavenging these ligands using soluble RAGE or overexpressing the dominant negative form of RAGE. However, it is important to notice that in other contexts, such as in diabetic conditions, blockade of RAGE signaling improves nerve regeneration by preventing maladaptive effects of RAGE signaling in inflammatory cells [16].
As indicated above, a decrease in sRAGE levels has been associated with the development of multiple pathologies, including AD, ALS and MS [95, 97–99, 122, 135, 136]. Moreover, increasing sRAGE levels by overexpression or direct administration exerts neuroprotective effects in mouse models of these diseases [4, 101, 123, 133]. Furthermore, sRAGE levels, and in particular cRAGE (generated by proteolytic cleavage), decline with age; while a higher level of sRAGE in plasma correlates with extreme longevity [141, 142]. Interestingly, Scavello et al. [143] observed that cRAGE levels inversely correlate with the age of the population and kept decreasing in long-lived individuals, while centenarians display increased levels of esRAGE (produced by alternative splicing). sRAGE prevents RAGE signaling by preventing the formation of signaling-competent RAGE complexes on the cell surface and/or by sequestering ligands [67]. However, as mentioned above, sRAGE potentially blocks the interaction of these ligands with other receptors as well. Thus, the development of therapeutic approaches using sRAGE requires a better understanding of its potential effects on signaling pathways activated by other receptors.
Other drawbacks regarding the use of sRAGE for therapeutic purposes include the limitations imposed by the production of recombinant proteins in enough quantities and at affordable costs, and the potential for the administered recombinant protein to induce an immunogenic reaction. Tae et al. [144] showed that in contrast to the sRAGE produced in insect Sf9 cells, sRAGE produced in mammalian cells [Chinese Hamster Ovary (CHO) cells] is modified by sialylated complex-type N-glycoforms which confer upon sRAGE higher bioactivity in cell assays and in an in vivo model of artery injury. While daily administration of high doses of sRAGE produced in Sf9 cells are required to achieve therapeutic effects, the recombinant protein produced in CHO cells induces beneficial effects after a single low dose administration [144]. In addition to influencing the biological activity of RAGE by modulating ligand interaction (see above, Section 1), glycosylation can also impact the immunogenicity of the recombinant protein [145]. Accordingly, recombinant glycoproteins produced in non-human cell lines or in human cell lines grown in media with animal supplements display glycans known to induce immunogenic reactions in humans [146]. Thus, the advancement of sRAGE therapeutics would benefit from a better understanding of the modulation of RAGE-ligand interaction by specific N-glycan structures and further advances in the field of protein glycoengineering. This can confer a certain degree of ligand specificity to sRAGE therapeutic approaches.
Another strategy to specifically target RAGE signaling is the use of RAGE blocking antibodies. This approach has the potential to confer high specificity and has proven to be effective in models of sepsis and diabetes-associated vascular and renal dysfunction [147–151]. The humanization and affinity optimization of these antibodies could further improve their therapeutic potential [152]. The use of single-domain antibodies, known as nanobodies, offers several advantages over conventional antibodies, including their small size (~15kDa), thermal stability, limited immunogenicity, and strong tissue penetration. Moreover, multiple strategies can be used to favor their penetration through the blood-brain barrier [153]. Nanobodies specific to the V-C1 domain of RAGE have been recently developed [154]. These nanobodies have been shown to block the interaction between S100B and RAGE V-C1 domain in a competitive ELISA [154], but further evaluation of their therapeutic potential in preclinical animal models is still required. Another alternative to the use of conventional immunoglobulins is the use of aptamers, also known as chemical antibodies. Aptamers are short single-stranded DNA or RNA oligonucleotides that adopt unique three-dimensional conformations which allow them to interact with specific targets with high affinity and specificity [155]. Aptamers have multiple advantages over conventional antibodies, including rapid production, efficient penetration into various tissues and less immunogenicity [156]. The group of Dr. Sho-Ichi Yamagishi developed a DNA-aptamer against RAGE that has proven to exert beneficial therapeutic effects in animal models of experimental diabetic nephropathy, sepsis and melanoma growth and metastasis [157–159]. The therapeutic potential of this aptamer for the treatment of neurodegenerative diseases has not been tested yet.
An alternate approach to specifically target RAGE signaling, which has attracted most of the attention, is the development of small-molecule inhibitors to prevent ligand binding. One of these inhibitors, TTP488 (also known as PF-04494700 or Azeliragon), has been evaluated in clinical trials for the treatment of AD. TTP488 is an orally bioavailable RAGE antagonist that was shown to prevent in vitro binding of multiple ligands (S100B, HMGB1, carboxymethyl-lysine and Aβ1–42) to sRAGE and to have beneficial effects in vivo in an AD mouse model, reducing the expression of inflammatory markers and amyloid deposition and improving performance on behavioral testing [160–162]. Phase 1 and 2 clinical trials of TTP488 confirmed that it is safe to target RAGE for therapeutic purposes and obtained promising beneficial results in AD patients [160–164]. A phase 3 study (STEADFAST Study) was later performed but was terminated due to lack of efficacy [165]. However, post hoc analysis of the data showed that TTP448 has a beneficial effect in AD patients with type 2 diabetes [166]. Supported by these data, a new phase 2 study was performed (Elevage study), but the results of the study are not currently available [167].
Another high-affinity RAGE-specific inhibitor was designed by Deane et al. [121]. This inhibitor, known as FPS-ZM1, prevents the binding of multiple ligands, including Aβ40, S100B, and HMGB1, to immobilized sRAGE [121]. Moreover, FPS-ZM1 displays no toxicity in mice, even at high doses, and crosses the BBB to gain access into the CNS [121]. Importantly, FPS-ZM1 was shown to exert beneficial therapeutic effects in animal models of AD, ALS and PD [114, 121, 128]. In addition, other small molecule inhibitors of RAGE have been developed, including 4,6-bis(4-chlorophenyl)pyrimidine analogs and pyrazole-5-carboxamide derivatives, which also had beneficial effects in an AD mouse model [168–170]. Other groups have designed and developed RAGE antagonistic peptides based on the structure of known RAGE ligands. Arumugam et al. [171] developed a peptide, referred to as RAP (RAGE antagonistic peptide), based on the structure of S100P. This peptide was shown to inhibit the interaction of RAGE with S100P, S100A4, and HMGB1 [171]. In a cell culture model, RAP prevents the motor neuron death induced by astrocytes isolated from hSOD1G93A ALS mice or by lumbar spinal cord extracts from early symptomatic hSOD1G93A mice [114]. RAP was also shown to have therapeutic effects after systemic in vivo administration, reducing growth and metastasis of pancreatic tumors and inhibiting glioma tumor growth [171]. Huttunen et al. [172] designed a peptide corresponding to the RAGE-binding region in HMGB1. This peptide blocks transendothelial migration of tumor cells in vitro and significantly suppressed the formation of lung metastases in an in vivo model [172].
Another group of small molecule inhibitors has been designed to act intracellularly to block RAGE signaling activation. Using a high-throughput screening assay, a small molecule library of 58,000 compounds was evaluated for its ability to inhibit the binding of DIAPH1 to RAGE intracellular domain. This study identified 13 molecules that display differential ability to inhibit RAGE signaling in vitro and in vivo, in a model of heart ischemia/reperfusion injury in diabetic mice [173]. Another approach to block RAGE signaling activation involves the use of cell-penetrating peptides designed to prevent the interaction of adaptor proteins with the intracellular domain of RAGE. Putranto et al. [174] developed a decoy peptide mimicking the phosphorylated intracellular domain of RAGE at Ser391. This residue is phosphorylated by PKCζ upon ligand binding and facilitates the recruitment of TIRAP and MyD88 and further downstream signaling [48]. After polyethylenimine (PEI) cationization for efficient delivery into the cell, the peptide prevented the cell death induced by an excess of S100B and the migration and invasion of glioma cells in cell culture models [174]. A similar effect is obtained using inhibitor peptides that bind to the TIR domain of TIRAP and MyD88, previously developed to prevent the interaction of these adaptor proteins to Toll-like receptors [174]. More recently, a TAT cell-penetrating peptide approach was used by Yan et al. to competitively inhibit the interaction of SLP76 with RAGE-ICD [49]. SLP76 was identified as an adaptor protein involved in RAGE downstream signaling activation in macrophages. SLP76 interacts with the intracellular domain of RAGE through its SAM domain, and blocking this interaction using a TAT-SAM fusion peptide inhibits the activation of proinflammatory pathways by AGE/RAGE signaling in RAW264.7 cells [49]. Moreover, tail vein injection of TAT-SAM exerted protective effects in a mouse model of sepsis [49]. Together, these results indicate that blocking the interaction of RAGE with specific adaptor proteins could be a valid therapeutic approach to inhibit specific signaling cascades activated by RAGE signaling.
4. Conclusions and future perspectives.
Extensive evidence supports the involvement of RAGE signaling in various pathological conditions, both inside and outside the nervous system. Increased RAGE expression is observed in multiple neurodegenerative diseases, where it is expressed by different cell types, including neurons, astrocytes, microglia, oligodendrocytes, vascular endothelial cells, and pericytes. The outcome of RAGE signaling in each cellular compartment is different and the final effect on the progression of the disease is likely defined by the interplay of the multiple cell type-specific responses. Both beneficial and detrimental effects of RAGE signaling have been described in multiple pathologies. These depend, at least in part, on the cell type considered and the pathophysiological context, including the level of antioxidant defenses, energy metabolism, and expression of downstream effectors. In addition, the outcome of RAGE signaling depends not only on the ligand considered, but also on its concentration and duration of the stimulus. Hence, the final outcome of RAGE signaling could vary during the progression of chronic diseases, where the cellular context and expression/accumulation of different RAGE ligands change at different disease stages. Thus, the timing of RAGE signaling inhibition could be crucial to define the final therapeutic effect.
The expression of multiple RAGE isoforms that can differentially modulate the signaling of the full-length receptor adds another level of complexity to RAGE signaling. The relative expression of these isoforms varies depending on the cell type considered and the physiological context, and there are fundamental species-specific differences in their regulation, an important factor to consider when performing preclinical studies in animal models.
RAGE is a pattern recognition receptor, and as such it binds multiple ligands. This promiscuity is also observed in its ligands, which bind to other pattern recognition receptors in addition to interacting with RAGE. As indicated above, the strategy used to target RAGE signaling could have profound consequences on the signaling of its ligands not only through RAGE but also through other receptors. A better understanding of this intricate regulatory network of pattern recognition receptors could aid in the development of successful therapeutic approaches.
In summary, the key to the success of therapeutic approaches targeting RAGE signaling depends on the development of different strategies to specifically counteract the detrimental effects of RAGE signaling while preserving potential beneficial effects in a cell type-specific manner.
Acknowledgments.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant R01NS100835]. This work used resources and facilities of the William S. Middleton Memorial Veterans Hospital (Madison, WI, USA).
Footnotes
Conflict of interest: The authors declare no competing interests.
References.
- [1].Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem, 1992; 267: 14998–5004. [PubMed] [Google Scholar]
- [2].Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem, 1992; 267: 14987–97. [PubMed] [Google Scholar]
- [3].Fritz G RAGE: a single receptor fits multiple ligands. Trends Biochem Sci, 2011; 36: 625–32. [DOI] [PubMed] [Google Scholar]
- [4].Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med, 2003; 9: 907–13. [DOI] [PubMed] [Google Scholar]
- [5].Candela P, Gosselet F, Saint-Pol J, Sevin E, Boucau MC, Boulanger E, Cecchelli R, Fenart L. Apical-to-basolateral transport of amyloid-beta peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J Alzheimers Dis, 2010; 22: 849–59. [DOI] [PubMed] [Google Scholar]
- [6].Yamamoto Y, Higashida H. RAGE regulates oxytocin transport into the brain. Commun Biol, 2020; 3: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sessa L, Gatti E, Zeni F, Antonelli A, Catucci A, Koch M, Pompilio G, Fritz G, Raucci A, Bianchi ME. The receptor for advanced glycation end-products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS One, 2014; 9: e86903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xie J, Mendez JD, Mendez-Valenzuela V, Aguilar-Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal, 2013; 25: 2185–97. [DOI] [PubMed] [Google Scholar]
- [9].Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl), 2005; 83: 876–86. [DOI] [PubMed] [Google Scholar]
- [10].Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, Ramsgaard L, Richards TJ, Loutaev I, Nawroth PP, Kasper M, Bierhaus A, Oury TD. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol, 2008; 172: 583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta, 2013; 1833: 101–9. [DOI] [PubMed] [Google Scholar]
- [12].Litwinoff E, Hurtado Del Pozo C, Ramasamy R, Schmidt AM. Emerging Targets for Therapeutic Development in Diabetes and Its Complications: The RAGE Signaling Pathway. Clin Pharmacol Ther, 2015; 98: 135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Malik P, Chaudhry N, Mittal R, Mukherjee TK. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochim Biophys Acta, 2015; 1850: 1898–904. [DOI] [PubMed] [Google Scholar]
- [14].Derk J, MacLean M, Juranek J, Schmidt AM. The Receptor for Advanced Glycation Endproducts (RAGE) and Mediation of Inflammatory Neurodegeneration. J Alzheimers Dis Parkinsonism, 2018; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rong LL, Trojaborg W, Qu W, Kostov K, Yan SD, Gooch C, Szabolcs M, Hays AP, Schmidt AM. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J, 2004; 18: 1812–7. [DOI] [PubMed] [Google Scholar]
- [16].Juranek JK, Geddis MS, Song F, Zhang J, Garcia J, Rosario R, Yan SF, Brannagan TH, Schmidt AM. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes, 2013; 62: 931–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Malherbe P, Richards JG, Gaillard H, Thompson A, Diener C, Schuler A, Huber G. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Brain Res Mol Brain Res, 1999; 71: 159–70. [DOI] [PubMed] [Google Scholar]
- [18].Schlueter C, Hauke S, Flohr AM, Rogalla P, Bullerdiek J. Tissue-specific expression patterns of the RAGE receptor and its soluble forms--a result of regulated alternative splicing? Biochim Biophys Acta, 2003; 1630: 1–6. [DOI] [PubMed] [Google Scholar]
- [19].Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J, 2003; 370: 1097–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Park IH, Yeon SI, Youn JH, Choi JE, Sasaki N, Choi IH, Shin JS. Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol Immunol, 2004; 40: 1203–11. [DOI] [PubMed] [Google Scholar]
- [21].Ding Q, Keller JN. Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci Lett, 2005; 373: 67–72. [DOI] [PubMed] [Google Scholar]
- [22].Harashima A, Yamamoto Y, Cheng C, Tsuneyama K, Myint KM, Takeuchi A, Yoshimura K, Li H, Watanabe T, Takasawa S, Okamoto H, Yonekura H, Yamamoto H. Identification of mouse orthologue of endogenous secretory receptor for advanced glycation end-products: structure, function and expression. Biochem J, 2006; 396: 109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. FASEB J, 2008; 22: 1572–80. [DOI] [PubMed] [Google Scholar]
- [24].Sterenczak KA, Willenbrock S, Barann M, Klemke M, Soller JT, Eberle N, Nolte I, Bullerdiek J, Murua Escobar H. Cloning, characterisation, and comparative quantitative expression analyses of receptor for advanced glycation end products (RAGE) transcript forms. Gene, 2009; 434: 35–42. [DOI] [PubMed] [Google Scholar]
- [25].Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J, 2009; 23: 1766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jules J, Maiguel D, Hudson BI. Alternative splicing of the RAGE cytoplasmic domain regulates cell signaling and function. PLoS One, 2013; 8: e78267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Peng Y, Horwitz N, Lakatta EG, Lin L. Mouse RAGE Variant 4 Is a Dominant Membrane Receptor that Does Not Shed to Generate Soluble RAGE. PLoS One, 2016; 11: e0153657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kalea AZ, See F, Harja E, Arriero M, Schmidt AM, Hudson BI. Alternatively spliced RAGEv1 inhibits tumorigenesis through suppression of JNK signaling. Cancer Res, 2010; 70: 5628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, Reinhart TA, Oury TD. Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J Biol Chem, 2004; 279: 50019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang L, Bukulin M, Kojro E, Roth A, Metz VV, Fahrenholz F, Nawroth PP, Bierhaus A, Postina R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem, 2008; 283: 35507–16. [DOI] [PubMed] [Google Scholar]
- [31].Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J, 2008; 22: 3716–27. [DOI] [PubMed] [Google Scholar]
- [32].Galichet A, Weibel M, Heizmann CW. Calcium-regulated intramembrane proteolysis of the RAGE receptor. Biochem Biophys Res Commun, 2008; 370: 1–5. [DOI] [PubMed] [Google Scholar]
- [33].Braley A, Kwak T, Jules J, Harja E, Landgraf R, Hudson BI. Regulation of Receptor for Advanced Glycation End Products (RAGE) Ectodomain Shedding and Its Role in Cell Function. J Biol Chem, 2016; 291: 12057–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Metz VV, Kojro E, Rat D, Postina R. Induction of RAGE shedding by activation of G protein-coupled receptors. PLoS One, 2012; 7: e41823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Di Maggio S, Gatti E, Liu J, Bertolotti M, Fritz G, Bianchi ME, Raucci A. The Mouse-Specific Splice Variant mRAGE_v4 Encodes a Membrane-Bound RAGE That Is Resistant to Shedding and Does Not Contribute to the Production of Soluble RAGE. PLoS One, 2016; 11: e0153832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Srikrishna G, Nayak J, Weigle B, Temme A, Foell D, Hazelwood L, Olsson A, Volkmann N, Hanein D, Freeze HH. Carboxylated N-glycans on RAGE promote S100A12 binding and signaling. J Cell Biochem, 2010; 110: 645–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Park SJ, Kleffmann T, Hessian PA. The G82S polymorphism promotes glycosylation of the receptor for advanced glycation end products (RAGE) at asparagine 81: comparison of wild-type rage with the G82S polymorphic variant. J Biol Chem, 2011; 286: 21384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wilton R, Yousef MA, Saxena P, Szpunar M, Stevens FJ. Expression and purification of recombinant human receptor for advanced glycation endproducts in Escherichia coli. Protein Expr Purif, 2006; 47: 25–35. [DOI] [PubMed] [Google Scholar]
- [39].Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, Freeze HH. N -Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem, 2002; 80: 998–1008. [DOI] [PubMed] [Google Scholar]
- [40].Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis, 2008; 29: 2035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Osawa M, Yamamoto Y, Munesue S, Murakami N, Sakurai S, Watanabe T, Yonekura H, Uchigata Y, Iwamoto Y, Yamamoto H. De-N-glycosylation or G82S mutation of RAGE sensitizes its interaction with advanced glycation endproducts. Biochim Biophys Acta, 2007; 1770: 1468–74. [DOI] [PubMed] [Google Scholar]
- [42].Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta, 2009; 1793: 993–1007. [DOI] [PubMed] [Google Scholar]
- [43].Chellappa RC, Lukose B, Rani P. G82S RAGE polymorphism influences amyloid-RAGE interactions relevant in Alzheimer’s disease pathology. PLoS One, 2020; 15: e0225487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett, 2003; 550: 107–13. [DOI] [PubMed] [Google Scholar]
- [45].Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem, 2008; 283: 34457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rai V, Maldonado AY, Burz DS, Reverdatto S, Yan SF, Schmidt AM, Shekhtman A. Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J Biol Chem, 2012; 287: 5133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim Y, Kim C, Son SM, Song H, Hong HS, Han SH, Mook-Jung I. The novel RAGE interactor PRAK is associated with autophagy signaling in Alzheimer’s disease pathogenesis. Mol Neurodegener, 2016; 11: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, Hibino T, Kataoka K, Huh NH. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One, 2011; 6: e23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yan Z, Luo H, Xie B, Tian T, Li S, Chen Z, Liu J, Zhao X, Zhang L, Deng Y, Billiar TR, Jiang Y. Targeting adaptor protein SLP76 of RAGE as a therapeutic approach for lethal sepsis. Nat Commun, 2021; 12: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim MJ, Vargas MR, Harlan BA, Killoy KM, Ball LE, Comte-Walters S, Gooz M, Yamamoto Y, Beckman JS, Barbeito L, Pehar M. Nitration and Glycation Turn Mature NGF into a Toxic Factor for Motor Neurons: A Role for p75(NTR) and RAGE Signaling in ALS. Antioxid Redox Signal, 2018; 28: 1587–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pehar M, Vargas MR, Robinson KM, Cassina P, England P, Beckman JS, Alzari PM, Barbeito L. Peroxynitrite transforms nerve growth factor into an apoptotic factor for motor neurons. Free Radic Biol Med, 2006; 41: 1632–44. [DOI] [PubMed] [Google Scholar]
- [52].Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med, 2003; 198: 1507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J, 2007; 26: 1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Son M, Porat A, He M, Suurmond J, Santiago-Schwarz F, Andersson U, Coleman TR, Volpe BT, Tracey KJ, Al-Abed Y, Diamond B. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood, 2016; 128: 2218–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu T, Xiang A, Peng T, Doran AC, Tracey KJ, Barnes BJ, Tabas I, Son M, Diamond B. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci U S A, 2019; 116: 23254–23263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Slowik A, Merres J, Elfgen A, Jansen S, Mohr F, Wruck CJ, Pufe T, Brandenburg LO. Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)--and amyloid beta 1–42-induced signal transduction in glial cells. Mol Neurodegener, 2012; 7: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ichiki T, Koga T, Okuno T, Saeki K, Yamamoto Y, Yamamoto H, Sakaguchi M, Yokomizo T. Modulation of leukotriene B4 receptor 1 signaling by receptor for advanced glycation end products (RAGE). FASEB J, 2016; 30: 1811–22. [DOI] [PubMed] [Google Scholar]
- [58].Pickering RJ, Tikellis C, Rosado CJ, Tsorotes D, Dimitropoulos A, Smith M, Huet O, Seeber RM, Abhayawardana R, Johnstone EK, Golledge J, Wang Y, Jandeleit-Dahm KA, Cooper ME, Pfleger KD, Thomas MC. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J Clin Invest, 2019; 129: 406–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yokoyama S, Kawai T, Yamamoto K, Yibin H, Yamamoto H, Kakino A, Takeshita H, Nozato Y, Fujimoto T, Hongyo K, Takahashi T, Nakagami F, Akasaka H, Takami Y, Takeya Y, Sugimoto K, Sawamura T, Rakugi H. RAGE ligands stimulate angiotensin II type I receptor (AT1) via RAGE/AT1 complex on the cell membrane. Sci Rep, 2021; 11: 5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, Taguchi A, Yan SF, Yan SD, Stern DM, Schmidt AM. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci, 2002; 59: 1117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].May O, Yatime L, Merle NS, Delguste F, Howsam M, Daugan MV, Paul-Constant C, Billamboz M, Ghinet A, Lancel S, Dimitrov JD, Boulanger E, Roumenina LT, Frimat M. The receptor for advanced glycation end products is a sensor for cell-free heme. FEBS J, 2021; 288: 3448–3464. [DOI] [PubMed] [Google Scholar]
- [62].Ma W, Rai V, Hudson BI, Song F, Schmidt AM, Barile GR. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell Immunol, 2012; 274: 72–82. [DOI] [PubMed] [Google Scholar]
- [63].Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry, 2007; 46: 6957–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem, 2007; 282: 31317–31. [DOI] [PubMed] [Google Scholar]
- [65].Rani SG, Sepuru KM, Yu C. Interaction of S100A13 with C2 domain of receptor for advanced glycation end products (RAGE). Biochim Biophys Acta, 2014; 1844: 1718–28. [DOI] [PubMed] [Google Scholar]
- [66].Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J Biol Chem, 2008; 283: 27255–69. [DOI] [PubMed] [Google Scholar]
- [67].Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, Fritz G. Structural basis for ligand recognition and activation of RAGE. Structure, 2010; 18: 1342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yatime L, Andersen GR. Structural insights into the oligomerization mode of the human receptor for advanced glycation end-products. FEBS J, 2013; 280: 6556–68. [DOI] [PubMed] [Google Scholar]
- [69].Wei W, Lampe L, Park S, Vangara BS, Waldo GS, Cabantous S, Subaran SS, Yang D, Lakatta EG, Lin L. Disulfide bonds within the C2 domain of RAGE play key roles in its dimerization and biogenesis. PLoS One, 2012; 7: e50736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Donato R S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol, 2001; 33: 637–68. [DOI] [PubMed] [Google Scholar]
- [71].Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem, 2000; 275: 40096–105. [DOI] [PubMed] [Google Scholar]
- [72].Kogel D, Peters M, Konig HG, Hashemi SM, Bui NT, Arolt V, Rothermundt M, Prehn JH. S100B potently activates p65/c-Rel transcriptional complexes in hippocampal neurons: Clinical implications for the role of S100B in excitotoxic brain injury. Neuroscience, 2004; 127: 913–20. [DOI] [PubMed] [Google Scholar]
- [73].Businaro R, Leone S, Fabrizi C, Sorci G, Donato R, Lauro GM, Fumagalli L. S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J Neurosci Res, 2006; 83: 897–906. [DOI] [PubMed] [Google Scholar]
- [74].Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, Feldman EL. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology, 2007; 148: 548–58. [DOI] [PubMed] [Google Scholar]
- [75].Villarreal A, Aviles Reyes RX, Angelo MF, Reines AG, Ramos AJ. S100B alters neuronal survival and dendrite extension via RAGE-mediated NF-kappaB signaling. J Neurochem, 2011; 117: 321–32. [DOI] [PubMed] [Google Scholar]
- [76].Schmidt A, Kuhla B, Bigl K, Munch G, Arendt T. Cell cycle related signaling in Neuro2a cells proceeds via the receptor for advanced glycation end products. J Neural Transm (Vienna), 2007; 114: 1413–24. [DOI] [PubMed] [Google Scholar]
- [77].Deane RJ. Is RAGE still a therapeutic target for Alzheimer’s disease? Future Med Chem, 2012; 4: 915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Toth C, Schmidt AM, Tuor UI, Francis G, Foniok T, Brussee V, Kaur J, Yan SF, Martinez JA, Barber PA, Buchan A, Zochodne DW. Diabetes, leukoencephalopathy and rage. Neurobiol Dis, 2006; 23: 445–61. [DOI] [PubMed] [Google Scholar]
- [79].Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature, 1996; 382: 685–91. [DOI] [PubMed] [Google Scholar]
- [80].Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol, 2001; 171: 29–45. [DOI] [PubMed] [Google Scholar]
- [81].Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y, Takeuchi M, Makita Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res, 2001; 888: 256–262. [DOI] [PubMed] [Google Scholar]
- [82].Miller MC, Tavares R, Johanson CE, Hovanesian V, Donahue JE, Gonzalez L, Silverberg GD, Stopa EG. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res, 2008; 1230: 273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Choi BR, Cho WH, Kim J, Lee HJ, Chung C, Jeon WK, Han JS. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp Mol Med, 2014; 46: e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci, 2008; 28: 3521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Origlia N, Capsoni S, Cattaneo A, Fang F, Arancio O, Yan SD, Domenici L. Abeta-dependent Inhibition of LTP in different intracortical circuits of the visual cortex: the role of RAGE. J Alzheimers Dis, 2009; 17: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Sturchler E, Galichet A, Weibel M, Leclerc E, Heizmann CW. Site-specific blockade of RAGE-Vd prevents amyloid-beta oligomer neurotoxicity. J Neurosci, 2008; 28: 5149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J, 2004; 23: 4096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I. RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J, 2009; 23: 2639–49. [DOI] [PubMed] [Google Scholar]
- [89].Fang F, Yu Q, Arancio O, Chen D, Gore SS, Yan SS, Yan SF. RAGE mediates Abeta accumulation in a mouse model of Alzheimer’s disease via modulation of beta- and gamma-secretase activity. Hum Mol Genet, 2018; 27: 1002–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Vodopivec I, Galichet A, Knobloch M, Bierhaus A, Heizmann CW, Nitsch RM. RAGE does not affect amyloid pathology in transgenic ArcAbeta mice. Neurodegener Dis, 2009; 6: 270–80. [DOI] [PubMed] [Google Scholar]
- [91].Meneghini V, Bortolotto V, Francese MT, Dellarole A, Carraro L, Terzieva S, Grilli M. High-mobility group box-1 protein and beta-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-kappaB axis: relevance for Alzheimer’s disease. J Neurosci, 2013; 33: 6047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J, 2010; 24: 1043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV. Human blood-brain barrier receptors for Alzheimer’s amyloid-beta 1– 40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest, 1998; 102: 734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol, 2000; 279: C1772–81. [DOI] [PubMed] [Google Scholar]
- [95].Li K, Dai D, Zhao B, Yao L, Yao S, Wang B, Yang Z. Association between the RAGE G82S polymorphism and Alzheimer’s disease. J Neural Transm (Vienna), 2010; 117: 97–104. [DOI] [PubMed] [Google Scholar]
- [96].Daborg J, von Otter M, Sjolander A, Nilsson S, Minthon L, Gustafson DR, Skoog I, Blennow K, Zetterberg H. Association of the RAGE G82S polymorphism with Alzheimer’s disease. J Neural Transm (Vienna), 2010; 117: 861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Emanuele E, D’Angelo A, Tomaino C, Binetti G, Ghidoni R, Politi P, Bernardi L, Maletta R, Bruni AC, Geroldi D. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol, 2005; 62: 1734–6. [DOI] [PubMed] [Google Scholar]
- [98].Liang F, Jia J, Wang S, Qin W, Liu G. Decreased plasma levels of soluble low density lipoprotein receptor-related protein-1 (sLRP) and the soluble form of the receptor for advanced glycation end products (sRAGE) in the clinical diagnosis of Alzheimer’s disease. J Clin Neurosci, 2013; 20: 357–61. [DOI] [PubMed] [Google Scholar]
- [99].Xu XY, Deng CQ, Wang J, Deng XJ, Xiao Q, Li Y, He Q, Fan WH, Quan FY, Zhu YP, Cheng P, Chen GJ. Plasma levels of soluble receptor for advanced glycation end products in Alzheimer’s disease. Int J Neurosci, 2017; 127: 454–458. [DOI] [PubMed] [Google Scholar]
- [100].Chen J, Mooldijk SS, Licher S, Waqas K, Ikram MK, Uitterlinden AG, Zillikens MC, Ikram MA. Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw Open, 2021; 4: e2033012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Son M, Oh S, Park H, Ahn H, Choi J, Kim H, Lee HS, Lee S, Park HJ, Kim SU, Lee B, Byun K. Protection against RAGE-mediated neuronal cell death by sRAGE-secreting human mesenchymal stem cells in 5xFAD transgenic mouse model. Brain Behav Immun, 2017; 66: 347–358. [DOI] [PubMed] [Google Scholar]
- [102].Shibata N, Hirano A, Hedley-Whyte ET, Dal Canto MC, Nagai R, Uchida K, Horiuchi S, Kawaguchi M, Yamamoto T, Kobayashi M. Selective formation of certain advanced glycation end products in spinal cord astrocytes of humans and mice with superoxide dismutase-1 mutation. Acta Neuropathol, 2002; 104: 171–8. [DOI] [PubMed] [Google Scholar]
- [103].Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, Sta M, Troost D, Aronica E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience, 2011; 179: 233–43. [DOI] [PubMed] [Google Scholar]
- [104].Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, Schmidt AM. Receptor for Advanced Glycation End Products and its Inflammatory Ligands are Upregulated in Amyotrophic Lateral Sclerosis. Front Cell Neurosci, 2015; 9: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kikuchi S, Shinpo K, Ogata A, Tsuji S, Takeuchi M, Makita Z, Tashiro K. Detection of N epsilon-(carboxymethyl)lysine (CML) and non-CML advanced glycation end-products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotroph Lateral Scler Other Motor Neuron Disord, 2002; 3: 63–8. [DOI] [PubMed] [Google Scholar]
- [106].Lee JY, Lee JD, Phipps S, Noakes PG, Woodruff TM. Absence of toll-like receptor 4 (TLR4) extends survival in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation, 2015; 12: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lee JD, McDonald TS, Fung JNT, Woodruff TM. Absence of Receptor for Advanced Glycation End Product (RAGE) Reduces Inflammation and Extends Survival in the hSOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Mol Neurobiol, 2020; 57: 4143–4155. [DOI] [PubMed] [Google Scholar]
- [108].MacLean M, Juranek J, Cuddapah S, Lopez-Diez R, Ruiz HH, Hu J, Frye L, Li H, Gugger PF, Schmidt AM. Microglia RAGE exacerbates the progression of neurodegeneration within the SOD1(G93A) murine model of amyotrophic lateral sclerosis in a sex-dependent manner. J Neuroinflammation, 2021; 18: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem, 2006; 97: 687–96. [DOI] [PubMed] [Google Scholar]
- [110].Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci, 2007; 10: 615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Serrano A, Donno C, Giannetti S, Peric M, Andjus P, D’Ambrosi N, Michetti F. The Astrocytic S100B Protein with Its Receptor RAGE Is Aberrantly Expressed in SOD1(G93A) Models, and Its Inhibition Decreases the Expression of Proinflammatory Genes. Mediators Inflamm, 2017; 2017: 1626204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Villarreal A, Seoane R, Gonzalez Torres A, Rosciszewski G, Angelo MF, Rossi A, Barker PA, Ramos AJ. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: implications for its role in the propagation of reactive gliosis. J Neurochem, 2014; 131: 190–205. [DOI] [PubMed] [Google Scholar]
- [113].Brambilla L, Martorana F, Guidotti G, Rossi D. Dysregulation of Astrocytic HMGB1 Signaling in Amyotrophic Lateral Sclerosis. Front Neurosci, 2018; 12: 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Liu L, Killoy KM, Vargas MR, Yamamoto Y, Pehar M. Effects of RAGE inhibition on the progression of the disease in hSOD1(G93A) ALS mice. Pharmacol Res Perspect, 2020; 8: e00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Song JS, Kang CM, Park CK, Yoon HK, Lee SY, Ahn JH, Moon HS. Inhibitory effect of receptor for advanced glycation end products (RAGE) on the TGF-beta-induced alveolar epithelial to mesenchymal transition. Exp Mol Med, 2011; 43: 517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kumar V, Fleming T, Terjung S, Gorzelanny C, Gebhardt C, Agrawal R, Mall MA, Ranzinger J, Zeier M, Madhusudhan T, Ranjan S, Isermann B, Liesz A, Deshpande D, Haring HU, Biswas SK, Reynolds PR, Hammes HP, Peperkok R, Angel P, Herzig S, Nawroth PP. Homeostatic nuclear RAGE-ATM interaction is essential for efficient DNA repair. Nucleic Acids Res, 2017; 45: 10595–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci U S A, 1996; 93: 5709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem, 2013; 288: 20745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Bartling B, Zunkel K, Al-Robaiy S, Dehghani F, Simm A. Gene doubling increases glyoxalase 1 expression in RAGE knockout mice. Biochim Biophys Acta Gen Subj, 2020; 1864: 129438. [DOI] [PubMed] [Google Scholar]
- [120].Rabbani N, Thornalley PJ. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem Biophys Res Commun, 2015; 458: 221–6. [DOI] [PubMed] [Google Scholar]
- [121].Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest, 2012; 122: 1377–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Ilzecka J Serum-soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand, 2009; 120: 119–22. [DOI] [PubMed] [Google Scholar]
- [123].Juranek JK, Daffu GK, Geddis MS, Li H, Rosario R, Kaplan BJ, Kelly L, Schmidt AM. Soluble RAGE Treatment Delays Progression of Amyotrophic Lateral Sclerosis in SOD1 Mice. Front Cell Neurosci, 2016; 10: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Santoro M, Maetzler W, Stathakos P, Martin HL, Hobert MA, Rattay TW, Gasser T, Forrester JV, Berg D, Tracey KJ, Riedel G, Teismann P. In-vivo evidence that high mobility group box 1 exerts deleterious effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model and Parkinson’s disease which can be attenuated by glycyrrhizin. Neurobiol Dis, 2016; 91: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Dalfo E, Portero-Otin M, Ayala V, Martinez A, Pamplona R, Ferrer I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol, 2005; 64: 816–30. [DOI] [PubMed] [Google Scholar]
- [126].Munch G, Luth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P. Crosslinking of alpha-synuclein by advanced glycation endproducts--an early pathophysiological step in Lewy body formation? J Chem Neuroanat, 2000; 20: 253–7. [DOI] [PubMed] [Google Scholar]
- [127].Teismann P, Sathe K, Bierhaus A, Leng L, Martin HL, Bucala R, Weigle B, Nawroth PP, Schulz JB. Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiol Aging, 2012; 33: 2478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Gasparotto J, Ribeiro CT, Bortolin RC, Somensi N, Rabelo TK, Kunzler A, Souza NC, Pasquali MAB, Moreira JCF, Gelain DP. Targeted inhibition of RAGE in substantia nigra of rats blocks 6-OHDA-induced dopaminergic denervation. Sci Rep, 2017; 7: 8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, Schulte C, Mustafa S, Synofzik M, Vukovic Z, Itohara S, Berg D, Teismann P. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain, 2012; 135: 3336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Viana SD, Valero J, Rodrigues-Santos P, Couceiro P, Silva AM, Carvalho F, Ali SF, Fontes-Ribeiro CA, Pereira FC. Regulation of striatal astrocytic receptor for advanced glycation end-products variants in an early stage of experimental Parkinson’s disease. J Neurochem, 2016; 138: 598–609. [DOI] [PubMed] [Google Scholar]
- [131].Ma L, Nicholson LF. Expression of the receptor for advanced glycation end products in Huntington’s disease caudate nucleus. Brain Res, 2004; 1018: 10–7. [DOI] [PubMed] [Google Scholar]
- [132].Kim J, Waldvogel HJ, Faull RL, Curtis MA, Nicholson LF. The RAGE receptor and its ligands are highly expressed in astrocytes in a grade-dependant manner in the striatum and subependymal layer in Huntington’s disease. J Neurochem, 2015; 134: 927–42. [DOI] [PubMed] [Google Scholar]
- [133].Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, Schmidt AM, Brown C, Stern A, LaFaille J, Chess L, Stern DM, Jiang H. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med, 2003; 9: 287–93. [DOI] [PubMed] [Google Scholar]
- [134].Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal Bianco A, Khademi M, Wallstrom E, Lobell A, Brundin L, Lassmann H, Harris RA. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J Leukoc Biol, 2008; 84: 1248–55. [DOI] [PubMed] [Google Scholar]
- [135].Sternberg Z, Weinstock-Guttman B, Hojnacki D, Zamboni P, Zivadinov R, Chadha K, Lieberman A, Kazim L, Drake A, Rocco P, Grazioli E, Munschauer F. Soluble receptor for advanced glycation end products in multiple sclerosis: a potential marker of disease severity. Mult Scler, 2008; 14: 759–63. [DOI] [PubMed] [Google Scholar]
- [136].Li K, Zhao B, Dai D, Yao S, Liang W, Yao L, Yang Z. A functional p.82G>S polymorphism in the RAGE gene is associated with multiple sclerosis in the Chinese population. Mult Scler, 2011; 17: 914–21. [DOI] [PubMed] [Google Scholar]
- [137].Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol, 2013; 94: 55–68. [DOI] [PubMed] [Google Scholar]
- [138].Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S, Okamoto H, Shimura S, Karasawa T, Yonekura H, Yamamoto H. Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol, 2011; 186: 3248–57. [DOI] [PubMed] [Google Scholar]
- [139].Gasiorowski K, Brokos B, Echeverria V, Barreto GE, Leszek J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol Neurobiol, 2018; 55: 1463–1476. [DOI] [PubMed] [Google Scholar]
- [140].Rong LL, Yan SF, Wendt T, Hans D, Pachydaki S, Bucciarelli LG, Adebayo A, Qu W, Lu Y, Kostov K, Lalla E, Yan SD, Gooch C, Szabolcs M, Trojaborg W, Hays AP, Schmidt AM. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J, 2004; 18: 1818–25. [DOI] [PubMed] [Google Scholar]
- [141].Geroldi D, Falcone C, Minoretti P, Emanuele E, Arra M, D’Angelo A. High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J Am Geriatr Soc, 2006; 54: 1149–50. [DOI] [PubMed] [Google Scholar]
- [142].Scavello F, Zeni F, Tedesco CC, Mensa E, Veglia F, Procopio AD, Bonfigli AR, Olivieri F, Raucci A. Modulation of soluble receptor for advanced glycation end-products (RAGE) isoforms and their ligands in healthy aging. Aging (Albany NY), 2019; 11: 1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Scavello F, Tedesco CC, Castiglione S, Maciag A, Sangalli E, Veglia F, Spinetti G, Puca AA, Raucci A. Modulation of soluble receptor for advanced glycation end products isoforms and advanced glycation end products in long-living individuals. Biomark Med, 2021; 15: 785–796. [DOI] [PubMed] [Google Scholar]
- [144].Tae HJ, Kim JM, Park S, Tomiya N, Li G, Wei W, Petrashevskaya N, Ahmet I, Pang J, Cruschwitz S, Riebe RA, Zhang Y, Morrell CH, Browe D, Lee YC, Xiao RP, Talan MI, Lakatta EG, Lin L. The N-glycoform of sRAGE is the key determinant for its therapeutic efficacy to attenuate injury-elicited arterial inflammation and neointimal growth. J Mol Med (Berl), 2013; 91: 1369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev, 2012; 28: 147–75. [DOI] [PubMed] [Google Scholar]
- [146].Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol, 2010; 28: 863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Wautier JL, Zoukourian C, Chappey O, Wautier MP, Guillausseau PJ, Cao R, Hori O, Stern D, Schmidt AM. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J Clin Invest, 1996; 97: 238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Flyvbjerg A, Denner L, Schrijvers BF, Tilton RG, Mogensen TH, Paludan SR, Rasch R. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes, 2004; 53: 166–72. [DOI] [PubMed] [Google Scholar]
- [149].Jensen LJ, Denner L, Schrijvers BF, Tilton RG, Rasch R, Flyvbjerg A. Renal effects of a neutralising RAGE-antibody in long-term streptozotocin-diabetic mice. J Endocrinol, 2006; 188: 493–501. [DOI] [PubMed] [Google Scholar]
- [150].Lutterloh EC, Opal SM, Pittman DD, Keith JC Jr., Tan XY, Clancy BM, Palmer H, Milarski K, Sun Y, Palardy JE, Parejo NA, Kessimian N. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care, 2007; 11: R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Christaki E, Opal SM, Keith JC Jr., Kessimian N, Palardy JE, Parejo NA, Tan XY, Piche-Nicholas N, Tchistiakova L, Vlasuk GP, Shields KM, Feldman JL, Lavallie ER, Arai M, Mounts W, Pittman DD. A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock, 2011; 35: 492–8. [DOI] [PubMed] [Google Scholar]
- [152].Finlay WJ, Cunningham O, Lambert MA, Darmanin-Sheehan A, Liu X, Fennell BJ, Mahon CM, Cummins E, Wade JM, O’Sullivan CM, Tan XY, Piche N, Pittman DD, Paulsen J, Tchistiakova L, Kodangattil S, Gill D, Hufton SE. Affinity maturation of a humanized rat antibody for anti-RAGE therapy: comprehensive mutagenesis reveals a high level of mutational plasticity both inside and outside the complementarity-determining regions. J Mol Biol, 2009; 388: 541–58. [DOI] [PubMed] [Google Scholar]
- [153].Ruiz-Lopez E, Schuhmacher AJ. Transportation of Single-Domain Antibodies through the Blood-Brain Barrier. Biomolecules, 2021; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Mohammed A, Zeng W, Mengist HM, Kombe Kombe AJ, Ou H, Yang Y, Dan Z, Xu Z, Ma H, Jin T. Generation, biochemical characterizations and validation of potent nanobodies derived from alpaca specific for human receptor of advanced glycation end product. Biochem Biophys Res Commun, 2021; 581: 38–45. [DOI] [PubMed] [Google Scholar]
- [155].Lipi F, Chen S, Chakravarthy M, Rakesh S, Veedu RN. In vitro evolution of chemically-modified nucleic acid aptamers: Pros and cons, and comprehensive selection strategies. RNA Biol, 2016; 13: 1232–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov, 2017; 16: 181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Matsui T, Higashimoto Y, Nishino Y, Nakamura N, Fukami K, Yamagishi SI. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes, 2017; 66: 1683–1695. [DOI] [PubMed] [Google Scholar]
- [158].Nakamura N, Matsui T, Ishibashi Y, Sotokawauchi A, Fukami K, Higashimoto Y, Yamagishi SI. RAGE-aptamer Attenuates the Growth and Liver Metastasis of Malignant Melanoma in Nude Mice. Mol Med, 2017; 23: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Koga Y, Sotokawauchi A, Higashimoto Y, Nishino Y, Hashizume N, Kakuma T, Akiba J, Tanaka Y, Matsui T, Yagi M, Yamagishi SI. DNA-Aptamer Raised against Receptor for Advanced Glycation End Products Improves Survival Rate in Septic Mice. Oxid Med Cell Longev, 2021; 2021: 9932311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord, 2011; 25: 206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, Mjalli AM. Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol, 2014; 14: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Galasko D, Bell J, Mancuso JY, Kupiec JW, Sabbagh MN, van Dyck C, Thomas RG, Aisen PS, Alzheimer’s Disease Cooperative S. Clinical trial of an inhibitor of RAGE-Abeta interactions in Alzheimer disease. Neurology, 2014; 82: 1536–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Walker D, Lue LF, Paul G, Patel A, Sabbagh MN. Receptor for advanced glycation endproduct modulators: a new therapeutic target in Alzheimer’s disease. Expert Opin Investig Drugs, 2015; 24: 393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J Prev Alzheimers Dis, 2018; 5: 149–154. [DOI] [PubMed] [Google Scholar]
- [165].Evaluation of the Efficacy and Safety of Azeliragon (TTP488) in Patients With Mild Alzheimer’s Disease (STEADFAST). In: ed.êds. Updated May 7, 2021, ClinicalTrials.gov Identifier (NCT number): NCT02080364. [Google Scholar]
- [166].Valcarce C, Dunn I, Burstein AH. Linking diabetes and Alzheimer’s disease through rage: a retrospective analysis of Azeliragon Phase 2 and Phase 3 studies. Alzheimer’s Association International Conference 2019, 2019; Volume 15: P1263 Abstract O4–11-04. [Google Scholar]
- [167].Study of Azeliragon in Patients With Mild Alzheimer’s Disease and Impaired Glucose Tolerance (Elevage). In: ed.êds. Updated May 13, 2021, ClinicalTrials.gov Identifier (NCT number): NCT03980730. [Google Scholar]
- [168].Han YT, Choi GI, Son D, Kim NJ, Yun H, Lee S, Chang DJ, Hong HS, Kim H, Ha HJ, Kim YH, Park HJ, Lee J, Suh YG. Ligand-based design, synthesis, and biological evaluation of 2-aminopyrimidines, a novel series of receptor for advanced glycation end products (RAGE) inhibitors. J Med Chem, 2012; 55: 9120–35. [DOI] [PubMed] [Google Scholar]
- [169].Kim SH, Han YT. Design, synthesis, and biological evaluation of pyrimidine-2-carboxamide analogs: investigation for novel RAGE inhibitors with reduced hydrophobicity and toxicity. Arch Pharm Res, 2015; 38: 1952–62. [DOI] [PubMed] [Google Scholar]
- [170].Han YT, Kim K, Choi GI, An H, Son D, Kim H, Ha HJ, Son JH, Chung SJ, Park HJ, Lee J, Suh YG. Pyrazole-5-carboxamides, novel inhibitors of receptor for advanced glycation end products (RAGE). Eur J Med Chem, 2014; 79: 128–42. [DOI] [PubMed] [Google Scholar]
- [171].Arumugam T, Ramachandran V, Gomez SB, Schmidt AM, Logsdon CD. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin Cancer Res, 2012; 18: 4356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res, 2002; 62: 4805–11. [PubMed] [Google Scholar]
- [173].Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, DeVita RJ, Ramasamy R, Shekhtman A, Schmidt AM. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci Rep, 2016; 6: 22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Putranto EW, Murata H, Yamamoto K, Kataoka K, Yamada H, Futami J, Sakaguchi M, Huh NH. Inhibition of RAGE signaling through the intracellular delivery of inhibitor peptides by PEI cationization. Int J Mol Med, 2013; 32: 938–44. [DOI] [PubMed] [Google Scholar]