

Abstract
Cancer is one of the most severe medical conditions in the world, causing millions of deaths each year. Chemotherapy and radiotherapy are critical for treatment approaches, but both have numerous adverse health effects. Furthermore, the resistance of cancerous cells to anticancer medication leads to treatment failure. The rising burden of cancer requires novel efficacious treatment modalities. Natural remedies offer feasible alternative options against malignancy in contrast to available synthetic medication. Selective killing of cancer cells is privileged mainstream in cancer treatment, and targeted therapy represents the new tool with the potential to pursue this aim. The discovery of innovative therapies targeting essential components of DNA damage signaling and repair pathways such as ataxia telangiectasia mutated and Rad3 related Checkpoint kinase 1 (ATR-CHK1)has offered a possibility of significant therapeutic improvement in oncology. The activation and inhibition of this pathway account for chemopreventive and chemotherapeutic activity, respectively. Targeting this pathway can also aid to overcome the resistance of conventional chemo- or radiotherapy. This review enlightens the anti-cancer role of natural products by ATR-CHK1 activation and inhibition. Additionally, these compounds have been shown to have chemotherapeutic synergistic potential when used in combination with other anticancer drugs. Ideally, this review will trigger interest in natural products targeting ATR-CHK1 and their potential efficacy and safety as cancer lessening agents.
Keywords: ATR-CHK1, Chemopreventive, Chemotherapeutic, MDR cancer, Natural products targeting ATR-CHK1
1. Introduction
Cancer is one of the world’s top causes of death, and the number of cases is increasing all the time, with an expected 21 million cases by 2030[1]. The absence of effective anticancer medicines continues to be a clinical issue. Chemotherapy and radiation are the two most used treatments for cancer, yet both have been linked to severe side effects. Multi drug resistance(MDR) is another problem. Natural products are the most suited candidate for anticancer drugs since they primarily target malignant development[2]. Natural products are demonstrated to be a rich source for identifying bioactive substances designed and implemented in the management of a number of illnesses and disorders, particularly cancer. The production of secure and effective medicines for the treatment and prevention of cancer, though, depends on the comprehensive characterization of natural product and herbal remedies and the discovery of their mechanisms involved [3]. With the advent of potential therapeutic methodologies, significant attempts have increased the effectiveness of natural anticancer medications [4–6]. A number of pharmacological actions, such as anti-bacterial, anti-spasmodic, chemopreventive, anticancer, and analgesic agents, have been attributed to isolated natural alkaloids and their analogues [7,8]. Besides this, it has been shown that C. papaya seeds are abundant in alkaloids that have anti-carcinogenic effects [9]. In various anti-cancer medications, like camptothecin and vinblastine, alkaloids from Rauwolfia vomitoria are a key ingredient [10,11]. It’s worth noting that the Rauwolfiaextract seems to affect the expression of multiple cyclins in conflicting ways. While the expression of cyclins D1 and H was noticeably reduced, that of cyclins A1 and A2 showed a significant rise [3].
Cell genomic stability is critical for cell proliferation and the effective transfer of genetic information to future generations [12]. DNA is constantly damaged by internal (reactive oxygen species, halted replication forks) and external (Ultraviolet, ionizing radiation, genotoxic agents) insults that challenge DNA integrity throughout the cell cycle. DNA damage response (DDR) and cell cycle checkpoints are intertwined signaling networks that arrest the cell cycle. Cells have developed DDR to find and resolve DNA abnormalities and stop the cell cycle from allowing additional time to resolve or cause apoptosis, if the damage is severe or persistent[13]. They restrict the transfer of genetic information to daughter cells in order to preserve genomic integrity [14–16]. Cell cycle checkpoints enable an orderly progression of cell cycle events and avoiding the development of genomic instability associated diseases such as cancer [17,18]. When DNA damage is detected, G1-S, S and G2-M checkpoints are activated. Cells recover from the checkpoint after DNA repair and move through the cell cycle[19].
The DDR pathways and ataxia telangiectasia mutated and Rad3 related Checkpoint kinase 1 (ATR–CHK1) are triggered by DNA double-strand break (DSB) and single-stranded DNA (ssDNA) respectively [20]. The Ataxia telangiectasia mutated Checkpoint kinase 2 (ATM-CHK2) pathway is commonly altered in tumors. ATR and CHK1 genetic abnormalities are rare. Oncoproteins (Human papillomavirus-16 oncoproteins, E6 and E7) and hypoxia (poor oxygenation is a crucial factor in the development of tumours, metabolism, angiogenesis, metastasis, proliferation, and cell death) result in increased replication stress (RS) and DSBs generation. To ensure that this damage is not transferred further, G1 checkpoints are dysregulated in most cancer cells; consequently, they rely on S and G2 checkpoints[15,21]. The dependency of cancer cells on S and G2 checkpoints for their survival and high level of replication stress (RS) make them dependent on ATR-CHK1. Chemotherapeutics that affects DNA replicating cells stimulate the ATR–CHK1. Therefore, the study of ATR–CHK1 targeting compounds is essential as anticancer agents[19,20,22]. Ataxia-telangiectasia mutated, and Rad3 related (ATR) and checkpoint kinase 1 (CHK1) controls cell cycle checkpoints and promote cell survival via interfering with cell-cycle advancement (S and G2 phases), providing the essential delay for DNA repair and promoting DNA replication restart [23–25]. ATR is a DDR core component, engaged during each S-phase and activates CHK1. It prevents premature entry into mitosis [26]. CHK1 was identified in fruit fly (Drosophila grapes), mice and humans that regulate the G2-M phase transformation in DDR. CHK1 regulates the cell cycle and transcription [27]. ATR signaling is triggered by DNA lesions that expose fragile single-stranded DNA (ssDNA). Replication protein A (RPA) coated ssDNA primarily activates ATR signaling in S-phase cells. ATR and ATR interacting protein (ATRIP) bind with the RPA-ssDNA complex for CHK1 activation. Activated CHK1 arrests S and G2-M cell cycle and prevents cells with damaged DNA from entering mitosis, thus enabling the DDR mechanism more time to repair DNA. After DNA repair, the checkpoint is terminated by deactivating ATR-CHK1 and promoting mitosis [19,21,22].
The importance of ATR-CHK1 in the DDR is discussed here, and the present state of natural chemicals targeting these pathways. We also give examples of these compounds participating in synthetic lethality and overcoming cancer resistance by affecting this pathway.
2. ATR and CHK1 in cancer therapy
2.1. ATR and CHK1 activation by DNA damage
The DNA damage response (DDR) is essential for protecting cells from the high amounts of oxidative damages that are subjected to cellular damages on a regular basis. Because it is produced intracellularly (reactive oxygen species) or as a consequence of typical ecological exposures, the overwhelming proportion of this damages is inevitable (UV rays exposure). The DDR is a complex system that includes cell cycle checkpoints, that avoid harm from being addressed through DNA replication or transferred on to new cells during mitosis, as well as the DNA repair mechanisms altogether.Ataxia telangiectasia mutated and Rad3 related (ATR) kinase is a PIKK family member with a structure comparable to ATM and DNA-PKcs, both of which play important parts in the DDR. Checkpoint kinase 1 (CHK1) is ATR’s primary phosphorylation target, and the activity of either of these enzymes is crucial for cellular growth and maintaining integrity of the genome in response to DNA damage and replicating distress[28,29]. In the earliest embryonic development, homozygous loss of ATR or CHK1 is fatal[30,31], underlining the significance of these protein kinases in the creation of kinase dead ATR (ATR-KD) cells, in which an inactivated form of ATR acts as a dominant negative inhibitor of native ATR function. Thus led to the discovery of ATR-KD cells which were sensitive to DNA damaging agents and did not arrest at the G2/M checkpoint, implying that ATR plays an important role in both DNA harm repair and cell cycle checkpoint regulation [32]. Replication stress, which is frequent in malignancies with active oncogenes and malfunctioning G1/S checkpoint regulation, is the primary activator of the ATR-CHK1 cascade (Fig. 1) [33]. Single stranded DNA (ssDNA) resulting from halted origin of replication, nucleotide excision repair (NER) intermediates, or resected DSBs that have been subjected to exonuclease digestion which activates ATR at the molecular level [34,35]. The ATR-interacting protein is required for TR recognition of the RPA-ssDNA complex (ATRIP). ATRIP is so important to ATR’s activity that individuals lacking ATR or its obligatory subunit exhibit no phenotypic changes [36,37]. ATR and ATRIP (ATR-interacting protein) form a stable complex that controls ATR localization and is necessary for ATR signalling in response to DNA damages and replicating stress [38]. Along with ATRIP, many other proteins are involved in the ATR pathway, such as Topoisomerase binding protein-1 (TopBP1), which is involved in both checkpoint signaling and the start of DNA replication [39]. ATR is brought by ATRIP, and TopBP1 by Rad9. TopBP1 can interact with contact surface on both ATRIP and ATR due to the formation and localization of such constituents at DNA damage regions. ATR activation is therefore encouraged by this interaction [38]. The extra localization of RAD9, RAD1, and HUS1 (which forms the hetero-trimeric ring-shaped complex known as 9–1–1) via RPA interaction with RAD17 is required for stimulation of the ATR-CHK1 cascade [40]. TOPBP1 is recruited by the 9–1–1 complexes, and it is assumed that TOPBP1 binding is a key step in ATR activation and subsequent phosphorylation processes [41]. Checkpoint kinase 1 (CHK1) is ATR kinase’s principal phosphorylation target, and it acts as an intermediate in many of the DNA repair and DNA checkpoint events that occur when ATR is activated at locations of DNA damage.
Fig. 1.
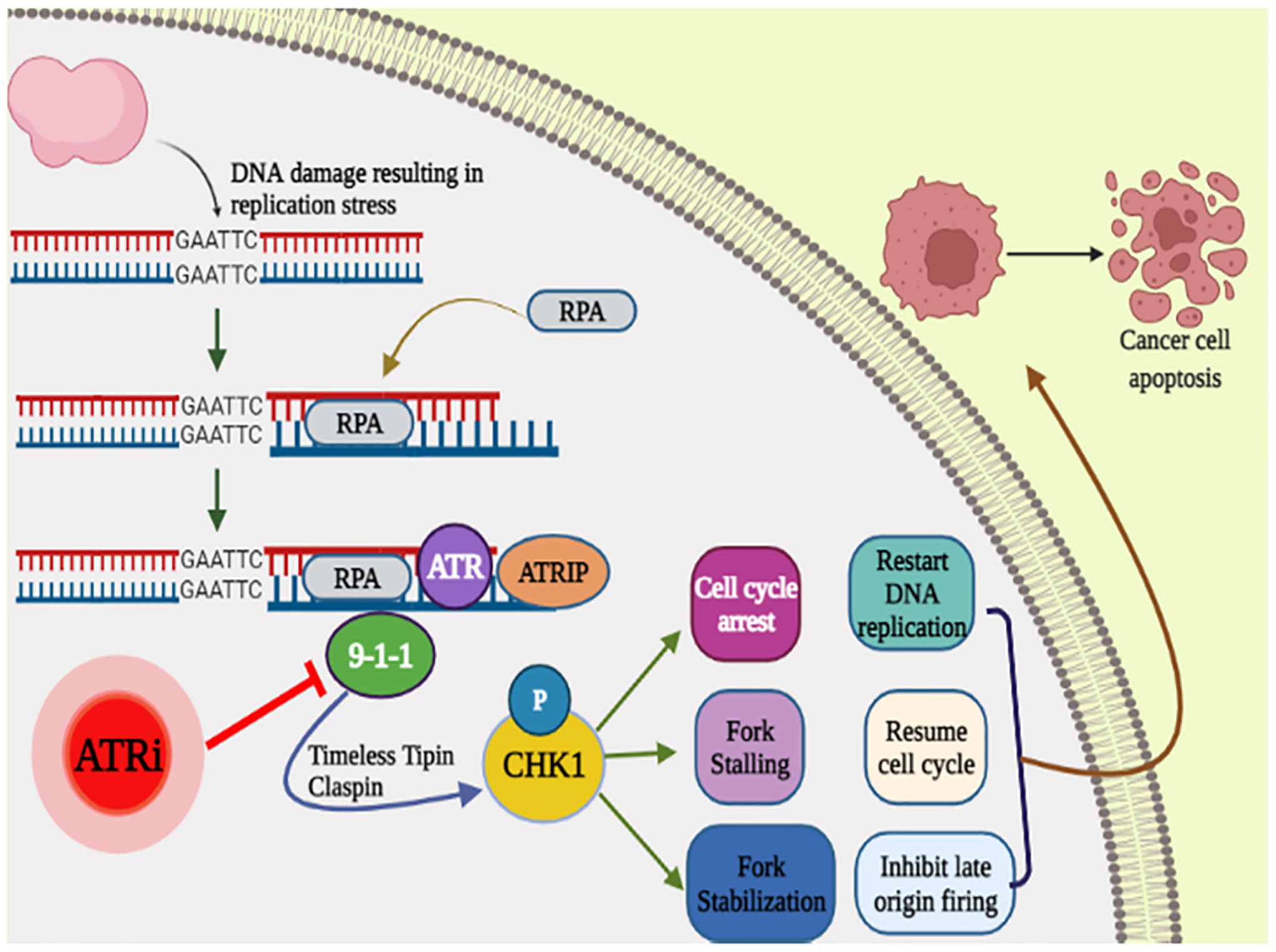
Replication checkpoints and DNA damage Anticancer medications cause problems with replication. The slowing or halting of replication fork advancement causes replication stress. The ATR–CHK1 pathway is activated when DNA synthesis is inhibited or damaged, resulting in checkpoint responses. DNA damages can cause a latency in entering S-phase (the G1 checkpoint), limit the replication of damaged DNA, or even prohibit the cell from entering mitosis (G2 checkpoint). Because PARP and checkpoint proteins suppress fork collapse, antagonists of these proteins might enhance replication stresses, genomic instability, and, as a result, cellular demise.
2.2. ATR and CHK1 signaling to DNA checkpoints
ATR signals to coordinate cell checkpoint regulation and DNA repair that is carried out by a vast number of ATR substrates, once activated and localized at the site of the DNA damage. The ATR-CHK1 sequence is important for both cell cycle regulation at the G2/M checkpoint and DNA replication. CHK1 is transiently present at the site of DNA damage and is triggered by ATR phosphorylation of two sites, Ser345 and Ser317, on CHK1[42,43]. Claspin acts as a “mediator” protein between ATR and CHK1, allowing ATR to phosphorylate CHK1 [44,45]. Claspin is recruited and loaded into the 9–1–1 complex at the site of Oxidative damages by RAD17, and is triggered when coupled to its phosphorylation state. ATR is required for RAD17 phosphorylation [45]. CHK1 is liberated from chromatin when it is triggered and phosphorylated, and it signals DNA degradation to the remainder of the nucleus. It’s still unclear if checkpoint responses are caused by such a subcellular re-distribution of stimulated CHK1 or enhanced catalytic properties [46]. Wee1 and the cell differentiation phase proteins (cdc) cdc25A and cdc25C are significant CHK1 targets. The function of cyclin-dependent kinase (CDK1/CDK2) is inhibited when cdc25 proteins and Wee1 are phosphorylated. Phosphorylation of cdc25A induces CDK2 suppression and S-phase arrest, while phosphorylation of cdc25C and Wee1 induces CDK1 suppression and G2/M arrest(Fig. 1)[47–50]. The ATR-CHK1 system controls cell cycle progression at the G2/M and intra-S cell cycle checkpoints via inhibiting CDK1,thus limiting immediate mitotic catastrophe or permanent genetic material loss [51].
2.3. Reduction of replication stress via ATR and CHK1 signaling
During replication stress and DNA damaging circumstances, ATR-CHK1 signaling slows replicating origin’s activation, thus lowering the rate of DNA replication. The exact biochemical mechanisms through which ATR performs this impact are unknown, although they seem to be closely linked to the events that activate and maintain the intra-S checkpoint [51,52]. Wee-1 stimulation induced by CHK-1 and cdc25A suppression results in CDK2 catalytic activity reduction and a slowdown of DNA synthesis via late replicating origin’s inhibition [53]. Replication fork development is stopped whenever cellular reproduction is stopped due to a lesion obstructing fork progression or an extremely limited number of dNTPs, and the stalled replication forks may stop (Fig. 2). The existence of functional ATR signalling through CHK1 is required for the stabilization of stalled replication forks, and their absence causes replication fork ‘collapse’. Nevertheless, it’s uncertain how CHK1 avoids or delays replicating fork collapse undergoing replication stress [54].
Fig. 2.
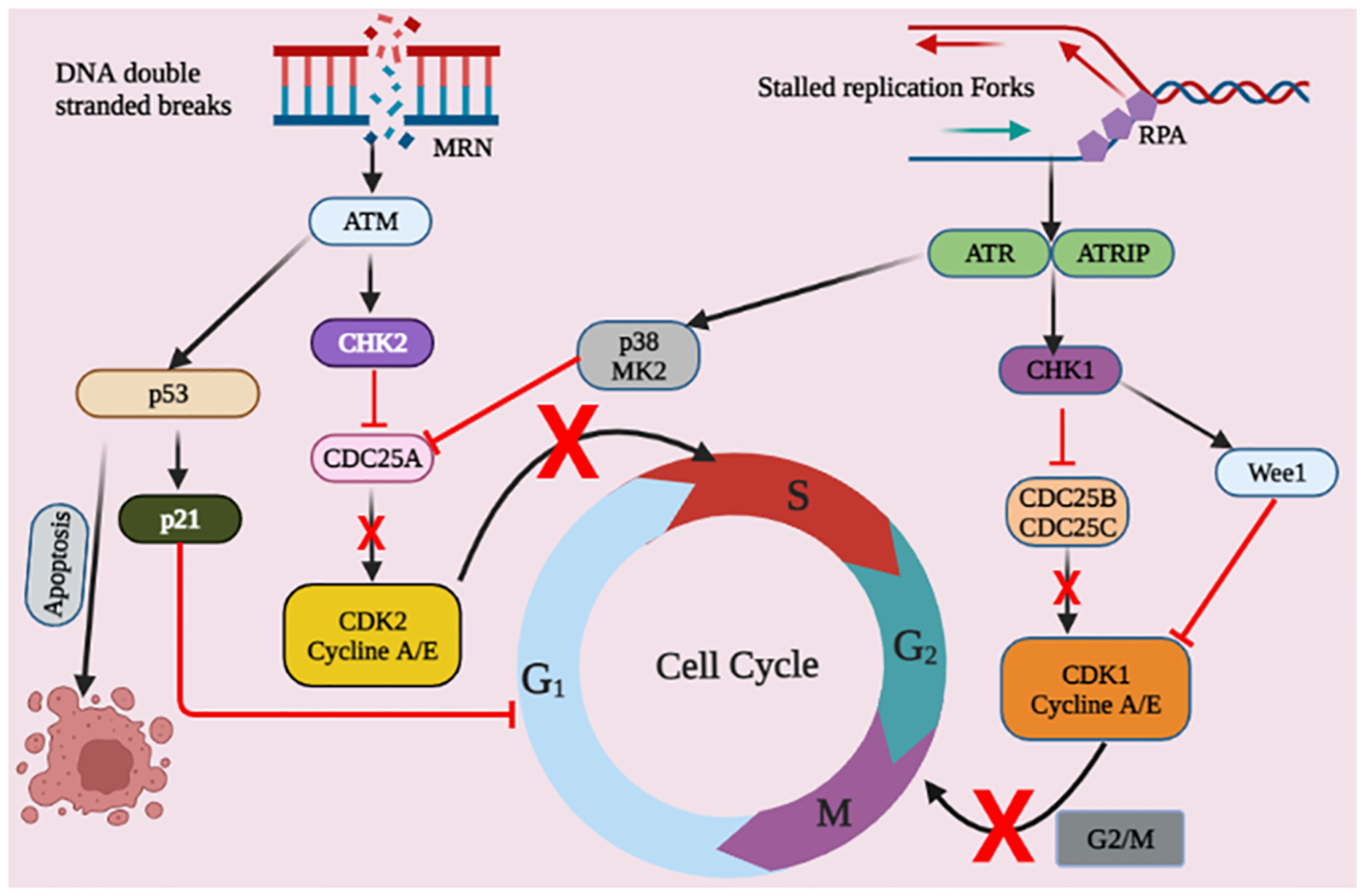
The DNA damage-induced checkpoint mechanisms are depicted in this schematic representation. DNA double-strand breakage or DNA single-strand breaks and replication stress stimulate the ATM/CHK2 and ATR/CHK1 mechanisms, accordingly. Cell cycle checkpoints are predominantly activated by ATM and ATR phosphorylation of p53, CHK2, CHK1, and p38/MK2. Activated p53 causes G1-phase inhibition and apoptosis in cells.The phosphorylation of CDC25 by CHK2 and CHK1 prevents CDK expression, halting cellular proliferation in S-phase or at the G2/M transition. Cancerous cells lacking the G1 checkpoint due to mutation or loss of p53 seem to be more reliant on the intra-S and G2/M checkpoints.
2.4. Repairing DNA via ATR and CHK1 signaling
ATR substrates that govern DNA repair, in contrast to the well-known regulation of cell cycle checkpoints and signaling to DNA replication by CHK1 kinase, are a new class of ATR substrates. ATR regulates inter-strand crosslink repair by targeting the Fanconi-anemia proteins (FANCI) and (FANCD2)[55]. FANCD2 monoubiquitination is aided by ATR phosphorylation, which enhances its localization to DNA damage foci [31]. ATR also regulates the recruitment of the NER protein XPA to DNA lesions by phosphorylating it [56]. The DNA repair through homologous recombination DNA repair (HRR) appears to be tightly regulated by ATR and CHK1. The major HRR regulatory protein BRCA1 was phosphorylated and triggered by ATR in early investigations [57]. RAD51 recombinase and BRCA2 are two important HRR proteins that CHK1 recruits and phosphorylates. In regard to treatment with DNA damaging hydroxyurea (HU), treated cells with coffee and NU6027, initial antagonists of ATR, or blockers of CHK1, have fewer RAD51 repair foci [58–60]. ATR’s function in connecting HRR with cell cycle checkpoints have subsequently been clarified by showing that ATR improves BRCA1-PALB2 interaction to enhance HRR, which is at least partly due to CDK suppression. As a result, ATR and CHK1 perform a key responsibility in maintaining DNA stability in the face of DNA-damaging insults, primarily via their participation in HRR and cell cycle checkpoints [15,61].
3. Mechanistic insights of natural products targeting ATR-CHK1 pathway
3.1. Chemopreventive effect by ATR-CHK1 activation
Chemopreventive action is attributed to ATR-CHK1 activation. This can potentially restrict tumour formation in its early stages by serving as a barrier to the proliferation of abnormal cells, primarily through p53 activation. This activation may result in the initiation of DNA repair and the G2 phase arrest [18]. The primary hypothesis is that ATR/CHK1 inhibitors, especially in p53-deficient cells, would improve the death of tumour cells by cytotoxic medicines or radiotherapy by disrupting cell cycle checkpoints [62,63]. The first CHK1 inhibitor with broad-spectrum effectiveness targeting the protein kinase C family is UCN-01 (7-hydroxystaurosporine). Moreover, since UCN-01 binds to alpha acidic-glycoprotein, which causes hyperglycemia, it lacks selectivity and has a lengthy half-life, which have limited its potential applications [64,65]. The ATP-competitive inhibitor of CHK1, CHK2, VEGFR-2, and VEGFR-3, XL844, is highly effective. When used with gemcitabine, XL844 prevents the degradation of CDC25A, overrides the S-phase checkpoints, and enhances DNA damage. As a result, XL844 increases the activity of gemcitabine in xenografts and in vitro. However, for reasons that are now unknown, the clinical study of XL844 (NCT00475917 and NCT00234481) was abruptly stopped[66,67]. The development of natural drugs that target ATR and CHK1 has permitted their roles to be thoroughly investigated in the preclinical and clinical context. Olaparib’s clinical success as the first DDR inhibitor certified by the FDA has bolstered attempts to scrutinize additional DDR-targeting medicines. Little progress has been made in screening several natural compounds for ATR-CHK1 inhibition regarding this promising approach. The published preclinical studies support the clinical applicability of natural ATR-CHK1 activators and inhibitors for targeted cancer treatment.
Rocaglamide-A (Roc-A) may directly affect cancer cell cycle advancement by activating ATR-CHK1 to potentiate Cdc25A (Fig. 2) deterioration in cancer cells. Cdc25A overexpression has been seen in various human malignancies with more severe illnesses and a bad prognosis. Roc-A triggers G1-S arrest in 13 malignant cell lines but not in normal cells with IC50s in the range of 50–100 nM [68,69]. With an IC50 of 20 M, Harmine promotes S and G2-M arrest in Hep3B cells with quite a low cytotoxicity in normal cells [70].
3.2. Chemotherapeutic effect by ATR-CHK1 inhibition
ATR-CHK1 inhibition results in profound G2 and S-M checkpoint defects that give rise to entering mitosis prematurely, resulting in cell death. The absence of ATR-CHK1 signaling is incompatible with life [18,20,23]. ATR-CHK1 inhibitors are not very hazardous, most likely because inhibition is neither continuous nor complete. While still synergizing with genotoxic therapy, ATR inhibition did not exacerbate the toxic effects of several genotoxic drugs[24]. Caffeine disrupted the G2-M and hastened the transition to mitosis, culminating in apoptosis in A549 cells (IC50 = 1.1 mM) [15,71]. Schisandrin B (Sch-B) was shown to inhibit ATR in A549 adenocarcinoma cells (IC50 = 7.25 μM) [72]. The apple peel flavonoid fraction (AF4) includes quercetin glycosides, epicatechin, chlorogenic acid, phloridzin and cyanidin 3-galactoside. AF4 pretreatment protects BEAS-2B cells against different carcinogens, including nicotine-derived nitrosamine ketones, via decreasing ATR-CHK1 signaling [73]. Triptolide from Tripterygium wilfordii induces DNA damage in A375. S2 melanoma cells by ATR-CHK1 inhibition[74]. Kaempferol promoted DNA damage in HL-60 cells with an IC50 of 75 μM [75]. Similarly, mangiferin inhibits cell cycle advancement at the G2-M phase in HL-60 with no toxicity to normal cells [76,77]. Protoapigenone (WYC02) and its synthetic derivative WYC0209 cure cancer by ATR inhibition and causing chromosomal breakage. On numerous cancer cell lines, the natural substance protoapigenone (WYC02) and its synthetic counterpart WYC0209 both demonstrated anticancer activity. In Chinese hamster ovary cells that are mitotically spreading, WYC02 results in chromosomal abnormality. It’s worth noting that when exposed to WYC02 and WYC0209 cancer cells did not show the typical DDR markers (WYCs). When Chk1 and the Fanconi anaemia group D2 protein (FANCD2) are activated, in particular, they may be able to suppress DDR, although Chk2 is not one of the molecular pathways that WYCs can affect [78].
3.3. Chemopreventive as well as chemotherapeutic effects
Curcumin and resveratrol are reported to show both chemopreventive and chemotherapeutic effect by ATR-CHK1 activation and inhibition, respectively. Treatment of MGC-803 gastric cancer cells with 20 μM curcumin led to activates ATR-CHK1[79]. It inhibits ATR-CHK1 in HeLa, HT1080 and H1299 cells with IC50 of 15 μM with no toxicity to normal cells [80,81]. Resveratrol causes ATR-CHK1 checkpoint activation and induced S and G2-M arrest in ovarian Ovcar-3, PA-1 and SKOV-3 cells. It does not affect the normal human foreskin fibroblast cells. As resveratrol as well continued to increase phospho-H2A.X (Ser139), which is identified to be phosphorylated by ATM/ATR in response to DNA damage, resveratrol induces phosphorylation of cell division cycle 25 C (Cdc25C) tyrosine phosphatase through the stimulation of checkpoint CHK1 and CHK2, that in turn have been stimulated via ATM (ataxia telangiectasia-Rad) [82]. It has shown similar behavior in TK6 and WTK1 B-lymphoblastoid cell lines with IC50 of 30 μM [83]. Resveratrol analogue 8-ADEQ treatment inhibits the HeLa cell proliferation (IC50: 8 μM) by ATR activation [84]. Rad51 overexpression is seen in many malignancies and is linked to higher DNA repair efficiency and resistance to chemotherapy[85]. ATR–CHK1 activation, G2 arrest, and other complex responses to DNA damage require Breast Cancer Gene 1 (BRCA1). BRCA2 promotes the loading of Rad51, displaces RPA and binds single-strand DNA at the regions of damage-forming filaments leading to strand invasion and subsequent recombination [86]. Resveratrol impedes the Rad51, BRCA1 and BRCA2 expression involved in ATR-CHK1 activation in the MCF-7 cell line (IC50:150 μmol / L)[87]. On numerous cancer cell lines, the natural compound protoapigenone (WYC02) and its synthesized analogue WYC0209 shown cytotoxic effects. In Chinese hamster ovary cells that are mitotically spreading, WYC02 results in chromosomal abnormality. It’s interesting to note that when exposed to WYC02 and WYC0209 (WYCs), cancer cells did not show the typical DDR markers. Additional research into the molecular mechanisms behind WYC activity suggested that they may be able to suppress DDR, namely when Chk1 and the Fanconi anaemia group D2 protein (FANCD2) are activated but not Chk2. WYCs prevented ATR-mediated DNA damage checkpoint and repair in this manner. In addition, therapy with WYCs boosted tumour susceptibility to interstrand cross-link-generating drugs both in vitro and in vivo when paired with the DNA cross-linking agent cisplatin. In situations of oncogene-driven replicative stress or tolerance to DNA-interfering agents, where the ATR checkpoint is constitutively active, the results specifically implicate WYCs in increasing tumour chemosensitivity [88].
3.4. MDR cancers
Cancer cells show genotoxic substances and chemo-radiation resistance by activating the ATR-CHK1 to enhance their DNA repair systems and facilitate survival. Cancer treatments activate ATR-CHK1 by generating massive DNA lesions. ATR-CHK1 inhibition was also reported to suppress p-glycoprotein (P-gp) levels. As a result, inhibiting ATR-CHK1 could improve the therapeutic efficacy of chemotherapeutic or DNA-damaging treatments while also overcoming resistance[14,27,89]. Protoapigenone (WYCs) 8 μmol/L and synthetic derivative (WYC0209) 2 μmol/L showed an anticancer effect in MDA-MB-231 by ATR-CHK1 inhibition. However, this study does not show the effect on drug transporter function[78]. Schisandrin B (Sch-B) exhibits ATR-dependent cytotoxic effects on A549 cells and also behaves as a P-gp inhibitor may be due to ATR-CHK1 inhibition [72,90,91].
3.5. Synthetic lethality
Synthetic lethality is defined as a mismatch between two genetic events that result in a deadly impact[25]. When ATR-CHK1 signaling is blocked, the cell-killing impact of chemotherapy or radiation increases and results in the synthetic lethality effect in cancer treatment [27]. A considerable proportion of human malignancies with p53 deficiency rely on the ATR-CHK1-dependent S and G2-M checkpoints for survival [14]. The p53-dependent response protects healthy cells against the adverse effects of DNA-damaging medicines in the event of ATR-CHK1 suppression. So, the combination of ATR-CHK1 inhibitors and DNA damaging agents should effectively target tumour cells with milder side effects to healthy cells, following the principle of synthetic lethality [23]. Cisplatin’s anticancer activity comes from its capacity to cause DNA damage, but this therapy’s efficacy is frequently reduced quickly due to cancer cell resistance [92]. Caffeine 2 mM increases apoptosis in both HTB182 and CRL5985 lung cancer cells in response to cisplatin 10 μM treatment through decreasing ATR-CHK1 activity[93]. Caffeine enhanced the cytotoxic effect of cisplatin in G2-arrested Chinese hamster ovary CHO/UV41 cells [94,95]. Protoapigenone (WYC02 and WYC0209) enhanced the A549 and MDA-MB-231 cancer cell sensitivity to cisplatin (4 μmol/ L) by inhibiting the G2-M checkpoint in the concentration of 0.5 and 0.1 μmol/ L, respectively [78]. In the same study, human xenograft MDA-MB-231 tumours in nude mice treated with 2 mg/kg cisplatin combined with 0.2 mg/kg WYC0209 i.p.Qid increase the tumour inhibitory effect more significant than that of cisplatin treatment alone and at the end, result in a decrease in tumour growth in mice [78]. Sch-B (30 μM) decreases cell survival percentage in UV-irradiated A549, HEK293T and HeLa cells by inhibiting S and G2-M phase checkpoints[72].
4. Clinical trials of ATR and CHK1 targets in cancer therapy
As evidenced by preclinical studies, logical incorporation of ATR, CHK1, and WEE1 inhibitors into cancer treatment must be limited to tumor cells with raised RS or in combination with agents which can stimulate RS. As a consequence, ATR, CHK1, and WEE1 blockers might increase cancerous cells responsiveness, potentially increasing therapeutic potential. In accordance with the foregoing, numerous cancers have high concentrations of ATR, CHK1, or WEE1, all of which would make excellent biomarkers candidates [96–98]. As a result of preclinical effectiveness, the bulk of such investigations involve ATR/CHK1/WEE1 antagonists in conjunction with standard radiotherapy and chemotherapy [99].
Two ATR blocker, VX-970 (previously owned by Merck KGaA, Darmstadt, Germany, from Vertex pharmaceuticals) and AZD6738 (Astrazeneca), as well as three novel CHK1 inhibitors, SRA737 (formerly as CCT245737, Sierra Oncology Inc., Vancouver, BC, Canada), MK8776 (known officially as SCH-900776, Merck and Co., Whitehouse Station) and LY2606368 (Prexasertib, Lilly Oncology, Indianapolis, IN, USA) are in the earliest stages of clinical investigation [100,101]. A current analysis of the clinical trials.gov global database reveals 23 active trials enrolling subjects for single individual and combination usage(https://www.clinicaltrials.gov/). Chemotherapy, radiation, and other specialized treatments are included in the combination research. In this section, we examine the minimal clinical data available from trials that have been completed or early outcomes have been discussed at international congresses.
The very first ATR antagonist to enter human anti-cancer therapeutic studies was VX-970 (VE-822), a powerful selective intravenous ATR blocker [102]. VX-970 monotherapy and in conjunction with carboplatin (CP) were explored in this first stage I trial, which included pharmacodynamic (PD) tests. In section A, individual patient cohorts administered VX-970 monotherapy QW; if second grade drug-related complications were reported, 3 + 3 cohorts were started. In part B of the trial, three patient groups received CP on day one (D1) and VX-970 on day 2 and day 9 in 3-week rotations. VX-970 was well tolerated in part A (n = 17), without any dose-limiting toxicities (DLT) observed. Outcomes included an individual with highly pre-treated K-ras wild type metastatic colorectal cancer who has had a full response (RECIST) maintained for less than twenty months. This individual was later confirmed by immunohistochemistry to be completely devoid of ATM. The highest outcome in 5 additional patients was stable disease, with a maximum period of responsiveness of 11 weeks. Variations in concentrations of phosphorylated-CHK1 in matched pre-dose/post-dose tumour samples in 3 patients were used to evaluate ATR suppression. After therapy with VX-970, the level of pCHK1 was reduced by more than 70%. For VX-970 monotherapy, the suggested phase 2 doses (RP2D) was 240 mg/m2 once weekly and 240 mg/m2 bi weekly. In section B, VX-970 were usually effectively tolerated in conjunction with CP, with most toxicities falling into the grade 1–2 range. Six patients had CP, dosage delays or reductions due to neutropenia and/or thrombocytopenia [103]. Clinical evidence supported the toxicity modelling that anticipated 5% WHO Grade 4 neutropenia and 1% thrombocytopenia at the RP2D, which turned out to be VX-970 90 mg/m2 + Carboplatin AUC5. Single individual with a somatic Y220C TP53 mutation and a germline BRCA1 mutation who had platinum-refractory, PARP inhibitor-resistant ovarian cancer had a RECIST limited response for 6 months, and eight additional individuals in part B had stable disease [103]. Following that, phase 1 trials evaluated at VX-970 in conjunction with gemcitabine (Gem) (NCT02157792), with the RP2D being VX-970 210 mg/m2 and Gem 1000 mg/m2, and VX-970 in conjunction with cisplatin (NCT02157792), with anti-tumour responses shown in platinum-refractory/resistant individuals [104,105]. VX-970’s ongoing advancement is unpredictable, as Vertex Pharmaceuticals sold the VX-970 ATR programme to Merck KGaA in late 2016, with Merck KGaA taking complete responsibilities for all research & innovation.
AZD6738 was the first easily accessible ATR blocker to reach clinical studies, and it’s being tested in a range of indications as a single drug and in combination. There have been no preliminary data released to date, and each trial is still recruiting participants [106]. Preliminary findings from CHK1 antagonist trials reveal that this family of medications is well tolerated, with the cardiac dose-limiting toxicity that halted the advancement of AZD7762 being comforting. Preliminary data from CHK1 blocker studies demonstrate that this category of medications is well tolerated, and encouragingly, the cardiac dose-limiting toxicity that halted advancement of AZD7762 has not been documented in studies connecting the more specific CHK1 suppressor, implying an off-target effect [107,108]. MK-8776, a powerful, selective CHK1 blocker that has reached phase 1 clinical trials, is one of these highly specific antagonists. MK-8776 was studied in patients with advanced solid malignancies in a phase 1 trial as a monotherapy and in combination with gemcitabine [109].
LY2603618 is the single known CHK1 inhibitor with published clinical trial outcomes till date. LY2603618 is a competitive CHK1 antagonist that has been tested in two phase 1 trials in conjunction with gemcitabine and has now progressed to phase 2. The overall performance rate, tolerability, and pharmacokinetics (PK) of LY2603618 and pemetrexed in individuals with NSCLC who had progressed ever since prior first-line therapy regimens were evaluated in this phase two trials [110–112]. There are presently 23 clinical studies testing ATR and CHK1 antagonists that have been filed. The field is constantly evolving, and the significant difficulties ahead will be identifying the small number of patients who might benefit from ATR/CHK1 inhibitor monotherapy, as well as determining the best schedule and dosages in conjunction with both conventional and novel chemotherapeutics to minimize toxicity while maximizing responses. The outcomes of the numerous ongoing investigations are anxiously awaited.
5. Future prospects
Although cancer’s hallmarks impede treatment, they can be efficiently used for selective cancer cell targeting to cure the patient eventually. The absence of in vivo and clinical studies and a lack of knowledge of their mechanisms of action make this a topic worthy of future investigation. The toxicity of medications is a significant barrier to chemotherapy in the treatment of cancer patients. Unfortunately, many known anticancer treatments also target multiplying normal cells. The bulk of the phytochemicals investigated, including curcumin, mangiferin, resveratrol, rocaglamide-A, and harmine, elicit G1-S arrest in malignant cells but do not affect the cell division cycle of proliferating healthy cells.
Cancer cells may be sensitized to anticancer therapies such as DNA-damaging methylating/alkylating substances and topoisomerase inhibitors that activate ATR via phytochemicals. Few studies have revealed synthetic lethality of natural chemicals in conjunction with chemo and radiation, and more research is needed. The suppression of DNA repair genes implies that these phytochemicals may assist to overcome medication resistance and offer a solid foundation for undertaking clinical trials with phytochemicals in conjunction with other therapeutic agents. Phytochemicals may be employed as an adjuvant drug for anticancer therapy by suppressing the G2-M checkpoint, particularly for targeting cancer cells resistant to chemo and radiation due to the lack of functioning p53. The majority of the evidence for anticancer action was gained in vitro. Unfortunately, limited solubility, fast elimination, and poor absorption have hampered their use as a medicinal agent. Nanotechnology approaches, liposomes, phytosomes, micelles, and natural adjuvants improved bioavailability significantly [113–115]. Although CHK1 inhibitors have shown promise in preclinical research on chemo- and radio-sensitization, clinical trial outcomes have been less striking. Additionally, both in preclinical research and in clinical trials, p53’s independence has been seen. As a result, it’s important to find additional factors and biomarkers that determine how well CHK1 inhibitors work. Since a lower dose of a CHK1 inhibitor is anticipated to be beneficial if the right individuals were chosen, the most significant advancement in the treatment of cancer with these inhibitors may be the creation of inhibitor-specific patient stratification criteria. In the future, a new generation of CHK1 inhibitors with lower toxicity is also required.ATR-CHK1 might be a new phytochemical target for cancer prevention and therapy. It is critical to do further research on natural chemicals that target ATR-CHK1 to identify new candidate molecules.
6. Conclusion
ATR is a basal kinase that plays a key role in DNA repair, cell cycle control, and apoptosis. It has a wide range of pharmacological functions. Knowing the role of ATR in replication stress and other aspects of the DDR has advanced significantly, although research into the mechanisms of ATR signaling is still ongoing. To sum up, ATR has become a desirable target for cancer therapies, and the ATRi area is fast growing, with a number of early phase clinical trials now in progress. This review has concluded that different compounds isolated from natural products have reported to target the ATR-CHK1 signaling pathway in cancer therapy. Additionally, these compounds have shown to have chemotherapeutic synergistic potential when used in combination with other anticancer drugs. Ideally, this review will trigger interest in natural products targeting ATR-CHK1 and their potential efficacy and safety as cancer lessening agents.(Table. 1).
Table 1.
Natural products targeting ATR/CHK1 signaling pathway as anticancer agent.
| Secondary metabolites | Class of compound | Sources | Cell lines | IC50in vitro | References |
|---|---|---|---|---|---|
| ATR-CHK1 activation | |||||
| Resveratrol | Stilbenoid (Polyphenol) | Vitis vinifera (fruit) | TK6 and WTK1 | 30 μM | [83] |
| Resveratrol | Ovcar-3, PA-1 | 50 μM | [82] | ||
| Resveratrol | SKOV-3 | 30 μM | [82] | ||
| Resveratrol analogue (E)− 8-acetoxy-2-[2-(3,4-diacetoxyphenyl) ethenyl]- quinazoline (8-ADEQ) | HeLa | 8 μM | [84] | ||
| Mangiferin | Flavonoid | Mangifera indica (leaves and bark) | HL-60 | 160 μM | [76] |
| Harmine | Tricyclic b-carboline alkaloid | Peganum harmala (seeds) | Hep3B | 20 μM | [70] |
| Rocaglamide-A (Roc-A) | Flavagline | Aglaia Sp. | HL-60, Jurkat-16, CEM, Molt-4 and DND41; Hut-78, L1236; HepG2 and Huh7; HT-29 and HCT116; PC3; MCF-7 | 50–100 nM | [68] |
| ATR-CHK1 inhibition | |||||
| Kaempferol | Flavonoid | berries, grapefruit, and Ginkgo biloba | HL-60 | 75 μM | [75] |
| Curcumin | Flavonoid | Curcuma longa (rhizome) | HEK293E | 493 nM | [81] |
| Curcumin | Flavonoid | Curcuma longa(rhizome) | HeLa, HT1080, H1299 | 15 μM | [81] |
| Protoapigenone (WYC02) | Flavonoid | Thelypteristorresiana (whole plant) | MDA-MB-231 | 8 μmol/L |
[78] |
| A549 | 12 μmol/L | [78] | |||
| Protoapigenone Synthetic derivative (WYC0209) | Flavonoid | MDA-MB-231 | 2 μmol/L | [78] | |
| A549 | 4 μmol/L | [78] | |||
| AF4 (apple peel flavonoid fraction) | Flavonoid | Malus domestica (peel) | BEAS-2B | 50 μg /mL | [73] |
| Triptolide | Diterpenoid triepoxide | Tripterygium wilfordii (whole plant) | A375. S2 | 30 nM | [74] |
| Caffeine | Alkaloid | Theobroma cacao(seeds) | A549 | 1.1 mM | [71] |
| Schisandrin B | Dibenzocyclooctadiene lignin | Schisandra chinensis (fruits) | A549 | 7.25 μM | [72] |
Human Cell lines: HL-60 =Human Promyelocytic Leukemia / acute myeloid leukemia; Jurkat-16, CEM, Molt-4 and DND41 = acute T cell leukemia; Hut-78 = T lymphoma; L1236 = Hodgkin lymphoma; A375. S2 = malignant melanoma; HEK293E = immortalized human embryonic kidney cells; A549 = lung adenocarcinoma; H1299 = nonsmall-cell lung carcinoma; BEAS-2B = Normal human bronchial epithelial cells; TK6 and WTK1 = B-lymphoblastoid cell lines; MCF-7, MDA-MB-231 = breast adenocarcinoma; Ovcar-3, PA-1, SKOV-3 = ovarian carcinoma; HeLa = human cervical carcinoma; HT1080 = fibrosarcoma; Hep3B, HepG2 and Huh7 = Human hepatocellular carcinoma; HT-29 and HCT116 = colorectal cancer; PC3 =prostate cancer.
Abbreviations:
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia mutated and Rad3 related
- BRCA1
Breast Cancer Gene 1
- cdc25A
cell-division cycle 25A
- CHK1
Checkpoint kinase 1
- DDR
DNA damage response
- DSB
DNA double-strand break
- MDR
Multi Drug Resistance
- P-gp
p-glycoprotein
- RPA
Replication protein A
- RS
replication stress
- ssDNA
single-stranded DNA
- Roc-A
Rocaglamide-A
- Sch-B
Schisandrin B
- WYC02
protoapigenone
- WYC0209
Protoapigenone synthetic derivative
- Rad51
RAD51 Recombinase
- ATR-KD
Ataxia telangiectasia mutated and Rad3 related kinase dead cells
- PARP
Poly ADP ribose polymerase
- CDK1/CDK2
Cyclin-dependent kinase1/ Cyclin-dependent kinase2
- FANCD2
Fanconi anemia complementation group D2
- UV
Ultraviolet
Footnotes
CRediT authorship contribution statement
Salman Ahmed, Waqas Alam, Khalaf F Alsharif, and Ashraf Albrakati have written the initial draft. Michael Aschner has reviewed the initial draft. Luciano Saso and Haroon Khan finalized the article and supervised the overall project.
Conflict of Interest Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
No data was used for the research described in the article.
References
- [1].Bray F, et al. , Global cancer transitions according to the Human Development Index (2008–2030): a population-based study, Lancet Oncol. 13 (8) (2012) 790–801. [DOI] [PubMed] [Google Scholar]
- [2].Mans DR, Rocha AB, Schwartsmann G, Anti-Cancer Drug Discovery and Development in Brazil: Targeted Plant Collection as A Rational Strategy to Acquire Candidate Anti-cancer Compounds, Oxford University Press, 2000, pp. 185–198. [DOI] [PubMed] [Google Scholar]
- [3].Bemis D, et al. , Anti-prostate cancer activity of a β-carboline alkaloid enriched extract from Rauwolfia vomitoria, Int. J. Oncol 29 (5) (2006) 1065–1073. [PubMed] [Google Scholar]
- [4].Ahmed S, et al. , Anticancer potential of furanocoumarins: mechanistic and therapeutic aspects, Int. J. Mol. Sci 21 (2020) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Choudhari AS, et al. , Phytochemicals in cancer treatment: from preclinical studies to clinical practice, Front. Pharmacol 2020. (1614) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ahmed S, et al. , Apoptosis induced by luteolin in breast cancer: mechanistic and therapeutic perspectives, Phytomedicine 59 (2019), 152883. [DOI] [PubMed] [Google Scholar]
- [7].Roy A, A review on the alkaloids an important therapeutic compound from plants, IJPB 3 (2) (2017) 1–9. [Google Scholar]
- [8].Barffour IK, Anti-hepatocellular carcinoma effect of an alkaloidal extract derived from zanthoxylum zanthoxyloides 2019, University of Cape Coast. [Google Scholar]
- [9].Chinonye II, et al. , Phytochemicals and antimicrobial properties of the root and leaf extract of Carica papaya, Int. J. Innov. Res. Dev 5 (8) (2016) 175–179. [Google Scholar]
- [10].Kumar NS, Devi P, The surprising health benefits of papaya seeds: a review, J. Pharmacogn. Phytochem 6 (1) (2017) 424–429. [Google Scholar]
- [11].Lok AS, et al. , Des-γ-carboxy prothrombin and α-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma, Gastroenterology 138 (2) (2010) 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huang R-X, Zhou P-K, DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer, Signal Transduct. Target. Ther 5 (1) (2020) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carusillo A, Mussolino C, DNA damage: from threat to treatment, Cells 9 (7) (2020) 1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ronco C, et al. , ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells, MedChemComm 8 (2) (2016) 295–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rundle S, et al. , Targeting the ATR-CHK1 axis in cancer therapy, Cancers 9 (5) (2017) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Panagopoulos A, Altmeyer M, The hammer and the dance of cell cycle control, Trends Biochem. Sci 46 (4) (2021) 301–314. [DOI] [PubMed] [Google Scholar]
- [17].Matthews HK, Bertoli C, de Bruin RAM, Cell cycle control in cancer, Nat. Rev. Mol. Cell Biol (2021). [DOI] [PubMed] [Google Scholar]
- [18].Weber AM, Ryan AJ, ATM and ATR as therapeutic targets in cancer, Pharmacol. Ther 149 (2015) 124–138. [DOI] [PubMed] [Google Scholar]
- [19].Benada J, Macurek L, Targeting the checkpoint to kill cancer cells, Biomolecules 5 (3) (2015) 1912–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Smith J, et al. , Chapter 3 - The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer, in: Vande Woude GF, Klein G (Eds.), Advances in Cancer Research, Academic Press, 2010, pp. 73–112. [DOI] [PubMed] [Google Scholar]
- [21].Smith HL, et al. , DNA damage checkpoint kinases in cancer, Expert Rev. Mol. Med 22 (2020), e2. [DOI] [PubMed] [Google Scholar]
- [22].Qiu Z, Oleinick NL, Zhang J, ATR/CHK1 inhibitors and cancer therapy, Radiother. Oncol 126 (3) (2018) 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yazinski SA, Zou L, Functions, regulation, and therapeutic implications of the ATR checkpoint pathway, Annu. Rev. Genet (2016) 50. [DOI] [PubMed] [Google Scholar]
- [24].Karnitz LM, Zou L, Molecular pathways: targeting ATR in cancer therapy, Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res 21 (21) (2015) 4780–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gorecki L, Andrs M, Korabecny J, Clinical candidates targeting the ATR–CHK1–WEE1 axis in cancer, Cancers 13 (2021) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saldivar JC, Cortez D, Cimprich KA, The essential kinase ATR: ensuring faithful duplication of a challenging genome, Nat. Rev. Mol. Cell Biol 18 (10) (2017) 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Y, Hunter T, Roles of Chk1 in cell biology and cancer therapy, Int. J. Cancer 134 (5) (2014) 1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bradbury A, et al. , Targeting ATR as cancer therapy: a new era for synthetic lethality and synergistic combinations? Pharmacol. Ther 207 (2020), 107450. [DOI] [PubMed] [Google Scholar]
- [29].Sun X, et al. , ATR kinase activity promotes antibody class switch recombination in B cells through cell cycle regulation without suppressing DSB resection and microhomology usage, J. Leukoc. Biol 110 (6) (2021) 1101–1112. [DOI] [PubMed] [Google Scholar]
- [30].Takai H, et al. , Aberrant cell cycle checkpoint function and early embryonic death in Chk1 −/− mice, Genes Dev 14 (12) (2000) 1439–1447. [PMC free article] [PubMed] [Google Scholar]
- [31].Brown EJ, Baltimore D, ATR disruption leads to chromosomal fragmentation and early embryonic lethality, Genes Dev 14 (4) (2000) 397–402. [PMC free article] [PubMed] [Google Scholar]
- [32].Cliby WA, et al. , Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. The, EMBO J 17 (1) (1998) 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zeman MK, Cimprich KA, Causes and consequences of replication stress, Nat. Cell Biol 16 (1) (2014) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].MacDougall CA, et al. , The structural determinants of checkpoint activation, Genes Dev 21 (8) (2007) 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Maŕechal A, Zou L, DNA damage sensing by the ATM and ATR kinases, Cold Spring Harb. Perspect. Biol 5 (9) (2013) a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cortez D, et al. , ATR and ATRIP: partners in checkpoint signaling, Science 294 (5547) (2001) 1713–1716. [DOI] [PubMed] [Google Scholar]
- [37].Ball HL, et al. , Function of a conserved checkpoint recruitment domain in ATRIP proteins, Mol. Cell. Biol 27 (9) (2007) 3367–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mordes DA, et al. , TopBP1 activates ATR through ATRIP and a PIKK regulatory domain, Genes Dev 22 (11) (2008) 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Garcia V, Furuya K, Carr AM, Identification and functional analysis of TopBP1 and its homologs, DNA Repair 4 (11) (2005) 1227–1239. [DOI] [PubMed] [Google Scholar]
- [40].Hussain RN, Radiation Biology of Uveal Melanoma 2020: The University of Liverpool; (United Kingdom: ). [Google Scholar]
- [41].Shiotani B, Zou L, ATR signaling at a glance, J. Cell Sci 122 (3) (2009) 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Walworth NC, Bernards R, rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint, Science 271 (5247) (1996) 353–356. [DOI] [PubMed] [Google Scholar]
- [43].Liu Q, et al. , Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint, Genes Dev 14 (12) (2000) 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- [44].Liu S, et al. , Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation, Mol. Cell. Biol 26 (16) (2006) 6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kumagai A, Dunphy WG, Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts, Mol. Cell 6 (4) (2000) 839–849. [DOI] [PubMed] [Google Scholar]
- [46].Smits VA, Gillespie DA, DNA damage control: regulation and functions of checkpoint kinase 1, FEBS J 282 (19) (2015) 3681–3692. [DOI] [PubMed] [Google Scholar]
- [47].Sørensen CS, Syljuåsen RG, Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication, Nucleic Acids Res 40 (2) (2012) 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen M-S, Ryan CE, Piwnica-Worms H, Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14–3-3 binding, Mol. Cell. Biol 23 (21) (2003) 7488–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dai Y, Grant S, New insights into checkpoint kinase 1 in the DNA damage response signaling network, Clin. Cancer Res 16 (2) (2010) 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lee J, Kumagai A, Dunphy WG, Positive regulation of Wee1 by Chk1 and 14–3-3 proteins, Mol. Biol. Cell 12 (3) (2001) 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cimprich KA, Cortez D, ATR: an essential regulator of genome integrity, Nat. Rev. Mol. Cell Biol 9 (8) (2008) 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nam EA, Cortez D, ATR signalling: more than meeting at the fork, Biochem. J 436 (3) (2011) 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sørensen CS, et al. , Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A, Cancer Cell 3 (3) (2003) 247–258. [DOI] [PubMed] [Google Scholar]
- [54].Paulsen RD, Cimprich KA, The ATR pathway: fine-tuning the fork, DNA Repair 6 (7) (2007) 953–966. [DOI] [PubMed] [Google Scholar]
- [55].Shigechi T, et al. , ATR–ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway, Cancer Res 72 (5) (2012) 1149–1156. [DOI] [PubMed] [Google Scholar]
- [56].Wu X, et al. , ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation, Oncogene 26 (5) (2007) 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tibbetts RS, et al. , Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress, Genes Dev 14 (23) (2000) 2989–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sørensen CS, et al. , The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair, Nat. Cell Biol 7 (2) (2005) 195–201. [DOI] [PubMed] [Google Scholar]
- [59].Peasland A, et al. , Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines, Br. J. Cancer 105 (3) (2011) 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Parsels LA, et al. , Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells, Mol. Cancer Ther 8 (1) (2009) 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Buisson R, et al. , Coupling of homologous recombination and the checkpoint by ATR, Mol. Cell 65 (2) (2017) 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ma CX, Janetka JW, Piwnica-Worms H, Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics, Trends Mol. Med 17 (2) (2011) 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vance S, et al. , Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1, Cell Cycle 10 (24) (2011) 4321–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dent P, et al. , CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle, Mol. Interv 11 (2) (2011) 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bunch RT, Eastman A, Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor, Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res 2 (5) (1996) 791–797. [PubMed] [Google Scholar]
- [66].Matthews DJ, et al. , Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo, Cell Cycle 6 (1) (2007) 104–110. [DOI] [PubMed] [Google Scholar]
- [67].Sha S-K, et al. , Cell cycle phenotype-based optimization of G2-abrogating peptides yields CBP501 with a unique mechanism of action at the G2 checkpoint, Mol. Cancer Ther 6 (1) (2007) 147–153. [DOI] [PubMed] [Google Scholar]
- [68].Neumann J, et al. , The natural anticancer compound rocaglamide selectively inhibits the G1-S-phase transition in cancer cells through the ATM/ATR-mediated Chk1/2 cell cycle checkpoints, Int. J. Cancer 134 (8) (2014) 1991–2002. [DOI] [PubMed] [Google Scholar]
- [69].Shen T, Huang S, The role of Cdc25A in the regulation of cell proliferation and apoptosis, Anti-Cancer Agents Med. Chem 12 (6) (2012) 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang L, et al. , Harmine suppresses homologous recombination repair and inhibits proliferation of hepatoma cells, Cancer Biol. Ther 16 (11) (2015) 1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sarkaria JN, et al. , Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine, Cancer Res 59 (17) (1999) 4375–4382. [PubMed] [Google Scholar]
- [72].Nishida H, et al. , Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response, Nucleic Acids Res 37 (17) (2009) 5678–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].George VC and Rupasinghe H, Apple flavonoids suppress carcinogen-induced DNA damage in normal human bronchial epithelial cells. Oxidative Medicine and Cellular Longevity, 2017 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chueh F-S, et al. , Triptolide induced DNA damage in A375. S2 human malignant melanoma cells is mediated via reduction of DNA repair genes, Oncol. Rep 29 (2) (2013) 613–618. [DOI] [PubMed] [Google Scholar]
- [75].Wu L-Y, et al. , Kaempferol induces DNA damage and inhibits DNA repair associated protein expressions in human promyelocytic Leukemia HL-60 cells, Am. J. Chin. Med 43 (02) (2015) 365–382. [DOI] [PubMed] [Google Scholar]
- [76].Peng Z, et al. , Mangiferin induces cell cycle arrest at G2/M phase through ATR-Chk1 pathway in HL-60 leukemia cells, Genet Mol. Res 14 (2) (2015) 4989–5002. [DOI] [PubMed] [Google Scholar]
- [77].Morozkina SN, et al. , Mangiferin as new potential anti-cancer agent and mangiferin-integrated polymer systems—a novel research direction, Biomolecules 11 (1) (2021) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wang H-C, et al. , Inhibition of ATR-dependent signaling by protoapigenone and its derivative sensitizes cancer cells to interstrand cross-link–generating agents in vitro and in vivo, Mol. Cancer Ther 11 (7) (2012) 1443–1453. [DOI] [PubMed] [Google Scholar]
- [79].Tong R, et al. , Curcumin-induced DNA demethylation in human gastric cancer cells is mediated by the DNA-damage response pathway, Oxid. Med. Cell. Longev 2020 (2020) 2543504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sharifi-Rad J, et al. , Turmeric and its major compound curcumin on health: bioactive effects and safety profiles for food, pharmaceutical, biotechnological and medicinal applications, Biotechnol. Med. Appl. Front. Pharmacol 11 (2020), 01021–01021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ogiwara H, et al. , Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor, Carcinogenesis 34 (11) (2013) 2486–2497. [DOI] [PubMed] [Google Scholar]
- [82].Tyagi A, et al. , Resveratrol causes Cdc2-tyr15 phosphorylation via ATM/ ATR–Chk1/2–Cdc25C pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells, Carcinogenesis 26 (11) (2005) 1978–1987. [DOI] [PubMed] [Google Scholar]
- [83].Gatz SA, et al. , Resveratrol modulates DNA double-strand break repair pathways in an ATM/ATR–p53- and –Nbs1-dependent manner, Carcinogenesis 29 (3) (2008) 519–527. [DOI] [PubMed] [Google Scholar]
- [84].Kim J-Y, et al. , Resveratrol analogue (E)-8-acetoxy-2-[2-(3, 4-diacetoxyphenyl) ethenyl]-quinazoline induces G2/M cell cycle arrest through the activation of ATM/ATR in human cervical carcinoma HeLa cells, Oncol. Rep 33 (5) (2015) 2639–2647. [DOI] [PubMed] [Google Scholar]
- [85].Gachechiladze M, et al. , RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies, Int. J. Cancer 141 (7) (2017) 1286–1294. [DOI] [PubMed] [Google Scholar]
- [86].Raimundo L, Calheiros J, Saraiva L, Exploiting DNA damage repair in precision cancer therapy: BRCA1 as a prime therapeutic target, Cancers 13 (14) (2021) 3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Leon-Galicia I, et al. , Resveratrol induces downregulation of DNA repair genes in MCF-7 human breast cancer cells, Eur. J. Cancer Prev 22 (1) (2013) 11–20. [DOI] [PubMed] [Google Scholar]
- [88].Wang H-C, et al. , Inhibition of ATR-dependent signaling by protoapigenone and its derivative sensitizes cancer cells to interstrand cross-link–generating agents in vitro and in vivoinhibition of ATR and FANCD2 activation by natural products, Mol. Cancer Ther 11 (7) (2012) 1443–1453. [DOI] [PubMed] [Google Scholar]
- [89].Li C-C, et al. , ATR-Chk1 signaling inhibition as a therapeutic strategy to enhance cisplatin chemosensitivity in urothelial bladder cancer, Oncotarget 7 (2) (2016) 1947–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Nasser MI, et al. , A comprehensive review on schisandrin B and its biological properties, Oxid. Med. Cell. Longev 2020 (2020) 2172740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Qiangrong P, et al. , Schisandrin B—A novel inhibitor of P-glycoprotein, Biochem. Biophys. Res. Commun 335 (2) (2005) 406–411. [DOI] [PubMed] [Google Scholar]
- [92].Florea A-M, Büsselberg D, Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects, Cancers 3 (1) (2011) 1351–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang G, et al. , The effect of caffeine on cisplatin-induced apoptosis of lung cancer cells, Exp. Hematol. Oncol 4 (2015), 5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Demarcq C, et al. , The role of cell cycle progression in cisplatin-induced apoptosis in Chinese hamster ovary cells. [PubMed]
- [95].Yazlovitskaya EM, Persons DL, Inhibition of cisplatin-induced ATR activity and enhanced sensitivity to cisplatin, Anticancer Res 23 (3B) (2003) 2275–2279. [PubMed] [Google Scholar]
- [96].Abdel-Fatah TM, et al. , Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer, Mol. Oncol 9 (3) (2015) 569–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ruiz S, et al. , A genome-wide CRISPR screen identifies CDC25A as a determinant of sensitivity to ATR inhibitors, Mol. Cell 62 (2) (2016) 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Music D, et al. , Expression and prognostic value of the WEE1 kinase in gliomas, J. Neuro-Oncol 127 (2) (2016) 381–389. [DOI] [PubMed] [Google Scholar]
- [99].Gorecki L, Andrs M, Korabecny J, Clinical candidates targeting the ATR–CHK1–WEE1 axis in cancer, Cancers 13 (4) (2021) 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Guzi TJ, et al. , Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening, Mol. Cancer Ther 10 (4) (2011) 591–602. [DOI] [PubMed] [Google Scholar]
- [101].Matthews TP, Jones AM, Collins I, Structure-based design, discovery and development of checkpoint kinase inhibitors as potential anticancer therapies, Expert Opin. Drug Discov 8 (6) (2013) 621–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Reaper PM, et al. , Selective killing of ATM-or p53-deficient cancer cells through inhibition of ATR, Nat. Chem. Biol 7 (7) (2011) 428–430. [DOI] [PubMed] [Google Scholar]
- [103].O’Carrigan B, et al. , Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies 2016, American Society of Clinical Oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Plummer ER, et al. , Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792). 2016, American Society of Clinical Oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Shapiro G, et al. , Abstract CT012: phase 1 trial of first-in-class ATR inhibitor VX-970 in combination with cisplatin (Cis) in patients (pts) with advanced solid tumors (NCT02157792). 2016, AACR. [Google Scholar]
- [106].Dillon M, et al. PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. in ASCO 2016: ASCO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sausville E, et al. , Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors, Cancer Chemother. Pharmacol 73 (3) (2014) 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Manic G, et al. , Trial watch: targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy, Mol. Cell. Oncol 2 (4) (2015) e1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Daud AI, et al. , Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors, J. Clin. Oncol 33 (9) (2015) 1060–1066. [DOI] [PubMed] [Google Scholar]
- [110].Doi T, et al. , Phase I study of LY2603618, a CHK1 inhibitor, in combination with gemcitabine in Japanese patients with solid tumors, Anti-Cancer Drugs 26 (10) (2015) 1043–1053. [DOI] [PubMed] [Google Scholar]
- [111].Calvo E, et al. , Phase I study of CHK1 inhibitor LY2603618 in combination with gemcitabine in patients with solid tumors, Oncology 91 (5) (2016) 251–260. [DOI] [PubMed] [Google Scholar]
- [112].Scagliotti G, et al. , Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer, Investig. N. Drugs 34 (5) (2016) 625–635. [DOI] [PubMed] [Google Scholar]
- [113].Nocito MC, et al. , Antitumoral activities of curcumin and recent advances to improve its oral bioavailability, Biomedicines 9 (10) (2021) 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Morozkina SN, et al. , Mangiferin as new potential anti-cancer agent and mangiferin-integrated polymer systems-a novel research direction, Biomolecules 11 (1) (2021) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Annaji M, et al. , Resveratrol-loaded nanomedicines for cancer applications, Cancer Rep 4 (3) (2021), e1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.