

SUMMARY
CRISPR-Cas systems must enact robust immunity against foreign genetic material without inducing cytotoxic autoimmunity. For type VI systems that use Cas13 nucleases and recognize RNA targets, immune activation requires extensive CRISPR RNA (crRNA) guide:target complementarity and a target-flanking motif. Here we report a third requirement shaping the immune response: expression of the target transcript exceeding a threshold. We found that endogenous non-essential transcripts targeted by crRNAs rarely elicited autoimmunity. Instead, autoimmune induction required over-expressing targeted transcripts above a threshold. A genome-wide screen confirmed target expression levels as a global determinant of cytotoxic autoimmunity and revealed that this threshold shifts with each guide:target pair. This threshold further ensured defense against a lytic bacteriophage yet allowed tolerance of a targeted beneficial gene expressed from an invading plasmid. These findings establish target expression levels as an additional criterion for immune defense by RNA-targeting CRISPR-Cas systems, preventing autoimmunity and distinguishing pathogenic and benign invaders.
Graphical Abstract

eTOC Blurb
Vialetto et al. report that immune induction by CRISPR-Cas13 systems intimately depends on the levels of the target transcript. This expression threshold allows the immune system to tolerate self-targeting and harmless infections yet robustly combat rapidly replicating invaders. The threshold also holds implications for programmable RNA silencing with Cas13.
INTRODUCTION
All cellular immune systems face the challenge of differentiating foreign entities from the host. Failure to recognize features associated with an invader exposes the host to a potentially fatal infection, while failure to ignore similar features in the host can trigger a catastrophic autoimmune response. Elucidating how immune systems make these decisions and the consequences of errors is critical not only for treating infectious and autoimmune diseases in higher eukaryotes but also for understanding the physiology and evolution of single-cell microbes under constant assault by mobile genetic elements and bacteriophages (Goldberg and Marraffini, 2015; Theofilopoulos, Kono and Baccala, 2017; Hampton, Watson and Fineran, 2020).
CRISPR-Cas systems, the only known adaptive immune systems in bacteria and archaea, face these same challenges (Barrangou et al., 2007; Makarova et al., 2015, 2020). These systems store fragments of invading genetic material as spacers situated between conserved repeats in CRISPR arrays (Barrangou et al., 2007; Jackson et al., 2017). To stem an infection, the arrays are transcribed and processed into individual CRISPR RNAs (crRNAs) that direct the system’s effector nucleases to complementary genetic sequences (Marraffini and Sontheimer, 2010a; van der Oost et al., 2014; Charpentier et al., 2015; Hille et al., 2018). The nature of the target genetic material (i.e., DNA or RNA) and the consequence of recognizing a target sequence vary widely across systems. For example, type II CRISPR-Cas systems recognize target sequences within double-stranded DNA, prompting the Cas9 effector nuclease to cleave the DNA target (Gasiunas et al., 2012; Jinek et al., 2012). Separately, type VI CRISPR-Cas systems recognize target sequences within RNA, prompting the Cas13 effector nuclease to non-specifically cleave cellular RNAs (Abudayyeh et al., 2016; East-Seletsky et al., 2016). Widespread RNA degradation induces growth arrest--a state called cellular dormancy--that prevents the replication and dissemination of the invader (Abudayyeh et al., 2016; Meeske, Nakandakari-Higa and Marraffini, 2019).
Beyond recognition of sequences complementary to the crRNA guide, Cas effector nucleases normally require an additional factor to undergo activation: an appropriate sequence flanking the target. This flanking sequence is called a protospacer-adjacent motif (PAM) for DNA-targeting systems as reviewed previously (Leenay and Beisel, 2017) and a protospacer-flanking sequence (PFS) for RNA-targeting systems (Marraffini and Sontheimer, 2010b; Abudayyeh et al., 2016; Meeske and Marraffini, 2018). The PAM is specifically recognized by the effector nuclease as part of target identification, whereas the PFS avoids extensive base pairing with the repeat-derived portion of the crRNA that otherwise blocks nuclease activation (Meeske and Marraffini, 2018). In either case, the flanking sequence allows the effector nuclease to differentiate the targeted invader from the spacer in the CRISPR array (or the antisense transcript of the CRISPR array for RNA-targeting systems), which is perfectly complementary to the crRNA guide. However, in the infrequent instance of a spacer being acquired from a chromosomal sequence, the genomic target would fulfill both requirements for nuclease activation, killing the cell or driving either mutation of the target or inactivation of the CRISPR-Cas system (Stern et al., 2010; Vercoe et al., 2013).
Here, we report that RNA-targeting type VI CRISPR-Cas systems take into account a third criterion for activation of a full immune response: minimal expression of the target transcript. This expression threshold is higher than most cellular transcripts under our experimental conditions, allowing the co-existence of a targeted endogenous transcript and an active immune system. The threshold also allows the immune system to tolerate a passive invader such as a plasmid yet mount a robust immune response to an actively replicating invader such as a lytic phage. Finally, the threshold may help explain why applying Cas13 in eukaryotes yields programmable gene silencing in some contexts (Abudayyeh et al., 2017; Cox et al., 2017) and non-specific RNA degradation in others (Wang et al., 2019; Buchman et al., 2020; Özcan et al., 2021; Xu et al., 2021).
RESULTS
CRISPR-Cas13 fails to induce dormancy when targeting selected endogenous transcripts
Cas13-induced immunity has been principally assessed through targeting transcripts expressed from plasmids or phages or from the genome with an inducible promoter (Abudayyeh et al., 2016, 2017; Meeske and Marraffini, 2018; Meeske, Nakandakari-Higa and Marraffini, 2019; Kiga et al., 2020). What has remained less clear is the impact of targeting native transcripts, particularly given that targeting such transcripts in eukaryotes has been largely associated with targeted gene silencing (Abudayyeh et al., 2017; Cox et al., 2017). To address this gap, we employed the Cas13 nuclease from the type VI-A CRISPR-Cas system in Leptotrichia shahii (LshCas13a) (Abudayyeh et al., 2016; Liu, Li, Wang, et al., 2017; Watanabe et al., 2019) in Escherichia coli as a simple model, paralleling prior studies showing that this nuclease could drive collateral RNA cleavage and dormancy (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Liu, Li, Ma, et al., 2017). We first verified Cas13a activity by targeting either of two different sites within a synthetic transcript constitutively expressed from a plasmid, which resulted in a 2,800-fold and 460-fold reduction in the transformation efficiency of the respective crRNA-encoding plasmid versus a non-targeting crRNA plasmid (Fig. 1A–B). To verify that the reduction in transformation depended on Cas13a endonuclease activity, we inactivated the HEPN domain of the nuclease using a previously characterized point mutation (R1278A) that abolishes RNA degradation (Abudayyeh et al., 2016). The resulting catalytically-dead Cas13a (dCas13a) yielded similar colony counts between targeting and non-targeting crRNAs, demonstrating that Cas13a RNase activity is responsible for the observed reduction in plasmid transformation (Fig. 1B). We further confirmed that targeting the synthetic transcript induces collateral activity based on the degradation of total RNA upon target expression (Fig. S1A–B).
Figure 1.

CRISPR-Cas13a is functional in E. coli yet fails to confer autoimmunity against selected endogenous targets. See also Fig. S1.
(A) Experimental setup for the plasmid transformation assay. A crRNA plasmid targeting either a plasmid-encoded or genomically-encoded transcript is transformed into E. coli cells expressing the L. shahii Cas13a (LshCas13a). The relative number of transformants compared to transformation of a non-targeting crRNA plasmid are quantified.
(B) Impact of targeting a plasmid-encoded transcript with LshCas13a. dLshCas13a: catalytically-dead Cas13a. Two different regions were targeted (T1, T2) within a transcript encoding a portion of the mRFP1 gene. The transcript was expressed from the strong constitutive promoter P8. Fold-reduction was calculated as the ratio of transformants with the non-targeting (NT) and targeting (T) crRNA plasmids.
(C) Impact of targeting different genomically-encoded transcripts with LshCas13a. Transcripts were targeted from genes considered essential or non-essential in E. coli MG1655 under standard growth conditions. gapA1 and gapA2 represent two different target locations within the gapA transcript.
Bars represent the mean of triplicate independent experiments (n = 3). Statistical significance was calculated by comparing the transformation fold-reduction for LshCas13a and dLshCas13a. ***: p < 0.001. **: p < 0.01. *: p < 0.05. ns: not significant.
After validating the targeting activity of LshCas13a in E. coli, we proceeded to assess the consequences of self-targeting. Previous studies of Cas13a in bacteria suggested that targeting leads to dormancy as long as the target transcript is present (Abudayyeh et al., 2016; Meeske and Marraffini, 2018), so we expected to observe cytotoxic autoimmunity for all the targets expressed under standard growth conditions. To assess self-targeting, we designed 12 crRNAs targeting mRNAs encoded by essential and non-essential genes in E. coli following current guide design rules (Meeske and Marraffini, 2018; Wessels et al., 2020) and repeated the transformation assay (Fig. 1A). Surprisingly, targeting yielded a measurable reduction in colony counts for only two targets: one in the essential gapA mRNA and another in the non-essential yfaP mRNA (Fig. 1C). Repeating the assay with dCas13a or without the nuclease revealed that the transformation reduction with the yfaP-targeting crRNA was due to cytotoxicity associated with the crRNA itself (Figs. 1C and S1C). For targets exhibiting negligible reduction in plasmid transformation (e.g. IrhA, rdgC and soxS), we observed no discernable degradation of total RNA and at most a modest reduction in growth in liquid culture for the rdgC-targeting crRNA (Fig. S1D–E). These results suggest other factors are at work that prevent cytotoxic autoimmunity.
Boosting target expression above a threshold enables Cas13-based autoimmunity
Given that transcript levels are generally lower when expressed from the chromosome versus a multicopy plasmid, we asked if levels of the target transcript play a role in the induction of Cas13 autoimmunity. We constitutively expressed a 72-nucleotide (nt) fragment of selected mRNA targets on a plasmid (Fig. 2A). Expressing these fragments without disrupting the endogenous locus resulted in a 280-fold to 1,000-fold reduction in colony counts compared to the non-targeting control, indicating that target expression levels impact the autoimmune response. The large reduction in colony counts was lost when using dCas13a (Fig. 2A), confirming the involvement of RNA cleavage.
Figure 2.

Cas13a-induced immunity requires target expression to exceed a threshold. See also Fig. S2.
(A) Impact of over-expressing genomic targets in E. coli. Targets include the sequence complementary to the crRNA guide along with the 20 nucleotides upstream and downstream and are expressed under the strong constitutive promoter P8.
(B) Quantified strength of different constitutive promoters in E. coli. Promoter strength was measured based on the fluorescence produced from a downstream gfp reporter.
(C) Impact of targeting the plasmid-encoded soxS transcript expressed from different constitutive promoters by Cas13a in E. coli.
(D) Impact of targeting the plasmid-encoded soxS transcript expressed from different constitutive promoters by Cas13a in E. coli. The genomic copy of soxS was intact in the E. coli strain.
Bars represent the mean of triplicate independent experiments (n = 3). Statistical significance in A was calculated by comparing the transformation fold-reduction for LshCas13a and dLshCas13a. Statistical significance in C and D was calculated by comparing the transformation fold-reduction to that of the weakest P1 promoter. ***: p < 0.001. **: p < 0.01. p < 0.05. ns: not significant.
Our observations suggested that a certain target expression level might need to be reached to elicit a cytotoxic autoimmune response. To explore this possibility, we expressed the plasmid-encoded synthetic transcript targeted at two locations (T1, T2) under eight different constitutive promoters (P1 to P8) (Table S1) exhibiting varying expression strengths (Fig. 2B–C). While the three weakest promoters yielded no reduction in transformation efficiency compared to the non-targeting control, we observed a strong reduction for the remaining promoters (~1,100-fold for T1, ~60-fold for T2). No reduction was observed with dCas13a for any of the promoters (Fig. S2). The transition between low and high transformation efficiencies was remarkably sharp and occurred for both targets between the same two promoters separated by only a 5.8-fold difference in transcriptional activity (Fig. 2C). We performed a similar analysis with the full-length soxS mRNA using the same set of constitutive promoters (Fig. 2D), where the transformation reduction occurred only with the strongest promoter. These results show that low expression of a target transcript can prevent cytotoxic autoimmunity.
A genome-wide screen establishes target expression levels as the principal determinant of cytotoxic autoimmunity
Thus far, our observations were based on a small number of endogenous targets. In order to determine to what extent an expression threshold influences targeting across the transcriptome and whether other factors (e.g., target position, sequence context) also impact the outcome of targeting, we performed a genome-wide screen. We designed a library of 25,597 crRNAs targeting all chromosomally-encoded mRNAs and rRNAs in E. coli (Fig. 3A). The guide sequences were selected following current design rules (e.g., PFS lacking complementarity to crRNA repeat tag) to reduce the inclusion of low-efficiency guides, and the targets were spaced across the entire length of each coding region (or transcribed region in the case of rRNAs). The library included 400 randomized guides as non-targeting controls lacking complementarity to any endogenous transcripts. We then transformed the crRNA library into E. coli cells with or without LshCas13a and cultured the transformed cells with antibiotic selection to deplete guides causing collateral activity and dormancy. Short-read sequencing was finally applied to measure the depletion of each guide compared to the no-LshCash13a control cultured under the same conditions (Fig. 3B). Under this setup, highly active guides would be heavily depleted within the library, while poorly active guides would be minimally depleted.
Figure 3.
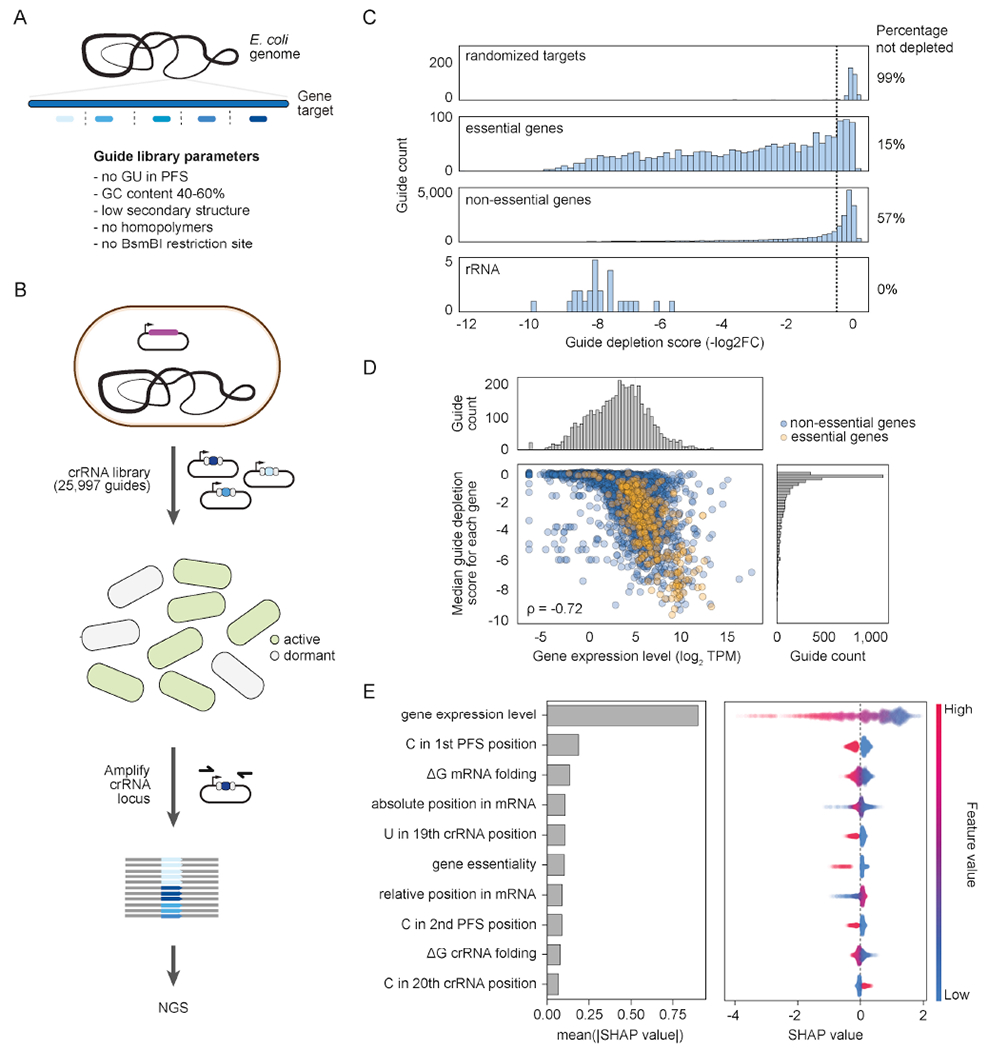
A genome-wide CRISPR-Cas13a screen reveals target expression levels as the main determinant of cytotoxic autoimmunity. See also Figs. S3 and S4.
(A) Design of crRNA guide library. Guide selection accounted for standard rules lending to efficient targeting and spanning the entire coding region of each target gene. Other parameters (i.e., homopolymers, BsmBI sites) were included to facilitate library synthesis and cloning. The resulting library included 25,470 guides targeting protein-coding genes, 127 guides targeting rRNAs, and 400 randomized guides as negative controls.
(B) Workflow for library screening. As part of the screen, cells with or without a LshCas13a plasmid (purple) are transformed with the crRNA plasmid library and cultured while selecting all present plasmids. Guide depletion is determined in comparison to the same workflow with the no-LshCas13a control.
(C) Distribution of depletion scores for different groups of guides within the library. The cutoff for no fitness defect is based on the range of depletion scores for a set of randomized guides.
(D) Correlation between median guide depletion score and the expression levels of the target gene. Expression levels were measured by RNA-seq analysis with E. coli cells harboring the no-LshCas13a control and subjected to the library workflow to a turbidity of ABS600 ≈ 0.5. Values for transcript levels and guide depletion are the average of duplicate independent experiments and screens, respectively, ρ: Spearman coefficient. See Fig. S3B for the correlation for a turbidity of ABS600 ≈ 0.8.
(E) SHAP values for the strongest predictors of guide depletion from the library. The left barplot indicates the average absolute contribution of each feature to the predicted depletion values, while the right beeswarm plot shows the impact of each feature on each individual prediction.
Initial analysis revealed that the extent of depletion depended on whether the target was associated with an essential or non-essential gene. This observation is in line with the idea that target transcript cleavage of an essential gene would be expected to cause a fitness defect even in the absence of widespread collateral cleavage. Indeed, compared to the set of randomized guides, 85% of guides targeting essential genes were depleted versus only 43% of guides targeting non-essential genes. The median depletion was also higher for guides targeting essential (2.9-fold) versus non-essential (0.3-fold) genes (Fig. 3C). The screen was validated by testing individual highly depleted guides using the transformation assay (Fig. S3A). The limited depletion of guides targeting non-essential genes further shows that guides, even when designed following standard rules, infrequently lead to widespread collateral cleavage and dormancy.
We also noticed that guides targeting rRNAs, the highest expressed RNAs in the cell, were strongly and consistently depleted in the library (Fig. 3C). We therefore analyzed in more detail how transcript levels contributed to guide depletion across the library. After performing transcriptomics analyses in middle and late exponential growth phases, we found a clear correlation between guide depletion and the levels of the targeted transcript (Figs. 3D and S3B). This correlation was stronger for transcript levels in middle exponential growth (Spearman coefficient = 0.72) than for late exponential growth (Spearman coefficient = 0.68), possibly because middle exponential growth dominates the screen. The observed correlation applied to both essential and non-essential genes (Fig. S3C). Here, guide depletion was consistently stronger for essential than non-essential genes with similar expression levels, further supporting the added growth defect when silencing essential genes (Fig. S3C–D). Translational strength predicted using the ribosome-binding site (RBS) calculator showed minimal correlation (Salis, 2011) (Fig. S3E), indicating that protection of the mRNA by translating ribosomes does not account for differences in guide depletion.
In order to determine which features of the guide:target pair most impact induction of cytotoxic autoimmunity, we subjected the dataset to machine learning (Fig. 3E). The resulting SHAP values (Lundberg et al., 2020) revealed that transcript levels were the strongest predictor of guide depletion. In contrast, gene essentiality had a modest effect on the predicted depletion for the average guide, which can be attributed to the small number of essential genes in E. coli. We also identified nucleotide preferences within the crRNA guide or PFS with predictive value, such as a C in the 1st or 2nd PFS position or a U in the 19th crRNA guide position (Figs. 3E and S4A–D). The predictive factors were not captured in an existing Cas13d design tool based on experiments in human cell culture (Wessels et al., 2020), which was unable to predict depletion scores for our bacterial experiments with Cas13a (Fig. S4E). Together, our results demonstrate that most self-targeting guides do not activate cytotoxic autoimmunity due to insufficient target expression, at least under our experimental conditions.
The target expression threshold depends on the selected guide
Although transcript levels were an important predictor of cytotoxic autoimmunity, we also found that guide features impacted the response. For example, depletion of the highly expressed tolB transcript (Fig. 4A), which was targeted by nine different guides within the library, varied between minimal depletion to depletion paralleling that of some guides targeting rRNAs (Figs. 3C and 4B). We tested each guide individually in the transformation assay and observed a strong correlation between the reduction in colony counts and the depletion score from the screen (Spearman coefficient = −0.87). Furthermore, some of the guides yielded an insignificant reduction in colony counts compared to the non-targeting control, underscoring the variability in the extent of autoimmunity for this one highly expressed mRNA depending on the guide sequence.
Figure 4.
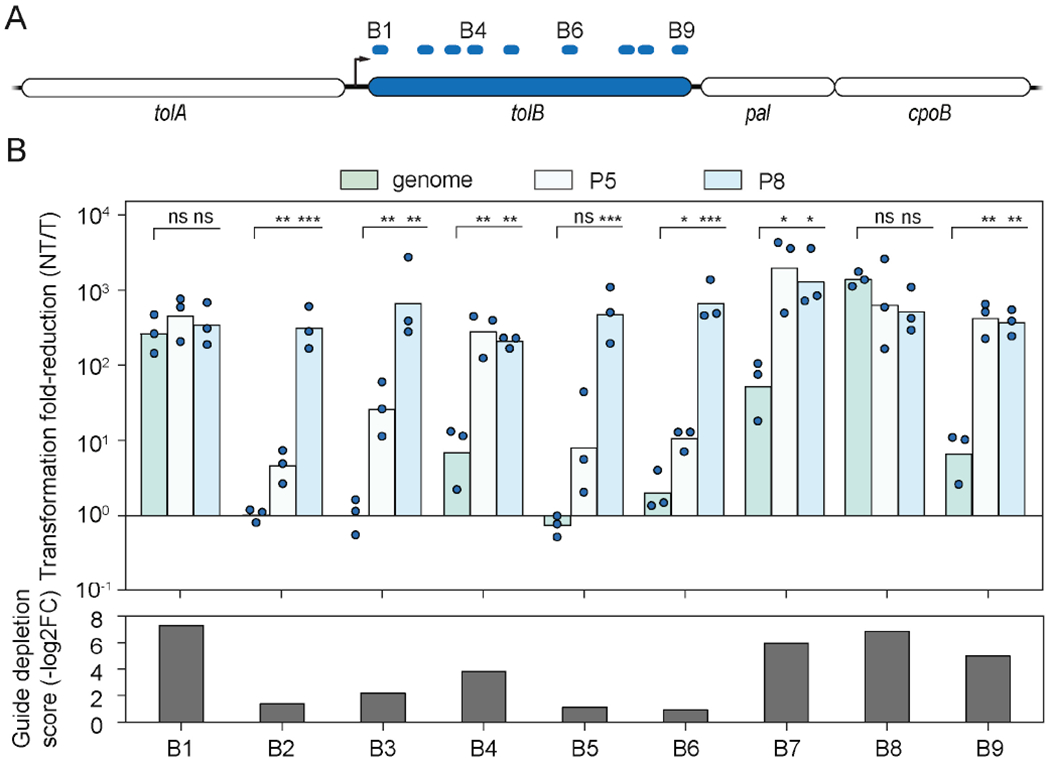
The target expression threshold varies between guide:target pairs associated with the same transcript. See also Fig. S5.
(A) Target locations within the tolB gene from the guide library. Targets are labeled B1 - B9.
(B) Impact of boosting tolB expression for each crRNA guide in E. coli. The tolB gene was expressed from one of two constitutive promoters (P5 or P8) on a plasmid. The genomic copy of tolB was always intact. Bottom: Average guide depletion score for each guide from the library screen. Bars represent the mean of triplicate independent experiments (n = 3). Statistical significance was calculated by comparing the transformation fold-reduction to that with only the genomic copy of tolB. ***: p < 0.001. **: p < 0.01. *: p < 0.05. ns: not significant.
Our initial data also suggested that boosting expression of the tolB mRNA might allow poorly performing guides to achieve cytotoxic autoimmunity. We therefore placed the tolB RBS and coding region under a medium or strong constitutive promoter on a plasmid, and we repeated the transformation assay (Fig. 4B). For all guides that displayed low efficiency when targeting the endogenous transcript, expressing tolB from the plasmid led to a strong reduction in plasmid transformation. Notably, guides that exhibited some reduction in colony counts required only the medium promoter, while guides exhibiting negligible reduction in colony counts required the strong promoter. Therefore, boosting target expression can elicit cytotoxic autoimmunity by otherwise poorly active crRNAs. Concurrently, the target expression threshold can vary between guides, even when targeting the same transcript.
Reducing Cas13a levels precludes cytotoxic autoimmunity
We demonstrated that the expression level of the target must exceed a threshold for Cas13a to induce dormancy. While thus far we focused on the properties of the target, the concentration of the Cas13a:crRNA complex could also be an influencing factor. We therefore explored the impact of altering levels of this complex on plasmid transformation. Specifically, we swapped the native promoter from L. shahii driving transcription of cas13a with a constitutive synthetic promoter with weaker expression (Pw). After replacing cas13a with deGFP and measuring fluorescence of the cells, we measured a 44-fold decrease in fluorescence for Pw compared to the native promoter (Fig. S5A). We then performed the transformation assay targeting the synthetic transcript expressed from the set of constitutive promoters. Remarkably, we did not observe any reduction in the plasmid transformation even when expressing the synthetic target from the strongest promoter (Fig. S5B).
To confirm that the nuclease is expressed, we introduced plasmids encoding the nuclease, targeting or non-targeting crRNA and a deGFP reporter to measure collateral cleavage activity in cell-free transcription-translation (TXTL) reactions (Marshall et al., 2018; Liao et al., 2019; Marshall, Beisel and Noireaux, 2020) (Fig. S5C). Monitoring reporter fluorescence over time, we found that the nuclease expressed from the Pw promoter decreased deGFP expression compared to the non-targeting control, albeit with slower kinetics than the construct with the native promoter. Nevertheless, these data indicate that Cas13a expressed under the Pw promoter is active. Overall, these results show that low levels of Cas13a nuclease can impair immune induction, even when target expression levels are high.
Infection by a lytic phage activates Cas13a-based immunity even for less efficient guides
Beyond self-targeting, the observed dependency on target transcript levels could impact the immune response to foreign invaders targeted by Cas13. We began with the lytic MS2 RNA bacteriophage that rapidly replicates and lyses E. coli as part of the infection cycle. We designed 6 crRNA guides (M1-M6) targeting within the rep and cp genes (Fig. 5A), as both genes are non-toxic when expressed in E. coli and thus can be expressed individually to determine the expression threshold for each crRNA. To determine each crRNA’s target expression threshold, we expressed rep or cp under a weak, medium, and strong promoter on a low-copy plasmid and measured the extent of plasmid interference (Fig. 5B). We found that the crRNAs were associated with a wide range of target expression thresholds for immune induction, with one exhibiting a low expression threshold, three exhibiting a higher expression threshold, and two exhibiting an expression threshold higher than the strongest promoter.
Figure 5.

Cas13a defends E. coli from a lytic bacteriophage even with guides exhibiting a higher immunity threshold. See also Fig. S6.
(A) ssRNA genome of the lytic MS2 bacteriophage and location of guides targeting cp and rep genes used for accessing targeting and phage defense.
(B) Immunity threshold for different LshCas13a guides targeting cp or rep genes expressed under different promoters on a plasmid. Bars represent the mean of triplicate experiments (n = 3). Statistical significance in B was calculated by comparing the transformation fold-reduction to that of the weakest P2 promoter. ***: p < 0.001. **: p < 0.01. *: p < 0.05. ns: not significant or average below the reference.
(C) LshCas13a-mediated defense against MS2 bacteriophage at different MOIs. Growth curves of E. coli with active or dead LshCas13a are compared over time. The lines and bars represent respectively the mean and standard deviation of three independent biological replicates.
We then infected E. coli cultures with MS2 phages at different MOIs and assessed defense through each of the six targeting crRNAs or a non-targeting crRNA (Fig. 5C). In the absence of MS2 phage, all cultures exhibited continual growth over the time course. Cultures with LshCas13a and crRNAs exhibiting at least some immunity in the transformation assay (M1-M4) halted growth and maintained turbidity, indicative of immunity-induced dormancy. In contrast, cultures with LshCas13a and the crRNAs that did not exhibit immunity in the transformation assay (M5, M6) or a non-targeting crRNA as well as all dLshCas13a controls showed a temporary drop in turbidity, indicative of successful phage infection. Halted growth for the highly active guides was surprising given the low MOIs. However, this can be attributed to LshCas13a conferring only partial immunity against MS2 infection in E. coli as evidenced by increased phage titers over time (Fig. S6A–B), which can be attributed in part to the high burst size of this phage (Grosjean and Fiers, 1982). Overall, crRNAs exhibiting even a high target expression threshold can confer defense against a pathogenic invader.
The target expression threshold allows tolerance of invaders conferring benefits to the host
Beyond pathogenic invaders, we considered benign invaders that do not rapidly replicate and instead generally maintain transcript levels. We reasoned that the target transcript would not elicit a robust immune response if expressed below the expression threshold, allowing the invader to persist. One notable scenario is a targeted gene that benefits the host cell, such as a virulence factor or an antibiotic resistance marker spread through mobile genetic elements (Frost et al., 2005; Partridge et al., 2018; Koonin et al., 2020). If targeting does not substantially affect levels of this transcript, then the host could benefit from its expression but not elicit an immune response.
To explore this scenario directly, we created a plasmid encoding two resistance genes: a kanamycin resistance gene targeted by two distinct crRNAs (our targeted beneficial gene) as well as a non-targeted hygromycin resistance gene (for plasmid selection). The kanamycin gene was placed downstream of a strong, medium, or weak constitutive promoter (Fig. 6A). We then assessed tolerance to and growth benefits from the kanamycin resistance gene in two separate steps: assessing tolerance by transforming the plasmid under hygromycin selection, and assessing the growth benefit by measuring growth of tolerant cells in the presence of kanamycin.
Figure 6.
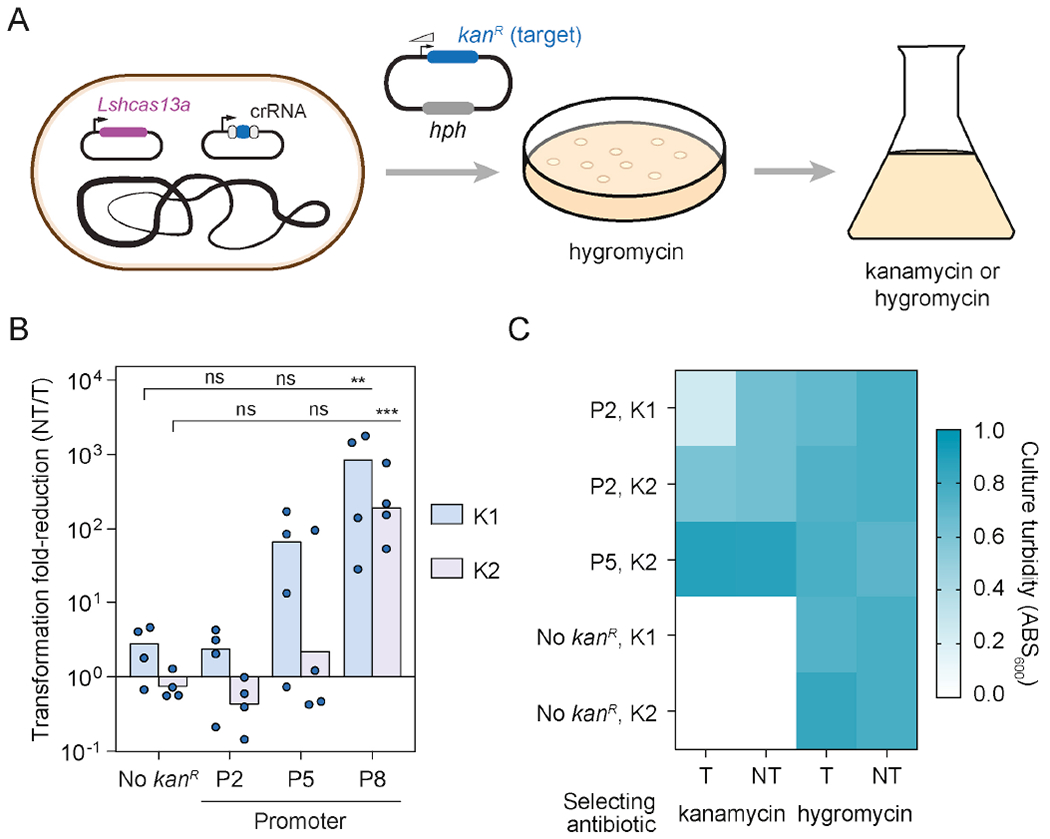
A plasmid transcript targeted by Cas13a and expressed under the threshold can be tolerated and can confer benefits to the host. See also Fig. S7.
(A) Experimental setup for evaluating tolerance for a plasmid-expressed beneficial gene. The two targets (K1, K2) fall within the kanR transcript, which is expressed under different constitutive promoters (P2, P5, P8).
(B) Impact of Cas13a-based targeting of the kanR transcript expressed under the different constitutive promoters. Bars represent the mean of quadruplicate independent experiments (n = 4). Statistical significance was calculated by comparing the transformation fold-reduction to that without the kanR gene. ***: p < 0.001. **: p < 0.01. *: p < 0.05. ns: not significant.
(C) Heat map comparing growth in the presence of kanamycin (10 μg/mL) or hygromycin (100 μg/mL) for constructs associated with a low transformation fold-reduction in B. Values represent the average turbidity after 12 hours of growth from 9 biological replicates. Constructs lacking the kanR gene serve as negative controls, while the non-targeting crRNA plasmid serves as a non-targeting (NT) control. See Fig. S7 for the same growth measurements with a higher concentration of kanamycin (50 μg/mL).
For the first step, high target expression induced an immune response while low target expression was tolerated (Fig. 6B), in line with other target transcripts expressed from plasmids (Figs. 2, 4). For the second step, we found that the tolerated crRNA:promoter combinations yielded substantial growth on 10 μg/mL kanamycin (Fig. 6C). One combination (K2 crRNA with P5-expressed target) maintained growth even in the presence of 50 μg/mL kanamycin (Fig. S7). In contrast, a plasmid lacking the kanamycin resistance gene did not yield any growth. For some combinations, growth on kanamycin was slower with the targeting versus non-targeting crRNA. As this same growth defect was not observed in the presence of hygromycin, the growth defect on kanamycin may be attributed to Cas13a-mediated gene silencing that sensitized the cells to kanamycin. Overall, these results show that the target expression threshold can allow tolerance of a benign invader, even allowing the targeted gene to provide benefits to the host cell.
DISCUSSION
A robust immune response by CRISPR nucleases normally requires two established criteria: complementarity between the crRNA guide and the target, and a PAM or PFS flanking the target. Here, we show that the expression levels of the target transcript represent a third criterion determining invader defense and cytotoxic autoimmunity by the RNA-targeting nuclease Cas13. The outcome depends on whether expression levels are above or below a specific threshold (Fig. 7). When target transcript levels are above the threshold, widespread collateral RNA cleavage leads to cell dormancy. When target transcript levels are below the threshold, cells escape dormancy. In that case, target expression can be either unperturbed or silenced. Gene silencing under the expression threshold is supported by two lines of evidence: extensive depletion of guides targeting essential genes but not non-essential genes in the library screen (Figs. 3C and S3C) and reduced growth on kanamycin but not hygromycin when targeting the kanR gene (Figs. 6C and S7). In both cases, silencing of the essential gene would reduce growth, even if the cells do not enter dormancy through collateral RNA cleavage. The target expression threshold also can vary between guide:target pairs, even when the targets are present in the same transcript. With sufficient expression of the target transcript, however, the threshold can be crossed to elicit a full immune response.
Figure 7.

Proposed model for the target expression threshold as a determinant of Cas13-based defense and autoimmunity. Any transcript with extensive complementarity to the crRNA guide flanked by a PFS can be targeted by Cas13. However, the resulting impact on the target gene and the host cell depends on a third factor: target expression. Exceeding a threshold of target expression potentially represents when a sufficient number of Cas13:crRNA complexes are activated to enact widespread RNA cleavage and cellular dormancy. Target expression under the threshold is tolerated and can lead to targeted gene silencing. Target expression above the threshold leads to cytotoxicity for self-targeting and immunity for invader targeting. The exact threshold is target-dependent, likely due to factors that influence guide performance (e.g., local secondary structure, GC content).
One simple biochemical explanation for the threshold is the level of activated Cas13:crRNA complexes. While each activated complex would lead to the same extent of collateral RNA cleavage, the concentration of these complexes would dictate how extensive RNA degradation is and the resulting impact on the cell. Higher levels would lead to widespread RNA cleavage that induces growth arrest and dormancy. Lower levels might lead to preferential cleavage of the target, resulting in specific silencing. For less efficient targets, the target transcript may be poorly recognized for multiple reasons associated with guide selection (e.g., GC content, crRNA or target folding) (Wessels et al., 2020), resulting in a negligible impact on target transcript levels. In some cases, we observed a sharp threshold, where immunity was fully activated with as little as a 5.8-fold change in promoter activity driving target expression. While the extent of collateral cleavage should scale with the concentration of activated Cas13:crRNA complexes, collateral cleavage could create a positive feedback loop. Specifically, degradation of key transcripts might reduce growth, leading to slowed dilution of activated Cas13:crRNA complexes (Elowitz et al., 2002), leading to further RNA degradation and growth arrest. This mechanism is supported by our findings that lowering Cas13a levels prevents autoimmune induction (Fig. S5), where the concentration of activated Cas13a:crRNA complexes could not be reached to initiate the feedback loop. However, there could be other possible explanations for the sharp threshold, such as cooperativity between Cas13 enzymes or how well activated complexes can diffuse through the dense cytoplasm of a bacterial cell. Future work could evaluate the link between collateral RNA cleavage and growth arrest as well as other factors that affect the threshold, such as expression levels of the crRNAs, the impact of multiplexed targeting, and the extent of collateral activity exhibited by different Cas13 nucleases (Abudayyeh et al., 2017; Xu et al., 2021).
The existence of a target expression threshold has major implications for self-targeting and invader defense (Fig. 7). For targets above the threshold, immunity resembles that associated with other CRISPR-Cas systems: cytotoxicity incompatible with cell survival when targeting the host’s own genetic material, or invader clearance or induced dormancy when targeting an invader. For targets below the threshold, immunity takes a different form. For self-targeting, targets under the threshold are tolerated, allowing the cells to persist with a limited impact on fitness. Indeed, in Flavobacterium columnare harboring a II-C and a VI-B system that share the same acquisition machinery, acquisition of self-targeting spacers and phage spacers was observed for the resident type VI system but not for the resident type II-C system (Hoikkala et al., 2021). However, self-targeting spacers have been observed less frequently in native arrays of type VI CRISPR-Cas systems and generally target close to prophage regions (Nobrega et al., 2020). For invader defense, the threshold ties the immune response to the threat level posed by the invader. For rapidly replicating invaders that pose a major threat (e.g. lytic phages), target transcripts will accumulate in the cell, crossing the threshold and induce a robust immune response. For invaders that pose a minor threat (e.g. lysogenic phages, mobile plasmids), target transcripts may be expressed at a sufficiently low level to not induce the immune response. If targeting leads to gene silencing of essential transcripts, it could block invader replication, subsequently clearing the invader while sparing the infected cell from dormancy. In this setup, induced dormancy would instead represent a back-up strategy if the target transcript continues to accumulate in the cell.
For invader defense, one ramification of the target expression threshold is that an Acr (Bondy-Denomy et al., 2013; Marino et al., 2020) that reduces the concentration of activated Cas13a complexes could rapidly interfere with the immune response. Accordingly, recent work reporting an Acr against Cas13a showed that the invading Acr-encoding phage could shut down CRISPR defenses and proliferate in the first wave of infection (Meeske et al., 2020). In contrast, Acrs that block DNA-targeting systems could allow only a second wave of infecting phages to proliferate, as the defenses would clear the first wave before the Acrs could fully inhibit the CRISPR defenses (Borges et al., 2018; Landsberger et al., 2018). The prior work on the Cas13a Acr attributed escape by the Acr-encoding phage to Cas13a degrading the phage transcripts but not the phage DNA, allowing the phage to eventually recover. However, we found that lowering the concentration of activated Cas13a complexes could allow phage transcripts to cross the original expression threshold without inducing dormancy. Therefore, both mechanisms could be at work for phages encoding Cas13 Acrs.
Beyond type VI CRISPR-Cas systems, type III systems also target RNA and can elicit collateral RNA cleavage that could also be impacted by a target expression threshold (Kazlauskiene et al., 2017; Niewoehner et al., 2017; Rostøl and Marraffini, 2019). For these systems, collateral RNA cleavage is induced through Cas10 within the activated effector complex synthesizing cyclic oligo adenylates (cOAs) from ATP. These small molecules then bind to different accessory proteins such as Csm6/Csx1 encoded by some type III CRISPR-Cas systems, which then begin non-specifically cleaving cellular RNAs. A key difference compared to Cas13 is that one activated type III effector complex can produce a large number of cOA molecules, potentially activating a large number of accessory proteins. This intermediate amplification step could effectively eliminate the threshold, leading to widespread collateral RNA cleavage and dormancy. However, depending on how quickly the cOAs are synthesized, diffuse, and turnover, a minimal threshold of activated effector complexes may be necessary to elicit widespread RNA cleavage. Furthermore, the existence of cOA-degrading anti-CRISPR proteins could offer a means to artificially raise the target expression threshold to the point where this mode of immunity is never activated over the course of the infection. Similar behaviors may be expected for Type VI CRISPR-Cas systems encoding Cas13b and the accessory protein Csx28 that appears to amplify the immune response (Smargon et al., 2017; VanderWal et al., 2021). Exploring the extent to which a target expression threshold exists for different type III and VI-B CRISPR-Cas systems could broaden our understanding of the target expression threshold, particularly when back-up defenses exist that enhance the immune response.
Finally, the target expression threshold may help resolve otherwise contradictory observations when implementing Cas13-based technologies in bacteria and in eukaryotic cells. In bacteria, Cas13 was previously thought to induce widespread RNA cleavage as long as the target transcript was present (Abudayyeh et al., 2016). In contrast, in eukaryotic cells, Cas13 was reported to function as a sequence-specific gene silencer with no obvious off-target effects (Abudayyeh et al., 2017; Konermann et al., 2018; Mahas, Aman and Mahfouz, 2019; Huynh et al., 2020). To resolve this clear discrepancy, Cas13 was proposed to function differently in bacteria and in eukaryotic cells. At the same time, there are emerging reports of Cas13 inducing collateral RNA cleavage in human cells, although cell-type specific factors were suggested as the underlying cause (Wang et al., 2019; Özcan et al., 2021; Wu et al., 2021). The target expression threshold helps unify these observations. In bacteria, target transcripts under threshold do not induce widespread collateral RNA cleavage and instead are unaffected by targeting or undergo silencing. Applying our proposed biochemical mechanism to eukaryotic cells, activated Cas13 complexes would be much less concentrated given the larger size of the cytoplasm and the distribution of transcripts in this cellular compartment that lend to a higher expression threshold. The few examples of collateral cleavage might represent scenarios where the concentration of activated Cas13 complexes or the target transcript is much higher or localized close to important cellular RNAs. Investigating Cas13 nucleases that exhibit less collateral activity (Abudayyeh et al., 2017; Xu et al., 2021) could pose a simple solution to avoid potentially catastrophic RNA cleavage. The target expression threshold therefore may impact not only invader defense in bacteria but also the application of Cas13 for programmable gene silencing in eukaryotic cells as well as for antivirals (Abudayyeh et al., 2017; Konermann et al., 2018; Abbott et al., 2020).
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Chase L. Beisel (chase.beisel@helmholtz-hiri.de).
Material availability
The following plasmids have been deposited to Addgene: pFT62 (#185832), pFT50-repeat (#185833), pFT62-pBAD (#185836), pFT62-soxS full (#185897), pFT62-tolB full (#185898), pBAD33_PJ23108_LsCas13a (#185899), pFT62-PJ23119-cp protein (#185900), pUA66-PJ23119 kanR + cloDF13 + hygR (#185902), pUA66-PJ23119 kanR + hygR (#185903), pUA66 + hygR (#185904). All the other plasmids will be provided by the lead contact upon reasonable request.
Data and code availability
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original images of the RNA gels have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All original code has been deposited at Mendeley and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E.coli MG1655 | Susan Gottesman’s lab (NIH) | N/A |
| E. coli MG1655 ΔaraBAD ΔCM PconAraFGH | This manuscript | N/A |
| E.coli K12 (CGSC 4401) | DSMZ | DSM 9037 (CGSC 4401) |
| E. coli bacteriophage MS2 | DSMZ | DSM 13767 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Chloramphenicol | Roth - Carl Roth | Cat# 3886.2 |
| Ampicillin | Roth - Carl Roth | Cat# K029.2 |
| Kanamycin | Roth - Carl Roth | Cat# T832.3 |
| Hygromycin B | Roth - Carl Roth | Cat# 1287.2 |
| BsmBI-v2 | New England BioLabs (NEB) | Cat# R0739L |
| Instant Sticky-end Ligase Master Mix | New England BioLabs (NEB) | Cat# M0370S |
| Q5® Site-Directed Mutagenesis Kit | New England BioLabs (NEB) | Cat# E0554S |
| Gibson Assembly® Master Mix | New England BioLabs (NEB) | Cat# E2611L |
| KAPA HiFi HotStart ReadyMix | Roche Diagnostic | Cat# 7958935001 |
| Agencourt® AMPure® XP, 60 mL | Beckman Coulter | Cat# A63881 |
| TURBO DNase | Thermo Fisher Scientific | Cat# AM2239 |
| myTXTL Sigma 70 Master Mix | Arbor Biosciences | Cat# 507005 |
| Critical Commercial Assays | ||
| Direct-zol™ RNA MiniPrep Plus kit | Zymo research | Cat# R2072 |
| ZymoPURE II Maxiprep Kit | Zymo Research | Cat# D4203 |
| ZymoPure II Plasmid Midiprep Kit | Zymo Research | Cat# D4200 |
| NextSeq 500/550 Mid Output Kit v2.5 (300 Cycles) | Illumina | Cat# 20024905 |
| Rybo-off rRNA depletion kit | Vazyme Biotech | Cat# N407-01 |
| NEBNext® Ultra™ II Directional RNA Library Prep Kit | New England BioLabs (NEB) | Cat# E7760L |
| NovaSeq 6000 S1 PE Reagent Kit (100 cycles) | Illumina | Cat# 20012865 |
| Deposited Data | ||
| Genome-wide screen | NCBI’s Gene Expression Omnibus | GEO Series accession number GSE179913 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179913) |
| Transcriptomic analysis | NCBI’s Gene Expression Omnibus | GEO Series accession number GSE179914 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179914) |
| Original gel images | Mendeley | 10.17632/97s4hpvztb.1 |
| Original code | Mendeley | doi:10.17632/d5vw9sjcrv.1 |
| E. coli K12 MG1655 genome | NCBI | NC_000913.3 |
| Oligonucleotides | ||
| For all oligos used in the study see Supplementary Table 3 (SI Tables Excel file, tab 2). | ||
| For oligos used for library experiment see Supplementary Table 4 (SI Tables Excel file, tab 3). | ||
| Recombinant DNA | ||
| For plasmids used in this study see Supplementary Table 2 (SI Tables Excel file, tab 1). | ||
| Software and Algorithms | ||
| Wessels et al. algorithm | Wessels et al., 2020 | https://gitlab.com/sanjanalab/cas13 |
| RNAfold version 2.4.12. | Vienna RNA Package (Lorenz et al., 2011) | https://www.tbi.univie.ac.at/RNA/index.html |
| BBMerge (version 38.69) | Bushnell et al., 2017 | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbmerge-guide/ |
| edgeR (version 3.28.0) | Robinson and Oshlack, 2010; Robinson et al., 2009 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| RBS calculator (version 1.0) | Salis, 2011 | https://github.com/hsalis/Ribosome-Binding-Site-Calculator-v1.0 |
| STAR (version 2.7.4a) | Dobin et al., 2013 | https://github.com/alexdobin/STAR/releases |
| HTSeq (version 0.9.1) | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.9.1/ |
| auto-sklearn (version 0.10.0) | Feurer et al., 2019 | https://automl.github.io/auto-sklearn/master/releases.html# |
| EcoCyc | Keseler et al., 2017 | https://ecocyc.org |
| TreeSHAP (version 0.36.0) | Lundberg et al., 2020 | https://github.com/slundberg/shap |
| Mixed-effects random forest (MERF) | Hajjem et al., 2014 | https://github.com/manifoldai/merf |
| Other | ||
| See Supplementary Table 1 for list of Anderson promoters used in this study | http://parts.igem.org/Promoters/Catalog/Anderson | N/A |
Experimental Model and Subject Details
Strains
The strains used are E. coli MG1655 (F−), E. coli MG1655 ΔaraBAD ΔCM PconAraFGH (F−) and E. coli MG1655 (F+). Cells were grown in LB medium at 37°C shaking at 220 rpm. Strains information is reported in Table S2.
Plasmids
The main plasmids used are pZ003 (LshCas13a), pFT50 (crRNA backbone), pFT50-repeat (crRNA backbone with repeat) and pFT62 (target backbone). Spacers were inserted into the backbone by digestion with BsmBI and ligation with Instant Sticky-end Ligase Master Mix (NEB, M0370S). Q5 mutagenesis or Gibson assembly were used to modify the target plasmid or the nuclease backbone. Plasmids used in this study are listed in Table S2, while all of the oligos and cloning details can be found in Table S3.
Method Details
Transformation-based targeting assays
The transformation assays were conducted in two ways: targeting a genomically-encoded transcript and a plasmid-encoded transcript. For genomically-encoded transcripts, biological replicates containing the nuclease plasmid were inoculated overnight in LB medium (10 g tryptone, 5 g yeast extract, and 10 g NaCl in 1 L of dH2O) with chloramphenicol (Cm, 34 μg/mL). After 16 h, the ABS600 was measured and the samples were normalized, back-diluted 1:50 in fresh LB with Cm, and grown until an ABS600 of 0.6 - 0.8. Cultures were placed on ice and made electrocompetent by washing the pellet twice with 10% glycerol. Then, 50 ng of the crRNA plasmids were transformed into 40 μL of competent cells using the E. coli 1 program on the MicroPulser Electroporator (Bio-rad). After 1 h of recovery in 500 μL of SOC medium (SOB medium: 20 g Tryptone, 5 g Yeast Extract, 0.5 g NaCl, 800 mL dH2O and 10 mL 250 mM KCl adjusted to pH 7. To SOB medium added 5ml 2M MgCl2, 20 ml of 1 M Glucose), 10-fold dilutions of the cultures in 1X PBS (10X PBS: 80 g NaCL, 2 g KCl, 17,7 g Na2HPO4*2 H2O, 2.72 g KH2PO4, fill up to 1 L with mqH2O, set pH to 7.4 and autoclave) were prepared and 5 μL spot dilutions were plated on Cm and ampicillin (Amp, 100 μg/mL) LB plates (prepared by mixing 1 L of LB medium with 18 g Agar) and incubated at 37°C for 16-18 h. The number of transformants has been calculated by counting the first row of countable colonies and multiplying the value by the dilution factor. Every targeting assay has been conducted with three biological replicates.
For the plasmid-encoded transcripts, cultures containing the nuclease plasmid and the target plasmid were transformed with the crRNA plasmid. The rest of the procedure matched that followed for the genome targeting assay. The synthetic sequence expressed on a plasmid (pFT62) contains part of the mRFP1 gene sequence, which does not match the E. coli genome. Fragments from mRNAs (target plus 20 bp upstream and downstream) were cloned in pFT62 in place of the synthetic target. Finally, to see if cloning an entire gene on a plasmid would change the targeting outcome, full gene sequences each with an RBS and stop codon were cloned in pFT62 through Gibson assembly.
Flow cytometry analysis
Plasmids expressing GFP under different Anderson promoters were cloned by Q5 mutagenesis to introduce the different promoters in the pUA66-PJ23119 GFP plasmid. Cells containing the nuclease plasmid and the GFP plasmid were inoculated overnight in LB medium with kanamycin (Kan, 50 μg/mL) and Cm, then normalized, back-diluted to ABS600 = 0.02 and cultured until ABS600 ≈ 0.8. Cells were then pelleted and resuspended in 1X PBS before being applied to an Accuri C6 Plus analytical flow cytometer (BD Biosciences). 30,000 events were obtained by gating on living cells, and the mean of the FL1-H values were quantified as GFP fluorescence. Final fluorescence values were obtained by subtracting the autofluorescence of cells not expressing GFP.
L-arabinose induction of Cas13a-mediated targeting in E. coli
The plasmid with the synthetic target under control of the arabinose-inducible promoter was cloned by introducing the PBAD promoter in pFT62 through Gibson assembly. The utilized strain, E. coli MG 1655 ΔaraBAD Pcon-araFGH, possesses mutations that yield a more linear response to arabinose induction (Afroz et al., 2015). To validate the inducible system, cells were transformed with the nuclease, the crRNA and the arabinose-inducible target plasmids. Three biological replicates were inoculated overnight in LB supplemented with Amp, Cm, and Kan as well as 0.2% glucose to reduce background expression from the PBAD promoter. The samples were then pelleted, resuspended in LB with antibiotics and then back-diluted to ABS600 = 0.01 with or without 0.2% L-arabinose. Culture turbidity was recorded over time on a Synergy Neo2 or H1 fluorescence microplate reader (BioTek) for 16 h by measuring ABS600 every 3 min.
Assessment of collateral RNA cleavage
Using the validated arabinose-inducible setup, collateral RNA cleavage was visualized on an agarose gel. Cells with the nuclease, crRNA, and target plasmids were grown overnight in Cm, Amp, Kan LB with 0.2% glucose, washed in LB with antibiotics to remove glucose, and back-diluted to ABS600 = 0.01. The cells were cultured until ABS600 ≈ 0.4, after which each sample was split in equal volumes with or without the inducer. After 1 h from induction, the ABS600 of each sample was measured, and the same number of cells (2250 ABS600×mL) was snap frozen on dry ice. The next day, total RNA was extracted using the Direct-zol RNA MiniPrep Plus kit (Zymo research, R2072). Then 1 μg of each RNA was mixed 2:3 with an RNA loading dye (2x) (for 50 mL: 625 μL Bromophenol blue 2%, 625 μL 2% Xylene Cyanol, 1800 μL of 0.5 M EDTA pH = 8.0, 46,821 mL Formamide), heated at 70°C for 10 min, placed on ice, and resolved on a 1% TBE gel at 120 V for 40 min. RiboRuler High Range RNA ladder (Thermo Fisher Scientific, SM1821) was used as a size marker.
To further evaluate the self-targeting crRNAs that did not reduce transformation of the target plasmid, we inoculated overnight cells constitutively expressing the targeting or NT crRNA plasmids and the Cas13a nuclease or an empty control plasmid. The inocula were back-diluted to ABS600 ≈ 0.02 and cultured for 4 h before extracting total RNA and resolving it on a gel as described above. In parallel, culture growth (ABS600) was measured in a plate reader at 37°C for 16 h.
Library design and validation
The reference genome and annotation of E. coli K12 MG1655 (NC_000913.3) was used for crRNA library design. First, all potential 32-nt guides were designed for protein-coding genes (limited to the CDS) and rRNAs with non-GU PFS and GC content between 40% and 60%, resulting in an average of 484 guides per gene. To reduce the size of the library, and considering the unknown effect of the targeting location within a gene on guide efficiency, each gene was divided into a maximum of 10 sections with equal length. Within each section, guides were filtered based on the strength of local secondary structure, defined as ΔG, in both repeat-guide sequence and the mRNA targeting region (including a region of 2 times length of crRNA before and after the target). ΔG was calculated as the energy difference between the unconstrained minimum free energy (MFE) structure and the constrained MFE structure with no base pairs, estimated using RNAfold from the Vienna RNA Package (Lorenz et al., 2011) version 2.4.12. Sequences (either guide or flanking primer sequences) containing BsmBI restriction sites or homopolymer stretches of more than four consecutive nucleotides were excluded to facilitate synthesis and cloning. The guide with the lowest secondary structure strength in each section was selected, resulting in a library of 25,997 guides, including 25,470 guides targeting protein-coding genes, 127 guides targeting rRNAs, and 400 randomized non-targeting guides as negative controls. A sequence containing a universal primer binding site and the BsmBI restriction site was added to the guides to amplify the oligo library and digest it before ligating it into the backbone (see Table S4). The library was synthesized by Twist Bioscience.
The base backbone pFT50 was slightly modified to insert the direct repeat before the GFP dropout site to be able to limit the insertion size to the spacer itself. A BsmBI restriction site present in pFT50 was also eliminated through Q5 mutagenesis.
Guide library cloning and verification
The library was amplified with Kapa Hifi polymerase (Roche Diagnostics, 7958935001) (20 ng DNA) for 10 cycles following the manufacturer’s instructions (Ta = 64°C; 30 s denaturation, 20 s annealing, 15 s extension) using primers SPCpr 349/350. 5 μL library (150 nM) and 5 μL of backbone (50 nM) were mixed in 25 μL of total reaction volume. The mixture was subjected to 50 cycles of BsmBI digestion (3 min at 42°C) and ligation (T4 ligase - 5 min at 16°C) with a final digestion at 55°C for 60 min to ensure complete removal of the backbone, followed by a 10-min heat inactivation at 80°C. The sample was then ethanol precipitated, and 5 μg were transformed into fresh electrocompetent Top10 cells (90 μL). The transformation was conducted with two separate batches of electrocompetent cells to ensure enough transformants were obtained. After recovering the two cultures in 500 μL of SOC medium shaking at 37°C for 1 h, the recovered cultures were back-diluted into 150 mL LB with Amp and cultured with shaking at 37°C for 12 h. The next day, plasmid DNA from the culture was isolated using the ZymoPURE II Maxiprep Kit (Zymo Research, D4203) and further purified by ethanol precipitation.
Guide library screen
Two replicates of E. coli MG1655 cells with or without the nuclease plasmid were inoculated overnight, then the next day the ABS600 was normalized and the cells back-diluted to ABS600 ≈ 0.1 in fresh LB with or without Cm. Once each culture reached ABS600 ≈ 0.8, cells were made electrocompetent by washing twice with 10% glycerol and finally resuspended in 480 μL 10% glycerol. For each sample, six separate transformations were conducted each with 1 μg of library DNA (40 μL/transformation). Transformed cells were then recovered in 500 μL SOC medium for 1 h with shaking at 37°C. The six reactions were combined to yield 3 mL of culture per condition. Serial dilutions of this culture were made, and 100 μL of 1:10,000 dilutions were plated for the targeting and no-Cas13a samples with the appropriate antibiotics (Cm and Amp, or Amp only), yielding a theoretical library coverage of ~9,500. The remaining culture was diluted 1:100 in LB with Amp and Cm to ABS600 = 0.06 and cultured for 12 h with shaking at 37°C. Finally, the library was isolated with the ZymoPure II Plasmid Midiprep Kit (Zymo Research, D4200).
Next-generation sequencing of the guide libraries
The guides sequences from the purified library DNA were amplified with Kapa Hifi polymerase using primers oEV-315/316 (NT1), oEV-317/318 (NT2), oEV-319/320 (T1), oEV-321/322 (T2). 10 ng of DNA were included in a 50-μL PCR reaction for 15 amplification cycles (15 s at 98°C, 30 s at 64°C, 30 s extension). The amplification products were purified using AMPure beads (Beckman Coulter, A63881) and further amplified with primers oEV-323/324 (NT1), oEV-325/326 (NT2), oEV-327/328 (T1), oEV-329/330 (T2) to add the appropriate indices and Illumina adaptors. For this reaction, the same settings were used with the only difference being the amount of input DNA (25 ng) and the number of cycles (10). The resulting amplification products were purified with AMPure beads and resolved on a gel to verify the presence of the correct amplicon. The samples were submitted for Sanger sequencing and Bioanalyzer 2100 analysis (Agilent Technologies) as a quality check. Finally, samples were submitted for next-generation sequencing at the NextSeq 500 sequencer (Illumina) with a 150 bp paired-ends kit (130 million reads, Illumina, 20024905) to obtain 1000-fold coverage. To increase the library diversity, 20% of phiX phage DNA was spiked-in.
To correlate transcript expression levels with guide depletion, we measured transcript levels in E. coli MG 1655 under conditions paralleling the library screen. Briefly, two independent replicates of cells containing the plasmid cBAD33 (empty backbone for nuclease plasmid) were cultured overnight and then normalized to ABS600 = 0.06 in LB with Cm and cultured to ABS600 ≈ 0.5 or ABS600 ≈ 0.8. At those growth points, cells were pelleted and snap-frozen for RNA extraction with the Direct-zol RNA MiniPrep Plus kit. The samples were also DNase-treated with TURBO DNase (Thermo Fisher Scientific, AM2239) and quality verified using a 5200 Fragment Analyzer System (Agilent Technologies). Finally, rRNA was removed with the Rybo-off rRNA depletion kit (Vazyme Biotech, N407-01), the samples were prepared with NEBNext® Ultra™ II Directional RNA Library Prep Kit (New England BioLabs) and sequenced on a NovaSeq 6000 (Illumina) with 50-bp paired-end reads.
The resulting NGS data were deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE179913 for the genome-wide screen and GSE179914 for transcriptomic analysis.
Screen analysis and machine learning model
For analysis of the genome-wide screen, after merging using BBMerge (version 38.69) (Bushnell, Rood and Singer, 2017) with parameters “qtrim2=t, ecco, trimq=20, -Xmx1g, mix=f”, paired-end sequence reads with a perfect match were assigned to crRNA sequences. After filtering guides for at least 1 count per million reads in at least 2 samples, the library sizes were normalized using the read counts for non-targeting guides with the trimmed mean of M-values (TMM) method in edgeR (Robinson, McCarthy and Smyth, 2009; Robinson and Oshlack, 2010) (version 3.28.0). Differential abundance of crRNAs between targeting samples and control samples lacking the Cas13a nuclease was assessed using the edgeR quasi-likelihood F test after fitting a generalized linear model. The translation initiation rate of each gene was predicted using RBS calculator (version 1.0) (Salis, 2011).
For RNA seq analysis, sequencing reads were aligned to the E. coli K12 MG 1655 genome (NC_000913.3) using STAR (Dobin et al., 2013) (version 2.7.4a) with parameters “-- alignIntronMax 1 --genomeSAindexNbases 10 --outSAMtype BAM SortedByCoordinate” and the count of reads mapping to each gene was obtained using HTSeq (Anders, Pyl and Huber, 2015) (version 0.9.1) with parameters “-i locus_tag -r pos --stranded reverse --nonunique none -t gene”, followed by calculating transcripts per million (TPM).
The machine learning regression model was developed with 144 features as predictors and the log2FC values of crRNAs from the genome-wide screen as targets using auto-sklearn version 0.10.0 (Feurer et al., 2019) with all possible estimators and preprocessors included and parameters ‘ensemble_size’: 1, ‘resampling_strategy’: ‘cv’, ‘resampling_strategy_arguments’: {‘folds’: 5}, ‘per_run_time_limit’: 360, and ‘time_left_for_this_task’: 3600. Features included gene expression level (log2 transformed TPM at OD ≈ 0.5), gene essentiality, gene id, gene length, (percent) targeting position in gene, delta G of repeat-crRNA and mRNA targeting region, and the one-hot-encoded PFS sequence and crRNA sequence. Gene essentiality information in LB Lennox medium was obtained from EcoCyc (Keseler et al., 2017). The optimal histogram-based gradient boosting model was evaluated using 10-fold cross-validation and interpreted using TreeSHAP version 0.36.0 (Lundberg et al., 2020). In order to further explore the contribution of guide features to depletion, a mixed effect random forest (MERF) (Hajjem, Bellavance and Larocque, 2014) was applied to remove the effects of gene expression level, gene length and gene essentiality using a random-effect linear regression model, with guide features used to predict the residual depletion using a fixed-effect random forest model. The contribution of the guide features to predictions of depletion by the fixed-effect random forest was investigated using TreeSHAP.
For the comparison of depletion of guides targeting non-essential genes and essential genes independently of their expression levels, essential genes were split into deciles based on their expression levels at OD ≈ 0.5. 100 sets of 30 non-essential genes with expression level in each corresponding decile were randomly selected and the median expression levels and guide depletion scores were compared to the values for essential genes in the same decile.
Targeting assay with lower nuclease expression
In order to look at targeting with varied nuclease expression, we used a plasmid expressing the nuclease under the constitutive promoter PJ23108 (Pw). At first, we assessed the relative promoter strength of Pw and the native promoter (Pnative) by cloning the gfp gene in the nuclease backbone in place of the nuclease itself through Gibson assembly. Each gfp plasmid was transformed with the target plasmid into E. coli MG1655 cells, and the ‘Flow cytometry analysis’ protocol was followed to measure fluorescence of the two constructs. We then performed the transformation-based targeting assay with the low expressed nuclease, the crRNA (T1) and different Anderson promoters (P1 to P8) cloned in front of the synthetic target sequence.
Cell-free transcription-translation assays
Plasmids encoding the nuclease, a crRNA, a target sequence, and deGFP were used to assess the targeting activity in a cell-free transcription-translation assay. The nuclease was either under the control of Pnative or Pw and the crRNA encoded either a targeting (T) spacer or non-targeting (NT) control. To assess differences in targeting activity when the nuclease is under the control of different promoters, the two nuclease constructs were added separately with either the targeting or the non-targeting crRNA and the targeted plasmid to myTXTL Sigma 70 Master Mix (Arbor Biosciences, 507005), with a final concentration of 2 nM, 1 nM and 0.5 nM, respectively. The samples were incubated for 2 h at 29°C, before a plasmid encoding deGFP was added to a final concentration of 0.5 nM. The samples were then incubated at 29°C for 16 h in a plate reader (BioTek Synergy Neo2) and fluorescence was measured every three minutes (excitation, emission: 485 nm, 528 nm). All shown data was produced using the Echo 525 Liquid Handler (Beckman Coulter). The assays were therefore scaled down to 3 μl reactions per replicate, with four replicates each. As part of the analysis, the background fluorescence from myTXTL mix and water samples was subtracted from all samples. Grubb’s test was performed using the values after 16 h to identify outliers between replicates (α = 0.1). If no outliers were identified, the first of the four replicates was discarded. The graph shows the average deGFP fluorescence over time together with the standard deviation.
Infection experiments with MS2 phage
For transcription-based targeting assays, the rep and cp gene target sequences with RBS and stop codon have been cloned in the pFT62 plasmid having different Anderson promoters. The targeting assays have been performed by transforming E. coli CGSC 4401 cells (F+) expressing the nuclease and differentially abundant target rep or cp gene transcripts with the correspondent targeting guides and by calculating the reduction in colony numbers compared to a non-targeting guide.
Prior to the infection experiments, MS2 phage concentration, measured as plaque forming units (PFU)/mL, was calculated with a plaque assay. Briefly, serial dilutions of the phage suspension are plated as 3 μL spots on LB agar plates with a bacterial overlay (E. coli cells without plasmids at OD ≈ 0.6 mixed with soft LB agar (7.5 g/L)) followed by incubation for 12-15 h at 37°C. The number of quantifiable plaques in one dilution are then used to calculate PFU/mLI. In order to assess Cas13a-based protection from MS2, E. coli cells containing the active or dead nuclease and the guide encoding plasmids have been grown overnight in selective LB medium, back-diluted to ABS600 = 0.05 and let grow until ABS600 ≈ 0.3. The samples have then been normalized to ABS600 = 0.3 and aliquoted in a 96 well plate (Thermo Scientific, 167008) together with different amounts of phages (MOI 0.001 and 0.05) or the respective volume of LB for the no-infection control. Cell growth has been recorded over 16 h by measuring ABS600 every three minutes in a plate reader at 37°C.
In order to evaluate phage amplification, E. coli CGSC 4401 cells expressing the active or dead nuclease plasmids and the M2 crRNA plasmid were grown overnight in selective medium, back-diluted to OD = 0.01 and infected at OD ≈ 0.1 with MS2 phages (MOI = 0.01 or 1.0). After 3 h, phage particles were isolated from the bacterial cultures by centrifugation, cell lysis with chloroform and recovery of the aqueous phase. Phage titres were measured with the plaque assay described above. In parallel, cell growth (ABS600) of the same cultures after infection were measured for 16 h in a plate reader at 37°C.
Tolerance to targeted kanR gene
E. coli colonies containing the nuclease and crRNA (K1, K2) plasmids were transformed with the targeted plasmid, which has a cloDF13 ori, different promoters in front of the kanR gene (P2, P5, P8), and a non-targeted hygromycin (Hyg, 100 μg/mL) resistance cassette. The plasmid was cloned in two steps by Gibson assembly with pUA66 as the backbone. K1 and K2 were targeting two different regions within the kanR transcript. The procedure is analogous to the transformation assay with a plasmid-encoded transcript, with the targeted plasmid selected on Hyg. The next day, colonies from the spot dilutions were counted, and colonies from the samples showing a negligible reduction in transformation compared to the non-targeting control were inoculated overnight in LB with Cm, Amp and Hyg. Then the cultures were washed to remove Hyg and back-diluted to ABS600 = 0.01 in either Hyg or Kan. The growth curves for the different conditions were measured over time on the microplate reader for 14 h at 37°C by measuring ABS600 every 3 min. For generating the heatmap, 12 h time points were selected to compare the different conditions and the resulting graph is the average of 9 biological replicates.
Data analysis and image visualization
Microsoft Excel was used to analyze the data, and GraphPad Prism was used to generate the bar plots and heatmaps. The graphs were then modified in Adobe Illustrator to construct the final figures. Transformation fold-reduction in the transformation assays was calculated as the ratio between non-targeting and targeting colonies.
Quantification and statistical analyses
See individual methods descriptions for quantification. Analysis of the library screen was performed using the programs BBMerge and edgeR, with differential abundance determined using the quasi-likelihood F test. RNA-seq analysis was conducted using the programs STAR and HTseq. Analysis of the machine learning was performed using TreeSHAP. These analyses were facilitated with custom code available on Mendeley (see key resources table). See the associated methods for details. All statistical analyses for the transformation assays and phage infection assays were performed in GraphPad Prism using a Welch’s t test assuming unequal variances. All correlations were calculated based on the Spearman coefficient. The number of replicates is specified in the associated figure legends. Each replicate represents a biological replicate of the specified experiment. P-values above 0.05 or average values lower than the reference average were considered non-significant. Statistical comparisons for the transformation assays relied on log values, which assumes the samples are normally distributed on a log scale. More detailed information related to the single experiments (e.g. n value) can be found in the figure legends.
Supplementary Material
Document S1. Figures S1–S7 and Supplementary Table S1.
Supplementary Table S4. Library design and oligos. Related to Figure 3.
HIGHLIGHTS.
Cas13-induced dormancy requires RNA target levels to exceed an expression threshold
The expression threshold can prevent cytotoxic autoimmunity for endogenous targets
The threshold shifts depending on the CRISPR RNA guide:target pair
The threshold allows cells to distinguish pathogenic and benign infections
ACKNOWLEDGMENTS
We thank Fani Ttofali and Chunyu Liao for providing initial plasmids from which constructs used in this work were generated. We also thank Tatjana Achmedov for technical advice and assistance. pZ003 (LshC2c2 locus) was a gift from Feng Zhang (Addgene plasmid # 70168 ; http://n2t.net/addgene:70168 ; RRID:Addgene_70168). DS-NMcas was a gift from George Church (Addgene plasmid # 48646 ; http://n2t.net/addgene:48646 ; RRID:Addgene_48646). This work was supported by the Joint Programming Initiative on Antimicrobial Resistance (01KI1824 to C.L.B.), the National Institutes of Health (1R35GM119561 to C.L.B.), a bayresq.net Bavarian research network grant (to L.B.), and the Defense Advanced Research Projects Agency Safe Genes program (HR0011-17-2-0042 to C.L.B.). The views, opinions and/or findings expressed should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
C.L.B. is a co-founder and member of the Scientific Advisory Board for Locus Biosciences as well as a member of the Scientific Advisory Board for Benson Hill. The other authors have no conflicts of interest to declare.
REFERENCES
- Abbott TR et al. (2020) ‘Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza’, Cell, 181(4), pp. 865–876.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO et al. (2016) ‘C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector’, Science, 353(6299), p. aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO et al. (2017) ‘RNA targeting with CRISPR-Cas13’, Nature, 550(7675), pp. 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afroz T et al. (2015) Trade-offs in engineering sugar utilization pathways for titratable control’, ACS synthetic biology, 4(2), pp. 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT and Huber W (2015) ‘HTSeq--a Python framework to work with high-throughput sequencing data’, Bioinformatics , 31(2), pp. 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R et al. (2007) ‘CRISPR provides acquired resistance against viruses in prokaryotes’, Science, 315(5819), pp. 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J et al. (2013) ‘Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system’, Nature, 493(7432), pp. 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL et al. (2018) ‘Bacteriophage Cooperation Suppresses CRISPR-Cas3 and Cas9 Immunity’, Cell, 174(4), pp. 917–925.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman AB et al. (2020) ‘Programmable RNA Targeting Using CasRx in Flies’, The CRISPR journal, 3(3), pp. 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B, Rood J and Singer E (2017) ‘BBMerge - Accurate paired shotgun read merging via overlap’, PloS one, 12(10), p. e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E et al. (2015) ‘Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity’, FEMS microbiology reviews, 39(3), pp. 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT et al. (2017) ‘RNA editing with CRISPR-Cas13’, Science, 358(6366), pp. 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A et al. (2013) ‘STAR: ultrafast universal RNA-seq aligner’, Bioinformatics , 29(1), pp. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A et al. (2016) ‘Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection’, Nature, 538(7624), pp. 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB et al. (2002) ‘Stochastic Gene Expression in a Single Cell’, Science, 297(5584), pp. 1183–1186. [DOI] [PubMed] [Google Scholar]
- Feurer M et al. (2019) ‘Auto-sklearn: Efficient and Robust Automated Machine Learning’, in Automated Machine Learning. Springer, pp. 113–134. [Google Scholar]
- Frost LS et al. (2005) ‘Mobile genetic elements: the agents of open source evolution’, Nature reviews. Microbiology, 3(9), pp. 722–732. [DOI] [PubMed] [Google Scholar]
- Gasiunas G et al. (2012) ‘Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria’, Proceedings of the National Academy of Sciences of the United States of America, 109(39), pp. E2579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GW and Marraffini LA (2015) ‘Resistance and tolerance to foreign elements by prokaryotic immune systems — curating the genome’, Nat Rev Immunol, 15(11), pp. 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H and Fiers W (1982) ‘Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes’, Gene, 18(3), pp. 199–209. [DOI] [PubMed] [Google Scholar]
- Hajjem A, Bellavance F and Larocque D (2014) ‘Mixed-effects random forest for clustered data’, Journal of statistical computation and simulation, 84(6), pp. 1313–1328. [Google Scholar]
- Hampton HG, Watson BNJ and Fineran PC (2020) The arms race between bacteria and their phage foes’, Nature, 577(7790), pp. 327–336. [DOI] [PubMed] [Google Scholar]
- Hille F et al. (2018) The Biology of CRISPR-Cas: Backward and Forward’, Cell, 172(6), pp. 1239–1259. [DOI] [PubMed] [Google Scholar]
- Hoikkala V et al. (2021) ‘Cooperation between Different CRISPR-Cas Types Enables Adaptation in an RNA-Targeting System’, mBio, 12(2), pp. e03338–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh N et al. (2020) ‘A versatile toolkit for CRISPR-Cas13-based RNA manipulation in Drosophila’, Genome Biol, 21, p. 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SA et al. (2017) ‘CRISPR-Cas: Adapting to change’, Science, 356(6333), p. eaal5056. [DOI] [PubMed] [Google Scholar]
- Jinek M et al. (2012) ‘A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity’, Science, 337(6096), pp. 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M et al. (2017) ‘A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems’, Science, 357(6351), pp. 605–609. [DOI] [PubMed] [Google Scholar]
- Keseler IM et al. (2017) The EcoCyc database: reflecting new knowledge about Escherichia coli K-12’, Nucleic acids research, 45(D1), pp. D543–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiga K et al. (2020) ‘Development of CRISPR-Cas13a-based antimicrobials capable of sequence-specific killing of target bacteria’, Nature communications, 11(1), p. 2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S et al. (2018) ‘Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors’, Cell, 173(3), pp. 665–676.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV et al. (2020) ‘Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire’, Nature reviews. Genetics, 21(2), pp. 119–131. [DOI] [PubMed] [Google Scholar]
- Landsberger M et al. (2018) ‘Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity’, Cell, 174(4), pp. 908–916.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT and Beisel CL (2017) ‘Deciphering, Communicating, and Engineering the CRISPR PAM’, Journal of molecular biology, 429(2), pp. 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C et al. (2019) ‘Modular one-pot assembly of CRISPR arrays enables library generation and reveals factors influencing crRNA biogenesis’, Nature communications, 10(1), p. 2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li X, Ma J, et al. (2017) The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a’, Cell, 170(4), pp. 714–726.e10. [DOI] [PubMed] [Google Scholar]
- Liu L, Li X, Wang J, et al. (2017) Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities’, Cell, 168(1-2), pp. 121–134.e12. [DOI] [PubMed] [Google Scholar]
- Lorenz R et al. (2011) ‘ViennaRNA Package 2.0’, Algorithms for molecular biology: AMB, 6, p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg SM et al. (2020) ‘From local explanations to global understanding with explainable AI for trees’, Nat Mach Intell., 2(1), pp. 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahas A, Aman R and Mahfouz M (2019) ‘CRISPR-Cas13d mediates robust RNA virus interference in plants’, Genome biology, 20(1), p. 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS et al. (2015) ‘An updated evolutionary classification of CRISPR-Cas systems’, Nature reviews. Microbiology, 13(11), pp. 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS et al. (2020) ‘Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants’, Nature reviews. Microbiology, 18(2), pp. 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ND et al. (2020) ‘Anti-CRISPR protein applications: natural brakes for CRISPR-Cas technologies’, Nat Methods, 17(5), pp. 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA and Sontheimer EJ (2010a) ‘CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea’, Nature reviews. Genetics, 11(3), pp. 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA and Sontheimer EJ (2010b) ‘Self versus non-self discrimination during CRISPR RNA-directed immunity’, Nature, 463(7280), pp. 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R et al. (2018) ‘Rapid and Scalable Characterization of CRISPR Technologies Using an E. coli Cell-Free Transcription-Translation System’, Molecular cell, 69(1), pp. 146–157.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R, Beisel CL and Noireaux V (2020) ‘Rapid Testing of CRISPR Nucleases and Guide RNAs in an Cell-Free Transcription-Translation System’, STAR protocols, 1(1), p.100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ et al. (2020) ‘A phage-encoded anti-CRISPR enables complete evasion of type VI-A CRISPR-Cas immunity’, Science, 369(6499), pp. 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ and Marraffini LA (2018) ‘RNA Guide Complementarity Prevents Self-Targeting in Type VI CRISPR Systems’, Molecular cell, 71(5), pp. 791–801.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ, Nakandakari-Higa S and Marraffini LA (2019) ‘Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage’, Nature, 570(7760), pp. 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner O et al. (2017) Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers’, Nature, 548(7669), pp. 543–548. [DOI] [PubMed] [Google Scholar]
- Nobrega FL et al. (2020) ‘Prophages are associated with extensive CRISPR-Cas autoimmunity’, Nucleic acids research, 48(21), pp. 12074–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J et al. (2014) ‘Unravelling the structural and mechanistic basis of CRISPR-Cas systems’, Nature reviews. Microbiology, 12(7), pp. 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- özcan A et al. (2021) ‘Programmable RNA targeting with the single-protein CRISPR effector Cas7-11’, Nature, 597(7878), pp. 720–725. [DOI] [PubMed] [Google Scholar]
- Partridge SR et al. (2018) ‘Mobile Genetic Elements Associated with Antimicrobial Resistance’, Clin Microbiol Rev., 31(4), pp. e00088–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ and Smyth GK (2009) ‘edgeR: A Bioconductor package for differential expression analysis of digital gene expression data’, Bioinformatics, 26(1), pp. 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD and Oshlack A (2010) ‘A scaling normalization method for differential expression analysis of RNA-seq data’, Genome biology, 11(3), p. R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostøl JT and Marraffini LA (2019) ‘Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR-Cas immunity’, Nature microbiology, 4(4), pp. 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis HM (2011) ‘The ribosome binding site calculator’, Methods in enzymology, 498, pp. 19–42. [DOI] [PubMed] [Google Scholar]
- Smargon AA et al. (2017) ‘Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28’, Molecular cell, 65(4), pp. 618–630.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A et al. (2010) ‘Self-targeting by CRISPR: gene regulation or autoimmunity?’, Trends Genet, 26(8), pp. 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Kono DH and Baccala R (2017) ‘The multiple pathways to autoimmunity’, Nature Immunol, 18(7), pp. 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderWal AR et al. (2021) ‘CRISPR-Csx28 forms a Cas13b-activated membrane pore required for robust CRISPR-Cas adaptive immunity’, BioRxiv [Preprint], doi: 10.1101/2021.11.02.466367. [DOI] [Google Scholar]
- Vercoe RB et al. (2013) ‘Cytotoxic Chromosomal Targeting by CRISPR/Cas Systems Can Reshape Bacterial Genomes and Expel or Remodel Pathogenicity Islands’, PLoS Genet, 9(4), p. e1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q et al. (2019) ‘The CRISPR- Cas13a Gene- Editing System Induces Collateral Cleavage of RNA in Glioma Cells’, Advanced Science, 6(20), p. 1901299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S et al. (2019) ‘Composition and Diversity of CRISPR-Cas13a Systems in the Genus’, Frontiers in microbiology, 10, p. 2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels H-H et al. (2020) ‘Massively parallel Cas13 screens reveal principles for guide RNA design’, Nature biotechnology, 38(6), pp. 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y et al. (2021) ‘Precise editing of FGFR3-TACC3 fusion genes with CRISPR-Cas13a provides a personalized therapeutic strategy for the treatment of human glioblastoma’, Mol. Ther, 29(11), pp. 3305–3318. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu C et al. (2021) ‘Programmable RNA editing with compact CRISPR–Cas13 systems from uncultivated microbes’, Nat Methods, 18(5), pp. 499–506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S7 and Supplementary Table S1.
Supplementary Table S4. Library design and oligos. Related to Figure 3.
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original images of the RNA gels have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All original code has been deposited at Mendeley and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E.coli MG1655 | Susan Gottesman’s lab (NIH) | N/A |
| E. coli MG1655 ΔaraBAD ΔCM PconAraFGH | This manuscript | N/A |
| E.coli K12 (CGSC 4401) | DSMZ | DSM 9037 (CGSC 4401) |
| E. coli bacteriophage MS2 | DSMZ | DSM 13767 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Chloramphenicol | Roth - Carl Roth | Cat# 3886.2 |
| Ampicillin | Roth - Carl Roth | Cat# K029.2 |
| Kanamycin | Roth - Carl Roth | Cat# T832.3 |
| Hygromycin B | Roth - Carl Roth | Cat# 1287.2 |
| BsmBI-v2 | New England BioLabs (NEB) | Cat# R0739L |
| Instant Sticky-end Ligase Master Mix | New England BioLabs (NEB) | Cat# M0370S |
| Q5® Site-Directed Mutagenesis Kit | New England BioLabs (NEB) | Cat# E0554S |
| Gibson Assembly® Master Mix | New England BioLabs (NEB) | Cat# E2611L |
| KAPA HiFi HotStart ReadyMix | Roche Diagnostic | Cat# 7958935001 |
| Agencourt® AMPure® XP, 60 mL | Beckman Coulter | Cat# A63881 |
| TURBO DNase | Thermo Fisher Scientific | Cat# AM2239 |
| myTXTL Sigma 70 Master Mix | Arbor Biosciences | Cat# 507005 |
| Critical Commercial Assays | ||
| Direct-zol™ RNA MiniPrep Plus kit | Zymo research | Cat# R2072 |
| ZymoPURE II Maxiprep Kit | Zymo Research | Cat# D4203 |
| ZymoPure II Plasmid Midiprep Kit | Zymo Research | Cat# D4200 |
| NextSeq 500/550 Mid Output Kit v2.5 (300 Cycles) | Illumina | Cat# 20024905 |
| Rybo-off rRNA depletion kit | Vazyme Biotech | Cat# N407-01 |
| NEBNext® Ultra™ II Directional RNA Library Prep Kit | New England BioLabs (NEB) | Cat# E7760L |
| NovaSeq 6000 S1 PE Reagent Kit (100 cycles) | Illumina | Cat# 20012865 |
| Deposited Data | ||
| Genome-wide screen | NCBI’s Gene Expression Omnibus | GEO Series accession number GSE179913 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179913) |
| Transcriptomic analysis | NCBI’s Gene Expression Omnibus | GEO Series accession number GSE179914 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179914) |
| Original gel images | Mendeley | 10.17632/97s4hpvztb.1 |
| Original code | Mendeley | doi:10.17632/d5vw9sjcrv.1 |
| E. coli K12 MG1655 genome | NCBI | NC_000913.3 |
| Oligonucleotides | ||
| For all oligos used in the study see Supplementary Table 3 (SI Tables Excel file, tab 2). | ||
| For oligos used for library experiment see Supplementary Table 4 (SI Tables Excel file, tab 3). | ||
| Recombinant DNA | ||
| For plasmids used in this study see Supplementary Table 2 (SI Tables Excel file, tab 1). | ||
| Software and Algorithms | ||
| Wessels et al. algorithm | Wessels et al., 2020 | https://gitlab.com/sanjanalab/cas13 |
| RNAfold version 2.4.12. | Vienna RNA Package (Lorenz et al., 2011) | https://www.tbi.univie.ac.at/RNA/index.html |
| BBMerge (version 38.69) | Bushnell et al., 2017 | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbmerge-guide/ |
| edgeR (version 3.28.0) | Robinson and Oshlack, 2010; Robinson et al., 2009 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| RBS calculator (version 1.0) | Salis, 2011 | https://github.com/hsalis/Ribosome-Binding-Site-Calculator-v1.0 |
| STAR (version 2.7.4a) | Dobin et al., 2013 | https://github.com/alexdobin/STAR/releases |
| HTSeq (version 0.9.1) | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.9.1/ |
| auto-sklearn (version 0.10.0) | Feurer et al., 2019 | https://automl.github.io/auto-sklearn/master/releases.html# |
| EcoCyc | Keseler et al., 2017 | https://ecocyc.org |
| TreeSHAP (version 0.36.0) | Lundberg et al., 2020 | https://github.com/slundberg/shap |
| Mixed-effects random forest (MERF) | Hajjem et al., 2014 | https://github.com/manifoldai/merf |
| Other | ||
| See Supplementary Table 1 for list of Anderson promoters used in this study | http://parts.igem.org/Promoters/Catalog/Anderson | N/A |