

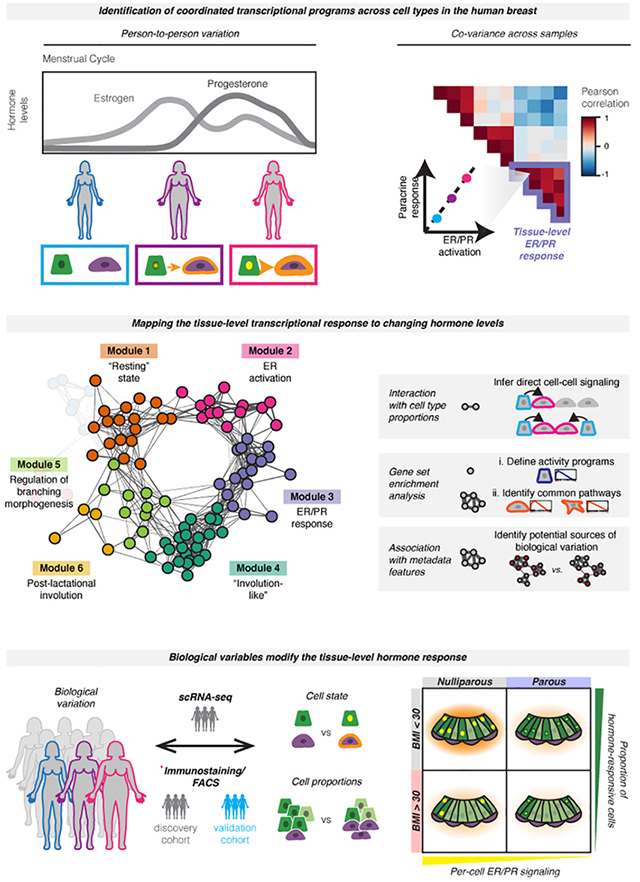
Summary
The rise and fall of estrogen and progesterone across menstrual cycles and during pregnancy regulates breast development and modifies cancer risk. How these hormones impact each cell type in the breast remains poorly understood, because they act indirectly through paracrine networks. Using single-cell analysis of premenopausal breast tissue, we reveal a network of coordinated transcriptional programs representing the tissue-level response to changing hormone levels. Our computational approach, DECIPHER-seq, leverages person-to-person variability in breast composition and cell state to uncover programs that co-vary across individuals. We use differences in cell-type proportions to infer a subset of programs that arise from direct cell-cell interactions regulated by hormones. Further, we demonstrate that prior pregnancy and obesity modify hormone responsiveness through distinct mechanisms: obesity reduces the proportion of hormone-responsive cells, whereas pregnancy dampens the direct response of these cells to hormones. Together, these results provide a comprehensive map of the cycling human breast.
Keywords: scRNA-seq, cell-cell interactions, human breast, hormone signaling, single-cell genomics, sample heterogeneity
Graphical Abstract
eTOC Summary
Estrogen and progesterone regulate breast development and modify cancer risk. Using singlecell analysis and leveraging person-to-person variability to identify gene programs that co-vary across individuals, Murrow et al. map the tissue-level response to ovarian hormones. Prior pregnancy and obesity modify hormone-responsiveness in the breast through distinct mechanisms.
Introduction
Coordinated interactions between cells are essential for the development and maintenance of normal tissue function, and dysregulation of cell-cell interactions is a key driver of disease. In the human breast, fluctuations in the levels of estrogen and progesterone with each menstrual cycle and during pregnancy control cell growth, survival, differentiation, and tissue morphology. The impact of these changes is profound: cumulative lifetime exposure to cycling hormones is a major modifier of breast cancer risk (Collaborative Group on Hormonal Factors in Breast Cancer, 2012), and the majority of breast tumors are estrogen-dependent. However, many of the effects of ovarian hormones within the breast are indirect. The estrogen and progesterone receptors (ER/PR) are expressed in only 10-15% of cells within the epithelium (Clarke et al., 1997). Thus, most of the changes that occur in response to hormone receptor activation are mediated by a complex cascade of paracrine signaling from hormone-responsive (HR+) cells to other cell types in the breast. Accordingly, cell-cell interactions between HR+ cells and other cell types are key to normal breast morphogenesis. However, due to a number of challenges inherent to hormone signaling and human breast biology, we lack a systems-level understanding of how different cell populations respond to cycling hormone levels.
A first challenge for understanding the tissue-level response to estrogen and progesterone is that there are major differences in glandular architecture and stromal composition and complexity between humans and model organisms like the mouse (Dontu and Ince, 2015; Parmar and Cunha, 2004). For example, while ER expression is restricted to the epithelium in humans, it is also expressed in the stroma in rodents (Mueller et al., 2002; Palmieri et al., 2004). Therefore, understanding the consequences of cellular crosstalk downstream of estrogen and progesterone requires studying these processes in humans or human models.
A second challenge is that the human breast is both heterogeneous across individuals and characterized by a highly dynamic microenvironment. There is a high degree of variability between individuals in terms of epithelial architecture (Russo et al., 1992), cell composition (Nakshatri et al., 2015; Rosenbluth et al., 2020), and hormone-responsiveness (Dunphy et al., 2020; Muenst et al., 2017; Tanos et al., 2013), and these differences likely impact both normal breast function and breast cancer susceptibility. Within individuals, the menstrual cycle and pregnancy/lactation/involution cycle are major drivers of epithelial remodeling, characterized by alternating periods of epithelial expansion and regression in response to changing hormone levels (Anderson et al., 1982; Jindal et al., 2014, Soderqvist et al., 1997, Russo et al., 1992). Histological analyses of paraffin-embedded human tissue sections have also identified cyclical alterations in epithelial architecture and stromal organization across the menstrual cycle (Ramakrishnan et al., 2002; Vogel et al., 1981) and broad remodeling following weaning (Lyons et al., 2011; O’Brien et al., 2010). However, little is known about how this underlying heterogeneity impacts cell state and the intercellular signaling networks that control tissue morphogenesis. As it enables unbiased analysis of cell types within the human mammary gland at single-cell resolution, single-cell RNA sequencing (scRNA-seq) is particularly well-suited to investigate this problem.
Here, we use scRNA-seq in a cohort of twenty-eight premenopausal reduction mammoplasty tissue specimens to trace the transcriptional changes that occur in the human breast downstream of hormone signaling. To provide insight into the cellular interactions that regulate breast tissue homeostasis, we develop DECIPHER-seq (Deconstructing Cell-cell Interactions using Phenotypic Heterogeneity in single-cell RNA sequencing data), a systematic computational approach that leverages the high degree of inter-sample transcriptional heterogeneity in the breast to identify coordinated interaction networks across cell types. Our approach was guided by two hypotheses. First, we predicted that if two cell types are acting together—via either direct cell-to-cell signaling or a response to shared microenvironmental/paracrine cues—the transcriptional signatures representing those cell-cell interactions should be correlated across samples. More specifically, since the effects of estrogen and progesterone on other cell types in the breast are controlled by paracrine signaling from HR+ luminal cells, we reasoned that hormone receptor activation in HR+ luminal cells would be correlated with transcriptional changes in other cell types, representing the downstream paracrine response. Second, we predicted that we could infer the types of cell-cell interactions that make up each pairwise correlation by incorporating information about: 1) the proportion of each cell type across samples, 2) the similarity of each transcriptional signature, and 3) enrichment of common biological pathways across signatures. Thus, we sought to use the inter-sample transcriptional variability and differences in cell type proportions present in the dataset as a type of “natural experiment” to understand how the behaviors of different cell types in the breast are coordinated at the tissue level.
Based on this approach, we identify a network of coordinated activity programs in HR+ cells and other cell types that represent the dynamic tissue-level response of the human breast to changing hormone levels. Using differences in cell-type proportions across samples, we infer a subset of activity programs that depend on direct cell-to-cell signaling, and find that these direct interactions primarily comprise signaling from HR+ cells to other cell types. Using these data, we generate hypotheses about how person-to-person variation at the tissue level is linked to specific biological mechanisms at the cellular level, and directly test these hypotheses using flow cytometry and immunostaining in an expanded cohort of samples. We find that paracrine signaling from HR+ cells to neighboring cell types depends on both the magnitude of the ER/PR transcriptional response and the overall abundance of HR+ cells in the tissue. Accordingly, we demonstrate that prior pregnancy and obesity both lead to decreased hormone responsiveness in the breast, but act through distinct mechanisms: pregnancy influences the magnitude of the ER/PR signaling response in HR+ luminal cells, whereas obesity reduces the proportion of HR+ cells and therefore downstream paracrine signaling. These changes are consistent with the protective effect of prior pregnancy and high body mass index (BMI) against premenopausal breast cancer. Overall, these results provide a comprehensive map of the cycling human breast and the dynamic cell-cell interactions that underlie normal breast function and breast cancer risk. More broadly, we describe a systematic approach to unravel the functional significance of person-to-personal variability in the human breast at the tissue level, by linking individual cell types’ transcriptional signatures to higher order modules of cell-cell interactions.
Results
Person-to-person variability in transcriptional cell state in the premenopausal human breast
To identify inter-individual differences in transcriptional cell state in the human breast, we performed scRNA-seq on 86,136 cells collected from 28 healthy premenopausal donors who underwent reduction mammoplasty surgery (Figure 1A, Figure S1A, and Table S1). To obtain an unbiased snapshot of the epithelium and stroma, we collected live (DAPI negative) singlet cells from all samples by fluorescence activated cell sorting (FACS) (Figure S1A-B, Table S2). For a subset of samples, we also collected purified epithelial cells or purified luminal and basal/myoepithelial cells (Figure S1A-B, Table S2). We used MULTI-seq barcoding and in silico genotyping for sample multiplexing to minimize technical variability between samples (Figure S1C, Table S2, and STAR Methods) (Heaton et al., 2020; McGinnis et al., 2019).
Figure 1. Sample-to-sample variability in transcriptional cell state in the premenopausal human breast.

(A) Single-cell transcriptional analysis links biological variables with person-to-person heterogeneity in transcriptional cell state. scRNA-seq workflow: Reduction mammoplasty samples were processed to epithelial-enriched tissue fragments, then to single cells, followed by MULTI-seq sample barcoding, library preparation using the 10X Chromium system, and sequencing.
(B) The major epithelial and stromal cell types in the breast were identified and visualized by UMAP dimensionality reduction and unsupervised clustering of twenty-eight samples reduction mammoplasty samples (GSE198732, Table S1).
(C) Density plots (arbitrary units, linear scale) highlighting the transcriptional cell state of hormone-responsive (HR+) luminal cells and basal/myoepthelial cells from each sample.
(D) Overview of conceptual approach: We hypothesized that hormone receptor activation would represent a major source of transcriptional variability in our dataset, and that hormone receptor activation in hormone-responsive (HR+) luminal cells would correlate with transcriptional changes in other cell types—representing the downstream paracrine response. Based on differences in hormone levels due to menstrual cycling (depicted, left) or hormonal contraceptive use, we predicted that gene expression programs representing ER/PR signaling in HR+ luminal cells and the downstream signaling response in other cell types would co-vary across samples (right).
(E) Using individual pairwise correlations between cell activities, DECIPHER-seq builds a tissue-level map of the cell-cell interactions present in the healthy human breast and identifies modules of transcriptional states that co-occur across the same sets of samples. In downstream analyses, we exclude modules driven by non-cell-type specific responses to shared signals, and uncover modules enriched for putative direct cell-cell signaling interactions. We define activity programs using gene set enrichment analysis, identify common pathways enriched across activity programs in a module, and uncover potential sources of biological variation by testing association with annotated metadata features.
Sorted basal and luminal cell populations were well-resolved by UMAP (Figure S1D). Unsupervised clustering identified one basal/myoepithelial cluster, two luminal clusters, and six stromal clusters (Figure 1B). Based on the expression of known markers, the two luminal clusters were annotated as hormone-responsive (HR+) and secretory luminal cells, and the six stromal clusters were annotated as fibroblasts, vascular endothelial cells, lymphatic endothelial cells (“lymphatic”), smooth muscle cells/pericytes (“vascular accessory”), lymphocytes, and macrophages (Figure 1B and Figure S1E-F). The luminal populations described here closely match those identified as “hormone-responsive/mature luminal” and “secretory/luminal progenitor” in previous scRNA-seq analyses of the human breast (Bhat-Nakshatri et al., 2021; Nguyen et al., 2018). Here, we use the nomenclature “hormone-responsive/HR+” and “secretory” to refer to these two luminal cell types. The HR+ cluster was enriched for the hormone receptors ESR1 and PGR (Figure S1G), and other known markers such as ANKRD30A (Figures S1E-F) (Nguyen et al., 2018). Consistent with previous studies demonstrating variable hormone receptor expression across the menstrual cycle (Battersby et al., 1992), expression of ESR1 and PGR transcripts were sporadic and often non-overlapping. Within the HR+ luminal cluster, 22% of the cells had detectable levels of ESR1 or PGR, with only 2% of hormone-responsive cells expressing both transcripts (Figure S1H).
Beyond identifying the major cell types, single-cell analysis resolved a high degree of person-to-person transcriptional variability in the human breast. Following batch-correction (Figure S2A) (Butler et al., 2018), cells from different individuals were represented across all cell-type clusters (cluster entropy = 0.93, STAR Methods) (Figure S1B). However, despite this mixing across cell types, individuals displayed distinct transcriptional signatures within individual cell type clusters (Figure 1C, Figure S1C). Because we used MULTI-seq to multiplex samples, we were able to confirm that this variation in cell state was not due to technical variation, as we directly compared cells from different samples that were multiplexed in the same batch to cells from matched samples that were run across multiple batches. Cells from the same sample were more similar to each other than cells from different samples, regardless of the batch/day of processing (Figure S2D-E, Table S2, and STAR Methods).
Inferring shared transcriptional responses and direct cell-to-cell signaling interactions in the human breast
Since estrogen and progesterone are master regulators of breast development, and the levels of these hormones fluctuate across the menstrual cycle, we predicted that ER/PR signaling and the downstream paracrine response would be a major source of transcriptional heterogeneity across samples in our dataset. Based on random sampling across the menstrual cycle and differences in hormonal contraceptive use, we would expect to identify samples with varying levels of ER/PR activation in hormone-responsive (HR+) luminal cells (Figure 1D). If these hormone-responsive cells are signaling to other cell types, such as basal cells, we would further expect to see a second activity program in those cells representing the downstream paracrine response. Finally, this “paracrine response” activity program should co-vary with the level of ER/PR activation across different samples (Figure 1D). Thus, we developed a computational pipeline, DECIPHER-seq, based on the hypothesis that inter-sample transcriptional variation contains meaningful information about how the behaviors of different cell types in the breast are coordinated at the tissue level, and that transcriptional signatures (“activity programs”) representing interactions between two cell types should correlate across samples. DECIPHER-seq uses individual pairwise correlations between activity programs to build a higher-order network map of coordinated cell-state changes in the human breast (Figure 1E).
The activities of two cell types can be coordinated in multiple ways. In the premenopausal breast, we expect the tissue-level response to hormones to lead to at least two types of coordinated interactions: direct cell-to-cell signaling interactions between HR+ cells and other cell types, and more complex downstream interactions involving cell-type-specific responses to a shared microenvironment. We predict that the first type of interaction would depend on the proportion of HR+ cells in the breast, whereas the second type of interaction would involve cell-type specific (e.g. transcriptionally distinct) activity programs that may be enriched for similar biological processes. Therefore, in downstream analyses, we infer modules that are enriched for direct cell-cell signaling interactions (i.e. modules containing links that depend on the proportion of one cell type across samples), and exclude modules driven by non-cell-type specific responses (i.e. modules containing transcriptionally similar activity programs) (Figure 1E). We also define individual activity programs and modules by performing gene set enrichment analysis, which allows us to infer higher-order functional interactions between multiple cell types. Finally, we uncover associations between annotated metadata features and sets of activity programs to infer potential sources of biological variation (Figure 1E). Known biology associated with paracrine signaling downstream of ER/PR activation provides a powerful “proof of concept” to establish that correlated changes in cell state can be used to identify biologically relevant cell-cell interactions.
To identify activity programs within cell types in the premenopausal breast, we performed non-negative matrix factorization (NMF) on each of the major cell type clusters in our dataset (Figure S3A). A similar approach was recently used by Pelka et al. to identify multicellular immune “hubs” in colorectal cancer (Pelka et al., 2021). We used integrative NMF (iNMF) (Gao et al., 2021; Welch et al., 2019), which successfully corrected for batch differences while retaining sample-to-sample transcriptional variability (Figures S4A-B), and adapted a consensus approach (Kotliar et al., 2019) to identify activity programs that were consistent across replicates (Figure S4C, STAR Methods). The main user-supplied parameter in NMF is the number of programs identified (rank, K). None of the three commonly used heuristics for guiding the choice of K identified an obvious “elbow” in our dataset (Figure S4D-E). We therefore developed a metric based on the goal of identifying the greatest number of robust (i.e. consistent across values of K) and unique (i.e. distinct from other programs at the same K) activity programs (Figure S5, STAR Methods). This approach identified distinct “blocks” of activity programs in multiple cell types that co-varied across samples (Figure 2A). To build a tissue-level map of these cell-cell interactions, we constructed a weighted network of coordinated activity programs based on the pair-wise Pearson correlations r (Figure S3B, Figure S6). Based on this analysis, we identified eight major modules comprising highly correlated transcriptional states across cell types in the breast (Figure 2A, S6D).
Figure 2. Inferring non-cell-type-specific transcriptional responses and direct cell-to-cell signaling interactions in the human breast.
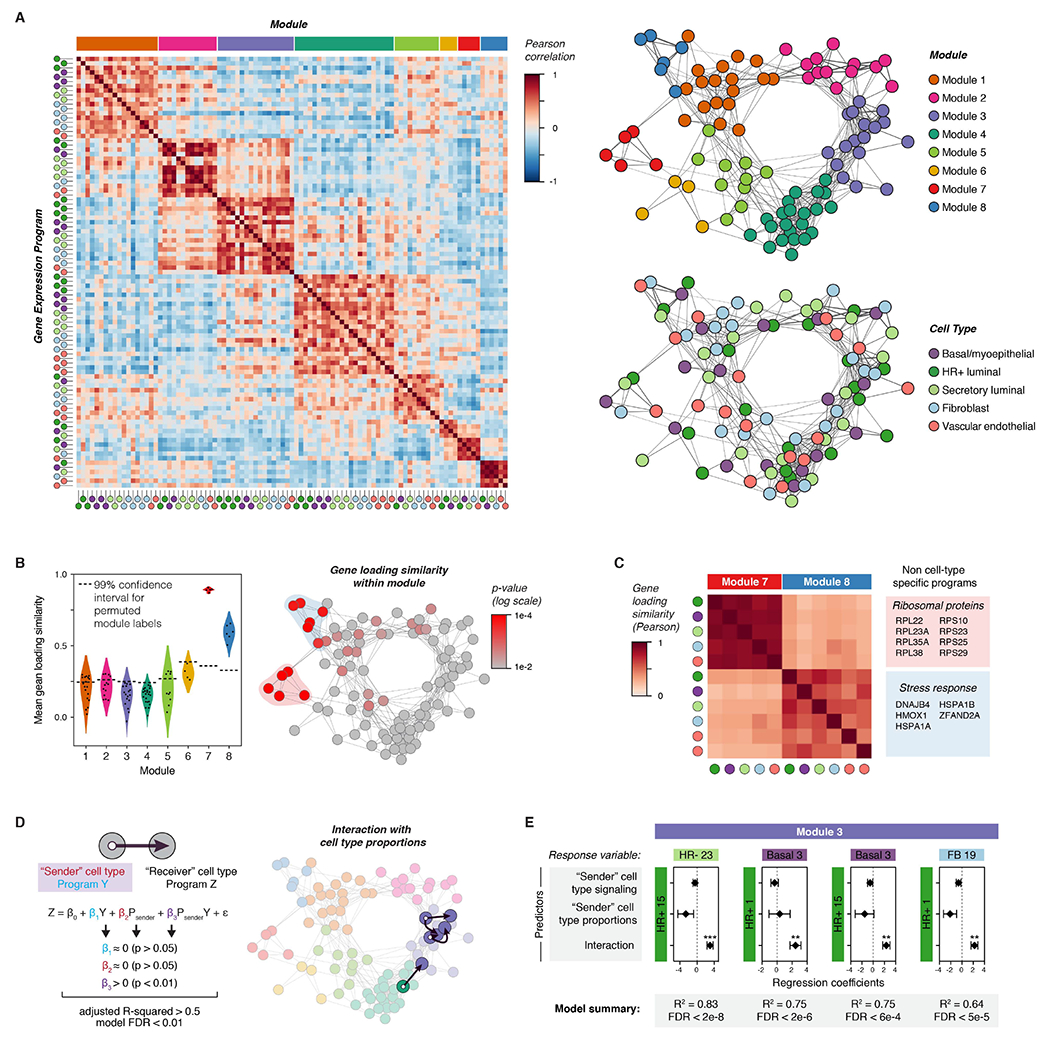
(A) Left: Heatmap depicting Pearson correlation coefficients between activity programs in the eight major modules identified by DECIPHER-seq. Right: Network graph of correlated activity programs in the human breast. Nodes represent distinct activity programs in the indicated cell types, and edges connect significantly correlated programs (Pearson correlation coefficient > 0, p < 0.05). Modules of correlated programs were identified using a Constant Potts Model for community detection.
(B) Left: Violin plot of the mean Pearson correlation between gene loadings for each activity program and all other activity programs in the same module (“gene loading similarity”). The horizontal dashed line represents the 99% confidence interval for permuted module labels. Right: Network graph of activity programs in the human breast, colored by the p-value for gene loading similarity for each program (log scale). P-values were calculated by permutation testing.
(C) Heatmap depicting Pearson correlation coefficients between gene loadings for the indicated activity programs. The colored boxes list the top-loading genes shared by all programs in the indicated modules.
(D) Network graph of activity programs in the human breast, with arrows highlighting inferred direct cell-cell interactions. We modeled each pairwise combination of activity programs as a linear response to the mean expression score of an activity program in a “sender” cell type (β1Y), the proportion of the “sender” cell type in the epithelium (β2 Psender), and an interaction term representing the combined effect of both terms (β3 PsenderY). Arrows highlight pairs where only the interaction term is significant, the model describes over 50% of the variation in the response variable, and the FDR-corrected p-value for the overall model is less than 0.01.
(E) Results from multiple linear regression analysis, depicting the four most significant (FDR < 0.01) inferred direct cell-cell interactions. For each pairwise combination, the response variable was modeled in response to three predictors: the expression score in a “sender” cell type (signaling), the proportion of the “sender” cell type, and an interaction term between both predictors. Points represent the regression coefficient for each predictor, error bars depict the standard error.
To exclude non-cell-type specific transcriptional responses—that are unlikely to be directly related to hormone signaling in the breast—we identified modules made up of activity programs with similar gene loadings. We found that modules 7 and 8 were highly enriched for activity programs with correlated gene loadings (Figure 2B, S7A). Programs in module 7 primarily consisted of ribosomal transcripts and genes involved in cellular respiration, whereas programs in module 8 consisted of stress response genes such as heat shock and chaperone proteins (Figure 2C, Figure S7C). We speculate that module 8 represents an artifact of tissue processing rather than biologically meaningful transcriptional variation, since prior studies have identified a similar signature in dissociated solid tissues (O’Flanagan et al., 2019). However, one advantage of DECIPHER-seq is that it describes cells as a combination of activity programs rather than forcing cells into distinct clusters. Thus, samples with high expression of “dissociation-related” activity programs still contain biologically meaningful signals from other programs and can be retained in the analysis.
Next, we inferred modules enriched for putative direct cell-cell signaling interactions by identifying interactions between two nodes that depended on both the magnitude of activity program expression in a “sender” cell type and the proportion of that sender cell type in the tissue (Figure 1E, Figure 2D). We reasoned that if one cell type was signaling to another, the activity program representing the transcriptional response in the “receiver” cell type should be sensitive to the proportion of sender cells in the tissue, particularly for direct interactions involving short-range signaling molecules. While this simplified model does not consider the effects of signal amplification, cooperation between signaling pathways, or higher-order interactions between more than two cell types, it identifies a subset of “high-confidence” direct cell-cell interactions that meet a set of simple criteria. We annotated putative direct cell-cell signaling interactions as those where the combined effects of signaling from a sender cell type and its proportion in a tissue described over 50% of the variation in activity program expression across samples in a second “receiver” cell type, and the individual effects of signaling and cell proportions were not significant (Figure 2D, STAR Methods). As the proportion of epithelial versus stromal cells in our samples may be influenced by tissue dissociation, we restricted our analysis to links between epithelial cell types as “sender” cells (HR+ luminal, secretory luminal, or basal cells) and all other cell types as “receivers”. We modeled each pairwise interaction as a linear response to three variables: signaling from a sender cell type (i.e. the mean expression score of an activity program in that cell type), the proportion of the sender cell type in the epithelium, and an interaction term representing the combined effects of signaling and cell proportions (Figure 2D). Consistent with our prediction about the nature of hormone signaling in the breast, four out of the five high-confidence direct cell-cell interactions (FDR < 0.01) were part of the same module (Module 3), and consisted of a link between HR+ luminal cells as the “sender” cell type and a second “receiver” cell type (Figure 2D-E).
ER/PR signaling and the downstream transcriptional response
We next performed marker and gene set enrichment analysis to define potential functions for activity programs within each module and identify common pathways upregulated across multiple activity programs in a module (STAR Methods, Table S3, Table S4). We first focused on Module 3 (Figure 3A, Figure S8A), as our previous analysis demonstrated that this module was highly enriched for putative direct cell-cell signaling interactions. Since estrogen and progesterone are master regulators of breast development that act via paracrine signaling from hormone-responsive (HR+) luminal cells to other cell types, we predicted that ER/PR signaling and the downstream paracrine response would represent a major source of direct cell-cell signaling signatures present in our dataset.
Figure 3. ER/PR signaling and the downstream response.
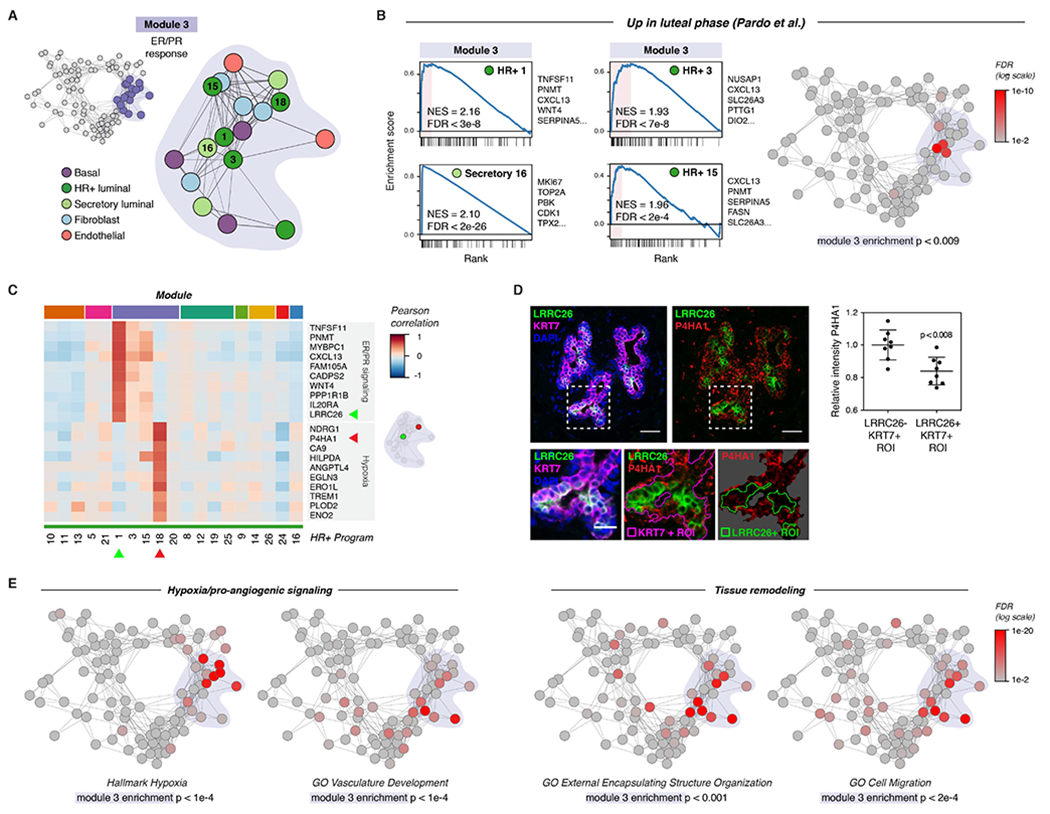
(A) Diagram highlighting activity programs in the “ER/PR response” module.
(B) Left: Gene set enrichment analysis of the indicated activity programs in the “ER/PR response” module, showing enrichment of genes upregulated during the luteal phase of the menstrual cycle (Pardo et al., 2014). The top five leading edge genes for each activity program are listed. Right: Network graph of activity programs, colored by the FDR for gene set enrichment of genes upregulated during the luteal phase of the menstrual cycle (log scale; Pardo et al., 2014). Overall enrichment of this gene set in the “ER/PR response” module was determined by permutation analysis.
(C) Heatmap of the top 10 marker genes for HR+ 1 and HR+ 8. Results depict the Pearson correlation between the expression score of the indicated activity programs and the normalized expression of the indicated genes across cells.
(D) Representative immunostaining for LRRC26, P4HA1, and KRT7 and quantification of the relative mean intensity of P4HA1 signal in LRRC26-/KRT7+ and LRRC26+/KRT7+ regions of interest. Data are represented as individual points, error bars indicate mean ± SEM of 8 regions from 3 samples with high ER/PR signaling. Scale bars 20 μm. Inset scale bar 10 μm.
(E) Network graph of activity programs, colored by the FDR for enrichment of the indicated gene sets in each activity program (log scale). Overall enrichment of gene sets within module 1 was determined by permutation analysis.
Consistent with this hypothesis, activity programs in Module 3—here annotated as the “ER/PR response” module—were highly enriched for genes previously found to be upregulated during the luteal phase of the menstrual cycle in a bulk RNA sequencing analysis (module enrichment p < 0.01; Figure 3B, Table S5) (Pardo et al., 2014). Activity program 1 in HR+ luminal cells (“ER/PR signaling”) was associated with high expression of the essential PR target genes WNT4 and TNFSF11 (RANKL) (Rajaram et al., 2015; Tanos et al., 2013), and enriched for transcripts in the Molecular Signatures Database Hallmark “early estrogen response” (p < 0.001) and “late estrogen response” (p < 0.01) gene sets (Figure 3C, Figure S8B-C) (Liberzon et al., 2015). Additional canonical hormone-responsive genes including TFF1, AREG, PGR, and VEGFA were highly expressed across multiple activity programs in this module (Figure S8D) (Aupperlee et al., 2013; Hyder et al., 2000; LaMarca and Rosen, 2007; Ribieras et al., 1998). Consistent with previous work demonstrating that STAT5 acts as a cofactor to mediate signaling downstream of PR activation in the breast, the ER/PR response module was also enriched for genes involved in IL-2/STAT5 signaling (module enrichment p < 1e-4; Figure S8E). Finally, gene set enrichment analysis identified a rare subpopulation of proliferative secretory luminal cells within the ER/PR response module (Figure 3B). This “proliferation” activity program (Secretory program 16) was highly enriched for cell-cycle related genes previously found to be upregulated during the luteal phase of the menstrual cycle (Figure 3B, Table S5) (Pardo et al., 2014).
Our analysis also revealed that high levels of ER/PR signaling in HR+ cells (HR+ 1) coincided with the emergence of a second transcriptional state in a distinct subpopulation of HR+ luminal cells (HR+ 18) (Figure 3C, S8F). Marker and gene set enrichment analysis demonstrated that HR+ program 18 was characterized by upregulation of a hypoxia gene signature and pro-angiogenic factors such as VEGFA and ANGPTL4 (Figure S8D, S8G). The identification of this “hypoxia” gene signature is consistent with a previous study using microdialysis of healthy human breast tissue which found that VEGF levels increased in the luteal phase of the menstrual cycle (Dabrosin, 2003). As estrogen response elements have been identified in the untranslated regions of VEGFA (Hyder et al., 2000), our results suggest that this increased expression may be, in part, a direct effect of hormone signaling to a subpopulation of HR+ cells.
To confirm these results in vivo, we performed marker analysis to identify genes specific to each cluster that could be used for immunostaining. We identified LRRC26 as a marker of the ER/PR signaling activity program HR+ 1 and P4HA1 as a marker of the hypoxia/pro-angiogenic activity program HR+ 18 (Figure 3C). In intact human tissue sections, we found that LRRC26 staining marked a distinct set of luminal cells from P4HA1 (Figure 3D). Moreover, these two subpopulations co-occurred within the same regions of the breast, demonstrating that they are unlikely to be an artifact of sample processing. Together, these results identify at least two diverging transcriptional states in HR+ cells in samples with high ER/PR signaling, one associated with signaling via RANK ligand and WNT4 to the surrounding epithelium and a second associated with a hypoxia-related/pro-angiogenic transcriptional signature.
We next expanded our analysis of gene activity programs to other epithelial lineages and stromal cell types in the “ER/PR response” module. Similar to program 18 in HR+ cells, multiple activity programs across other cell types in this module were enriched for transcripts involved in hypoxia and blood vessel remodeling including VEGFA and ANGPTL4 (Figure 3E, S8D, S8G). The ER/PR response module was also enriched for genes involved in tissue remodeling, cell migration, and ECM organization (Figure 3E, S8H), consistent with previously reported morphological changes in the breast epithelium (Ramakrishnan et al., 2002) and alterations in stromal organization and ECM composition (Ferguson et al., 1992; Hallberg et al., 2010) across the menstrual cycle. Stromal cell types in this module were characterized by upregulation of ECM and matrix remodeling proteins including collagens (COL3A1, COL1A2), the crosslinking enzyme LOXL2, and the cytokine TGFB3 (Figure S8I). Together, these results identify distinct transcriptional signatures for ER/PR activation in HR+ luminal cells and the downstream paracrine response in other cell types.
Coordinated changes in signaling states across cell types in the breast
Next, we used a similar approach to analyze the remaining five major modules—annotated here as “Resting state”, “Estrogen receptor (ER) activation”, “Involution-like”, “Post-lactational involution”, and “Regulation of branching morphogenesis”—each made up of highly interconnected transcriptional states across cell types in the breast (Figure 4A). The “Resting state” module (Module 1, Figure S9A) consisted of gene expression programs that were negatively correlated with ER/PR signaling (HR+ program 1) in HR+ luminal cells (Figure 4B). Activity programs in this module were enriched for pathways involved in RNA processing and transport (Figure S9B). The “ER activation” module (Module 2, Figure S9C), consisted of activity programs linked to both the “Resting state” and “ER/PR response” modules (Figure 4A). This module was enriched for genes involved in the unfolded protein response (UPR) and endoplasmic reticulum stress (Figure 4C, Figure S9D-E), as well as the response to estrogen (Figure S9D). Prior work has shown that estrogen receptor activation leads to a rapid “anticipatory” activation of the UPR in the absence of accumulation of unfolded proteins (Andruska et al., 2015). In keeping with this, expression of canonical estrogen receptor target genes such as PGR, AREG, TFF1, and TFF3 was most closely associated with HR+ cell activity programs in this “ER activation” module as well as the “ER/PR response” module (Figure 4D).
Figure 4. Coordinated changes in signaling states across cell types in the breast.
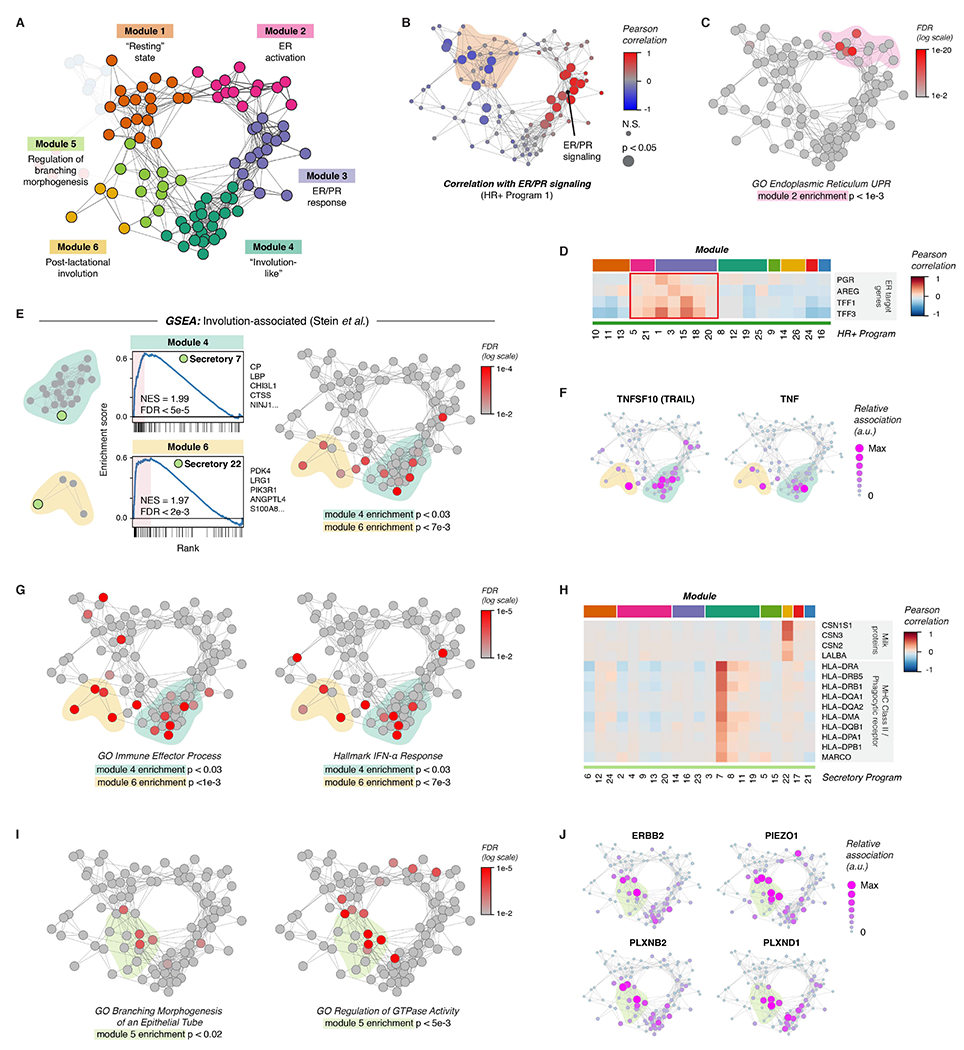
(A) Network diagram highlighting Modules 1-6.
(B) Network graph of activity programs in the human breast, colored by the Pearson correlation of each program’s mean expression score across samples with ER/PR signaling (HR+ activity program 1). Significant positive and negative correlations as identified by bootstrap resampling are represented by larger nodes.
(C) Network graph of activity programs, colored by the FDR for enrichment of genes in the GO Biological Process set “Endoplasmic Reticulum Unfolded Protein Response” (log scale). Overall enrichment of this gene set within module 2 was determined by permutation analysis.
(D) Heatmap of selected estrogen receptor (ER) target genes. Results depict the Pearson correlation between the expression score of the indicated activity programs and the normalized expression of ER target genes across cells.
(E) Left: Gene set enrichment analysis of the indicated activity programs in the “Involution-associated” and “Post-lactational involution” modules, showing enrichment of genes previously shown to be upregulated during the post-lactational involution in the mouse (Stein et al., 2004). The top five leading edge genes for each activity program are listed. Right: Network graph of activity programs, colored by the FDR for enrichment of genes upregulated in the Stein gene set (log scale). Overall enrichment of this gene set in each of the indicated modules was determined by permutation analysis.
(F) Network graph of activity programs, depicting the relative association of the indicated marker genes with each activity program (arbitrary units, linear scale).
(G) Network graph of activity programs, colored by the FDR for enrichment of the indicated gene sets in each activity program (log scale). Overall enrichment of gene sets within the indicated modules was determined by permutation analysis.
(H) Heatmap of selected genes including milk proteins, MHC Class II molecules, and the phagocytic receptor MARCO. Results depict the Pearson correlation between the expression score of the indicated activity programs and the normalized expression of the indicated genes across cells.
(I) Network graph of activity programs, colored by the FDR for enrichment of the indicated gene sets in each activity program (log scale). Overall enrichment of gene sets within module 5 was determined by permutation analysis.
(J) Network graph of activity programs, depicting the relative association of the indicated marker genes with each activity program (arbitrary units, linear scale).
Gene set enrichment analysis of the “Post-lactational involution” module (Module 6, Figure S10A) and “Involution-like” module (Module 4, Figure S10D) uncovered transcriptional signatures in secretory luminal cells that were similar to those that have been described during post-lactational involution in the mouse (Figure 4E, Figure S10E, Table S6) (Stein et al., 2004). Activity programs in both modules were characterized by high expression of death receptor ligands such as TNFSF10 (TRAIL) and TNF (Figure 4F) and of genes involved in the immune response, including interferon-response genes (Figure 4G). We annotated Module 6 as related to post-lactational involution, since activity program expression in secretory luminal cells within this module (secretory program 22) was highly associated with expression of milk proteins (Figure 4H) and genes involved in lactation (Figure S10B). Moreover, activity programs across all cell types in this module were more highly expressed in parous versus nulliparous samples (Figure S10C). This “Post-lactational involution” module was also enriched for genes involved in the acute phase response, complement proteins, and defense response, consistent with pathways that have been previously described as upregulated during post-lactational involution in the mouse (Figure S10F) (Stein et al., 2004). Since prior studies in human tissue samples have shown that differences in lobular area and epithelial architecture between parous and nulliparous women persist for up to 18 months following weaning (Jindal et al., 2014), we speculate that activity programs in this module may be associated with the time since weaning, although more complete patient data would be required to formally test this hypothesis.
The “Involution-like” signature (Module 4) in secretory luminal cells was characterized by expression of major histocompatibility complex class II (MHC-II) molecules and the phagocytic receptor MARCO (Figure 4H), suggesting that these cells play a role as non-professional phagocytes in the clearance of apoptotic cells, similar to what has been described during post-lactational involution (Monks et al., 2008). As previous data have demonstrated that the fraction of apoptotic cells in the mammary epithelium peaks between the late luteal and early follicular phases of the menstrual cycle, this module may represent the response to falling hormone levels at the end of the menstrual cycle (Anderson et al., 1982). TGFB3 is a major signaling molecule involved in post-lactational involution that enhances phagocytosis by mammary epithelial cells (Fornetti et al., 2016) suggesting that TGFB3 secreted by cells in response to ER/PR signaling (Figure S8I) activates a subset of secretory luminal cells that go on to express “involution-like” markers.
Finally, we annotated Module 5 (Figure S10G) as associated with “Regulation of branching morphogenesis” based on enrichment for the gene ontology (GO) term “branching morphogenesis of an epithelial tube” (Figure 4I, S10H). Consistent with the critical role of Rac and Rho GTPases in mammary branching (Ewald, 2008), the GO term “regulation of GTPase activity” was also highly enriched across this module (Figure 4I). Activity programs in this module were also associated with genes involved in cell motility, mechanotransduction, and invasion—including ERBB2, PIEZO1, PLXNB2, and PLXND1 (Figure 4J)—that have been previously described as important for epithelial remodeling (Gay et al., 2011; Stewart et al., 2021; Worzfeld et al., 2012).
Together, these results demonstrate how the underlying sample-to-sample variability in the breast can be used to infer functional connections between cell types in cell-cell interaction networks. Using DECIPHER-seq, we provide a comprehensive, systems-level view of the transcriptional changes that underlie normal breast morphogenesis.
The ER/PR signaling response of HR+ luminal cells is reduced in parous women
Previous epidemiologic analyses have demonstrated that prior pregnancy is highly protective against ER+/PR+ breast cancer (Fortner et al., 2019), and decreased hormone responsiveness following pregnancy is one proposed mechanism for this effect (Britt et al., 2007). Supporting this, previous studies demonstrated decreased expression of the PR effector WNT4 following pregnancy (Meier-Abt et al., 2014; Muenst et al., 2017). Moreover, in an explant culture model, estrogen consistently induced expression of the ER target gene AREG only in nulliparous women (Dunphy et al., 2020). As our network analysis suggested that activity programs in the “ER/PR response” module were dependent on both the magnitude of signaling from HR+ luminal cells and their proportion in the tissue (Figure 2D-E), we hypothesized that decreased hormone responsiveness could be caused by either: 1) a change in the magnitude of paracrine signals produced by each HR+ luminal cell, and/or 2) a reduction in the overall proportion of HR+ luminal cells leading to a “dilution” of paracrine signals following ER/PR activation. It has been difficult to distinguish between these mechanisms using bulk tissue-level analyses. By individually probing the single-cell transcriptional landscape of the HR+ luminal cell population and downstream cell types, scRNA-seq provided a means to directly interrogate whether parity influences the per-cell hormone signaling response of HR+ luminal cells.
To quantify variation in ER/PR signaling in HR+ luminal cells, we first measured the similarity between each sample’s single-cell distribution across HR+ activity program 1 (ER/PR signaling). Hierarchical clustering identified two sets of samples, representing those with high or low ER/PR signaling (Figure 5A). Based on this, we found that while the levels of hormone signaling in HR+ luminal cells varied between nulliparous women—likely reflecting differences in hormone levels across the menstrual cycle or due to hormonal contraceptive use—per-cell ER/PR signaling in HR+ luminal cells was significantly reduced in parous women (p < 0.02, Mann-Whitney test; Figure 5B) and did not depend on other biological variables such as age and body mass index (Figure S11A). Equal numbers of individuals from each cohort were using hormonal contraceptives (n = 4 out of 11 nulliparous or parous individuals, Table S1). For women not using hormonal contraceptives (n = 7 out of 11 nulliparous or parous individuals), we modeled the expected number of samples with high ER/PR signaling based on a binomial distribution using average menstrual cycle phase lengths (Bull et al., 2019). The number of nulliparous samples with high ER/PR signaling was consistent with the expected number of samples in the luteal phase (2 of 7 samples, P = 0.24), whereas the number of parous samples with high hormone signaling was significantly lower than expected based on the average length of the follicular and luteal phases of the menstrual cycle (0 of 7 samples, P = 0.02) (Figure 5C). These results remained consistent when we used a model accounting for previously reported differences in the relative lengths of the follicular versus luteal phases in parous women (Figure S11B) (Barrett et al., 2014). Thus, the decreased per-cell ER/PR signaling seen in HR+ luminal cells from parous women cannot be explained by differences in hormonal contraceptive use or random sampling across the menstrual cycle.
Figure 5. The ER/PR signaling response of HR+ luminal cells is reduced in parous women.
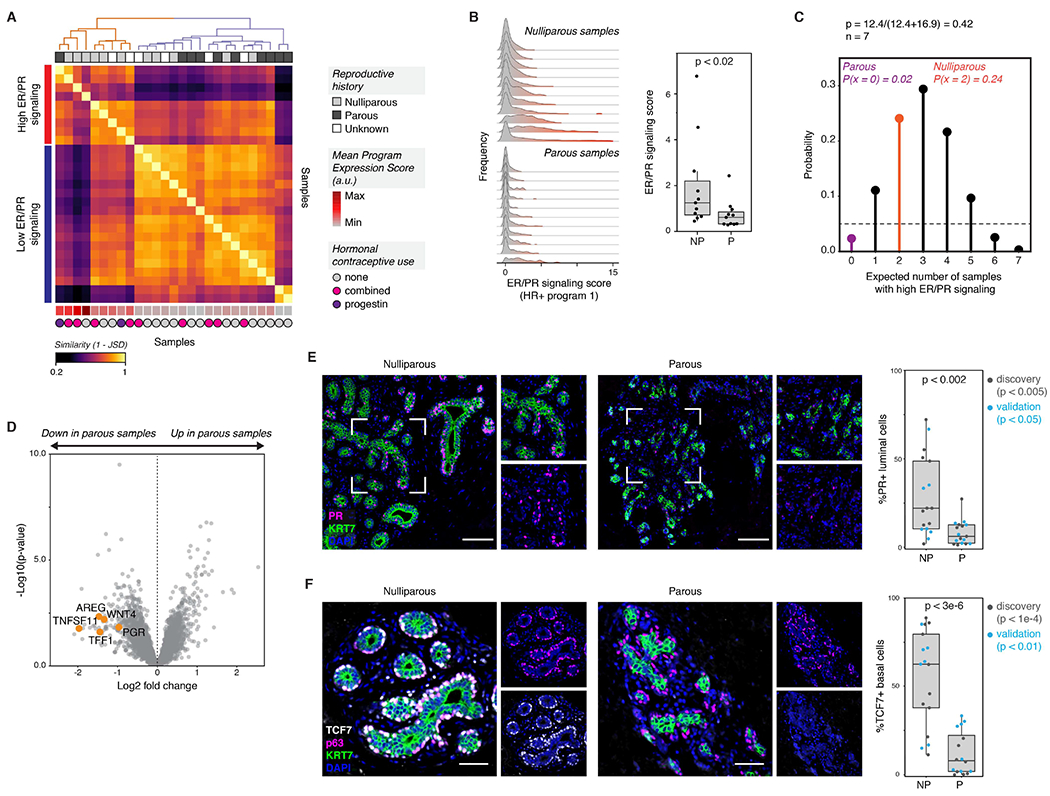
(A) Heatmap showing the similarity between each sample’s single-cell expression score distribution across HR+ activity program 1 (ER/PR signaling), measured as (1 - Jensen-Shannon distance). Hierarchical clustering (complete linkage) identifies two sets of samples representing high or low expression of the “ER/PR signaling” gene program. The mean expression score for HR+ activity program 1 is annotated at the bottom of the heatmap (arbitrary units, linear scale).
(B) Ridge plots depicting the distribution of HR+ program 1 (ER/PR signaling) expression in HR+ luminal cells across nulliparous (NP) versus parous (P) samples, and quantification of the average expression score for HR+ program 1 (n = 22 samples, p < 0.02, Mann-Whitney test). Data are represented as individual points; box indicates the median and interquartile range (IQR) for 11 nulliparous and 11 parous samples; whiskers extend from Q1 - 1.5IQR to Q3 + 1.5IQR.
(C) Binomial probability distribution for the expected number of samples with high ER/PR signaling. The binomial probability of high ER/PR signaling is modeled as the average length of the luteal phase of the menstrual cycle, in days, divided by the average total length of the menstrual cycle (p = 0.42) (Bull et al., 2019).
(D) Volcano plot highlighting the differential expression of canonical hormone-responsive genes between parous and nulliparous “pseudo-bulk” samples in HR+ luminal cells. Dots represent individual genes.
(E) Immunostaining for PR and KRT7, and quantification of the percentage of PR+ cells within the KRT7+ luminal compartment for nulliparous (NP) versus parous (P) samples (n = 34 samples, p < 0.002, Mann-Whitney test). Results are shown for a subset of the original cohort of sequenced samples (“discovery set”, n=19 samples, p < 0.005) and a second independent cohort of samples (“validation” set, n = 15 samples, p < 0.05). Scale bars 100 μm. Data are represented as individual points; box indicates the median and interquartile range (IQR) for the combined dataset (N = 17 nulliparous samples and 17 parous samples); whiskers extend from Q1 - 1.5IQR to Q3 + 1.5IQR.
(F) Immunostaining for TCF7, p63, and KRT7, and quantification of the percentage of TCF7+ cells within the p63+ basal/myoepithelial cell compartment for nulliparous (NP) versus parous (P) samples (n = 33 samples, p < 3e-6, Mann-Whitney test). Results are shown for a subset of the original cohort of sequenced samples (“discovery set”, n=18 samples, p < 1e-4) and a second independent cohort of samples (“validation” set, n = 15 samples, p < 0.01). Scale bars 50 μm. Data are represented as individual points; box indicates the median and interquartile range (IQR) for the combined dataset (N = 17 nulliparous samples and 16 parous samples); whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR.
To identify differentially expressed genes between nulliparous and parous women with high sensitivity, we generated a “pseudo-bulk” dataset of aggregated HR+ luminal cells from each sample (STAR Methods) and confirmed that parous women had decreased expression of the canonical hormone-responsive genes AREG, WNT4, PGR, TNFSF11 (RANKL), and TFF1 (Figure 5D, Table S7). The progesterone receptor itself is an ER target gene (Kastner et al., 1990). Staining for the progesterone receptor (PR) confirmed that PR expression was reduced in luminal cells in parous samples in both our original sequenced cohort of samples (“discovery” set, p < 0.005) and a second independent cohort of samples (“validation” set, p < 0.05) (combined p < 0.002, Mann-Whitney test; Figure 5E). This reduction in PR expression was not due to broad changes in the lobular architecture of parous women, as our results were consistent when we restricted our analysis to either lobular (terminal ductal lobular units, TDLUs) or ductal regions of the epithelium (Figure S11C).
Finally, we confirmed that paracrine signaling downstream of PR activation was reduced in parous samples by assessing the effects of one of these genes, WNT4. As WNT4 from HR+ luminal cells has been shown to signal to basal cells (Rajaram et al., 2015), we performed co-immunostaining for the WNT effector TCF7 and basal/myoepithelial cell marker p63 and found that TCF7 expression was markedly decreased in parous samples (overall p < 3e-6, “discovery” set p < 1e-4, “validation” set p < 0.01, Mann-Whitney test; Figure 5F). Again, this decrease was not due to differences in lobular architecture, as TCF7 staining was reduced in both ducts and TDLUs in parous samples (Figure S11D). Together, these data demonstrate that ER/PR signaling is a source of transcriptional variation among HR+ luminal cells, that transcription along this axis (HR+ activity program 1) is reduced in women with prior history of pregnancy, and that these transcriptional changes in HR+ cells coincide with a reduction in downstream paracrine signaling to basal/myoepithelial cells.
Parity and body mass index influence epithelial cell proportions
Based on our previous finding that paracrine signaling from HR+ luminal cells to other epithelial cell types is strongly influenced by the proportion of HR+ cells in the epithelium (Figure 2D-E), we next asked whether the architectural changes associated with parity would contribute to systematic changes in epithelial cell proportions, and thus influence hormone-responsiveness across samples. The breast undergoes a major expansion of the mammary epithelium during pregnancy, followed by a regression back towards the pre-pregnant state after weaning in a process called involution. Following involution, the epithelial architecture remains distinct from that of women without prior pregnancy, consisting of larger TDLUs containing greater numbers of acini. At the same time, individual acini are reduced in size (Russo et al., 1992).
We focused our initial analysis on the 63,583 cells in the live/singlet and epithelial sort gates to get an unbiased view of how the epithelial composition of the breast changes with pregnancy. The proportion of basal/myoepithelial cells in the epithelium was approximately two-fold higher in women with prior history of pregnancy (parous) relative to women without prior pregnancy (nulliparous) (Figure 6A and Figure S12A; FDR < 0.02, Wald test with post hoc multiple-comparisons test). This effect remained significant when we controlled for menstrual cycle stage and/or exogenous hormones (i.e hormonal contraceptive use) using our previously identified “ER/PR signaling” score (Table S8). We confirmed these results in an expanded cohort of samples using three additional methods. First, we measured basal cell proportions by flow cytometry analysis of EpCAM and CD49f. Consistent with scRNA-seq clustering results, parity was associated with an increase in the average proportion of EpCAM−/CD49f+ basal cells from about 15% to about 40% of the epithelium (Figure 6B; overall p < 3e-5, “discovery” set p < 0.008, “validation” set p < 0.008, Mann-Whitney test). The proportion of basal cells did not vary with other discriminating factors such as BMI or hormonal contraceptive use, but was weakly associated with age (R2 = 0.20, p < 0.04, Wald test) (Figure S12C). To determine the relative effect of each factor, we performed multiple linear regression analysis and found that the basal cell fraction positively correlated with pregnancy history (p < 2e-05, Wald test), but not age (p = 0.17, Wald test) (Figure S12D, Table S9; R2 = 0.77, p < 8e-6).
Figure 6. Reproductive history and body mass index are associated with epithelial cell proportions.
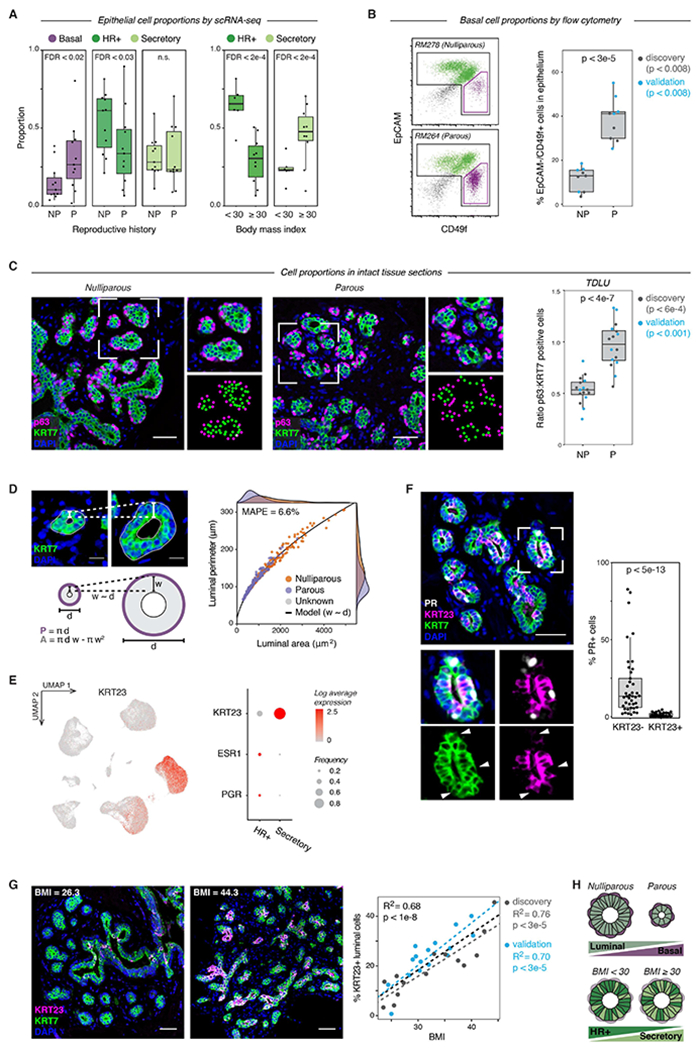
(A) Quantification of the proportion of the indicated cell types by scRNA-seq for nulliparous versus parous samples (n = 22 samples, Wald test) and obese versus non-obese samples (n = 16 samples, Wald test). Data are represented as individual points; box indicates the median and interquartile range (IQR) (Left: N = 11 nulliparous and 11 parous samples. Right: N = 6 samples with BMI < 30 and 10 samples with BMI ≥ 30); whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR.
(B) Representative flow cytometry analysis of the percentage of EpCAM−/CD49f+ basal cells within the Lin- epithelial population, and quantification of the percentage of basal cells in nulliparous (NP) versus parous (P) women (n = 18 samples; p < 3e-5, Mann-Whitney test). Results are shown for a subset of the original cohort of sequenced samples (“discovery set”, n=9 samples, p < 0.008) and a second independent cohort of samples (“validation” set, n = 9 samples, p < 0.008). Data are represented as individual points; box indicates the median and interquartile range (IQR) for the combined dataset (N = 9 nulliparous and 9 parous samples); whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR.
(C) Immunostaining for the basal/myoepithelial marker p63 and pan-luminal marker KRT7 in terminal ductal lobular units (TDLUs), and quantification of the ratio of p63+ basal cells to KRT7+ luminal cells in nulliparous (NP) versus parous (P) women (n = 32 samples; p < 4e-7, Mann-Whitney test). Results are shown for a subset of the original cohort of sequenced samples (“discovery set”, n=17 samples, p < 6e-4) and a second independent cohort of samples (“validation” set, n = 15 samples, p < 0.001). Data are represented as individual points; box indicates the median and interquartile range (IQR) for the combined dataset (N = 16 nulliparous and 16 parous samples); whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR. Scale bars 50 μm.
(D) Two-dimensional geometric model of the relative space available for basal cells (luminal perimeter, P) and luminal cells (luminal area, A) within individual acini. Acini were modeled as hollow circles with a shell thickness (w) proportional to their diameter (d). Dots represent measurements of individual acini from TDLUs in parous (n=158 acini from 15 samples) or nulliparous (n=164 acini from 16 samples) specimens as indicated. Line represents results from geometric model (mean absolute percentage error = 6.6%). Scale bars 15 μm.
(E) Left: UMAP depicting log normalized expression of KRT23 in reduction mammoplasty samples (GSE198732). Right: Dot plot depicting the log normalized mean and frequency of KRT23, ESR1, and PGR expression across luminal cell types.
(F) Co-immunostaining of PR, KRT23, and the pan-luminal marker KRT7, and quantification of the percentage of PR+ cells within the KRT23− and KRT23+ luminal cell populations (n = 41 samples; p < 5e-13, Mann-Whitney test). Data are represented as individual points; box indicates the median and interquartile range (IQR) for 41 samples; whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR. Scale bar 50 μm.
(G) Co-immunostaining of KRT23 and KRT7 and linear regression analysis of the percentage of KRT23+ luminal cells versus BMI (n = 30 samples; R2 =0.68, p < 1e-8, Wald test). Scale bars 50 μm. Results are shown for a subset of the original cohort of sequenced samples (“discovery set”, n=14 samples; R2 =0.76, p < 3e-5) and a second independent cohort of samples (“validation” set, n = 16 samples; R2 =0.70, p < 3e-5). Data are represented as individual points; dotted lines represent the best-fit lines for the discovery cohort (light grey), validation cohort (blue), and combined cohort (dark grey).
(H) Summary of changes in epithelial cell proportions with prior pregnancy and obesity (BMI ≥ 30).
Dissociation of tissue for scRNA-seq or FACS may affect measurements of cell composition. We therefore performed two further analyses to confirm these findings in intact tissue. First, we reanalyzed two previously published microarray datasets of total RNA isolated from core needle biopsies from either premenopausal (n = 71 parous/42 nulliparous) or postmenopausal (n = 79 parous/30 nulliparous) women (Peri et al., 2012; Santucci-Pereira et al., 2019), and confirmed a significant increase in the basal/myoepithelial markers KRT5, KRT14, and TP63 relative to luminal markers in parous samples (Figure S12E). Second, we performed immunostaining and confirmed an approximately 2-fold increase in the ratio of p63+ basal cells to KRT7+ luminal cells in intact tissue sections (Figure 6C; overall p < 4e-7, “discovery” set p < 6e-4, “validation” set p < 0.001, Mann-Whitney test). Immunostaining demonstrated that this change in epithelial proportions was specific to TDLUs rather than ducts (Figure 6C, Figure S12F). We hypothesized that the increased frequency of basal/myoepithelial cells observed in parous women could be explained, in part, by changes in TDLU architecture following pregnancy. To test this, we performed a morphometric comparison of TDLUs between parous and nulliparous samples in our dataset. Consistent with previous reports (Russo et al., 1992), we observed a marked decrease in the average diameter of individual acini in parous women (Figure S12G; p < 4e-5, Mann-Whitney test). Additionally, we found that the average thickness of the luminal cell layer increased in proportion to acinus diameter (Figure S12H; R2 = 0.75, p < 3e-16) and was thus higher in nulliparous women (Figure S12I; p < 7e-7, Mann-Whitney test). These results were independent of ER/PR signaling, and thus cannot be explained by differences in menstrual cycle stage (Table S10).
To determine how these parameters influence the relative proportions of each cell type, we implemented a simple geometric model (Figure 6D, STAR Methods). When normalized to cross-sectional area (for luminal cells) or perimeter (for basal cells), there was no change in mean luminal cell density or basal cell coverage between parous versus nulliparous samples (Figure S12J). Across all samples, the number of basal or luminal cells per acinus was proportional to the space available for each cell type (Figure S12K). Geometric modeling accurately predicted the relationship between the luminal area and outer perimeter for individual acini (mean absolute percentage error loss = 6.6%) and demonstrated that as individual acini increased in size, the space available for luminal cells (luminal area) increased at a faster rate than the space available for basal cells (luminal perimeter) (Figure 6D). Thus, the observed differences in epithelial cell proportions between parous and nulliparous samples are not due to a change in basal/myoepithelial coverage, but rather a change in the overall morphology of the luminal layer (e.g. thickness, diameter) and relative surface area of individual acini in parous women.
While parity was associated with a decreased overall proportion of luminal cells in the epithelium, the proportions of individual HR+ and secretory subtypes within the luminal compartment were highly variable. Consistent with previous work (Meier-Abt et al., 2014; Muenst et al., 2017), we observed reduced frequencies of HR+ luminal cells in parous women. However, the proportion of secretory luminal cells was not associated with parity (Figure 6A). Together, these data suggested that additional factors influence the relative proportion of HR+ versus secretory cells within the luminal compartment. Therefore, we performed linear regression analysis to test for the effects of parity, BMI, age, and hormonal contraceptive use on the proportions of HR+ versus secretory luminal cells. We found that the relative proportion of HR+ luminal cells versus secretory luminal cells was reduced in obese (BMI ≥ 30) women (Figure 6A, Figure S12B; FDR < 0.0002, Wald test with post hoc multiple-comparisons test) and did not vary significantly with other discriminating factors such as age, reproductive history, or hormonal contraceptive use (Figure S13A; Wald test with post hoc multiple-comparisons test). On a continuous scale, every 12 units of BMI was associated with a 2-fold reduction in the proportion of HR+ cells in the luminal compartment (Figure S13B; FDR < 0.001, Wald test with post hoc multiple-comparisons test). Similar to our previous results, this effect remained significant when we controlled for ER/PR signaling (Table S11).
One limitation of this dataset derived from reduction mammoplasty tissue was that all samples classified as non-obese were from nulliparous women less than 24 years old, whereas obese samples were more likely to be from parous and older age women (Table S1, Figure S13C). Therefore, we performed scRNA-seq analysis on an independent set of breast core biopsies from healthy premenopausal women who donated tissue to the Komen Tissue Bank (KTB) (Figure S13D-E; Table S2). In contrast with the reduction mammoplasty cohort, the KTB cohort consisted of older (37-47 years) parous samples with BMI in the normal or overweight range (BMI 20.7-28.3) (Table S1, Figure S13C). Using the reduction mammoplasty cohort as a training set, we accurately predicted the proportion of HR+ luminal cells in the KTB cohort as a function of BMI with a mean absolute percentage error of 14.8% (Figure S13F).
We next attempted to measure the relative proportion of the hormone-responsive luminal lineage in situ by performing immunostaining for ER and PR. As in our previous analysis, we included samples from both our original sequenced cohort of samples (“discovery” set) and a second independent cohort of samples (“validation” set). The “validation” set was well-balanced across age and BMI, overcoming a limitation of the “discovery” set (Figure S12G). There was a weak trend toward decreased expression of ER and/or PR with increasing BMI, but the change was not statistically significant in the “validation” set or the combined cohort (Figure S13H). Consistent with the heterogeneous ESR1 and PGR transcript expression we observed in scRNA-seq data (Figure S1H), ER and PR protein expression by immunostaining was variable and partly non-overlapping, ranging from 11-71% overlap (Figure S13H, bottom panel). We hypothesized that the variability in hormone receptor staining was due to changes in ER/PR expression, stability, and nuclear localization that have all been previously observed based on hormone receptor activation status (Battersby et al., 1992; Métivier et al., 2003; Petz and Nardulli, 2000). Based on this, we predicted that ER transcript and protein levels would co-vary across samples due to the overall proportion of HR+ luminal cells and their hormonal microenvironment, but would be stochastically expressed in individual cells at any one time due to fluctuations in mRNA and protein expression, localization, and stability. To test this, we performed co-immunostaining and RNA-FISH and confirmed that although ER transcript and protein levels correlate across tissue sections (R2 = 0.60, p < 0.01), they do not correlate on a per-cell basis (p = 0.63, Wilcoxon signed-rank test)—on average, only 31% of cells expressing ESR1 transcript also expressed ER protein (Figure S13I). Expression of ESR1 or PGR transcript was highly specific for cells in the HR+ luminal cluster, although the sensitivity of each transcript for the HR+ cluster was low and varied across individuals (Figure S13J). Thus, these data demonstrate that immunostaining or RNA-FISH for nuclear hormone receptors underestimates the fraction of cells in the HR+ lineage and that lack of ER/PR expression cannot be used to reliably define a cell as part of the secretory versus HR+ luminal cell lineages.
On the basis of these results, we sought to identify another marker to distinguish between the luminal lineages, and identified keratin 23 (KRT23) as highly enriched in the secretory luminal cell cluster (Figure 6E), as was also reported by a previous scRNA-seq study (Nguyen et al., 2018). Immunohistochemistry for KRT23 and PR or ER confirmed that these proteins are expressed in mutually exclusive luminal populations (Figure 6F, and Figure S13K-L). KRT23 thus represents a discriminatory marker between the two luminal populations. Staining in an expanded cohort of intact tissue sections confirmed that the proportion of KRT23+ secretory luminal cells increased by about 20% for every 10-unit increase in BMI (Figure 6G; overall R2 = 0.68, p < 1e-8; “discovery” set R2 = 0.76, p < 3e-5; “validation” set R2 = 0.70, p < 3e-5). Using multiple regression analysis, we confirmed that the proportion of KRT23-positive cells in the luminal compartment was significantly associated with BMI, but not parity or age (Figure S13M). Together, these data demonstrate that there are two independent effects of reproductive history and body weight on cell proportions in the mammary epithelium: parity affects the ratio of basal to luminal cells whereas BMI affects the ratio of HR+ versus secretory luminal cells (Figure 6H).
Biological variables impact coordinated changes in signaling states across cell types in the breast
Finally, we used the cell-cell interaction network identified by DECIPHER-seq to better understand how biological variables—such as BMI, parity, and hormonal contraceptive use—affect cell-cell interactions in the breast. Based on the above results, we propose that parity and BMI affect the hormone responsiveness of the breast through two distinct mechanisms: parity decreases the per-cell ER/PR signaling response in HR+ luminal cells, whereas BMI indirectly affects hormone signaling by reducing the proportion of HR+ luminal cells in the mammary epithelium (Figure 7A). Consistent with this, both prior pregnancy and increasing body mass index were negatively associated with activity programs across the “ER/PR response” module and positively correlated with programs in the “resting state” module (Figure 7B, Figure S14A). To confirm these results in intact tissue sections, we performed immunostaining for PR as a measure of ER activation in HR+ luminal cells, and for TCF7 as a measure of the downstream paracrine response (WNT activation) in basal cells. As expected, we found that PR expression in the hormone-responsive (KRT23-) luminal cell subpopulation was not significantly different between non-obese and obese women (p = 0.17, Mann-Whitney test; Figure 7C), but that WNT signaling in basal cells was markedly reduced in obese samples (p < 3e-5, Mann-Whitney test; Figure 7D). We confirmed these results using multiple linear regression to simultaneously test the effects of prior pregnancy and obesity. Whereas PR expression in hormone-responsive (KRT23−) cells was dependent on parity but not obesity, downstream WNT signaling in basal cells was dependent on both variables (Figure 7E).
Figure 7. Biological variables are linked to predicted tissue states.
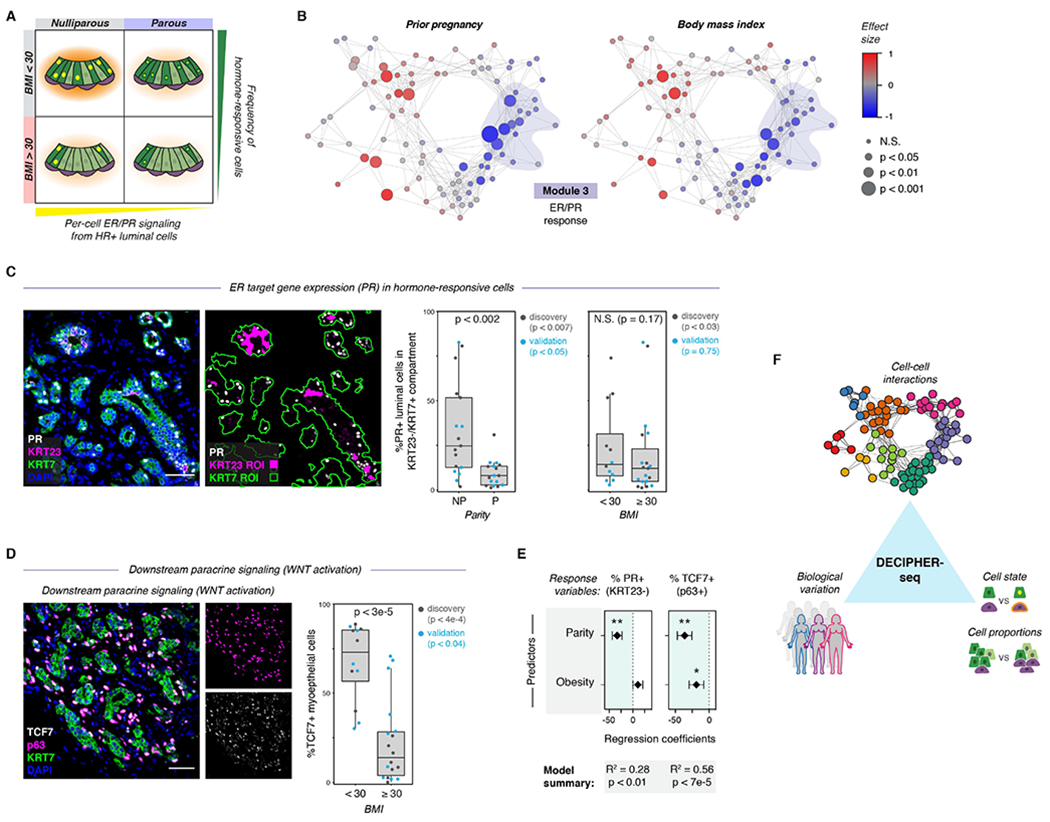
(A) Schematic depicting the model for how parity and obesity impact hormone signaling in the breast through distinct mechanisms. Parity affects per-cell ER/PR signaling in HR+ luminal cells, and obesity (BMI ≥ 30) leads to a reduction in the proportion of hormone-responsive (HR+) luminal cells in the epithelium.
(B) Network graph of activity programs in the human breast, colored by the effect size of prior pregnancy (Wilcoxon effect size) or body mass index (Pearson correlation coefficient) on each activity program (linear scale). Significant positive and negative associations are represented by larger nodes (prior pregnancy: Mann-Whitney test; BMI: Wald test).
(C) Co-immunostaining of the estrogen receptor target gene PR, KRT23, and the pan-luminal marker KRT7, and quantification of the percentage of PR+ cells in the KRT23−/KRT7+ luminal cell population for nulliparous (NP) versus parous (P) samples (n = 34 samples, p < 0.002, Mann-Whitney test) or non-obese (BMI <30) versus obese (BMI ≥ 30) samples (n = 31 samples, p = 0.17, Mann-Whitney test). Results are shown for a subset of the original cohort of sequenced samples (“discovery” set) and a second independent cohort of samples (“validation” set). Data are represented as individual points; boxes indicate the median and interquartile range (IQR) for the combined datasets (Left: N = 17 nulliparous and 17 parous samples. Right: N = 12 samples with BMI < 30 and 19 samples with BMI ≥ 30); whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR. Scale bar 50 μm.
(D) Immunostaining forTCF7, p63, and KRT7, and quantification of the percentage of TCF7+ cells within the p63+ basal/myoepithelial cell compartment for non-obese (BMI <30) versus obese (BMI ≥ 30) samples (n = 30 samples, p < 3e-5, Mann-Whitney test). Results are shown for a subset of the original cohort of sequenced samples (“discovery set”, n=14 samples, p < 4e-4) and a second independent cohort of samples (“validation” set, n = 16 samples, p < 0.04). Data are represented as individual points; box indicates the median and interquartile range (IQR) for the combined dataset (N = 12 samples with BMI < 30 and 18 samples with BMI ≥ 30); whiskers extend from Q1 − 1.5IQR to Q3 + 1.5IQR. Scale bar 50 μm.
(E) Results from multiple linear regression analysis, with prior pregnancy (parity) and obesity (BMI ≥ 30) as predictors and the percentage of PR+ cells in the KRT23−/KRT7+ luminal cell population or of TCF7+ cells in the p63+ basal cell compartment as response variables. Points represent the regression coefficient for each predictor, error bars depict the standard error.
(F) Summary of results. DECIPHER-seq predicts how specific changes in transcriptional cell state and cell type proportions influence cell-cell interactions in the human breast, and links specific sources of biological variation (e.g. Parity, BMI) to the overall signaling state of the tissue.
Second, we took advantage of the different dynamics of serum estrogen and progestin/progesterone in donors using combined hormonal contraceptives versus those undergoing natural menstrual cycles (Figure S14B) to ask whether activity program expression in the “Involution-like” module (Module 4) was influenced by the hormonal microenvironment. The natural menstrual cycle is characterized by an initial rise in estrogen levels during the follicular phase of the menstrual cycle followed by a combined surge of estrogen and progesterone during the luteal phase. In contrast, following oral contraceptive use, estrogen and progestin levels rise simultaneously, reach peak concentrations in the blood about 2 hours following ingestion, and return fairly rapidly to a steady state level over the following 22 hours (Figure S14B) (Hampson, 2020). We found that activity programs in the “involution-like” module were highly correlated with the use of combined (estrogen/progestin) oral contraceptives (Figure S14C-D). These results suggest that the “involution-like” phenotype is influenced by hormone levels and dynamics (since exogenous hormones are associated with increased expression across this module), but does not require the precise sequential estrogen/progesterone dynamics observed during natural menstrual cycles (since estrogen and progesterone rise simultaneously upon oral contraceptive ingestion). Overall, these results demonstrate how sample-to-sample variation in the breast can be used to predict how specific changes in transcriptional cell state and cell type proportions influence cell-cell interactions in a tissue, and to understand some of the sources of biological variation (e.g. metadata factors) that control the overall state of the tissue (Figure 7F).
Discussion
In this study, we leverage inter-sample transcriptional variation in the breast to identify a set of highly correlated “activity programs” representing the in situ response to hormone receptor activation in HR+ cells and the effects of downstream paracrine signaling in other cell types. We uncover additional correlated programs representing the dynamic response of the breast to changing hormone levels (e.g. “involution-like”). Furthermore, we show that person-to-person heterogeneity in hormone-responsiveness in the breast is directly linked to two factors known to be correlated with premenopausal breast cancer risk—reproductive history and body mass index.
Cumulative lifetime hormone exposure is a major determinant of breast cancer risk (Collaborative Group on Hormonal Factors in Breast Cancer, 2012). Here, we mapped the coordinated changes in cell state that occur in response to paracrine signaling from HR+ luminal cells. Notably, many of these changes closely mimic those seen during the pregnancy/involution cycle that have been linked to a transient increased breast cancer risk following pregnancy (Lyons et al., 2011; O’Brien et al., 2010; Schedin et al., 2007). First, we identify a proliferative gene signature in secretory luminal cells that is highly correlated with hormone signaling in HR+ luminal cells, consistent with previous studies demonstrating that TNFSF11 (RANKL) and WNT control progesterone-mediated epithelial proliferation (Joshi et al., 2015). Second, we identify previously uncharacterized subpopulations of HR+ and secretory luminal cells in the cycling premenopausal breast with transcriptional signatures closely matching that described for post-lactational involution (Clarkson et al., 2004; Stein et al., 2004), including upregulation of immune mediators, MHC class II molecules, and the phagocytic receptor MARCO. This idea that the menstrual cycle mimics a miniature pregnancy/involution cycle is consistent with studies showing that the fraction of apoptotic cells in the epithelium peaks between the late luteal and early follicular phases (Anderson et al., 1982). We also observe upregulation of hypoxic gene signatures in multiple epithelial and stromal cell types that are highly correlated with hormone signaling in HR+ cells. A previous study identified these same pathways as highly enriched during post-lactational involution in the mouse. More importantly from the perspective of breast cancer risk, this “hypoxia/pro-angiogenic” signature identified breast cancers with increased metastatic activity (Stein et al., 2009), suggesting that these pathways can be co-opted by cancer cells to support a permissive tumor microenvironment. Thus, we speculate that many of the same mechanisms underlie both the short-term increased breast cancer risk following pregnancy and the lifetime increased risk due to total number of menstrual cycles.
Pregnancy has two opposing effects on breast cancer risk: although breast cancer risk is increased for the first 5-10 years following pregnancy, it also has a pronounced long-term protective effect, with up to a 50% reduction in ER/PR+ breast cancer risk for women with multiple full-term pregnancies at a young age (Britt et al., 2007). The cellular basis for this long-term protective effect remains an area of active research. Our analysis revealed that parity is associated with a stark increase in the proportion of basal and/or myoepithelial cells within the breast epithelium, as well as decreased hormone signaling in HR+ luminal cells. While the precise role of myoepithelial cells during cancer progression remains an active area of research (Risom et al., 2022), previous work has described two properties of myoepithelial cells consistent with a tumor-protective effect: they are resistant to malignant transformation (Koren et al., 2015; Proia et al., 2011) and may also act as a dynamic barrier to prevent tumor cell invasion (Sirka et al., 2018; Sternlicht et al., 1997). Thus, our data support the notion that pregnancy protects against breast cancer risk through multiple mechanisms: by decreasing the relative frequency of luminal cells—the tumor cell-of-origin for most breast cancer subtypes (Keller et al., 2012; Melchor et al., 2014; Molyneux et al., 2010), by reducing the overall hormone-responsiveness of HR+ cells and subsequent pro-tumorigenic microenvironmental changes, and by suppressing progression to invasive carcinoma (Sirka et al., 2018; Sternlicht et al., 1997).
Finally, we found that paracrine signaling from HR+ cells to other cell types depends on both the magnitude of signaling from HR+ cells and the overall proportion of HR+ cells in the epithelium. Prior pregnancy and obesity are specifically associated with a reduced risk of ER+/PR+ breast cancer in premenopausal women (Fortner et al., 2019; Premenopausal Breast Cancer Collaborative Group et al., 2018), and our data are consistent with a model that these biological variables lead to reduced paracrine signaling downstream of estrogen and progesterone via two distinct mechanisms. First, parity leads to a reduced per-cell hormone signaling response in HR+ luminal cells. Second, we identify a marked decrease in the ratio of HR+ cells relative to secretory luminal cells with increasing BMI. Both changes are associated with reduced paracrine signaling across the ER/PR response module.
Several potential mechanisms could account for the decreased hormone signaling response observed in HR+ luminal cells in parous women. Previous studies have identified small reductions in the levels of estrogen metabolites in the urine of parous women, which may be indicative of lower serum levels of estradiol (Barrett et al., 2014). Since progesterone receptor expression is induced downstream of estrogen receptor activation, lower levels of serum estradiol could lead to reduced signaling through both ER and PR. A second possibility is that structural differences in the mammary epithelium of parous women, such as increased lobular density (Russo et al., 1992) or alterations in vascularization could lead to decreased access of hormones to HR+ luminal cells. Finally, changes in the differentiation state or epigenetic remodeling of HR+ luminal cells following pregnancy could lead to a direct change in the ability of these cells to respond to hormone. Interestingly, recent work has shown that matrix stiffness and/or compressive stress is required for maintenance of ER expression in explant cultures, via H3K27me3-dependent epigenetic regulation (Munne et al., 2022), and previous work has shown that parity is associated with decreased mammographic density (Vachon et al., 2000). Further studies are required to determine whether the decreased hormone response of HR+ luminal cells in parous women is a result of these or other processes.
A key insight of our computational approach is that a subset of “high confidence” direct cell-cell interactions can be inferred based on their dependence on the proportion of one cell type in the tissue. Because the DECIPHER-seq workflow corrects for batch effects while maintaining meaningful biological variation and optimizes both the granularity and robustness of identified activity programs, it has the potential to be flexibly adapted to a broad range of preexisting single-cell datasets, or across datasets from multiple sources. Further, we find that the coordinated activity programs in our dataset naturally self-organize into a cycle, precisely as we would expect based on hormone fluctuations across menstrual cycles. This raises the intriguing possibility that a similar computational approach could reveal cyclical cellular programs in other tissue types in the body, such as circadian rhythms, feeding cycles, or the response to wounding. While we focus on single-cell transcriptional data in this study, integrative NMF has also been applied to multi-omic datasets containing spatial or epigenetic data together with transcriptional information (Welch et al., 2019, Gao et al. 2021).
In summary, using scRNA-seq of a unique cohort of 28 healthy premenopausal women, we provide a comprehensive, systems-level view of the cellular and transcriptional variation within the human breast, which profoundly affects the response to hormones and may impact breast cancer risk. As the human breast is one of the only human organs that undergoes repeated cycles of morphogenesis and involution, this study serves as a roadmap for deeper interrogation of the cell state changes associated with hormone dynamics. Finally, it provides a foundation for future systems-level studies dissecting how the paracrine communication networks downstream of hormone signaling are altered during ER+/PR+ breast cancer progression.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zev Gartner (zev.gartner@ucsf.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNA-seq data (raw FASTQ files, processed gene expression and barcode count matrices, and de-identified patient metadata) have been deposited at the Gene Expression Omnibus (GSE198732) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All original code has been deposited at Zenodo and Github and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC-EpCAM | Stem Cell Technologies | 60136FI |
| APC-CD49f | BioLegend | 313616 |
| Biotin-CD2 | BD Biosciences | 555325 |
| Biotin-CD3 | BD Biosciences | 55338 |
| Biotin-CD16 | BD Biosciences | 555405 |
| Biotin-CD64 | BD Biosciences | 555526 |
| Biotin-CD31 | Invitrogen | MHCD31154 |
| Biotin-CD45 | BioLegend | 304004 |
| p63 | Cell Signaling Technology | 13109 |
| KRT7 | Abcam | AB68459 |
| KRT23 | Abcam | AB156569 |
| ER | Fisher Scientific | RM9101S |
| PR | Cell Signaling Technology | 8757 |
| TCF7 | Cell Signaling Technology | 2203 |
| P4HA1 | Thermo Fisher | PA5-55353 |
| LRRC26 | Thermo Fisher | PA5-63285 |
| Biological samples | ||
| Human breast specimens from reduction mammoplasty surgeries | CHTN and Kaiser Permanente Northern California | Table S1 |
| Human breast specimens from core biopsies | Komen Tissue Bank | Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| BV785-Streptavidin | BioLegend | 405249 |
| Collagenase Type 3 | Worthington | CLS-3 |
| Collagenase Type 2 | Worthington | CLS-2 |
| Hyaluronidase | Sigma Aldrich | H3506 |
| RPMI with HEPES | Corning | 10-041-CV |
| Amphotericin B | Lonza | 17-836E |
| Gentamicin | Lonza | 17-518 |
| Dispase | Stem Cell Technologies | 07913 |
| DNase I | Stem Cell Technologies | 07900 |
| MEGM | Lonza | CC-3150 |
| MEBM | Lonza | CC-3151 |
| Lab Vision Ultra-V Block | Thermo Fisher | TA-125-UB |
| UltraVision LP Detection System | Thermo Fisher | TL-060-HL |
| Vectashield HardSet Mounting Media with DAPI | Vector Labs | H-1400 |
| FITC-TSA | Akoya Biosciences | NEL701A001KT |
| Cy3-TSA | Akoya Biosciences | NEL744001KT |
| Cy5-TSA | Akoya Biosciences | NEL745E001KT |
| Probe Hs-ESR1 | ACD Bio | 310301 |
| Critical commercial assays | ||
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10X Genomics | PN-120237 |
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| Chromium Single Cell A Chip Kit | 10X Genomics | PN-120236 |
| Chromium Single Cell B Chip Kit | 10X Genomics | PN-1000153 |
| Chromium i7 Multiplex Kit | 10X Genomics | PN-120262 |
| MULTI-seq Lipid-Modified Oligos | Millipore Sigma | LMO001-100RXN |
| Bioanalyzer High Sensitivity DNA Kit | Agilent | 5067-4626 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Q32851 |
| RNAscope Multiplex Fluorescent Reagent Kit V2 | ACD Bio | 323100 |
| Deposited data | ||
| Raw data and processed scRNA-seq UMI counts and barcode matrices of reduction mammoplasty breast specimens | This study | GEO: GSE198732 |
| Raw data and processed scRNA-seq UMI counts and barcode matrices of Komen Tissue Bank breast specimens | This study | GEO: GSE198732 |
| Software and algorithms | ||
| CellRanger v3.0.2 | 10x Genomics | Github: https://github.com/10XGenomics/cellranger |
| MULTI-seq | McGinnis et al., 2019b | Github: https://github.com/chris-mcginnis-ucsf/MULTI-seq |
| SoupOrCell | Heaton et al., 2020 | Github: https://github.com/wheaton5/souporcell |
| Seurat v3.1.5 | Stuart et al., 2019; Hafemeister and Satija, 2019 | Github: https://github.com/satijalab/seurat |
| DoubletFinder | McGinnis et al., 2019a | Github: https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| LIGER | Gao et al., 2021; Welch et al., 2019 | Github: https://github.com/welch-lab/liger |
| DECIPHER-seq computational workflow | This study | Github: https://github.com/lmurrow/DECIPHER-seq and https://doi.org/10.5281/zenodo.6596414 |
| ape | Desper and Gascuel, 2002 | Github: https://github.com/emmanuelparadis/ape |
| Leidenalg | Traag et al., 2011 | Github: https://github.com/vtraag/leidenalg |
| wTO | Gysi et al., 2018 | Github: https://github.com/cran/wTO |
| fgsea | Korotkevich et al., 2019 | Github: https://github.com/ctlab/fgsea |
| DESeq2 | Love et al., 2014 | Github: https://github.com/mikelove/DESeq2 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tissue samples
Reduction mammoplasty tissue samples were obtained from the Cooperative Human Tissue Network (CHTN, Vanderbilt University Medical Center, Nashville, TN) and Kaiser Permanente Northern California (KPNC, Oakland, CA). Core biopsy samples were provided by the Susan G. Komen Tissue Bank (KTB). Tissues were obtained as de-identified samples and all subjects provided written informed consent. When possible, medical reports or other patient data were obtained with personally identifiable information redacted. Use of breast tissue specimens to conduct the studies described above were approved by the UCSF Committee on Human Research under Institutional Review Board protocols 16-18865 and 10-01532. Donor information for all tissue specimens and their use in scRNA-seq, flow cytometry, and immunostaining experiments is detailed in Table S1.
METHOD DETAILS
Tissue processing
A portion of each sample was fixed in formalin and paraffin-embedded using standard procedures. The remainder was dissociated mechanically and enzymatically to obtain epithelial-enriched tissue fragments. Tissue was minced, followed by enzymatic dissociation with 200 U/mL collagenase type III (Worthington CLS-3, samples RM108 - RM203) or collagenase type II (Worthington CLS-2, samples RM216 - RM314) and 100 U/mL hyaluronidase (Sigma H3506) in RPMI 1640 with HEPES (Corning 10-041-CV) plus 10% (v/v) dialyzed FBS, penicillin, streptomycin, amphotericin B (Lonza 17-836E), and gentamicin (Lonza 17-518) at 37 °C for 16 h. For KTB samples, the resulting cell suspension containing single cells and stroma was frozen and maintained at −180 °C until use. For reduction mammoplasty samples, the cell suspension was centrifuged at 400 × g for 10 min and resuspended in RPMI 1640 plus 10% FBS. Digested tissue fragments enriched for epithelial cells and closely-associated stroma were collected after serial filtration through 150 μm and 40 μm nylon mesh strainers. Following centrifugation, tissue fragments and filtrate were frozen and maintained at −180 °C until use.
Dissociation to single cells
The day of sorting, epithelial-enriched tissue fragments from the 150 μm fraction, or total banked material for the KTB samples, were thawed and digested to single cells by trituration in 0.05% trypsin for 2 min, followed by trituration in 5 U/mL dispase (Stem Cell Technologies 07913) plus 1 mg/mL DNase I (Stem Cell Technologies 07900) for 2 min. Single-cell suspensions were resuspended in HBSS supplemented with 2% FBS, filtered through a 40 μm cell strainer, and pelleted at 400 × g for 5 min. The pellets were resuspended in 10 mL of complete mammary epithelial growth medium with 2% v/v FBS without GA-1000 (MEGM; Lonza CC-3150). Cells were incubated at 37 °C for 1 h, rotating on a hula mixer, to regenerate surface antigens.
MULTI-seq sample barcoding (Batches 3, 4, and KTB)
Single-cell suspensions were pelleted at 400 x g for 5 min and washed once with 10 mL mammary epithelial basal medium (MEBM; Lonza CC-3151). For each sample, one million cells were aliquoted, washed a second time with 200 μL MEBM, and resuspended in 90 μL of a 200 nM solution containing equimolar amounts of anchor lipid-modified oligonucleotides (LMOs) and sample barcode oligonucleotides in phosphate buffered saline (PBS). Following a 5-minute incubation on ice with anchor-LMO/barcode, 10 uL of 2 μM co-anchor LMO in PBS was added to each sample (for a final concentration of 200 nM), and wells were mixed by gentle pipetting and incubated for an additional 5 min on ice. Following incubation, cells were washed twice in 200 μL PBS with 1% BSA and pooled together into a single 15 mL conical tube containing 10 mL PBS/1% BSA. All subsequent steps were performed on ice.
Sorting for scRNA-seq
Cells were pelleted at 400 x g for 5 min and resuspended in PBS/1% BSA at a concentration of 1 million cells per 100 μL, and incubated with primary antibodies. Cells were stained with Alexa 488-conjugated anti-CD49f to isolate basal/myoepithelial cells, PE-conjugated anti-EpCAM to isolate luminal epithelial cells, and biotinylated antibodies for lineage markers CD2, CD3, CD16, CD64, CD31, and CD45 to remove hematopoietic (CD16/CD64-positive), endothelial (CD31-positive), and leukocytic (CD2/CD3/CD45-positive) lineages by negative selection (Lin−). Sequential incubation with primary antibodies was performed for 30 min on ice in PBS/1% BSA, and cells were washed with cold PBS/1% BSA. Biotinylated primary antibodies were detected with a streptavidin-Brilliant Violet 785 conjugate. After incubation, cells were washed once and resuspended in PBS/1% BSA plus 1 ug/mL DAPI for live/dead discrimination. Cell sorting was performed on a FACSAria II cell sorter. Live/singlet (DAPI−), luminal (DAPI−/Lin−/CD49f−/EpCAM+), basal/myoepithelial (DAPI−/Lin−/CD49f−/EpCAM−), or total epithelial (pooled luminal and basal/myoepithelial) cells were collected for each sample as specified in table S2 and resuspended in PBS/1% BSA at a concentration of 1000 cells/μL. For Batch 4, an aliquot of MULTI-seq barcoded cells were separately stained with biotinylated-CD45/streptavidin-Brilliant Violet 785 to enrich for immune cells, and sorted CD45+ cells were pooled with the Live/singlet fraction as specified in Table S2.
Antibodies and dilutions used (μL/million cells) were as follows: FITC-EpCAM (1.5 μL, Stem Cell Technologies 60136FI, clone VU1D9), APC-CD49f (4 μL, BioLegend 313616, clone GoH3), Biotin-CD2 (8 μL; BD 555325, clone RPA-2.10), Biotin-CD3 (8 μL; BD 55338, clone HIT3a), Biotin-CD16 (8 μL; BD 555405, clone 3G8), Biotin-CD64 (8 μL; BD 555526, clone 10.1), Biotin-CD31 (4 μL; Invitrogen MHCD31154, clone MBC78.2), Biotin-CD45 (1 μL; BioLegend 304004, clone HI30), BV785-Streptavidin (1 μL; BioLegend 405249).
scRNA-seq library preparation
cDNA libraries were prepared using the 10X Genomics Single Cell V2 (CG00052 Single Cell 3’ Reagent Kit v2: User Guide Rev B) or Single Cell V3 (CG000183 Single Cell 3’ Reagent Kit v3: User Guide Rev B) standard workflows as specified in Table S2. Library concentrations were quantified using high sensitivity DNA Bioanalyzer chips (Agilent, 5067-4626) and Qubit dsDNA HS Assay Kit (Thermo Fisher Q32851). Individual libraries were sequenced on a lane of a HiSeq4500 or NovaSeq, as specified in table S2, for an average of ~150,000 reads/cell.
Expression library pre-processing
Cell Ranger (10x Genomics) was used to align sequences, filter data and count unique molecular identifiers (UMIs). Data were mapped to the human reference genome GRCh37 (hg19). The resulting sequencing statistics are summarized in Table S2. For samples run across multiple 10X lanes, the cellranger aggr pipeline (10X Genomics) was used to normalize read depth across droplet microfluidic lanes (see “sort gate” information in Table S2).
Cell calling
For V2 experiments, cell-associated barcodes were defined using Cell Ranger. For V3/MULTI-seq experiments, cells were defined as barcodes associated with ≥600 total RNA UMIs and ≤20% of reads mapping to mitochondrial genes. We manually selected 600 RNA UMIs and 20% mitochondrial genes to exclude low-quality cell barcodes.
MULTI-seq barcode library pre-processing
Raw barcode FASTQs were converted to barcode UMI count matrices as described previously (McGinnis et al., 2019b). Briefly, FASTQs were parsed to discard reads where: 1) the first 16 bases of read 1 did not match a list of cell barcodes generated as described above, and 2) the first 8 bases of read 2 did not align with any reference barcode with less than 1 mismatch. Duplicated UMIs, defined as reads with the same cell barcode where bases 17-28 (V3 chemistry) of read 2 exactly matched, were removed to produce a final barcode UMI count matrix.
Sample demultiplexing
Barcode UMI count matrices were used to classify cells using the MULTI-seq classification suite (McGinnis et al., 2019b). In Batch 3, sample RM192 was poorly labeled for the lane of cells from the epithelial cell sort gate. Therefore, to reduce spurious doublet calls in this dataset, we manually set UMI counts which were <10 for this barcode to zero. For all experiments, raw barcode reads were log2-transformed and mean-centered, the top and bottom 0.1% of values for each barcode were excluded, and a probability density function (PDF) was constructed for each barcode. Next, all local maxima were computed for each PDF, and the negative and positive maxima were selected. To define a threshold between these two maxima, we iterated across 0.02-quantile increments and chose the quantile maximizing the number of singlet classifications, defined as cells surpassing the threshold for a single barcode. Multiplets were defined as cells surpassing two or more thresholds, and unlabeled cells were defined as cells surpassing zero thresholds. Unclassified cells were removed and the procedure was repeated until all remaining cells were classified.
To classify cells that were identified as unlabeled by MULTI-seq, we used the SoupOrCell pipeline (Heaton et al., 2020) to assign cells to different individuals based on single nucleotide polymorphisms (SNPs). For each dataset, we set the number of clusters (k) to the total number of samples in that experiment. To avoid local minima, SoupOrCell restarts clustering multiple times and takes the solution that minimizes the loss function. For Batch 3, we chose the number of restarts that produced less than a 1.5% misclassification rate between MULTI-seq and SoupOrCell singlet sample classifications (Live/singlet: 30 restarts/1.2% mismatch rate; Epithelial: 75 restarts/1.5% mismatch rate). SoupOrCell classification performed more poorly across parameters for Batch 4 (Live/singlet plus CD45+: 50 restarts/8.1 % mismatch rate, 75 restarts/4.8% mismatch rate; Epithelial: 50 restarts/8.6% mismatch rate, 75 restarts/14.9% mismatch rate, 100 restarts/4.1% mismatch rate). Therefore, for these datasets we used sample classifications that were consistent across two restarts (Live/singlet plus CD45+: consistent calls across 50 and 75 restarts/0.4% overall mismatch rate; Epithelial: consistent calls across 50 and 100 restarts/1% overall mismatch rate) to identify high-confidence singlets.
Dataset integration and cell type identification
Cell type identification was performed using the Seurat package (version 3.1.5) in R (Stuart et al., 2019). To identify and remove doublets formed from cells from the same sample that would not be identified by MULTI-seq or SoupOrCell, we filtered each lane to remove cells with greater than 20% of reads mapping to mitochondrial genes and ran DoubletFinder (version 2.0) on each data subset (McGinnis et al., 2019a), using parameters identified by the ‘paramSweep_v3’ function. Aggregated data for singlet cells for each batch was filtered to remove cells that had fewer than 200 genes and genes that appeared in fewer than 3 cells. Cells with a Z score of 4 or greater for the total number of genes expressed were presumed to be doublets and removed from analysis. The remaining cells were log transformed and scaled to a total of 1e4 molecules per cell, and the top 2000 most variable genes based on variance stabilizing transformation were identified for each batch (Hafemeister and Satija, 2019). Data from all four batches were integrated using the standard workflow and default parameters from Seurat v3 (Stuart et al., 2019). This data integration workflow identifies pairwise correspondences between cells across datasets and uses these anchors to transform datasets into a shared expression space. Following dataset integration, the resulting batch-corrected expression matrix was scaled, and principal component (PC) analysis was performed using the identified integration genes. The top 28 statistically significant PCs as determined by visual inspection of elbow plots were used as an input for UMAP visualization and k-nearest neighbor (KNN) modularity optimization-based clustering using Seurat’s ‘FindNeighbors’ and ‘FindClusters’ functions.
PC analysis of individual cell types
To perform principal component analysis on individual cell types, we subset out each cluster from the integrated dataset and repeated the standard workflow from Seurat v3 to identify integration genes specific to this cell type. The resulting batch-corrected expression matrices were scaled, and PC analysis was performed using the identified integration genes.
Activity program identification in each cell type
To identify gene expression signatures, or “activity programs”, within individual cell types, we subset raw counts data from each of the five most abundant cell type clusters (HR+ luminal cells, secretory luminal cells, basal/myoepithelial cells, fibroblasts, and endothelial cells) and performed matrix factorization. We chose to perform matrix factorization independently on each cell type rather than on the combined dataset, as preliminary analyses demonstrated that the number of gene programs identified for each cell type was highly dependent on the relative sizes of each cluster in the combined dataset. To correct for batch differences between samples run on different days, we used the LIGER package in R to perform integrative NMF (iNMF) (Gao et al., 2021; Welch et al., 2019), and performed subsequent gene set enrichment analyses on shared, rather than batch-specific, gene loadings for each activity program. Activity program expression in cells from the same sample run across different batches was more similar than program expression in cells from different samples processed in the same batch, demonstrating that this approach successfully corrected for batch differences while retaining sample-to-sample transcriptional variability (Figures S4A-B). To avoid identification of gene signatures dominated by highly-expressed transcripts, we normalized the raw counts matrix for each cell based on its total expression, multiplied by a scale factor of 1e4, and log-transformed and scaled the result without centering. The resulting datasets (one for each cell type) were decomposed using the ‘online_iNMF’ function from LIGER (Gao et al., 2021). Online iNMF uses an online learning algorithm to iteratively cycle through the data in small mini-batches, greatly increasing convergence times for large datasets. We performed 10 complete passes (‘max.epochs’ parameter) through each dataset, and chose the mini-batch size (‘miniBatch_size’) by rounding down to the nearest 500 from the smallest batch size in that cell type (HR+ luminal cells: 1000, Secretory luminal cells: 2000, Basal cells: 500, Fibroblasts: 500, Endothelial cells: 500).
Since solutions to NMF are non-unique, we adapted a consensus matrix factorization approach from (Kotliar et al., 2019) to identify activity programs that were consistent across multiple replicates. For each cell type, we ran 20 replicates of iNMF on the same normalized dataset with the same choice of rank K, starting from different random seeds. We row normalized the resulting 20 shared gene loading matrices (W, each of dimension Kprograms X Ngenes) to have an L2 norm of one. Following normalization, we combined the shared gene loading matrices from each matrix into a 20Kprograms X Ngenes dimensional matrix, where each row represents the gene loading from one activity program in one replicate. Next, we filtered out programs with a high mean Euclidean distance from their 6 nearest neighbors (30% of replicates), using the third quartile plus 1.5 times the interquartile range (q0.75 + 1.5·IQR) as an outlier threshold. After filtering outlier programs, we grouped the rows of the resulting matrix using k-means clustering, with the number of clusters set to the chosen iNMF rank K. Next, we collapsed each group of shared gene loadings to a single consensus vector by taking the median value for each gene across activity programs in that cluster, to produce a final KPrograms X Ncells consensus program matrix, W. We performed the same row normalization on the batch-specific gene loading matrices, filtered programs identified as outliers in the shared gene loading matrix, and collapsed groups of batch gene loadings into a consensus vector by taking the median value for each gene across programs in that cluster to produce consensus batch matrices Vbatch, each of dimension KPrograms X Ngenes. Finally, we solve for the consensus cell expression score matrix H (Xcells X Kprograms), by using non-negative least squares initialized with the consensus shared (W) and batch-specific (Vbatch) gene loading matrices.
A key parameter in matrix factorization is the choice of rank K. This parameter determines the granularity of identified activity programs. Three commonly used heuristics for guiding the optimum choice of K are: 1) minimizing the Frobenius reconstruction error of the final solution (Kotliar et al., 2019), 2) maximizing the median Kullback-Leibler (KL) divergence of activity program loadings across cells relative to a uniform distribution (Welch et al., 2019), and 3) estimating the “dimensionality” of the dataset via elbow plot of the proportion of variance explained across principal components (Kotliar et al., 2019). We propose a metric for choosing an optimum K, based on the goal of identifying the greatest number of activity programs that are robust (i.e. consistent across multiple choices of K) and unique (i.e. distinct from other programs at a particular choice of K). First, we perform consensus iNMF as described above over a range of ranks, with the sweep range guided by the heuristics described above. Here, we chose a range of 2 to 40 for all cell types. Next, we use the ‘fastme.bal’ function in the ‘ape’ R package to build a balanced minimum evolution phylogenetic tree based on the correlation matrix of the gene loadings for activity programs across all ranks (Desper and Gascuel, 2002). For each cell type, we partitioned the resulting phylogenetic tree into clusters using an empirical distance threshold to define distinct groups of activity programs (Prosperi et al., 2011) (Figure S5A-B). To identify partitions, we first artificially rooted each tree by taking the median of the activity programs at K = 2. Next, we identified clusters by performing a depth-first search starting from this artificial root, stopping at sub-trees where the median value of the pairwise patristic distance between all programs in that sub-tree was below an empirically determined threshold of 0.3 (see Figure S5B). To filter out “outlier” activity programs that are expressed in only rare contaminating cells (e.g. a “fibroblast-like” gene signature in HR+ luminal cells), we calculated the maximum expression score for each activity program divided by the mean expression score for the next 50 highest-scoring cells, and removed programs where this ratio was greater than 5 (Figure S5C). We also removed subtrees with fewer than 5 total activity programs. Finally, we plotted the number of subtrees identified at each K (excluding outlier programs), weighted by the total number of programs in each subtree. We choose the optimum K (Kopt) as the saturation point in this curve, representing the point at which increasing the granularity of matrix factorization does not identify activity programs that comprise major new subtrees (Figure S5D).
Network clustering of correlated activity programs
To identify sets of activity programs that co-varied across samples, we first decomposed each cell type into a set of distinct gene expression signatures, or “activity programs”, using consensus iNMF with Kopt chosen for each cell type as described above. We then quantified the average expression of each gene program in each sample and constructed a weighted network of coordinated gene expression programs based on the pair-wise Pearson correlations between gene programs. To account for correlations driven by outlier samples, we used bias-corrected and accelerated bootstrap resampling to estimate confidence intervals associated with each correlation coefficient. The resulting Pearson correlation matrix was transformed into a weighted adjacency matrix by setting all Pearson correlation coefficients with p-values greater than 0.05 (based on the null hypothesis r = 0) to zero (Figure S6A-B). We identified modules of highly correlated gene expression programs using a Constant Potts Model for community detection in signed graphs in the ‘leidenalg’ package in python (Figure S6B) (Traag et al., 2011). We ran this algorithm at a range of resolutions from 0.001 to 0.4 and chose the resolution that maximized overall modularity. To filter out isolated links and modules, we calculated the signed weighted topological overlap (wTO) between activity programs in each module (Gysi et al., 2018) and filtered nodes with low wTO and modules containing fewer than four nodes (Figure S6C). In contrast to Pearson correlation values which consider each pair of nodes in isolation, wTO is based on the similarity of two activity programs’ correlation values with all other programs in the network. We calculated the mean wTO between each node and all other nodes in the same module, and compared this to the value calculated for nodes in randomly selected modules of equal size. We determined p-values for each node’s mean wTO by determining the fraction of permutation trials where the mean wTO of nodes from “random” modules was greater than the mean wTO of nodes from tested modules, and removed nodes where p > 0.01. Community detection results remained unchanged after this filtering step (Figure S6B, S6D). For visualization, we use positive edges to create a force-directed layout. Consistent with our goal of choosing the rank K that captured the greatest number of unique activity programs (see above), the overall organization of modules into cell-cell interaction networks remained highly robust to the choice of rank at values of K ≥ Kopt, whereas the network structure at K ≤ Kopt had much sparser connections between modules (Figure S6E).
Fluorescent Immunohistochemistry
For immunofluorescent staining, formalin-fixed paraffin-embedded tissue sections were deparaffinized and rehydrated using standard methods. Endogenous peroxides were blocked using 3% hydrogen peroxide in PBS, and antigen retrieval was performed in 0.1 M citrate buffer pH 6.0. Sections were blocked for 5 min at room temperature using Lab Vision Ultra-V block (Thermo TA-125-UB) and rinsed with TNT wash buffer (1X Tris-buffered saline with 5 mM Tris-HCI and 0.5% TWEEN-20). Primary antibody incubations were performed for 1 hour at room temperature or overnight at 4°C. Sections were washed three times for 5 min each with TNT wash buffer, incubated with Lab Vision UltraVision LP Detection System HRP Polymer (Thermo Fisher TL-060-HL) for 15 min at room temperature, washed, and incubated with one of three colors of tyramide signal amplification amplification (TSA) reagent at a 1:50 dilution. After TSA, antibody complexes were removed by boiling in citrate buffer, followed by blocking and incubation with additional primary antibodies as above. Finally, sections were rinsed with deionized water and mounted using Vectashield HardSet Mounting Media with DAPI (Vector H-1400). Immunofluorescence was analyzed by spinning disk confocal microscopy using a Zeiss Cell Observer Z1 equipped with a Yokagawa spinning disk and running Zeiss Zen Software.
Antibodies, TSA reagents, and dilutions used are as follows: p63 (1:2000; CST 13109, clone D2K8X), KRT7 (1:4000; Abcam AB68459, clone EPR1619Y), KRT23 (1:2000; Abcam AB156569, clone EPR10943), ER (1:4000; Thermo Scientific RMM-9101-S, clone SP1), PR (1:3000; CST 8757, clone D8Q2J), TCF7 (1:2000; CST 2203, clone C63D9), P4HA1 (1:9000; Thermo PA5-55353), LRRC26 (1:2000; Thermo PA5-63285), FITC-TSA (2 min; Akoya Biosciences NEL701A001KT), Cy3-TSA (3 min; Akoya Biosciences NEL744001KT), Cy5-TSA (7 min; Akoya Biosciences NEL745E001KT).
Morphometric analysis and geometric modeling
Formalin-fixed paraffin-embedded tissue sections were immunostained for the pan-luminal marker KRT7, counterstained with DAPI and imaged as described above. Images containing lobular tissue were acquired randomly, and the area and perimeter of the KRT7-positive luminal layer of each acinus was analyzed in ImageJ. To reduce noise and remove small gaps in KRT7 fluorescence, we applied a closing filter from the MorphoLibJ plugin with a 2-pixel (1.33 μm) radius disk (Legland et al., 2016). The resulting image was smoothed by applying a Gaussian filter with sigma 5 pixels (3.33 μm), and binarized using the default thresholding algorithm in ImageJ. Finally, individual acini with visible lumens were manually selected and the area (A), perimeter (P), and circularity of the KRT7-positive region was measured for each structure. To estimate the average diameter (d) and luminal thickness (w) of each acinus, we used area and perimeter measurements to fit a circle containing a hollow lumen to each structure. Based on these results, we implemented a geometric model in which each acinus was represented as a hollow circle with shell thickness that was linearly related to diameter (d). Since basal cells form a monolayer along the luminal surface, we represented the space available for basal cells as the outer perimeter of the luminal layer, and the space available for luminal cells as the area of the luminal layer. To estimate the linear relationship between w and d, we performed linear regression analysis using measurements from all structures.
RNA FISH analysis of ESR1 transcripts
Combined RNA FISH and immunofluorescence analysis of estrogen receptor transcript (RNAscope Probe Hs-ESR1; ACD 310301) and protein (anti-ER; Thermo RMM-9101-S, clone SP1) was performed using the RNAscope in situ hybridization kit (RNAscope Multiplex Fluorescent Reagent Kit V2, ACD 323100) according to the manufacturer’s instructions and fluorescent immunohistochemistry protocol outlined above with the following modifications. Immunostaining for ER was performed prior to in situ hybridization, using the hydrogen peroxide and antigen retrieval solutions supplied with the RNAscope kit and the mildest recommended conditions. After ER immunostaining and tyramide signal amplification, in situ hybridization for ESR1 was performed according to the manufacturer’s instructions, followed by immunostaining for KRT7 as described above. For all RNA FISH experiments, we used positive (PPIB) and negative controls (DAPB) to verify staining conditions and probe specificity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of sample-to-sample heterogeneity
Cluster entropy: To measure how well-mixed cells from different samples were across cell type clusters, we quantified the normalized relative cluster entropy for our dataset, weighted by cluster size (Barkas et al., 2019). A cluster entropy value of 1 represents complete intermixing of samples across clusters.
Similarity scores/alignment: To measure transcriptional variation in cell state within cell types between cells from the same versus different batches and/or samples, we measured the pairwise alignment between each sample/batch (Butler et al., 2018), where batches consisted of sets of samples processed on the same day (Table S2). This “similarity score” examines the local neighborhood of each cell in a particular sample/batch, asks how many of its k nearest neighbors (in PC or iNMF space) belong to a second sample/batch, and averages this over all cells. We chose k to be 1% of the total number of cells within a cluster. The result was normalized by the expected number of cells from each sample/batch. For repeat measurements, samples run across multiple batches were highly similar. For Figure S2E, we calculated the pairwise similarity score between each sample/batch using the first 14 principal components for each cell type (See also figure S4E depicting the standard deviation of each principal component). For Figures S4A and S4B, we calculated the pairwise similarity score between each sample/batch using all iNMF components for each cell type (at Kopt, see text below for optimization of K).
Testing for changes in cell type proportions
We modeled the detected number of each cell type in each sample as a random count variable using a quasi-Poisson process to allow for overdispersion, with the condition being tested (e.g. parity, BMI, obesity) as a predictor and the total number of detected epithelial or luminal cells in each sample as an offset variable (Haber et al., 2017). To account for uncertainty due to variable numbers of profiled cells in each sample, we used bootstrap resampling to estimate confidence intervals associated with detection of each cell type (Cao et al., 2019). Results from 1000 bootstrap replicates were pooled using the ‘mice::pool’ function in R, and the model was fit using a quasi-Poisson generalized linear model from the ‘stats’ R package. Tests for statistical significance were performed using a Wald test on the regression coefficient. Multiple hypothesis correction was controlled using the false discovery rate. For the Komen Tissue Bank (KTB) data set, a quasi-Poisson model was trained on the reduction mammoplasty cohort as described above, and the ‘predict’ function in the ‘stats’ R package was used to predict the proportion of HR+ luminal cells in the KTB samples based on BMI.
Identification of non-cell-type specific programs
To identify transcriptionally similar activity programs representing non-cell-type specific responses, we calculated the Pearson correlation of gene loadings between activity programs using pairwise complete observations (i.e. excluding genes that are not expressed in either cell type). We defined each node’s “mean gene loading similarity” as the mean correlation between the tested node and all other nodes in the same module. To determine p-values for each node’s gene loading similarity, we compared this value to that calculated for nodes in randomly selected modules of equal size. The reported p-values represent the fraction of permutation trials where the mean gene loading similarity for nodes from “random” modules was greater than the mean gene loading similarity for nodes in tested modules.
Inferring direct cell-cell interactions
To infer modules enriched for putative direct cell-cell signaling interactions, we identified links between nodes that depended on both the magnitude of activity program expression in a “sender” cell type and the proportion of that “sender” cell type in the tissue. Since the proportion of epithelial versus stromal cells in our samples was highly dependent on tissue dissociation conditions, we restricted this analysis to links between epithelial cell types as “sender” cells (HR+ luminal, secretory luminal, or basal cells) and other cell types as “receivers”. We modeled activity program expression in the “receiver” cell type as a linear response to three predictors: activity program expression Y in the “sender” cell type (i.e. “signaling” from that cell type), the proportion Psender of the “sender” cell type in the epithelium, and an interaction term representing the combined effects of signaling and cell proportions (Signaling × Proportions). For links between two epithelial cell types, we tested both directions as “sender” versus “receiver” nodes. To infer high-confidence direct cell-cell signaling interactions, we identified pairwise combinations of activity programs where a) the individual effects of Y and Psender were not significant (p > 0.05), b) there was a positive interaction effect between Y and Psender (Signaling × Proportions; p < 0.01 and β > 0), c) the adjusted R-squared for the overall model was at least 0.5, and d) the false discovery rate-corrected p-value for the overall model was less than 0.05.
Gene set enrichment analysis
To identify marker genes statistically associated with each gene program, we used ordinary least squares regression of each gene’s normalized (z-scored) expression against the activity program expression score for each program in each cell type, after filtering genes not expressed in that cell type (Kotliar et al., 2019). This results in a vector of regression coefficients representing the strength of the relationship between a cell’s expression score for a particular activity program and its scaled expression of each gene (e.g. see Figure 4F). The resulting ranked gene lists (Table S3) were analyzed by gene set enrichment analysis, using the ‘fgsea’ package in R (Korotkevich et al., 2019).
Enrichment of gene sets within modules
To identify gene sets enriched across activity programs in a module, we first calculated the false discovery rate (FDR) for each gene set in each node. We performed false discovery rate correction for Hallmark and GO Biological Process gene sets separately, as many of the pathways in each database are highly related. For all gene sets enriched across at least 5 activity programs in our network, we calculated the number of activity programs in each module that were significantly enriched for each gene set (FDR < 0.01), and compared this value to randomly selected modules of equal size. We determined p-values for enrichment of gene sets in each module by determining the fraction of permutation trials where the number of significantly enriched nodes from “random” modules was greater than number of significantly enriched nodes from tested modules.
Sample-to-sample variability in ER/PR signaling
To quantify variation in expression of the “ER/PR signaling” gene program in HR+ luminal cells (HR+ gene program 1), we performed the following workflow. First, we used the cell loadings across HR+ gene program 1 for each sample to compute kernel density estimations using the ‘density’ function in the ‘stats’ R package. We excluded sample RM172 from this analysis as it had fewer than 50 HR+ luminal cells; thus, the resulting kernel density estimation was highly sensitive to individual outliers. Second, we used the ‘JSD’ function in the ‘philentropy’ R package (Drost, H.G., 2018) to measure the pairwise Jensen-Shannon divergence between samples. Third, we converted this to a distance metric (Jensen-Shannon Distance, JSD) by taking the square root and performed hierarchical clustering using the ‘hclust’ function in the ‘stats’ R package, using ‘ward.D2’ linkage. The similarity between samples was plotted on a heatmap as (1-JSD).
Pseudo-bulk differential gene expression analysis
To identify genes differentially expressed between samples from parous and nulliparous individuals in specific cell types, we constructed pseudo-bulk datasets consisting of the summed raw read counts across all single HR+ luminal cells for each batch and sample. We restricted our analysis to samples/batches that had at least 100 HR+ luminal cells. Each dataset was then randomly down-sampled to the lowest library size, and differential expression analysis was performed using DESeq2 (version 1.18.1) to test for genes differentially expressed between samples from parous and nulliparous individuals, using batch as a covariate (Love et al., 2014). As certain samples were sequenced across more than one batch (table S2), replicates of the same sample from different batches were combined using the ‘collapseReplicates’ function. False discovery rate corrected p-values were calculated using the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995).
Supplementary Material
Table S1. Donor information for reduction mammoplasty (RM) and Komen Tissue Bank (KTB) samples and their use in scRNA-seq, flow cytometry, and immunostaining experiments. Related to Figure 1.
Table S2. Summary statistics for sequencing of twenty-eight reduction mammoplasty samples (RM) and seven Komen Tissue Bank core biopsy samples (KTB). Related to Figure 1.
Table S3. Association between gene expression and activity program expression score across cells. Related to Figures 3–4.
Table S4. List of the top 20 gene sets enriched in each cell-cell interaction module. Related to Figures 3–4.
Table S5. Genes found to be upregulated in the human breast during the luteal phase of the menstrual cycle by Pardo et al. Related to Figure 3.
Table S6. List of genes found to be upregulated during post-lactational involution in the mouse by Stein et al. 2004, with names of human orthologs used for gene set enrichment analysis. Related to Figure 4.
Table S7. Canonical hormone-responsive genes differentially expressed in “pseudo-bulk” HR+ luminal cells from nulliparous versus parous samples. Related to Figure 5.
Table S8. Multiple linear regression analysis of the percent basal/myoepithelial cells in the epithelium (quantified by scRNA-seq clustering) in response to parity and mean ER/PR signaling score. Related to Figure 6.
Table S9. Multiple linear regression analysis of the percent EpCAM−/CD49+ basal/myoepithelial cells in the epithelium (quantified by flow cytometry) in response to age and parity. Related to Figure 6.
Table S10. Multiple linear regression analysis of the indicated morphometric measurements in response to parity and mean ER/PR signaling score. Related to Figure 6.
Table S11. Multiple linear regression analysis of the percent hormone-responsive (HR+) cells in the luminal compartment (quantified by scRNA-seq clustering) in response to BMI and mean ER/PR signaling score. Related to Figure 6.
Highlights.
Single-cell analysis of the human breast maps the tissue-level response to hormones
DECIPHER-seq identifies gene programs that co-vary across individuals
Dependency on cell-type proportions predicts direct cell-cell interactions
Prior pregnancy and obesity modify hormone-responsiveness through distinct mechanisms
Acknowledgments
We thank Drs. Tom Norman and Jonathan Weissman for technical support and for generously providing access to equipment and computing resources. Sequencing was performed in the Center for Advanced Technology at UCSF. Tissue samples were provided by the Cooperative Human Tissue Network (CHTN), which is funded by the National Cancer Institute. Other investigators may have received specimens from the same subjects. Samples from the Susan G. Komen Tissue Bank at the IU Simon Cancer Center were used in this study. We thank contributors, including Indiana University who collected samples used in this study, as well as donors and their families, whose help and participation made this work possible. This research was supported by grants from the Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 and W81XWH-13-1-0221), NIH (U01CA199315 and DP2 HD080351-01), the NSF (MCB-1330864), and the UCSF Center for Cellular Construction (DBI-1548297), an NSF Science and Technology Center, to Z.J.G. Z.J.G is a Chan-Zuckerberg BioHub Investigator. L.M.M is a former Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2239-15).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Z.J.G. and C.S.M. hold patents related to the MULTI-seq barcoding method. Z.J.G. is an equity holder in Scribe Biosciences and Provenance bio and a member of the scientific advisory board of Serotiny Bio. C.S.M is a consultant for ImYoo. Since January 10, 2022 L.M.M is an employee of Genentech, a member of the Roche group.
Diversity and Inclusion Statement
We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
References
- Anderson TJ, Ferguson DJ, Raab GM, 1982. Cell turnover in the “resting” human breast: influence of parity, contraceptive pill, age and laterality. Br. J. Cancer 46, 376–382. doi: 10.1038/bjc.1982.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ, 2015. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene 34, 3760–3769. doi: 10.1038/onc.2014.292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aupperlee MD, Leipprandt JR, Bennett JM, Schwartz RC, Haslam SZ, 2013. Amphiregulin mediates progesterone-induced mammary ductal development during puberty. Breast Cancer Res 15, R44–15. doi: 10.1186/bcr3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkas N, Petukhov V, Nikolaeva D, Lozinsky Y, Demharter S, Khodosevich K, Kharchenko PV, 2019. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat Meth 16, 695–698. doi: 10.1038/s41592-019-0466-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett ES, Parlett LE, Windham GC, Swan SH, 2014. Differences in ovarian hormones in relation to parity and time since last birth. Fertil. Steril 101, 1773–80.e1. doi: 10.1016/j.fertnstert.2014.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battersby S, Robertson BJ, Anderson TJ, King RJ, McPherson K, 1992. Influence of menstrual cycle, parity and oral contraceptive use on steroid hormone receptors in normal breast. Br. J. Cancer 65, 601–607. doi: 10.1038/bjc.1992.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031 .x [DOI] [Google Scholar]
- Bhat-Nakshatri P, Gao H, Sheng L, Storniolo AMV, Nakshatri H, 2021. A single-cell atlas of the healthy breast tissues reveals clinically relevant clusters of breast epithelial cells. Cell Reports Medicine 2, 100219. doi: 10.1016/j.xcrm.2021.100219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt K, Ashworth A, Smalley M, 2007. Pregnancy and the risk of breast cancer. Endocr Relat Cancer 14, 907–933. doi: 10.1677/ERC-07-0137 [DOI] [PubMed] [Google Scholar]
- Bull JR, Rowland SP, Scherwitzl EB, Scherwitzl R, Danielsson KG, Harper J, 2019. Real-world menstrual cycle characteristics of more than 600,000 menstrual cycles. NPJ Digit Med 2, 83–8. doi: 10.1038/s41746-019-0152-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RB, Howell A, Potten CS, Anderson E, 1997. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Research 57, 4987–4991. [PubMed] [Google Scholar]
- Clarkson RWE, Wayland MT, Lee J, Freeman T, Watson CJ, 2004. Gene expression profiling of mammary gland development reveals putative roles for death receptors and immune mediators in post-lactational regression. Breast Cancer Res 6, R92–109. doi: 10.1186/bcr754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Lin Y, Ormerod JT, Yang P, Yang JYH, Lo KK, 2019. scDC: single cell differential composition analysis. BMC Bioinformatics 20, 721–12. doi: 10.1186/s12859-019-3211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Group on Hormonal Factors in Breast Cancer, 2012. Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. The Lancet Oncology 13, 1141–1151. doi: 10.1016/S1470-2045(12)70425-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrosin C, 2003. Variability of vascular endothelial growth factor in normal human breast tissue in vivo during the menstrual cycle. J. Clin. Endocrinol. Metab 88, 2695–2698. doi: 10.1210/jc.2002-021584 [DOI] [PubMed] [Google Scholar]
- Desper R, Gascuel O, 2002. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J Comput Biol 9, 687–705. doi: 10.1089/106652702761034136 [DOI] [PubMed] [Google Scholar]
- Dontu G, Ince TA, 2015. Of Mice and Women: A Comparative Tissue Biology Perspective of Breast Stem Cells and Differentiation. J Mammary Gland Biol Neoplasia 20, 51–62. doi: 10.1007/s10911-015-9341-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost HG, 2018, Philentropy: information theory and distance quantification with R. Journal of Open Source Software. 3, 26. doi: 10.21105/joss.00765. [DOI] [Google Scholar]
- Dunphy KA, Black AL, Roberts AL, Sharma A, Li Z, Suresh S, Browne EP, Arcaro KF, Ser-Dolansky J, Bigelow C, Troester MA, Schneider SS, Makari-Judson G, Crisi GM, Jerry DJ, 2020. Inter-Individual Variation in Response to Estrogen in Human Breast Explants. J Mammary Gland Biol Neoplasia 25, 51–68. doi: 10.1007/s10911-020-09446-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald AJ, 2008. Collective Epithelial Migration and Cell Rearrangements Drive Mammary Branching Morphogenesis. Developmental Cell 14, 570–581. doi: 10.1016/j.devcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson JE, Schor AM, Howell A, Ferguson MW, 1992. Changes in the extracellular matrix of the normal human breast during the menstrual cycle. Cell Tissue Res. 268, 167–177. doi: 10.1007/BF00338066 [DOI] [PubMed] [Google Scholar]
- Fornetti J, Flanders KC, Henson PM, Tan A-C, Borges VF, Schedin P, 2016. Mammary epithelial cell phagocytosis downstream of TGF-β3 is characterized by adherens junction reorganization. Cell Death Differ. 23, 185–196. doi: 10.1038/cdd.2015.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortner RT, Sisti J, Chai B, Collins LC, Rosner B, Hankinson SE, Tamimi RM, Eliassen AH, 2019. Parity, breastfeeding, and breast cancer risk by hormone receptor status and molecular phenotype: results from the Nurses’ Health Studies. Breast Cancer Res 21, 40–9. doi: 10.1186/s13058-019-1119-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Liu J, Kriebel AR, Preissl S, Luo C, Castanon R, Sandoval J, Rivkin A, Nery JR, Behrens MM, Ecker JR, Ren B, Welch JD, 2021. Iterative single-cell multi-omic integration using online learning. Nat. Biotechnol 39, 1000–1007. doi: 10.1038/s41587-021-00867-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay CM, Zygmunt T, Torres-Vázquez J, 2011. Diverse functions for the semaphorin receptor PlexinD1 in development and disease. Dev. Biol 349, 1–19. doi: 10.1016/j.ydbio.2010.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysi DM, Voigt A, Fragoso T. de M., Almaas E, Nowick K, 2018. wTO: an R package for computing weighted topological overlap and a consensus network with integrated visualization tool. BMC Bioinformatics 19, 392–16. doi: 10.1186/s12859-018-2351-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, Tirosh I, Beyaz S, Dionne D, Zhang M, Raychowdhury R, Garrett WS, Rozenblatt-Rosen O, Shi HN, Yilmaz O, Xavier RJ, Regev A, 2017. A single-cell survey of the small intestinal epithelium. Nature Publishing Group 551, 333–339. doi: 10.1038/nature24489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, Satija R, 2019. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296–15. doi: 10.1186/s13059-019-1874-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg G, Andersson E, Naessén T, Ordeberg GE, 2010. The expression of syndecan-1, syndecan-4 and decorin in healthy human breast tissue during the menstrual cycle. Reprod. Biol. Endocrinol 8, 35. doi: 10.1186/1477-7827-8-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson E, 2020. A brief guide to the menstrual cycle and oral contraceptive use for researchers in behavioral endocrinology. Horm Behav 119, 104655. doi: 10.1016/j.yhbeh.2019.104655 [DOI] [PubMed] [Google Scholar]
- Heaton H, Talman AM, Knights A, Imaz M, Gaffney DJ, Durbin R, Hemberg M, Lawniczak MKN, 2020. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat Meth 17, 615–620. doi: 10.1038/s41592-020-0820-1 [DOI] [PubMed] [Google Scholar]
- Hyder SM, Nawaz Z, Chiappetta C, Stancel GM, 2000. Identification of functional estrogen response elements in the gene coding for the potent angiogenic factor vascular endothelial growth factor. Cancer Research 60, 3183–3190. [PubMed] [Google Scholar]
- Jindal S, Gao D, Bell P, Albrektsen G, Edgerton SM, Ambrosone CB, Thor AD, Borges VF, Schedin P, 2014. Postpartum breast involution reveals regression of secretory lobules mediated by tissue-remodeling. Breast Cancer Res 16, 1–14. doi: 10.1186/bcr3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PA, Waterhouse PD, Kannan N, Narala S, Fang H, Di Grappa MA, Jackson HW, Penninger JM, Eaves C, Khokha R, 2015. RANK Signaling Amplifies WNT-Responsive Mammary Progenitors through R-SPONDIN1. STEMCR 5, 31–44. doi: 10.1016/j.stemcr.2015.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, Chambon P, 1990. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. The EMBO Journal 9, 1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, Smith AE, Prat A, Perou CM, Gilmore H, Schnitt S, Naber SP, Garlick JA, Kuperwasser C, 2012. Defining the cellular precursors to human breast cancer. Proc. Natl. Acad. Sci. U.S.A 109, 2772–2777. doi: 10.1073/pnas.1017626108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O, Cardiff RD, Bentires-Alj M, 2015. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature Publishing Group 525, 114–118. doi: 10.1038/nature14669 [DOI] [PubMed] [Google Scholar]
- Kotliar D, Veres A, Nagy MA, Tabrizi S, Hodis E, Melton DA, Sabeti PC, 2019. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife 8, 507. doi: 10.7554/eLife.43803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Sergushichev A, 2019. Fast gene set enrichment analysis. bioRxiv, October 22, 2019. doi: 10.1101/060012 [DOI] [Google Scholar]
- LaMarca HL, Rosen JM, 2007. Estrogen regulation of mammary gland development and breast cancer: amphiregulin takes center stage. Breast Cancer Res 9, 304–3. doi: 10.1186/bcr1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legland D, Arganda-Carreras I, Andrey P, 2016. MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32, 3532–3534. doi: 10.1093/bioinformatics/btw413 [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P, 2015. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. doi: 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Wu D, Pal B, Bouras T, Asselin-Labat M-L, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, Visvader JE, 2010. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res 12, R21. doi: 10.1186/bcr2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–21. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons TR, O’Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan A-C, Schedin P, 2011. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med 17, 1109–1115. doi: 10.1038/nm.2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS, Werb Z, Chow ED, Gartner ZJ, 2019. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat Meth 30, 1. doi: 10.1038/s41592-019-0433-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, Gartner ZJ, 2019a. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337.e4. doi: 10.1016/j.cels.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Abt F, Brinkhaus H, Bentires-Alj M, 2014. Early but not late pregnancy induces lifelong reductions in the proportion of mammary progesterone sensing cells and epithelial Wnt signaling. Breast Cancer Res 16, 209. doi: 10.1186/bcr3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor L, Molyneux G, Mackay A, Magnay F-A, Atienza M, Kendrick H, Nava-Rodrigues D, López-García MÁ, Milanezi F, Greenow K, Robertson D, Palacios J, Reis-Filho JS, Smalley MJ, 2014. Identification of cellular and genetic drivers of breast cancer heterogeneity in genetically engineered mouse tumour models. The Journal of Pathology 233, 124–137. doi: 10.1002/path.4345 [DOI] [PubMed] [Google Scholar]
- Métivier R, Penot G, Hübner MR, Reid G, Brand H, Kos M, Gannon F, 2003. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763. doi: 10.1016/s0092-8674(03)00934-6 [DOI] [PubMed] [Google Scholar]
- Molyneux G, Geyer FC, Magnay F-A, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, Reis-Filho JS, Smalley MJ, 2010. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7, 403–417. doi: 10.1016/j.stem.2010.07.010 [DOI] [PubMed] [Google Scholar]
- Monks J, Smith-Steinhart C, Kruk ER, Fadok VA, Henson PM, 2008. Epithelial cells remove apoptotic epithelial cells during post-lactation involution of the mouse mammary gland. Biol. Reprod 78, 586–594. doi: 10.1095/biolreprod.107.065045 [DOI] [PubMed] [Google Scholar]
- Mueller SO, Clark JA, Myers PH, Korach KS, 2002. Mammary gland development in adult mice requires epithelial and stromal estrogen receptor alpha. Endocrinology 143, 2357–2365. doi: 10.1210/endo.143.6.8836 [DOI] [PubMed] [Google Scholar]
- Muenst S, Mechera R, Däster S, Piscuoglio S, Ng CKY, Meier-Abt F, Weber WP, Soysal SD, 2017. Pregnancy at early age is associated with a reduction of progesterone-responsive cells and epithelial Wnt signaling in human breast tissue. Oncotarget 8, 22353–22360. doi: 10.18632/oncotarget.16023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munne PM, Martikainen L, Räty I, Bertula K, Nonappa, Ruuska J, Ala-Hongisto H, Peura A, Hollmann B, Euro L, Yavuz K, Patrikainen L, Salmela M, Pokki J, Kivento M, Väänänen J, Suomi T, Nevalaita L, Mutka M, Kovanen P, Leidenius M, Meretoja T, Hukkinen K, Monni O, Pouwels J, Sahu B, Mattson J, Joensuu H, Heikkilä P, Elo LL, Metcalfe C, Junttila MR, Ikkala O, Klefström J, 2021. Compressive stress-mediated p38 activation required for ERα + phenotype in breast cancer. Nature Communications 12, 6967–17. doi: 10.1038/s41467-021-27220-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakshatri H, Anjanappa M, Bhat-Nakshatri P, 2015. Ethnicity-Dependent and -Independent Heterogeneity in Healthy Normal Breast Hierarchy Impacts Tumor Characterization. Sci Rep 5, 13526–14. doi: 10.1038/srep13526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QH, Pervolarakis N, Blake K, Ma D, Davis RT, James N, Phung AT, Willey E, Kumar R, Jabart E, Driver I, Rock J, Goga A, Khan SA, Lawson DA, Werb Z, Kessenbrock K, 2018. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nature Communications 9, 2028. doi: 10.1038/s41467-018-04334-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flanagan CH, Campbell KR, Zhang AW, Kabeer F, Lim JLP, Biele J, Eirew P, Lai D, McPherson A, Kong E, Bates C, Borkowski K, Wiens M, Hewitson B, Hopkins J, Pham J, Ceglia N, Moore R, Mungall AJ, McAlpine JN, CRUK IMAXT Grand Challenge Team, Shah SP, Aparicio S, 2019. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 20, 210–13. doi: 10.1186/s13059-019-1830-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J, Lyons T, Monks J, Lucia MS, Wilson RS, Hines L, Man Y-G, Borges V, Schedin P, 2010. Alternatively activated macrophages and collagen remodeling characterize the postpartum involuting mammary gland across species. Am. J. Pathol 176, 1241–1255. doi: 10.2353/ajpath.2010.090735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri C, Saji S, Sakaguchi H, Cheng G, Sunters A, O’Hare MJ, Warner M, Gustafsson J-A, Coombes RC, Lam EW-F, 2004. The expression of oestrogen receptor (ER)-beta and its variants, but not ERalpha, in adult human mammary fibroblasts. J. Mol. Endocrinol 33, 35–50. doi: 10.1677/jme.0.0330035 [DOI] [PubMed] [Google Scholar]
- Pardo I, Lillemoe HA, Blosser RJ, Choi M, Sauder CAM, Doxey DK, Mathieson T, Hancock BA, Baptiste D, Atale R, Hickenbotham M, Zhu J, Glasscock J, Storniolo AMV, Zheng F, Doerge RW, Liu Y, Badve S, Radovich M, Clare SE, Susan G Komen for the Cure Tissue Bank at the IU Simon Cancer Center, 2014. Next-generation transcriptome sequencing of the premenopausal breast epithelium using specimens from a normal human breast tissue bank. Breast Cancer Res 16, R26. doi: 10.1186/bcr3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar H, Cunha GR, 2004. Epithelial-stromal interactions in the mouse and human mammary gland in vivo. Endocr Relat Cancer 11, 437–458. doi: 10.1677/erc.1.00659 [DOI] [PubMed] [Google Scholar]
- Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, Chao SX, Zollinger DR, Lieb DJ, Reeves JW, Fuhrman CA, Hoang ML, Delorey T, Nguyen LT, Waldman J, Klapholz M, Wakiro I, Cohen O, Albers J, Smillie CS, Cuoco MS, Wu J, Su M-J, Yeung J, Vijaykumar B, Magnuson AM, Asinovski N, Moll T, Goder-Reiser MN, Applebaum AS, Brais LK, DelloStritto LK, Denning SL, Phillips ST, Hill EK, Meehan JK, Frederick DT, Sharova T, Kanodia A, Todres EZ, Jané-Valbuena J, Biton M, Izar B, Lambden CD, Clancy TE, Bleday R, Melnitchouk N, Irani J, Kunitake H, Berger DL, Srivastava A, Hornick JL, Ogino S, Rotem A, Vigneau S, Johnson BE, Corcoran RB, Sharpe AH, Kuchroo VK, Ng K, Giannakis M, Nieman LT, Boland GM, Aguirre AJ, Anderson AC, Rozenblatt-Rosen O, Regev A, Hacohen N, 2021. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20. doi: 10.1016/j.cell.2021.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S, de Cicco RL, Santucci-Pereira J, Slifker M, Ross EA, Russo IH, Russo PA, Arslan AA, Belitskaya-Lévy I, Zeleniuch-Jacquotte A, Bordas P, Lenner P, Åhman J, Afanasyeva Y, Johansson R, Sheriff F, Hallmans G, Toniolo P, Russo J, 2012. Defining the genomic signature of the parous breast. BMC Med Genomics 5, 46. doi: 10.1186/1755-8794-5-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petz LN, Nardulli AM, 2000. Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol. Endocrinol 14, 972–985. doi: 10.1210/mend.14.7.0493 [DOI] [PubMed] [Google Scholar]
- Premenopausal Breast Cancer Collaborative Group, Schoemaker MJ, Nichols HB, Wright LB, Brook MN, Jones ME, O’Brien KM, Adami H-O, Baglietto L, Bernstein L, Bertrand KA, Boutron-Ruault M-C, Braaten T, Chen Y, Connor AE, Dorronsoro M, Dossus L, Eliassen AH, Giles GG, Hankinson SE, Kaaks R, Key TJ, Kirsh VA, Kitahara CM, Koh W-P, Larsson SC, Linet MS, Ma H, Masala G, Merritt MA, Milne RL, Overvad K, Ozasa K, Palmer JR, Peeters PH, Riboli E, Rohan TE, Sadakane A, Sund M, Tamimi RM, Trichopoulou A, Ursin G, Vatten L, Visvanathan K, Weiderpass E, Willett WC, Wolk A, Yuan J-M, Zeleniuch-Jacquotte A, Sandler DP, Swerdlow AJ, 2018. Association of Body Mass Index and Age With Subsequent Breast Cancer Risk in Premenopausal Women. JAMA Oncol 4, e181771. doi: 10.1001/jamaoncol.2018.1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, Gilmore H, Tung N, Naber SP, Schnitt S, Lander ES, Kuperwasser C, 2011. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 8, 149–163. doi: 10.1016/j.stem.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosperi MCF, Ciccozzi M, Fanti I, Saladini F, Pecorari M, Borghi V, Di Giambenedetto S, Bruzzone B, Capetti A, Vivarelli A, Rusconi S, Re MC, Gismondo MR, Sighinolfi L, Gray RR, Salemi M, Zazzi M, De Luca A, ARCA collaborative group, 2011. A novel methodology for large-scale phylogeny partition. Nature Communications 2, 321–10. doi: 10.1038/ncomms1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram RD, Buric D, Caikovski M, Ayyanan A, Rougemont J, Shan J, Vainio SJ, Yalcin-Ozuysal O, Brisken C, 2015. Progesterone and Wnt4 control mammary stem cells via myoepithelial crosstalk. The EMBO Journal 34, 641–652. doi: 10.15252/embj.201490434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R, Khan SA, Badve S, 2002. Morphological changes in breast tissue with menstrual cycle. Mod. Pathol 15, 1348–1356. doi: 10.1097/01.MP.0000039566.20817.46 [DOI] [PubMed] [Google Scholar]
- Ribieras S, Tomasetto C, Rio MC, 1998. The pS2/TFF1 trefoil factor, from basic research to clinical applications. Biochim. Biophys. Acta 1378, F61–77. doi: 10.1016/s0304-419x(98)00016-x [DOI] [PubMed] [Google Scholar]
- Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, McCaffrey EF, Greenwald NF, Rivero-Gutiérrez B, Strand SH, Varma S, Kong A, Keren L, Srivastava S, Zhu C, Khair Z, Veis DJ, Deschryver K, Vennam S, Maley C, Hwang ES, Marks JR, Bendall SC, Colditz GA, West RB, Angelo M, 2022. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 185, 299–310.e18. doi: 10.1016/j.cell.2021.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth JM, Schackmann RCJ, Gray GK, Selfors LM, Li CM-C, Boedicker M, Kuiken HJ, Richardson A, Brock J, Garber J, Dillon D, Sachs N, Clevers H, Brugge JS, 2020. Organoid cultures from normal and cancer-prone human breast tissues preserve complex epithelial lineages. Nature Communications 11, 1711–14. doi: 10.1038/s41467-020-15548-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J, Rivera R, Russo IH, 1992. Influence of age and parity on the development of the human breast. Breast Cancer Res. Treat 23, 211–218. doi: 10.1007/BF01833517 [DOI] [PubMed] [Google Scholar]
- Santucci-Pereira J, Zeleniuch-Jacquotte A, Afanasyeva Y, Zhong H, Slifker M, Peri S, Ross EA, de Cicco RL, Zhai Y, Nguyen T, Sheriff F, Russo IH, Su Y, Arslan AA, Bordas P, Lenner P, Åhman J, Eriksson ASL, Johansson R, Hallmans G, Toniolo P, Russo J, 2019. Genomic signature of parity in the breast of premenopausal women. Breast Cancer Res 21, 1–19. doi: 10.1186/s13058-019-1128-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedin P, O’Brien J, Rudolph M, Stein T, Borges V, 2007. Microenvironment of the Involuting Mammary Gland Mediates Mammary Cancer Progression. J Mammary Gland Biol Neoplasia 12, 71–82. doi: 10.1007/s10911-007-9039-3 [DOI] [PubMed] [Google Scholar]
- Sirka OK, Shamir ER, Ewald AJ, 2018. Myoepithelial cells are a dynamic barrier to epithelial dissemination. J. Cell Biol 217, 3368–3381. doi: 10.1083/jcb.201802144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderqvist G, Isaksson E, Schoultz, von B, Carlström K, Tani E, Skoog L, 1997. Proliferation of breast epithelial cells in healthy women during the menstrual cycle. Am. J. Obstet. Gynecol 176, 123–128. doi: 10.1016/s0002-9378(97)80024-5 [DOI] [PubMed] [Google Scholar]
- Stein T, Morris JS, Davies CR, Weber-Hall SJ, Duffy M-A, Heath VJ, Bell AK, Ferrier RK, Sandilands GP, Gusterson BA, 2004. Involution of the mouse mammary gland is associated with an immune cascade and an acute-phase response, involving LBP, CD14 and STAT3. Breast Cancer Res 6, R75–91. doi: 10.1186/bcr753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T, Salomonis N, Nuyten DSA, van de Vijver MJ, Gusterson BA, 2009. A mouse mammary gland involution mRNA signature identifies biological pathways potentially associated with breast cancer metastasis. J Mammary Gland Biol Neoplasia 14, 99–116. doi: 10.1007/s10911-009-9120-1 [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Kedeshian P, Shao ZM, Safarians S, Barsky SH, 1997. The human myoepithelial cell is a natural tumor suppressor. Clin. Cancer Res. 3, 1949–1958. [PubMed] [Google Scholar]
- Stewart TA, Hughes K, Stevenson AJ, Marino N, Ju AL, Morehead M, Davis FM, 2021. Mammary mechanobiology - investigating roles for mechanically activated ion channels in lactation and involution. J. Cell. Sci 134. doi: 10.1242/jcs.248849 [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, 2019. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye J-F, Raffoul W, Fiche M, Dougall W, Schneider P, Yalcin-Ozuysal O, Brisken C, 2013. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med 5, 182ra55–182ra55. doi: 10.1126/scitranslmed.3005654 [DOI] [PubMed] [Google Scholar]
- Traag VA, Van Dooren P, Nesterov Y, 2011. Narrow scope for resolution-limit-free community detection. Phys Rev E Stat Nonlin Soft Matter Phys 84, 016114. doi: 10.1103/PhysRevE.84.016114 [DOI] [PubMed] [Google Scholar]
- Vachon CM, Kuni CC, Anderson K, Anderson VE, Sellers TA, 2000. Association of mammographically defined percent breast density with epidemiologic risk factors for breast cancer (United States). Cancer Causes Control 11,653–662. doi: 10.1023/a:1008926607428 [DOI] [PubMed] [Google Scholar]
- Vogel PM, Georgiade NG, Fetter BF, Vogel FS, McCarty KS, 1981. The correlation of histologic changes in the human breast with the menstrual cycle. Am. J. Pathol 104, 23–34. [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ, 2019. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 177, 1873–1887.e17. doi: 10.1016/j.cell.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worzfeld T, Swiercz JM, Looso M, Straub BK, Sivaraj KK, Offermanns S, 2012. ErbB-2 signals through Plexin-B1 to promote breast cancer metastasis. J Clin Invest 122, 1296–1305. doi: 10.1172/JCI60568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Donor information for reduction mammoplasty (RM) and Komen Tissue Bank (KTB) samples and their use in scRNA-seq, flow cytometry, and immunostaining experiments. Related to Figure 1.
Table S2. Summary statistics for sequencing of twenty-eight reduction mammoplasty samples (RM) and seven Komen Tissue Bank core biopsy samples (KTB). Related to Figure 1.
Table S3. Association between gene expression and activity program expression score across cells. Related to Figures 3–4.
Table S4. List of the top 20 gene sets enriched in each cell-cell interaction module. Related to Figures 3–4.
Table S5. Genes found to be upregulated in the human breast during the luteal phase of the menstrual cycle by Pardo et al. Related to Figure 3.
Table S6. List of genes found to be upregulated during post-lactational involution in the mouse by Stein et al. 2004, with names of human orthologs used for gene set enrichment analysis. Related to Figure 4.
Table S7. Canonical hormone-responsive genes differentially expressed in “pseudo-bulk” HR+ luminal cells from nulliparous versus parous samples. Related to Figure 5.
Table S8. Multiple linear regression analysis of the percent basal/myoepithelial cells in the epithelium (quantified by scRNA-seq clustering) in response to parity and mean ER/PR signaling score. Related to Figure 6.
Table S9. Multiple linear regression analysis of the percent EpCAM−/CD49+ basal/myoepithelial cells in the epithelium (quantified by flow cytometry) in response to age and parity. Related to Figure 6.
Table S10. Multiple linear regression analysis of the indicated morphometric measurements in response to parity and mean ER/PR signaling score. Related to Figure 6.
Table S11. Multiple linear regression analysis of the percent hormone-responsive (HR+) cells in the luminal compartment (quantified by scRNA-seq clustering) in response to BMI and mean ER/PR signaling score. Related to Figure 6.
Data Availability Statement
Single-cell RNA-seq data (raw FASTQ files, processed gene expression and barcode count matrices, and de-identified patient metadata) have been deposited at the Gene Expression Omnibus (GSE198732) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All original code has been deposited at Zenodo and Github and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC-EpCAM | Stem Cell Technologies | 60136FI |
| APC-CD49f | BioLegend | 313616 |
| Biotin-CD2 | BD Biosciences | 555325 |
| Biotin-CD3 | BD Biosciences | 55338 |
| Biotin-CD16 | BD Biosciences | 555405 |
| Biotin-CD64 | BD Biosciences | 555526 |
| Biotin-CD31 | Invitrogen | MHCD31154 |
| Biotin-CD45 | BioLegend | 304004 |
| p63 | Cell Signaling Technology | 13109 |
| KRT7 | Abcam | AB68459 |
| KRT23 | Abcam | AB156569 |
| ER | Fisher Scientific | RM9101S |
| PR | Cell Signaling Technology | 8757 |
| TCF7 | Cell Signaling Technology | 2203 |
| P4HA1 | Thermo Fisher | PA5-55353 |
| LRRC26 | Thermo Fisher | PA5-63285 |
| Biological samples | ||
| Human breast specimens from reduction mammoplasty surgeries | CHTN and Kaiser Permanente Northern California | Table S1 |
| Human breast specimens from core biopsies | Komen Tissue Bank | Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| BV785-Streptavidin | BioLegend | 405249 |
| Collagenase Type 3 | Worthington | CLS-3 |
| Collagenase Type 2 | Worthington | CLS-2 |
| Hyaluronidase | Sigma Aldrich | H3506 |
| RPMI with HEPES | Corning | 10-041-CV |
| Amphotericin B | Lonza | 17-836E |
| Gentamicin | Lonza | 17-518 |
| Dispase | Stem Cell Technologies | 07913 |
| DNase I | Stem Cell Technologies | 07900 |
| MEGM | Lonza | CC-3150 |
| MEBM | Lonza | CC-3151 |
| Lab Vision Ultra-V Block | Thermo Fisher | TA-125-UB |
| UltraVision LP Detection System | Thermo Fisher | TL-060-HL |
| Vectashield HardSet Mounting Media with DAPI | Vector Labs | H-1400 |
| FITC-TSA | Akoya Biosciences | NEL701A001KT |
| Cy3-TSA | Akoya Biosciences | NEL744001KT |
| Cy5-TSA | Akoya Biosciences | NEL745E001KT |
| Probe Hs-ESR1 | ACD Bio | 310301 |
| Critical commercial assays | ||
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10X Genomics | PN-120237 |
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| Chromium Single Cell A Chip Kit | 10X Genomics | PN-120236 |
| Chromium Single Cell B Chip Kit | 10X Genomics | PN-1000153 |
| Chromium i7 Multiplex Kit | 10X Genomics | PN-120262 |
| MULTI-seq Lipid-Modified Oligos | Millipore Sigma | LMO001-100RXN |
| Bioanalyzer High Sensitivity DNA Kit | Agilent | 5067-4626 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Q32851 |
| RNAscope Multiplex Fluorescent Reagent Kit V2 | ACD Bio | 323100 |
| Deposited data | ||
| Raw data and processed scRNA-seq UMI counts and barcode matrices of reduction mammoplasty breast specimens | This study | GEO: GSE198732 |
| Raw data and processed scRNA-seq UMI counts and barcode matrices of Komen Tissue Bank breast specimens | This study | GEO: GSE198732 |
| Software and algorithms | ||
| CellRanger v3.0.2 | 10x Genomics | Github: https://github.com/10XGenomics/cellranger |
| MULTI-seq | McGinnis et al., 2019b | Github: https://github.com/chris-mcginnis-ucsf/MULTI-seq |
| SoupOrCell | Heaton et al., 2020 | Github: https://github.com/wheaton5/souporcell |
| Seurat v3.1.5 | Stuart et al., 2019; Hafemeister and Satija, 2019 | Github: https://github.com/satijalab/seurat |
| DoubletFinder | McGinnis et al., 2019a | Github: https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| LIGER | Gao et al., 2021; Welch et al., 2019 | Github: https://github.com/welch-lab/liger |
| DECIPHER-seq computational workflow | This study | Github: https://github.com/lmurrow/DECIPHER-seq and https://doi.org/10.5281/zenodo.6596414 |
| ape | Desper and Gascuel, 2002 | Github: https://github.com/emmanuelparadis/ape |
| Leidenalg | Traag et al., 2011 | Github: https://github.com/vtraag/leidenalg |
| wTO | Gysi et al., 2018 | Github: https://github.com/cran/wTO |
| fgsea | Korotkevich et al., 2019 | Github: https://github.com/ctlab/fgsea |
| DESeq2 | Love et al., 2014 | Github: https://github.com/mikelove/DESeq2 |